
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic olefin metathesis in blood†
Igor
Nasibullin
 a,
Hiromasa
Yoshioka
a,
Akari
Mukaimine
a,
Akiko
Nakamura
a,
Yuriko
Kusakari
a,
Tsung-Che
Chang
*a and
Katsunori
Tanaka
*ab
a,
Hiromasa
Yoshioka
a,
Akari
Mukaimine
a,
Akiko
Nakamura
a,
Yuriko
Kusakari
a,
Tsung-Che
Chang
*a and
Katsunori
Tanaka
*ab
aBiofunctional Synthetic Chemistry Laboratory, Cluster for Pioneering Research RIKEN, Wako-shi, Saitama 351-0198, Japan. E-mail: chang.t.ac@m.titech.ac.jp; kotzenori@riken.jp
bDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, Meguro-ku, Tokyo 152-8552, Japan
First published on 27th September 2023
Abstract
The direct synthesis of drugs in vivo enables drugs to treat diseases without causing side effects in healthy tissues. Transition-metal reactions have been widely explored for uncaging and synthesizing bioactive drugs in biological environments because of their remarkable reactivity. Nonetheless, it is difficult to develop a promising method to achieve in vivo drug synthesis because blood cells and metabolites deactivate transition-metal catalysts. We report that a robust albumin-based artificial metalloenzyme (ArM) with a low loading (1–5 mol%) can promote Ru-based olefin metathesis to synthesize molecular scaffolds and an antitumor drug in blood. The ArM retained its activity after soaking in blood for 24 h and provided the first example of catalytic olefin cross metathesis in blood. Furthermore, the cyclic-Arg-Gly-Asp (cRGD) peptide-functionalized ArM at lower dosages could still efficiently perform in vivo drug synthesis to inhibit the growth of implanted tumors in mice. Such a system can potentially construct therapeutic drugs in vivo for therapies without side effects.
Introduction
All therapeutic drugs have side effects,1 some of which are serious, resulting in permanent damage to the body, or even life-threatening. A straightforward method to solve this problem is to directly synthesize therapeutic drugs at disease sites to avoid unwanted side effects against healthy tissues. If this goal can be achieved, numerous effective drugs known to have harmful side effects can be utilized again for disease treatment, thereby advancing the fields of drug discovery and life sciences. To minimize the burden on the body and maximize therapeutic effects, a method that is biocompatible and exhibits robust catalytic ability to produce the necessary amounts of drugs in vivo is desirable.One strategy for minimizing the adverse effects of drugs is based on the concept of prodrugs, which are inactive derivatives of drugs that undergo an enzymatic and/or chemical transformation to generate the active forms.2 In the literature, numerous studies have demonstrated the controllable production of drugs in biological systems via prodrug activation with natural enzymes or in the disease microenvironment (e.g., acidic pH).2 Because such enzymes or microenvironments are widely distributed in healthy tissues, this approach has off-target side effects.3 With advances in bio-orthogonal click-to-release chemistry over the past several decades, many researchers have shifted their focus to developing prodrug uncaging strategies. With this approach, the relevant functional groups of drugs (e.g., amine, hydroxyl, or acid groups) are masked using abiotic small molecules, which can then be cleaved via an external trigger,4 improving the specificity of drug release. This strategy, however, is not applicable for regulating the bioactivity of drugs that do not contain the aforementioned functional groups.
Various transition-metal catalysts have been widely explored for uncaging prodrugs because they display remarkable catalytic reactivity in myriad chemical transformations. Such transformations include deprotection reactions such as deallylation (Ru, Pd),5–10 depropargylation (Pd, Au, Pt),11–14 azide reduction (Fe, Ru),15,16 pentynoyl amide cleavage (Pt),13 2-alkynylbenzamide decaging (Au),17 and ring-closing metathesis (RCM)-triggered 1,4-elimination (Ru).18 In addition to uncaging prodrugs, some studies have recently demonstrated the use of transition-metal-catalyzed bond formation reactions to synthesize drugs, including Suzuki–Miyaura coupling (Pd),19 alkyne hydroamination (Au),20 azide–alkyne cycloaddition (Cu),21,22 olefin metathesis (Ru),23,24 and transfer hydrogenation (Pd).25 Because of biocompatibility, most of the aforementioned examples of transition-metal-mediated reactions are limited to use in cell culture environments and microorganisms. The examples of reactions that work in live mammals (in vivo)7–9,22,24,25 have been limited because of the highly complex composition in the bloodstream of the body, where hundreds of different serum proteins, complex metabolites (e.g., glutathione (GSH)), and numerous blood cells will quickly deactivate transition-metal catalysts (Fig. 1A). The majority of these in vivo examples have involved nanocarriers with encapsulated transition metals because the large surface-area-per-volume ratio of nanocarriers enables the loading of greater amounts of metal catalysts and thereby enhances the reaction rates. However, they have been used in excess rather than in catalytic quantities to produce the required amounts of desired products in vivo, indicating that the blood environment substantially hindered the reactivity of metallic nanocarriers. Notably, Völker and Meggers have developed a Ru complex that can catalyze the deallylation of a substrate in blood serum.5 Although 10 mol% of the Ru complex could afford the product in 30% yield, blood serum is much less complex than blood because it does not contain any white/red blood cells or platelets. A method that implements highly efficient catalytic organometallic reactions in blood has not yet been reported.
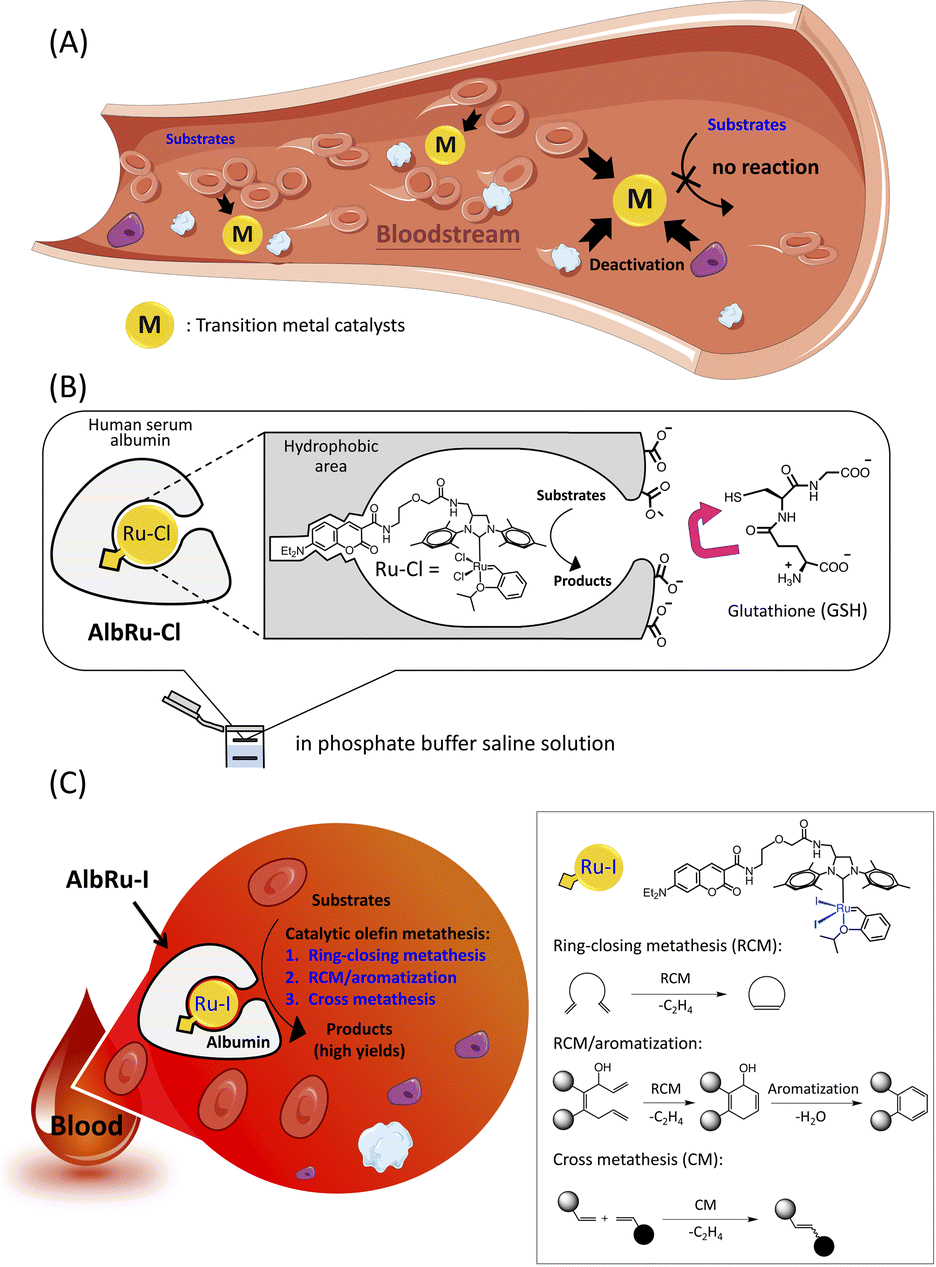 | ||
| Fig. 1 Catalytic olefin metathesis in blood. (A) A primary requirement for applying transition-metal catalyzed reactions in blood is to protect their activity to avoid rapid deactivation by numerous serum proteins, metabolites, and blood cells. (B) A Ru–Cl ruthenium complex encased by human serum albumin to form a biocompatible artificial metalloenzyme (ArM) (AlbRu–Cl). (C) The albumin-based Ru–I-containing ArM (AlbRu–I) can catalyze ring-closing metathesis (RCM), sequential RCM/aromatization, and olefin cross metathesis (CM) reactions in blood. | ||
Artificial metalloenzymes (ArMs) are the result of inserting transition-metal complexes into protein scaffolds of interest, which can impart enhanced biofunctionality to the anchored metal catalysts and facilitate non-natural reactions under mild conditions.26 Previously, our group has developed a biocompatible ArM23 in which a Hoveyda–Grubbs complex, Ru–Cl, is anchored into a hydrophobic binding pocket of human serum albumin (HSA) (AlbRu–Cl) through the interaction of a coumarin moiety with the cavity (Fig. 1B). The negatively charged surface of HSA prevented the charged GSH from entering the metal-binding site, enabling the bound Ru catalyst to be protected even in the presence of GSH at concentrations as high as 20 mM in phosphate buffered saline (PBS) solution. Moreover, we recently reported that a cancer-targeting glycosylated AlbRu–Cl could mediate Ru-based olefin metathesis for synthesis of a drug in mice to induce tumor growth inhibition; however, a high dose of the glycosylated AlbRu–Cl (116 mg kg−1) was required.24 This indicates that the catalytic reactivity of the ArM in vivo should be improved. Building upon these results, we propose an albumin-based Ru–I-containing ArM (AlbRu–I) that can efficiently manipulate olefin metathesis in blood (Fig. 1C). We found that just 1–5 mol% of AlbRu–I can catalyze various substrates in blood in substantial yields, including RCM, sequential RCM/aromatization, and the first example of olefin cross-metathesis reactions. Importantly, after being soaked in blood for 24 h, AlbRu–I still exhibited outstanding catalytic reactivity, highlighting its robust stability in blood. In particular, the cyclic-Arg-Gly-Asp (cRGD) peptide-functionalized AlbRu–I at lower dosages (20 and 40 mg kg−1) could efficiently perform in vivo drug synthesis to inhibit the growth of implanted tumors in mice, highlighting the significant potential of the ArM for future therapeutic applications.
Results and discussion
Catalytic activity investigation of a Ru–I-based ArM (AlbRu–I) in blood
Olefin metathesis is one of the most efficient methods for building new carbon–carbon double bonds. Davis and coworkers reported the first example of the modification of proteins in an aqueous buffer solution using olefin cross metathesis by the Hoveyda–Grubbs second-generation catalyst.27 Ward and coworkers conducted groundbreaking research on in-cell RCM by assembling a Ru-based ArM in Escherichia coli,28 while Schunck and Mecking very recently used a Ru catalyst to perform cross metathesis of unsaturated fatty acids in microalgae.29 Although Ru-based olefin metathesis has been applied to live microbes, these examples were strictly carried out in a thiol-free area to avoid deactivation of the Ru by GSH such as the periplasm of E. coli or the lipid vesicles of microalgae. We previously found that AlbRu–Cl can catalytically convert substrates via RCM in the presence of GSH in PBS solution. Although AlbRu–Cl was able to convert a prodrug via sequential RCM/aromatization in a blood mixture of blood/PBS/1,4-dioxane (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1), a high loading of it (10 mol%) was required to achieve a good conversion yield (46%).24 The capacity of AlbRu–Cl to catalyze other types of olefin metathesis for various substrates in blood solution has not yet been investigated. First, we examined the capacity of AlbRu–Cl with a low loading (1 mol%) to catalyze RCM for model substrate 1 in the same blood mixture solution (Fig. 2A). However, the conversion yield of 2 was unsatisfactory (11%, entry 1). To improve the RCM reactivity of AlbRu–Cl in blood, we tested longer reaction times (entry 2) or a greater loading amount of AlbRu–Cl (entry 3); however, neither test showed a substantial increase in the RCM yield in blood.
:4:1), a high loading of it (10 mol%) was required to achieve a good conversion yield (46%).24 The capacity of AlbRu–Cl to catalyze other types of olefin metathesis for various substrates in blood solution has not yet been investigated. First, we examined the capacity of AlbRu–Cl with a low loading (1 mol%) to catalyze RCM for model substrate 1 in the same blood mixture solution (Fig. 2A). However, the conversion yield of 2 was unsatisfactory (11%, entry 1). To improve the RCM reactivity of AlbRu–Cl in blood, we tested longer reaction times (entry 2) or a greater loading amount of AlbRu–Cl (entry 3); however, neither test showed a substantial increase in the RCM yield in blood.
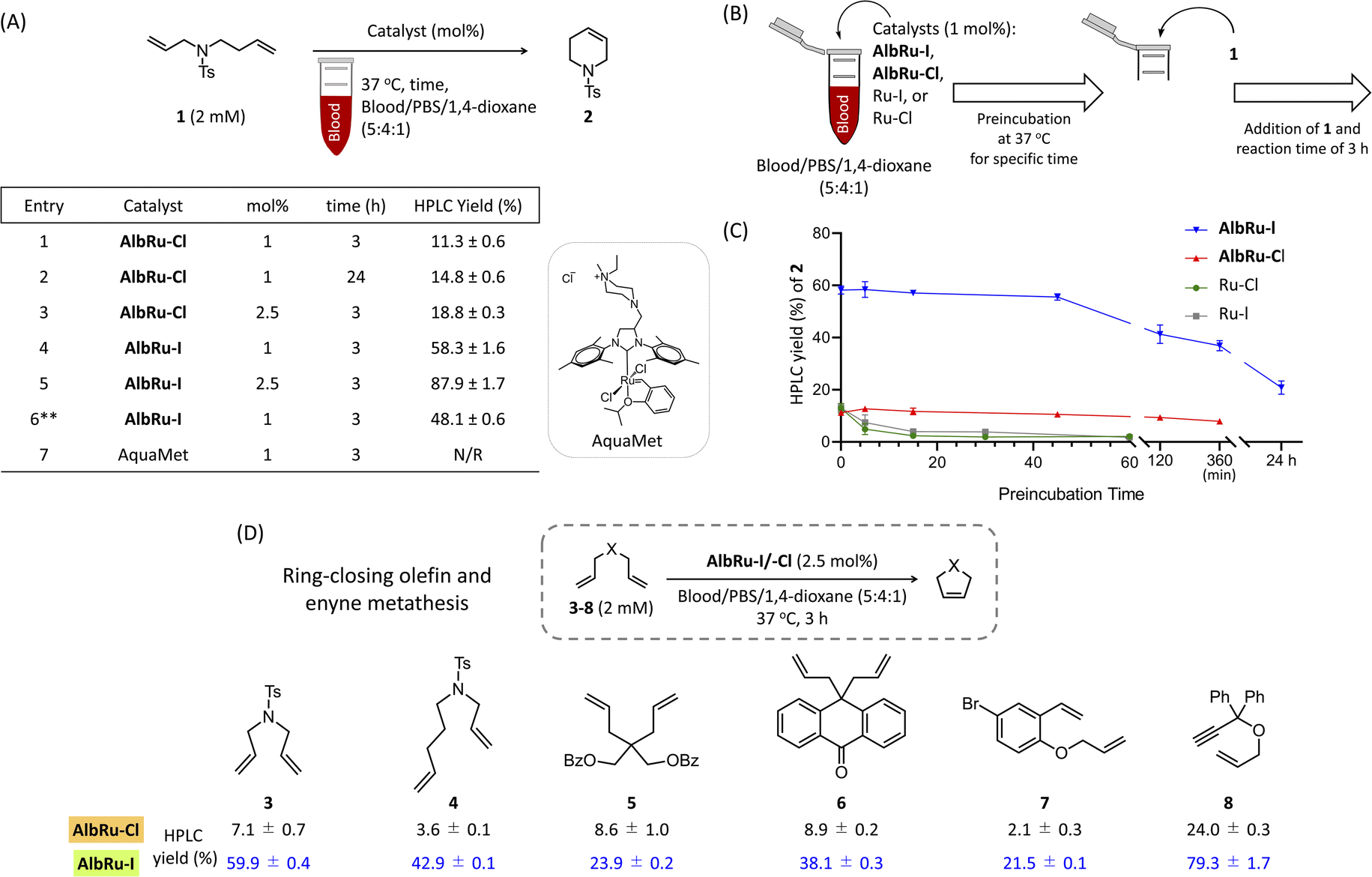 | ||
| Fig. 2 Catalytic activity investigation of Ru-based ArMs for ring-closing metathesis in blood. (A) Reactivity studies of substrate 1 (2 mM) with AlbRu–Cl/–I (1 or 2.5 mol%) and AquaMet catalyst (1 mol%) in a mixture of blood/PBS/1,4-dioxane (5:4:1) (**8:1:1). (B) To determine the time and extent of Ru protection, a series of experiments were run as illustrated. The indicated Ru catalysts (1 mol%) were preincubated in blood for a specific time (0, 5, 15, 30, and 60 min for Ru–I/Cl; 0, 5, 15, 45, 120, and 360 min for AlbRu–Cl; and 0, 5, 15, 45, 120, and 360 min, and 24 h for AlbRu–I), followed by reaction initiation using 1 (2 mM). After a reaction time of 3 h, the yield of 2 was measured. (C) Graphical representation of stability results of the Ru catalysts in blood. (D) Substrate scope for testing ring-closing olefin and enyne metathesis. Given HPLC yields were determined by HPLC analysis (peak retention times relative to product standards, followed by MS analysis for confirmation, and calculation of resultant yields based on product standard curves). Error bars represent the s.d. of three independent measurements. The blood used here was commercially available sheep blood. Abbreviations: Ts, 4-toluenesulfonyl; N/R, no reaction. | ||
We drew inspiration from the results of Skowerski and coworkers30—specifically, their finding that steric hindrance in the proximity of the Ru center as a result of bulky iodides can stabilize the active intermediates—and prepared another Ru-based ArM (AlbRu–I) with Ru–I as an anchored metal catalyst. Surprisingly, AlbRu–I exhibited dramatically improved catalytic activity for RCM, affording the desired 2 in good yield (58%, entry 4) compared with the yield achieved with AlbRu–Cl (11%, entry 1). As expected, increasing the amount of AlbRu–I to 2.5 mol% resulted in an excellent conversion yield of 2 (88%, entry 5). In addition, the catalytic RCM for substrate 1 was tested in a mixture that contained 80% blood, and AlbRu–I still showed high activity (entry 6). A highly water-soluble Ru complex, AquaMet catalyst,31 has been shown to effectively catalyze RCM in aqueous media. However, as expected by the report of Schwaneberg,32 the reactivity of the AquaMet catalyst in the blood mixture was completely abolished (entry 7). The results in Fig. 2A clearly demonstrate the robust catalytic activity of AlbRu–I because even using a tiny amount of ArMs was found to achieve RCM in excellent yields in blood.
In the next experiment, we focused on the approximate duration of the stability of Ru-based ArMs (AlbRu–I/–Cl) in blood. As depicted in Fig. 2B, a series of experiments were carried out in which the AlbRu–I/–Cl and free-in-solution catalysts (Ru–I/–Cl) as controls were preincubated in blood for a specific time. Substrate 1 was then added to the mixture, and the yield of 2 was measured after a 3 h reaction. As expected, the Ru–I/–Cl catalysts were deactivated after a few minutes of preincubation in the blood mixture (green and gray lines, Fig. 2C). By contrast, both AlbRu–I/–Cl still gave substantial yields of 2 even after 6 h of preincubation (blue and red lines, Fig. 2C). Importantly, although AlbRu–I was used in a very small amount (1 mol%), it still produced a high yield of 2 (21%) after 24 h of preincubation in the blood mixture, demonstrating its exceptional biocompatibility and excellent catalytic activity for RCM. In comparison with other known ArMs and metallic nanocarriers, AlbRu–I is the first to demonstrate that it can carry out a highly efficient catalytic organometallic reaction in such a challenging biological environment.
The AlbRu–I/–Cl catalysts were further investigated with a small substrate scope to confirm the remarkable reactivity of AlbRu–I in blood. Under the standard conditions corresponding to Fig. 2D, 2.5 mol% of ArMs and 2 mM of substrates were used in the mixture of blood/PBS/1,4-dioxane (5:4:1). This result clearly demonstrates that, compared with AlbRu–Cl, AlbRu–I showed a substantially greater activity toward the RCM over substrates 3–7, resulting in substantial yields (21–60%). Notably, AlbRu–I could also catalyze ring-closing enyne metathesis of 8 in excellent yield (79%). The overall results in Fig. 2 clearly show that AlbRu–I is a capable catalyst for promoting olefin metathesis in blood.
Substrate scope for the AlbRu–I in blood
Encouraged by these promising results, we shifted to investigating the applicability of AlbRu–I with various substrates in blood (Fig. 3). These substrates were divided into three groups for testing: (1) RCM; (2) sequential RCM/aromatization; and (3) olefin cross metathesis. AlbRu–I catalyzed RCM for substrates 11–17 in good yields (15–64%) (Fig. 3A), whereas substrates 9 and 10 gave relatively low yields (7–8%). These results can be explained by the products with five- or six-membered rings being more stable. Substrate 18 resulted in the lowest yield, likely because of its structural effect. Collectively, the results in Fig. 2D and 3A show that AlbRu–I exhibits strong potential to be used in the synthesis of carbocyclic molecules in blood for various applications.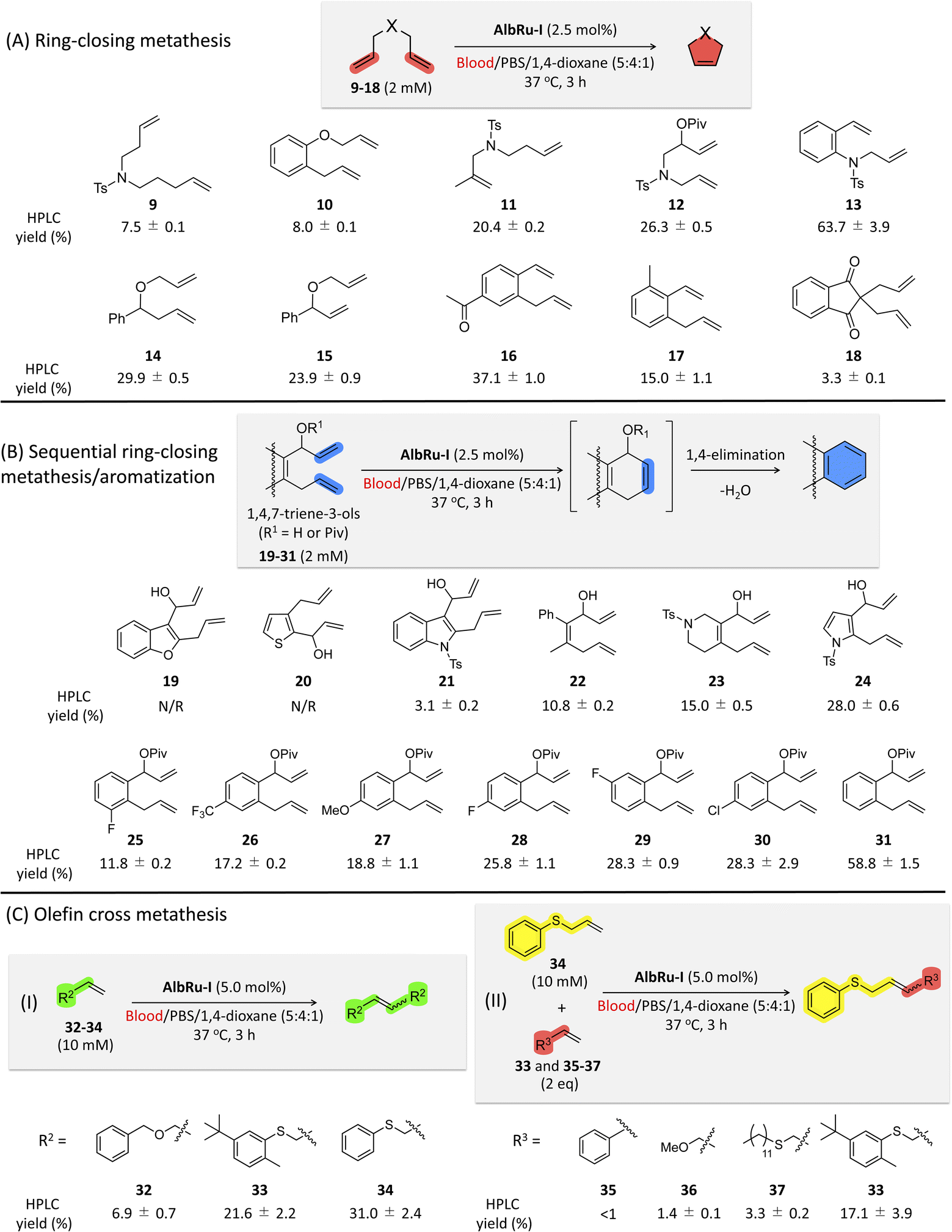 | ||
| Fig. 3 Substrate scope for AlbRu–I in blood. (A) Ring-closing metathesis (RCM). (B) Sequential RCM/aromatization. (C) Olefin cross-metathesis. Reaction conditions: substrates (2 mM for (A) and (B) and 10 mM for (C)) and AlbRu–I (2.5 mol% for (A) and (B) and 5.0 mol% for (C)) were used in a mixture of blood/PBS/1,4-dioxane (5:4:1). Incubations were carried out in triplicate at 37 °C for 3 h. Given HPLC yields were determined by HPLC analysis (peak retention times relative to product standards, followed by MS analysis for confirmation, and calculation of resultant yields based on product standard curves). Abbreviations: Piv, pivaloyl; Ts, 4-toluenesulfonyl; N/R, no reaction. | ||
Previous studies conducted by Ward and coworkers have demonstrated that, in RCM reactions of 1,4,7-trien-3-ols through Ru-based ArMs, spontaneous aromatization can proceed via 1,4-elimination to produce phenyl moieties.18,33,34 Investigating sequential RCM/aromatization reactions in blood is especially important because many bioactive drugs contain at least one phenyl moiety in their structure. Heterocyclic precursors 19–24 were prepared for the RCM/aromatization (Fig. 3B). Even though AlbRu–I did not work with substrates 19 and 20, the carbazole 21, biphenyl 22, tetrahydroisoquinoline 23, and indole 24 precursors gave substantial yields in the blood mixture (3–28%). In particular, a 28% yield of the indole product from 24 was obtained, which is highly promising for drug design because myriad bioactive compounds contain an indole moiety. We previously reported that the allylic hydroxyl groups of 31 protected with a pivalate group could facilitate 1,4-elimination in the RCM/aromatization process to afford naphthalene in aqueous solution.24 Therefore, in the present work, we tested a set of substituted naphthalene precursors 25–31 (Fig. 3B). As a result, AlbRu–I catalyzed the RCM/aromatization for substrates 25–30 to generate the different substituted naphthalenes in good yields (12–28%). In particular, substrate 31 produced naphthalene in blood in an excellent yield (59%). The results in Fig. 3B offer a useful technique for producing various heterocyclic and naphthalene-related chemicals in blood. Although the aforementioned reaction yields were not high, we note that these Ru-based reactions were carried out in blood, not in an organic solvent or aqueous buffer solution.
The reactivity of olefin cross metathesis in blood warrants investigation because it can facilitate the creation of complex molecules by linking two alkene fragments. After a detailed investigation of the cross-metathesis conditions (see Fig. S83†), larger amounts of AlbRu–I (5 mol%) and substrates 32–37 (10–20 mM) were used to afford higher product yields (Fig. 3C). Initially, olefin homodimerization of 32–34 catalyzed by AlbRu–I was tested in blood before olefin cross metathesis was investigated. Although dimerization of 32 gave a lower yield (7%), the exciting outcome in blood encouraged us to investigate further (Fig. 3C-I). Under the sulfur-assisted olefin metathesis mechanism reported by Davis and coworkers,27 the homodimerization yields of 33 and 34 were increased dramatically, with the 31% yield of 34 being particularly noteworthy. Importantly, AlbRu–I successfully mediated the cross metathesis of 34 with either 33, 35, 36, or 37 in blood (Fig. 3C-II). Although 35–37 gave lower yields of products (1–3%) because of their lower reactivity, these examples show the first demonstration of the feasibility of carrying out olefin cross metathesis in blood. Moreover, another substrate containing a sulfur moiety, 33, achieved a substantial yield (17%), clearly demonstrating the first example of implementing catalytic olefin cross metathesis in blood. The results in Fig. 3C represent a new avenue for constructing complex molecules in blood via olefin cross metathesis.
In vivo drug synthesis against tumour growth
As mentioned in the introduction, the direct synthesis of bioactive drugs in vivo is an important vision for future drug discovery and other biomedical applications. For localized in vivo drug synthesis to avoid off-target effects, a suitable targeting system was needed to direct the biocatalysts to specific disease sites within the body. As shown in Fig. 4A, a cyclic-Arg-Gly-Asp (cRGD) pentapeptide is frequently used as a drug delivery system through an interaction with the overexpression of integrin in cancer cells.35 Recently, we revealed that after injection into mice, the cRGD-conjugated human serum albumin (cRGD)HSA could rapidly and specifically accumulate into tumors derived from SW620 colon cancer cells after just 4 hours.36 For the following cancer-targeting studies, the cRGD-linked ArMs bound with Ru–Cl/–I ((cRGD)AlbRu–Cl and (cRGD)AlbRu–I) were used as the biocatalysts for localized in vivo drug synthesis (Fig. 4A). In the literature, combretastatin-A4 (CA-4) and its derivatives containing drug 39 showed a high-affinity tubulin ligand with anticancer characteristics and inhibited tumor growth in vivo through anti-angiogenic mechanisms37 (Fig. 4B). As mentioned earlier in the introduction, we have reported that a cancer-targeting glycosylated AlbRu–Cl can mediate RCM/aromatization for synthesizing 39 from the prodrug 38 in mice to induce tumor growth inhibition;24 however, a high dose of the glycosylated AlbRu–Cl (116 mg kg−1) was necessary. To improve the efficiency of in vivo drug synthesis, the last stage of this study was to investigate using lower dosages of (cRGD)AlbRu–I to produce 39 from 38 to treat subcutaneous SW620-xenografted mice (Fig. 4C). | ||
| Fig. 4
In vivo drug synthesis against SW620 tumor growth in mice. (A) Schematic illustration of cancer-targeting (cRGD)AlbRu. (B) Investigation of the catalytic reactivity of RCM/aromatization by (cRGD)AlbRu (2.5 mol%) in a mixture of blood/PBS/1,4-dioxane (5:4:1) for transformation of prodrug 38 (2 mM) into drug 39. Given HPLC yields were determined by HPLC analysis (peak retention times relative to product standards, followed by MS analysis for confirmation, and calculation of resultant yields based on product standard curves). Error bars represent the s.d. of three independent measurements. (C) To highlight the biocatalytic reactivity of the (cRGD)AlbRu–I, the objective was to apply in vivo drug synthesis via intravenous administration to treat subcutaneous SW620-xenografted mice. Tumors were initially implanted in mice and developed over 1 day before therapy. Dosages were applied in daily injections spread out over 8 days. (D) Effects of tumor therapy on tumor volume changes of xenograft mice subjected to the following treatments: saline (black), drug 39 (32.5 mg kg−1, orange), prodrug 38 (58 mg kg−1) + (cRGD)AlbRu–Cl (20 or 40 mg kg−1, gray or green, respectively), and prodrug 38 (58 mg kg−1) + (cRGD)AlbRu–I (20 or 40 mg kg−1, red or blue, respectively). Data in (D) are represented as mean value ± SD, n = 3 biologically independent samples. | ||
Before moving on to animal experiments, the in vitro data in Fig. 4B showed that, compared with (cRGD)AlbRu–Cl, (cRGD)AlbRu–I again demonstrated excellent catalytic activity in blood because 2.5 mol% of it could produce drug 39 in substantial yield (40%), indicating great potential for producing a high concentration of 39 from 38 even if a lower dosage of (cRGD)AlbRu–I is used in vivo. Likewise, based on the results of cell-based experiments (see Fig. S88 and S89†), it was clearly demonstrated that (cRGD)AlbRu–I with a low concentration (0.5 μM) could still effectively perform drug 39 synthesis to induce a significant suppression in SW620 cancer cell growth, whereas (cRGD)AlbRu–Cl failed to convert 38 under the same setting conditions.
With these promising in vitro results, as shown in Fig. 4D, six groups of mice were arranged to receive the indicated compounds [saline, 39 only, co-treatment with (cRGD)AlbRu–Cl (20 or 40 mg kg−1) and 38, or co-treatment with (cRGD)AlbRu–I (20 or 40 mg kg−1) and 38] via intravenous administration every day for 8 consecutive days. As a control group, a saline solution was used to replace compounds in the treatment protocol. With the treatment groups, (cRGD)AlbRu was first administered, followed by prodrug 38. As a result, co-treatment with (cRGD)AlbRu–Cl (20 or 40 mg kg−1) and 38 (Fig. 4D, gray and green lines, respectively) showed the same rate of tumor growth as the saline group (Fig. 4D, black line). This illustrated that the in vivo reactivity of (cRGD)AlbRu–Cl at these dosages was insufficient to synthesize the required amount of drug, resulting in failure in tumor inhibition. In contrast, co-treatment with (cRGD)AlbRu–I (20 or 40 mg kg−1) and 38 (Fig. 4D, red and blue lines, respectively) resulted in a dose-dependent depreciation in the rate of tumor growth compared to the saline group. These results demonstrated that, compared with (cRGD)AlbRu–Cl, (cRGD)AlbRu–I at lower dosages was still able to exhibit robust catalytic reactivity to mediate RCM/aromatization for progressing in vivo synthesis of 39 for cancer treatment. Moreover, a notable finding was that combining (cRGD)AlbRu–I (20 or 40 mg kg−1) with 38 resulted in a stronger suppression of tumor growth than treatment with drug 39 alone (Fig. 4D, orange line). This would suggest that cancer targeting is also critical for the cancer treatment by co-treatment of (cRGD)AlbRu–I with 38 because localized drug synthesis can be achieved to generate high concentrations of drug in specific disease sites, leading to the enhancement of therapy effects.
Conclusions
This study significantly advances the research fields of Ru-based olefin metathesis under biological conditions and ArMs. Overall, just 1–5 mol% AlbRu–I could catalyze three types of olefin metathesis reactions in blood to construct carbocyclic, heterocyclic, phenyl rings, and olefin dimerization in substantial yields. AlbRu–I also showed robust stability for 24 h in blood, expanding the biocompatibility of ArMs and opening the door for the development of general metal-based ArMs for catalytic reactions in blood. Moreover, the cancer-targeting AlbRu–I at a low dosage was able to elicit significant tumor growth inhibition in mice through localized synthesis of an antitumor drug, highlighting the significant potential of the ArM for future therapeutic applications. Since applications of click-to-release chemistry are limited to drug release, this promising metallic system offers a new avenue for building bioactive drugs and functional molecules in vivo, enabling the development of innovative drug therapies without side effects.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: I. N. and K. T.; methodology: I. N., T.-C. C., and K. T.; investigation: I. N., T.-C. C., H. Y., A. M., A. N. and Y. K.; visualization: T.-C. C., I. N., and K. T.; funding acquisition: K. T.; project administration: K. T.; supervision: K. T.; writing – original draft: T.-C. C. and K. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of RIKEN and approved by the Animal Ethics Committee of RIKEN (W2019-2-049). This work was financially supported by the AMED Grant JP15KM0908001 (K. T.), a research grant from the Astellas Foundation (K. T.), Mizutani Foundation for Glycoscience (K. T.), and JSPS KAKENHI Grant Numbers JP21H02065 (K. T.) and JP21K19042 (K. T.). HRMS (EI), and DQF-COSY were acquired by the Molecular Structure Characterization Unit (RIKEN CSRS, Wako).Notes and references
- O. Osanlou, M. Pirmohamed and A. K. Daly, Adv. Pharmacol., 2018, 83, 155 CrossRef CAS.
- V. Abet, F. Filace, J. Recio, J. Alvarez-Builla and C. Burgos, Eur. J. Med. Chem., 2017, 127, 810 CrossRef CAS PubMed.
- K. M. Huttunen, H. Raunio and J. Rautio, Pharmacol. Rev., 2011, 63, 750 CrossRef CAS PubMed.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077 RSC.
- T. Völker and E. Meggers, ChemBioChem, 2017, 18, 1083 CrossRef PubMed.
- M. I. Sánchez, C. Penas, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2014, 5, 1901 RSC.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS.
- T. L. Bray, M. Salji, A. Brombin, A. M. Pérez-López, B. Rubio-Ruiz, L. C. A. Galbraith, E. E. Patton, H. Y. Leung and A. Unciti-Broceta, Chem. Sci., 2018, 9, 7354 RSC.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, ACS Nano, 2018, 12, 12814 CrossRef CAS PubMed.
- Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, Nat. Commun., 2018, 9, 1943 CrossRef PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaría and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548 CrossRef PubMed.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. de Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior and G. J. L. Bernardes, J. Am. Chem. Soc., 2020, 142, 10869 CrossRef CAS.
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Pérez-López, V. Sebastián, P. Martín-Duque, M. Arruebo, J. Santamaría and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864 CrossRef CAS.
- R. Huang, C.-H. Li, R. Cao-Milán, L. D. He, J. M. Makabenta, X. Zhang, E. Yu and V. M. Rotello, J. Am. Chem. Soc., 2020, 142, 10723 CrossRef CAS PubMed.
- J. Chen, K. Li, J. S. L. Shon and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 142, 4565 CrossRef PubMed.
- K. Vong, T. Yamamoto, T.-C. Chang and K. Tanaka, Chem. Sci., 2020, 11, 10928 RSC.
- V. Sabatino, J. G. Rebelein and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 17048 CrossRef CAS PubMed.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212 RSC.
- T.-C. Chang, K. Vong, T. Yamamoto and K. Tanaka, Angew. Chem., Int. Ed., 2021, 60, 12446 CrossRef CAS PubMed.
- S. Li, L. Wang, F. Yu, Z. Zhu, D. Shobaki, H. Chen, M. Wang, J. Wang, G. Qin, U. J. Erasquin, L. Ren, Y. Wang and C. Cai, Chem. Sci., 2017, 8, 2107 RSC.
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2019, 58, 6987 CrossRef CAS PubMed.
- S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Nat. Catal., 2019, 2, 780 CrossRef CAS.
- I. Nasibullin, I. Smirnov, P. Ahmadi, K. Vong, A. Kurbangaleva and K. Tanaka, Nat. Commun., 2022, 13, 39 CrossRef CAS.
- Z. Du, C. Liu, H. Song, P. Scott, Z. Liu, J. Ren and X. Qu, Chem, 2020, 6, 2060 CAS.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120 CrossRef CAS PubMed.
- Y. A. Lin, J. M. Chalker and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 16805 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661 CrossRef CAS PubMed.
- N. S. Schunck and S. Mecking, Angew. Chem., Int. Ed., 2022, 61, e202211285 CrossRef CAS PubMed.
- A. Tracz, M. Matczak, K. Urbaniak and K. Skowerski, Beilstein J. Org. Chem., 2015, 11, 1823 CrossRef CAS PubMed.
- T. K. Olszewski, M. Bieniek and K. Skowerski, Org. Process Res. Dev., 2020, 24, 125 CrossRef CAS.
- A. R. Grimm, D. F. Sauer, M. D. Davari, L. Zhu, M. Bocola, S. Kato, A. Onoda, T. Hayashi, J. Okuda and U. Schwaneberg, ACS Catal., 2018, 8, 3358 CrossRef CAS.
- A. Samanta, V. Sabatino, T. R. Ward and A. Walther, Nat. Nanotechnol., 2020, 15, 914 CrossRef CAS PubMed.
- S. Fischer, T. R. Ward and A. D. Liang, ACS Catal., 2021, 11, 6343 CrossRef CAS PubMed.
- Y. Cheng and Y. Ji, Eur. J. Pharm. Sci., 2019, 128, 8 CrossRef CAS PubMed.
- P. Ahmadi, K. Muguruma, T.-C. Chang, S. Tamura, K. Tsubokura, Y. Egawa, T. Suzuki, N. Dohmae, Y. Nako and K. Tanaka, Chem. Sci., 2021, 12, 12266 RSC.
- M. Su, J. Huang, S. Liu, Y. Xiao, X. Qin, J. Liu, C. Pi, T. Luo, J. Li, X. Chen and Z. Luo, Sci. Rep., 2016, 6, 28139 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, compound data, and NMR spectra. See DOI: https://doi.org/10.1039/d3sc03785a |
| This journal is © The Royal Society of Chemistry 2023 |