
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modular aldol approach for internal fluorescent molecular rotor chalcone surrogates for DNA biosensing applications†
Ryan E.
Johnson
a,
Makay T.
Murray
b,
Lucas J.
Bycraft
a,
Stacey D.
Wetmore
 *b and
Richard A.
Manderville
*a
*b and
Richard A.
Manderville
*a
aDepartment of Chemistry & Toxicology, University of Guelph, Guelph, Ontario N1G 2W1, Canada. E-mail: rmanderv@uoguelph.ca
bDepartment of Chemistry & Biochemistry, University of Lethbridge, Lethbridge, Alberta T1K 3M4, Canada
First published on 11th April 2023
Abstract
Fluorescent molecular rotors (FMRs) are critical tools for probing nucleic acid structure and function. Many valuable FMRs have been incorporated into oligonucleotides, although the methods of doing so can be cumbersome. Development of synthetically simple, high yielding modular methods to fine-tune dye performance is crucial to expand the biotechnological applications of oligonucleotides. Herein, we report the utility of 6-hydroxy-indanone (6HI) with a glycol backbone to serve as a handle for on-strand aldehyde capture as a modular aldol approach for site-specific insertion of internal FMR chalcones. Aldol reactions with aromatic aldehydes containing N-donors proceed in high yield to create modified DNA oligonucleotides, which in the duplex match the stability of the fully paired canonical B-form with strong stacking interactions between the planar probe and the flanking base pairs, as evidenced by molecular dynamics (MD) simulations. The FMR chalcones possess remarkable quantum yields (Φfl up to 76%) in duplex DNA, coupled with large Stokes shifts (Δν up to 155 nm), light-up emissions (Irel up to 60-fold) that span the visible region (λem 518–680 nm) with brightness up to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 cm−1 M−1. The library also contains a FRET pair and dual emission probes, suitable for ratiometric sensing. The ease of aldol insertion coupled with the excellent performance of the FMR chalcones permits their future wide-spread use.
480 cm−1 M−1. The library also contains a FRET pair and dual emission probes, suitable for ratiometric sensing. The ease of aldol insertion coupled with the excellent performance of the FMR chalcones permits their future wide-spread use.
Introduction
Fluorescent probes are critical tools for sensing the structure, dynamics, and binding interactions of nucleic acids.1–3 Fluorescent molecular rotors (FMRs), such as thioflavin-T (ThT),4 and cyanine dyes,5 are extensively utilized for DNA sensing6–8 because they possess segmental mobility, with low intrinsic fluorescence in the unbound state, and with large emission intensity upon DNA binding due to enhanced dye rigidity.9,10 They are commercially available, or readily synthesized, making them simple to employ.11 However, because they are free in solution, they can suffer from false readouts, slow response times, and cannot provide site-specific information.Internal FMRs remediate label-free drawbacks due to site-specific attachment to nucleic acids. Additionally, internal reporters can provide direct fluorescent signaling for metal binding,12 hydrogen-bonding,13,14 hybridization,15–17 changes in pH,18 topology switching,19,20 and protein binding21–25 with fast response times that are resistant to false readouts.26 As such, they represent a powerful tool in life sciences. Internal FMR probes are either canonical (base-pairing capable) or non-canonical (lack of base-pairing, enhanced π-stacking).1 Canonical probes may be further divided into fluorescent nucleobase analogs (FBAs), which are capable of directly base-pairing with a complementary nucleobase,12–15 and conjugates, in which the FMR is attached to a native nucleobase by a flexible tether.23–25 For the nucleic acid community to widely adopt internal FMR probes for biosensing strategies, synthetically simple, high yielding modular approaches are needed. In this way, tunable FMR probe libraries can be easily created and surveyed to arrive at desirable probes that are amenable to high-throughput DNA preparation and analysis. FBAs often involve complicated multi-step syntheses to generate a single probe,12–14 which lacks post-synthetic fluorophore tuning/modularity should performance be inadequate. Non-canonical surrogates16,27,28 and conjugates23–25 are conducive to modularity, but the typical strategies are cumbersome, requiring the synthesis of each FMR dye and internal DNA handle (Fig. 1).
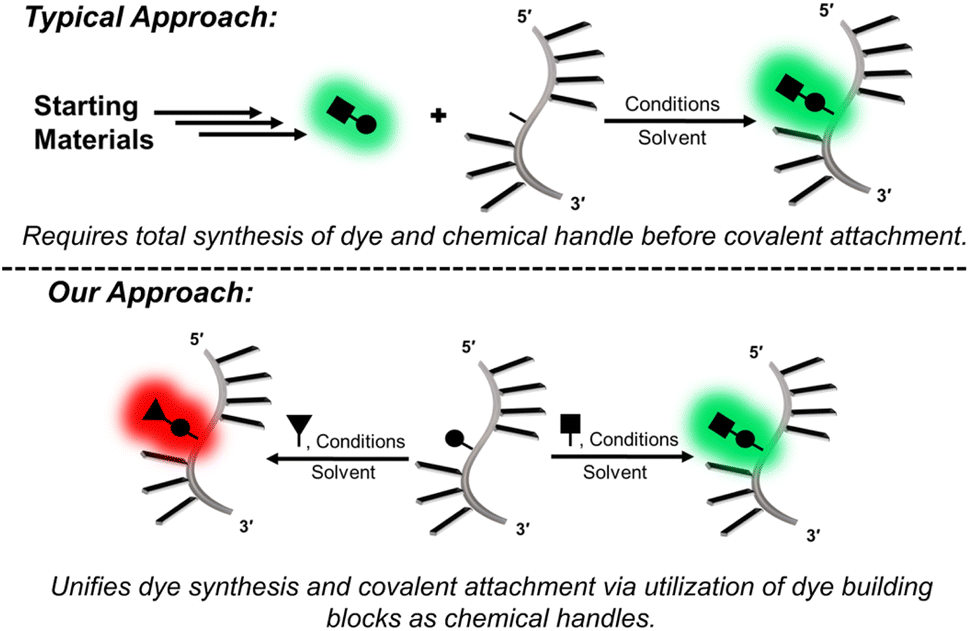 | ||
| Fig. 1 Conceptual comparison of modular approaches for the covalent insertion of FMRs into oligonucleotides. | ||
Our modular approach unifies dye synthesis and covalent attachment via utilization of dye building blocks as chemical handles (Fig. 1).29,30 In this design, the push–pull fluorophore is divided into the donor and acceptor components and assembled on the DNA strand using DNA-compatible chemistry. Initial efforts employed a 4-bromoaniline (4BAn) handle containing an (S)-glycol backbone for on-strand Suzuki–Miyaura cross-coupling reactions31 with arylboronic acids containing electron-withdrawing substituents to create push–pull biaryls (Fig. 2A).29 We also utilized an (S)-glycol-3-formylindole (3FI) building block for on-strand Knoevenagel condensations with heterocyclic quaternary salts for insertion of cyanine-indole hemicyanines (Fig. 2B).30 However, in these designs the donor component with rotatable bonds served as the handle and it's close proximity to neighboring bases of the single-stranded DNA (SS) led to a restricted environment with high background signal with minimal light-up fluorescence upon hybridization. Additionally, both linear dye types caused duplex destabilization and the probe libraries lacked diversity.
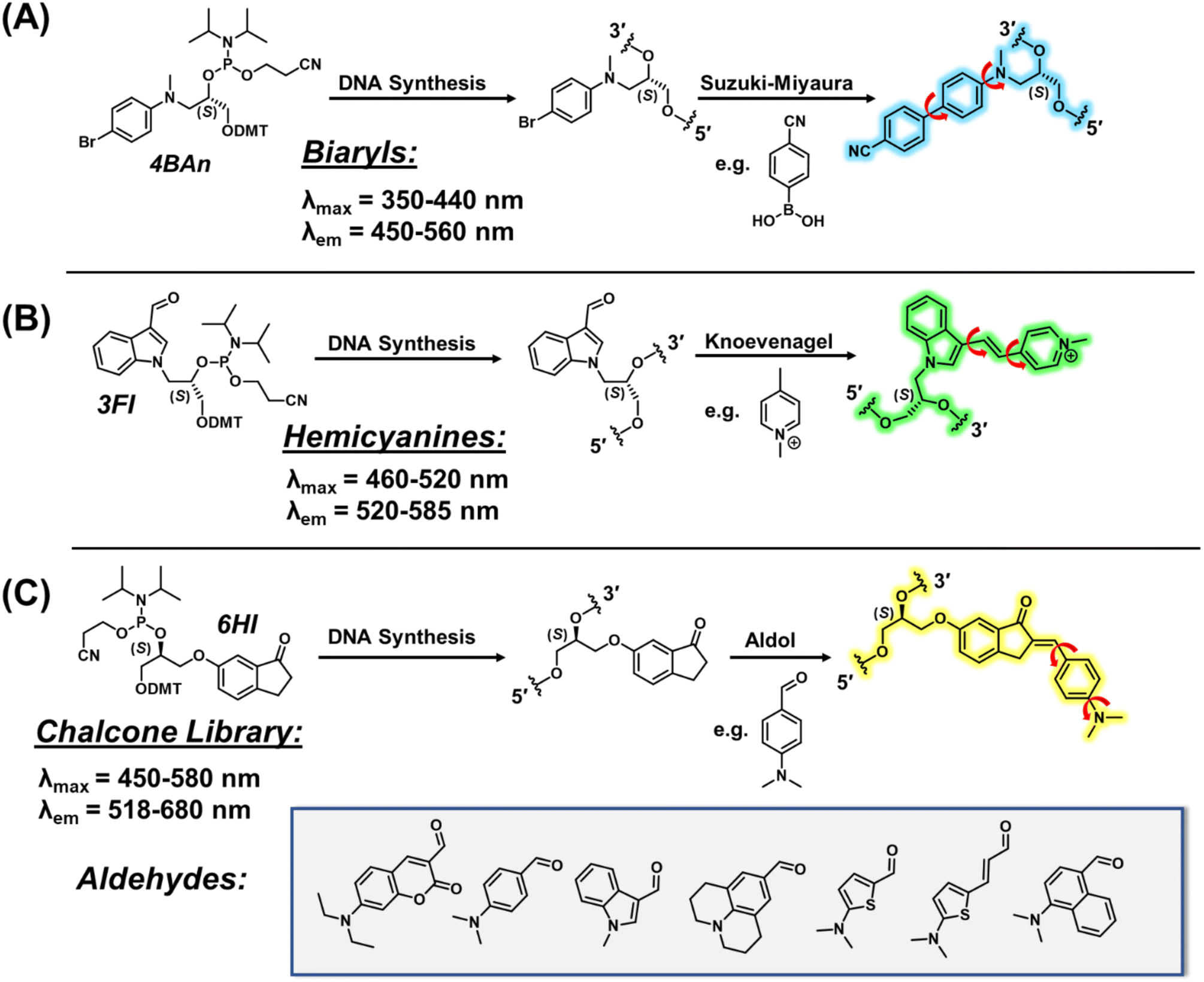 | ||
| Fig. 2 Modular approaches for internal FMR nucleobase surrogates using DNA-compatible on-strand reactions. (A) Suzuki–Miyaura cross-coupling to generate push–pull biaryls. (B) Knoevenagel condensation to afford cyanine-indole hemicyanines. (C) This work using aldol condensation to create chalcone FMR probes. Red arrows in the dye products indicates rotatable bonds. | ||
Based on these observations, we sought to reduce background fluorescence via distancing of rotatable bonds from neighboring bases in the SS. This suggested reversal of probe orientation with attachment of the rigid acceptor as the internal DNA handle. To achieve this goal, we selected 6-hydroxy indanone (6HI) as a modular template due to its ability to create fluorescent chalcones through aldol condensation, which should possess fewer rotatable bonds in the SS stack (Fig. 2C). Furthermore, it was anticipated that this strategy, which permits condensation with the vast variety of aromatic aldehydes used for the creation of fluorescent chemosensors for analyte detection,32 would afford highly diverse probe libraries, unlike our previous efforts using on-DNA Suzuki–Miyaura cross-coupling (Fig. 2A)29 and Knoevenagel condensations (Fig. 2B).30 Chalcones are unexplored as DNA fluorescent probes, but the free dyes exhibit brightness comparable to commercial fluorophores along with FMR character.33–35
Herein, we demonstrate, to the best of our knowledge, the first example of an internal label which is capable of aldehyde capture to generate a donor–acceptor FMR library. Aldol reactions proceed in high yield with relative synthetic simplicity for universal appeal. Notably, the resultant FMR chalcones are highly diverse and efficient, with excellent brightness and turn-on fluorescence for monitoring binding interactions by the modified oligonucleotides. Advantages of FMR chalcones over other available state-of-the-art internal nucleobase probes is discussed.
Results and discussion
Synthesis and on-strand aldehyde capture
We first focused on attaching the commercially available N,N-dimethyl-4-formyl-aniline (An6HI) with the exocyclic amine donor substituent. The 6HI-glycol phosphoramidite (Fig. 1C), was easily accessible (2 steps from 6HI and the known DMT-(S)-glycidol, see ESI† for experimental, Scheme S1, Fig. S1–S3 for NMR and Fig. S6, S7 for HRMS) and compatible with standard solid-phase oligonucleotide synthesis (coupling times, reagents and cleavage from the solid support and deprotection using 30% NH4OH for 2 h at 65 °C). The 6HI handle was initially inserted into the G7-site (X) of NarI12 (5′-CTCGGC-X-CCATC-3′), which was previously utilized as a substrate for 4BAn29 and 3FI,30 and places the dye in a CXC sequence context. As an authentic standard for the on-strand reaction, the entire An6HI phosphoramidite (Fig. S4, S5 for NMR and Fig. S8, S9† for HRMS) was also synthesized and incorporated into the G7-site of NarI12 using standard solid-phase DNA synthesis. In addition, the free chalcone was prepared through aldol condensation of 6-methoxy-indanone (6MI) with the aniline aldehyde, which afforded a single product with the chalcone in the E-configuration, as evidenced by NMR spectroscopy (Fig. S10†). Chalcones can undergo photochemical E/Z-isomerization when treated with UV light (345 nm).36 For example, the An7HI isomer is obtained exclusively as the E-isomer and generates 10% of the Z-isomer following 2 h irradiation. The chemical shift of the olefinic proton is diagnostic of isomer formation and resonates at 7.55 ppm for the E-isomer of An7HI in CDCl3, which shifts to 6.98 ppm for the Z-isomer.36 For An6MI, the olefinic proton resonates at 7.65 ppm in CDCl3 (Fig. S10†), confirming E-isomer formation. Throughout our studies samples were stored in the dark under refrigeration and were not treated with strong UV light for extended periods of time.Two methods were used for on-strand aldol synthesis involving an enamine approach through piperidine addition (Method 1 (M1)) and an enolate strategy involving treatment with NaOH (Method 2 (M2)). For NarI-An6HI synthesis, M1 produced 45% yield of product (reaction yields obtained via HPLC), while ∼90% was obtained using M2 with almost complete loss of the NarI-6HI starting material, as evidenced by HPLC analysis (Fig. 3A). Both methods provided clean HPLC traces for conversion of NarI-6HI into NarI-An6HI. The product identity was confirmed by ESI-MS analysis and through separate solid-phase synthesis using the intact An6HI phosphoramidite. The aldol methods were then extended to a wide range on N-donor aromatic aldehydes (Fig. 2C), which varied in donor strength, ring size, polarity, and extent of conjugation to generate a library of chalcones with broad color palette (Fig. 4A and B). The aldehyde library also contained the fluorescent 7-(diethylamino) coumarin aldehyde (Cou), which serves as a donor for the creation of dual-emitting probes for ratiometric detection strategies.37,38 The entire chalcone library was generated using the milder enamine approach with yields ranging from 35–90%. For the harsher enolate method, high yields of An6HI, Nap6HI, Th6HI and Ind6HI were obtained, while Ju6HI, ETh6HI and Cou6HI (Fig. 3B) were sensitive to NaOH and could only be obtained using M1 (see ESI† for HPLC chromatograms and ESI-MS of modified oligonucleotides).
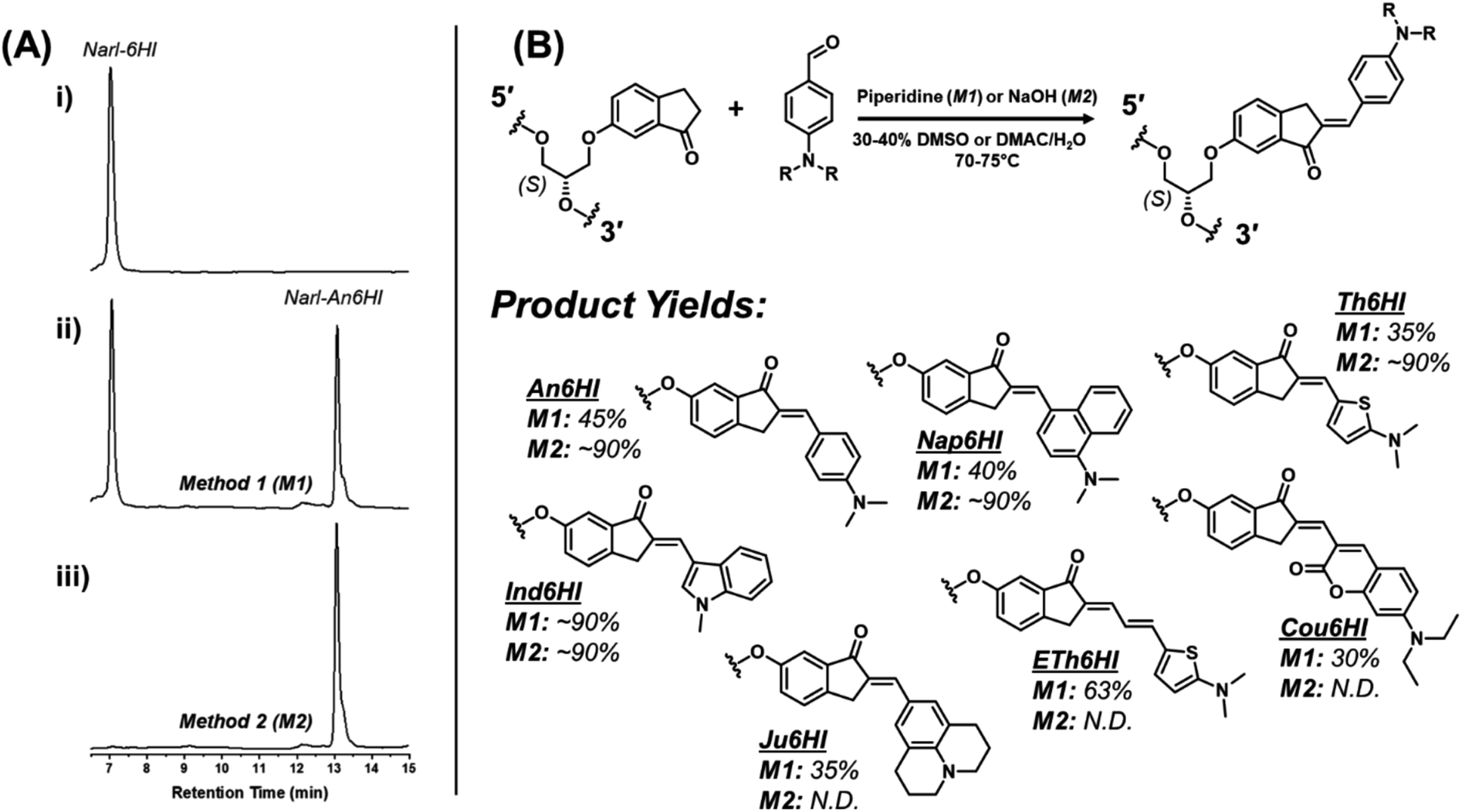 | ||
| Fig. 3 (A) RP-HPLC chromatograms for on-strand aldol reactions post ethanol precipitation: (i) chromatogram of NarI starting material; (ii) chromatogram following reaction under conditions of Method 1 (M1) to generate the NarI-An6HI product; (iii) chromatogram following reaction under conditions of Method 2 (M2) to generate the NarI-An6HI product. See ESI† for full experimental details. (B) General scheme for the on-strand aldol condensation with respective structures and yields from the described synthetic methods (M1/M2). | ||
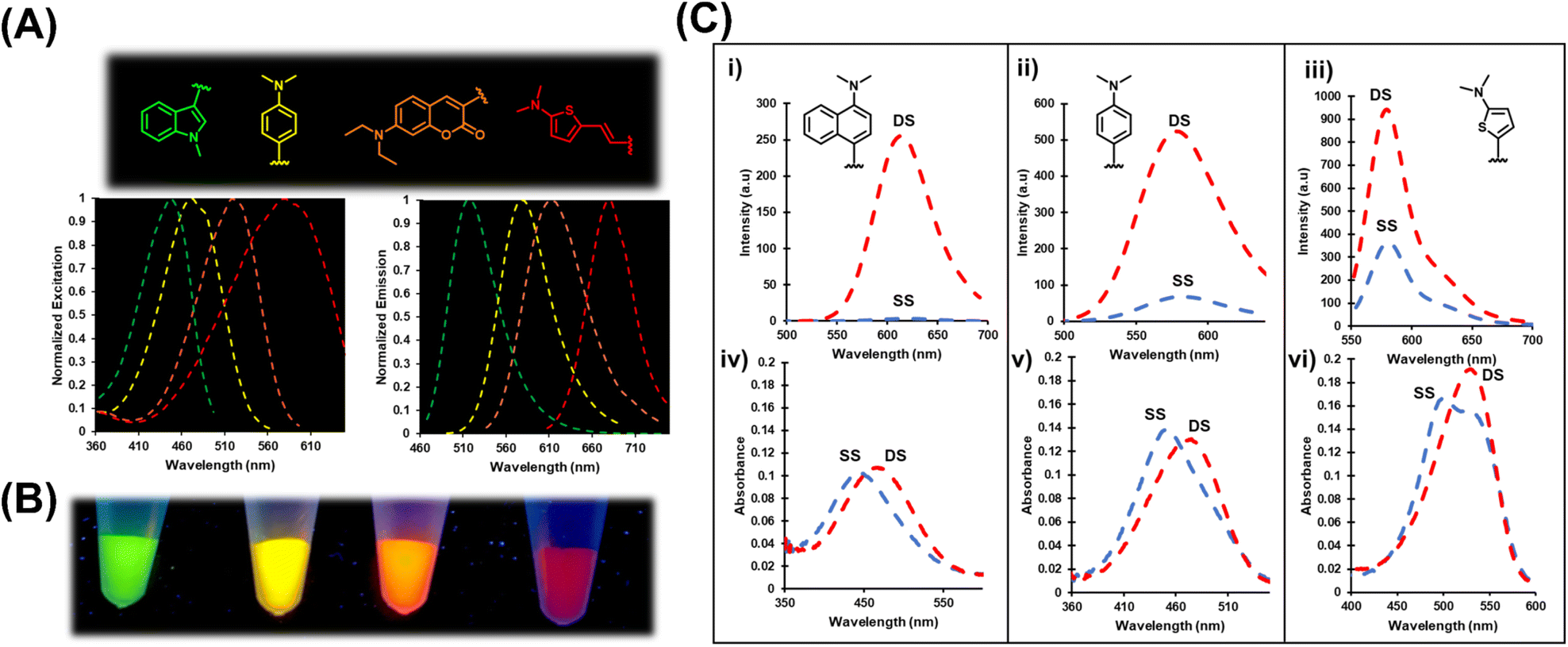 | ||
| Fig. 4 (A) Normalized excitation and emission spectra generated by four representative chalcone FMRs spanning the visible region. (B) DNA stock solutions containing internal Ind (green), An (yellow), Nap (orange) and ETh (red) probes were illuminated with ultraviolet light and photographed. (C) Emission (i–iii) and UV-vis (iv–vi) response of Nap, An and Th FMRs to duplex hybridization N = THF within NarI12. All experiments carried out at 5 μM duplex in 100 mM NaCl, 50 mM phosphate buffer at pH 7.0 at room temperature. | ||
Photophysical properties of free chalcones
Members of the library were isolated as free dyes through aldol condensations with 6MI to measure their photophysical properties in MeOH, 75:25 glycerol:MeOH and 1,4-dioxane (Table S1, ESI†). In MeOH the chalcones have molar absorption coefficients ranging from 20000 cm−1 M−1 for Nap6MI to a high of 50000 cm−1 M−1 for Th6MI. The probes are weakly emissive in MeOH and in 75:25 glycerol:MeOH the chalcones exhibit red spectral shifts in both excitation (Δλex, up to 45 nm) and emission (Δλem, up to 32 nm) maxima with turn-on (Irel) fluorescence from a low of 5.6-fold for Ind6MI to a high of 17-fold for Ju6MI, which confirms their FMR character. In aprotic dioxane the chalcones display blue spectral shifts compared to their maxima in MeOH (Δλex, up to −27 nm; Δλem, up to −55 nm). Quantum yields (Φfl) for the probes in dioxane ranged from 0.03 for Ind6MI to 0.16 for Ju6MI for Irel values ranging from 4-fold to 166-fold compared to their emission intensity in MeOH. Thus, the probes possess FMR character and positive solvatochromism, which can both act to increase their fluorescence intensity. The two factors can be distinguished based on spectral shifts, with blue shifting of λem suggesting that positive solvatochromism is contributing to their light-up response.
Chalcone performance in NarI12
The optical response of the internal probes to hybridization within the G7 site of NarI12 was initially assessed by pairing Ind6HI, An6HI, Nap6HI and Th6HI with the model tetrahydrofuran (THF) abasic site (Table 1, Fig. 4). This served as a test for FMR character, because the confined intrahelical vacant space restricts probe rotation to mimic a medium of elevated viscosity.15 The duplex (DS) thermal melting temperatures (Tm) with the chalcones opposite THF closely matched the Tm of 64 °C for the native NarI12 duplex containing a G:C base-pair (Table 1). For comparison, the native NarI12 DS with G paired opposite THF has a Tm of 45 °C. We previously obtained Tm values ∼53 °C for linear biaryl surrogates paired opposite THF,29 and ranges of 51–61 °C for cyanine-indole dyes.30 Clearly, the bent chalcones fit better within the DS with more favorable stacking interactions to match the thermal stability of the fully paired canonical duplex.| 5′-CTC-GGC-X-CCA-TC-3′ | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 3′-GAG-CCG-N-GGT-AG-5′ | |||||||||
| Xa | N | T m (ΔTm) (°C) | λ ex (Δλex) (nm) | ε max (cm−1 M−1) | λ em (Δλem) (nm) | I rel | Δνe (nm) | Φ fl | B (cm−1 M−1) |
| a See Fig. 3B for donor abbreviations. b T m values of NarI12 (5′-CTCGGC-X-CCATC) DS (5 μM) measured at 260 nm in 50 mM sodium phosphate buffer, pH 7, with 0.1 M NaCl, heating rate of 0.5 °C min−1, errors are ±1 °C; ΔTm = Tm (modified DS with probe (X) opposite N = abasic site (THF) or C) − 64 °C (Tm of native NarI12 DS containing X = G paired with N = C). c Values in brackets represent wavelength shifts in DS relative to SS. d Relative emission intensity of the chalcone probes in the DS versus SS. e Stokes shift in nm for the probes in the NarI12 DS. f Fluorescence quantum yield of chalcone probes in the NarI12 DS with N = C measured by the comparative method using either fluorescein (Φs = 0.95) in 0.1 M NaOH or rhodamine 101 (Φs = 1.0) in absolute ethanol as fluorescent standards, errors are ±5% obtained from three measurements via titration of labeled NarI12 into excess complementary oligo. g Probe brightness (Φfl·εmax) in the NarI12 DS with N = C. | |||||||||
| Ind | THF | 65.7 (1.7) | 448 (4) | — | 515(−7) | 7 | 67 | — | — |
| Ind | C | 64.7 (0.7) | 448 (0) | 24000 |
514 (−8) | 5 | 66 | 0.09 | 2160 |
| An | THF | 62.1 (−1.9) | 472 (−2) | — | 578 (−4) | 8 | 106 | — | — |
| An | C | 62.7 (−1.3) | 471 (−2) | 24000 |
578 (−4) | 7 | 107 | 0.49 | 11760 |
| Ju | C | 63.4 (−0.6) | 508 (−10) | 23000 |
607 (−4) | 16 | 99 | 0.15 | 3450 |
| Nap | THF | 63.6 (−0.4) | 473 (32) | — | 614 (−2) | 75 | 141 | — | — |
| Nap | C | 62.1 (−1.9) | 472 (31) | 16000 |
613 (−3) | 60 | 141 | 0.27 | 4320 |
| Th | THF | 64.4 (0.4) | 532 (−7) | — | 577 (−4) | 3 | 45 | — | — |
| Th | C | 64.3 (0.1) | 533 (−6) | 31000 |
579 (−2) | 3 | 46 | 0.42 | 13020 |
| ETh | C | 61.9 (−2.1) | 581 (−31) | 28000 |
677 (−3) | 8 | 96 | 0.13 | 3640 |
| Cou | C | 59.6 (−4.4) | 521 (0) | 29000 |
611 (−6) | 2 | 90 | 0.34 | 9860 |
The four probes displayed light-up emission upon hybridization, with Irel values ranging from 3-fold for Th6HI to an impressive 75-fold for Nap6HI (Table 1 and Fig. 4C) with only minor blue-shifts in λem (−2 to −6 nm) suggesting that rotor character was mainly responsible for their turn-on emission. The probes also displayed bathochromic (red) shifts in their absorbance maxima upon hybridization (Fig. 4C (iv–vi)). The Th6HI analog provided the clearest example of this concept (Fig. 4C (vi)), in which two well defined absorbances were present in the SS (stacked at ∼545 nm vs. unstacked at ∼490 nm). Hybridization caused loss of the 490 nm absorbance to create a single peak at ∼545 nm. Interestingly, the bulky donor Nap6HI probe exhibited little amount of “stacked” structure in the SS (Fig. 4C (iv)). Consequently, this probe provided the weakest SS background fluorescence (Fig. 4C (i)) and the greatest Irel upon DS formation.
The entire chalcone library within NarI12 was hybridized to the complementary strand with the probe opposite the natural C base. Changing the opposing base from THF to C had minimal impact on duplex stability (Tm). Photophysical parameters for the probes in the DS, including molar absorption coefficients (estimated from values for the unmodified duplex (X = G)) and Φfl values afforded brightness (εmax·Φfl) values for the chalcone library. The brightest probe was the thiophene analog Th6HI (13020 cm−1 M−1) followed by An6HI (11760 cm−1 M−1) and Cou6HI (9860 cm−1 M−1). The best light-up probe was clearly Nap6HI (Irel = 60-fold), followed by Ju6HI (16-fold), with An6HI and ETh6HI (8-fold) tied for third (Table 1).
The impact of An6HI and Ind6HI on the NarI12 DS was analyzed using circular dichroism (CD) measurements (Fig. S17†) and molecular dynamics (MD) simulations (Fig. 5). The native NarI12 DS displayed roughly equal positive (275 nm) and negative CD bands (250 nm) for B-form DNA with MD simulations providing standard B-DNA features (Fig. 5A)39 including long-lasting interstrand hydrogen bonds (93–99% occupancy, Table S7†), average base–base rise (Table S8†) and average helical twist (Table S9†). When G7 is replaced with An6HI or Ind6HI, the CD spectra maintained positive CD intensity at 275 nm but displayed diminished intensity of the negative CD band at 250 nm and this feature was more pronounced for NarI12 containing Ind6HI than An6HI (Fig. S17†). These spectral changes suggested more A-like character for the modified DS structures.31 MD simulations suggest that both An6HI and Ind6HI intercalate between the flanking base pairs and displace the opposing C (Fig. 5B and S44†), predominantly into the major groove. Stacking with the flanking base pairs is more persistent for Ind6HI (Fig. S47D†), while steric clashes between the terminal methyl groups in An6HI and the deoxyribose of the extrahelical C result in greater dynamics of An6HI opposite C (Fig. S46D†), which marginally disrupts stacking with the flanking base pairs (Fig. 5B). In addition, there is a reduction in the canonical helical twist angle in the base pairs flanking the dye (54.0° (An6HI), 47.9° (Ind6HI) versus 71.3° in native NarI12, Table S9†). The loss in helical twist allows the dye to stack effectively with the flanking base pairs (Fig. 5D and S47†). The remainder of the helix maintains canonical B-DNA structural features, including persistent hydrogen bonding in the base pairs flanking the dye (Table S7†). Thus, MD simulations predict chalcone-mediated local DS perturbations with loss in helical twist consistent with more A-like character (Fig. S17†).39
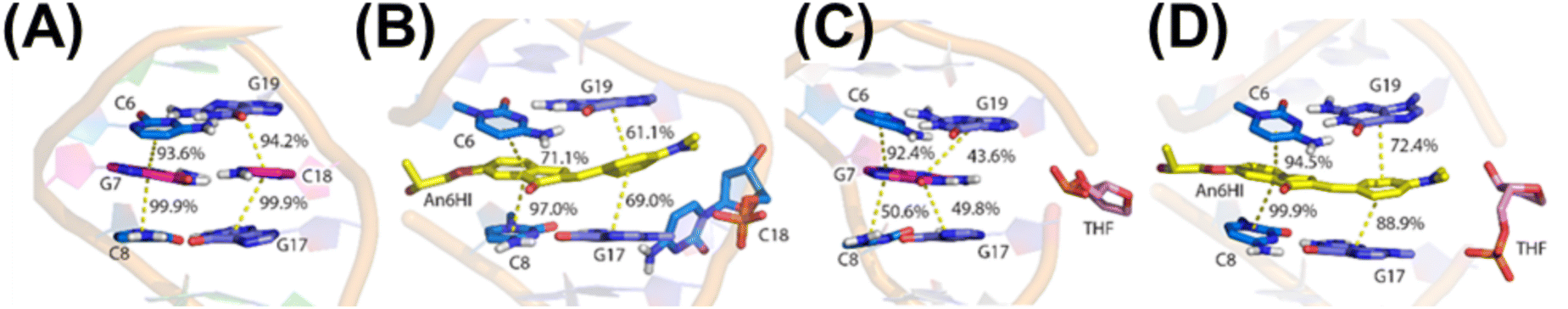 | ||
| Fig. 5 Representative MD structures of NarI12 DS containing (A) canonical G (magenta) opposite C (magenta), (B) An6HI (yellow) opposite C (light blue), (C) canonical G opposite THF (pink), and (D) An6HI (yellow) opposite THF (pink). Base–base stacking occupancies are provided. All images are side views from the major groove. Water, ions, and nonpolar hydrogen atoms are hidden for clarity. | ||
When G7 in the NarI12 DS is opposite THF, the model sugar adopts an extrahelical position (Fig. 5C and S48†), which elongates the backbone and increases the base pair step proximal to the THF site (Table S8†). This distortion is accompanied by kinking of the backbone (by up to 123.3°), which is consistent with previous reports of a significant decrease in Tm for duplexes containing a THF residue.40
The incorporation of THF into DNA generates an intrahelical void for accommodation of the dye. Indeed, as found when canonical G7 resides opposite THF, the THF moiety adopts an extrahelical position opposite the FMR probes, which permits the probe to span the interstrand distance unperturbed (Fig. 5D and S44†). As noted with the probes paired opposite C, a reduction in the helical twist angle occurs in the region surrounding the dye (52.3° (An6HI), 49.1° (Ind6HI), 71.3° for native, Table S9†). A complete discussion of the computational results can be found in the ESI.†
The impact of oligonucleotide length on light-up emission upon hybridization was tested with the Nap6HI probe inserted at G7, C13 or A17 of NarI22 (i.e. 5′-CTC-GGC-G7-CCA-TC-C13-TT-A17-CGA-GC-3′). For Nap6HI at G7 within NarI22 and NarI12, direct comparison of its light-up emission response was monitored as a function of temperature using fluorescent thermal melting experiments (Fig. S18†). The native NarI22 DS possesses a Tm of ∼78 °C and thus, the emission of Nap6HI displayed a sharp increase in intensity from 80–70 °C, which was then followed by a linear increase in dye fluorescence as the DS was cooled to 20 °C. In NarI12, Nap6HI displayed an Irel of 50-fold upon hybridization at 37 °C, which was reduced to ∼4-fold for the dye within NarI22. The melting data demonstrated that emission of the probe within the SS was 10-fold greater within the longer 22mer sequence. Clearly, oligonucleotide folding into secondary structures to increase dye rigidity is more favorable for the longer 22mer compared to the 12mer. Melting curves for the Nap6HI probe at C13 and A17 of NarI22 (Fig. S19 and S20†), led to similar conclusions.
Ratiometric response to duplex formation
The modular approach also permitted the selection of a FRET pair which can be used to increase signal upon hybridization.41–43 The Ind6HI probe can serve as the donor to the brightly emissive Th6HI acceptor (Fig. 6A). Placement of the acceptor (Th6HI, X) and donor (Ind6HI, N) into complementary strands, with separation of three base-pairs afforded a FRET efficiency of 85% (Fig. 6A). This FRET platform was able to enhance the fluorescent intensity response of the single Th6HI in the DS from 3-fold (Table 1) to 18-fold with λex at 425 nm, and 40-fold with λex at 405 nm. Ratiometric detection via a single probe possessing dual emission is also a powerful technique utilized to help eliminate microenvironment interferences and improve quantitative measurements.44 Within NarI12, both Cou6HI (Fig. 6B) and ETh6HI (Fig. S21†) provided dual emission. For Cou6HI, preferential excitation at 415 nm gave rise to emission at ∼490 nm for the coumarin donor, and internal charge transfer (ICT) emission at ∼611 nm for the fully conjugated chalcone (Fig. 6B). Upon hybridization, only ICT fluorescence at 611 nm was responsive, with little to no change in coumarin intensity at 490 nm, which serves to calibrate probe response (Fig. 6B). Similar results were observed for DNA binding by a free coumarin–hemicyanine, which also displayed preferential enhancement of ICT emission.45 Reasoning for such a response was attributed to an increase in dye planarity upon stacking interactions.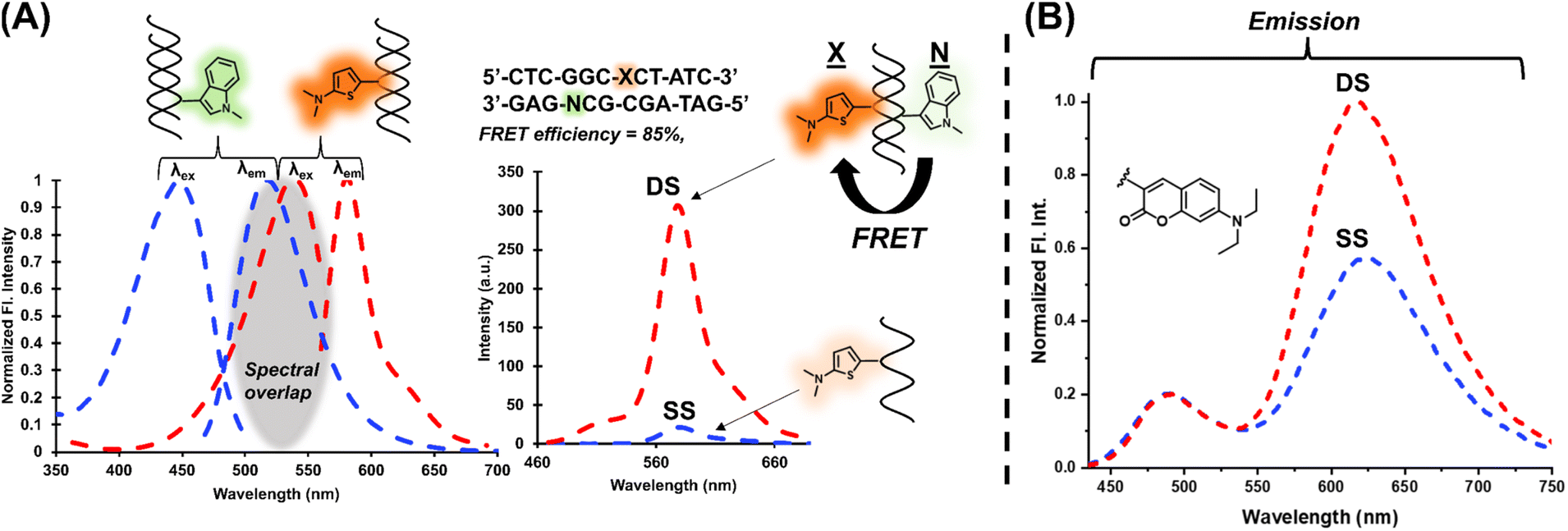 | ||
| Fig. 6 (A) Normalized fluorescence spectra for NarI12 duplexes displaying the spectral overlap between Ind6HI (donor) and Th6HI (acceptor) and FRET response of Th6HI (X, flanked by Cs and opposing C) upon excitation of Ind6HI (N, flanking G and C with opposing G) at 425 nm upon hybridization. Experiment carried out with 5 μM duplex in 100 mM NaCl, 50 mM phosphate buffer at pH 7.0. (B) Ratiometric response of Cou6HI within NarI12 (flanked by Cs and opposing C) to hybridization with full-length complementary DNA. Excitation at 415 nm with 5 μM duplex in 100 mM NaCl, 50 mM phosphate buffer at pH 7.0. | ||
Impact of flanking sequences
The DNA base pairs flanking the fluorescent probe within the duplex can have significant impact on its photophysical properties.27–29 To address this issue, the An6HI phosphoamidite was used to prepare three 12mer sequences by solid-phase synthesis. Two of the sequences were directly related to NarI12, but the flanking Cs were replaced by As or Ts to place the probe in AXA and TXT sequence contexts, while the third sequence was the NarI12 complementary strand, which placed An6HI in a GXG sequence. Upon DS formation, An6HI was brightest in TXT, displayed a significant blue Δλem shift (−11 nm) in AXA, and these changes differed from its response in CXC (Table 1) and in GXG. These findings prompted the synthesis of the corresponding AXA and TXT 12mers containing Ind6HI, Th6HI and Nap6HI to establish trends for chalcone response to flanking bases. These probes were inserted into the strands in high yield using M2 (i.e., Fig. 3B).All four probes were brightest in TXT, but the response of Ind6HI was a clear standout (Table 2). This probe was dim in CXC (Table 1, Φfl = 0.09), but provided bright green fluorescence with Φfl = 0.76 for brightness of 17480 cm−1 M−1 coupled with Irel = 4-fold in TXT (Fig. 7A). We are unaware of an internal nucleobase fluorescent probe with greater quantum efficiency in duplex DNA. The probe also displayed excellent brightness in AXA (11250 cm−1 M−1, Table 2 and Fig. 7A), and displayed a 10 nm Δλem shift to the blue, suggesting probe positioning in a less polar microenvironment within the AXA sequence context, as noted for An6HI. Thus, the emission of Ind6HI is highly sensitive to the nature of flanking bases, while Th6HI remained bright regardless of the flanking sequence (13020 cm−1 M−1 (CXC), 12160 cm−1 M−1 (AXA) and 16450 cm−1 M−1 (TXT)). Ind is a much weaker donor than Th and it is speculated that π-stacking of the 6HI acceptor with electron-deficient flanking Ts increases acceptor strength to promote ICT for Ind6HI for enhanced emission. Conversely, in AXA the Ind donor is flanked by Ts to reduce donor strength and cause a blue-shift due to the positive solvatochromism of the probe. The Th6HI probe undergoes ICT regardless of the flanking bases. Compared to probe performance in NarI12 (CXC, Table 1), the Irel values were diminished in TXT and AXA, as the probes were brighter in the SS. This was especially the case for Nap6HI, which displayed a 5-fold light-up response in TXT, which was reduced to 2-fold in AXA, compared to 60-fold in CXC (Table 1).
| 5′-CTC-GGN-X-NCA-TC-3′ | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 3′-GAG-CCY-C-YGT-AG-5′ | |||||||||
| Xa | NXN/YCY | T m (ΔTm) (°C) | λ ex (Δλex) (nm) | ε max (cm−1 M−1) | λ em (Δλem) (nm) | I rel | Δνe (nm) | Φ fl | B (cm−1 M−1) |
| a See Fig. 3B for donor abbreviations. b T m values of 12mer (5′-CTCGGN-X-NCATC, N = A or T) DS (5 μM) measured at 260 nm in 50 mM sodium phosphate buffer, pH 7, with 0.1 M NaCl, heating rate of 0.5 °C min−1, errors are ±1 °C; ΔTm = Tm (modified DS with probe (X) opposite C) – Tm of native 12mer DS containing X = G paired with C. c Values in brackets represent wavelength shifts in DS relative to SS. d Relative emission intensity of the chalcone probes in the DS versus SS. e Stokes shift in nm for the probes in the 12mer DS. f Fluorescence quantum yield of chalcone probes in the 12mer DS measured by the comparative method using either fluorescein in 0.1 M NaOH or rhodamine 101 in absolute ethanol as fluorescent standards, errors are ±5% obtained from three measurements at different DS concentrations. g Probe brightness (Φfl·εmax) in the 12mer DS. | |||||||||
| Ind | TXT/ACA | 57.3 (−0.2) | 450 (3) | 23000 |
516 (−4) | 4 | 66 | 0.76 | 17480 |
| Ind | AXA/TCT | 55.4 (−0.8) | 445 (−2) | 25000 |
509 (−10) | 3 (485 nm) | 64 | 0.45 | 11250 |
| An | TXT/ACA | 54.5 (−3.0) | 472 (−12) | 32500 |
577 (−2) | 2 | 105 | 0.46 | 14950 |
| An | AXA/TCT | 54.2 (−2.0) | 463 (−2) | 22000 |
566 (−11) | 6 (545 nm) | 103 | 0.49 | 10780 |
| Nap | TXT/ACA | 55.7 (−1.8) | 466 (20) | 17000 |
612 (−1) | 5 | 146 | 0.30 | 5100 |
| Nap | AXA/TCT | 52.6 (−3.6) | 458 (22) | 15000 |
613 (−4) | 2 | 155 | 0.18 | 2700 |
| Th | TXT/ACA | 57.6 (0.1) | 531 (0) | 35000 |
579 (1) | 2 | 48 | 0.47 | 16450 |
| Th | AXA/TCT | 57.8 (1.6) | 524 (−7) | 32000 |
574 (−4) | 4 | 50 | 0.38 | 12160 |
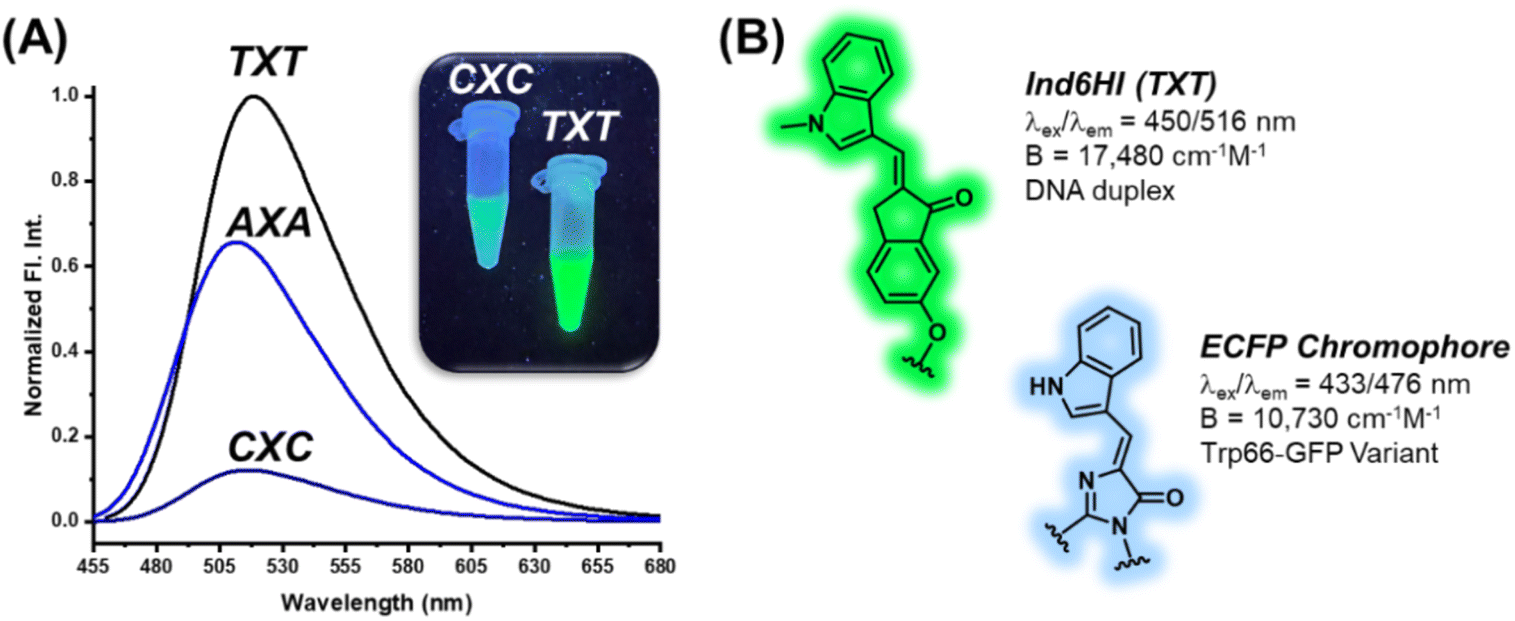 | ||
| Fig. 7 (A) Normalized fluorescence emission spectra for 12mer duplexes displaying the impact of flanking sequence on emission intensity for the Ind6HI probe that were acquired using 5 μM duplex in 100 mM NaCl, 50 mM phosphate buffer at pH 7.0, insert displays Ind6HI within CXC versus TXT duplexes that were illuminated with ultraviolet light and photographed. (B) Comparison of Ind6HI structure, λex/λem and brightness in TXT duplex DNA to the enhanced cyan fluorescent protein (ECFP) tryptophan (Trp66) chromophore within a protein matrix. | ||
The finding that Ind6HI can serve as the donor to Th6HI in a FRET pair (Fig. 6A) correlates with the wide-spread utility of the enhanced cyan fluorescent protein (ECFP) variant to serve as the donor to yellow fluorescent proteins (YFPs) for cellular imaging.46 The ECFP chromophore is a natural tryptophan (Trp66) derivative,47 which is strikingly similar in structure to the Ind6HI probe (Fig. 7B). Site-directed mutagenesis to vary the amino acids interacting with the ECFP chromophore has been used to improve brightness and the characteristics of ECFP for FRET measurements.46 Our finding that the Ind6HI donor possesses excellent brightness within TXT, while the Th6HI acceptor remains bright regardless of flanking sequence, will aid in optimization of this new FRET pair for biosensing applications.
Biosensing application
The ability of FMR probes to respond to changes in their microenvironment provides a direct path towards their use in aptasensor biosensing platforms.26 To highlight the potential use of FMR chalcones for DNA biosensing applications, a hybridization platform involving use of the quinine binding (MN4) aptamer48 was employed. The MN4 aptamer is a 36mer containing a dynamic triangular-shaped hydrophobic three-way junction (3WJ), which was initially selected to bind cocaine.49 However, the aptamer exhibits ligand binding promiscuity and interacts with other alkaloids, such as quinine, with higher affinity.48 Our interest in MN4 and other 3WJ DNA aptamers, stems from our recent finding that phenolic merocyanine dyes bind selectively to 3WJs to provide an impressive turn-on fluorescence response.50 The free dyes can be utilized to detect alkaloid binding through host-guest platforms, in which alkaloid binding displaces the merocyanine dye from the 3WJ to elicit a turn-off fluorescence response.50,51To employ an FMR chalcone for quinine detection using MN4, the aptamer was split into two fragments by removing three loop residues from stem 2 to afford an 11mer and 22mer sequence. The An6HI probe was inserted into the G9 position (X) of the 11mer, with flanking A and G bases with the probe opposite C (Fig. 8A). The split aptamer approach is used to reduce background signal, as the two aptamer fragments are only loosely assembled without target, but it also results in reduced target affinity, which can compromise the sensitivity of the aptamer sensor platform.52
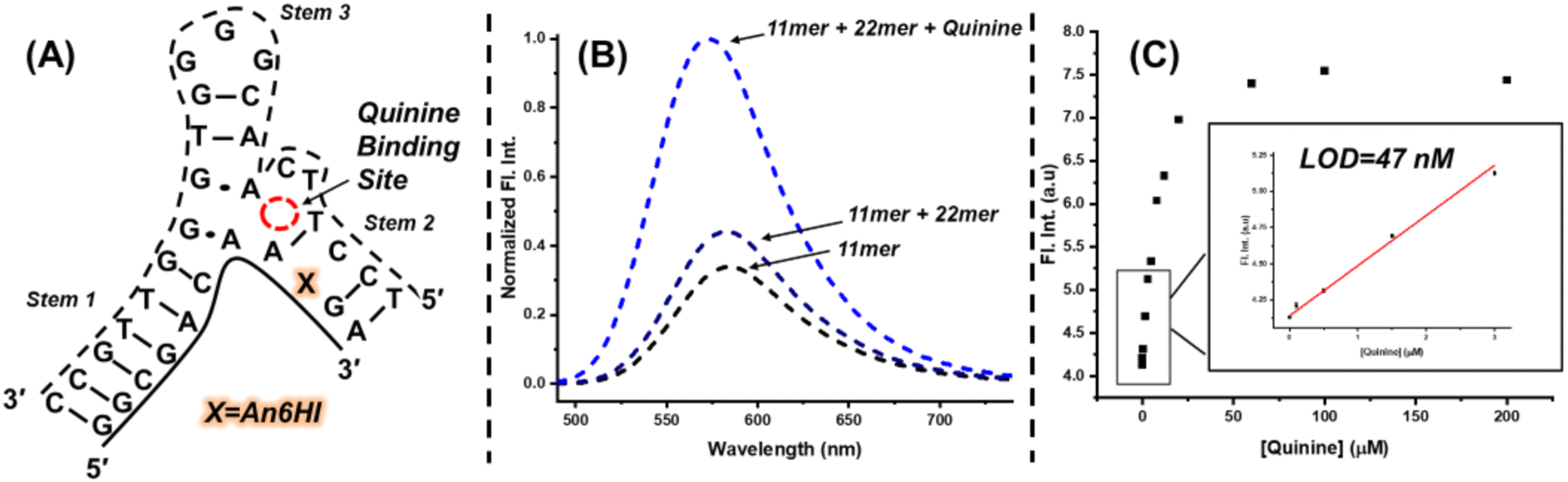 | ||
| Fig. 8 (A) Sequence and general structure of the An6HI modified MN4 split aptamer. (B) Fluorescence emission spectra (λex = 475 nm) of the 11mer An6HI MN4 split aptamer (4 μM) in absence and presence of the 22mer (5 μM), and quinine (200 μM). Solution buffered at pH 7 using 15 mM sodium phosphate and 30 mM sodium chloride. (C) Detection of quinine using the MN4 split aptamer platform with An6HI. Conditions: 5 μM 11mer labelled with An6HI, 6 μM 22mer, buffered at pH 7 using 15 mM sodium phosphate and 30 mM sodium chloride. Measurements performed in triplicate. | ||
Under optical conditions it was found that mixing the modified 11mer (4 μM) with a slight excess of the unmodified 22mer (5 μM) provided only a modest increase in the emission intensity of the internal An6HI probe (Fig. 8B). However, addition of excess quinine (200 μM) afforded a 3-fold turn-on emission response compared to the 11mer/22mer mixture in the absence of target. From quinine titrations a limit of detection (LOD) of 47 nM was determined in the buffer solution (Fig. 8C). For comparison, we previously obtained an LOD of 130 nM for label-free detection of quinine through dye displacement using the intact 36mer MN4 aptamer in buffer. That the internal split aptamer platform outperforms the label-free assay using the full MN4 aptamer is quite remarkable.52 Furthermore, the internal probe provides the highly desirable turn-on emission response. Turn-off fluorescent platforms are not advantageous for trace analyte detection due to their inability to provide a detectable signal change in contrast to the high background emission.53
Chalcone performance and comparison to state-of-the-art
The synthesis and performance of the seven chalcones reported herein represents their first usage as internal FMR probes within DNA oligonucleotides. Evaluation of recently published internal nucleobase probes (Fig. 9), which are regarded as state-of-the-art, highlights the many significant advantages of our simple modular aldol strategy to create internal FMR chalcones.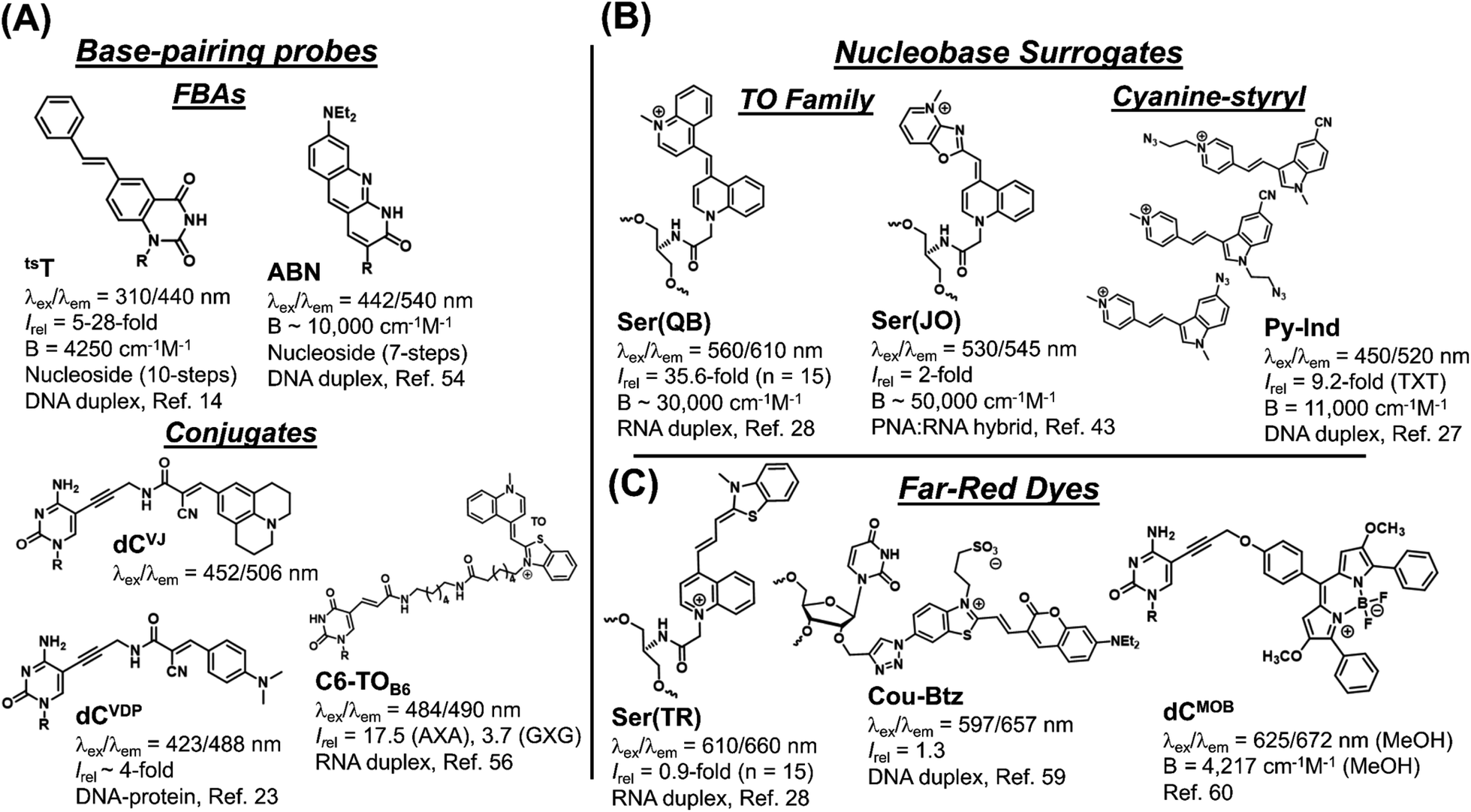 | ||
| Fig. 9 Recently reported internal fluorescent nucleic acid probes that are regarded as state-of-the-art. (A) Base-pairing probes that include fluorescent base analogs (FBAs) that have base-pairing capability and conjugates in which the fluorescent reporter is attached via a non-emissive linker to a natural base. (B) Nucleobase surrogates, which lack base-pairing ability, and include the thiazole orange (TO) family and cyanine-styryls. (C) Far-red emitting probes that are exemplified by a member of the TO family of nucleobase surrogates and base-pairing conjugates. | ||
The main limitation of FBAs (Fig. 9A) involves their challenging synthesis. The trans-stilbene annulated uracil derivative (tsT) involves a 10-step synthesis,14 while the ABN nucleoside was prepared in 7-steps with an overall yield of 0.8% and it lacks FMR character.54 The tsT probe possesses FMR properties, but undergoes excitation at 310 nm, which is discernible from DNA absorbance, but less so from protein, which limits its biosensing applications for studying DNA–protein interactions. Nucleobase conjugates permit modularity and examples include dCVJ and dCVDP (Fig. 9A), synthesized by the Hocek laboratory.23 The nucleosides were converted into triphosphates for enzymatic incorporation into oligonucleotides. This strategy lacks universal appeal because it is technically challenging, involving the tedious nucleoside triphosphate synthesis.55 Polymerase synthesis then imposes limits on what structures will serve as suitable substrates for enzymatic incorporation, as dCVJ was too bulky.23 Peptide coupling strategies are more commonly employed, as exemplified by the thiazole orange (TO) conjugate ThC6-TOB6 (Fig. 9A), in which TO is connected to amino-C6-dT (C6) via a hexanoic linker (TOB6).56 This design can limit site-specific information because TO is not spatially fixed and the long linker separating it from the oligonucleotide can permit intermolecular interactions57 in addition to the desirable TO back-folding for intramolecular intercalation with DS DNA or RNA.
For nucleobase surrogates (Fig. 9B) the Seitz laboratory has meticulously optimized the performance of the TO family (forced intercalation (FIT)-probes), while the Wagenknecht laboratory favours cyanine-styryls because the probes are photochemically more stable with larger Stokes shifts than TO. The TO dye also binds SS DNA, and to decrease fluorescence background the Seitz laboratory has inserted them into protein nucleic acid (PNA).16 The FIT-probes also fluoresce with higher intensity upon hybridization with RNA than with DNA.16 The best turn-on probe is the quinoline blue (Ser(QB)) derivative,28 while the oxazolopyridine analogue (Ser(JO)) lacks the turn-on features of Ser(QB), but is very bright in a PNA:RNA hybrid. Compared to the FMR chalcones, FIT-probes are brighter because they possess larger molar absorption coefficients. However, they are less efficient with very narrow Stokes shifts.
For modular synthesis of DNA containing cyanine-styryls a “click” approach has recently been presented, which involves the synthesis of a glycol-based ethynyl handle, which is then reacted with each dye azide via click chemistry.27 However, the ethynyl handle requires synthesis as the H-phosphonate making it tedious to synthesize the ethynyl-modified oligonucleotides using both phosphoramidite and H-phosphonate chemistry. Focus was placed on screening pyridinium (shown in Fig. 9B) and quinolinium styryl-indole dyes with azide groups at different positions. Unexpectedly, no responses were reported for three TO derivatives that were inserted into DNA using the click approach, which severely limits its utility.27
The performance of three recently reported far-red emitting probes (Fig. 9C) showcases the competitive advantage of our modular aldol approach. Fluorophores that emit in the far-red or NIR are in high demand to minimize autofluorescence interference for cellular bioimaging applications.58 The three far-red emitting probes were created using typical approaches with total synthesis of the far-red dye prior to oligonucleotide attachment to an internal handle. Thus, out of necessity, the probes are large with the thiazole red (Ser(TR)) FIT-probe being non-responsive to hybridization,28 while the Cou-Btz analogue displayed a modest turn-on response (1.3-fold).59 The BODIPY dCMOB analogue displayed changes in emission lifetime to protein binding (increase by ∼2.1 ns),60 but this requires sensitive instrumentation to accurately measure small lifetime changes in the nanosecond regime. In our aldol method, the acceptor is placed within the π-stack of the SS prior to donor attachment. This keeps the dye manageably small and able to respond to hybridization with an 8-fold light-up for ETh6HI at 677 nm in a CXC sequence (Table 1), which is encouraging for an internal far-red emitting probe.
Overall, the FMR chalcones out-perform the fluorescent readout of most state-of-the-art internal probes that are generally far more difficult to synthesize (Fig. 9). The only real exception are the brighter FIT-probes which have been incorporated into PNA, and flanked by LNA in RNA strands to enhance quantum efficiency for cellular RNA imaging through hybridization. FMR chalcones already possess high quantum efficiency in DNA, which is far more cost effective and user amenable in comparison to RNA and PNA synthesis. While not discussed in this analysis, the Kool laboratory has developed an in situ modular approach that utilizes an internal abasic (AP) site as the handle, which is created through base-excision repair, and reacted with fluorescent probes to create an oxime bond.61 This molecular biology approach is ideal for imaging DNA repair in cells and tissues, but it appears to be a free-dye to duplex platform making it challenging to make a direct comparison to our hybridization results using the aldol modular approach.
Experimental
All experimental procedures, HPLC chromatographs, MS, NMR spectra, detailed computational methods are found in the ESI.†Conclusions
Our goal is to develop internal FMR probes that are easy to employ in DNA biosensing applications and can be made amenable to high-throughput synthesis and analysis to keep pace with the growing demand to generate large volumes of oligonucleotides as therapeutics and biosensors. Our modular aldol approach has a small number of high yielding steps to create the chalcone-modified oligonucleotide that is readily separated from excess neutral aromatic aldehyde by ethanol precipitation. The chalcone surrogates effectively stack with the flanking base pairs to match the thermal stability of the fully paired canonical duplex. They have remarkable Φfl values (up to 76%) in duplex DNA, with excellent light-up emission upon hybridization with broad colour palette. For the chalcones and aldol modular approach our results reported herein for the seven internal FMR probes just scratch the surface. Aromatic aldehydes with various donor groups and substituents are critical building blocks for the construction of highly diverse and functional fluorescent chemosensors. Furthermore, the internal chalcone probes are structurally related to the chromophores within the fluorescent protein (FP) family that have been extensively utilized by the molecular biology community for cellular staining applications. This community has worked hard to enhance their properties and we can use this wealth of knowledge to optimize the performance of the internal chalcones. Our method paves the way for the larger biosensor community to incorporate numerous aldehydes into nucleic acids to expand their biotechnological applications. Current efforts are focused on tuning the nature of substituents to enhance dye brightness, optimizing the performance of the Ind6HI/Th6HI FRET pair, and exploring the properties of O-donor chalcones derived from phenolic aldehydes.Data availability
Supporting data for this manuscript has been uploaded as part of the ESI.†Author contributions
Ryan E. Johnson: investigations, data curation, writing – original draft. Makay T. Murray: investigations, data curation, writing. Lucas J. Bycraft: data curation. Stacey D. Wetmore: supervision, funding acquisition, writing – review & editing. Richard A. Manderville: supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (grant 2016-04568 to S. D. W. and 2018-400136 to R. A. M.) S. D. W. also thanks computational resources provided by the Digital Research Alliance of Canada (the Alliance), and the University of Lethbridge Board of Governors Research Chair Program for supporting this research. R. A. M. also thanks the Canada Foundation for Innovation (CFI) for funding an upgrade to an in-lab DNA synthesizer.References
- W. Xu, W. K. M. Chan and E. T. Kool, Fluorescent Nucleobases as Tools for Studying DNA and RNA, Nat. Chem., 2017, 9, 1043–1055 CrossRef CAS PubMed.
- B. Y. Michel, D. Dziuba, R. Benhida, A. P. Demchenko and A. Burger, Probing of Nucleic Acid Structures, Dynamics, and Interactions with Environment-Sensitive Fluorescent Labels, Front. Chem., 2020, 8, 112 CrossRef CAS.
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Rational Design of Small Molecule Fluorescent Probes for Biological Applications, Org. Biomol. Chem., 2020, 18, 5747–5763 RSC.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, Thioflavin T as an Efficient Inducer and Selective Fluorescent Sensor for the Human Telomeric G-Quadruplex DNA, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef CAS PubMed.
- H. Zipper, H. Brunner, J. Bernhagen and F. Vitzthum, Investigations on DNA Intercalation and Surface Binding by SYBR Green I, Its Structure Determination and Methodological Implications, Nucleic Acids Res., 2004, 32, e103 CrossRef PubMed.
- Y. Du and S. Dong, Nucleic Acid Biosensors: Recent Advances and Perspectives, Anal. Chem., 2017, 89, 189–215 CrossRef CAS PubMed.
- P. S. Deore, M. D. Gray, A. J. Chung and R. A. Manderville, Ligand-Induced G-Quadruplex Polymorphism: A DNA Nanodevice for Label-Free Aptasensor Platforms, J. Am. Chem. Soc., 2019, 141, 14288–14297 CrossRef CAS PubMed.
- Y. Li, R. Su, H. Li, J. Guo, N. Hildebrandt and C. Sun, Fluorescent Aptasensors: Design Strategies and Applications in Analyzing Chemical Contamination of Food, Anal. Chem., 2022, 94, 193–224 CrossRef CAS.
- M. A. Haidekker and E. A. Theodorakis, Ratiometric Mechanosensitive Fluorescent Dyes: Design and Applications, J. Mater. Chem. C, 2016, 4, 2707–2718 RSC.
- S.-C. Lee, J. Heo, H. C. Woo, J.-A. Lee, Y. H. Seo, C.-L. Lee, S. Kim and O.-P. Kwon, Fluorescent Molecular Rotors for Viscosity Sensors, Chem. Eur. J., 2018, 24, 13706–13718 CrossRef CAS PubMed.
- Y. Du, B. Li and E. Wang, “Fitting” Makes “Sensing” Simple: Label-Free Detection Strategies Based on Nucleic Acid Aptamers, Acc. Chem. Res., 2013, 46, 203–213 CrossRef CAS PubMed.
- O. P. Schmidt, G. Mata and N. W. Luedtke, Fluorescent Base Analogue Reveals T-HgII-T Base Pairs Have High Kinetic Stabilities That Perturb DNA Metabolism, J. Am. Chem. Soc., 2016, 138, 14733–14739 CrossRef CAS PubMed.
- D. D. Burns, K. L. Teppang, R. W.Lee, M. E. Lokensgard and B. W. Purse, Fluorescence Turn-On Sensing of DNA Duplex Formation by a Tricyclic Cytidine Analogue, J. Am. Chem. Soc., 2017, 139, 1372–1375 CrossRef CAS PubMed.
- A. Karimi, R. Börner, G. Mata and N. W. Luedtke, A Highly Fluorescent Nucleobase Molecular Rotor, J. Am. Chem. Soc., 2020, 142, 14422–14426 CrossRef CAS PubMed.
- R. W. Sinkeldam, A. J. Wheat, H. Boyaci and Y. Tor, Emissive Nucleobases as Molecular Rotors, ChemPhysChem, 2011, 12, 567–570 CrossRef CAS PubMed.
- F. Hövelmann and O. Seitz, DNA Stains as Surrogate Nucleobases in Fluorogenic Hybridization Probes, Acc. Chem. Res., 2016, 49, 714–723 CrossRef.
- A. Heidari, A. Ghorbani-Choghamarani, M. Hajjami and R. H. E. Hudson, Fluorescent Biaryl Uracils with C5-Dihydro- and Quinazolinone Heterocyclic Appendages in PNA, Molecules, 2020, 25, 1995 CrossRef CAS.
- G. Mata and N. W. Luedtke, Fluorescent Probe for Proton-Coupled DNA Folding Revealing Slow Exchange of i-Motif and Duplex Structures, J. Am. Chem. Soc., 2015, 137, 699–707 CrossRef CAS PubMed.
- A. A. Tanpure and S. G. Srivatsan, Conformation-sensitive Nucleoside Analogues as Topology-specific Fluorescence Turn-on Probes for DNA and RNA G-quadruplexes, Nucleic Acids Res., 2015, 43, e149 CrossRef PubMed.
- R. A. Manderville and S. D. Wetmore, C-Linked 8-Aryl Guanine Nucleobase Adducts: Biological Outcomes and Utility as Fluorescent Probes, Chem. Sci., 2016, 7, 3482–3493 RSC.
- T. Z. Cservenyi, A. J. Van Riesen, F. D. Berger, A. Desoky and R. A. Manderville, A Simple Molecular Rotor for Defining Nucleoside Environment within a DNA Aptamer-Protein Complex, ACS Chem. Biol., 2016, 11, 2576–2582 CrossRef CAS.
- M. D. Gray, P. S. Deore, A. J. Chung, A. J. Van Riesen, R. A. Manderville, P. S. Prabhakar and S. D. Wetmore, Lighting Up the Thrombin-Binding Aptamer G-Quadruplex with an Internal Cyanine-Indole-Quinolinium Nucleobase Surrogate. Direct Fluorescent Readout for Thrombin Binding without Topology Switching, Bioconjugate Chem., 2020, 31, 2596–2606 CrossRef CAS.
- D. Dziuba, R. Pohl and M. Hocek, Polymerase Synthesis of DNA Labelled with Benzylidene Cyanoacetamide-based Fluorescent Molecular Rotors: Fluorescent Light-up Probes for DNA-binding Proteins. Chem, Commun, 2015, 51, 4880–4882 CAS.
- P. Güixens-Gallardo and M. Hocek, Acetophenyl-thienyl-aniline-Linked Nucleotide for Construction of Solvatochromic Fluorescence Light-Up DNA Probes Sensing Protein-DNA Interactions, Chem.–Eur. J., 2021, 27, 7090–7093 CrossRef PubMed.
- M. Hocek, Enzymatic Synthesis of Base-Functionalized Nucleic Acids for Sensing, Cross-linking, and Modulation of Protein–DNA Binding and Transcription, Acc. Chem. Res., 2019, 52, 1730–1737 CrossRef CAS PubMed.
- S. B. Ebrahimi, D. Samanta, H. F. Cheng, L. I. Nathan and C. A. Mirkin, Forced Intercalation (FIT)-Aptamers, J. Am. Chem. Soc., 2019, 141, 13744–13748 CrossRef CAS PubMed.
- J. Gebhard, L. Hirsch, C. Schwechheimer and H.-A. Wagenknecht, Hybridization-Sensitive Fluorescent Probes for DNA and RNA by a Modular “Click” Approach, Bioconjugate Chem., 2022, 33, 1634–1642 CrossRef CAS PubMed.
- F. Hövelmann, I. Gaspar, J. Chamiolo, M. Kasper, J. Steffen, A. Ephrussi and O. Seitz, LNA-Enhanced DNA FIT-Probes for Multicolour RNA Imaging, Chem. Sci., 2016, 7, 128–135 RSC.
- T. W. Manning, A. J. Van Riesen and R. A. Manderville, Screening Internal Donor-Acceptor Biaryl Nucleobase Surrogates for Turn-On Fluorescence Affords an Aniline-Carboxythiophene Probe for Protein Detection by G-Quadruplex DNA, Bioconjugate Chem., 2021, 32, 1791–1801 CrossRef CAS PubMed.
- R. E. Johnson, A. J. Van Riesen and R. A. Manderville, On-Strand Knoevenagel Insertion of a Hemicyanine Molecular Rotor Loop Residue for Turn-On Fluorescence Detection of Pb-Induced G-Quadruplex Rigidity, Bioconjugate Chem., 2021, 32, 2224–2232 CrossRef CAS PubMed.
- A. Omumi, D. G. Beach, M. Baker, W. Gabryelski and R. A. Manderville, Postsynthetic Guanine Arylation of DNA by Suzuki-Miyaura Cross-Coupling, J. Am. Chem. Soc., 2011, 133, 42–50 CrossRef CAS.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Recent Development of Chemosensors Based on Cyanine Platforms, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- T.-B. Ren, W. Xu, Q.-L. Zhang, X.-X. Zhang, S.-Y. Wen, H.-B. Yi, L. Yuan and X.-B. Zhang, Enhancing the Anti-Solvatochromic Two-Photon Fluorescence for Cirrhosis Imaging by Forming a Hydrogen-Bond Network, Angew. Chem. Int. Ed., 2018, 57, 7473–7477 CrossRef CAS.
- E. Colucci-Guyon, A. S. Batista, S. D. S. Oliveira, M. Blaud, I. C. Bellettini, B. S. Marteyn, K. Leblanc, P. Herbomel and R. Duval, Ultraspecific Live Imaging of the Dynamics of Zebrafish Neutrophil Granules by a Histopermeable Fluorogenic Benzochalcone Probe, Chem. Sci., 2019, 10, 3654–3670 RSC.
- C. Wang, T. Wang, M. Zhao, F. Dai, Z. Niu, W. Zhang and Y. Ma, A Simple Chalcone Molecular Rotor for Specific Fluorescence Imaging of Mitochondrial Viscosity Changes in Living Cells, Dyes Pigm., 2021, 194, 109593 CrossRef CAS.
- M. V. Sigalov, B. A. Shainyan and I. V. Sterkhova, Photoinduced Intramolecular Bifurcate Hydrogen Bond: Unusual Mutual Influence on the Components, J. Org. Chem., 2017, 82, 9075–9086 CrossRef CAS PubMed.
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Coumarin-Based Small-Molecule Fluorescent Chemosensors, Chem. Rev., 2019, 119, 10403 CrossRef CAS PubMed.
- R. E. Johnson, J. M. van der Zalm, A. Chen, I. J. Bell, T. J. Van Raay, M. S. Al-Abdul-Wahid and R. A. Manderville, Unraveling the Chemosensing Mechanism by the 7-(Diethylamino) coumarin-hemicyanine Hybrid: A Ratiometric Fluorescent Probe for Hydrogen Peroxide, Anal. Chem., 2022, 94, 11047–11054 CrossRef CAS.
- H.-L. Ng, M. L. Kopka and R. E. Dickerson, The Structure of a Stable Intermediate in the A ↔ B DNA Helix Transition, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 2035–2039 CrossRef CAS.
- T. J. Matray and E. T. Kool, Selective and Stable DNA Base Pairing without Hydrogen Bonds, J. Am. Chem. Soc., 1998, 120, 6191–6192 CrossRef CAS PubMed.
- C. Holzhauser and H.-A. Wagenknecht, DNA and RNA “Traffic Lights”: Synthetic Wavelength-Shifting Fluorescent Probes Based on Nucleic Acid Base Substitutes for Molecular Imaging, J. Org. Chem., 2013, 78, 7373–7379 CrossRef CAS PubMed.
- M. S. Wranne, A. F. Füchtbauer, B. Dumat, M. Bood, A. H. El-Sagheer, T. Brown, H. Gradén, M. Grøtli and L. M. Wilhelmsson, Toward Complete Sequence Flexibility of Nucleic Acid Base Analogue FRET, J. Am. Chem. Soc., 2017, 139, 9271–9280 CrossRef CAS.
- G. Fang, J. Chamiolo, S. Kankowski, F. Hövelmann, D. Friedrich, A. Löwer, J. C. Meier and O. Seitz, A Bright FIT-PNA Hybridization Probe for the Hybridization State Specific Analysis of a C → U RNA Edit via FRET in a Binary System, Chem. Sci., 2018, 9, 4794–4800 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Small Molecule-based Ratiometric Fluorescence Probes for Cations, Anions, and Biomolecules, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- P. S. Deore, D. S. Coman and R. A. Manderville, A Coumarin–Hemicyanine Hybrid as a Ratiometric Fluorescent Sensor of Microenvironment Proticity. Chem, Commun, 2019, 55, 3540–3543 CAS.
- M. A. Rizzo, G. H. Springer, B. Granada and D. W. Piston, An Improved Cyan Fluorescent Protein Variant Useful for FRET, Nat. Biotechnol., 2004, 22, 445–449 CrossRef CAS PubMed.
- K. S. Sarkisyan, I. V. Yampolsky, K. M. Solntsev, S. A. Lukyanov, K. A. Lukyanov and A. S. Mishin, Tryptophan-Based Chromophore in Fluorescent Proteins can be Anionic, Sci. Rep., 2012, 2, 608 CrossRef PubMed.
- O. Reinstein, M. Yoo, C. Han, T. Palmo, S. A. Beckham, M. C. J. Wilce and P. E. Johnson, Quinine Binding by the Cocaine-binding Aptamer. Thermodynamic and Hydrodynamic Analysis of High-affinity Binding of an Off-target Ligand, Biochemistry, 2013, 52, 8652–8662 CrossRef CAS PubMed.
- M. S. Stojanovic, P. de Prada and D. W. Landry, Fluorescent Sensors Based on Aptamer Self-Assembly, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS PubMed.
- A. J. Van Riesen, J. Le, S. Slavkovic, Z. R. Churcher, A. A. Shoara, P. E. Johnson and R. A. Manderville, Visible Fluorescent Light-up Probe for DNA Three-Way Junctions Provides Host–Guest Biosensing Applications, ACS Appl. Bio Mater., 2021, 4, 6732–6741 CrossRef CAS PubMed.
- A. J. Van Riesen, B. Kalnitsky, A. A. Shoara, S. Slavkovic, Z. R. Churcher, P. E. Johnson and R. A. Manderville, Tuning the DNA Binding Properties of Phenolic Hemicyanine Dyes for Host-Guest Fluorescent Aptasensor Applications, Dyes Pigm., 2023, 209, 110936 CrossRef CAS.
- H. Yu, J. Canoura, B. Guntupalli, X. Lou and Y. Xiao, A Cooperative-Binding Split Aptamer Assay for Rapid, Specific and Ultra-Sensitive Fluorescence Detection of Cocaine in Saliva, Chem. Sci., 2017, 8, 131–141 RSC.
- J. Zhou and H. Ma, Design Principles of Spectroscopic Probes for Biological Applications, Chem. Sci., 2016, 7, 6309–6315 RSC.
- G. N. Samaan, M. K. Wyllie, J. M. Cizmic, L.-M. Needham, D. Nobis, K. Ngo, S. Andersen, S. W. Magennis, S. F. Lee and B. W. Purse, Single-Molecule Fluorescence Detection of a Tricyclic Nucleoside Analogue, Chem. Sci., 2021, 12, 2623–2628 RSC.
- K. Burgess and D. Cook, Synthesis of Nucleoside Triphosphates, Chem. Rev., 2000, 100, 2047–2059 CrossRef CAS PubMed.
- P. Klimkowski, S. De Ornellas, D. Singleton, A. H. El-Sagheer and T. Brown, Design of Thiazole Orange Oligonucleotide Probes for Detection of DNA and RNA by Fluorescence and Duplex Melting, Org. Biomol. Chem., 2019, 17, 5943–5950 RSC.
- T. Inoue, Y. Sugiura, J. Saitoh, T. Ishiguro and M. Otsuka, Fluorescence Property of Oxazole Yellow-Linked Oligonucleotide. Triple Helix Formation and Photocleavge of Double-Stranded DNA in the Presence of Spermine, Bioorg. Med. Chem., 1999, 7, 1207–1211 CrossRef CAS PubMed.
- Y. W. Jun, H. R. Kim, Y. J. Reo, M. Dai and K. H. Ahn, Addressing the Autofluorescence Issue in Deep Tissue Imaging by Two-Photon Microscopy: The Significance of Far-Red Emitting Dyes, Chem. Sci., 2017, 8, 7696–7704 RSC.
- A. Eördögh, J. Steinmeyer, K. Peewasan, U. Schepers, H.-A. Wagenknecht and P. Kele, Polarity Sensitive Bioorthogonally Applicable Far-Red Emitting Labels for Postsynthetic Nucleic Acid Labeling by Copper-Catalyzed and Copper-Free Cycloaddition, Bioconjugate Chem., 2016, 27, 457–464 CrossRef PubMed.
- M. Kuba, R. Pohl, T. Kraus and M. Hocek, Nucleotides Bearing Red Viscosity-Sensitive Dimethoxy-Bodipy Fluorophore for Enzymatic Incorporation and DNA Labeling, Bioconjugate Chem., 2023, 34, 133–139 CrossRef CAS PubMed.
- Y. W. Jun, E. M. Harcourt, L. Xiao, D. L. Wilson and E. T. Kool, Efficient DNA Fluorescence Labeling via Base Excision Trapping. Nat, Commun, 2022, 13, 5043 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, HPLC, MS, NMR spectra, detailed computational methods. See DOI: https://doi.org/10.1039/d3sc00772c |
| This journal is © The Royal Society of Chemistry 2023 |