
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural and dimensional control of porphyrin capsules using Group 15 tris(3-pyridyl) linkers†
Álvaro
García-Romero
 a,
Daniel
Miguel
a,
Dominic S.
Wright
b,
Celedonio M.
Álvarez
a and
Raúl
García-Rodríguez
*a
a,
Daniel
Miguel
a,
Dominic S.
Wright
b,
Celedonio M.
Álvarez
a and
Raúl
García-Rodríguez
*a
aGIR MIOMeT-IU Cinquima-Química Inorgánica Facultad de Ciencias, Universidad de Valladolid, Campus Miguel Delibes, 47011 Valladolid, Spain. E-mail: raul.garcia.rodriguez@uva.es
bChemistry Department, Cambridge University, Lensfield Road, Cambridge CB2 1EW, UK
First published on 6th June 2023
Abstract
While supramolecular chemistry involving organic and metallo-organic host assemblies is a well-established and important field with applications in gas-storage, drug-delivery and the regio- and stereo-control of organic reactions, the use of main group elements in this setting (beyond the second row of the p-block) has been little explored. In this paper we show how periodic trends in the p-block can provide the means for systematic size and structural control in an important class of supramolecular porphyrin-based capsules. The formation of molecular and extended 2D capsule arrangements between the heavier Group 15 tris(3-pyridyl) linkers Sb(3-py)3 and Bi(3-py)3 and the metallo-porphyrins MTPP (M = Zn, Mg; TPP = tetraphenylporphyrin, 3-py = 3-pyridyl) is the first study involving heavier Group 15 pyridyl linkers. The increase in C–E bond length in the E(3-py)3 linkers moving down Group 15 (from E = P, to Sb, to Bi) can be used to alter the dimensions and structural preference of the capsules, as can oxidation of the Group 15 bridgehead atoms themselves. The subtle changes in the dimensions and Lewis acidity of the encapsulates have a dramatic effect on the rate and selectivity of the catalytic oxidative cleavage of organic diols and catalytic oxidation of α-hydroxyketones. By providing simple tools for modulating the chemical and steric properties of the capsules this work should have direct applications for the tuning of the activity and specificity of a range of catalytic systems based on main-group-based capsules of this type.
Introduction
Nature represents a perennial source of inspiration for researchers for the design and function of molecules. Enzymes exhibit exquisite selectivity in substrate binding and reactivity through the shape and electronic complementarity of the binding pocket around the active site. To mimic the chemical control and abilities of enzymes, researchers have designed various capsule-like structures with well-defined cavities reminiscent of the enzymatic active site.1–3 These systems offer the possibility to achieve specific encapsulation, binding, or controlled passage of guest molecules.4 As a result, they have attracted strong research interest across multiple research areas, with applications in drug delivery,5,6 sensing and imaging,7,8 gas storage,9–12 molecular separation,13,14 and catalysis.15–18 Confinement within the cavity can lead to enhanced reactivity of the guest or unique reaction selectivity, or conversely, the stabilization of a reactive guest molecule.19–21 In particular, since the 1990s, there has been increasing interest in the study of the application of molecular capsules as nanoreactors for a number of chemical reactions.17,20,22–27 The microenvironment within the capsules (i.e., the shape, size, and chemical properties of the cavity) is key, and thus, full realization of these applications requires very fine structural control over the capsule architecture to tailor the cavity for specific host–guest interactions. Despite the vast number of capsules that have been described over the years and the efforts towards the design and control of these attractive systems,28–32 the use of relatively heavy p-block elements as a means to achieve fine structural control is surprisingly very limited.In this context, capsules based on tris(3-pyridyl)phosphine have emerged as a novel way to obtain encapsulated transition metal catalysts.25–27 The first capsule-forming example {[P(3-py)3]·(ZnTPP)3} (3-py = 3-pyridyl, TPP = tetraphenylporphyrin) was described in 2001,33 and the single-crystal X-ray structure was reported in 2013 by Reek and co-workers (Fig. 1).34 These capsules are of interest not only for the aesthetic aspects of their supramolecular arrangements, but also for their confinement of single-site Pd- and Rh-based catalysts, which results in rate acceleration and the unprecedented selective hydroformylation of terminal and internal alkenes.33,35–40 The binding of substrates to the constrained metal centres in these capsules restricts their possible reaction pathways, leading to greater regioselectivity than that of the unencapsulated catalysts. Studies of capsules based on P(3-py)3 with various substituted porphyrins have demonstrated that small distortions in the capsule shape have a critical impact on selectivity.34,36,37,41
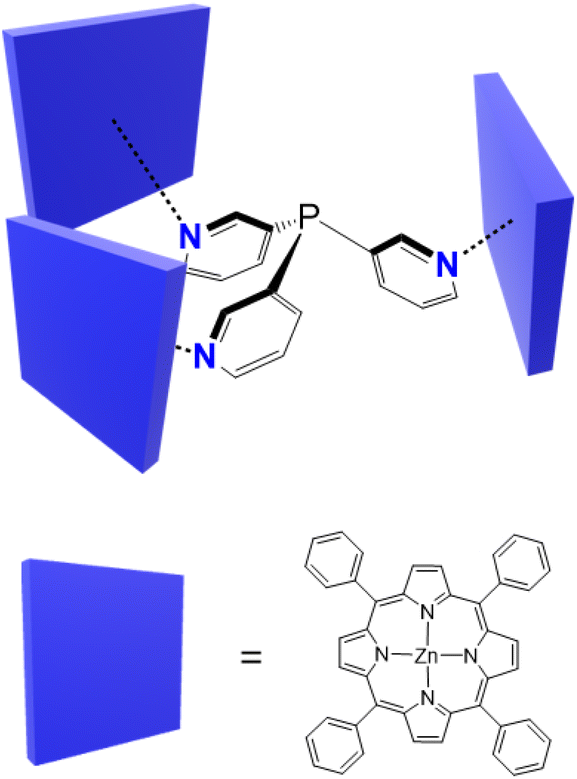 | ||
| Fig. 1 First example of a {[P(3-py)3]·(ZnTPP)3} assembly. | ||
In addition to varying the nature of and substituents present on the porphyrins, another potential way of modifying the dimensions and coordination characteristics of the capsules is changing the bridgehead atom present in the tris(3-pyridyl) linker unit. We have very recently developed synthetic approaches to metallic or semi-metallic tris-pyridyl ligands,42–47 including the heavier Group 15 ligands E(3-py)3 [E = Sb (1), Bi (2)] (Fig. 2a),48 and shown that the latter can be employed in the construction of a series of hybrid MOF arrangements with Cu+ and Ag+ (=M+) salts, in which the ligands adopt a tetradentate N,N,N,E-bonding mode.48 As shown in Fig. 2b, these MOFs consist of E4M4 cubane units in which the anions are encapsulated.
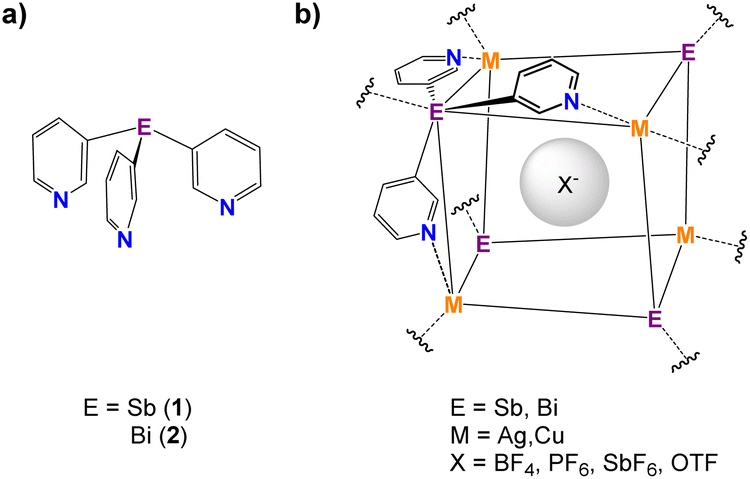 | ||
| Fig. 2 (a) Antimony- and bismuth-based tris(3-pyridyl) ligands. (b) The E4M4 unit in the MOFs formed by coordination of 1 and 2 with Cu+ and Ag+ salts. | ||
Having illustrated that the new heavy Group 15 ligands can be employed as linkers in supramolecular assemblies, we turn our attention here to their use in capsule formation with metalloporphyrins. The periodic effects of changing the bridgehead atom on the E–C bond length and C–E–C bond angle,49 we reasoned, should have a direct impact on the microenvironment of capsules of this type and provide a valuable toolkit for tailoring their cavities for specific host–guest and catalysis chemistry. In the current study, we show how the bridgehead atom can be used to modulate the properties of these capsules and control their size and shape.
Results and discussion
Effect of the bridgehead atom
To evaluate whether capsule formation was possible using 1 and 2, we first explored their coordination with zinc tetraphenylporphyrin (ZnTPP). The addition of ZnTPP to a solution of ligands 1 or 2 in CDCl3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) resulted in a large upfield shift of the 1H NMR signals of the pyridyl ring due to the porphyrin ring-current effect, which was indicative of axial binding of the pyridine nitrogen to the Zn(II) metalloporphyrin. Slow diffusion of n-hexane into a CHCl3 solution of ligands 1 or 2 (1 equiv.) and ZnTPP (3 equiv.) over a week gave dark purple hexagonal crystals of {[E(3-py)3]·(ZnTPP)3} (E = Sb (1·Zn), Bi (2·Zn)) in 62 and 58% yields, respectively (Scheme 1). The room-temperature 1H NMR spectra of 1·Zn and 2·Zn in CDCl3 indicated a 1:3 ratio of 1 or 2 to ZnTPP. Further confirmation of the formation of 1:3 complexes was provided by ESI-TOF high-resolution mass-spectrometry, which showed the expected [M + H]+ peak at m/z 2390.4940 (calcd 2390.4957) for 1·Zn and 2478.5664 (calcd 2478.5719) for 2·Zn (see ESI†).
:1) resulted in a large upfield shift of the 1H NMR signals of the pyridyl ring due to the porphyrin ring-current effect, which was indicative of axial binding of the pyridine nitrogen to the Zn(II) metalloporphyrin. Slow diffusion of n-hexane into a CHCl3 solution of ligands 1 or 2 (1 equiv.) and ZnTPP (3 equiv.) over a week gave dark purple hexagonal crystals of {[E(3-py)3]·(ZnTPP)3} (E = Sb (1·Zn), Bi (2·Zn)) in 62 and 58% yields, respectively (Scheme 1). The room-temperature 1H NMR spectra of 1·Zn and 2·Zn in CDCl3 indicated a 1:3 ratio of 1 or 2 to ZnTPP. Further confirmation of the formation of 1:3 complexes was provided by ESI-TOF high-resolution mass-spectrometry, which showed the expected [M + H]+ peak at m/z 2390.4940 (calcd 2390.4957) for 1·Zn and 2478.5664 (calcd 2478.5719) for 2·Zn (see ESI†).
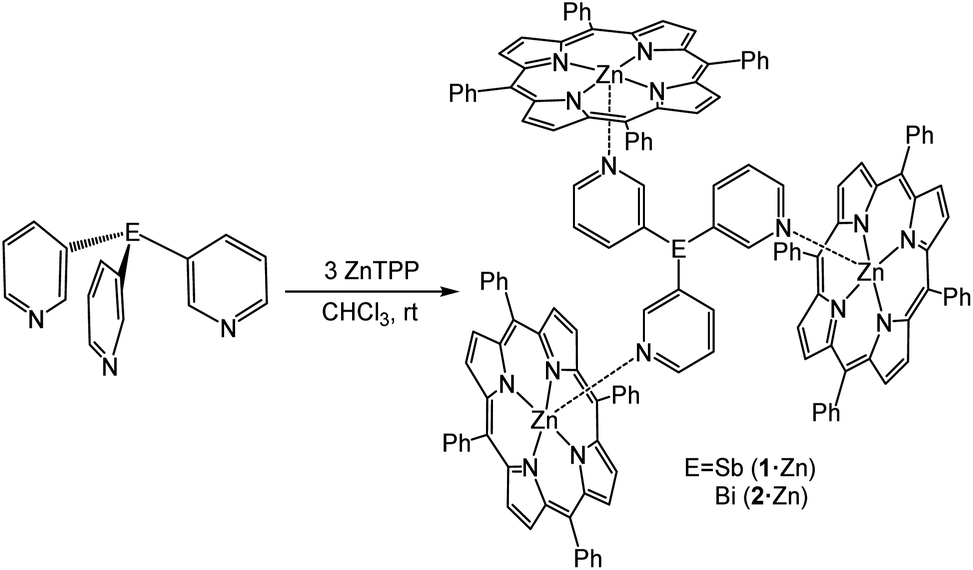 | ||
| Scheme 1 Synthesis of the capsules {[Sb(3-py)3]·(ZnTPP)3} (1·Zn) and {[Bi(3-py)3]·(ZnTPP)3} (2·Zn). | ||
Crystallographic analysis of 1·Zn and 2·Zn confirms the formation of capsules for both, but reveals that bulk samples contain mixtures of crystals in which two different conformations of the capsules are present. In crystals of the ‘closed’ (or ‘in’) conformation the porphyrin rings are approximately perpendicular to each other, forming a roughly cylindrical arrangement, while in the ‘open’ conformation one of the porphyrin units is displaced downwards with respect to the other two (see Fig. 3 and 6 and later discussion). These two conformations therefore allow different accessibility to the cavities. In previous structural studies of {[P(3-py)3]·(Zn-porphyrin)3} systems, only the closed conformation has been identified.34,40 In theory the rotation of all three pyridyl rings about the E–Cpy bonds could lead to a conformation in which the three porphyrins are pointed down, in which E is de-encapsulated (the “out” conformer, Fig. 3). However, this possibility can be discarded in the case of ZnTPP units due to the steric hindrance between the three porphyrin rings, although it is possible for smaller building blocks such as Zn(II) salphens.38 The open conformer that we have characterised here is the first reported observation of this form.
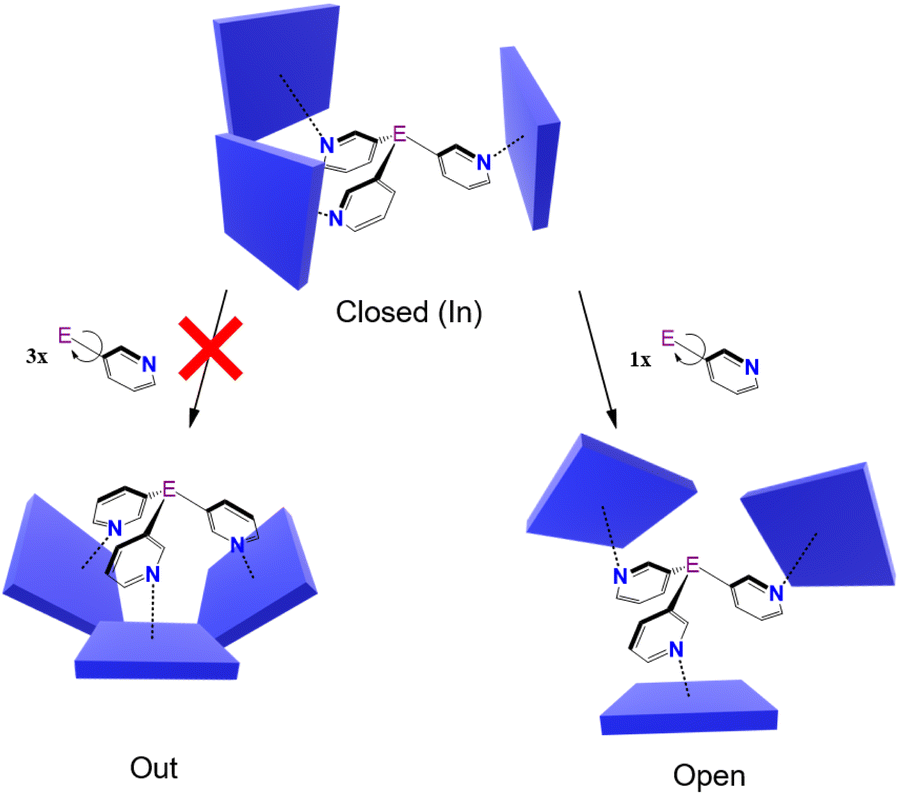 | ||
| Fig. 3 Possible conformations of capsules of the type [(E(3-py)3)·(ZnTPP)3]. | ||
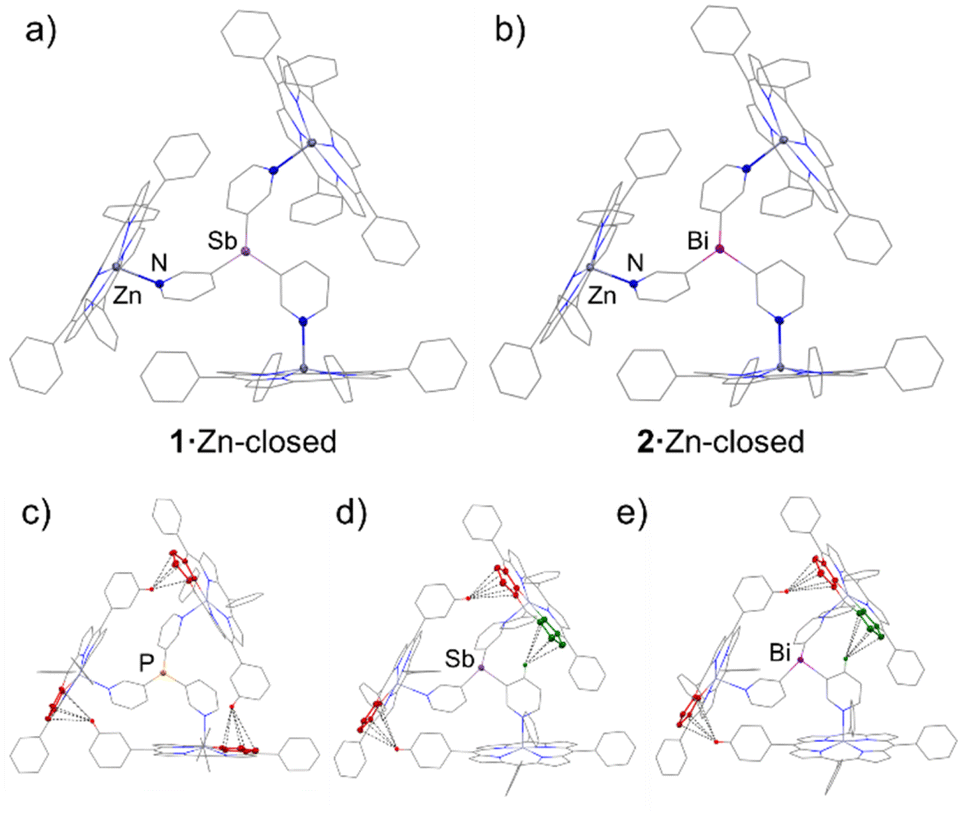 | ||
| Fig. 4 X-ray structures of the complexes 1·Zn-closed (a) and 2·Zn-closed (b). In both structures, the ligand coordinates three porphyrins through N–Zn interactions. The ligands are fenced by the three porphyrins, leading to the formation of capsule-type structures. Solvent molecules and H-atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): 1·Zn, Sb–Cpy range 2.146(5)–2.158(5); Npy–Zn range 2.149(4)–2.194(4); Cpy–Sb–Cpy range 95.4(2)–96.0(2). 2·Zn, Bi–Cpy range 2.235(6)–2.261(5); Npy–Zn range 2.149(5)–2.184(5); Cpy–Bi–Cpy range 93.0(2)–94.2(2). Colour key: C (grey), Zn (dark grey), N (blue), P (orange), Sb (light purple), Bi (purple). X-ray structures of {[P(3-py)3]·(ZnTPP)3} (c), 1·Zn (d) and 2·Zn (e) showing the capsules with close intramolecular CH–π interactions between ZnTPP building blocks highlighted in red and interactions between C–H pyridyl and ZnTPP highlighted in green. CH–π interaction lengths (Å): {[P(3-py)3]·(ZnTPP)3} 2.725, 2.893 and 2.960;341·Zn 2.971, 3.051 and 2.978 (CHpy–π); 2·Zn 2.991, 3.025 and 2.978 (CHpy–π). | ||
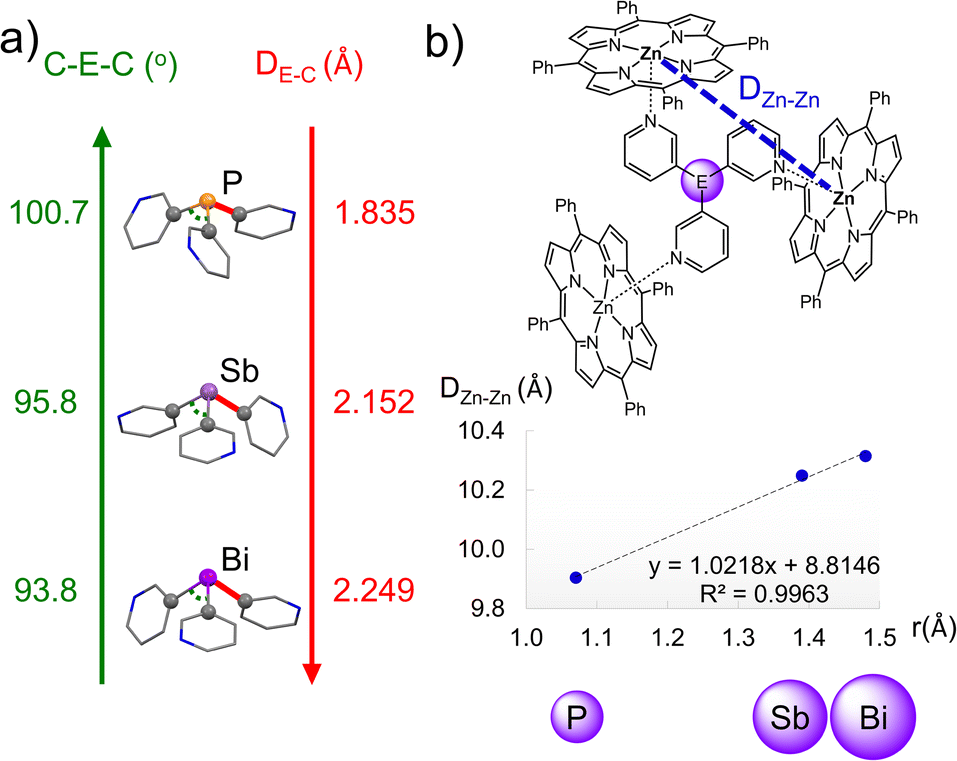 | ||
| Fig. 5 (a) Trends in the average C–E–C angles (green) and E–C distances (red) in the X-ray structures of the capsules (E = P, Sb and Bi). Colour key: C (grey), N (blue), P (orange), Sb (light purple), Bi (purple). (b) Linear correlation between the covalent radii of the bridgehead atom and the effective size of the capsule (DZn–Zn) in the X-ray structures of {[P(3-py)3]·(ZnTPP)3}, 1·Zn-closed and 2·Zn-closed. | ||
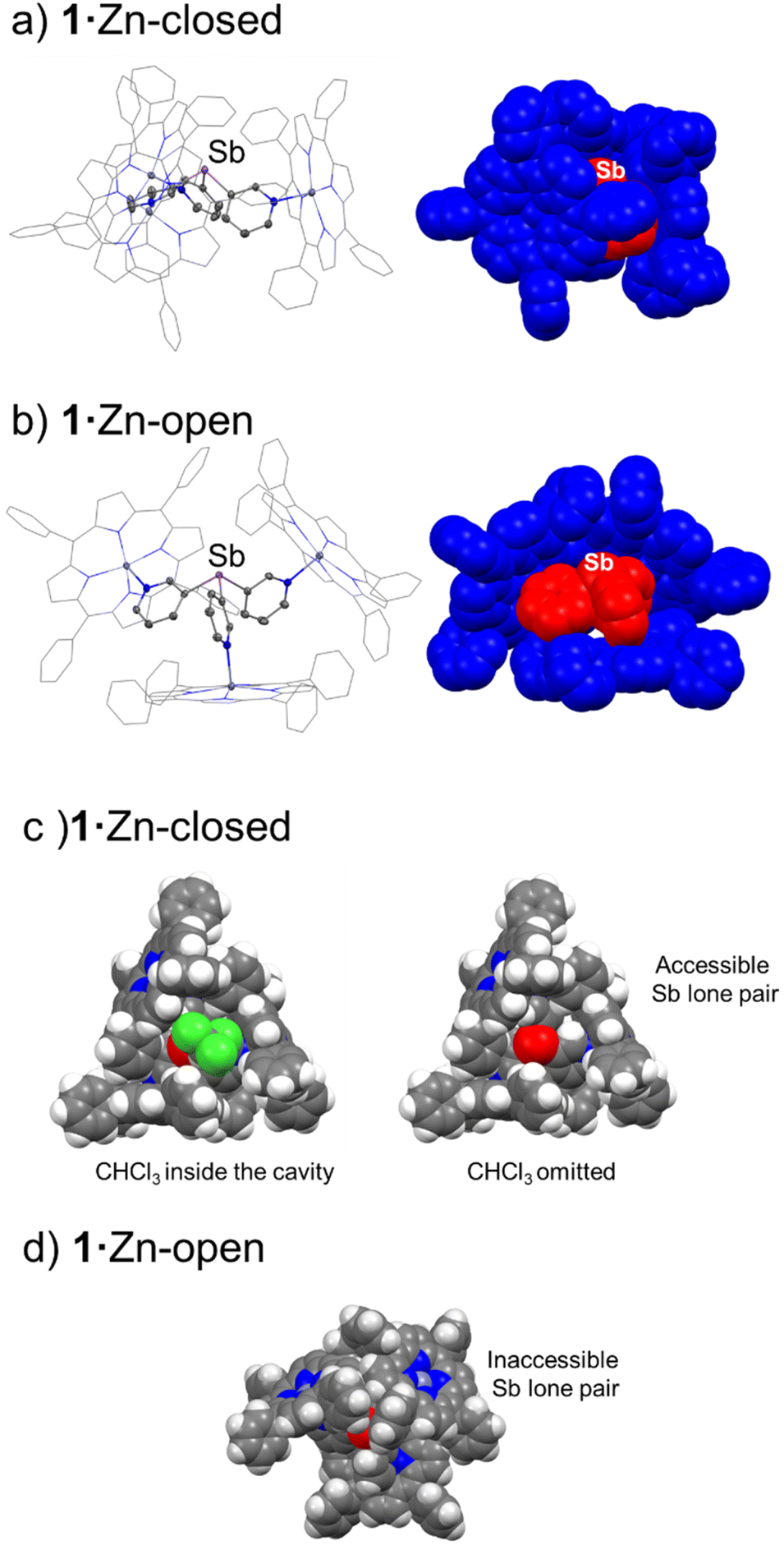 | ||
| Fig. 6 Solid-state structures of the closed-capsule conformer (a) and open-capsule conformer (b) of 1·Zn. In the space-filling views of the X-ray structures (a and b, right), ligand 1 is indicated in red, and the three ZnTPP moieties in blue. Displacement ellipsoids at 50% probability. H-atoms are omitted for clarity. Colour key: C (grey), Zn (dark grey), N (blue), Sb (light purple). (c) Left: space-filling top view of the 1·Zn-closed conformer, showing one molecule of CHCl3 present in the cavity; right: the same view with the molecule of CHCl3 omitted, illustrating the accessibility of the antimony lone pair. (d) 1·Zn-open with the bridgehead Sb atom highlighted in red. Colour key: H (white), C (grey), N (blue), Sb (red), Cl (green). | ||
1·Zn-closed and 2·Zn-closed are isostructural in the solid state. The ligands 1 and 2 link the porphyrin-bound Zn2+ centres via their three 3-Py N-donor atoms, leading to bimetallic capsule arrangements (Fig. 4a and b). These arrangements are very similar to that found for [(P(3-py)3)·(ZnTPP)3] by Reek, in which the ligand is laterally flanked by the three porphyrins.34 There are only minor geometric differences in the conformation of the porphyrin units due to the different geometric profiles of the heavier Group 15 ligands; one of the porphyrin arms is slightly tilted in 1·Zn-closed and 2·Zn-closed compared to {[P(3-py)3]·(ZnTPP)3}. Despite their overall similarity, changing the bridging ligand has a direct impact on the size of the capsules. The reduction in the average C–E–C angle upon descending Group 15 (100.7, 95.8 and 93.8° for P, Sb and Bi, respectively) is offset by the increase in average E–C bond length (1.835, 2.152 and 2.249 Å for P, Sb and Bi, respectively) (Fig. 5a), but results overall in an increase in the size of the capsules from P to Sb to Bi. The average Zn⋯Zn distances in {[P(3-py)3]·(ZnTPP)3}, 1·Zn and 2·Zn (9.905, 10.249 and 10.315 Å, respectively) correlate directly with the covalent radii of the Group 15 elements50 of the bridgehead atom in the E(3-py)3 ligands, which illustrates the capability of ligands 1 and 2 to modulate the cavity size (Fig. 5b – see also the discussion in the ESI†) of the “effective size” of the ligands within the capsules and the calculated volumes of the capsules.
The importance of attractive CH⋯π interactions between adjacent ZnTPP building blocks in {[P(3-py)3]·(ZnTPP)3} (Fig. 4c, red) has been noted previously.26,34 These interactions help to preserve the integrity and rigidity of the capsule, which are critical to their catalytic selectivity and activity since these will favour or disfavour alternative transition states.34Fig. 4d and e show the three shortest CH⋯π distances found in 1·Zn-closed (range 2.971–3.051 Å) and 2·Zn-closed (2.978–3.025 Å). Unlike in {[P(3-py)3]·(ZnTPP)3}, only two of these involve adjacent ZnTPP fragments (Fig. 4d and e, red), while the third CH⋯π interaction involves a CH group of the pyridyl ring and a ZnTPP unit (Fig. 4d and e, green). It is clear that due to the larger size of the capsules for 1·Zn and 2·Zn, these CH⋯π interactions are lost or become weaker as compared with {[P(3-py)3]·(ZnTPP)3} (range 2.725–2.960 Å), which could lead to more flexible capsules.
Fig. 6 compares the structures of the open and closed conformers of 1·Zn. A similar open conformer was also found for the bismuth capsule 2·Zn (see ESI Fig. S58†). As a result of the different arrangement of the ZnTPP units in the closed conformers, the lone pair of the bridgehead E atom is accessible for coordination (despite its poor donor properties, particularly for Sb) while in the open conformation, the orientation of two of the ZnTPP units blocks the lone pairs on the Sb and Bi centre (Fig. 6c right and Fig. 6d). The importance of the supramolecular conformation in this system is illustrated by the top-view of the space-filling representation of the structure of 1·Zn-closed, in which a CHCl3 molecule (from crystallization) is located in the cavity of the capsule (Fig. 6c, left). The long Sb⋯Cl contact to the CHCl3 molecule [3.752(2) cf. ΣVDW (Sb−Cl) = 3.81]51 reflects the Lewis acidity of the Sb(III) and contributes to the stabilization of the CHCl3 within the cavity, illustrating the effects of the heavy pnictogen atom in host–guest interactions. In contrast, in the top view of a molecule of 1·Zn-open, the lone pair of the antimony atom is completely blocked by the phenyl groups of the two upward-pointing porphyrins, preventing the possibility of solvent encapsulation (Fig. 6d). Additionally, variable-temperature NMR experiments in CDCl3 showed that the building blocks of the capsules 1·Zn and 2·Zn are in fast exchange on the NMR timescale even at −60 °C (similar to previous observations for the {[P(3-py)3]·(ZnTPP)3} capsule).36 Therefore, no specific conclusions regarding the conformational preferences of the capsules in solution could be made (see ESI†).
DFT computational analysis in which free relaxation of the structure in gas phase was allowed using the APFD functional indicated that the open conformation is always the most stable for these capsules. However, its stability with respect to the closed conformer depends on the nature of E, and increases in the order Bi ≈ Sb > P, with the Bi-and Sb-based capsules favouring the open conformer by 50.46 and 50.42 kJ mol−1, respectively, compared to 45.35 kJ mol−1 for P. The greater preference for the open conformers of the Sb- and Bi-based capsules is probably steric in origin, reflecting the larger distance between the porphyrin units at their peripheries.
To obtain further insights into the thermodynamics of the formation of the supramolecular capsules 1·Zn and 2·Zn in solution, NMR titration experiments of the ligands E(3-py)3 [E = Sb(1), Bi(2)] and ZnTPP in CDCl3 were carried out. The titration curves could be fitted to a 1:3 model to give the association constants K1 [(9.2 ± 1.9) × 103 M−1], K2 [(1.6 ± 0.3) × 103 M−1] and K3 [(2.5 ± 0.6) × 102 M−1] for Sb(3-py)3 to ZnTPP. Very similar results were obtained for Bi(3-py)3: K1 [(6.4 ± 1.5) × 103 M−1], K2 [(1.6 ± 0.3) × 103 M−1] and K3 [(3.8 ± 0.9) × 102 M−1] (see ESI† for further information). The titration studies show the expected decrease in binding affinities for systems lacking positive cooperativity. This is in contrast to the tendency of P(3-py)3 in the assembly {[P(3-py)3]·(ZnTPP)3}, for which a positive cooperative effect was observed and was suggested to be the result of CH–π interactions.34,36 The lack of cooperativity in our larger Sb and Bi capsules could be the result of the fewer and weaker CH–π interactions between the porphyrin moieties observed in the solid-state structures of the closed conformers. This may also be affected by the greater preference for the open conformation of the Sb and Bi-based capsules. Additionally, the size of the capsules 1·Zn and 2·Zn in solution was studied through 1H DOSY experiments in CDCl3. These studies yielded hydrodynamic radii of 7.49 and 7.50 Å for 1·Zn and 2·Zn, respectively, which are about 80% of the crystallographic ones, in line with reports in the literature (see ESI†).52,53
Effect of changing the metalloporphyrin unit
Inspired by these previous studies, we next investigated the effect of changing the metalloporphyrin unit on the supramolecular assemblies. No example of a capsule of this type containing a linker other than P(3-Py)3 has been structurally characterized previously, and all previous examples have employed Zn-containing metalloporphyrins. We reasoned that magnesium tetraphenylporphyrin (MgTPP) might be an ideal building block to obtain expanded supramolecular arrangements, since both axial positions at the Mg2+ centre are potentially available for coordination.54–57Slow diffusion of n-hexane into a CHCl3 solution of the ligands P(3-py)3, 1 or 2 (2 equiv.) and (MgTPP) (3 equiv.) over a week gave dark-purple hexagonal crystals of {[E(3-py)3]2(MgTPP)3}n (E = P, Sb (1·Mg), Bi (2·Mg)) in 58%, 56% and 72% yield, respectively (Scheme 2).
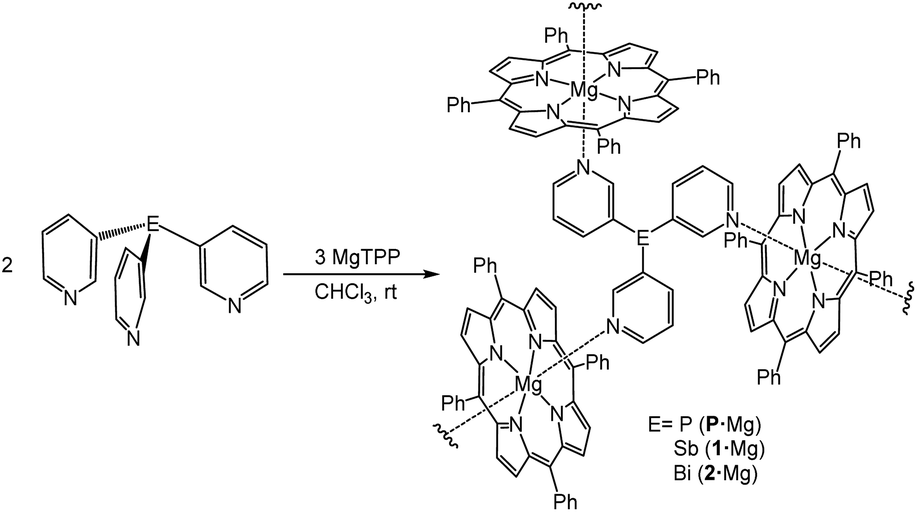 | ||
| Scheme 2 Synthesis of the complexes {[P(3-py)3]2·(MgTPP)3}n (P·Mg), {[Sb(3-py)3]2·(MgTPP)3}n (1·Mg) and {[Bi(3-py)3]2·(MgTPP)3}n (2·Mg). | ||
Complexes P·Mg, 1·Mg and 2·Mg have analogous structures in the solid-state in which the ligand molecules link three Mg2+ centres using the three 3-Py N-donor atoms and each Mg2+ centre is coordinated by two N-donor atoms of different ligand molecules, leading to a 2D polymeric arrangement formed by a sheet network consisting of E6Mg6 (E = P, Sb, Bi) ring units. Fig. 7a and b shows the structure of 1·Mg (see ESI† for the solid-state structures of P·Mg and 2·Mg). Surprisingly, the integrity of the monomeric capsule structure is still preserved in the polymeric arrangement (Fig. 7a, inset), forming a hybrid structure in which the capsules are assembled into an expanded 2D network using both axial positions of the magnesium metalloporphyrin (Fig. 7a). The presence of six-coordinate Mg2+ instead of five-coordinate Zn2+ in the porphyrin core also leads to greater M–N bond lengths of the E(3-py)3 N-atoms (range 2.149(4)–2.194(4) Å for Zn derivatives vs. range 2.324(3)–2.374(2) Å for Mg derivatives). Consequently, there is an increase in the capsule size compared to the corresponding discrete Zn-based capsules, as revealed by the larger M⋯M distances in the capsules. The Mg⋯Mg distances in these larger capsules (10.3184(6), 10.4650(3) and 10.4872(2) Å for P·Mg, 1·Mg and 2·Mg, respectively) again correlate with the covalent radii of the bridgehead atoms of the ligands (see ESI Fig. S65†), as was the case for the discrete Zn-based capsules (see Fig. 5b). These results demonstrate that capsule assemblies can be combined with the ability of the metalloporphyrin to produce extended materials by using this diaxial bonding coordination strategy, and opens a new area of extended capsule structures of this type.
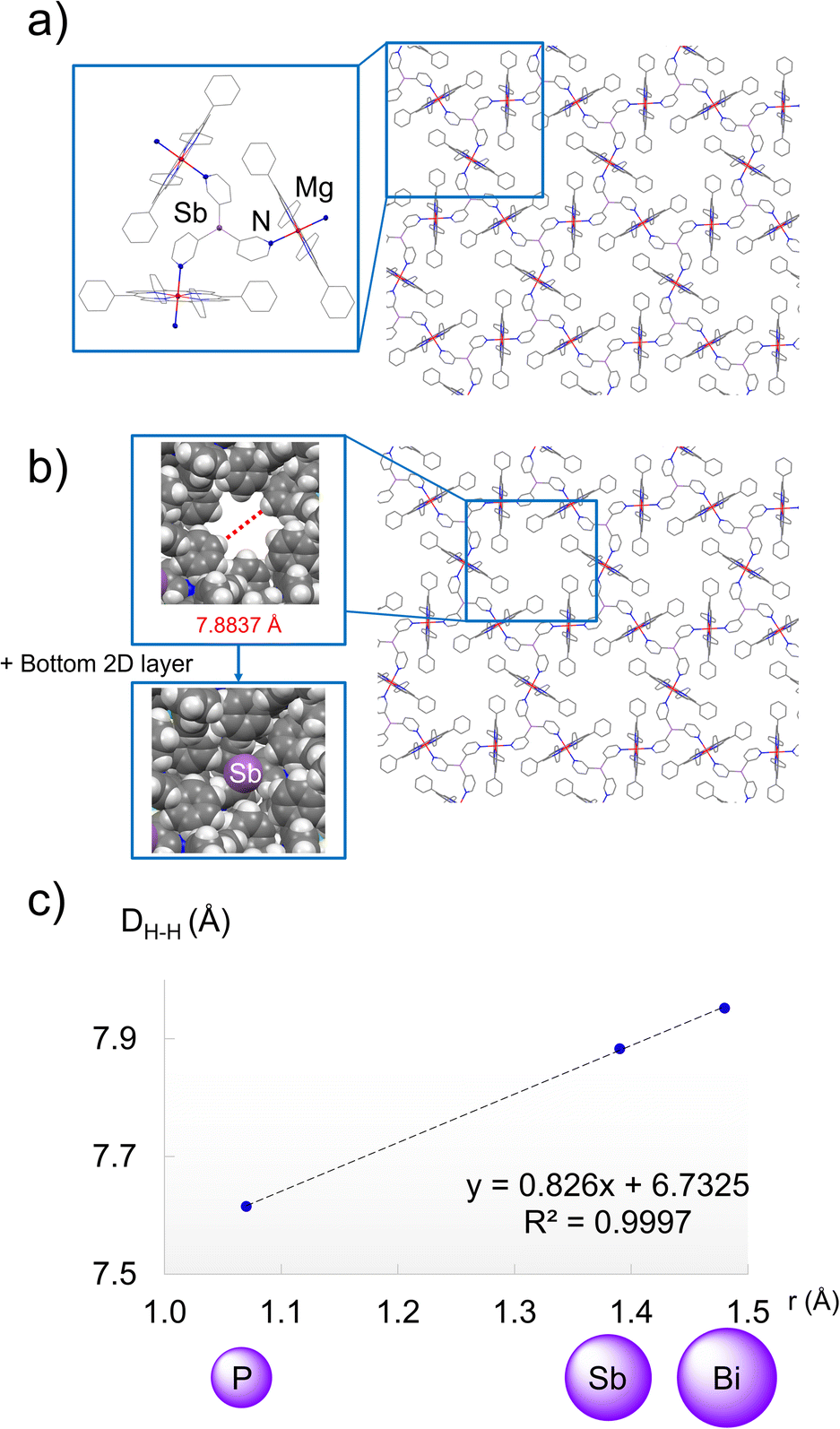 | ||
| Fig. 7 (a) X-ray structure showing the 2D polymeric arrangement of 1·Mg. Pyridyl coordination at both axial positions of MgTPP results in an extended structure. The inset shows the capsule that results from the coordination of 1 to three MgTPP units. H atoms and CHCl3 solvent molecules are omitted for clarity. Selected bond lengths (Å) and angles (°): 1·Mg, Sb–Cpy 2.155(4); Npy–Zn 2.330(2); Cpy–Sb–Cpy 94.6(2). Colour key: C (grey), N (blue), Sb (light purple), Mg (red). (b) X-ray structure of the 2D polymeric structure of 1·Mg showing the 2-D network structure consisting of Sb6Mg6 ring units, which leads to the formation of hexagonal voids in the structure (inset). (c) Linear correlation between the covalent radii of the bridgehead atom and the effective size of the hexagonal voids (DH–H) in the X-ray structures of P·Mg, 1·Mg and 2·Mg. Colour key: C (grey), N (blue), Sb (light purple), Mg (red). | ||
The solid-state structure of these Mg complexes is formed by stacked 2D layers that are offset such that the bridgehead atom of a ligand is located in the middle of the E6Mg6 ring units of the adjacent layers, leading to the formation of hexagonal voids between the layers (Fig. 7b, insets). The sizes of these hexagonal voids (Fig. 7b for 1·Mg) also correlate directly with the covalent radii of the bridgehead atom of the E(3-py)3 ligands (7.6156(4), 7.8837(3) and 7.9525(2) Å for P·Mg, 1·Mg and 2·Mg, respectively, Fig. 7b and c), further illustrating the capability of the bridgehead to modulate the supramolecular structures of these porous assemblies.
Calculations using the software Olex2 (ref. 58) indicate that the solvent-accessible volume in the fully desolvated structures of the complexes P·Mg, 1·Mg and 2·Mg are 45.4, 46.1 and 46.1%, respectively. In order to investigate the permanent porosity of these materials, P·Mg and 2·Mg were initially treated at 150 °C for 12 h under high vacuum prior to BET and DR measurements to remove lattice solvent molecules. The results of N2 (77 K) adsorption show SBET (BET surface area) values lower than 5 m2 g−1, while slightly higher values were observed for CO2 (273 K) adsorption, with SDR (DR surface area) in the range of 41–76 m2 g−1. Therefore, although the materials could be expected to have microporosity, the treatment necessary to measure this (e.g., full desolvation of the structure) leads to a low microporosity material (see ESI†).
1H NMR studies of P·Mg, 1·Mg and 2·Mg in CDCl3 show large characteristic upfield shifts in the proton signals of the pyridyl ring in the E(3-py)3 (E = P, Sb, Bi) ligand, which are indicative of the coordination of the pyridine nitrogen to the Mg(II)-TPP metalloporphyrin. The aggregation state of the Mg(II) complexes in solution was studied via1H DOSY experiments, which indicated the collapse of the polymeric structures observed in the solid state for all three compounds, and suggested the presence of small aggregates in solution (see ESI† for details). However, the formation of discrete capsules was observed by mass spectrometry (ESI-TOF, see ESI†).
Effect of the oxidation of the bridgehead
Ligands Cl2SbV(3-py)3 (3) and Cl2BiV(3-py)3 (4) were easily obtained by the oxidation of 1 and 2, respectively, with 1 equiv. of SO2Cl2 in DCM. This enables the change from a pyramidal coordination node in the E(III) ligands 1 and 2 to a trigonal planar coordination node in the E(V) ligands 3 and 4, providing a way to modulate the coordination mode of the antimony and bismuth ligands. Compounds 3 and 4 were characterized using elemental analysis, NMR, analytical techniques and single-crystal X-ray diffraction studies (see ESI†). As expected on the basis of VSEPR theory, in the solid-state structures of molecules 3 and 4, the more electronegative Cl atoms are axial while the donor-pyridine units lie in the trigonal plane.With the new ligands 3 and 4 in hand, we studied their coordination with ZnTPP and MgTPP to evaluate the effect of the change in the coordination vectors on capsule formation. Attempts to coordinate the bismuth(V)-based ligand 4 to zinc tetraphenylporphyrin (ZnTPP) or magnesium tetraphenylporphyrin (MgTPP) were unfruitful, leading in both cases to the formation of complex mixtures along with the formation of a black precipitate (presumably of Bi). Reaction with the Sb(V) ligand 3 was more successful. Slow diffusion of n-hexane into a CHCl3 solution of 3 (1 equiv.) and zinc tetraphenylporphyrin (ZnTPP) (3 equiv.) gave dark-purple hexagonal crystals of [{Cl2SbV(3-py)3}(ZnTPP)3] (3·Zn) in 57% yield (Fig. 8a).
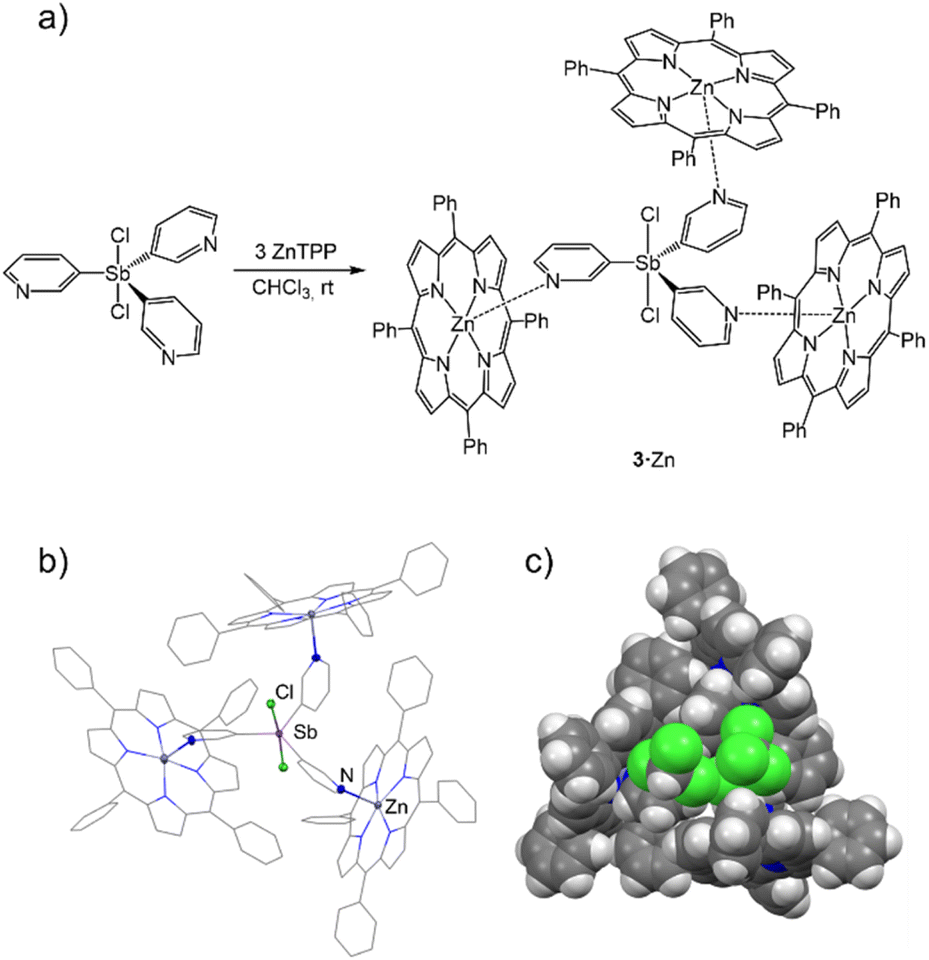 | ||
| Fig. 8 (a) Synthesis of 3·Zn. (b) X-ray structure of the complex 3·Zn. The ligand coordinates three porphyrins through N–Zn interactions. As a result of the coordination, the ligand is fenced by the three porphyrins, leading to the formation of a capsule-type structure. The three CHCl3 molecules located in the unit cell are omitted for clarity. (c) Space-filling view of the compound 3·Zn, in which two CHCl3 molecules are accommodated in the capsule cavity. Displacement ellipsoids at 50% probability. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): 3·Zn, Sb–Cpy range 2.109(4)–2.119(4); Npy–Zn range 2.178(4)–2.228(4); Sb–Cl 2.423(1) and 2.460(1); Cpy–Sb–Cpy range 115.6(2)–128.3(2). Colour key: C (grey), Zn (dark grey), N (blue), Sb (light purple), Cl (green). | ||
Complex 3·Zn has a similar structure to 1·Zn and 2·Zn in the solid state, in which the ligand molecules link Zn2+ centres using the three 3-Py donor-N atoms, leading to the formation of a capsule arrangement (Fig. 8b). The change in the geometry at the Sb centre results in an increase in the C–Sb–C angles (range 115.6(2)–128.3(2)° vs. range 95.4(2)–96.0(2)° for 3·Zn and 1·Zn-closed, respectively). This leads to a change in not only the shape but also the size of the capsule (average DZn⋯Zn 10.462 vs. 10.249 Å for 3·Zn and 1·Zn-closed, respectively). The increased size of the capsule arrangement is illustrated by the presence of two CHCl3 molecules inside the cavity instead of the one observed in the CHCl3 solvate of 1·Zn (cf.Fig. 6c and 8c).
In contrast to the Sb(III) ligand 1, the reaction of ligand 3 and magnesium tetraphenylporphyrin (MgTPP) did not form a 3D capsule arrangement, instead giving the 1D coordination polymer [{Cl2SbV(3-py)3}(MgTPP)]n (3·Mg). Although its formation was observed regardless of the stoichiometry used (3:MgTPP ratios of 1:2 or 1:3), better yields (34%) were obtained using the required 1:1 stoichiometry (see ESI†). Fig. 9 shows the 1D polymeric structure of 3·Mg in the solid state. The Sb(V) ligands 3 link the Mg2+ centres in a polymeric arrangement using two 3-Py N donor atoms (the third remains uncoordinated) (Fig. 9b). The change in geometry of the linker from pyramidal in Sb(III) 1 to trigonal planar in Sb(V) 3 precludes the formation of the hybrid 2D capsule arrangement observed for 1·Mg, resulting in the formation of the 1D zig–zag structure of 3·Mg.
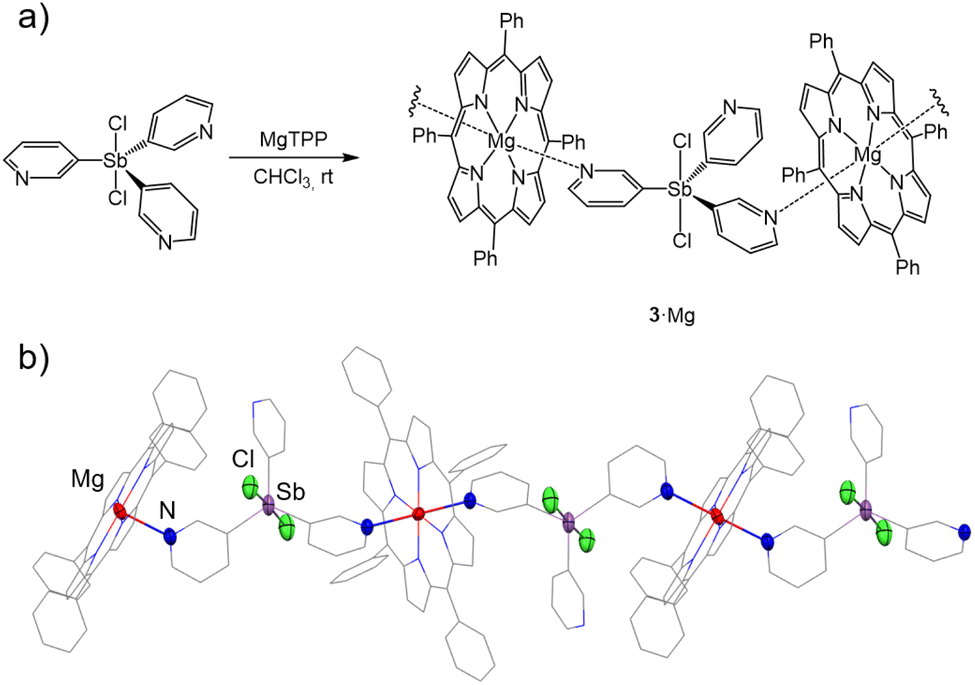 | ||
| Fig. 9 (a) Synthesis of 3·Mg. (b) X-ray structure of the 1D polymeric structure of 3·Mg through N⋯Mg interactions. Displacement ellipsoids at 50% probability. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): 3·Mg, Sb–Cpy range 2.102(3)–2.123(3); Npy–Zn 2.380(2) and 2.385(2); Sb–Cl 2.4348(9) and 2.4426(9); Cpy–Sb–Cpy range 119.4(1)–120.6(1). Colour key: C (grey), Cl (green), N (blue), Sb (light purple) and Mg (red). | ||
Preliminary catalytic studies
In recent years, there has been dramatic progress in organo-pnictogen catalysis and in particular growing interest in catalytic redox reactions.49,59 To the best of our knowledge the catalytic redox behaviour of organo-bismuth and -antimony compounds has not been explored in a confined environment. We decided to study the effect of the encapsulation of the bismuth center in the catalytic oxidative cleavage of 1,2-diols, which constitutes one of the first bismuth redox catalytic systems developed.Barton and, more recently, Cornella, reported the bismuth redox catalytic behavior of several bismuthines, showing that catalytic amounts of the bismuth(III)-containing species are able to catalyze the oxidative cleavage of 1,2-diol in the presence of N-bromo-succinimide (NBS) as a stoichiometric oxidant.60–62 With this in mind, we evaluated the catalytic behavior of our tris(3-pyridyl)bismuthine (2) in the oxidative cleavage of two well-studied substrates, pinacol (Me2C(OH)C(OH)Me2) and benzopinacol (Ph2C(OH)C(OH)Ph2), using the reaction conditions established by Barton.61 When the reaction of pinacol was carried out in the absence of BiPy3 (2) (either with or without ZnTPP metalloporphyrin), formation of Me2CO was not observed, proving the necessity of bismuth as a catalyst, as previously reported60–62 (see ESI†). In contrast, the presence of 1 mol% of the ligand 2 produces the cleavage of pinacol, yielding acetone (Table 1, entry 1). Under the same conditions, Ph2C(OH)C(OH)Ph2 also gave the corresponding oxidative cleavage product benzophenone (entry 3), albeit at a slower rate, which parallels early observations by Barton. Having demonstrated catalysis in the presence of 2, we next explored the effect of its encapsulation. It is found that employing 2·Zn has a direct impact in the catalytic activity. In the case of pinacol the reaction rate was strongly decreased (entry 2) while for Ph2C(OH)C(OH)Ph2, it was slightly increased (entry 4). Although the reason for this difference in rate is unclear at this stage, the effect is obviously not only steric in origin.
|
|
||||
|---|---|---|---|---|
| Entry | Bi catalyst | Yield (%) | ||
| R = Me | R = Ph | |||
| a Oxidative cleavage of 1,2-diols in the presence of 2 or 2·Zn. Reaction conditions: 1,2-diol (0.14 mmol), NBS (0.14 mmol), K2CO3 (1.4 mmol), 1.5 ml of CD3CN at 25 °C. b Reaction conditions: 1,1,2,2-tetramethyl-1,2-ethanediol (0.07 mmol), 1,1,2,2-tetraphenyl-1,2-ethanediol (0.07 mmol), NBS (0.14 mmol), K2CO3 (1.4 mmol), 1.5 ml of CD3CN at 25 °C. Yields were determined by 1H NMR after 90 min. | ||||
| 1 | 2 | 65 | — | |
| 2 | 2·Zn | 16 | — | |
| 3 | 2 | — | 31 | |
| 4 | 2·Zn | — | 38 | |
| Competitiveb | 5 | 2 | 76 | 20 |
| 6 | 2·Zn | 18 | 63 | |
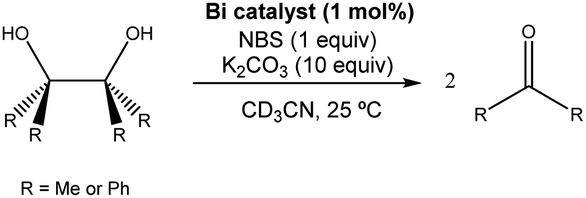
To evaluate the selectivity of the unencapsulated and encapsulated catalyst, competitive oxidative cleavage experiments were performed in the presence of both substrates. The competitive reaction in the presence of ligand 2 leads to benzophenone and acetone in a ratio of 0.26 (entry 5). The same reaction performed in the presence of capsule 2·Zn leads to inversion in the selectivity with a product ratio of 3.50 (entry 6). Thus, the preferred product is dependent upon the encapsulation of the catalyst.
A similar effect was observed in the catalytic oxidation of α-hydroxyketones to the corresponding α-diketones using our tris(3-pyridyl) stibine (1) and the antimony capsule 1·Zn as catalysts under aerobic oxidation conditions (air), using similar conditions to those previously reported for organoantimony-catalyzed reactions.63 While the oxidation of benzoin (PhCOCH(OH)Ph) and acetoin (MeCOCH(OH)Me) to the corresponding diketones could be accomplished using 1 as the catalyst (10 mol%), the use of the capsule 1·Zn instead of 1 dramatically inhibits the reaction of acetoin but not the reaction of benzoin, echoing the observations in the oxidative cleavage of diols using 2 and 2·Zn (see ESI†). Moreover, the use of stoichiometric Bi(3-py)3 (2) does not result in product formation in the oxidation of α-hydroxyketones, in agreement with previously published observations for BiPh3, which points to an antimony-bridgehead-specific catalytic reaction.49,63
Conclusions
This study has shown how changing the bridgehead atom or oxidation of the bridgehead of the E(3-Py)3 ligands can be used as a strategy to modulate the size and shape of supramolecular capsules formed by coordination to metalloporphyrins. Previous studies, restricted to the use of P(3-py)3 and Zn(II) metalloporphyrins, had shown the potential of these systems to form well-defined catalytic cavities and achieve reactivity in confined environments. In our study, we have reported the formation of the first capsules based on heavier Group 15 E(3-Py)3 ligands [E = Sb (1), Bi (2)]. The use of the E(3-Py)3 linker in conjunction with the appropriate metalloporphyrin M-TPP enables the formation of discrete molecular capsules (M = Zn(II)) or the assembly of these capsules into extended structures (M = Mg(II)). We have also demonstrated that encapsulation of 1 and 2 can have a dramatic effect on the rate and selectivity of the catalytic oxidative cleavage of organic diols and catalytic oxidation of α-hydroxyketones. In ongoing studies, we are exploring the way in which the size of the macrocycles and the main group bridgehead atoms present in the linker can affect reactivity and selectivity in a number of other catalytic reactions.Data availability
ESI† is available, including experimental details, NMR and mass-spectrometry data, DFT calculations and X-ray single-crystal structure details. Crystallographic data for compounds have been deposited at the Cambridge Crystallographic Database under access numbers 2195694–2195704.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Spanish Ministry of Science and Innovation (MCIN) (RG-R, PID2021-124691NB-I00, funded by MCIN/AEI/10.13039/501100011033/FEDER, UE) for funding. A. G.-R. acknowledges the University of Valladolid and Santander Bank for a Fellowship. We thank Dr Angel E. Lozano for insightful discussions and generous support and assistance.Notes and references
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC
.
- W. B. Motherwell, M. J. Bingham and Y. Six, Tetrahedron, 2001, 57, 4663–4686 CrossRef CAS
- A. J. Kirby, Angew. Chem., Int. Ed. Engl., 1996, 35, 707–724 CrossRef CAS
- N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Chem. Soc. Rev., 2015, 44, 9–25 RSC
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715–14729 CrossRef CAS PubMed
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS
- M. M. Deegan, M. R. Dworzak, A. J. Gosselin, K. J. Korman and E. D. Bloch, Chem.–Eur. J., 2021, 27, 4531–4547 CrossRef CAS PubMed
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed
- J. Trouvé and R. Gramage-Doria, Chem. Soc. Rev., 2021, 50, 3565–3584 RSC
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H. C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC
-
E. P. W. N. M. Van Leeuwen, Supramolecular Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed
- S. S. Nurttila, P. R. Linnebank, T. Krachko and J. N. H. Reek, ACS Catal., 2018, 8, 3469–3488 CrossRef CAS PubMed
- H. Takezawa and M. Fujita, Bull. Chem. Soc. Jpn., 2021, 94, 2351–2369 CrossRef CAS
-
G. Lloyd and R. S. Forgan, Reactivity in Confined Spaces, Royal Society of Chemistry, UK, 2021 Search PubMed
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS PubMed
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC
-
U. H. Brinker and J.-L. Mieusset, Molecular Encapsulation: Organic Reactions in Constrained Systems, John Wiley & Sons, Ltd, Chichester, 2010 Search PubMed
-
J. N. H. Reek and S. Pullen, in Supramolecular Catalysis: New Directions and Developments, ed. P. W. N. M. van Leeuwen and M. Raynal, Wiley-VCH, Weinheim, Germany, 2002, pp. 255–270 Search PubMed
- L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC
- T. Tateishi, M. Yoshimura, S. Tokuda, F. Matsuda, D. Fujita and S. Furukawa, Coord. Chem. Rev., 2022, 467, 214612 CrossRef CAS
- C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed
- M. M. Conn and J. Rebek, Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS
- V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271–4274 CrossRef CAS PubMed
- V. Bocokić, A. Kalkan, M. Lutz, A. L. Spek, D. T. Gryko and J. N. Reek, Nat. Commun., 2013, 4, 2670 CrossRef PubMed
- M. Kuil, T. Soltner, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2006, 128, 11344–11345 CrossRef CAS PubMed
- V. F. Slagt, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 1526–1536 CrossRef CAS PubMed
- L. J. Jongkind, J. A. A. W. Elemans and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 2696–2699 CrossRef CAS PubMed
- I. Jacobs, A. C. T. van Duin, A. W. Kleij, M. Kuil, D. M. Tooke, A. L. Spek and J. N. H. Reek, Catal. Sci. Technol., 2013, 3, 1955–1963 RSC
- T. Besset, D. W. Norman and J. N. H. Reek, Adv. Synth. Catal., 2013, 355, 348–352 CAS
- X. Wang, S. S. Nurttila, W. I. Dzik, R. Becker, J. Rodgers and J. N. H. Reek, Chem.–Eur. J., 2017, 23, 14769–14777 CrossRef CAS PubMed
- L. J. Jongkind and J. N. H. Reek, Chem.–Asian J., 2020, 15, 867–875 CrossRef CAS PubMed
- A. J. Plajer, A. L. Colebatch, F. J. Rizzuto, P. Proehm, A. D. Bond, R. García-Rodríguez and D. S. Wright, Angew. Chem., Int. Ed., 2018, 57, 6648–6652 CrossRef CAS PubMed
- Á. García-Romero, A. J. Plajer, L. Álvarez-Miguel, A. D. Bond, D. S. Wright and R. García-Rodríguez, Chem.–Eur. J., 2018, 24, 17019–17026 CrossRef PubMed
- J. E. Waters, G. Berger, A. J. Peel, R. García-Rodríguez, A. D. Bond and D. S. Wright, Chem.–Eur. J., 2021, 27, 12036–12040 CrossRef CAS PubMed
- E. S. Yang, A. J. Plajer, Á. García-Romero, A. D. Bond, T. K. Ronson, C. M. Álvarez, R. García-Rodríguez, A. L. Colebatch and D. S. Wright, Chem.–Eur. J., 2019, 25, 14003–14009 CrossRef CAS PubMed
- Á. García-Romero, J. M. Martín-Álvarez, A. L. Colebatch, A. J. Plajer, D. Miguel, C. M. Álvarez and R. García-Rodríguez, Dalton Trans., 2021, 50, 13059–13065 RSC
- Á. García-Romero, A. J. Plajer, D. Miguel, D. S. Wright, A. D. Bond, C. M. Álvarez and R. García-Rodríguez, Inorg. Chem., 2020, 59, 7103–7116 CrossRef
- Á. García-Romero, J. M. Martín-Álvarez, D. Miguel, D. S. Wright, C. M. Álvarez and R. García-Rodríguez, Inorg. Chem., 2021, 60, 19206–19218 CrossRef PubMed
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC
- M. Marchini, A. Luisa, G. Bergamini, N. Armaroli, B. Ventura, M. Baroncini, N. Demitri, E. Iengo and P. Ceroni, Chem.–Eur. J., 2021, 27, 16250–16259 CrossRef CAS PubMed
- V. McKee, O. C. Choon and G. A. Rodley, Inorg. Chem., 1984, 23, 4242–4248 CrossRef CAS
- G. Wu, A. Wong and S. Wang, Can. J. Chem., 2003, 81, 275–283 CrossRef CAS
- C. H. Devillers, A. K. D. Dimé, H. Cattey and D. Lucas, C. R. Chim., 2013, 16, 540–549 CrossRef CAS
- N. Amiri, M. Hajji, T. Roisnel, G. Simonneaux and H. Nasri, Res. Chem. Intermed., 2018, 44, 5583–5595 CrossRef CAS
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed
- M. Magre, J. Kuziola, N. Nöthling and J. Cornella, Org. Biomol. Chem., 2021, 19, 4922–4929 RSC
- D. H. R. Barton, W. B. Motherwell and A. Stobie, J. Chem. Soc., Chem. Commun., 1981, 1232–1233 RSC
- D. H. R. Barton, J.-P. Finet, W. B. Motherwell and C. Pichon, Tetrahedron, 1986, 42, 5627–5636 CrossRef CAS
- S. Yasuike, Y. Kishi, S.-I. Kawara and J. Kurita, Chem. Pharm. Bull., 2005, 53, 425–427 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR and mass-spectrometry data, DFT calculations and X-ray single-crystal structure details. CCDC 2195694–2195704. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02151c |
| This journal is © The Royal Society of Chemistry 2023 |