
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical halogen-bonding assisted generation of vinyl and sulfur-centered radicals: stereoselective catalyst-free C(sp2)–S bond forming reactions†
Helena F.
Piedra
 and
Manuel
Plaza
*
and
Manuel
Plaza
*
Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles”, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: plazamanuel@uniovi.es
First published on 7th December 2022
Abstract
The combination of photochemistry and halogen bonding interactions has risen in the last few years as a powerful synthetic tool for the creation of radical intermediates under mild conditions. In the formation of carbon-centered radicals, this reactivity has been to date restricted to the employment of aryl and alkyl halides as precursors. We now envisioned that the halogen-bonding initiated formation of highly reactive vinyl radicals would be a feasible process for the photochemical cross-coupling between thiols and alkenyl halides under basic conditions. The reaction shows indeed a very broad functional group tolerance, is stereoselective, simple and scalable. In-depth mechanistic studies point at the formation of vinyl and sulfur-centered radicals as the intermediates of the reaction and DFT calculations support the pre-formation of a halogen-bonding complex as the initiator of the photochemical transformation. Synthetic applications were developed to extend the utility of this methodology.
1 Introduction
The development of new methodologies for the construction of C(sp2)–S bonds is an important task in organic chemistry.1 In particular, the synthesis of vinyl sulfides is particularly appealing due to their presence in biologically active molecules2 and building blocks in organic synthesis.3 Classical thermal approaches for the preparation of these compounds typically implicate the use of either an alkenyl halide or an alkyne and a sulfur precursor (commonly, a thiol or a disulfide species) as the partner reagents for the synthesis of the desired cross-coupling products (Scheme 1a).4 However, the unavoidable, mandatory employment of metal catalysts, strong oxidants, harsh conditions or starting materials that are difficult to synthesize hampers the sustainability and simplicity of these methodologies. Therefore, the exploration of more environmentally benign, greener strategies that would forge vinyl sulfides in a modular way starting from easily accessible precursors and under operationally simple reaction conditions stands as a big challenge for the field. | ||
| Scheme 1 Synthetic strategies for the synthesis of vinyl sulfides from alkenyl halides. (DABCO = 1,4-diazabicyclo[2.2.2]octane; HB = halogen-bonding.) | ||
In the last few years, photochemical approaches for the construction of C–C and C–heteroatom bonds have risen as powerful synthetic tools in the context of green and sustainable chemistry.5 In this context, the exploitation of radical chemistry strategies stands as an alternative to classical methodologies thanks to the intrinsic reactivity of these highly reactive open-shell intermediates, which has revolutionized the way chemists embrace nowadays the retrosynthetic analysis of molecules.6 In particular, photochemical reactions destined to the preparation of sulfur-containing molecules have recently received a lot of attention.7 In this context, König and Ananikov reported recently the synthesis of vinyl thioethers starting from alkynes and thiols through an elegant metal-free photocatalytic transformation.8 However, to the best of our knowledge, to date only one example has been reported for the photochemical construction of the previous molecules starting from alkenyl halides (Scheme 1b).9 This transformation, although promoted by visible light, requires long heating times (24 hours) and the employment of an expensive iridium-based photocatalyst and DABCO as a sacrificial donor to regenerate the photoredox catalytic cycle. A major drawback associated with this reaction is the lack of stereocontrol in the formation of the final disubstituted olefins, where the observed E/Z diastereomeric ratios seem to depend exclusively on the nature of each particular substrate.
Despite its apparent simplicity, the direct generation of vinyl radicals from alkenyl halides under visible-light photocatalytic processes stands as a very challenging approach. Indeed, direct irradiation of a solution of an alkenyl halide with high-energy γ-rays10a or a laser pulse of low frequency UV light10b (266 nm) is known to cause a scission of the C(sp2)–Br bond, generating the corresponding carbon centered vinyl radical or cation species respectively. Rather than direct irradiation, photochemical approaches for the creation of vinyl radicals normally rely on a SET or energy transfer event from a photocatalyst.11 However, the high reduction potentials associated with the alkenyl halides, as well as the high instability of the vinyl radicals, are additional challenges that make the photocatalytic generation of vinyl radicals from the corresponding alkenyl halides a rather difficult strategy. Taking all of this into consideration, it is not surprising that the creation of these highly reactive vinyl radicals through photochemical transformations only promoted by visible-light irradiation in the absence of a photocatalyst starting from the corresponding alkenyl halides is still not reported.
Common activation modes in supramolecular photochemistry through the formation of aggregates via non-covalent interactions typically implicate the formation of electron donor–acceptor (EDA) complexes,12 H-bonds,13 π-effects14 or ionic interactions.15 This synergistic prearrangement between the photosubstrate and an appropriate host molecule allows photochemical reactions to be carried out with an exquisite control in the reactivity and stereoselectivity towards the formation of the desired target molecules. Interestingly, in the last few years, a type of n–σ interactions known as halogen-bonding interactions16 has been exploited in photochemical processes for the generation of alkyl17 and aryl18 (or heteroaryl) carbon-centered radicals. These transformations consist of the formation of a halogen-bonding complex due to the interaction between an acceptor molecule that presents a halogen atom with a σ-orbital hole and a donor nucleophilic species. This complex can potentially be excited upon visible light irradiation and a photoinduced electron transfer (PET) is responsible for the generation of a radical ion pair, which after fragmentation creates two different radical species arising from each of the starting molecules. Different strategies and reactivities have been exploited thanks to this kind of supramolecular interactions in the context of photochemistry.17,18 In light of all of the above, we wondered whether the employment of alkenyl halides as acceptor molecules in the reaction with nucleophilic thiolate species could potentially form a halogen-bonding complex. Direct irradiation at a suitable wavelength would potentially end up in the formation of highly reactive vinyl and sulfur-centered radicals, whose rapid recombination would forge the corresponding vinyl sulfide compound in a simple and elegant manner (Scheme 1c).
2 Results and discussion
Motivated by our initial hypothesis, we started to run a set of reactions in order to find optimal conditions for the photochemical halogen-bonding assisted cross-coupling between the alkenyl bromide 1a and the thiol 2a (Table 1).|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yield of 3aa | d.r. trans/cis |
| a Incomplete conversion. | |||
| 1 | Dark, 72 h | — | — |
| 2 | No base, 12 h | <5% | — |
| 3 | Ar, 2 h | 93% | 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 4 | 1 eq. 2a | 69% | 8:1 |
| 5 | K2CO3 as the base | 95% | 8:1 |
| 6 | NaOH as the base, 1 h | 96% |
8:1 |
| 7 | DMF instead of DMSO | 96% | 8:1 |
| 8 | MeCN instead of DMSO | 84% | 8:1 |
| 9 | X = I | 22% | 8:1 |
| 10 | X = Cl | 95% | 6:1 |
| 11 | NaOH as the base, 1 h, 427 nm | 96% | 6:1 |
| 12 | NaOH as the base, 1 h, 390 nm | 78% | 3:1 |
| 13 | NaOH as the base, 1 h, 456 nm | 35%a | 8:1 |

Much to our pleasure, initial experiments showed that the desired vinyl sulfide 3a was obtained with an excellent yield and high diastereomeric ratio (8:1 trans/cis) when cesium carbonate was employed as the base and a 440 nm lamp was used as the irradiation source. Control experiments revealed that no reaction occurred in the absence of light or a base. Moreover, the employment of an inert argon atmosphere could be avoided, increasing the operational simplicity of our transformation. Other different polar solvents (DMF, acetonitrile) also afforded the formation of the desired vinyl sulfide 3a. The employment of other bases (K2CO3, KOH, Na2CO3) was tolerated, but the shortest reaction time was secured by the usage of NaOH (reaction completed in less than 1 hour). Interestingly, when an alkenyl chloride was employed, the reaction also proceeded towards the formation of the expected vinyl sulfide, although a worse diastereomeric ratio was obtained (Table 1, entry 10). On the other hand, a low yield was obtained when the corresponding vinyl iodide was used under the standard conditions, which was most likely due to decomposition of the unstable starting material (Table 1, entry 9). It is important to note that the employment of a suitable light source was not only crucial for the photoexcitation of the HB complex, but also had an impact on the in situ trans/cis isomerization due to the absorbance of the obtained vinyl sulfides. In this regard, we noticed that the employment of shorter irradiation wavelengths (366, 390 and 427 nm) failed to provide better stereoselectivity in the transformation than when the 440 nm lamp was used (d.r.: 8:1 trans/cis). This is in agreement with the absorbance profile of the cis (3aa′) and trans (3aa) isomers of the vinyl sulfide (see the ESI† for details). Finally, we noticed that if a less powerful 456 nm lamp was used, the reaction did not reach full conversion even after 1.5 hours of irradiation. All in all, the unprecedented short reaction times and operational simplicity are noteworthy in comparison to previously reported alternative methodologies for the synthesis of vinyl sulfides. With the optimized reaction conditions at hand, we started to evaluate the scope of our transformation. The results are summarized in Scheme 2. It is worth mentioning that the irradiation time was set to 1 h to ensure full conversion, although in some cases the reaction was finished even after 20 minutes (for example, for the case of compounds 3da and 3ka). Longer irradiation periods at 440 nm did not affect the trans/cis ratio of the final products due to the lack of absorbance of the vinyl sulfides employing that lamp. First, we tested the impact of different substituents at the aryl ring of the vinyl bromides 1. In this regard, it was observed that the presence of different electron-withdrawing and electron-donating groups was well tolerated (compounds 3aa–3la). It is noteworthy that in all of the cases, good to excellent yields and diastereomeric ratios in favor of the trans isomer of the final products 3 were obtained. Remarkably, when a 2-furyl heterocyclic substituent at the alkenyl moiety was employed, the reaction also proceeded smoothly to afford the compound 3ma, albeit with a moderate stereoselectivity. Importantly, when the reaction to forge the compound 3aa was run in a 1 mmol scale, the reaction proceeded equally well (95% yield), which proved that our transformation is scalable in case larger amounts of the vinyl sulfides were needed. Next, we examined different substitution patterns at the thiols 2. Once again, the obtention of the desired vinyl sulfides was successful (compounds 3ab–3af). It must be pointed out that the presence of fluorine and chlorine atoms at the thiol unit, which could potentially present halogen-bonding interactions with other thiolate molecules, was compatible with the reaction conditions. As expected, when alkenyl bromides and thiols bearing at the same time different substituents were tested in our reaction, the cross-coupling products were obtained (compounds 3gc–3nb). Unfortunately, when an alkyl substituent was attached to the alkenyl bromide, the reaction failed to result in the formation of the vinyl sulfide 3qa. This fact may be attributed to the lack of absorptive properties of the HB complex derived from the vinyl bromide 3q and the thiolate anionic form of 2a. However, and much to our pleasure, the usage of alkylic cyclic and linear thiols was compatible with our transformation in the formation of the molecules 3ah and 3ni. Interestingly, a set of vinyl sulfides could be obtained derived from the employment of biologically active molecules. In this context, the obtention of the compound 3oc is especially interesting, not only because it is derived from a naturally occurring cinnamaldehyde, but also because it illustrates the ability of the reaction to perform also with 1-bromo-1,3-butadienes, and therefore to gain access to 1,3-butadienylsulfanes. Interestingly, the employment of molecules which present enolizable hydrogens in the strongly basic reaction media could be adapted to our methodology if a weaker base (K2CO3) and longer irradiation times were used. With this protocol, the vinyl sulfides derived from L-cysteine 3al and thioglycolic acid 3an could be prepared. Finally, other appealing vinyl sulfides derived from biologically active scaffolds such as thiosalicylic acid and vanillin gave correspondingly the compounds 3am and 3pb.
 | ||
| Scheme 2 Substrate scope for the C(sp2)–S bond forming photochemical reaction. aAlso 95% yield when the reaction was run in a 1 mmol scale. bK2CO3 as the base (1.5 eq.) and 2.5 h (compounds 3af, 3an and 3ja) and 5 h (compound 3al) of irradiation time were needed. | ||
A set of different mechanistic studies were carried out to support the radical nature of the intermediates involved in the transformation (Scheme 3). We hypothesized that the formation of a vinyl radical derived from the alkenyl bromides 1 is due to a PET at the irradiated HB complex. Thus, we performed different control experiments orientated to validate this fact. Two different structures are normally speculated for a vinyl radical: a linear σ-type or a bent π-type.19 To gain insight into the nature of the vinyl radicals formed in our reaction, we performed a stereoconvergence experiment in which two different reactions, one starting from the trans vinyl bromide 1a, and from the corresponding cis isomer 1a′ respectively were evaluated. We observed that the stereogenic information of the alkenyl bromide did not affect the stereoselectivity of the reaction and the vinyl sulfide 3a was furnished with an 8:1 trans/cis diastereomeric ratio regardless of the stereochemistry of the starting bromide (Scheme 3a). This observation indicates that the formation of a linear sp-hybridized vinyl radical 5 should be preferred, either through its direct formation from the vinyl bromide precursor, or due to a fast interconversion of the two different bent π-type radicals 4 and 6 (which could arise from each of the precursors 1a and 1a′, respectively) towards the linear σ-type one in each of the reactions. This conclusion is consistent with previous reports and experiments oriented to elucidate the structure of vinyl radicals in photochemical reactions.19 This stereoconvergent nature of our transformation should be highlighted, since this concept has been previously exploited in photochemical reactions by other research groups.20 Inhibition experiments showed that the reaction was shut down if two equivalents of a radical scavenger such as TEMPO were added to the reaction media, giving additional support to a radical mechanism (Scheme 3b). Importantly, intermolecular radical-clock probe experiments employing the vinyl bromide 1a, thiol 2e and compound 7 as the radical trapping agent afforded not only the formation of the expected cross-coupling product 3ae as the major reaction product, but also a small amount (13% yield) of the product 8 (Scheme 3c). This molecule is most likely forged from an addition of the sulfur-centered radical arising from 2e to 4, followed by a ring opening of the cyclopropane to give a linear chain, which provides additional support to the presence of radical species being formed within our reaction.6,21 Given that the photochemical catalyst-free cross-coupling reaction between aryl halides and thiols had been previously reported under similar conditions to the ones of our work,22 we designed a competition experiment in which the vinyl bromide 1c and the aryl bromide 9 were made to react with one equivalent of the thiol 2e (Scheme 3d). Considering the high reactivity vinyl radicals,23 it was expected that our reaction would be much faster than the corresponding competing cross-coupling of the thiol with the aryl bromide. Indeed, we observed that the formation of the compound 3ce outcompeted the one of the compound 10, showcasing once again the high efficiency of our transformation. Finally, light on/off experiments and quantum yield measurements were run (Scheme 3e). Indeed, in the reaction between 1d and 2a, it was observed that the reaction only proceeded during the irradiation times, showing no conversion in the periods in which the light had been turned off. Additionally, we calculated quantum yield for the same reaction (see the ESI† for a detailed description) and its value is below the unit (φ = 0.1). The combination of these two last experiments proves the lack of radical chain propagation processes within our transformation.24
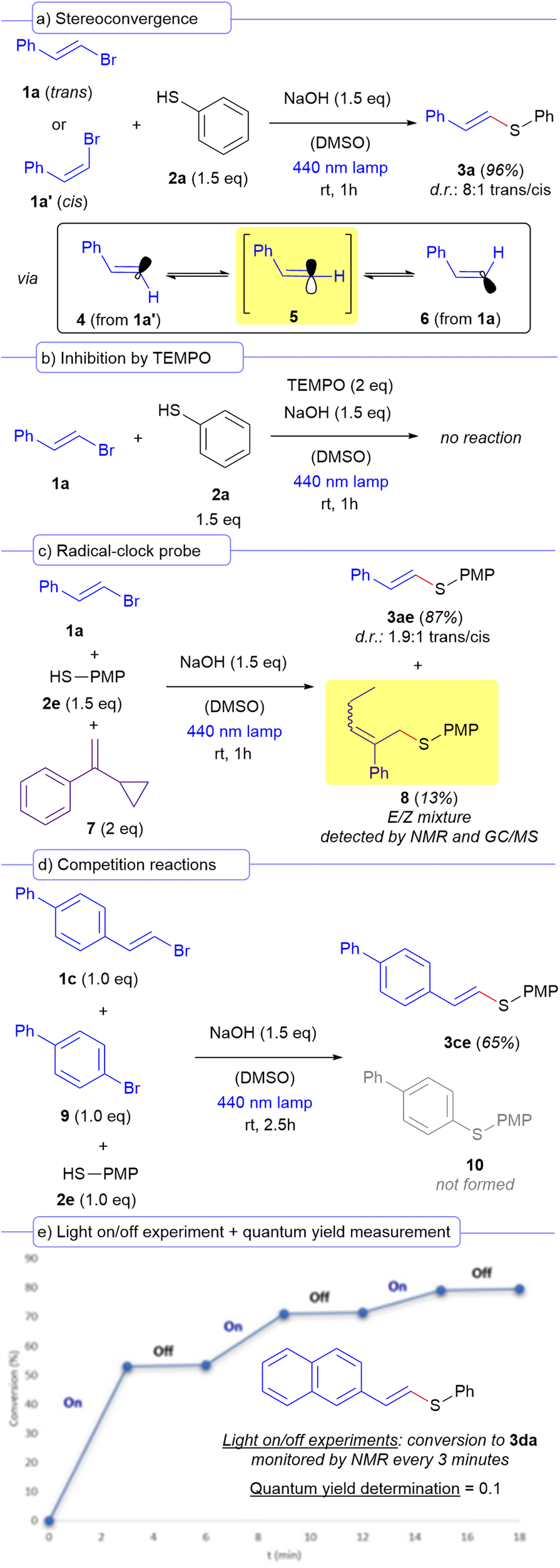 | ||
| Scheme 3 Set of mechanistic experiments. PMP = para-methoxyphenyl. | ||
A set of different mechanistic studies were also performed in order to provide evidence for the formation of the halogen-bonding complex. First, we performed UV-Vis measurements of different solutions in DMSO of 1a, 2a, 2a + Cs2CO3 and a mixture of 1a + 2a + Cs2CO3 in equimolar amounts (see Section 3 of the ESI† for details). Satisfyingly, comparison of the UV-Vis absorption profile of this last mixture with each of the individual reaction components pointed at the formation of a halogen-bonding complex due to the identification of a new charge-transfer band at higher wavelengths than the one observed for the individual starting materials. This explains the selective excitation of the halogen-bonding complex at 440 nm, which is believed to be the driving force of our transformation.
Further support to the formation of the halogen-bonding complex was found by performing NMR titration experiments (Scheme 4). In this regard, when we prepared solutions in d6-DMSO of increasing concentrations of sodium thiophenolate maintaining the same quantity of the vinyl bromide 1a (see Section 8 of the ESI† for a detailed description), we observed a significative increasing downfield shift in the 13C-NMR signals of the vinylic carbons of the starting alkenyl bromide 1a. For instance, we noticed that the signal of the carbon C1 (marked with a red dot, initially at 108.07 ppm) was increasingly deshielded to 108.95 ppm (almost a 1 ppm shift) when a 1:10 1a/NaSPh was present in the solution. This increase in the chemical shift is most likely due to the formation of a complex between 1a and the thiolate of 2a.25 This strategy has been employed in various recent studies for the structural identification of halogen-bonding complexes in solution.17a,18c
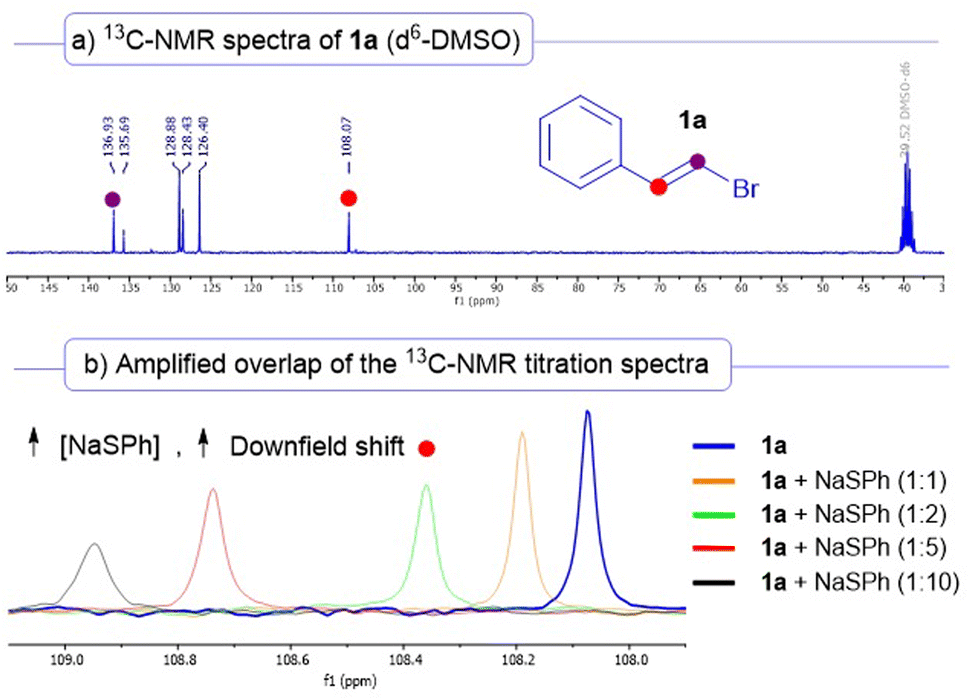 | ||
| Scheme 4 (a) 13C-NMR spectra recorded for the compound 1a in d6-DMSO. (b) Overlap of the 13C-NMR spectra obtained in the titration experiments. Different ratios of the acceptor 1a and electron-donor (NaSPh) were employed to prepare the d6-DMSO solutions (from 1:1 to 1:10). | ||
Finally, preliminary DFT calculations were run (ωB97x-D3/Def2TZVPP level). Delightfully, a halogen-bonding aggregate between 1a and the anionic form of 2a could be identified (see the ESI† for details) as a minimum in the potential energy surface (Scheme 5a). The halogen bond interaction between the bromine and sulfur atom presents a great directionality, displaying a dihedral angle of 173.4° between the Csp2–Br–S bonds, which results in a nearly linear structure for the halogen-bonding complex. Indeed, in the electrostatic potential surface it is observed that the positively charged bromine σ-hole zone is located at almost the center of the C–Br axis (Scheme 5b). This linear geometry with dihedral angles between 160° and 180° for the HB complex is in agreement with previously reported observations on parent complexes.26 Furthermore, we found that in the halogen-bonding complex the distance between the bromine and sulfur atoms was 3.21 Å, which is shorter than the sum of the van der Waals radii of the sulfur and bromine atoms (3.65 Å). This fact supports the existence of a non-covalent weak interaction between these atoms, which is most likely due to the halogen bond formation.
 | ||
| Scheme 5 (a) HB complex between 1a and the anionic form of 2a calculated at the ωB97x-D3/Def2TZVPP. (b) Calculated electrostatic potential (red = negative electrostatic potential) on the 0.0004 au isodensity surface. | ||
In light of our DFT calculations and mechanistic studies, and previous well-established reports on the photochemical halogen-bonding promoted generation of carbon centered radicals,17,18 we propose the mechanism described in Scheme 6. First, a thiolate anion is formed in the basic reaction media from the corresponding thiol 2. Next, the formation of a halogen-bonding complex between this molecule and the alkenyl halide 1 is hypothesized to occur. Subsequent photoinduced electron transfer (PET) followed by fragmentation would afford the radical species 12 and 13. Recombination of these intermediates eventually affords the formation of the vinyl sulfides 3 in a stereoselective manner where the trans isomer product prevails in most of the cases.
 | ||
| Scheme 6 Mechanistic proposal for the photochemical halogen-bonding assisted cross-coupling reaction between alkenyl halides and thiolates. | ||
In order to prove the usefulness of our reaction, we searched for synthetic applications of our compounds. Given the importance of vinyl sulfoxides and sulfones in naturally occurring molecules27 and as valuable building blocks in organic synthesis,28 we looked for conditions to oxidize the vinyl sulfides to both of these molecules (Scheme 7a). In this regard, starting from the vinyl sulfide 3aa, we optimized a procedure29 which employs mCPBA as an oxidant for the obtention of the corresponding vinyl sulfoxide 14, which was delightfully furnished in our case with a 100:1 trans/cis diastereomeric ratio. On the other hand, the overoxidation of the same vinyl sulfide to the sulfone 4 could be accomplished under reflux conditions using an aqueous solution of hydrogen peroxide.30 Next, we decided to evaluate the effect of the substitutions in the alkenyl bromide (Scheme 7b). In this context, the synthesis of trisubstituted vinyl sulfides31 from the vinyl bromide precursor 16 could be accomplished albeit with no stereoselectivity (d.r.: 1:1 E/Z). It is known that phenyl substituents at the position of a vinyl radical induce a stabilization effect,32 which could explain the different behaviour of the starting material 16 in our transformation. However, when a tetrasubstituted alkenyl bromide 18 was employed, the reaction failed to provide the desired coupling product and a complex mixture was obtained. This may be attributed to the higher stability of the vinyl radical, which is potentially delocalized at the olefin and therefore fails to provide chemical selectivity in the coupling with the sulfur-centered radical arising from 2a. As a last synthetic application, we wondered if our trans-configured substrates 3 could potentially be converted into the corresponding cis isomers via a photochemical E/Z isomerization process (Scheme 7c). This class of reactions has been extensively studied in the last few years.33 However, we realized that to date there were no reports on the photochemical isomerization of vinyl sulfides. Therefore, we decided to carry out an extensive optimization (see the ESI† for a detailed description of the optimization studies) to find the best conditions for this reaction to take place. We found that when we employed the ruthenium complex 19 as the photosensitizer (ET = 205 kJ mol−1), a photostationary state is reached after 12 hours in which we could invert the stereogenic information in favor of the cis isomer 3a′ (from 8:1 to 1:5 trans/cis d.r.). These preliminary results are intended to be a proof of concept within the context of this work, but are currently being in-depth investigated in our research group to extend this methodology to the photochemical isomerization of other sulfur-containing olefins.
 | ||
| Scheme 7 Synthetic applications of the vinyl sulfides. mCPBA = meta-chloroperbenzoic acid. | ||
3 Conclusions
In conclusion, we have developed a novel, simple, sustainable and general synthetic methodology for the photochemical catalyst-free construction of vinyl sulfides. The activation of the vinyl halides through halogen-bonding interactions with a thiolate anion would form a HB complex, which is believed to be the key to this transformation. In-depth mechanistic studies support the participation of vinyl and sulfur centered radicals as the intermediates of this reaction, which are expected to be generated via PET after irradiation of the HB complex. The reaction was proven to be general, functional-group tolerant, operationally simple and scalable. Moreover, it could be applied to the structural modification of biologically relevant molecules. Eventually, synthetic applications were found which enhance the utility of our transformation in diversity-oriented synthesis.Data availability
All of the experimental and computational data is available and has been included in the ESI† of this publication.Author contributions
H. F. Piedra conducted most of the reactions, experimental mechanistic studies and full characterization of the compounds. M. Plaza conceptualized and directed the project, performed the DFT calculations and wrote the manuscript. Both authors contributed to scientific discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by an FICYT (Principality of Asturias) “Margarita Salas Jóven” postdoctoral grant to M. P. (AYUD/2021/58397) is gratefully acknowledged. We wish to thank Prof. Carlos Valdés for his assistance with the DFT calculations and helpful discussions during the development of this work.Notes and references
- For some recent reviews on the importance of C–S bond construction, see:
(a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110 CrossRef CAS PubMed
; (b) P. Annamalai, K.-C. Liu, S. S. Badsara and C.-F. Lee, Chem. Rec., 2021, 21, 3674 CrossRef CAS PubMed
-
(a) X.-S. Liu, Z. Tang, Z. Li, M. Li, L. Xu and L. Liu, Nat. Commun., 2021, 12, 7298 CrossRef CAS PubMed
- For some recent examples, see:
(a) Z. Nie, H. Lv, T. Yang, M. Su, W. Luo, Q. Liu and C. Guo, Adv. Synth. Catal., 2022, 14, 364 Search PubMed
- For some examples, see:
(a) P. K. Samanta, R. Biswas, S. N. Bhaduri, S. Ray and P. Biswas, Microporous Mesoporous Mater., 2021, 323, 111198 CrossRef CAS
- For a recent special issue on photocatalytic processes, see: P. Melchiorre, Chem. Rev., 2022, 122, 1483 CrossRef CAS PubMed
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed
- For some recent reviews, see:
(a) D. Yang, Q. Yan, E. Zhu, J. Lv and W.-M. He, Chin. Chem. Lett., 2022, 33, 1798 CrossRef CAS
- J. V. Burykina, N. S. Shlapakov, E. G. Gordeev, B. König and V. P. Ananikov, Chem. Sci., 2020, 11, 10061 RSC
- M. L. Czyz, G. K. Weragoda, R. Monaghan, T. U. Connell, M. Brzozowski, A. D. Scully, J. Burton, D. W. Lupton and A. Polyzos, Org. Biomol. Chem., 2018, 16, 1543 RSC
-
(a) G. W. Eastland, Y. Kurita and M. C. R. Symons, J. Chem. Soc., Perkin Trans. 2, 1984, 1843 RSC
- S. K. Pagire, T. Föll and O. Reiser, Acc. Chem. Res., 2020, 53, 782 CrossRef CAS PubMed
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed
-
(a) F. Burg and T. Bach, J. Org. Chem., 2019, 84, 8815 CrossRef CAS PubMed
- J.-H. Deng, J. Luo, Y.-L. Mao, S. Lai, Y.-N. Gong, D.-C. Zhong and T.-B. Lu, Sci. Adv., 2020, 6, eaax9976 CrossRef CAS PubMed
- For a recent review on supramolecular photochemistry: Y. Sempere, M. Morgenstern, T. Bach and M. Plaza, Photochem. Photobiol. Sci., 2022, 21, 719 CrossRef CAS PubMed
- Reviews on the importance of halogen-bonding interactions:
(a) R. Hein and P. D. Beer, Chem. Sci., 2022, 13, 7098 RSC
- For some key recent references on the generation of alkyl radical through photochemical halogen-bonding assisted reactions, see:
(a) C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686 CrossRef CAS PubMed
- For important recent references on the generation of aryl radical through photochemical halogen-bonding initiated transformations, see:
(a)
K. Matsuo, E. Yamaguchi and A. Itoh, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-glvkg;
(b) N. Sundaravelu, A. Nandy and G. Sekar, Org. Lett., 2021, 23, 3115 CrossRef CAS PubMed
- T. P. M. Goumans, K. v. Alem and G. Lodder, Eur. J. Org. Chem., 2008, 435 CrossRef CAS
- Selected examples on photochemical stereoconvergent transformations:
(a) H. Cho, H. Suematsu, P. H. Oyala, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2022, 144, 4550 CrossRef CAS PubMed
-
(a) J. Großkopf, M. Plaza, A. Seitz, S. Breitenlechner, G. Storch and T. Bach, J. Am. Chem. Soc., 2021, 143, 21241 CrossRef PubMed
- B. Liu, C. H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS PubMed
-
Free Radicals in Organic Chemistry, ed. J. Fossey, D. Lefort and J. Sorba, Wiley and Masson, 1995 Search PubMed
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC
- D. L. Widner, E. R. Robinson, A. B. Perez, H. G. Vang, R. A. Thorson, Z. L. Driscoll, S. M. Giebel, C. W. Berndt, E. Bosch, E. D. Speetzen and N. P. Bowling, Eur. J. Org. Chem., 2017, 2017, 5739 CrossRef CAS
-
(a) L. Maugeri, E. M. G. Jamieson, D. B. Cordes, A. M. Z. Slawin and D. Philp, Chem. Sci., 2017, 8, 938 RSC
-
(a) S. Y. Woo, J. H. Kim, M. K. Moon, S. H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473 CrossRef CAS PubMed
- For selected examples, see:
(a) Y. Sun, J. Jiang, X. Guo, J. Wen and X. Zhang, Org. Chem. Front., 2019, 6, 1438 RSC
- Y. Li and A. Studer, Org. Lett., 2017, 19, 666 CrossRef CAS PubMed
- H.-Y. Tu, B.-L. Hu, C.-L. Denga and X.-G. Zhang, Chem. Commun., 2015, 51, 15558 RSC
- For an alternative methodology to access trisubstituted vinyl sulfides, see: S. Ni, L. Zhang, W. Zhang, H. Mei, J. Han and Y. Pan, J. Org. Chem., 2016, 81, 9470 CrossRef CAS PubMed
- B. Branchi, C. Galli and P. Gentili, Eur. J. Org. Chem., 2002, 2844 CrossRef CAS
- For a recent review on photochemical E/Z isomerizations, see: T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05556b |
| This journal is © The Royal Society of Chemistry 2023 |