
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCationic tetranuclear macrocyclic CaCo3 complexes as highly active catalysts for alternating copolymerization of propylene oxide and carbon dioxide†
Haruki
Nagae‡
 a,
Saki
Matsushiro
a,
Jun
Okuda
*b and
Kazushi
Mashima
*a
a,
Saki
Matsushiro
a,
Jun
Okuda
*b and
Kazushi
Mashima
*a
aDepartment of Chemistry, Graduate School of Engineering Science, Osaka University Toyonaka, Osaka 560-8531, Japan. E-mail: mashima@chem.es.osaka-u.ac.jp
bInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, D-52062 Aachen, Germany
First published on 12th July 2023
Abstract
We found that a cationic hetero tetranuclear complex including a calcium and three cobalts exhibited high catalytic activity toward alternating copolymerization of propylene oxide (PO) and carbon dioxide (CO2). The tertiary anilinium salt [PhNMe2H][B(C6F5)4] was the best additive to generate the cationic species while maintaining polymer selectivity and carbonate linkage, even under 1.0 MPa CO2. Density functional theory calculations clarified that the reaction pathway mediated by the cationic complex is more favorable than that mediated by the neutral complex by 1.0 kcal mol−1. We further found that the flexible ligand exchange between Ca and Co ions is important for the alternating copolymerization to proceed smoothly.
Introduction
Alternating copolymerization of carbon dioxide (CO2) and epoxide to produce polyalkylenecarbonates (PACs) has attracted much interest because not only are PACs biodegradable and biocompatible materials (Fig. 1a), but they also contain significant amounts of CO2 upon treatment with commercially available inexpensive comonomers such as propylene oxide (PO), for which the content of CO2 reaches 43% wt of the resulting polypropylenecarbonate (PPC).1–3 Thus, such an efficient CO2 capturing system has prompted many researchers to develop more efficient catalysts, including mononuclear salen complexes,3j,4 mononuclear porphyrin and corrole complexes,5 homo- and hetero-dinuclear complexes,6–8 and hetero-multinuclear complexes,9 with improved catalytic activity, polymer selectivity, and carbonate linkage. Among these complexes, dinuclear complexes together with tetranuclear complexes supported by alkoxide bridged skeletons (Fig. 1b) are investigated most often because of their advantages for catalysing the alternating copolymerization, even under lower CO2 pressure.6–9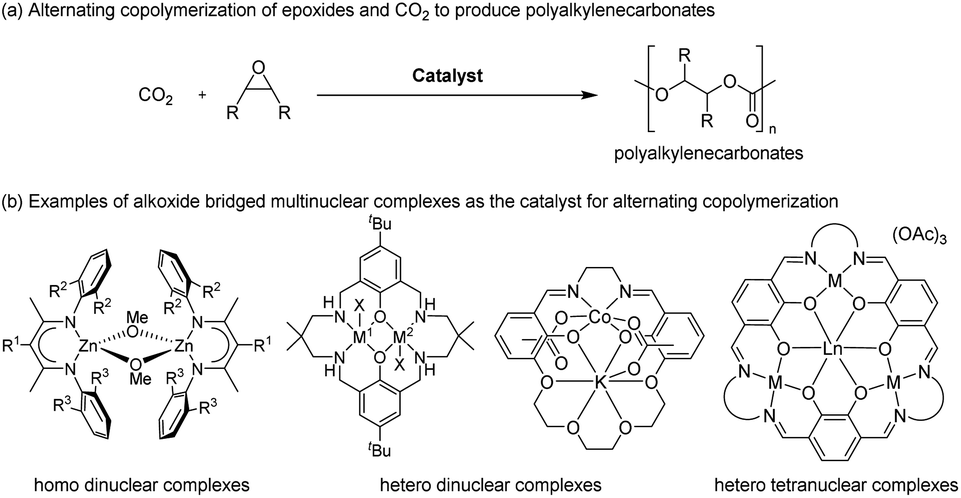 | ||
| Fig. 1 Multinuclear complexes catalysed alternating copolymerization of epoxide and CO2. | ||
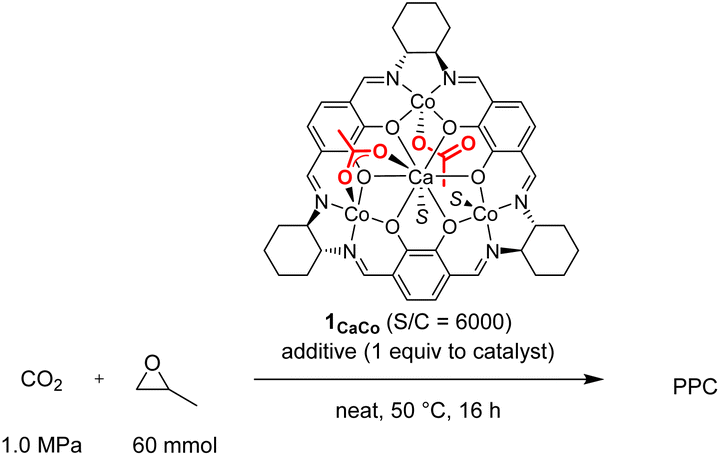
A notable feature of the hetero-multinuclear complex active for alternating copolymerization is that two metal ions exist in proximal positions to cooperatively activate both epoxides and CO2, for which the turnover limiting step is established to be a ring-opening step by nucleophilic attack of the carbonate species to the coordinating epoxide (Scheme 1a).10 These multinuclear complexes, however, are only effective for reactive epoxides such as cyclohexene oxide (CHO), and do not work well for less reactive epoxides such as PO. In 2020, Williams reported the first example of an aryloxide-bridged hetero dinuclear Co(III)K(I) complex-catalysed alternating copolymerization of PO and CO2, though the catalyst system requires high CO2 pressure (2–3 MPa).8g In this context, we found a new hetero tetranuclear complex including a Ca ion and three divalent Co ions by which alternating copolymerization of PO proceeded selectively under the lowest CO2 pressure (1.0 MPa) in hetero-multinuclear complexes (Scheme 1b). In addition, we report the remarkable additive effects of tertiary anilinium salts with non-coordinating anions, such as [PhNMe2H][B(C6F5)4], to improve the catalytic activity of the tetranuclear complex by generating the corresponding cationic hetero tetranuclear complex (Scheme 1c).
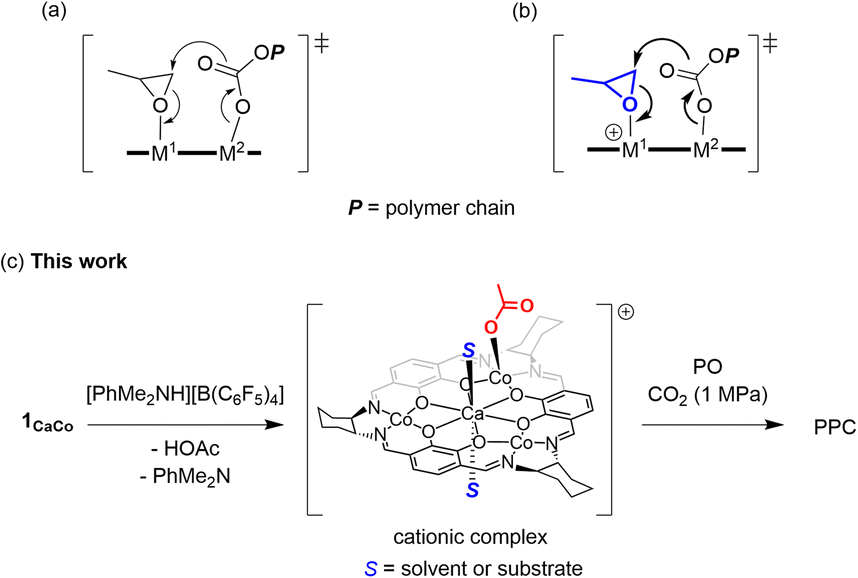 | ||
| Scheme 1 Key transition state structures of (a) neutral multinuclear catalysts, (b) cationic multinuclear catalysts. (c) This work: alternating copolymerization of PO and CO2 by cationic multinuclear species in the presence of anilinium salt. | ||
Results and discussion
First, we synthesized macrocyclic tetranuclear CaM3 complexes 1CaM through the formation of a macrocyclic mononuclear calcium complex, 1Ca, which was prepared by treating Ca(OAc)2·H2O and three equivalents of 1,4-diformyl-2,3-dihydroxylbenzene, followed by the addition of three equivalents of (1R,2R)-(−)-1,2-cyclohexanediamine in MeOH at 70 °C. Complex 1Ca had three vacant salen motifs around the calcium ion as characterized by its 1H NMR spectrum and mass spectrum. In the 1H NMR spectrum in CD3CN, a singlet signal of imine protons and a singlet signal of aromatic protons were observed at 8.43 and 6.87 ppm, respectively, and the integral ratio of the imine and aromatic signals was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, consistent with the condensation of two aldehydes of 1,4-diformyl-2,3-dihydroxylbenzene with two amine moieties of (1R,2R)-(−)-1,2-cyclohexanediamine to form a macrocyclic ligand. In high resolution mass spectroscopy, the molecular ion peak (m/z = 771.3186) was observed for [1Ca + H]+ (C42H47N6O6Ca: m/z (calcd) = 771.3183). Various divalent first-row transition metal ions (M = Mn, Fe, Co, Ni, Cu, and Zn) were introduced to 1Ca to give the corresponding macrocyclic tetranuclear complexes 1CaM (Scheme 2);11 it is noteworthy that tetranuclear complexes including Mn, Fe, and Co were inaccessible by the previously reported one-step template method using lanthanide ions and divalent first-row transition metal ions such as Ni, Cu, and Zn.12 The structure of complex 1CaZn was determined by a preliminary X-ray single crystal analysis, where one of two acetate ligands of 1CaZn bridged Ca and Zn ions in a κ2-fashion, and the other acetate ligand coordinated onto the Zn ion in a κ1-fashion (Fig. S2†).
:1, consistent with the condensation of two aldehydes of 1,4-diformyl-2,3-dihydroxylbenzene with two amine moieties of (1R,2R)-(−)-1,2-cyclohexanediamine to form a macrocyclic ligand. In high resolution mass spectroscopy, the molecular ion peak (m/z = 771.3186) was observed for [1Ca + H]+ (C42H47N6O6Ca: m/z (calcd) = 771.3183). Various divalent first-row transition metal ions (M = Mn, Fe, Co, Ni, Cu, and Zn) were introduced to 1Ca to give the corresponding macrocyclic tetranuclear complexes 1CaM (Scheme 2);11 it is noteworthy that tetranuclear complexes including Mn, Fe, and Co were inaccessible by the previously reported one-step template method using lanthanide ions and divalent first-row transition metal ions such as Ni, Cu, and Zn.12 The structure of complex 1CaZn was determined by a preliminary X-ray single crystal analysis, where one of two acetate ligands of 1CaZn bridged Ca and Zn ions in a κ2-fashion, and the other acetate ligand coordinated onto the Zn ion in a κ1-fashion (Fig. S2†).
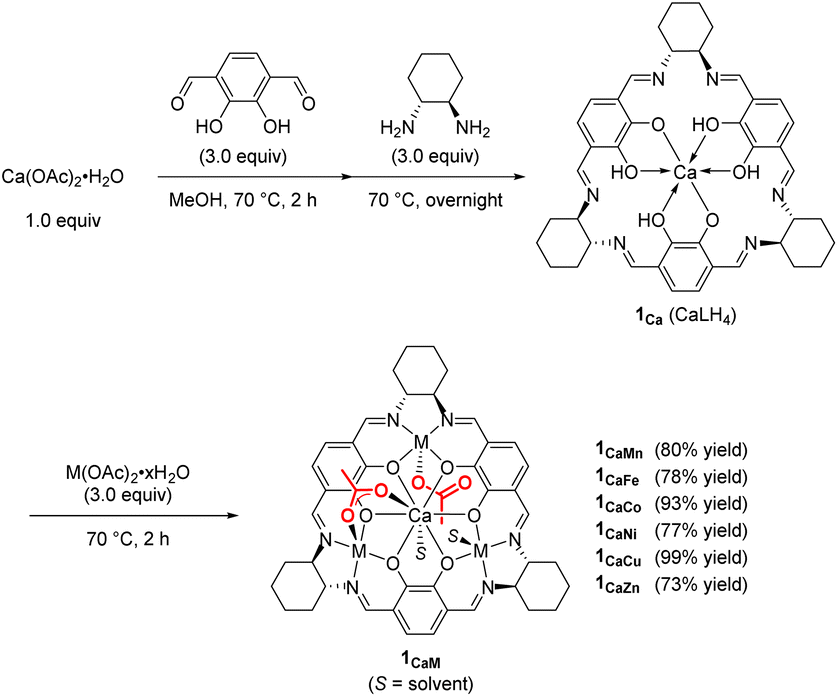 | ||
| Scheme 2 Preparation of hetero tetranuclear complexes 1CaM. | ||
Alternating copolymerization mediated by CaM3 complexes
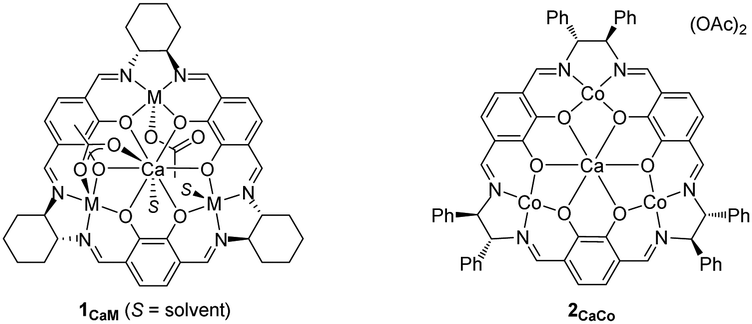
We tested the catalytic performance of these newly prepared tetranuclear complexes 1CaM (0.01 mmol) for alternating copolymerization of propylene oxide (PO; 60 mmol, neat) and CO2 (1.0 MPa) at 50 °C for 18 h, and found that only complex 1CaCo exhibited catalytic activity. The turnover number (TON) of 1CaCo reached 240 to afford poly(propylenecarbonate) (PPC) with high polymer selectivity (PS, 92%) and high carbonate linkage (CL, >99%) in a bimodal manner due to the inevitable contamination of a small amount of water, whose molecular weights and polydispersity indexes (PDI) were Mn = 4.9 kg mol−1 (PDI = 1.07) and 11.6 kg mol−1 (PDI = 1.08), respectively (Table 1, entry 1).13 In sharp contrast, other 1CaM complexes of Mn, Fe, Ni, Cu, and Zn were inactive (Table 1, entries 2–6). A notable finding was that the catalytic activity of the tetranuclear CaCo3 complexes was sensitive to the ligand backbone: complex 2CaCo having a 1,2-diphenylethylene tether between two nitrogen atoms exhibited slightly higher catalytic activity than 1CaCo, though with lower polymer selectivity (entry 7).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | TONa | PSa [%] | CLa [%] | M n [kg mol−1] | PDIb |
| a Determined by 1H NMR. n.d. = not detected. b Determined by GPC analysis using polystyrene standards. | ||||||
| 1 | 1CaCo | 240 | 92 | >99 | 4.9/11.6 | 1.07/1.08 |
| 2 | 1CaMn | n.d. | — | — | — | — |
| 3 | 1CaFe | n.d. | — | — | — | — |
| 4 | 1CaNi | n.d. | — | — | — | — |
| 5 | 1CaCu | n.d. | — | — | — | — |
| 6 | 1CaZn | n.d. | — | — | — | — |
| 7 | 2CaCo | 370 | 75 | >99 | 8.3/19.6 | 1.06/1.06 |
|
||||||

With the best tetranuclear complex 1CaCo in hand, we looked into the additive effects of protic cocatalysts for alternating copolymerization of PO and CO2 (Table 2). N,N-Dimethylanilinium tetrakis(pentafluorophenyl)borate, [PhMe2NH][B(C6F5)4], dramatically improved the TON value to 1840 without decreasing the PS and CL (entries 2 vs. 1), while other anilinium salts, such as [PhMeNH2][B(3,5-(CF3)2C6H3)4] (entry 3, TON = 600, PS = 99%, CL = 87%), [PhNH3][B(3,5-(CF3)2C6H3)4] (entry 4, TON = 560, PS = 99%, CL = 50%), and [Ph2MeNH(OEt2)][B(3,5-(CF3)2C6H3)4] (entry 5, TON = 790, PS = 83%, CL = 63%), resulted in only moderate catalytic activity and lower CL. In contrast, the addition of trialkyl ammonium salt, [nBu3NH][PF6] was ineffective for increasing the catalytic activity (entry 6, TON = 110, PS = 77%, CL = >99%). A strong proton source, [(Et2O)2H][B(3,5-(CF3)2C6H3)4], enhanced the TON value to 1470; however, the PS and CL decreased to 83% and 31%, respectively (entry 7). When we used tritylium tetrakis(pentafluorophenyl)borate, [Ph3C][B(C6F5)4], the TON value was high (1340) but the PS and the CL were lower than that for [PhMe2NH][B(C6F5)4] (entry 8), and when 2 equiv. of [Ph3C][B(C6F5)4] was added, only homo polymerization of PO proceeded with a high TON value (2910), and no PPC was obtained (entry 9).14 In the absence of 1CaCo, [PhMe2NH][B(C6F5)4] acted as a catalyst for the homo polymerization of PO (entry 10). The addition of quaternary ammonium salts, such as [nBu4N]Cl, or iminium salts, such as [PPN]Cl, both of which are frequently used as cocatalysts to improve the nucleophilicity of reaction intermediates, alkoxide species and carbonate species, by generating anionic active species,5,15 resulted in lower catalytic activity of 1CaCo and a lower PS (TON = 460, PS = 17% for [nBu4N]Cl, entry 11; TON = 380, PS = 34% for [PPN]Cl, entry 12). On the basis of the above results, we drew three conclusions regarding the first example of the alternating copolymerization of epoxide and CO2 catalyzed by cationic hetero-multinuclear complex:16 (i) the catalytically active cationic species was in situ-generated through removal of the AcO moiety bound to metals by an acidic proton source or the trityl cation, while a dicationic species was inactive for alternating copolymerization but active for ring-opening polymerization of PO (Scheme 3), (ii) the acidity of anilinium salts is quite important for generating the catalytically active cationic species: low acidic ammonium salts are unable to produce the cationic species, while strong acids, such as [Ph2MeNH][B(3,5-(CF3)2C6F3)4] and [(Et2O)2H][B(3,5-(CF3)2C6F3)4], promoted homo polymerization of PO, and (iii) N,N-dimethylaniline suppressed the ring-opening polymerization of PO to keep the CL level high. Accordingly, we selected [PhMe2NH][B(C6F5)4] as the best additive.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | TONa | PSa [%] | CLa [%] | M n [kg mol−1] | PDIb |
| a Determined by 1H NMR. b Determined by GPC analysis using polystyrene standards. c 2 equiv. of [Ph3C][B(C6F5)4] to catalyst 1CaCo was added. d No catalyst was added. | ||||||
| 1 | — | 240 | 92 | >99 | 4.9/11.6 | 1.07/1.08 |
| 2 | [PhMeNH2][B(C6F5)4] | 1840 | 95 | 98 | 40.0/85.7 | 1.11/1.04 |
| 3 | [PhMeNH2][B(3,5-(CF3)2C6H3)4] | 600 | 99 | 87 | 5.9 | 1.30 |
| 4 | [PhNH3][B(3,5-(CF3)2C6H3)4] | 560 | 99 | 50 | — | — |
| 5 | [Ph2MeNH(OEt2)][B(3,5-(CF3)2C6H3)4] | 790 | 83 | 63 | 8.4/22.1 | 1.13/1.10 |
| 6 | [nBu3NH][PH6] | 110 | 77 | >99 | — | — |
| 7 | [(Et2O)2H][B(3,5-(CF3)2C6H3)4] | 1470 | 83 | 31 | 6.9 | 1.51 |
| 8 | [Ph3C][B(C6F5)4] | 1340 | 87 | 84 | 21.1/50.1 | 1.14/1.06 |
| 9 | [Ph3C][B(C6F5)4]c | 2910 | 99 | <1 | 29.6 | 1.45 |
| 10d | [PhMe2NH][B(C6F5)4] | 2880 | 96 | <1 | 12.5 | 1.58 |
| 11 | [PPN]Cl | 460 | 17 | >99 | — | — |
| 12 | [nBu4N]Cl | 380 | 34 | >99 | — | — |
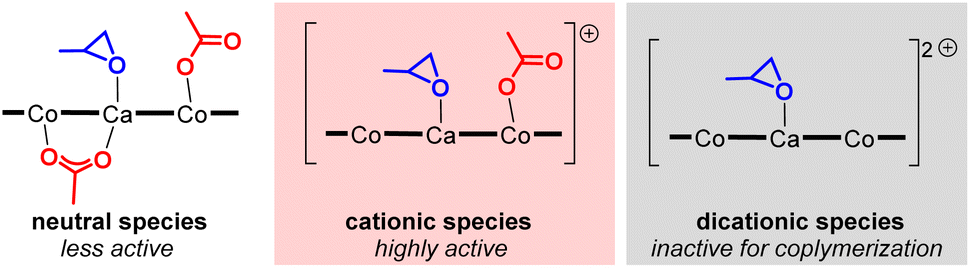 | ||
| Scheme 3 Catalytic species derived from complex 1CaCo. | ||
Plausible reaction mechanism
We propose a plausible mechanism as outlined in Scheme 4. Treatment of less catalytically active neutral species A with [PhMe2NH][B(C6F5)4] affords a highly active cationic species B, from which one of two acetate moieties of A is liberated to release acetic acid. In the initiation stage, a coordinating PO is ready to open its epoxy ring due to activation by electronically positive cationic species B, and nucleophilic attack of the remaining acetate moiety gives cobalt-alkoxide species C. The cobalt-alkoxide species C isomerized to calcium-alkoxide species C′, and then, a CO2 molecule coordinates to the Co center to produce intermediate D, where the alkoxide moiety reacts with CO2 to generate carbonate species E. Following ring-opening reaction of PO and CO2 insertion propagates the polycarbonate chain.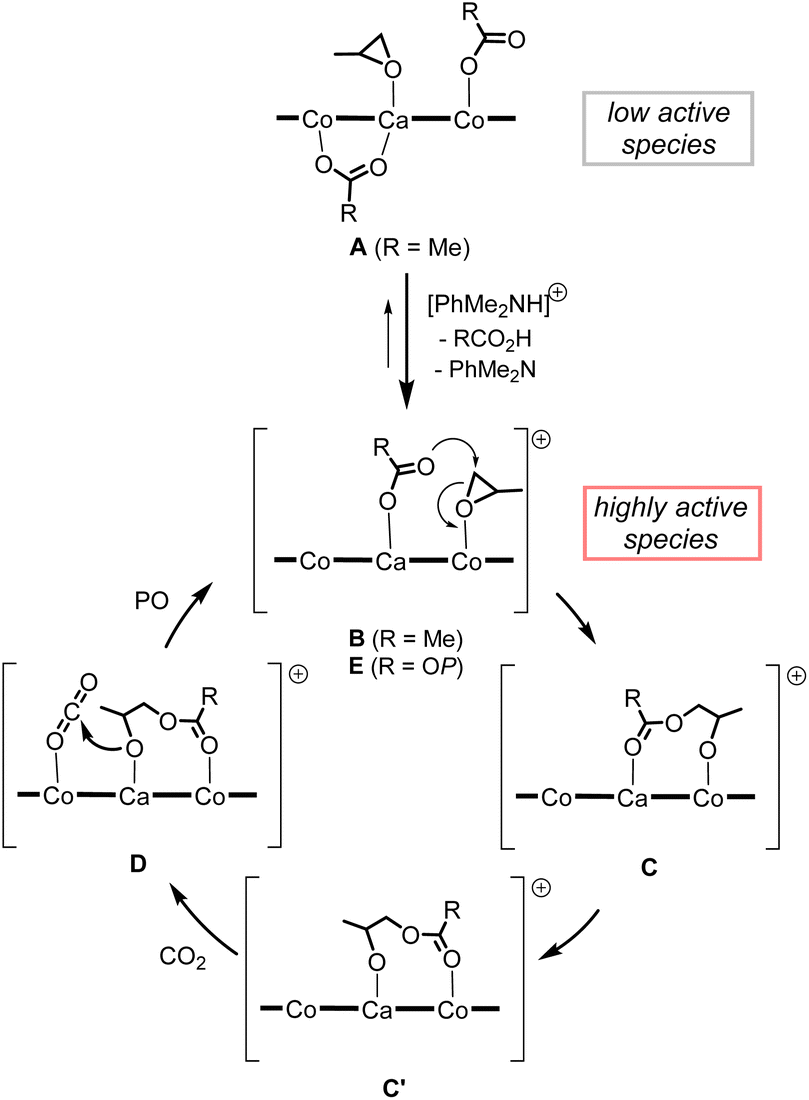 | ||
| Scheme 4 Plausible reaction mechanism of alternating copolymerization of PO and CO2 mediated by complex 1CaCo with anilinium borate. | ||
To clarify the additive effects of [PhNMe2H][B(C6F5)4], we performed a reaction pathway search based on density functional theory calculations. Because of the high calculation cost of our very large complexes, we calculated a model reaction using simplified Ca(II)Co(II)3 complexes and ethylene oxide (EO) using simple function and basis (B3LYP function with a basis-set of 6-31G(d,p) for H, C, N, O and LANL2DZ for Ca and Co) and the IEFPCM option with ε = 12.42 (ε of ethylene oxide).17,18 The structure of a neutral complex A1 was constructed based on the crystal structure of 1CaZn, and the structure of a cationic complex B1 is constructed by removing one κ1-acetate ligand of A1, in which an ethylene oxide (EO) coordinating to a Co ion was moved to a Ca ion to occupy a coordination site of a large Ca ion.
In the cationic pathway shown in Scheme 5, model complex B1 is stabilized by the dissociation of an EO and forms B1-EO; further isomerization by the transfer of coordinating EO on Ca ions to Co ions affords complex B2-EO. Subsequent nucleophilic attack of the acetate ligand leads to a ring-opening reaction of EO through a transition state TSBC2-EO to give Co-alkoxide intermediate C2-EO, and the activation energy of this step is 24.7 kcal mol−1. The formation of Ca-alkoxide C1-EO from B1-EO through B1′-EO and TSBC1-EO requires slightly higher activation energy (25.3 kcal mol−1) than TSBC2-EO. Next, the coordination of CO2 on the Co-alkoxide species generates unstable intermediate D2-EO, while the coordination of CO2 on Ca-alkoxide species affords much more stable intermediate D1-EO, suggesting that the CO2 insertion proceeds via the Ca-alkoxide intermediate D1-EO. The CO2 insertion from D1-EO goes barrierlessly to afford E1-EO, which is then isomerized to E1′-EO to start the propagation. Without the dissociation of an EO, the ring-opening step to give both Ca- and Co-alkoxides (TSBC1 and TSBC2) requires much higher energies than TSBC2-EO, and thus these pathways are eliminated.
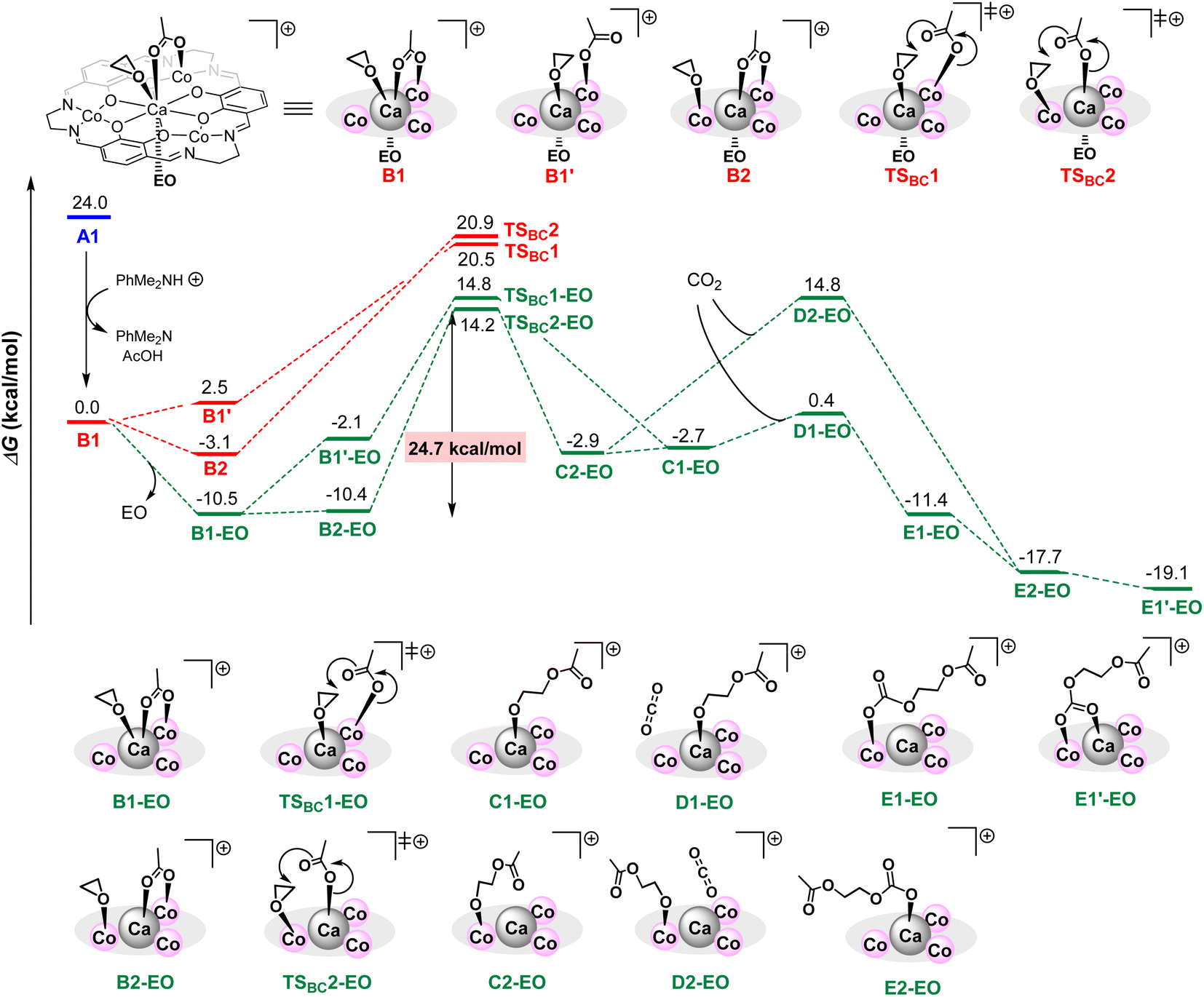 | ||
| Scheme 5 Energy diagram for alternating copolymerization by cationic CaCo3 complexes. | ||
On the other hand, the activation barrier of the most favorable neutral pathway is 25.7 kcal mol−1 for A2-EO→TS2AF-EO, which is 1.0 kcal mol−1 higher than that of the cationic pathway (Scheme 6). Dissociation of one EO from A1 affords A1-EO, and the coordination mode change of the κ1-acetate ligand to the κ2-coordination mode and the transfer of coordinating EO on Ca ions to Co ions yields the most stable intermediate A2-EO. Like the cationic pathway, transition state TSAF2-EO, which generates Co-alkoxide intermediate F2-EO, has lower activation energy than TSAF1-EO for generating the Ca-alkoxide intermediate F1-EO. In addition, CO2 coordination onto Ca-alkoxide species G1-EO is favored compared with CO2 coordination onto Co-alkoxide species G2-EO by 4.5 kcal mol−1.
 | ||
| Scheme 6 Energy diagram for alternating copolymerization by neutral CaCo3 complexes. | ||
According to these results, we concluded that in the CaCo3 tetranuclear catalyst, an electronically positive cationic species strongly activates epoxide, allowing for smooth progression of the ring-opening step. Furthermore, flexible ligand exchange between Ca and Co ions is important to promote the CO2 insertion step to give PPC with high CL. Though the energy difference of activation barriers is small, just looking at the number of the difference in the activation energies of the cationic and neutral pathways, the reaction rate of the cationic pathway was estimated to be 5.4 times faster than that of the neutral pathway, consistent with the experimental results on catalytic activities of complex 1CaCo (TON = 240) and 1CaCo with [PhNMe2H][B(C6F5)4] (TON = 1840): cationic species advanced the alternating copolymerization 7.7 times faster than neutral species.
Conclusions
We found new tetranuclear macrocyclic catalyst 1CaCo for alternating copolymerization of CO2 and PO, and remarkable additive effects of [PhMe2NH][B(C6F5)4] to improve the catalytic activity of 1CaCo while maintaining high polymer selectivity and high carbonate linkage, even under 1.0 MPa of CO2. On the basis of control experiments, using 1 or 2 equiv. of [Ph3C][B(C6F5)4], together with density functional theory calculations, we concluded that the reaction of 1CaCo with [PhMe2NH][B(C6F5)4] afforded catalytically active cationic species, which strongly activated PO as a Lewis acid to reduce the activation energy. In addition, flexible ligand exchange between Ca and Co ions is important to reduce the energy for CO2 insertion steps to obtain PPC with high CL.Data availability
The authors declare that all data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
H. N. conducted DFT calculations, and he wrote the manuscript with the assistance of K. M. and J. O. S. M. conducted most of the experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI 20K152770 as a Grant-in-Aid for Young Scientists and 21K18205 as a Grant-in Aid for Challenging Research. DFT calculation was achieved through the use of PC cluster for large-scale visualization at the Cybermedia Center, Osaka University. We thank Dr Tobias Schindler for his experimental contribution in the early stage of the project.Notes and references
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287 CrossRef CAS.
- (a) D. J. Darensbourg, Salen metal complexes as catalysts for the synthesis of polycarbonates from cyclic ethers and carbon dioxide, in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer-Verlag, Berlin, Germany, 2012, Advances in Polymer Science, 245, pp. 1−27 Search PubMed; (b) G. A. Luinstra and E. Borchardt, Material properties of poly(propylene carbonates), in Synthetic Biodegradable Polymers, ed. B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus and T. A. Zevaco, Springer-Verlag, Berlin, Germany, 2012, Advances in Polymer Science, 245, pp. 29−48 Search PubMed.
- (a) C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS; (b) A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS; (c) H. Cao, S. Liu and X. Wang, Green Chem. Eng., 2022, 3, 111–124 CrossRef; (d) J. Huang, J. C. Worch, A. P. Dove and O. Coulembier, ChemSusChem, 2020, 13, 469–487 CrossRef CAS PubMed; (e) B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC; (f) C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS; (g) S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC; (h) G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed; (i) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (j) D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS; (k) H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS; (l) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- (a) D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335–6342 CrossRef CAS PubMed; (b) Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS; (c) R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194–196 CrossRef CAS; (d) K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS PubMed; (e) E. K. Noh, S. J. Na, S. S, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS; (f) S. S, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306–7309 CrossRef.
- (a) T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 8–13 CrossRef CAS; (b) S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS; (c) H. Sugimoto, H. Ohshima and S. Inoue, Stud. Surf. Sci. Catal., 2004, 153, 247–250 CrossRef CAS; (d) S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef; (e) H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS; (f) C. E. Anderson, S. I. Vagin, W. Xia, H. Jin and B. Rieger, Macromolecules, 2012, 45, 6840–6849 CrossRef CAS; (g) K. Nakano, K. Kobayashi, T. Ohkawara, H. Imoto and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 8456–8459 CrossRef CAS PubMed; (h) X. Jiang, F. Gou and H. Jing, J. Catal., 2014, 313, 159–167 CrossRef CAS; (i) J. Deng, M. Ratanasak, Y. Sako, H. Tokuda, C. Maeda, J. Y. Hasegawa, K. Nozaki and T. Ema, Chem. Sci., 2020, 11, 5669–5675 RSC.
- (a) W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS; (b) L. Grefe and E. Mejía, Tetrahedron, 2021, 98, 132433 CrossRef CAS.
- (a) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS; (b) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed; (c) K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC; (d) M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS; (e) A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC; (f) M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed; (g) M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245–1255 RSC; (h) C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS; (i) K. Nishioka, H. Goto and H. Sugimoto, Macromolecules, 2012, 45, 8172–8192 CrossRef CAS; (j) P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC; (k) Y. Liu, W. M. Ren, W. P. Zhang, R. R. Zhao and X. B. Lu, Nat. Commun., 2015, 6, 8594 CrossRef CAS PubMed; (l) A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS; (m) A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed; (n) J. Li, Y. Liu, W. M. Ren and X. B. Lu, J. Am. Chem. Soc., 2016, 138, 11493–11496 CrossRef CAS PubMed; (o) M. Hatazawa, R. Takahashi, J. Deng, H. Houjou and K. Nozaki, Macromolecules, 2017, 50, 7895–7900 CrossRef CAS; (p) L. S. Huang, C. Y. Tsai, H. J. Chuang and B. T. Ko, Inorg. Chem., 2017, 56, 6141–6151 CrossRef CAS PubMed.
- (a) P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC; (b) J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed; (c) J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC; (d) A. C. Deacy, C. B. Durr and C. K. Williams, Dalton Trans., 2019, 49, 223–231 RSC; (e) G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC; (f) A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS; (g) A. C. Deacy, E. Moreby, A. Phanopoulos and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 19150–19160 CrossRef CAS; (h) W. Lindeboom, D. A. X. Fraser, C. B. Durr and C. K. Williams, Chem.–Eur. J., 2021, 27, 12224–12231 CrossRef CAS PubMed; (i) N. V. Reis, A. C. Deacy, G. Rosetto, C. B. Durr and C. K. Williams, Chem.–Eur. J., 2022, 28, e202104198 CrossRef CAS PubMed.
- (a) H. Nagae, R. Aoki, S. N. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS PubMed; (b) H. Asaba, T. Iwasaki, M. Hatazawa, J. Deng, H. Nagae, K. Mashima and K. Nozaki, Inorg. Chem., 2020, 59, 7928–7933 CrossRef CAS PubMed; (c) A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2021, 60, 13372–13379 CrossRef CAS; (d) H. Nagae, S. Y. Akebi, S. Matsushiro, K. Sakamoto, T. Iwasaki, K. Nozaki and K. Mashima, Macromolecules, 2022, 55, 9066–9073 CrossRef CAS; (e) Q. Yao, Y. Wang, B. Zhao, X. Zhu, Y. Luo, D. Yuan and Y. Yao, Inorg. Chem., 2022, 61, 10373–10382 CrossRef CAS PubMed.
- (a) M. W. Lehenmeier, C. Bruckmeier, S. Klaus, J. E. Dengler, P. Deglmann, A. K. Ott and B. Rieger, Chem.– Eur. J., 2011, 17, 8858–8869 CrossRef CAS PubMed; (b) A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS; (c) T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728–10735 CrossRef CAS PubMed; (d) H. Shao, Y. Reddi and C. J. Cramer, ACS Catal., 2020, 10, 8870–8879 CrossRef CAS; (e) A. Roznowska, K. Dyduch, B. Y. Lee and A. Michalak, J. Mol. Model., 2020, 26, 113 CrossRef CAS; (f) J. Deng, M. Ratanasak, Y. Sako, H. Tokuda, C. Maeda, J. Y. Hasegawa, K. Nozaki and T. Ema, Chem. Sci., 2020, 11, 5669–5675 RSC; (g) A. C. Deacy, A. Phanopoulos, W. Lindeboom, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2022, 144, 17929–17938 CrossRef CAS.
- W. T. S. Huck, F. C. J. M. van Veggel and D. N. Reinhoudt, Recl. Trav. Chim. Pays-Bas, 1995, 114, 273 CrossRef CAS.
- (a) S. Akine and T. Nabeshima, Dalton Trans., 2009, 9226, 10395–10408 RSC; (b) H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2009, 253, 1458–1475 CrossRef CAS; (c) T. Nabeshima, Bull. Chem. Soc. Jpn., 2010, 83, 969–991 CrossRef CAS; (d) S. Akine, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 25–54 CrossRef CAS.
- The regioregularity of the resulting PPC was random, in which the ratio of HH:HT(TH):TT = 1:2:1 was observed in 1H NMR spectroscopy.
- Y. Sarazin and J. F. Carpentier, Chem. Rev., 2015, 115, 3564–3614 CrossRef CAS PubMed.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed.
- S. Elmas, M. A. Subhani, W. Leitner and T. E. Müller, Beilstein J. Org. Chem., 2015, 11, 42–49 CrossRef.
- (a) B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (c) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS; (d) H. F. Schaefer, Mod. Theor. Chem., 1977, 3, 1–27 Search PubMed; (e) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS; (f) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (g) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (h) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (i) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- J. González-Fabra, F. Castro-Gómez, W. M. C. Sameera, G. Nyman, A. W. Kleij and C. Bo, Catal. Sci. Technol., 2019, 9, 5433–5440 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00974b |
| ‡ Present address: Interdisciplinary Research Center for Catalytic Chemistry (IRC3), National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 5, 1-1-1 Higashi, Japan. |
| This journal is © The Royal Society of Chemistry 2023 |