
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and comparison of iso-structural f-block metal complexes (Ce, U, Np, Pu) featuring η6-arene interactions†
Jesse
Murillo
ab,
Conrad A. P.
Goodwin
 b,
Lauren
Stevens
bc,
Skye
Fortier
*a,
Andrew J.
Gaunt
*b and
Brian L.
Scott
c
b,
Lauren
Stevens
bc,
Skye
Fortier
*a,
Andrew J.
Gaunt
*b and
Brian L.
Scott
c
aDepartment of Chemistry and Biochemistry, University of Texas at El Paso, El Paso, Texas 79968, USA. E-mail: asfortier@utep.edu
bChemistry Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: gaunt@lanl.gov
cMaterials Physics and Applications Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
First published on 20th June 2023
Abstract
Reaction of the terphenyl bis(anilide) ligand [{K(DME)2}2LAr] (LAr = {C6H4[(2,6-iPr2C6H3)NC6H4]2}2−) with trivalent chloride “MCl3” salts (M = Ce, U, Np) yields two distinct products; neutral LArM(Cl)(THF) (1M) (M = Np, Ce), and the “-ate” complexes [K(DME)2][(LAr)Np(Cl)2] (2Np) or ([LArM(Cl)2(μ-K(X)2)])∞ (2Ce, 2U) (M = Ce, U) (X = DME or Et2O) (2M). Alternatively, analogous reactions with the iodide [MI3(THF)4] salts provide access to the neutral compounds LArM(I)(THF) (3M) (M = Ce, U, Np, Pu). All complexes exhibit close arene contacts suggestive of η6-interactions with the central arene ring of the terphenyl backbone, with 3M comprising the first structurally characterized Pu η6-arene moiety. Notably, the metal–arene bond metrics diverge from the predicted trends of metal–carbon interactions based on ionic radii, with the uranium complexes exhibiting the shortest M–Ccentroid distance in all cases. Overall, the data presents a systematic study of f-element M-η6-arene complexes across the early actinides U, Np, Pu, and comparison to cerium congeners.
Introduction
Moving towards a more detailed understanding of chemical behaviour and bonding trends within the 5f-block elements has been of keen interest since the proposal of the actinide (An) concept by Seaborg in the later part of the 1930's.1 Once thought to predominantly engage in metal–ligand bonding chiefly electrostatic in nature, decades of concerted advances in the syntheses and characterization of actinide-containing molecular complexes have demonstrated the ability of actinide metal ions to form covalent bonds. These interactions are generally intermediate between that of transition metals (highly directional based on metal–ligand orbital overlap), and lanthanides (non-directional).2–10 In several cases, actinide–ligand interactions are not purely electrostatic in nature and are engendered by the availability of 6d and 5f orbitals to participate in bonding, especially for the early actinide members (An = U, Np, Pu).10–23Of note, f-block metal complexes which contain metal–arene interactions have proven to be highly valuable for understanding bonding and the role of valence orbitals for lanthanides and actinides.24–32 Among this class of molecules, those which feature neutral arene coordinating motifs are of particular interest as these may exhibit covalent participation of the metal in the form of π/δ/φ type interactions.31,33–37 Such complexes have potential to provide new key insight into f-block bonding modes as has been the case in other areas of the periodic table; for example, the seminal discovery of bis(benzene)chromium, Cr(η6-C6H6)2, by E. O. Fischer, which revolutionized the understanding of transition metal chemistry.38
A challenge to overcome in establishing a suite of analogous metal–arene molecules across the f-block is the “hard” Lewis acidic character of the f-block metal ions that causes interactions with “soft” arene donor substituents to be difficult to form and have often required the use of hard donor atom substituents to act as an “anchor-point” and facilitate binding of the arene to the metal.35,36,39–50 A handful of examples exist which do coordinate neutral arene species by coordinating strongly electron withdrawing groups to the actinide.51–57 Indeed, despite their challenging synthetic nature, several milestone works have successfully accessed and isolated actinide–arene complexes. Notably, the work of Meyer and co-workers to form the tris(aryloxide) uranium(III) complex, [{(Ad,MeArO)3mes}U], has illustrated the utility of the tethered arene strategy and led to the formation of low-valent (2+) uranium complexes, which show remarkable reactivity and are stabilized by unique actinide–arene bonding motifs.35,39–41,58,59 Later work by Arnold and co-workers extended this strategy to a transuranium element, neptunium, by the formation of several complexes featuring Np-η6-arene interactions, facilitated by the trans-calix[2]benzene[2]pyrrole ligand platform.21
We recently employed a terphenyl bis(anilide) ligand system [LAr] (LAr = {C6H4[(2,6-iPr2C6H3)NC6H4]2}2−),46 which features aromatic substituents in the ligand backbone that, upon complexation with Ln/An metal ions, binds in a κ2:η6-fashion. We found this ligand platform to have remarkable versatility.46,47,60 Given that the [LAr] ligand platform has been demonstrated to coordinate both Dy and U, in addition to the architectural enforcement of a metal-η6-arene interaction upon complexation, we felt this ligand system would serve as an excellent foundation for forming iso-structural complexes of f-block elements, including transuranium species, allowing us to gain insight into metal–arene bonding interactions and elucidate periodic trends. Based upon ionic radii alone, we expected to observe a metal–arene distance trend U > Ce ∼ Np > Pu if a purely electrostatic bonding model adequately described the bonding. Conversely, if deviations from that trend were observed, then that may be suggestive of metal–ligand covalent interactions more pronounced in some of the f-metal complexes than others.
Results and discussion
The reaction of the dipotassium salt of the ligand, [{K(DME)2}2LAr], with 1 equiv. of “MCl3” at room temperature in THF formed a deep brown/red turbid solution (M = U, Np) or a bright yellow/orange turbid solution (M = Ce) (Scheme 1). The U and Np trivalent chloride metal precursors were formed by the in situ reduction of tetravalent UCl4 and NpCl4(DME)2 in THF using 1.1 equiv. of potassium graphite (KC8), forming presumed MCl3(THF)n adducts, while the Ce source was commercially purchased CeCl3. Drying the reaction mixture and extracting with Et2O, presents a dark red solution (M = Np, U) or a vivid yellow solution (M = Ce), which upon workup and storage at −35 °C provides crystals suitable for single-crystal X-ray diffraction (SC-XRD) and were identified as LArM(Cl)(THF) (1M) (M = Ce, Np) (Fig. 1 and S1†) isolated in relatively low yields (20% for 1Np and 11% for 1Ce). For M = U, the Et2O extract failed to give the analogous 1U complex, but instead formed the contact polymer [LArM(Cl)2{μ-K(Et2O)2}]∞ (2U), isolated in 28% yield. Attempts to generate and isolate the Pu congener, 1Pu, using in situ generated PuCl3(THF)xvia reduction of PuCl4(DME)2, to enable a systematic U/Np/Pu comparison, were unsuccessful and yielded intractable products.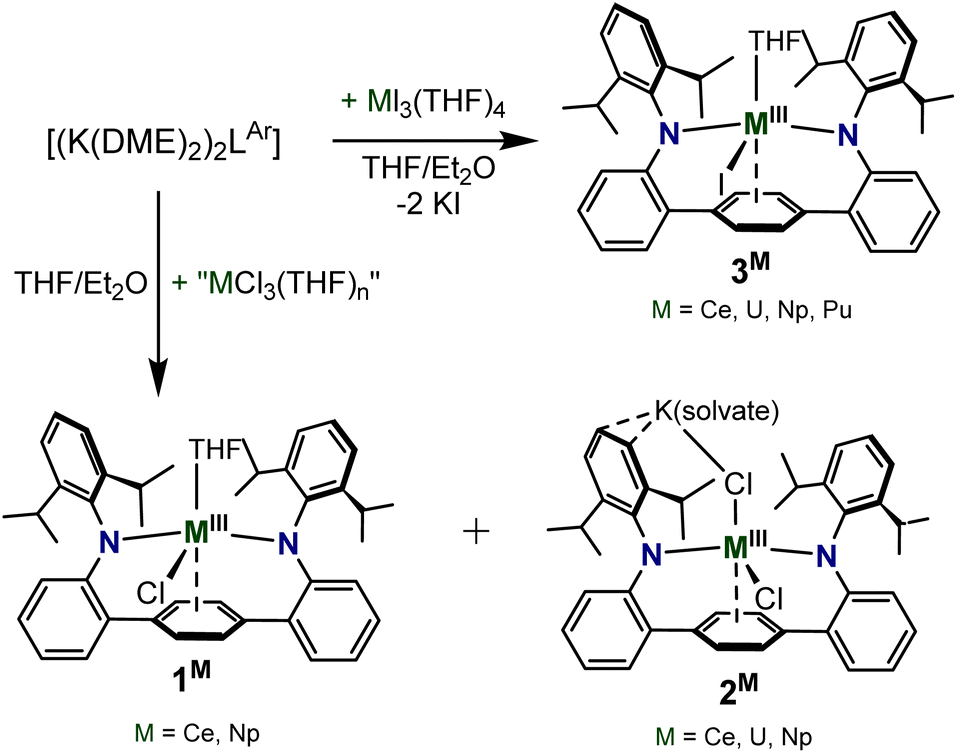 | ||
| Scheme 1 Syntheses of 1M, 2M, and 3M. | ||
 | ||
| Fig. 1 ORTEP of the solid-state molecular structure of 1Np. Hydrogen atoms and co-crystalized Et2O molecule are removed for clarity. Ellipsoids are shown at the 30% probability level. Shown for connectivity purposes only. | ||
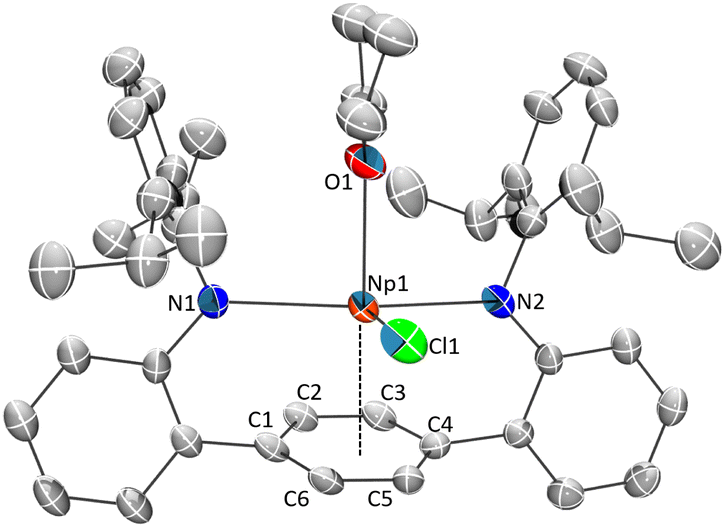
The Et2O insoluble products from the reactions are highly soluble in DME (DME = 1,2-dimethoxyethane) or THF to give dark red/brown solutions (M = Np, U), or a dark orange solution (M = Ce). These solutions, after workup and storage at −35 °C for several days, give X-ray quality single-crystals identified as the “-ate” complex [K(DME)2(LAr)Np(Cl)2] (2Np) or the contact polymer [LArM(Cl)2{μ-K(X)2}]∞ (2Ce) (X = DME or Et2O) (Scheme 1) in 20% and 10% yields respectively. In the case of the uranium reaction, the THF extract produced intractable products, although on one attempt we were able to isolate the 1-D polymeric complex [LArU(Cl)2(THF){μ-K(THF)4}]∞, where the uranium metal ions are bridged by Cl–K–Cl contacts (Fig. S2†). In the case of 2Np, the complex exists as a discrete molecular species in the solid-state, where the potassium ion is contacting the apical chloride and is coordinated by two DME molecules, while one non-coordinated DME molecule is located in the lattice (Fig. 2). In complexes 2Ce and 2U the potassium ions are coordinated by a mix of DME and Et2O solvates that act as bridging moieties, forming 1-D polymeric species (see Fig. S3 and S4† for extended structures). For 2Ce and 2U, disordered hexane molecules fill the void space within the crystal lattice. Although the solid-state arrangement differs, the anionic component of the 2M series is homologous and are shown in Fig. 2.
 | ||
| Fig. 2 ORTEP of the solid-state molecule structures of 2Ce·2Hex (left), 2U·2Hex (centre), and 2Np·DME (right). Hydrogen atoms and co-crystalized solvents are removed for clarity. Ellipsoids are shown at the 50% probability level. | ||
The 1H NMR spectra of 1M and 2M span a large range of chemical shifts (characteristic of the metal ion electronic configurations – Ce3+(4f1), U3+(5f3), Np3+(5f4)), showing proton resonances within the range from −30.65 to +42.22 ppm for the six complexes (Fig. S8–S12†). In accordance with low-symmetry molecular environments in solution, the complicated spectra are typical of previously reported complexes of [LAr], some of which display dozens of unique resonances in their 1H NMR spectra.46,47,60 For all complexes, except for 2U, resonances for the protonated ligand, H2LAr, can be seen in the 1H NMR spectra. The quantity of H2LAr varies among the measured spectra but is consistently observed by 1H NMR spectroscopy. The majority of 1H NMR spectra were collected on isolated crystalline material of the complexes, which suggests that the formation of H2LAr may be taking place in solution. However, the presence of small amounts of H2LAr as a crystalline by-product cannot be definitively discounted.
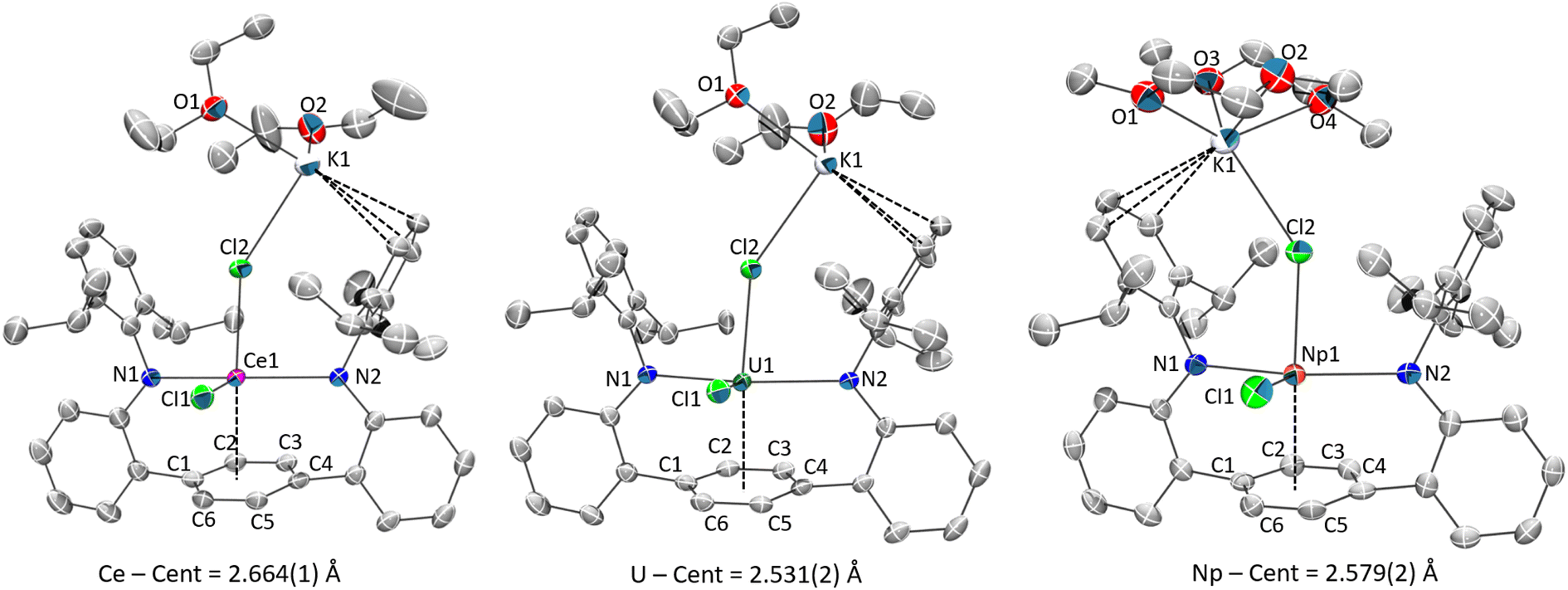
Single-crystal X-ray structures 1M and 2M show a common coordination geometry, with all complexes displaying a pseudo four-coordinate, see-saw type geometry around the metal centre (Fig. 1, 2 and S1†). The [LAr] ligand binds in a κ2-mode via the anilide N-donor atoms, which are approximately in the trans configuration (Tables 1, S4†). With respect to key structural parameters, only minor disparities between the neutral 1M and “-ate” 2M complexes are observed, although rigorous metrical comparisons of 1Np are not possible due to the poor quality of X-ray data for that complex. A significant shortening of the M–Cl bond in the neutral complex 1Ce is seen as compared to 2Ce, when taking the average M–Cl bonding value for 2Ce and the replacement of the apical Cl− anion with a neutral THF molecule in 1Mvs.2M (Tables 1 and S4†). Given the broad similarities in the metal–ligand interactions, and the superior quality SC-XRD data for the 2M series, we will discuss in detail only 2M structures in the following section.
| Complex | M–N bond distances (Å) (N1/N2) | M–cent distance (Å) | M–Carene range (Å) | M–Cl distance (Å) (Cl1/Cl2) | Cl2–M–Cl1 bond angle (°) | N–M–N bond angle (°) | Metal ionic radius (Å) |
|---|---|---|---|---|---|---|---|
| 2Ce·2Hex | 2.470 (2)/2.525 (2) | 2.664 (1) | 2.936 (2)–3.092 (2) avg. 3.01 | 2.669 (1)/2.687 (1) | 101.90 (3) | 156.36 (7) | 1.01 |
| 2U·2Hex | 2.452 (3)/2.509 (3) | 2.530 (1) | 2.834 (4)–2.959 (4) avg. 2.89 | 2.670 (1)/2.689 (1) | 101.39 (4) | 157.8 (1) | 1.025 |
| 2Np·DME | 2.494 (4)/2.483 (4) | 2.579 (2) | 2.911 (5)–2.966 (5) avg. 2.93 | 2.627 (1)/2.674 (1) | 101.09 (4) | 156.3 (1) | 1.01 |
Selected bond metrics for the 2M series are shown in Table 1. It is important to note here that although the anionic core of these structures is similar, they differ in their extended structures with 2Ce and 2U being polymeric while 2Np is a discrete molecule. Additionally, the potassium coordinated, and non-coordinated lattice solvents differ in identity and relative amounts across the series. While these differences likely influence bonding metrics, we point out that trends found in the 2M series are mimicked in the 1M and 3M (vide infra) complexes. With these distinctions in mind, we next discuss in more detail bonding features discovered in the 2M series.
The M–N bond distances for all complexes are within the expected range for amide-M(III) bonds of their type, though they tend towards the longer end of the reported ranges.21,47,61–64 Additionally, there are no clear statistically meaningful differences in the M–N metrics as a function of f-metal identity. On the other hand, subtle yet significant distinctions can be observed with respect to the M–Carene interactions among the 2M compounds. Curiously, these disparities do not seem to trend with a purely electrostatic model based on metal ionic radius.
The metal–arene centroid distance, M–Ccent, among the series is shortest in 2U at 2.530 (1) Å and longest in 2Ce at 2.664 (1) Å (Δ = 0.134 Å), despite the reported six-coordinate ionic radius of Ce3+ being smaller than U3+ (Δ = 0.015 Å).65 Compound 2Np has an intermediate value at 2.579 (2) Å which is 0.085 Å shorter than in 2Ce and 0.049 Å longer than in 2U. For context, Np3+ has an ionic radii essentially identical to Ce3+ (both 1.01 Å) and therefore equally smaller than U3+ (Δ = 0.015 Å).65 These differences are noteworthy because, if following an ionic bonding model, the 2U complex should possess slightly longer M–Ccent contacts than in the 2Ce and 2Np complexes (not accounting for steric congestion changes and lattice packing effects). Similarly, the range of the metal–arene carbon bonds in 2U are shorter than in the Ce and Np analogues, a fact reflected in the average M–Carene bond distances.
In general, the internal C–C bonds of the η6-coordinated ring display alternating short-long bond distances for all 2M (Table S5†) along with subtle out-of-plane distortions across the ipso-substituents of the central ring (C1–Ccent–C4; 7.8, 8.7, and 7.6° for M = Ce, Np, U, respectively) (Fig. S36†). These features are consistent with previous similar observations in some other metal–arene complexes.66–68
When switching the metal precursor source to the well-defined trivalent iodide starting material MI3(THF)4 (M = U, Np, Pu)69,70 or commercially sourced CeI3, reactions with [{K(DME)2}2LAr] in THF consistently yield the monomeric neutral LArM(I)(THF) (3M) complex. Gratifyingly, the 3M series facilitated inclusion of Pu, as 3Pu (Fig. 3), which to the best of our knowledge is the first reported plutonium complex containing an η6-coordinated arene ring. These complexes are neutral in character and lack the potassium ion found in 2M. The lack of the “-ate” complex formation in the case of the MI3(THF)4 reactions we attribute to the larger ionic radius of the I− ligand, which makes the formation of the neutral complex preferred, aiding in the isolation of the 3M structural analogues for all the metal ions studied here.
 | ||
| Fig. 3 ORTEP rendering of the solid-state molecular structures of 3Pu·THF0.66Et2O0.33. Hydrogen atoms and co-crystalized solvents are removed for clarity. Ellipsoids are shown at the 50% probability level. Complexes 3Ce, 3U and 3Np are structurally analogous and can be found in the ESI for this document (S5–S7†). | ||
A useful comparison can be made between the 2M and 3M series given their many similarities; however, one should bear in mind the caveat that structural metrics between 2M and 3M may be affected by the differences in crystal packing systems, differences in the coordinating and non-coordinating solvents in the solid-state, as well as the presence of the contacting potassium ion in all of 2M, which is absent in 3M. Despite these differences, the coordination number and overall geometry about the metal centres in 3M is comparable to that of 2M; however, the coordinated THF molecule replaces the apical chloride ligand of the 2M series commensurate with charge balance differences between the ‘-ate’ 2M and neutral 3M. For the 3M series, all the M–[LAr] contact distances are like those in 2M, with only a subtle contraction in bond distances observed. With regards to the M–Ccent values, going from 2Ce to 3Ce we see a decrease in the distance (ΔCe–Ccent = −0.018 Å) similar to that seen going from 2Np to 3Np (ΔNp–Ccent = −0.018 Å), while going from 2U to 3U sees a modest increase in the distance (ΔU–Ccent = +0.007 Å). Importantly, the M–Ccent trend seen in 2M complexes, where the trend of M–arene distances deviates from that predicted by ionic radius, is mirrored in the 3M series, but with the comparison now extending to include a Pu3+ complex.
As with the 2M series, the uranium complex 3U has the shortest M–arene bond distances of the 3M series (Table 2). For instance, the U–Ccent value (2.538 (1) Å) in 3U is 0.108 Å shorter than the Ce–Ccent in 3Ce, slightly less than the magnitude of the M–Ccent difference between 2U and 2Ce of 0.134 Å. This is due to the contraction of the Ce–Ccent distance from 3Ce (2.646 (1) Å) compared to 2Ce (2.664 (1) Å). Moreover, the U–Ccent value in 3U is 0.023 Å shorter than the Np–Ccent value in 3Np, narrowing the difference of 0.049 Å between 2U and 2Np. The Np–Ccent value in 3Np is 0.086 Å shorter than the Ce–Ccent value in 3Ce. This tracks closely to a difference of 0.085 Å between 2Np and 2Ce. Finally, consideration of the 3Pu metrics shows that the value Pu–Ccent is 0.073 Å shorter than the Ce–Ccent value in 3Ce, 0.036 Å longer than U–Ccent value in 3U, and 0.013 Å longer than the Np–Ccent value in 3Np.
| Complex | M–N bond distances (Å) (N1/N2) | M–cent distance (Å) | M–Carene range (Å) | M–I distance (Å) | N–M–N bond angle (°) | Metal ionic radius (Å) |
|---|---|---|---|---|---|---|
| 3Ce·Et2O | 2.509 (1)/2.441 (1) | 2.646 (1) | 2.943 (2)–3.047 (2) avg. 2.99 | 3.0810 (5) | 156.32 (6) | 1.01 |
| 3U·THF0.8Et2O0.2 | 2.440 (1)/2.489 (1) | 2.538 (1) | 2.876 (1)–2.935 (1) avg. 2.90 | 3.0534 (7) | 154.01 (5) | 1.025 |
| 3Np·Pent | 2.489 (8)/2.457 (7) | 2.561 (4) | 2.878 (8)–2.962 (9) avg. 2.91 | 3.0288 (9) | 153.7 (3) | 1.01 |
| 3Pu·THF0.66Et2O0.33 | 2.469 (4)/2.428 (4) | 2.574 (1) | 2.902 (4)–2.970 (5) avg. 2.93 | 3.0276 (7) | 153.97 (14) | 1.00 |
All told, there are two patterns which emerge across the M–Ccent distances in 2M and 3M: (a) all of the actinide arene centroid interactions are shorter than the corresponding cerium interactions despite similarities in ionic radii, and (b) the actinide arene–centroid distance appears to increase from U to Np to Pu, counter to the trend expected based on ionic radii alone, albeit with the acknowledgement that only the 3M series can compare across all three actinide elements studied here (U, Np, Pu). Additionally, this trend is chiefly observed for the M–Carene interaction and not consistently with any of the other M–ligand contacts. To visualize this trend with respect to the M–Namideversus M–Ccent distances, we have compiled them into graphical form in Fig. 4. For additional context, we have also compared the M-halide distances and M-arene distances for other molecular actinide series, which span relevant atoms of the 5f block (Fig. S39 and S40†). This break in observed bonding distances with predicted ionic radii/electrostatic interactions for the actinides is known, especially in respect to enhanced covalency in An3+vs. Ln3+ systems, but we are not aware of documented instances of increasing metal–ligand bond length from U3+ to Np3+ to Pu3+,18,71–77 and such trends have not been examined for neutral arene interactions across those metals. The trend we observe here could be a result of covalency or steric effects, or both.
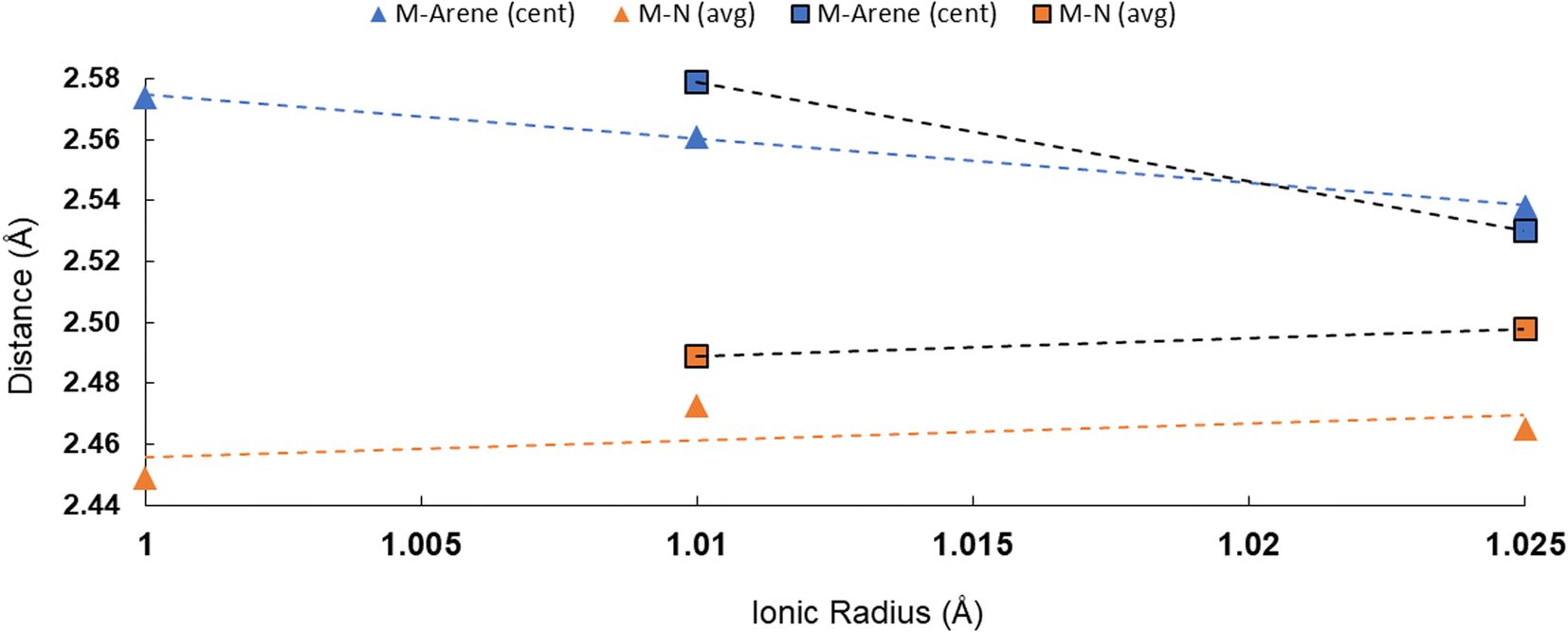 | ||
| Fig. 4 Plot of metal ionic radius versus M–ligand (Namide and Ccent) bond distances for 2M and 3M complexes. Dotted lines shown were generated by linear trend line fits. Ionic radii values are for 6-coordinate An3+ species.65 | ||
Turning to discussion of the 2M and 3M structures in the broader context of previously reported literature, the most noteworthy feature of these complexes is the metal–arene η6-interactions present. Examples of f-element interactions with formally neutral arenes for these metal ions are reported, with a handful of structurally verified reports for uranium,30,36,41,42,46,47,51–53,55,56,78–81 six for cerium,82–87 one for neptunium21 and none for plutonium at the time of writing. It should be noted that this type of interaction is also known for several rare-earth and lanthanide compounds, though here we focus on comparisons to similar f-element complexes (Ce, U, Np).
With respect to comparison against other uranium complexes, 2U contains an average U–Carene bond distance of 2.89 Å and a U–Ccent value of 2.538 (1) Å, which is in close agreement with 3U which displays an average U–Carene bond distance of 2.90 Å and a U–Ccent of 2.5384 (7) Å. These values are slightly shorter than our previously reported LArUIII(I)(DME) complex (avg. U–Carene = 2.92 Å, U–Ccent = 2.56 Å).46 Compared to other U3+ tethered-arene systems, the bis(arene) sandwich complex IU(NHAriPr6)2, displays average U–Ccent values 2.78 and 2.79 Å,42 while the bidentate LUIII(I) (L = trans-calix[2]benzene[2]pyrrolide) contains a U–Ccent = 2.67 Å.43 Although 2U displays shorter U–Ccent distances than the bis(arene) complexes noted above, the mono–arene U3+ complex, κ3:η6-[(Ad,MeArO)3mes]U has significantly shorter average U–Carene distances of = 2.75 Å and U–Ccent of 2.35 Å.41
Complexes 2Ce and 3Ce contain M–Carene interactions with average metal–arene contact distances of Ce–Carene = 3.01 and 2.99 Å, respectively, with Ce–Ccent = 2.664 (1) and 2.646 (1) Å. These Ce3+η6-arene interactions are intermediate compared to the unsupported terminal arene interactions of Ce(mes){N(C6F5)2}3 (avg. Ce–Carene = 3.15 Å, Ce–Ccent = 2.82 Å),84 and Ce(C6H5Me)(GaCl4)3 (avg. Ce–Carene = 2.950 Å, Ce–Ccent = 2.61 Å).82 Compared to the Ce2+ quadruple decker complex, [K(2.2.2-crypt)]2[{(KL3Ce)(μ-η6:η6-C7H8)}2Ce], in which the Ce2+ centres are bridged by anionic toluene moieties supported by δ-bonding interactions, the Ce–Carene interactions are significantly shorter (avg. Ce1–Carene = 2.69 Å and Ce2–Carene = 2.64 Å) than in the case of 2Ce and 3Ce.83
Complexes 1Np, 2Np and 3Np represent rare examples of formally neutral η6-arenes bound to neptunium. To our knowledge, only one such other example is reported in the literature consisting of the complexes LNpIII(Cl) and [(L)NpIII2Cl4(THF)3] where (L = trans-calix[2]benzene[2]pyrrolide).21 In this reported case, the Np–arene interactions were suggested to be key constituents which allow for the formation of an intermediate putative Np2+ complex, as supported by spectroscopic data. The Np η6-arene interactions in 2Np and 3Np (Np–Ccent = 2.579 (2) and 2.561 (4) Å, respectively) are slightly shorter than those of LNpIII(Cl) (Np–Ccent = 2.60 Å) and [(L)NpIII2Cl4(THF)3] (Np–Ccent = 2.63 Å).
UV-vis-NIR spectra were measured on solutions (THF or toluene) of 1M, 2M and 3M. All complexes share broad absorption features in the UV-vis region (Fig. S17–S25†) that extend out to 500–700 nm and are characteristic of charge transfer (CT) activity, and is consistent with those reported for other complexes of the [LAr] ligand platform.46,47,60 It is important to note, as mentioned earlier, the presence of small amounts of H2LAr impurities may have minor contributions to the absorption bands in the UV-vis region of these spectra. The near-IR (NIR) absorption features of 2U and 3U (Fig. 5a) are comparable with minor shifts in peak location and are quite similar to the U3+ complex, LArU(I)(DME), where the bands are consistent with 5f–5f transitions for U3+.46
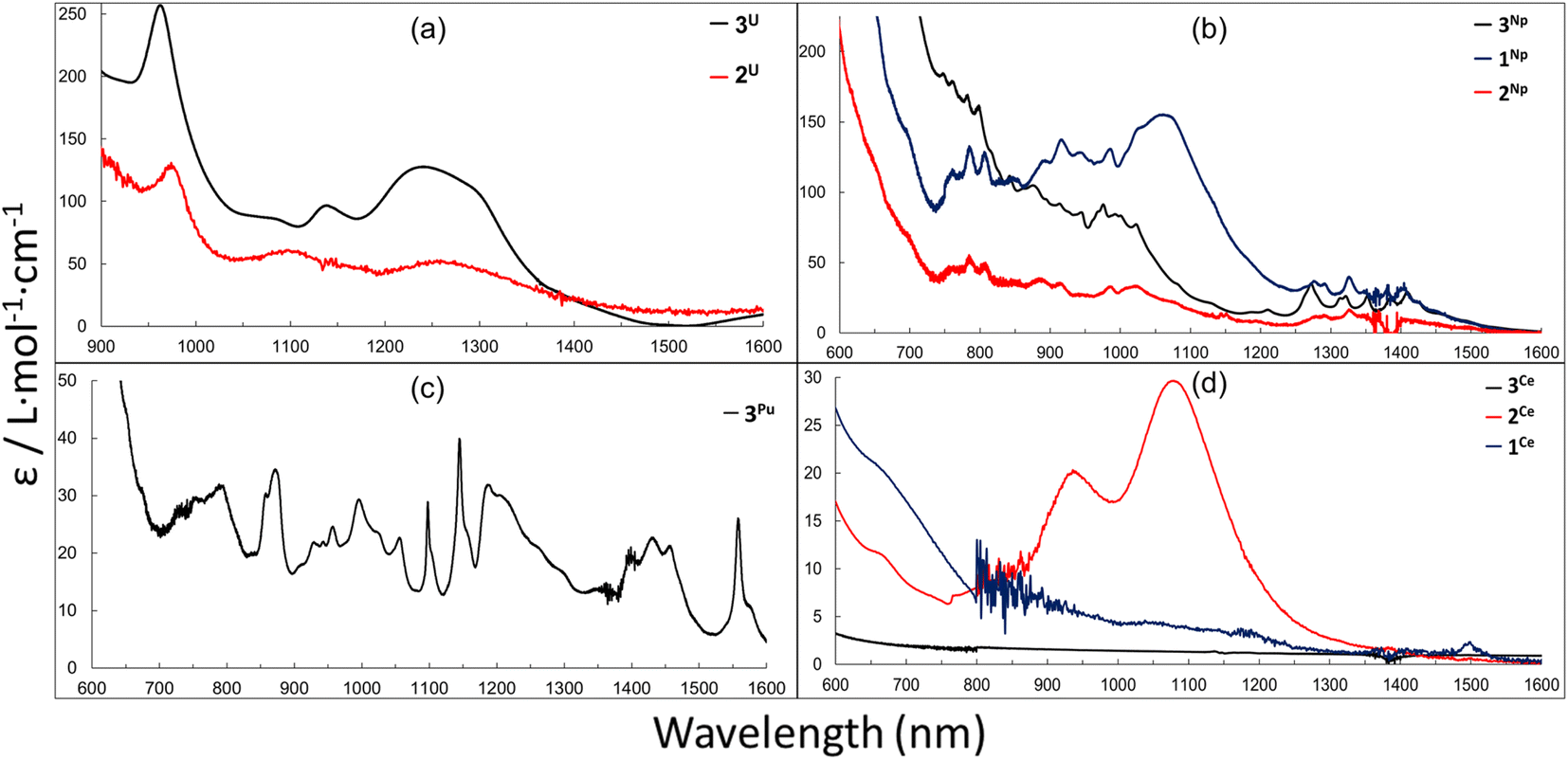 | ||
| Fig. 5 (a): NIR spectra for 2U (4.6 mM in Tol) and 3U (3.0 mM in THF). (b): vis-NIR spectra for the 1Np, 2Np and 3Np series. (0.69 mM in Tol, 0.50 mM in Tol and 2.1 mM in THF respectively). (c): vis-NIR spectra for 3Pu (1.0 mM in THF). (d): vis-NIR spectra of 3Ce, 2Ce and 1Ce (9.51 mM, 5.86 mM and 2.32 mM respectively) in toluene. | ||
Complexes 1Np, 2Np and 3Np display a series of weak absorptions observed in the NIR region from 700–1355 nm, consistent with Laporte–forbidden 5f–5f transitions and typical of many neptunium complexes in the trivalent oxidation state.75,88,89 The three spectra are qualitatively similar, although the broad feature centred around 1069 nm present in 1Np is largely absent in the spectra of 2Np and 3Np (Fig. 5b). The more intense nature of the bands in all three Np spectra in the ∼700–1100 nm region may also be consistent with 5f–6d transitions and/or 5f–6d/5f/5f transitions.
Complex 3Pu contains a number of broad and sharp features with low absorption coefficients (ε/L mol−1 cm−1 > 50) between 550–1600 nm (Fig. 5c), which is consistent with 5f–5f transitions often observed for other Pu3+ complexes.69,75,90,91 The absence of more intense transitions in the ∼700–1100 nm region (in contrast to the Np spectra) could be because the increasing 5f–6d energy gap as the actinide series is traversed means that those transitions shift to higher energy and are either mixed with the charge-transfer region or outside the spectral window. Interestingly, 2Ce displays two weak, broad features in the NIR region centred at 937 and 1080 nm, while the spectra of the neutral complexes 1Ce and 3Ce are silent in the NIR region (Fig. 5d). We have yet to attribute these features and it is possible that they arise from impurities from synthesis or instability in solution.
Conclusions
Through the use of terphenyl bis(anilide) ligand [{K(DME)2}2LAr], (LAr = {C6H4[(2,6-iPr2C6H3)NC6H4]2}2−), we installed tethered η6-arene interactions onto various trivalent 4f and 5f-block ions to isolate complex types 1M, 2M and 3M. These series include the formation of rare, neptunium η6-arene complexes 1Np, 2Np and 3Np as well as a first structurally documented plutonium η6-arene interaction in 3Pu. Taken together, the structural analysis and the UV-vis-NIR studies are consistent with the M3+ assignments for the metal oxidation state in the complexes with the coordinated η6-arene possessing neutral character.Of particular note, structural analyses of the reported complexes show a preference for shorter M–Carene bonds for the U3+ complex over the Np3+, Pu3+ and Ce3+ complexes, despite U3+ having the larger reported ionic radius. Notwithstanding the longer M–Carene bonds for the Np3+ and Pu3+ complexes relative to U3+, both are still notably shorter than in the Ce3+ congeners. These bond metrics fail to adhere to structural trends predicted by a purely electrostatic model. This possibly indicates enhanced metal–arene orbital overlap in the case of the actinide ions as compared to cerium, which would be expected due to the greater ability of these elements to participate in covalent bonding interactions over their 4f-counterparts. Furthermore, this structural data points to an interesting phenomenon in these complexes that the U–Carene bonds trend shorter than the Np–Carene and Pu–Carene bonds, counter to almost all other homologous series of U, Np and Pu complexes which exhibit shortening of the actinide ligands bond lengths from uranium across to plutonium within the trivalent oxidation state.
Complexes 1M, 2M and 3M represent an underexplored area of f-block chemical research, in which closely related complexes spanning the lanthanides and actinides can be evaluated for structural and electronic trends. Especially pertinent is the presence of the η6-arene interactions, which serve as an unusual model to probe the nature of bonding among the f-block and expose any underlying periodicity. We anticipate potential for further reactivity studies on the reported complexes, including redox examinations to assess the ability for these complexes to support high and low-valent metal species, along with electronic structure analysis.
Author contributions
J. M. led and performed lanthanide and actinide synthetic experimental work, compound characterization data collection, and was principally responsible for manuscript drafting. C. A. P. G. and L. S. assisted with transuranium experimental work. S. F. and A. J. G. were the project principal investigators. S. F. conceptualized the synthesis of reported compounds and assisted in experimental work. A. J. G. supervised development of transuranium synthetic strategies and experimental work. B. L. S. supervised the single crystal X-ray data collection and assisted in structure refinements. All authors contributed to the manuscript writing, editing, and review process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Transuranium work was conducted at Los Alamos National Laboratory (LANL) for which A. J. G., J. M. and B. L. S. acknowledge the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, Heavy Element Chemistry Program at LANL (DE-AC52-06NA25396). J. M. also thanks the LANL G. T. Seaborg Institute for Graduate Summer Student and Postdoctoral Fellowships, provided the Laboratory Directed Research and Development (LDRD) programs. C. A. P. G. was supported by a J. R. Oppenheimer Distinguished Postdoctoral Fellowship under LANL-LDRD funding (20180703PRD1). S. F. is grateful to the Welch Foundation (AH-1922-20200401) and the UTEP NSF-PREM program (DMR-1827745) for financial support of this work. Partial crystallographic support was made possible through the NSF-MRI program (S. F.; CHE-1827875).Notes and references
- G. T. Seaborg, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1994, vol. 18, pp. 1–27 Search PubMed.
- D. Freedman, J. H. Melman, T. J. Emge and J. G. Brennan, Inorg. Chem., 1998, 37, 4162–4163 CrossRef CAS.
- J. L. Krinsky, S. G. Minasian and J. Arnold, Inorg. Chem., 2011, 50, 345–357 CrossRef CAS PubMed.
- C. J. Burns and B. E. Bursten, Comments Inorg. Chem., 1989, 9, 61–93 CrossRef CAS.
- D. L. Clark, J. C. Gordon, P. J. Hay and R. Poli, Organometallics, 2005, 24, 5747–5758 CrossRef CAS.
- G. R. Choppin, J. Alloys Compd., 2002, 344, 55–59 CrossRef CAS.
- T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- A. Formanuik, A.-M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. L. McInnes and D. P. Mills, Nat. Chem., 2017, 9, 578–583 CrossRef CAS PubMed.
- M. W. Löble, J. M. Keith, A. B. Altman, S. C. E. Stieber, E. R. Batista, K. S. Boland, S. D. Conradson, D. L. Clark, J. Lezama Pacheco, S. A. Kozimor, R. L. Martin, S. G. Minasian, A. C. Olson, B. L. Scott, D. K. Shuh, T. Tyliszczak, M. P. Wilkerson and R. A. Zehnder, J. Am. Chem. Soc., 2015, 137, 2506–2523 CrossRef PubMed.
- N. L. Eatough and H. T. Hall, Inorg. Chem., 1970, 9, 417–418 CrossRef CAS.
- S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125–12136 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, D. E. Schwarz, D. K. Shuh, G. L. Wagner, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2012, 134, 5586–5597 CrossRef CAS.
- R. M. Diamond, K. Street and G. T. Seaborg, J. Am. Chem. Soc., 1954, 76, 1461–1469 CrossRef CAS.
- E. Lu, S. Sajjad, V. E. J. Berryman, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Nat. Commun., 2019, 10, 634 CrossRef CAS PubMed.
- T. Vitova, I. Pidchenko, D. Fellhauer, P. S. Bagus, Y. Joly, T. Pruessmann, S. Bahl, E. Gonzalez-Robles, J. Rothe, M. Altmaier, M. A. Denecke and H. Geckeis, Nat. Commun., 2017, 8, 16053 CrossRef CAS PubMed.
- A. Kerridge, Chem. Commun., 2017, 53, 6685–6695 RSC.
- A. J. Gaunt, S. D. Reilly, A. E. Enriquez, B. L. Scott, J. A. Ibers, P. Sekar, K. I. M. Ingram, N. Kaltsoyannis and M. P. Neu, Inorg. Chem., 2008, 47, 29–41 CrossRef CAS.
- J. T. Brewster II, D. N. Mangel, A. J. Gaunt, D. P. Saunders, H. Zafar, V. M. Lynch, M. A. Boreen, M. E. Garner, C. A. P. Goodwin, N. S. Settineri, J. Arnold and J. L. Sessler, J. Am. Chem. Soc., 2019, 141, 17867–17874 CrossRef PubMed.
- J. Su, T. Cheisson, A. McSkimming, C. A. P. Goodwin, I. M. DiMucci, T. Albrecht-Schönzart, B. L. Scott, E. R. Batista, A. J. Gaunt, S. A. Kozimor, P. Yang and E. J. Schelter, Chem. Sci., 2021, 12, 13343–13359 RSC.
- M. S. Dutkiewicz, J. H. Farnaby, C. Apostolidis, E. Colineau, O. Walter, N. Magnani, M. G. Gardiner, J. B. Love, N. Kaltsoyannis, R. Caciuffo and P. L. Arnold, Nat. Chem., 2016, 8, 797–802 CrossRef CAS PubMed.
- J. J. Shephard, V. E. J. Berryman, T. Ochiai, O. Walter, A. N. Price, M. R. Warren, P. L. Arnold, N. Kaltsoyannis and S. Parsons, Nat. Commun., 2022, 13, 5923 CrossRef CAS PubMed.
- M. S. Dutkiewicz, C. A. P. Goodwin, M. Perfetti, A. J. Gaunt, J.-C. Griveau, E. Colineau, A. Kovács, A. J. Wooles, R. Caciuffo, O. Walter and S. T. Liddle, Nat. Chem., 2022, 14, 342–349 CrossRef CAS PubMed.
- W. A. King, T. J. Marks, D. M. Anderson, D. J. Duncalf and F. G. N. Cloke, J. Am. Chem. Soc., 1992, 114, 9221–9223 CrossRef CAS.
- M. N. Bochkarev, Chem. Rev., 2002, 102, 2089–2118 CrossRef CAS PubMed.
- O. Walter, Chem.–Eur. J., 2019, 25, 2927–2934 CrossRef CAS PubMed.
- N. Kaltsoyannis, Chem.–Eur. J., 2018, 24, 2815–2825 CrossRef CAS PubMed.
- J. Murillo, R. Bhowmick, K. L. M. Harriman, A. Gomez-Torres, J. Wright, R. W. Meulenberg, P. Miró, A. Metta-Magaña, M. Murugesu, B. Vlaisavljevich and S. Fortier, Chem. Sci., 2021, 12, 13360–13372 RSC.
- J. Murillo, R. Bhowmick, K. L. M. Harriman, A. Gomez-Torres, J. Wright, P. Miró, A. Metta-Magaña, M. Murugesu, B. Vlaisavljevich and S. Fortier, Chem. Commun., 2022, 58, 9112–9115 RSC.
- F. Y. T. Lam, J. A. L. Wells, T. Ochiai, C. J. V. Halliday, K. N. McCabe, L. Maron and P. L. Arnold, Inorg. Chem., 2022, 61, 4581–4591 CrossRef CAS PubMed.
- P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2004, 126, 14533–14547 CrossRef CAS PubMed.
- M. P. Kelley, I. A. Popov, J. Jung, E. R. Batista and P. Yang, Nat. Commun., 2020, 11, 1558 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC.
- H. S. La Pierre, A. Scheurer, F. W. Heinemann, W. Hieringer and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7158–7162 CrossRef CAS.
- S. M. Franke, B. L. Tran, F. W. Heinemann, W. Hieringer, D. J. Mindiola and K. Meyer, Inorg. Chem., 2013, 52, 10552–10558 CrossRef CAS.
- I. A. Popov, B. S. Billow, S. H. Carpenter, E. R. Batista, J. M. Boncella, A. M. Tondreau and P. Yang, Chem.–Eur. J., 2022, 28, e202200114 CrossRef CAS PubMed.
- D. Seyferth, Organometallics, 2002, 21, 2800–2820 CrossRef CAS.
- S. C. Bart, F. W. Heinemann, C. Anthon, C. Hauser and K. Meyer, Inorg. Chem., 2009, 48, 9419–9426 CrossRef CAS.
- D. P. Halter, H. S. La Pierre, F. W. Heinemann and K. Meyer, Inorg. Chem., 2014, 53, 8418–8424 CrossRef CAS PubMed.
- H. S. La Pierre, H. Kameo, D. P. Halter, F. W. Heinemann and K. Meyer, Angew. Chem., Int. Ed., 2014, 53, 7154–7157 CrossRef CAS PubMed.
- B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. McCracken, M. P. Shores, J. M. Boncella and A. L. Odom, J. Am. Chem. Soc., 2018, 140, 17369–17373 CrossRef CAS PubMed.
- P. L. Arnold, J. H. Farnaby, R. C. White, N. Kaltsoyannis, M. G. Gardiner and J. B. Love, Chem. Sci., 2014, 5, 756–765 RSC.
- P. L. Arnold, J. H. Farnaby, M. G. Gardiner and J. B. Love, Organometallics, 2015, 34, 2114–2117 CrossRef CAS.
- M. Suvova, K. T. P. O'Brien, J. H. Farnaby, J. B. Love, N. Kaltsoyannis and P. L. Arnold, Organometallics, 2017, 36, 4669–4681 CrossRef CAS.
- S. Fortier, J. R. Aguilar-Calderon, B. Vlaisavljevich, A. J. Metta-Magaña, A. G. Goos and C. E. Botez, Organometallics, 2017, 36, 4591–4599 CrossRef CAS.
- M. Yadav, A. J. Metta-Magaña and S. Fortier, Chem. Sci., 2020, 11, 2381–2387 RSC.
- C. J. Inman, A. S. P. Frey, A. F. R. Kilpatrick, F. G. N. Cloke and S. M. Roe, Organometallics, 2017, 36, 4539–4545 CrossRef CAS.
- M. D. Straub, E. T. Ouellette, M. A. Boreen, R. D. Britt, K. Chakarawet, I. Douair, C. A. Gould, L. Maron, I. Del Rosal, D. Villarreal, S. G. Minasian and J. Arnold, J. Am. Chem. Soc., 2021, 143, 19748–19760 CrossRef CAS.
- J. Murillo and S. Fortier, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2018, pp. 1–19, Search PubMed.
- M. Cesari, U. Pedretti, Z. Zazzetta, g. Lugli and W. Marconi, Inorg. Chim. Acta, 1971, 5, 439–444 CrossRef CAS.
- F. A. Cotton and W. Schwotzer, Organometallics, 1985, 4, 942–943 CrossRef CAS.
- F. A. Cotton and W. Schwotzer, Organometallics, 1987, 6, 1275–1280 CrossRef CAS.
- G. C. Campbell, F. A. Cotton, J. F. Haw and W. Schwotzer, Organometallics, 1986, 5, 274–279 CrossRef CAS.
- F. A. Cotton, W. Schwotzer and C. Q. Simpson II, Angew. Chem., Int. Ed. Engl., 1986, 25, 637–639 CrossRef.
- D. Baudry, E. Bulot, P. Charpin, M. Ephritikhine, M. Lance, M. Nierlich and J. Vigner, J. Organomet. Chem., 1989, 371, 155–162 CrossRef CAS.
- V. Paprocki, P. Hrobárik, K. L. M. Harriman, M. S. Luff, T. Kupfer, M. Kaupp, M. Murugesu and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 13109–13115 CrossRef CAS PubMed.
- D. P. Halter, F. W. Heinemann, L. Maron and K. Meyer, Nat. Chem., 2018, 10, 259–267 CrossRef CAS PubMed.
- D. P. Halter, F. W. Heinemann, J. Bachmann and K. Meyer, Nature, 2016, 530, 317–321 CrossRef CAS PubMed.
- K. L. M. Harriman, J. Murillo, E. A. Suturina, S. Fortier and M. Murugesu, Inorg. Chem. Front., 2020, 7, 4805–4812 RSC.
- S. D. Daniel, J.-S. M. Lehn, J. D. Korp and D. M. Hoffman, Polyhedron, 2006, 25, 205–210 CrossRef CAS.
- X. Xin and C. Zhu, Dalton Trans., 2020, 49, 603–607 RSC.
- E. M. Broderick, P. S. Thuy-Boun, N. Guo, C. S. Vogel, J. Sutter, J. T. Miller, K. Meyer and P. L. Diaconescu, Inorg. Chem., 2011, 50, 2870–2877 CrossRef CAS PubMed.
- S. M. Mansell, B. F. Perandones and P. L. Arnold, J. Organomet. Chem., 2010, 695, 2814–2821 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- S. G. Kukolich, J. Am. Chem. Soc., 1995, 117, 5512–5514 CrossRef CAS.
- S. M. Hubig, S. V. Lindeman and J. K. Kochi, Coord. Chem. Rev., 2000, 200–202, 831–873 CrossRef CAS.
- P. Le Maguères, S. V. Lindeman and J. K. Kochi, Organometallics, 2001, 20, 115–125 CrossRef.
- C. A. P. Goodwin, M. T. Janicke, B. L. Scott and A. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 20680–20696 CrossRef CAS.
- K. Izod, S. T. Liddle and W. Clegg, Inorg. Chem., 2004, 43, 214–218 CrossRef CAS PubMed.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- C. Tamain, M. Autillo, D. Guillaumont, L. Guérin, R. E. Wilson and C. Berthon, Inorg. Chem., 2022, 61, 12337–12348 CrossRef CAS.
- M. Autillo, R. E. Wilson, M. Vasiliu, G. F. de Melo and D. A. Dixon, Inorg. Chem., 2022, 61, 15607–15618 CrossRef CAS.
- T. Radoske, J. März, M. Patzschke, P. Kaden, O. Walter, M. Schmidt and T. Stumpf, Chem. – Eur. J., 2020, 26, 16853–16859 CrossRef CAS PubMed.
- C. A. P. Goodwin, S. R. Ciccone, S. Bekoe, S. Majumdar, B. L. Scott, J. W. Ziller, A. J. Gaunt, F. Furche and W. J. Evans, Chem. Commun., 2022, 58, 997–1000 RSC.
- R. Kloditz, S. Fichter, S. Kaufmann, T. S. Brunner, P. Kaden, M. Patzschke, T. Stumpf, P. W. Roesky, M. Schmidt and J. März, Inorg. Chem., 2020, 59, 15670–15680 CrossRef CAS PubMed.
- D. D. Schnaars, A. J. Gaunt, T. W. Hayton, M. B. Jones, I. Kirker, N. Kaltsoyannis, I. May, S. D. Reilly, B. L. Scott and G. Wu, Inorg. Chem., 2012, 51, 8557–8566 CrossRef CAS PubMed.
- D. Pividori, M. E. Miehlich, B. Kestel, F. W. Heinemann, A. Scheurer, M. Patzschke and K. Meyer, Inorg. Chem., 2021, 60, 16455–16465 CrossRef CAS PubMed.
- W. G. Van der Sluys, C. J. Burns, J. C. Huffman and A. P. Sattelberger, J. Am. Chem. Soc., 1988, 110, 5924–5925 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, W. R. Hillman and J. W. Ziller, Organometallics, 2005, 24, 4676–4683 CrossRef CAS.
- P. L. Arnold, C. J. Stevens, J. H. Farnaby, M. G. Gardiner, G. S. Nichol and J. B. Love, J. Am. Chem. Soc., 2014, 136, 10218–10221 CrossRef CAS.
- M. Gorlov, L. L. Hussami, A. Fischer and L. Kloo, Eur. J. Inorg. Chem., 2008, 2008, 5191–5195 CrossRef.
- R. P. Kelly, L. Maron, R. Scopelliti and M. Mazzanti, Angew. Chem., Int. Ed., 2017, 56, 15663–15666 CrossRef CAS PubMed.
- H. Yin, A. J. Lewis, P. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 8234–8243 CrossRef CAS PubMed.
- G. B. Deacon, T. Feng, C. M. Forsyth, A. Gitlits, D. C. R. Hockless, Q. Shen, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 2000, 961–966 RSC.
- A. N. Selikhov, A. V. Cherkasov, K. A. Lyssenko and A. A. Trifonov, Organometallics, 2022, 41, 820–828 CrossRef CAS.
- M. E. Hossain, Z. Guo, J. Wang, G. B. Deacon, P. C. Junk, D. Diether and R. Anwander, Eur. J. Inorg. Chem., 2022, 2022, e202101009 CrossRef CAS.
- S. A. Pattenaude, N. H. Anderson, S. C. Bart, A. J. Gaunt and B. L. Scott, Chem. Commun., 2018, 54, 6113–6116 RSC.
- A. J. Myers, M. L. Tarlton, S. P. Kelley, W. W. Lukens and J. R. Walensky, Angew. Chem., Int. Ed., 2019, 58, 14891–14895 CrossRef CAS PubMed.
- C. J. Windorff, J. M. Sperling, T. E. Albrecht-Schönzart, Z. Bai, W. J. Evans, A. N. Gaiser, A. J. Gaunt, C. A. P. Goodwin, D. E. Hobart, Z. K. Huffman, D. N. Huh, B. E. Klamm, T. N. Poe and E. Warzecha, Inorg. Chem., 2020, 59, 13301–13314 CrossRef CAS.
- L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248–2256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2238465–2238473. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02194g |
| This journal is © The Royal Society of Chemistry 2023 |