
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNaphthopyran molecular switches and their emergent mechanochemical reactivity
Molly E.
McFadden
 ,
Ross W.
Barber
,
Anna C.
Overholts
and
Maxwell J.
Robb
*
,
Ross W.
Barber
,
Anna C.
Overholts
and
Maxwell J.
Robb
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrobb@caltech.edu
First published on 24th August 2023
Abstract
Naphthopyran molecular switches undergo a ring-opening reaction upon external stimulation to generate intensely colored merocyanine dyes. Their unique modularity and synthetic accessibility afford exceptional control over their properties and stimuli-responsive behavior. Commercial applications of naphthopyrans as photoswitches in photochromic ophthalmic lenses have spurred an extensive body of work exploring naphthopyran–merocyanine structure–property relationships. The recently discovered mechanochromic behavior of naphthopyrans has led to their emergent application in the field of polymer mechanochemistry, enabling advances in the design of force-responsive materials as well as fundamental insights into mechanochemical reactivity. The structure–property relationships established in the photochemical literature serve as a convenient blueprint for the design of naphthopyran molecular force probes with precisely tuned properties. On the other hand, the mechanochemical reactivity of naphthopyran diverges in many cases from the conventional photochemical pathways, resulting in unexpected properties and opportunities for deeper understanding and innovation in polymer mechanochemistry. Here, we highlight the features of the naphthopyran scaffold that render it a powerful platform for the design of mechanochromic materials and review recent advances in naphthopyran mechanochemistry.
1 Introduction
1.1 Polymer mechanochemistry
Since the seminal report of site-specific mechanochemical activation of covalent bonds in 2005,1 polymer mechanochemistry has quickly grown into a burgeoning area of research.2,3 Mechanophores are force sensitive molecules or moieties that respond selectively to mechanical perturbation leading to a productive chemical transformation.4 Polymer chains are essential for transducing mechanical force to covalently linked mechanophores. On the one hand, the ability to control the force responsivity of molecules has revolutionized the design of smart materials capable of autonomically responding to mechanically dynamic environments.5 Recent research has also unveiled remarkable capabilities for tailoring the macroscopic mechanical properties of materials by controlling the molecular level force sensitivity of individual mechanophores within a polymer network.6,7 On the other hand, mechanical force can be leveraged as an external stimulus to effect desired chemical reactions with spatial and temporal precision.8–10 In an ever-growing collection of more than 100 different mechanophores developed to date,11 judicious structural design has enabled a diverse range of force-responsive functions including catalyst activation,12,13 conductivity switching,14 mechanically triggered chemiluminescence,15 and cargo release,16–18 among many others.19,20 Among these various categories, mechanochromic mechanophores, which change or produce color in response to force, have been widely developed as molecular force probes for enabling detection of stress and/or strain in polymeric materials (Fig. 1).20–24 Mechanical stress typically causes degradation of polymeric materials resulting, in part, from nonspecific chain scission.25–28 While this microscopic damage is often difficult to detect, mechanochromic mechanophores have enabled the visualization of single bond breaking events providing important mechanistic insights into fracture mechanics.29 Additionally, because optical changes are straightforward to monitor and characterize, mechanochromic mechanophores have facilitated wide-ranging fundamental investigations of polymer mechanochemistry.30–33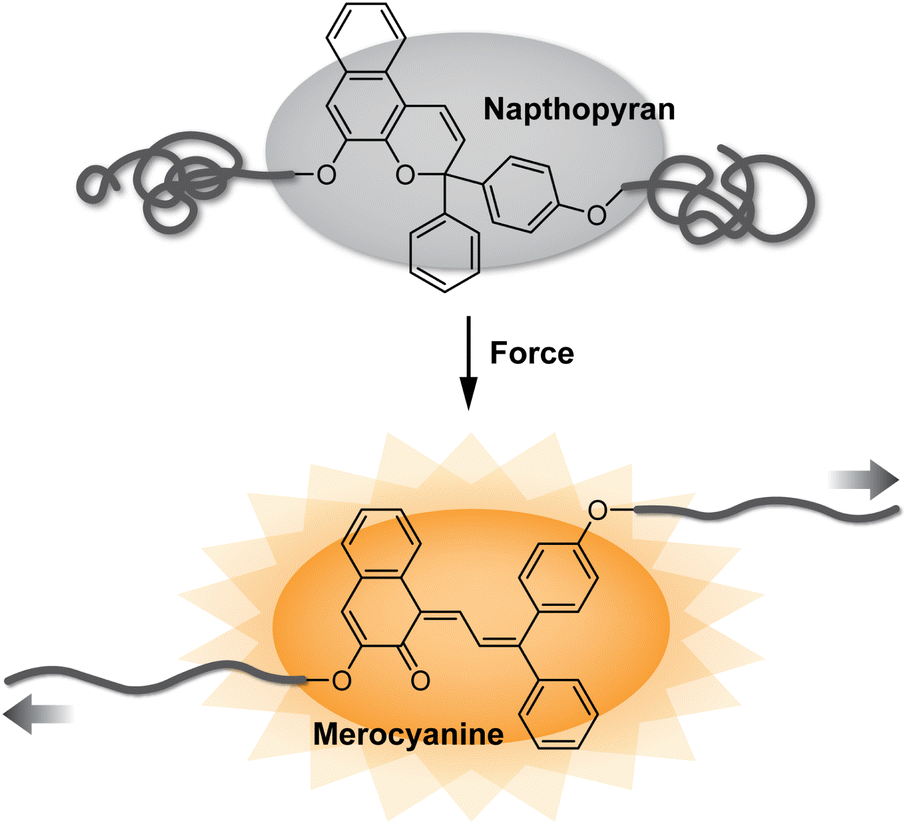 | ||
| Fig. 1 Mechanophores covalently incorporated into polymer chains undergo productive chemical reactions in response to mechanical force. The mechanochromic transformation of naphthopyran is illustrated. | ||
Recent research has showcased naphthopyran to be an effective and highly modular mechanophore for not only designing diverse mechanochromic materials, but also revealing unusual reactivity unique to mechanochemical activation pathways. Naphthopyrans undergo a ring-opening reaction in response to ultraviolet (UV) light or mechanical force to produce a highly colored merocyanine dye.34,35 Due to their synthetic accessibility and modularity coupled with the highly tunable properties of their merocyanine dyes, naphthopyrans have previously found widespread commercial application in photochromic ophthalmic lenses.36–38 These same qualities make naphthopyrans an ideal mechanophore scaffold for constructing mechanochromic materials, developing mechanochemical structure–property relationships, and studying fundamental aspects of mechanochemical reactivity. In this Perspective, we begin by briefly discussing mechanochromic mechanophores more broadly to provide context for the (mechano)chemistry of naphthopyran. Next, we describe the synthesis of naphthopyrans and review their photochemistry with an emphasis on the extensive and generalizable structure–property relationships of their merocyanine dyes. Finally, we discuss the mechanochemical reactivity of naphthopyrans, highlighting notable recent discoveries that demonstrate their unique potential as an emergent class of diverse molecular force probes.
1.2 Mechanochromic mechanophores
Mechanochromic mechanophores function as molecular force sensors, signaling critical stress and strain in materials through a convenient visual output. The color changes that accompany activation of a mechanochromic mechanophore identify locations in a material that have experienced a critical level of force and may serve as a warning for impending catastrophic failure.39 Alternatively, these mechanophores have been leveraged to provide mechanistic insights into the chain scission events that typically characterize degradation of polymeric materials subjected to mechanical loads.29,40,41 The development of mechanophores that exhibit force dependent changes in color, or multicolor mechanochromism, represents a grand challenge for being able to not only locate, but remotely quantify the stress state of a material through simple visual analysis. We will revisit the concept of force dependent multicolor mechanochromism below, including progress that has been made toward achieving this goal enabled by naphthopyran mechanochemistry. This section is not intended to be comprehensive, but rather provide context and highlight the breadth of structures and strategies that exist for covalent mechanophores that complement naphthopyran mechanochemistry. For further reading, we refer to several recently published reviews.20–24A wide range of mechanochromic mechanophores have been developed that rely on many different reaction mechanisms and produce a variety of dyes, each with their own unique properties and limitations. The Otsuka group has developed a series of scissile mechanophores that generate a pair of stable, colored free radical species following force-induced bond homolysis,42–47 as exemplified by diarylbibenzothiophenone (DABBT) illustrated in Scheme 1.45 Mechanical dissociation is thermally reversible and the lifetime of the radical products depends strongly on the environment.42,44,47,48 In some cases, only partial recombination occurs.49 Polymers that display colors spanning the visible spectrum can be accessed by blending a small library of structurally distinct radical-type mechanophores that individually produce pink, green, or blue colored radicals upon mechanochemical activation.45 Mechanochemical retro-cycloaddition reactions, and in particular retro-[4 + 2] cycloadditions, have also been leveraged to design mechanochromic mechanophores useful for stress sensing. For example, Göstl and Sijbesma have developed mechanophores that produce π-extended anthracene derivatives through a mechanochemical retro-Diels–Alder reaction of anthracene–maleimide adducts (see Scheme 1).50 The extended π system of the anthracene products was cleverly employed to both enhance the fluorescence quantum yield and shift the absorption into the visible range. The color of the anthracene products can be adjusted by substitution of the extended π system with various arenes,41,51 although like the radical-type mechanophores, this requires the synthesis of discrete congeners and not simply peripheral substitution. Another notable and unique class of mechanophores has been pioneered by Xia and coworkers with the development of polyladderene mechanochemistry (Scheme 2).14,52,53 Upon mechanochemical activation, polyladderene unzips through a multistep radical process transforming the colorless insulating polymer into semiconducting polyacetylene with a deep purple color.14 While polyacetylene typically suffers from oxidative degradation, a fluorinated ladderene derivative was recently demonstrated to provide a significantly more stable conjugated product after mechanochemical activation.53
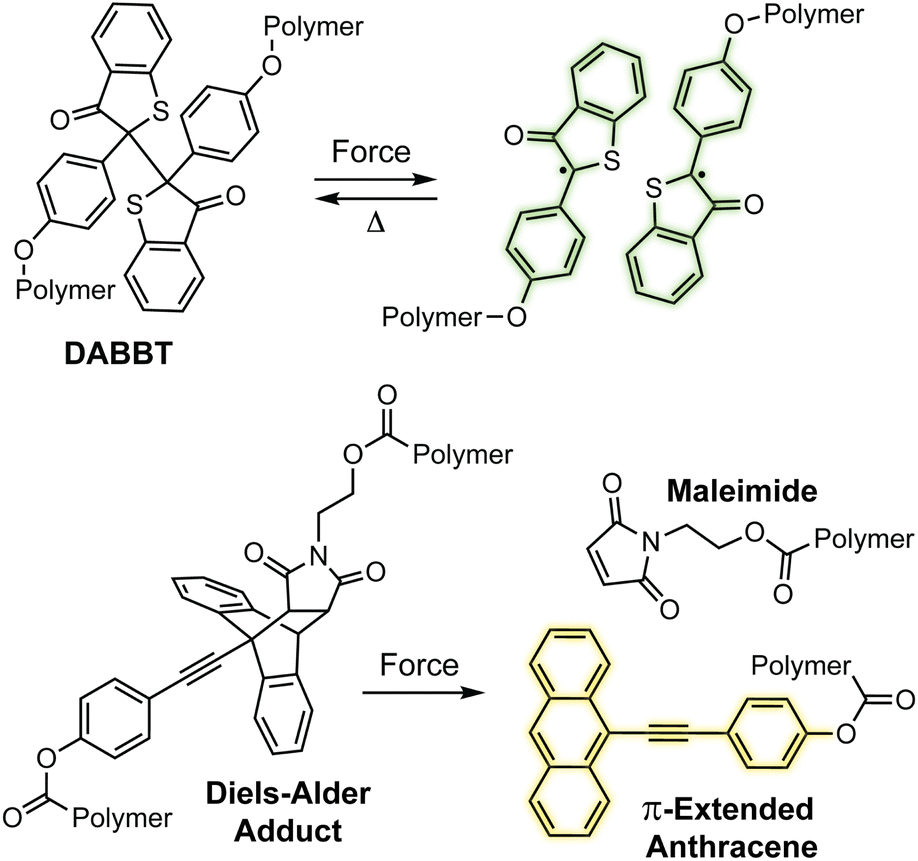 | ||
| Scheme 1 Examples of mechanochromic mechanophores that generate color upon mechanochemical activation via homolysis (top) or a retro-Diels–Alder reaction (bottom). | ||
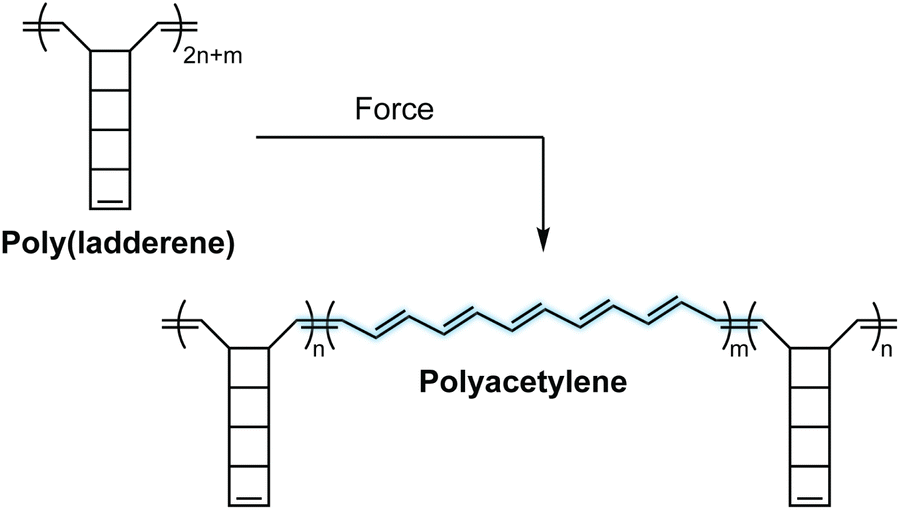 | ||
| Scheme 2 Poly(ladderene) undergoes a force-induced unzipping reaction to produce conjugated polyacetylene. | ||
Spiropyran is arguably the most ubiquitous mechanochromic mechanophore and also one of the most reactive (Scheme 3).54,55 One of the earliest papers in the field from Moore and coworkers in 2007 described the mechanochemical 6π electrocyclic ring-opening reaction of spiropyran leading to the generation of a highly colored merocyanine dye.56 Mechanochemical activation was achieved in solution upon ultrasonication of linear polymers incorporating spiropyran near the chain center where cavitation-induced elongational forces are maximized.3,57 In 2009, Sottos and coworkers demonstrated the mechanochemical activation of spiropyran in polymeric materials upon uniaxial tension and compression, establishing a new modality for stress sensing in polymers and opening the door to an entire domain of research innovation.39 Since these initial breakthroughs, many additional mechanochromic mechanophores—including naphthopyran—have been developed that rely on a mechanochemical ring-opening reaction to produce a highly colored conjugated dye such as spirothiopyran,58,59 rhodamine,60,61 and oxazine62,63 (Scheme 3). Unlike some of the other mechanophores described earlier, these mechanophores are typically non-scissile and thermally or photochemically reversible, enabling multiple cycles of mechanical activation in elastomeric materials. All of these mechanophores are also prominent photochromic compounds, establishing an interesting parallel between their photochemical and mechanochemical reactivity for which a mechanistic basis remains to be better understood. Unlike the quinoidal merocyanines typically derived from naphthopyrans, the mechanophores shown in Scheme 3 produce dyes that are usually zwitterionic. Furthermore, in addition to UV light and mechanical force, their ring-opening reactions are triggered by various other stimuli including heat, metal ions, electrical potential, pH changes, or polar environments, reducing the specificity of their stimuli-responsivity that can present practical challenges.64–66 For spiropyran, while synthetic modification can often prove challenging, certain substitution patterns have been shown to enhance selectivity for mechanochemical activation, but at the same time rendering a merocyanine product that is exceptionally thermally transient.67
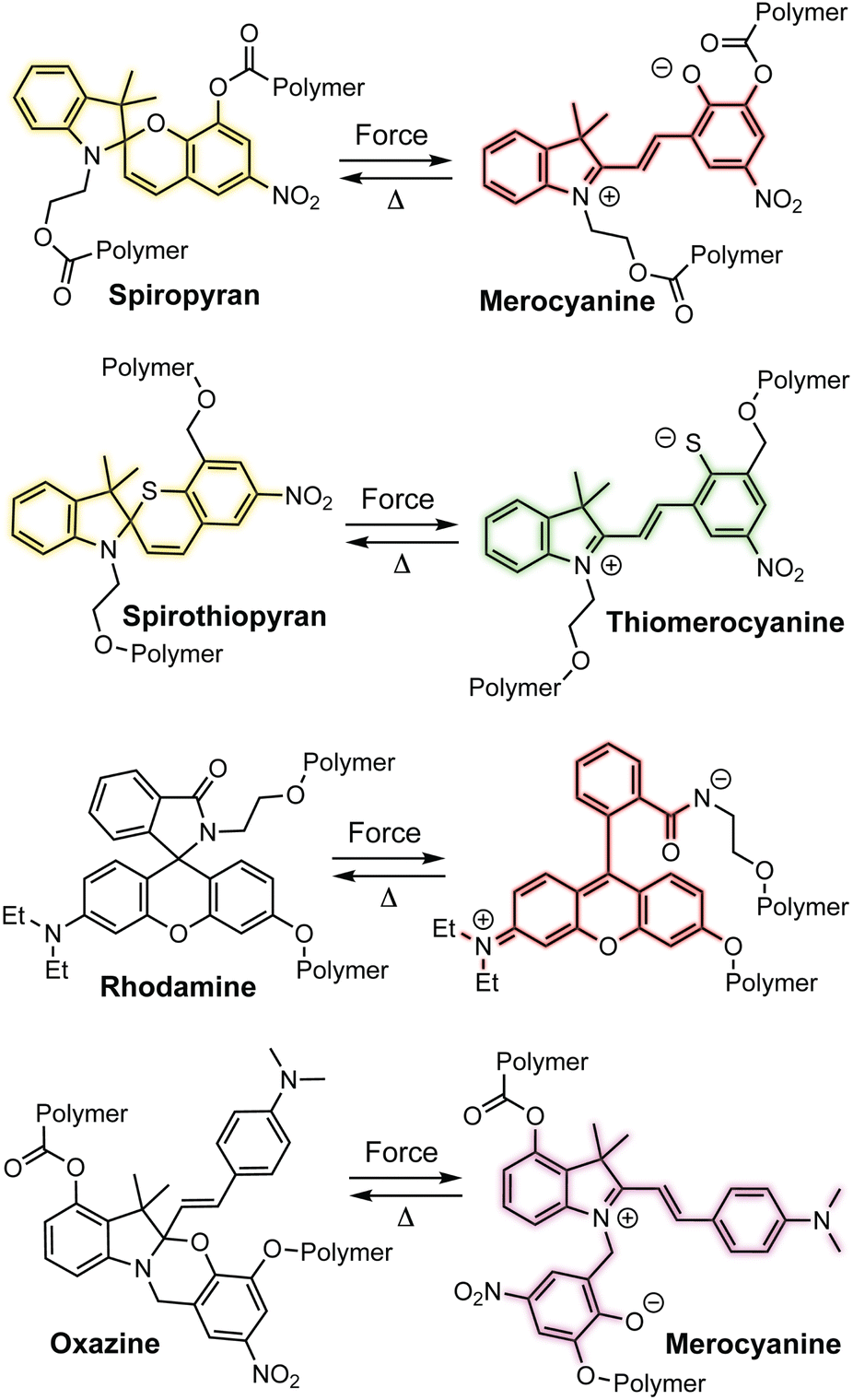 | ||
| Scheme 3 Examples of mechanochromic mechanophores that undergo ring-opening reactions upon mechanochemical activation to produce thermally reversible colored dyes. | ||
Although structurally related, naphthopyrans offer excellent fatigue resistance, stimulus specificity, synthetic modularity, and an extensive structure–property range, distinguishing them from spiropyran and related spiroheterocyclic mechanophores.36,37 Naphthopyrans and their corresponding merocyanine dyes typically exhibit a high degree of chemical stability as well as selective reactivity to mechanical force in the absence of UV light, making it an excellent scaffold for force sensing applications. Dye formation from the molecular switches shown in Scheme 3 is facilitated by a conjugated amine, whereby the pair of electrons on the nitrogen atom assists the ring-opening reaction and extends the length of the resulting conjugated π system.64,68 In contrast, naphthopyran-derived merocyanines are stabilized by extended conjugation afforded by the naphthalene core, which retains aromaticity in the quinoidal from.34,38,69 This feature significantly reduces the susceptibility of naphthopyran to spontaneous coloration, for example in polar environments, without sacrificing the stability of the merocyanine dye.70 We discuss the properties of naphthopyrans in more detail in the following sections.
1.3 Naphthopyran molecular switches
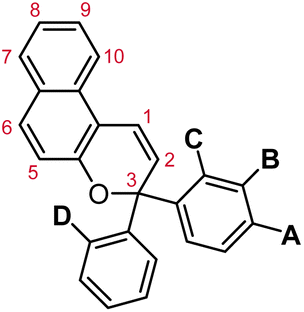
Naphthopyrans, or benzo-annulated benzopyrans (chromenes), are a class of molecular switches that undergo a reversible 6π electrocyclic ring-opening reaction upon photoirradiation with UV light or under extrinsic force, resulting in conversion of the colorless naphthopyran into a colored merocyanine dye (Scheme 4). There are three constitutional isomers of naphthopyran with differing position and orientation of the pyran ring about the naphthalene core (Fig. 2).69 However, only the angular 2H-naphtho[1,2-b]pyran and 3H-naphtho[2,1-b]pyran regioisomers display appreciable photochromic activity near room temperature. As a result, the linear 2H-naphthopyran congener has received comparatively little attention in the literature. We will typically refer to the two angular variants by their simpler familial names of 2H-naphthopyrans and 3H-naphthopyrans. As mentioned above, naphthopyran photoswitches have been widely developed commercially for use as reversible coloring agents in plastic ophthalmic lenses,36,37,71 with other notable applications in textiles,72 anti-counterfeiting measures,73 and cosmetics.74 Naphthopyrans have also been studied as conductivity switches,75–81 logic gates,82–84 molecular actuators,85 chemodosimeters,86 solar cell sensitizers,87 and artificial models of photo-excitable neurons.88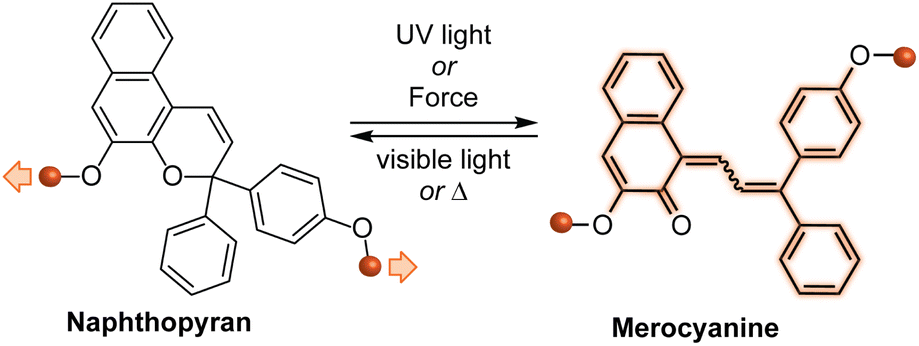 | ||
| Scheme 4 Ring-opening reaction of a naphthopyran molecular switch based on the 3H-naphtho[2,1-b]pyran skeleton to reversibly generate a colored merocyanine dye. | ||
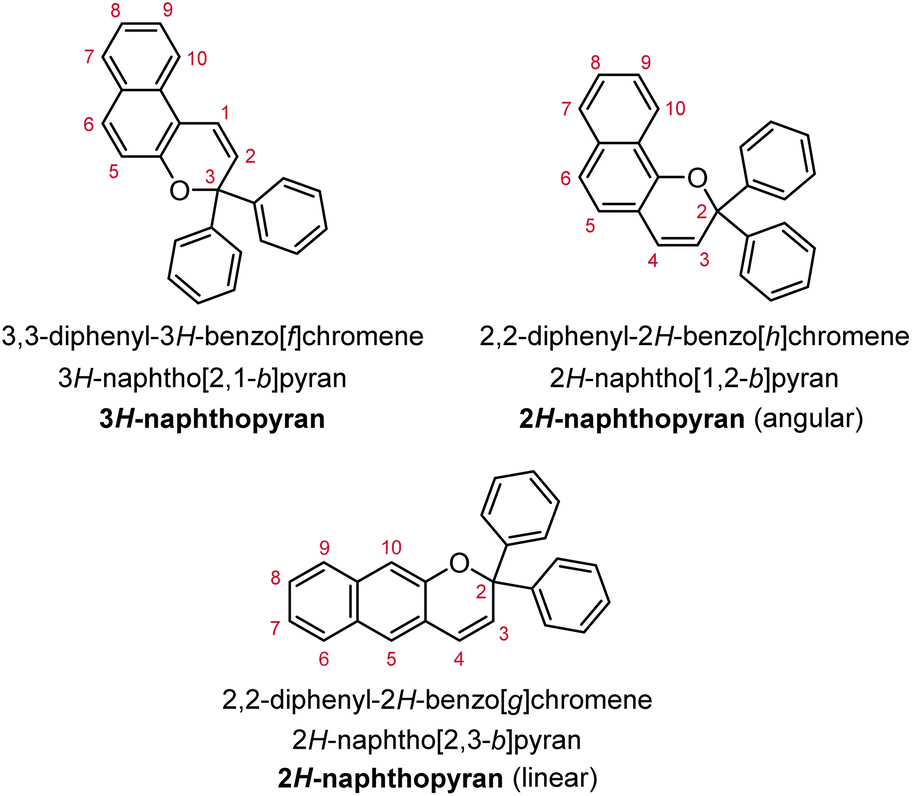 | ||
| Fig. 2 Molecular scaffolds depicting the three diaryl naphthopyran regioisomers with atomic positions identified. The different naming conventions are illustrated for each structure. | ||
The photochemistry of naphthopyran has been extensively studied since the photoinduced ring-opening reaction was first mentioned by Becker and Michl in 1966.34 Exactly 50 years later in 2016,35 it was discovered that the ring-opening reaction of naphthopyran can be accomplished with mechanical force applied across the labile C–O pyran bond. Naphthopyran has since emerged as a highly accessible and versatile mechanophore scaffold aided by the extensive structure–property relationships developed in the photochemical arena, which serve as a convenient and useful blueprint for the design of molecular force probes with an impressively broad range of properties. In a relatively short timeframe, research has illustrated that naphthopyran mechanophores can be scissile or non-scissile and generate merocyanine dyes with precisely tunable colors and lifetimes ranging from highly transient to permanent. Beyond the development of materials with a binary mechanochromic response, studies of naphthopyran have deepened insights into fundamental aspects of mechanochemical reactivity, elucidated reaction pathways that diverge significantly from the photochemical transformations, and expanded the mechanochromic toolbox toward the design of materials with multicolor mechanochromic behavior.
2 Synthesis of naphthopyrans
Naphthopyrans benefit from modular construction through straightforward synthetic routes in which structural diversity can be built relatively quickly and easily. Facile modification of the naphthopyran scaffold enables access to merocyanine dyes that display a wide range of properties with highly tunable color and lifetimes. Unless mentioned otherwise, all of the synthetic methods described below are applicable to the formation of both 2H-naphthopyrans and 3H-naphthopyrans.2.1 Acid-catalyzed reactions of propargyl alcohols and naphthols
The most straightforward and robust method of synthesizing naphthopyrans relies on the thermal rearrangement of naphthyl propargyl ethers (Scheme 5).89 These substrates are typically generated in situ via the condensation of a naphthol with a propargyl alcohol under acidic conditions. An ideal catalyst for this transformation is para-toluenesulfonic acid (TsOH) due to its solubility in organic solvents.75,76,90–95 Heterogeneous catalysis using acidic alumina is also exceedingly common, enabling efficient syntheses on large scale.35,96–101 The mechanism follows initial protonation of the propargyl alcohol and loss of water to generate a propargyl cation that is attacked by the nucleophilic naphthol. Next, a Claisen rearrangement and subsequent enolization generates the intermediate allenyl naphthol species, which undergoes a 1,5-hydrogen shift to produce the merocyanine form of the naphthopyran. Finally, a thermal 6π electrocyclization closes the ring to form the colorless naphthopyran.102 Notably, a wide range of substituents on both the naphthol and propargyl alcohol coupling partners are tolerated due to the relatively mild conditions. However, because the reaction mechanism proceeds via the intermediacy of the merocyanine, substituents that strongly stabilize the merocyanine form can decrease the yield of the naphthopyran.103 In our experience, photoirradiation with visible light helps to facilitate the final ring-closure in some cases.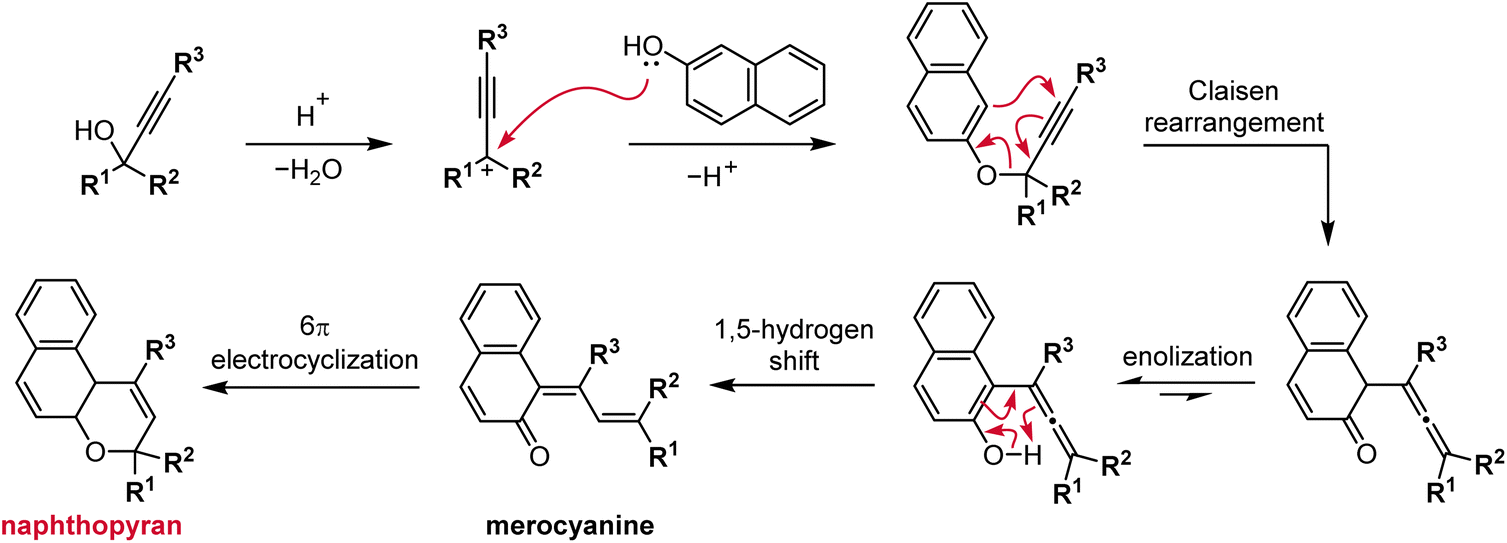 | ||
| Scheme 5 Typical synthesis of naphthopyrans via acid-catalyzed condensation of naphthols and diaryl propargyl alcohols that proceeds through a multistep pathway. The synthesis of a 3H-naphtho[2,1-b]pyran is illustrated. | ||
The propargyl alcohol and naphthol building blocks are either commercially abundant, or readily prepared and/or modified using robust chemistry, which reinforces the utility of this synthetic approach. Many inexpensive 1-naphthols and 2-naphthols are commercially available. The propargyl alcohol coupling partners can often be acquired from commercial sources or efficiently synthesized by treating the appropriate benzophenone with an alkali metal acetylide.75,99,104,105 More specialized benzophenones can also frequently be prepared in one step by Friedel–Crafts acylation of an arene with an acyl halide. The modularity of this strategy has enabled the construction of diverse libraries of naphthopyran derivatives displaying a wide range of substitution patterns, both on the naphthalene core and on the peripheral aryl groups, from a relatively small collection of simple yet highly customizable building blocks.
While generally robust, this synthetic route can involve the formation of undesired side products. If naphthol condensation does not occur rapidly after acidic dehydration, interception of the propargyl carbocation by adventitious water results in Meyer–Schuster rearrangement to produce a terminal α,β-unsaturated carbonyl compound (Scheme 6, blue arrows).106 Electron-rich diaryl propargyl alcohols form relatively stable carbocations and are particularly susceptible to this rearrangement side reaction.99 Additionally, the synthesis of 2H-naphthopyrans is complicated by the nucleophilicity at the 4-position para to the hydroxyl group. Nucleophilic attack at this carbon center instead of the oxygen produces a deeply colored dye that exhibits no photoswitching activity (Scheme 6, red arrows).99,107 The yield of this side product increases when particularly electron-rich propargyl alcohols are employed or if the nucleophilicity of the naphthol is diminished due to steric or electronic effects.
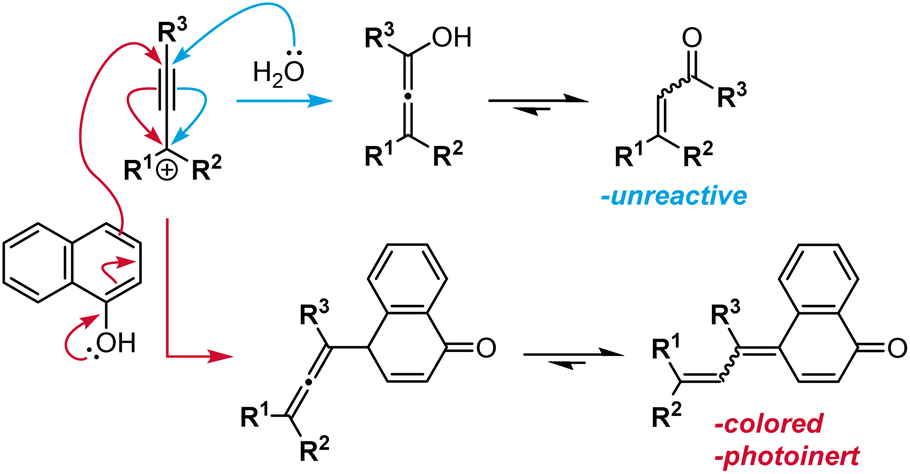 | ||
| Scheme 6 Side reactions encountered in the synthesis of naphthopyrans via the acid-catalyzed route including the Meyer–Schuster rearrangement (top pathway, blue arrows) and conjugate addition to the propargyl cation at the 4-position of the 1-naphthol (bottom pathway, red arrows). | ||
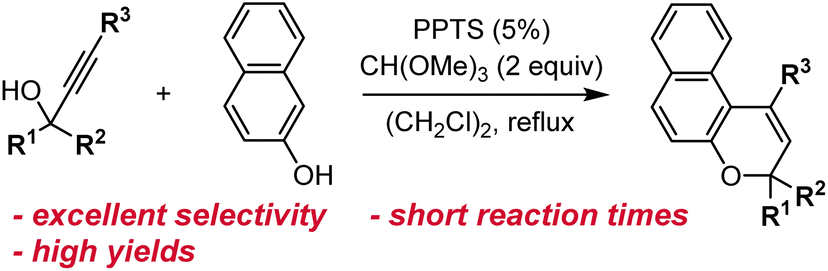 | ||
| Scheme 7 Reaction conditions for the acid-catalyzed synthesis of naphthopyrans developed by Zhao and Carreira.108 | ||
2.2 Alternative synthetic routes
Other methods have historically been used to synthesize substituted naphthopyrans, including Grignard addition to unsubstituted benzocoumarins,121–124 reduction of substituted naphthopyranones,124,125 and addition of a lithiated naphthol to cinnamaldehyde derivatives.124,126,127 However, these routes involve the use of harsh reagents, which limit the scope of accessible naphthopyrans, or suffer from low yields and have largely been rendered obsolete by improved protocols like the ones discussed above. Additionally, the starting materials are often not commercially available and require multistep syntheses. Although there may be niche applications for which these alternative methods prove useful, they can generally be regarded as inferior to the more modular, functional group tolerant, and straightforward synthetic approaches discussed above.One notable exception is a protocol involving the treatment of a naphthol with a substituted cinnamaldehyde derivative in the presence of titanium tetraethoxide, affording the naphthopyran in one operational step (Scheme 8).90,96,124,128 The mechanism involves displacement of one of the ethoxide ligands on titanium by the naphthol and coordination of the cinnamaldehyde, which is followed by a pericyclic rearrangement and elimination of the titanium alkoxide to produce the merocyanine. Thermal electrocyclization then produces the naphthopyran similar to the mechanism in the acid-catalyzed route. This reaction is remarkably robust with broad functional group tolerance and has proven to be more efficient than the acid-catalyzed conditions for some heterocycle-fused naphthopyrans and related chromenes.90,129
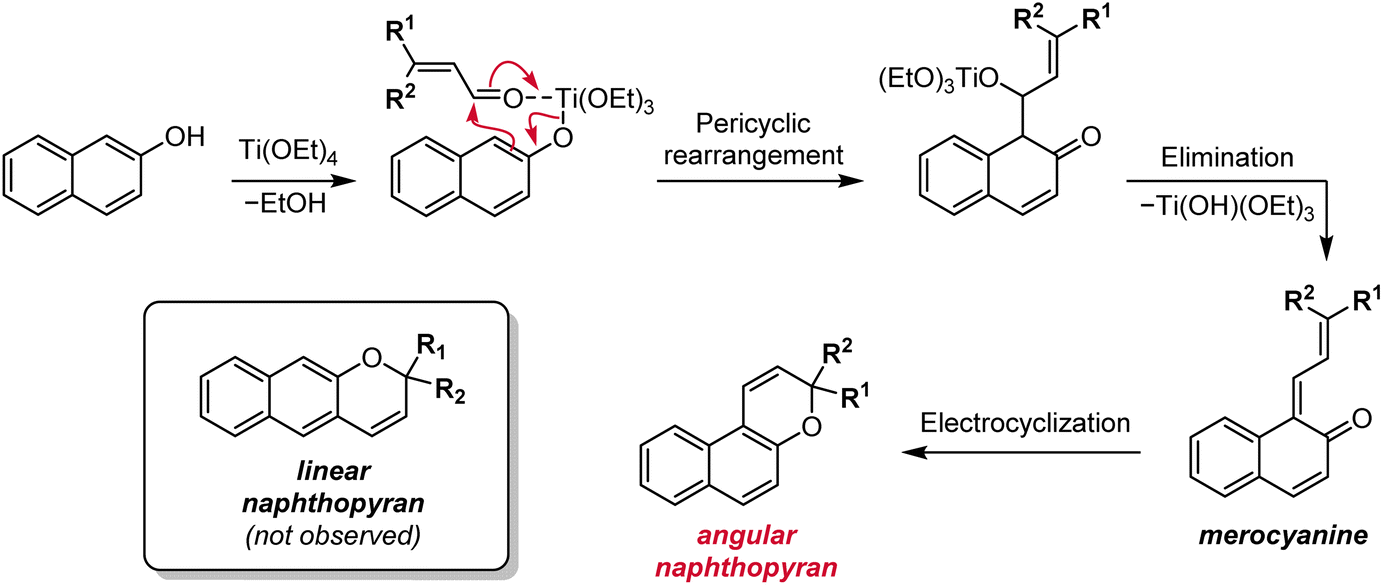 | ||
| Scheme 8 Alternative titanium-mediated synthesis of angular naphthopyrans from naphthols and substituted cinnamaldehydes. | ||
Other methods for synthesizing naphthopyrans have also been developed including those that use environmentally benign solvents like water or solid-state reactions that eliminate solvents altogether. Naphthopyrans were attained in good yields by heating an aqueous mixture of propargyl alcohol and naphthol starting materials with catalytic β-cyclodextrin hydrate.130 In a solid state protocol, grinding a mixture of propargyl alcohol, naphthol, TsOH, and silica gel at room temperature with a mortar and pestle produced the corresponding naphthopyrans, albeit in poor to moderate yields (Scheme 9).109 Substantial improvements were achieved using ball-milling and catalytic InCl3 leading to naphthopyran formation in excellent yields with minimal side products.131 Notably, the exclusion of a Brønsted acid catalyst lends this method to the construction of naphthopyrans containing acid-sensitive functional groups. The authors also demonstrated the solid state synthesis of a wide variety of propargyl alcohol starting materials in very good yields by ball-milling a terminal alkyne and a benzophenone in the presence of KOH and 18-crown-6, circumventing the use of potentially hazardous pyrophoric alkali metal acetylide species.131
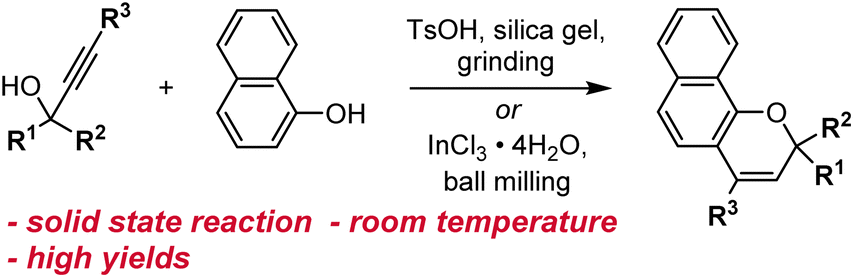 | ||
| Scheme 9 Solvent-free reaction conditions for the synthesis of naphthopyrans via grinding or ball-milling. | ||
2.3 Post-synthetic modification of naphthopyrans
Naphthopyrans generally tolerate a wide range of reaction conditions that include acids or bases,94,132,133 strong nucleophiles94,134 or electrophiles,134,135 organolithium species,133 and reducing agents.136 Thus, naphthopyran libraries can also be constructed via a divergent synthetic strategy in which a naphthopyran substrate is derivatized at a later stage in the synthesis. The most versatile of these strategies are those in which halide or triflate groups on the naphthopyran core or peripheral aryl rings serve as functional handles for cross-coupling reactions. Notably, Suzuki,79,94,137,138 Sonogashira,139 and Buchwald–Hartwig87,140 couplings have been used to install aryl/vinyl, alkynyl, or amine substituents, respectively. Further diversity is afforded by copper catalyzed Ullman-type132 or Grignard coupling.94 Appropriate cross-coupling handles have also been installed via selective halogenation137 or lithium–halogen exchange reactions.133The robustness of naphthopyran also enables its covalent incorporation into polymers necessary for the construction of mechanochromic materials using a wide variety of synthetic approaches. Naphthopyrans are generally tolerant of organic radicals, conveniently allowing naphthopyran-based initiators and monomers to be used in the synthesis of polymers via common radical polymerization techniques.33,134,135,138,141–144 Additionally, naphthopyrans have been covalently incorporated into siloxane networks via Pt-catalyzed hydrosilylation reactions,9,35,143–146 and in organic/inorganic composites via a sol–gel method using a Sn catalyst.134 Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reactions have also been successfully employed for the preparation of naphthopyran-containing polymers.134,147
3 Structure–activity relationships for naphthopyran-derived merocyanine dyes
Becker and Michl first alluded to the photochromism of naphthopyrans in 1966 in a seminal report on photochromic benzopyrans (chromenes),34 with formal structural and experimental details for naphthopyran provided in a subsequent patent.148 Whereas the benzene ring of benzopyrans undergoes complete loss of aromaticity in its merocyanine form, the naphthalene core maintains partial aromaticity in the ring-opened state of the analogous naphthopyran. This additional stabilization enables naphthopyrans to produce comparatively intense coloration with significantly improved fatigue resistance.34,38,69 In contrast to closely related spiropyran,149 naphthopyrans exhibit little to no thermochromism37,69 because the thermal barrier of the ring-opening reaction96,150 is significantly greater than that of the reverse ring-closing reaction.151,152 These properties, combined with its synthetic accessibility and modularity, have made naphthopyran a particularly robust and practical photoswitch for a wide range of applications. Given its industrial relevance, studies of naphthopyran photochemistry are abundant with numerous detailed investigations of merocyanine structure–property relationships toward the development of naphthopyrans as commercial photochromic switches.36,37,71 Since the same merocyanine dyes are typically produced upon mechanochemical activation of naphthopyran, the structure–property relationships established in the photochemical literature serve as an invaluable resource for designing mechanochromic mechanophores with predictable and highly tunable absorption and reversion properties.143,145 Furthermore, mechanistic studies of photochemically and thermally mediated naphthopyran ring-opening reactions (vide infra) are convenient points of comparison to the closely related, but also often divergent mechanically mediated processes, deepening fundamental insights into the nature of mechanical force-driven chemical transformations. In the next section, we will highlight some key developments in naphthopyran photochemistry and prominent structure–property relationships that have proved useful in the design of mechanochromic mechanophores. More exhaustive reviews dedicated to the topic are available for the interested reader.36–38,69,1533.1 Photochemical ring-opening and alkene isomerization reactions
The photochemical 6π electrocyclic ring-opening reaction of naphthopyran and possible olefin photoisomerization reactions generate several different stereoisomers of the merocyanine dye (Scheme 10).69,151,152,154–156 Rotation about the C–C single bond connecting the two newly formed alkenes from a cisoid to transoid conformation occurs rapidly to reduce steric interactions between the aryl substituents and the naphthalenone core. While the cis-cisoid form is a frequently predicted intermediate in computational studies150,157,158 and has been observed by transient spectroscopy for benzopyran,159 this species has not been experimentally observed for naphthopyrans.150,156 This experimental observation is consistent with the low barrier for bond rotation of ∼5 kcal mol−1 predicted computationally.150 The resulting cis-transoid (CT) species, wherein the exocyclic alkene is in the cis configuration, can revert back to the colorless naphthopyran thermally or with visible light irradiation via the cis-cisoid intermediate. Alternatively, cis-to-trans isomerization of this exocyclic alkene occurs with further UV irradiation, converting the CT form to the much more thermally stable trans-transoid (TT) merocyanine. While thermal trans-to-cis isomerization does occur, photochemical isomerization of the exocyclic alkene with visible light is significantly more efficient.93,150,152,155,160 We note that although the cis-transoid (CT) and trans-transoid (TT) or similar nomenclature is used broadly in the naphthopyran literature, later we will frequently refer to these merocyanine isomers simply by the configuration of the exocyclic alkene as being either cis or trans, respectively, because the cisoid and transoid descriptors are of little practical relevance. Evidence that naphthopyran derived merocyanine dyes assume a charge-neutral quinoidal form in the ground state is derived from NMR data,103,154,161 X-ray crystallography (for especially thermally stable species),103,117,162 and solvatochromic behavior.90,151,162–164 These merocyanines exhibit positive solvatochromism, or bathochromic shifts in λmax with increasing solvent polarity, reflecting a weakly polar ground state with minimal charge delocalization that is characteristic of a quinoidal structure.160,163,165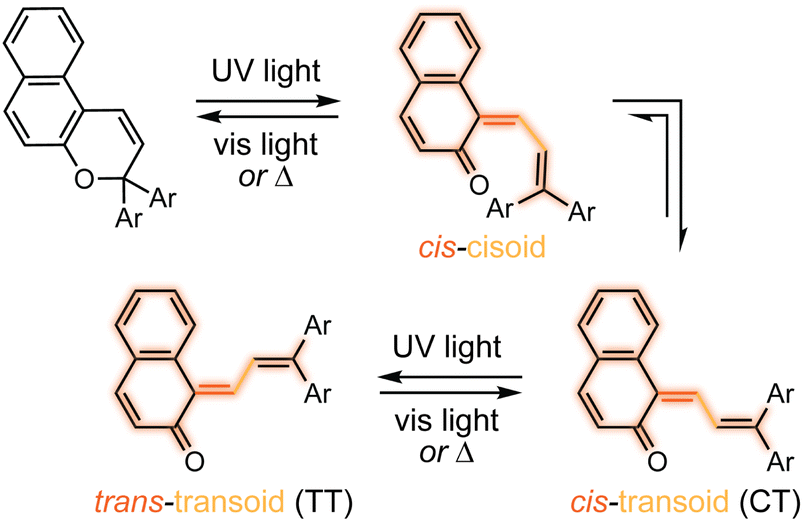 | ||
| Scheme 10 Exemplary photochemical ring-opening reaction of a 3H-naphthopyran and alkene isomerization to generate principal cis-transoid (CT) and trans-transoid (TT) isomeric merocyanine products. | ||
Merocyanine stereoisomers with different stereochemical configuration of the exocyclic alkene (i.e., CT and TT forms) exhibit modest differences in absorption with values of λmax typically within ∼10 nm.103,116,120,151,166 However, comparisons of the extinction coefficients between the CT and TT forms are often complicated by short lifetimes that render isolation difficult. In the case of 3,3-diphenyl-3H-naphtho[2,1-b]pyran, the extinction coefficients of the CT and TT isomers were determined to be 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 and 16900 M−1 cm−1 in toluene, respectively, after careful isolation of each species by low temperature chromatography.116 Extinction coefficients have been determined in other cases based on structural isolation via low temperature chromatography120,167 or with especially stable merocyanines,103,117,162,167 or estimated by fitting experimental absorption profiles to kinetic models117,152,168–171 or with computational simulations.103,117,172 Notably, determination of extinction coefficients for the TT stereoisomer is more prone to error because this isomer typically comprises only a small fraction of the photochemically generated merocyanine mixture.152 Given the challenges in characterizing individual stereoisomers, these species are often assumed to have a similar extinction coefficient to simplify kinetic analysis.80,94,118,162,173 To our knowledge, the photophysical changes between stereoisomers with varying configuration at the distal olefin for merocyanines containing dissimilar peripheral aryl groups have not been determined, but these are likely even less pronounced than the relatively subtle differences in absorption arising from isomerization of the exocyclic alkene.
100 and 16900 M−1 cm−1 in toluene, respectively, after careful isolation of each species by low temperature chromatography.116 Extinction coefficients have been determined in other cases based on structural isolation via low temperature chromatography120,167 or with especially stable merocyanines,103,117,162,167 or estimated by fitting experimental absorption profiles to kinetic models117,152,168–171 or with computational simulations.103,117,172 Notably, determination of extinction coefficients for the TT stereoisomer is more prone to error because this isomer typically comprises only a small fraction of the photochemically generated merocyanine mixture.152 Given the challenges in characterizing individual stereoisomers, these species are often assumed to have a similar extinction coefficient to simplify kinetic analysis.80,94,118,162,173 To our knowledge, the photophysical changes between stereoisomers with varying configuration at the distal olefin for merocyanines containing dissimilar peripheral aryl groups have not been determined, but these are likely even less pronounced than the relatively subtle differences in absorption arising from isomerization of the exocyclic alkene.
Careful analysis of reaction kinetics using UV-vis absorption spectroscopy has elucidated information about the photochemical reaction mechanism, as well as the quantum yields and extinction coefficients for naphthopyrans and the corresponding merocyanines. However, photochemical activation produces complex mixtures of several isomeric species,150,174 including several with overlapping absorption spectra. Analytical175 or even numerical80,115,133,171,176 solutions to such kinetic problems are complicated, and often assumptions must be introduced to simplify analysis.115,171,176 Mechanistic studies of naphthopyran photochemistry are dramatically simplified by monitoring reaction progress using NMR spectroscopy, which allows the evolution of each isomeric species to be monitored separately. Particularly elegant studies have employed fluorinated naphthopyrans to track each individual species using 19F NMR spectroscopy with excellent resolution.115,133,154,174,177–180 These studies support that UV photoirradiation of typical naphthopyrans results first in the nearly instantaneous formation of the CT merocyanine isomer; additional irradiation with UV light causes cis-to-trans isomerization of the exocyclic alkene to generate the TT merocyanine isomer.81,181
Greater mechanistic detail for the photochemical ring-opening reaction of naphthopyran has been revealed using transient spectroscopy techniques and supported by computation. Here, we briefly discuss findings for the simple photoswitch 3,3-diphenyl-3H-naphtho[2,1-b]pyran (Ar = Ph in Scheme 10). It should be noted that while changes in solvent and temperature alter the lifetimes of the individual states,160 the overall photochemical pathway is largely unaffected by solvent polarity,151,156 which is a notable distinction from closely related spiropyran.68,70,149 Photochemical ring-opening reactions of naphthopyran proceed via a singlet excited state that has a picosecond lifetime and is structurally similar to the ring-closed naphthopyran (Fig. 3).150,156,182–185 Transition to the ground state proceeds over an insignificant activation barrier (∼1 kcal mol−1) and through a conical intersection, at which point scission of the pyran C–O bond occurs to produce the CT merocyanine isomer in a concerted process.150,156 The essentially barrierless excited state reaction proceeds so rapidly that it competes even with vibrational relaxation,156,158,186–189 resulting in mode-dependent behavior wherein the reaction proceeds from any of several distinct vibronic states accessed from excitation sources of distinct wavelengths.190 While the quantum yield of the ring-opening reaction is typically high (∼0.8),150–152 a fraction of the excited naphthopyran species undergoes internal conversion back to the ground state.156
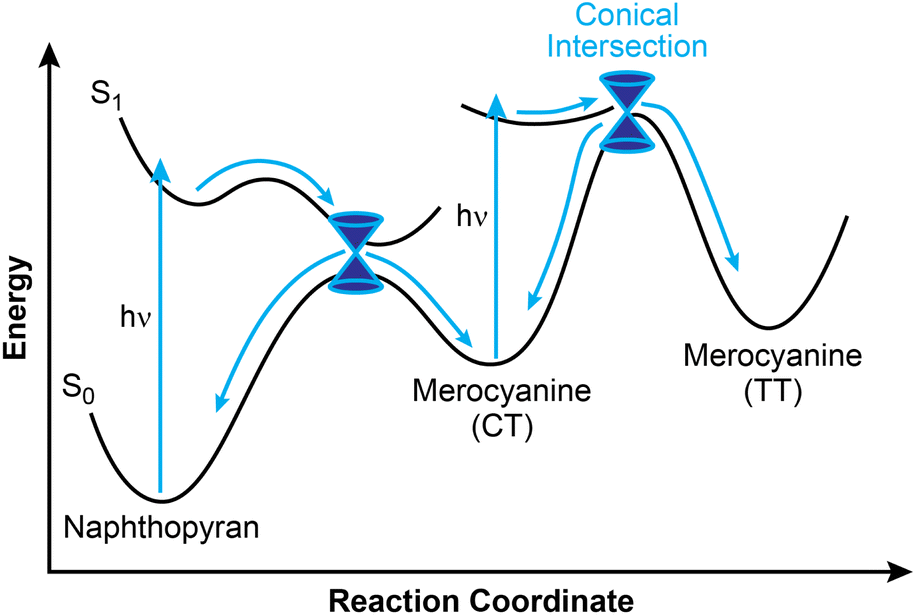 | ||
| Fig. 3 Representative reaction pathway for the photochemical ring-opening reaction of naphthopyran. | ||
The cis-to-trans isomerization of the exocyclic alkene to generate the TT isomer also occurs through a singlet excited state and transition to the ground state through a conical intersection, but only after a second photon absorption, rather than directly from the excited state of naphthopyran.150,156,166 Consistent with observations from NMR spectroscopy noted above, this assertion is further supported by theoretical modeling as well as flash photolysis experiments demonstrating minimal formation of the TT merocyanine isomer after brief exposure to UV light.80,155,191 Theoretical modeling suggests that isomerization proceeds through either a single twist about the exocyclic olefin, or through a “bicycle pedal” motion consisting of concerted rotation about both olefins of the merocyanine bridge.192 While the single twist pathway is favored for 3,3-diphenyl-3H-naphtho[2,1-b]pyran, the concerted dual-isomerization pathway is promoted in polar solvents or with certain electronic substituents. Excitation of the CT isomer can also lead to photoenolization to form the relatively unstable allenyl naphthol species.150,166,174,179 Notably, however, internal conversion to the ground state is extremely rapid for the excited CT merocyanine isomer, resulting in inefficient photoisomerization to either the TT isomer or the allene, consistent with the typically limited accumulation of these species observed experimentally.166
Whereas spectroscopic and computational investigations150,156,158 support a rapid and concerted electrocyclic ring-opening pathway upon photochemical activation of naphthopyran, there is evidence that this mechanism is not conserved when other stimuli are used. While inefficient, the thermal ring-opening reaction has been experimentally investigated via the racemization of chiral naphthopyrans.96,193 Both solvent and substituent effects on the thermal activation barrier indicate a polar, heterolytic ring-opening mechanism in direct contrast to the transient absorption studies performed on the photochemical transformations of naphthopyran156 and closely related benzopyran,159 which again reveal no evidence of a zwitterionic intermediate.
3.2 Thermal reversion of naphthopyran-derived merocyanine dyes
Thermal transformations play an important role in naphthopyran–merocyanine equilibria. As noted above, the ring-opening reaction of naphthopyran is thermally accessible, but the process is extremely inefficient. Experimental96 and computational150,157,194–196 studies indicate that the thermal activation barrier for ring opening is ∼25–30 kcal mol−1. By comparison, the barrier to ring closure from the CT merocyanine isomer has been estimated to be in the range 11–21 kcal mol−1.150–152,195 Thus, thermal reversion of the merocyanine dye is comparatively rapid such that the accumulation of colored products (i.e. thermochromism) is not typically observed. Therefore, electrocyclization of the merocyanine151 is the most kinetically relevant thermally mediated process in the context of naphthopyran chemistry. The rate of thermal reversion depends strongly on the regiochemistry and geometry of the merocyanine, which will be the focus of this section.As discussed earlier, the stereochemical configuration of the exocyclic alkene significantly impacts the kinetic stability of the merocyanine and therefore its tendency to revert to the ring-closed naphthopyran form. Cis and trans merocyanines exhibit similar thermodynamic stability, ∼10–14 kcal mol−1 higher in energy than the naphthopyran form.117,150 However, electrocyclization occurs exclusively from the cis merocyanine isomers, necessitating trans-to-cis isomerization of the exocyclic alkene prior to ring-closure.152,155,160,181 While trans-to-cis isomerization of the exocyclic alkene is possible thermally, this process is impeded by a substantial barrier of ∼28 kcal mol−1 (ref. 150) and thus proceeds significantly slower than the thermal ring-closing reaction of cis merocyanines.80,93,118,133,151,152,154,155,197 In some cases, trans merocyanines have even proven stable enough to isolate and characterize under ambient conditions.93,103,117,160,162,178 Isomerization of the exocyclic alkene occurs much more efficiently upon irradiation with visible light as discussed earlier. Therefore, while naphthopyrans are generally considered to be T-type (i.e. thermally reversible) photoswitches,36,37,71 P-type (i.e. photochemically reversible) behavior is observed for some species if extended exposure to UV light generates an appreciable amount of thermally persistent trans merocyanine. Thermal cis-to-trans isomerization of the exocyclic olefin is uncommon and has only been observed in exceptional cases.133
Reversion kinetics for mixtures of merocyanine stereoisomers are reliably determined using multiple methods, but most often by fitting time-dependent absorption data to a biexponential decay function.94,112,114,115,118,152,154 The smaller rate constant obtained is attributed to conversion of the trans stereoisomer. Thus, if little to no trans merocyanine isomer is present, fitting with a monoexponential decay function is sufficient. The enthalpy and entropy of activation for isomerization and ring-closure for several merocyanine derivatives has also been determined by Eyring-type analysis of reversion kinetics using both UV-vis absorption and NMR spectroscopy methods.133,151,171,198,199 The rates of naphthopyran formation typically differ by several orders of magnitude from cis and trans merocyanine dyes with different stereochemical configurations of the exocyclic alkene.80,118,133,150,152,154,155,168,181,197 In contrast, the rates of thermal ring-closure have been shown to vary only modestly for merocyanine isomers with different configuration of the distal disubstituted olefin.81,115,200
Naphthopyrans and their corresponding merocyanines are somewhat susceptible to environmental effects, although to a significantly lesser degree than other molecular switches like spiropyrans. Polar solvents destabilize the quinoidal merocyanine and increase the rate of thermal reversion,90,163,164,201,202 although the activation barrier for this process is typically invariant across solvents.90,151,202 Interestingly, the thermal ring-opening reaction of naphthopyran, which again is thought to proceed through a polar, heterolytic mechanism,96 is also faster in polar solvents that may facilitate polarization of the pyran C–O bond.90,164 Nevertheless, thermochromism is extremely weak. Relatively viscous solvents have also been found to slow the rate of thermal merocyanine reversion via interactions with the alkylidene bridge.124,203,204 Similarly, the rate of thermal merocyanine reversion is slower in polymeric matrices36,38,112,114,145,150,161,199 and ceramic coatings205,206 in which conformational freedom is restricted. The photochromism of naphthopyran is more efficient in polymeric materials with low glass transition temperatures and flexural moduli compared to rigid or glassy matrices.36 However, naphthopyrans do exhibit photochromism in the crystalline state, which is rare for photochromic compounds.162,199 The resulting merocyanines are thermally stable, but revert to the colorless ring-closed naphthopyran upon irradiation with visible light.
The thermal reversion behavior of merocyanine dyes derived from 2H-naphthopyrans and 3H-naphthopyrans is markedly different. Merocyanines produced from 2H-naphthopyrans are typically more stable and longer-lived than those arising from 3H-naphthopyrans due to reduced steric crowding in both the cis and trans stereoisomers (Scheme 11).207 As a result of the increased steric demand, merocyanines derived from 3H-naphthopyran typically recyclize at rates roughly two orders of magnitude faster than those derived from 2H-naphthopyrans.90,152,155 The reduced steric interactions in merocyanines derived from 2H-naphthopyrans also results in greater planarization of the diene portion of the chromophore resulting in better conjugation with the quinone and visible absorption features that are bathochromically shifted compared to merocyanine dyes generated from analogous 3H-naphthopyrans (Fig. 4).208 Merocyanines generated from 3H-naphthopyrans typically display only one broad absorption peak in the visible region, whereas 2H-naphthopyrans produce merocyanine dyes that exhibit two distinct visible absorption features. The more intense absorption peak is commonly bathochromically shifted by approximately 45 nm compared to that of similarly substituted 3H-naphthopyrans.208 The merocyanine dyes generated from 3H-naphthopyrans range from red, orange, yellow, blue, or purple depending on substitution. While 2H-naphthopyran-derived merocyanines span a similar color palette, their unique absorption properties also enable access to commercially desirable neutral hues including brown and grey.37,209–212 We turn our attention to the relationships between merocyanine structure, absorption, and lifetime in the next section.
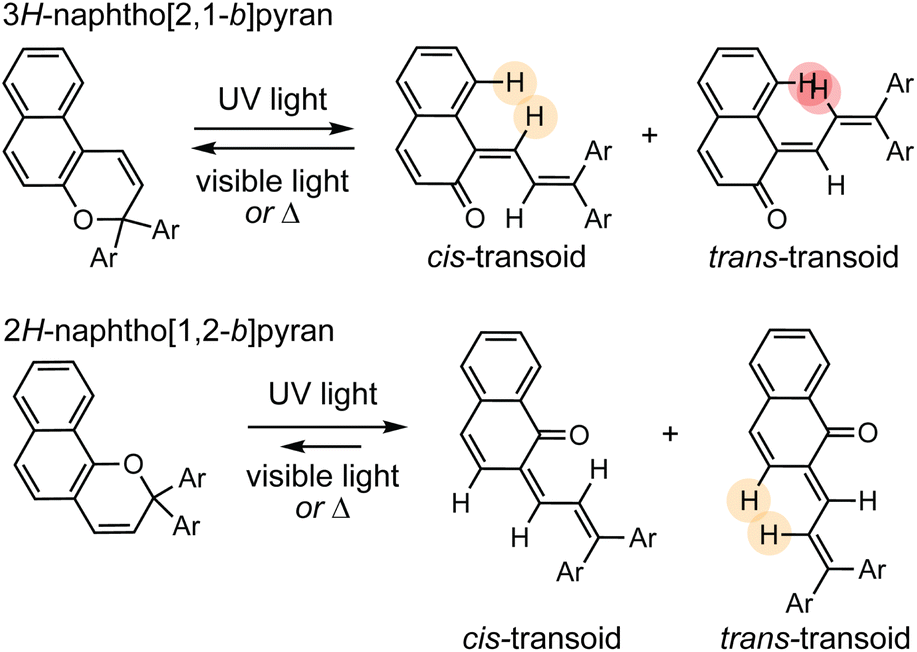 | ||
| Scheme 11 The ring-opening reactions of 3H- and 2H-naphthopyrans generate merocyanine isomers with varying degrees of steric demand, which strongly influences absorption properties and reversion kinetics. | ||
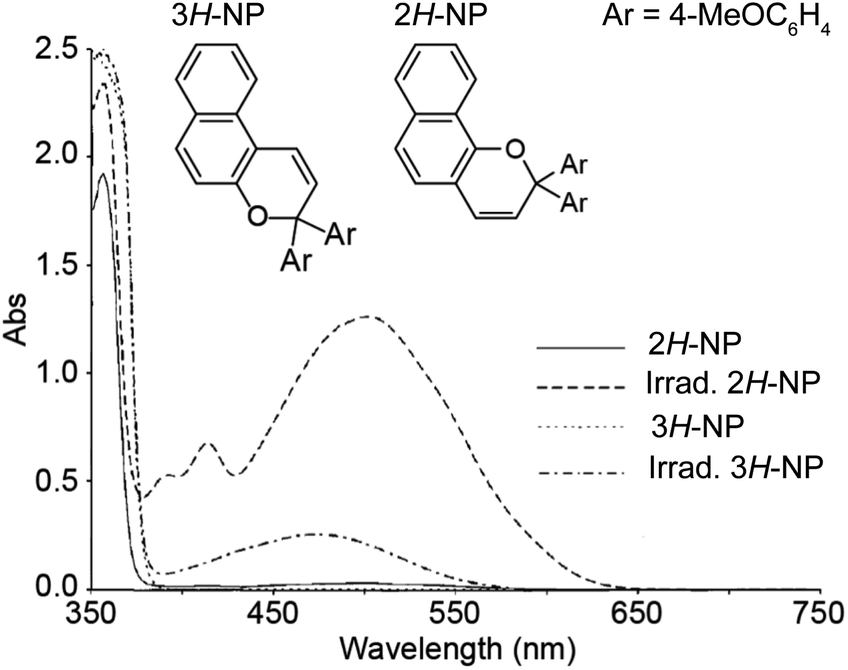 | ||
| Fig. 4 UV-vis absorption spectra of 2,2-bis(4-methoxyphenyl)-2H-naphtho[1,2-b]pyran (2H-NP) and 3,3-bis(4-methoxyphenyl)-3H-naphtho[2,1-b]pyran (3H-NP) in toluene before and after irradiation with UV light. Adapted with permission from ref. 38. Copyright 2006, Elsevier. | ||
3.3 Structural features affecting merocyanine color, intensity, and lifetime
Commercial interest in the development of naphthopyran derivatives as photoswitches for use in ophthalmic lenses has culminated in a diverse and extensive library of structures producing merocyanine dyes that exhibit a wide range of colors and thermal reversion behavior (Fig. 5).36,37,71 In the following sections, we highlight some of the most important structure–property relationships for the naphthopyran–merocyanine system that serves as a convenient and useful reference in the context of designing mechanochromic materials. | ||
| Fig. 5 Naphthopyrans have been developed extensively as UV light-responsive coloring (i.e., photochromic) agents in plastic ophthalmic lenses. | ||
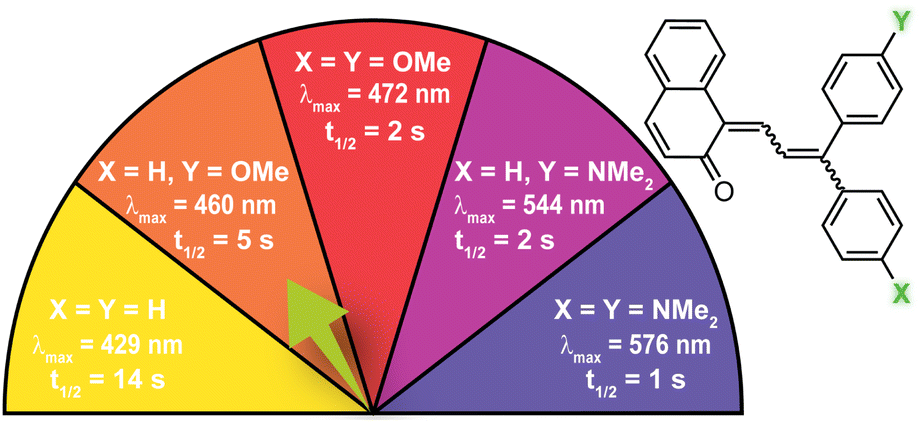 | ||
| Fig. 6 Electron-donating para-substituents result in bathochromically shifted (longer wavelength) merocyanine absorption and increase the rate of thermal reversion. Values shown were measured in toluene at 20 °C.37 | ||
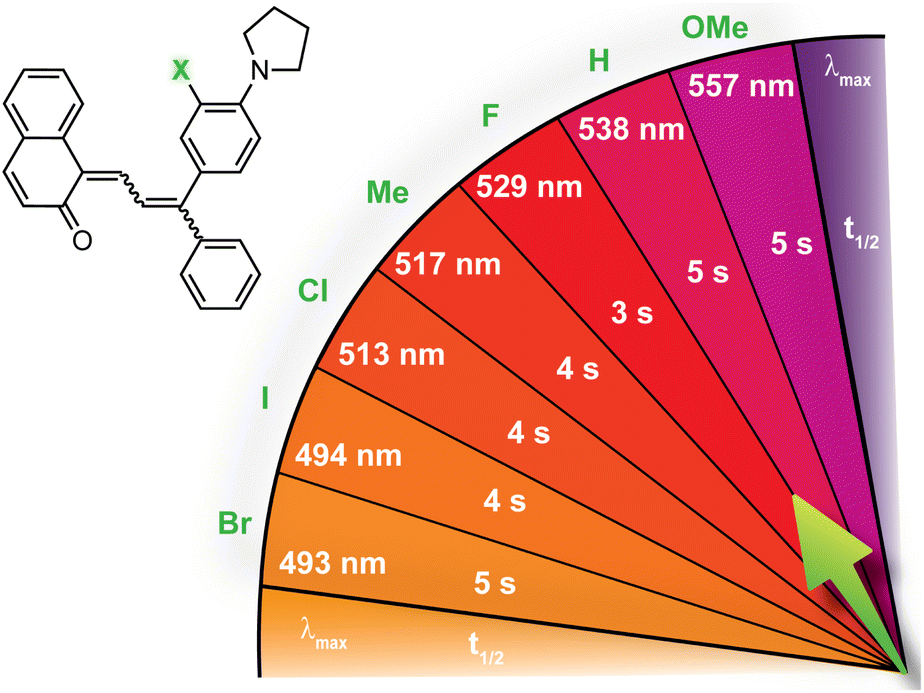 | ||
| Fig. 7 Merocyanine color can be finely tuned by modulating the steric demand of the group adjacent to a para-pyrrolidine substituent. The λmax and half-life (t1/2) are given for merocyanines with various meta-substituents (X). Values shown were measured in toluene at 20 °C.214 | ||
98
|
|
|||
|---|---|---|---|
| Entry | X | λ max (nm) | t 1/2 (s) |
| 1 | H | 538 | 5 |
| 2 | F | 554 | 40 |
| 3 | Cl | 554 | 741 |
| 4 | Br | 554 | 1024 |
| 5 | I | 553 | 1167 |
| 6 | OMe | 555 | 351 |
| 7 | Me | 555 | 639 |
 101
101
|
|
|||
|---|---|---|---|
| Entry | X | λ max (nm) | t 1/2 (s) |
| 1 | H | 538 | 5 |
| 2 | F | 394, 501 | 21 |
| 3 | Cl | 403, 480 | 44 |
| 4 | OMe | 399, 519 | 73 |
| 5 | Me | 410, 507 | 104 |
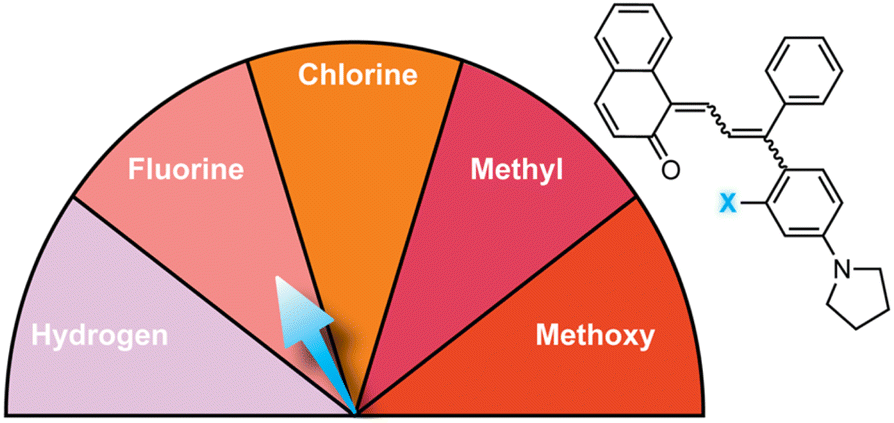
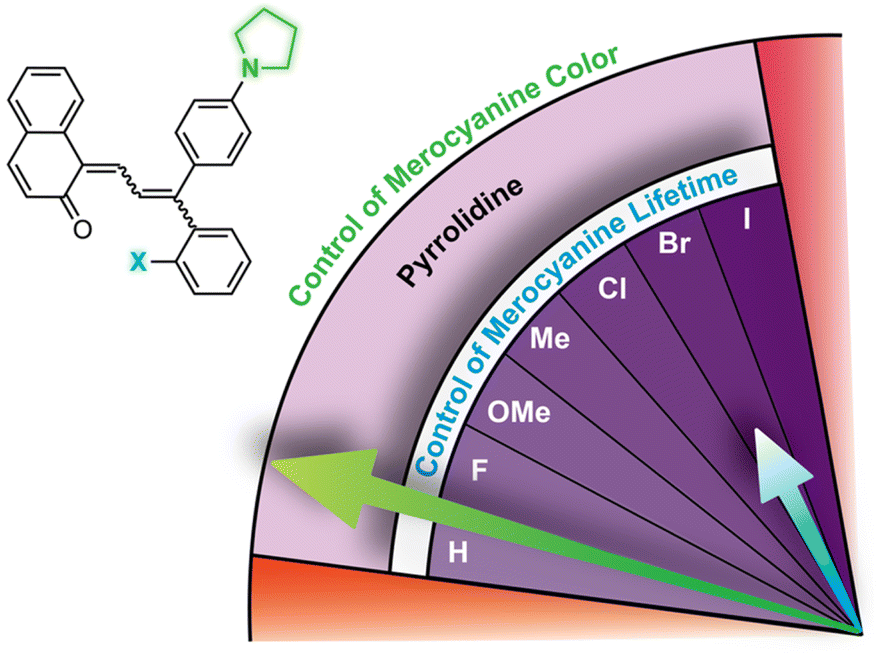
Substitution at the ortho-aryl position has a moderate effect on the absorption properties of merocyanines. If the ortho-substituent is located on a different aryl ring than one bearing a para-pyrrolidine group, a moderate bathochromic shift in λmax of 15–17 nm is observed regardless of the size or electronic character (Table 1).98 In this case, the pyrrolidine-substituted ring adopts a coplanar conformation with the merocyanine bridge to maximize conjugation with the rest of the π system. The ortho-substituted ring rotates out of the plane to minimize steric interactions, reducing its influence on merocyanine color. If the two substituents are on the same aryl ring, however, the absorption is hypsochromically shifted relative to the merocyanine without an ortho-aryl substituent due to less effective conjugation resulting from similar out-of-plane rotation (Table 2).101 An additional visible absorption peak around 400 nm is also observed for merocyanines with this substitution pattern. Increasing the size of the ortho-substituent generally leads to greater hypsochromic shifts, although electron-donating ortho-aryl substituents can counteract this effect. For example, the ortho-chlorinated merocyanine absorbs most strongly at 480 nm (Table 2, entry 3), while the merocyanine with a similarly sized ortho-methyl substituent has a λmax of 507 nm (Table 2, entry 5). The interplay between these steric and electronic factors has been harnessed to precisely modulate the color and reversion behavior of naphthopyran-derived merocyanines for photochromic applications.
|
|
||
|---|---|---|
| Entry | R | λ max (nm) |
| 1 | 5-OMe | 435 |
| 2 | 6-OMe | 423 |
| 3 | 7-OMe | 435 |
| 4 | 8-OMe | 477 |
| 5 | 9-OMe | 432 |
| 6 | H | 436 |
 | ||
| Scheme 12 Amine substituents can stabilize 3H-naphthopyran-derived merocyanines through resonance. | ||
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | λ max (nm) | t 1/2 (s) |
| 1 | H | H | 412, 508 | >1800 |
| 2 | Me | H | 416, 496 | 178 |
| 3 | Me | Me | 421, 494 | 66 |
| 4 | Ph | Ph | 418, 508 | 20 |
| 5 | CO2Me | H | 492 | 2 |
| 6 | CO2Me | Me | 416, 492 | 4 |
| 7 | CO2Me | Ph | 416, 506 | 7 |
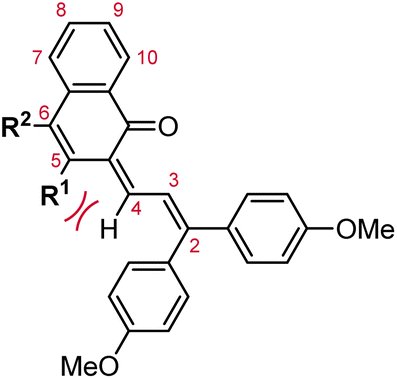
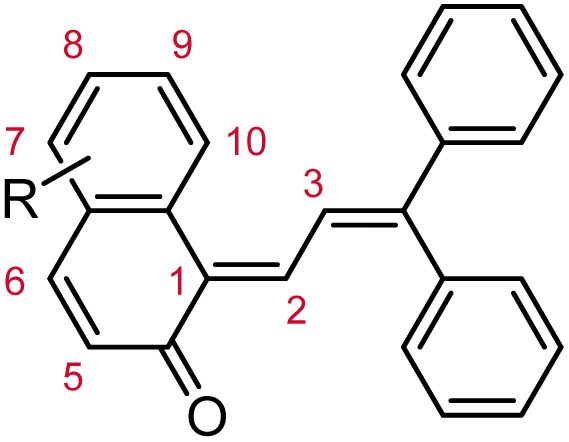
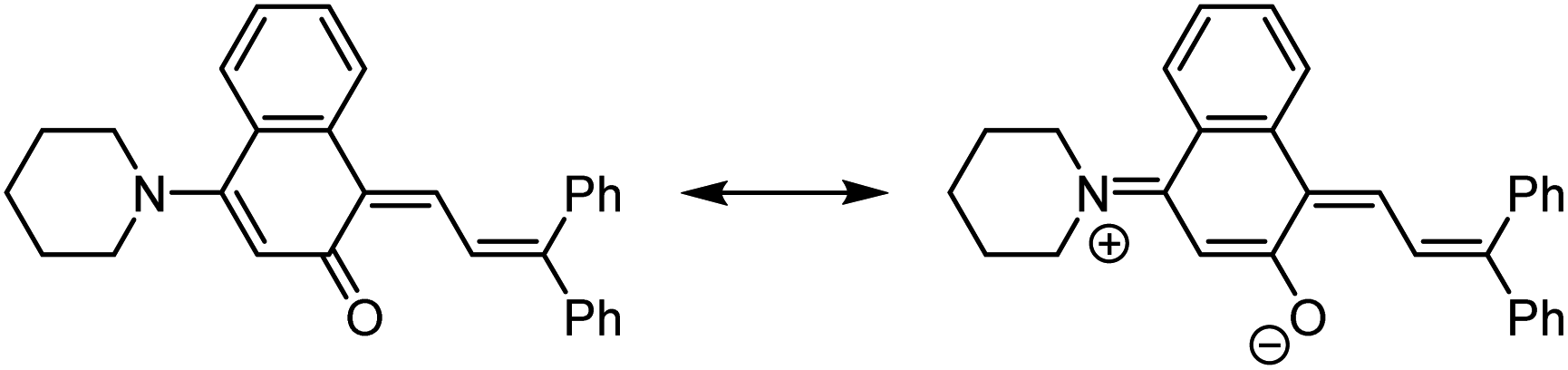
Commercial interests in the development of photochromic ophthalmic lenses have driven efforts to design naphthopyrans that generate fast-fading merocyanine dyes to achieve photoswitches with rapid switching behavior when transitioning between sunlight and indoor environments. For example, the persistence of merocyanines derived from 3H-naphthopyrans is significantly reduced by introducing bulky substituents at the 2- or 10-positions, which sterically impede the formation of the more thermally stable trans stereoisomer (Scheme 13a).137,185 These types of naphthopyrans produce merocyanine dyes with sub-millisecond half-lives (t1/2). Alternatively, the presence of an alkoxy group at the 10-position of a 3H-naphthopyran enables a hydrogen bonding interaction in the merocyanine between the ether oxygen and the hydrogen atom on the exocyclic alkene that favors the cis stereoisomer and, in several cases, nearly completely suppresses photochemical accumulation of the trans merocyanine (Scheme 13b).94,185
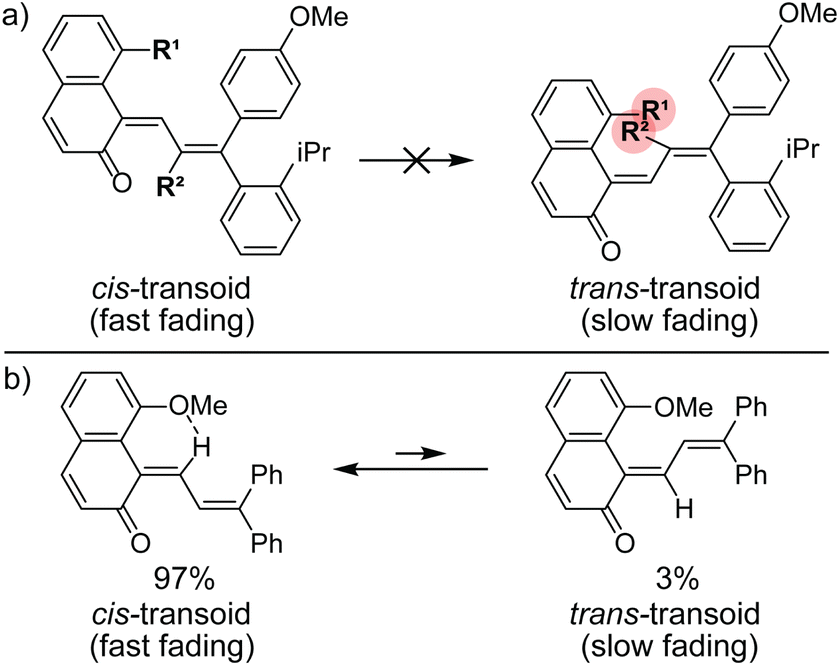 | ||
| Scheme 13 (a) Substituents at the 2- and 10-positions of 3H-naphthopyrans result in severe steric repulsion in the trans merocyanine. (b) Hydrogen bond acceptors at the 10-position of 3H-naphthopyrans stabilize the cis merocyanine stereoisomer. | ||
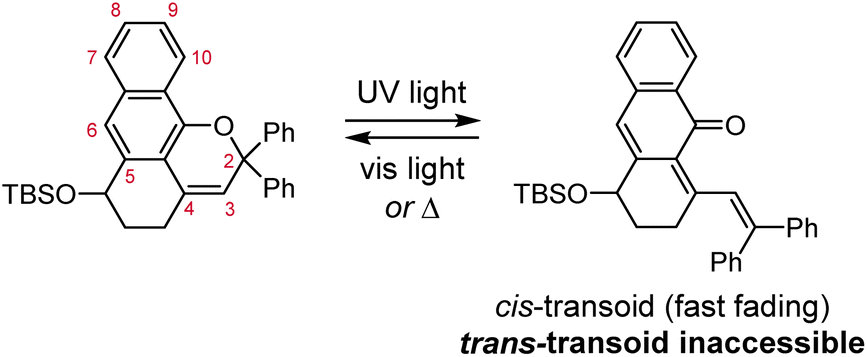 | ||
| Scheme 14 Ring fusion at the 4- and 5-positions of 2H-naphthopyrans prevents isomerization of the cis merocyanine isomer to the more thermally stable trans configuration.220 | ||
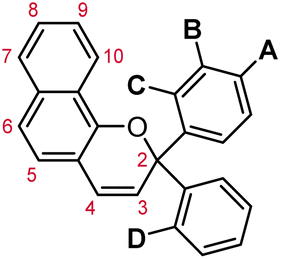
Tables 5 and 6 summarize the privileged substitution patterns discussed in Section 3.3 and their impact on the thermal reversion rate and absorption properties of merocyanine dyes derived from 3H-naphthopyran and 2H-naphthopyrans, respectively. Beyond those discussed here, many other structural attributes of naphthopyrans have been studied and identified for controlling the properties of their merocyanine dyes. Combining substitution of the peripheral aryl groups with variations in the naphthopyran core structure vastly expands the scope of accessible thermal reversion and absorption properties. The immense range of properties available to naphthopyran-derived merocyanine dyes as showcased in this section, coupled with the synthetic modularity and accessibility of naphthopyrans, lead to their description as “the most industrially important type of photochromic colorant”.37 These features not only make naphthopyran a privileged photoswitch, but in combination with their emergent mechanochemical activity, provide a highly useful platform for the design of mechanochromic polymers with an equally vast array of properties and force-responsive functions.
|
|
||||
|---|---|---|---|---|
| Position | Substituent property | Effect on reversion | Shift in λmax | Section |
| A | More e− donating | Faster | Bathochromic | 3.3.1 |
| B | Larger size | — | Hypsochromic (if A = pyrrolidine) | 3.3.1 |
| C | Larger size | Slower | Hypsochromic (if A = pyrrolidine) | 3.3.2 |
| D | Larger size | Slower | Bathochromic | 3.3.2 |
| 2 | Larger size | Faster | — | 3.3.4 |
| 5 | More e− donating | Slower | — | 3.3.3 |
| 6 | More e− donating | Slower | Hypsochromic | 3.3.3 |
| 8 | More e− donating | Slower | Bathochromic | 3.3.3 |
| 10 | Larger size | Faster | — | 3.3.4 |
|
|
||||
|---|---|---|---|---|
| Position | Substituent property | Effect on reversion | Shift in λmax | Section |
| A | More e− donating | Faster | Bathochromic | 3.3.1 |
| B | Larger size | — | Hypsochromic (if A = pyrrolidine) | 3.3.1 |
| C | Larger size | Slower | Hypsochromic (if A = pyrrolidine) | 3.3.2 |
| D | Larger size | Slower | Bathochromic | 3.3.2 |
| 5 | More e− donating | Faster | — | 3.3.4 |
| 6 | More e− donating | Slower | — | 3.3.3 |
4 The emergent mechanochemistry of naphthopyran
4.1 Discovery of the mechanochemical ring-opening reaction of naphthopyran
The mechanically triggered ring-opening reaction of naphthopyran was first described by Moore and Sottos in 2016.35 The regiochemistry of polymer attachment on the naphthopyran scaffold was found to be a critical parameter for achieving mechanochemical activation and merocyanine formation. Three different 3H-naphthopyran regioisomers were investigated, each with a polymer attached at the para-position of one of the peripheral aryl groups and another at either the 5-, 8-, or 9-position of the naphthopyran (Fig. 8a). Density functional theory (DFT) calculations performed using the constrained geometries simulates external force (CoGEF) method,223 which is a reliable and straightforward tool for predicting mechanochemical reactions,11 indicated that only the regioisomer with polymer attachment at the 5-position of the naphthopyran (NP5) would undergo a productive ring-opening reaction to generate the merocyanine dye. Regioisomers with pulling points at the 8- and 9-position of the naphthopyran were predicted to undergo nonproductive bond scission elsewhere in the structure upon mechanical elongation. These structure–mechanochemical activity predictions were validated by experiments performed on elastomeric poly(dimethyl siloxane) (PDMS) materials covalently incorporating each of the naphthopyran regioisomers as crosslinkers. While each material exhibited the expected photochromism upon irradiation with UV light, only the sample crosslinked with the naphthopyran regioisomer attached at the 5-position displayed mechanochromic behavior, producing an orange-yellow merocyanine dye under uniaxial tension consistent with the DFT predictions (Fig. 8b). Furthermore, no mechanochromic response was observed in control experiments performed on an analogous sample incorporating a similar monofunctional naphthopyran, in which mechanical force is not transferred across the pyran C–O bond, supporting the mechanochemical nature of the transformation.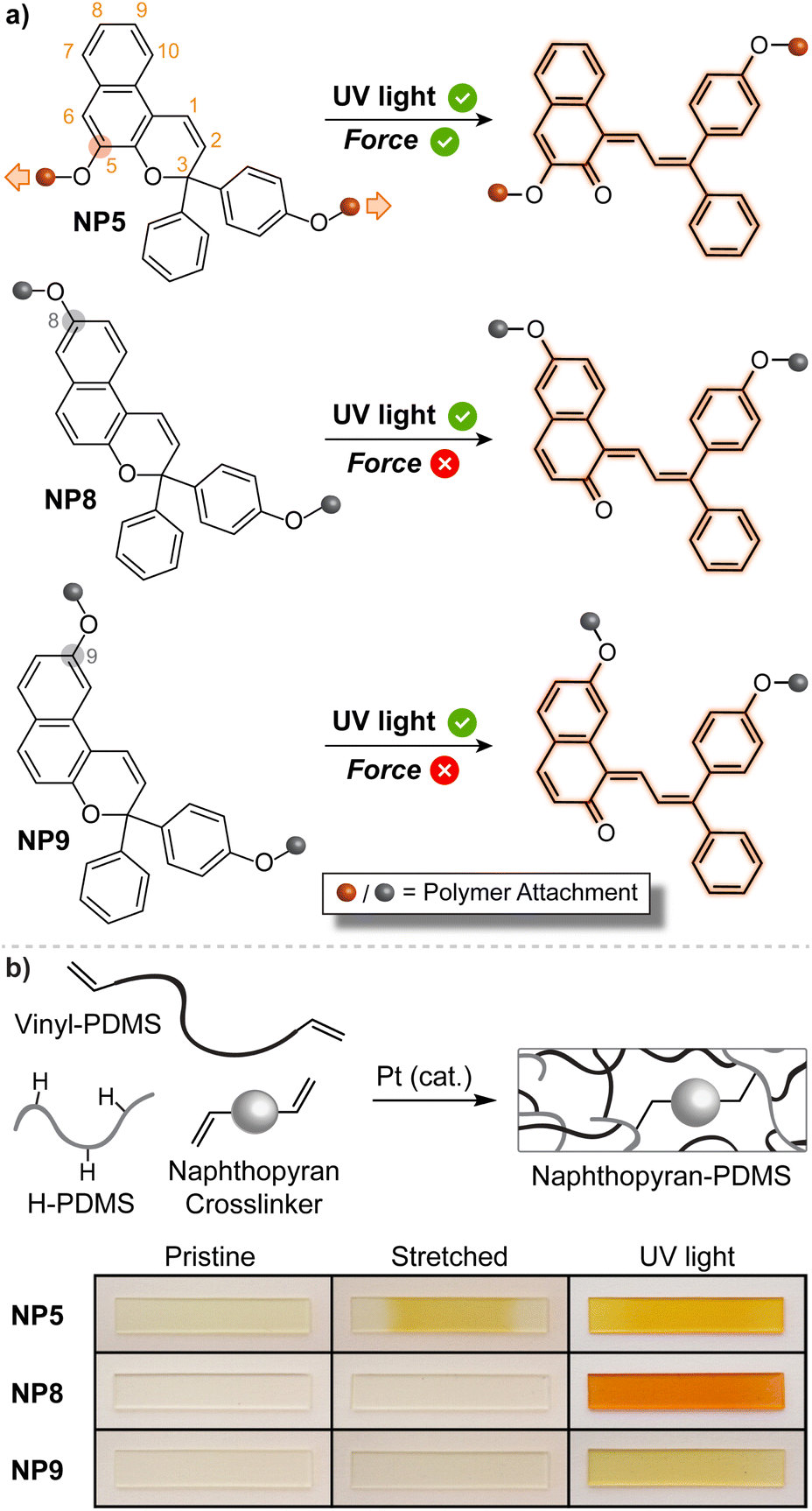 | ||
| Fig. 8 (a) Regioisomer specific mechanochemical activity of 3H-naphthopyran. Three naphthopyran regioisomers with varying polymer attachment positions all exhibit photochromic behavior, but only regioisomer NP5 is mechanochemically active due to effective force distribution across the labile C–O pyran bond. (b) Photographs of polydimethylsiloxane samples covalently crosslinked with the three different naphthopyran regioisomers subjected to uniaxial tension or UV light irradiation. Reproduced with permission from ref. 35. Copyright 2016, American Chemical Society. | ||
The regioisomer-specific mechanochemical reactivity of the three different naphthopyran congeners suggested that a pulling geometry providing sufficient alignment between the pyran C–O bond and the direction of externally applied mechanical force along the reaction coordinate is critical for achieving the mechanochemical ring-opening reaction of naphthopyran. Indeed, the angle between the pyran C–O bond and the external force vector at maximum elongation prior to bond rupture was calculated to be relatively narrow for the NP5 regioisomer and significantly wider for the other two mechanochemically unreactive naphthopyran regioisomers studied. Such geometric factors have emerged as one of the most important determinants for mechanochemical reactivity. A similar geometrical analysis was extended to furan–maleimide Diels–Alder adducts, further establishing important molecular design rules based on regiochemistry and stereochemistry and their effects on the efficiency of mechanochemical coupling.224
4.2 Structure–mechanochemical activity relationships
The discovery of a mechanochemical reaction pathway for naphthopyran-to-merocyanine conversion opened up exciting new opportunities in polymer mechanochemistry and enabled the design of diverse mechanochromic polymers by leveraging the extensive structure–property relationships developed for naphthopyran photoswitches. In 2020, our group capitalized on the modularity of the naphthopyran scaffold to design a series of mechanochromic mechanophores that produce merocyanine dyes with highly tunable color and fading kinetics after mechanochemical activation in polymeric materials (Fig. 9).145 Strategic structural modifications to the naphthopyran mechanophore produced dramatic differences in both the color and thermal reversion behavior of the corresponding merocyanine dyes generated upon tensile activation of PDMS elastomers. Similar to the original approach described above, 3H-naphthopyran crosslinkers bearing esters with terminal vinyl groups were covalently incorporated into PDMS materials using hydrosilylation chemistry.35,54 The color of the mechanically generated merocyanine dyes was varied from orange-yellow to purple upon the introduction of an electron donating pyrrolidine substituent at the para-position of one of the peripheral aryl groups. Likewise, the lifetime of the merocyanine dyes was modulated by varying the electronic character of the polymer attachment at the 5-position of the naphthopyran mechanophores using either a weakly electron donating alkyl linker or a more strongly electron donating alkoxy linker, which produced a faster and slower fading merocyanine, respectively. In addition, incorporation of an ortho-fluoro substituent on one of the peripheral aryl rings was found to significantly increase the lifetime of the merocyanine dyes produced upon mechanical activation according to the steric effects described previously in Section 3.3.2. Ultimately, this study demonstrated the generality of the design principles established for naphthopyran photoswitches, harnessing judicious yet straightforward structural modifications for the construction of naphthopyran-based mechanochromic polymers that display both fast-fading and persistent coloration upon mechanical activation with a range of accessible absorption properties. As discussed later in Section 4.4, the fine control of these features uniquely afforded by the naphthopyran framework was further exploited to design polymeric materials that exhibit desirable multicolor mechanochromic and complex stimuli-responsive behavior.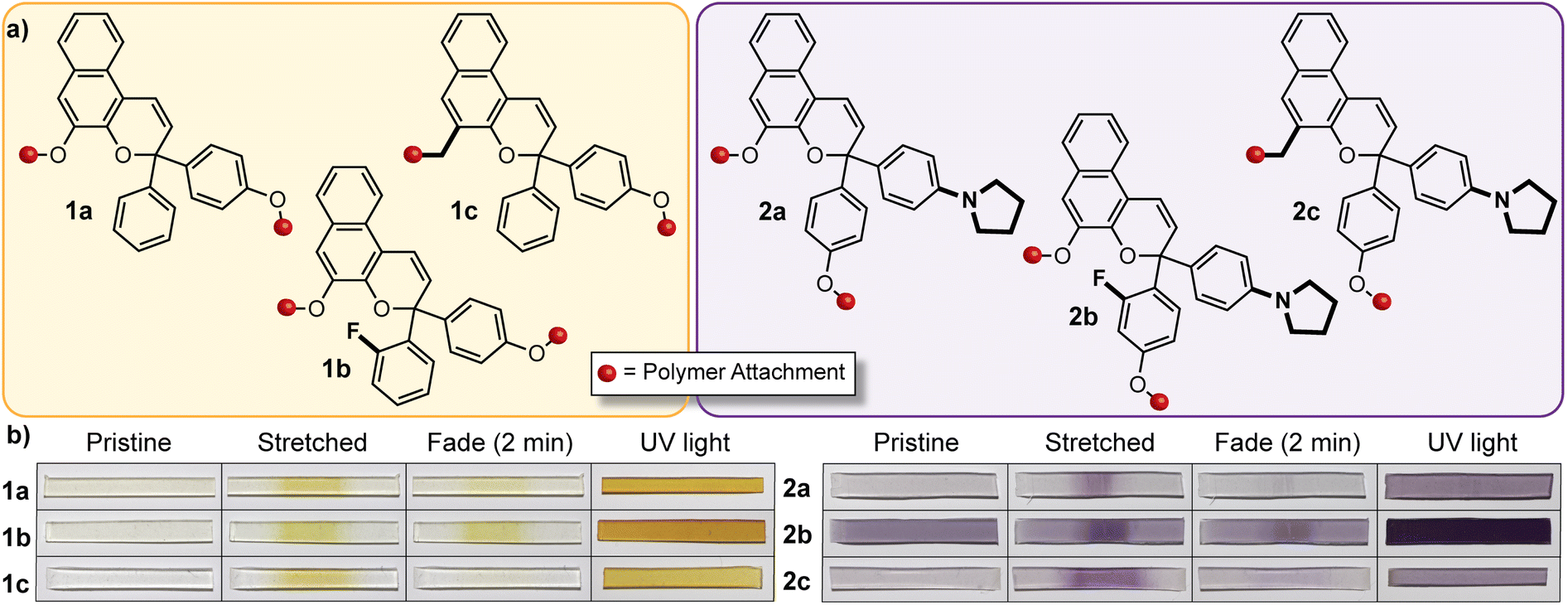 | ||
| Fig. 9 (a) Different 3H-naphthopyran mechanophores with varying substitution affecting merocyanine color and reversion rate. (b) Photographs of polydimethylsiloxane samples covalently incorporating the naphthopyran crosslinkers before and after being subjected to uniaxial tension and subsequent stress relaxation, or UV light irradiation, demonstrating the modularity of the naphthopyran mechanophore for designing mechanochromic materials. Adapted from ref. 145. | ||
In addition to the 3H-naphthopyran framework examined in earlier studies, our group has found that 2H-naphthopyrans are also competent mechanophores leading to the mechanically triggered generation of merocyanine dyes with comparatively longer lifetimes and bathochromically shifted absorption. For 2H-naphthopyran, polymer attachment at the 9-position was found to effectively couple external mechanical force with the selective scission of the labile pyran C–O bond leading to the desired ring-opening reaction (Fig. 10).143 Notably, mechanochemical activation of both 2H-naphthopyran and 3H-naphthopyran mechanophores incorporated near the center of poly(methyl acrylate) polymers was demonstrated for the first time in solution using ultrasonication, which generates elongational forces on linear polymers maximized near the chain midpoint as a result of acoustic cavitation.3,57 While accumulation of the merocyanine dye derived from the 2H-naphthopyran mechanophore was observed at ∼15 °C in solution, cooling the reaction to −13 °C was required to observe mechanochemical activation of the 3H-naphthopyran mechanophore, reflecting the significant differences in the thermal lifetimes of the resulting merocyanine dyes. UV-vis absorption spectra recorded after the ultrasound-induced mechanochemical activation of each mechanophore closely matched the spectra obtained after photochemical activation, indicating that similar merocyanine products are generated using mechanical force and UV light. It is interesting to note, however, that photochemical activation with UV light generates a fraction of merocyanine that is thermally persistent, in contrast to the product generated mechanochemically, which reverts completely back to the naphthopyran. These results are consistent with the photochemical generation of the more thermally stable trans merocyanine stereoisomer, whereas the less stable isomer with cis geometry of the exocyclic alkene generally appears to be produced exclusively using mechanical force. Indeed, the mechanochemical cis to trans isomerization of alkenes is uncommon,14,225,226 although an alternative mechanism for accessing trans merocyanine products from related 2H-naphthopyrans in force-coupled pathways has been postulated.147 Mechanochemical activation of the 2H-naphthopyran mechanophore was also successfully demonstrated in crosslinked PDMS materials, generating a red merocyanine dye under tension that was significantly longer lived following stress relaxation than the yellow merocyanine dye produced from the similarly substituted 3H-naphthopyran mechanophore (see Fig. 10). Building upon the earlier results on substituent effects, this study identified naphthopyran isomerism as an additional structural handle for modulating the mechanochromic properties of naphthopyran mechanophores, demonstrating that the diverse range of structure–property relationships developed for naphthopyran photoswitches is translatable to mechanochromic behavior.
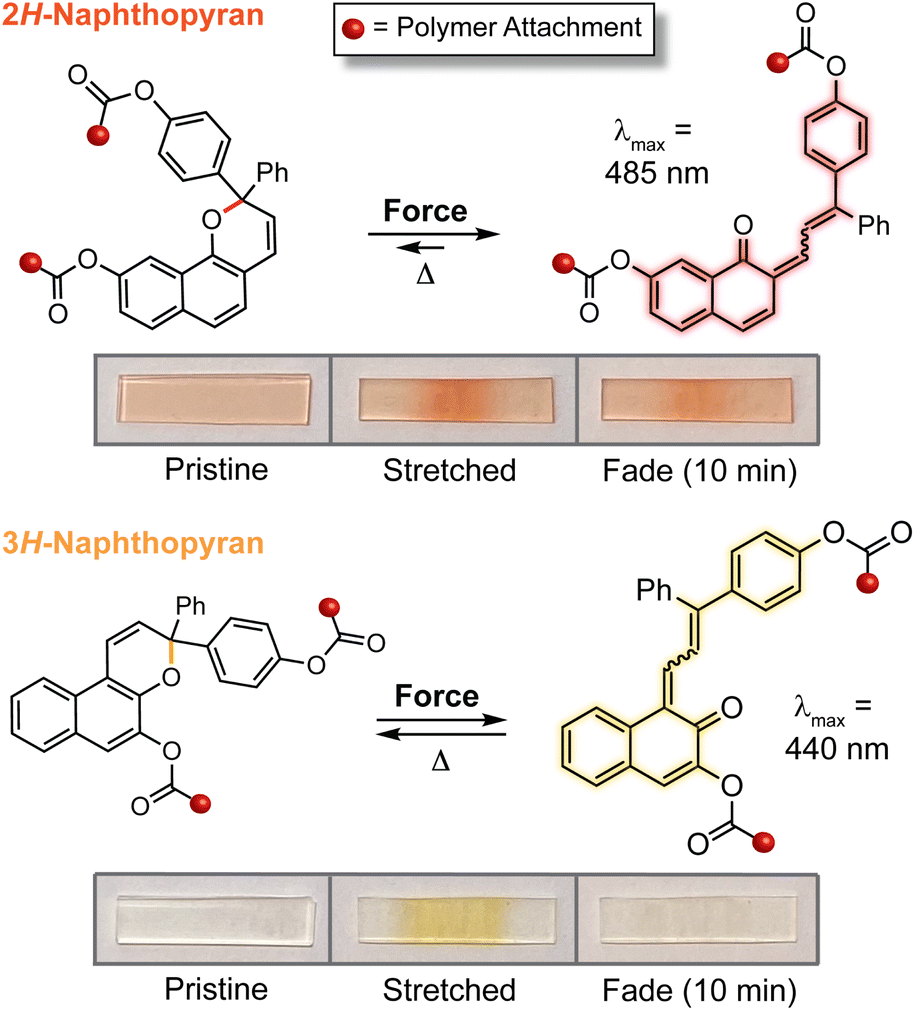 | ||
| Fig. 10 Isomeric 2H- and 3H-naphthopyran mechanophores generate merocyanine dyes with distinct coloration and thermal stability. Photographs illustrate PDMS materials covalently incorporating the different naphthopyran crosslinkers with 2H-naphthopyran generating a red merocyanine that reverts relatively slowly compared to the more transient yellow merocyanine produced from a similarly substituted 3H-naphthopyran mechanophore. | ||
4.3 Anomalous mechanochemical reactivity of naphthopyran
Mechanical force is a unique stimulus capable of promoting unusual reaction pathways and enabling the formation of unexpected products. For example, mechanical force has been shown to facilitate reactions that formally override conventional orbital symmetry rules including the disrotatory electrocyclic ring-opening reaction of cis-disubstituted benzocyclobutene227 and the conrotatory ring-opening reactions of gem-dihalocyclopropanes.228,229 Under sufficiently large forces, distortion of the potential energy surface fundamentally changes the reaction landscape.230–232 In the preceding sections, we described how the structure–activity relationships established for naphthopyran photoswitches translate to mechanochemical reactions of naphthopyran useful toward the design of mechanochromic polymers with a diverse set of highly tunable properties. In the following sections, we describe three exemplary cases in which the mechanochemical reactivity of naphthopyran diverges from its photochemical reactivity, leading to reaction pathways and products that are fundamentally unique. This anomalous reactivity has enabled the formation of an unusual permanent merocyanine dye and the design of multimodal mechanophores that exhibit complex mechanochromic behavior beyond the typical binary response including force-dependent multicolor mechanochromism from a single mechanophore.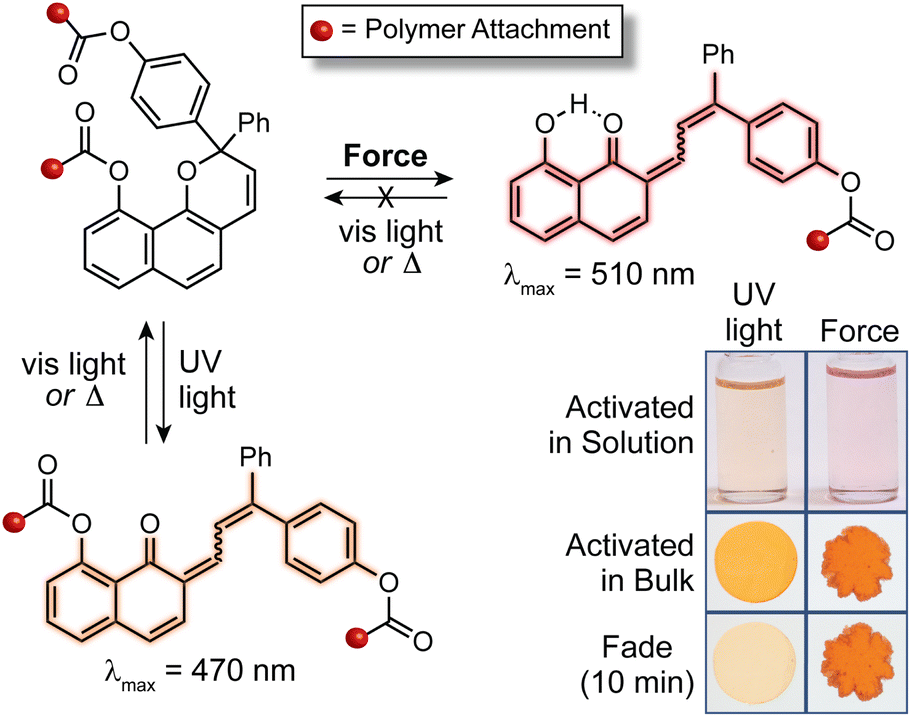 | ||
| Fig. 11 In contrast to the reversible photochemical reaction, mechanochemical activation of a 2H-naphthopyran mechanophore with polymer attachment via an ester linkage at the 10-position generates a persistent merocyanine dye stabilized by an intramolecular hydrogen bonding interaction that is established upon scission of the C(O)–O ester bond. | ||
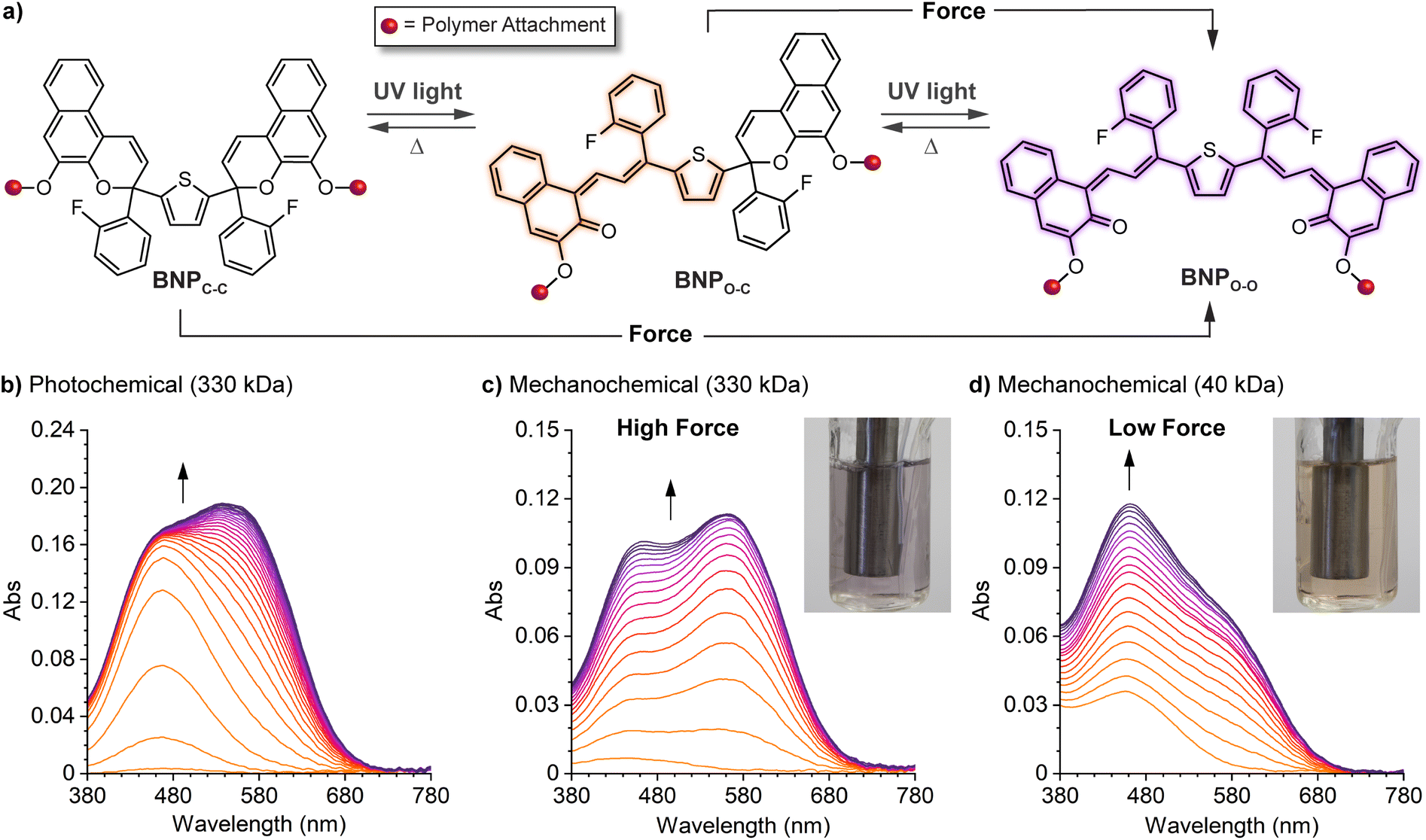 | ||
| Fig. 12 Force-dependent multicolor mechanochromism from a bis-naphthopyran (BNP) mechanophore. (a) Unlike the sequential ring-opening reactions observed upon photochemical activation, mechanical force directly converts the BNP mechanophore to the purple bis-merocyanine species, which thermally reverts to the yellow monomerocyanine. Changing the amount of force delivered to the mechanophore biases the merocyanine distribution resulting in force-dependent coloration. (b) UV-vis absorption spectra of a 330 kDa poly(methyl acrylate) polymer with a chain-centered BNP unit illustrating sequential-ring opening behavior upon irradiation with UV light. Ultrasound-induced mechanochemical activation of (c) the same 330 kDa polymer, and (d) an analogous 40 kDa polymer that experiences lower force, illustrating non-sequential ring opening behavior. Insets show photographs of the final sonicated solutions. Adapted with permission from ref. 135. Copyright 2019, American Chemical Society. | ||
Further insights were provided by a detailed kinetic analysis, which confirmed that mechanochemical activation of the BNP mechanophore occurs via a mechanistically distinct pathway compared to the photochemical process. In contrast to the photochemical pathway that proceeds via two sequential ring-opening reactions, the data support a non-sequential mechanochemical ring-opening mechanism in which the colorless BNP mechanophore is effectively converted directly to the purple bis-merocyanine product and formation of the yellow mono-merocyanine occurs primarily, if not exclusively, from thermal reversion of the bis-merocyanine. Biased by external force, the system achieves a dynamic equilibrium and the distribution of the two different merocyanine products is dictated by the balance between the forward rate of mechanochemical activation, which is force-dependent, and the rate of thermal electrocyclization, which is invariant with force (see Fig. 12a). Therefore, the relative distribution of the two distinctly colored merocyanine dyes is predictably modulated by varying the magnitude of force applied to the BNP mechanophore, resulting in a gradient change in the overall visible absorption. The direct conversion of BNP to the bis-merocyanine species may reflect unusual dynamic effects observed in the ultrasound-induced mechanochemical transformations of other mechanophores whereby the momenta resulting from extrinsic force lead to flyby trajectories that completely bypass reactive intermediates.241 We speculate that if the barriers associated with the two ring-opening reactions were sufficiently different, such as in the case of an asymmetric bis-naphthopyran mechanophore, then each ring-opening reaction may exhibit a discreet force threshold for activation leading to force-dependent multicolor mechanochromism that is not kinetically derived. Alternatively, the apparent mechanochemical reaction mechanism may be different in solid materials subjected to slower strain rates. In this case, it is reasonable to consider that strain could dominate the ring-opening reactivity and still lead to a similar multicolor mechanochromic response that more closely mirrors the sequential ring-opening behavior in the photochemical reaction pathway. Investigations of the solid state mechanochemical reactivity of bis-naphthopyran are currently underway in our laboratory that should shed light on some of these questions and advance the development of multimodal mechanophores toward quantitative visual force sensing applications. We also recently described another bis-naphthopyran mechanophore based on the scissile 2H-naphthopyran scaffold described above (refer to Section 4.3.3).147 Experiments suggested that the mechanically unveiled β-hydroxy ketone subunit confers remarkable thermal stability to bis-merocyanine isomers with a trans exocyclic alkene on the other merocyanine subunit and implicated the formation of an unusual trans merocyanine isomer as the product of mechanochemical activation.
In a notable deviation from the kinetic behavior of the BNP mechanophore, the absorption spectra evolved during the ultrasound-induced mechanochemical activation of naphthodipyran (Fig. 13c). At early reaction times, only the shorter wavelength absorption features of the monomerocyanine were observed, with the longer wavelength absorption features characteristic of the dimerocyanine emerging later. The kinetic behavior observed for the naphthodipyran mechanophore reports colorimetrically on temporal exposure to ultrasound-induced mechanical force and is effectively indistinguishable from a sequential ring-opening mechanism. This behavior may, in fact, indicate substantial differences in the barriers of the first and second ring-opening reactions, which occur in sequence as opposed to directly as in the case of the BNP mechanophore discussed earlier. Alternatively, the temporal evolution of the absorption spectra may reflect kinetic limitations in the trapping reaction of BF3 with the highly transient dimerocyanine species, requiring two separate chain-pulling events to form the thermally stable dimerocyanine adduct (NDPO–O·nBF3). Fundamental questions remain about how mechanical force is able to promote the dual ring-opening reaction of naphthodipyran in a process that diverges from the photochemical pathway. From a practical perspective, we envision that the newly unveiled multimodal reactivity of naphthodipyran, in combination with the unusual mechanochemical activation kinetics, will enable the development of additional mechanophores and force-responsive polymers that exhibit multicolor mechanochromism.
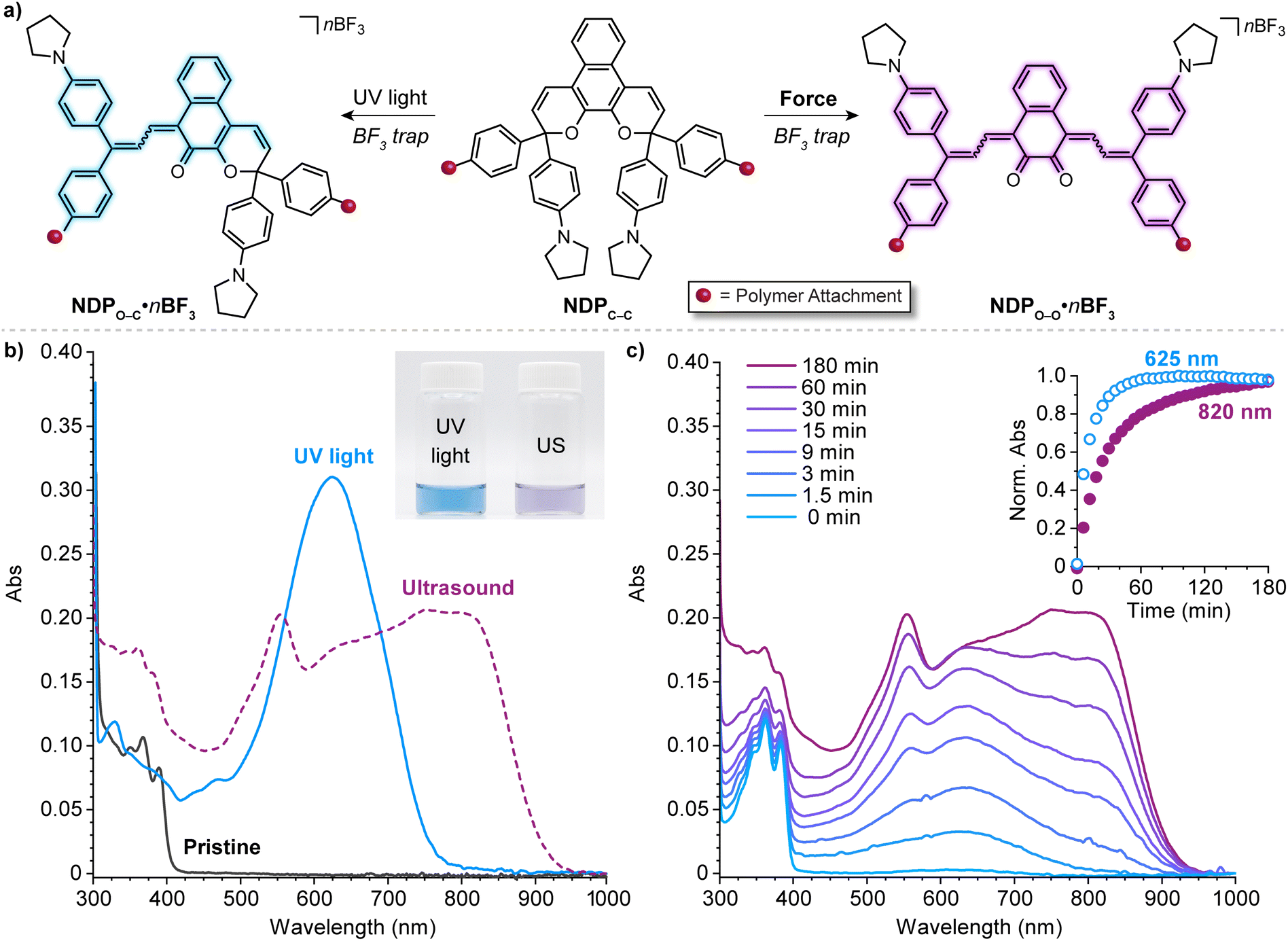 | ||
| Fig. 13 (a) Divergent photochemical and mechanochemical reactivity of a 3H-naphthodipyran (NDP) moiety incorporated near the center of a poly(methyl acrylate) chain. (b) UV-vis-near-infrared absorption spectra before and after photoirradiation of the polymer with UV light or ultrasound-induced mechanochemical activation. Inset shows a photograph of the two solutions after activation. (c) Absorption spectra recorded during ultrasonication of the polymer and time-dependent absorption profiles at 625 and 820 nm characteristic of monomerocyanine NDPO–C·nBF3 and dimerocyanine NDPO–O·nBF3, respectively. Adapted with permission from ref. 146. Copyright 2022, American Chemical Society. | ||
4.4 Design of more complex stimuli-responsive materials
The design of materials with multicolor mechanochromic and more complex stimuli-responsive behavior has also been achieved by blending distinct mechanophores with complementary reactivity and/or properties. The Otsuka and Moore groups teamed up to develop a polymer/silica composite material capable of displaying different colors under varying degrees of mechanical stimulation as well as orthogonal responses to light and solvent swelling (Fig. 14a).134 A naphthopyran mechanophore was incorporated into the soft polymer-rich domain while a radical-type diarylbibenzofuranone (DABBF) mechanophore was incorporated into a hard silica-rich domain. The design leverages the different colors of the merocyanine (orange-yellow) and stable free radical (blue) products that result from mechanochemical activation of naphthopyran and DABBF, respectively, as well as the disparate thermal reversion kinetics, which are also impacted by the substantial differences in mobility afforded by the hard and soft domains in the composite. The material turned blue upon gentle kneading in a mortar, which is characteristic of the mechanical activation of the more labile DABBF mechanophore at the polymer/silica interface. Stronger kneading caused the material to turn a greenish hue, reflecting the combined appearance of both the orange-yellow naphthopyran-derived merocyanine and the blue-colored radical species derived from the DABBF mechanophore. The authors also demonstrated that each colored product could be selectively bleached using either visible light to revert the merocyanine dye or upon the introduction of solvent to promote radical recombination, further modulating the appearance of the material with different stimuli following initial mechanical activation. This study illustrated that the combination of two different mechanophores with complementary reactivity and properties in a judiciously designed matrix could achieve more sophisticated stimuli-responsive behavior and ultimately increase the capacity of information encoded in a material via mechanical deformation.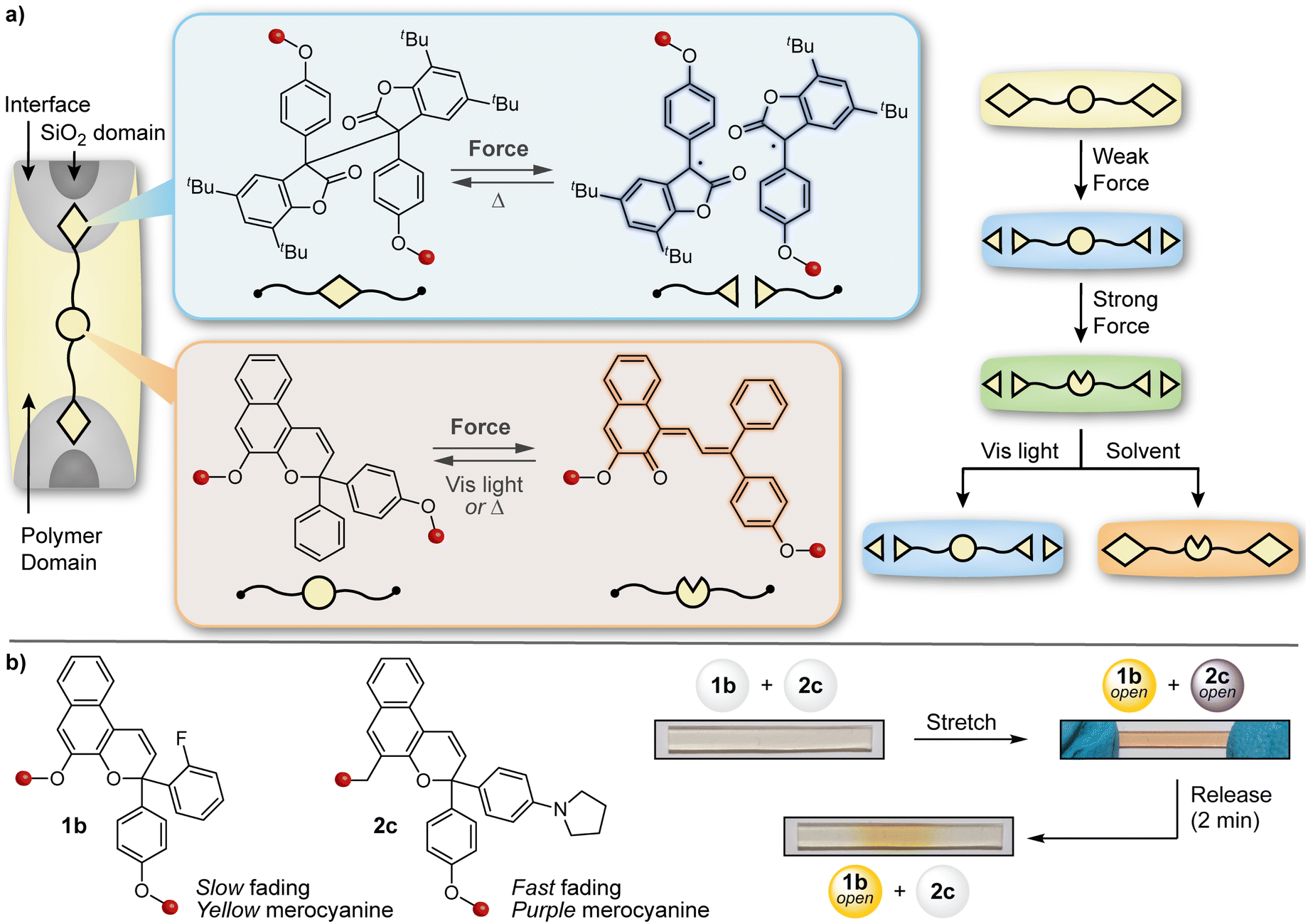 | ||
| Fig. 14 Complex force-responsive materials incorporating a naphthopyran mechanophore. (a) Blending a diarylbibenzofuranone and naphthopyran mechanophore into different phases of a silica/polymer composite material enables multicolor mechanochromism under different stimulation conditions. (b) Multicolor mechanochromism is achieved in polydimethylsiloxane elastomers by blending two naphthopyran mechanophores that generate merocyanine dyes with distinct colors and reversion kinetics. Portions of the figure were adapted from ref. 145. | ||
The broad structure–property range accessible to naphthopyrans lends itself to a similar strategy of blending complementary mechanophores to achieve more complex force and stimuli-responsive function. By leveraging two different naphthopyran mechanophores that produce distinctly colored merocyanine dyes with substantially different thermal reversion kinetics, our group designed PDMS elastomers capable of distinguishing between different stress states with temporal resolution (Fig. 14b).145 Specifically, elastomeric films were prepared that incorporated two different naphthopyran mechanophores, one that generates a fast-fading purple merocyanine dye and another that forms a more thermally persistent orange-yellow merocyanine dye (refer to Section 4.2). Mechanical activation of the material in tension caused the film to turn a red-orange color resulting from the combined appearance of both merocyanine dyes. Upon stress relaxation, however, rapid reversion of the purple merocyanine dye caused the color to immediately be transformed to the orange-yellow color of the more persistent merocyanine bearing an ortho-fluoro substituent on the peripheral aryl group. In another demonstration, blending a non-photochromic spiropyran mechanophore67 with a naphthopyran crosslinker that undergoes a ring-opening reaction in response to both UV light and mechanical force resulted in a polymeric material displaying visually orthogonal reactivity under mechanochemical and photochemical stimulation. Under tension, the material turned blue-purple due to the dominant appearance of the spiropyran-derived merocyanine dye; upon stress relaxation, the color of the film quickly disappeared due to the rapid thermal reversion of both merocyanine dyes. Photoirradiation of the same film with UV light, however, selectively triggered the ring-opening reaction of naphthopyran causing the material to become orange-yellow in color. These examples capitalize on the highly tunable and modular properties of naphthopyran mechanophores and illustrate the multicolor mechanochromic and more complex stimuli-responsive properties that are accessible to materials for multimodal sensing applications by leveraging simple blends of complementary molecular switches.
5 Conclusions and outlook
Since the seminal discovery of the photoswitching properties of chromenes by Becker and Michl in 1966,34 this broad class of molecular switches has attracted substantial research interest. In particular, intensive research and development of benzo-annulated chromenes, or naphthopyrans, has been driven by their commercial applications as reversible coloring agents in photochromic plastic ophthalmic lenses.36 Due to their modularity, synthetic accessibility, thermal stability, and expansive structure–property range, naphthopyrans have been credited as “the most industrially important type of photochromic colorant”.37 Fifty years after the initial discovery of the photochromic activity of naphthopyran, discovery of the mechanochemical ring-opening reaction of naphthopyran has precipitated renewed interest in this important molecular switch as a new class of mechanochromic mechanophores.35In this Perspective, we have highlighted the emergent mechanochemical activity of naphthopyran, contextualized by its development as a photoswitch with an extensive literature record that serves as an invaluable resource for the design of mechanochromic polymers with a myriad array of highly tunable properties. In just the past seven years, studies of naphthopyran mechanochemistry have provided important new insights into structure–mechanochemical activity relationships, elucidated anomalous reactivity that diverges from typical photochemical reaction pathways, and contributed broadly to the design and development of mechanochromic polymers and materials with diverse and often remarkable properties. The field of polymer mechanochemistry has exploded in research activity and interest in recent years because it represents a fundamentally different way to control and harness chemical reactivity. In the context of mechanochromism, mechanophores function as molecular force probes to enable the molecular-level visualization of critical stress and/or strain in polymers with applications in both basic and applied science. As new mechanophores are developed, focus is expanding to include the design of molecular force probes that exhibit complex, non-binary responses that enable the visual quantification of stress in materials. Naphthopyrans have proven to be an exceptional motif for constructing these types of multimodal mechanophores. As the field matures, naphthopyrans are privileged for their decades of industrial development as photoswitches that may prove fruitful in future translation of their mechanochromic function to commercial applications. Naphthopyran mechanophores have already played an important role in the development of the field and we look forward to seeing the impact of this unique and highly capable mechanophore platform on the bright and colorful future of polymer mechanochemistry.
Author contributions
M. E. M. wrote the first draft of the manuscript with contributions from R. W. B. and A. C. O. M. J. R. provided direction and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from an NSF CAREER award (CHE-2145791) and the Rose Hills Foundation is gratefully acknowledged. M. E. M. and A. C. O. were supported by NSF Graduate Research Fellowships (DGE-1745301) and Barbara J. Burger Graduate Fellowships from Caltech. Support from an Institute Fellowship at Caltech (A. C. O.) is also gratefully acknowledged. M. J. R. gratefully acknowledges the Alfred P. Sloan Foundation for a Sloan Research Fellowship and the Camille and Henry Dreyfus Foundation for a Camille Dreyfus Teacher-Scholar Award.References
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS
.
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed
- J. F. Patrick, M. J. Robb, N. R. Sottos, J. S. Moore and S. R. White, Nature, 2016, 540, 363–370 CrossRef CAS PubMed
- S. Wang, H. K. Beech, B. H. Bowser, T. B. Kouznetsova, B. D. Olsen, M. Rubinstein and S. L. Craig, J. Am. Chem. Soc., 2021, 143, 3714–3718 CrossRef CAS PubMed
- S. Wang, Y. Hu, T. B. Kouznetsova, L. Sapir, D. Chen, A. Herzog-Arbeitman, J. A. Johnson, M. Rubinstein and S. L. Craig, Science, 2023, 380, 1248–1252 CrossRef CAS PubMed
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, Nat. Commun., 2014, 5, 4899 CrossRef PubMed
- G. Kim, V. M. Lau, A. J. Halmes, M. L. Oelze, J. S. Moore and K. C. Li, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10214–10222 CrossRef CAS
- A. C. Overholts, W. Granados Razo and M. J. Robb, Nat. Chem., 2023, 15, 332–338 CrossRef CAS PubMed
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed
- P. Michael and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef CAS PubMed
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559–562 CrossRef CAS PubMed
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, ACS Cent. Sci., 2021, 7, 1216–1224 CrossRef CAS
- S. Huo, P. Zhao, Z. Shi, M. Zou, X. Yang, E. Warszawik, M. Loznik, R. Göstl and A. Herrmann, Nat. Chem., 2021, 13, 131–139 CrossRef CAS PubMed
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, Nat. Rev. Mater., 2020, 6, 84–98 CrossRef
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC
- C. Calvino, L. Neumann, C. Weder and S. Schrettl, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 640–652 CrossRef CAS
- R. W. Barber, M. E. McFadden, X. Hu and M. J. Robb, Synlett, 2019, 30, 1725–1732 CrossRef CAS
- M. Stratigaki and R. Göstl, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS
- H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2021, 42, 2000573 CrossRef CAS PubMed
- H. Staudinger and H. F. Bondy, Ber. Dtsch. Chem. Ges., 1930, 63, 724–730 CrossRef
- H. Staudinger and H. F. Bondy, Ber. Dtsch. Chem. Ges., 1930, 63, 734–736 CrossRef
- H. Staudinger and W. Heuer, Ber. Dtsch. Chem. Ges., 1934, 67, 1159–1164 CrossRef
- J. Sohma, Prog. Polym. Sci., 1989, 14, 451–596 CrossRef CAS
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS PubMed
- T. A. Kim, M. J. Robb, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2018, 51, 9177–9183 CrossRef CAS
- Y. Lin, M. H. Barbee, C. C. Chang and S. L. Craig, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS PubMed
- M. E. McFadden, A. C. Overholts, S. K. Osler and M. J. Robb, ACS Macro Lett., 2023, 12, 440–445 CrossRef CAS PubMed
- R. S. Becker and J. Michl, J. Am. Chem. Soc., 1966, 88, 5931–5933 CrossRef CAS
- M. J. Robb, T. A. Kim, A. J. Halmes, S. R. White, N. R. Sottos and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 12328–12331 CrossRef CAS PubMed
- S. N. Corns, S. M. Partington and A. D. Towns, Color. Technol., 2009, 125, 249–261 CAS
- A. Towns, Phys. Sci. Rev., 2020, 5, 20190085 Search PubMed
-
J. D. Hepworth and B. M. Heron, in Functional Dyes, Elsevier Science, 2006, pp. 85–135 Search PubMed
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS
- J. M. Clough, C. Creton, S. L. Craig and R. P. Sijbesma, Adv. Funct. Mater., 2016, 26, 9063–9074 CrossRef CAS
- C. Baumann, M. Stratigaki, S. P. Centeno and R. Göstl, Angew. Chem., Int. Ed., 2021, 60, 13287–13293 CrossRef CAS PubMed
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2015, 54, 6168–6172 CrossRef CAS PubMed
- F. Verstraeten, R. Göstl and R. P. Sijbesma, Chem. Commun., 2016, 52, 8608–8611 RSC
- K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 556–560 CrossRef CAS PubMed
- K. Ishizuki, H. Oka, D. Aoki, R. Goseki and H. Otsuka, Chem.–Eur. J., 2018, 24, 3170–3173 CrossRef CAS PubMed
- S. Kato, K. Ishizuki, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 12, 28 Search PubMed
- H. Sakai, T. Sumi, D. Aoki, R. Goseki and H. Otsuka, ACS Macro Lett., 2018, 7, 1359–1363 CrossRef CAS PubMed
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS PubMed
- T. Watabe, D. Aoki and H. Otsuka, Macromolecules, 2022, 55, 5795–5802 CrossRef CAS
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370–375 RSC
- C. Baumann and R. Göstl, Synlett, 2022, 33, 875–878 CrossRef CAS
- J. Yang, M. Horst, S. H. Werby, L. Cegelski, N. Z. Burns and Y. Xia, J. Am. Chem. Soc., 2020, 142, 14619–14626 CrossRef CAS
- B. R. Boswell, C. M. F. Mansson, J. M. Cox, Z. Jin, J. A. H. Romaniuk, K. P. Lindquist, L. Cegelski, Y. Xia, S. A. Lopez and N. Z. Burns, Nat. Chem., 2021, 13, 41–46 CrossRef CAS PubMed
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS PubMed
- G. R. Gossweiler, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2015, 137, 6148–6151 CrossRef CAS
- S. L. Potisek, D. A. Davis, N. R. Sottos, S. R. White and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 13808–13809 CrossRef CAS
- P. A. May and J. S. Moore, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed
- R. Yao, X. Li, N. Xiao, W. Weng and W. Zhang, Nano Res., 2021, 14, 2654–2658 CrossRef CAS
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS
- M. Wu, Y. Li, W. Yuan, G. De Bo, Y. Cao and Y. Chen, J. Am. Chem. Soc., 2022, 144, 17120–17128 CrossRef CAS PubMed
- H. Qian, N. S. Purwanto, D. G. Ivanoff, A. J. Halmes, N. R. Sottos and J. S. Moore, Chem, 2021, 7, 1080–1091 CAS
- Q. Qi, G. Sekhon, R. Chandradat, N. M. Ofodum, T. Shen, J. Scrimgeour, M. Joy, M. Wriedt, M. Jayathirtha, C. C. Darie, D. A. Shipp, X. Liu and X. Lu, J. Am. Chem. Soc., 2021, 143, 17337–17343 CrossRef CAS PubMed
- V. I. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC
- S. V. Paramonov, V. Lokshin and O. A. Fedorova, J. Photochem. Photobiol., C, 2011, 12, 209–236 CrossRef CAS
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CrossRef CAS PubMed
- S. Swansburg, E. Buncel and R. P. Lemieux, J. Am. Chem. Soc., 2000, 122, 6594–6600 CrossRef CAS
-
B. Van Gemert, in Organic Photochromic and Thermochromic Compounds, Springer, Boston, MA, 2002, pp. 111–140 Search PubMed
-
J. Hobley, M. J. Lear and H. Fukumura, in Photochemistry of Organic Molecules in Isotropic and Anisotropic Media, CRC Press, 2003 Search PubMed
-
A. Towns, in Applied Photochemistry, Springer International Publishing, 2016, pp. 227–279 Search PubMed
- S. Ghosh, J. Hall and V. Joshi, J. Text. Inst., 2018, 109, 723–729 CrossRef CAS
- K. Jin, X. Ji, T. Yang, J. Zhang, W. Tian, J. Yu, X. Zhang, Z. Chen and J. Zhang, Chem. Eng. J., 2021, 406, 126794 CrossRef CAS
-
X. Blin and J. C. Simon, Fr. Pat., FR2845910A1, 2004 Search PubMed
- C. Moustrou, N. Rebière, A. Samat, R. Guglielmetti, A. E. Yassar, R. Dubest and J. Aubard, Helv. Chim. Acta, 1998, 81, 1293–1302 CrossRef CAS
-
A. Yassar, N. Rebière-Galy, M. Frigoli, C. Moustrou, A. Samat, R. Guglielmetti and A. Jaafari, in Synthetic Metals, Elsevier, 2001, vol. 124, pp. 23–27 Search PubMed
- A. Yassar, H. Jaafari, N. Rebière-Galy, M. Frigoli, C. Moustrou, A. Samat and R. Guglielmetti, Eur. Phys. J.: Appl. Phys., 2002, 18, 3–8 CrossRef CAS
- A. Yassar, F. Garnier, H. Jaafari, N. Rebière-Galy, M. Frigoli, C. Moustrou, A. Samat and R. Guglielmetti, Appl. Phys. Lett., 2002, 80, 4297–4299 CrossRef CAS
- M. Frigoli, C. Moustrou, A. Samat and R. Guglielmetti, Eur. J. Org Chem., 2003, 2003, 2799–2812 CrossRef
- M. Frigoli, V. Pimienta, C. Moustrou, A. Samat, R. Guglielmetti, J. Aubard, F. Maurel and J.-C. Micheau, Photochem. Photobiol. Sci., 2003, 2, 888–892 CrossRef CAS PubMed
- F. Ortica, P. Smimmo, G. Favaro, U. Mazzucato, S. Delbaere, D. Venec, G. Vermeersch, M. Frigoli, C. Moustrou and A. Samat, Photochem. Photobiol. Sci., 2004, 3, 878–885 CrossRef CAS
- P. L. Gentili, A. L. Rightler, B. M. Heron and C. D. Gabbutt, Dyes Pigm., 2016, 135, 169–176 CrossRef CAS
- P. L. Gentili, A. L. Rightler, B. M. Heron and C. D. Gabbutt, Chem. Commun., 2016, 52, 1474–1477 RSC
- P. L. Gentili, M. S. Giubila and B. M. Heron, ChemPhysChem, 2017, 18, 1831–1841 CrossRef CAS
- H. Wu, A. Huang, Q. Liao, P. Lin, W. Yao, Q. Li and Z. Li, ACS Mater. Lett., 2023, 5, 753–761 CrossRef CAS
- Z. M. Dong, H. Ren, J. N. Wang and Y. Wang, Microchem. J., 2020, 155, 104676 CrossRef CAS
- V. M. Mwalukuku, J. Liotier, A. J. Riquelme, Y. Kervella, Q. Huaulmé, A. Haurez, S. Narbey, J. A. Anta and R. Demadrille, Adv. Energy Mater., 2023, 13, 2203651 CrossRef CAS
- P. L. Gentili, M. S. Giubila, R. Germani and B. M. Heron, Dyes Pigm., 2018, 156, 149–159 CrossRef CAS
- I. Iwai and J. Ide, Chem. Pharm. Bull., 1962, 10, 926–933 CrossRef CAS PubMed
- J.-L. Pozzo, A. Samat, R. Guglielmetti, R. Dubest and J. Aubard, Helv. Chim. Acta, 1997, 80, 725–738 CrossRef CAS
- C. D. Gabbutt, T. Gelbrich, J. D. Hepworth, B. M. Heron, M. B. Hursthouse and S. M. Partington, Dyes Pigm., 2002, 54, 79–93 CrossRef CAS
- E. N. Ushakov, V. B. Nazarov, O. A. Fedorova, S. P. Gromov, A. V. Chebun’kova, M. V. Alfimov and F. Barigelletti, J. Phys. Org. Chem., 2003, 16, 306–309 CrossRef CAS
- X. Sallenave, S. Delbaere, G. Vermeersch, A. Saleh and J. L. Pozzo, Tetrahedron Lett., 2005, 46, 3257–3259 CrossRef CAS
- Y. Inagaki, Y. Kobayashi, K. Mutoh and J. Abe, J. Am. Chem. Soc., 2017, 139, 13429–13441 CrossRef CAS PubMed
- H. Kuroiwa, Y. Inagaki, K. Mutoh and J. Abe, Adv. Mater., 2019, 31, 1805661 CrossRef
- G. Harié, A. Samat, R. Guglielmetti, I. van Parys, W. Saeyens, D. de Keukeleire, K. Lorenz and A. Mannschreck, Helv. Chim. Acta, 1997, 80, 1122–1132 CrossRef
- S. Aiken, O. D. C. C. de Azevedo, K. Chauhan, T. Driscoll, P. I. Elliott, C. D. Gabbutt and B. M. Heron, J. Org. Chem., 2020, 85, 952–966 CrossRef CAS PubMed
- B. M. Heron, C. D. Gabbutt and A. C. Instone, Heterocycles, 2003, 60, 843 CrossRef
- C. D. Gabbutt, B. M. Heron, A. C. Instone, D. A. Thomas, S. M. Partington, M. B. Hursthouse and T. Gelbrich, Eur. J. Org Chem., 2003, 2003, 1220–1230 CrossRef
- C. D. Gabbutt, B. M. Heron, S. B. Kolla, C. Kilner, S. J. Coles, P. N. Horton and M. B. Hursthouse, Org. Biomol. Chem., 2008, 6, 3096–3104 RSC
- C. D. Gabbutt, B. M. Heron and A. C. Instone, Tetrahedron, 2006, 62, 737–745 CrossRef CAS
- G. Harie, A. Samat and R. Guglielmetti, Mol. Cryst. Liq. Cryst., 1997, 297, 263–268 CrossRef CAS
- S. Aiken, K. Booth, C. D. Gabbutt, B. M. Heron, C. R. Rice, A. Charaf-Eddin and D. Jacquemin, Chem. Commun., 2014, 50, 7900–7903 RSC
-
B. Van Gemert and M. P. Bergomi, US Pat., US5066818A, 1991 Search PubMed
- C. D. Gabbutt, J. D. Hepworth, B. M. Heron, S. M. Partington and D. A. Thomas, Dyes Pigm., 2001, 49, 65–74 CrossRef CAS
- S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS
- L. M. Carvalho, A. M. S. Silva, C. I. Martins, P. J. Coelho and A. M. F. Oliveira-Campos, Tetrahedron Lett., 2003, 44, 1903–1905 CrossRef CAS
- W. Zhao and E. M. Carreira, Org. Lett., 2003, 5, 4153–4154 CrossRef CAS
- K. Tanaka, H. Aoki, H. Hosomi and S. Ohba, Org. Lett., 2000, 2, 2133–2134 CrossRef CAS PubMed
- K. Guo and Y. Chen, J. Phys. Org. Chem., 2010, 23, 207–210 CrossRef CAS
- S. Aiken, B. Allsopp, K. Booth, C. D. Gabbutt, B. M. Heron and C. R. Rice, Tetrahedron, 2014, 70, 9352–9358 CrossRef CAS
- K. Guo and Y. Chen, J. Mater. Chem., 2010, 20, 4193–4197 RSC
- S. Delbaere, G. Vermeersch, M. Frigoli and G. H. Mehl, Org. Lett., 2010, 12, 4090–4093 CrossRef CAS PubMed
- S. Han and Y. Chen, J. Mater. Chem., 2011, 21, 4961–4965 RSC
- S. Delbaere, J.-C. Micheau, Y. Teral, C. Bochu, M. Campredon and G. Vermeersch, Photochem. Photobiol., 2001, 74, 694 CrossRef CAS PubMed
- W. Zhao and E. M. Carreira, Chem.–Eur. J., 2007, 13, 2671–2688 CrossRef CAS
- M. Frigoli, J. Marrot, P. L. Gentili, D. Jacquemin, M. Vagnini, D. Pannacci and F. Ortica, ChemPhysChem, 2015, 16, 2447–2458 CrossRef CAS PubMed
- P. J. Coelho, M. A. Salvador, M. M. Oliveira and L. M. Carvalho, J. Photochem. Photobiol., A, 2005, 172, 300–307 CrossRef CAS
- W. Zhao and E. M. Carreira, Org. Lett., 2006, 8, 99–102 CrossRef CAS PubMed
- X. Lu, Q. Dong, X. Dong and W. Zhao, Tetrahedron, 2015, 71, 4061–4069 CrossRef CAS
- W. D. Cotterill and R. Livingstone, J. Chem. Soc., 1970, 1758–1764 CAS
- R. Livingstone, D. Miller and S. Morris, J. Chem. Soc., 1960, 5148–5152 RSC
- J. Cottam and R. Livingstone, J. Chem. Soc., 1964, 5228–5231 RSC
-
J. D. Hepworth and B. M. Heron, in Progress in Heterocyclic Chemistry, Elsevier Ltd, 2005, vol. 17, pp. 33–62 Search PubMed
- H. J. Kabbe, Synthesis, 1978, 1978, 886–887 CrossRef
- J. J. Talley, Synthesis, 1983, 10, 845–847 CrossRef
- H. G. Heller, J. R. Levell, D. E. Hibbs, D. S. Hughes and M. B. Hursthouse, Mol. Cryst. Liq. Cryst., 1997, 297, 123–130 CrossRef CAS
- G. Sartori, G. Casiraghi, L. Bolzoni and G. Casnati, J. Org. Chem., 1979, 44, 803–805 CrossRef CAS
- J.-L. Pozzo, V. A. Lokshin and R. Guglielmetti, J. Chem. Soc., Perkin Trans. 1, 1994, 2591–2595 RSC
- A. Ghatak, S. Khan and S. Bhar, Adv. Synth. Catal., 2016, 358, 435–443 CrossRef CAS
- Y.-W. Dong, G.-W. Wang and L. Wang, Tetrahedron, 2008, 64, 10148–10154 CrossRef CAS
- M. Naga Venkata Sastry, S. Claessens, P. Habonimana and N. De Kimpe, J. Org. Chem., 2010, 75, 2274–2280 CrossRef PubMed
- S. Delbaere, J.-C. Micheau, M. Frigoli and G. Vermeersch, J. Org. Chem., 2005, 70, 5302–5304 CrossRef CAS PubMed
- T. Kosuge, X. Zhu, V. M. Lau, D. Aoki, T. J. Martinez, J. S. Moore and H. Otsuka, J. Am. Chem. Soc., 2019, 141, 1898–1902 CrossRef CAS PubMed
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed
- C. D. Gabbutt, B. M. Heron, D. A. Thomas, M. E. Light and M. B. Hursthouse, Tetrahedron Lett., 2004, 45, 6151–6154 CrossRef CAS
- K. Arai, Y. Kobayashi and J. Abe, Chem. Commun., 2015, 51, 3057–3060 RSC
- C. J. Blackwell, C. D. Gabbutt, J. T. Guthrie and B. M. Heron, Dyes Pigm., 2012, 95, 408–420 CrossRef CAS
- M. Frigoli, C. Moustrou, A. Samat and R. Guglielmetti, Helv. Chim. Acta, 2000, 83, 3043–3052 CrossRef CAS
- R. Demadrille, C. Moustrou, A. Samat and R. Guglielmetti, Heterocycl. Commun., 1999, 5, 123–126 CAS
- W. Sriprom, M. Néel, C. D. Gabbutt, B. M. Heron and S. Perrier, J. Mater. Chem., 2007, 17, 1885–1893 RSC
- N. Malic, J. A. Campbell and R. A. Evans, Macromolecules, 2008, 41, 1206–1214 CrossRef CAS
- S. K. Osler, M. E. McFadden and M. J. Robb, J. Polym. Sci., 2021, 59, 2537–2544 CrossRef CAS
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 7925–7929 CrossRef CAS PubMed
- B. A. Versaw, M. E. McFadden, C. C. Husic and M. J. Robb, Chem. Sci., 2020, 11, 4525–4530 RSC
- M. E. McFadden, S. K. Osler, Y. Sun and M. J. Robb, J. Am. Chem. Soc., 2022, 144, 22391–22396 CrossRef CAS PubMed
- S. K. Osler, M. E. McFadden, T. Zeng and M. J. Robb, Polym. Chem., 2023, 14, 2717–2723 RSC
-
R. S. Becker, US Pat., US3567605A, 1971 Search PubMed
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC
- S. Brazevic, S. Nizinski, R. Szabla, M. F. Rode and G. Burdzinski, Phys. Chem. Chem. Phys., 2019, 21, 11861–11870 RSC
- H. Görner and A. K. Chibisov, J. Photochem. Photobiol., A, 2002, 149, 83–89 CrossRef
- G. Ottavi, G. Favaro and V. Malatesta, J. Photochem. Photobiol., A, 1998, 115, 123–128 CrossRef CAS
- S. Helmy and J. Read de Alaniz, Adv. Heterocycl. Chem., 2015, 117, 131–177 CrossRef CAS
- S. Delbaere, B. Luccioni-Houze, C. Bochu, Y. Teral, M. Campredon and G. Vermeersch, J. Chem. Soc., Perkin Trans. 2, 1998, 2, 1153–1157 RSC
- S. Jockusch, N. J. Turro and F. R. Blackburn, J. Phys. Chem. A, 2002, 106, 9236–9241 CrossRef CAS
- T. T. Herzog, G. Ryseck, E. Ploetz and T. Cordes, Photochem. Photobiol. Sci., 2013, 12, 1202–1209 CrossRef CAS PubMed
- S. Brazevic, M. Sikorski, M. Sliwa, J. Abe, M. F. Rode and G. Burdzinski, Dyes Pigm., 2022, 201, 110249 CrossRef CAS
- A. Migani, P. L. Gentili, F. Negri, M. Olivucci, A. Romani, G. Favaro and R. S. Becker, J. Phys. Chem. A, 2005, 109, 8684–8692 CrossRef CAS PubMed
- B. H. Strudwick, C. O'Bryen, H. J. Sanders, S. Woutersen and W. J. Buma, Phys. Chem. Chem. Phys., 2019, 21, 11689–11696 RSC
- J. Hobley, V. Malatesta, K. Hatanaka, S. Kajimoto, S. L. Williams and H. Fukumura, Phys. Chem. Chem. Phys., 2002, 4, 180–184 RSC
- J. C. Crano, T. Flood, D. Knowles, A. Kumar and B. Van Gemert, Pure Appl. Chem., 1996, 68, 1395–1398 CrossRef CAS
- J. Hobley, V. Malatesta, R. Millini, W. Giroldini, L. Wis, M. Goto, M. Kishimoto and H. Fukumura, Chem. Commun., 2000, 1339–1340 RSC
- F. Ortica, A. Romani, F. Blackburn and G. Favaro, Photochem. Photobiol. Sci., 2002, 1, 803–808 CrossRef CAS
- G. Harié, A. Samat, R. Guglielmetti, D. De Keukeleire, W. Saeyens and I. Van Parys, Tetrahedron Lett., 1997, 38, 3075–3078 CrossRef
- G. Such, R. A. Evans, L. H. Yee and T. P. Davis, J. Macromol. Sci., Part C: Polym. Rev., 2003, 43, 547–579 CrossRef
- S. Brazevic, M. Baranowski, M. Sikorski, M. Rode and G. Burdzinski, ChemPhysChem, 2020, 21, 1402–1407 CrossRef CAS PubMed
- G. Favaro, A. Romani and R. S. Becker, Photochem. Photobiol., 2000, 72, 632–638 CrossRef CAS
- M. Maafi and R. G. Brown, Int. J. Chem. Kinet., 2005, 37, 717–727 CrossRef CAS
- M. Maafi and R. G. Brown, Int. J. Chem. Kinet., 2006, 38, 421–430 CrossRef CAS
- M. Maafi, Molecules, 2008, 13, 2260–2302 CrossRef CAS PubMed
- S. Delbaere, G. Vermeersch and J. C. Micheau, J. Photochem. Photobiol., C, 2011, 12, 74–105 CrossRef CAS
- R. M. Christie, J. D. Hepworth, C. D. Gabbutt and S. Rae, Dyes Pigm., 1997, 35, 339–346 CrossRef CAS
- C. Lenoble and R. S. Becker, J. Photochem., 1986, 33, 187–197 CrossRef CAS
- S. Delbaere and G. Vermeersch, Tetrahedron Lett., 2003, 44, 259–262 CrossRef CAS
- M. Maafi and R. G. Brown, Int. J. Chem. Kinet., 2005, 37, 162–174 CrossRef CAS
- M. Maafi and R. G. Brown, Int. J. Chem. Kinet., 2006, 38, 431–438 CrossRef CAS
- J. Berthet, S. Delbaere, D. Levi, P. Brun, R. Guglielmetti and G. Vermeersch, J. Chem. Soc., Perkin Trans. 2, 2002, 2118–2124 RSC
- J. Berthet, P. J. Coelho, L. M. Carvalho, G. Vermeersch and S. Delbaere, J. Photochem. Photobiol., A, 2009, 208, 180–185 CrossRef CAS
- S. Delbaere, J. C. Micheau and G. Vermeersch, Org. Lett., 2002, 4, 3143–3145 CrossRef CAS
- S. Delbaere, J.-C. Micheau and G. Vermeersch, J. Org. Chem., 2003, 68, 8968–8973 CrossRef CAS
- D. Venec, S. Delbaere, J. C. Micheau, M. Frigoli, C. Moustrou, A. Samat and G. Vermeersch, J. Photochem. Photobiol., A, 2006, 181, 174–179 CrossRef CAS
-
R. G. Brown, M. Maafi, P. Foggi and L. Bussotti, in Femtochemistry and Femtobiology: Ultrafast Events in Molecular Science, 2004, pp. 283–286 Search PubMed
- J. Aubard, F. Maurel, G. Buntinx, O. Poizat, G. Levi, R. Guglielmetti and A. Samat, Mol. Cryst. Liq. Cryst., 2000, 345, 215–220 CrossRef CAS
- B. Moine, G. Buntinx, O. Poizat, J. Rehault, C. Moustrou and A. Samat, J. Phys. Org. Chem., 2007, 20, 936–943 CrossRef CAS
- S. Brazevic, M. Sliwa, Y. Kobayashi, J. Abe and G. Burdzinski, J. Phys. Chem. Lett., 2017, 8, 909–914 CrossRef CAS PubMed
- P. L. Gentili, A. Romani, R. S. Becker and G. Favaro, Chem. Phys., 2005, 309, 167–175 CrossRef CAS
- P. L. Gentili, E. Danilov, F. Ortica, M. A. J. Rodgers and G. Favaro, Photochem. Photobiol. Sci., 2004, 3, 886–891 CrossRef CAS PubMed
- G. Favaro, A. Romani and R. S. Becker, Photochem. Photobiol., 2001, 74, 378–384 CrossRef CAS
- R. S. Becker, E. Dolan and D. E. Balke, J. Chem. Phys., 1969, 50, 239–245 CrossRef CAS
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed
- M. Maafi and R. G. Brown, Int. J. Chem. Kinet., 2007, 39, 472–479 CrossRef CAS
- S. Brazevic, S. Nizinski, M. Sliwa, J. Abe, M. F. Rode and G. Burdzinski, Int. J. Mol. Sci., 2020, 21, 7825 CrossRef CAS PubMed
-
A. Mannschreck, K. Lorenz and M. Schinabeck, in Organic Photochromic and Thermochromic Compounds: Volume 2: Physicochemical Studies, Biological Applications, and Thermochromism, ed. J. C. Crano and R. J. Guglielmetti, Springer US, Boston, MA, 2002, pp. 261–295 Search PubMed
- B. Gierczyk, M. F. Rode and G. Burdzinski, Sci. Rep., 2022, 12, 10781 CrossRef CAS PubMed
- F. Castet, R. Méreau and D. Liotard, J. Chem. Educ., 2014, 91, 924–928 CrossRef CAS
- F. Maurel, S. Delbaere, S. L. Truong, T. Couesnon, R. Dubest, G. Vermeersch and J. Aubard, J. Phys. Org. Chem., 2007, 20, 944–952 CrossRef CAS
- B. Luccioni-Houzé, M. Campredon, R. Guglielmetti and G. Giusti, Mol. Cryst. Liq. Cryst., 1997, 297, 161–165 CrossRef
- M. Rumi, T. J. White and T. J. Bunning, J. Phys. Chem. B, 2016, 120, 12755–12767 CrossRef CAS PubMed
- M. R. di Nunzio, P. L. Gentili, A. Romani and G. Favaro, J. Phys. Chem. C, 2010, 114, 6123–6131 CrossRef CAS
- G. Pan, W. Jingqiang, A. Zhu, M. Yangfu, F. Meigong and Y. Side, Sci. China, Ser. B: Chem., 2001, 44, 276–282 CrossRef CAS
- P. Fan, G. Pan, J. Wei, Y. Ming, A. Zhu, M. G. Fan and W. M. Hung, Mol. Cryst. Liq. Cryst., 2000, 344, 283–288 CrossRef CAS
- G. Favaro, F. Ortica, A. Romani and P. Smimmo, J. Photochem. Photobiol., A, 2008, 196, 190–196 CrossRef CAS
- Y. Goto, K. Sugita, T. Takahashi, Y. Ohga and T. Asano, Chem. Lett., 2003, 32, 618–619 CrossRef CAS
- K. Sugita, Y. Goto, M. Ono, K. Yamashita, K. Hayase, T. Takahashi, Y. Ohga and T. Asano, Bull. Chem. Soc. Jpn., 2004, 77, 1803–1806 CrossRef CAS
- M. Zayat and D. Levy, J. Mater. Chem., 2003, 13, 727–730 RSC
- R. Pardo, M. Zayat and D. Levy, J. Mater. Chem., 2005, 15, 703–708 RSC
- B. Van Gemert, A. Kumar and D. B. Knowles, Mol. Cryst. Liq. Cryst., 1997, 297, 131–138 CrossRef CAS
- A. Kumar, B. Van Gemert and D. B. Knowles, Mol. Cryst. Liq. Cryst., 2000, 344, 217–222 CrossRef CAS
-
Y.-P. Chan and P. Jean, US Pat., US6207084B1, 2001 Search PubMed
-
D. A. Clarke, B. M. Heron, C. D. Gabbutt, J. D. Hepworth, S. M. Partington and S. N. Corns, WO Pat., WO2000018755A1, 2000 Search PubMed
-
D. A. Clarke, B. M. Heron, C. D. Gabbutt, J. D. Hepworth, S. M. Partington, and S. N. Corns, WO Pat., WO1998042693A2, 1998 Search PubMed
-
B. Van Gemert, WO Pat., WO1996014596A1, 1996 Search PubMed
- B. Van Gemert, M. Bergomi and D. Knowles, Mol. Cryst. Liq. Cryst., 1994, 246, 67–73 CrossRef CAS
- C. D. Gabbutt, B. M. Heron, A. C. Instone, P. N. Horton and M. B. Hursthouse, Tetrahedron, 2005, 61, 463–471 CrossRef CAS
- Z. Wang, Q. Meng, Z. Zhang, D. Fu and W. Zhang, Tetrahedron, 2011, 67, 2246–2250 CrossRef CAS
- C. M. Sousa, J. Berthet, S. Delbaere, A. Polónia and P. J. Coelho, J. Org. Chem., 2017, 82, 12028–12037 CrossRef CAS PubMed
- C. M. Sousa, J. Berthet, S. Delbaere and P. J. Coelho, Dyes Pigm., 2019, 169, 118–124 CrossRef CAS
- C. M. Sousa and P. J. Coelho, Eur. J. Org Chem., 2020, 2020, 985–992 CrossRef CAS
- P. J. Coelho, C. J. R. Silva, C. Sousa and S. D. F. C. Moreira, J. Mater. Chem. C, 2013, 1, 5387–5394 RSC
- C. M. Sousa, J. Berthet, S. Delbaere and P. J. Coelho, J. Org. Chem., 2012, 77, 3959–3968 CrossRef CAS PubMed
- C. M. Sousa, P. J. Coelho, G. Vermeersch, J. Berthet and S. Delbaere, J. Photochem. Photobiol.,
A, 2010, 216, 73–78 CrossRef CAS
- A. M. Gorelik, O. V. Venidiktova, O. I. Kobeleva, T. M. Valova and V. A. Barachevsky, Dyes Pigm., 2021, 184, 108833 CrossRef CAS
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS
- R. Stevenson and G. De Bo, J. Am. Chem. Soc., 2017, 139, 16768–16771 CrossRef CAS PubMed
- M. Radiom, P. Kong, P. Maroni, M. Schäfer, A. F. M. Kilbinger and M. Borkovec, Phys. Chem. Chem. Phys., 2016, 18, 31202–31210 RSC
- W. Huang, Z. Zhu, J. Wen, X. Wang, M. Qin, Y. Cao, H. Ma and W. Wang, ACS Nano, 2017, 11, 194–203 CrossRef CAS
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed
- J. M. Lenhardt, M. T. Ong, R. Choe, C. R. Evenhuis, T. J. Martinez and S. L. Craig, Science, 2010, 329, 1057–1060 CrossRef CAS PubMed
- J. Wang, T. B. Kouznetsova, Z. Niu, M. T. Ong, H. M. Klukovich, A. L. Rheingold, T. J. Martinez and S. L. Craig, Nat. Chem., 2015, 7, 323–327 CrossRef CAS PubMed
- M. T. Ong, J. Leiding, H. Tao, A. M. Virshup and T. J. Martínez, J. Am. Chem. Soc., 2009, 131, 6377–6379 CrossRef CAS PubMed
- J. Ribas-Arino, M. Shiga and D. Marx, Angew. Chem., Int. Ed., 2009, 48, 4190–4193 CrossRef CAS
- A. G. Roessler and P. M. Zimmerman, J. Phys. Chem. C, 2018, 122, 6996–7004 CrossRef CAS
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS
- W. Zhao and E. M. Carreira, J. Am. Chem. Soc., 2002, 124, 1582–1583 CrossRef CAS PubMed
- D. Kim, M. S. Kwon and C. W. Lee, Polym. Chem., 2022, 13, 5177–5187 RSC
- F. Ortica, C. Moustrou, J. Berthet, G. Favaro, A. Samat, R. Guglielmetti, G. Vermeersch and U. Mazzucato, Photochem. Photobiol., 2003, 78, 558–566 CrossRef CAS
- P. J. Coelho, M. A. Salvador, B. M. Heron and L. M. Carvalho, Tetrahedron, 2005, 61, 11730–11743 CrossRef CAS
- C. D. Gabbutt, B. M. Heron, A. C. Instone, S. B. Kolla, K. Mahajan, P. J. Coelho and L. M. Carvalho, Dyes Pigm., 2008, 76, 24–34 CrossRef
- S. Aiken, C. D. Gabbutt, B. M. Heron and S. B. Kolla, Dyes Pigm., 2015, 113, 239–250 CrossRef CAS
- J. A. Odell and A. Keller, J. Polym. Sci., Part B: Polym. Phys., 1986, 24, 1889–1916 CrossRef CAS
- Y. Liu, S. Holm, J. Meisner, Y. Jia, Q. Wu, T. J. Woods, T. J. Martinez and J. S. Moore, Science, 2021, 373, 208–212 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |