
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of atropisomeric indolosesquiterpenoids via N–N bond formation: dixiamycins A and B†‡
Rhituparna
Nandi§
 a,
Sovan
Niyogi§
b,
Sourav
Kundu§
a,
Vipin R.
Gavit
a,
Mintu
Munda
a,
Ranjit
Murmu
b and
Alakesh
Bisai
*ab
a,
Sovan
Niyogi§
b,
Sourav
Kundu§
a,
Vipin R.
Gavit
a,
Mintu
Munda
a,
Ranjit
Murmu
b and
Alakesh
Bisai
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal Bypass Road, Bhopal 462 066, Madhya Pradesh, India. E-mail: alakesh@iiserkol.ac.in; alakeshb@gmail.com
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur Campus, Kalyani, Nadia, 741 246, West Bengal, India
First published on 20th June 2023
Abstract
N–N dimeric indolosesquiterpene alkaloids constitute a class of under-investigated architecturally intriguing natural products. Herein, we report the first chemical oxidation approach to the asymmetric total syntheses of these atropisomeric indolosesquiterpenoids through N–N bond formation. Specifically, dixiamycins A (1a) and B (1b) were prepared through a Cu(I)-mediated aerobic dehydrogenative dimerization from the naturally occurring monomer xiamycin A methyl ester (2b); this preparation also represents the first total synthesis of dixiamycin A (1a). The monomer xiamycin A methyl ester (2b) was synthesized via a late-stage Buchwald Pd(II)-mediated aerobic dehydrogenative C–N bond formation.
Introduction
Pentacyclic indolosesquiterpene alkaloids (e.g., 1–3; Fig. 1), a novel and growing class of architecturally complex secondary metabolites, were first isolated from a range of Streptomyces species in 2010, exhibit important biological activities such as antimicrobial, antiviral, antitumor, immunomodulatory, and enzyme inhibitory activities, and are commonly referred to as the xiamycin family of alkaloids.1 In 2012, the atropisomeric indolosesquiterpenoid natural products dixiamycins A (1a) and B (1b) were isolated independently by Zhang and Hertweck.2,3 These indole alkaloids each have a rare N–N linkage that generates an important class of atropisomers, featuring axial chirality about the N–N axis where the N atoms are sp3-hybridized. Prior to this finding, the monomers of these dimers, i.e., xiamycin A (2a) and its methyl ester (2b), were isolated by Hertweck and coworkers from Streptomyces sp. GT2002/1503 (ref. 3a) and HKI0595,3b which are endophytes from the mangrove plants Bruguiera gymnorrhiza3a and Kandelia candel,3b respectively. Alkaloids 2a–b have been reported to display anti-HIV and antibiotic activities.3a Structurally, these alkaloids include a pentacyclic framework with four contiguous stereogenic centers at the periphery of a trans-decalin scaffold to which is attached a carbazole unit.4,5 Oridamycin A (3a) is a diastereomer of xiamycin A (2a) and has been isolated from Streptomyces sp. KS84.4b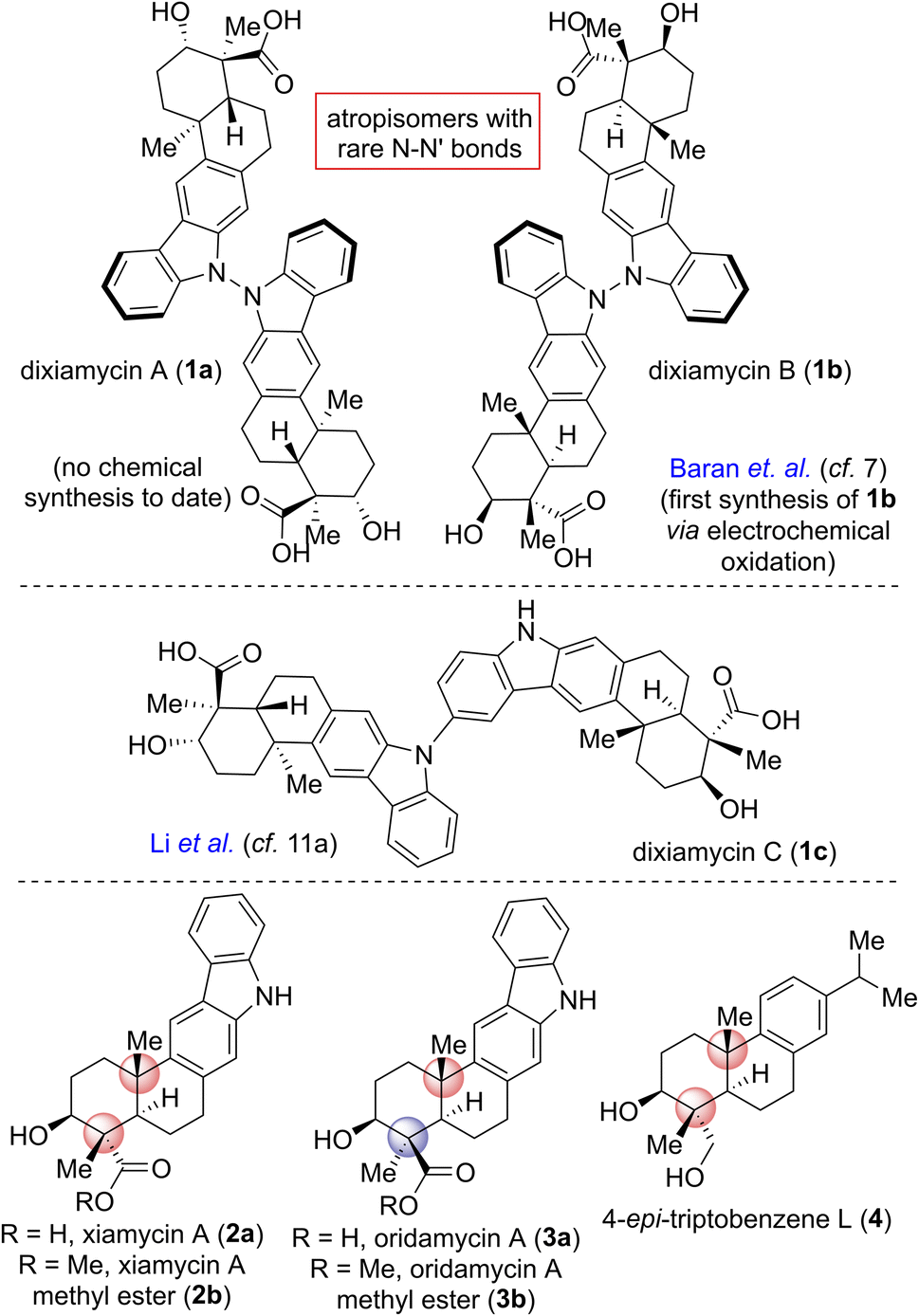 | ||
| Fig. 1 Indolosesquiterpene alkaloids 1–3. | ||
Importantly, two out of the four stereogenic centers are synthetically challenging all-carbon quaternary centers. The new uninvestigated N–N dimeric forms of xiamycin A are nearly an order of magnitude more potent than their monomeric scaffolds.2 The emerging recognition of the biological activity of these indolosesquiterpenoids has drawn considerable attention from the synthetic community, which has produced several syntheses of these alkaloids. The most efficient approach to the atropo-diastereomers of xiamycin A would be to effect a direct oxidation to form an N–N bond from the monomer as per the proposed biosynthesis following a single-electron transfer (SET) mechanism (Scheme 1). However, given that there has been rather limited success in achieving a direct N–N bond formation of two carbazole units with chemical oxidants (such as the use of stoichiometric amounts of oxidants I2,6a KMnO4,6b Ag2O,6c or dichromate6d), the synthesis of dixiamycins A (1a) and B (1b), bearing sensitive functionalities, remains a formidable challenge. In this regard, an electrochemical oxidation approach reported by Baran and coworkers elegantly addressed the total synthesis of dixiamycin B (1b).7
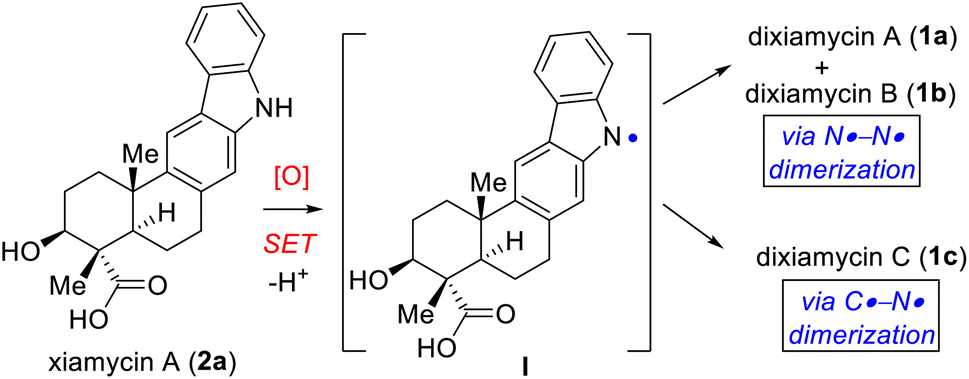 | ||
| Scheme 1 Biosynthesis of dixiamycins A (1a), B (1b) and C (1c). | ||
Based on the biosynthesis proposal (Scheme 1) and a recent report on aerobic oxidation by Stahl and coworkers,8 we envisioned that a suitably protected xiamycin A might be made to engage, under Cu(I)-catalysis, in an oxidative N–N bond formation to provide both 1a and 1b. Our efforts began with identifying a practical synthesis of xiamycin A methyl ester (2b). Prior elegant approaches to monomeric indolosesquiterpene alkaloids have been independently developed by Baran (cyclization of a carbazole-anchored epoxy ether),7 Krische (TiCl4-promoted Friedel–Crafts cyclization),9 Trotta (radical-induced polyene cyclization),10 Li11 (6π-electrocyclization/aromatization and indole C2–H bond activation/Heck annulation), Sarpong12 (from (R)-carvone, using a photoinduced benzannulation sequence to forge the carbazole core), Dethe13 (oxidative Heck/aromatization for carbazole synthesis) and our group (following δCsp3–H activation of the pentacyclic skeleton of indolosesquiterpene alkaloid).14
Herein, we report the total synthesis of both N–N atropo-diastereomers of indolosesquiterpene alkaloids, namely dixiamycins A (1a) and B (1b), via a nature-inspired aerobic oxidation. Impressively, this work represents, to the best of our knowledge, the first total synthesis of dixiamycin A (1a), clearly consistent with nature's oxidative approach to dixiamycins. Moreover, the currently developed unified practical asymmetric approach to naturally occurring abietane diterpenoids (via a key epoxy-ene cyclization15–18) and the congeners of the xiamycin family (via a late-stage Buchwald oxidative C–N bond formation19) would enable access to these natural products in significant quantities.
Result and discussion
On the basis of their structural similarity to naturally occurring diterpenoids, such as 4-epi-triptobenzene L (4), we envisioned a unified approach to the xiamycins (Scheme 2). Retrosynthetically, we imagined accessing highly functionalized o-bromo nitroarene 7 as an advanced intermediate for our synthesis. Thus, a late-stage Buchwald oxidative C–N bond formation of 2-phenyl acetanilide 6 could construct the carbazole moiety of the xiamycins. o-Bromo nitroarene 7 could be synthesized from o-bromo isopropylarene 8 using an ipso-nitration, which in turn could be synthesized from 13 using Sharpless asymmetric epoxidation followed by Lewis-acid-assisted epoxy-ene cyclization (via intermediate 4).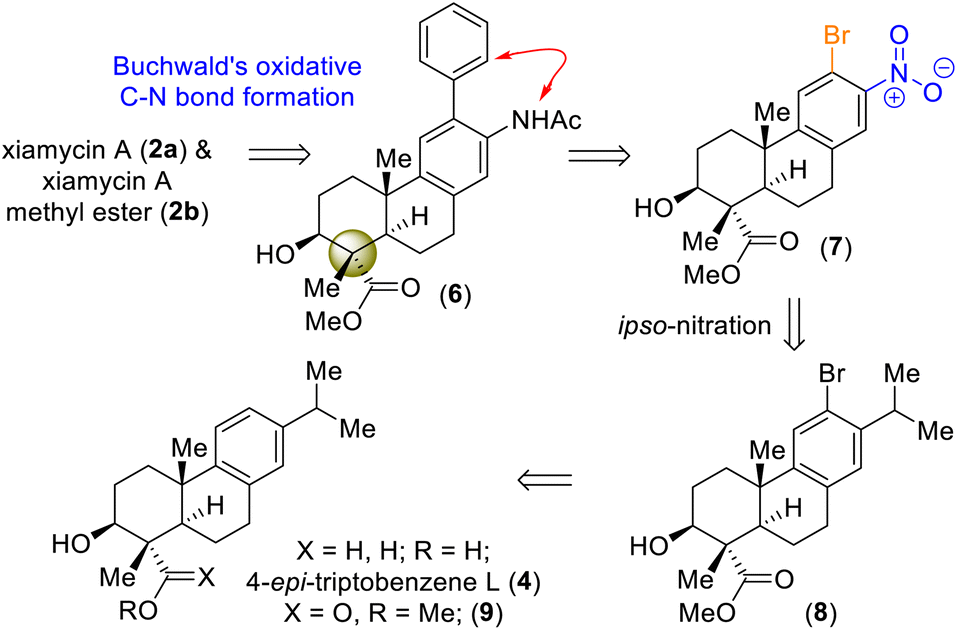 | ||
| Scheme 2 Hypothesized accessing of atropisomers of dixiamycins A (1a) and B (1b) and retrosynthetic plan (via 4-epi-triptobenzene L (4)). | ||
Our synthesis commenced with a Cu-catalyzed Csp3–Csp3 bond formation between functionalized geranyl acetate 10 and 3-isopropylbenzyl bromide 11, followed by TBS deprotection using TBAF to access allylic alcohol 13 (Scheme 3). Sharpless asymmetric epoxidation15 of 13 with TBHP in the presence of Ti(OiPr)4 and (+)-DET afforded epoxy-alcohol 14 with 95% ee in 92% yield. Treatment of 14 under Lewis-acid-assisted epoxy-ene cyclization completed the total synthesis of naturally occurring 4-epi-triptobenzene L (4) bearing a trans-decalin motif and four contiguous stereogenic centers in 63% yield (Scheme 3).16 The synthesized 4-epi-triptobenzene L (4) contained all the requisite stereogenic centers for the indolosesquiterpene alkaloids in place.17 Furthermore, a chemoselective oxidation of the 1,3-diol in the presence of PIDA and TEMPO followed by Pinnick oxidation and methylation with dimethyl sulfate (Me2SO4) furnished ester 9 in 76% yield (Scheme 3). Next, using the protocol described above, a mass of more than 10 g of compound 9 was prepared.
 | ||
| Scheme 3 Total syntheses of 4-epi-triptobenzene L (4) via polyene cyclization. | ||
Next, aromatic electrophilic bromination of 9 with N-bromo succinimide (NBS) in acetonitrile afforded 8 in 92% yield (Scheme 4). Thus, the stage was set for the ipso-nitration,18 which was investigated under various conditions. Gratifyingly, the ipso-nitration of the isopropyl group of 8 was realized with fuming HNO3 at −40 °C. Carrying out a nitration of the hydroxyl group afforded nitrite derivative 15 in 69% yield, with the identity of this derivative confirmed by X-ray analysis of its Suzuki–Miyaura coupling product (after recrystallization). Reduction of the nitro groups of 16 (10 mol% Pd–C under 1 atmosphere of H2 gas) followed by the treatment with Ac2O afforded acetanilide 6 in 92% yield over 2 steps (Scheme 4). At this point we were in a position to test the Buchwald19 oxidative C–N bond formation of acetanilide 6 to craft the pentacyclic core of xiamycin A (Scheme 4). Since molecular oxygen is a quintessential oxidizing agent that performs several dehydrogenative processes in the presence of substoichiometric amounts of high-valence transition-metal complexes and has important implications for the large-scale preparation of pharmaceuticals and fine chemicals, we shifted our attention to a transition-metal-free process under an oxygen atmosphere.
 | ||
| Scheme 4 Synthesis of a pentacyclic framework under oxidative C–N bond formation. | ||
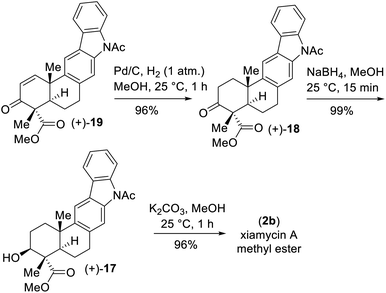
Our initial attempt using a combination of 10 mol% Pd(OAc)2 and 10 mol% Cu(OAc)2 in toluene at 110 °C under an oxygen atmosphere was disappointing as acetanilide 6 provided a multitude of spots on the corresponding TLC with no expected product. We did isolate a 32% yield of pentacyclic ketone 18 and 19% yield of enone 19 along with 31% yield of the starting material acetanilide 6 (Scheme 4). Thus, a quick optimization using different solvents, oxidants and Pd(II) source was carried out; it revealed that DMSO was a good choice, where complete consumption of starting acetanilide 6 was observed. Interestingly, we observed a three-fold oxidative process of 6 in the presence of 10 mol% Pd(OAc)2 in dimethylsulfoxide (DMSO) under an oxygen atmosphere (1 atm), without the use of a transition-metal based oxidant,20 to afford an 83% isolated yield of enone 19 (Scheme 4).19,21 The same efficiency was observed when the oxidative reaction was carried out using 10 mol% Pd(TFA)2 under an O2 atmosphere (1 atm) in DMSO (85% yield of isolated enone 19). In the presence of Pd and O2, the secondary alcohol of 17 was proposed to have oxidized to ketone 18 (Scheme 5). Next, in a one-pot reaction, ketone 18 oxidized to corresponding α,β-unsaturated enone 19. Here, the reaction may have proceeded via a C- and/or O-bound Pd-enolate (see, 17a–c in Scheme 5). A β-hydride elimination was thought to have occurred via C-bound enolate to result in enone 19 (Scheme 5).
 | ||
| Scheme 5 Plausible mechanism involving overoxidation. | ||
Since the secondary alcohol was responsible for the overoxidation under Pd(II)-catalysis, it was protected with an acetate group. Hence, compound 16, having an aromatic nitro group as well as a nitrite functionality, was reduced under hydrogenation conditions followed by a reaction with acetic anhydride to afford a 97% yield of acetanilide 20 (Scheme 6). Gratifyingly, 2-phenyl acetanilide 20 in the presence of 10 mol% Pd(OAc)2 in dimethylsulfoxide (DMSO) under an oxygen atmosphere afforded N,O-bis-acetylated pentacyclic core 21 in 87% isolated yield, without the use of a transition-metal-based oxidant (Scheme 6).20
 | ||
| Scheme 6 Total synthesis of xiamycin A (2a) via Buchwald's oxidative C–N bond formation. | ||
Next, deacetylation of compound 21 upon treatment with K2CO3 in MeOH completed the synthesis of xiamycin A methyl ester (2b) in 96% yield. Furthermore, saponification of 2b using a mixture of KOH and LiOH in a MeOH/H2O mixture at reflux completed the synthesis of xiamycin A (2a), thus setting the stage for the dimerization via the key dehydrogenative N–N-bond-forming reaction.22
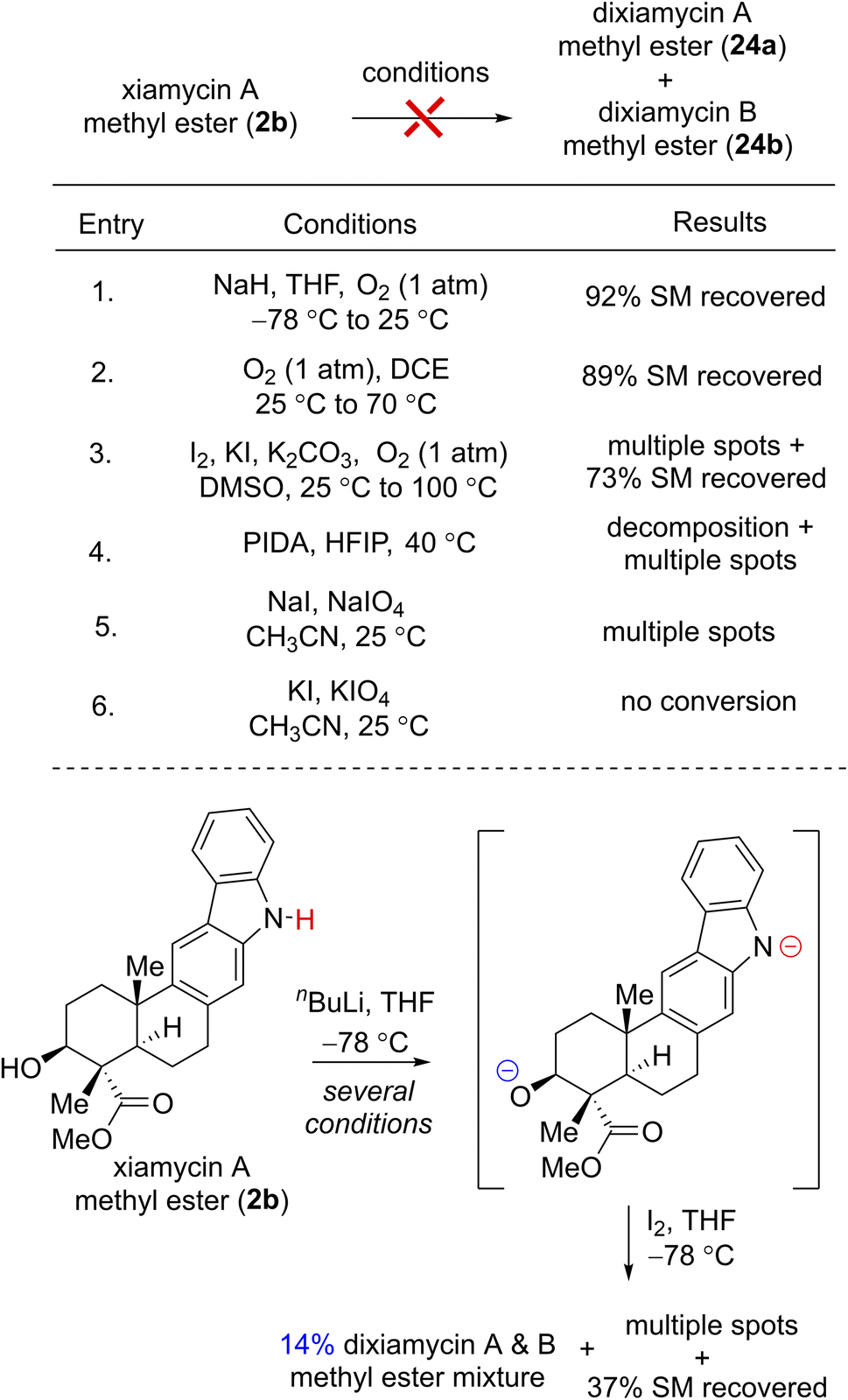
To investigate the chemical feasibility of the biosynthetic hypothesis, we next pursued the key dehydrogenative N–N-bond-forming dimerization of xiamycin A (2a) and xiamycin A methylester (2b) using various oxidants such as I2,6a KMnO4,6b Ag2O,6c dichromate,6d PIDA, KI or atmospheric O2. Each of these conditions, however, simply led to decomposition or no conversion. Interestingly, while using I2 and nBuLi at −78 °C to 25 °C for 72 h,23 xiamycin A methylester (2b) afforded 14% yields of dixiamycin A and B methyl esters (24a and 24b) (dr ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 37% yield of recovered starting material along with multiple TLC spots, probably arising from oxidative C–N and C–C bond formations (Table 1). This result was indeed encouraging, and thus a variety of other oxidative conditions were tested to effect a chemoselective N–N bond formation for dixiamycins A (1a) and B (1b).
:1), 37% yield of recovered starting material along with multiple TLC spots, probably arising from oxidative C–N and C–C bond formations (Table 1). This result was indeed encouraging, and thus a variety of other oxidative conditions were tested to effect a chemoselective N–N bond formation for dixiamycins A (1a) and B (1b).
|
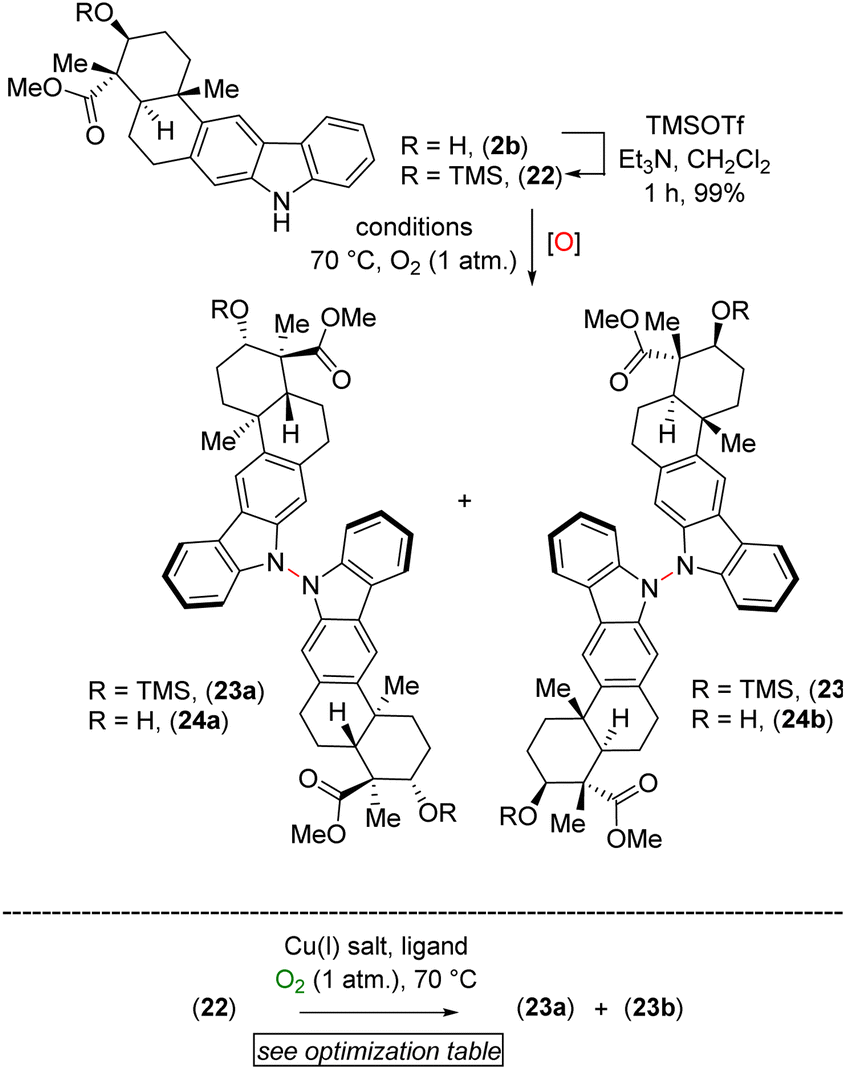
After testing various conditions for this goal of chemoselective N–N bond formation, we turned our attention to aerobic oxidation employing Cu(I)-catalysis as reported by Stahl and coworkers.8 In this regard, our attempt at using natural product xiamycin A (2a) under the Stahl conditions (20 mol% Cu(I)Br·Me2S and 40 mol% DMAP in dichloroethane at reflux for 17 h)8 led to decomposition. We found compound 2a to be a challenging substrate due to its several functional groups. To meet this challenge, we decided to use xiamycin A methyl ester TMS ether (22) as a starting material where the carboxylic acid and secondary alcohol of xiamycin A 2a were each in a protected form. Using the Cu(I)-catalyzed oxidative coupling conditions for 22 afforded promising N–N dimeric compounds 23a and 23b.
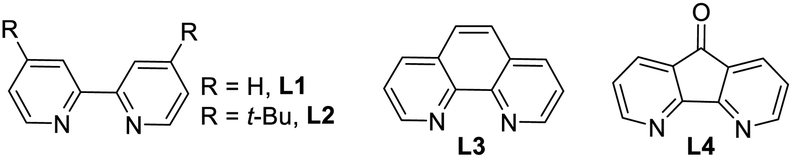
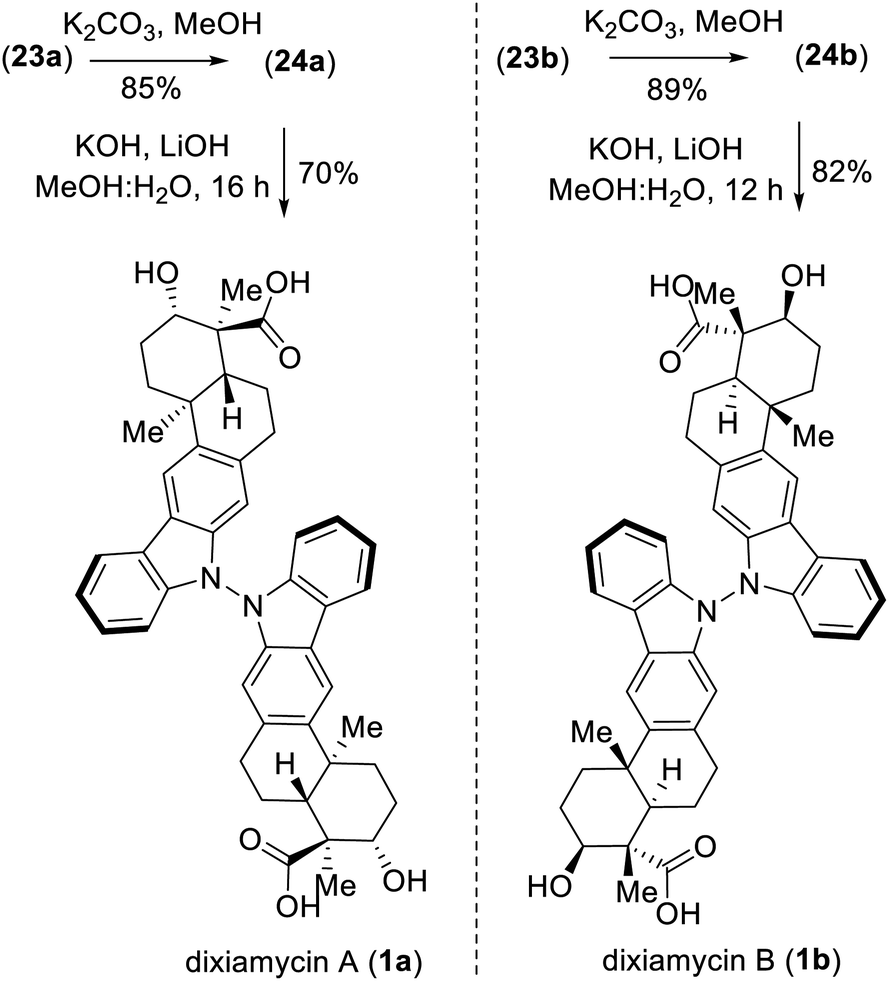
A detailed optimization of the Cu(I)-catalyzed aerobic oxidation of 22 is shown in Table 2. Following optimization, we found that treatment of 22 with 20 mol% Cu(I)Br and 40 mol% DMAP in DCE at reflux gave a 68% yield (ca. ∼1.6:1 mixture) of the TMS ethers of dixiamycins A methyl ester (23a) and B methyl ester (23b) (entry 6). Other bidentate ligands (L1–L4) were found to be inferior to the DMAP ligand, affording the products in only 23–51% yields along with decomposition side products (entries 2–5). Note that the TMS protecting group in 22 survived under the reaction condition at 70 °C. These N–N atropo-diastereomers were found to be separable using column chromatography. Following treatment with K2CO3 in MeOH, the atropo-diastereomers were smoothly converted to the corresponding methyl esters of dixiamycins A and B, i.e., 24a–b (Scheme 7). Furthermore, saponification of 24a–b with KOH and LiOH in MeOH/H2O at 80 °C for 12–16 h completed the total syntheses of dixiamycins A (1a) and B (1b) without an event (Scheme 7). The atropo-diastereomeric natures of dixiamycins A (1a) and B (1b) were confirmed using a detailed HPLC analysis and their retention times were found to differ [tR of dixiamycin A (1a) = 8.8 min and tR of dixiamycin B (1b) = 9.8 min] (see ESI‡ for detailed HPLC analysis).
|
|
|||||
|---|---|---|---|---|---|
| S. no. | Condition | Solvent | Time | 23a/23b | 22 |
| a All the reactions were carried out in the presence of an O2 balloon (1 atm). b Yields are reported as isolated yields after column chromatography. | |||||
| 1 | 30 mol% Cu(I)I:L1 | DCE | 48 h | 19% (∼1:1) |
62% |
| 2 | 20 mol% Cu(I)Br:L1 | DCE | 48 h | 42% (∼1.1:1) |
39% |
| 3 | 20 mol% Cu(I)Br:L2 | DCE | 48 h | 51% (∼1.2:1) |
31% |
| 4 | 20 mol% Cu(I)Br:L3 | DCE | 48 h | 23% (∼1.1:1) |
56% |
| 5 | 20 mol% Cu(I)Br:L4 | DCE | 48 h | 46% (∼1.2:1) |
32% |
| 6 | 20 mol% Cu(I)Br | DCE | 17 h | 68% (∼1.6:1) |
— |
| 40 mol% DMAP | |||||
| 7 | 20 mol% Cu(I)Br | MeCN | 17 h | 49% (1.6:1) |
— |
| 40 mol% DMAP | |||||
|
|||||
 | ||
| Scheme 7 Total syntheses of dimeric indolosesquiterpene alkaloids 1a–b. | ||
To demonstrate the versatility of our synthesis, TMS protection was used as an orthogonal protecting group. In a one-pot operation (Scheme 8), 2b was treated with TMSOTf in DCE followed by application of our optimal N–N-bond-forming conditions and subsequent TMS deprotection using K2CO3 in MeOH; this procedure afforded a direct approach to dixiamycins A and B methyl esters in 66% yield in a ca. 1.6:1 ratio (Scheme 8). In addition, Cu(I)-catalyzed aerobic oxidation of xiamycin A methyl ester (2b) afforded atropo-diastereomers 24a–b in 42% yield upon prolonged heating (72 h) along with 31% recovered 2b, consistent with the hypothesis of nature's oxidative approach to the dixiamycins.
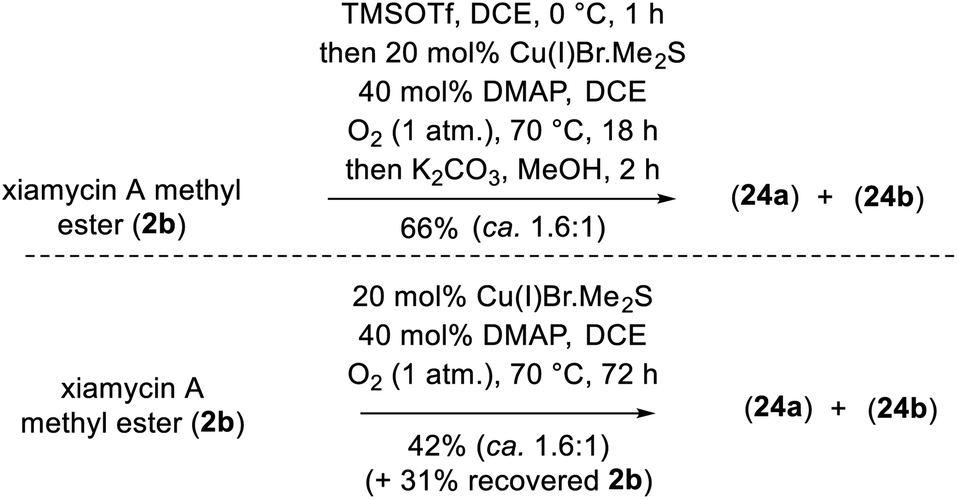 | ||
| Scheme 8 A direct conversion of xiamycin A methyl ester (2b) into dimeric indolosesquiterpene alkaloids 1a–b. | ||
Conclusions
In conclusion, a total synthesis of N–N atropo-diastereomers of indolosesquiterpene alkaloids, namely dixiamycins A (1a) and B (1b), was developed. This synthesis involved a key Cu(I)-catalyzed aerobic oxidation of xiamycin A methyl ester (2b), culminating in the first total synthesis of dixiamycin A (1a) bearing the rare N–N bond. The monomer of this approach, xiamycin A methyl ester (2b), was synthesized from a naturally occurring diterpenoid, namely 4-epi-triptobenzene L (4), following key Buchwald oxidative C–N-bond-forming reactions to craft the pentacyclic core of indolosesquiterpene alkaloid 2b. Thus, this synthesis featured the demonstration of two key aerobic oxidations, namely Pd(II)-(oxidative C–N bond formation) and Cu(I)-catalyzed (oxidative N–N bond formation) processes for the syntheses of, respectively, monomeric and dimeric indolosesquiterpene alkaloids. Our study also confirmed the high stability of the dixiamycins A (1a) and B (1b) under elevated temperature and basic conditions, suggesting that N–N atropo-diastereomers of the xiamycins could represent viable starting points for potential use in pharmaceuticals and agrochemicals.Data availability
Data supporting this article have been uploaded as ESI.‡Author contributions
A. B. conceived and supervised this project. R. N. investigated the key oxidative N–N bond-formation leading to dixiamycins A and B. R. N., S. N., and S. K. investigated the catalytic polyene cyclization and synthesized all the starting materials. S. N., M. M, V. R. G, and R. M. investigated the oxidative C–N bond-formation to craft the pentacyclic core of xiamycin A methyl ester. A. B. and R. N. wrote the original draft of the manuscript which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the SERB [CRG/2019/000113], [SCP/2022/000486] and STARS, MHRD [2023/0753] is gratefully acknowledged. RN thanks the INSPIRE for a research fellowship. SK, VRG, MM, SN, and RM thank the CSIR for research fellowships. AB is a SERB-STAR Fellow and sincerely acknowledges the SERB [STR/2020/000061] for generous support. We sincerely thank Prof. Richmond Sarpong, University of California, Berkeley, CA for insightful discussion on the oxidative N–N bond formation to access the atropo-diastereomers of dixiamycins A and B.Notes and references
- H. Li, Q. Zhang, S. Li, Y. Zhu, G. Zhang, H. Zhang, X. Tian, S. Zhang, J. Ju and C. Zhang, J. Am. Chem. Soc., 2012, 134, 8996–9005 CrossRef CAS PubMed.
- Q. Zhang, A. Mándi, S. Li, Y. Chen, W. Zhang, X. Tian, H. Zhang, H. Li, W. Zhang, S. Zhang, J. Ju, T. Kurtán and C. Zhang, Eur. J. Org. Chem., 2012, 5256–5262 CrossRef CAS.
- Isolation: (a) L. Ding, J. Münch, H. Goerls, A. Maier, H. H. Fiebig, W. H. Lin and C. Hertweck, Bioorg. Med. Chem. Lett., 2010, 20, 6685–6687 CrossRef CAS PubMed; (b) L. Ding, A. Maier, H. H. Fiebig, W. H. Lin and C. Hertweck, Org. Biomol. Chem., 2011, 9, 4029–4031 RSC.
- (a) S. H. Kim, T. K. Q. Ha, W. K. Oh, J. Shin and D. C. Oh, J. Nat. Prod., 2016, 79, 51–58 CrossRef CAS PubMed; (b) K. Takada, H. Kajiwara and N. Imamura, J. Nat. Prod., 2010, 73, 698–701 CrossRef CAS PubMed; (c) Q. Zhang, H. Li, L. Yu, Y. Sun, Y. Zhu, H. Zhu, L. Zhang, S.-M. Li, Y. Shen, C. Tian, A. Li, H.-W. Liu and C. Zhang, Chem. Sci., 2017, 8, 5067–5077 RSC.
- (a) Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 10293–10297 CrossRef CAS PubMed; (b) M. Baunach, L. Ding, T. Bruhn, G. Bringmann and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 9040–9043 CrossRef CAS PubMed.
- (a) F. D. Chattaway and H. Ingle, J. Chem. Soc., Trans., 1895, 67, 1090–1095 RSC; (b) W. H. Perkin and S. H. Tucker, J. Chem. Soc., Trans., 1921, 119, 216–225 RSC; (c) G. E. K. Branch and J. F. A. Smith, J. Am. Chem. Soc., 1920, 42, 2405–2413 CrossRef CAS; (d) J. McLintock and S. H. Tucker, J. Chem. Soc., 1927, 1214–1221 RSC.
- B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef CAS PubMed.
- (a) M. C. Ryan, J. R. Martinelli and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 9074–9077 CrossRef CAS PubMed; (b) C. A. Mulrooney, X. Li, E. S. DiVirgilio and M. C. Kozlowski, J. Am. Chem. Soc., 2003, 125, 6856–6857 CrossRef CAS PubMed.
- J. Feng, F. Noack and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 12364–12367 CrossRef CAS PubMed.
- (a) A. Trotta, Org. Lett., 2015, 17, 3358–3361 CrossRef CAS PubMed; (b) A. Trotta, J. Org. Chem., 2017, 82, 13500–13516 CrossRef CAS PubMed.
- (a) Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, P. Yang, D. J. Edmonds, J. Zhong and A. Li, Nat. Commun., 2015, 6, 6096–7003 CrossRef CAS PubMed; (b) Y. Sun, P. Chen, D. Zhang, M. Baunach, C. Hertweck and A. Li, Angew. Chem., Int. Ed., 2014, 53, 9012–9016 CrossRef CAS PubMed.
- M. Pfaffenbach, I. Bakanas, N. R. O'Connor, J. L. Herrick and R. Sarpong, Angew. Chem., Int. Ed., 2019, 58, 15304–15308 CrossRef CAS PubMed.
- D. H. Dethe and M. Shukla, Chem. Commun., 2021, 57, 10644–10646 RSC.
- M. Munda, R. Nandi, V. R. Gavit, S. Kundu, S. Niyogi and A. Bisai, Chem. Sci., 2022, 13, 11666–11671 RSC.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
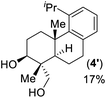
- Lewis acid promoted epoxy-ene cyclization of 14 gave 4-epi-triptobenzene L in 63% yield along with another regioisomer 4′ in 17% yield
 .
. - The characterization data for 4 is fully consistent with the isolation report as well as a recent total synthesis by Li and Carter. See, (a) H. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, Y. Jia and D. Li, J. Nat. Prod., 1999, 62, 1522–1525 CrossRef CAS PubMed; (b) D. Li and R. G. Carter, Org. Lett., 2018, 20, 5546–5549 CrossRef PubMed.
- ipso-Nitration: (a) R. C. Hahn and M. B. Groen, J. Am. Chem. Soc., 1973, 95, 6128–6129 CrossRef CAS; (b) M. W. Galley and R. C. Hahn, J. Am. Chem. Soc., 1974, 96, 4337–4339 CrossRef CAS.
- (a) W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560–14561 CrossRef CAS PubMed . For a related reference, see; ; (b) J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 16184–16186 CrossRef CAS PubMed.
- ‘transition-metal free’ means that ‘free of transition-metal based oxidant’, and not “Pd-free C–N bond forming process”.
- T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef CAS PubMed.
- Enone 19 could be transformed to xiamycin A methyl ester (2b) in a stepwise manner in 90% yield over 3 steps (see ESI‡ for the details)
 .
. - For the use of iodine in oxidative C–N bond formation, see; A. Bisai, S. P. West and R. Sarpong, J. Am. Chem. Soc., 2008, 130, 7222–7223 CrossRef CAS PubMed.


Footnotes |
| † This work is dedicated respectfully to Professor Goverdhan Mehta, FRS, on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2218312. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc07119c |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |