
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual-responsive ratiometric indicator designed for in vivo monitoring of oxidative stress and antioxidant capacity†
Majun
Yang
a,
Weida
Zhu
b,
Yilin
Lv
a,
Bin
Jiang
a,
Chenxia
Jiang
c,
Xiaobo
Zhou
a,
Guo
Li
 a,
Yuling
Qin
a,
Qi
Wang
*a,
Ziwei
Chen
*b and
Li
Wu
*a
a,
Yuling
Qin
a,
Qi
Wang
*a,
Ziwei
Chen
*b and
Li
Wu
*a
aSchool of Public Health, Nantong Key Laboratory of Public Health and Medical Analysis, Nantong University, 9 Seyuan Road, Nantong, 226019, P. R. China. E-mail: wangqi@ntu.edu.cn; drchenzv@163.com; wuli8686@ntu.edu.cn
bDepartment of Cardiovascular Medicine, The Affiliated Hospital of Nantong University, 20 Xisi Road, 226001 Nantong, China
cDepartment of Pathology, The Affiliated Hospital of Nantong University, 20 Xisi Road, 226001 Nantong, P. R. China
First published on 17th October 2023
Abstract
The imbalance between oxidative stress and antioxidant capacity is strongly associated with the development of numerous degenerative diseases, including cardiovascular diseases, diabetes, neurodegenerative diseases, and cancer. Therefore, monitoring oxidative stress and antioxidant capacity in vivo is crucial for maintaining cellular homeostasis and the stability of the organism's internal environment. Here, we present the findings of our study on DQ1, a dual-responsive indicator designed specifically for imaging H2O2 and NAD(P)H, which are critical indicators of oxidative stress and antioxidant capacity. DQ1 facilitated the colorimetric and fluorescence detection of H2O2 and NAD(P)H in two well-separated channels, exhibiting a detection limit of 1.0 μM for H2O2 and 0.21 nM for NAD(P)H, respectively. Experiments conducted on living cells and zebrafish demonstrated that DQ1 could effectively detect changes in H2O2 and NAD(P)H levels when exposed to exogenous hypoxic conditions and chemical stimuli. Furthermore, the effectiveness of the as-fabricated indicator was investigated in two distinct mouse models: evaluating H2O2 and NAD(P)H levels in myocardial cell dysfunction during acute myocardial infarction and liver tissue damage under trichloroethylene stress conditions. In vivo experiments demonstrated that the levels of the two cardiac biomarkers increase progressively with the development of myocardial infarction, eventually reaching a steady state after 7 days when the damaged cells in the infarcted region become depleted. Moreover, during 14 continuous days of exposure to trichloroethylene, the two biomarkers in liver tissue exhibited a sustained increase, indicating a significant enhancement in intracellular oxidative stress and antioxidant capacity attributed to the mouse liver's robust metabolic capacity. The aforementioned studies underscore the efficacy of DQ1 as a valuable tool for scrutinizing redox states at both the single-cell and biological tissue levels. It presents significant potential for investigating the dynamic alternations in oxidative stress and antioxidant capacity within disease models as the disease progresses, thereby facilitating a more profound comprehension of these processes across various disease models.
Introduction
Reactive oxygen species (ROS) are chemical molecules that are endogenously generated by living organisms during physiological cellular metabolism. They may also arise as a result of environmental factors, such as exposure to air pollutants or ozone.1 The presence of ROS in low to moderate concentrations plays crucial roles in various physiological processes within cells. However, excessive ROS levels can cause cellular damage to lipids, proteins, and DNA as well as other cellular components.2,3 To mitigate the deleterious effects of ROS, aerobic organisms have developed intricate antioxidant defense systems. These systems comprise a combination of enzymatic and nonenzymatic antioxidants that collaborate synergistically to counteract excessive ROS and maintain cellular homeostasis.4 However, under certain pathological conditions or exposure to high levels of environmental pollutants, the antioxidant systems can become overwhelmed, resulting in the accumulation of ROS and an increase in oxidative stress.5 The disparity between the generation of ROS and the ability of antioxidants to counteract them is referred to as “redox imbalance”.6 Redox imbalance is implicated in the pathogenesis and progression of a wide range of pathological conditions, including but not limited to cancer,7 neurological disorders (such as Alzheimer's and Parkinson's disease),8 atherosclerosis (the buildup of plaque in arteries),9 hypertension,10 ischemia/reperfusion injury (damage caused by the interruption and restoration of blood flow),11 diabetes,12 and idiopathic pulmonary fibrosis,13 among others. Therefore, monitoring in vivo oxidative stress and antioxidant capacity shows potential for developing therapeutic interventions for these conditions.ROS, including hydrogen peroxide (H2O2), superoxide anion (O2˙−), hydroxyl radical (˙OH), singlet oxygen (1O2), hypochlorous acid (HClO), and peroxynitrite anion (ONOO−), are generated through normal cellular metabolism from molecular oxygen.14 H2O2, a prominent ROS, exhibits relatively stable properties and fulfills diverse crucial functions in cellular signaling, differentiation, migration, immune response, and pathogen defense, as well as the maintenance of redox homeostasis within the body.15 When the intracellular production of H2O2 exceeds its clearance capacity, it can result in an elevation of cellular oxidative stress, leading to cellular damage. The 1,4-dihydro-nicotinamide adenine dinucleotide (NADH) and its phosphate ester form (NADPH) are indispensable antioxidant molecules within cells, serving as a crucial electron donor in a variety of biochemical reactions. These reactions encompass the metabolic pathways of glycolysis, the tricarboxylic acid cycle, oxidative phosphorylation in mitochondria, and the electron transport chain.16 Moreover, NAD(P)H is involved in various intracellular antioxidant reactions. For example, it participates in the reaction catalyzed by glutathione reductase, serving as a reducing agent to convert oxidized glutathione (GSSG) to its reduced form (GSH).17 Additionally, NAD(P)H is involved in the thioredoxin reductase reaction by reducing oxidized thioredoxin (Trx-S2) to its reduced form (Trx-SH).18 These antioxidants collaborate within the intricate “antioxidant network” to uphold the metabolic equilibrium of the endogenous body. It is noteworthy that H2O2 and NAD(P)H coexist in various physiological processes, thereby engaging in intricate signaling and oxidative pathways. The elevation of intracellular NAD(P)H levels may trigger a state of reducing stress, leading to the generation of a significant amount of H2O2 through cascade biological reactions. Excessive intracellular NAD(P)H can lead to an increase in electron transfer to oxygen, resulting in elevated ROS production under the catalysis of H2O2.19 Additionally, excessive H2O2 production occurs during oxidative phosphorylation due to the accumulation of NAD(P)H, which serves as the electron donor in mitochondrial respiration. All of this emphasizes the significance of H2O2 and NAD(P)H as compelling biomarkers for assessing cytopathological alterations in cells associated with both exogenous and endogenous stimuli.
As two crucial biomarkers in cell metabolism, fluorescent probes designed for H2O2 and NAD(P)H detection in single-cell assays should possess high sensitivity, non-toxicity to living cells, stability, rapid response time, and ease of use. Furthermore, the opposing properties of H2O2's oxidizing nature and NAD(P)H's reducing nature present a significant challenge for scientists attempting to concurrently detect both biomarkers using a single probe due to their conflicting combination. Fluorescent probes have been previously reported for the separate detection of H2O2 and NAD(P)H. However, none have achieved simultaneous real-time visualization of both bioactive substances.20–30 Other fluorescent probes possessing dual recognition capabilities for the simultaneous detection of oxidizing and reducing substances have been reported in the current literature. For instance, Kang et al. have developed a fluorescent probe that facilitates the investigation of regulatory and control mechanisms involved in oxidative and reductive reactions within cells. This innovative tool has enabled real-time imaging of changes in concentration of both exogenous and endogenous ˙OH and Cys in cells.31 In 2017, Yue et al. designed a probe that employs a reversible fluorescence reaction with Cys, enabling the visualization of the conversion process from Cys to SO2 in living cells.32 In 2022, Jing et al. developed Mito-CM-CD, a mitochondria-targeted fluorescent probe that enables real-time dynamic monitoring of ONOO− and GSH.33 However, to date, no fluorescent probe capable of simultaneously detecting H2O2 and NAD(P)H has been reported. H2O2 and NAD(P)H are two essential components of the oxidative–reductive system, and their simultaneous monitoring is imperative for gaining a comprehensive understanding of the physiological imbalance between oxidation and reduction in cells and tissues under pathological conditions. It is also essential for comprehending the mechanisms by which imbalanced conditions impact the body.
Herein, we present the discovery of DQ1, the first di-quinolinium derivative that exhibits rapid reactivity towards H2O2 and NAD(P)H under physiological conditions. This indicator was effectively employed to differentiate various levels of H2O2 and NAD(P)H in PC12 cells under chemical stimuli and hypoxic conditions. We utilized indicator DQ1 to assess the fluctuations in H2O2 and NAD(P)H levels in zebrafish models under rotenone stimulation and hypoxic conditions. Next, the indicator DQ1 was employed to examine the alterations in H2O2 and NAD(P)H levels within cardiac cells in a mouse model of myocardial infarction at different stages of the disease. Moreover, DQ1 was further utilized to investigate the fluctuations of the two biomarkers within the hepatic cells of mice exposed to acute trichloroethylene toxicity, a prevalent chemical pollutant. Through the establishment of the aforementioned mouse models, the tracking and monitoring of oxidative stress and antioxidant capacity within the body of mice under endogenous diseases or exogenous substance stimulation was successively achieved. This offers a valuable research tool for the enhanced diagnosis and treatment of diseases under conditions of redox imbalance.
Results and discussion
Design and synthesis of the indicator
Due to their high stability, excellent photophysical properties, and versatile organelle-targeting capabilities, cyanine dyes have been widely employed in the development of fluorescent probes.34 However, it remains a challenge to design dual-responsive probes based on this cyanine framework that can simultaneously detect both oxidative and reductive species. In this study, by employing two specific reactive groups, a dual-responsive probe (DQ1) was developed. DQ1 allowed for the simultaneous detection of H2O2 and NAD(P)H levels under physiological conditions while exhibiting distinct fluorescence emission signals. The design of such a molecule involved the utilization of two acceptors (2A-design, as shown in Fig. 1a), with electron-deficient heterocycle-quinolinium being selected. DQ1 consisted of two quinoline salts connected by a polymethine chain. 3-position substituted quinolinium is well known for its propensity to undergo two-electron reduction reactions with NAD(P)H analogs. Upon examination of NAD(P)H, the transfer of a hydride from NAD(P)H to the acceptor moiety of quinolinium led to the formation of a novel acceptor–π–donor (A–π–D) system with cyanine characteristics (Fig. 1a). On the other hand, 4-methylquinoline reacted with (4-bromomethylphenyl)boronic acid, leading to the formation of a highly electron-withdrawing quaternary ammonium salt moiety. Upon encountering H2O2, the boronic ester underwent oxidation, facilitating an intramolecular 1,6-rearrangement elimination reaction of the compound. This resulted in the formation of a novel weak acceptor–π–acceptor (A′–π–A) system of the cyanine type, where the 4-position substituted quinolinium product acted as a weak electron acceptor (Fig. 1a). In the presence of both H2O2 and NAD(P)H, the two acceptors' (A–π–A) structure transformed into a novel weak acceptor–π–donor system (A′–π–D), representing a weaker acceptor–donor interaction. Moreover, the presence of quaternary nitrogen atoms with a positive charge ensured the water-solubility of the aromatic ring structure, making it more suitable for biological applications. | ||
| Fig. 1 (a) Chemical structure and reaction mechanism of DQ1 with H2O2 and NADH. Counter-anions are omitted for clarity. (b) Schematic illustration of dual-responsive indicator DQ1 for the selective detection of H2O2 and NAD(P)H in living cells. | ||
The synthesis of DQ1 was achieved through a three-step process illustrated in Fig. S1.† First, quaternization of 4-methylquinoline with 2-(4-(bromomethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane was carried out under reflux conditions in toluene. This was followed by a Knoevenagel condensation of the quaternized quinolinium with 3-quinolinecarboxaldehyde to obtain the conjugated di-quinolinium 2. Finally, the pure product was precipitated in trichloromethane through a reaction with methyl trifluoromethanesulphonate. Each step was facile, yielding pure products without the necessity of chromatography. The isolated compounds were characterized utilizing HPLC, NMR, HRMS, and UV-vis spectroscopic techniques (Fig. S2–S10†).
Sensing properties of the as-obtained indicator DQ1
The spectroscopic evaluation of indicator DQ1 and its response to H2O2 and NADH was conducted under physiological conditions in a piperazine-1,4-bisethanesulfonic acid (PIPES) buffer at a temperature of 25 °C. As depicted in Fig. 2a and d (inset photographs), the free DQ1 indicator (20 μM) presented a grey-green color accompanied by a faint absorption peak at 586 nm and a pronounced absorption peak at 340 nm. After the addition of excess H2O2 to the DQ1 solution, a faint yellow color was observed and a new absorbance peak rapidly emerged at 370 nm. Simultaneously, the previously observed weak absorption at 586 nm disappeared. Upon the addition of NAD(P)H, the solution underwent a color change to a deep blue hue and displayed a prominent absorption peak at 586 nm. Subsequently, the changes in fluorescence emission spectra of DQ1 were examined. Fluorescence emission spectra revealed that the addition of H2O2 (0–300 μM) resulted in a significant 3.72-fold enhancement of the fluorescence signal at 505 nm for the DQ1 probe (20 μM), as depicted in Fig. 2b. The primary cause of this phenomenon was attributed to the reaction between H2O2 and DQ1, which led to the oxidation of arylboronic ester and triggered a 1,6-rearrangement elimination reaction. This transformation converted the A–π–A structure of DQ1 into the A′–π–A structure, thereby altering the electron arrangement within the molecule and consequently leading to an enhancement in the fluorescence intensity of the probe. The fluorescent H2O2 titration experiment exhibited a robust linear response (R2 = 0.992) in the detection of H2O2 concentrations ranging from 25 to 300 μM in the PIPES buffer (Fig. 2b, inset). The determined detection limit of DQ1 for H2O2 was 1.0 μM. This limit was determined by measuring the emission intensity at 505 nm and calculating it using the regression equation (3σ/k). In terms of NADH detection, upon the addition of NADH (0–24 μM), DQ1 exhibited a significant enhancement in fluorescence signal at 648 nm, resulting in an approximately 941-fold increase in emission intensity at a concentration of 24 μM for NADH detection. The mechanism underlying this phenomenon was attributed to the two-electron reduction reactions occurring between quinolinium and NADH. The transfer of hydride from NADH to quinolinium led to the formation of a novel donor–π–acceptor system. The titration experiment demonstrated that DQ1 exhibited a highly linear response (R2 = 0.993) in detecting NADH within the concentration range of 2–24 μM. Moreover, DQ1 showed an exceptionally low NAD(P)H detection limit. Using the regression equation (3σ/k), DQ1 exhibited a calculated detection limit of 0.21 nM when measuring emission intensity at 648 nm towards NADH, enabling it to monitor intracellular changes in NADH levels sensitively (Fig. 2e, inset). | ||
| Fig. 2 (a and d) UV-vis and (b and e) fluorescence spectra of DQ1 (20 μM): before (blue) and after (red) reaction with H2O2 (0–300 μM, 2 h) and NADH (0–24 μM, 10 min). Photographs (inset, a and d) depict the color change of DQ1 (20 μM, left) in the presence of H2O2 and NADH (right). Diagrams (inset, b and e): linear plot of emission changes against H2O2 and NADH concentration. (c) DQ1 reports H2O2, NADH, and H2O2/NADH with three different sets of fluorescence signals: blue-black; light blue-red; and blue-red. (f) Fluorescence intensity changes at 505 nm (interfering substances for H2O2 detection) and 648 nm (interfering substances for NADH detection). (1) H2O2; (2) blank; (3) K+; (4) Ca2+; (5) Fe3+; (6) Mg2+; (7) ˙OH; (8) O2˙−; (9) 1O2; (10) ONOO−; (11) ClO−; (12) H2S; (13) blank; (14) Na+; (15) K+; (16) Ca2+; (17) Fe2+; (18) HSO3−; (19) pyruvate; (20) glucose; (21) tyrosine; (22) ascorbic acid (VC); (23) cysteine; (24) FADH2; (25) GSH; (26) NADPH; (27) NADH. λex: 370 nm for H2O2 and 590 nm for NADH, each datum was acquired after various analytes addition at 37 °C. | ||
The spectral variations in the presence of both analytes simultaneously were subsequently investigated. As universally acknowledged, the coexistence of H2O2 and NADH during cellular metabolism plays a pivotal role in maintaining intracellular redox homeostasis.35 NADH possesses potent reducing properties and is capable of transferring electrons to other molecules, such as H2O2, O2−, and OH˙.36 For instance, the reaction between NADH and H2O2 resulted in the production of NAD+ (oxidized form of NADH) and water. In contrast, H2O2 is a ROS that possesses strong oxidizing properties. To mitigate the interference of both substances on detection results, separate investigations to evaluate DQ1's detection capabilities for H2O2 or NADH in the presence of each other were conducted. Fig. S11a† demonstrates that the absorption spectrum of the DQ1 solution, following sequential reactions with H2O2 and NADH, only exhibited a discernible enhancement at 350 nm. This represents the absorption peak specific to NADH. As shown in Fig. S11a,† after the addition of H2O2, there was an increase in the absorption spectrum of DQ1 at 370 nm. Further addition of NADH did not cause any significant change in the fluorescence emission at 505 nm. Interestingly, in contrast to the previous results, when DQ1 reacted with NADH, the absorption spectrum exhibited a significant increase at 586 nm. Upon further titration with H2O2, there were no significant changes observed in both absorption and fluorescence spectra (Fig. S11b, d and e†). Finally, when we simultaneously added H2O2 and NADH to the solution of DQ1, the fluorescence spectrum of the DQ1 solution shows robust emission intensity at both 505 nm and 650 nm (Fig. S11c†). This suggested that the probe exhibited minimal interference from coexisting substances during the detection of H2O2 or NADH.
The detection efficacy of DQ1 under different pH conditions for H2O2 and NADH was further investigated. As illustrated in Fig. S12,† the fluorescence intensity of DQ1 exhibited consistent stability upon the introduction of H2O2 within the pH range of 4–6. However, within the pH range of 6–10, the fluorescence intensity demonstrated an increase upon the addition of H2O2. Remarkably, as the alkalinity increased, the fluorescence enhancement became more pronounced, indicating that DQ1 exhibited higher sensitivity towards H2O2 and had a greater propensity to react with it under alkaline conditions. In contrast to H2O2, variations in pH did not significantly affect the detection of NADH (Fig. S12†). Moreover, time-dependent fluorescence spectra revealed that the fluorescence emission peaks at 505 nm and 650 nm attained their maximum intensities after 2 hours and 10 minutes, respectively, upon introduction of H2O2 (300 μM) and NADH (30 μM). Photostability experiments indicated that DQ1 maintained stable fluorescence emission even after 3 hours of exposure to UV irradiation at both 370 nm and 590 nm (Fig. S13†).
Selectivity is a crucial attribute that must be considered when evaluating the stability of fluorescent probes for cellular detection. As depicted in Fig. 2f, the emission response of DQ1 remained unaltered by a variety of other biologically relevant agents, including K+, Ca2+, Fe3+, Mg2+, ˙OH, O2˙−, 1O2, ONOO−, CIO−, and H2S. This demonstrated the high chemoselectivity of DQ1 towards H2O2. Similarly, the detection of NAD(P)H by DQ1 remained unaffected by Na+, K+, Ca2+, Fe3+, Mg2+, Fe2+, pyruvate, glucose, fructose, tyrosine, leucine, alanine and other biologically relevant factors (Fig. S13c†). This finding emphasized the remarkable chemical selectivity of DQ1 in detecting H2O2 or NAD(P)H. In summary, the indicator DQ1 provided a rapid kinetic profile, an exceptionally low detection limit, good stability, and specificity. These features enable it to be used to exclusively quantify H2O2 and NAD(P)H in living cells.
Bioimaging of H2O2 and NADH in living cells
Prior to conducting live cell imaging of H2O2 and NAD(P)H, the cytotoxicity of DQ1 in PC12 cells using a conventional MTT assay was evaluated. As illustrated in Fig. S14,† PC12 cells were exposed to varying concentrations of DQ1 (ranging from 0 to 100 mM) for a duration of 24 hours, and the cell viability remained consistently above 70%. This high level of cell viability indicated that DQ1 demonstrated excellent biocompatibility. This guaranteed its suitability for biological imaging. Colocalization studies targeting specific organelles were performed in PC12 cells using Mito-Tracker Green, Lyso-Tracker Green, and DAPI (4′,6-diamidino-2-phenylindole, a fluorescent stain that binds to DNA) as co-staining probes. As depicted in Fig. S15,† PC12 cells stained with Mito-Tracker Green, Lyso-Tracker Green, and DQ1 exhibited robust emissions in the green and red channels, respectively. The high Pearson's colocalization coefficient (P = 0.89) between Mito-Tracker Green and DQ1 indicated a predominant localization of DQ1 in the mitochondria of PC12 cells, while the low colocalization with Lyso-Tracker (P = 0.65) and DAPI (P = 0.30) suggested minimal presence in lysosomes and nuclei, respectively.To explore the feasibility of utilizing DQ1 for real-time monitoring of NAD(P)H and H2O2 levels in vivo, experiments using various chemical compounds known to stimulate or inhibit the secretion of H2O2 and NAD(P)H were conducted. As DQ1 employed two distinct fluorescence channels for the detection of H2O2 and NAD(P)H, the intracellular levels of H2O2 and NADH were measured by quantifying fluorescence intensities in the blue channel (representing H2O2) and red channel (representing NAD(P)H), respectively, using fluorescence microscopy. In this study, glucose, lactate, pyruvate, rotenone, and H2O2 were used as stimuli on PC12 cells individually, with the aim of inducing changes in the levels of these two analytes. The effect of glucose on H2O2 and NADH expression levels involved the participation of glucose dehydrogenase (GDH). GDH catalyzed the oxidation of β-D-glucose to β-D-gluconic-1,5-lactone while concomitantly reducing NAD+ to NADH and generating H2O2.37 The resulting NADH and H2O2 subsequently underwent a reaction with DQ1, leading to the formation of DQ1H and an increase in fluorescent emissions. As illustrated in Fig. 3a, following pre-treatment with glucose (5 mM, 10 mM) for a duration of 30 minutes, PC12 cells exhibited significantly enhanced fluorescence signals in both the blue and red channels (Fig. S16a†). The two analytes exhibited a simultaneous twofold increase (Fig. 3b). The inhibition of electron transfer in the cell respiratory chain by rotenone was a well-known phenomenon, resulting in the accumulation of H2O2 and NAD(P)H within cells.38 Therefore, rotenone was employed to stimulate PC12 cells and assess the detection capability of DQ1 for the variations in the two analytes. As shown in Fig. 3a, PC12 cells stimulated with rotenone (5 μM) exhibited significantly amplified fluorescence intensities in both the blue and red channels when compared to untreated PC12 cells. With the increase in rotenone concentration (0–5 μM), there was a sustained elevation in fluorescence observed in both fluorescent channels (Fig. S16d†). Lactate and pyruvate functioned as a circulating redox buffer, maintaining the balance of the NAD+/NADH ratio in cells and tissues. The disparity between lactate and pyruvate concentrations can exert a substantial influence on the levels of H2O2 and NADH in viable cells.39 In cellular metabolism, excessive pyruvate generated from hyperactive glycolysis is frequently converted into lactic acid via fermentation. The process of anaerobic glycolysis involves the utilization of two NAD(P)H molecules by lactate dehydrogenase (LDH). Therefore, DQ1 was employed to examine the alternations in H2O2 and NAD(P)H levels influenced by excessive pyruvate and lactate. Upon incubation of PC12 cells with exogenous lactate (5 mM and 10 mM), intracellular H2O2 and NADH levels were observed to increase, as depicted in Fig. 3a and S16b.† This was attributed to the conversion of lactate to pyruvate, resulting in a steady increment in fluorescence intensity within the H2O2 and NAD(P)H channels. In contrast, the fluorescence signals of PC12 cells exhibited a gradual decrease when treated with increasing concentrations of pyruvate (ranging from 0 to 5 μM) (Fig. S16c†). The H2O2 stimulation experiment demonstrated a significant increase in fluorescence intensity in both the blue and red channels of PC12 cells following incubation with H2O2 (100 μM, Fig. 3a and S16e†). The redox state in PC12 cells subjected to incubation with varying concentrations of NADH was also investigated. As illustrated in Fig. S16f,† the fluorescence intensity in the red channel exhibited a substantial enhancement with rising NADPH concentrations (0, 5, 10 μM), signifying an ongoing elevation in intracellular reducing pressure. Conversely, the fluorescence in the blue channel remained constant, indicating that oxidative stress remained nearly unchanged. This experiment underscores the probe's capability to effectively differentiate between oxidative and reducing pressures within live cells through two distinct fluorescence channels.
 | ||
| Fig. 3 Fluorescence images of H2O2 and NAD(P)H in PC12 cells stained with DQ1 (20 μM) under different conditions. (a) Fluorescence images of PC12 cells with the addition of glucose (10 mM, 30 min), rotenone (5 μM, 30 min), H2O2 (100 μM, 30 min), lactate (10 mM, 30 min) and pyruvate (5 μM, 30 min) as stimuli and under hypoxia stimulation. Cell images in the blue channel were acquired with λex: 362–396 nm, λem: 432–480 nm and in the red channel with λex: 540–580 nm, λem: 600–660 nm. Scale bars: 30 μm for the whole image. (b) Summarized data of mitochondrial mean fluorescence intensities (MFI) for the H2O2 and NAD(P)H channels, along with the corresponding ratio of MFIH2O2/MFINAD(P)H, for different treatments. Error bars: standard deviation (SD). | ||
The aforementioned experiments demonstrated the high sensitivity of DQ1 in detecting variations of H2O2 and NAD(P)H induced by exogenous compounds. Furthermore, additional validation of DQ1's detection ability for changes in intracellular H2O2 and NAD(P)H induced by endogenous factors was also performed. Specifically, cellular hypoxia was utilized as an acute model of hypoxia-induced cellular stress. Acute hypoxia stimulation was employed to mimic an external stressor on PC12 cells, thereby establishing a biological model for further validating the application of DQ1 in assessing oxidative stress and antioxidant capacity induced by endogenous diseases. Initially, PC12 cells were cultured in a hypoxic chamber with an oxygen concentration of approximately 1%, and subsequently divided into four groups based on varying durations of hypoxia exposure (2, 4, 6, 8 hours). Subsequently, DQ1 (20 μM) was added to each group of cells and incubated for 30 minutes. The fluorescence intensities of the blue and red channels were measured to evaluate the expression levels of H2O2 and NAD(P)H in PC12 cells under various hypoxic conditions. Fig. 3a and b illustrate that the fluorescence intensities of both the H2O2 and NAD(P)H channels exhibit a gradual increase in response to prolonged cellular hypoxia. Specifically, the fluorescence intensity in the H2O2 channel gradually increased after 4 hours of hypoxia, while the fluorescence intensity in the NAD(P)H channel continued to escalate at a relatively rapid pace. This implied that following 4 hours of hypoxia, the cellular secretion of H2O2 reached a gradual equilibrium while the organism continued to generate elevated concentrations of NAD(P)H in order to counteract the excess H2O2.
To validate the efficacy of the cellular hypoxia model, the extent of cellular stress using MDA (malondialdehyde), SOD (superoxide dismutase), LDH (lactate dehydrogenase), and ATP assay kits was evaluated. The detection results indicated that following a 4 hour period of hypoxia, cellular levels of MDA, ATP, and LDH exhibited an upward trend, while SOD levels demonstrated a downward trend (Fig. S17†). The findings suggested the successful establishment of the hypoxia model.
Tracking the variation of H2O2 and NAD(P)H in zebrafish induced by stimuli of H2O2, rotenone, and hypoxia
To investigate the detection capabilities of the DQ1 towards the organism's stress response elicited by external stimuli, DQ1 was further used to observe the levels of H2O2 and NAD(P)H in larval zebrafish. Zebrafish skin exhibited remarkable permeability, particularly in larvae younger than 10 days old when the blood–brain barrier was not yet fully developed.40 This characteristic rendered it an excellent model for real-time monitoring of the organism's oxidative and reductive stress status using fluorescent probes. As previously mentioned, the inhibitory effect of rotenone on electron transport in the respiratory chain led to the accumulation of NAD(P)H. Therefore, H2O2 and rotenone were further employed as stimuli to respectively induce oxidative stress and elevate the antioxidant NAD(P)H levels. Three-day-old zebrafish were consecutively exposed to H2O2 (20, 50 μM) and rotenone (2, 5 μM), followed by the addition of the DQ1 probe (20 μM) and co-incubation for a duration of 30 minutes. Fluorescence changes in the blue and red channels were monitored using fluorescence microscopy. Fig. 4a demonstrates a gradual increase in the average fluorescence intensity in both H2O2 and NAD(P)H channels upon the addition of H2O2 and rotenone. This observation suggested that the utilization of DQ1 effectively eliminated interference from other biomaterials, enabling real-time monitoring of oxidative and antioxidative substances within zebrafish.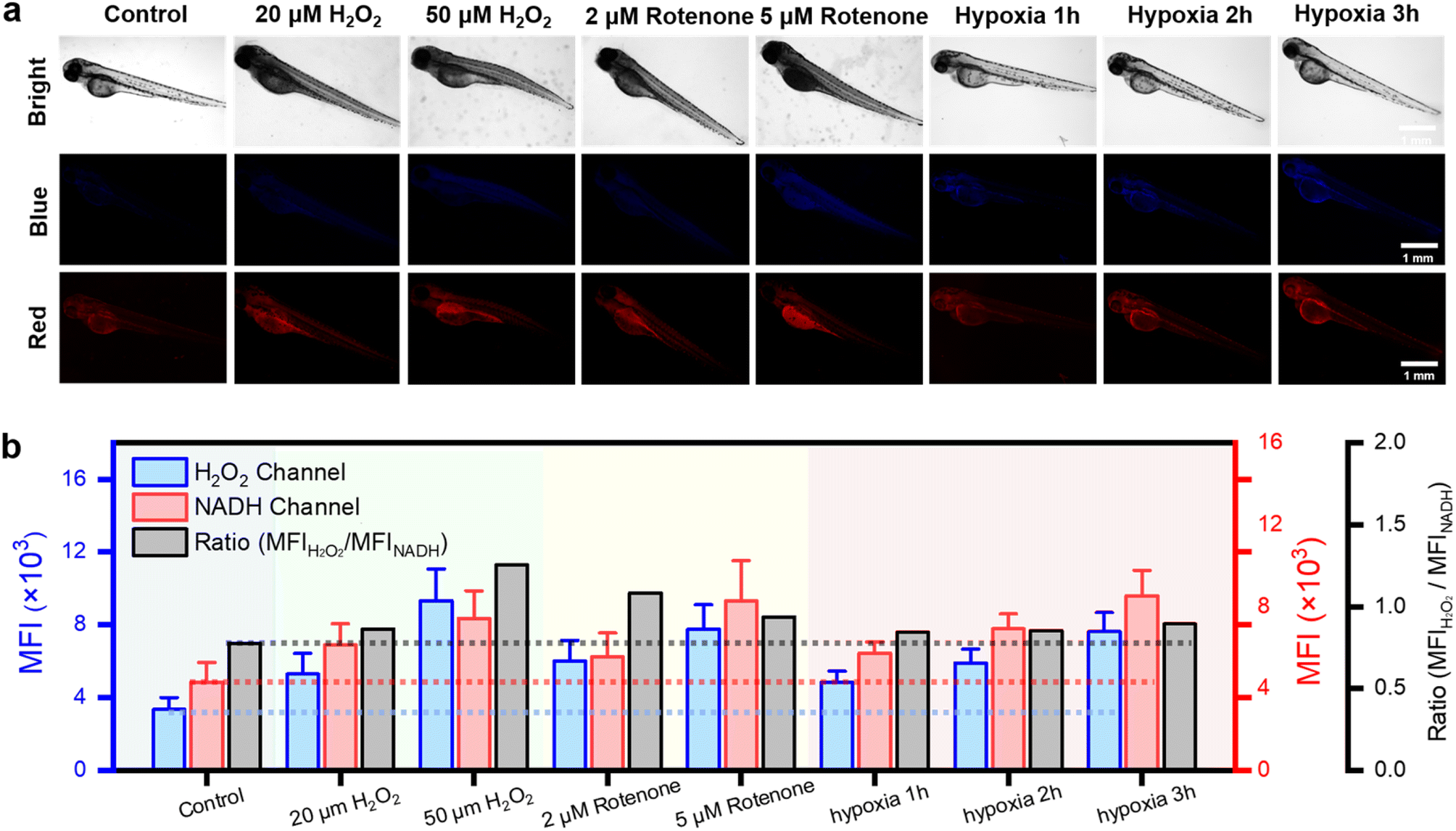 | ||
| Fig. 4 In vivo fluorescence imaging of 3 day-old zebrafish under H2O2, rotenone, normoxic and hypoxic conditions. (a) Fluorescence images of normoxic (21% O2), H2O2 (20, 50 μM), rotenone (2, 5 μM) and hypoxic (1.6% O2, 1, 2, 3 h) larval zebrafish stained with the DQ1 (20 μM) in the channels of H2O2 and NAD(P)H, respectively. Zebrafish images in the blue channel were acquired with λex: 362–396 nm, λem: 432–480 nm and in the red channel with λex: 540–580 nm, λem: 600–660 nm. Scar bar: 1 mm. (b) A histogram for H2O2 and NAD(P)H level changes of zebrafish under H2O2, rotenone, normoxic and hypoxic conditions. | ||
Next, the changes in H2O2 and NAD(P)H levels within zebrafish under different hypoxic conditions were investigated. The zebrafish were initially subjected to hypoxic conditions with an oxygen concentration of approximately 1% and then divided into three groups based on the duration of exposure to hypoxia (1–3 hours). Subsequently, after incubation with DQ1 (20 μM) for 30 minutes, the samples were imaged using a fluorescence microscope. As the duration of hypoxia increased, Fig. 4a demonstrated a continuous intensification in the blue channel fluorescence intensity of H2O2, reaching approximately 2.5 times the initial intensity after 3 hours. Similarly, the red channel fluorescence of NAD(P)H demonstrated a significant increase with prolonged exposure to hypoxia, reaching approximately twice the initial intensity after 3 hours (Fig. 4a and b). DQ1 exhibited a high sensitivity in detecting both H2O2 and NAD(P)H during zebrafish hypoxia monitoring. Moreover, the experiment revealed a significant elevation of oxidative stress and NAD(P)H content in zebrafish as the duration of hypoxia was prolonged.
Unveiling oxidative and reductive stress in mice models
Encouraged by the exceptional sensing performance of probe DQ1 for H2O2 and NAD(P)H in zebrafish, the application of DQ1 was further extended to more sophisticated mouse models. Within these murine models, the dynamic changes in H2O2 and NAD(P)H levels within myocardial tissue at various time points following acute myocardial infarction (AMI) were investigated. Additionally, the alterations of these two substances throughout the process of hepatic injury induced by exposing the mice to exogenous trichloroethylene (TCE) stimuli were also examined.AMI is a grave cardiovascular ailment characterized by the partial or complete obstruction of coronary arteries, leading to impaired blood circulation and localized necrosis of the myocardium due to prolonged ischemia and hypoxia.41 Pathological investigations of acute myocardial infarction had revealed that ischemia in myocardial cells, which was triggered by the occurrence of acute myocardial infarction, led to mitochondrial dysfunction. This disruption hindered the normal functioning of the mitochondrial respiratory chain by interrupting electron transfer, leading to subsequent disturbances in redox reactions. As a result, a substantial quantity of reactive oxygen species, such as H2O2, was generated. Furthermore, during acute myocardial infarction, the process of oxidative phosphorylation in mitochondria was hindered, thereby impeding the normal oxidation of NAD(P)H into NAD(P)+ and resulting in its accumulation within the cells.42 As acute myocardial infarction progressed, the accumulation of NAD(P)H in myocardial cells gradually increased until necrosis of the myocardial cell occurred. Therefore, the accurate assessment of H2O2 and NAD(P)H played a crucial role in the management of acute myocardial infarction, as it allowed for precise evaluation of myocardial cell injury.
In the mouse model of AMI, left anterior descending coronary artery ligation was employed to construct the infarction model. The experimental mice were categorized into five groups based on the duration of the obstruction: normal, 1, 3, 5, and 7 days. On the first, third, fifth, and seventh days following left anterior descending coronary artery ligation, the probe DQ1 (1 mM, 200 μL) was administered via tail vein injection in each group of mice. After a 12 hour incubation period to allow sufficient reaction between the probe and H2O2 and NAD(P)H in myocardial cells, the mice were humanely euthanized and their hearts were excised (Fig. 5a). The mouse hearts were cryopreserved for 20 minutes and subsequently sectioned into frozen slices. To validate the successful establishment of the AMI mouse model, the gold standard of myocardial infarction detection, 2,3,5-triphenyltetrazolium chloride (TTC) staining at a concentration of 2% was initially employed to identify areas of tissue damage resulting from ischemia. The principle underlying the TTC staining method laid in the significantly reduced dehydrogenase content within the infarcted tissue compared to normal heart tissue, resulting in an inability to convert TTC into insoluble and stable red triphenylformazan (TTF). As a result, a lighter staining area was observed, confirming the successful establishment of myocardial infarction.43 As depicted in Fig. S18,† TTC-stained myocardial infarction slices collected at 1, 3, 5, and 7 days post left anterior descending coronary artery ligation exhibited distinct areas of lighter staining, thereby confirming the successful establishment of the myocardial infarction model. The potential in vivo toxicities of DQ1 were evaluated through intravenous administration at three time points: 1, 7, and 14 days post-administration (Fig. S19†). No significant morphological changes were observed in the heart, liver, spleen, lung and kidney tissues of the tested groups compared to the control group based on H&E staining images. This implied that the administration of DQ1 did not elicit any discernible toxic effects on the macroscopic appearance of the examined organs throughout the duration of the study.
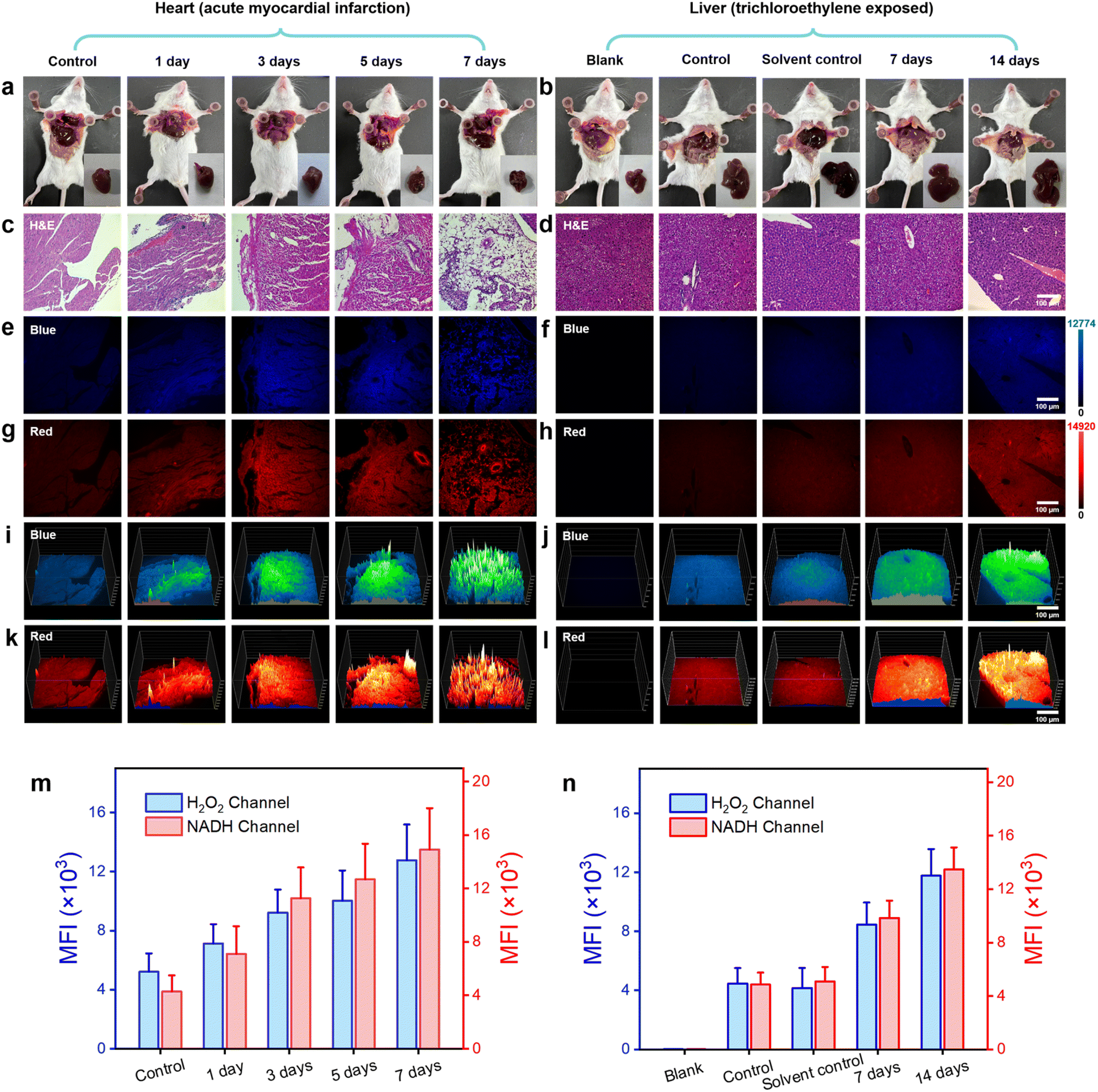 | ||
| Fig. 5 H&E and DQ1-staining histological examination of cardiac and hepatic tissues. (a and b) Photographs of anatomical mice. (c–h) Images of H&E and DQ1 (5 μM) stained histological sections of mouse heart tissues at different time stages following acute myocardial infarction (1, 3, 5, 7 days) and liver tissues exposed to trichloroethylene (0, 7, 14 days). (i–l) 3D strength surface for the above histological sections in three-dimensional space. Scale bar: 100 μm. (m and n) Histograms for H2O2 and NAD(P)H level changes of heart and liver tissues after acute myocardial infarction and trichloroethylene exposure, respectively. All fluorescent signals were selected from surviving cardiomyocytes and liver cells. Error bars: SD. Experimental times: n = 5. | ||
Furthermore, histological staining on mouse myocardial tissue was performed using hematoxylin and eosin (H&E) as well as the DQ1 probe. Subsequently, the stained samples were visualized under fluorescence microscopy. As the duration of myocardial infarction increased, H&E tissue staining revealed a progressive deepening in the degree of myocardial cell damage. By the 7th day, there were scarcely any structurally intact myocardial cells remaining (Fig. 5b and S20†). Fluorescence microscopy imaging revealed a consistent increase in the fluorescence intensity of the H2O2 channel and NAD(P)H channel in myocardial tissue following AMI in mice, with a slight decrease observed on the 7th day (Fig. 5c, d and S21†). The fluorescence imaging results revealed potential mechanisms of myocardial cell injury, including the following: during myocardial ischemia, a lack of oxygen and nutrients led to cellular metabolism and energy production suppression. This metabolic injury led to an upregulation of H2O2 production within the cell as a result of the accumulation of metabolic byproducts and mitochondria damage, which could subsequently result in ROS generation. Additionally, the cell might increase NAD(P)H production to supplement the ATP demand resulting from insufficient energy supply. This phenomenon could be confirmed by examining both the appearance and fluorescence images of heart sections in both obstructed and normal areas (Fig. S22†). The elevation of H2O2 and NAD(P)H levels exacerbated their interaction, thereby intensifying cellular oxidative stress and disrupting energy metabolism to a greater extent. This process persisted until the 7th day of AMI, by which time the myocardial cells were nearly entirely compromised. Fluorescence imaging of DQ1 on paraffin sections of AMI tissue revealed the changes in myocardial cell damage during the process of myocardial infarction, including structural destruction, oxidative stress, and abnormal energy metabolism (Fig. 5g). To achieve a more comprehensive observation of fluorescence changes, a 3D quantitative analysis was performed. The results demonstrated a gradual increase in fluorescence intensity in both the blue and red channels over time, which is consistent with the findings of qualitative experiments (Fig. 5e and f). Therefore, DQ1 was successfully utilized to continuously monitor the levels of reactive oxygen species and antioxidant substances in myocardial tissue following endogenous myocardial injury. These findings contributed to a more comprehensive understanding of the pathophysiological mechanisms underlying myocardial infarction.
In addition to monitoring endogenous diseases-induced redox imbalance, DQ1 can also serve as a tool for assessing organ damage caused by exogenous pollutants. TCE, also known as trichloroethene, is a widely used industrial organic solvent and environmental pollutant extensively employed in sectors such as electronics and hardware. TCE presents a significant threat to human health when associated with contaminated food, water sources, or PM 2.5 gas particles. Prolonged exposure to TCE can lead to elevated levels of oxidative stress in exposed individuals, resulting in various conditions such as TCE-induced drug rash-like dermatitis, hepatocellular and renal tumors, and cardiovascular and neurological impairments, among others.44 Investigations have revealed that populations exposed to TCE manifest significant accumulation of ROS and notable depletion of antioxidant enzymes within the liver, kidneys, heart, and other organs. This leads to lipid peroxidation, disruption of biomembrane structure and function, protein denaturation, DNA degradation, and related effects.45 Therefore, it is of significant importance to track and monitor the levels of ROS and NAD(P)H in the livers of TCE-exposed mice for elucidating the pathological processes underlying organ damage induced by TCE.
The TCE exposure model was established through subcutaneous injection of TCE into the dorsal region of mice, followed by observation of resultant liver damage. At the outset, the mice were categorized into five groups: blank group, control group, solvent control group, TCE-treated 7 day group, and TCE-treated 14 day group. Each of these groups consisted of five mice. The solvent control group was administered with a mixture of acetone and olive oil (100 μL) in a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, along with an equivalent amount of Freund's complete adjuvant (FCA). The TCE-treated groups were administered a subcutaneous injection of 100 μL of a mixture containing TCE, acetone, and olive oil in a volume ratio of 5:3:2 in the dorsal region. On the 7th and 14th days post-injection, the control group, solvent control group, TCE-treated 7 day group, and TCE-treated 14 day group of mice received intravenous administration of the DQ1 probe (1 mM, 200 μL) via the tail vein. After a 12 hour waiting period, the mice were humanely euthanized and their livers were harvested (Fig. 5a). Subsequently, paraffin embedding was performed on the liver samples to obtain sections for further analysis.
:2, along with an equivalent amount of Freund's complete adjuvant (FCA). The TCE-treated groups were administered a subcutaneous injection of 100 μL of a mixture containing TCE, acetone, and olive oil in a volume ratio of 5:3:2 in the dorsal region. On the 7th and 14th days post-injection, the control group, solvent control group, TCE-treated 7 day group, and TCE-treated 14 day group of mice received intravenous administration of the DQ1 probe (1 mM, 200 μL) via the tail vein. After a 12 hour waiting period, the mice were humanely euthanized and their livers were harvested (Fig. 5a). Subsequently, paraffin embedding was performed on the liver samples to obtain sections for further analysis.
Firstly, paraffin-embedded liver sections from each group of mice were subjected to H&E staining to evaluate the pathological damage induced by TCE stimulation. The H&E-stained sections of the control group mice demonstrated preserved and typical liver cell morphology, characterized by cytoplasm that appeared dense and homogeneous (Fig. 5b). In contrast, the TCE-stimulated group exhibited cytoplasmic loosening, sinusoidal compression, and cellular swelling, which were consistent with the pathological manifestations associated with TCE stimulation. Subsequently, malondialdehyde (MDA) and superoxide dismutase (SOD) levels were quantified in hepatic homogenates of mice using MDA and SOD assay kits (WST-8). The experimental results indicated that the MDA content in the liver increased significantly, reaching 12 nmol mg−1 after 7 days and 15 nmol mg−1 after 14 days, which represented a remarkable increase of approximately 160% and 226%, respectively, compared to the normal liver (Fig. S23a†). Meanwhile, the SOD level decreased by approximately 42% and 54%, respectively, to 0.75 U mg−1 (7 days) and 0.6 U mg−1 (14 days), from an initial value of 1.3 U mg−1 (Fig. S23b†). The successful establishment of the TCE sensitization model was confirmed by measuring MDA, an oxidative substance, and SOD, an antioxidant enzyme, in liver cells. Fluorescence imaging was subsequently conducted on the liver paraffin sections using a fluorescence microscope. The fluorescence imaging results revealed no significant difference in fluorescence intensity of H2O2 and NAD(P)H channels between the control group and the solvent control group, indicating that the solvent used for the TCE injection preparation did not interfere with the detection of H2O2 and NAD(P)H by DQ1. The results depicted in Fig. 5c, d and S24† indicated a significant increase in the fluorescence intensity of both blue and red channels within liver sections from TCE-exposed mice as exposure time was prolonged. The liver sections exposed for 7 days exhibited an average fluorescence intensity that was approximately twice as high as that of normal mice, whereas the liver sections exposed for 14 days showed an average fluorescence intensity that was approximately three times higher than that of normal mice (Fig. 5h and S24†). The progressively increasing mean fluorescence intensity indicated a significant rise in the levels of H2O2 and NAD(P)H in the hepatic tissue of mice exposed to TCE. Interestingly, the increase in both H2O2 and NAD(P)H exhibited a proportional relationship with the duration of exposure. The experiment of 3D quantitative analysis was conducted in a similar manner. The results were in accordance with the temporal variations observed in fluorescence imaging, as depicted in Fig. 5e and f. This study suggested that exposure to TCE resulted in oxidative stress in the hepatic cells of mice, leading to structural damage and a peroxidation effect. However, the increased levels of NAD(P)H did not demonstrate a significant antioxidant imbalance effect. This might be attributed to TCE's stimulating effect surpassing the body's regulatory capacity, leading to liver damage.
Live subject statement: All animal experiments were performed in compliance with the relevant laws and institutional guidelines for the Care and Use of Research Animals established by the Nantong University Animal Studies Committee, and the experiments have been approved by the committee (S20210228-010).
The results of these mouse experiments suggested that, in the presence of pathological or stress conditions, DQ1 demonstrated a heightened sensitivity towards detecting H2O2 and the reducing agent NAD(P)H in mice. This serves as a valuable tool for monitoring changes in oxidative stress and antioxidant capacity within the body under pathological conditions.
Conclusions
In summary, we have created a novel dual-responsive indicator named DQ1, composed of two adjustable quinolinium units. DQ1 has demonstrated exceptional selectivity and sensitivity in the measurement of H2O2 and NAD(P)H under physiological conditions, exhibiting a robust “turn-on” absorbance at wavelengths of 370 nm and 586 nm. The presence of the target species resulted in significant enhancements in fluorescence emission at 505 nm (approximately 3.4-fold) and 648 nm (approximately 940-fold). The fluorescence intensities of DQ1 exhibited a proportional relationship with increasing concentrations of H2O2 (25–300 μM) and NAD(P)H (2–24 μM). This linear correlation between the concentration of analytes and the response of indicators facilitates precise quantification of these target species. With its exceptional optical performance, the DQ1 indicator emerges as an ideal candidate for in situ bioimaging of H2O2 and NAD(P)H levels. The DQ1 assay, as expected, not only facilitated the detection of intracellular H2O2 and NAD(P)H expression levels at micro and nanomolar concentrations but also sensitively monitored alterations in intracellular H2O2 and NAD(P)H concentrations induced by external stimuli, such as glucose, lactate, rotenone, pyruvate, and hypoxia. Subsequently, DQ1 was employed to monitor the fluctuations of H2O2 and NAD(P)H in living zebrafish exposed to oxidative stress induced by H2O2, rotenone, and hypoxia stimulation. The DQ1 consistently exhibited a sensitive detection capability for both biomarkers under external stimuli. Finally, the DQ1 was employed to investigate the fluctuations of H2O2 and NAD(P)H levels in myocardial tissue following acute myocardial infarction, as well as to examine alterations in these species during exogenous TCE stimulation-induced liver injury in mice. Our findings have revealed that H2O2 induces discernible oxidative damage in tissues under both endogenous and exogenous stimuli. Although the levels of NAD(P)H increase simultaneously with H2O2 elevation, the extent of tissue or organ damage surpasses their self-repair capacity. Consequently, the body is incapable of mitigating oxidative stress in dysfunctional cells. All these findings have confirmed the successful development of a reliable indicator for precise and selective measurement of H2O2 and NAD(P)H levels, which are crucial indicators of oxidants and antioxidants involved in intracellular oxidative stress and antioxidant capacity. The DQ1 probe developed in this study has the potential to serve as a precise and noninvasive imaging research tool for the further establishment of comprehensive metabolomics models across various diseases.Data availability
Synthetic procedures, characterisation of optical properties, details of fluorescence cells imaging, NMR, HPLC and MALDI-TOF/MS spectra for the products can be found in the ESI.†Author contributions
Q. W., Y. L. Q. and L. W. conceived the project. M. J. Y. performed the experimental work. W. D. Z. established the myocardial infarction model of mice. L. Y. L. and B. J. carried out the computational work. C. X. J. provided tissue slices. X. B. Z. and G. L. contributed to the biological experiments. Z. W. C. analyzed the data. The manuscript was written by Q. W. with contributions from M. J. Y. and commented on by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (32171452, 62005132, 62105165), Excellent Youth Foundation of Jiangsu Scientific Committee (BK20220060), Jiangsu Specially-Appointed Professor (06200048, 06200053), “Postgraduate Research & Practice Innovation Program of Jiangsu Province” (KYCX22 3375), Sichuan Association of Thousand Talent Plan Experts, and the Large Instruments Open Foundation of Nantong University.Notes and references
- J. T. Hiltermann, T. S. Lapperre, L. van Bree, P. A. Steerenberg, J. J. Brahim, J. K. Sont, P. J. Sterk, P. S. Hiemstra and J. Stolk, Free Radical Biol. Med., 1999, 27, 1448–1454 CrossRef CAS PubMed.
- E. R. Stadtman, Curr. Med. Chem., 2004, 11, 1105–1112 CrossRef CAS PubMed.
- G. Bartosz, Cell Biol. Int., 2000, 24, 764 CrossRef.
- J. Styskal, H. Van Remmen, A. Richardson and A. B. Salmon, Free Radical Biol. Med., 2012, 52, 46–58 CrossRef CAS PubMed.
- A. Cieslar-Pobuda, J. Yue, H. C. Lee, M. Skonieczna and Y. H. Wei, Oxid. Med. Cell. Longevity, 2017, 2017, 5047168 Search PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- J. D. Hayes, A. T. Dinkova-Kostova and K. D. Tew, Cancer Cell, 2020, 38, 167–197 CrossRef CAS PubMed.
- O. Hwang, Exp. Neurobiol., 2013, 22, 11–17 CrossRef PubMed.
- A. J. Kattoor, N. V. K. Pothineni, D. Palagiri and J. L. Mehta, Curr. Atheroscler. Rep., 2017, 19, 42 CrossRef PubMed.
- K. K. Griendling, L. L. Camargo, F. J. Rios, R. Alves-Lopes, A. C. Montezano and R. M. Touyz, Circ. Res., 2021, 128, 993–1020 CrossRef CAS PubMed.
- N. S. Dhalla, A. K. Shah, A. Adameova and M. Bartekova, Biomedicines, 2022, 10, 1473 CrossRef CAS PubMed.
- P. Zhang, T. Li, X. Wu, E. C. Nice, C. Huang and Y. Zhang, Front. Med., 2020, 14, 583–600 CrossRef PubMed.
- C. Estornut, J. Milara, M. A. Bayarri, N. Belhadj and J. Cortijo, Front. Pharmacol, 2021, 12, 794997 CrossRef CAS PubMed.
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed.
- L. A. Sena and N. S. Chandel, Mol. Cell, 2012, 48, 158–167 CrossRef CAS PubMed.
- H. Zhang, D. Ryu, Y. Wu, K. Gariani, X. Wang, P. Luan, D. D'Amico, E. R. Ropelle, M. P. Lutolf, R. Aebersold, K. Schoonjans, K. J. Menzies and J. Auwerx, Science, 2016, 352, 1436–1443 CrossRef CAS PubMed.
- S. C. Lu, Biochim. Biophys. Acta, 2013, 1830, 3143–3153 CrossRef CAS PubMed.
- S. Gromer, S. Urig and K. Becker, Med. Res. Rev., 2004, 24, 40–89 CrossRef CAS PubMed.
- M. Velayutham, C. Hemann and J. L. Zweier, Free Radical Biol. Med., 2011, 51, 160–170 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- X. Jiao, Y. Li, J. Niu, X. Xie, X. Wang and B. Tang, Anal. Chem., 2018, 90, 533–555 CrossRef CAS PubMed.
- Y. Wen, F. Huo and C. Yin, Chin. Chem. Lett., 2019, 30, 1834–1842 CrossRef CAS.
- M. A. Fomin, R. I. Dmitriev, J. Jenkins, D. B. Papkovsky, D. Heindl and B. König, ACS Sens., 2016, 1, 702–709 CrossRef CAS.
- L. Guan, W. Hu, H. Zuo, H. Sun, Y. Ai, M. Q. He, C. Ma, M. Ding and Q. Liang, Chem. Commun., 2023, 59, 1617–1620 RSC.
- K. Ma, H. Yang, X. Wu, F. Huo, F. Cheng and C. Yin, Angew Chem. Int. Ed. Engl., 2023, 62, e202301518 CrossRef CAS PubMed.
- X. Pan, Y. Zhao, T. Cheng, A. Zheng, A. Ge, L. Zang, K. Xu and B. Tang, Chem. Sci., 2019, 10, 8179–8186 RSC.
- A. Podder, S. Koo, J. Lee, S. Mun, S. Khatun, H. G. Kang, S. Bhuniya and J. S. Kim, Chem. Commun., 2019, 55, 537–540 RSC.
- A. Podder, V. P. Murali, S. Deepika, A. Dhamija, S. Biswas, K. K. Maiti and S. Bhuniya, Anal. Chem., 2020, 92, 12356–12362 CrossRef CAS PubMed.
- Y. Tian, W. L. Jiang, W. X. Wang, G. J. Mao, Y. Li and C. Y. Li, Biomaterials, 2021, 271, 120736 CrossRef CAS PubMed.
- Y. Zhao, K. Wei, F. Kong, X. Gao, K. Xu and B. Tang, Anal. Chem., 2019, 91, 1368–1374 CrossRef CAS PubMed.
- Z. Kang, J. Jiang, Q. Tu, S. Liu, Y. Zhang, D. E. Wang, J. Wang and M. S. Yuan, J. Am. Chem. Soc., 2023, 145, 507–515 CrossRef CAS PubMed.
- Y. Yue, F. Huo, P. Ning, Y. Zhang, J. Chao, X. Meng and C. Yin, J. Am. Chem. Soc., 2017, 139, 3181–3185 CrossRef CAS PubMed.
- C. Jing, Y. Wang, X. Song, X. Li, Y. Feng, M. Kou, G. Zhang, W. Dou and W. Liu, Sens. Actuators, B, 2022, 365, 131847 CrossRef CAS.
- H. A. Shindy, Dyes Pigm., 2017, 145, 505–513 CrossRef CAS.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- J. A. Imlay, Nat. Rev. Microbiol., 2013, 11, 443–454 CrossRef CAS PubMed.
- S. Ferri, K. Kojima and K. Sode, J. Diabetes Sci. Technol., 2011, 5, 1068–1076 CrossRef PubMed.
- H. Pelicano, L. Feng, Y. Zhou, J. S. Carew, E. O. Hileman, W. Plunkett, M. J. Keating and P. Huang, J. Biol. Chem., 2003, 278, 37832–37839 CrossRef CAS PubMed.
- H. Wu, Y. Wang, M. Ying, C. Jin, J. Li and X. Hu, Signal Transduction Targeted Ther., 2021, 6, 242 CrossRef CAS PubMed.
- B. P. Eliceiri, A. M. Gonzalez and A. Baird, Methods Mol. Biol., 2011, 686, 371–378 CrossRef CAS PubMed.
- K. Thygesen, J. S. Alpert, A. S. Jaffe, B. R. Chaitman, J. J. Bax, D. A. Morrow and H. D. White, J. Am. Coll. Cardiol., 2018, 72, 2231–2264 CrossRef PubMed.
- D. B. Zorov, C. R. Filburn, L. O. Klotz, J. L. Zweier and S. J. Sollott, J. Exp. Med., 2000, 192, 1001–1014 CrossRef CAS PubMed.
- E. Gao and W. J. Koch, Methods Mol. Biol., 2013, 1037, 299–311 CrossRef PubMed.
- W. A. Chiu, J. Jinot, C. S. Scott, S. L. Makris, G. S. Cooper, R. C. Dzubow, A. S. Bale, M. V. Evans, K. Z. Guyton, N. Keshava, J. C. Lipscomb, S. Barone Jr, J. F. Fox, M. R. Gwinn, J. Schaum and J. C. Caldwell, Environ. Health Perspect., 2013, 121, 303–311 CrossRef PubMed.
- S. Khan, S. Priyamvada, S. A. Khan, W. Khan, N. Farooq, F. Khan and A. N. Yusufi, Food Chem. Toxicol., 2009, 47, 1562–1568 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04081j |
| This journal is © The Royal Society of Chemistry 2023 |