
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTherapeutic efficacy of 211At-radiolabeled 2,6-diisopropylphenyl azide in mouse models of human lung cancer†
Yudai
Ode
 a,
Ambara R.
Pradipta
*a,
Peni
Ahmadi‡
b,
Akihiro
Ishiwata
b,
Akiko
Nakamura
b,
Yasuko
Egawa
b,
Yuriko
Kusakari
b,
Kyohei
Muguruma
b,
Yang
Wang
c,
Xiaojie
Yin
c,
Nozomi
Sato
c,
Hiromitsu
Haba
c and
Katsunori
Tanaka
*ab
a,
Ambara R.
Pradipta
*a,
Peni
Ahmadi‡
b,
Akihiro
Ishiwata
b,
Akiko
Nakamura
b,
Yasuko
Egawa
b,
Yuriko
Kusakari
b,
Kyohei
Muguruma
b,
Yang
Wang
c,
Xiaojie
Yin
c,
Nozomi
Sato
c,
Hiromitsu
Haba
c and
Katsunori
Tanaka
*ab
aDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro, Tokyo 152-8552, Japan. E-mail: pradipta.a.aa@m.titech.ac.jp; tanaka.k.dg@m.titech.ac.jp
bBiofunctional Synthetic Chemistry Laboratory, Cluster for Pioneering Research, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
cNuclear Chemistry Research Team, RIKEN Nishina Center for Accelerator-Based Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 27th June 2023
Abstract
Targeted α-particle therapy (TAT) is an attractive alternative to conventional therapy for cancer treatment. Among the available radionuclides considered for TAT, astatine-211 (211At) attached to a cancer-targeting molecule appears very promising. Previously, we demonstrated that aryl azide derivatives could react selectively with the endogenous acrolein generated by cancer cells to give a diazo compound, which subsequently forms a covalent bond with the organelle of cancer cells in vivo. Herein, we synthesized 211At-radiolabeled 2,6-diisopropylphenyl azide (ADIPA), an α-emitting molecule that can selectively target the acrolein of cancer cells, and investigated its antitumor effect. Our results demonstrate that a single intratumor or intravenous administration of this simple α-emitting molecule to the A549 (human lung cancer) cell-bearing xenograft mouse model, at a low dose (70 kBq), could suppress tumor growth without inducing adverse effects. Furthermore, because acrolein is generally overproduced by most cancer cells, we believe ADIPA is a simple TAT compound that deserves further investigation for application in animal models and humans with various cancer types and stages.
Introduction
The significant improvement in cancer research within the last decades has contributed to an increase in cancer survival. However, the currently available treatment methods still have some challenges, particularly for treating advanced and metastatic cancer. Recently, targeted α-particle therapy (TAT) has been introduced as a potential approach to overcome these difficulties.1,2 The concept of TAT is based on unique properties of α-particles. α-Particles tend to have more energy but travel a shorter distance (50–80 μm) in human tissue than other forms of radiation.3 Therefore, α-particles can kill cancer cells by slicing through both strands of their DNA, but the damage is limited to only a small number of cells near the particles' source.Astatine-211 (211At),4 one of several elements that emits an α-particle when it decays, is of interest as a radionuclide for TAT. 211At decays with a half-life of 7.21 hours in two branches (ESI, Fig. S1†), either by α-emission to bismuth-207 (207Bi) or by electron capture (EC) to polonium-211 (211Po).5 After the EC, 211Po promptly decays by α-emission to the stable lead-207 (207Pb). This process demonstrates that each decay will yield one α particle. In addition, the EC branch to 211Po will deliver characteristic polonium X-rays in the range of 70–90 keV.6 This process enables simple quantification, and the energy of the X-rays is in the field where it can be imaged with a γ camera. Furthermore, being a halogen, 211At can often be incorporated into biomolecules of interest by adapting radioiodination chemistry.7 Therefore, by attaching 211At to molecules designed to target specific cancer cells, we could develop an anticancer therapy that should cause minor damage to surrounding healthy tissue compared with the effects of current chemotherapy and radiation therapy.8,9
meta-[211At]-Benzylguanidine was synthesized as an analog of meta-[131I]-benzylguanidine, a known radiotracer for neuroendocrine tumors (ESI, Fig. S2†).10,11 Likewise, [211At]-L-phenylalanine12,13 and [211At]-α-methyl-L-tyrosine14 were synthesized as analogs of amino acids that have a high binding affinity for the cancer-specific L-type amino acid transporter 1 (LAT1). Other groups have also reported the combination of 211At with RGD peptide15,16 or gold nanoparticles.17 Meanwhile, our group has previously utilized 211At attached to trastuzumab,18 a humanized anti-HER2 monoclonal antibody, as α-emitting cancer-targeting molecules.19 Principally, these 211At-labeled compounds were expected to target a specific cancer through a mechanism similar to that of their parent compounds. However, these previously reported TAT compounds are designed based on molecules targeting a particular type of cancer. Namely, a specific 211At-labeled compound needs to be prepared for each different cancer treatment. Also, astatination may work efficiently with some cancer-targeting compounds but not all. Therefore, we thought it might be helpful to develop a simple TAT compound that could target various types of cancer and be used generally without being affected by cancer subtypes.
Previously, we described the 1,3-dipolar cycloaddition between 2,6-diisopropylphenyl azide 1 and acrolein that proceeds under physiological conditions and without a catalyst (Fig. 1a).20 The reaction gives intermediate triazoline 2, which immediately transforms into triazole derivative 3 (27%) and α-diazocarbonyl derivative 4 (53%). The two bulky isopropyl substituents are essential to enhance the reactivity of the aryl azide towards acrolein.20,21 Significantly, the azide-acrolein 1,3-dipolar cycloaddition is highly chemoselective. Under similar conditions, no discernible products were found when the aryl azide was reacted with α- or β-substituted acrolein and activated olefins (ESI, Fig. S3a†).22
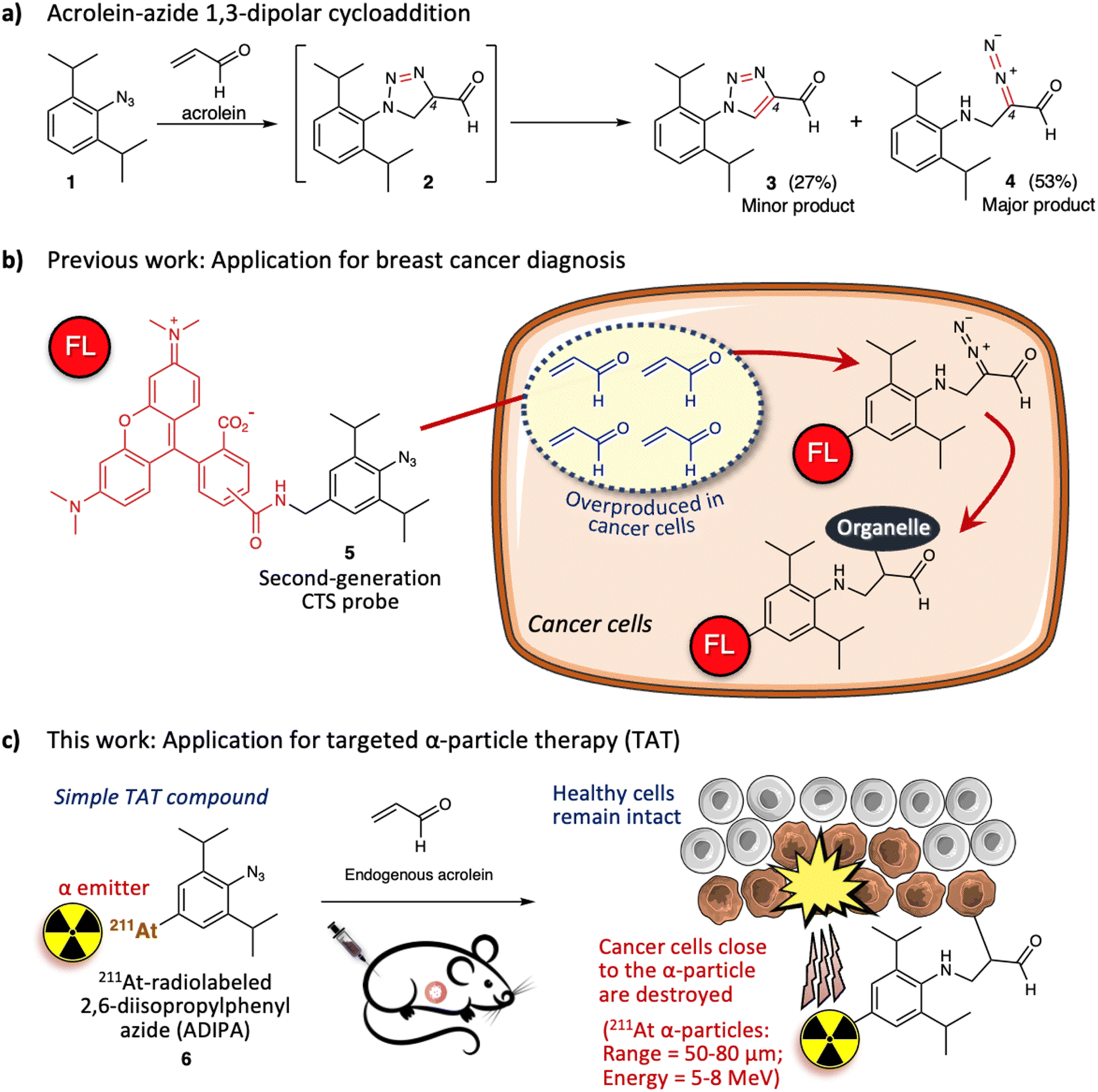 | ||
| Fig. 1 (a) 2,6-Diisopropylphenyl azide 1 reacts with acrolein through 1,3-dipolar cycloaddition to give intermediate triazoline 2, which oxidizes or rearranges to the minor product triazole 3 (27%) or major product α-diazocarbonyl 4 (53%), respectively. (b) Utilization of the second-generation click-to-sense (CTS) probe 5 for cancer diagnosis based on the reaction with endogenous acrolein. FL = 5(6)-carboxytetramethylrhodamine (TAMRA). (c) Utilization of 211At-radiolabeled 2,6-diisopropylphenyl azide (ADIPA) 6 for targeted α-particle therapy (TAT). | ||
Acrolein is a toxic substance that can be exposed to humans through environmental factors (e.g., combustion of organic substances, engine exhaust) and dietary intake (e.g., fried foods through overheating cooking oil).23,24 Concurrently, acrolein can also be produced endogenously within the human body.25–27 The high accumulation of acrolein in the human body is often linked with various diseases such as cardiovascular disease, Alzheimer's disease, and cancer.28–30 Subsequently, direct measurement of acrolein in biological systems is becoming essential to provide information for diagnosis and therapeutic purposes. Previously, by attaching 5(6)-carboxytetramethyl rhodamine (TAMRA) fluorophore to the 2,6-diisopropylphenyl azide 1, we designed a second-generation click-to-sense (CTS) probe 5 (Fig. 1b),31 which can be used to detect acrolein in live cells with high selectivity and sensitivity. Furthermore, by utilizing the CTS probe, we revealed that acrolein is generally overproduced in cancer cells but is negligible in healthy cells (ESI, Fig. S3b†).32 Thus, we realized that acrolein could be used as a new cancer marker.33–37
We also examined the mechanism for visualization of acrolein in cancer cells and found that endocytosis mediates the cellular uptake of the CTS probe.38 In the case of healthy cells, the CTS probe is inert and is secreted via exocytosis back into the extracellular environment. However, in the case of cancer cells, when the CTS probe encounters the intracellular acrolein, the 1,3-dipolar cycloaddition reaction occurs to give the diazo derivative, which further reacts with the nearest organelle of the cancer cells. Consequently, the TAMRA fluorophore could be covalently attached to the organelle within the cancer cells (Fig. 1b). Furthermore, by utilizing the CTS probe, we also found that human cancer tissue (e.g., breast cancer tissue) generates a high level of acrolein.32 By taking advantage of this phenomenon, we demonstrated that the 1,3-dipolar cycloaddition between endogenous acrolein and the CTS probe could be utilized as a selective and sensitive method to distinguish cancerous tissues from healthy tissues obtained from patients with breast cancer (ESI, Fig. S4†).39,40 Considering all of the above, in this work we aimed to develop 211At-radiolabeled 2,6-diisopropylphenyl azide (ADIPA) 6 (Fig. 1c) and sought to selectively target and destroy cancer cells without damaging healthy cells. We envision the mechanism of this ADIPA 6 to be similar to the CTS probe 5, where it would react selectively with the endogenous acrolein of cancer cells to anchor the α emitter 211At within cancer cells. Furthermore, since acrolein is generated in most cancer cells, we anticipate that this simple molecule, ADIPA 6, can target various cancers.
Results and discussion
From a structural perspective, the properties of astatine are similar to iodine. Thus, the reactions used for organoiodine compounds are typically applicable for organoastatine compounds. However, given the tendency of the carbon–halogen bond energy to decrease from the lighter to the heavier halogens, astatine has been mostly limited to bonding only to sp2 carbons (preferentially aromatic carbons) rather than sp3 carbons as the bond energies of sp3 carbons are too weak to provide sufficient stability for biomedical applications.41 Also, unlike the other halogens, astatine (0) is not diatomic. Therefore, in most cases, electrophilic At+ species or nucleophilic At− species need to be prepared by adding an oxidant [e.g., N-chlorosuccinimide (NCS)] or a reductant (e.g., sodium ascorbate), respectively, to the astatine (0) solution (vide infra). Considering these characteristics, we first attempted to introduce the 211At group into the 2,6-diisopropylphenyl azide 1 using organotin chemistry (Fig. 2a).42,43 Thus, we prepared the tributyltin precursor 7 by adapting an established method.44 Herein, the weak carbon–metal bond properties make the tin group a susceptible functional group to be substituted by the electrophilic At+ species. Accordingly, astatination of the tributyltin precursor 7 was performed via electrophilic destannylation with 211At in the presence of NCS in methanol solution at ambient temperature (see ESI† for the detailed procedure). The reaction proceeded rapidly to give the desired ADIPA 6 in 88% radiochemical yield (RCY) after purification using reversed-phase high-performance liquid chromatography (RP-HPLC) equipped with a γ detector. The HPLC purification was needed to remove the generated organotin waste and provide the final compound to be used in vivo. Moreover, because astatine does not have a stable isotope, the HPLC analysis was helpful to identify the synthesized organoastatine compound by comparing the peak retention time with that of the 4-iodo-2,6-diisopropylphenyl azide 8, a non-radioactive analog compound. As shown in Fig. 2b, the peak retention time of ADIPA 6 (red line) and the analog compound 8 (blue line) are identical.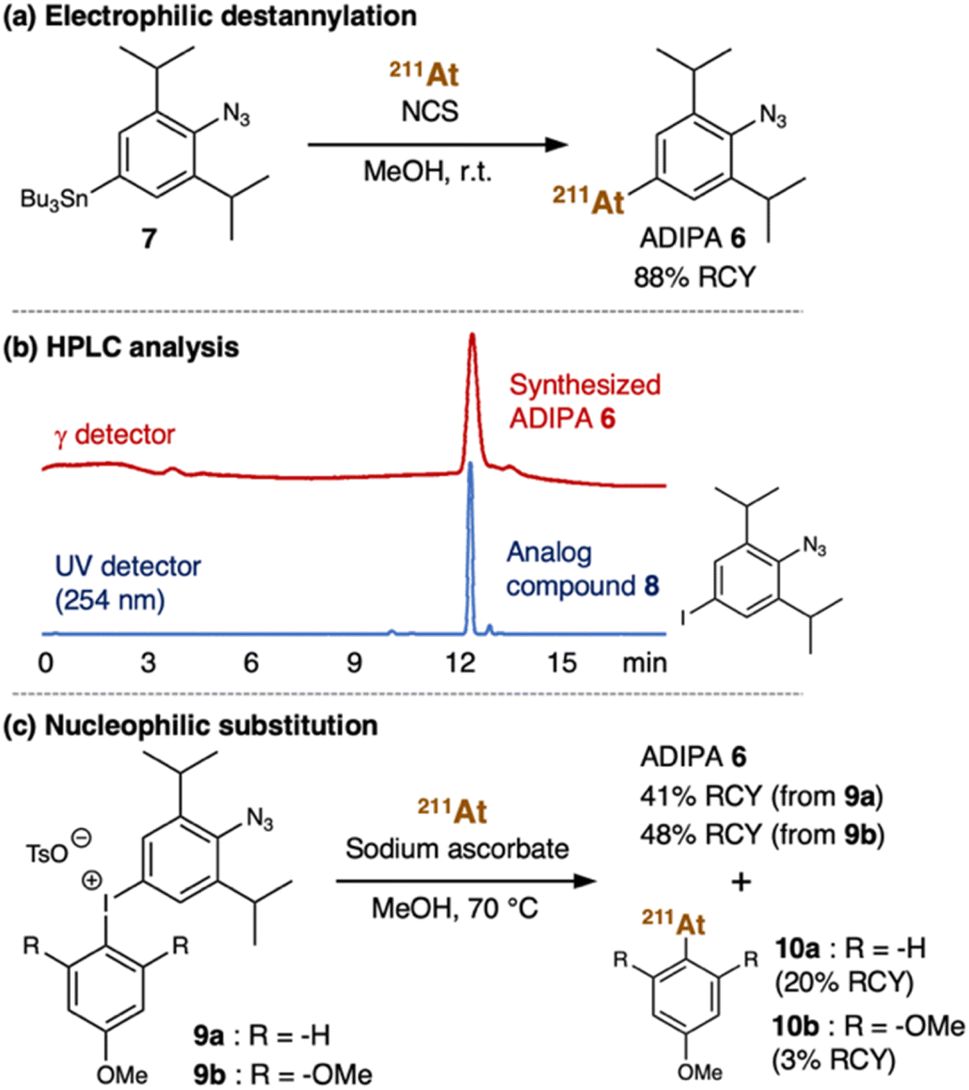 | ||
| Fig. 2 (a) Electrophilic destannylation of tributyltin precursor 7 with At+ species gave ADIPA 6 in 88% RCY. (b) HPLC analysis of synthesized ADIPA 6 (red line, monitored by the γ detector) and analog compound 8 (blue line, monitored by UV detector at 254 nm) shows identical retention time (12 minutes). (c) Nucleophilic substitution of aryliodonium salts 9a or 9b with At− species gave ADIPA 6 and side products 10a or 10b, respectively. | ||
For comparative purposes, we also considered aryliodonium salt as an attractive precursor. However, in the literature, there is only a limited example of an aryliodonium salt nucleophilic substitution reaction using At− species.45 Nonetheless, we prepared the aryliodonium salt precursor 9a (see ESI† for the detailed procedure) which we used for the reaction with 211At in the presence of sodium ascorbate in methanol at 70 °C (Fig. 2c). Under these conditions, the reaction proceeded smoothly to give the desired ADIPA 6, albeit in a moderate yield (41% RCY) after purification with RP-HPLC. Unfortunately, we also found that the reaction had a regioselectivity problem, where side product 10a was also obtained in 20% RCY (ESI, Fig. S6†). Thus, we thought that an electronic nature in the trimethoxybenzene group, such as in the aryliodonium salt precursor 9b, might be retarding the nucleophilic attack and enhancing regioselectivity. Accordingly, we prepared the aryliodonium salt precursor 9b and utilized it to react with 211At under similar conditions (Fig. 2c). This time we found that the unwanted nucleophilic addition product 10b could be suppressed down to 3% RCY. However, the desired compound's moderate yield was not much improved as the reaction from precursor 9b gave the ADIPA 6 only in 48% RCY. Although we do not have sufficient data to explain the reason, we thought that the methoxy groups of precursor 9b might cause steric hindrance that interferes with the reaction on the phenyl azide side.
Henceforth, considering the reaction outcomes, we decided to utilize ADIPA 6 prepared from precursor 7 (Fig. 2a). The prepared ADIPA 6 was stable after being incubated at 37 °C for 15 hours in both PBS and mouse serum, with approximately 76% and 85% of the tracer retained, respectively (ESI, Fig. S8†). Additionally, we found that ADIPA 6 has a high rate of cellular uptake, with a 35% binding rate to human lung cancer (A549) cells after just 10 minutes of incubation, which increased to 47% after 60 minutes (ESI, Fig. S9†). In agreement with our hypothesis (vide supra), this cellular uptake assay demonstrated that ADIPA 6 rapidly reacts with the intracellular acrolein to anchor the α-emitter 211At within A549 cells.
These achievements encouraged us to utilize the reaction between the synthesized ADIPA 6 and endogenous acrolein of cancer cells in the A549 cell-bearing xenograft mouse model. Our previous study estimated that approximately 10 μM of acrolein could be generated in A549 cancer tissues (1 × 1 cm) implanted into a nude mouse.20 In addition, we have previously utilized the reaction between endogenous acrolein and aryl azide derivatives as an effective method to treat cancer through selective prodrug activation in the mouse model (ESI, Fig. S12†).20,46 Considering the above, we anticipated that ADIPA 6 would react with the acrolein of cancer cells in the mouse model to give the intended anticancer effect (Fig. 1c). Accordingly, we prepared the A549 cell-bearing xenograft nude mouse model (see ESI, Fig. S13†), where one group was treated with ADIPA 6, and the other groups were treated with Na[211At] or vehicle [PBS (10% DMSO, 0.5% Tween 80)] as controls (Fig. 3a).
 | ||
| Fig. 3 (a) A549 cell-bearing xenograft nude mice were treated with vehicle [PBS (10% DMSO, 0.5% Tween 80)], Na[211At], and ADIPA 6. The compounds were administered intratumorally or intravenously in a single-dose injection (70 kBq). Subsequently, the (b) tumor growth levels (see also ESI Tables S1 and S2†), (c) body weight changes, and (d) survival rates were observed (n = 6). (e) Representative photos were taken on day 0 and day 16. The arrow shows the location of the tumor. See ESI Fig. S16 and S17a† for other images. (f) Biodistribution of ADIPA 6 in the xenograft mice 18 hours after the corresponding compounds were administered intratumorally or intravenously (n = 3). Data are represented as mean value ± SD. P values were determined using a two-way ANOVA with Tukey's correction for multiple comparisons between groups. n.s. = not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. | ||
The appropriate dose of 211At for animal experiments has been reported previously, with the maximum tolerated (non-lethal) amount in the mouse being 70 kBq g−1 of body weight (1.4 MBq per 20 g of body weight).47 In the first trial, we intratumorally treated the mouse model (female, 20 to 23 g range of body weight) with a single-dose injection of 211At compounds and observed tumor growth (see ESI† for the detailed procedure). We found that treatment with vehicle (10 μL, n = 6) or Na[211At] [10 μL, 200 kBq (about 10 kBq g−1 of body weight, which is lower than the maximum tolerated amount), n = 6] did not retard tumor growth, and we observed an increase in tumor size (approximately 1800 mm3) in 26 days (Fig. 3b, dashed black line and dashed blue line). On the other hand, treatment with ADIPA 6 [10 μL, 70 kBq (approximately 3.5 kBq g−1 of body weight, which is even much lower than the maximum tolerated amount), n = 6] inhibited tumor growth (Fig. 3b, solid red line, tumor size = approximately 700 mm3 at day 26). Namely, a single-dose of intratumorally administered ADIPA 6 reduced tumor growth to less than two-fifths of the other groups treated with vehicle or Na[211At]. Furthermore, considering that the treatment was performed only once with a 70 kBq single-dose injection, we noticed that utilizing ADIPA 6 to retard tumor growth was promising. To examine whether the administration route could affect the reactivity and biodistribution of ADIPA 6, we also applied the intravenous injection to a different mouse group. As a result, the group that received ADIPA 6 intravenously (100 μL, 70 kBq, n = 6) showed similar tumor inhibition to the intratumorally treated group, at least until day 19, before the tumor size increased again (Fig. 3b, solid orange line, tumor size = 1000 mm3 at day 26. See also ESI, Table S1†). Furthermore, there was no significant body weight decrease in any of the experimental groups (Fig. 3c). We also did not observe inflammation on the mouse bodies in the experimental groups (Fig. 3e, photo of post treatment and ESI, Fig. S16 and S17a†).
Concurrently, the survival rate of the Na[211At]-treated group (Fig. 3d, dashed blue line) was close to the survival rate of the vehicle-treated group (Fig. 3d, dashed black line), where less than 50% of mice in these groups survived to day 31 post treatment. Therefore, we assume that tumor treatment with Na[211At] was inadequate. In contrast, the mouse group treated intratumorally with 70 kBq of ADIPA 6 remained alive at day 40 post treatment (Fig. 3d, solid red line), which demonstrates the potential of this method. Also, the mouse group treated intravenously with 70 kBq of ADIPA 6 remained alive at day 31 post treatment (Fig. 3d, solid orange line). Given this phenomenon, we then examined in vivo biodistribution of the compounds (Fig. 3f). We administered Na[211At] (10 μL, 200 kBq, n = 3, intratumorally), ADIPA 6 (10 μL, 70 kBq, n = 3, intratumorally), and ADIPA 6 (100 μL, 70 kBq, n = 3, intravenously) into three different mouse groups. After 18 hours, the mice were sacrificed, and then radioactivity in each dissected organ was measured. The mouse group treated with Na[211At] showed a low, if not negligible, radioactive accumulation level in the tumor and other organs but high-level radioactive accumulation in the thyroid (Fig. 3f, blue bar).19 This result indicates that, although Na[211At] was administered intratumorally, the astatine did not remain in tumor tissues. Somehow, it immediately moved and ended up mainly in the thyroid and marginally in the stomach and urine. Although there is minimal information about the chemical form of 211At in the thyroid, it is possible that astatine could be firmly bound in the mouse thyroid by forming stable complexes with proteins such as thyroglobulin.48
In the case of ADIPA 6 administered intratumorally, to our satisfaction, a prominent radioactive accumulation was observed in the tumor (Fig. 3f, red bar). At the same time, only a marginal amount was observed in the thyroid, stomach, and urine. Therefore, we assumed that the observed radioactive accumulation in tumor tissues occurred due to the reaction between ADIPA 6 and the endogenous acrolein of cancer cells in the mouse models. Although intravenous administration produces lower tumoral radioactive accumulation (Fig. 3f, orange bar), we found that intravenously administered 70 kBq of ADIPA 6 could retard the tumor growth, at least until day 19 (Fig. 3b, solid orange line, see also ESI, Tables S1 and S2†). This result might be caused by the efficient reactivity of ADIPA 6 with the acrolein of cancer cells in vivo. Thus, although intravenous administration delivered a smaller amount of ADIPA 6 to the tumor area than intratumoral administration, it could still react with acrolein to covalently bond to the organelle, tether 211At in cancer cells, and then selectively kill cancer cells.
Furthermore, in order to demonstrate that the observed therapeutic effect of ADIPA 6 is indeed triggered by 1,3-dipolar cycloaddition with acrolein, we prepared two different groups of the cancer-bearing xenograft mouse model as controls (see ESI, Fig. S14†). One group was intratumorally treated with 3,5-diisopropylphenyl astatine 11 (10 μL, 70 kBq, n = 6), which lacks an azide moiety. Another group was intratumorally treated with 2,6-diisopropylphenyl azide 1 (5 μL, 120 nmol, 1.2 mg kg−1, n = 6) to scavenge the endogenous acrolein before administering ADIPA 6 (100 μL, 70 kBq, n = 6) through intravenous injection. For both the 3,5-diisopropylphenyl astatine 11 treated mouse group (ESI, Fig. S14, dashed green line, and photos in ESI, Fig. S17b†) and the 2,6-diisopropylphenyl azide 1 and ADIPA 6 co-treated mouse group (ESI, Fig. S14, dashed grey line, and photos in ESI, Fig. S17c†), we observed that the tumor growth was not inhibited (similar to the Na[211At]-treated group, see Fig. 3b, dashed blue line, and ESI, Fig. S17a†). As a result, less than 20% of these groups survived to day 38 post treatment (ESI, Fig. S14d†).
Finally, we also treated the cancer-bearing xenograft mouse with four different concentrations (60 kBq, 100 kBq, 160 kBq, and 300 kBq) of ADIPA 6 and found that the therapeutic effect works in a dose-dependent manner (see ESI† for the detailed procedure). The tumor growth difference between the 60 kBq and 300 kBq ADIPA 6 treated groups was significant starting at day 28 post treatment (ESI, Fig. S15, and Table S3†), with approximately a two-fold difference between the 60 kBq and 300 kBq treated group at day 31 post treatment (see also ESI, Fig. S18 and S19† for representative photos). These results indicate that ADIPA 6 reacted selectively with the acrolein of cancer cells through 1,3-dipolar cycloaddition to target cancer tissues in the mouse model. Therefore, because ADIPA 6 is selectively attached by covalent bonding to the organelle of cancer cells (see Fig. 1), a single-dose administration of low radioactive doses (i.e., between 60 to 300 kBq) ADIPA 6 was enough to retard the tumor growth.
Conclusions
Herein, we have developed an in vivo targeted α-therapy by utilizing the reaction between 211At-radiolabeled 2,6-diisopropylphenyl azide (ADIPA) 6 and endogenous acrolein of cancer cells. The results demonstrate that this simple molecule can treat a xenograft model of human lung cancer in a dose-dependent manner. When a single-dose of 70 kBq ADIPA 6 was intratumorally or intravenously administered into the mouse model, cancer growth was inhibited, no inflammation was observed, and the treated group survived longer than the control groups. Moreover, biodistribution analysis shows that the ADIPA 6 accumulated mainly in tumor tissues. Namely, ADIPA 6 reacted effectively with endogenous acrolein to covalently bind to the organelle, kill cancer cells, and leave healthy cells intact. It is noteworthy that the dose applied in the current study was far below the maximum tolerated quantity of α-emissions for clinical application. Finally, because acrolein is overproduced in cancer cells, including cancer tissues obtained from patients with cancer, we anticipate that this method deserves further application in mouse models with various cancer types and stages.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: A. R. P., K. T.; funding acquisition: A. R. P., K. T.; investigation: O. Y., A. R. P., P. A., A. I., A. N., Y. E., Y. K., K. M.; formal analysis: O. Y., A. R. P., P. A.; resources: Y. W., X. Y., N. S., H. H.; visualization; A. R. P., O. Y.; writing—original draft preparation: A. R. P.; writing—review and editing: A. R. P., O. Y., K. T.; supervision: A. R. P., H. H., K. T.Ethical statement
All animal experiments were approved by RIKEN's Animal Ethics Committee (W2019-2-049).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Hiroshi Mizuma, PhD from the Department of Functional Brain Imaging, National Institutes for Quantum Science and Technology (QST), for helpful guidance in the statistical analysis. All animal procedures were performed following the Guidelines for Care and Use of Laboratory Animals of RIKEN and approved by the Animal Ethics Committee of RIKEN (W2019-2-049). This work was financially supported by JSPS KAKENHI [Grant Numbers JP21H02065, JP21K19042, JP21K05269, JP18H05503], the Naito Foundation, and the Canon Foundation.References
- G. Vaidyanathan and M. R. Zalutsky, Curr. Rad., 2008, 1, 177 CrossRef CAS.
- S. Lindegren, P. Albertsson, T. Back, H. Jensen, S. Palm and E. Aneheim, Cancer Biother. Radiopharm., 2020, 35, 425–436 CrossRef.
- B. J. Allen, C. Raja, S. Rizvi, Y. Li, W. Tsui, D. Zhang, E. Song, C. F. Qu, J. Kearsley, P. Graham and J. Thompson, Phys. Med. Biol., 2004, 49, 3703–3712 CrossRef CAS PubMed.
- D. R. Corson, K. R. MacKenzie and E. Serge, Phys. Rev., 1940, 58, 672–678 CrossRef CAS.
- R. H. Larsen, B. W. Wieland and M. R. Zalutsky, Appl. Radiat. Isot., 1996, 47, 135–143 CrossRef CAS.
- M. K. Schultz, M. Hammond, J. T. Cessna, P. Plascjak, B. Norman, L. Szajek, K. Garmestani, B. E. Zimmerman and M. Unterweger, Appl. Radiat. Isot., 2006, 64, 1365–1369 CrossRef CAS PubMed.
- K. Berei and L. Vasáros, in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd., 2009, pp. 787–819, DOI:10.1002/9780470682531.pat0014.
- S. W. Hadley, D. S. Wilbur, M. A. Gray and R. W. Atcher, Bioconjugate Chem., 1991, 2, 171–179 CrossRef CAS PubMed.
- F. Guérard, C. Maingueneau, L. Liu, R. Eychenne, J.-F. Gestin, G. Montavon and N. Galland, Acc. Chem. Res., 2021, 54, 3264–3275 CrossRef.
- G. Vaidyanathan and M. R. Zalutsky, Bioconjugate Chem., 1992, 3, 499–503 CrossRef CAS PubMed.
- H. Sudo, A. B. Tsuji, A. Sugyo, K. Nagatsu, K. Minegishi, N. S. Ishioka, H. Ito, K. Yoshinaga and T. Higashi, Transl. Oncol., 2019, 12, 879–888 CrossRef.
- S. Watanabe, M. A.-U. Azim, I. Nishinaka, I. Sasaki, Y. Ohshima, K. Yamada and N. S. Ishioka, Org. Biomol. Chem., 2019, 17, 165–171 RSC.
- Y. Shirakami, T. Watabe, H. Obata, K. Kaneda, K. Ooe, Y. Liu, T. Teramoto, A. Toyoshima, A. Shinohara, E. Shimosegawa, J. Hatazawa and K. Fukase, Sci. Rep., 2021, 11, 12982 CrossRef CAS.
- K. Kaneda-Nakashima, Z. Zhang, Y. Manabe, A. Shimoyama, K. Kabayama, T. Watabe, Y. Kanai, K. Ooe, A. Toyoshima, Y. Shirakami, T. Yoshimura, M. Fukuda, J. Hatazawa, T. Nakano, K. Fukase and A. Shinohara, Cancer Sci., 2021, 112, 1132–1140 CrossRef CAS PubMed.
- K. Ogawa, T. Takeda, K. Mishiro, A. Toyoshima, K. Shiba, T. Yoshimura, A. Shinohara, S. Kinuya and A. Odani, ACS Omega, 2019, 4, 4584–4591 CrossRef CAS.
- K. Ogawa, H. Echigo, K. Mishiro, S. Hirata, K. Washiyama, Y. Kitamura, K. Takahashi, K. Shiba and S. Kinuya, Mol. Pharm., 2021, 18, 3553–3562 CrossRef CAS.
- H. Kato, X. Huang, Y. Kadonaga, D. Katayama, K. Ooe, A. Shimoyama, K. Kabayama, A. Toyoshima, A. Shinohara, J. Hatazawa and K. Fukase, J. Nanobiotechnol., 2021, 19, 223 CrossRef CAS PubMed.
- H. K. Li, Y. Morokoshi, K. Nagatsu, T. Kamada and S. Hasegawa, Cancer Sci., 2017, 108, 1648–1656 CrossRef CAS PubMed.
- K. Fujiki, Y. Kanayama, S. Yano, N. Sato, T. Yokokita, P. Ahmadi, Y. Watanabe, H. Haba and K. Tanaka, Chem. Sci., 2019, 10, 1936–1944 RSC.
- A. R. Pradipta, P. Ahmadi, K. Terashima, K. Muguruma, M. Fujii, T. Ichino, S. Maeda and K. Tanaka, Chem. Sci., 2021, 12, 5438–5449 RSC.
- S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed.
- A. R. Pradipta, M. Taichi, I. Nakase, E. Saigitbatalova, A. Kurbangalieva, S. Kitazume, N. Taniguchi and K. Tanaka, ACS Sens., 2016, 1, 623–632 CrossRef CAS.
- J. F. Stevens and C. S. Maier, Mol. Nutr. Food Res., 2008, 52, 7–25 CrossRef CAS PubMed.
- A. Moghe, S. Ghare, B. Lamoreau, M. Mohammad, S. Barve, C. McClain and S. Joshi-Barve, Toxicol. Sci., 2015, 143, 242–255 CrossRef CAS PubMed.
- M. M. Anderson, S. L. Hazen, F. F. Hsu and J. W. Heinecke, J. Clin. Invest., 1997, 99, 424–432 CrossRef CAS.
- P. J. O'Brien, A. G. Siraki and N. Shangari, Crit. Rev. Toxicol., 2005, 35, 609–662 CrossRef PubMed.
- J.-M. Lü, P. H. Lin, Q. Yao and C. Chen, J. Cell. Mol. Med., 2010, 14, 840–860 CrossRef.
- N. Y. Calingasan, K. Uchida and G. E. Gibson, J. Neurochem., 1999, 72, 751–756 CrossRef CAS PubMed.
- R. Shi, T. Rickett and W. Sun, Mol. Nutr. Food Res., 2011, 55, 1320–1331 CrossRef CAS.
- K. Muguruma, A. R. Pradipta, Y. Ode, K. Terashima, H. Michiba, M. Fujii and K. Tanaka, Bioorg. Med. Chem., 2020, 28, 115831 CrossRef CAS PubMed.
- A. R. Pradipta, H. Michiba, A. Kubo, M. Fujii, T. Tanei, K. Morimoto, K. Shimazu and K. Tanaka, Bull. Chem. Soc. Jpn., 2022, 95, 421–426 CrossRef CAS.
- T. Tanei, A. R. Pradipta, K. Morimoto, M. Fujii, M. Arata, A. Ito, M. Yoshida, E. Saigitbatalova, A. Kurbangalieva, J.-I. Ikeda, E. Morii, S. Noguchi and K. Tanaka, Adv. Sci., 2019, 6, 1801479 CrossRef PubMed.
- K. Uchida, M. Kanematsu, Y. Morimitsu, T. Osawa, N. Noguchi and E. Niki, J. Biol. Chem., 1998, 273, 16058–16066 CrossRef CAS PubMed.
- S. Kato, G. C. Post, V. M. Bierbaum and T. H. Koch, Anal. Biochem., 2002, 305, 251–259 CrossRef CAS PubMed.
- K. Zarkovic, K. Uchida, D. Kolenc, L. Hlupic and N. Zarkovic, Free Radic. Res., 2006, 40, 543–552 CrossRef CAS PubMed.
- K. Uchida, M. Kanematsu, K. Sakai, T. Matsuda, N. Hattori, Y. Mizuno, D. Suzuki, T. Miyata, N. Noguchi, E. Niki and T. Osawa, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4882–4887 CrossRef CAS PubMed.
- R. A. Alarcon, Med. Hypotheses, 2012, 79, 522–530 CrossRef CAS PubMed.
- A. R. Pradipta, M. Fujii, T. Tanei, K. Morimoto, K. Shimazu, S. Noguchi and K. Tanaka, Bioorg. Med. Chem., 2019, 27, 2228–2234 CrossRef CAS PubMed.
- A. R. Pradipta, T. Tanei, K. Morimoto, K. Shimazu, S. Noguchi and K. Tanaka, Adv. Sci., 2020, 7, 1901519 CrossRef CAS PubMed.
- A. Kubo, T. Tanei, A. R. Pradipta, K. Morimoto, M. Fujii, Y. Sota, T. Miyake, N. Kagara, M. Shimoda, Y. Naoi, Y. Motoyama, E. Morii, K. Tanaka and K. Shimazu, Eur. J. Surg. Oncol., 2022, 48, 1520–1526 CrossRef PubMed.
- F. Guérard, J. F. Gestin and M. W. Brechbiel, Cancer Biother. Radiopharm., 2013, 28, 1–20 CrossRef PubMed.
- R. A. Milius, W. H. McLaughlin, R. M. Lambrecht, A. P. Wolf, J. J. Carroll, S. J. Adelstein and W. D. Bloomer, Int. J. Rad. Appl. Instrum. A, 1986, 37, 799–802 CrossRef CAS PubMed.
- M. J. Adam and D. S. Wilbur, Chem. Soc. Rev., 2005, 34, 153–163 RSC.
- G. Vaidyanathan, D. J. Affleck, K. L. Alston, X.-G. Zhao, M. Hens, D. H. Hunter, J. Babich and M. R. Zalutsky, Bioorg. Med. Chem., 2007, 15, 3430–3436 CrossRef CAS PubMed.
- F. Guérard, Y.-S. Lee, K. Baidoo, J.-F. Gestin and M. W. Brechbiel, Chem.–Eur. J., 2016, 22, 12332–12339 CrossRef.
- A. R. Pradipta and K. Tanaka, Chem. Commun., 2021, 57, 9798–9806 RSC.
- L. M. Cobb, A. Harrison and S. A. Butler, Hum. Toxicol., 1988, 7, 529–534 CrossRef CAS PubMed.
- J. G. Hamilton and M. H. Soley, Proc. Natl. Acad. Sci. U. S. A., 1940, 26, 483–489 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02513f |
| ‡ Present address for P. A.: Research Center for Vaccine and Drug, Research Organization for Health, National Research and Innovation Agency (BRIN), Indonesia. |
| This journal is © The Royal Society of Chemistry 2023 |