
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalyst-mediated, pot-economical total synthesis of latanoprost†
Genki
Kawauchi
 ,
Yurina
Suga
,
Shunsuke
Toda
and
Yujiro
Hayashi
*
,
Yurina
Suga
,
Shunsuke
Toda
and
Yujiro
Hayashi
*
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: yujiro.hayashi.b7@tohoku.ac.jp
First published on 1st August 2023
Abstract
The enantioselective total synthesis of latanoprost, an antiglaucoma agent, has been accomplished with excellent diastereo- and enantioselectivities in a pot-economical manner using six reaction vessels. An enantioselective Krische allylation was conducted in the first pot. In the second pot, olefin metathesis, silyl protection, and hydrogenolysis proceeded efficiently. In the third pot, an organocatalyst-mediated Michael reaction proceeded with excellent diastereoselectivity. The fourth pot involved a substrate-controlled Mukaiyama intramolecular aldol reaction and elimination of HNO2 to afford a methylenecyclopentanone, also with excellent diastereoselectivity. The fifth pot involved a Michael reaction of vinyl cuprate. In the sixth pot, three reactions, a cis-selective olefin metathesis, diastereoselective reduction, and deprotection, afforded latanoprost. Nearly optically pure latanoprost was obtained, and the total yield was 24%.
Introduction
Prostaglandins are an important class of molecules with potent biological activities, and many related drugs have been developed.1 Many methods have been developed for the synthesis of prostaglandins,2 including Corey's famous syntheses via the Corey lactone3 and Noyori's three-component coupling process.4 Because of the importance of prostaglandins, the development of new synthetic methods for prostaglandins is still a significant topic in synthetic organic chemistry. Latanoprost (1) is an antiglaucoma agent and an analog of the prostaglandin PGF2α.5 As it is a blockbuster drug developed by Pharmacia, it is one of the important targets in the synthesis of prostaglandins.Recently, the field of organocatalysis has developed rapidly6 and organocatalyst-mediated reactions have been successfully employed in the synthesis of prostaglandins. A proline-mediated aldol reaction of succinaldehyde was a key step in Aggarwal's synthesis.7 An organocatalytic Baeyer–Villiger oxidation was used by Peng and Chen,8 while Oger and Galano employed an organocatalyst-mediated intramolecular Michael reaction of a formyl-enal derivative.9
We propose the importance of “pot economy” because one-pot operations are efficient methods for making several bonds and can generate complex molecules in a single reaction vessel with several sequential reactions.10 Moreover, one-pot operations circumvent purification steps via in situ quenching, thereby minimizing chemical waste and saving time. Based on this concept, our group has investigated the synthesis of drugs and natural products in a small number of pots.11
Our group also has an interest in the organocatalyst-mediated synthesis of prostaglandins.12 In 2013, we reported the three-pot synthesis of prostaglandin E1 methyl ester.12a Recently we reported a one-pot, 152-minute synthesis of the Corey lactone,12d in which the key step was a formal asymmetric [3 + 2] cycloaddition reaction of ethyl 4-oxo-2-pentenoate and an α,β-unsaturated aldehyde catalyzed by diphenylprolinol silyl ether13 (eqn (1)). We synthesized latanoprost12e and clinprost12f based on this strategy.
Results and discussion
Our new idea for the synthesis of latanoprost was to use 4-nitro-3-butene-2-one instead of ethyl 4-oxo-2-pentenoate (eqn (2), Scheme 1): the ethyl ester was changed to a nitro group, which is not only a good electron-withdrawing group but also an excellent leaving group. The expected reactions were as follows: the Michael reaction of 4-nitrobut-3-ene-2-one and the aldehyde would proceed via an enamine intermediate to afford keto aldehyde A, according to our diphenylprolinol silyl ether-mediated Michael reaction of aldehydes and nitroalkenes.13 A substituted cyclopentanone would be synthesized by a subsequent intramolecular aldol reaction. As NO2 is a good leaving group, a methylenecyclopentanone, which is a key intermediate of prostaglandin F2α reported by Stork and Isobe,14 would be formed by an E1cB reaction.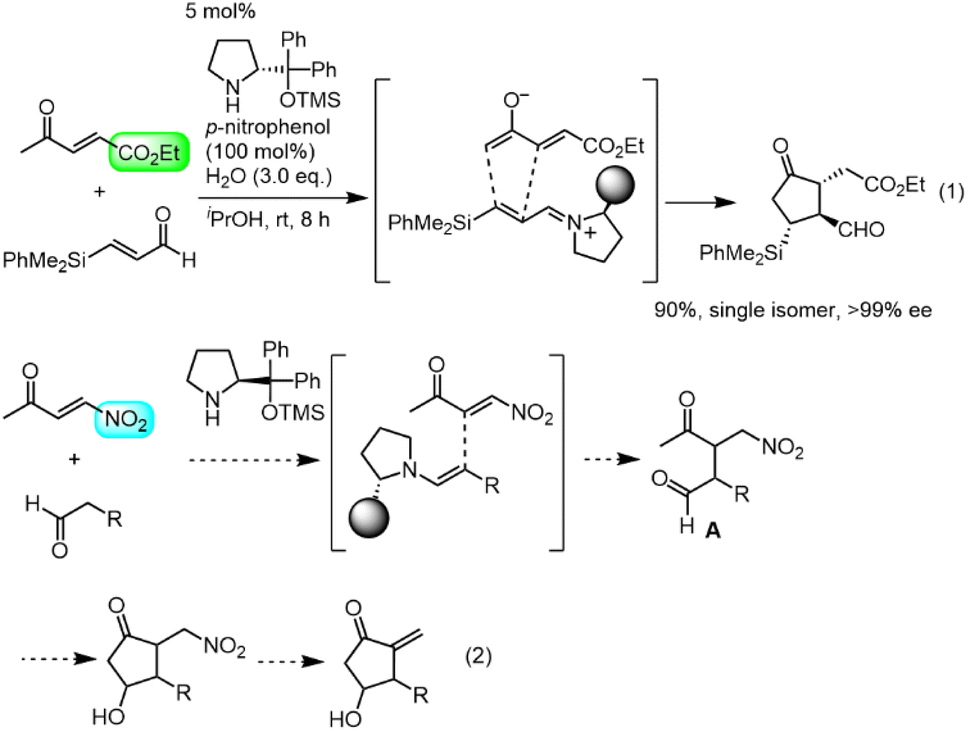 | ||
| Scheme 1 New idea for the synthesis of methylenecyclopentanone. | ||
We examined the reaction of 4-nitrobut-3-en-2-one and 3-phenylpropanal as a model reaction (eqn (3)). Although the first Michael reaction proceeded, the second aldol reaction did not proceed under many different conditions. This was because the keto aldehyde A′ and the generated product underwent facile elimination of HNO2 and/or H2O. Next, we investigated the Mukaiyama aldol reaction15 in the second step. As it is difficult to prepare a silyl enol ether from A′ in the presence of an aldehyde, 4-nitro-2-siloxybuta-1,3-diene 6 was selected as the nitroalkene in the first step. Based on this reasoning, our retrosynthetic analysis is shown in Scheme 2.
 | (3) |
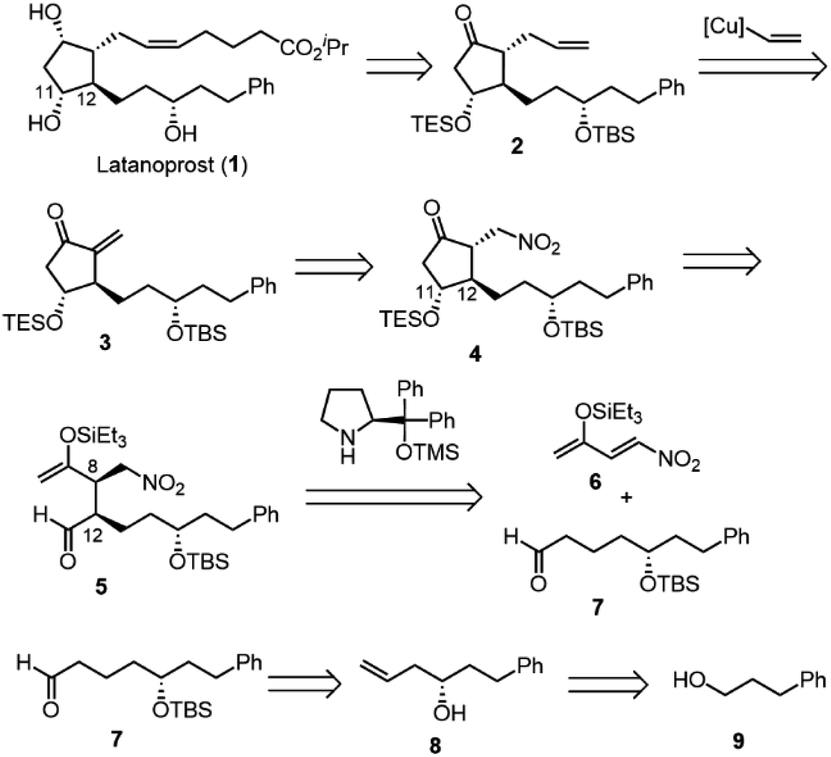 | ||
| Scheme 2 The retrosynthesis of latanoprost (1). | ||
Latanoprost (1) would be synthesized from alkene 2via a cis-selective olefin metathesis, stereoselective reduction of the ketone, and deprotection. 2 would be prepared by a 1,4-addition of vinyl cuprate into methylenecyclopentanone 3. 3 would be synthesized by the elimination of the nitro group from 4, which would be prepared from 5 by an intramolecular Mukaiyama aldol reaction. An organocatalyst-mediated Michael reaction of 6 and 7 would afford 5. 7 would be prepared from 8, which would be synthesized by a Krische allylation from alcohol 9.
There are several concerns with this retrosynthesis. One is the reactivity of 6 as a Michael acceptor. Nitroalkene 6 has an electron-donating group, which would decrease its reactivity as a Michael acceptor. The other concern is the diastereoselectivity at C11 and C12. The C12 position has a chance to epimerize during the Mukaiyama aldol reaction. It was also a concern whether high diastereoselectivity at C11 would be obtained in the Mukaiyama aldol reaction.
Our synthesis commenced with Krische allylation16 of 3-phenylpropanol (9) to afford the allyl alcohol 10 in 88% yield with 96% ee (Scheme 3). Olefin metathesis of 10 with acrolein catalyzed by the Grubbs second generation catalyst17 proceeded to afford 11 in 86% yield. Alcohol protection with tert-butyldimethylsilyl chloride (TBSCl) provided 12. Hydrogenolysis using Pd/C gave aldehyde 7 in 94% yield.
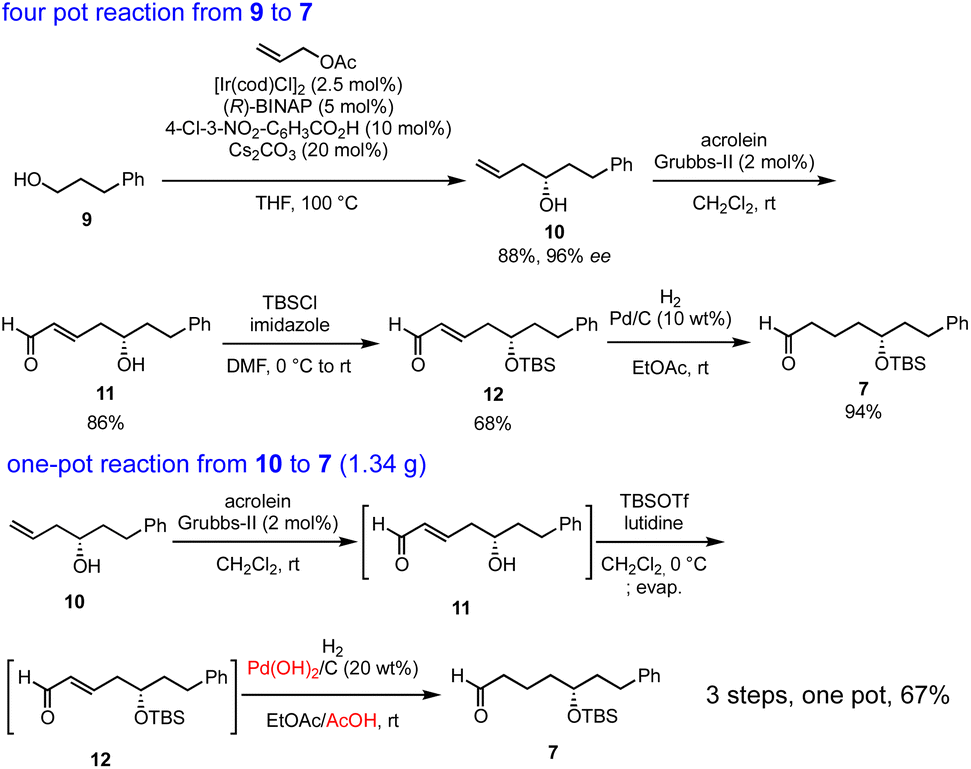 | ||
| Scheme 3 Four-pot reaction from 9 to 7 and one-pot reaction from 10 to 7. | ||
The transformation from 10 into 7 could be conducted in a single vessel. After the olefin metathesis, the addition of TBSOTf and lutidine afforded 12. After evaporation and the addition of EtOAc, AcOH, and Pd(OH)2/C, hydrogenolysis proceeded under an H2 atmosphere to afford 7 in 67% yield over three steps in one pot. The use of Perlman's catalyst18 under acidic conditions (AcOH) is key to the success of the one-pot reaction. The reaction proceeded on a gram scale. Notably, the yield in the one-pot reaction (67%, 10 → 7) was higher than that of the stop-and-go method (55%, three steps).
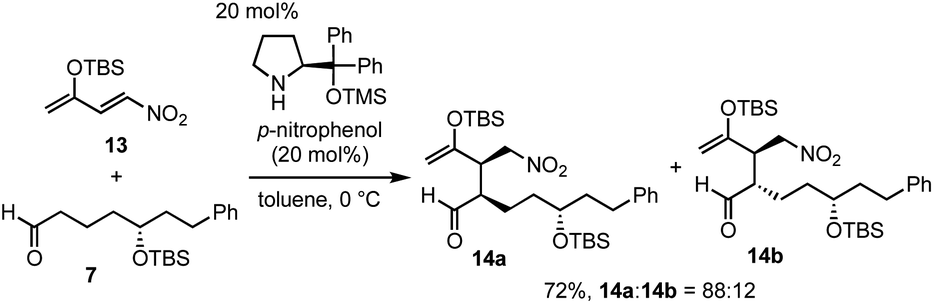
Next was one of the key reactions. First, a nitroalkene with tert-butyldimethylsilyl enol ether 13 was used as a Michael acceptor. Despite our concern about the decrease in the reactivity of 13 as a Michael acceptor (vide supra), the reaction of nitroalkene 13 and aldehyde 7 proceeded efficiently using 20 mol% of the catalyst in the presence of p-nitrophenol (eqn (4)). The reaction was completed within 45 minutes at 0 °C to afford the Michael product 14 in 72% yield with good diastereoselectivity (dr = 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12).
:12).
 | (4) |
We then investigated the intramolecular aldol reaction. We found that the aldol product 15 was unstable. Thus, after the treatment of the Michael product 14 with a Lewis acid, the aldol product 15 was converted into methylene-cyclopentanone 16 using NaF and Et3N in the same reaction vessel. The yield and diastereoselectivity of 16 were determined (Table 1). Several Lewis acids are known to catalyze the Mukaiyama aldol reaction. The reaction did not proceed in the presence of Sc(OTf)319 (entry 1). A combination of trimethylsilyl chloride (TMSCl) and SnCl220 or trityl trifluoromethanesulfonate (TrOTf)21 gave a complex mixture (entries 2 and 3). A combination of TrCl and SnCl222 afforded the product 16 in 20% yield, along with the deprotected alcohol 17 in 51% yield (entry 4). Me2AlCl23 afforded 16 in 46% yield with a good diastereoselectivity (dr = 6:1) and alcohol 17 in 10% (entry 5). To increase the yield of 16, we tried to suppress the deprotection of the TBS group, but there was no success.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Lewis acid | X [mol%] | Temp. [°C] | Time [h] | Yieldb [%] | drc |
|
a Unless otherwise shown, reactions were performed by employing 14 (0.20 mmol) and a Lewis acid (0.040 mmol) in CH2Cl2 (4.0 mL) at the indicated temperature and time.
b Isolated yield of 16.
c The diastereomer ratio (C11:C12) was determined by 1H-NMR analysis.
d NR = no reaction.
e CM = complex mixture.
f
17 was obtained in 51% yield.
g
17 was obtained in 10% yield.
|
||||||
| 1 | Sc(OTf)3 | 20 | −78 to 23 | 13 | NRd | |
| 2 | TMSCl + SnCl2 | 20 + 20 | −78 to 0 | 4 | CMe | |
| 3 | TrOTf | 20 | −78 | 1 | CMe | |
| 4f | TrCl + SnCl2 | 20 + 20 | −20 | 1 | 20 | 3:1 |
| 5g | Me2AlCl | 70 | −78 | 2 | 46 | 6:1 |
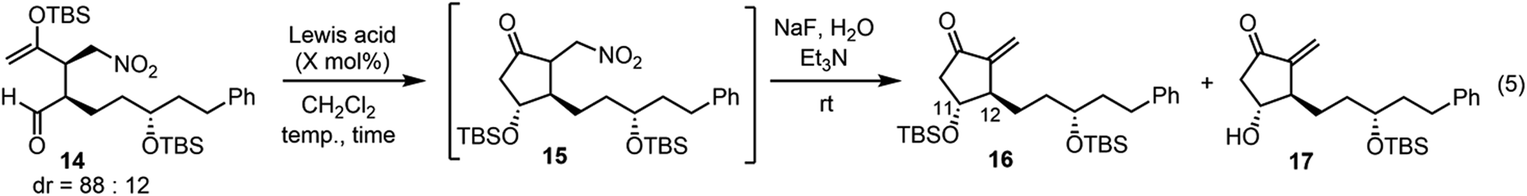
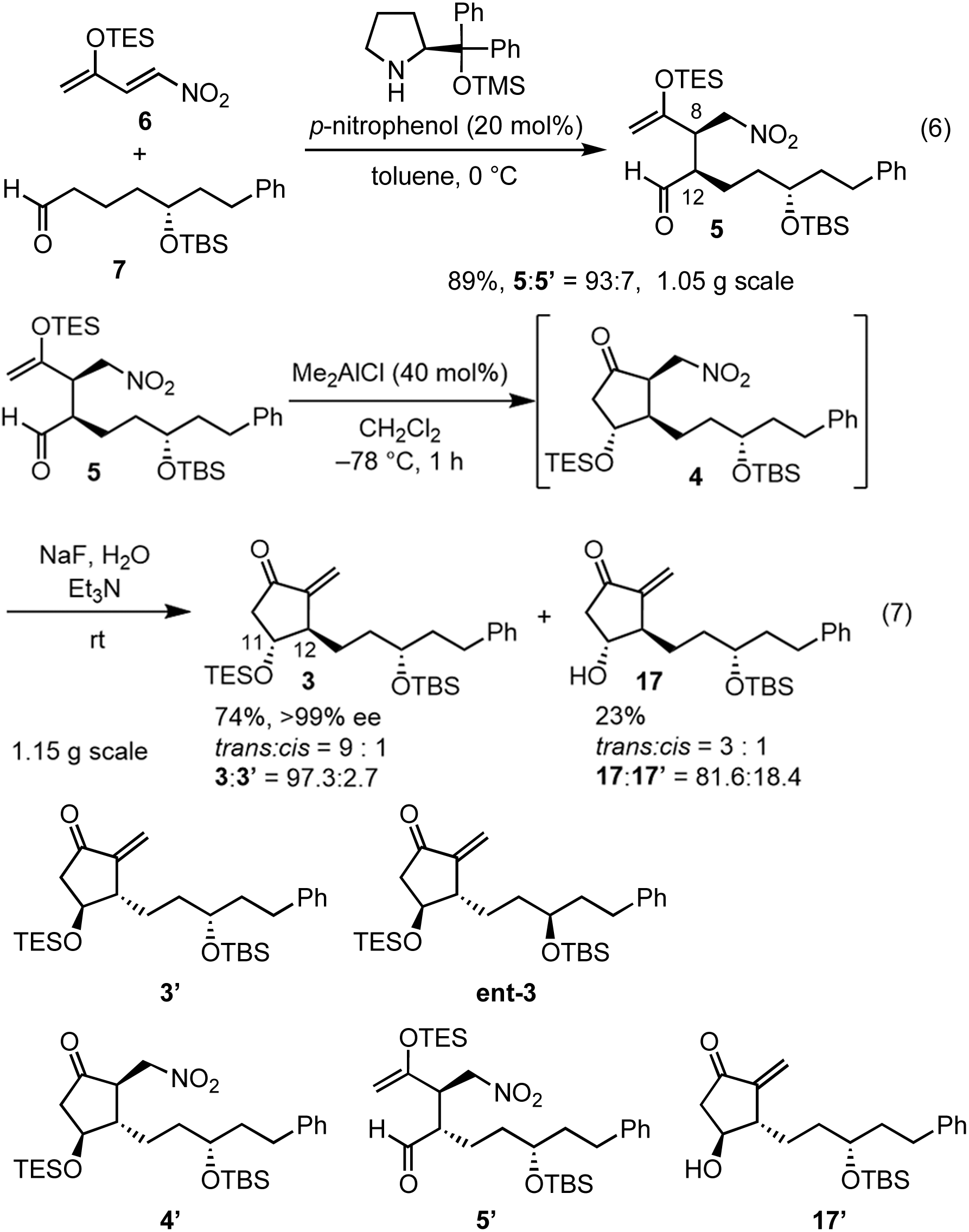
In the Mukaiyama aldol reaction, triethylsilyl enol ethers are more reactive than tert-butyldimethylsilyl enol ethers. Thus, we examined the reaction of the nitroalkene triethylsilyl enol ether 6. The first Michael reaction of 6 and 7 proceeded with a much higher yield (89%) and diastereoselectivity (5:5′ = 93:7, eqn (6), Fig. 1) than those of the reaction using the TBS enol ether 13 (eqn (4)). The aldol reaction and elimination of HNO2 proceeded efficiently using sequential treatment with Me2AlCl followed by NaF and Et3N to afford 3 in good yield (74%) along with alcohol 17 in 23% yield (eqn (7)). 3 possesses good diastereoselectivity: the trans:cis selectivity is 9:1, and the diastereomer ratio of 3:3′ is excellent (97.3:2.7). We also synthesized the enantiomer of 3 (ent-3), and prepared the racemic (±)-3 by mixing 3 and ent-3. The HPLC analysis of 3 and racemic (±)-3 using a chiral phase column indicated that the optical purity of 3 is over 99%.24
 | ||
| Fig. 1 The other intermediates 3′, ent-3, 4′, 5′ and 17′. | ||
It was found that the diastereoselectivity of 17:17′ (81.6:18.4) is lower than that of 3:3′. As the epimerization from 5 to 5′ would proceed during the next Mukaiyama aldol reaction, the selectivity of 5:5′ would be much worse. Even though, the ratio of 3:3′ is higher than those of 5:5′ and 17:17′, which is synthetically useful. As 4 and 4′ are diastereomers, the reaction speed of the deprotection of the TES group would be different. It is very difficult to check the diastereoselectivity of 4:4′ because of the facile elimination of HNO2. The deprotection from 4′ would be faster than that from 4. Thus, kinetic resolution would occur to afford the higher diastereoselectivity in 3 with lower diastereoselectivity in 17 than in the parent 5.
It should be noted that TES enol ether 6 is superior to its TBS counterpart in terms of yield and selectivity in both the organocatalyst-mediated Michael reaction and the Mukaiyama aldol reaction. Both reactions proceeded efficiently on a gram scale. The stereochemistry at C12 was controlled by the diphenylprolinol silyl ether. At this stage, we could not definitively determine the stereochemistry at C11 by NMR analysis. However, we continued the total synthesis, hoping that 3 would possess the correct configuration (vide infra).
Next, we investigated the Michael addition of a vinyl anion to 3. Stork and Isobe reported the Michael reaction of a similar methylenecyclopentanone with a dialkenyl cuprate bearing a long alkyl chain.14 We found that the choices of the copper reagent and additive were important for the success of the reaction, as the siloxy elimination was a side reaction to afford products analogous to 18 and 19 (Fig. 2). For instance, when TMSCl was employed as an additive, 18 and 19 were generated at about 20% and 28%, respectively.24 The addition product 2 was obtained in good yield (83%) as a single isomer on a gram scale when [CuI(PBu3)]425 and vinyl lithium in the presence of BF3·OEt226 were employed (eqn (8)).
 | ||
| Fig. 2 The side products in the Michael reaction of 3. | ||
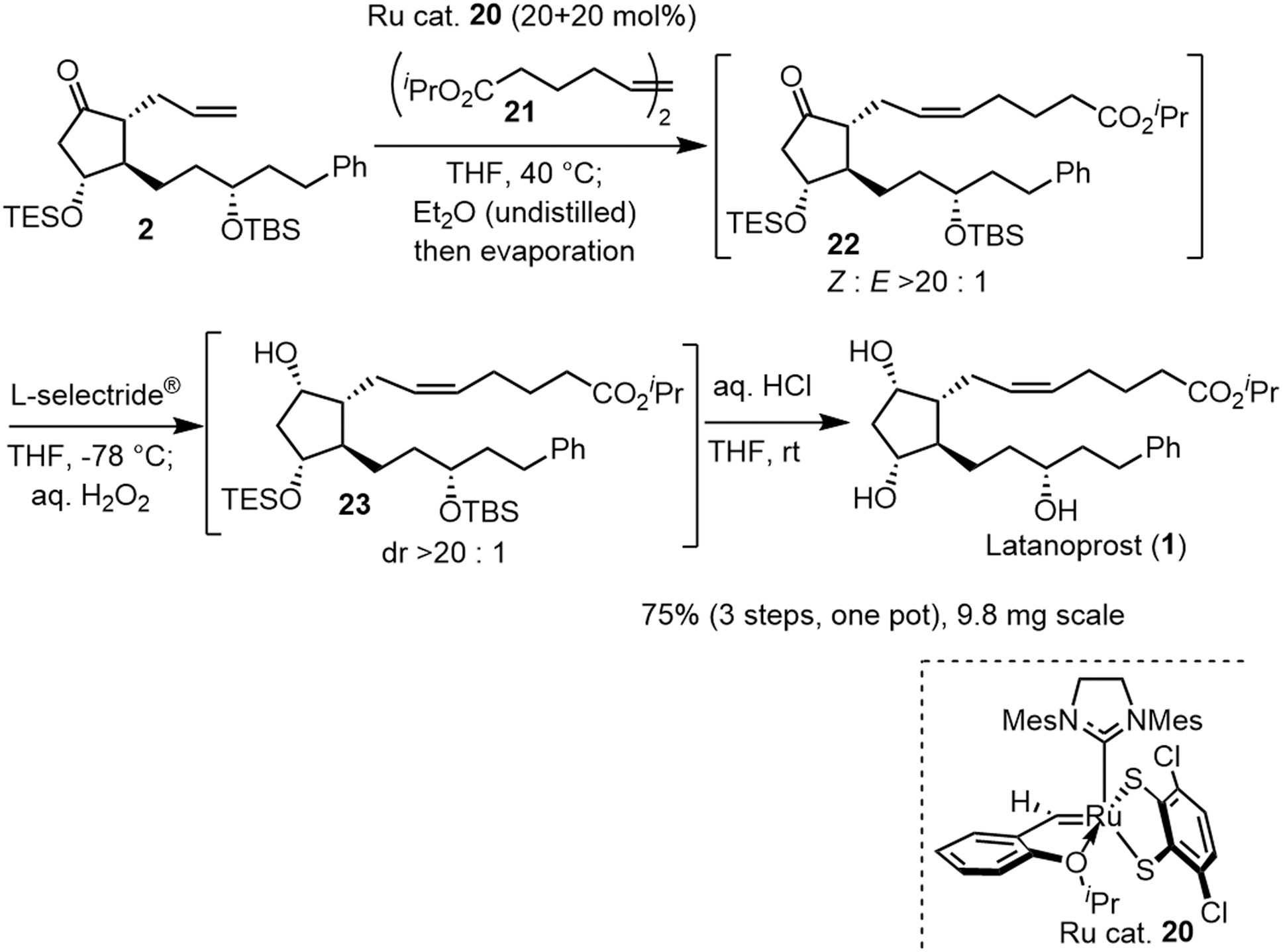
The last three steps (2 → 1) were olefin metathesis, reduction, and deprotection, which could be conducted in a single vessel (Scheme 4). First, the cis-selective olefin metathesis27 proceeded when 2 was treated with alkene 2127c in the presence of the Ru catalyst 20, which was developed by Grubbs and coworkers and used in the prostaglandin synthesis to afford 22 with excellent diastereoselectivity. The next reaction was the stereoselective reduction of the ketone employing L-selectride®. After olefin metathesis, the remaining Ru catalyst was deactivated by the addition of undistilled Et2O.27a Then, after evaporation, the reduction proceeded efficiently with the addition of L-selectride® and THF in the same reaction vessel. The addition of aqueous H2O2 decomposed the remaining L-selectride®. With the further addition of aqueous HCl, deprotection of the two silyl groups afforded latanoprost (1). This was a one-pot reaction, and the yield of 1 from 2 was 75%. As latanoprost possesses very potent biological activity, the final step and purification must be carried out with great care. For the safety of the experimenters, the reaction was conducted on a 9.8 mg scale. The physical properties of the synthetic latanoprost (1) were identical in all respects to those in the reported data,5g which confirmed the stereochemistry at C11 generated by the Mukaiyama aldol reaction (5 → 4).
 | ||
| Scheme 4 One-pot reaction from 2 to latanoprost (1). | ||
Conclusions
In summary, this is a highly diastereo- and enantioselective, six-pot total synthesis of latanoprost (1) with a total yield of 24% (Scheme 5). The first pot consists of a Krische allylation. The second pot involves three reactions: olefin metathesis, TBS protection, and hydrogenolysis. The third pot involves an organocatalyst-mediated Michael reaction of an aldehyde and nitroalkene. The fourth pot involves an intramolecular Michael reaction and an E1cB reaction with the elimination of HNO2. The fifth pot reaction is a Michael addition of vinyl cuprate. The sixth pot involves olefin metathesis, reduction, and deprotection. The present synthesis possesses several noteworthy features: (1) the stereochemistry at C15 was controlled by an asymmetric allylation developed by Krische and coworkers. (2) The stereochemistry at C12 was controlled by an organocatalytic reaction developed by our group. (3) A key substituted cyclopentanone was synthesized by an organocatalyst-mediated Michael reaction and a substrate-controlled intramolecular Mukaiyama aldol reaction, both of which proceeded with high diastereoselectivity. (4) The α-side chain was introduced to the methylenecyclopentanone via vinyl addition, followed by the cis-selective olefin metathesis. (5) This is the total synthesis of latanoprost with the fewest number of pots. (6) Nearly optically pure latanoprost was obtained. | ||
| Scheme 5 Six-pot total synthesis of latanoprost (1). | ||
Data availability
General information, detailed experimental procedures, characterization data for compounds, and NMR, HPLC, IR spectra are available in the ESI.†Author contributions
G. K., Y. S., and S. T. performed the experiments. Y. H. conceived the concept and prepared the manuscript with feedback from G. K., Y. S., and S. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Minoru Isobe for the information on the Michael reaction of dialkenyl cuprate. We also thank the reviewers for their excellent suggestions. This work was supported by JSPS KAKENHI Grant Number JP19H05630.Notes and references
- C. D. Funk, Science, 2001, 294, 1871 CrossRef CAS PubMed.
- Reviews, see; (a) S. Das, S. Chandrasekhar, J. S. Yadav and R. Grée, Chem. Rev., 2007, 107, 3286 CrossRef CAS PubMed; (b) H. Peng and F.-E. Chen, Org. Biomol. Chem., 2017, 15, 6281 RSC; recent selected syntheses (c) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (d) T. Kim, S. I. Lee, S. Kim, S. Y. Shim and D. H. Ryu, Tetrahedron, 2019, 75, 130593 CrossRef CAS; (e) R. Kučera, F. W. Goetzke and S. P. Fletcher, Org. Lett., 2020, 22, 2991 CrossRef PubMed; (f) K. Zhu, M. Jiang, B. Ye, G.-T. Zhang, W. Li, P. Tang, Z. Huang and F. Chen, Chem. Sci., 2021, 12, 10362 RSC; (g) F. Zhang, J. Zeng, M. Gao, L. Wang, G.-Q. Chen, Y. Lu and X. Zhang, Nat. Chem., 2021, 13, 692 CrossRef CAS PubMed; (h) X. Yi, X. Long, Y. Chen, X. Cen, P. Tang and F. Chen, Chem. Commun., 2022, 58, 6000 RSC; (i) L. Cunningham, S. Mishra, L. Matthews and S. P. Fletcher, Org. Lett., 2022, 24, 8886 CrossRef CAS PubMed.
- (a) E. J. Corey and X. M. Cheng, The Logic of Chemical Synthesis, Wiley, New York, 1995 Search PubMed; (b) E. J. Corey, N. M. Wesinshenker, T. K. Schaaf and W. Huber, J. Am. Chem. Soc., 1969, 91, 5675 CrossRef CAS PubMed; (c) E. J. Corey, T. K. Schaaf, W. Huber, U. Koelliker and N. M. Wesinshenker, J. Am. Chem. Soc., 1970, 92, 397 CrossRef CAS PubMed; (d) E. J. Corey, S. M. Albonico, U. Koelliker, T. K. Schaaf and R. K. Varma, J. Am. Chem. Soc., 1971, 93, 1490 CrossRef CAS PubMed; (e) E. J. Corey and H. E. Ensley, J. Am. Chem. Soc., 1975, 97, 6908 CrossRef CAS PubMed; (f) E. J. Corey and T. P. Loh, J. Am. Chem. Soc., 1991, 113, 8966 CrossRef CAS.
- (a) R. Noyori and M. Suzuki, Angew. Chem., Int. Ed. Engl., 1984, 23, 847 CrossRef; (b) M. Suzuki, A. Yanagisawa and R. Noyori, J. Am. Chem. Soc., 1988, 110, 4718 CrossRef CAS.
- (a) S. S. Patel and C. M. Spencer, Drugs Aging, 1996, 9, 363 CrossRef CAS PubMed; (b) C. M. Perry, J. K. McGavin, C. R. Culy and T. Ibbotson, Syntheses, Drugs Aging, 2003, 20, 597 CrossRef CAS PubMed; (c) I. Józef, H. István, I. Józefiné and B. Resul, WO9300329, KABI PHARMACIA AB, 1993; (d) B. Resul, J. Stjernschantz, K. No, C. Liljebris, G. Selen, M. Astin, M. Karlsson and L. Z. Bito, J. Med. Chem., 1993, 36, 243 CrossRef CAS PubMed; (e) I. Obadalová, T. Pilarčík, M. Slavíková and J. Hájíček, Chirality, 2005, 17, S109 CrossRef PubMed; (f) J. G. Martynow, J. Jóźwik, W. Szelejewski, O. Achmatowicz, A. Kutner, K. Wiś-niewski, J. Winiarski, O. Zegrocka-Stendel and P. Gołębiewski, Eur. J. Org. Chem., 2007, 689 CrossRef CAS; (g) G. Zanoni, A. D'Alfonso, A. Porta, L. Feliciani, S. P. Nolan and G. Vidari, Tetrahedron, 2010, 66, 7472 CrossRef CAS; (h) K. Vijendhar, B. Srinivas and S. Boodida, J. Chem. Sci., 2015, 127, 2023 CrossRef CAS; (i) S. A. Sasane, N. B. Bhise, G. P. Shingh, A. Joseph and G. G. Shenoy, Synth. Commun., 2019, 49, 2350 CrossRef CAS; formal total syntheses (j) M. Harikrishna, H. R. Mohan, P. K. Dubey, M. Shankar and G. V. Subbaraju, Asian J. Chem., 2011, 23, 3121 CAS; (k) M. L. Contente, P. Zambelli, S. Galafassi, L. Tamborini, A. Pinto, P. Conti, F. Modinari and D. Romano, J. Mol. Catal. B: Enzym., 2015, 114, 7 CrossRef CAS.
- Selected reviews on organocatalysis: (a) T. D. Beeson, M. Benohoud, J. W. Bode, S. Chen, M. Christmann, P.-C. Chiang, D. A. DiRocco, Y. C. Fan, T. Furuta, P. García-García, S. Hatakeyama, Y. Hayashi, J. Jia, T. Kawabata, N. J. Kerrigan, O. Kwon, Y. Liu, D. W. C. MacMillan, N. Mase, P. Melchiorre, S. Mukherjee, A. Piisola, P. M. Pihko, T. A. Ramirez, T. Rovis, E. C. Salo, Y. Shi, A. D. Smith, K. Suzuki, H. Takikawa, X.-W. Wang, Y. Wang, A. J. B. Watson, O. A. Wong, P. A. Woods and S. M. Yliniemelä-Sipari, Asymmetric Organocatalysis, ed. B. List, Thieme, Stuttgart, 2012, vol. 1 Search PubMed; (b) H. Jiang, Ł. Albrecht, G. Dickmeiss, K. L. Jensen and K. A. Jørgensen, Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2013, pp. 33–50 Search PubMed.
- (a) G. Coulthard, W. Erb and V. K. Aggarwal, Nature, 2012, 489, 278 CrossRef CAS PubMed; (b) S. Prévost, K. Thai, N. Schützenmeister, G. Coulthard, W. Erb and V. K. Aggarwal, Org. Lett., 2015, 17, 504 CrossRef PubMed; (c) H. Baars, M. J. Classen and V. K. Aggarwal, Org. Lett., 2017, 19, 6008 CrossRef CAS PubMed; (d) A. Pelss, N. Gandhamsetty, J. R. Smith, D. Mailhol, M. Silvi, A. Watson, I. Perez-Powell, S. Prévost, N. Schützenmeister, P. Moore and V. K. Aggarwal, Chem. - Eur. J., 2018, 24, 9542 CrossRef CAS PubMed; (e) S. Bennett, G. Coulthard and V. K. Aggarwal, Chem. Rec., 2020, 20, 936 CrossRef CAS PubMed.
- (a) K. Zhu, S. Hu, M. Liu, H. Peng and F.-E. Chen, Angew. Chem., Int. Ed., 2019, 58, 9923 CrossRef CAS PubMed; (b) K. Zhu, M. Jiang, B. Ye, G.-T. Zhang, W. Li, P. Tang, Z. Huang and F. Chen, Chem. Sci., 2021, 12, 10362 RSC.
- J. Revol-Cavalier, V. Bultel-Poncé, A. Guy, T. Durand, C. Oger and J.-M. Galano, Org. Lett., 2020, 22, 7455 CrossRef CAS PubMed.
- (a) Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC; (b) Y. Hayashi, J. Org. Chem., 2021, 86, 1 CrossRef CAS PubMed; (c) Y. Hayashi, Acc. Chem. Res., 2021, 54, 1385 CrossRef CAS PubMed.
- (a) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304 CrossRef CAS PubMed; (b) H. Ishikawa, M. Honma and Y. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 2824 CrossRef CAS PubMed; (c) Y. Hayashi and S. Ogasawara, Org. Lett., 2016, 18, 3426 CrossRef CAS PubMed; (d) Y. Hayashi, D. Sakamoto and D. Okamura, Org. Lett., 2016, 18, 4 CrossRef CAS PubMed; (e) Y. Hayashi, S. Koshino, K. Ojima and E. Kwon, Angew. Chem., Int. Ed., 2017, 56, 11812 CrossRef CAS PubMed; (f) T. Terunuma and Y. Hayashi, Nat. Commun., 2022, 13, 7503 CrossRef CAS PubMed.
- (a) Y. Hayashi and S. Umemiya, Angew. Chem., Int. Ed., 2013, 52, 3450 CrossRef CAS PubMed; (b) S. Umemiya, D. Sakamoto, G. Kawauchi and Y. Hayashi, Org. Lett., 2017, 19, 1112 CrossRef CAS PubMed; (c) G. Kawauchi, S. Umemiya, T. Taniguchi, K. Monde and Y. Hayashi, Chem. - Eur. J., 2018, 24, 8409 CrossRef CAS; (d) N. Umekubo, Y. Suga and Y. Hayashi, Chem. Sci., 2020, 11, 1205 RSC; (e) N. Umekubo and Y. Hayashi, Eur. J. Org. Chem., 2020, 6221 CrossRef CAS; (f) N. Umekubo and Y. Hayashi, Org. Lett., 2020, 22, 9365 CrossRef CAS PubMed.
- Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed.
- G. Stork and M. Isobe, J. Am. Chem. Soc., 1975, 97, 4745 CrossRef CAS PubMed.
- (a) T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 2, 1011 CrossRef; (b) J. Matsuo and T. Mukaiyama, Angew. Chem., Int. Ed., 2013, 52, 9109 CrossRef CAS PubMed; (c) T. Mukaiyama, Organic reactions, ed. W. G. Dauben, G. A. Boswell, Jr., S. Danishefsky, H. W. Gschwend, R. F. Heck, R. F. Hirshchmann, A. S. Kende, L. A. Paquette, G. H. Posner, B. M. Trost, R. Bittman and B. Weinstein, Wiley, New York, 1982, vol 28, pp. 203–331 Search PubMed.
- I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 6340 CrossRef CAS PubMed.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed.
- W. M. Perlman, Tetrahedron Lett., 1967, 8, 1663 CrossRef.
- M. P. Doyle, K. Kundu and A. E. Russell, Org. Lett., 2005, 7, 5171 CrossRef CAS PubMed.
- N. Iwasawa and T. Mukaiyama, Chem. Lett., 1987, 16, 463 CrossRef.
- S. Kobayashi, M. Murakami and T. Mukaiyama, Chem. Lett., 1985, 14, 1535 CrossRef.
- S. Kobayashi and T. Mukaiyama, J. Organomet. Chem., 1990, 382, 39 CrossRef.
- Y. Naruse, J. Ukai, N. Ikeda and H. Yamamoto, Chem. Lett., 1985, 14, 1451 CrossRef.
- See the ESI† in detail.
- J. Hood and R. B. Layton, Can. J. Chem., 1970, 48, 1626 CrossRef.
- Y. Yamamoto, Angew. Chem., Int. Ed., 1986, 25, 947 CrossRef.
- (a) S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461 CrossRef CAS PubMed; (b) M. J. Koh, R. K. M. Khan, S. Torker, M. Yu, M. S. Mikus and A. H. Hoveyda, Nature, 2015, 517, 181 CrossRef CAS PubMed; (c) C. Xu, X. Shen and A. H. Hoveyda, J. Am. Chem. Soc., 2017, 139, 10919 CrossRef CAS PubMed; (d) J. Li, T. S. Ahmed, C. Xu, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2019, 141, 154 CrossRef CAS PubMed; (e) J. Li, B. M. Stoltz and R. H. Grubbs, Org. Lett., 2019, 21, 1013 Search PubMed; (f) K. C. Nicolaou, K. K. Pulukuri, S. Rigol, Z. Peitsinis, R. Yu, S. Kishigami, N. Cen, M. Aujay, J. Sandoval, N. Zepeda and J. Gavrilyuk, J. Org. Chem., 2019, 84, 365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02978f |
| This journal is © The Royal Society of Chemistry 2023 |