
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSignificance of the connecting position between Zn(II) porphyrin and Re(I) bipyridine tricarbonyl complex units in dyads for room-temperature phosphorescence and photocatalytic CO2 reduction: unexpected enhancement by triethanolamine in catalytic activity†
Yusuke
Kuramochi
 *ab,
Yuto
Suzuki
a,
Somyo
Asai
c,
Tomohiro
Suzuki
c,
Hiroki
Iwama
d,
Motoko S.
Asano
*c and
Akiharu
Satake
*ab
*ab,
Yuto
Suzuki
a,
Somyo
Asai
c,
Tomohiro
Suzuki
c,
Hiroki
Iwama
d,
Motoko S.
Asano
*c and
Akiharu
Satake
*ab
aDepartment of Chemistry, Graduate School of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8621, Japan. E-mail: kuramochiy@rs.tus.ac.jp; asatake@rs.tus.ac.jp
bDepartment of Chemistry, Faculty of Science Division II, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8621, Japan
cDivision of Molecular Science, School of Science and Technology, Gunma University, 1-5-1 Tenjin-cho, Kiryu, Gunma 376-8515, Japan. E-mail: motoko@gunma-u.ac.jp
dDepartment of Applied Chemistry, Faculty of Science Division I, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8621, Japan
First published on 18th July 2023
Abstract
We synthesized three new dyads composed of a Zn porphyrin and fac-Re(bpy)(CO)3Br (bpy = 2,2′-bipyridine) units, ZnP-nBpy![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ReBr (n = 4, 5, and 6), in which the porphyrin is directly connected at the meso-position through the 4-, 5-, or 6-position of the bpy. We investigated the relationships between the connecting positions and the photophysical properties as well as catalytic activity in the CO2 reduction reaction. The dyad connected through the 6-position, ZnP-6BpyReBr, showed obvious phosphorescence with a lifetime of 280 μs at room temperature, in N,N-dimethylacetamide (DMA), whereas the other two dyads showed almost no phosphorescence under the same conditions. The photocatalytic CO2 reduction reactions in DMA using 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole as the electron donor and the three dyads ZnP-nBpyReBr selectively produced CO with similar initial rates, but the durabilities were low. The addition of triethanolamine (TEOA) suppressed the decomposition of dyads, improving their durabilities and reaction efficiencies. In particular, ZnP-5BpyReBr was remarkably improved—it gave the highest durability and reaction efficiency among the three dyads; the reaction quantum yield reached 24%. The reason for this significant activity is no accumulation of electrons on the Zn porphyrin in ZnP-5BpyReBr, which would be caused by dual interactions of TEOA with the Re and Zn ions in the dyad. As the highest catalytic activity was observed in ZnP-5BpyReBr among the three dyads, which had no room-temperature phosphorescence (RTP), the catalytic activities and RTP properties are considered independent, but they are greatly influenced by the connecting positions on the bpy ligand in ZnP-nBpyReBr.
ReBr (n = 4, 5, and 6), in which the porphyrin is directly connected at the meso-position through the 4-, 5-, or 6-position of the bpy. We investigated the relationships between the connecting positions and the photophysical properties as well as catalytic activity in the CO2 reduction reaction. The dyad connected through the 6-position, ZnP-6BpyReBr, showed obvious phosphorescence with a lifetime of 280 μs at room temperature, in N,N-dimethylacetamide (DMA), whereas the other two dyads showed almost no phosphorescence under the same conditions. The photocatalytic CO2 reduction reactions in DMA using 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole as the electron donor and the three dyads ZnP-nBpyReBr selectively produced CO with similar initial rates, but the durabilities were low. The addition of triethanolamine (TEOA) suppressed the decomposition of dyads, improving their durabilities and reaction efficiencies. In particular, ZnP-5BpyReBr was remarkably improved—it gave the highest durability and reaction efficiency among the three dyads; the reaction quantum yield reached 24%. The reason for this significant activity is no accumulation of electrons on the Zn porphyrin in ZnP-5BpyReBr, which would be caused by dual interactions of TEOA with the Re and Zn ions in the dyad. As the highest catalytic activity was observed in ZnP-5BpyReBr among the three dyads, which had no room-temperature phosphorescence (RTP), the catalytic activities and RTP properties are considered independent, but they are greatly influenced by the connecting positions on the bpy ligand in ZnP-nBpyReBr.
Introduction
The global warming problem and fuel resource shortage necessitates research on artificial photosynthetic systems that use solar energy to reduce CO2 to carbon resources. In constructing an artificial photosynthesis system, it is essential to develop a photosensitizer that efficiently captures dilute sunlight,1 and a catalyst that promotes the multielectron reduction reaction and suppresses H2 evolution.2 As a catalyst, metal complexes are promising candidates for CO2 reduction due to their multiple accessible redox states, high activation energies against proton reduction, and various molecular design possibilities on the combination of appropriate metal ions and ligands. Among them, Re(I) diimine carbonyl complexes are advantageous as catalysts for CO2 reduction because the Re complexes almost exclusively produce CO without the formation of H2 even in aqueous solutions.2b After the first report appeared describing the use of fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine) by Lehn and co-workers in the 1980s,3 solvents containing triethanolamine (TEOA) have often been used in many photocatalytic systems that employ Re complexes. While TEOA acts as a sacrificial reducing reagent, TEOA still acts as an effective additive when use is made of stronger and more efficient reducing reagents, where TEOA does then not act as a reducing reagent. In particular, the mixed solvent of DMF–TEOA (DMF = N,N-dimethylformamide) or DMA–TEOA (DMA = N,N-dimethylacetamide)4 behaves as a “magic solvent” for photocatalytic CO2 reduction when employing Re complexes. Ishitani and co-workers found that fac-[Re(bpy)(CO)3(DMF)]+ is converted to the TEOA–CO2 adduct (Re–CO2–TEOA) through the TEOA adduct (Re–TEOA) in DMF–TEOA under a CO2 atmosphere (Fig. 1).5 Thus, CO2 is efficiently captured by the Re complex, with the assistance of TEOA, facilitating the photocatalytic CO2 reduction even under low-concentration CO2.5a The similar insertion reactions of CO2 into the M–OR (R = alkyl or aryl groups) bonds have been observed6 not only in Re complexes7 but also in the various other metal complexes,8–11 and are expected to be utilized in a wide range of fields such as CO2 storage/release as well as CO2 reduction reaction.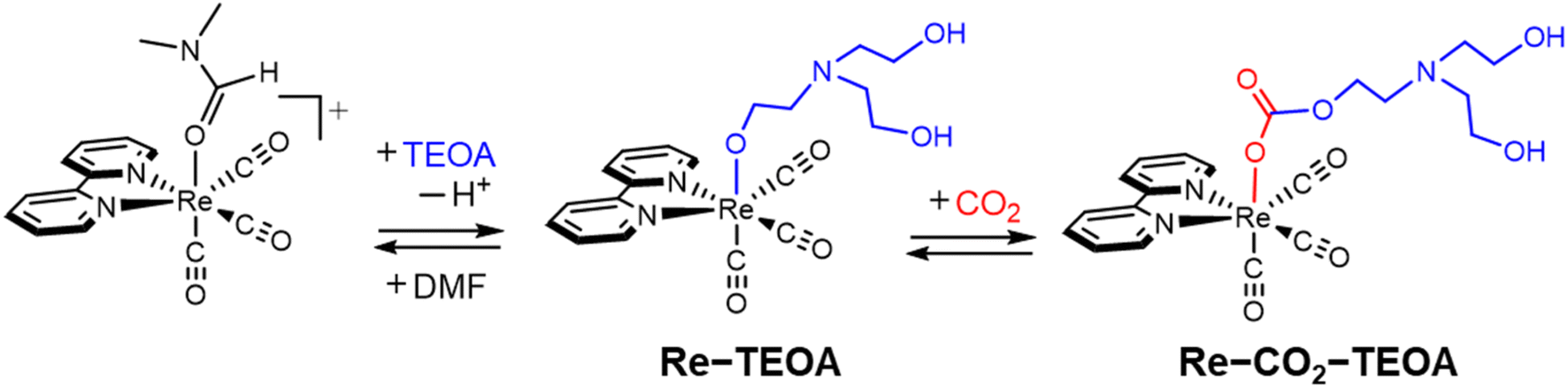 | ||
| Fig. 1 Formation of the Re–CO2–TEOA adduct through the Re–TEOA adduct in a CO2-saturated DMF–TEOA solution.5a | ||
The Re complex also acts as a photosensitizer because it exhibits absorption in the near-ultraviolet to visible region; however, its durability is low. On the other hand, combination with a suitable photosensitizer, such as a Ru(II) diimine complex ([Ru(N^N)3]2+), improves the efficiency and durability of photocatalytic CO2 reduction.2b The Ru complex has relatively strong absorption bands around 450 nm and can efficiently drive photoinduced electron transfer reactions by excitation with visible light. When a Ru diimine complex was connected with a Re complex (a Ru–Re complex), the photocatalytic CO2 reduction selectively produced CO in the presence of 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]-imidazole (BIH) as an electron donor—the turnover number (TONCO) was > 2000 and the reaction quantum yield (ΦCO) was 45%.12 Furthermore, when the Ru–Re complex was combined with a semiconductor photocatalyst, then two-step photoexcitation of the Ru complex and the semiconductor selectively produced CO from CO2 using water as the electron donor.13 The Ru diimine complexes have been utilized as the photosensitizer by combining other metal complex catalysts, showing high catalytic activity and durability in not only CO2 reduction reaction but also oxygen and hydrogen evolutions from water.14 However, the absorption ability of the Ru diimine complex was insufficient for the full utilization of sunlight on the semiconductors.15 Therefore, the accumulation of photosensitizers16 and/or the development of photosensitizers that absorb visible light more strongly are essential.
In natural photosynthetic systems, networks of chlorophylls efficiently capture dilute photons from the sun and utilize the absorbed solar energy to produce chemical energy.17 Zn porphyrins are chlorophyll analogues—they have strong absorption bands in the visible region—and are used as photosensitizers in the photocatalytic CO2 reduction.18–20 However, the use of Zn porphyrins as photosensitizers is often limited due to their poor stability under the photocatalytic reduction conditions because the accumulation of more than two electrons on the Zn porphyrins leads to hydrogenation, which finally results in color bleaching.19c,21
Recently, we reported that photocatalytic CO2 reduction using a dyad (ZnP-phenRe, Fig. 2a) in which fac-Re(phen)(CO)3Br (phen = 1,10-phenanthroline) is directly linked to Zn tris(4-tert-butylphenyl)porphyrin selectively produced CO with a high reaction quantum yield (ΦCO = 8%) and a high durability (TONCO > 1400) in the presence of phenol as proton source and BIH as electron donor.22 The high durability is achieved by efficient electron transfer from the porphyrin to the adjacent Re complex where electrons are consumed by CO2 reduction, thus suppressing the formation of two-electron reduced porphyrin that causes hydrogenation of the CC bonds of the porphyrin skeleton. The high reaction quantum yield would be contributed to by the initial electron transfer from BIH through the long-lived excited triplet state (T1) of porphyrin, enabling the excited species to be fully utilized in the electron transfer.22a The efficient electron transfer from the T1 is evidenced by the fact that the room-temperature phosphorescence (RTP) of the Zn porphyrin unit is remarkably quenched by a trace amount of BIH. In general, the phosphorescence from Zn porphyrin is not observed at room temperature, but only at low temperature. Thus, the unique RTP from the Zn porphyrin unit in ZnP-phenRe would be imposed by the spin–orbital coupling by the heavy atoms of the Re complex.
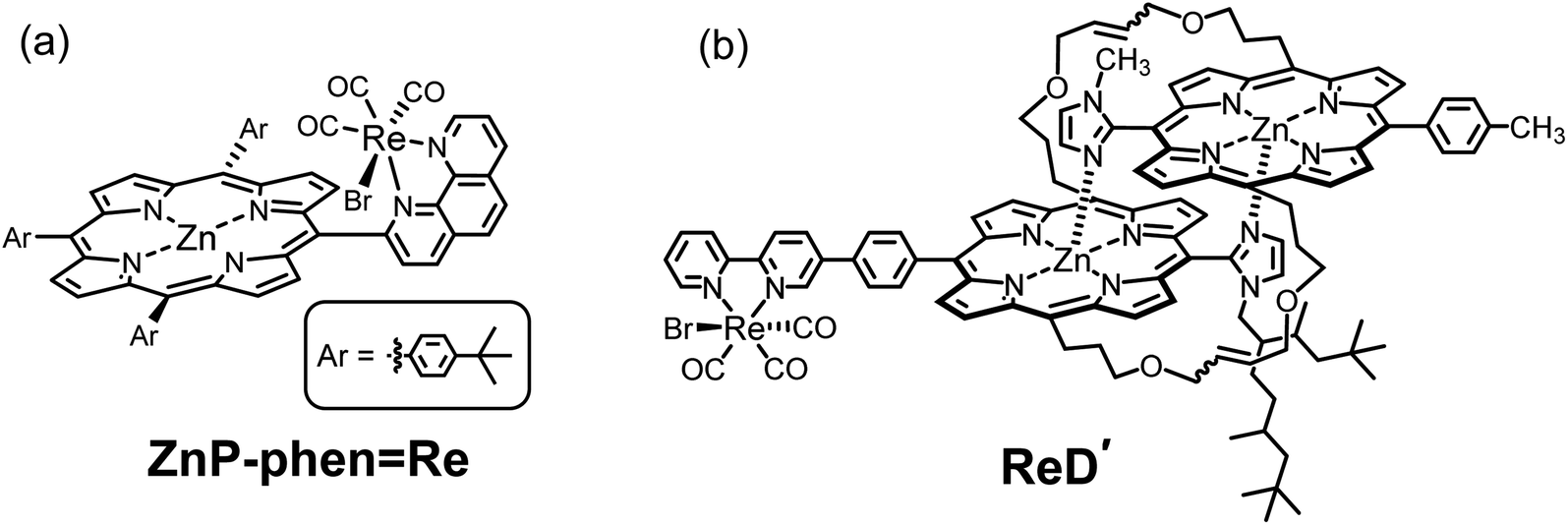 | ||
| Fig. 2 Structures of (a) a previously reported dyad (ZnP-phenRe)22 and (b) the special-pair mimic porphyrin dimer (ReD′).23 | ||
In another paper, we reported that a slipped-cofacial porphyrin dimer (ReD′, Fig. 2b) mimicking the natural special pair in photosynthesis exhibits high catalytic activity even under irradiation with strong light.23 This is attributed to the intramolecular electron transfer from the lowest excited singlet state (S1) of the porphyrin dimer to the Re complex unit, rather than the electron transfer through the T1 of the porphyrin, resulting in no accumulation of the one-electron reduced species (OERS) on the porphyrin that causes the inner-filter effect.24,25 However, ReD′ requires a high concentration of BIH, yet the reaction quantum yield is still lower (2%) than that of ZnP-phenRe under weak light irradiation because a rapid back-electron transfer process generally competes with the catalytic reaction in the intramolecular charge-separated state.
ZnP-phenRe exhibits unique properties: RTP from the Zn porphyrin and excellent activity for photocatalytic CO2 reduction. However, it is unknown how the spatial distance and connecting positions between the Zn porphyrin and the Re complex units affect the RTP and the photocatalytic activity. Although the phen ligand was reported to show interesting properties, systematic modification of the phen part does not seem to be easy. The bpy ligand is used instead of the phen because the structure of bpy can be easily synthesized from a pyridyl precursor by a Pd-catalyzed coupling reaction, and various substituents can be introduced at the bpy part. Furthermore, in a fac-Re(diimine)(CO)3Br-type complex the bpy ligand exhibits similar photocatalytic activity for CO2 reduction to the phen ligand.26
We have synthesized three new dyads, ZnP-nBpyReBr (n = 4, 5, and 6), in which fac-Re(bpy)(CO)3Br (bpy = 2,2′-bipyridine) is directly connected with Zn(II) tris(2,4,6-trimethylphenyl)porphyrin at the meso-position through the 4, 5, or 6-position of the bpy (Fig. 3). The photochemical properties of ZnP-nBpyReBr were investigated, including RTP and photocatalytic activity for CO2 reduction. When using ZnP-nBpyReBr in DMA containing BIH as the electron donor, the photocatalytic CO2 reduction had comparable initial production rates of CO as achieved with ZnP-phenRe. The catalytic activities of ZnP-nBpyReBr were remarkably enhanced in the presence of TEOA, whereas ZnP-phenRe was rather deactivated with TEOA. The reaction quantum yield reached the high value of ΦCO = 24% in ZnP-5BpyReBr; it showed little RTP. The reaction quantum yield of ZnP-5BpyReBr was more than twice that of the other dyads. ZnP-5BpyReBr suppressed the electron accumulation at the porphyrin unit during the photocatalytic CO2 reduction with unexpected specific assistance of TEOA.
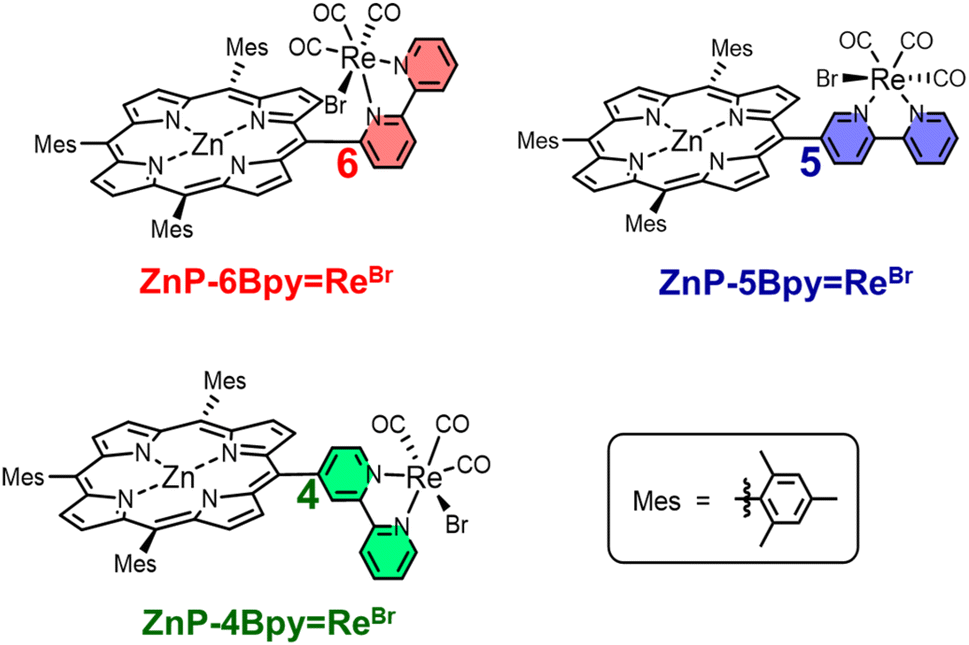 | ||
| Fig. 3 Structures of three dyads, ZnP-nBpyReBr (n = 4, 5, and 6). | ||
Results and discussion
Synthesis
The synthetic route is shown in Scheme 1. Porphyrins in which a bpy group is directly connected at the meso-position have been synthesized by various procedures.27–29 We selected a synthetic route in which the bromopyridyl-substituted porphyrins are first synthesized and then a pyridyl group is introduced by reaction with the porphyrins using Pd-catalyzed Migita–Kosugi–Stille coupling.30 Three types of bromopyridyl-substituted porphyrins were synthesized by the Lindsey method31 in the presence of pyrrole and 2,4,6-trimethylbenzaldehyde using 2-bromo-pyridine derivatives with a formyl group at the 4-, 5-, or 6-position. Zinc metalation of the free-base porphyrins was performed prior to the subsequent coupling reaction to prevent coordination of the palladium ion to the porphyrin center. The coupling reaction using 2-(tributylstannyl)pyridine in the presence of Pd(PPh3)4 followed by purification with silica gel and anhydrous K2CO3/flash silica gel (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 w/w)32 afforded pure porphyrins with a bipyridyl group, ZnP-nBpy. The reaction of ZnP-nBpy with one equivalent of Re(CO)5Br followed by recrystallization from CHCl3/ethanol afforded ZnP-nBpyReBr.
:9 w/w)32 afforded pure porphyrins with a bipyridyl group, ZnP-nBpy. The reaction of ZnP-nBpy with one equivalent of Re(CO)5Br followed by recrystallization from CHCl3/ethanol afforded ZnP-nBpyReBr.
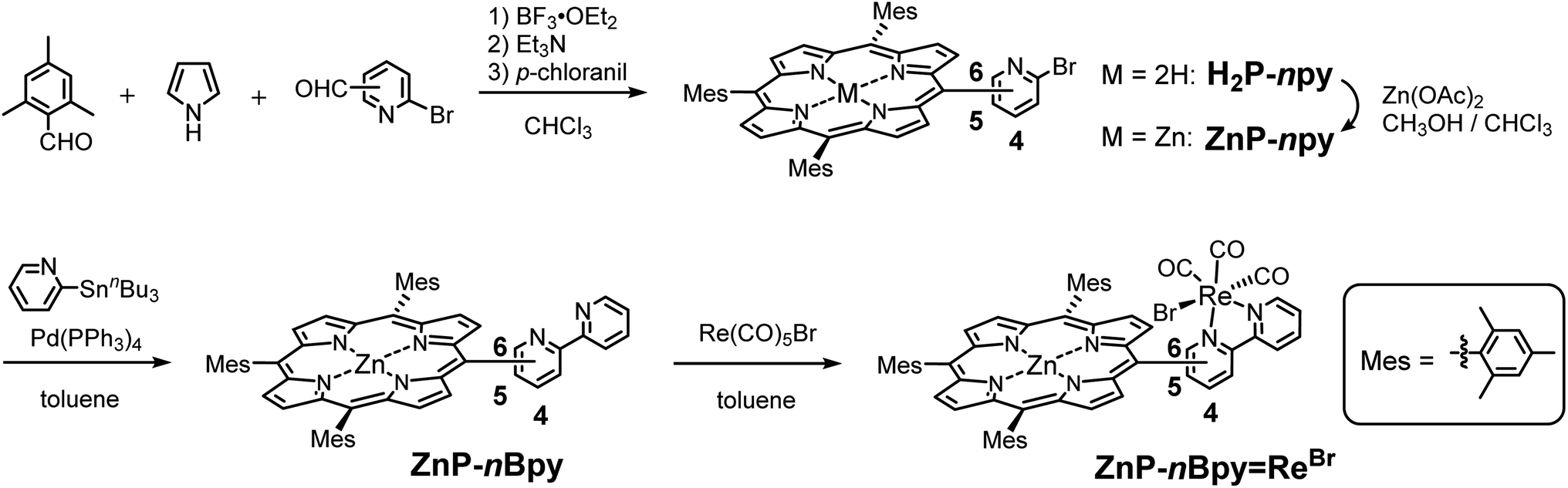 | ||
| Scheme 1 Synthetic routes of ZnP-nBpyReBr (n = 4, 5, and 6). | ||
All signals of the 1H NMR spectra were assigned using 2D NMR (1H–1H correlation spectroscopy (COSY)). Judging from the three CO signals in 13C NMR spectra (Fig. S41, S46, and S51†) and the patterns of the CO stretching bands in the infrared (IR) spectra (KBr pellets) (Fig. S44, S49, and S54†),33 the facial Re complexes are obtained as shown in Fig. 3.
We also prepared ZnP-5BpyReMeCN and ZnP-6BpyReMeCN by replacing the bromide ion with acetonitrile using silver ions in the Re complex unit (Scheme 2). The substitutions of acetonitrile ligands with DMA in DMA solutions were confirmed by the observed changes in the stretching vibrations of CO in the IR spectra (Fig. S65†).5a
 | ||
| Scheme 2 Synthesis of ZnP-nBpyReMeCN and ligand substitution of acetonitrile to DMA. | ||
Photophysical properties
The UV-vis absorption spectra of the dyads in DMA show intense Soret bands at approximately 430 nm and Q-bands at >540 nm (Fig. 4a). Upon comparing the spectra of ZnP-nBpy and ZnP-nBpyReBr, we note that the introduction of the Re tricarbonyl complex broadens the Soret bands and slightly red-shifts the Q-bands (Table 1 and Fig. S66†). This behavior was also observed in ZnP-phenRe. Judging from the largest full width at half maxima (FWHM) of the Soret bands (Fig. 4a inset and Table 1), the electronic interaction of ZnP-6BpyReBr is stronger (FWHM = 24 nm) than that of ZnP-4BpyReBr (FWHM = 17 nm) and ZnP-5BpyReBr (FWHM = 18 nm). On the other hand, broadening of the absorption band occurs regardless of the distance between the Zn and Re atoms (Fig. S67†), suggesting that the electronic interaction is mediated via the bipyridine moieties. The solvent-substituted dyads, ZnP-5BpyReDMA and ZnP-6BpyReDMA, exhibited nearly identical absorption spectrum shapes to their corresponding Br-substituted dyads (Fig. S68†). The charge of the Re complex unit seems to have little effect on the absorption of the porphyrin unit.
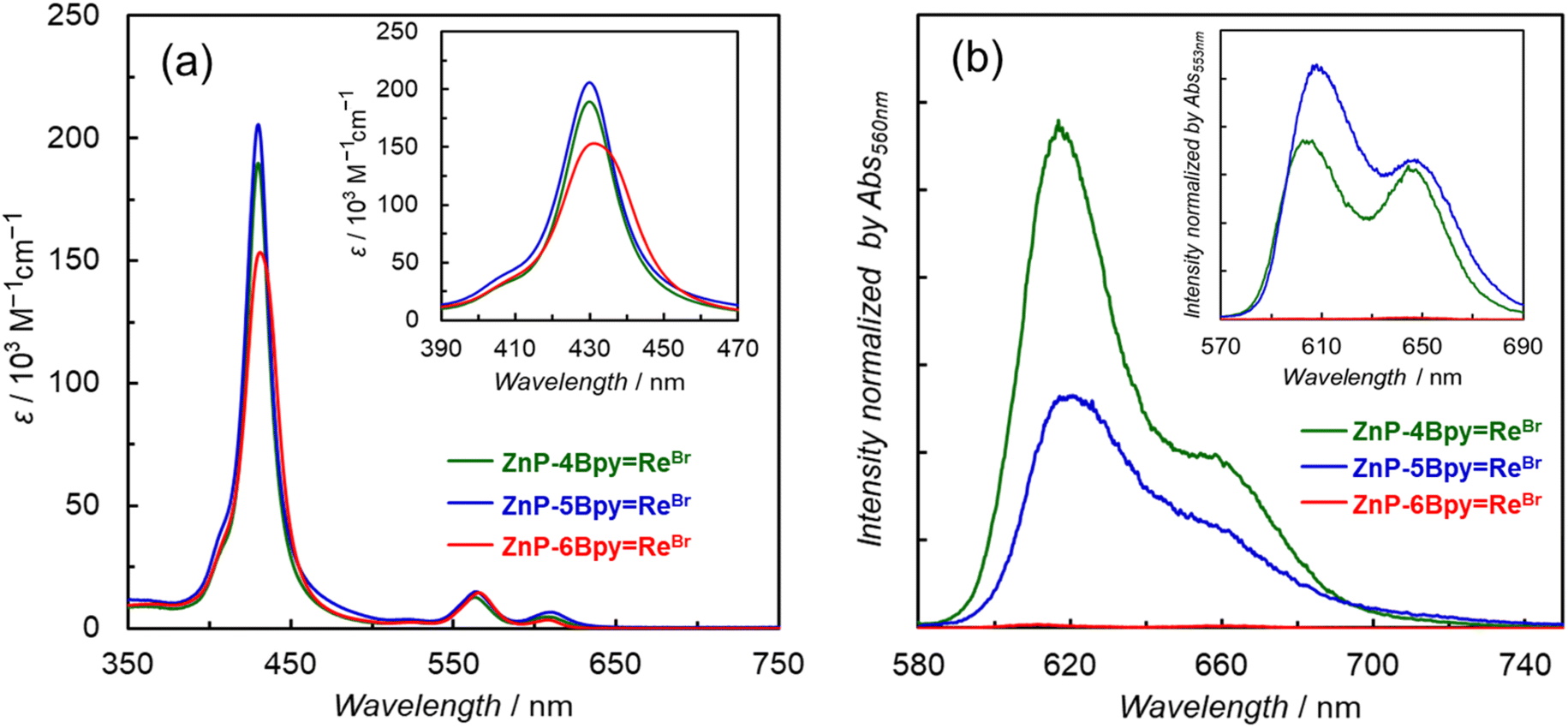 | ||
| Fig. 4 (a) UV-vis absorption spectra of ZnP-nBpyReBr (n = 4, 5, and 6) in DMA. Inset shows magnification of the Soret bands. (b) Fluorescence spectra of ZnP-nBpyReBr (n = 4, 5, and 6) in Ar-saturated DMA (λex = 560 nm) at 298 K. Inset shows spectra in Ar-saturated toluene (λex = 553 nm) at 298 K. The fluorescence spectra were normalized with absorbance at the excitation wavelength. | ||
| λ abs/nm (ε/104 M−1 cm−1) | FWHM/nm | λ em /nm | Φ f | ||
|---|---|---|---|---|---|
| DMA | Toluene | ||||
| a The solutions were degassed by bubbling with Ar gas. Excited at 560 nm in DMA and 553 nm in toluene. b In DMA. c The values in parentheses are the ratio to the fluorescence quantum yield of the corresponding porphyrin before the introduction of the Re complex. | |||||
| ZnP-4Bpy | 428 (50), 561 (1.7), 600 (0.52) | 7.8 | 606 | 3.6% | 3.3% |
|
ZnP-4BpyReBr
|
430 (19), 563 (1.3), 608 (0.45) | 17 | 617 | 2.0% (0.56) | 1.8% (0.55) |
| ZnP-5Bpy | 429 (44), 562 (1.7), 602 (0.65) | 9.4 | 605 | 2.6% | 3.6% |
|
ZnP-5BpyReBr
|
430 (21), 564 (1.5), 609 (0.66) | 18 | 621 | 1.1% (0.42) | 2.2% (0.61) |
| ZnP-6Bpy | 428 (42), 561 (1.5), 600 (0.44) | 8.2 | 605 | 4.2% | 3.6% |
|
ZnP-6BpyReBr
|
431 (15), 566 (1.5), 608 (0.36) | 24 | 611 | ∼0.02% (∼0.004) | ∼0.03% (∼0.009) |
|
ZnP-phenRe
|
430 (20), 565 (2.1), 606 (0.89) | 29 | 610 | ∼0.06% | ∼0.04% (∼0.01) |
Fluorescence spectra of the dyads are shown in Fig. 4b. Herein, DMA can coordinate to the central Zn ion of the porphyrin, affecting the spectral shape and wavelength.22b The fluorescence of the porphyrin is almost completely quenched in ZnP-6BpyReBr, while the fluorescence quenching due to the introduction of the Re complex is moderate in ZnP-4BpyReBr and ZnP-5BpyReBr (Table 1 and Fig. S69†). The fluorescence lifetimes were measured in polar DMA (Fig. S70–S73†) and less polar toluene, to estimate the radiative and nonradiative rate constants together with the fluorescence quantum yields (Table 2). Upon comparing ZnP-6Bpy with ZnP-6BpyReBr, we note that the nonradiative rate constants are remarkably enhanced by the introduction of the Re complex regardless of the solvent (knr = 3.8 × 108 s−1 for ZnP-6Bpy and ∼1 × 1011 s−1 for ZnP-6BpyReBr in DMA), suggesting that there is a rapid intersystem crossing (ISC) induced by the Re complex. A large knr value was also observed in ZnP-phenRe. In ZnP-4BpyReBr and ZnP-5BpyReBr, the nonradiative rate constants were also enhanced by introduction of the Re complex, although the extent of enhancement is not as great as in ZnP-phenRe and ZnP-6BpyReBr. In all the dyads, the knr values are independent of the solvent polarity, indicating that the main nonradiative process from the S1 of the porphyrin is the ISC on the porphyrin, which is imposed by the Re complex, rather than the photoinduced electron transfer from the S1 of the Zn porphyrin unit to the Re complex unit as observed in ReD′ (Fig. 2).23 The nonradiative rate constant from the S1 of the porphyrin of ZnP-6BpyReBr is approximately two orders of magnitude greater than for the other two dyads (1.3 × 109 and 1.5 × 109 s−1 for ZnP-4BpyReBr and ZnP-5BpyReBr in DMA, respectively), suggesting that the Zn porphyrin connected through the 6-position of the bpy is significantly influenced by spin–orbit coupling by the Re complex, whereas the porphyrins connected through the 4- and 5-positions are similarly and moderately affected.
| In DMA | In toluene | |||||
|---|---|---|---|---|---|---|
| τ f/ns | k r/109 s−1 | k nr/109 s−1 | τ f/ns | k r/109 s−1 | k nr/109 s−1 | |
| a Excited at 406 nm (FWHM 70 ps) in DMA and in toluene. b Radiative rate constant: kr = Φf/τf. Nonradiative rate constant: knr = 1/τf − kr. | ||||||
| ZnP-4Bpy | 2.2 | 0.016 | 0.44 | 2.1 | 0.016 | 0.46 |
|
ZnP-4BpyReBr
|
0.76 | 0.026 | 1.3 | 0.57 | 0.032 | 1.7 |
| ZnP-5Bpy | 2.5 | 0.010 | 0.39 | 2.5 | 0.014 | 0.39 |
|
ZnP-5BpyReBr
|
0.68 | 0.016 | 1.5 | 0.67 | 0.033 | 1.5 |
| ZnP-6Bpy | 2.5 | 0.017 | 0.38 | 2.5 | 0.014 | 0.39 |
|
ZnP-6BpyReBr
|
∼0.01 | ∼0.02 | ∼100 | ∼0.01 | ∼0.03 | ∼100 |
|
ZnP-phenRe
|
∼0.02 | ∼0.03 | ∼50 | ∼0.02 | ∼0.03 | ∼70 |
ZnP-phenRe showed phosphorescence from the Zn porphyrin in Ar-saturated DMA even at room temperature;22aZnP-6BpyReBr showed similar RTP. Fig. 5a shows the emission spectra at room temperature with delay time > 90 μs after the excitation pulse. The emission lifetimes were estimated to be 230 and 280 μs for ZnP-phenRe and ZnP-6BpyReBr, respectively (Fig. 5b). Observation of the phosphorescence spectrum for Zn porphyrin at room temperature is extremely rare in solution.
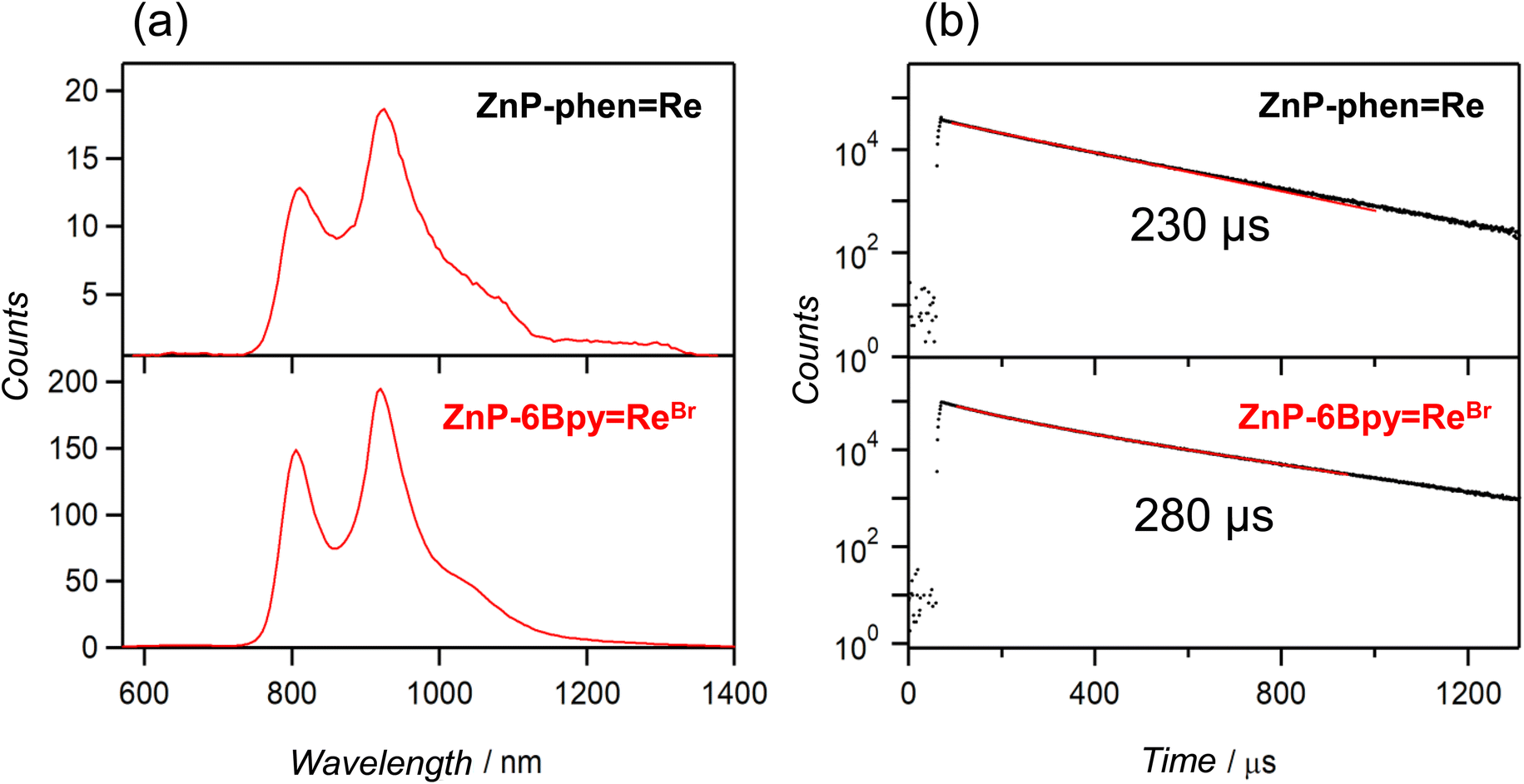 | ||
| Fig. 5 (a) Phosphorescence spectra of ZnP-phenRe and ZnP-6BpyReBr in Ar-saturated DMA (λex = 405 nm) at room temperature. The spectra were recorded with delay time > 90 μs after the excitation pulse. (b) Phosphorescence decay profiles (semilog plot) of ZnP-phenRe and ZnP-6BpyReBr in Ar-saturated DMA (λex = 405 nm) at room temperature, monitored at 800 and 920 nm. The decay profiles were fitted with one component (red lines). | ||
In contrast to ZnP-6BpyReBr, ZnP-4BpyReBr showed very weak phosphorescence, while phosphorescence from ZnP-5BpyReBr was hardly detected in DMA (Fig. 6a and S74†). The phosphorescence lifetimes of ZnP-4BpyReBr and ZnP-5BpyReBr were preliminarily estimated to be several tens of microseconds—this is an order of magnitude smaller than for ZnP-6BpyReBr. Considering that almost no phosphorescence was observed, the shorter lifetimes would originate from competitive rapid thermal deactivation processes.
To investigate the electron transfer process through the T1 of the porphyrin, quenching experiments with the addition of BIH were carried out. As observed in ZnP-phenRe,22a the phosphorescence of ZnP-6BpyReBr was efficiently quenched by the addition of BIH (Fig. 6b). The Stern–Volmer constant was estimated to be KSV = 11000 M−1. On the other hand, compared with the value of ZnP-phenRe (KSV = 180000 M−1),22a the value of 11000 M−1 for ZnP-6BpyReBr is almost an order of magnitude smaller. Considering that the phosphorescence lifetimes of ZnP-phenRe (230 μs) and ZnP-6BpyReBr (280 μs) are similar, the smaller Stern–Volmer constant in ZnP-6BpyReBr would come from the smaller quenching rate constants (kq) due to the slower electron transfer rate from BIH to the excited porphyrin (vide infra).
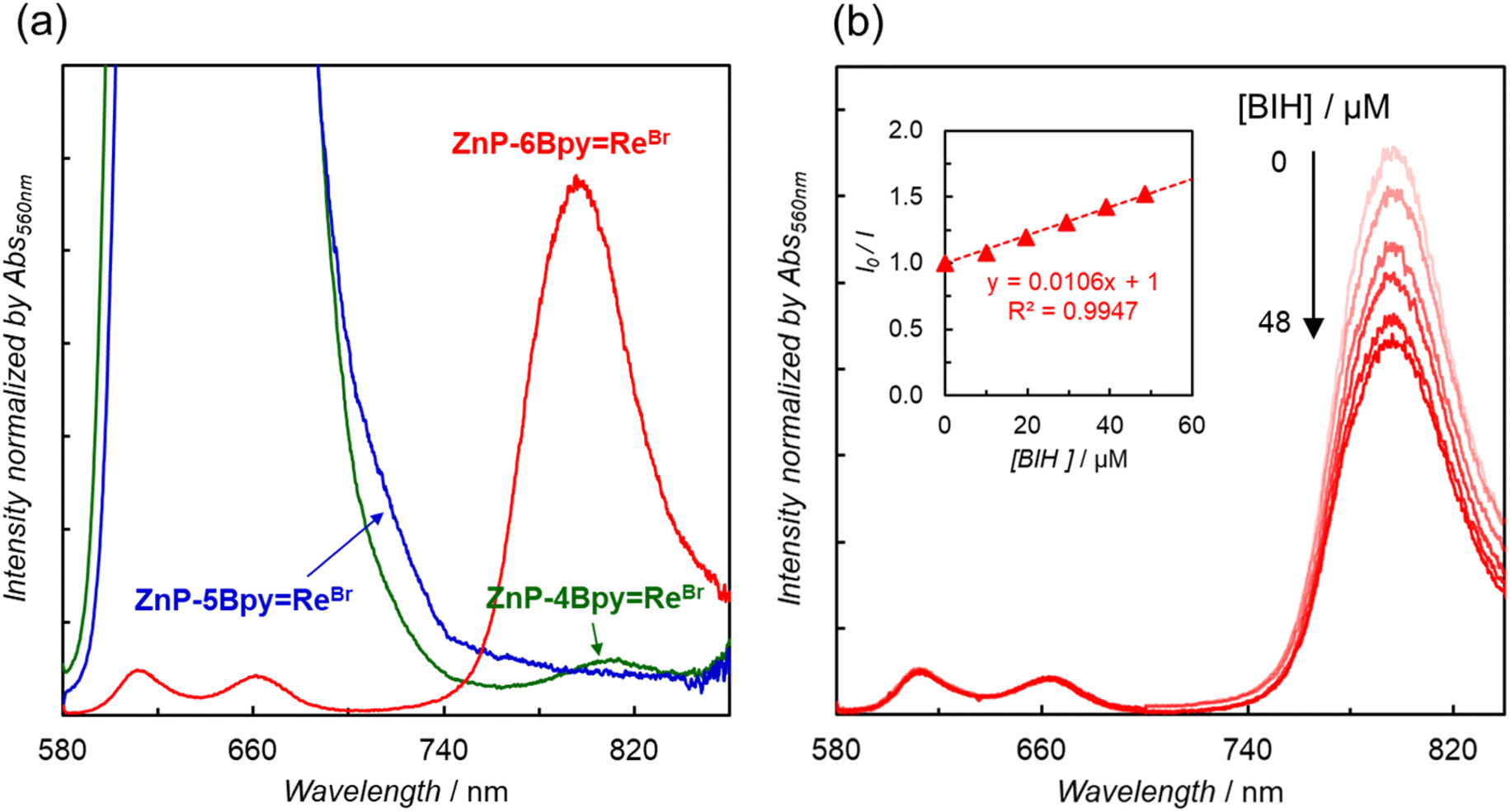 | ||
| Fig. 6 (a) Comparison of emission spectra of ZnP-nBpyReBr in Ar-saturated DMA (λex = 560 nm) at 298 K. (b) Emission spectra of ZnP-6BpyReBr in the presence of various amounts of BIH in Ar-saturated DMA (λex = 560 nm) at 298 K. Inset shows Stern–Volmer plot of the emission. | ||
We also performed emission measurements on ZnP-5BpyReDMA and ZnP-6BpyReDMA (Fig. S75†), showing that the fluorescence intensities remained unchanged, and in the case of ZnP-6BpyReDMA, the RTP was observed. This suggests that even in the dyads with the solvent-substituted cationic Re complexes, the intramolecular photoinduced electron transfer is a minor process, contrasting with previous studies19c,e,34,35 and observation in ReD′.23 In our system, the porphyrin and Re complex are in close proximity. This proximity effect may destabilize charge-separated states and lead to intersystem crossing. To clarify these details, further time-resolved absorption and infrared spectroscopic measurements are currently underway.
Electrochemical properties
Cyclic voltammograms (CVs) of the ZnP-nBpy series show that first reduction waves are reversible in Ar-saturated DMA (Fig. S76,† dotted lines), indicating that the OERS of the Zn porphyrin is stable in DMA. The redox potentials obtained from the differential pulse voltammograms (DPVs) of the dyads and the corresponding porphyrins are summarized in Table S1.† The first reduction peaks of the ZnP-nBpyReBr series appear at −1.68 V (n = 4), −1.64 V (n = 5), and −1.76 V (n = 6), which are on the more positive side than the reduction peaks (−1.84 to −1.87 V) of the ZnP-nBpy series. The first reduction potentials of the ZnP-nBpyReBr series were rather similar to that of fac-Re(bpy)(CO)3Br (−1.73 V). Density functional theory (DFT) calculations with consideration of the solvent (Fig. S77†) indicate that the lowest unoccupied molecular orbitals (LUMOs) and spin density plots for the OERS are abundantly distributed on the Re complex (Fig. S78 and S79†), indicating that the first reductions of the dyads occur mainly on the Re complex units of the ZnP-nBpyReBr series in the electrochemical measurements. The reduction potentials of the ZnP-nBpy series are more negative (−1.84 to −1.87 V) than that of the porphyrin unit of ZnP-phenRe (−1.79 V).22a The reduction potential of Zn meso-tetramesitylporphyrin (ZnTMP) is reported to be 180 mV more negative than that of Zn meso-tetraphenylporphyrin (ZnTPP), and the substituent effect of the tert-butyl groups is small (ΔEred = 20 mV in Fig. S80c†).36 Thus, the negative shifts arise from the substituents at the meso-positions of the porphyrin unit: the ZnP-nBpyReBr series has mesityl groups while the porphyrin unit of ZnP-phenRe has 4-tert-butylphenyl groups.
Assuming that the energy levels of the T1 of the ZnP-nBpyReBr series are similar to in ZnP-phenRe and that the reduction potential of the porphyrin moiety remains the same before and after introduction of the Re complex moiety, the electron transfer from BIH to the T1 is thermodynamically unfavorable (Fig. S81†). However, the phosphorescence quenching experiment gave a Stern–Volmer constant in ZnP-6BpyReBr that reached KSV = 11000 M−1; this value is much greater than values recorded for the well-used [Ru(N^N)3]2+.12 This originates from the long T1 lifetime (τp) of the Zn porphyrin, which allows the unfavorable electron transfer process. The kq value for ZnP-6BpyReBr (3.9 × 107 M−1 s−1) was smaller than that for ZnP-phenRe (7.8 × 108 M−1 s−1). This would be attributed to the more negative shift of the reduction potential of the Zn porphyrin unit due to the mesityl substituents.37
Photocatalytic CO2 reduction in DMA and additives suppressing dissociation of the Re complex
Photocatalytic CO2 reductions were carried out in DMA containing the dyad and BIH as the electron donor (Fig. 7). Here, 0.05 M of BIH was used to almost completely quench the T1 (99.8% in ZnP-6BpyReBr). Irradiation of a CO2-saturated DMA solution containing ZnP-phenRe and BIH showed a linear increase in CO formation with high durability, as observed previously.22a Almost no activity difference was observed between ZnP-4BpyReBr, ZnP-5BpyReBr, and ZnP-6BpyReBr. Although the three dyads showed similar initial reaction rates for CO formation to that of ZnP-phenRe, their activities were lost within 1 h and TONCO was <20 even after 8 h.
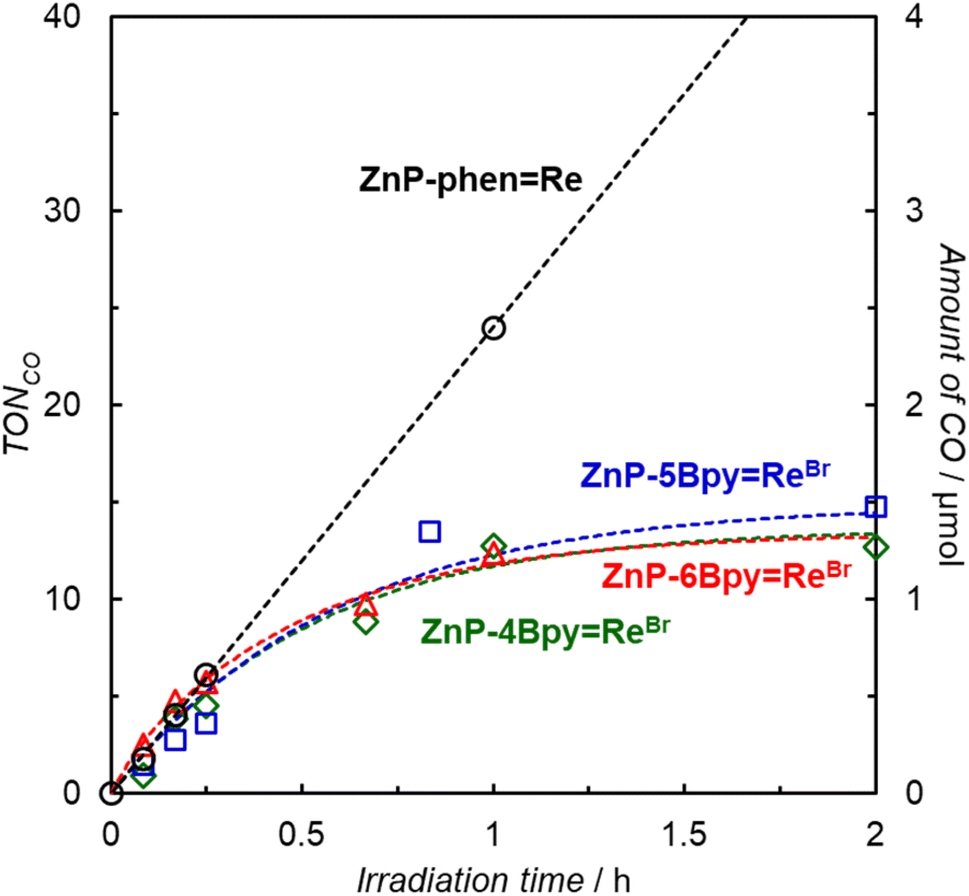 | ||
| Fig. 7 Time dependence of CO formation (0–2 h) during irradiation at 420 nm by LED lamps (input power: 30 mW), in a merry-go-round irradiation apparatus, of CO2-saturated DMA solutions containing the dyad (0.05 mM) and BIH (0.05 M). | ||
In the emission quenching experiments, we found that the fluorescence of ZnP-6BpyReBr increased during irradiation in the presence of BIH under a CO2 atmosphere (Fig. 8a). No increase in the fluorescence was observed in ZnP-phenRe or in the absence of BIH. The rate of fluorescence increase in ZnP-6BpyReBr was slower under a CO2 atmosphere than under an Ar atmosphere. Differential UV-vis absorption spectra of ZnP-6BpyReBr during the irradiation show the appearance of a sharp peak in the Soret band corresponding to ZnP-6Bpy (Fig. 8a inset). Thus, it is thought that the reduced ZnP-6BpyReBr leads to dissociation of the Re tricarbonyl unit to revive the fluorescence quenched by the Re complex. The phen ligand of ZnP-phenRe is structurally more rigid, with two nitrogen atoms fixed in the same direction, giving a more stable Re complex structure and high durability for the photocatalytic CO2 reduction. Fig. 8b shows the fluorescence spectral changes in the presence of phenol and TEOA. Surprisingly, phenol partially suppresses dissociation of the Re tricarbonyl unit and TEOA almost completely suppresses the dissociation. The significant suppression of the decomposition by the additives was not observed under an Ar atmosphere. It has been reported that a proton promotes the reaction of the reduced Re complex with CO2,38 and TEOA coordinates to the Re complex in the ground state to form an adduct with CO2.5 Otherwise, TEOA can reversibly react with CO2 to form a zwitterionic adduct which acts as a good proton source (Fig. S82†).8a Thus, TEOA might also contribute to the protonation of Re–COO− to Re–COOH. In any case, the additives might change the electron distribution of the reduced Re complex upon reaction with CO2, thus suppressing the dissociation of the Re tricarbonyl unit. Several deactivation pathways of Re diimine carbonyl complexes have been reported to involve the formation of Re formate complex,3a Re dimeric species,39 and so on.25a To the best of our knowledge, the dissociation of the Re tricarbonyl moiety from the bpy ligand after reduction has not been reported as the catalytic deactivation pathway for the CO2 reduction reactions. During the measurements of the emission spectra of the DMA-substituted cationic complex (ZnP-5BpyReDMA and ZnP-6BpyReDMA), increases in fluorescence were observed under dilution conditions (Fig. S83†). On the other hand, no such spectral changes were observed with the Br-substituted complex (ZnP-5BpyReBr and ZnP-6BpyReBr). These results suggest that dissociation of Re ion occurs in the solvent-substituted cationic complex, lacking the Br ion. Since the fluorescence spectral changes were suppressed in the presence of TEOA (Fig. S84†). TEOA stabilizes the solvent-substituted species, probably by coordination to the cationic Re ion.
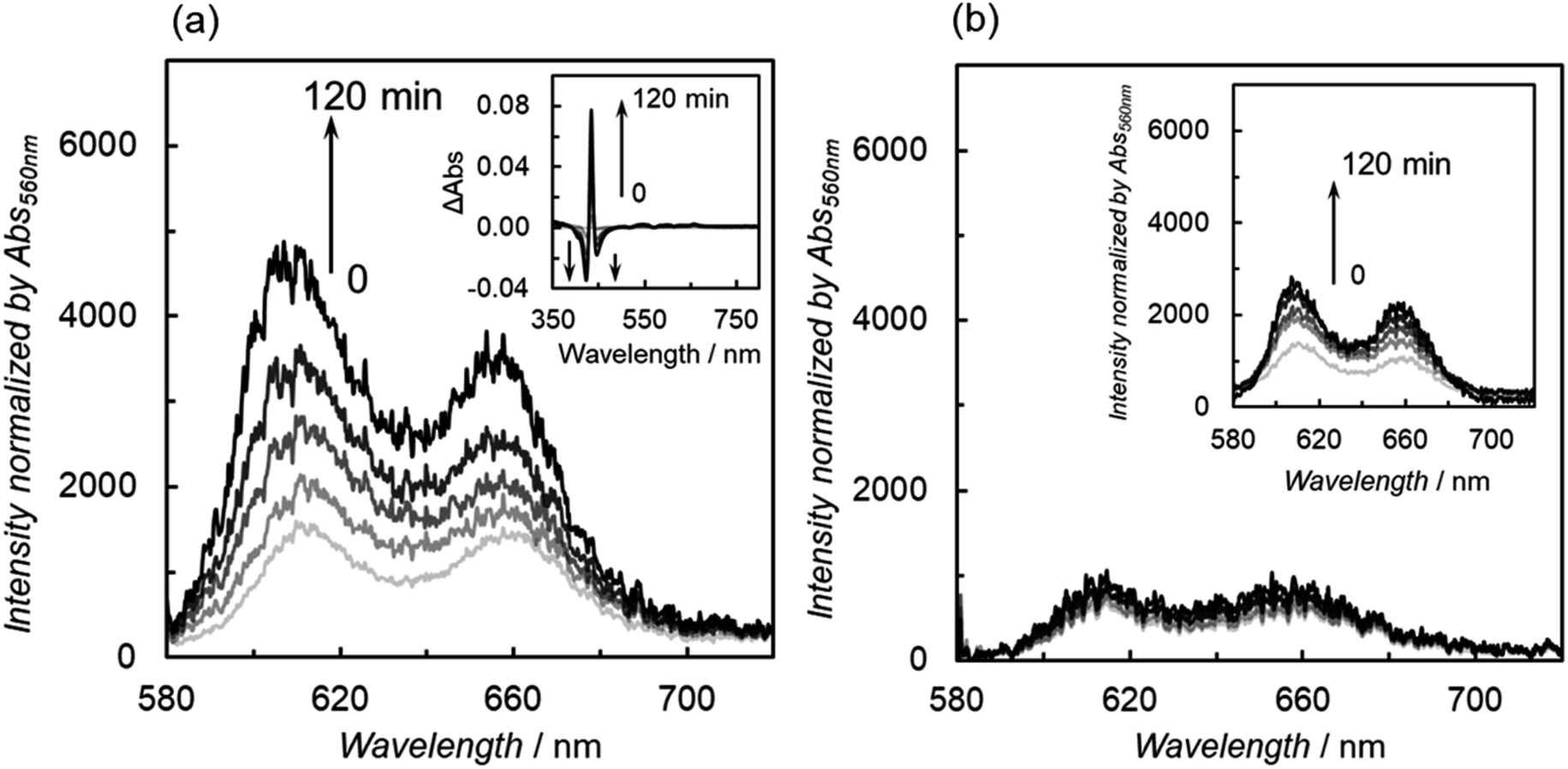 | ||
| Fig. 8 (a) Fluorescence spectral changes (0–120 min) of the CO2-saturated DMA containing ZnP-6BpyReBr (3 μM) in the presence of 50 μM of BIH during irradiation at 560 nm. Inset shows differential UV-vis absorption spectra during the irradiation. (b) Fluorescence spectral changes (0–120 min) of the CO2-saturated DMA–TEOA (4:1 v/v) containing ZnP-6BpyReBr (3 μM) in the presence of 50 μM of BIH during irradiation at 560 nm. Inset shows fluorescence spectral change in the presence of 6.7 mM phenol. | ||
Photocatalytic CO2 reduction in DMA–TEOA
Because the additives improved the stability of ZnP-6BpyReBr, the photocatalytic CO2 reductions were carried out in the presence of phenol (Fig. S85†) and TEOA (Fig. 9). In particular, the addition of TEOA remarkably improved the catalytic activities of the ZnP-nBpyReBr series in terms of reaction rate and durability. Herein, the photocatalytic CO2 reductions selectively produced CO without forming detectable amounts of CH4 and HCOOH, but with a trace amount of H2 (Fig. S86†). This improvement would result from the suppression of the Re dissociation by TEOA. Fig. 9 shows that only ZnP-phenRe is deactivated in the presence of TEOA. Previously we reported that TEOA promotes the formation of chlorins and decreases the catalytic durability in ZnP-phenRe.23 Furthermore, the chlorins are formed by the reaction of TEOA with the excited porphyrin at the β-pyrrole position.40 Thus, the lack of inactivation of the ZnP-nBpyReBr series would be attributed to the low reactivity of the porphyrins to TEOA, which would be caused by the mesityl groups having high steric hindrance and electron donating. Upon comparing the initial reaction rates, we note that all dyads in the ZnP-nBpyReBr series surpass the previous ZnP-phenRe system under optimal conditions. In particular, ZnP-5BpyReBr unexpectedly exhibits a higher initial reaction rate (twice as high) and higher durability than achieved with ZnP-4BpyReBr and ZnP-6BpyReBr; the turn over frequency (TOFCO) reaches 590 h−1. However, even in ZnP-5BpyReBr the CO formation stopped within 5 h and further addition of BIH to the reaction mixture did not restart the photocatalytic reaction. The UV-vis absorption spectra after 17 h showed degradation of the porphyrins (Fig. S87†), indicating that the photocatalysts decomposed during the irradiation. The UV-vis absorption spectral change of ZnP-5BpyReBr during relatively strong light irradiation at 420 nm showed the formations of the OERS of the porphyrin units at ca. 450 and 700 nm41 and chlorins at ca. 610 nm42 at the initial stage (Fig. S88†). Thus, the photocatalyst would undergo further reduction from chlorin and faded during the irradiation.
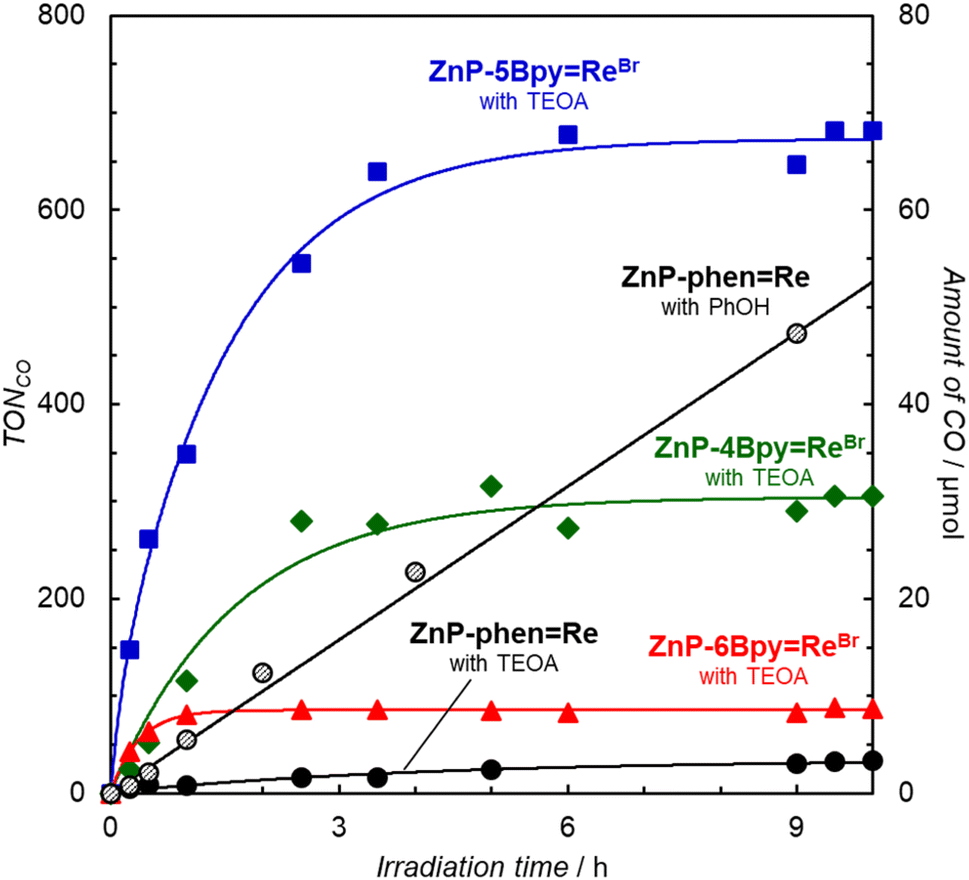 | ||
| Fig. 9 Time dependence of CO formation during irradiation at 420 nm by LED lamps (input power: 30 mW), in a merry-go-round irradiation apparatus, of CO2-saturated DMA–TEOA (4:1 v/v) solutions containing the dyad (0.05 mM) and BIH (0.05 M). ZnP-4BpyReBr (green-filled diamond), ZnP-5BpyReB (blue-filled square), ZnP-6BpyReBr (red-filled triangle), ZnP-phenRe (black-filled circle). Plot of ZnP-phenRe (0.05 mM) in CO2-saturated DMA containing BIH (0.05 M) and phenol (0.1 M) shown as black open circles. | ||
As it is thought that the accumulation of two electrons on the porphyrin causes the decomposition,21 we carried out the photocatalytic CO2 reduction using weaker light to prevent the electron accumulation rate from exceeding the CO2 reduction reaction rate. Fig. 10a shows the time-course comparison of CO production using ZnP-5BpyReBr when the input power of the apparatus was changed from 30 mW to 0.5 mW. As a control experiment, the reaction using a mixed system of ZnTMP and fac-Re(bpy)(CO)3Br was carried out with weak light (0.5 mW). In the mixed system, CO production was mainly observed but the reaction stopped when the TONCO reached approximately 100. On the other hand, in ZnP-5BpyReBr the CO production linearly increased to TONCO ≈ 1000, corresponding to a quantitative amount of added BIH (1000 equivalents). Further addition of 500 equivalents of BIH restarted the reaction, with the same reaction rate, and the reaction continued until all BIH was consumed. A plot of TONCOversus the product of input power and irradiation time shows that the reaction proceeds more efficiently with weak light irradiation (Fig. 10b).
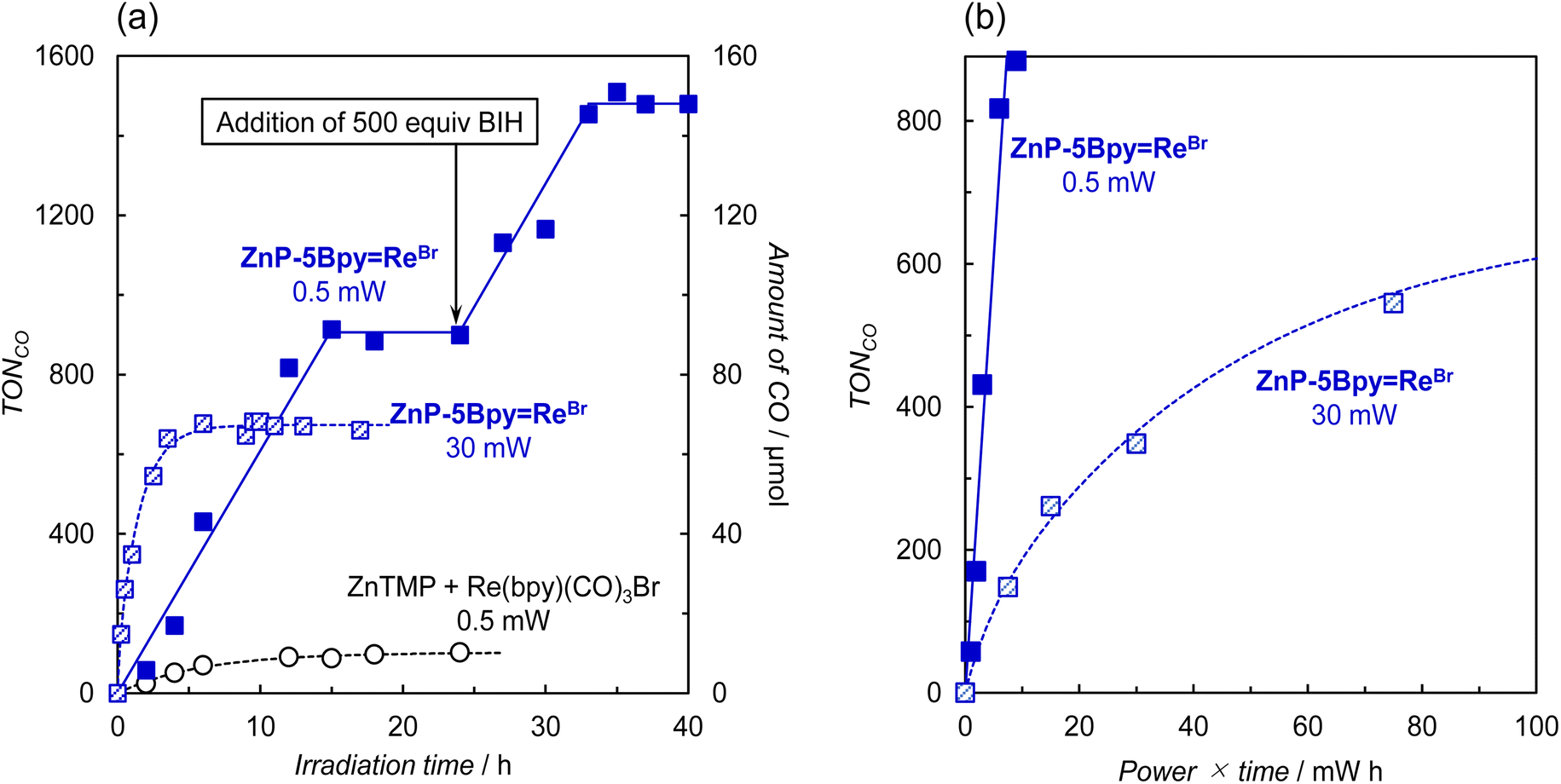 | ||
| Fig. 10 (a) Time dependence of CO formation during irradiation at 420 nm by LED lamps (input power: 0.5 and 30 mW), in a merry-go-round irradiation apparatus, of CO2-saturated DMA–TEOA (4:1 v/v) solutions containing ZnP-5BpyReBr (0.05 mM), and a mixed system of ZnTMP (0.05 mM) and fac-Re(bpy)(CO)3Br (0.05 mM) in the presence of BIH (0.05 M). (b) Plots of TONCOversus product of input power and irradiation time. | ||
Initial photoinduced electron transfer process from BIH
The photocatalytic CO2 reduction using ZnP-5BpyReBr showed the highest reaction quantum yield with high durability, but ZnP-5BpyReBr showed no RTP. This indicates that there is no direct correlation between the phosphorescence and the catalytic activity. On the other hand, the time course of CO production showed that the photocatalytic CO2 reduction proceeded at a constant rate until all BIH in the system was consumed (Fig. 10a). Thus, the reaction proceeds efficiently even under dilute BIH conditions, indicating that the electron transfer from BIH proceeds through a long-lived excited state, that is, the T1 of the porphyrin. In fact, the plots of ΦCO of ZnP-5BpyReBr against the initial concentration of BIH (Fig. 11) show that the ΦCO is almost constant and well fitted with the simulation curve assuming KSV = 1200 M−1, where the value of kq is similar to that of ZnP-6BpyReBr (3.9 × 107 M−1 s−1) and the phosphorescence lifetime assuming 30 μs. A similar independent plot of ΦCOversus [BIH] is obtained for ZnP-phenRe (Fig. S89†). Thus, in ZnP-5BpyReBr, even though no RTP was observed, the quantitative electron transfer from BIH to the T1 of the porphyrin occurs, allowing the efficient photocatalytic CO2 reduction.
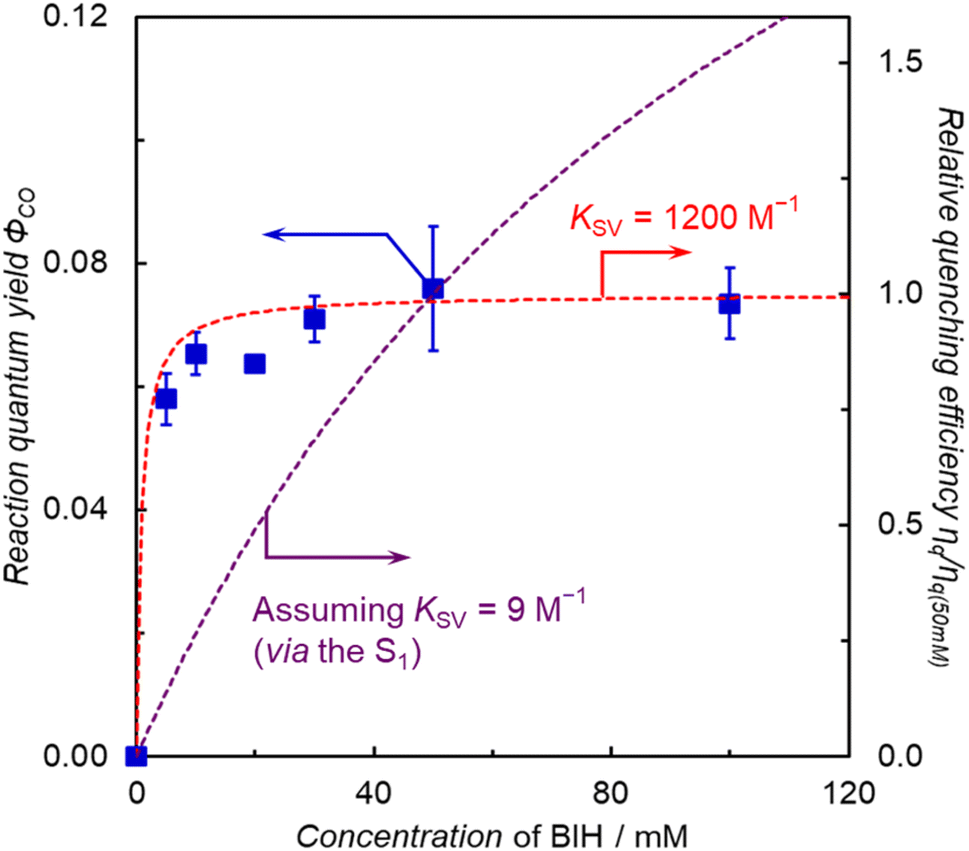 | ||
| Fig. 11 Plot of the ΦCOversus the initial concentration of BIH in the photocatalytic CO2 reduction (Xe lamp, 420 nm, 3.6 × 10−8 einstein per s) using DMA–TEOA (4:1 v/v) solutions containing ZnP-5BpyReBr (0.05 mM). The quenching efficiency relative to ηq at [BIH] = 50 mM was calculated using ηq = [BIH]KSV/(1 + [BIH]KSV). | ||
Light intensity dependence of the reaction quantum yield
In Fig. 10, the initial reaction rates are not proportional to the input power. We reported earlier that the reaction quantum yield (ΦCO) of ZnP-phenRe significantly decreases as the light intensity increases, due to an inner-filter effect by the OERS of the porphyrin unit which has strong absorption at the excitation wavelength.23Fig. 12 shows the plots of ΦCO with respect to the light intensity. The ZnP-nBpyReBr series showed similar light intensity dependences. The ΦCO converges under very low light intensity (<3 × 10−10 einstein per s) and finally reaches 24% in ZnP-5BpyReBr. Upon comparing the ΦCO at a low light intensity (3.2–3.5 × 10−9 einstein per s), we note that the value of ZnP-5BpyReBr is twice higher than the values of the other dyads (Table 3).
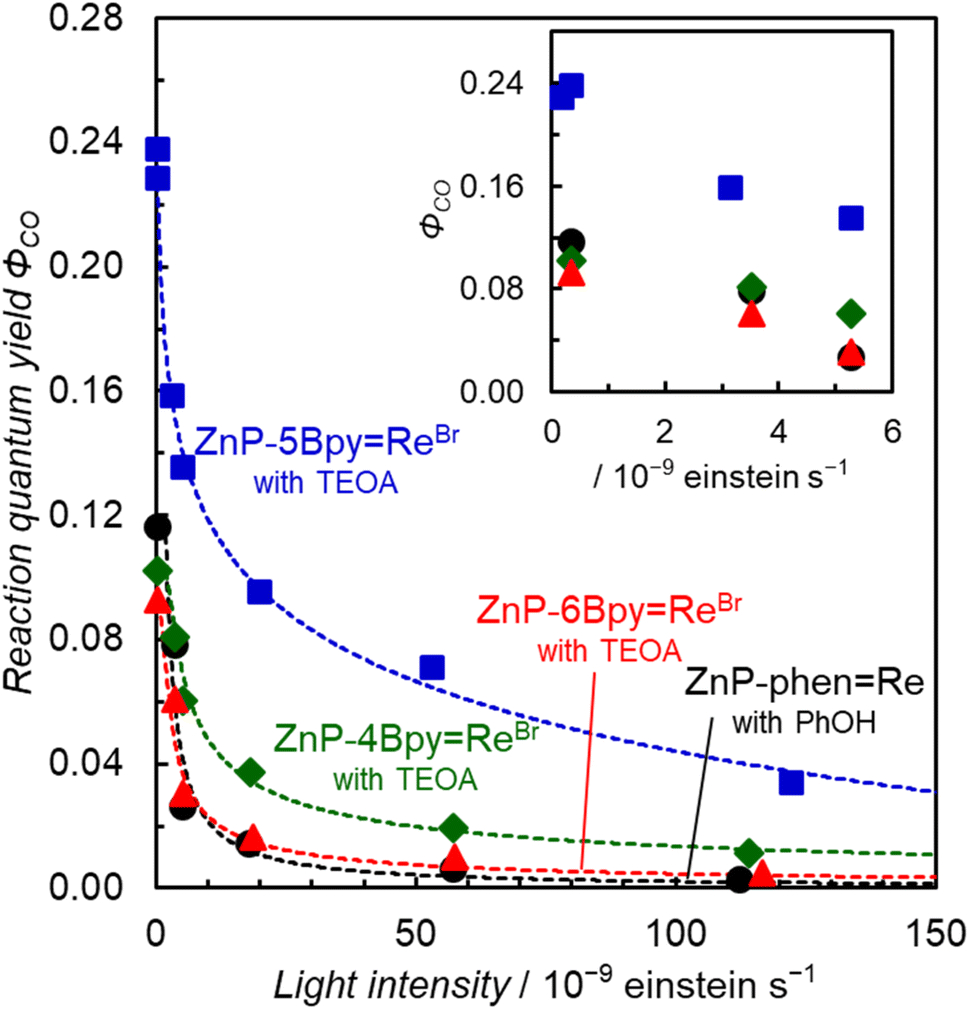 | ||
| Fig. 12 Dependence of ΦCO on the irradiation light intensity (Xe lamp, λex = 420 nm) for the dyads ZnP-4BpyReBr (green diamond), ZnP-5BpyReBr (blue square), and ZnP-6BpyReBr (red triangle) (0.05 mM) in CO2-saturated DMA–TEOA (4:1 v/v) solutions containing BIH (0.05 M), and for ZnP-phenRe (black circle) (0.05 mM) in CO2-saturated DMA containing BIH (0.05 M) and phenol (0.1 M). Inset shows magnification of CO formation dependence at low light intensity. | ||
ReBr series with TEOA and ZnP-phenRe with phenola
| Light intensity/einstein per s |
ZnP-4BpyReBr
|
ZnP-5BpyReBr
|
ZnP-6BpyReBr
|
ZnP-phenRe
|
|
|---|---|---|---|---|---|
|
a Irradiated at 420 nm (Xe lamp, λex = 420 nm) to CO2-saturated DMA–TEOA (4:1 v/v) containing the ZnP-nBpyReBr series (0.05 mM) and BIH (0.05 M). In ZnP-phenRe, phenol (0.1 M) was present, not TEOA.
|
|||||
| Φ CO | 3.2–3.5 × 10−9 | 8% | 16% | 6% | 8% |
| 3 × 10−10 | 10% | 24% | 9% | 11% | |
Fig. 13 shows the UV-vis absorption spectral changes during weak light irradiation at 420 nm. The formations of the OERS of the porphyrin units at ca. 450 and 700 nm41 and chlorins at ca. 610 nm42 were observed in ZnP-4BpyReBr, ZnP-6BpyReBr, and ZnP-phenRe (Fig. 13a, c and d). Chlorins are formed by two-electron reduction of porphyrin.19c Indications are therefore that the accumulation of two or more electrons occurs in ZnP-4BpyReBr, ZnP-6BpyReBr, and ZnP-phenRe. On the other hand, ZnP-5BpyReBr undergoes hardly any change during the irradiation (Fig. 13b). Thus, the reduced species tend to accumulate on the porphyrin in ZnP-4BpyReBr, ZnP-6BpyReBr, and ZnP-phenRe, whereas ZnP-5BpyReBr showed less electron accumulation, resulting in the low light intensity dependence and the high ΦCO.
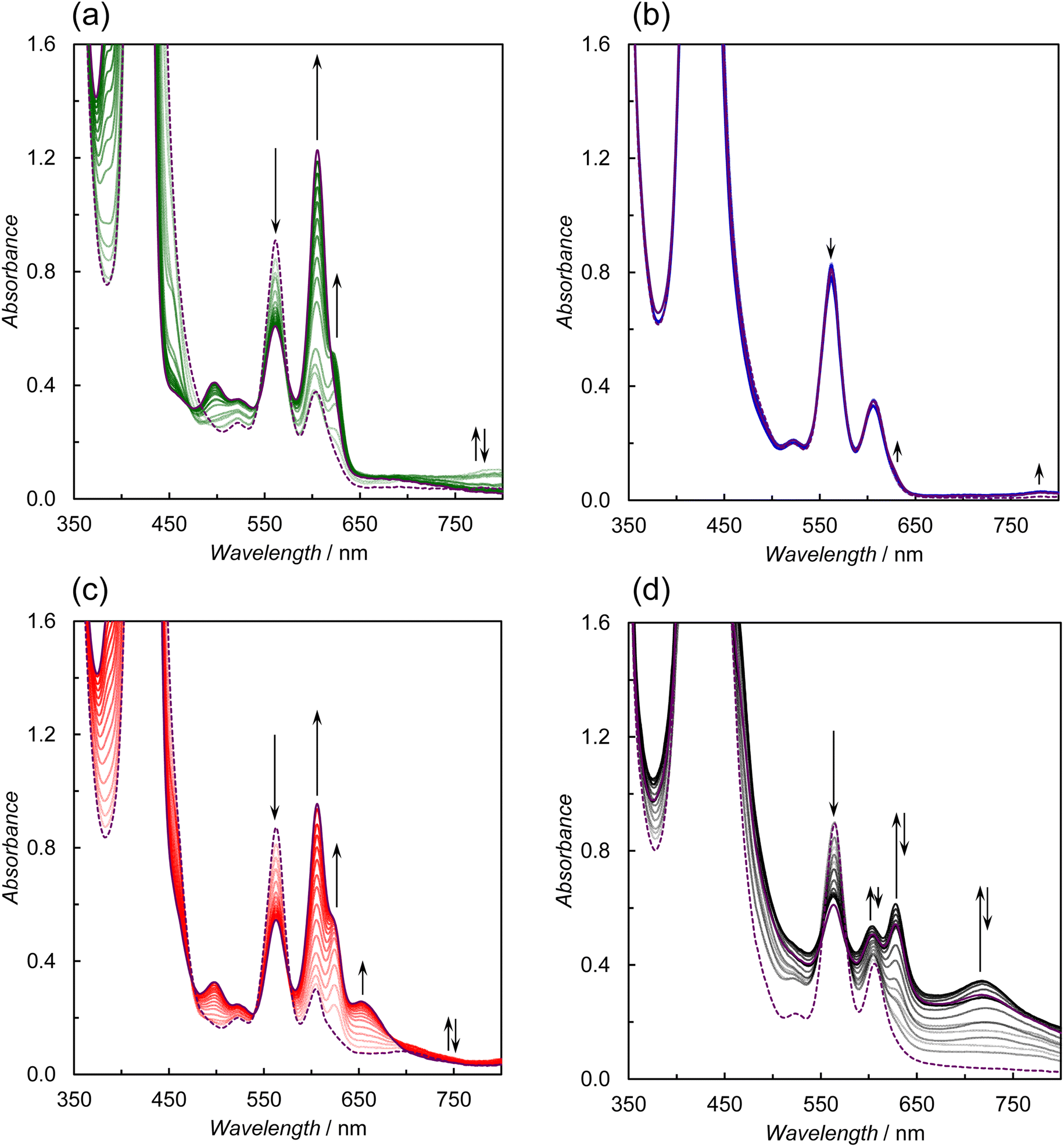 | ||
| Fig. 13 UV-vis absorption spectral changes during irradiation (0–60 min) at 420 nm (Xe lamp, 5.0 × 10−9 einstein per s) of CO2 saturated DMA–TEOA (4:1 v/v) solutions containing BIH (0.05 M) and the dyad (0.05 mM) (a) ZnP-4BpyReBr, (b) ZnP-5BpyReBr, (c) ZnP-6BpyReBr, and of (d) a CO2-saturated DMA solution containing BIH (0.05 M) and ZnP-phenRe (0.05 mM) in the presence of phenol (0.1 M). Purple dotted and solid lines show the spectra before and after the irradiation, respectively. | ||
Mechanistic insight into the photocatalytic reaction in the presence of TEOA
It has also been reported that the catalysis of CO2 reduction in the fac-Re(bpy)(CO)3X-type complex (X = Cl−, Br−, l−) commences with the dissociation of a halogen anion upon reduction (Fig. S90†).2b,43 No induction periods in the time-course profiles of CO formation were observed in the ZnP-nBpyReBr series (Fig. 7 and 9), which suggests that efficient reduction of the Re(bpy)(CO)3Br unit occurs to dissociate the halogen ligand in the three dyads. Considering that the cationic solvent-substituted Re complexes, [Re(bpy)(CO)3(solvent)]+, exhibit more positive reduction potentials than the neutral fac-Re(bpy)(CO)3X-type complexes,43 the reduction of the Re complex unit is also expected to occur efficiently under the catalytic cycle (Fig. S90†). Increasing the concentration of Br ions decreased the catalytic activity (Fig. S91†), supporting the proposed reaction mechanism that the solvent-substituted species after Br dissociation is the active species.
The ΦCO of ZnP-5BpyReBr is double that of the other three dyads in DMA–TEOA (Table 3). Furthermore, despite the high catalytic activity, the UV-vis absorption spectra of ZnP-5BpyReBr show almost no change during the photocatalytic reaction (Fig. 13b), indicating that the electron consumption by the CO2 reduction reaction is accelerated, particularly in ZnP-5BpyReBr. Considering that the catalytic activities were similar among the three dyads in the absence of TEOA (Fig. 7), the higher ΦCO in ZnP-5BpyReBr would be attributed to TEOA. As mentioned in the Introduction, the ground state of [Re(bpy)(CO)3(solvent)]+ can capture CO2 in the presence of TEOA (Fig. 1). We calculated the spin densities of the OERS of the ZnP-nBpyReBr series with and without the bromide ion, and their TEOA and CO2 adducts (Fig. S79†). However, there was no significant difference in the spin density among the three dyads. This suggests that there is another activity-enhancing factor besides TEOA.
From the molecular model optimized by the DFT calculation, TEOA would coordinate both the Re complex and the Zn porphyrin only in ZnP-5BpyRe (Fig. 14). If TEOA is bound via the Zn porphyrin, as shown in Fig. 14, it is expected that CO2 is quickly captured and the reduction reaction proceeds; then, after the CO2 reduction reaction it is possible that the next CO2 is captured and so the reaction proceeds. During the catalytic reaction, it is thought that the bromide ion of the Re complex is substituted by the solvent molecule (Fig. S90†),2b,38 and the coordination of TEOA to the Re complex occurs in the solvent-substituted complex. Thus, the binding ability of TEOA to the DMA-substituted dyads was investigated to verify the hypothesis of the multidentate coordination (Fig. 14).
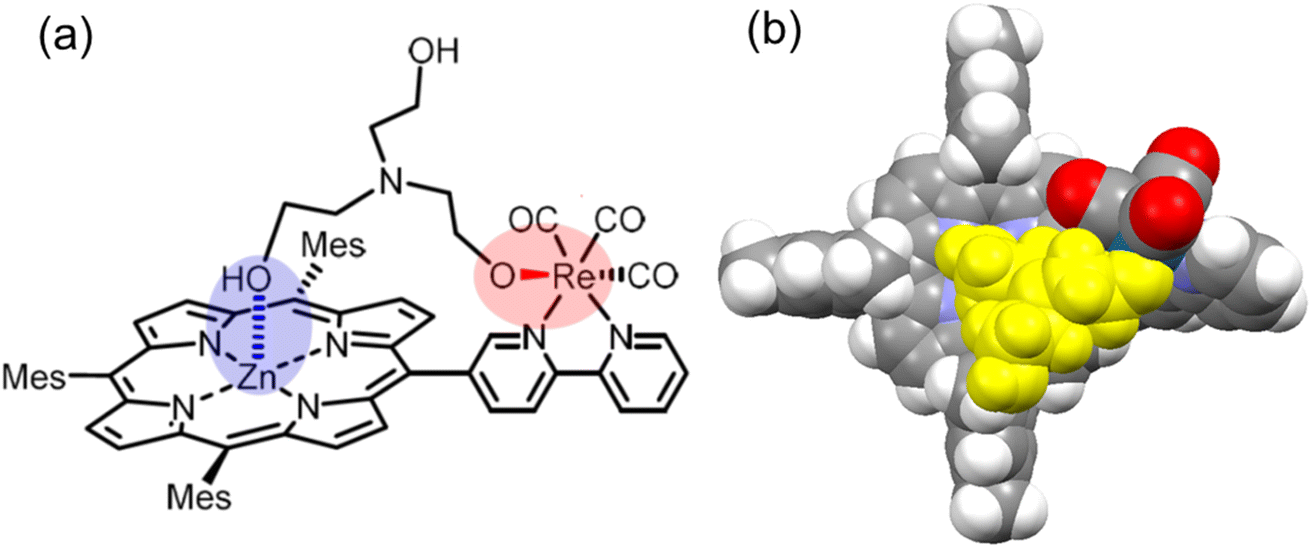 | ||
| Fig. 14 (a) Plausible structure and (b) molecular model (optimized by DFT calculation) of a TEOA adduct of ZnP-5BpyRe (without the bromide ion). TEOA is colored in yellow. | ||
The formation of the DMA-substituted dyads, ZnP-5BpyReDMA and ZnP-6BpyReDMA, was monitored with CO stretching bands of IR spectra (Fig. S65†). We confirmed that the substitution reaction with DMA was completed before the addition of TEOA, because TEOA irreversibly reacts with the acetonitrile-substituted Re complex to afford the corresponding iminoester complex (Fig. S92†).5b TEOA was added to the resulting DMA solutions: DMA/TEOA = 400:1 (v/v). The formation of the Re–TEOA structure in Fig. 1 was monitored with the CO stretching bands' shift to lower wavenumbers in IR spectra (Fig. S93†). The CO stretching band at 2000–2050 cm−1 of the thermodynamically equilibrated mixture was separated by curve fitting (Fig. 15). The absorption band profiles were well fitted with two Gaussian-shape functions, corresponding to the DMA-substituted dyad and the TEOA-coordinated dyad (ZnP-5BpyReTEOA or ZnP-6BpyReTEOA). The absorption intensities as the band areas in ZnP-5BpyRe and ZnP-6BpyRe were analyzed as 4:6 and 8:2 mixtures of the DMA- and TEOA-coordinated dyads, respectively. The equilibrium constants assuming K = [ZnP-nBpyReTEOA][DMA]/[ZnP-nBpyReDMA][TEOA]5 were estimated to be 1000 and 110 for ZnP-5BpyReDMA and ZnP-6BpyReDMA, respectively, indicating the stronger TEOA coordination to the Re complex unit in ZnP-5BpyReDMA than in ZnP-6BpyReDMA.
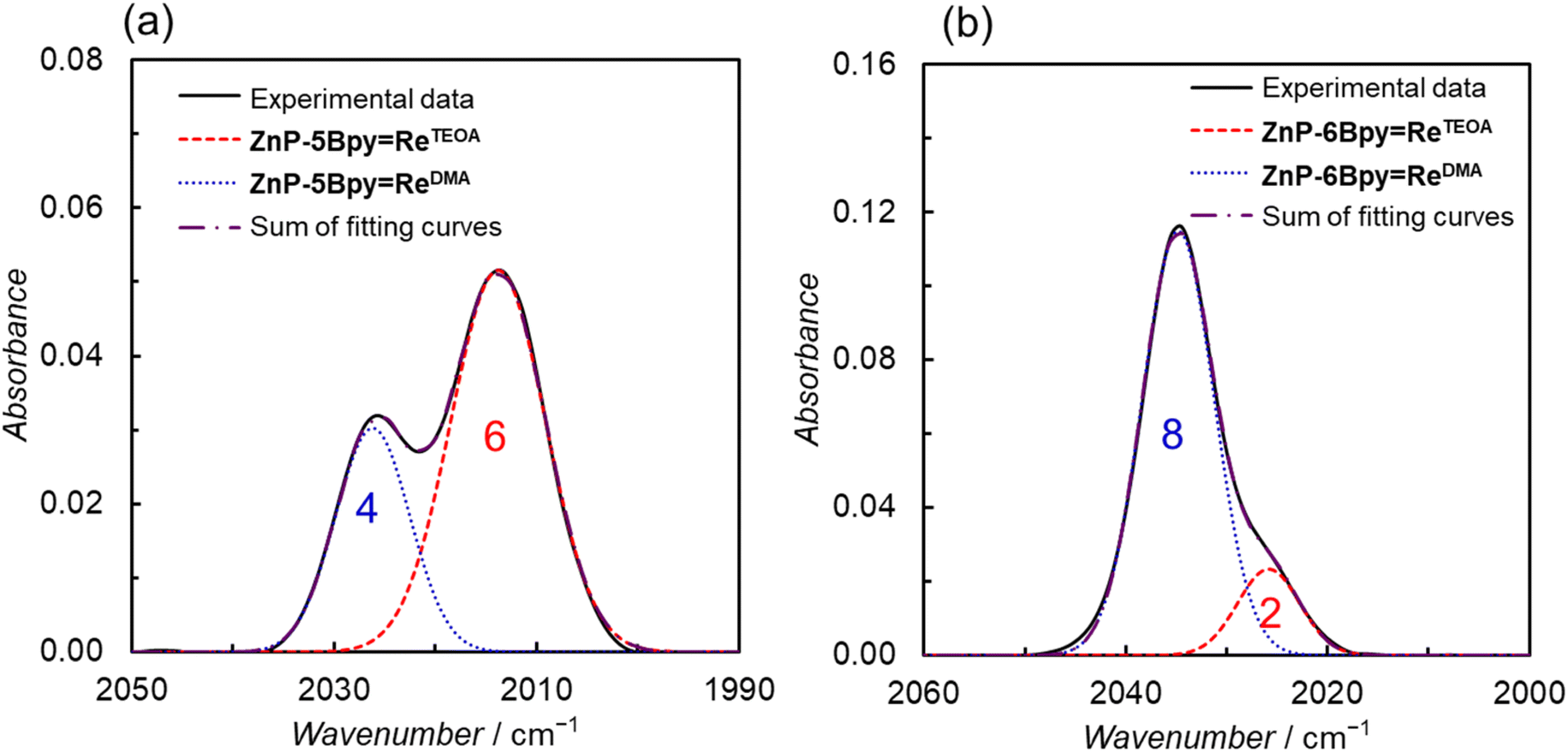 | ||
| Fig. 15 IR spectra (solid line) of the equilibrated mixture of (a) ZnP-5BpyReDMA and ZnP-5BpyReTEOA, and (b) ZnP-6BpyReDMA and ZnP-6BpyReTEOA in DMA–TEOA (400:1 v/v) with the fitting curves (dotted lines). Concentrations of the dyads: 9 mM. | ||
We then attempted to verify that the Zn porphyrin unit contributes to the TEOA coordination in ZnP-5BpyReDMA through the 1H NMR spectra in DMF-d7. However, the addition of one equivalent of TEOA hardly showed signal shifts because the binding ability of TEOA to the dyad was too weak in the presence of the competing coordinating DMF. The contribution of the Zn porphyrin unit was verified through UV-vis absorption titration experiments. In general, the Soret and Q-bands of Zn porphyrin are significantly red-shifted by axial coordination of the Zn porphyrin. Upon the addition of TEOA, no spectral change was observed in DMA because DMA coordinates to the Zn porphyrin unit. Thus, TEOA was added to ZnP-5BpyReDMA and ZnP-6BpyReDMA dissolved in dichloromethane, which is a noncoordinating solvent against Zn porphyrin (Fig. S94†).
As control experiments, we also carried out titrations using ZnP-5BpyReBr and ZnP-5Bpy (Scheme 1). During the titration of TEOA, all the dyads showed red-shifted Soret and Q-bands with isosbestic points (Fig. S95†). Spectral change converged with the addition of ca. 6 mM TEOA for ZnP-5BpyReDMA, but did not converge without the addition of >150 mM TEOA for the other dyads (Fig. S96†). The apparent binding constants assuming Kapp = [ZnP-TEOA]/[ZnP][TEOA] (ZnP = Zn porphyrin unit) were estimated using Benesi–Hildebrand plots (Fig. S97†).44 Results showed approximately one order of magnitude larger value for ZnP-5BpyReDMA (Kapp = 330 M−1) than for the other three dyads under Ar atmosphere (Table 4). This indicates that after dissociation of the bromide ion a TEOA molecule interacts on both the Re complex and Zn porphyrin units in ZnP-5BpyReDMA, as shown in Fig. 14.
| K app/M−1, under Ar | K app/M−1, under CO2 | |
|---|---|---|
| a K app = [ZnP–TEOA]/[ZnP][TEOA]. | ||
| ZnP-5Bpy | 31 | — |
|
ZnP-5BpyReBr
|
30 | 48 |
|
ZnP-5BpyReDMA
|
330 | 760 |
|
ZnP-6BpyReDMA
|
48 | — |
In order to confirm whether TEOA can coordinate properly to the Zn ion even in DMA–TEOA (4:1 v/v), the titration experiments with DMA were carried out using ZnP-5BpyReBr and ZnP-5BpyReDMA in dichloromethane (Fig. S98†). Based on the apparent binding constants of DMA coordination, we estimated that 67% of the Zn porphyrin was coordinated with TEOA in Ar-saturated DMA–TEOA (4:1 v/v). This suggests that TEOA can sufficiently coordinate with the Zn ion under catalytic reaction conditions. It should be noted that since deprotonation of the OH groups of TEOA is unlikely to occur in dichloromethane, the actual proportion of the TEOA-coordinated Zn porphyrin would be greater than 67% in DMA–TEOA (4:1 v/v), where the OH groups of TEOA are more readily deprotonated.
The titration experiments under CO2 atmosphere for ZnP-5BpyReDMA and ZnP-5BpyReBr showed that the binding constant for ZnP-5BpyReDMA increased significantly to Kapp = 760 M−1, compares to that under Ar atmosphere (Table 4 and Fig. S99†). This suggests that the CO2 insertion into the Re–OR bond occurs, enhancing the binding of TEOA. The binding constant of ZnP-5BpyReBr was also found to be larger under CO2 atmosphere than under Ar atmosphere. While it has been reported that CO2 is captured by insertion into the Zn–methanol bonds,10 the titration experiments using ZnTMP with TEOA showed similar binding constants (Kapp ∼ 20 M−1) under both Ar and CO2 atmospheres. Therefore, it is speculated that the enhanced interaction under CO2 is mainly due to the interaction around the Re complex.
The IR spectra and results of UV-vis experiments suggest that TEOA strongly binds to the Re complex unit in ZnP-5BpyReDMA by the dual interaction of both the Re complex and the Zn porphyrin units. The tightly bound TEOA would facilitate the CO2 capture in the ground state of the dyad, resulting in the reaction with CO2 immediately after the reduction of the Re complex unit to prevent the accumulation of the OERS of the porphyrin (Fig. S100†).45 A detailed reaction mechanism involving TEOA and ZnP-5BpyRe has not been established yet—further time-resolved spectroscopic measurements are still required.
Conclusions
We systematically synthesized three Zn porphyrin = Re complex dyads, ZnP-4BpyReBr, ZnP-5BpyReBr, and ZnP-6BpyReBr, to investigate the effect of the connecting positions between the Re complex and the Zn porphyrin units on photophysical properties and the catalytic activity of CO2 reduction. Although RTP was observed only in ZnP-6BpyReBr, there was no significant difference in the catalytic activity and durability of the three dyads when only BIH was used (as electron donor) without additives. The durability of the present three dyads was inferior to that of the previously reported dyad ZnP-phenRe as a result of the dissociation of the Re complex moiety during the reduction reaction. The addition of TEOA suppressed the Re dissociation and remarkably improved the activity and durability in the CO2 reduction reaction. The initial reaction rate of ZnP-5BpyReBr was unexpectedly more than twice that of the other dyads. The reaction quantum yield of ZnP-5BpyReBr reached 24%—the highest yet recorded for the photocatalytic CO2 reduction using porphyrins as photosensitizers. The higher ΦCO value originated from no electron accumulation at the porphyrin unit during the photocatalytic reaction as well as acceleration of the CO2 reduction. The IR and UV-vis titration experiments suggested a dual interaction of TEOA via the Zn porphyrin and the Re complex units in ZnP-5BpyReDMA, allowing a stronger interaction of TEOA to the Re complex. Although ZnP-5BpyReBr exhibited no RTP, the dependence of ΦCO on the BIH concentration suggests that the catalytic reaction is mediated via the T1 of the Zn porphyrin unit. This therefore indicates that the RTP is not an essential property for porphyrin to act as a good triplet photosensitizer for the photocatalytic CO2 reduction.
We reconfirmed that, when using porphyrin as photosensitizer, the balance of the electron relay cycle on the photosensitizer and the catalytic cycle on the catalyst2b is crucial to achieving high durability for CO2 reduction. The prompt catalytic reaction, with the assistance of the coordination of TEOA, plays an important role in the high durability of the photosensitizer. In particular, a dual interaction via the Zn porphyrin further promotes the catalytic reaction. The enhancement of the coordination of TEOA has been also observed in other metal complex catalysts.6–11 The present dual interaction of TEOA via a coordination scaffold suggests new opportunities to achieve enhanced catalytic performance by capturing CO2 with high efficiency, and subsequently the further development of practical photocatalytic systems using a wide range of organic dyes that include porphyrin-based photosensitizers.
Experimental section
General procedure
All chemicals and solvents were of commercial reagent quality and were used without further purification unless otherwise stated. Syntheses of fac-Re(bpy)(CO)3Br and BIH were carried out according to the literature.5a,12 DMA and TEOA were dried over molecular sieves of size 4A and distilled under reduced pressure. Dry toluene was prepared by distillation over benzophenone/Na. Toluene used for fluorescence quantum yields and lifetimes was dealt with conc. H2SO4 after simple distillation, and washed with H2O and Na2CO3 aq. After dried over CaCl2, the toluene was distilled on CaH2. Dry dichloromethane was dried over molecular sieves of size 4A. Pyrrole was prepared by distillation over CaH2. The reactions were monitored on silica-gel 60 F254 TLC plates (Merck). Silica-gels utilized for column chromatography were purchased from Kanto Chemical Co. Inc.: Silica-Gel 60N (Spherical, Neutral) 63–210 μm and (Flash) 40–50 μm. Alumina used for column chromatography were purchased from Merck (aluminum oxide 90 standardized). 1H and 13C NMR, distortionless enhancement by polarization transfer 135 (DEPT 135), 1H–1H COSY spectra were recorded by using JEOL JNM-ECS300 (300 MHz), JNM-ECZ400S (400 MHz), JNM-ECA500II (500 MHz), and Bruker AVANCE NEO 400 (400 MHz). Chemical shifts were recorded in parts per million (ppm) relative to tetramethylsilane. UV-vis absorption spectra were collected using square cells (optical path length = 10 mm) on JASCO V-650 and V-660 spectrometers. Steady-state emission spectra were collected on Hitachi F-4500 spectrophotometer or Horiba FluoroMAX 4 and corrected for the response of the detector system. The fluorescence intensities were normalized at the absorbance of the excitation wavelength. Fluorescence quantum yields were determined by integrated ratios of the fluorescence spectra relative to that of ZnTPP (Φf = 3.3% in toluene).46 Their values were corrected by refractive indexes of the used solvents. Time-resolved emission signals for fluorescence lifetimes were obtained by a pico-second fluorescence measurement system consisting of Chromex 250IS imaging spectrograph and a charge-coupled device (CCD) streak scope Hamamatsu model C4334, with excitation by a light pulser Hamamatsu model PLP-1-040 (406 nm, full width at half-maximum (fwhm) = 70 ps).47 Hi-resolution MALDI-TOF mass spectra were collected on JEOL JMS S-3000 with dithranol or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) as a matrix. FTIR spectra were recorded in KBr or acetonitrile using a JASCO FT/IR-4600. CV and DPV were measured using an ALS-H/CHI Model 612E electrochemical analyzer (BAS) in a micro-cell equipped with a glassy carbon working electrode (ϕ 1.6 mm) and a Pt counter-electrode. The micro-cell was connected via a Luggin capillary with a reference electrode of Ag/AgNO3 (10 mM in DMA). Tetrabutylammonium hexafluorophosphate (nBu4NPF6) recrystallized from ethyl acetate was used as a supporting electrolyte. Ferrocene was used as an external standard, and all potentials were referenced to the ferrocene/ferrocenium couple (E1/2 = +0.099 V vs. Ag/AgNO3). UV-vis spectral changes during irradiation were obtained using an Asahi Spectra PRA-201 apparatus equipped with a Xe lamp (Asahi Spectra, MAX-350).5,10,15-Trimesityl-20-(2-bromopyridin-6-yl)-21H,23H-porphyrin (H2P-6py)
In a 1 L flask were placed 2,4,6-trimethylbenzaldehyde (542 mg, 3.7 × 10−3 mol), 6-bromo-2-pyridinecarboxaldehyde (226 mg, 1.2 × 10−3 mol), pyrrole (327 mg, 4.9 × 10−3 mol), and CHCl3 (500 mL). The solution was degassed by bubbling with Ar gas, and BF3OEt2 (270 mg, 1.9 × 10−3 mol) was added. After the reaction mixture was stirred for 1 h at room temperature, triethylamine (370 mg, 3.7 × 10−3 mol) and p-chloranil (900 mg, 3.7 × 10−3 mol) were added and stirring was continued for 19 h. The crude product obtained by evaporation of the solvent was purified with flash silica gel columns (CHCl3:hexane = 1:1). The second band was collected and the solvent was evaporated to dryness, giving the title compound as a purple solid (171 mg, 17% based on 6-bromo-2-pyridinecarboxaldehyde). TLC (silica gel, CHCl3) Rf = 0.6, (alumina, CHCl3) Rf = 0.8; MALDI-TOF-mass (dithranol) m/z [M + H]+ 820.3019, calcd for [C52H47BrN5]+ 820.3009; 1H NMR (400 MHz, CDCl3) δ/ppm = 8.78 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.71 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.64 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.62 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.08 (dd, J = 4.7 and 3.6 Hz, 1H, py), 7.90 (d, J = 4.7 Hz, 1H, py), 7.89 (d, J = 3.6 Hz, 1H, py), 7.25 (s, 6H, mesityl), 2.62 (s, 9H, CH3), 1.84 (s, 18H, CH3), −2.58 (s, 2H, NH); 13C NMR (100 MHz, CDCl3) δ/ppm = 161.94 (C), 141.02 (C), 139.54 (C), 138.36 (C), 138.17 (C), 137.88 (C), 136.95 (CH), 130.65 (CH), 129.55 (CH), 127.89 (CH), 126.96 (CH), 118.95 (C), 118.47 (C), 115.19 (C), 21.80 (CH), 21.59 (CH).
5,10,15-Trimesityl-20-(2-bromopyridin-5-yl)-21H,23H-porphyrin (H2P-5py)
In a 2 L flask were placed 2,4,6-trimethylbenzaldehyde (1.9 g, 13 × 10−3 mol), 6-bromo-3-pyridinecarboxaldehyde (800 mg, 4.3 × 10−3 mol), pyrrole (1.2 g, 17 × 10−3 mol), and CHCl3 (1.7 L). The solution was degassed by bubbling with N2 gas, and BF3OEt2 (1.3 mL, 11 × 10−3 mol) diluted with CHCl3 (50 mL) was added through a dropping funnel over 4 h. After the reaction mixture was further stirred for 5 h at room temperature, triethylamine (2.1 mL, 15 × 10−3 mol) and p-chloranil (3.2 g, 13 × 10−3 mol) were added and stirring was continued for 20 h. The resulting solution was filtered through a Celite pad and the filtrate was evaporated to dryness in vacuo. The crude product was purified with an alumina column (CHCl3), and silica gel columns twice (CHCl3:hexane = 2:1) and flash silica gel columns twice (CHCl3:hexane = 2:1 → CHCl3), giving the title compound as a purple solid (380 mg, 14% based on 6-bromo-3-pyridinecarboxaldehyde). TLC (silica gel, CHCl3) Rf = 0.6, (alumina, CHCl3) Rf = 0.8; MALDI-TOF-mass (dithranol) m/z [M + H]+ 820.2969, calcd for [C52H47BrN5]+ 820.3009; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.20 (d, J = 2.5 Hz, 1H, py), 8.76 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.71 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.67 (s, 4H, β-pyrrole), 8.31 (dd, J = 2.5 and 8.0 Hz, 1H, py), 7.85 (d, J = 8.0 Hz, 1H, py), 7.28 (s, 4H, mesityl), 7.27 (s, 2H, mesityl), 2.61 (s, 6H, CH3), 2.60 (s, 3H, CH3), 1.86 (s, 18H, CH3), −2.55 (s, 2H, NH); 13C NMR (100 MHz, CDCl3) δ/ppm = 153.74 (CH), 143.30 (CH), 142.08 (C), 139.63 (C), 139.59 (C), 139.55 (C), 138.30 (C), 138.15 (C), 138.09 (C), 138.06 (C), 137.65 (C), 130.71 (CH), 128.09 (CH), 128.03 (CH), 126.46 (CH), 118.91 (C), 118.77 (C), 112.87 (C), 21.98 (C), 21.92 (C), 21.68 (C).
5,10,15-Trimesityl-20-(2-bromopyridin-4-yl)-21H,23H-porphyrin (H2P-4py)
In a 1 L flask were placed 2,4,6-trimethylbenzaldehyde (880 mg, 5.9 × 10−3 mol), 2-bromo-4-pyridinecarboxaldehyde (370 mg, 2.0 × 10−3 mol), pyrrole (530 mg, 7.9 × 10−3 mol), and CHCl3 (790 mL). The solution was degassed by bubbling with Ar gas, and BF3OEt2 (550 mg, 3.9 × 10−3 mol) was added. After the reaction mixture was stirred for 3 h at room temperature, triethylamine (600 mg, 5.9 × 10−3 mol) and p-chloranil (1.5 g, 5.9 × 10−3 mol) were added and stirring was continued for 3 h. The crude product obtained by evaporation of the solvent was purified with alumina columns twice (CHCl3) and a flash silica gel column (CHCl3:hexane = 1:1 → CHCl3), giving the title compound as a purple solid (156 mg, 10% based on 2-bromo-4-pyridinecarboxaldehyde). TLC (silica gel, CHCl3) Rf = 0.5, (alumina, CHCl3) Rf = 0.8; MALDI-TOF-mass (dithranol) m/z [M + H]+ 820.2981, calcd for [C52H47BrN5]+ 820.3009; 1H NMR (400 MHz, CDCl3) δ/ppm = 8.75 (d, J = 4.8 Hz, 3H, β-pyrrole and py), 8.72 (d, J = 4.8 Hz, 2H, β-pyrrole), 8.66 (s, 4H, β-pyrrole), 8.35 (d, J = 1.3 Hz, 1H, py), 8.11 (dd, J = 1.3 and 4.8 Hz, 1H, py), 7.28 (s, 4H, mesityl), 7.27 (s, 2H, mesityl), 2.62 (s, 6H, CH3), 2.61 (s, 3H, CH3), 1.86 (s, 18H, CH3), −2.60 (s, 2H, NH); 13C NMR (100 MHz, CDCl3) δ/ppm = 153.55 (C), 148.28 (CH), 141.40 (C), 139.49 (C), 138.13 (C), 138.06 (C), 138.02 (C), 133.32 (CH), 130.66 (CH), 128.63 (CH), 127.98 (CH), 127.94 (CH), 119.13 (C), 118.82 (C), 113.55 (C), 21.89 (CH), 21.81 (CH), 21.60 (CH).
Zinc(II) 5,10,15-trimesityl-20-(2-bromopyridin-6-yl)-21H,23H-porphyrin (ZnP-6py)
A saturated methanolic solution of zinc(II) acetate (270 mg, 1.5 × 10−3 mol) was added to a solution of H2P-6py (120 mg, 1.5 × 10−4 mol) in CHCl3 (30 mL). The mixture was stirred overnight at room temperature. The resulting solution was diluted with CHCl3 and washed with water. The organic layer was dried over anhydrous Na2SO4 and evaporated to dryness. The further purification was carried out with a flash silica gel column (CHCl3:hexane = 1:1), giving the titled compound as a purple solid (130 mg, 95%). TLC (silica gel, CHCl3) Rf = 0.5; MALDI-TOF-mass (dithranol) m/z [M]+ 881.1969, calcd for [C52H44BrN5Zn]+ 881.2072; 1H NMR (400 MHz, CDCl3) δ/ppm = 8.83 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.79 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.72 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.70 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.20 (dd, J = 1.5 and 6.8 Hz, 1H, py), 7.93 (dd, J = 6.8 and 8.0 Hz, 1H, py), 7.89 (dd, J = 1.5 and 8.0 Hz, 1H, py), 7.27 (s, 6H, mesityl), 2.63 (s, 9H, CH3), 1.96 (s, 3H, CH3), 1.83 (s, 15H, CH3); 13C NMR (100 MHz, CDCl3) δ/ppm = 162.81 (C), 150.28 (C), 149.86 (C), 149.83 (C), 149.55 (C), 140.76 (C), 139.40 (C), 139.03 (C), 138.98 (C), 137.56 (C), 136.80 (CH), 131.51 (CH), 131.46 (CH), 131.34 (CH), 131.18 (CH), 128.97 (CH), 127.76 (CH), 126.77 (CH), 119.75 (C), 119.30 (C), 116.08 (C), 21.77 (CH), 21.57 (CH).
Zinc(II) 5,10,15-trimesityl-20-(2-bromopyridin-5-yl)-21H,23H-porphyrin (ZnP-5py)
In a 200 mL flask, a methanolic solution saturated with zinc(II) acetate (2.2 g, 12 × 10−3 mol) was added to a solution of H2P-5py (490 mg, 6.0 × 10−4 mol) in CHCl3 (120 mL). The mixture was stirred overnight at room temperature. The resulting solution was diluted with CHCl3 and washed with water. The organic layer was dried over anhydrous Na2SO4 and evaporated to dryness. The further purification was carried out with a flash silica gel column (CHCl3:hexane = 1:2). The collected fraction was evaporated, giving the titled compound as a purple solid (430 mg, 81%). TLC (silica gel, CHCl3) Rf = 0.5; MALDI-TOF-mass (dithranol) m/z [M]+ 881.2034, calcd for [C52H44BrN5Zn]+ 881.2072; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.01 (s, 1H, py), 8.80 (d, J = 4.4 Hz, 2H, β-pyrrole), 8.72 (m, 6H, β-pyrrole), 8.33 (d, J = 7.9 Hz, 1H, py), 7.85 (d, J = 7.9 Hz, 1H, py), 7.27 (s, 6H, mesityl), 2.63 (s, 9H, CH3), 1.87 (s, 3H, CH3), 1.83 (s, 15H, CH3); 13C NMR (100 MHz, CDCl3 + 1drop of CD3OD) δ/ppm = 153.31 (CH), 150.15 (C), 149.96 (C), 149.70 (C), 149.14 (C), 143.30 (CH), 139.39 (C), 139.32 (C), 139.28 (C), 138.98 (C), 137.35 (C), 131.20 (CH), 131.07 (CH), 131.03 (CH), 130.69 (CH), 127.60 (CH), 126.04 (CH), 119.03 (C), 118.85 (C), 21.71 (CH), 21.43 (CH).
Zinc(II) 5,10,15-trimesityl-20-(2-bromopyridin-4-yl)-21H,23H-porphyrin (ZnP-4py)
A saturated methanolic solution of zinc(II) acetate (350 mg, 1.9 × 10−3 mol) was added to a solution of H2P-4py (160 mg, 1.9 × 10−4 mol) in CHCl3 (38 mL). The mixture was stirred overnight at room temperature. The resulting solution was diluted with CHCl3 and washed with water. The organic layer was dried over anhydrous Na2SO4 and evaporated to dryness. The further purification was carried out with a flash silica gel column (CHCl3:hexane = 1:1), giving the titled compound as a purple solid (170 mg, 99%). TLC (silica gel, CHCl3) Rf = 0.4; MALDI-TOF-mass (dithranol) m/z [M]+ 881.2040, calcd for [C52H44BrN5Zn]+ 881.2072; 1H NMR (400 MHz, CDCl3) δ/ppm = 8.81 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.75 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.72 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.71 (d, J = 4.7 Hz, 2H, β-pyrrole), 8.55 (dd, J = 5.0, 0.7 Hz, 1H, py), 8.32 (dd, J = 1.5, 0.7 Hz, 1H, py), 8.09 (dd, J = 5.0, 1.5 Hz, 1H, py), 7.28 (s, 4H, mesityl), 7.27 (s, 2H, mesityl), 2.63 (s, 6H, CH3), 2.62 (s, 3H, CH3), 1.85 (s, 3H, CH3), 1.84 (s, 3H, CH3), 1.83 (s, 6H, CH3), 1.82 (s, 6H, CH3); 13C NMR (100 MHz, CDCl3) δ/ppm = 154.38 (C), 150.40 (C), 150.17 (C), 149.89 (C), 148.42 (C), 147.69 (CH), 140.77 (C), 139.31 (C), 139.28 (C), 138.88 (C), 137.69 (C), 137.65 (C), 133.32 (C), 131.67 (CH), 131.55 (CH), 131.47 (CH), 130.96 (CH), 128.46 (CH), 127.81 (CH), 119.81 (C), 119.56 (C), 114.37 (C), 21.89 (CH), 21.79 (CH), 21.57 (CH).
Zinc(II) 5,10,15-trimesityl-20-(2,2′-bipyridin-6-yl)-21H,23H-porphyrin (ZnP-6Bpy)
In a 200 mL Schlenk tube were placed ZnP-6py (150 mg, 1.7 × 10−4 mol), 2-(tributylstannyl)pyridine (120 μL, 3.4 × 10−4 mol), Pd(PPh3)4 (42 mg, 3.6 × 10−5 mol), and dry toluene (57 mL). The reaction mixture was degassed by freeze–thaw cycles and stirred at 105 °C overnight. The solvent of the resulting mixture was evaporated after addition of water (50 mL). The residue was dissolved in CHCl3 and washed with water, and the organic layer was dried over anhydrous Na2SO4. The crude product obtained by evaporation of the solvent was purified with a flash silica gel column (CHCl3) and an anhydrous K2CO3/flash silica gel (1:9 w/w) column (CHCl3) to remove organotin impurities.32 Further purification was carried out by reprecipitation with CHCl3/hexane, giving the titled compound as a purple solid (98 mg, 64%). TLC (silica gel, CHCl3) Rf = 0.2, (silica gel, CHCl3:acetone = 4:1) Rf = 0.4; MALDI-TOF-mass (dithranol) m/z [M + H]+ 881.3202, calcd for [C57H49N6Zn]+ 881.3305; 1H NMR (500 MHz, CDCl3) δ/ppm = 8.92 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.80 (dd, J = 1.1 and 7.7 Hz, 1H, bpy), 8.79 (d, J = 4.9 Hz, 1H, bpy), 8.78 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.73 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.71 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.58 (d, J = 7.7 Hz, 1H, bpy), 8.25 (dd, J = 1.1 and 7.7 Hz, 1H, bpy), 8.20 (t, J = 7.7 Hz, 1H, bpy), 7.71 (td, J = 7.7 and 1.1 Hz, 1H, bpy), 7.32 (ddd, J = 1.1, 4.9 and 7.7 Hz, 1H, bpy), 7.26 (s, 6H, mesityl), 2.63 (s, 3H, CH3), 2.62 (s, 6H, CH3), 1.93 (s, 3H, CH3), 1.88 (s, 6H, CH3), 1.79 (s, 9H, CH3); 13C NMR (125 MHz, CDCl3) δ/ppm = 160.99 (C), 156.67 (C), 154.81 (C), 150.14 (C), 149.94 (C), 149.82 (C), 149.31 (CH), 139.42 (C), 139.35 (C), 139.31 (C), 139.12 (C), 139.05 (C), 137.49 (C), 137.09 (CH), 135.52 (CH), 131.85 (CH), 131.32 (CH), 131.20 (CH), 131.10 (CH), 130.17 (CH), 127.72 (CH), 123.91 (CH), 121.99 (CH), 119.45 (CH), 119.31 (C), 119.04 (C), 118.23 (C), 21.78 (CH), 21.55 (CH).
Zinc(II) 5,10,15-trimesityl-20-(2,2′-bipyridin-5-yl)-21H,23H-porphyrin (ZnP-5Bpy)
In a 100 mL Schlenk tube were placed ZnP-5py (95 mg, 1.1 × 10−4 mol), 2-(tributylstannyl)pyridine (72 μL, 2.2 × 10−4 mol), Pd(PPh3)4 (25 mg, 2.2 × 10−5 mol), and dry toluene (36 mL). The reaction mixture was degassed by freeze–thaw cycles and stirred at 105 °C overnight. The solvent of the resulting mixture was evaporated after addition of water (40 mL). The residue was dissolved in CHCl3 and washed with water, and the organic layer was dried over anhydrous Na2SO4. The crude product obtained by evaporation of the solvent was purified with a flash silica gel column (CHCl3) and an anhydrous K2CO3/flash silica gel (1:9 w/w) column (CHCl3) to remove organotin impurities.32 Further purification was carried out by reprecipitation with CHCl3/hexane, giving the titled compound as a purple solid (61 mg, 64%). TLC (silica gel, CHCl3) Rf = 0.2, (silica gel, CHCl3:acetone = 4:1) Rf = 0.4; MALDI-TOF-mass (dithranol) m/z [M + H]+ 881.3335, calcd for [C57H49N6Zn]+ 881.3305; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.47 (s, 1H, bpy), 8.87 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.82–8.79 (m, 2H, bpy), 8.80 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.72 (s, 4H, β-pyrrole), 8.68 (d, J = 7.4 Hz, 1H, bpy), 8.66 (d, J = 7.4 Hz, 1H, bpy), 7.96 (t, J = 7.4 Hz, 1H, bpy), 7.43 (t, J = 7.4 Hz, 1H, bpy), 7.27 (s, 6H, mesityl), 2.62 (s, 9H, CH3), 1.87 (s, 3H, CH3), 1.85 (s, 3H, CH3), 1.84 (s, 12H, CH3); 13C NMR (100 MHz, CDCl3) δ/ppm = 156.38 (C), 154.96 (C), 153.15 (CH), 150.18 (C), 150.07 (C), 149.89 (C), 149.78 (CH), 149.52 (C), 141.97 (CH), 139.38 (C), 139.34, (C) 139.15 (C), 139.09 (C), 137.53 (C), 137.25 (CH), 136.83 (C), 131.58 (CH), 131.41 (CH), 131.31 (CH), 131.20 (CH), 128.83 (CH), 127.75 (CH), 127.01 (CH), 123.98 (CH), 121.56 (CH), 119.18 (CH), 115.41 (C), 21.87 (CH3) 21.79 (CH3) 21.55 (CH3).
Zinc(II) 5,10,15-trimesityl-20-(2,2′-bipyridin-4-yl)-21H,23H-porphyrin (ZnP-4Bpy)
In a 100 mL Schlenk tube were placed ZnP-4py (100 mg, 1.2 × 10−4 mol), 2-(tributylstannyl)pyridine (77 μL, 2.3 × 10−4 mol), Pd(PPh3)4 (28 mg, 2.4 × 10−5 mol), and dry toluene (39 mL). The reaction mixture was degassed by freeze–thaw cycles and stirred at 105 °C overnight. The solvent of the resulting mixture was evaporated after addition of water (40 mL). The residue was dissolved in CHCl3 and washed with water, and the organic layer was dried over anhydrous Na2SO4. The crude product obtained by evaporation of the solvent was purified with a flash silica gel column (CHCl3) and an anhydrous K2CO3/flash silica gel (1:9 w/w) column (CHCl3) to remove organotin impurities.32 Further purification was carried out by reprecipitation with CHCl3/hexane, giving the titled compound as a purple solid (44 mg, 43%). TLC (silica gel, CHCl3) Rf = 0.2, (silica gel, CHCl3:acetone = 4:1) Rf = 0.3; MALDI-TOF-mass (dithranol) m/z [M + H]+ 881.3333, calcd for [C57H49N6Zn]+ 881.3305; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.27 (s, 1H, bpy), 9.03 (d, J = 4.8 Hz, 1H, bpy), 8.84 (d, J = 5.0 Hz, 2H, β-pyrrole), 8.75 (d, J = 5.0 Hz, 2H, β-pyrrole), 8.71 (d, 1H, bpy), 8.70 (s, 4H, β-pyrrole), 8.63 (d, J = 4.8 Hz, 1H, bpy), 8.17 (d, J = 4.8 Hz, 1H, bpy), 7.92 (t, J = 7.8 Hz, 1H, bpy), 7.33 (dd, J = 7.8, 4.8 Hz, 1H, bpy), 7.27 (s, 6H, mesityl), 2.62 (s, 9H, CH3), 1.84 (m, 18H, CH3); 13C NMR (100 MHz, CDCl3) δ/ppm = 156.56 (C), 154.34 (C), 152.82 (C), 150.11 (C), 149.90 (CH), 149.87 (CH), 149.41 (C), 148.92 (C), 147.36 (CH), 143.24 (CH), 139.39 (C), 139.34 (C), 139.30 (C), 137.42 (C), 137.16 (CH), 131.51 (CH), 131.27 (CH), 131.11 (CH), 131.07 (CH), 129.58 (CH), 127.71 (CH), 126.58 (CH), 123.93 (CH), 121.99 (CH), 121.87 (CH), 119.01 (C), 118.95 (C), 116.54 (C), 21.93 (CH3), 21.86 (CH3), 21.82 (CH3), 21.56 (CH3).
Dyad connected via the 6-position of the Bpy ligand (ZnP-6Bpy![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) ReBr)
ReBr)
In a 200 mL Schlenk tube were placed ZnP-6Bpy (49 mg, 5.5 × 10−5 mol) and bromopentacarbonylrhenium(I) (23 mg, 5.5 × 10−5 mol), and toluene (37 mL). The reaction mixture was stirred at 90–95 °C overnight. The solvent of the resulting mixture was evaporated and the residue was purified with a flash silica gel column (CHCl3→CHCl3:acetone = 4:1). Further purification was carried out by recrystallization with CHCl3/hexane followed by CHCl3/ethanol, giving the titled compound as needle purple crystals (32 mg, 47%). TLC (silica gel, CHCl3) Rf = 0.1, (silica gel, CHCl3:acetone = 4:1) Rf = 0.6; MALDI-TOF-mass (DCTB) m/z [M]+ 1228.1670, calcd for [C60H48BrN6O3ReZn]+ 1228.1792; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.01 (d, J = 5.5 Hz, 1H, bpy), 8.99 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.81 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.72 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.71 (s, 2H, β-pyrrole), 8.70 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.68 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.66 (d, J = 7.8 Hz, 1H, bpy), 8.53 (d, J = 8.0 Hz, 1H, bpy), 8.48 (d, J = 7.8 Hz, 1H, bpy), 8.32 (t, J = 7.8 Hz, 1H, bpy), 8.29 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.16 (td, J = 8.0, 1.8 Hz, 1H, bpy), 7.51 (dd, J = 8.0, 5.5 Hz, bpy), 7.29 (s, 1H, mesityl), 7.26 (s, 3H, mesityl), 7.24 (s, 1H, mesityl), 7.22 (s, 1H, mesityl), 2.63 (s, 3H, CH3), 2.61 (s, 3H, CH3), 2.60 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.94 (s, 3H, CH3), 1.89 (s, 3H, CH3), 1.79 (s, 3H, CH3), 1.76 (s, 3H, CH3), 1.74 (s, 3H, CH3); 1H NMR (500 MHz, DMF-d7) δ/ppm = 9.16 (m, 1H, bpy), 9.04 (d, J = 8.0 Hz, 1H, bpy), 8.93 (dd, J = 5.4, 1.8 Hz, 1H, bpy), 8.84 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.55–8.44 (m, 7H, β-pyrrole and bpy × 2), 8.43 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.35 (td, J = 8.0, 1.8 Hz, 1H, bpy), 8.32 (d, J = 4.6 Hz, 1H, β-pyrrole), 7.68 (ddd, J = 8.0, 5.4, 1.6 Hz, 1H, bpy), 7.24 (s, 1H, mesityl), 7.21 (s, 1H, mesityl), 7.20 (s, 1H, mesityl), 7.19 (s, 1H, mesityl), 7.17 (s, 2H, mesityl), 2.47 (s, 3H, CH3), 2.46 (s, 3H, CH3), 2.43 (s, 3H, CH3), 1.88 (s, 3H, CH3), 1.71 (s, 3H, CH3), 1.70 (s, 3H, CH3), 1.68 (s, 3H, CH3), 1.66 (s, 3H), 1.56 (s, 3H, CH3); 13C NMR (125 MHz, DMF-d7) δ/ppm = 199.03 (CO), 191.93 (CO), 189.13 (CO), 164.82 (C), 158.29 (C), 155.73 (C), 153.09 (CH), 150.30 (C), 150.20 (C), 149.92 (C), 149.83 (C), 149.73 (C), 149.38 (C), 149.29 (C), 149.27 (C), 148.09 (CH), 140.04 (CH), 139.67 (C), 139.61 (C), 139.43 (C), 139.25 (C), 138.98 (C), 138.76 (C), 138.68 (C), 138.59 (C), 137.44 (C), 137.38 (C), 137.34 (CH), 136.44 (CH), 132.54 (CH), 132.09 (CH), 131.22 (CH), 130.95 (CH), 130.83 (CH), 130.62 (CH), 130.55 (CH), 130.44 (CH), 127.84 (CH), 127.79 (CH), 127.73 (CH), 127.66 (CH), 127.30 (CH), 125.66 (CH), 123.71 (CH), 123.64 (CH), 119.36 (C), 118.81 (C), 118.76 (C), 117.54 (C), 21.45 (CH3), 21.26 (CH3), 21.16 (CH3), 21.06 (CH3), 20.85 (CH3), 20.81 (CH3); FT-IR (KBr) νCO = 1895, 1923, 2021 cm−1, (acetonitrile) νCO = 1894, 1919, 2021 cm−1.
Dyad connected via the 5-position of the Bpy ligand (ZnP-5BpyReBr)
In a 200 mL Schlenk tube were placed ZnP-5Bpy (57 mg, 6.4 × 10−5 mol) and bromopentacarbonylrhenium(I) (26 mg, 6.5 × 10−5 mol), and toluene (43 mL). The reaction mixture was stirred at 90–95 °C overnight. The solvent of the resulting mixture was evaporated and the residue was recrystallized with CHCl3/hexane followed by CHCl3/ethanol, giving the titled compound as needle brown crystals (44 mg, 55%). TLC (silica gel, CHCl3) Rf = 0.2, (silica gel, CHCl3:acetone = 4:1) Rf = 0.7; MALDI-TOF-mass (DCTB) m/z [M]+ 1228.1768, calcd for [C60H48BrN6O3ReZn]+ 1228.1792; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.89 (s, 1H, bpy), 9.20 (d, J = 5.7 Hz, 1H, bpy), 8.93 (d, J = 4.9 Hz, 1H, β-pyrrole), 8.89 (d, J = 4.9 Hz, 2H, β-pyrrole), 8.81 (m, 2H, β-pyrrole, bpy), 8.75 (s, 4H, β-pyrrole), 8.54 (d, J = 8.3 Hz, 1H, bpy), 8.45 (d, J = 8.1 Hz, 1H, bpy), 8.15 (dd, J = 8.1, 7.8 Hz, 1H, bpy), 7.60 (dd, J = 7.8, 5.7 Hz, 1H, bpy), 7.27 (m, 6H, mesityl), 2.62 (s, 9H, CH3), 1.88 (s, 3H, CH3), 1.87 (s, 3H, CH3), 1.85 (s, 9H, CH3), 1.80 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ/ppm = 197.06 (CO), 196.55 (CO), 189.21 (CO), 158.60 (C), 156.57 (CH), 156.15 (C), 154.51 (CH), 154.06 (C), 153.60 (CH), 150.77 (C), 150.52 (C), 150.32 (C), 149.75 (C), 149.29 (C), 148.79 (C), 143.22 (CH), 139.49 (C), 139.44 (C), 139.20 (C), 139.07 (C), 138.86 (C), 138.82 (C), 138.75 (C), 138.65 (C), 138.65 (C), 137.75 (C), 132.66 (CH), 132.08 (CH), 131.92 (CH), 131.86 (CH), 131.70 (CH), 131.58 (CH), 131.38 (CH), 130.32 (CH), 128.02 (CH), 127.93 (CH), 127.87 (CH), 127.77 (CH), 127.15 (CH), 125.98 (CH), 125.82 (CH), 123.64 (CH), 121.18 (CH), 120.33 (C), 120.09 (C), 119.92 (C), 111.25 (C), 21.92 (CH3), 21.83 (CH3), 21.75 (CH3), 21.57 (CH3); FT-IR (KBr) νCO = 1900, 1921, 2022 cm−1, (acetonitrile) νCO = 1902, 1918, 2023 cm−1.
Dyad connected via the 4-position of the Bpy ligand (ZnP-4BpyReBr)
In a 200 mL Schlenk tube were placed ZnP-4Bpy (44 mg, 5.0 × 10−5 mol) and bromopentacarbonylrhenium(I) (20 mg, 5.0 × 10−5 mol), and toluene (33 mL). The reaction mixture was stirred at 90–95 °C overnight. The solvent of the resulting mixture was evaporated and the residue was purified with flash silica gel columns (CHCl3 → CHCl3:acetone = 4:1). Further purification was carried out by recrystallization with CHCl3/hexane followed by CHCl3/ethanol, giving the titled compound as needle dark-green crystals (25 mg, 41%). TLC (silica gel, CHCl3) Rf = 0.3, (silica gel, CHCl3:acetone = 4:1) Rf = 0.8; MALDI-TOF-mass (DCTB) m/z [M]+ 1228.1757, calcd for [C60H48BrN6O3ReZn]+ 1228.1792; 1H NMR (500 MHz, CDCl3) δ/ppm = 9.43 (d, J = 5.6 Hz, 1H, bpy), 9.24 (d, J = 5.6 Hz, 1H, bpy), 9.03 (d, J = 1.7 Hz, 1H, bpy), 8.87 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.83 (d, J = 4.6 Hz, 2H, β-pyrrole), 8.80 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.75–8.72 (m, 4H, β-pyrrole), 8.38 (dd, J = 5.6, 1.7 Hz, 1H, bpy), 8.29 (d, J = 8.3 Hz, 1H, bpy), 8.02 (t, J = 8.3 Hz, 1H, bpy), 7.59 (dd, J = 8.3, 5.6 Hz, 1H, bpy), 7.29 (s, 2H, mesityl), 7.28 (s, 2H, mesityl), 7.26 (s, 2H, mesityl), 2.63 (s, 9H, CH3), 1.87 (s, 3H, CH3), 1.85 (s, 3H, CH3), 1.84 (s, 9H, CH3), 1.82 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ/ppm = 196.98 (CO), 196.94 (CO), 189.36 (CO), 156.33 (C), 155.24 (C), 153.75 (C), 153.68 (CH), 151.27 (CH), 150.77 (C), 150.60 (C), 150.33 (C), 150.13 (C), 149.79 (C), 148.20 (C), 147.96 (C), 139.33 (C), 139.24 (C), 139.16 (C), 139.08 (C), 138.93 (CH), 137.87 (C), 138.65 (C), 138.57 (C), 137.98 (C), 137.87 (C), 137.83 (C), 137.78 (C), 132.29 (CH), 132.21 (CH), 132.03 (CH), 131.95 (CH), 131.73 (CH), 131.63 (CH), 130.95 (CH), 130.29 (CH), 129.13 (CH), 128.47 (CH), 128.33 (CH), 127.98 (CH), 127.92 (CH), 127.88 (CH), 127.83 (CH), 127.20 (CH), 125.39 (CH), 123.51 (CH), 120.48 (C), 120.16 (C), 119.97 (C), 113.20 (C), 21.91 (CH3), 21.86 (CH3), 21.80 (CH3), 21.57 (CH3); FT-IR (KBr) νCO = 1902, 1920, 2021 cm−1.
Acetonitrile-substituted ZnP-6BpyRe (ZnP-6BpyReMeCN)
In a 50 mL flask were placed ZnP-6BpyReBr (10 mg, 8.1 × 10−6 mol), AgBF4 (1.9 mg, 9.7 × 10−6 mol), and acetonitrile (4 mL). The reaction mixture was stirred at reflux for 4 h, and then the solvent was evaporated to dryness. The crude product was dissolved in CHCl3 (50 mL) and washed with water (50 mL × 3). The organic layer was passed through Phase Separator paper (Whatman). After evaporating the solvent, the residue was purified with a flash silica gel column (CHCl3 → CHCl3:acetonitrile = 10:1), giving a purple-green solid (18 mg, 90%). TLC (silica gel, CHCl3:acetonitrile = 10:1) Rf = 0.3; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.02 (dd, J = 8.2, 1.4 Hz, 1H, bpy), 8.89 (d, J = 8.2, 1.1 Hz, 1H, bpy), 8.86–8.84 (m, 1H, bpy), 8.83 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.79 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.77–8.71 (m, 5H, β-pyrrole), 8.62 (dd, J = 8.2, 1.4 Hz, 1H, bpy), 8.54 (t, J = 8.2 Hz, 1H, bpy), 8.50 (d, J = 4.6 Hz, 1H, β-pyrrole), 8.36 (ddd, J = 8.2, 7.6, 1.4 Hz, 1H, bpy), 7.62 (ddd, J = 7.6, 5.4, 1.4 Hz, 1H, bpy), 7.32 (s, 1H, mesityl), 7.30 (s, 2H, mesityl), 7.28 (m, 3H, mesityl), 2.65 (s, 3H, CH3), 2.64 (s, 3H, CH3), 2.64 (s, 3H, CH3), 2.34 (s, 3H, coordinated acetonitrile), 2.01 (s, 3H, CH3), 1.88 (s, 3H, CH3), 1.86 (s, 6H, CH3), 1.84 (s, 3H, CH3), 1.80 (s, 3H, CH3); 19F NMR (375 MHz, CDCl3) δ/ppm = −152 (BF4−); FT-IR (acetonitrile) νCO = 1921, 1941, 2034 cm−1.
Acetonitrile-substituted ZnP-5BpyRe (ZnP-5BpyReMeCN)
In a 50 mL flask were placed ZnP-5BpyReBr (7.0 mg, 5.7 × 10−6 mol), AgBF4 (1.3 mg, 6.8 × 10−6 mol), and acetonitrile (4 mL). The reaction mixture was stirred at reflux for 4 h, and then the solvent was evaporated to dryness. The crude product was dissolved in CHCl3 (50 mL) and washed with water (50 mL × 3). The organic layer was passed through Phase Separator paper (Whatman). After evaporating the solvent, the residue was purified with a flash silica gel column (CHCl3 → CHCl3:acetonitrile = 10:1), giving a purple-green solid (12 mg, 86%). TLC (silica gel, CHCl3:acetonitrile = 10:1) Rf = 0.3; 1H NMR (400 MHz, CDCl3) δ/ppm = 9.70 (s, 1H, bpy), 9.07 (d, J = 5.2 Hz, 1H, bpy), 9.04–8.98 (m, 2H, bpy and β-pyrrole), 8.97–8.87 (m, 3H, bpy and β-pyrrole), 8.81 (d, J = 8.8 Hz, 1H, bpy), 8.79–8.70 (m, 5H, β-pyrrole), 8.38 (t, J = 8.0 Hz, 1H, bpy), 7.73 (t, J = 6.5 Hz, 1H, bpy), 7.30 (s, 1H, mesityl), 7.28 (s, 5H, mesityl), 2.63 (s, 9H, CH3), 2.42 (s, 3H, coordinated acetonitrile), 1.90 (s, 3H, CH3), 1.87 (s, 3H, CH3), 1.85 (s, 6H, CH3), 1.83 (s, 3H, CH3), 1.81 (s, 3H, CH3); 19F NMR (375 MHz, CDCl3) δ/ppm = −152 (BF4−); FT-IR (acetonitrile) νCO = 1926, 1940, 2037 cm−1.
Quenching experiments
The Stern–Volmer relationship (eqn (1)) was obtained by the plots of the relative emission band intensity (I0/I) versus the concentration of BIH:| I0/I = 1 + KSV[BIH] = 1 + kqτ[BIH] | (1) |
Photocatalytic CO2 reduction
In glass tubes (8.0 mL, i.d. = 10 mm), 1.4 mL of CO2-saturated solutions containing BIH was added by 0.6 mL of Ar-saturated solutions containing the photocatalysts, and the reaction solutions were bubbled through septum caps with CO2 gas for 20 min. Photo-irradiations to determine TON were carried out using a merry-go-round irradiation apparatus (Iris-MG, Cell Systems) equipped with LED lamps at 298 K. Reaction quantum yields (ΦCO) and UV-vis absorption spectral changes during photoirradiation were determined with an Asahi Spectra PRA-201 apparatus equipped with a Xe lamp (Asahi Spectra, MAX-350). The irradiation of very weak light (<5 × 10−10 einstein per s) was carried out with a Hitachi F-4500 spectrophotometer and the number of the incident photons was determined by a standard chemical actinometer using K3[Fe(C2O4)3].48 Reaction quantum yields were calculated according to ΦCO = (amounts of CO)/(incident photons). In 7 mL quartz cubic cells (7.0 mL, optical path length = 10 mm), 2.4 mL of CO2-saturated solutions containing BIH was added by 0.6 mL of Ar-saturated solutions containing the photocatalysts, and the reaction solutions were bubbled through septum caps with CO2 gas for 20 min. The gaseous reaction products (CO and H2) were quantified with a gas chromatography system (GC-2014, Shimadzu Science) equipped with a Shincarbon column (i.d. 3.0 mm × 3.0 m, Shinwa Chemical Industries) and a thermal conductivity detector (TCD). The product (HCOOH) in the solutions was analyzed with a capillary electrophoresis system (Otsuka Electronics Co. CAPI-3300I).Computational methods
DFT calculations were carried out using the Gaussian 09 package of programs.49 Each structure was fully optimized using the B3LYP functional using the 6-31G(d) basis set for all atoms except Re and the standard double-ζ type LANL2DZ basis set with the effective core potential of Hay–Wadt for Re. The stationary points were verified using the vibrational analysis. The calculations were carried out by using the polarizable continuum model (PCM) with default parameter for DMA.Equilibrium constants of TEOA in DMA
ZnP-nBpyReMeCN (n = 5, 6) was added to Ar-saturated DMA (dyad concentration 11 mM) and the ligand substitutions from acetonitrile to DMA were monitored by IR spectroscopy. An Ar-saturated DMA–TEOA (79:1 v/v) solution (60 μL) was added to equilibrated DMA solutions of the dyads (240 μL) and the resulting mixtures were bubbled with Ar gas for 10 min. The IR spectra were monitored. The concentrations of ZnP-nBpyReDMA and ZnP-nBpyReTEOA were estimated based on the deconvolution of the absorption bands in Fig. 15, which were obtained by curve fittings as a linear combination of the Gaussian functions with OriginPro software. The equilibrium constants in DMA were defined as K = [ZnP-nBpyReTEOA][DMA]/[ZnP-nBpyReDMA][TEOA].5
Apparent binding constants of TEOA in dichloromethane
Ar-saturated DMA solutions containing ZnP-nBpyReMeCN (n = 5, 6) were allowed to stand for 2 h prior to collecting samples of solutions (20 μL) and drying in vacuo. Solid samples were dissolved in Ar- or CO2-saturated dichloromethane to prepare dichloromethane solutions containing the dyads (3 μM). Solutions of ZnP-5BpyReBr and ZnP-5Bpy were prepared by dissolving in Ar- or CO2-saturated dichloromethane to be 3 μM. Titrations were carried out in square cells (optical path length 10 mm) by adding three dichloromethane solutions containing TEOA (0.1, 1.0, and 10 M) via septum caps. The apparent binding constants of TEOA to the Zn porphyrins in dichloromethane were defined as Kapp = [TEOA-coordinated Zn porphyrin]/[Zn porphyrin][TEOA]. The values of Kapp were estimated from spectral data using Benesi–Hildebrand plot assuming the formation of a 1:1 complex of TEOA and Zn porphyrins (Fig. S96†).
Time-resolved near-infrared emission measurements
Near-infrared time-resolved emission spectra and lifetimes were measured by a lab-made system controlled by a Labview-based program. Samples were excited at 406 nm pulses with an appropriate duration time (100 ns to 100 μs) which were prepared by combination of an ORBIS 405LX laser and a Stanford Research Systems (SRS) digital pulse generator Model DG535. After dispersed by a Horiba spectrometer MicroHR, emission signals were detected on a Hamamatsu R5509-43 photomultiplier by the single-photon-counting method. SRS Multichannel scaler SRS430 was used for signal counting and accumulation. Two dimensional time-wavelength data sets were acquired to a PC and emission spectra for an appropriate window were reconstructed from the 2D dataset.Data availability
All the data supporting this article have been included in the ESI.†Author contributions
Y. K. designed the dyads, conceived the idea of the dual interaction of TEOA, performed a part of the titration experiments, and wrote the manuscript. Y. S. synthesized and characterized the dyads, and performed most of photocatalytic experiments. S. A. and T. S. performed the time-resolved emission measurements. H. I. contributed to the synthesis of a portion of the dyads. M. S. A. supervised the time-resolved emission measurements and analyzed the data, and provided suggestions on writing the manuscript. A. S. supervised the project, and provided suggestions on the experiments and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP22H02186 and partially supported by JP20K05679. We thank Prof. Akihiko Kudo (Tokyo University of Science) for help with capillary electrophoresis analyses, and Mr Ken Kobori (Gunma University) for his assistance in setting up the time-resolved near IR emission measurement system.References
- (a) H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, The Water Oxidation Bottleneck in Artificial Photosynthesis: How Can We Get Through It? An Alternative Route Involving a Two-Electron Process, ChemSusChem, 2011, 4, 173–179 CAS; (b) J.-H. Alstrum-Acevedo, M. K. Brennanman and T. J. Meyer, Chemical Approaches to Artificial Photosynthesis. 2, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS PubMed.
- (a) Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y. Q. Lan, From Molecular Metal Complex to Metal-Organic Framework: The CO2 Reduction Photocatalysts with Clear and Tunable Structure, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS; (b) Y. Kuramochi, O. Ishitani and H. Ishida, Reaction Mechanisms of Catalytic Photochemical CO2 Reduction Using Re(I) and Ru(II) Complexes, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS; (c) Y. Yamazaki, H. Takeda and O. Ishitani, Photocatalytic Reduction of CO2 Using Metal Complexes, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS; (d) N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Molecular Polypyridine-based Metal Complexes as Catalysts for the Reduction of CO2, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- (a) J. Hawecker, J.-M. Lehn and R. Ziessel, Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2′-Bipyridine)tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS; (b) J. Hawecker, J.-M. Lehn and R. Ziessel, Efficient Photochemical Reduction of CO2 to CO by Visible Light Irradiation of Systems Containing Re(bipy)(CO)3X or Ru(bipy)32+-Co2+ Combinations as Homogeneous Catalysts, J. Chem. Soc. Chem. Commun., 1983, 536–538 RSC.
- Y. Kuramochi, M. Kamiya and H. Ishida, Photocatalytic CO2 Reduction in N,N-dimethylacetamide/Water as an Alternative Solvent System, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed.
- (a) T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, Photocatalytic Reduction of Low Concentration of CO2, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed; (b) T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, CO2 Capture by a Rhenium(I) Complex with the Aid of Triethanolamine, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- Y. Yamazaki, M. Miyaji and O. Ishitani, Utilization of Low-Concentration CO2 with Molecular Catalysts Assisted by CO2-Capturing Ability of Catalysts, Additives, or Reaction Media, J. Am. Chem. Soc., 2022, 144, 6640–6660 CrossRef CAS PubMed.
- (a) A. V. Müller, L. A. Faustino, K. T. de Oliveira, A. O. T. Patrocinio and A. S. Polo, Visible-Light-Driven Photocatalytic CO2 Reduction by Re(I) Photocatalysts with N-Heterocyclic Substituents, ACS Catal., 2023, 13, 633–646 CrossRef; (b) K. H. Chen, N. Wang, Z. W. Yang, S. M. Xia and L. N. He, Tuning of Ionic Second Coordination Sphere in Evolved Rhenium Catalyst for Efficient Visible-Light-Driven CO2 Reduction, ChemSusChem, 2020, 13, 6284–6628 CrossRef CAS PubMed; (c) M. Abdellah, A. M. El-Zohry, L. J. Antila, C. D. Windle, E. Reisner and L. Hammarstrom, Time-Resolved IR Spectroscopy Reveals a Mechanism with TiO2 as a Reversible Electron Acceptor in a TiO2-Re Catalyst System for CO2 Photoreduction, J. Am. Chem. Soc., 2017, 139, 1226–1232 CrossRef CAS PubMed; (d) R. D. Simpson and R. G. Bergman, A Dramatic Difference in the Reactivities of Alkoxido- and Aryloxidorhenium Complexes in Insertion Reactions, Angew. Chem., Int. Ed., 1992, 31, 220–223 CrossRef; (e) D. J. Darensbourg, B. L. Mueller, C. J. Bischoff, S. S. Chojnacki and J. H. Reibenspies, Investigations into the Steric Influences on the Reaction Mechanism of CO2 Insertion into Metal-Oxygen Bonds. COS Activation as a Model for CO2, Inorg. Chem., 1991, 30, 2418–2424 CrossRef CAS.
- Ru(II) complex: (a) R. N. Sampaio, D. C. Grills, D. E. Polyansky, D. J. Szalda and E. Fujita, Unexpected Roles of Triethanolamine in the Photochemical Reduction of CO2 to Formate by Ruthenium Complexes, J. Am. Chem. Soc., 2020, 142, 2413–2428 CrossRef CAS PubMed; (b) N. Queyriaux, W. B. Swords, H. Agarwala, B. A. Johnson, S. Ott and L. Hammarstrom, Mechanistic Insights on the Non-Innocent Role of Electron Donors: Reversible Photocapture of CO2 by Ru(II)-Polypyridyl Complexes, Dalton Trans., 2019, 48, 16894–16898 RSC; (c) P. Vadivelu and C. H. Suresh, Metal- and Ligand-Assisted CO2 Insertion into Ru-C, Ru-N, and Ru-O Bonds of Ruthenium(II) Phosphine Complexes: A Density Functional Theory Study, Inorg. Chem., 2015, 54, 502–512 CrossRef CAS PubMed; (d) Y. Arikawa, T. Nakamura, S. Ogushi, K. Eguchi and K. Umakoshi, Fixation of Atmospheric Carbon Dioxide by Ruthenium Complexes Bearing an NHC-based Pincer Ligand: Formation of a Methylcarbonato Complex and Its Methylation, Dalton Trans., 2015, 44, 5303–5305 RSC.
- Mn(I) complex: (a) H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, CO2 Capture by Mn(I) and Re(I) Complexes with a Deprotonated Triethanolamine Ligand, Chem. Sci., 2019, 10, 3080–3088 RSC; (b) D. J. Darensbourg, W.-Z. Lee, A. L. Phelps and E. Guidry, Kinetic Study of the Insertion and Deinsertion of Carbon Dioxide into fac-(CO)3(dppe)MnOR Derivatives, Organometallics, 2003, 22, 5585–5588 CrossRef CAS; (c) S. K. Mandal, D. M. Ho and M. Orchin, Reaction of Electrophiles with Manganese(I) and Rhenium(I) Alkoxide Complexes: Reversible Absorption of Atmospheric Carbon Dioxide, Organometallics, 1993, 12, 1714–1719 CrossRef CAS.
- Zn(II) complex: (a) C. A. Phipps, D. T. Hofsommer, C. D. Zirilli, B. G. Duff, M. S. Mashuta, R. M. Buchanan and C. A. Grapperhaus, Metal–Ligand Cooperativity Promotes Reversible Capture of Dilute CO2 as a Zn(II)-Methylcarbonate, Inorg. Chem., 2023, 62, 2751–2759 CrossRef CAS PubMed; (b) M. Kato and T. Ito, Facile Carbon Dioxide Uptake by Zinc(II)-Tetraazacycloalkane Complexes. 1. Syntheses, Characterizations, and Chemical Properties of (Monoalkyl carbonato)(tetraazacycloalkane)zinc(II) Complexes, Inorg. Chem., 1985, 24, 504–508 CrossRef CAS.
- Ni(II) and Pd(II) complexes: L. M. Martinez-Prieto, P. Palma and J. Campora, Monomeric Alkoxide and Alkylcarbonate Complexes of Nickel and Palladium Stabilized with the iPrPCP Pincer Ligand: A Model for the Catalytic Carboxylation of Alcohols to Alkyl Carbonates, Dalton Trans., 2019, 48, 1351–1366 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, Substantial Improvement in the Efficiency and Durability of a Photocatalyst for Carbon Dioxide Reduction Using a Benzoimidazole Derivative as an Electron Donor, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, Photoelectrochemical Reduction of CO2 Coupled to Water Oxidation Using a Photocathode with a Ru(II)–Re(I) Complex Photocatalyst and a CoOX/TaON Photoanode, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed.
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Dinuclear Metal Complexes: Multifunctional Properties and Applications, Chem. Soc. Rev., 2020, 49, 765–838 RSC.
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Visible Light-Driven Water Oxidation Using a Covalently-Linked Molecular Catalyst–Sensitizer Dyad Assembled on a TiO2 Electrode, Chem. Sci., 2016, 7, 1430–1439 RSC.
- (a) F. Kuttassery, H. Kumagai, R. Kamata, Y. Ebato, M. Higashi, H. Suzuki, R. Abe and O. Ishitani, Supramolecular Photocatalysts Fixed on the Inside of the Polypyrrole Layer in Dye Sensitized Molecular Photocathodes: Application to Photocatalytic CO2 Reduction Coupled with Water Oxidation, Chem. Sci., 2021, 12, 13216–13232 RSC; (b) R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, Photoelectrochemical CO2 Reduction Using a Ru(II)-Re(I) Supramolecular Photocatalyst Connected to a Vinyl Polymer on a NiO Electrode, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS PubMed.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell, Oxford, 2001 Search PubMed.
- Recent reviews: (a) E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Porphyrins and Phthalocyanines as Biomimetic Tools for Photocatalytic H2 Production and CO2 Reduction, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC; (b) L. Zou, R. Sa, H. Lv, H. Zhong and R. Wang, Recent Advances on Metalloporphyrin-Based Materials for Visible-Light-Driven CO2 Reduction, ChemSusChem, 2020, 13, 6124–6140 CrossRef CAS PubMed; (c) B. M. Pirzada, A. H. Dar, M. H. Shaikh and A. Qurashi, Reticular-Chemistry-Inspired Supramolecule Design as a Tool to Achieve Efficient Photocatalysts for CO2 Reduction, ACS Omega, 2021, 6, 29291–29324 CrossRef CAS PubMed.
- (a) P. Lang, M. Pfrunder, G. Quach, B. Braun-Cula, E. G. Moore and M. Schwalbe, Sensitized Photochemical CO2 Reduction by Hetero–Pacman Compounds Linking a ReI Tricarbonyl with a Porphyrin Unit, Chem.–Eur. J., 2019, 25, 4509–4519 CrossRef CAS PubMed; (b) C. Matlachowski, B. Braun, S. Tschierlei and M. Schwalbe, Photochemical CO2 Reduction Catalyzed by Phenanthroline Extended Tetramesityl Porphyrin Complexes Linked with a Rhenium(I) Tricarbonyl Unit, Inorg. Chem., 2015, 54, 10351–10360 CrossRef CAS PubMed; (c) C. D. Windle, M. W. George, R. N. Perutz, P. A. Summers, X. Z. Sun and A. C. Whitwood, Comparison of Rhenium–Porphyrin Dyads for CO2 Photoreduction: Photocatalytic Studies and Charge Separation Dynamics Studied by Time-Resolved IR Spectroscopy, Chem. Sci., 2015, 6, 6847–6864 RSC; (d) C. D. Windle, M. V. Campian, A.-K. Duhme-Klair, E. A. Gibson, R. N. Perutz and J. Schneider, CO2 Photoreduction with Long-Wavelength Light: Dyads and Monomers of Zinc Porphyrin and Rhenium Bipyridine, Chem. Commun., 2012, 48, 8189–8191 RSC; (e) K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, Electron Transfer from the Porphyrin S2 State in a Zinc Porphyrin-Rhenium Bipyridyl Dyad having Carbon Dioxide Reduction Activity, J. Phys. Chem. C, 2009, 113, 11667–11673 CrossRef CAS; (f) R. Miyatani and Y. Amao, Photochemical Synthesis of Formic Acid from CO2 with Formate Dehydrogenase and Water-Soluble Zinc Porphyrin, J. Mol. Catal. B: Enzym., 2004, 27, 121–125 CrossRef CAS.
- (a) D.-I. Won, J.-S. Lee, Q. Ba, Y.-J. Cho, H.-Y. Cheong, S. Choi, C. H. Kim, H.-J. Son, C. Pac and S. O. Kang, Development of a Lower Energy Photosensitizer for Photocatalytic CO2 Reduction: Modification of Porphyrin Dye in Hybrid Catalyst System, ACS Catal., 2018, 8, 1018–1030 CrossRef CAS; (b) Y. Kou, S. Nakatani, G. Sunagawa, Y. Tachikawa, D. Masui, T. Shimada, S. Takagi, D. A. Tryk, Y. Nabetani, H. Tachibana and H. Inoue, Visible Light-Induced Reduction of Carbon Dioxide Sensitized by a Porphyrin-Rhenium Dyad Metal Complex on p-Type Semiconducting NiO as the Reduction Terminal End of an Artificial Photosynthetic System, J. Catal., 2014, 310, 57–66 CrossRef CAS.
- Y. Kuramochi and A. Satake, Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction, Catalysts, 2023, 13, 282 CrossRef CAS.
- (a) Y. Kuramochi, Y. Fujisawa and A. Satake, Photocatalytic CO2 Reduction Mediated by Electron Transfer via the Excited Triplet State of Zn(II) Porphyrin, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed; (b) Y. Kuramochi and A. Satake, Photocatalytic CO2 Reductions Catalyzed by meso-(1,10-Phenanthrolin-2-yl)-Porphyrins Having a Rhenium(I) Tricarbonyl Complex, Chem.–Eur. J., 2020, 26, 16365–16373 CrossRef CAS PubMed.
- Y. Kuramochi, R. Sato, H. Sakuma and A. Satake, Photocatalytic CO2 Reduction Sensitized by Special-Pair Mimic Porphyrin Connected with Rhenium(I) Tricarbonyl Complex, Chem. Sci., 2022, 13, 9861–9879 RSC.
- (a) J. Rohacova and O. Ishitani, Rhenium(I) Trinuclear Rings as Highly Efficient Redox Photosensitizers for Photocatalytic CO2 Reduction, Chem. Sci., 2016, 7, 6728–6739 RSC; (b) V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, Visible-Light Photoredox Catalysis: Selective Reduction of Carbon Dioxide to Carbon Monoxide by a Nickel N-Heterocyclic Carbene–Isoquinoline Complex, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed; (c) Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Development of Highly Efficient Supramolecular CO2 Reduction Photocatalysts with High Turnover Frequency and Durability, Faraday Discuss., 2012, 155, 115–127 RSC; (d) H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, Efficient Photocatalytic CO2 Reduction using [Re(bpy)(CO)3{P(OEt)3}]+, J. Photochem. Photobiol. A., 1996, 96, 171–174 CrossRef CAS.
- (a) S. Meister, R. O. Reithmeier, M. Tschurl, U. Heiz and B. Rieger, Unraveling Side Reactions in the Photocatalytic Reduction of CO2: Evidence for Light-Induced Deactivation Processes in Homogeneous Photocatalysis, ChemCatChem, 2015, 7, 690–697 CrossRef CAS; (b) H. Takeda, K. Koike, H. Inoue and O. Ishitani, Development of an Efficient Photocatalytic System for CO2 Reduction Using Rhenium(I) Complexes Based on Mechanistic Studies, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- P. Kurz, B. Probst, B. Spingler and R. Alberto, Ligand Variations in [ReX(diimine)(CO)3] Complexes: Effects on Photocatalytic CO2 Reduction, Eur. J. Inorg. Chem., 2006, 2006, 2966–2974 CrossRef.
- (a) A. Satake, Y. Katagami, Y. Odaka, Y. Kuramochi, S. Harada, T. Kouchi, H. Kamebuchi and M. Tadokoro, Synthesis of DoubleBridged Cofacial Nickel Porphyrin Dimers with 2,2′-Bipyridyl Pillars and Their Restricted Coordination Space, Inorg. Chem., 2020, 59, 8013–8024 CrossRef CAS PubMed; (b) Y. Ohkoda, A. Asaishi, T. Namiki, T. Hashimoto, M. Yamada, K. Shirai, Y. Katagami, T. Sugaya, M. Tadokoro and A. Satake, D 3h-Symmetric Porphyrin-Based Rigid Macrocyclic Ligands for Multicofacial Multinuclear Complexes in a One-Nanometer-Sized Cavity, Chem.–Eur. J., 2015, 21, 11745–11756 CrossRef CAS PubMed; (c) Y. Tomohiro, A. Satake and Y. Kobuke, Synthesis of BipyridyleneBridged Bisporphyrin by Nickel-Mediated Coupling Reaction: ON–OFF Control of Cofacial Porphyrin Unit by Reversible Complexation, J. Org. Chem., 2001, 66, 8442–8446 CrossRef CAS PubMed.
- (a) D. N. Tritton, G. B. Bodedla, G. Tang, J. Zhao, C.-S. Kwan, K. C.-F. Leung, W.-T. Wong and X. Zhu, Iridium Motif Linked Porphyrins for Efficient Light-driven Hydrogen Evolution via Triplet state Stabilization of Porphyrin, J. Mater. Chem. A, 2020, 8, 3005–3010 RSC; (b) P. A. Angaridis, E. Ferentinos, G. Charalambidis, K. Ladomenou, V. Nikolaou, S. Biswas, G. D. Sharma and A. G. Coutsolelos, Pyridyl vs. Bipyridyl Anchoring Groups of Porphyrin Sensitizers for Dye Sensitized Solar Cells, RSC Adv., 2016, 6, 22187–22203 RSC; (c) T. Nakamura, H. Ube, M. Shiro and M. Shionoya, A Self-Assembled Multiporphyrin Cage Complex through Three Different Zinc(II) Center Formation under Well-Balanced Aqueous Conditions, Angew. Chem., Int. Ed., 2013, 52, 720–723 CrossRef CAS; (d) Z. Fang and R. Breslow, Metal Coordination-Directed Hydroxylation of Steroids with a Novel Artificial P-450 Catalyst, Org. Lett., 2006, 8, 251–254 CrossRef CAS PubMed; (e) M. Linke-Schaetzel, C. E. Anson, A. K. Powell, G. Buth, E. Palomares, J. D. Durrant, T. S. Balaban and J.-M. Lehn, Dynamic Chemical Devices: Photoinduced Electron Transfer and its Ion-triggered Switching in Nanomechanical Butterfly-type Bis(porphyrin)terpyridines, Chem.–Eur. J., 2006, 12, 1931–1940 CrossRef CAS PubMed; (f) K. Mizuno, M. Kurihara, S. Takagi and H. Nishihara, Novel Intramolecular Electron-transfer Equilibrium in a Spatially Interaction Binary System of a Metal-free Porphyrin and a Ruthenium Trichloro Terpyridyl Complex, Chem. Lett., 2003, 788–789 CrossRef CAS; (g) K. F. Cheng, C. M. Drain and K. Grohmann, Porphyrins Linked Directly to the 5,5′ Positions of 2,2′-Bipyridine: A New Supramolecular Building Block and Switch, Inorg. Chem., 2003, 42, 2075–2083 CrossRef CAS PubMed; (h) K. Phillips-McNaughton and J. T. Groves, Zinc-Coordination Oligomers of Phenanthrolinylporphyrins, Org. Lett., 2003, 5, 1829–1832 CrossRef CAS PubMed; (i) J.-P. Collin, A. Harriman, V. Heitz, F. Odobel and J.-P. Sauvage, Photoinduced Electron- and Energy-Transfer Processes Occurring within Porphyrin-Metal-Bisterpyridyl Conjugates, J. Am. Chem. Soc., 1994, 116, 5679–5690 CrossRef CAS.
- Y. Yamada and K. Tanaka, Metal-Induced Structural Switching of a Folded Quinone-Sandwiched Porphyrin, J. Inorg. Organomet. Polym., 2013, 23, 180–185 CrossRef CAS.
- (a) M. Kosugi, K. Sasazawa, Y. Shimizu and T. Migita, Reactions of Allyltin Compounds III. Allyltion of Aromatic Halides with Allyltributyltin in the Presence of Tetrakis(triphenylphosphine)palladium(0), Chem. Lett., 1977, 301–302 CrossRef CAS; (b) D. Milstein and J. K. Stille, A General, Selective, and Facile Method for Ketone Synthesis from Acid Chlorides and Organotin Compounds Catalyzed by Palladium, J. Am. Chem. Soc., 1978, 100, 3636–3638 CrossRef CAS.
- J. S. Lindsey and R. W. Wagner, Investigation of the Synthesis of Ortho-substituted Tetraphenylporphyrins, J. Org. Chem., 1989, 54, 828–836 CrossRef CAS.
- D. C. Harrowven, D. P. Curran, S. L. Kostiuk, I. L. Wallis-Guy, S. Whiting, K. J. Stenning, B. Tang, E. Packard and L. Nanson, Potassium Carbonate–Silica: A Highly Effective Stationary Phase for the Chromatographic Removal of Organotin Impurities, Chem. Commun., 2010, 46, 6335–6337 RSC.
- S. Sato, T. Morimoto and O. Ishitani, Photochemical Synthesis of mer-[Re(bpy)(CO)3Cl], Inorg. Chem., 2007, 46, 9051–9053 CrossRef CAS PubMed.
- A. Gabrielsson, F. Hartl, H. Zhang, J. R. Lindsay Smith, M. Towrie, A. Vlcek and R. N. Perutz, Ultrafast Charge Separation in a Photoreactive Rhenium-Appended Porphyrin Assembly Monitored by Picosecond Transient Infrared Spectroscopy, J. Am. Chem. Soc., 2006, 128, 4253–4266 CrossRef CAS PubMed.
- T. Gatti, P. Cavigli, E. Zangrando, E. Iengo, C. Chiorboli and M. T. Indelli, Improving the Efficiency of the Photoinduced Charge-Separation Process in a Rhenium(I)–Zinc Porphyrin Dyad by Simple Chemical Functionalization, Inorg. Chem., 2013, 52, 3190–3197 CrossRef CAS PubMed.
- C.-I. Lin, M.-Y. Fang and S.-H. Cheng, Substituent and Axial Ligand Effects on the Electrochemistry of Zinc Porphyrins, J. Electroanal. Chem., 2002, 531, 155–162 CrossRef CAS.
- We tried to synthesize a porphyrin with three mesityl groups and one phen group. The Lindsey method was not applicable to the phen-substituted porphyrins because 1,10-phenanthroline-2-carboxaldehyde would precipitate upon protonation with trifluoroacetic acid and BF3 (see ref. 23). Thus, the condensation of 1,10-phenanthroline-2-carboxaldehyde and mesitylaldehyde with pyrrole was carried out in propionic acid under air, but only trace amounts of the porphyrin could be obtained due to steric hindrance of the mesityl group.
- (a) Y. Kou, Y. Nabetani, R. Nakazato, N. V. Pratheesh, T. Sato, S. Nozawa, S. Adachi, H. Tachibana and H. Inoue, Mechanism of the Photoreduction of Carbon Dioxide Catalyzed by the Benchmarking Rhenium Dimethylbipyridine Complexes; Operando Measurements by XAFS and FT-IR, J. Catal., 2022, 405, 508–519 CrossRef CAS; (b) Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, Direct Detection of Key Reaction Intermediates in Photochemical CO2 Reduction Sensitized by a Rhenium Bipyridine Complex, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- (a) A. V. Müller, L. A. Faustino, K. T. de Oliveira, A. O. T. Patrocinio and A. S. Polo, Visible-Light-Driven Photocatalytic CO2 Reduction by Re(I) Photocatalysts with N-Heterocyclic Substituents, ACS Catal., 2023, 13, 633–646 CrossRef; (b) E. E. Benson and C. P. Kubiak, Structural Investigations into the Deactivation Pathway of the CO2 Reduction Electrocatalyst Re(Bpy)(CO)3Cl, Chem. Commun., 2012, 48, 7374–7376 RSC.
- Y. Harel and J. Manassen, Photoreduction of Tetraphenylporphyrins by Amines in the Visible. Photochemical Syntheses of Reduced Tetraphenylporphyrins and the Mechanism of Photoreduction, J. Am. Chem. Soc., 1978, 100, 6228–6234 CrossRef CAS.
- (a) H. Yamaguchi, A. Soeta, H. Toeda and K. Itoh, Raman Scattering Study on Electrochemical Reduction Products of Magnesium, Zinc and Copper Tetraphenylporphines, J. Electroanal. Chem. Interfacial Electrochem., 1983, 159, 347–359 CrossRef CAS; (b) J. G. Lanese and G. S. Wilson, Electrochemical Studies of Zinc Tetraphenylporphin, J. Electrochem. Soc., 1972, 119, 1039–1043 CrossRef CAS; (c) G. L. Closs and L. E. Closs, Negative Ions of Porphin Metal Complexes, J. Am. Chem. Soc., 1963, 85, 818–819 CrossRef CAS.
- M. Taniguchi and J. S. Lindsey, Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins, Chem. Rev., 2017, 117, 344–535 CrossRef CAS PubMed.
- F. P. A. Johnson, M. W. George, F. Hartl and J. J. Turner, Electrocatalytic Reduction of CO2 Using the Complexes [Re(bpy)(CO)3L]n (n = + 1, L = P(OEt)3, CH3CN; n = 0, L = Cl−, OTf−; bpy = 2,2′-Bipyridine; OTf− = CF3SO3) as Catalyst Precursors: Infrared Spectroelectrochemical Investigation, Organometallics, 1996, 15, 3374–3387 CrossRef CAS.
- K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley-Interscience, New York, 1987 Search PubMed.
- K. Kamogawa, Y. Shimoda, K. Miyata, K. Onda, Y. Yamazaki, Y. Tamaki and O. Ishitani, Mechanistic Study of Photocatalytic CO2 Reduction Using a Ru(II)-Re(I) Supramolecular Photocatalyst, Chem. Sci., 2021, 12, 9682–9693 RSC.
- D. J. Quimby and F. R. Longo, Luminescence Studies on Several Tetraarylporphins and Their Zinc Derivatives, J. Am. Chem. Soc., 1975, 97, 5111–5117 CrossRef CAS.
- M. S. Asano, T. Morita, T. Miwata and K. Nomura, Observation of Intramolecular Interaction in Fluorescent Star-Shaped Polymers: Evidence for Energy Hopping between Branch Chains, J. Phys. Chem., 2020, 124, 11510–111518 CrossRef CAS PubMed.
- Y. Kuramochi, T. Yoshida, K. Kodama, M. Kiriyama, S. Yoshioka, S. Hirota and A. Satake, Efficient Photochemical Reduction of Quinone into Hydroquinone Promoted by Imidazolyl N-H Proton, Chem. Lett., 2018, 47, 1343–1345 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02430j |
| This journal is © The Royal Society of Chemistry 2023 |