
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical complexity for targeted function in heterometallic titanium–organic frameworks
Javier
Castells-Gil†
 ab,
Neyvis
Almora-Barrios†
a,
Belén
Lerma-Berlanga
ac,
Natalia M.
Padial
a and
Carlos
Martí-Gastaldo
*a
ab,
Neyvis
Almora-Barrios†
a,
Belén
Lerma-Berlanga
ac,
Natalia M.
Padial
a and
Carlos
Martí-Gastaldo
*a
aInstituto de Ciencia Molecular, Universidad de Valencia, C/Catedrático José Beltrán 2, 46980, Paterna, Spain. E-mail: carlos.marti@uv.es
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
cInstituto de Tecnología Química (UPV-CSIC), Universidad Politècnica de València-Consejo Superior de Investigaciones Científicas, Avda. de los Naranjos s/n, 46022 Valencia, Spain
First published on 27th May 2023
Abstract
Research on metal–organic frameworks is shifting from the principles that control the assembly, structure, and porosity of these reticular solids, already established, into more sophisticated concepts that embrace chemical complexity as a tool for encoding their function or accessing new properties by exploiting the combination of different components (organic and inorganic) into these networks. The possibility of combining multiple linkers into a given network for multivariate solids with tunable properties dictated by the nature and distribution of the organic connectors across the solid has been well demonstrated. However, the combination of different metals remains still comparatively underexplored due to the difficulties in controlling the nucleation of heterometallic metal-oxo clusters during the assembly of the framework or the post-synthetic incorporation of metals with distinct chemistry. This possibility is even more challenging for titanium–organic frameworks due to the additional difficulties intrinsic to controlling the chemistry of titanium in solution. In this perspective article we provide an overview of the synthesis and advanced characterization of mixed-metal frameworks and emphasize the particularities of those based in titanium with particular focus on the use of additional metals to modify their function by controlling their reactivity in the solid state, tailoring their electronic structure and photocatalytic activity, enabling synergistic catalysis, directing the grafting of small molecules or even unlocking the formation of mixed oxides with stoichiometries not accessible to conventional routes.
Introduction
The field of metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), has been continuously expanding during the last 25 years owing to their rich chemical and structural diversity.1 MOFs are hybrid materials obtained by the self-assembly of inorganic nodes and organic linkers into periodic structures with high surface areas and porosities. Here, the large number of conceivable metal-linker combinations provide a vast chemical and structural landscape allowing access to a virtually unlimited number of structures with different functionalities. Presently, the field of MOFs is shifting from the principles that control this assembly into more sophisticated concepts that embrace chemical complexity as a tool for encoding new functions and properties that exploit the combination or synergy of different components into these networks. This has led to the development of the multivariate (MTV) approach that enables the introduction of multiple types of functional groups in the structure of the MOF leading to the formation of so-called MTV-MOFs.2The concept of multivariance in reticular solids was originally used to account for the incorporation of a variable number of independent organic linkers with equivalent connection points but bearing different chemical functionalities.2 By choosing an appropriate MOF network, it has been possible to introduce up to 8 different functional organic groups of varying size and ratios by one-pot synthesis.2,3 With this approach, it has been possible to study charge transfer phenomena between linkers with different functional groups in MTV-MOFs, also allowing to quantify their proximity within the structure;4 create new asymmetric MOF catalysts capable of carrying out sequential alkene epoxidation/epoxide ring-opening reactions;5 and even the selective generation of mesopores in a MOF structure by selective linker thermolysis/ozonolysis.6–8 Nevertheless, this approach is by no means limited to the conventional one-pot synthetic approaches. MTV-MOFs can also be synthesised via post-synthetic ligand exchange reactions. Unlike the one-pot synthetic approach, this methodology relies on the diffusion of the new linker into the framework, enabling the formation of core–shell MOF structures9,10 or even, in some rare cases, of core–shell MOF heterostructures when using ligands with different geometry or anchoring groups.11
However, the organic approach to multivariance mostly relies on the use of linkers with unaltered length and connectivity. Their relative proportion in the reaction mixture controls their incorporation to the framework provided negligible effect of the chemistry of their substituents over nucleation and crystal growth. In turn, the extension of this concept to the synthesis of mixed-metal MOFs is limited by the difficulties in combining metals of different ionic radii, polarizing power, acidity, or coordination geometries that shall be compatible with the formation of multi-metallic nodes respectful with the connectivity of the targeted framework. This is particularly exacerbated for frameworks based on titanium. Ti-MOFs have attracted significant attention due to the intrinsic photoactivity of this metal and its high polarizing power, ideal for assembling frameworks with exceptional stability. Nonetheless, the challenging chemistry of Ti(IV) in solution has limited the development of crystalline, porous titanium frameworks for an even more limited number of mixed-metal systems.
In this perspective, we provide an overview of the recent progress in controlling and analysing metal variance in reticular solids formed by two or more different metals in the same framework. This general introduction is used to set particular focus on the comparatively underexplored titanium–organic frameworks. Built upon the controlled distribution of these metals at the cluster level, we outline the possibilities offered by heterobimetallic titanium frameworks to access advanced functions in applications as solid-state reactivity, catalysis, photocatalysis or their use as precursors for the formation of mixed oxides compositions inaccessible to simple combinations of single metal phases. All these possibilities demonstrate the importance of controlling cluster composition at an atomic level to unleash its potential for directing framework function.
Mixed-metal or heterometallic frameworks
Whereas the introduction of chemical complexity by means of linker variation has a minimum impact over framework assembly, the effect of metal combination over the assembly of polynuclear secondary building units (SBUs) with predefined extension points are more difficult to control and not as versatile. This is arguably the reason for which organic multivariance in MOFs is more often approached by direct synthesis whereas inorganic multivariance has imposed the development of alternative routes.12,13Direct synthesis or one-pot reactions seem to be well adapted to rod-type SBUs. The synthesis of MOF-74 is compatible with the formation of multimetallic alloys from the combination of 2, 4, 6, 8 or even 10 metals (M2+ = Mg, Ca, Sr, Ba, Mn, Fe, Co, Ni, Zn, and Cd) in a single M3O3(CO2)3 framework by reaction of different metals at variable stoichiometries (Fig. 1a).14 MOF-74 is also amenable to doping with Fe3+ ions,15 or the synthesis of bimetallic combinations at variable ratio by using mechanochemistry.16 This is also the case of ZnPF-1 (Fig. 1b), with a rod-shaped SBU that can be adapted to the incorporation of multimetallic arrangements (M2+ = Zn, Ca, Mn, Co) with variable coordination geometries during synthesis.17,18 The mononuclear inorganic nodes characteristic of electrically conductive MOFs as M3(hexaiminotryphenylene)2 (M2+ = Co, Ni, Cu),19 are also adequate to produce mixed metal alloys with metal incorporation ratios controlled by their relative stochiometries in solution (Fig. 1c).
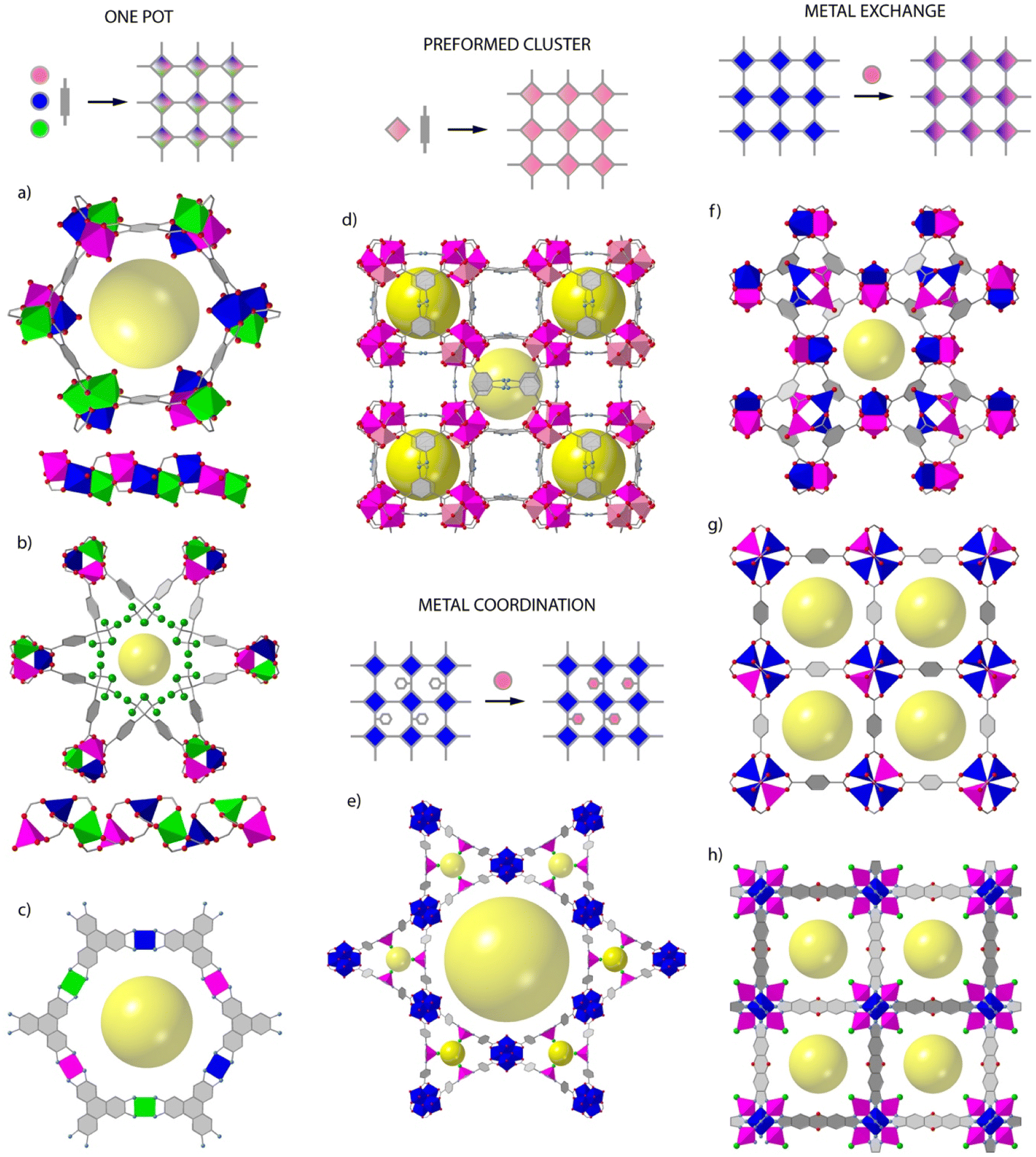 | ||
| Fig. 1 1Overview of the different routes toward the synthesis of mixed-metal frameworks. One pot synthesis compatible with the assembly of rod-type SBUs as (a) MOF-74 and (b) ZnPF-1 or mononuclear nodes as (c) M3(HITP)2. Use of pre-formed clusters exemplified by the synthesis of (d) MIL-127 or PCN-250 from bimetallic FeIII2MII(μ3-O) SBUs. Structure of PCN-222 exemplifying the use of metalloligands as connectors for the combination of metal units in distant positions in the framework. Metal exchange reaction of MOFs for the formation of mixed-metal SBUs in: (f) HKUST-1, (g) MOF-5 and (h) MFU-4l. | ||
This same concept can be also extended to other archetypical frameworks based on polynuclear SBUs. However, the difficulties in controlling the assembly of mixed-metal nodes by one pot synthesis can prevent the formation of the material, result in the segregation of homometallic phases, or lead to an inhomogeneous distribution of metals and/or domains in the framework, all associated to the preferential incorporation of metals with adequate charge and ionic radius. This can be partly circumvented by controlling the reactivity of metals in solution,20 or by using the SBU approach, in which the use of heterometallic clusters as starting precursors ensure the implantation of heterometallic nodes in the final framework. This route is compatible with the synthesis of multimetallic versions of the MIL-127 (ref. 21) and PCN-250 (ref. 22) families by one pot reaction of FeIII2MII(μ3-O) preformed clusters (Fig. 1d). Another alternative for introducing additional metals to a pre-formed framework is the use of metalloligands as connectors. Linkers incorporating 2,2′-bipyridine,23–25 porphyrins,26 1′-bi-2-naphthol27 or β-diketiminates28 as chelating groups can facilitate the incorporation of metals as Ir3+, Ru3+, Pd2+, Ti4+, Ni2+, Cu2+, Fe2+ or Co2+ in satellite positions that do not interfere with the formation of the SBUs of the corresponding frameworks (Fig. 1e). This approach does not allow for close electronic proximity between different metal sites, but it is compatible with a good number of architectures and permits dispersing single catalytic sites in a porous matrix.
The use of post-synthetic metalation reactions is arguably more versatile in terms of the compositional complexity of SBUs and the range of frameworks accessible. Here, preformed MOF crystals are reacted in a metal solution for the transmetallation of the metal nodes and the formation of the corresponding mixed-metal material.29 Metal exchange reactions are controlled by thermodynamic and kinetic factors that are influenced by the structure of the MOF or the solvent used.30 As a rule of thumb, softer metals with more labile coordination bonds as Zn(II) are more likely to be exchanged more quantitatively and in shorter reaction times. For example, the Zn analogue of HKUST-1 (Fig. 1f),31 PMOF-2 (ref. 31) or PCN-921,32 all based on Zn2(COO)4 paddlewheel SBUs can be partly replaced in days or months with Cu2+ ions to generate mixed-metal frameworks with exchange efficiencies from 50 to 100%. However, the extent of metal exchange can be compromised for other SBUs with higher connection points or different coordination environments. This is exemplified by MOF-5 Zn4O(COO)3 (Fig. 1g),33 ZIF-8 Zn-(MeIm) and ZIF-71 Zn-(Cl2Im)34 or MFU-4l ZnZn4Cl4(BTDD6) (Fig. 1h),35 which are based on carboxylate, imidazolate or bisbenzotriazolate linkers and show Zn exchange levels below 20% with first row transition metal ions as Ti4+, V3+/2+, Cr3+/2+, Mn2+, Fe2+, Co2+, or Ni2+. Only MFU-4l shows quantitative exchange of non-central Zn2+ sites with Co2+ in 20 hours. These examples help to outline the main pros and cons of this method. The use of mild conditions and single crystals as starting precursors are ideal to respect structural integrity and investigate the factors that govern metal exchange alongside with the formation of inhomogeneous domains in the final material. The limitations imposed by the framework and constituting SBUs to quantitative metalation reactions is arguably the main limitation towards controllable synthesis of multimetallic frameworks.
The distribution of metal cations in heterometallic MOFs is not obvious and sometimes difficult to anticipate experimentally. The use of computational methods can be useful in this regard. A combination of computational techniques as systematic density functional theory (DFT) or reverse Monte Carlo (RMC), have been used on several bimetallic families of MOFs like MOF-5 and UiO-66,36,37 MOF-74,38,39 or ZIF-8 (ref. 40) to predict the geometrical arrangements of metal cations, the most favourable configurations, and the impact of the distribution on their properties. Simulation bounded with characterisation techniques are the key to unravelling the precise role of several factors that have been proposed to control full metal node exchange: including ionic radii and preferential coordination geometries,36,38,41,42 or solvent effects.43,44
All these approaches to produce mixed-metal MOFs display different advantages and disadvantages but we argue gaining control over the distribution of two or more metals across the framework is crucial for the rational use of metal variance in the design of multimetallic frameworks with targeted function. In this context, the use of advanced characterization techniques that helps probing the formation of heterometallic building units will be imperative to decipher metal arrangements and control chemical complexity both required to establish future directions.
Deciphering chemical complexity in heterometallic MOFs: local structure and metal distribution
The characterization techniques used for probing the effect of metal mixing in the properties of the resulting MOFs shall cover not only the identification and quantification of the different metals in the structure, but also their local structure, specific coordination environment, oxidation state, or global arrangement into the framework, all relevant to establish clear composition-to-function relationships.The presence and relative ratio of metals can be qualitatively confirmed by using energy dispersive X-ray (EDX) analysis coupled to scanning or transmission microscopies for a simple mapping of metal distribution with spatial resolution in the μm or nm scale for selected areas and limited to the surface of the crystal analysed. Other techniques as Atomic Absorption Spectroscopy (AAS),45 Inductively Coupled Plasma-Mass Spectroscopy (ICP-MS)46 or Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES)47 are better suited to determine metal concentrations and ratios quantitatively. They rely on the detection of optical or mass spectra of atoms or small molecular fragments in the gas state. A non-destructive alternative would be X-Ray Fluorescence (XRF),48 but it requires careful calibration for meaningful results.
Powder X-ray diffraction (PXRD) can be also used to account for the incorporation of different metals. The continuous shift of the position of characteristic reflections according to Vegard's law is sometimes regarded as an experimental proof of miscible mixing of metals at variable compositions (Fig. 2a).40 In turn, deviations from linearity can be associated to metal clustering or compositional inhomogeneity in the solid. When it comes to determining the position and relative occupation of different metals in the framework, the low sensitivity of X-rays can hinder the distinction between elements of similar electronic density imposing the use of neutron diffraction to quantify relative occupation factors and coordination environments in mixed-metal MOFs. In such cases, neutron diffraction becomes very useful in the characterisation of heterometallic titanium–organic frameworks, as the negative scattering length of titanium atoms provides an excellent contrast between Ti ions and other transition metals, allowing for an accurate quantification of relative occupancies.
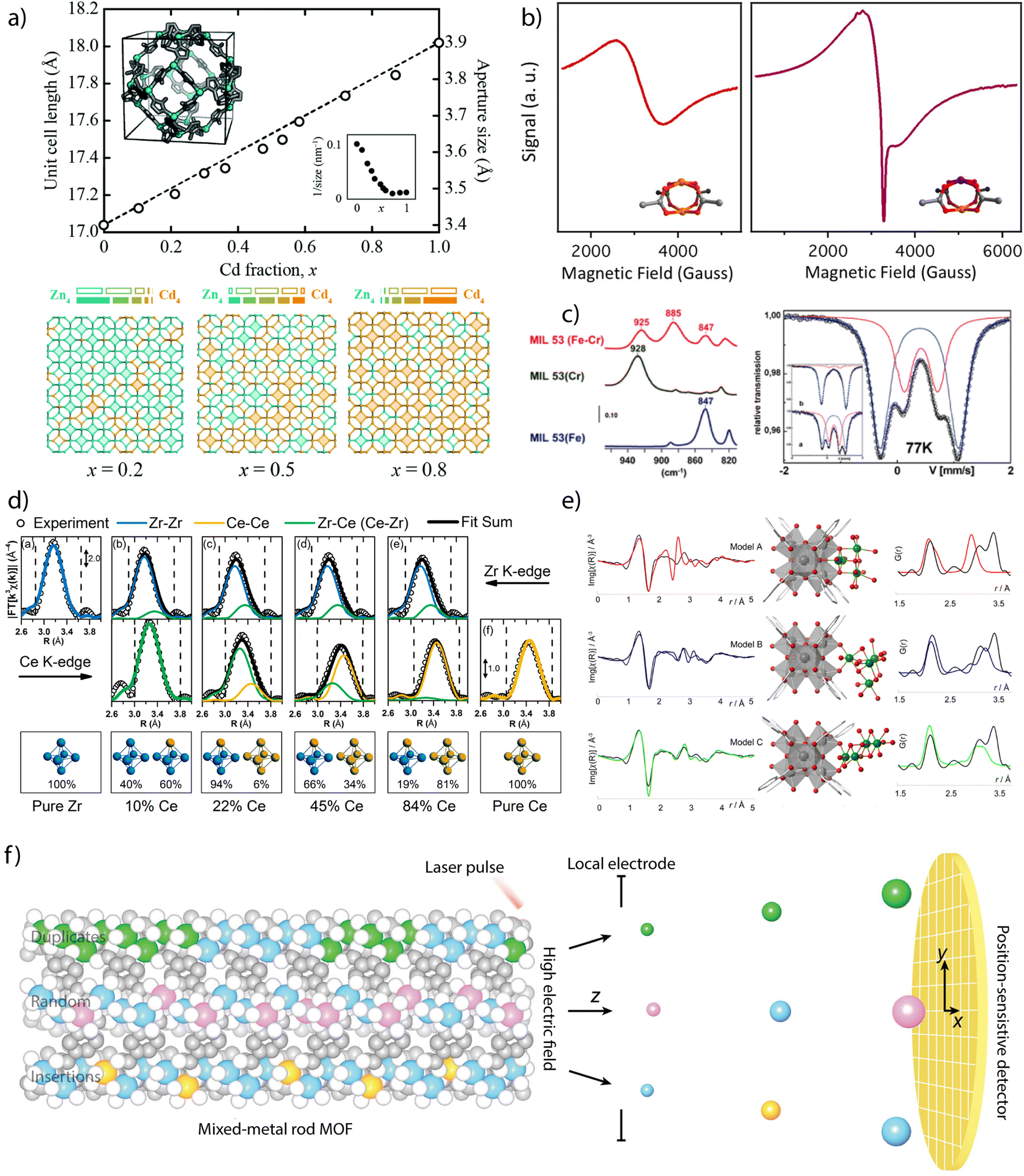 | ||
| Fig. 2 Examples of chemical characterisation of mixed-metal MOFs. (a) Dependence of the unit cell parameters, approximate aperture size and metal distribution as a function of the composition for the Zn1−xCdx ZIF-8 solid solution. (b) Identification of Cu–Rh heterometallic units in Cu/Rh-HKUST-1 via EPR spectroscopy. (c) FT-IR (left) and Mössbauer spectroscopy (right) measurements on heteromentallic Cr/Fe-MIL-53 showing the appearance of new signals due to the mixing of both cations in the structural unit. (d) EXAFS derived data showing the compositional dependence Zr–Zr, Ce–Zr and Ce–Ce distances in heterometallic Zr1−xCex UiO-66 solid solutions. (e) Local characterisation of Ni4-clusters incorporated to Zr6 nodes in NU-1000 by a combination of EXAFS and Pair Distribution Function (PDF) analyses. (f) Use of atom probe tomography for direct analysis of the type of metal sequences in mixed-metal MOFs. Reproduced from ref. 40, 54, 20, 61 and 68. | ||
Other techniques as Electronic Magnetic Resonance (EPR) or Mössbauer spectroscopy can be also used to confirm the presence of metals with different oxidation states or local environments. EPR has been used to confirm the incorporation of paramagnetic metals to frameworks as MIL-47(V4+/V3+),49 MIL-53(V4+/V3+),50 DUT-8 (Cu2+/Ni2+)51 or DUT-5 (V3+/Al3+).52 The introduction of these metals results in the appearance of characteristic signals not present in the featureless EPR spectra of the homometallic frameworks, that can be also used to provide information on the coordination symmetry and local environment of the paramagnetic metals. EPR has been particularly used for analysing mixed-metal analogues of HKUST-1. In this case, the antiferromagnetic coupling of Cu2(COO)4 dimers for a silent EPR spectra, characteristic of a S = 0 ground state, can be modified by introducing diamagnetic metals as Zn2+,53 Rh2+,54 or Ti4+.42 This results in the formation of active S = ½ or 1 ground states, from either uncompensated or dipolar couplings in the heterometallic dimers, and the corresponding signals in their EPR spectra (Fig. 2b). This technique is also particularly useful in the case of titanium frameworks for the detection of photoinduced Ti3+ species upon irradiation with UV-visible light.41,55,56 Though restricted to57 Fe elements, Mössbauer spectroscopy can also offer valuable structural information as exemplified by recent studies with bimetallic MIL-101(Cr/Fe) at variable iron contents, where it was used to rule out the formation of segregated oxide/hydroxide iron phases. FT-IR spectroscopy can be also helpful to analyse heterometallic arrangements. The changes in the position of the δ(OH)bending mode of the bridging μ2-OH linkers in the chains of MIL-53 relative to the presence of either Cr3+, Fe3+ or a combination of both (Fig. 2c), were used to confirm the presence of mixed Cr–Fe chains in the solid.20
X-ray absorption spectroscopy (XAS) is another tool to investigate short-range order of selected metals although it often requires synchrotron radiation.57 The sharp edges at the photo-ionization energies of atomic core levels are unique for each element and thus can provide compositional information. Also, edge energy positions are slightly shifted by the oxidation state of metals and their chemical binding state. The XAS spectrum can be divided into two energy regions and their corresponding techniques: the near edge that is associated to X-ray Absorption Near Edge Structure (XANES), and the high energy range for Extended X-ray Absorption Fine Structure (EXAFS). The position of the edge in the XANES spectrum can be used to obtain information of the oxidation state and coordination geometry of different metals.25 This technique has been widely used to correlate changes in the oxidation state or coordination geometry of metals as Pd,58 Ir,59 Co60 or Fe61,62 with the catalytic activity of the corresponding mixed-metal MOFs. These examples are based on metallolinkers in which the metal analysed is not forming part of the metal-oxo clusters that dictate the topology of the framework. The quantitative fitting of the EXAFS spectrum can be used to obtain information on the changes triggered by metal substitution in SBUs related to intermetallic distances or multiplicity and chemical identity of neighbours. The presence of second metal species in close vicinity can have a significant influence in the spectrum. In cases in which the disorder in the nearest metal ion shells is not detrimental and the different contributions to the spectrum can be assigned, the identification of the nearest metal shell is also possible. This technique has been used to demonstrate the coexistence of bimetallic CeZr5 and homometallic Zr6 or Ce6 clusters in mixed-metal UiO-66 phases prepared by direct synthesis at variable metal ratios (Fig. 2d)63 as well as the coexistence of Ti and Co in the heterometallic units of MUV-101(Co).64 It is also helpful to determine the local environment of the metals incorporated to SBUs by metal-exchange reactions. For example, the formation of vanadium clusters, Cu–O dimers or Zn–O–Zr sites have been demonstrated by EXAFS analysis of MFU-4l,65 NU-1000 (ref. 66) or MOF-808 (ref. 67) after reaction with different metal precursors.
The integration of EXAFS analysis with Pair Distribution Function (PDF) and computational simulations is another powerful tool to analyse in detail longer-range local structural changes. PDF analysis is based on total scattering experiments, typically using a high-energy X-ray synchrotron source or neutrons. It analyses both Bragg, and diffuse scattering signals simultaneously, providing powerful information at local and longer-range scales. In contrast to EXAFS spectroscopy, PDF is not an element sensitive technique and provides “in one shot” the entire structural information of a given material. The experimental data can be correlated with computational and structural models. This approach was used to study the structural changes affecting the zirconia nodes in different MOFs after atomic layer (ALD) deposition of copper or nickel (Fig. 2d). ALD of Cu in NU-901 and 907 results in the formation of fcc Cu0 nanoparticles of 1.5–0.9 nm, depending on the pore size of the host.68 In turn, ALD of Ni in NU-1000 leads to the selective tethering of NiOxHy clusters at the smallest pores available to enable the formation of bimetallic Ni/Zr-oxo nanowires by building a bridge between the Zr6 clusters in the MOF.69 More recently, PDF has been used to monitor the changes to the local structure in the bimetallic nodes (Fe2Co and Fe2Ni) of the PCN-250 family with temperature, and the influence of node chemistries in the formation of metal nanoparticles.70 Additionally, and in an analogous way as described for neutron diffraction, neutron PDF experiments can provide unique local structural information about the Ti ions in the structure of a MOF. Due to its negative scattering length, the peaks corresponding to Ti–O distances appear as negative peaks in the neutron PDF, allowing to discern easily between the local structures of Ti and the second metal.
These examples demonstrate how the increasing level of complexity in multimetallic frameworks imposes the use of more advanced techniques to provide precise information on not only the presence or relative ratio of metals, but also details on their oxidation state, coordination environment or local structure. The recent introduction of Atom Probe Tomography (APT) to decipher metal sequences in real space and at the atomic scale in MOFs (Fig. 2f).71 The detection efficiency of APT enabled the identification of four types of metal sequences in MOF-74 crystals prepared from different metal combinations (Co/Cd, Co/Pb, and Co/Mn) at different temperatures (120 and 85 °C). Random sequences, short duplicates of two to four metals and more than four, as well as sequences of insertions of a single metal into duplicates of another metal were identified. This is an excellent example of how the development of new techniques might be crucial not only to design but demonstrate experimentally unprecedented levels of complexity in molecular frameworks for targeted sequence-to-function selection.
Heterometallic titanium–organic frameworks
The combination of Ti(IV) with other metals to produce heterometallic titanium–organic frameworks can offer important advantages compared to their homometallic counterparts. This can enable the targeted modification of the adsorption properties, structural response, electronic structure, or chemical reactivity alongside with the appearance of new features from the synergistic interaction of the different metals. However, compared to other mixed-metal MOFs, the examples involving titanium are still scarce. Scheme 1 gives the reader a chronological overview of the early and recent developments in heterometallic titanium frameworks since the first examples reported in 2013. The low number of pure and heterometallic Ti-MOFs reported to date is arguably due to the high reactivity of this metal in solution, which challenges the assembly of crystalline frameworks with intrinsic porosity in favor of amorphous oxides.72 In this regard, the use of high-throughput synthetic techniques is gaining importance to control this chemistry by providing a systematic and thorough exploration of the chemical space to find the optimal synthetic conditions in which these materials can be formed.73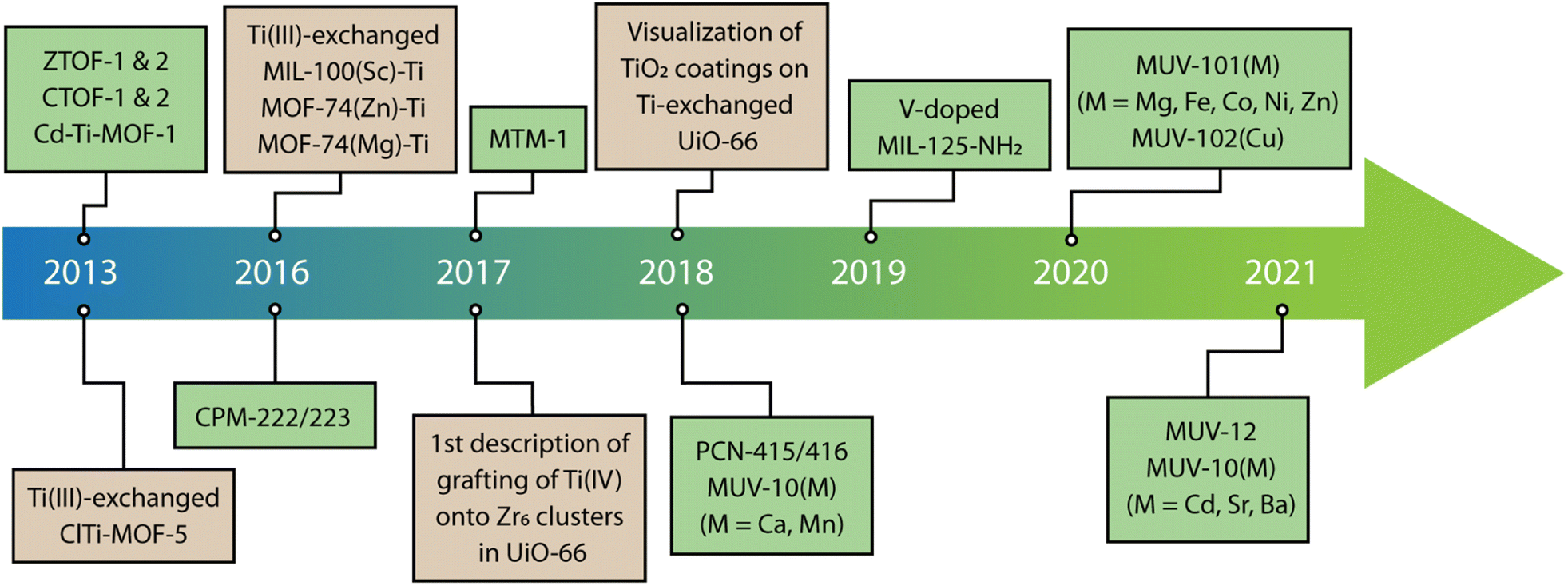 | ||
| Scheme 1 Chronology of the development of heterometallic titanium–organic frameworks via indirect (metal-exchange) and direct (solvothermal synthesis) routes. | ||
The synthesis of heterometallic titanium–organic frameworks is commonly achieved via either of the two following pathways: (i) a direct synthetic pathway that involves the mixing of both metals in a one-pot reaction; and (ii) an indirect pathway consisting in a post-synthetic metal exchange. The latter arouse as a promising way to introduce Ti(IV) in the structure of pre-formed iconic MOFs as UiO-66,74 MOF-5,33 or MIL-100.75 However, more recent works have shown how this methodology can suffer from undesired grafting to the surface of the cluster76 or the formation of TiO2 coatings onto the surface of the crystals.77
This is most likely due to the differences in charge or ionic radii and coordination environment between Ti4+ and the metal to be exchanged. For instance, transmetalation reactions between Ti4+ or Ti3+ and lower valence M2+ metals require the introduction of counterions to counterbalance the excess of positive charge. This in turn also involves a structural reorganization in the metallic unit in order to accommodate the counterions in the structure, resulting in lower exchange.33 Whereas other MOFs with highly charged metals as Zr4+ or Hf4+ may be good candidates, these ions display comparatively higher ionic radii and a different coordination environment than Ti4+. Hence, the transmetallation reaction between Ti4+ and Zr4+ in UiO-66 requires a structural reorganization to accommodate Ti4+ in the Zr6 cluster that is likely to impose a huge activation barrier for the metal exchange to occur. As result, Ti4+ ions are often grafted to linker vacancy defect sites in the cluster or anchored to the surface of the MOF particles as TiO2.76,77 It should be noted that higher metal exchange values have been achieved by using Ti3+ with other M3+-based MOFs such as MIL-100.75 However, Ti3+ ions are not stable and rapidly oxidize to Ti4+ upon exposure to oxygen in ambient conditions, which compromises the stability of the resulting framework.
Compared to indirect modification, de novo synthesis can provide better control for homogenous metal distributions across the framework, which can be found either in well-defined crystallographic positions or disordered between equivalent crystallographic sites (e.g., solid solution). Though this route often leads to heterobimetallic Ti-M SBU, there are also examples of heterometallic Ti MOFs built from the combination of homometallic Ti and M clusters.78–81
Perhaps one of the most recurrent SBUs in reticular chemistry is the trinuclear unit of formula [M3(μ3-O)(O2C)6X3] (X = F−, Cl−, O2−, OH−, OH2), which is present in frameworks as MIL-88,82 MIL-100,83 and MIL-101,84 to cite a few. The importance of this SBU relies on the fact that it can accommodate different metals in different oxidation states, where the excess positive charge can be balanced with different X− anions in the axial positions of the metallic ions (Fig. 3a). One of the earliest examples of heterometallic Ti frameworks based on this cluster are the CTOF-1 and CTOF-2 series (CTOF = Cobalt–Titanium Organic Framework), built from Co(II), Ti(IV) and terephthalate linkers.85 Both CTOF-1 and CTOF-2 featured Co and Ti in the SBU in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Co:Ti ratio and displayed the same structure and topology as the MOF family known as MIL-88B. More recently, we reported the formation of the MUV-101(M) (M = Mg, Fe, Co, Ni) family, based on heterobimetallic [TiIVM2(μ3-O)(O2C)6X3] (X = OH−, OH2) clusters.42,64 In this case, the linker used was trimesic acid, which in combination with this SBU naturally leads to the formation of the MIL-100 structure with mtn topology. Compared to the CTOF family, this SBU permits combining Ti(IV) with other 1st row transition metals into a persistent node compatible with the formation of isoreticular frameworks (Fig. 3a).
:1 Co:Ti ratio and displayed the same structure and topology as the MOF family known as MIL-88B. More recently, we reported the formation of the MUV-101(M) (M = Mg, Fe, Co, Ni) family, based on heterobimetallic [TiIVM2(μ3-O)(O2C)6X3] (X = OH−, OH2) clusters.42,64 In this case, the linker used was trimesic acid, which in combination with this SBU naturally leads to the formation of the MIL-100 structure with mtn topology. Compared to the CTOF family, this SBU permits combining Ti(IV) with other 1st row transition metals into a persistent node compatible with the formation of isoreticular frameworks (Fig. 3a).
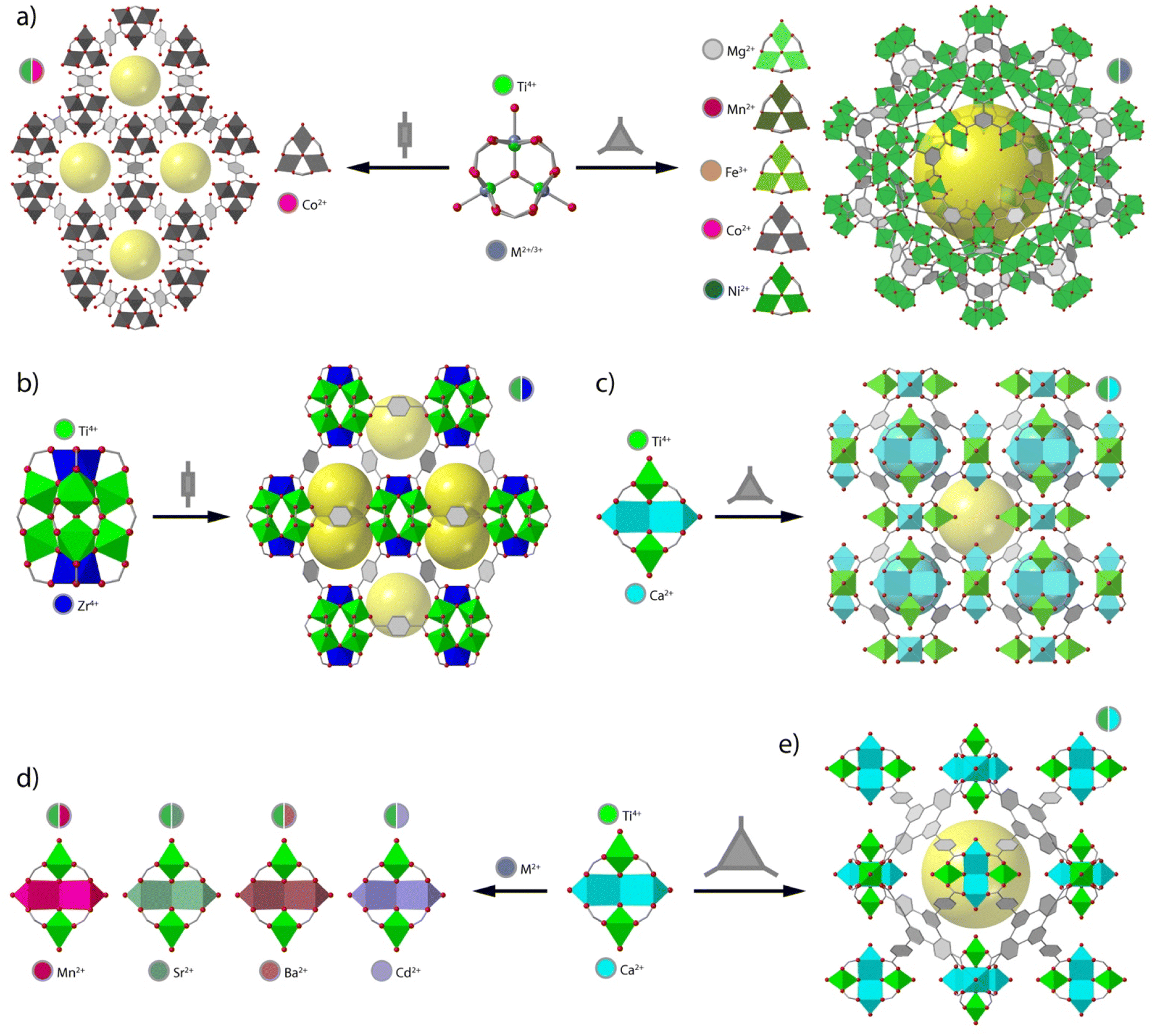 | ||
| Fig. 3 (a) Heterobimetallic titanium frameworks built from trinuclear clusters with no preferential coordination environments for a statistical distribution of metals across the framework. CTOF-1 (left) and MUV-101(M) (right) families and their corresponding mixed-metal node combinations. Examples of bimetallic titanium frameworks with distinctive coordination environments for each metal in (b) PCN-415 and (c) MUV-10(Ca). (d) Diversification of metal node chemistry in Ti2Ca2 clusters and (e) isoreticular expansion of the MUV-10 framework. | ||
It is worth noting that in this trigonal cluster, all metals display octahedral coordination environments with no preference for one site or another. Though compliant with the formation of mixed-metal nodes, this can lead to the formation solid solutions and the possibility of short-range ordering from the creation of monometallic domains,86 which can in turn affect the framework properties. In contrast, the use of different metals with preference for distinct coordination environments leads to heterometallic MOFs in which the metallic ions occupy well-defined crystallographic independent sites. This was first highlighted in 2013 with the synthesis of the heterometallic ZTOF-1 and ZTOF-2 (ZTOF = Zinc–Titanium–Organic Framework), constructed from Ti(IV), Zn(II) and 2-hydroxyterephthalic acid (H2bdc-OH) and 3-hydroxy-2,7-naphthalenedicarboxylic acid (H2ndc-OH), respectively.78,79 The preference of Ti(IV) and Zn(II) for octahedral and tetrahedral coordination geometries, respectively, allowed the identification of both metals with single-crystal X-ray diffraction. However, ZTOF-1 and ZTOF-2 are built from different Zn6Ti2 and Zn3Ti2 nodes, thus preventing the use of this clusters in the design of alternative frameworks by reticular design.
One of the first examples of heterometallic Ti-based MOF with a persistent SBU was reported in 2018 by Zhou and co-workers.87 This material, named as PCN-415 (Fig. 3b), displayed heterometallic Ti8Zr2 This SBU is composed of a central Ti8(μ-O)4 cube in which the Ti(IV) atoms sit at the vertices. The two remaining Zr(IV) atoms sit on opposite faces of the Ti8 cube and are linked to the Ti ions through 8 μ3-O2− ions generating the [Ti8Zr2(μ-O)4(μ3-O)8]16+ core. This inorganic node is bridged by 16 terephthalate connectors to generate a porous structure with fcu topology. Furthermore, the authors succeeded in the synthesis of PCN-416 by employing 2,6-naphthalen-dicarboxylic acid (H2ndc) as linker. Later that year, our group developed a series of heterometallic Ti2M2, named MUV-10(M) (M = Ca, Mn; MUV = Material from University of Valencia) by combining Ti(IV), Ca(II) or Mn(II) and trimesic acid (Fig. 3c).41 This resulted in a porous cubic structure with tetranuclear SBUs of formula [Ti2Ca2(μ3-O)2(O2C)8(H2O)4] where the Ti(IV) ions occupy the positions with an octahedral coordination geometry whereas the Ca atoms are in a trigonal prismatic geometry. The assembly of the Ti2M2 units with the trimesate linkers gave rise to a porous cubic 3,8-connected framework with the topology. Surprisingly, these materials showed excellent chemical stability in H2O in a wide range of pH values, even despite the presence of soft Ca(II) or Mn(II) sites in the structure, which highlights the importance of Ti(IV)–O bonds in endowing the framework with superior chemical stability. Brozek and co-workers recently used this same cluster to synthesise an isostructural family of frameworks by incorporating different Lewis acid ions (Sr2+, Ba2+, and Cd2+) to the Ti2M2 cluster for combination of open metal sites and photoactive Ti centers (Fig. 3d).88 Based on this same cluster, we have also succeeded in isolating MUV-12(Ca), an isoreticular expanded version of the MUV-10 framework, by using 1,3,5-tris(4-carboxyphenyl)benzene (H3btb) as a linker.89 MUV-12 is doubly interpenetrated as result of the framework expansion and concomitant increase of the pore size, and represents one of the first successful examples of isoreticular expansion in titanium frameworks (Fig. 3e).
Effect of additional metal in controlling function
The combination of two or more metals in a MOF structure has emerged as an appealing approach to endow these frameworks with additional functions beyond linker modification. Our focus in this article is to highlight the possibilities offered by titanium frameworks. Should the reader feel more curious about the state-of-the-art of the properties and applications of mixed-metal MOFs, we recommend some of the excellent reviews that have been published recently.12,13,97Metal-induced topological transformations
The different metal-exchange chemistries associated to the presence of a highly polarizing metal ion like Ti4+ with softer metals in these heterobimetallic clusters can be used to modify the reactivity of the framework with metals in solution. We recently demonstrated this possibility for MUV-10(Ca).42 It was found that the combination of hard (Ti) and soft (Ca) metals in the SBU enabled a controlled metal exchange in the soft positions that triggers either metal exchange or different structural transformations upon the formation of heterobimetallic nodes of variable connectivity, all controlled by the identity of the metal in solution (Fig. 4). This behaviour was rationalized by calculating the energy balance for the replacement of Ca2+ sites with other 1st row transition metal ions. The formation of an isostructural cluster is only thermodynamically favourable for Mn2+, whereas it is disfavoured for Co2+, Ni2+, Fe2+, or Zn2+. Our DFT calculations showed that these metals have a preference for the octahedral coordination geometry and are responsible for the gradual transformation of the Ti2Ca2 units into heterometallic [TiIVMII2(μ3-O)(O2C)6(OH2)3] units, with the subsequent topological transformation for the formation of MUV-101(M) mtn frameworks. On the other hand, when using Cu(II), the preference for a square planar geometry of Cu(II) ions, leads to the transformation of MUV-10(Ca) into MUV-102(Cu). This material is an isostructural analogue of HKUST-1 but built from heterometallic paddlewheel [TiIVCuII(O2C)4(O)(OH2)] units for the formation of a 4,3-connected tbo net. In both cases, this dynamic transformation could be followed and controlled with time and temperature, allowing for the formation of hierarchical micro/mesoporous MOF composites at different reaction times.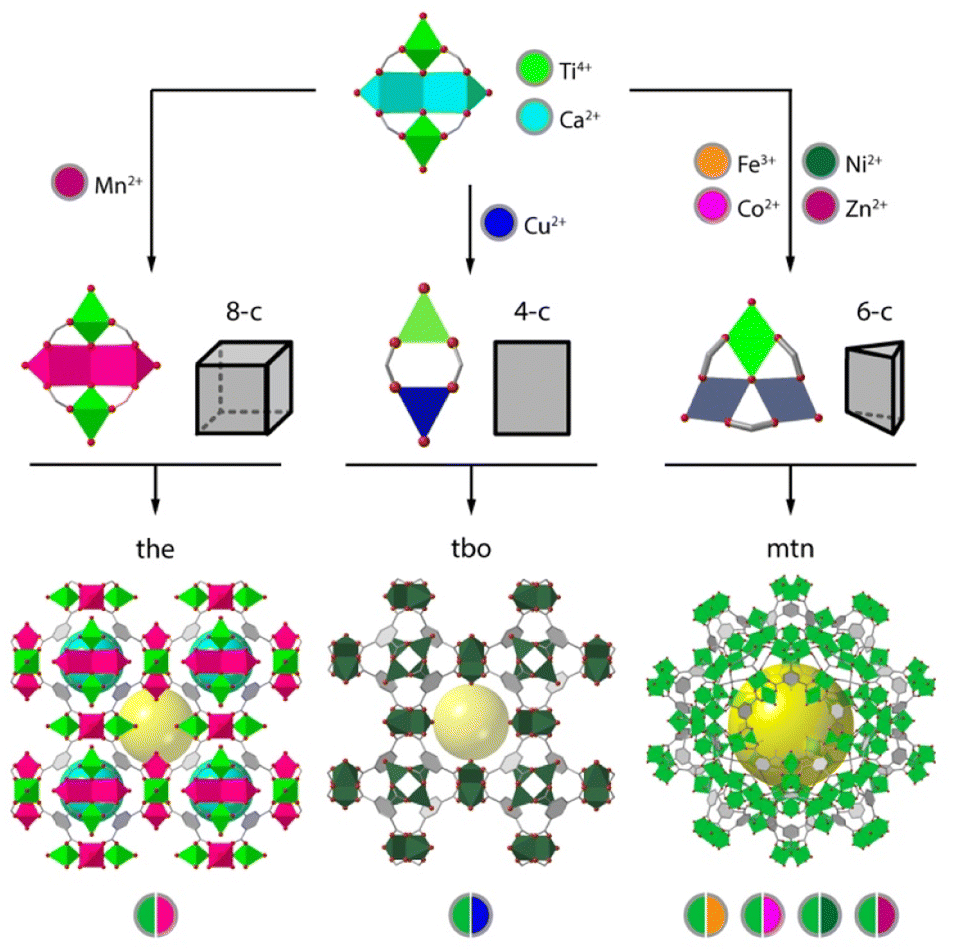 | ||
| Fig. 4 Metal-exchange reactions with the Ti2Ca2 clusters in MUV-10 lead to heterobimetallic titanium frameworks of varying connectivity and chemical composition, which results in the formation of the corresponding net topologies all fixed by the identity of the incoming metal. | ||
Engineering of electronic structure and photoactivity
The application of homometallic Ti-MOFs as MIL-125,55 PCN-415,87 NTU-9,90 COF-69,91 MIL-100(Ti)92 and MUV-11 (ref. 93) in photocatalysis has been motivated by the suitability of their optical band gaps, both experimental and computational, with visible-light photoactivity. However, this is a simplistic approximation as not all of them render good photocatalytic activities due to relative changes in the population of the density of states in the conduction band (CB) or lowest unoccupied crystalline orbital (LUCO), which can be dominated by titanium. This is the case for NTU-9 and MUV-11,93 whose LUCOs are located simultaneously at the organic linker and the metal for more favourable ligand-to-ligand transition, that results in neither spatial separation nor stable excited organic radicals for poor photocatalytic performance. This intrinsic limitation can be circumvented by functionalizing the organic linker with substituents to maximize light absorption or incorporating a co-catalyst as exemplified by MIL-125.94–96In this context, the combination of titanium with different metals can give rise to changes in the electronic structure of the framework. From an experimental point of view, changes in the optical band gap can be estimated via analysis of the absorption spectrum of MOFs in the UV-vis-NIR region obtained by diffuse reflectance spectroscopy (DRS). However, the dual nature of MOFs that lie between molecular and extended solids, makes it difficult to determine the band gap with this methodology in some cases. Brozek, Hendon and co-workers came up with a simple methodology to accurately estimate the band gap from DRS measurements depending on the nature of the electronic transition.88 For systems with partially filled d-orbitals, the use of the Tauc plot used for semiconductors is preferred due to the high number of overlapping electronic transitions. For a meaningful analysis though, the electronic structure and density of states (DOS) must be computed first to determine the type of electronic transitions. In turn, for MOFs with empty d-orbitals it is preferable to use Gaussian curves to fit the DRS spectra. This type of analysis fits better with the theoretical predictions made for close-shell metals with more localised transitions and is more adequate for assigning accurate band gap energies.
In titanium frameworks, the reduction of the optical bandgap is of particular importance for increasing light absorption and photocatalytic activity with visible light. We demonstrated this concept with the isostructural MUV-10(Ca) and MUV-10(Mn) MOFs.41 The substitution of closed-shell Ca2+ with Mn2+ ions triggered a reduction of the optical bandgap from 3.26 to 2.56 eV. An analysis of the DOS of both frameworks suggested that the narrowing of the band gap was due to the contribution of Mn 3d orbitals to the valence band (VB) or lowest occupied crystal orbital (LUCO) of the solid, which improved the visible light absorption of MUV-10(Mn) (Fig. 5a). In turn, the LUCO of MUV-10(Ca) is dominated by the contribution of Ti 3d orbitals for a band gap similar to TiO2, which is only active under UV light. The narrowing of the band gap is the main responsible for the boost of photocatalytic activity of MUV-10(Mn) in the H2 evolution reaction (HER) without additional co-catalysts for a total H2 production of near 6500 μmol g−1 after 24 hours. Based on this framework, Brozek and Hendon recently proposed that the substitution of alkaline earth metals in the Ti2M2 cluster (M = Cd, Sr, and Ba) was an effective way to stabilize the frontier orbital energies of these frameworks compared to MUV-10(Ca).88 As shown in Fig. 5b, the Ti-based orbitals are stabilized by the larger networks created by the softer orbitals in Sr2+ and Ba2+ metals. Alongside with the possibility of creating open metal sites at the photoactive Ti centres, these works anticipate how these heterometallic MUV-10(M) frameworks are excellent platforms to explore new possibilities in heterogeneous photoredox reactivity.
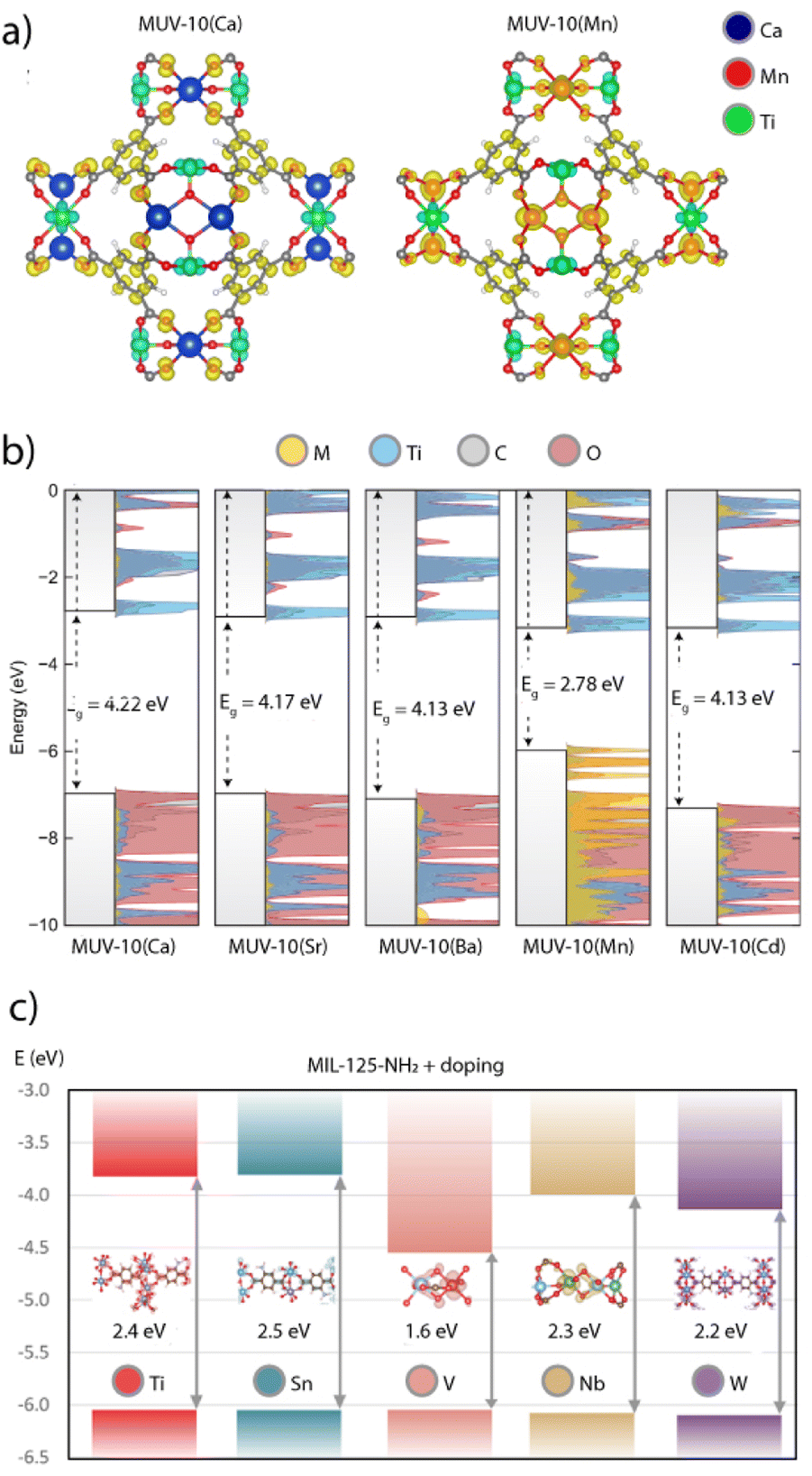 | ||
| Fig. 5 (a) Electron density in the VB (yellow) and CB (red) of MUV-10(Ca) and MUV-10(Mn) frameworks. (b) Density functional theory DOS calculations for all MUV-10(M) derivatives showing the impact of metal node modification in their theoretical band gaps. (c) Calculated impact of metal doping in the theoretical band gap of MIL-125-NH2. Adapted from ref. 41, 86 and 89. | ||
Syzgantseva and colleagues employed a similar method to reduce the bandgap of MOFs by metal node substitution. By using computational methods, they examined how the electronic structures of MIL-125-NH2 and UiO-66-NH2 might be affected by various metal dopants at varying concentrations.98 The electron affinity of the dopant, and the absolute position of the CB edge or highest occupied crystal orbital (HOCO) of the associated oxides, were used as the two main selection factors for choosing the dopant. The authors investigated the substitution of Ti with V, Sn, W, and Nb in MIL-125-NH2 and the replacement of Zr with Ta, Nb, Y, or W in UiO-66-NH2. The substitution of Ti4+ with V5+ led to a strong localization effect of the electronic density on the dopant as result of the injection of V electronic states near the HOCO edge. This is supported by a drastic reduction in the band gap of approximately 0.8 eV when 12.5% of Ti sites are replaced with V, which decreases continuously with higher amounts of V, down to a minimum of 1.1 eV for 50% of Ti replacement. Compared to V, replacing Ti with other metals results in comparatively modest or even negligible localization effects for much smaller band gap decreases (Fig. 5c).
A similar effect was observed in MUV-101(Co), an isostructural analogue of MIL-100 based on heterobimetallic [TiCo2(μ3-O)(O2C)6(OH2)3] units. Compared to the Ti3 nodes in MIL-100(Ti),92 the presence of Co2+ in the TiCo2 cluster endows a narrower band gap of 2.7 eV for visible-light activity.99 This caused a dramatic increase of the photocatalytic H2 production rate via hydrolysis of ammonia borane for TOF values of 113.7 mol gcat−1 min−1, far superior to the performance of P25 and MIL-125 under the same conditions. Based on computational data, the authors suggested that this increase was possibly due to the presence of a mixed-valence state involving Co2+/3+ centers that was photogenerated by a metal-to-metal charge transfer (MMCT) mechanism. This possibility is certainly interesting from a catalytic standpoint, but further experimental and computational work needs to be carried out in this and other heterometallic Ti-MOFs to better understand the role of Ti4+ sites and its interaction with other metallic ions in a periodic model.
Dual-metal synergistic catalysis and mixed oxide precursors
The interplay between different metal combinations can also give rise to the appearance of interesting features not accesible to single metal nodes.100 The activity of the heterobimetallic titanium frameworks MUV-101 towards the degradation of nerve agent simulants is a good example of this.64 Compared to other TiM2 clusters (M = Mg, Co, Ni), only the TiFe2 nodes in MUV-101(Fe) were capable of hydrolysing the nerve agent simulant DIFP (DIFP = diisopropyl-fluorophosphate) in pure water and in the absence of basic co-catalysts such as N-ethyl-morpholine. Surprisingly, its homometallic analogues MIL-100(Fe) and MIL-100(Ti) as well as a physical mixture of them also showed little or no catalytic activity, with MIL-100(Fe) only being able to degrade ca. 8% after 24 hours. These results led to propose a dual-metal catalytic reaction mechanism by which the DIFP molecules firstly bind to the Ti(IV) ions acting as Lewis acid sites. This immobilizes the DIFP molecule and activates the P(V) centre towards a nucleophilic attack (Fig. 6a). The second step of the mechanism involves the hydrolysis of a water molecule via the FeIII–OH sites of an adjacent SBU. These moieties act as Brønsted basic sites, thus generating hydroxyl anions that would attack the P(V) site for the hydrolysis of the P–F bond and formation of a non-toxic phosphate and hydrofluoric acid. This mechanism was supported by DFT calculations and reminds that intrinsic to bimetallic enzymes, where the combination of Ti4+ Lewis acid and Fe3+–OH Brønsted base sites leads to a lower energy barrier for more efficient degradation of organophosphates in the absence of a base. Very recently, we have used this same control over cluster chemistry to control Brønsted acidity in heterobimetallic titanium frameworks (Fig. 6b).101 Their use as catalysts for the aminolysis of epoxides confirm that only the TiFe2 nodes display an intrinsic activity comparable to other benchmark MOF catalysts indirectly modified with strong acids. Whereas the Lewis acidity intrinsic to Ti4+ or Fe3+ sites is present in all simple (Ti3, Fe3) and bimetallic (TiFe2, TiCo2, TiNi2) nodes, only the Brønsted acidity of the TiFe2 cluster might enable a dual-metal mechanism for protonation of the epoxide to facilitate the nucleophilic attack by the amine.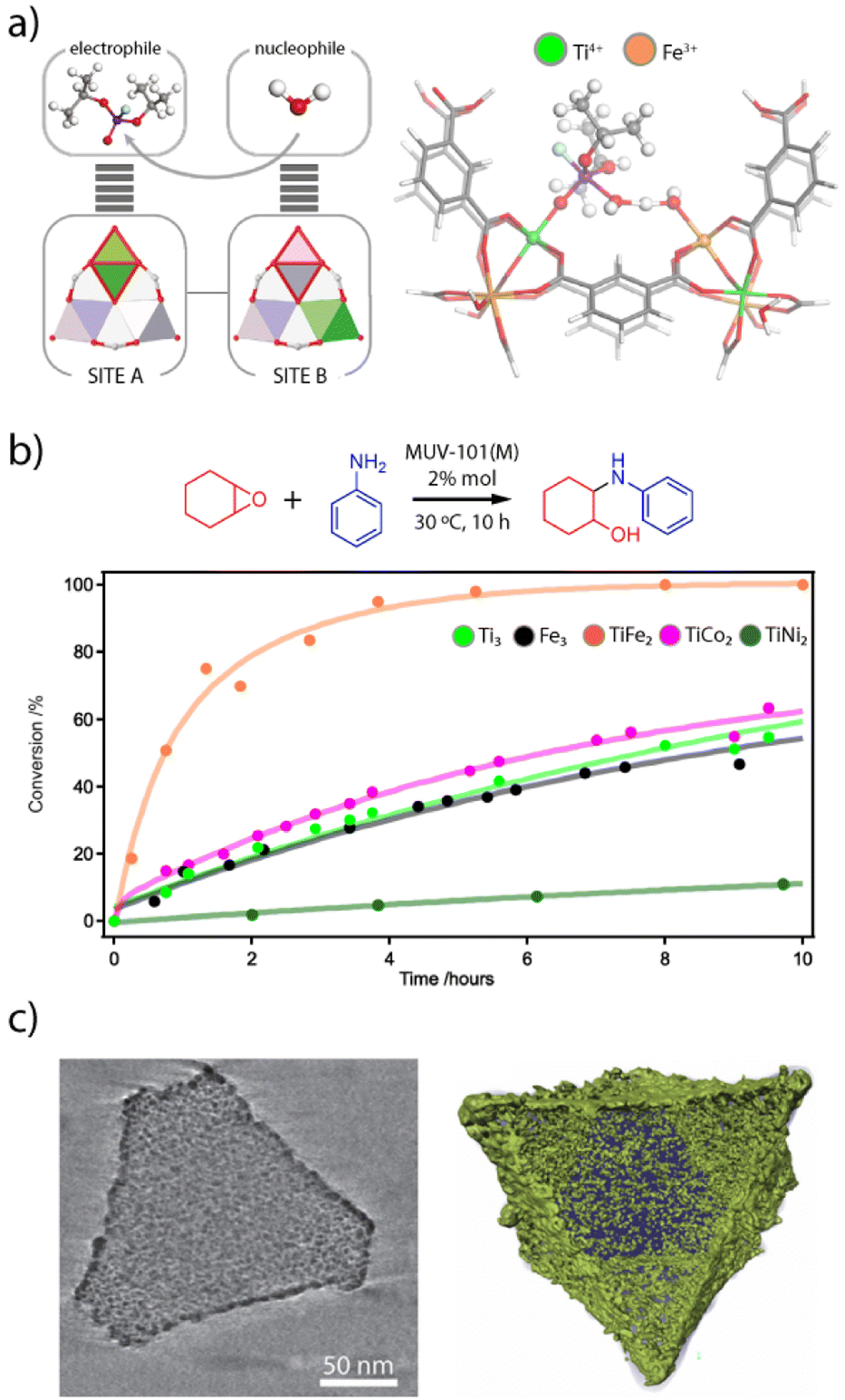 | ||
| Fig. 6 (a) Scheme illustrating the cooperative activation of DIFP and water molecules at neighbouring metal sites in MUV-101(Fe) (left). Proposed reaction mechanism for the synergetic degradation of nerve agents with TiFe2 clusters (right). (b) Changes in the catalytic activity of M3 and TiM2 nodes for the aminolysis of epoxides. c) Reconstructed tomogram and surface-rendered 3D reconstruction of a representative TiFe/C catalyst prepared by pyrolysis of MUV-101(Fe). Metal oxide NPs are represented in green. Adapted from ref. 83, 93 and 97. | ||
This compositional control over cluster chemistry can be also used to generate new mixed oxides by calcination of these frameworks. MOFs have long been used as single-source precursors for the generation of nanostructured oxides and carbon@oxide composites via heat treatment as it enables control of the particle size and shape.102,103 In the case of heterometallic MOFs, it has been observed that homogenous metal distributions in the framework are necessary to produce oxides with homogeneous metal distributions.104 Moreover, the particle size, phase, and composition of the oxide can be controlled using this technology by adjusting factors such as temperature, atmosphere, and different metal ratios, also allowing for the generation of mixed oxides with unprecedented stoichiometries. This is again the case of MUV-101(Fe). In collaboration with Gascón and co-workers, we demonstrated that the controlled pyrolysis of this material in inert atmosphere led to the formation of mixed Ti–Fe oxide nanoparticles (TiFe/C) with homogeneous metal distribution supported by a porous carbon matrix (Fig. 6c).105 An extensive characterisation via PXRD, PDF, X-ray photoelectron spectroscopy and TEM computed tomography revealed that the Ti–Fe oxide nanoparticles adopt a highly defective titanomaghemite structure of formula FeIII0.55,□0.45[FeIII0.41TiIV0.48,□1.11]O0.8(OH)3.2 with an unprecedented Ti:Fe ratio, and average nanoparticle (NPs) sizes of 2.2 and 4.5 nm, for the NPs inside and outside the porous carbon matrix. The presence of Ti4+ sites and the large number of vacancies helped promoting the direct hydrogenation of CO2 to CO at low temperatures (375 °C) with unprecedented selectivity (>99%) and no sign of deactivation after several days on stream. The pyrolysis of MIL-100(Fe) or MIL-100(Ti) separately or in a physical mixture, led in turn to the formation of TiO2 and Fe3C. This emphasises how crucial it is to have uniform metal distributions to achieve a homogeneous integration of titanium into the maghemite structure, thus highlighting the potential of heterometallic MOFs as precursors to mixed metal oxides that cannot be prepared by conventional synthetic methods.
Metal-controlled grafting for framework modification
Heterometallic nodes based on metals with different chemistries can also enable the functionalization of the framework in specific sites by exploiting their different affinity for different binding groups. We demonstrated this possibility with the Ti2Ca2 clusters in MUV-10 and its expanded isoreticular analogue MUV-12.89 The possibility of generating open vacant Ti4+ and soft Ca2+ sites with different polarizing power and acidities was used to direct the grafting of diamine 1,3-diamino-propane (DAP) only to the last (Fig. 7). This enabled the selective functionalization of the framework for the combination of Ti4+ Lewis acid centres and –NH2 Brønsted basic sites as confirmed by XPS and CO2 adsorption measurements. The increase in porosity of MUV-12 also allowed for an additional modification of free –NH2 groups by alkylation reaction to produce alkylammonium salts, which are the co-catalysts typically used to tackle epoxide apertures by CO2 cycloaddition reaction.106 In MUV-12-DAP(CH3I), the cooperative action of free Ti4+ Lewis acid sites and –N+(CH3)3 salts in the same pore led to the complete conversion of propylene oxide into the corresponding carbonate at room temperature and atmospheric pressure in the absence of additional catalysts, while maintaining its activity over several consecutive cycles. This example illustrates how heterometallic nodes can be also used to control the positioning and distribution of functional molecules in MOFs to combine multiple functionalities for synergistic interaction.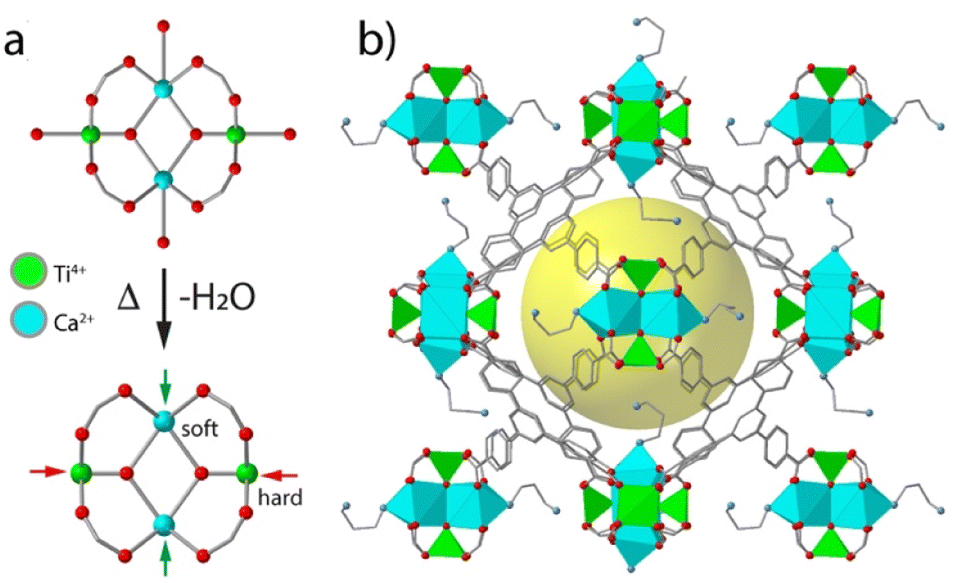 | ||
| Fig. 7 (a) Scheme illustrating the generation of hard and soft open metal sites in the heterometallic Ti2Ca2 clusters by thermal desolvation of MUV-10. (b) Simulated structure of MUV-12-DAP showing the selective grafting of diamines to vacant Ca2+ sites. Adapted from ref. 87. | ||
Conclusions and perspectives
The principles controlling the assembly and modification of MOFs have enabled to accelerate the design of over 90000 materials for unrivalled chemical and structural versatilities.107 The field is now shifting from the concepts established to control the assembly of default topologies by suitable choice of binary combinations of organic and inorganic blocks, into new strategies and characterization techniques that intend to implement and visualize the compositional changes associated to the use of multiple combinations of these units for a fixed framework.
When it comes to mixed-metal inorganic blocks, this multivariance or chemical complexity is often considered random in nature, but recent works anticipate the potential of this approach to access new functions provided the integration of these elements can be controlled not only in average ratio but with specific spatial distributions.108 In other words, when it comes to dictating the function of the framework, the way in which elements are arranged can be as important as which elements or in which proportion they are combined. After summarizing general aspects on the synthesis and analysis of metal variance in mixed-metal MOFs, we have used recent advances in the chemistry of titanium–organic frameworks to outline how cluster chemistry can be used to control framework function in aspects that include solid-state reactivity, electronic structure, dual-metal catalysis, or the design of mixed oxide precursors among others. Provided some of its intrinsic properties as redox versatility, photoactivity or robust coordination bonds, the combination of titanium with other transition metal ions might offer unrivalled possibilities to unleash the potential of chemical complexity in accessing mixed valency to enable charge delocalization for conducting photoredox catalysts.
Author contributions
All authors contributed to the discussion, participated in the writing of the original draft, and revised the manuscript. J. C.-G. and N. A.-B. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EU (ERC-2021-COG-101043428), the Generalitat Valenciana (PROMETEU/2021/054, SEJIGENT/2021/059 & MFA/2022/026) and the Spanish government (CEX2019-000919-M & PID2020-118117RB-I00). J.C.-G. thanks the GVA for an APOSTD postdoctoral grant (CIAPOS/2021/272). B. L.-B. thanks the Spanish government and Next Generation EU funding for a postdoctoral grant (MS21-127). N. M. P. thanks La Caixa Foundation for a Postdoctoral Junior Leader–Retaining Fellowship (LCF/BQ/PR20/11770014).Notes and references
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- J. Jia, L. Gutiérrez-Arzaluz, O. Shekhah, N. Alsadun, J. Czaban-Jóźwiak, S. Zhou, O. M. Bakr, O. F. Mohammed and M. Eddaoudi, J. Am. Chem. Soc., 2020, 142, 8580–8584 CrossRef CAS PubMed.
- Q. Xia, Z. Li, C. Tan, Y. Liu, W. Gong and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8259–8266 CrossRef CAS PubMed.
- L. Feng, S. Yuan, L.-L. Zhang, K. Tan, J.-L. Li, A. Kirchon, L.-M. Liu, P. Zhang, Y. Han, Y. J. Chabal and H.-C. Zhou, J. Am. Chem. Soc., 2018, 140, 2363–2372 CrossRef CAS PubMed.
- V. Guillerm, H. Xu, J. Albalad, I. Imaz and D. Maspoch, J. Am. Chem. Soc., 2018, 140, 15022–15030 CrossRef CAS PubMed.
- J. Geary, A. H. Wong and D. J. Xiao, J. Am. Chem. Soc., 2021, 143, 10317–10323 CrossRef CAS PubMed.
- R. A. Dodson, A. P. Kalenak and A. J. Matzger, J. Am. Chem. Soc., 2020, 142, 20806–20813 CrossRef CAS PubMed.
- B. Lerma-Berlanga, C. R. Ganivet, N. Almora-Barrios, S. Tatay, Y. Peng, J. Albero, O. Fabelo, J. González-Platas, H. García, N. M. Padial and C. Martí-Gastaldo, J. Am. Chem. Soc., 2021, 143, 1798–1806 CrossRef CAS PubMed.
- D. Yu, Q. Shao, Q. Song, J. Cui, Y. Zhang, B. Wu, L. Ge, Y. Wang, Y. Zhang, Y. Qin, R. Vajtai, P. M. Ajayan, H. Wang, T. Xu and Y. Wu, Nat. Commun., 2020, 11, 927 CrossRef CAS PubMed.
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Angew. Chem., Int. Ed., 2019, 58, 15188–15205 CrossRef CAS PubMed.
- S. Abednatanzi, P. G. Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. V. D. Voort and K. Leus, Chem. Soc. Rev., 2019, 48, 2535–2565 RSC.
- L. J. Wang, H. Deng, H. Furukawa, F. Gándara, K. E. Cordova, D. Peri and O. M. Yaghi, Inorg. Chem., 2014, 53, 5881–5883 CrossRef CAS PubMed.
- V. Rubio-Giménez, J. C. Waerenborgh, J. M. Clemente-Juan and C. Martí-Gastaldo, Chem. Mater., 2017, 29, 6181–6185 CrossRef.
- G. Ayoub, B. Karadeniz, A. J. Howarth, O. K. Farha, I. Đilović, L. S. Germann, R. E. Dinnebier, K. Užarević and T. Friščić, Chem. Mater., 2019, 31, 5494–5501 CrossRef CAS.
- C. Castillo-Blas, V. A. de la Peña-O’Shea, I. Puente-Orench, J. R. de Paz, R. Sáez-Puche, E. Gutiérrez-Puebla, F. Gándara and Á. Monge, Sci. Adv., 2017, 3, e1700773 CrossRef PubMed.
- C. Castillo-Blas, N. López-Salas, M. C. Gutiérrez, I. Puente-Orench, E. Gutiérrez-Puebla, M. L. Ferrer, M. A. Monge and F. Gándara, J. Am. Chem. Soc., 2019, 141, 1766–1774 CrossRef CAS PubMed.
- T. Chen, J.-H. Dou, L. Yang, C. Sun, N. J. Libretto, G. Skorupskii, J. T. Miller and M. Dincă, J. Am. Chem. Soc., 2020, 142, 12367–12373 CrossRef CAS PubMed.
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J. M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2012, 48, 10237–10239 RSC.
- S. Wongsakulphasatch, F. Nouar, J. Rodriguez, L. Scott, C. L. Guillouzer, T. Devic, P. Horcajada, J.-M. Grenèche, P. L. Llewellyn, A. Vimont, G. Clet, M. Daturi and C. Serre, Chem. Commun., 2015, 51, 10194–10197 RSC.
- D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723–5729 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- K. Manna, T. Zhang, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed.
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 124, 10453–10456 CrossRef.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
- N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501–3509 CrossRef CAS PubMed.
- C. K. Brozek and M. Dinča, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef CAS.
- X. Song, S. Jeong, D. Kim and M. S. Lah, CrystEngComm, 2012, 14, 5753–5756 RSC.
- Z. Wei, W. Lu, H.-L. Jiang and H.-C. Zhou, Inorg. Chem., 2013, 52, 1164–1166 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed.
- D. Denysenko, T. Werner, M. Grzywa, A. Puls, V. Hagen, G. Eickerling, J. Jelic, K. Reuter and D. Volkmer, Chem. Commun., 2011, 48, 1236–1238 RSC.
- F. Trousselet, A. Archereau, A. Boutin and F.-X. Coudert, J. Phys. Chem. C, 2016, 120, 24885–24894 CrossRef CAS.
- C. Castillo-Blas and F. Gándara, Isr. J. Chem., 2018, 58, 1036–1043 CrossRef CAS.
- J. D. Howe, C. R. Morelock, Y. Jiao, K. W. Chapman, K. S. Walton and D. S. Sholl, J. Phys. Chem. C, 2017, 121, 627–635 CrossRef CAS.
- R. M. Marti, J. D. Howe, C. R. Morelock, M. S. Conradi, K. S. Walton, D. S. Sholl and S. E. Hayes, J. Phys. Chem. C, 2017, 121, 25778–25787 CrossRef CAS.
- A. Sapnik, H. Geddes, E. Reynolds, H. H.-M. Yeung and A. Goodwin, Chem. Commun., 2018, 54, 9651–9654 RSC.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, J. Albero, A. R. Ruiz-Salvador, J. González-Platas, H. García and C. Martí-Gastaldo, Angew. Chem., Int. Ed., 2018, 57, 8453–8457 CrossRef CAS PubMed.
- N. M. Padial, B. Lerma-Berlanga, N. Almora-Barrios, J. Castells-Gil, I. da Silva, M. de la Mata, S. I. Molina, J. Hernández-Saz, A. E. Platero-Prats, S. Tatay and C. Marti-Gastaldo, J. Am. Chem. Soc., 2020, 142, 6638–6648 CrossRef CAS PubMed.
- C. K. Brozek, L. Bellarosa, T. Soejima, T. V. Clark, N. López and M. Dincă, Chem. - Eur. J., 2014, 20, 6871–6874 CrossRef CAS PubMed.
- L. Bellarosa, C. K. Brozek, M. García-Melchor, M. Dincă and N. López, Chem. Mater., 2015, 27, 3422–3429 CrossRef CAS.
- F. Gul-E-Noor, B. Jee, M. Mendt, D. Himsl, A. Pöppl, M. Hartmann, J. Haase, H. Krautscheid and M. Bertmer, J. Phys. Chem. C, 2012, 116, 20866–20873 CrossRef CAS.
- X. Feng, P. Ji, Z. Li, T. Drake, P. Oliveres, E. Y. Chen, Y. Song, C. Wang and W. Lin, ACS Catal., 2019, 9, 3327–3337 CrossRef CAS.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- H. Depauw, I. Nevjestić, J. D. Winne, G. Wang, K. Haustraete, K. Leus, A. Verberckmoes, C. Detavernier, F. Callens, E. D. Canck, H. Vrielinck and P. V. D. Voort, Chem. Commun., 2017, 53, 8478–8481 RSC.
- K. Leus, M. Vandichel, Y.-Y. Liu, I. Muylaert, J. Musschoot, S. Pyl, H. Vrielinck, F. Callens, G. B. Marin, C. Detavernier, P. V. Wiper, Y. Z. Khimyak, M. Waroquier, V. V. Speybroeck and P. V. D. Voort, J. Catal., 2012, 285, 196–207 CrossRef CAS.
- I. Nevjestić, H. Depauw, P. Gast, P. Tack, D. Deduytsche, K. Leus, M. V. Landeghem, E. Goovaerts, L. Vincze, C. Detavernier, P. V. D. Voort, F. Callens and H. Vrielinck, Phys. Chem. Chem. Phys., 2017, 19, 24545–24554 RSC.
- M. Mendt, S. Ehrling, I. Senkovska, S. Kaskel and A. Pöppl, Inorg. Chem., 2019, 58, 4561–4573 CrossRef CAS PubMed.
- H. Depauw, I. Nevjestić, G. Wang, K. Leus, F. Callens, E. D. Canck, K. D. Buysser, H. Vrielinck and P. V. D. Voort, J. Mater. Chem. A, 2017, 5, 24580–24584 RSC.
- B. Jee, K. Eisinger, F. Gul-E-Noor, M. Bertmer, M. Hartmann, D. Himsl and A. Pöppl, J. Phys. Chem. C, 2010, 114, 16630–16639 CrossRef CAS.
- K. Metavarayuth, O. Ejegbavwo, G. McCarver, M. L. Myrick, T. M. Makris, K. D. Vogiatzis, S. D. Senanayake, O. M. Manley, A. M. Ebrahim, A. I. Frenkel, S. Hwang, T. Rajeshkumar, J. D. Jimenez, K. Chen, N. B. Shustova and D. A. Chen, J. Phys. Chem. Lett., 2020, 11, 8138–8144 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- A. Cadiau, N. Kolobov, S. Srinivasan, M. G. Goesten, H. Haspel, A. V. Bavykina, M. R. Tchalala, P. Maity, A. Goryachev, A. S. Poryvaev, M. Eddaoudi, M. V. Fedin, O. F. Mohammed and J. Gascon, Angew. Chem., Int. Ed., 2020, 59, 13468–13472 CrossRef CAS.
- S. Bordiga, F. Bonino, K. P. Lillerud and C. Lamberti, Chem. Soc. Rev., 2010, 39, 4885–4927 RSC.
- Y. Song, X. Feng, J. S. Chen, C. Brzezinski, Z. Xu and W. Lin, J. Am. Chem. Soc., 2020, 142, 4872–4882 CrossRef CAS PubMed.
- X. Feng, Y. Song, Z. Li, M. Kaufmann, Y. Pi, J. S. Chen, Z. Xu, Z. Li, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 11196–11203 CrossRef CAS PubMed.
- T. Zhang, K. Manna and W. Lin, J. Am. Chem. Soc., 2016, 138, 3241–3249 CrossRef CAS PubMed.
- W. Shi, Y. Quan, G. Lan, K. Ni, Y. Song, X. Jiang, C. Wang and W. Lin, J. Am. Chem. Soc., 2021, 143, 16718–16724 CrossRef CAS PubMed.
- K. Manna, T. Zhang, M. Carboni, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2014, 136, 13182–13185 CrossRef CAS PubMed.
- K. A. Lomachenko, J. Jacobsen, A. L. Bugaev, C. Atzori, F. Bonino, S. Bordiga, N. Stock and C. Lamberti, J. Am. Chem. Soc., 2018, 140, 17379–17383 CrossRef CAS PubMed.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, R. Gil-San-Millán, M. Romero-Ángel, V. Torres, I. da Silva, B. C. J. Vieira, J. C. Waerenborgh, J. Jagiello, J. A. R. Navarro, S. Tatay and C. Martí-Gastaldo, Chem, 2020, 6, 3118–3131 CAS.
- R. J. Comito, Z. Wu, G. Zhang, J. A. Lawrence, M. D. Korzyński, J. A. Kehl, J. T. Miller and M. Dincă, Angew. Chem., Int. Ed., 2018, 57, 8135–8139 CrossRef CAS PubMed.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef CAS PubMed.
- J. Zhang, B. An, Z. Li, Y. Cao, Y. Dai, W. Wang, L. Zeng, W. Lin and C. Wang, J. Am. Chem. Soc., 2021, 143, 8829–8837 CrossRef CAS PubMed.
- M. R. Mian, L. R. Redfern, S. M. Pratik, D. Ray, J. Liu, K. B. Idrees, T. Islamoglu, L. Gagliardi and O. K. Farha, Chem. Mater., 2020, 32, 3078–3086 CrossRef CAS.
- A. E. Platero-Prats, A. B. League, V. Bernales, J. Ye, L. C. Gallington, A. Vjunov, N. M. Schweitzer, Z. Li, J. Zheng, B. L. Mehdi, A. J. Stevens, A. Dohnalkova, M. Balasubramanian, O. K. Farha, J. T. Hupp, N. D. Browning, J. L. Fulton, D. M. Camaioni, J. A. Lercher, D. G. Truhlar, L. Gagliardi, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2017, 139, 10410–10418 CrossRef CAS PubMed.
- Z. Chen, Z. Chen, O. K. Farha and K. W. Chapman, J. Am. Chem. Soc., 2021, 143, 8976–8980 CrossRef CAS PubMed.
- Z. Ji, T. Li and O. M. Yaghi, Science, 2020, 369, 674–680 CrossRef CAS PubMed.
- H. Assi, G. Mouchaham, N. Steunou, T. Devic and C. Serre, Chem. Soc. Rev., 2017, 46, 3431–3452 RSC.
- I. G. Clayson, D. Hewitt, M. Hutereau, T. Pope and B. Slater, Adv. Mater., 2020, 32, 2002780 CrossRef CAS PubMed.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed.
- L. Zou, D. Feng, T.-F. Liu, Y.-P. Chen, S. Yuan, K. Wang, X. Wang, S. Fordham and H.-C. Zhou, Chem. Sci., 2016, 7, 1063–1069 RSC.
- J. G. Santaclara, A. I. Olivos-Suarez, A. G. Nelson, D. Osadchii, M. A. Nasalevich, M. A. van der Veen, F. Kapteijn, A. M. Sheveleva, S. L. Veber, M. V. Fedin, A. T. Murray, C. H. Hendon, A. Walsh and J. Gascon, Chem. Mater., 2017, 29, 8963–8967 CrossRef CAS.
- M. S. Denny, L. R. Parent, J. P. Patterson, S. K. Meena, H. Pham, P. Abellan, Q. M. Ramasse, F. Paesani, N. C. Gianneschi and S. M. Cohen, J. Am. Chem. Soc., 2018, 140, 1348–1357 CrossRef CAS PubMed.
- K. Hong, W. Bak and H. Chun, Inorg. Chem., 2013, 52, 5645–5647 CrossRef CAS PubMed.
- K. Hong and H. Chun, Chem. Commun., 2013, 49, 10953–10955 RSC.
- W. Xuan, C. Ye, M. Zhang, Z. Chen and Y. Cui, Chem. Sci., 2013, 4, 3154–3159 RSC.
- C. Wang, C. Liu, X. He and Z.-M. Sun, Chem. Commun., 2017, 53, 11670–11673 RSC.
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284–286 RSC.
- G. Ferey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef CAS PubMed.
- G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- K. Hong, W. Bak, D. Moon and H. Chun, Cryst. Growth Des., 2013, 13, 4066–4070 CrossRef CAS.
- Q. Liu, H. Cong and H. Deng, J. Am. Chem. Soc., 2016, 138, 13822–13825 CrossRef CAS PubMed.
- S. Yuan, J.-S. Qin, H.-Q. Xu, J. Su, D. Rossi, Y. Chen, L. Zhang, C. Lollar, Q. Wang, H.-L. Jiang, D. H. Son, H. Xu, Z. Huang, X. Zou and H.-C. Zhou, ACS Cent. Sci., 2017, 4, 105–111 CrossRef PubMed.
- K. Fabrizio, K. A. Lazarou, L. I. Payne, L. P. Twight, S. Golledge, C. H. Hendon and C. K. Brozek, J. Am. Chem. Soc., 2021, 143, 12609–12621 CrossRef CAS PubMed.
- E. López-Maya, N. M. Padial, J. Castells-Gil, C. R. Ganivet, A. Rubio-Gaspar, F. G. Cirujano, N. Almora-Barrios, S. Tatay, S. Navalón and C. Martí-Gastaldo, Angew. Chem., Int. Ed., 2021, 60, 11868–11873 CrossRef PubMed.
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC.
- B. Bueken, F. Vermoortele, D. E. P. Vanpoucke, H. Reinsch, C.-C. Tsou, P. Valvekens, T. D. Baerdemaeker, R. Ameloot, C. E. A. Kirschhock, V. V. Speybroeck, J. M. Mayer and D. D. Vos, Angew. Chem., Int. Ed., 2015, 54, 13912–13917 CrossRef CAS PubMed.
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, I. da Silva, D. Mateo, J. Albero, H. García and C. Martí-Gastaldo, Chem. Sci., 2019, 10, 4313–4321 RSC.
- N. M. Padial, J. Castells-Gil, N. Almora-Barrios, M. Romero-Angel, I. da Silva, M. Barawi, A. García-Sánchez, V. A. P. O'Shea and C. Martí-Gastaldo, J. Am. Chem. Soc., 2019, 141, 13124–13133 CrossRef CAS PubMed.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CrossRef CAS.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. Darras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- B. G. Diamond, L. I. Payne and C. H. Hendon, Commun. Chem., 2023, 6, 67 CrossRef CAS PubMed.
- A. M. Rice, G. A. Leith, O. A. Ejegbavwo, E. A. Dolgopolova and N. B. Shustova, ACS Energy Lett., 2019, 4, 1938–1946 CrossRef CAS.
- M. A. Syzgantseva, C. P. Ireland, F. M. Ebrahim, B. Smit and O. A. Syzgantseva, J. Am. Chem. Soc., 2019, 141, 6271–6278 CrossRef CAS PubMed.
- Y. Wang, H. Lv, E. S. Grape, C. A. Gaggioli, A. Tayal, A. Dharanipragada, T. Willhammar, A. K. Inge, X. Zou, B. Liu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 6333–6338 CrossRef CAS PubMed.
- D. Y. Osadchii, A. I. Olivos-Suarez, A. Szécsényi, G. Li, M. A. Nasalevich, I. A. Dugulan, P. S. Crespo, E. J. M. Hensen, S. L. Veber, M. V. Fedin, G. Sankar, E. A. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542–5548 CrossRef CAS.
- A. Rubio-Gaspar, S. Navalón, S. Tatay, F. G. Cirujano, C. Fernández-Conde, N. M. Padial and C. Martí-Gastaldo, J. Am. Chem. Soc., 2023, 145, 3855–3860 CrossRef CAS PubMed.
- S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef.
- L. Chen, H.-F. Wang, C. Li and Q. Xu, Chem. Sci., 2020, 11, 5369–5403 RSC.
- J.-S. M. Lee, Y. Fujiwara, S. Kitagawa and S. Horike, Chem. Mater., 2019, 31, 4205–4212 CrossRef CAS.
- J. Castells-Gil, S. Ould-Chikh, A. Ramírez, R. Ahmad, G. Prieto, A. R. Gómez, L. Garzón-Tovar, S. Telalovic, L. Liu, A. Genovese, N. M. Padial, A. Aguilar-Tapia, P. Bordet, L. Cavallo, C. Martí-Gastaldo and J. Gascón, Chem Catal., 2021, 1, 364–382 CrossRef CAS.
- J. Liang, R.-P. Chen, X.-Y. Wang, T.-T. Liu, X.-S. Wang, Y.-B. Huang and R. Cao, Chem. Sci., 2017, 8, 1570–1575 RSC.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Nat. Commun., 2020, 11, 4068 CrossRef CAS PubMed.
- W. Xu, B. Tu, Q. Liu, Y. Shu, C.-C. Liang, C. S. Diercks, O. M. Yaghi, Y.-B. Zhang, H. Deng and Q. Li, Nat. Rev. Mater., 2020, 5, 764–779 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |