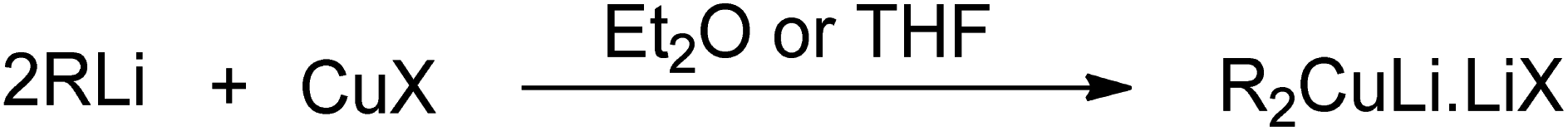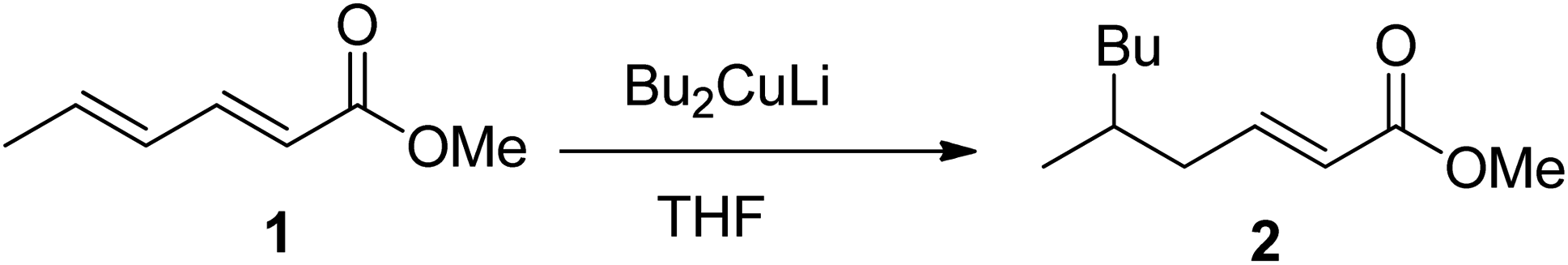DOI:
10.1039/D3RA07359A
(Review Article)
RSC Adv., 2023,
13, 35172-35208
Gilman reagent toward the synthesis of natural products
Received
29th October 2023
, Accepted 19th November 2023
First published on 4th December 2023
Abstract
With the ever-increasing scope of organocuprates, a well-established Gilman reagent has been considered as an unprecedented synthetic tool in modern organic chemistry. The broad research profile of the Gilman reagent (R2CuLi in THF or Et2O) is owing to its propensity to carry out three kinds of reactions, i.e., epoxide ring opening reactions, 1,4-conjugate addition reactions, and SN2 reactions in a regioselective manner. This review examines the applications of Gilman reagent in the total synthesis of both abundant and scarce natural products of remarkable synthetic pharmaceutical profile reported since 2011. The presented insights will be of a vital roadmap to general organic synthesis and it will contribute to the development of new natural products and their analogues in future drug discovery.
Introduction
Carbon–carbon bond formation is a preliminary step for most of the synthetic transformations in organic chemistry. In this regard, the role of organocopper reagents is at the forefront. These reagents are prepared by reacting organometallics (RMgX, RLi, or RZnX) with copper salts (CuBr, CuI, or CuCN) in the presence of solvents, such as THF, DMS, DCM, or dry ether. The use of various additives, such as HMPA (hexamethyl phosphoramide), TMSCl (trimethylsilyl chloride), and some Lewis acids, along with these reagents has also been reported in the literature.1–3 Gilman reagent (R2CuLi) is the first organocuprate compound, discovered by an American chemist, Henry Gilman, in 1952, which was prepared by reacting 2 equivalents of alkyl or aryl lithium with 1 equivalent of Cu salt in the presence of TMS or diethyl ether as solvent (Fig. 1).4 On dissolving in diethyl ether, this lithium diorganocuprate reagent is expected to exist in a dimeric, eight-membered ring form.7
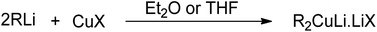 |
| | Fig. 1 Preparation of Gilman reagent. | |
Gilman reagent is usually employed in three kinds of reactions. In the first one (conjugate addition), it acts as an alkylating agent on treatment with α,β-unsaturated carbonyl compound (enone) to give the 1,4-addition product.5 Secondly, it is used in the regioselective epoxide (or aziridine) ring opening reactions. Thirdly, Gilman reagent is a good nucleophile and undergoes SN2 reaction (cross-coupling) with primary and secondary alkyl halides or alcohols and exhibits applications in Corey House reactions.5,10
With an appreciation for the inscrutable synthetic chemistry of Gilman reagent, Lipshutz et al. performed an extensive methodological study by applying a series of Gilman tests (using combinations of various alkyl or aryl halides along with copper salt) and determined the exact composition of the Gilman reagent.3 According to their spectroscopic observation, properties of the Gilman reagent are directly affected by the kind of CuX (here X = Cl, Br, I, OTf, SCN, or CN).4–6 Following the inventive study of the conjugate addition of Gilman reagent, Bertz et al. studied the reactive profile of iodo-Gilman reagent (R2CuLi·LiI) and cyano-Gilman reagent (R2CuLi·LiCN). The phenyl Gilman reagent (Ph2CuLi & PhLi) in the presence of ether or THF as a solvent has also been reported as a higher-order catalyst.7–11 The mechanistic study of Gilman reagent in THF at low temperatures has been conducted, and the formation of π complexes of Me2CuLi not only with α,β-unsaturated carbonyl compounds but also with aldehydes and ketones has been confirmed as a stable synthetic intermediate using spectroscopic analyses.12,13 The d orbital of copper in Gilman reagent (acting as a base) interacts with the π* of the carbonyl carbons (π acids) to form the d–π complexes. The optimum temperature reported for the formation of these complexes is −78 °C.14,15 Moreover, Yamamoto et al. reported the synthesis of the 1,6-addition product by reacting Bu2CuLi with methyl sorbate in 82% yield, concluding that the electronic properties of Gilman reagent as a nucleophile greatly affect its regioselectivity (Fig. 2).16,17
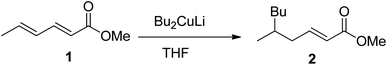 |
| | Fig. 2 1,6-Conjugate addition by using Gilman reagent. | |
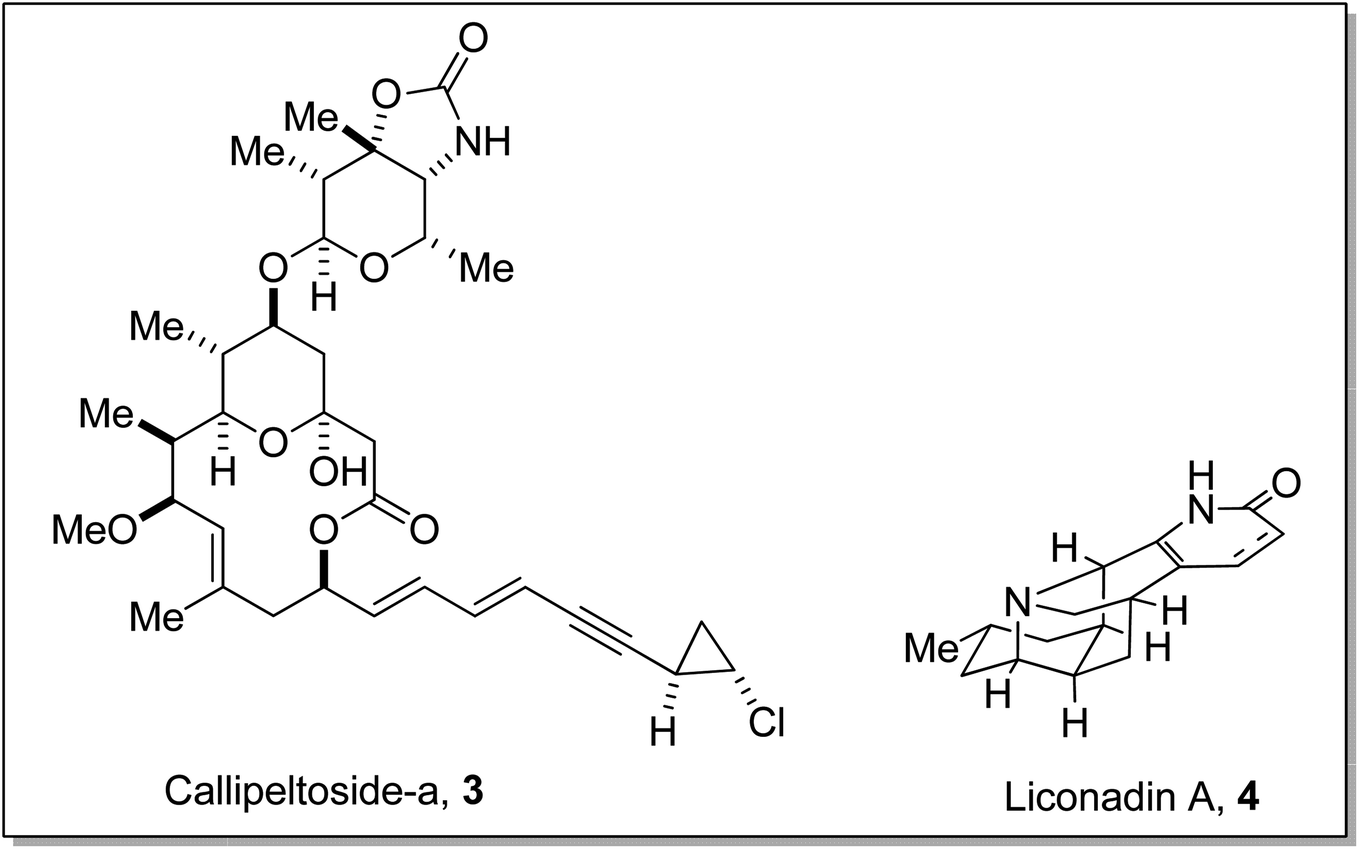
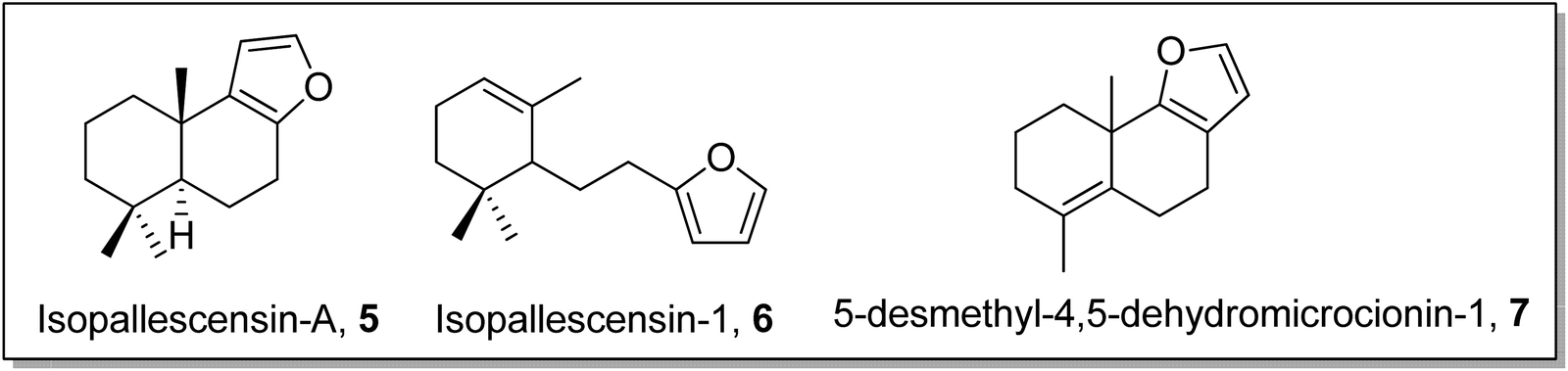
Gilman reagent has been extensively employed in the synthesis of a number of architecturally unique natural products, such as the C1 to C9 framework of callipeltoside-a 3 (isolated from a marine sponge Callipelta sp), which is an anti-HIV agent, liconadin A 4 (a Lycopodium alkaloid) (Fig. 3)18,19 and furosesquiterpenes i.e., isopallescensin-A 5, isopallescensin-1 6, 5-desmethyl-4,5-dehydromicrocionin-1 7, which are best known for their diverse biological activities (Fig. 4).20
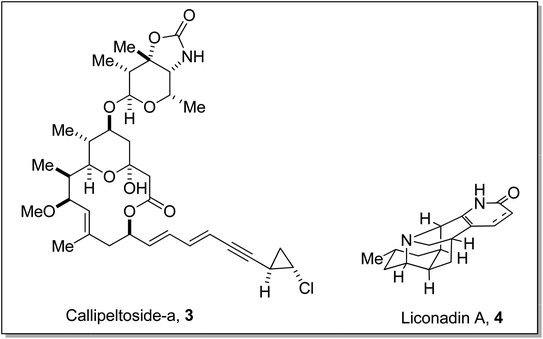 |
| | Fig. 3 Structures of callipeltoside-a 3 and liconadin A 4. | |
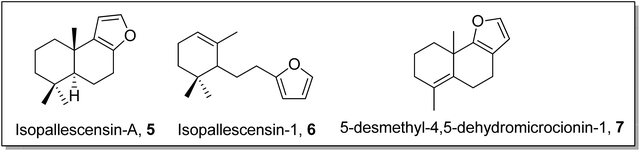 |
| | Fig. 4 Structures of isopallescensin-A 5, isopallescensin-1 6, and 5-desmethyl-4,5-dehydromicrocionin-1 7. | |
Inspired by the fact that methyl-substituted stereogenic centers are present in various natural products possessing a high pharmaceutical profile, Gilman reagent finds its application as an effective methyl source via conjugate addition and plays remarkable roles in the synthesis of these natural products. Further, Gilman reagent is used as a regioselective epoxide ring opening agent to produce 1,2 diols or 1,3 diols. In total synthesis, keeping control of the diastereomeric ratio of these diols, this reagent has been found to be helpful in the development of asymmetric centers with the desired stereochemistry of natural products. Herein, the synthetic compilation based on the employment of Gilman reagent in the synthesis of medicinally important natural products has been reported.
Review of literature
Synthesis of alkaloid-based natural products
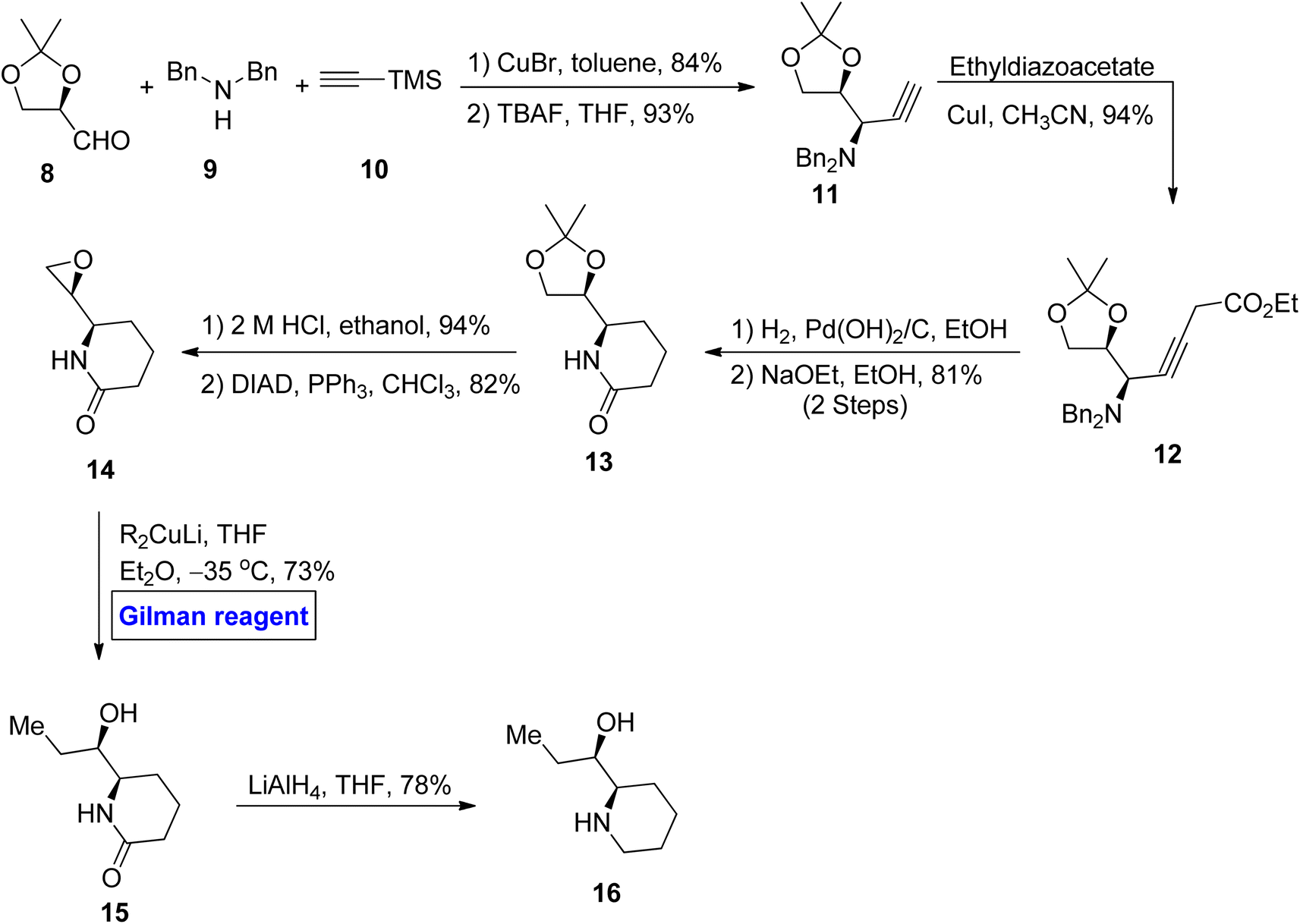
Piperidine alkaloid. Piperidine ring-based natural products are well-known for possessing glycosidase inhibitory effects as well as anti-diabetic, anti-cancer, and anti-obesity activities.21 (+)-α-Conhydrine 15 is an example of a 2-(hydroxyalkyl)-piperidine ring containing alkaloid.22 Considering the interesting features of this class of natural products, various synthetic methods have been designed regarding the synthesis of (+)-α-conhydrine and its enantiomers. In continuation of this work, Deshmukh et al. in 2012 performed the synthesis of (+)-β-conhydrine 16 with a sequence of 8 steps in 26% overall yield (Scheme 1).23 In their synthetic route, aldehyde 8, dibenzylamine 9, and terminal alkyne 10 were allowed to react in the presence of copper bromide as a catalyst and toluene as a solvent, accompanied by deprotection of the TMS group in the presence of THF to give terminal alkyne 11 in 93% yield (in 2 steps). The terminal alkyne 11 was then reacted with ethyl diazoacetate in the presence of copper iodide as a catalyst to acquire compound 12 (in 94% yield), which underwent further hydrogenolysis and subsequent treatment with NaOEt, resulting in compound 13 in 81% yield. In the subsequent step, ketal group deprotection and conversion of diol into epoxide gave lactam 14 in 82% yield. Further, regioselective ring opening of epoxide was performed via the application of a well-suited Gilman reagent in the presence of tetrahydrofuran and diethyl ether at −35 °C that resulted in compound 15 in 73% yield. In the last step, the reduction was performed by using LAH to furnish the desired β-(+) conhydrine 16 in 78% yield.
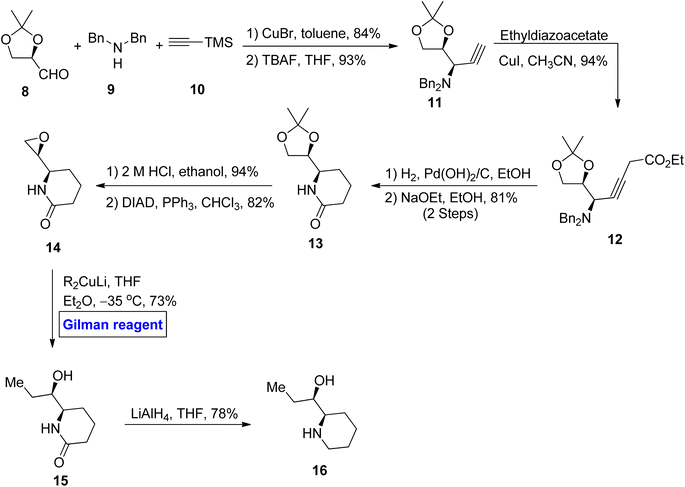 |
| | Scheme 1 Synthesis of β-(+) conhydrine 16. | |
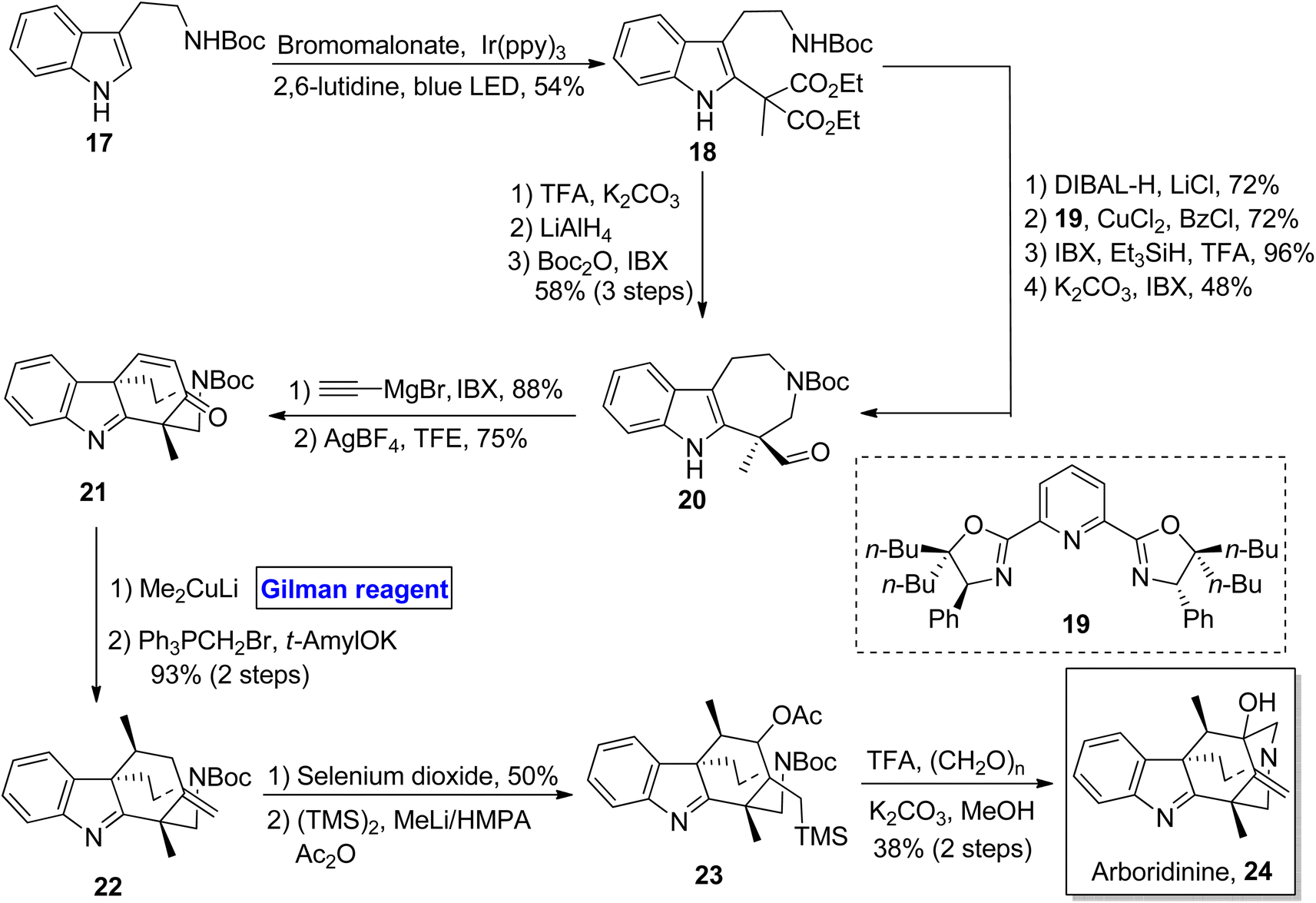
Indole alkaloid. Arboridinine 24 belongs to the class of indole alkaloids isolated from Kopsia plants (usually found in Malaysia).24 Its unprecedented structure consists of a tetracyclic indolenine cage, and it shares its structural features with many other families of alkaloids, making it a unique and interesting scaffold.25 As a part of the synthetic challenge for many synthetic endeavors, Gan et al. in 2018 designed an efficient, scalable, and enantioenriched synthetic scheme towards the synthesis of arboridinine 24 based on a 16 steps sequence using Boc protected tryptamine 17 as a starting material (Scheme 2).26 Compound 17 was photo-irradiated in the presence of bromomalonate (under suitable conditions) to result in indole 18 in 54% yield. From indole 18, the aldehyde 20 was prepared by using two routes. In the first one, compound 18 was deprotected by using TFA and potassium carbonate, followed by a sequence of reduction and subsequent oxidation, which furnished aldehyde 20 in 58% yield over 3 steps. The second route, which was more scalable, involved the treatment of compound 18 with DIBAL-H, further reacting with 1,3-diol and ligand 19 in the presence of CuCl2 to give the intermediate, which proceeded further via a sequence of oxidation and reduction under suitable conditions. In the following step, the aldehyde 20 was cyclized to afford the α,β-unsaturated ketone 21. In order to prepare precursor 28 for aza-Prins cyclization, 1,4-conjugate addition of compound 21 was performed by using Me2CuLi (Gilman reagent) with the successful installation of methyl group and subsequent treatment with Wittig reagent, resulting in alkene 22 in 93% yield (over 2 steps). The alkene 22 was subjected to oxidation via selenium dioxide and subsequent 1,4-addition of the TMS group to afford precursor 23. The last step involved the aza-Prins cyclization and Boc deprotection, concluding the total synthesis of arboridinine 24 (38% yield).
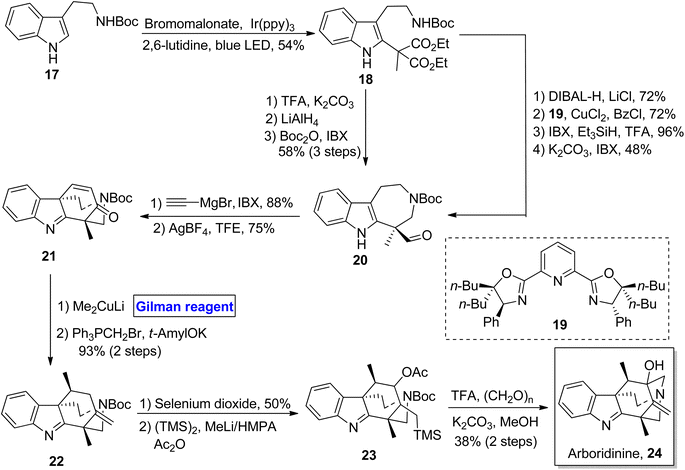 |
| | Scheme 2 Total synthesis of arboridinine 24. | |
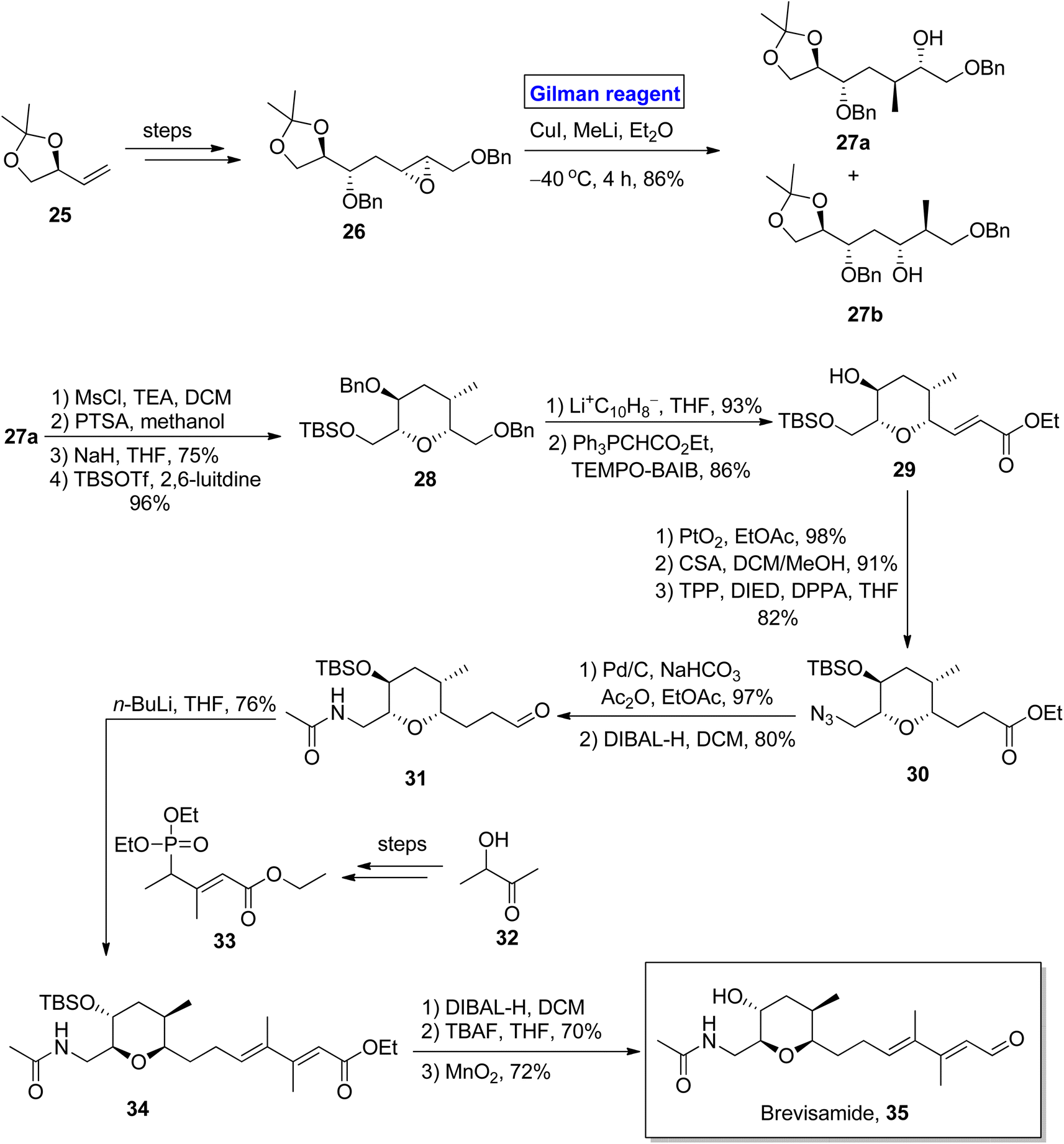
(−)-Brevisamide was isolated from Karenia brevis, a dinoflagellate, by Wright and colleagues for the first time.27 It is a brevenal and exhibits antagonist effects against brevetoxins. Brevetoxins are neurotoxic substances responsible for the death of fish and other marine organisms.28 Owing to the impressive biological profile of brevisamide, 5 groups of researchers have reported its total synthesis. In the continuation of this study, Yadav et al. in 2013, also presented a valuable and stereoselective approach towards its synthesis starting from an easily available starting material, i.e., 2,3-O-isopropylidene glyceraldehyde 25 (Scheme 3).29 The key steps entail the Sharpless epoxidation, ring opening of epoxide by using Gilman reagent, intramolecular SN2 cyclization, Wittig olefination, and HWE reaction. The compound 25 was modified (in a few steps) into epoxide 26, which upon treatment with Gilman reagent, i.e., CuI, MeLi, and diethyl ether as a solvent, resulted in a regioselective ring opening to give 27a and 27b in 86% combined yield. The compound 27a was mesylated in the presence of MsCl, and its acetonide group was removed by using PTSA to give the intermediate, which was subjected further to an SN2 cyclization in the presence of NaH and subsequent treatment with TBSOTf in the presence of 2,6-lutidine to give compound 28 in 96% yield. The benzyl group deprotection of compound 28, its chemoselective oxidation, and Wittig olefination furnished compound 29 with 86% yield. Then, compound 29 underwent a series of protection and deprotection steps under the given conditions to provide compound 31 (via intermediate 30) with 80% yield. In the following step, the HWE reaction of compound 31 with compound 33 (obtained from compound 32) using n-BuLi and THF afforded compound 34 with 76% yield. Deprotection via treatment with DIBAL-H, TBAF, and MnO2 in consecutive steps completed the total synthesis of brevisamide 35 with 72% yield.
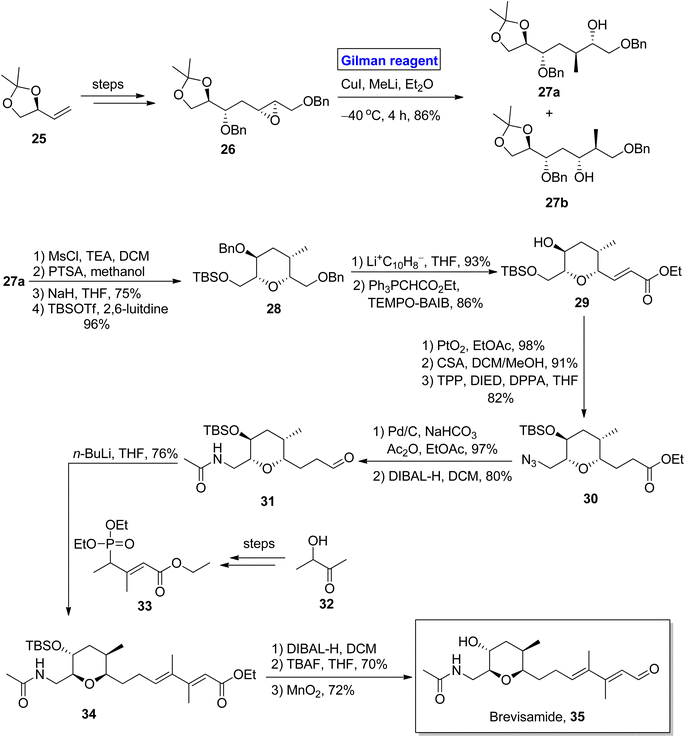 |
| | Scheme 3 Total synthesis of brevisamide 35. | |
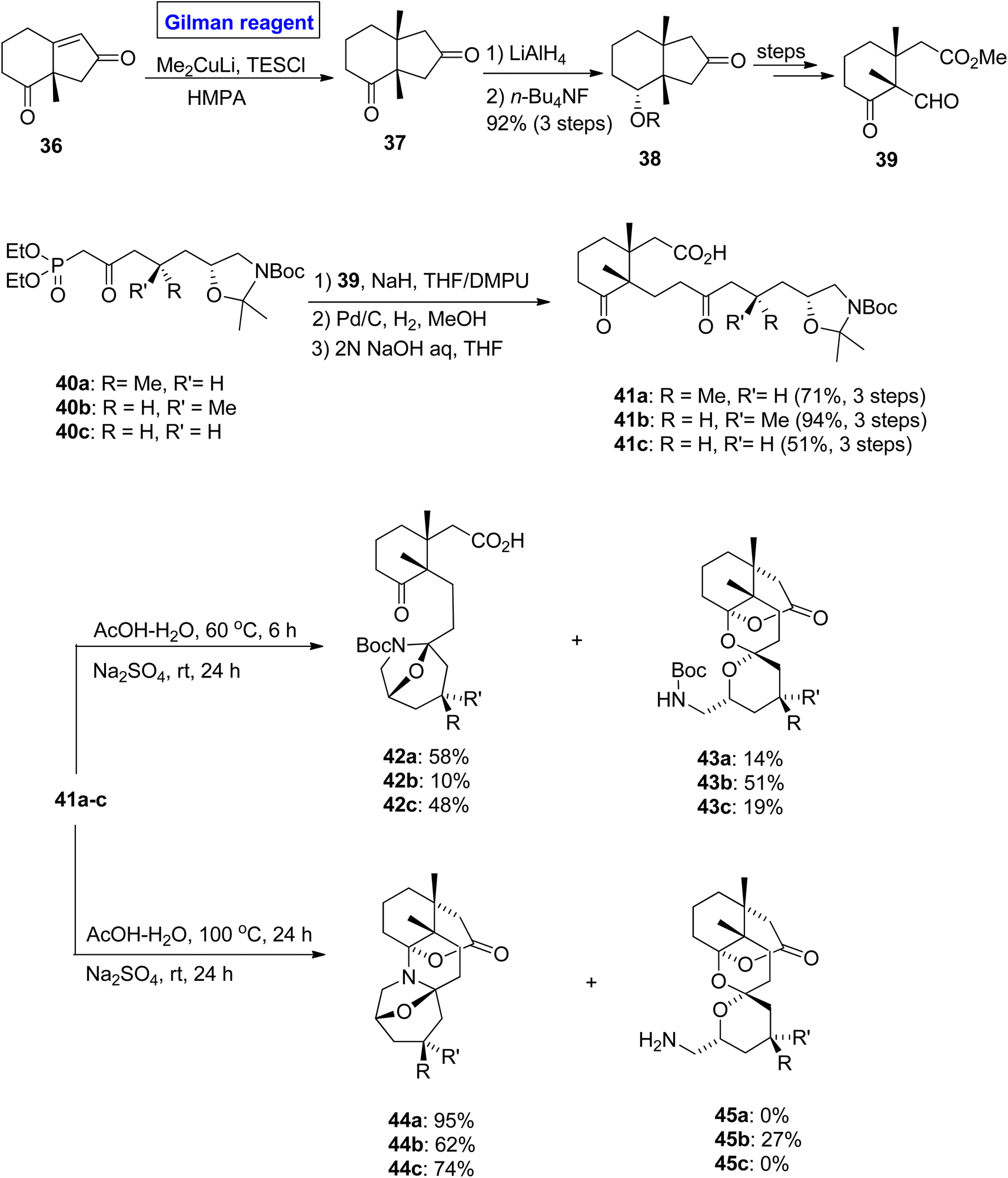
Zoanthamine alkaloid. Zoanthamine alkaloids are marine alkaloids, which contain a heptacyclic ring system. It exhibits potent biological activities such as anti-inflammatory, cytotoxic, anti-bacterial, and anti-osteoporotic activities, thus having an effective pharmacological profile.30,31 Many attempts have been made towards the synthesis of its pentacyclic ring system (based on 8 chiral centers including 3 carbon quaternary and 2 aminal functionalities).32–34 To accomplish this challenging total synthesis, Nakajam et al. in 2011 studied and disclosed the stereochemical effects of a methyl group at the C-4 position towards the bisaminal cyclization in zoanthamine alkaloids (Scheme 4).35 In their strategy, the enone 36 (for 1,4-conjugate addition) was treated with Gilman reagent, i.e., Me2CuLi, in the presence of TESCl and hexamethyl phosphoramide to obtain compound 37. After 1,4-conjugate addition, the compound 37 was immediately reduced in the presence of LiAlH4 and desilylated by using quaternary ammonium salt to synthesize ketone 38 (with 92% yield), which was modified over a few steps to obtain aldehyde 39. In the next step, the HWE reaction of ketophosphonates 40a–c and aldehyde 39 by treating it with DMPU in THF afforded precursors 41a–c in moderate to good yield. In the last step of the synthesis, cyclization was performed with a slight variation in conditions. The treatment of compounds 41a–c with AcOH in water at T = 60 °C for 6 hours, accompanied by the addition of sodium sulphate with 24 hours of stirring at room temperature, provided a diastereomeric mixture of monoaminals 42a–c (10 to 58% yield) and spiroketals 43a–c (14 to 51% yield). In another case, the treatment of compounds 41a–c with AcOH at T = 100 °C for 12 hours, followed by the addition of sodium sulfate affected the mode of cyclization and furnished the bisaminal product 44a and 44c (in 95% and 74% yield, respectively), along with a diastereomeric mixture (from 41b) of bisaminal product 44b and spiroketal 45b (with % yield = 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27).
:27).
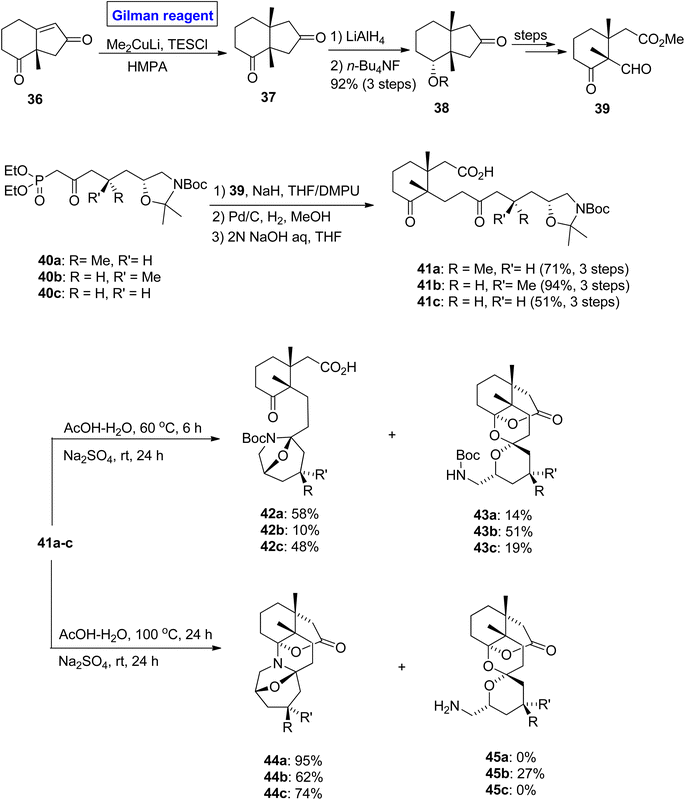 |
| | Scheme 4 Synthesis of precursor 41a–c and its cyclization. | |
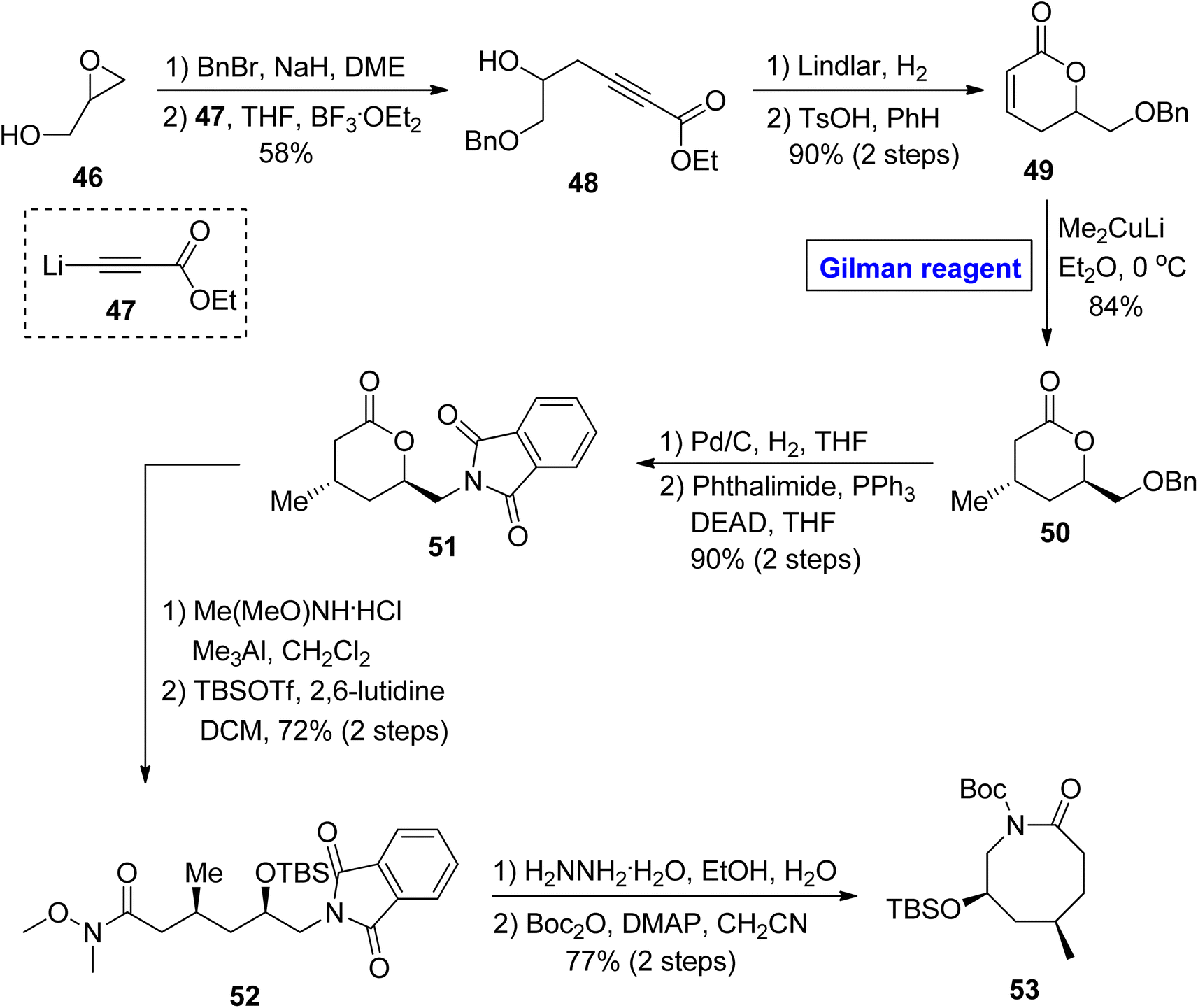
In continuation of synthetic studies on zoanthenol, Bagdanoff et al. in 2016, performed an enantioselective and facile synthesis of caprolactam 53 (as a precursor for zoanthenol) (Scheme 5).36 The synthesis commenced with the benzyl protection of glycidol 46, accompanied by a reaction with ethyl propiolate 47 under the given conditions, affording compound 48 with a 58% yield. In the next step, the reduction of compound 48 followed by cyclization provided lactone 49 (in 90% yield), which was subjected to a 1,4-conjugate addition reaction by employing the well-established Gilman reagent (Me2CuLi) in diethyl ether at 0 °C to furnish compound 50 in 84% yield. The compound 51 (achieved after the palladium-catalyzed reduction of compound 50 and subsequent Mitsunobu reaction)37 was subjected to Weinreb amide reaction, accompanied by treatment with hydrazine hydrate and subsequently cyclized under given conditions to provide the targeted caprolactam 53 (via intermediate 52) in 77% yield.
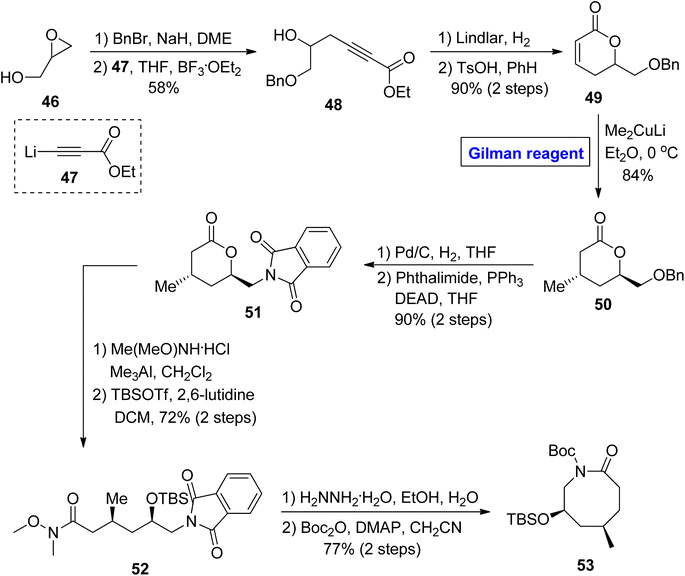 |
| | Scheme 5 Synthesis of caprolactam 53 toward the synthesis of zoanthenol. | |
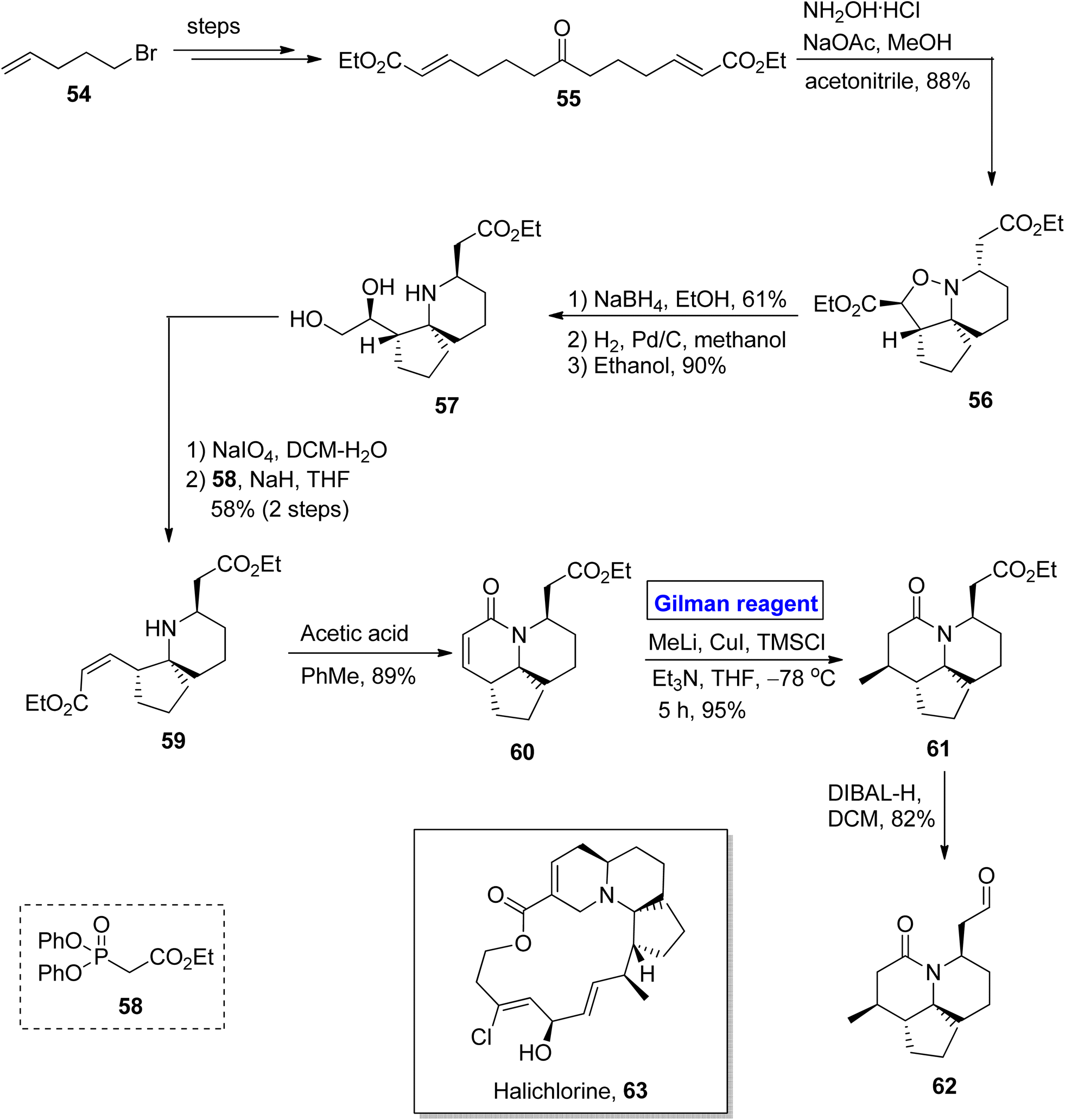
Quinolizidine alkaloid. Halichlorine 63 belongs to the class of quinolizidine alkaloids. It was isolated by Uemura and colleagues in 1966 from Halichondria okadai, a marine sponge found in Japan.38 Halichlorine 63 is a suppressor of NF-κB activation and inhibits the expression of vascular cell adhesion molecule 1 (VCAM-1) (which is related to inflammation and tumor cell growth). Halichlorine is also a hypertensive agent as it suppresses L-type calcium channels in smooth muscles.39 Owing to the biochemical potential of this heterocyclic scaffold, various organic chemists have reported its total synthesis, among which the Clive group presented a remarkable route in 2009. Pioneering this work, Gignoux et al. devised a concise and two directional strategy for the synthesis of intermediate 62 towards the synthesis of halichlorine 63 in 2012 (Scheme 6).40 Their synthetic route was based on a 12-step sequence, providing 13.2% overall yield and comprising easily available starting materials (ethyl formate 54). The ketodiester 55 (after modification from bromide 54 in a few steps) was cyclized under the given conditions to give isoxazolidine 56, which was then selectively reduced by using NaBH4 in ethanol and subsequently hydrogenated to give diol 57 in 90% yield. The oxidation of diol 57 was followed by a reaction with phosphonate 58 in the presence of sodium hydride, which furnished compound 59 with 58% yield. In the following step, compound 59 reacted with acetic acid in the presence of toluene to give bicyclic lactam 60 in 89% yield. In order to install a methyl group at the C14 position from the upper side, a well-suited Gilman reagent was used. To accomplish this task, compound 60 was treated with methyl lithium, copper iodide, and TMSCl in the presence of triethyl amine and tetrahydrofuran at −78 °C for five hours, resulting in compound 61 with the desired stereochemistry in 95% yield. In the last step, oxidation was performed in the presence of DIBAL-H and CH2Cl2 to furnish aldehyde 62 (in 82% yield), a well-designed intermediate toward the synthesis of halichlorine 63.
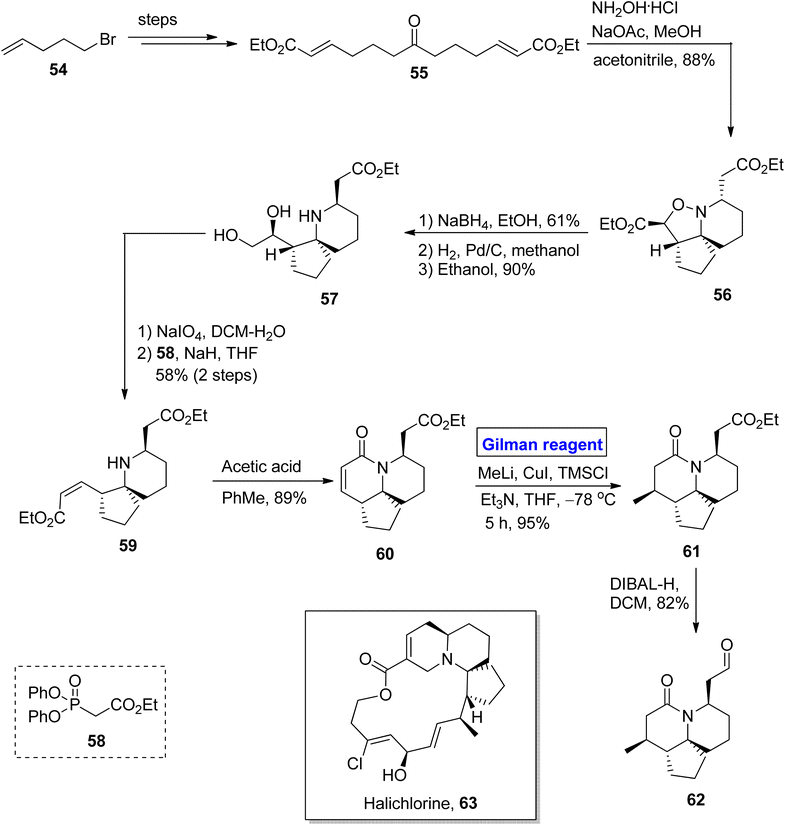 |
| | Scheme 6 Synthesis of intermediate 62 towards the synthesis of halichlorine. | |
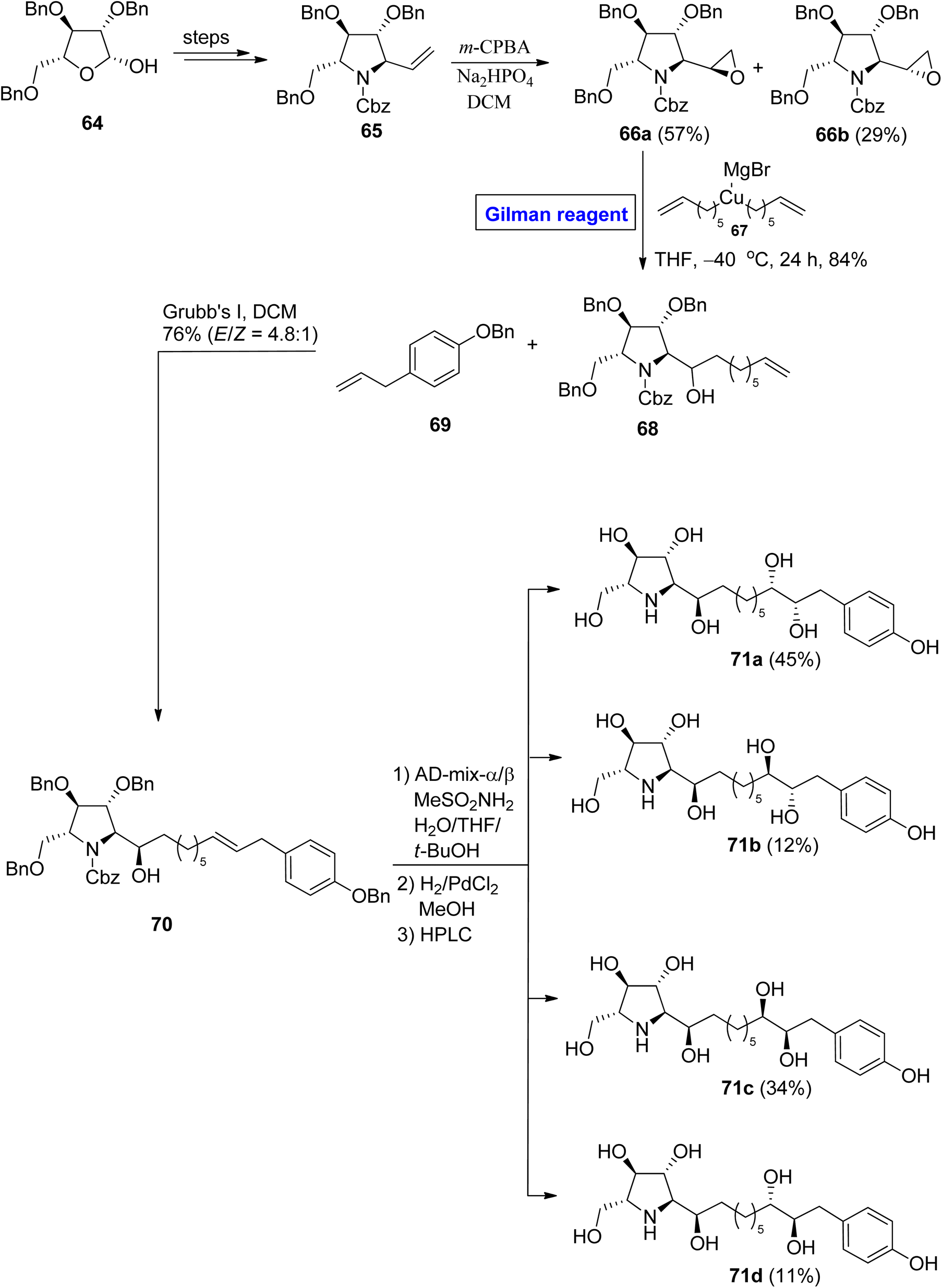
Glyphaeaside alkaloids. Glyphaeaside alkaloids were isolated from Glyphaea brevis.41 Structurally, these classes of alkaloids are based on a piperidine iminosugar along with a polyhydroxylated alkyl side chain that ends at the phenyl group and are classified into three types, i.e., A, B, and C. Among these, glyphaeaside C exhibits maximum potential as an inhibitor of snail β-mannosidase and β-glucosidase. It also shows a slight inhibitory effect for α-glucosidase.42 In 2021, Byatt et al. attempted the synthesis and structural elucidation of glyphaeaside C by employing well-suited Gilman reagent43 starting from compound 64, which was modified over a few steps to give vinyl pyrrolidine 65 (Scheme 7). The epoxidation of compound 65 in the presence of m-CPBA and Na2HPO4 provided epoxide (66a and 66b) in 57% and 29% yield, respectively. The epoxide 66a proceeded for ring opening reaction via Gilman reagent (in situ generated from compound 67) in THF at −40 °C for 24 hours to furnish compound 68 in 84% yield. In the next step, compound 68 was made to react with compound 69 in the presence of Grubb's first-generation catalyst, which resulted in a stereoisomeric pair of compounds 70 in 76% yield (E/Z = 4.8:1). The compound 70 was first treated with an AD mixture and MeSO2NH2 and then underwent hydrogenolysis. After chromatographic separation, compounds 71a to 71d were achieved in quantitative yield. In a structural analysis of the synthesized derivatives, the compound 71a was found to be the enantiomer of glyphaeaside C.
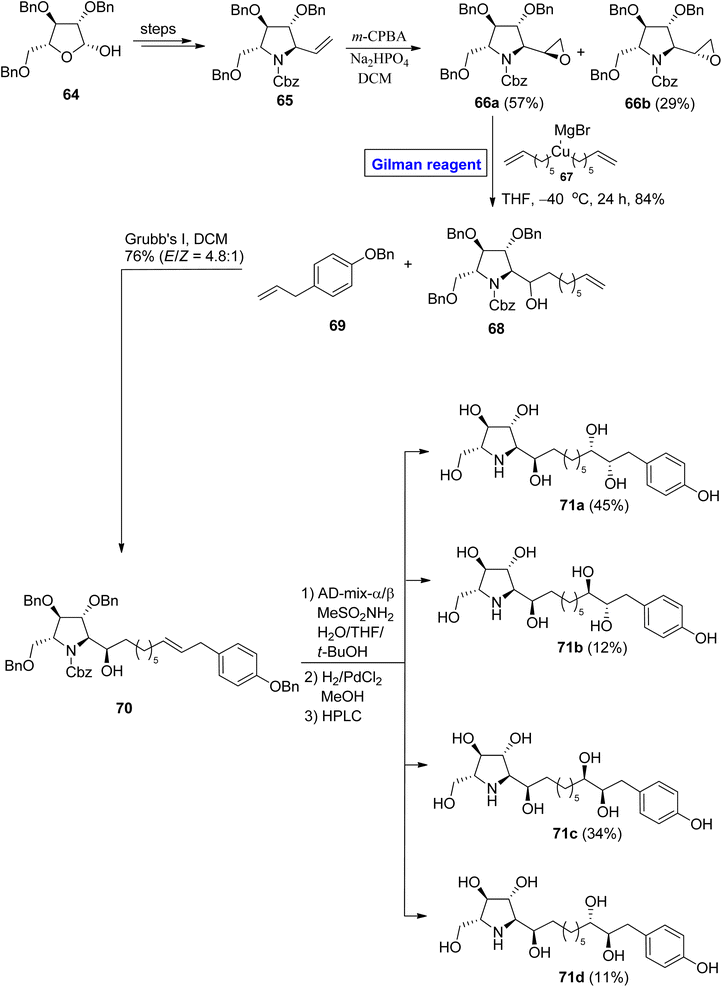 |
| | Scheme 7 Synthesis of 71a–71d towards the synthesis of glyphaeaside C. | |
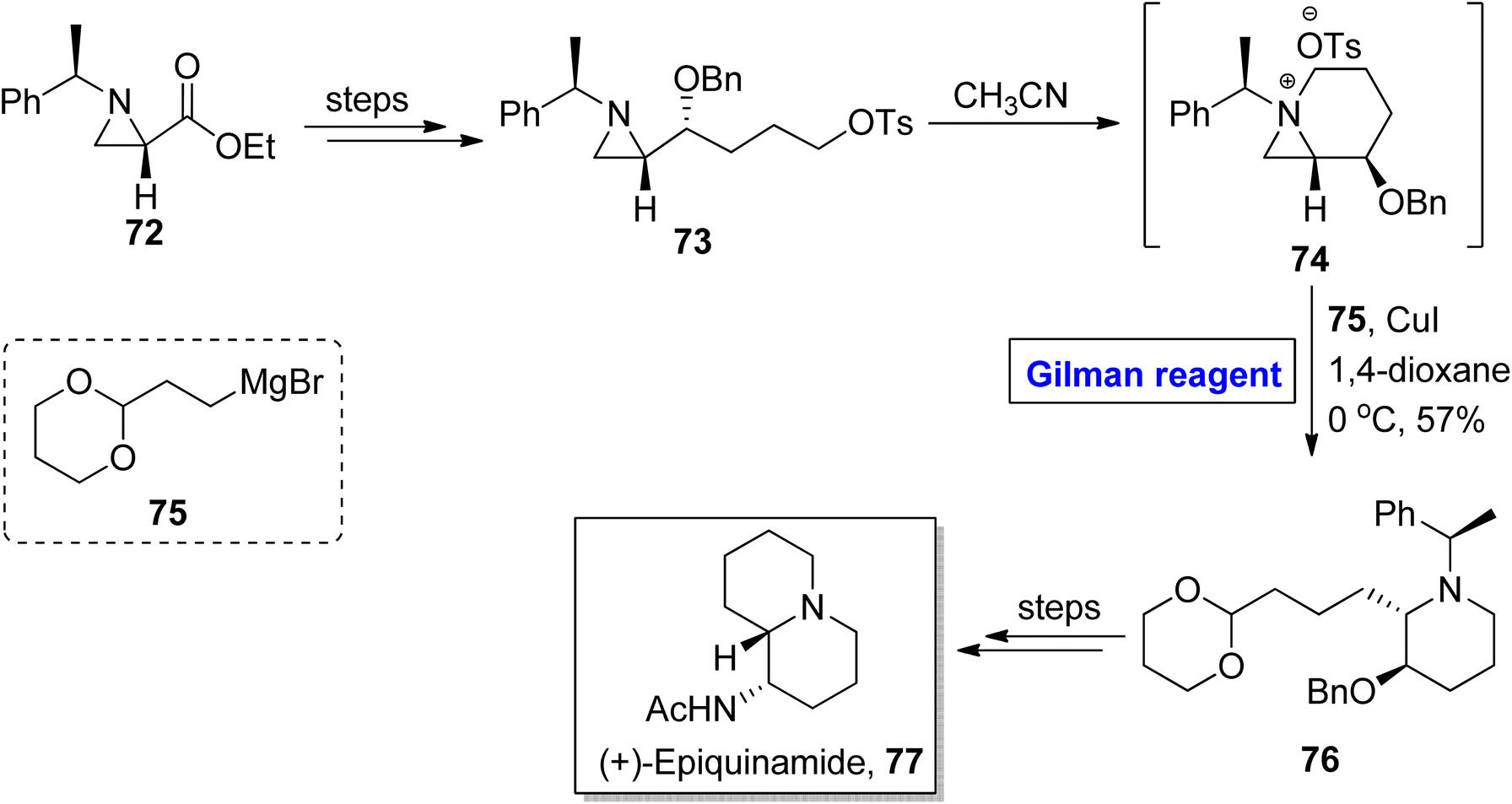
Quinolizidine alkaloid. (+)-Epiquinamide 77, a quinolizidine alkaloid, was isolated for the first time in 2003 from Epipedobates tricolor (a poisonous frog).44 This naturally scarce natural product is renowned as a nicotinic agonist.45 Owing to this fact, Yadav et al. in 2019 disclosed an efficient methodology for the synthesis of (+)-epiquinamide 7 by employing Gilman reagent-induced aziridinium ion ring opening reaction as the key step (Scheme 8).46,47 In their methodology, compound 73 (modified from aziridine carboxylate 72) was treated with methyl cyanide to provide aziridinium intermediate 74, which underwent ring opening reaction via treatment with reagent 75 in the presence of copper iodide and 1,4-dioxane as a solvent (for the in situ generation of Gilman reagent). The temperature was adjusted to 0 °C and the synthesis of compound 76 was achieved in 57% yield. It took a few steps for the modification of compound 76 into the desired natural product 77.
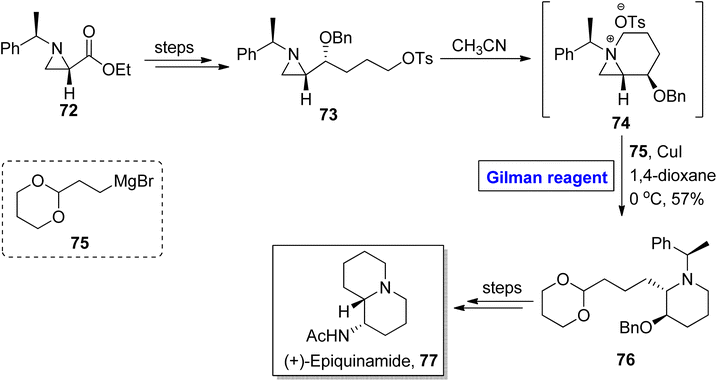 |
| | Scheme 8 Total synthesis of (+)-epiquinamide 77. | |
Synthesis of terpenoid-based natural products
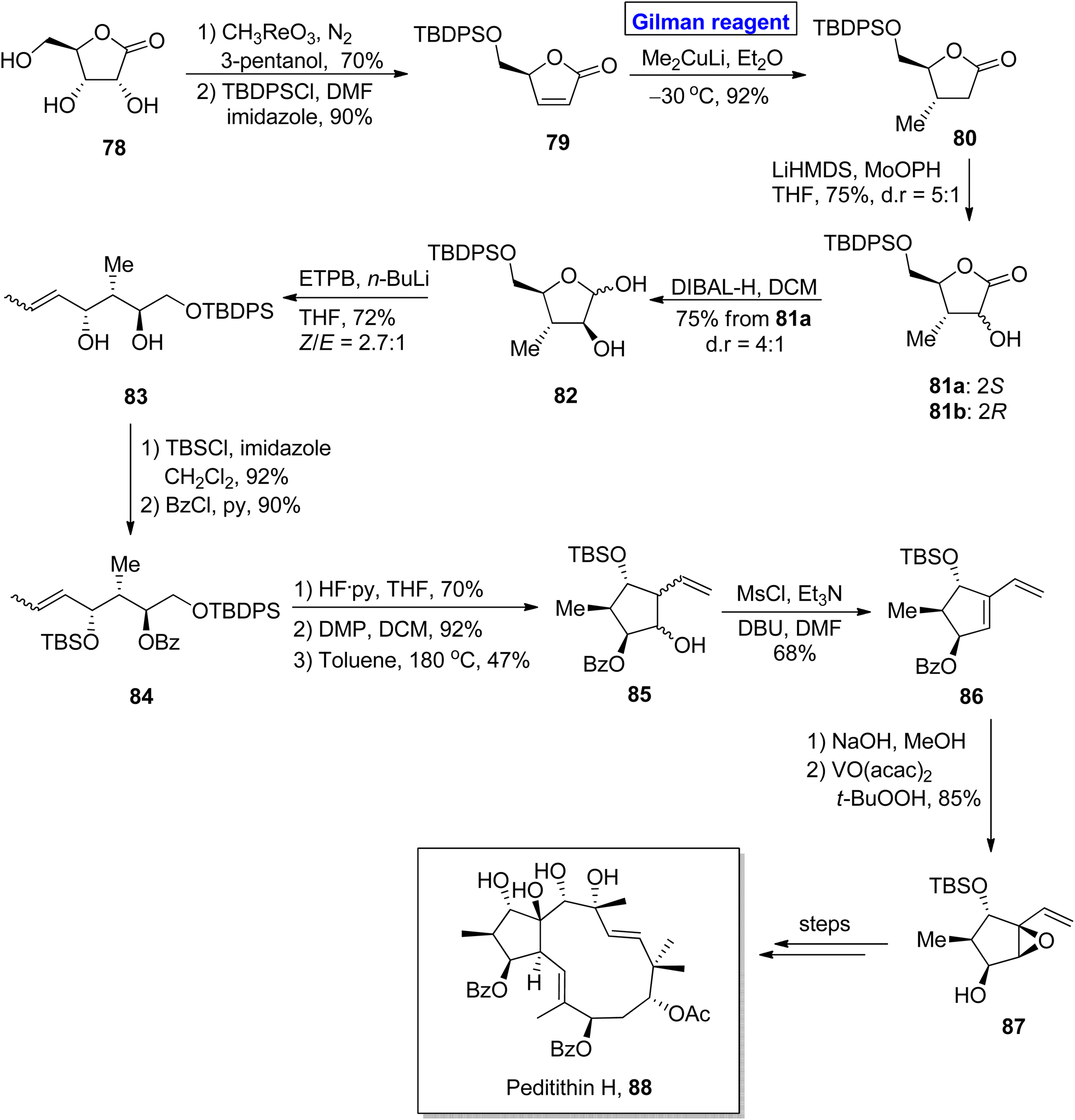
Diterpenoids. Peditithins B–H belongs to the class of Euphorbiaceae diterpenoids, isolated from Pedilanthus tithymaloides. These are P-glycoprotein (Pgp) inhibitors and effective anti-cancer agents.48,49 Methylcyclopentane is a fascinating heterocycle and is incorporated in many diterpenoid-based natural products. Structurally, peditithins H constitute substituted methylcyclopentane as a core, having 5 continuous stereocenters.50 Various synthetic strategies for the synthesis of methylcyclopentane constituting 3 or 4 stereocenters have been reported. Nonetheless, a synthetic strategy for the synthesis of 5 stereocenters based methylcyclopentane was reported recently. Ni et al. in 2020 performed this challenging task and presented an efficient and facile route for the synthesis of methylcyclopentane 87 (containing five stereocenters) as a building block for the synthesis of peditithins H (Scheme 9).51 In their synthetic procedure with 14 steps, the easily available lactone 78 was transformed into α,β-unsaturated compound 79 under given conditions. Next, the installation of a methyl group with the desired stereochemistry was accomplished via 1,4-conjugate addition using a well-suited Gilman reagent. Thus, the treatment of compound 79 with lithium dimethyl cuprate in the presence of diethyl ether at −30 °C resulted in lactone 80 with a 92% yield. In the next step, hydroxylation of lactone 80 by using Vedejs reagent gave a diastereomeric mixture, i.e., compound 81a and 81b in 75% yield with dr = 5:1. After chromatographic separation, 81a was reduced to give hemiacetal 82, which was subjected to Wittig olefination to furnish compound 83 in 72% yield. Upon TBDPS protection of compound 83, compound 84 was subjected further to TBS group removal and subsequent oxidation, accompanied by intramolecular carbonyl ene reaction by raising the temperature to 180 °C in toluene, which afforded compound 85 in 47% yield. After mesylation of homoallyl alcohol 85, the diene 86 was subjected to benzoyl group deprotection and subsequent epoxidation by using a suitable reagent to complete the synthesis of methylcyclopentane 87 with 85% yield.
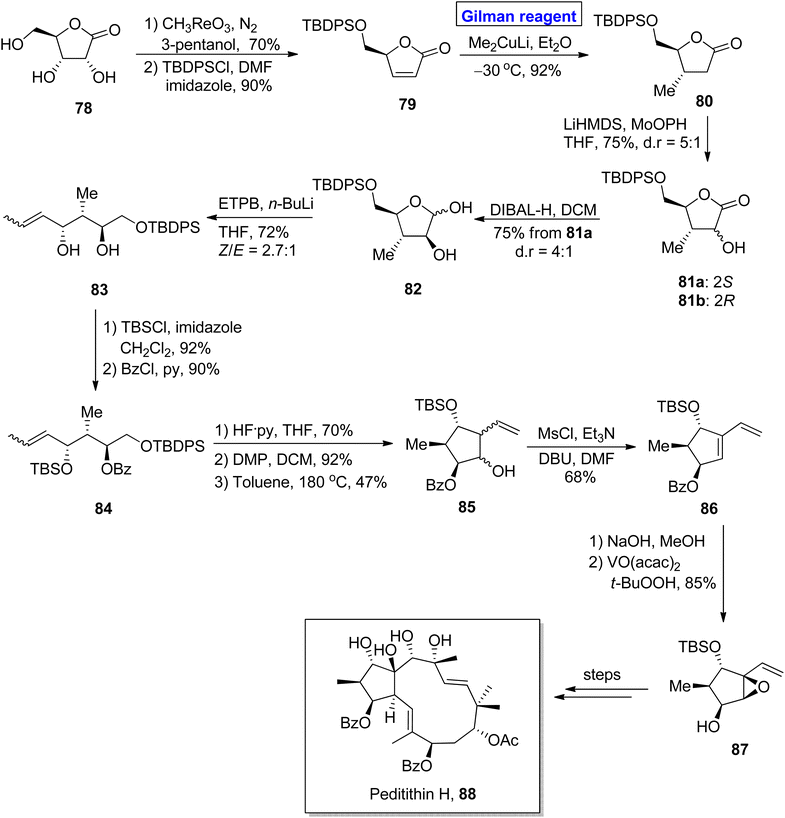 |
| | Scheme 9 Synthesis of intermediate 87 towards the synthesis of peditithin H 88. | |
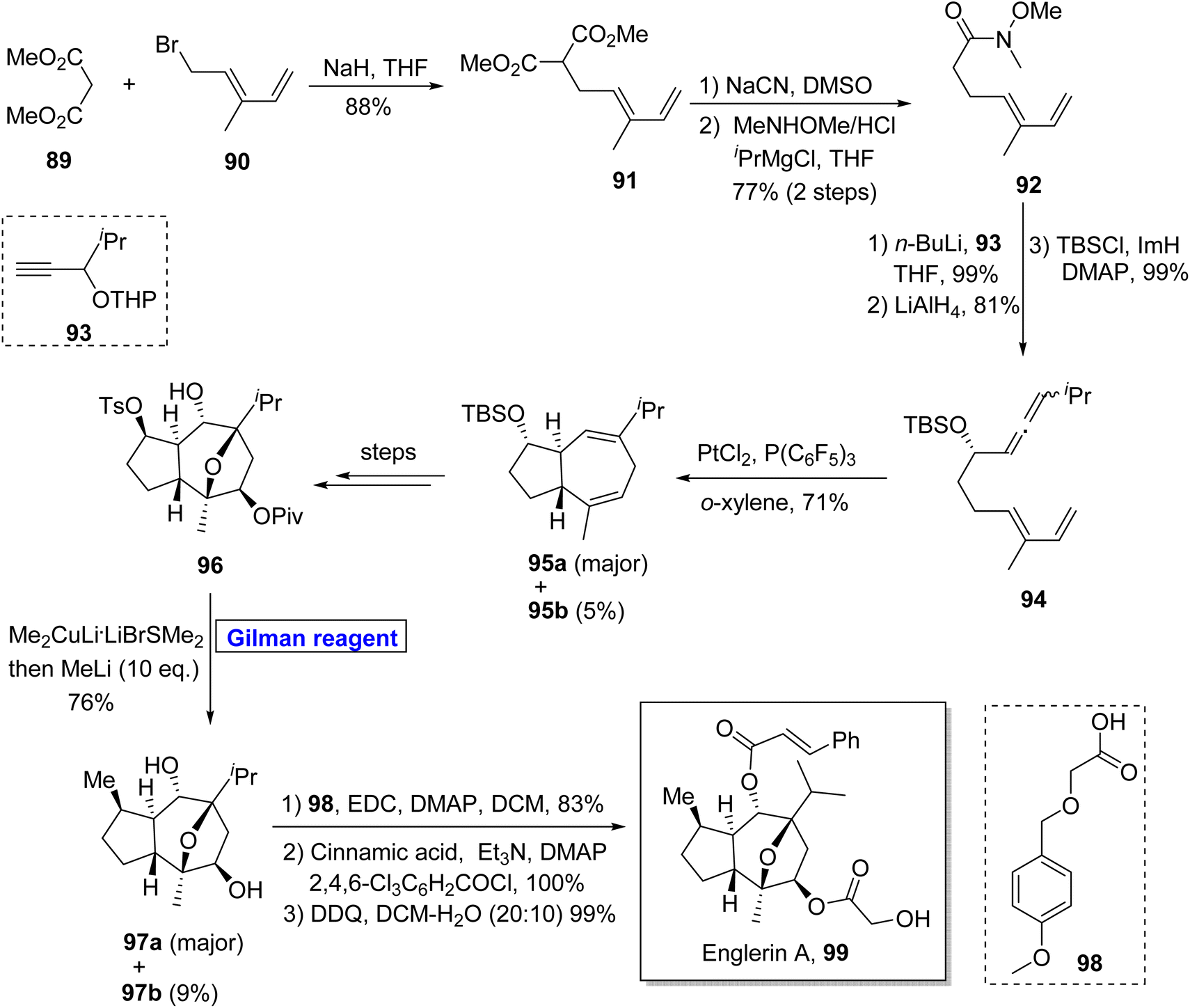
Sesquiterpenes. (−)-Englerin A 99 belongs to the class of guaiane sesquiterpene, isolated from Phyllanthus engleri. It activates the TRPC channel and exhibits potential anti-cancer activities against renal tumor cells.52 Owing to its impressive biological potential, Ma and colleagues reported the chiral pool approach for the synthesis of (−)-englerin A 99. Echavarren and colleagues reported the synthesis of an intermediate towards the synthesis of (−)-englerin A based on the Sharpless epoxidation.53 Pioneering this work, Nelson et al. in 2016 reported the most rapid and efficient methodology for the synthesis of this natural product, overcoming all the drawbacks of previous approaches (Scheme 10).54 Their synthetic scheme was based on a 17-step sequence and 11% overall yield starting from easily available starting materials by employing the Gilman reagent in the key step. In the first step, dimethyl malonate 89 and dienyl bromide 90 were allowed to react in the presence of sodium hydride to give diester 91 (in 88% yield), which underwent decarboxylation to result in amide 92 with 77% yield. The compound 92 was then treated with butyl lithium and after subsequent treatment with LiAlH4 and silyl protection, afforded allenediene 94 (99% yield). In the following step, allenediene 94 was treated with PtCl4 and P(C6F5)3 in the presence of o-xylene to give a diastereomeric mixture and 95a as a major product in 71% yield. Over a few steps, compound 96 was obtained, which was treated with Gilman reagent, i.e., Me2CuLi·LiBrSMe2, and further addition of methyl lithium furnished compound 97a (as a major product with 76% yield) and 97b (9% yield). The esterification of compound 97a using 98 under given conditions and further modification over two steps completed the total synthesis of our desired (−)-englerin A 99 in 99% yield.
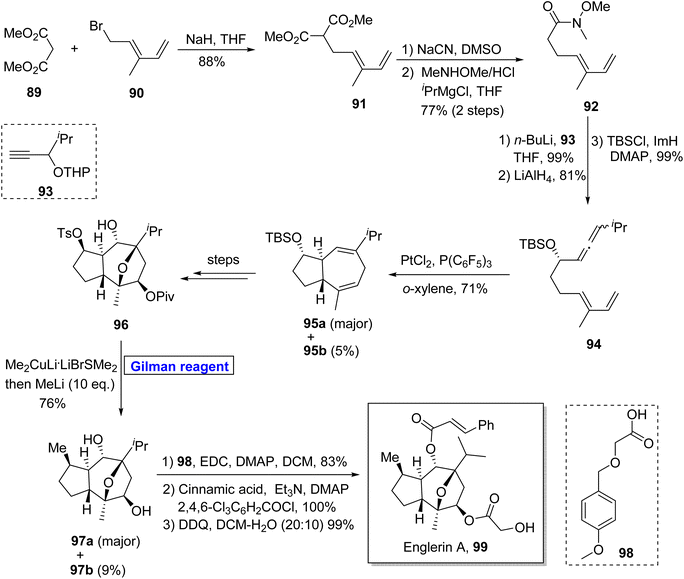 |
| | Scheme 10 Total synthesis of englerin A 99. | |
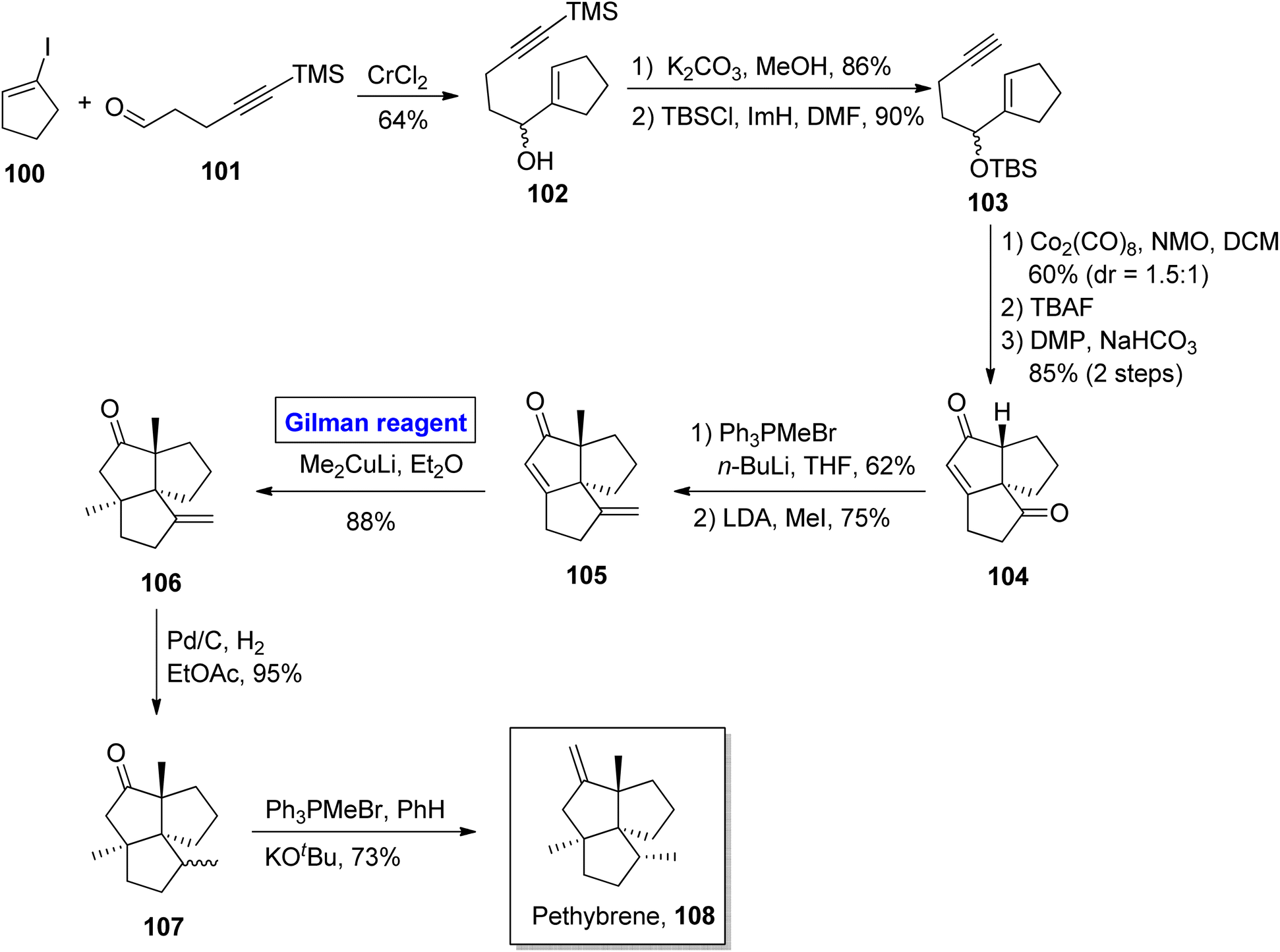
Pethybrene 108 is a sesquiterpene isolated from plants of Petasites hybridus. Its structural framework is based on fused triquinanes with a unique substitutional pattern.55 This heterocycle exists in various isomeric forms and rapidly undergoes rearrangement under acidic conditions, which makes it a continuing source of attraction for many synthetic endeavors.56 Jee & Lee in 2021 presented its total synthesis in 13 steps by employing the Pauson–Khand reaction, 1,4-conjugate addition by Gilman reagent, and Wittig olefination as key steps (Scheme 11).57 Their synthesis commenced with the NHTK coupling reaction of iodocyclopentene 100 and aldehyde 101 in the presence of CrCl2 to give compound 102 with a 64% yield. The silyl group deprotection of compound 102 and further treatment with TBSCl resulted in silyl ether 103 in 90% yield. The Pauson–Khand reaction of silyl ether 103 and subsequent oxidation provided diketone 104 (with 85% yield), which was subjected to Wittig olefination and methylenation to obtain enone 105. In the next step, 1,4-conjugate addition of compound 105 was performed by employing a well-suited Gilman reagent, i.e., lithium dimethyl cuprate in diethyl ether, to give compound 106 with 88% yield. The hydrogenation of compound 106 resulted in a separable, diastereomeric mixture of compound 107 (93% yield), which was (after separation) subjected to Wittig olefination to furnish our desired pethybrene 108.
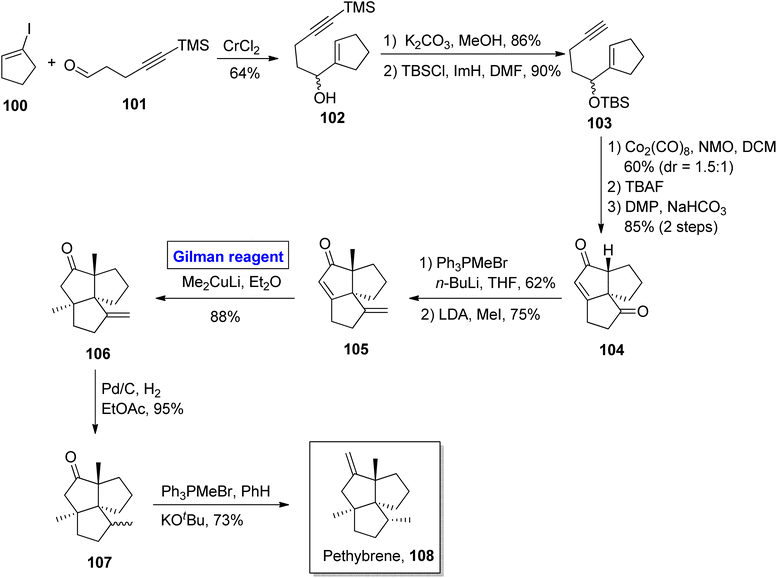 |
| | Scheme 11 Total synthesis of pethybrene 108. | |
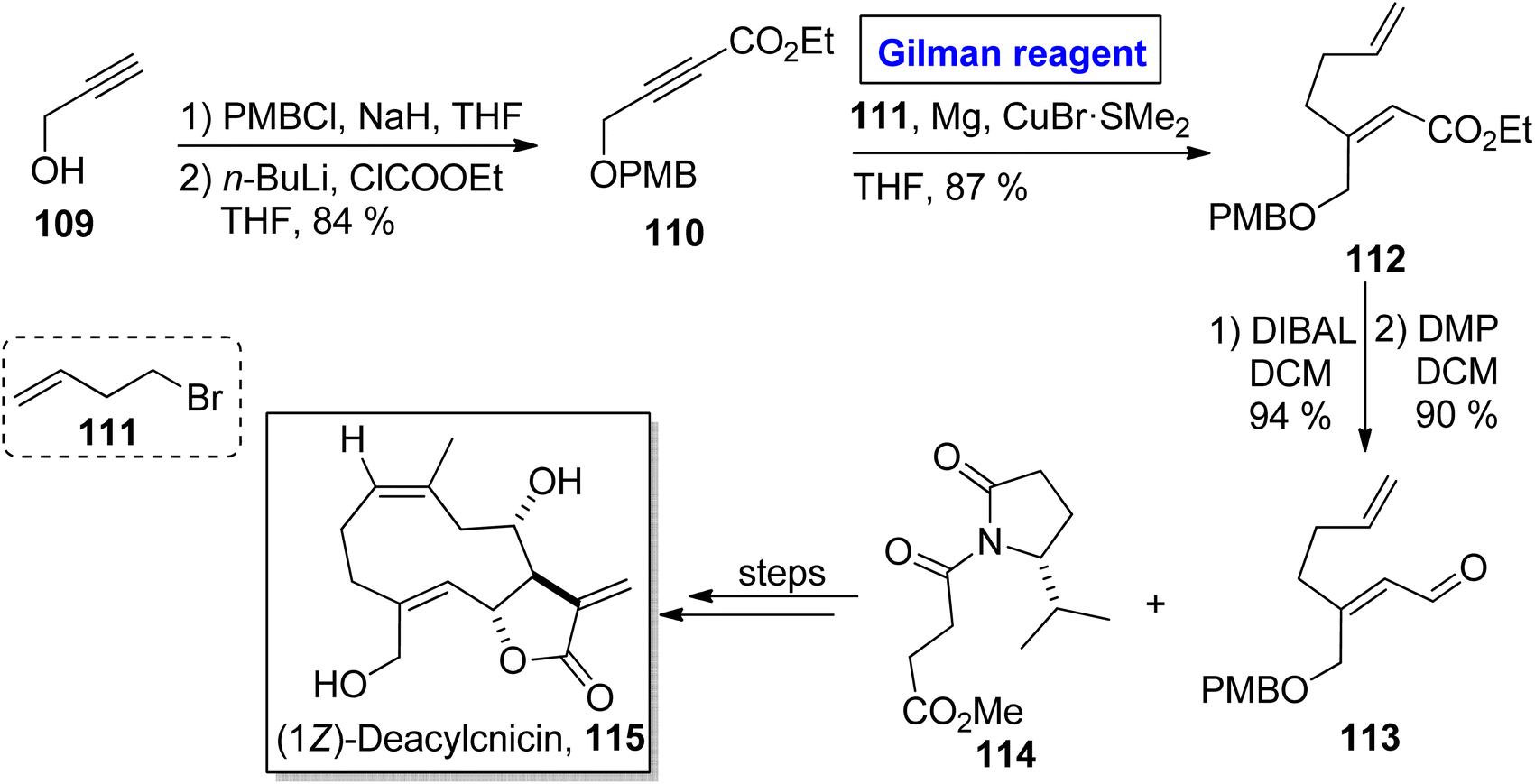
One of the germacranolide sesquiterpene, cnicin, is a 10-membered lactone isolated for the first time from the leaves of Cnicus benedictus (in 1959) by Sorm and colleagues.58 It exhibits a wide range of biological activities such as anti-bacterial, anti-myeloma, and cytostatic activities with allelopathic effects. Cnicin also shows inhibitory effects against Trypanosoma brucei with IC50 = 0.4 μM.59,60 Since the inception of this natural product, many synthetic pathways have been devised and reported in the literature. In 2022, Kimura and Usuki reported the synthetic procedure towards (1Z)-deacylcnicin 115, which was based on a 15-step sequence with 3.3% overall yield by consuming the easily available starting material, i.e., 2-propyn-1-ol 109 (Scheme 12).61 In the first step of their synthesis, compound 109 (after PMB group protection) was subjected to esterification under given conditions to afford compound 110 (in 84% yield). In order to perform the conjugate addition reaction of ester 110, it was reacted with compound 111, in the presence of Mg and CuBr·SMe2 (for in situ generation of Gilman reagent) in THF to furnish compound 112 in 87% yield. The sequence of DIBAL-induced reduction and DMP-induced oxidation of compound 112 provided compound 113 (in 90% yield), which underwent further reaction with compound 114 and took several steps for accomplishing the synthesis of (1Z)-deacylcnicin 115.
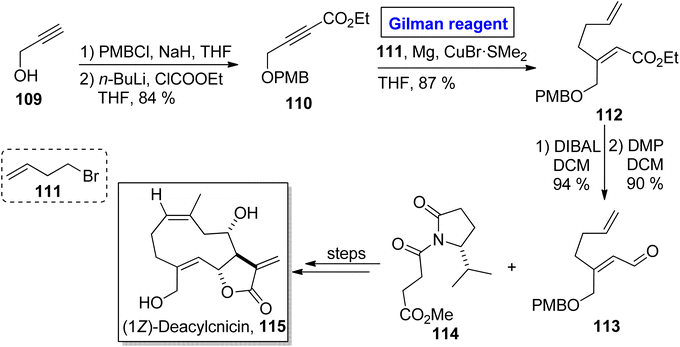 |
| | Scheme 12 Total synthesis of (1Z)-deacylcnicin 115. | |
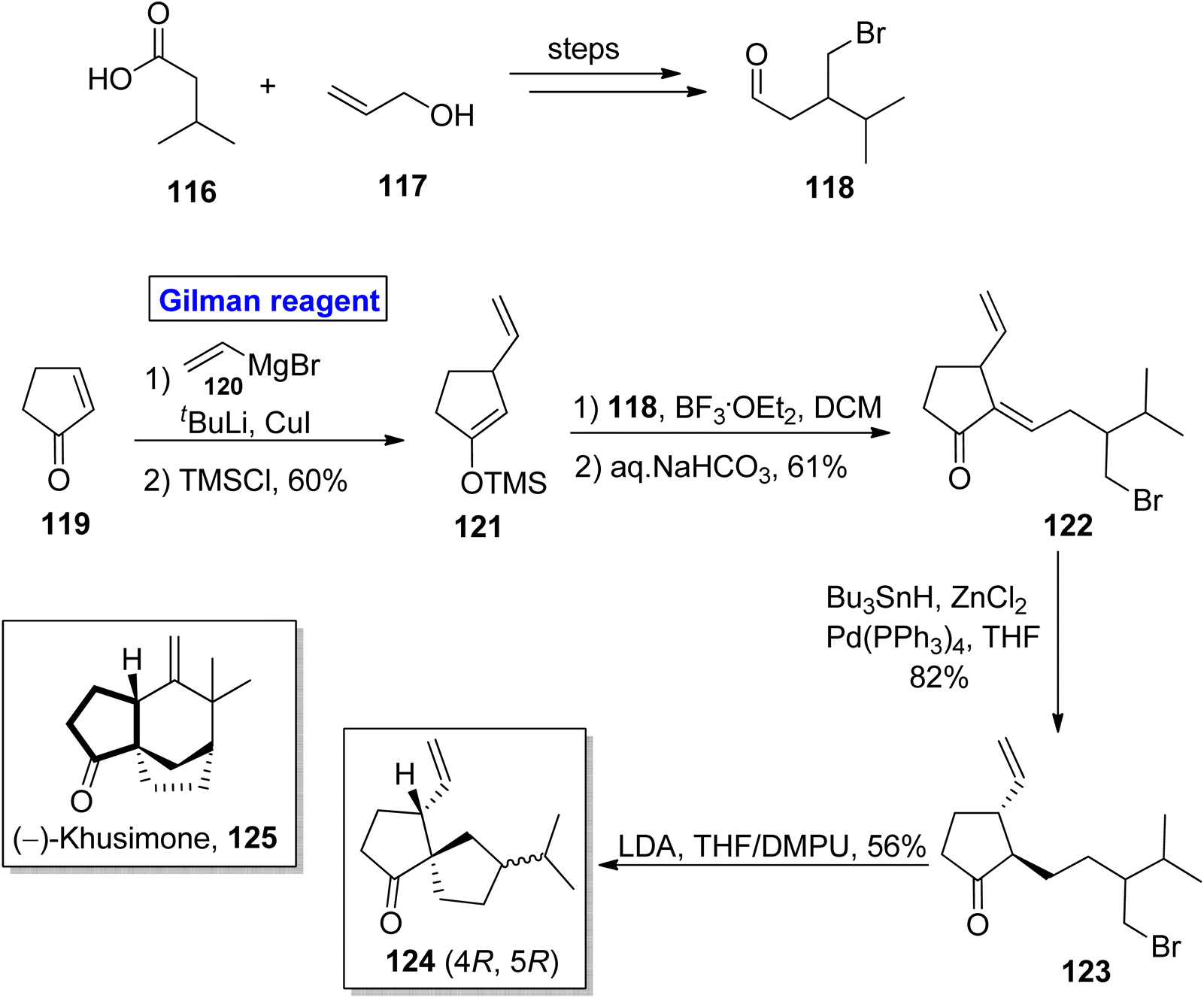
Norsesquiterpene. A tricyclic norsesquiterpene, (−)-khusimone 125 was firstly isolated from Vetiveria zizanoides L. (vetiver oil) by Umrani and colleagues.62 Due to its aromatic nature, it is widely used as a perfume, and its unique structural features have grasped the attention of many researchers.63 In 2013, Kraft and Denizot performed the facile synthesis of the analogue 124 in 10 steps by utilizing the easily available cyclopent-2-en-1-one 119 (Scheme 13).64 To accomplish this task, compound 119 was treated with Gilman reagent for nucleophilic substitution reaction in the presence of compound 120, tertiary butyl lithium, and copper iodide, after which silylation was carried out to provide compound 121 in 60% yield. For aldol condensation, the compound 121 reacted with the compound 118 (from isovaleric acid 116 and allyl alcohol 117) in the presence of BF3.·OEt2 and DCM and the subsequent treatment with aqueous sodium bicarbonate provided enone 122 in 61% yield. The palladium catalyzed reduction of enone 122 under given conditions accompanied by LDA induced cyclization (of compound 123) in THF/DMPU furnished the target compound 124 in 56% yield.
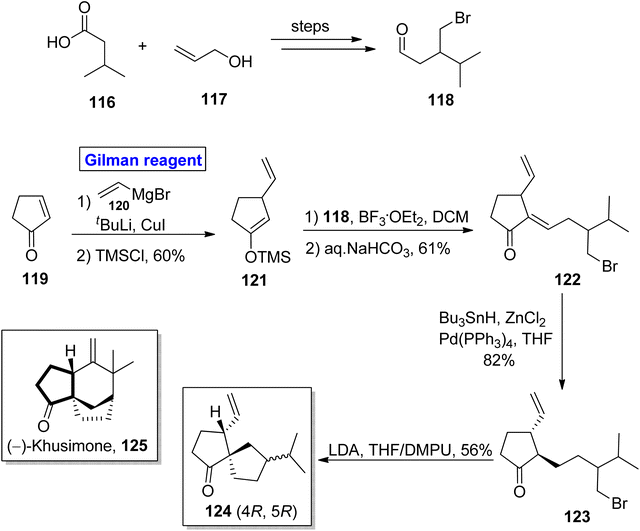 |
| | Scheme 13 Synthesis of analogue 124. | |
Synthesis of macrolide-based natural products
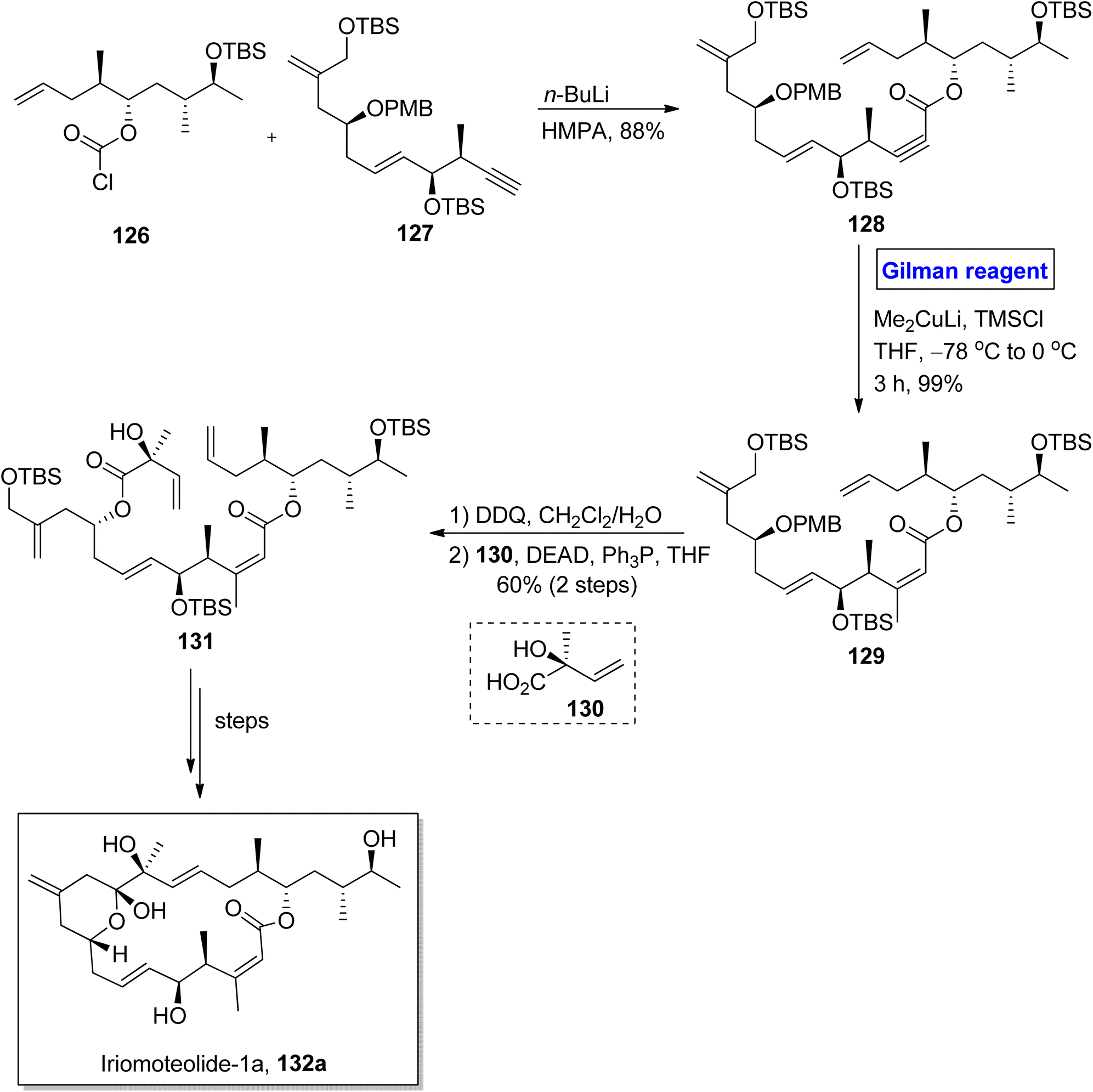
Iriomoteolide-1a 132a and Iriomoteolide 132b are twenty-membered cytotoxic macrolides. These were isolated independently by Tsuda and colleagues from the dinoflagellate of Amphidinium sp. found in Japan.65 Both heterocyclic scaffolds are structurally similar with slight differences. In iriomoteolide-1b 132a, there is a six-membered hemiketal ring with a methylene group at the eleventh carbon. Iriomoteolide-1a is specified for effective cytotoxic effects against human lymphocyte DG-75 cell line (with IC50 = 2 ng mL−1) and EBV-infected human lymphocyte (with IC50 = 3 ng mL−1).66 The unique molecular architecture of medicinally important iriomoteolide 1a has always been the target of many synthetic chemists. In this effort, Huang et al. in 2012 performed its total synthesis and elucidated its stereochemistry (Scheme 14).67 Their synthesis commenced with the reaction between chloroformate 126 and compound 127 in the presence of butyl lithium and HMPA to get compound 128 with 88% yield. The compound 128 was subjected to conjugate addition by using Gilman reagent, i.e., Me2CuLi in the presence of TMSCl and THF. The temperature was adjusted between −78 °C to 0 °C, and it took almost 3 hours to achieve compound 129 in 99% yield. In the next step, the selective deprotection of compound 129 and the subsequent Mitsunobu reaction37 in the presence of chiral compound 130 furnished compound 131 with a 60% yield. Over a few steps, compound 131 was converted into the desired iriomoteolide-1a 132a with required stereochemistry.
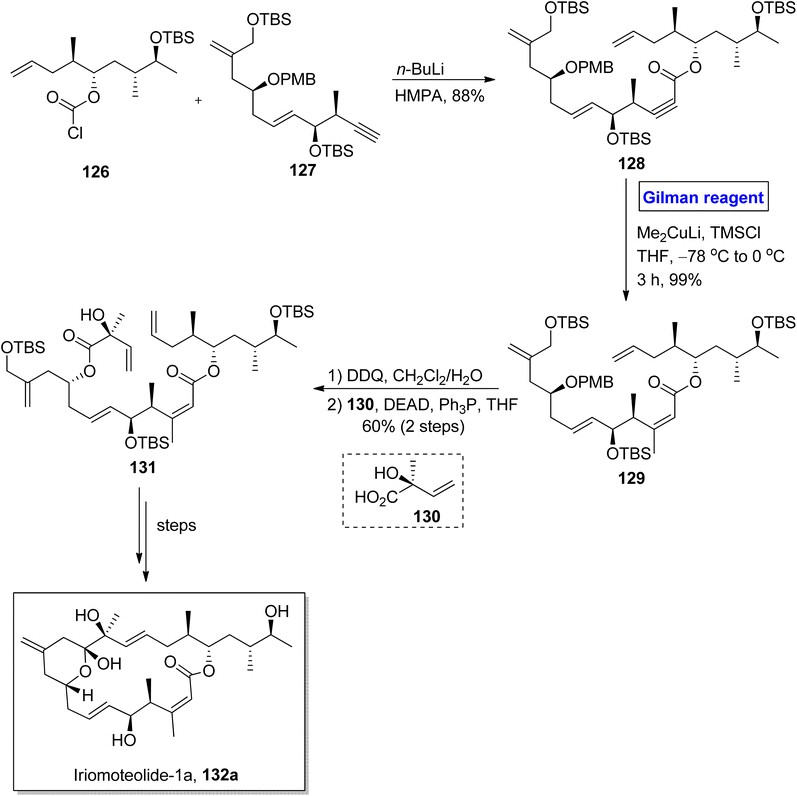 |
| | Scheme 14 Synthesis elucidated structure of iriomoteolide-1a 132a. | |
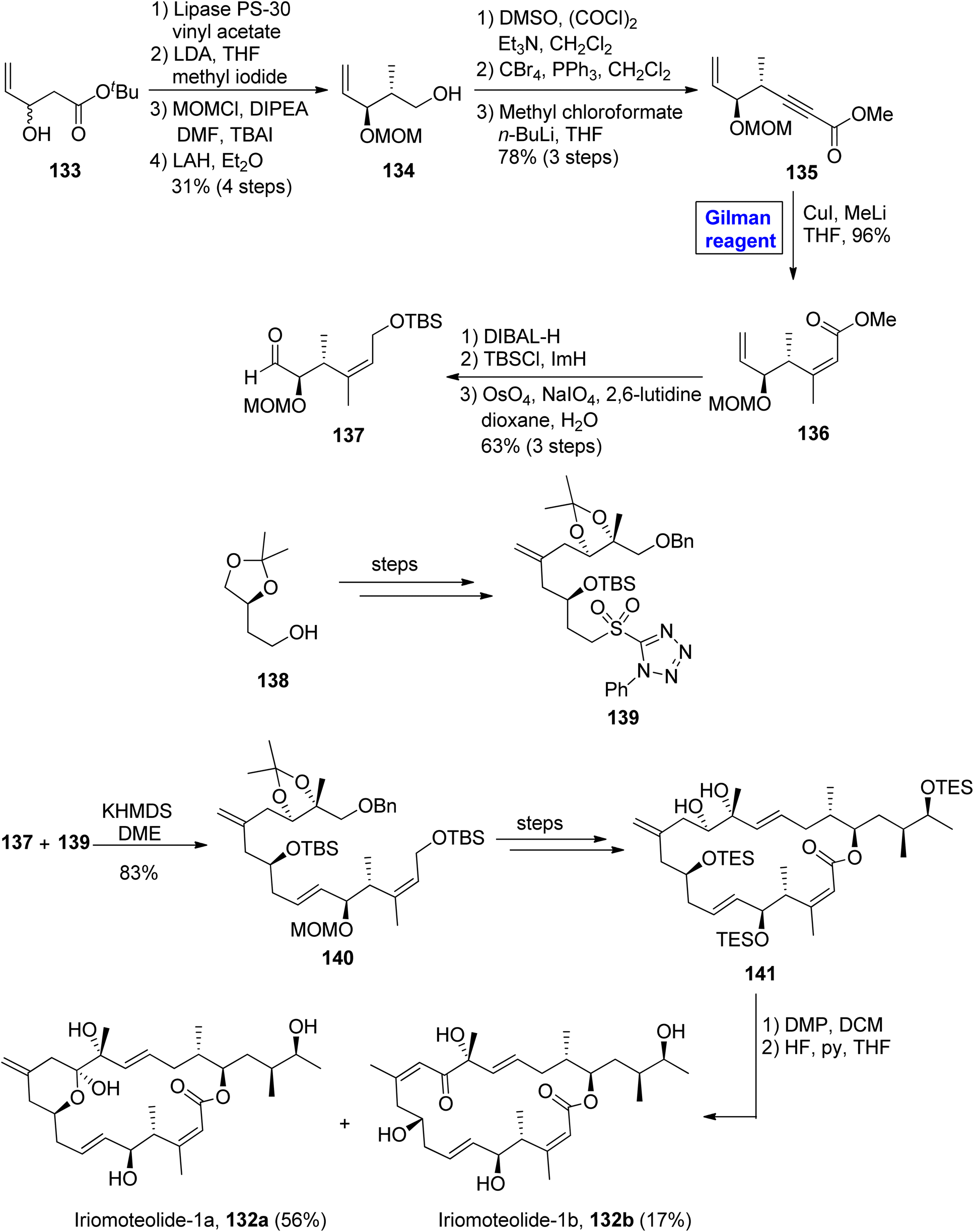
Iriomoteolide-1b 132b constitutes a cis double bond between the eleventh and twelfth carbons in conjugation with a ketone group at the thirteenth carbon. Iriomoteolide-1b 132b is also effective against the human lymphocyte DG-75 cell line (with IC50 = 900 ng mL−1).68 The interesting biological profile of iriomoteolide-1a 132a and Iriomoteolide-1b 132b urged Gosh and Yuan to perform their total synthesis and structural elucidation by employing the Gilman reagent, Julia Kocienski olefination, and Yamaguchi macrolactonization in key steps (Scheme 15).69 However, these synthesized structures did not show cytotoxic activity in biological evaluation. Their methodology involved the synthesis of fragment 137 and fragment 139, which later reacted together and were modified to compound 141. The synthesis of fragment 137 commenced with the treatment of racemic alcohol 133 with lipase PS-30 catalyst in the presence of vinyl acetate to obtain the enantioenriched product, which underwent reaction with lithium diisopropylamide and methyl iodide, accompanied by MOM protection and the subsequent reduction of the ether group in the presence of lithium aluminium hydride, furnished alcohol 134 with 31% yield. Further, the Swern oxidation of compound 134, followed by Corey Fuchs homologation and the subsequent reaction with methyl chloroformate and butyl lithium in THF, resulted in alkynyl ester 135 in 78% yield. In order to perform carbocupration, a well-suited Gilman reagent (CuI and MeLi in THF) was added to achieve the single isomer of olefin 136 in excellent yield (96%). In the next step, compound 136 was reduced by using DIBAL-H, accompanied by silyl protection and oxidative cleavage in the subsequent step to get the desired fragment 137 in 63% yield. The Julia Kocienski reaction of fragment 137 and sulfone 139 (modified from compound 138) resulted in compound 140 with an 83% yield. After a few steps, the compound 141 was oxidized by treating with DMP in DCM, and subsequently, deprotection was performed to get our desired mixture of iriomoteolide-1a 132a and iriomoteolide-1b 132b, which were latter on separated by column chromatography in 56% and 17% yields, respectively.
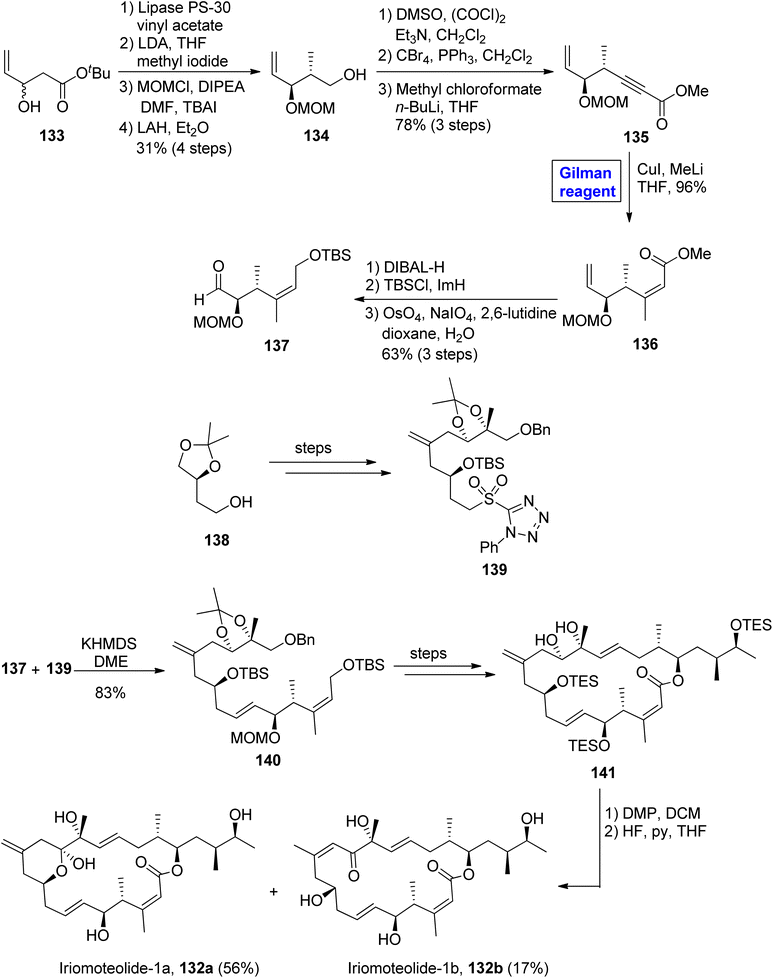 |
| | Scheme 15 Synthesis of proposed structures of iriomoteolide-1a 132a and iriomoteolide-1b 132b. | |
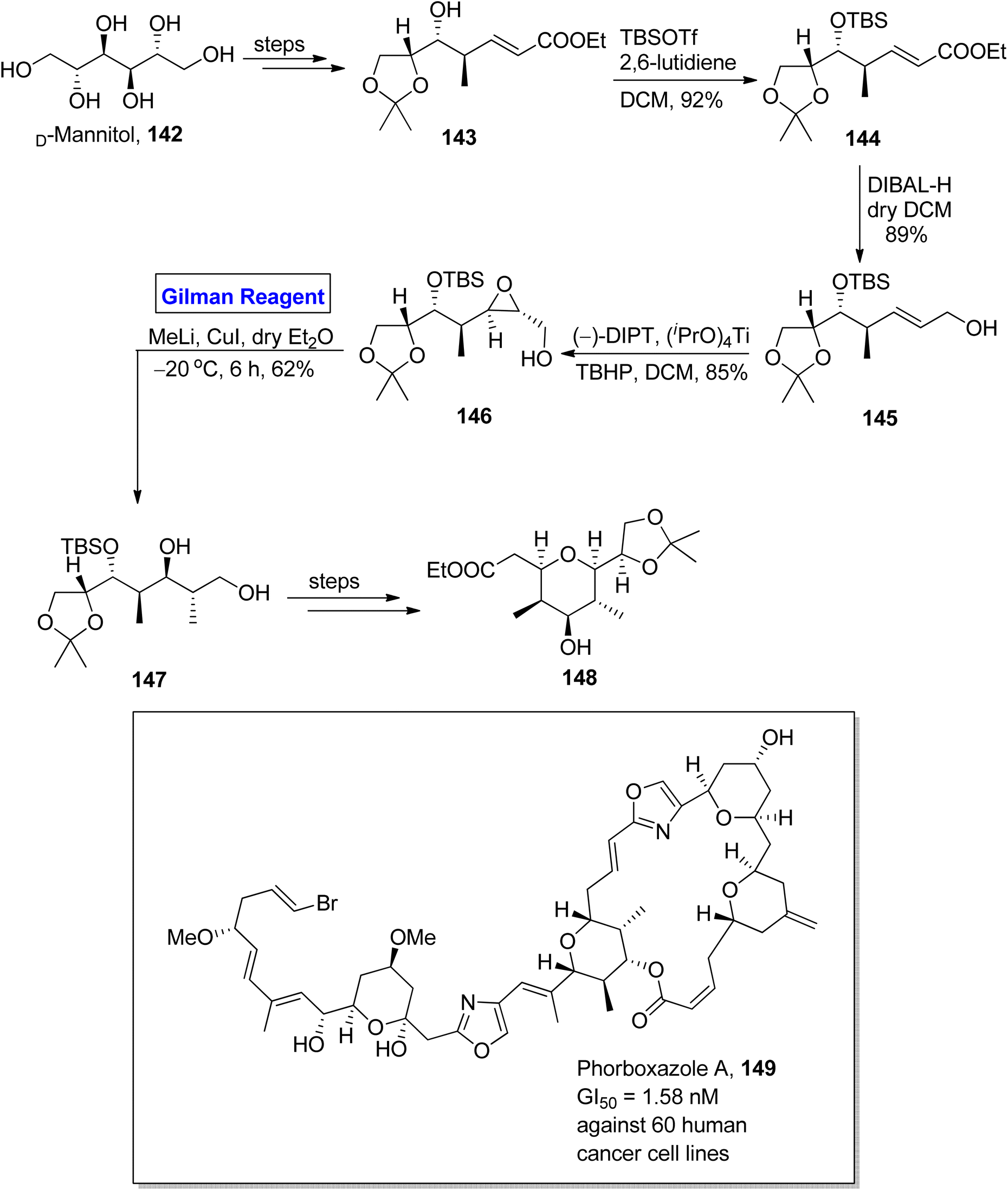
Phorboxazole A 149 is a 21-membered macrolide, which was isolated for the first time by Searl and Molinski from an Indian marine sponge.70 It exhibits cytotoxic activity against 60 human cancer cell lines, i.e., GI50 = 1.58 nM. It also shows anti-cancer activity against Candida albicans and anti-fungal activity against Saccharomyces carlsbergensis.71 Raju et al. in 2015 performed the facile and well-organized synthesis of C20–C27 and C31–C39 fragments of phorboxazole A by using an inexpensive and easily available starting material, i.e., D-mannitol (Scheme 16).72 The synthesis of the C31–C39 fragment 148 entailed aldol condensation, Sharpless asymmetric epoxidation, and Gilman reagent-promoted epoxide ring opening reaction as key steps. The D-mannitol was transformed into compound 143 (in a few steps), which was subjected to silyllation in the presence of TBSOTf and 2,6-lutidine to furnish compound 144 in 92% yield. The reduction of compound 144 by using DIBAL-H in DCM resulted in alcohol 145 in 89% yield. In the next step, the Sharpless asymmetric epoxidation of alcohol 145 by using diisopropyltartrate, titanium isopropoxide, and 2-phenylpropan-2-yl hydroperoxide in DCM yielded compound 146 in 85% yield. In order to conduct a regioselective epoxide ring opening, Gilman reagent was used, and the reacting compound 146 was made to react with methyl lithium and copper iodide in dry diethyl ether by maintaining the temperature up to −20 °C. After 6 hours, compound 147 was achieved with a 62% yield. The compound 147 was modified over a few steps to furnish fragment 148 successfully in quantitative yield.
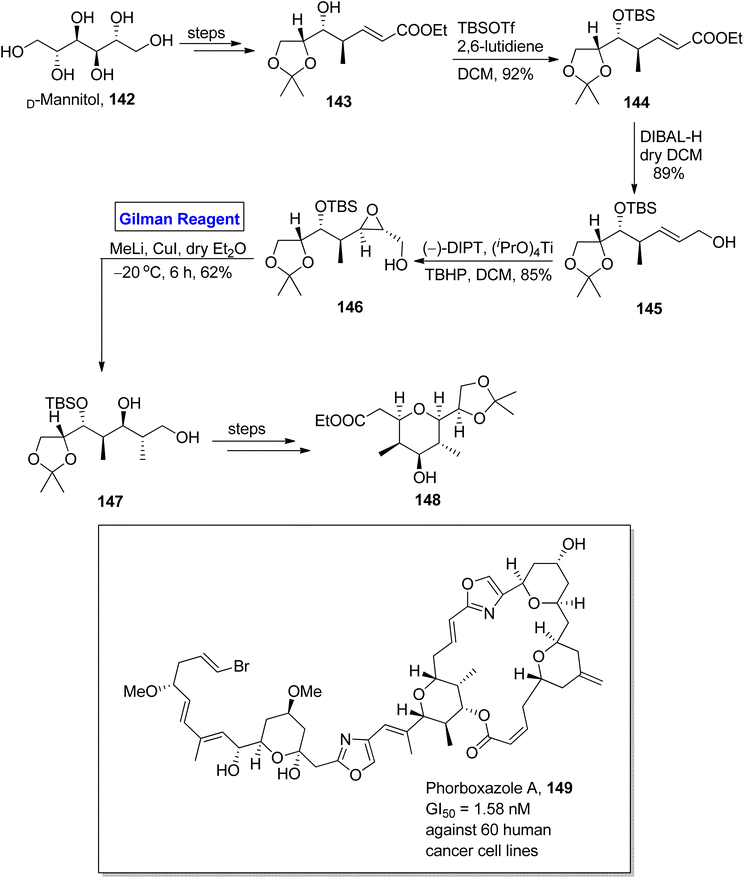 |
| | Scheme 16 Synthesis of the C20–C27 fragment 148 of phorboxazole A. | |
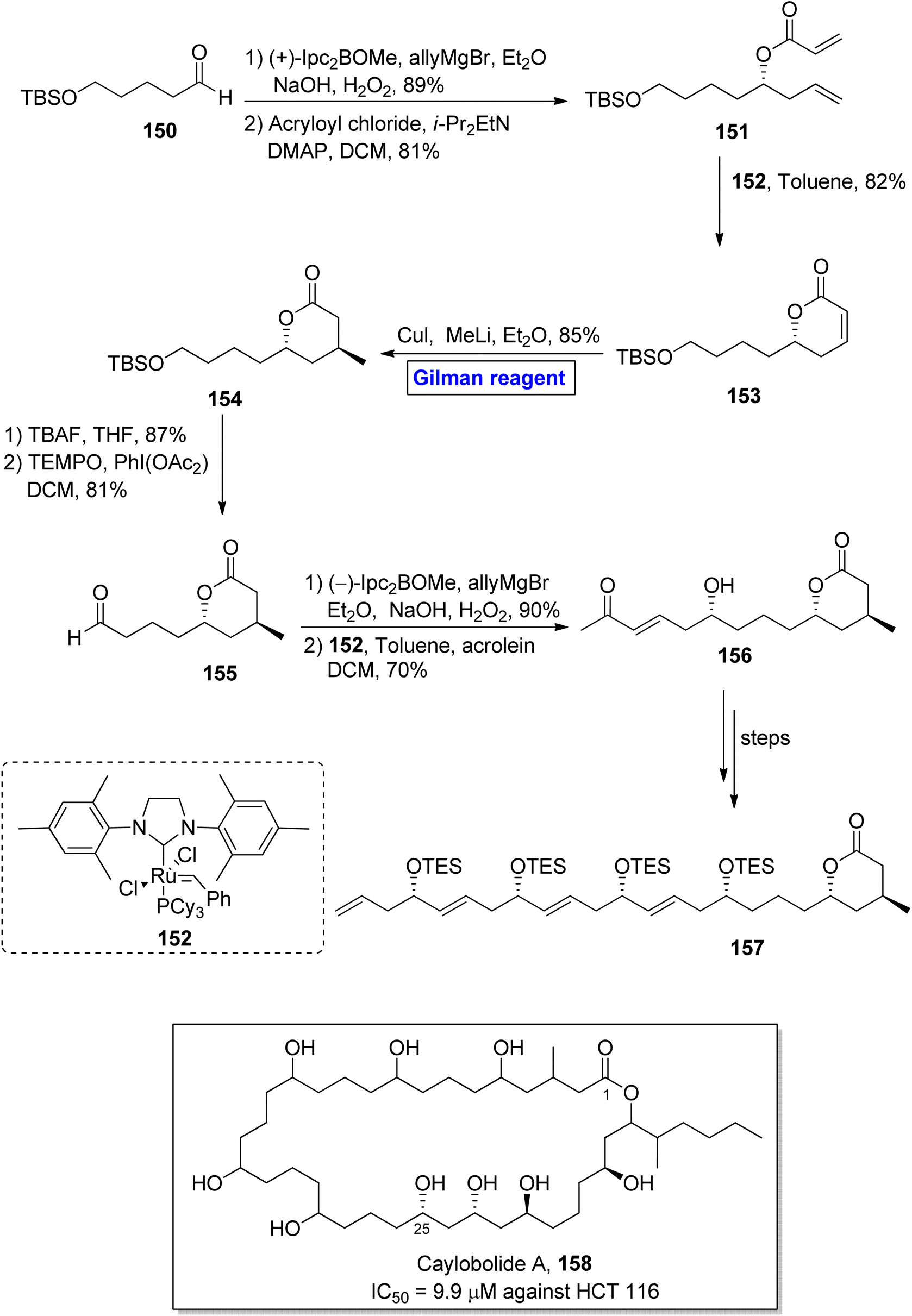
Caylobolide A 158 is a 36-membered, lactone-based macrolide. It was isolated for the first time from Lyngbya majuscule, a marine cayanobacteria, by MacMillan and Molinski.73 Caylobolide A 158 has 256 possible diastereomeric structural configurations due to its eight undetermined stereocenters. It exhibited cytotoxic activities against the human colon cancer cell line (HCT 116) with IC50 = 9.9 μM.74 These features make the synthesis of this fascinating heterocycle a challenging task to achieve. In 2013, Joarder et al. performed the enantioselective synthesis of a 24-carbon-containing intermediate 157 towards the total synthesis of caylobolide A 158 (Scheme 17).75 The key steps in their synthetic path employed Gilman reagent-induced conjugate addition reaction, ruthenium-catalyzed cross metathesis, and boron-catalyzed allylation reactions. In the first step of their synthesis, the compound 150 was treated with (+)-Ipc2BOMe, allylMgBr, and Et2O, followed by the subsequent oxidation under basic conditions, resulting in the intermediate (in 89% yield), which was subjected to esterification with acryloyl chloride, providing compound 151 in 81% yield. Further, the cross metathesis reaction of compound 151 in the presence of catalyst 152 furnished lactenone 153 with 82% yield. In order to perform the regioselective conjugate addition reaction, a well-suited Gilman reagent (CuI and MeLi) was used in the presence of diethyl ether, resulting in lactone 154 in 85% yield. In the following step, desilylation of lactone 154 and subsequent oxidation by using TEMPO and PhI(OAc2) in DCM provided aldehyde 155 in 81% yield. The reaction of aldehyde 155 with (−)-Ipc2BOMe, allylMgBr, and Et2O with a subsequent cross metathesis reaction by using catalyst 152 resulted in aldehyde 156 (70% yield), which after modification over a few steps, furnished the desired intermediate 157, leading towards the synthesis of caylobolide A 158.
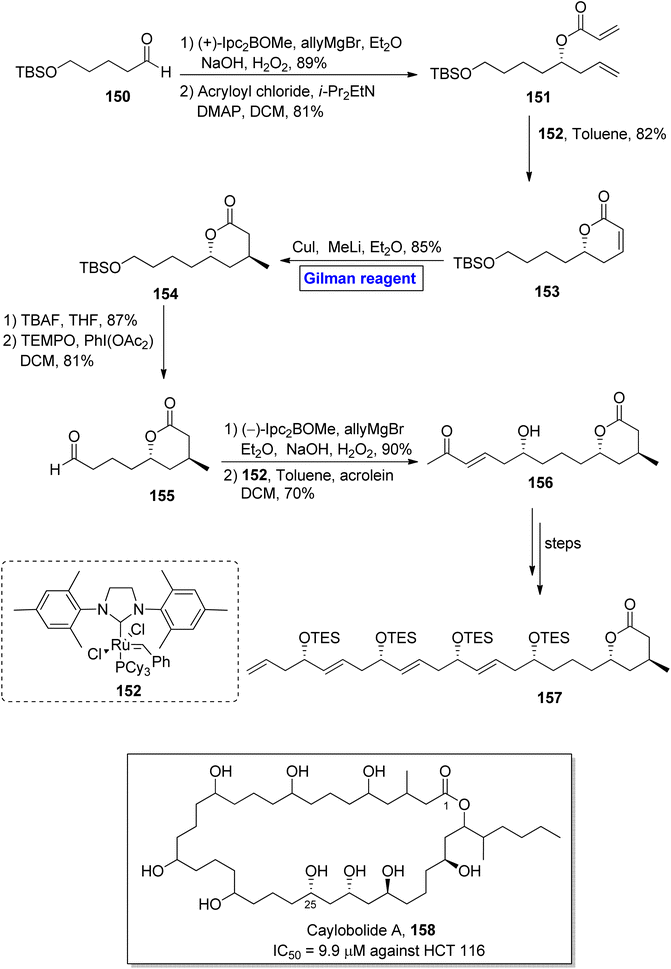 |
| | Scheme 17 Synthesis of intermediate 157 towards the total synthesis of caylobolide A 158. | |
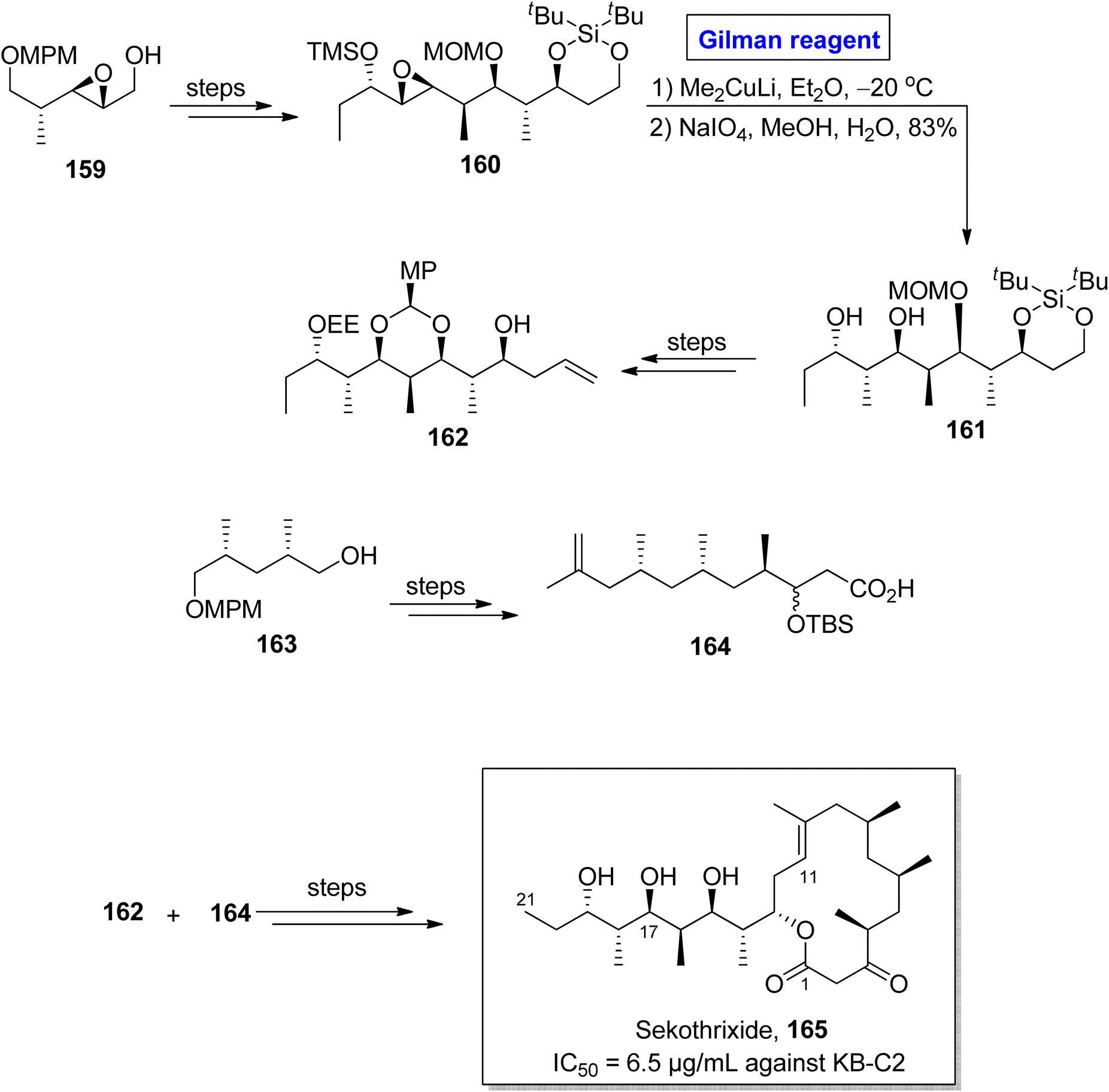
Sekothrixide 165 belongs to the class of macrolide, and it is renowned for its cytocidal activity against KB-C2 cells (that are usually colchicine resistant) with IC50 of 6.5 μg mL−1.76 It was isolated by Seto and colleagues from Saccharothrixide sp. and its structure is based on a fourteen-membered ketolide, having a lengthy side chain, along with seven consecutive chiral centers.77 The interesting pharmacological profile of sekothrixide 165 prompted Terayama et al. to perform its first total synthesis in 2014 by employing Gilman reagent induced-epoxide ring opening reaction (Scheme 18).78 In their methodology, the compound 160 (obtained from the modification of epoxy alcohol 159 over a few steps) was allowed to react with Me2CuLi in Et2O by keeping the temperature at −20 °C, accompanied by treatment with NaIO4 to furnish diol 161 in 83% yield. Next, compound 162 (from compound 161) and compound 164 (from compound 163) were reacted and modified over a series of steps to complete the total synthesis of sekothrixide 165.
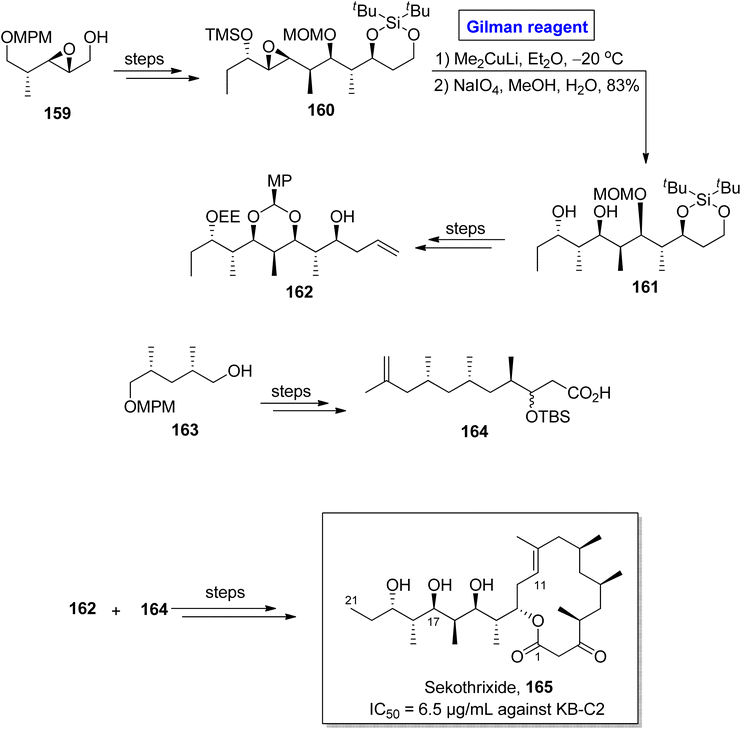 |
| | Scheme 18 Total synthesis of sekothrixide 165. | |
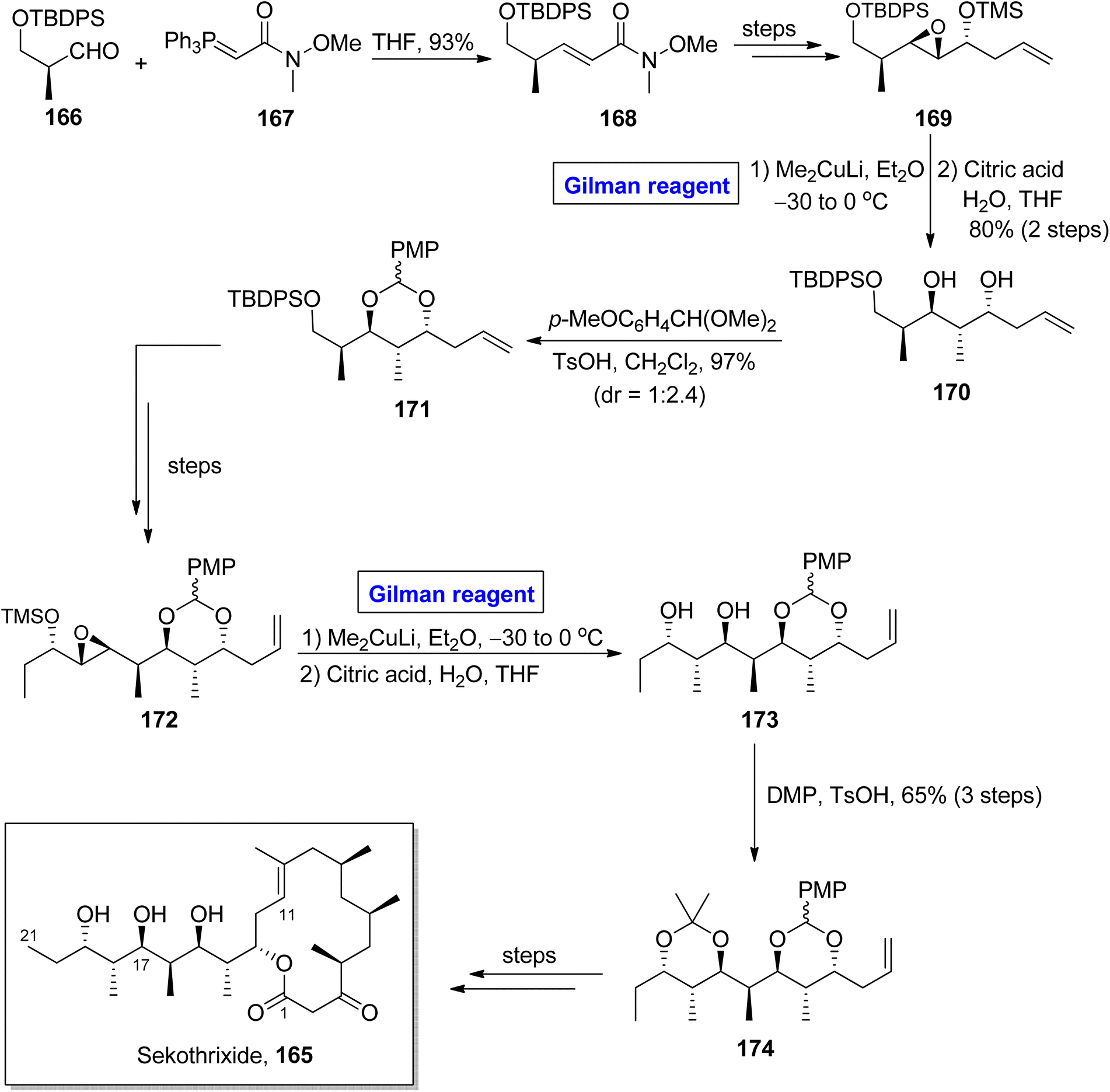
Katsumi et al. performed the total synthesis of sekothrixide two times, as their first synthetic route contained an extensive number of steps and resulted in a low yield of the target molecule. So, they moved towards a more practical and quick method.79,80 In 2019, they performed its total synthesis again, in 26 steps, by using 3-silyloxy-2-methylaldehyde 166 (optically active) as the starting material (Scheme 19).81 The key reaction in their synthetic scheme for the synthesis of the C11 to C21 fragment 174 of sekothrixide, entailed a regioselective ring opening reaction of the TBS-protected epoxy secondary alcohol by employing Gilman reagent. Initially, compound 166 and compound 167 were reacted in the presence of THF to give compound 168 (in 93% yield), which was modified into compound 169 over a few steps. The treatment of epoxide 169 with dimethyl copper lithium in diethyl ether (Gilman reagent) by maintaining the temperature between −30 to 0 °C and the subsequent acid-prompted deprotection resulted in 1,3 diol 170 in 80% yield. After this, benzylidene acetal protection of diol 170 provided compound 171 (in 97% yield and dr = 1:2.4), which over a few steps was transformed into intermediate 172. Further, in order to perform regioselective epoxide ring opening, the Gilman reagent protocol was used, which was assisted by THF deprotection, thus accomplishing the synthesis of diol 173. The synthesis of the C11 to C21 fragment 174 of sekothrixide 165 was completed by the treatment of compound 173 with DMP and TsOH, affording compound 174 in a 65% yield. Over a few steps, the compound 174 was modified into sekothrixide 165.
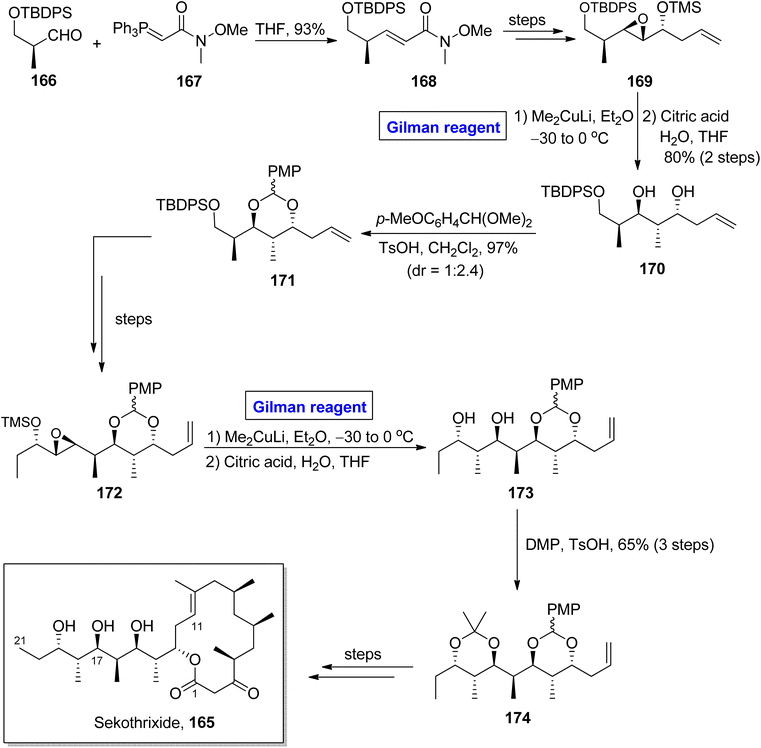 |
| | Scheme 19 Synthesis of the C11 to C21 fragment of sekothrixide 165. | |
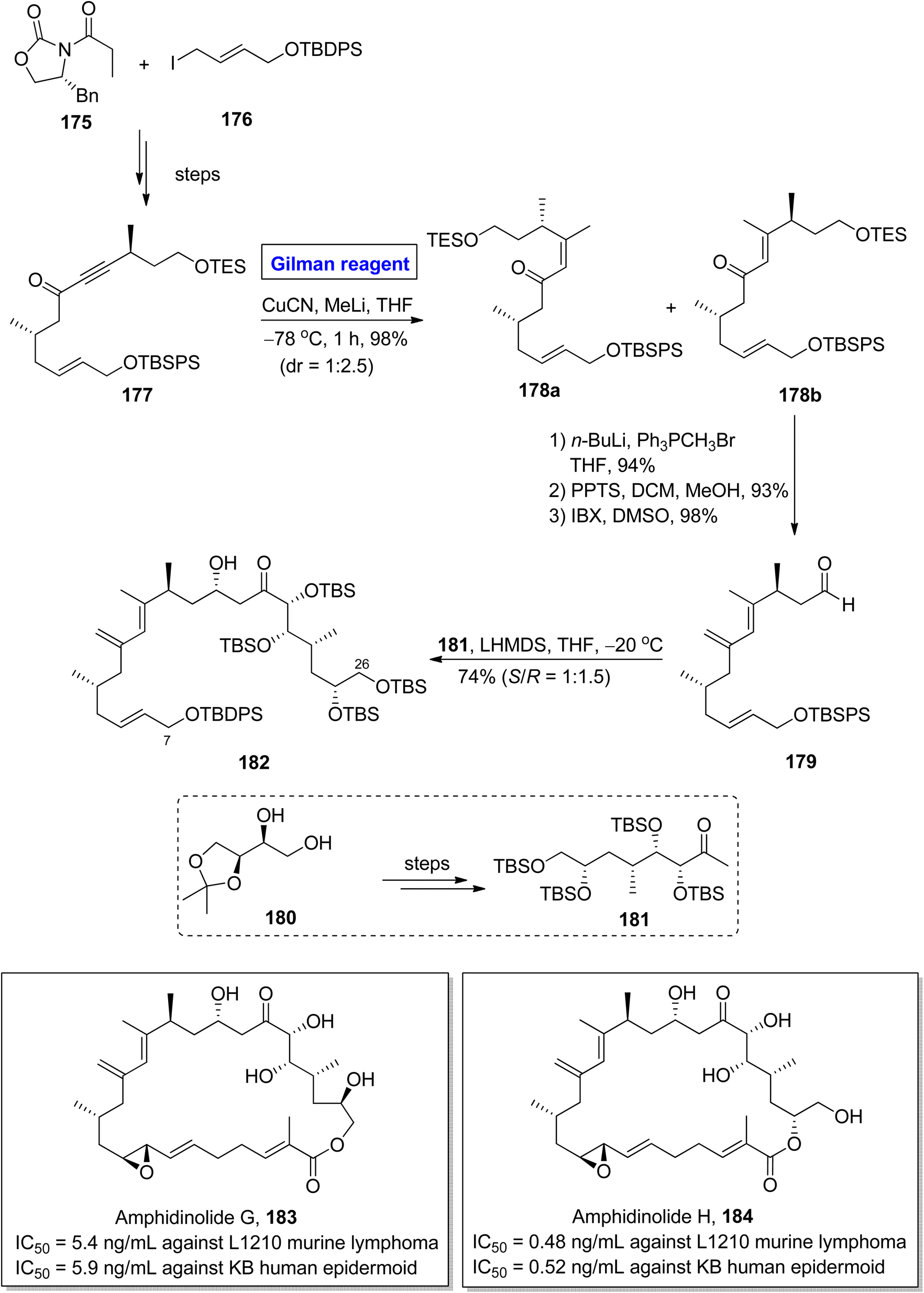
Amphidinolide G 183 and amphidinolide H 184 were isolated from Amphiscolops sp. of Okinawan flatworm.82 The molecular framework of both these compounds constitutes cis diene units, allyl epoxide, 5 hydroxyl groups along with 9 asymmetric centers.83 Amphidinolide G 183 is a potential drug candidate against L1210 murine lymphoma (IC50 = 5.4 ng mL−1) and KB human epidermoid (IC50 = 5.9 ng mL−1). Amphidinolide H 184 also shows cytotoxic activity against both L1210 murine lymphoma (IC50 = 0.48 ng mL−1) and KB human epidermoid (IC50 = 0.52 ng mL−1).84 The fascinating molecular architecture and biological potential of both these compounds made them a challenging synthetic target. In this effort, Hara et al., in 2011 developed a new approach towards the synthesis of the C7–C26 fragment of both amphidinolide G 183 and amphidinolide H 184 (Scheme 20).85 The key steps entailed Sharpless asymmetric dihydroxylation, conjugate addition reaction by Gilman reagent, and aldol coupling reaction. In their methodology, the ketone 177 (obtained from the starting oxazolidinone 175 and allyl iodide 176) was treated with copper cyanide and methyl lithium (Gilman reagent) in THF at −78 °C for 1 hour to give an isomeric mixture of compounds 178a and 178b in 98% yield and dr = 1:2.5. After chromatographic separation, the compound 178b was subjected to Wittig olefination, followed by TES group deprotection and subsequent IBX oxidation affording aldehyde 179 in 98% yield. Lastly, the aldol coupling reaction of aldehyde 179 and ketone 181 (prepared from 180) in the presence of LHMDS at −20 °C furnished fragment 182 in a 74% yield (S/R = 1:1.5).
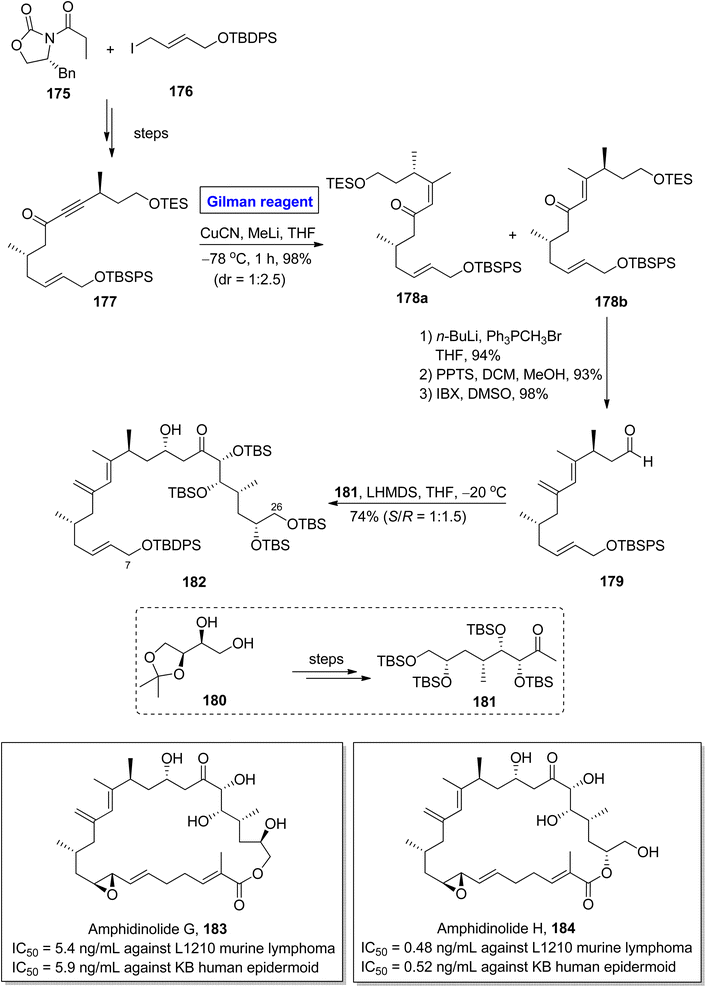 |
| | Scheme 20 Synthesis of the C7–C26 fragment 182 of amphidinolide G 183 and H 184. | |
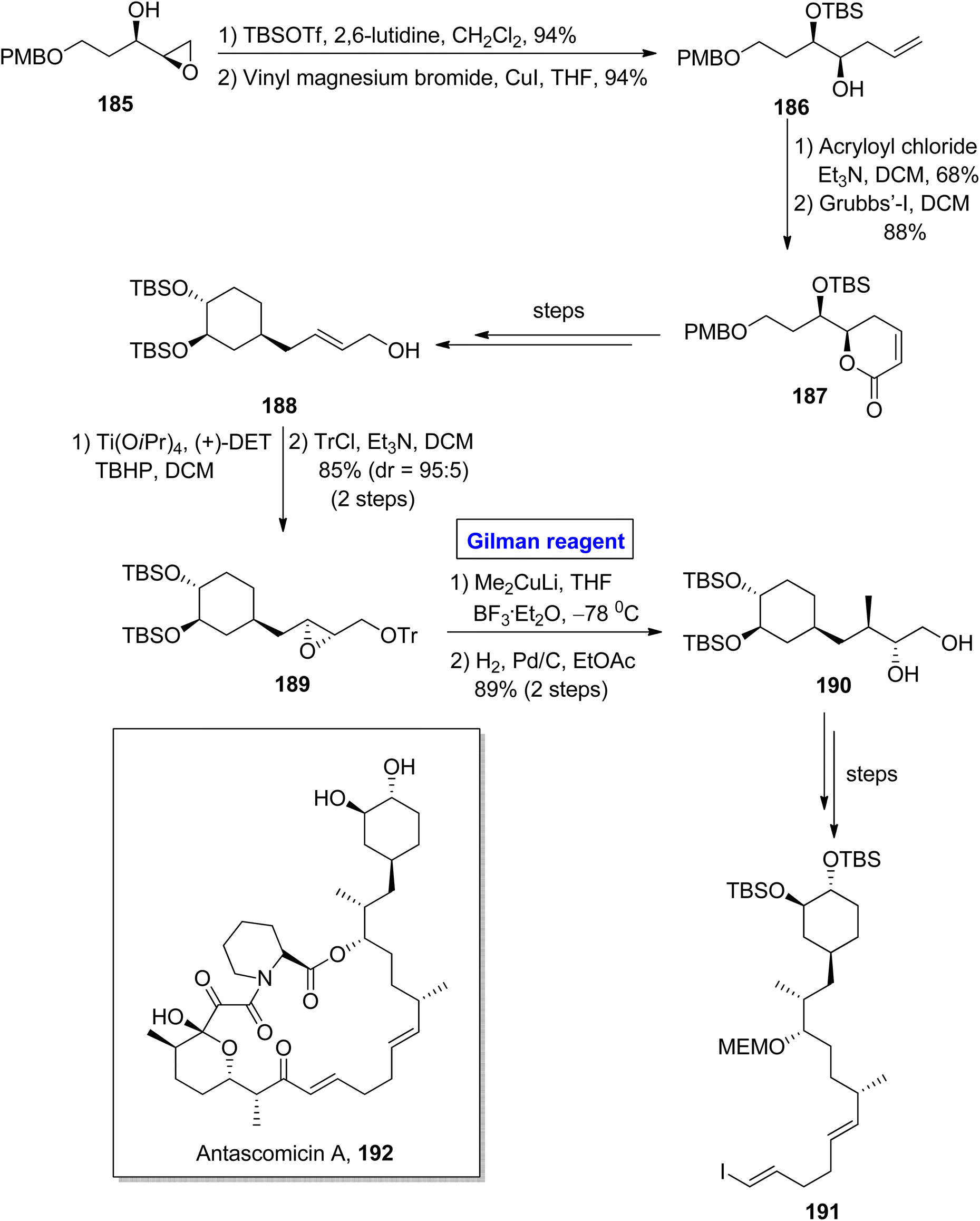
Antascomicins belong to the class of macrolides and show structural resemblance with rapamycin and ascomycin and were isolated firstly from Micromonospora by Fehr and colleagues in 1996.86 This class of natural products can bind with FKBP12 and stimulate neuronal growth. For this reason, antascomicins are expected to be used as potential drug candidates for the treatment of neurodegenerative disorders.87 Vakiti et al. reported the facile synthesis of the C17–C34 fragment 191 of antascomicins A 192 in 2014 (Scheme 21).88 The main steps in their synthetic scheme involved Sharpless asymmetric epoxidation, regioselective epoxide opening reaction via Gilman reagent, and Julia olefination reaction. In the first step, the epoxy alcohol 185 (after TBSOTf protection) was allowed to react with vinyl magnesium bromide and copper iodide in THF to acquire homoallylic alcohol 186 in 94% yield. The treatment of compound 186 with acryloyl chloride and triethyl amine in DCM and further reaction with Grubb's first-generation catalyst in the subsequent step provided ether 187 in 88% yield. The compound 187 (over a few steps) was converted into compound 188, which proceeded further for Sharpless asymmetric epoxidation and subsequent protection of the alcoholic group, resulting in compound 189 in 85% yield (dr = 95:5). For the ring opening reaction of compound 189, well-suited Gilman reagent was used by reaction with lithium dimethyl cuprate in the presence of THF and catalytic amount of BF3· Et2O. The temperature was adjusted at −78 °C, and after completion of the ring-opening reaction, hydrogenation was performed with tosyl group deprotection, which resulted in diol 190 in 89% yield. A few steps later, the diol 190 was converted into the desired fragment 191, which completed the synthetic process.
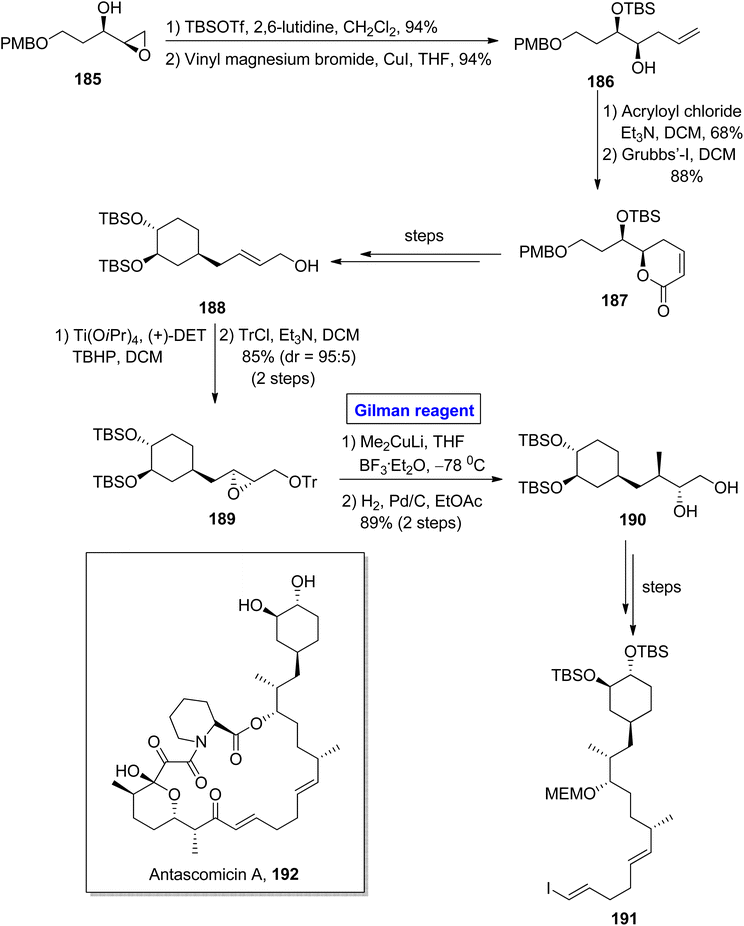 |
| | Scheme 21 Synthesis of the C17–C34 fragment 191 towards the synthesis of antascomicin A 192. | |
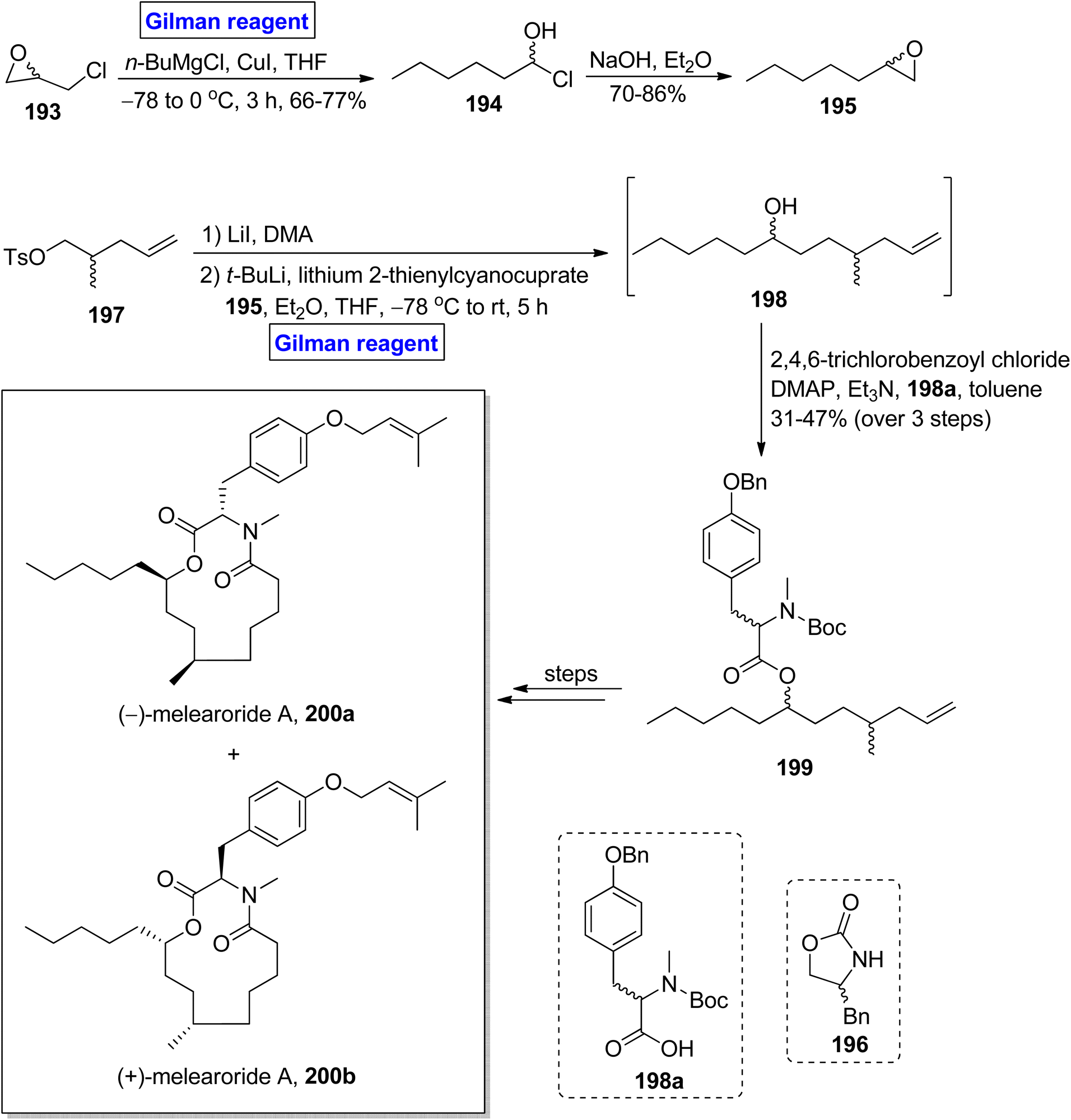
(−)-Melearoride A 200a is a thirteen-membered macrolide, which was first isolated from Penicillum meleagrinum (in 2016) and exhibits anti-fungal activities (particularly against Candida albicans).89,90 Reed et al. in 2019 accomplished the total synthesis of (−)-melearoride A 200a and its non-natural isomer (+)-melearoride A 200b based on a 13-step sequence in 4.3% and 1% overall yield (Scheme 22).91 The key steps entailed Evans alkylation, Gilman reagent-induced epoxide ring-opening reaction, ring-closing metathesis, and Mitsunobu reaction.37 Their synthetic scheme commenced with the in situ generation of Gilman reagent via the treatment of epichlorohydrin 193 with butyl magnesium chloride and copper iodide in tetrahydrofuran to achieve compound 194 (in 66 to 77% yield), which underwent treatment with sodium hydroxide and diethyl ether to provide compound 195 in 70–86% yield. Compound 197 (prepared from compound 196) was subjected to Evans alkylation, accompanied by the reaction with compound 195 in the presence of tertiary butyl lithium, lithium 2-thienylcyanocuprate (Gilman reagent) in a mixture of diethyl ether and tetrahydrofuran at −78 °C to room temperature. It took 5 hours to obtain alcohol 198, which was then proceeded for esterification with acid 198a in the presence of 2,4,6-trichlorobenzoyl chloride, DMAP, Et3N in toluene, which resulted in ester 199 in 31 to 47% yield (in 3 steps). A few steps latter, the ester 199 was utilized for the successful synthesis of (−)-melearoride A 200a and its non-natural isomer 200b.
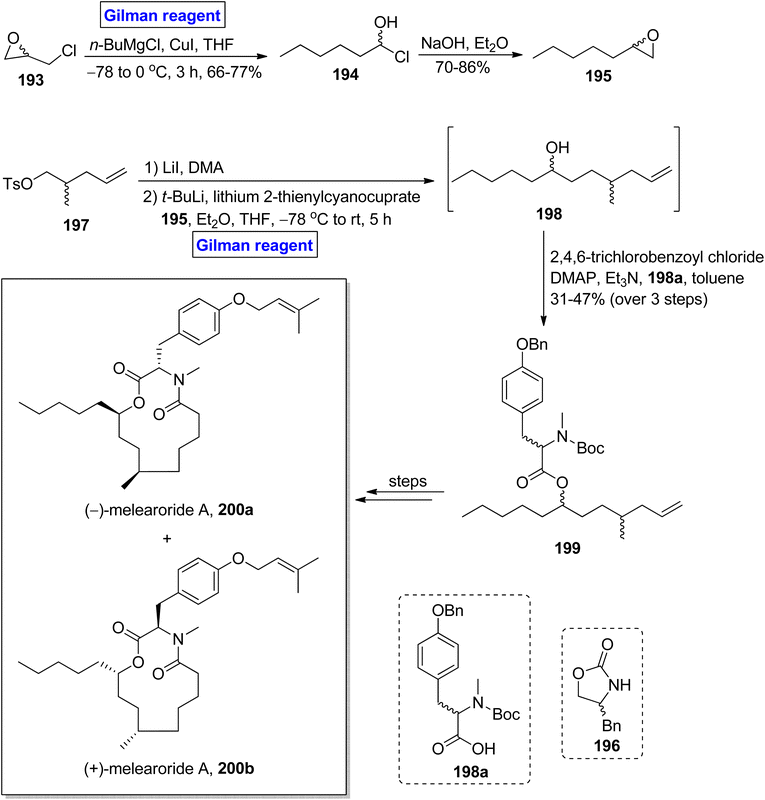 |
| | Scheme 22 Total synthesis of (−)-melearoride A 200a. | |
Synthesis of polyketide-based natural products
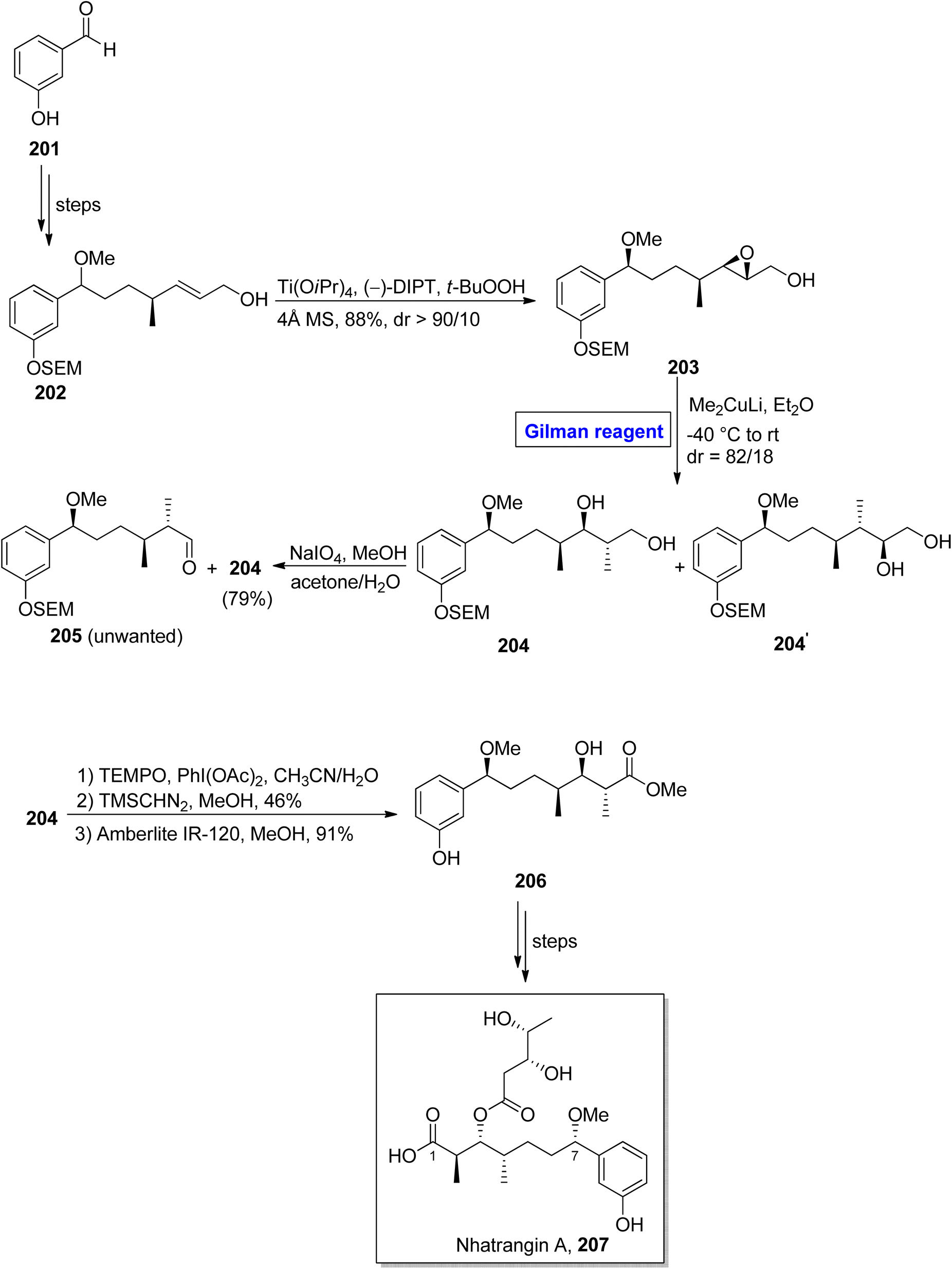
Nhatrangin A 207 belongs to the polyketide class of natural products. In 2010, it was isolated for the first time from a marine cyanobacterium, i.e., Lyngbya majuscula, found in Vietnam.92 The purification of nhatrangin A 207 after its isolation from its natural source did not result in productive yield. Aplysiatoxin is also a natural compound, well-known for its potassium channel inhibition and anti-viral and anti-cancer activities.93 Nhatrangin A 207 and aplysiatoxin have the same biosynthetic origin and have structural similarities with each other. Owing to these facts, nhatrangin A 1 is supposed to be a potential drug candidate and its total synthesis has been previously reported by Kamal, Yadav, and Dias research groups.94 In continuation of this work, Feuillastre et al. reported the enantioselective synthesis of the C1–C7 fragment 206 of nhatrangin A 207 in 2020 (Scheme 23).95 It was a 14-step sequenced methodology, starting from the easily available achiral 3-hydroxybenzaldehyde, and the overall yield was 13%. The main steps in their scheme were Sharpless asymmetric epoxidation and Gilman reagent-induced regioselective epoxide ring opening reaction. In their synthetic path, the 3-hydroxybenzaldehyde 201 was converted into allylic alcohol 202 in a number of steps. The allylic alcohol 202 was subjected to Sharpless asymmetric epoxidation under given conditions, which resulted in compound 203 in 88% yield (dr > 90/10). In order to perform the epoxide ring-opening reaction, the compound 203 was allowed to react with lithium dimethyl cuprate in diethyl ether by maintaining the temperature between −40 °C to room temperature, which furnished a diastereomeric mixture of diol 204 (with dr = 82/18). Subsequently, the treatment of this mixture with NaIO4 in a suitable solvent afforded diol 204 (with 79% yield), which was subjected to selective oxidation and suitable deprotection under given conditions to complete the synthesis of fragment 206 in 91% yield.
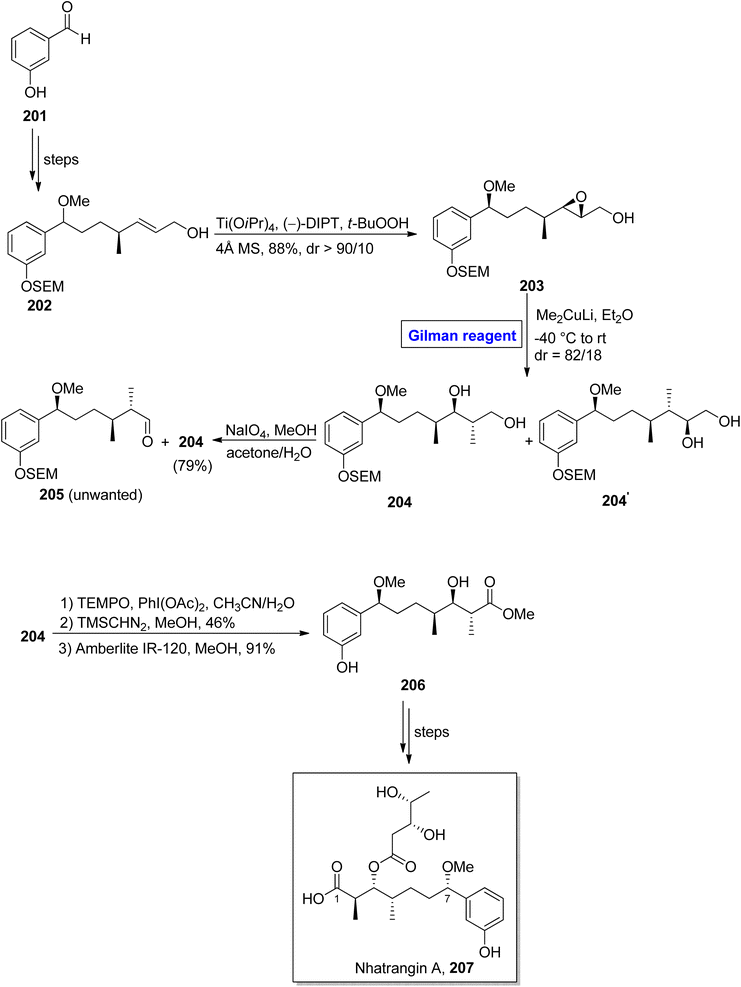 |
| | Scheme 23 Synthesis of C1–C7 fragment 9 of nhatrangin A 207. | |
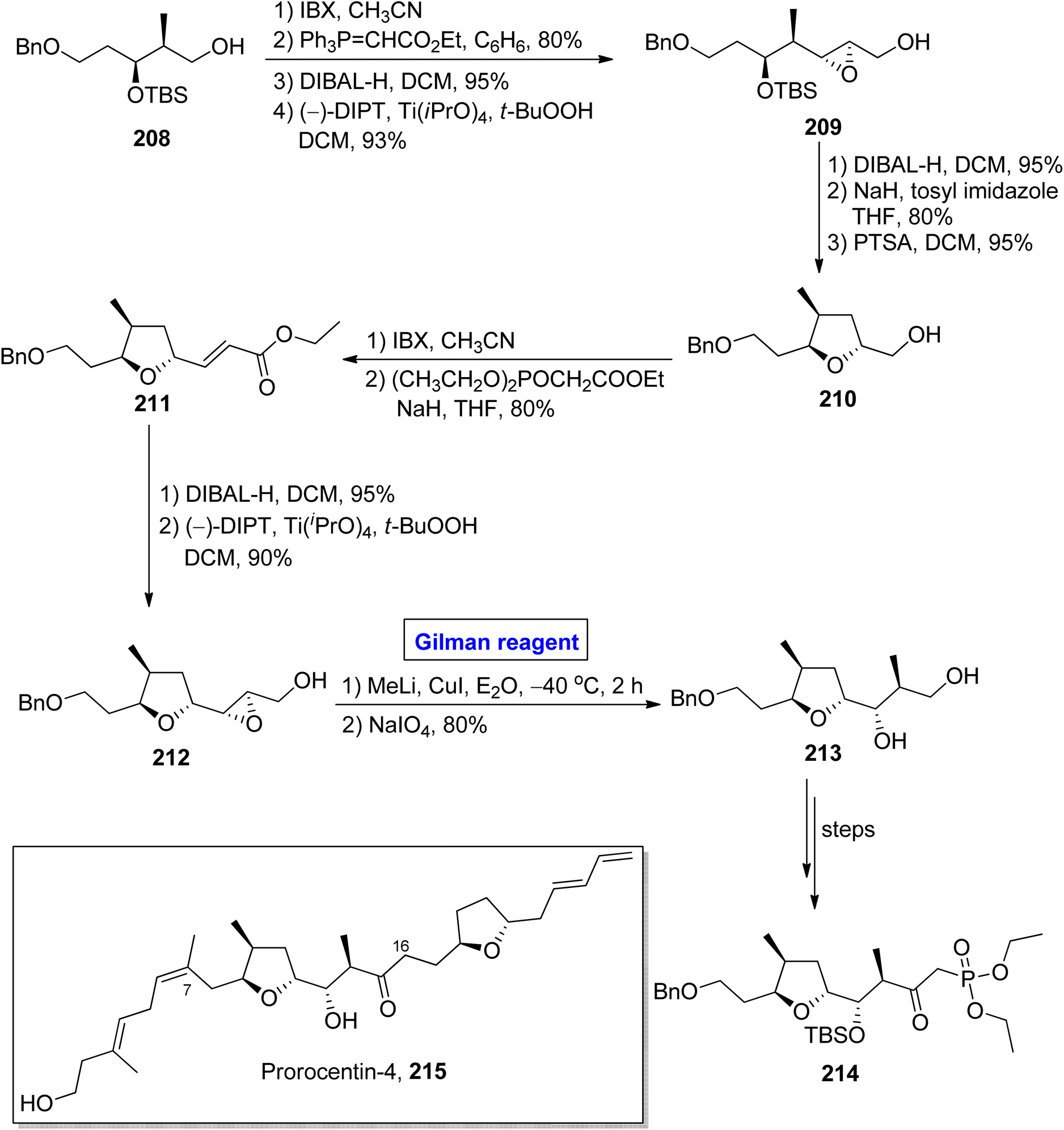
One of the linear polyketides, prorocentin-4 215, was isolated from Prorocentrum sp. of dinoflagellates, which are found in tropical oceans all over the world.96 It is a potential cytotoxic agent and is present in okadic acid-synthesizing organisms. Its structure contains two tetrahydrofuran rings and seven asymmetric centers.97 By focusing on these features, AnkiReddy et al. disclosed the synthesis of the C1–C23 fragment of prorocentin-4 215 in 2018 (Scheme 24).98 The synthetic path that they adopted is based on the synthesis of three main fragments (C1–C6, C7–C16, and C17–C23). However, the synthesis of the middle fragment C7–C16 214 entailed the employment of the Wittig reaction, Sharpless asymmetric epoxidation, and epoxide ring opening reaction by Gilman reagent as key steps. Their synthesis of this fragment 214 commenced with the easily available starting alcohol 208 that was oxidized by using IBX and then subjected to Wittig reaction to result in the intermediate in 80% yield. The DIBAL-H induced reduction of the intermediate and Sharpless asymmetric epoxidation in sequence, providing the epoxy alcohol 209 in 93% yield. The synthesis of the tetrahydrofuran ring in compound 210 (in 95% yield) was achieved by DIBAL-H-induced reduction of compound 209 followed by the treatment with tosyl imidazole and NaH in THF and subsequent reaction with PTSA in DCM. In the next step, IBX oxidation of compound 210 and subsequent HWE reaction provided ester 211 in 80% yield. Again, DIBAL-H induced reduction and Sharpless asymmetric epoxidation in sequence were employed for ester 211 to furnish diol 212 in 90% yield. Further, in order to carry out the epoxide ring-opening reaction, a well-suited Gilman reagent was employed. Thus, the compound 211 was treated with methyl lithium and copper iodide in diethyl ether, followed by the addition of NaIO4 to get ether 213 (in 80% yield), which was then converted into the desired fragment 214 over a few steps.
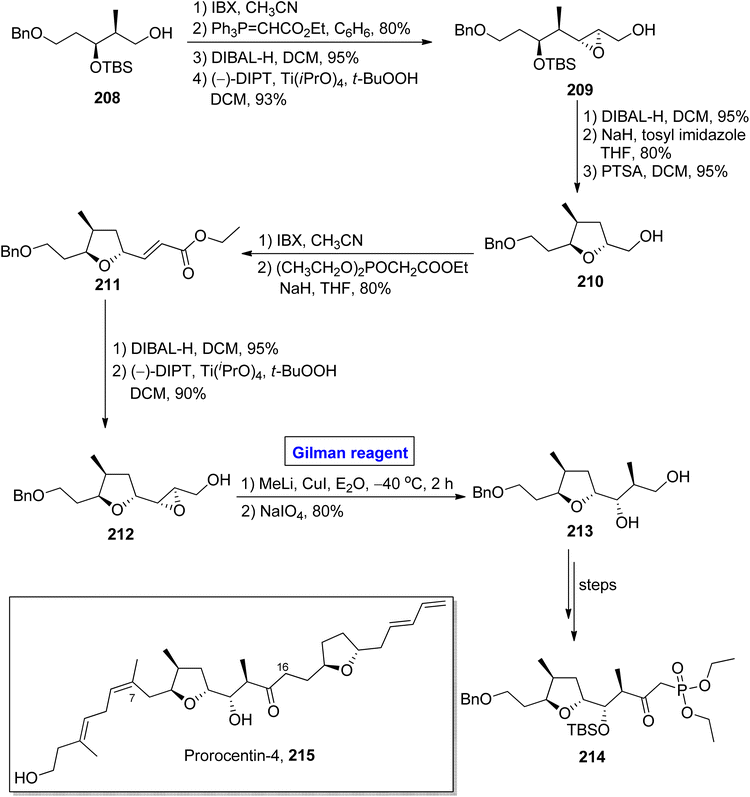 |
| | Scheme 24 Synthesis of the C7–C16 fragment of prorocentin-4 215. | |
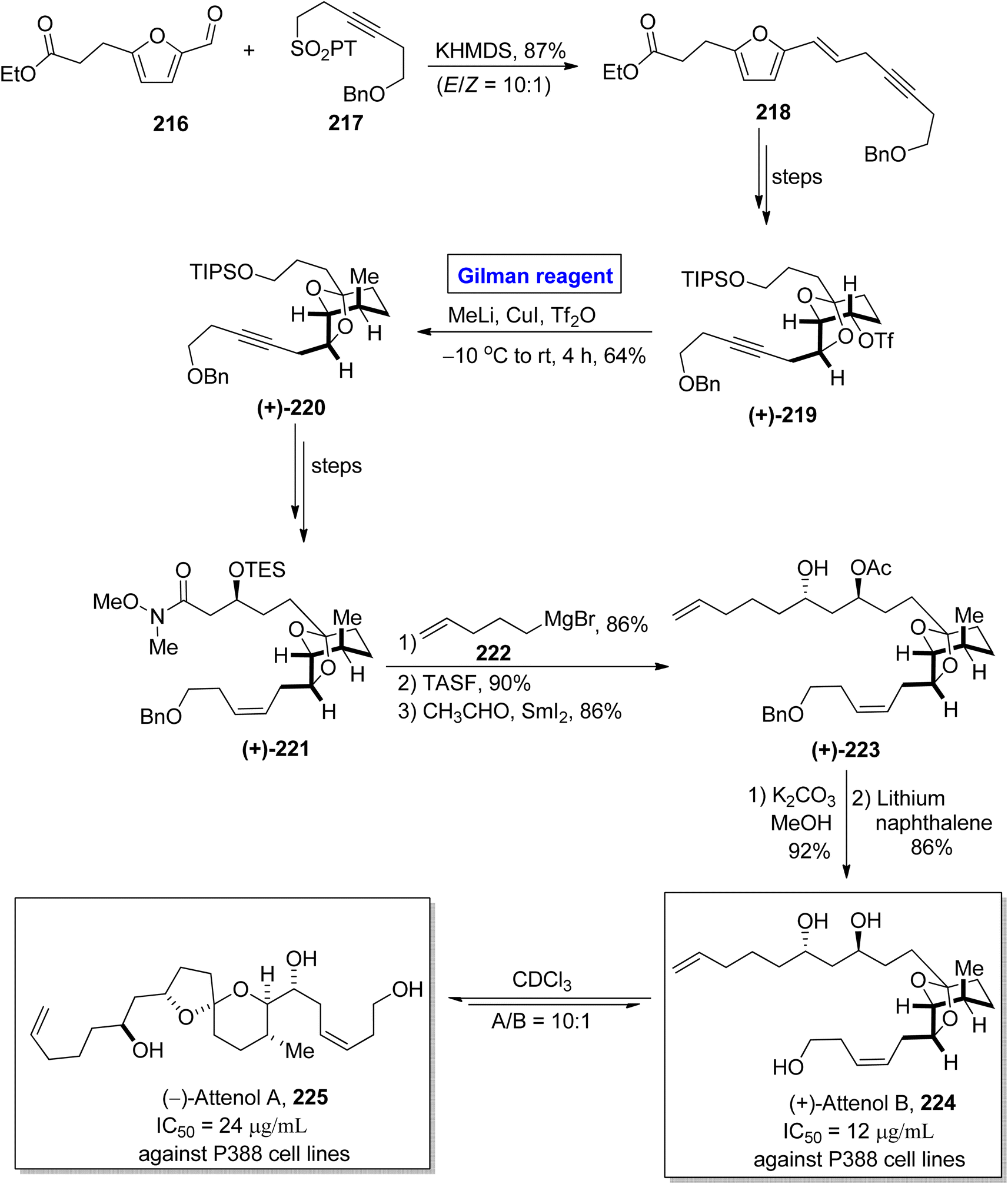
The synthesis of scarce natural products has engaged the attention of many organic chemists. One of the Chinese natural products, i.e., attenol A and attenol B were isolated from Pinna attenuate by Uemura and colleagues.99 These are novel ethereal compounds having rare natural existence. Attenol A is made up of a [5,6]-spiroketal ring embellished with three OH groups on two unsaturated flanking chains. (+)-Attenol B is a minor isomer of attenol A and exists under acidic conditions. These two polyketides are cytotoxic. The IC50 values of (−)-attenol A 225 and (+)-attenol B 224 against P388 cell lines are 24 μg mL−1 and 12 μg mL−1 respectively.100 Owing to the distinctive structural features with a great pharmacological profile, many synthetic approaches toward the synthesis of these two compounds have been reported in the literature. In continuation of this effort, Ren et al. in 2015 disclosed a novel synthetic route towards the synthesis of (+)-attenol B 224 and its isomer 225 as well by employing Gilman reagent (Scheme 25).101 Their synthesis commenced with the Julia–Kocienski reaction of aldehyde 216 and sulfone 217 in the presence of KHMDS to afford eyne 218 in 87% yield (with E/Z = 10:1). After a few steps, triflate 219 (obtained from compound 218) was subjected to a bimolecular nucleophilic substitution reaction (SN2) by reacting with methyl lithium and copper iodide (Gilman reagent) in the presence of Tf2O. The temperature was maintained between −10 °C to room temperature. After 4 hours, the alkyne 220 was obtained with a 64% yield. The compound 220 was converted into compound 221 over a few steps. The treatment of amide 221 with Grignard reagent, accompanied by the desilylation and Evans Tishchenko reaction in sequence, provided the diol derivative 223 in 86% yield. In the following two steps, the deacetylation (in the presence of potassium carbonate and methanol) and debenzylation (in the presence of lithium and naphthalene) furnished the desired (+)-attenol B 224 in 86% yield. The synthesis of (−)-attenol A 225 was achieved in 91% yield (A/B = 10:1) by isomerizing (+)-attenol B 224 in the presence of CDCl3.
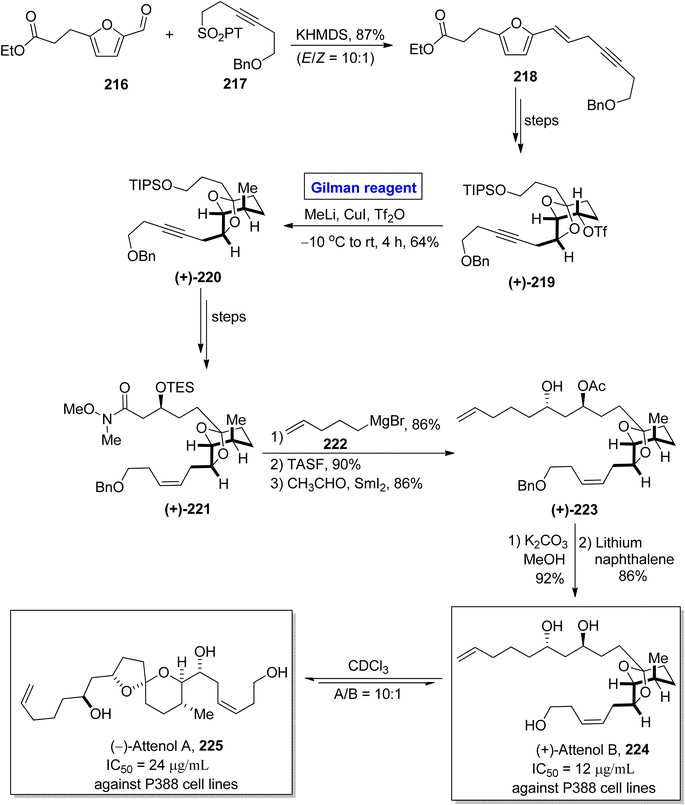 |
| | Scheme 25 Total synthesis of (+)-attenol B 224 and (−)-attenol A 225. | |
Synthesis of amino acid-based natural products
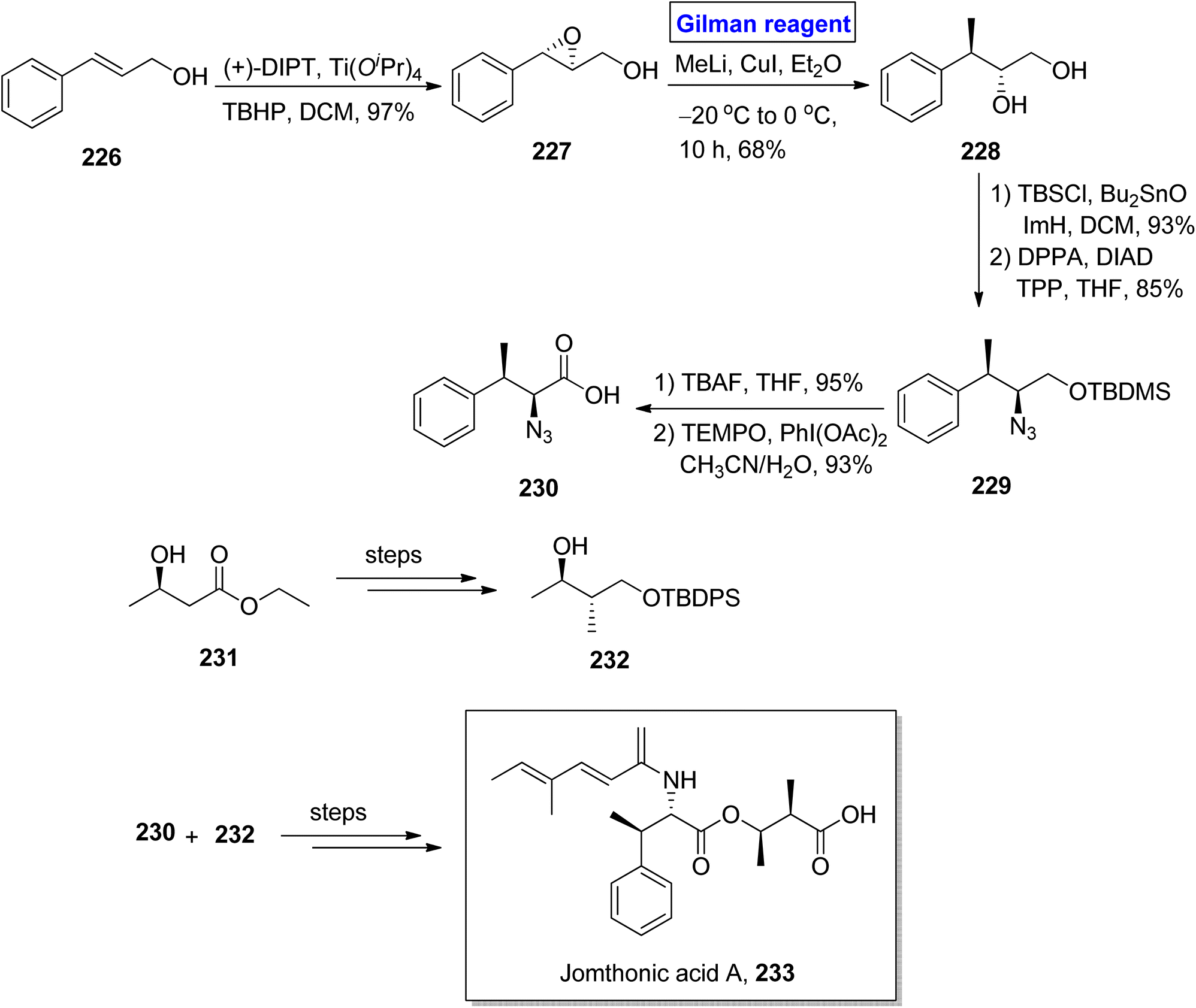
Jomthonic acid A 233 belongs to the class of amino acids isolated from Streptomyces sp. of soil antinomycete.102 Its sophisticated structure is based on fragment 230 and fragment 232, having 4 stereogenic centers. It shows antiatherogenic and antidiabetic activities against St-13 mice and exhibits preadipocyte differentiation inhibition with IC50 = 2–50 μM. Their fascinating structure and medicinally important pharmaceutical profile make them a valuable synthetic target.103,104 Dumpala et al. in 2020 presented a concise and efficient synthetic route towards the first total synthesis of this heterocyclic scaffold 233, with 8.0% overall yield (Scheme 26).105 The main steps entailed Sharpless epoxidation, regioselective epoxide ring opening by Gilman reagent, Mitsunobu reaction,37 Yamaguchi esterification, and amide coupling reaction. To accomplish this task, the easily available cinnamyl alcohol 226 was converted into epoxide 227 via Sharpless asymmetric epoxidation under given conditions with 97% yield. To perform the epoxide ring opening, the well-suited Gilman reagent was applied by reacting epoxide 227 with methyl lithium and copper iodide in diethyl ether as solvent at −20 °C to 0 °C in 10 hours, which resulted in diol 228, with 68% yield. The compound 228, after silyl protection, was subjected to Mitsunobu conditions to afford compound 229 with 85% yield. After this, silyl deprotection of silyl ether 229 and subsequent oxidation in the presence of PhI(OAc)2 and TEMPO provided compound 230 with 93% yield. Later on, fragment 230 and fragment 232 (obtained from compound 231) were reacted together and (after some modification) in a few steps, which completed the total synthesis by providing jomthonic acid A 233.
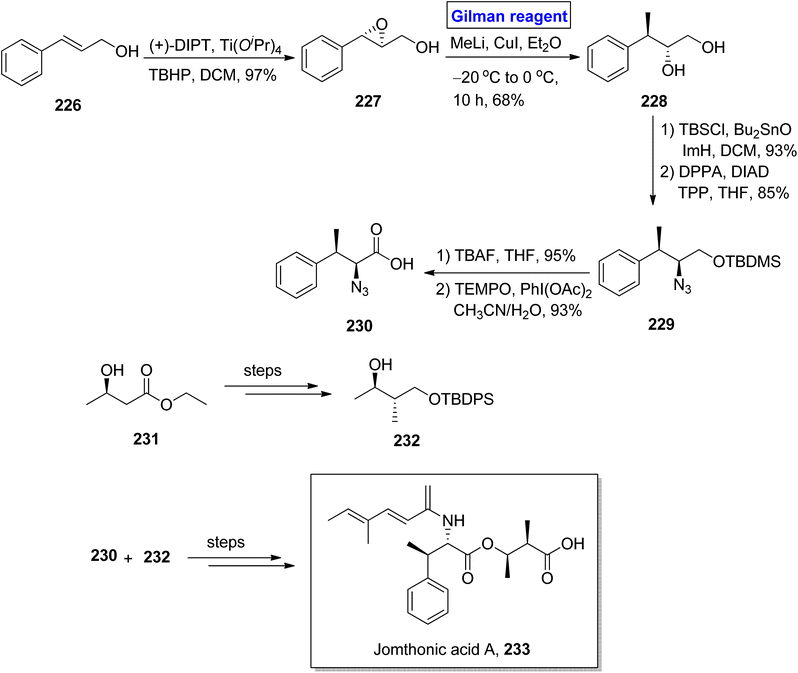 |
| | Scheme 26 Synthesis of jomthonic acid A 233. | |
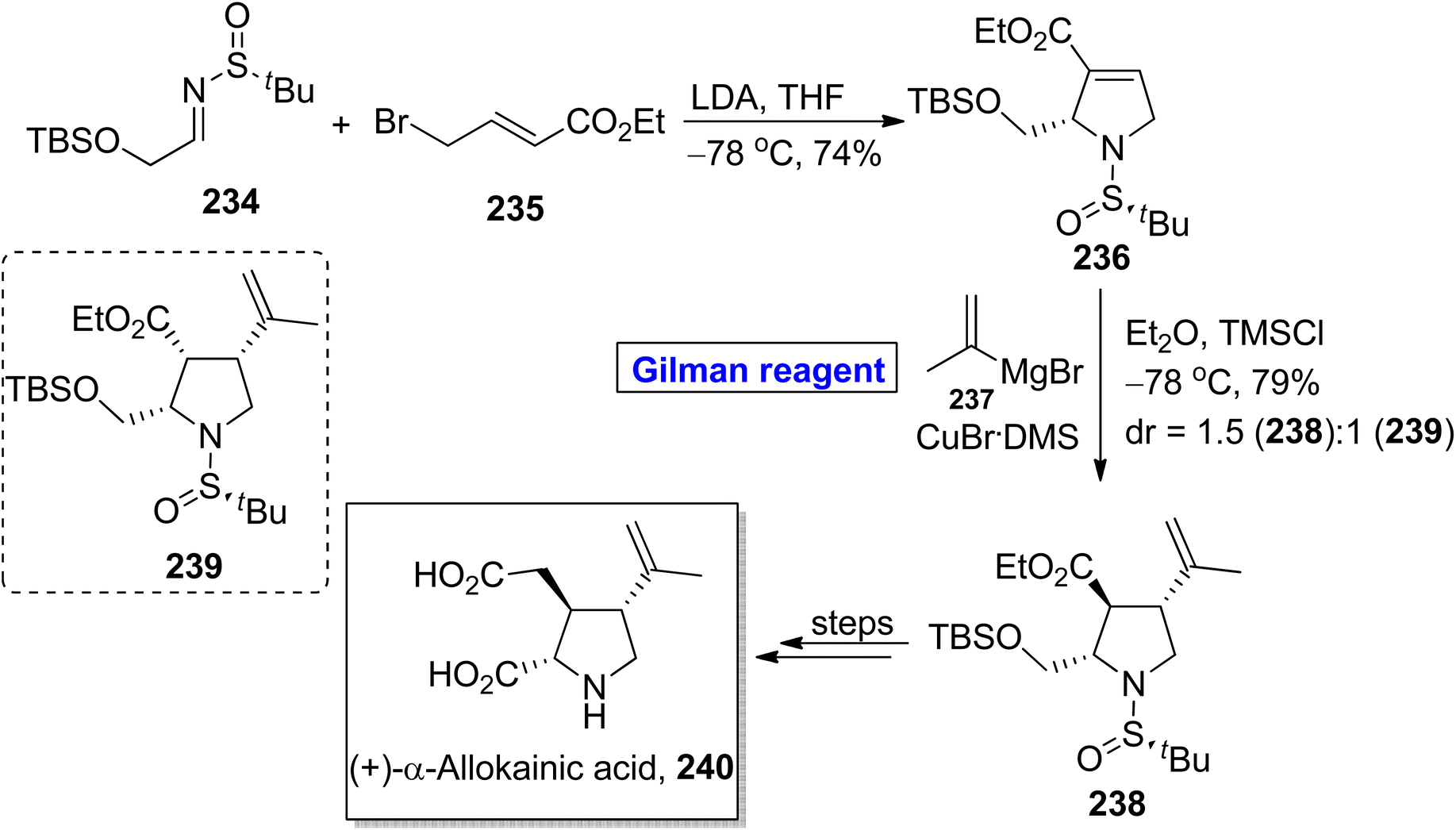
Kainic acid is a kainoid based natural product (having 3 stereocenters) with a propenyl group attached at the C4 position. This remarkable naturally occurring amino acid is a glutamate receptor neurotransmitter, therefore, it is an intriguing synthetic target.106,107 In 2019, Chogii et al. proposed an efficient and simple synthetic route towards the synthesis of (+)-α-allokainic acid 240 (Scheme 27).108 In their synthetic scheme, the imine 234 and compound 235 were made to react in the presence of LDA and THF to provide pyrrolidine 236 (in 74% yield), which underwent further reaction with Gilman reagent via treatment with compound 237 in the presence of diethyl ether and TMSCl at −78 °C to furnish a mixture of compound 238 (in 79% yield) and compound 239 (dr = 1.5:1). The compound 238 was modified in a few steps to complete the synthesis of (+)-α-allokainic acid 240.
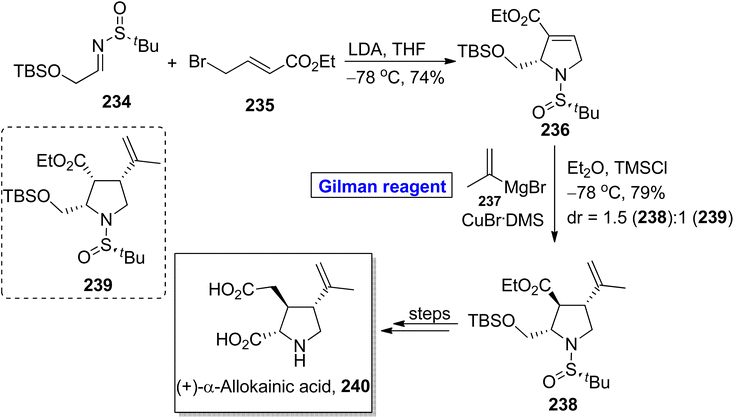 |
| | Scheme 27 Synthetic approach towards (+)-α-allokainic acid 240. | |
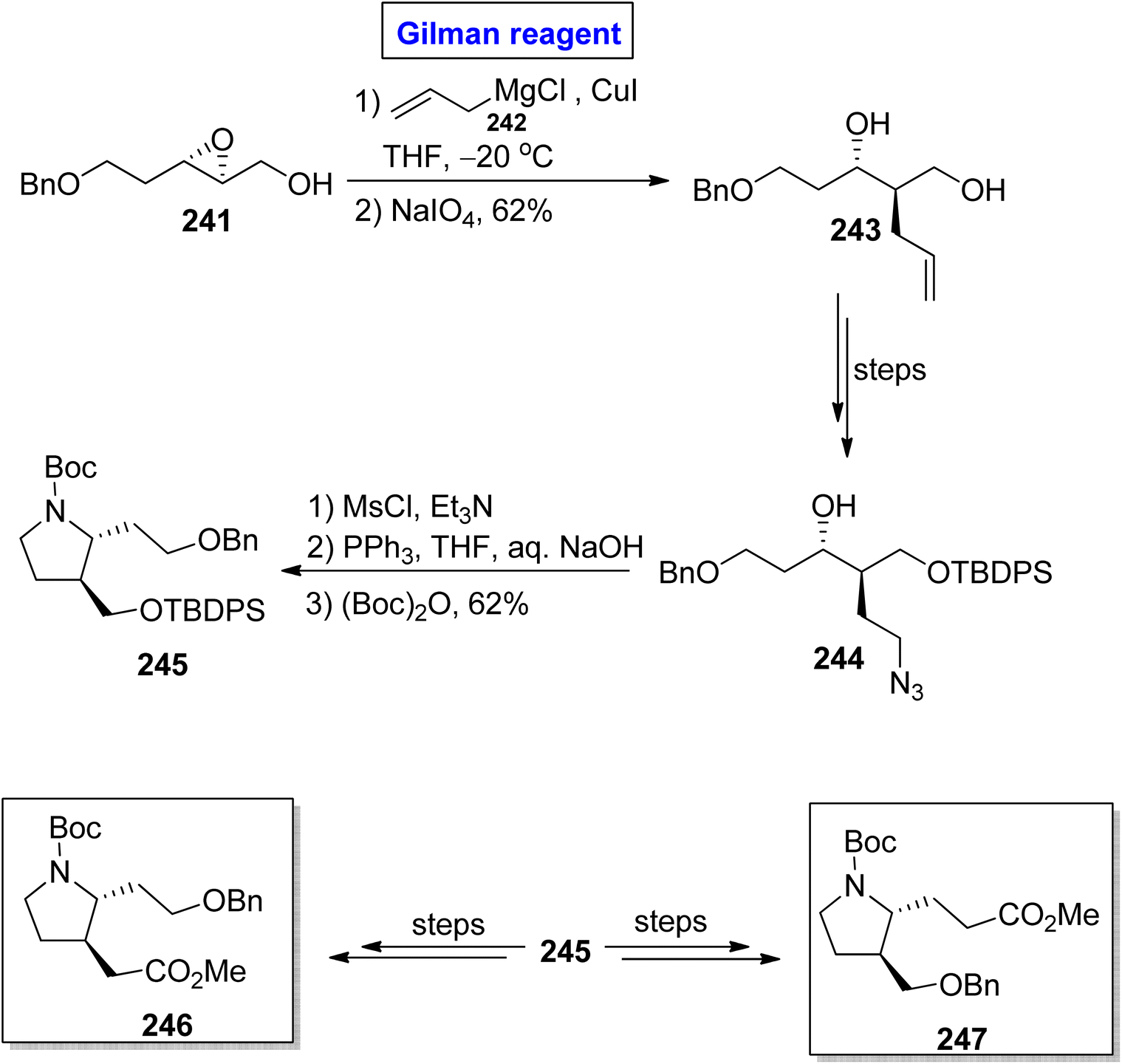
β-Proline and β-homoproline have the ability to form interactions with collagen and are expected to exhibit anti-thrombotic activity.109,110 This concept prompted Basu et al. in 2014 to synthesize substituted β2,3-proline 246 and β3-homoproline 247 monomers as well as their dimers via the Gilman reagent synthetic protocol and investigating their conformational studies (Scheme 28).111 In the first step, the epoxide 241 was subjected to a ring-opening reaction via treatment with allyl magnesium chloride 242 and copper iodide in THF (for in situ generation of Gilman reagent). In order to avoid the unwanted 1,2-diol, NaIO4 was added, which resulted in diol 243 (in 62% yield) and was then transformed into alcohol 244 over a few steps. The mesylation of alcohol 244, subsequent reduction (with PPh3 in THF), and Boc group protection provided the precursor 245 in 62% yield. Later on, compound 245 was utilized for the synthesis of compound 246 and compound 247 over a few steps.
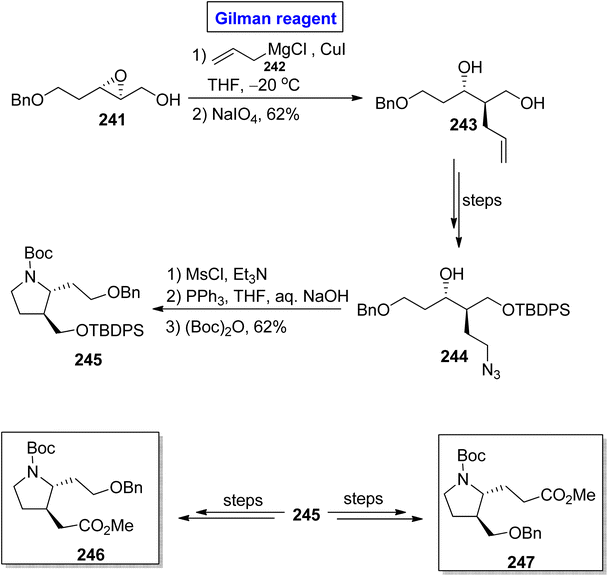 |
| | Scheme 28 Synthesis of substituted β2,3-proline 246 and β3-homoproline 247. | |
Synthesis of peptide-based natural products
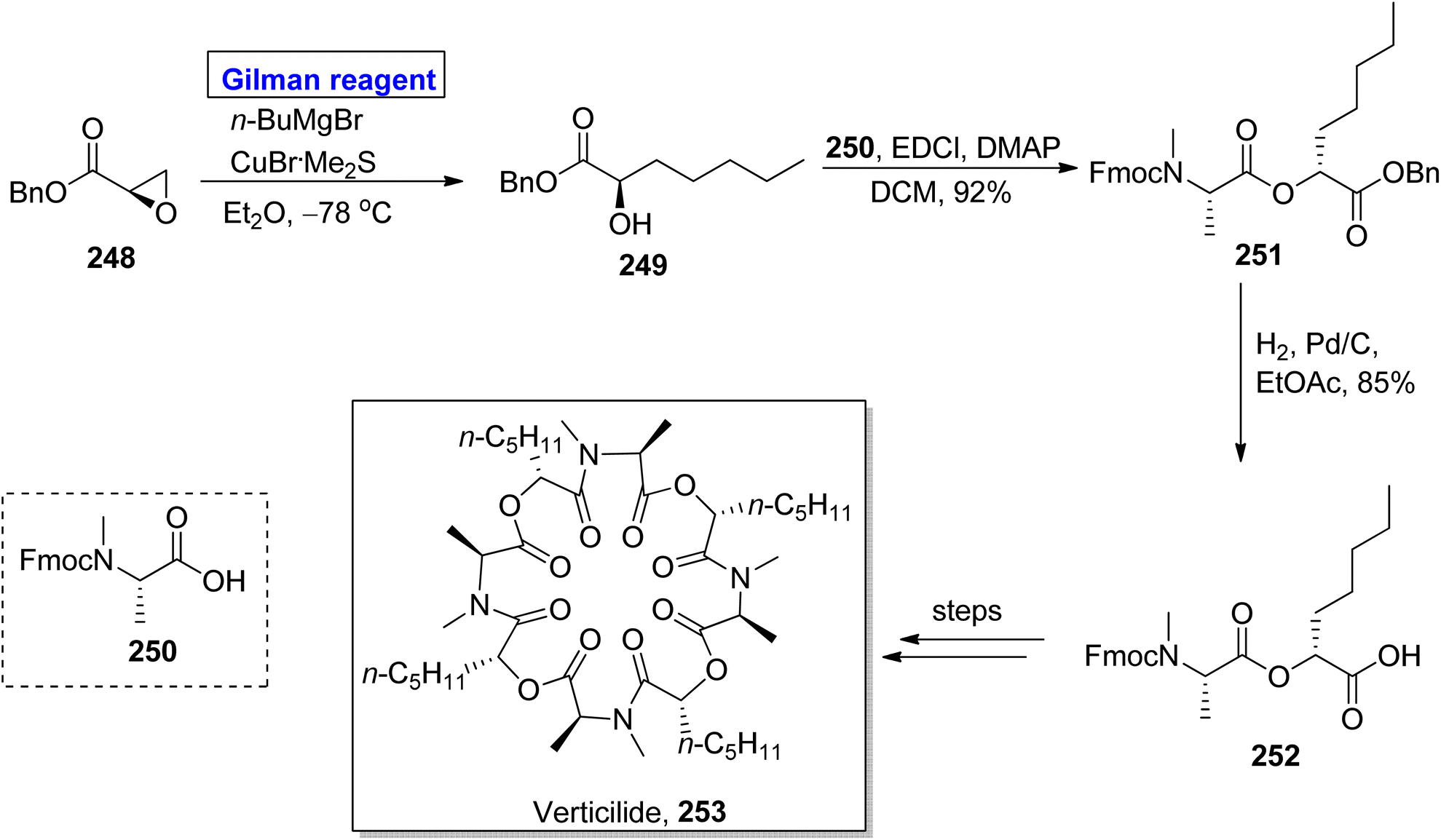
Verticilide 253, a cyclic depsipeptide, was isolated from Acremonium variecolor (a fungus).112 It is a novel insecticide and a selective inhibitor of ryanodine binding and is structurally based on a 24-membered macromolecular ring consisting of two types of repeating units, i.e., N-methyl alanine and 2-hydroxyheptanoic acid.113 The first total synthesis of verticilide 253 was reported by Watanabe et al. in 2020. The same group presented its total synthesis by adopting two different (economical and safe) approaches for the macrocyclization of fragment 252 (Scheme 29).114 The names of these two techniques are hydrophobic anchor molecule process and solid phase peptide synthesis. In their synthetic scheme, fragment 252 was synthesized by employing Gilman reagent. Their synthesis was initiated with the ring opening of epoxy benzyl ester 248 (easily available starting material) by treating it with butyl magnesium bromide, copper bromide, and dimethyl sulphide in diethyl ether as a solvent at −78 °C to give N-methyl alanine 249 in quantitative yield. Esterification of compound 249 with compound 250 in the presence of EDCl, DMAP, and DCM furnished compound 251 (in 92% yield), which was subjected to hydrogenation to acquire fragment 252 in 85% yield. Further steps involving the macrocyclization of fragment 252 by two different approaches completed the synthesis of verticilide 253.
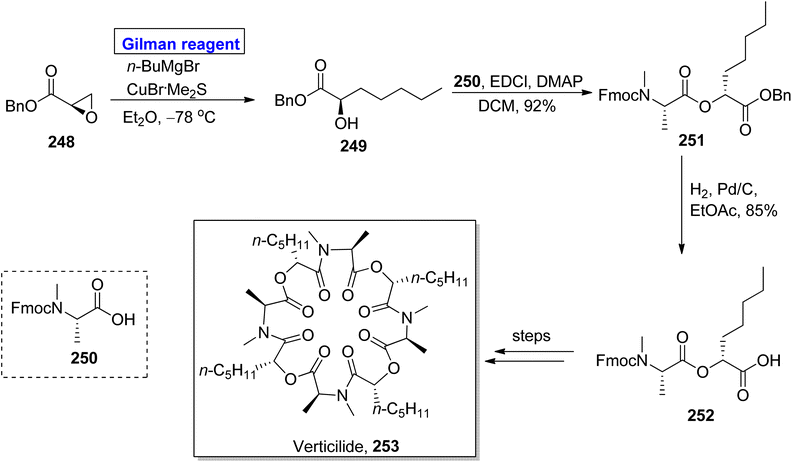 |
| | Scheme 29 Synthesis of intermediate 252 towards the synthesis of verticilide 253. | |
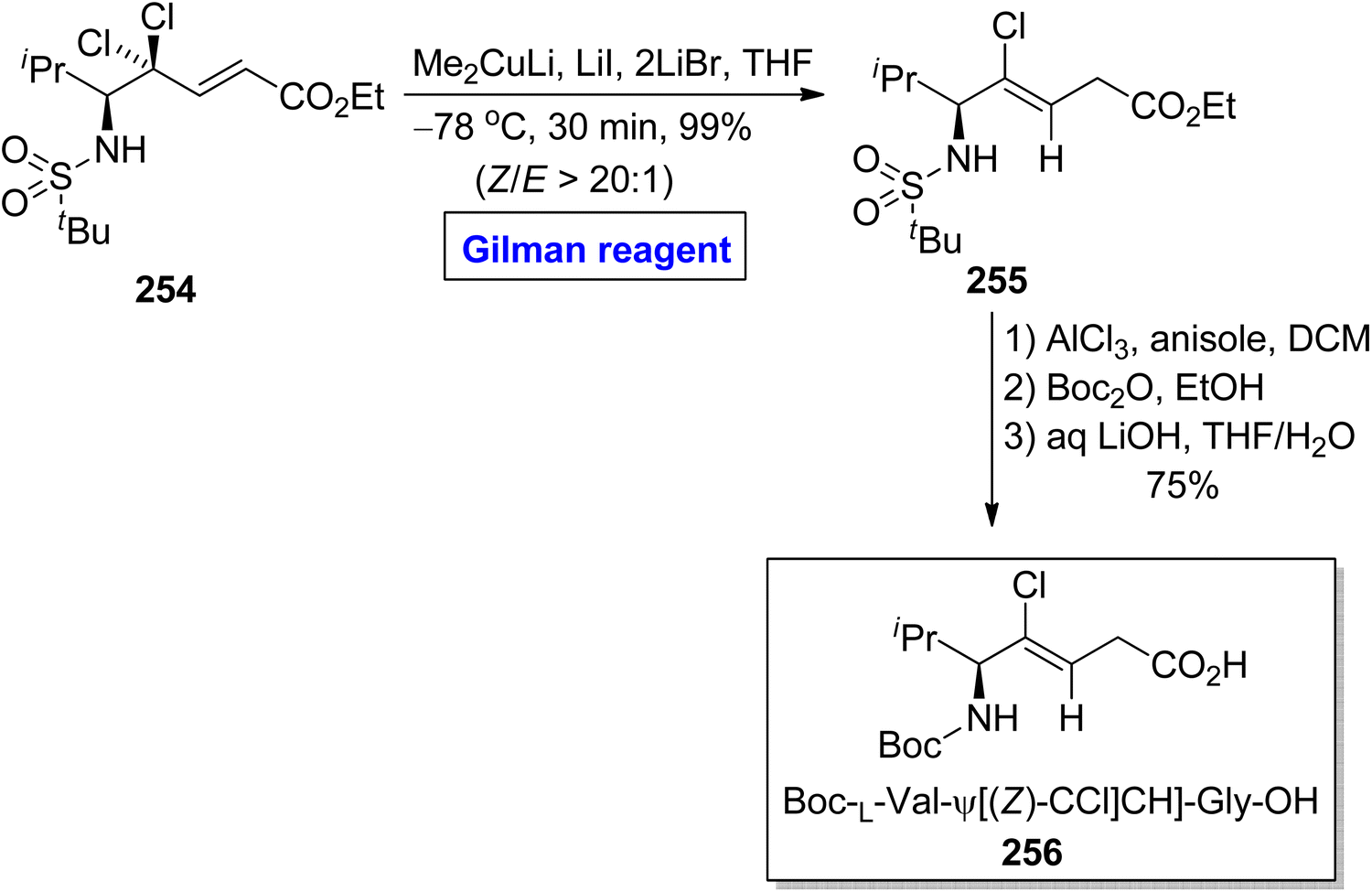
Peptides are medicinally and biologically important heterocyclic compounds. Owing to the rare bioavailability, synthetic chemists are working on replacing the amide group of peptides with alkene-based structures, which resemble peptides with improved biomimetic properties.115,116 Among these, chloroalkenes are considered the best synthetic intermediate as these are the core motifs of many natural products.117 In continuation of this work, Kobayakawa and Tamamura performed the synthesis of (Z)-chloroalkene-based dipeptide isosteres 256 in 2016 by employing the Gilman reagent (Scheme 30).118 To perform this task, the compound 254 was allowed to react with lithium dimethyl cuprate, lithium iodide, and lithium bromide in THF at −78 °C, which provided compound 255 in 99% yield (E/Z > 20:1). The compound 255 was further converted into compound 256 via selective protection and deprotection steps.
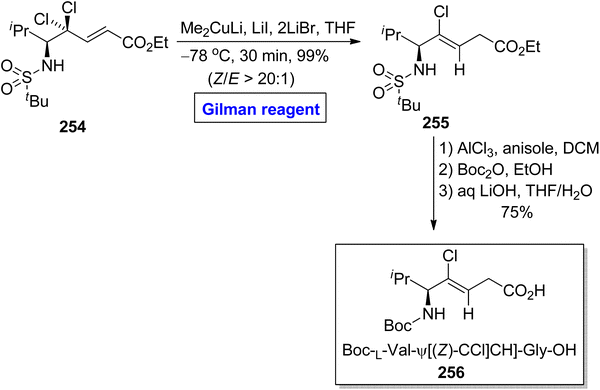 |
| | Scheme 30 Synthesis of L-Val-Gly based chloroalkene dipeptide 256. | |
Synthesis of nucleoside-based natural products
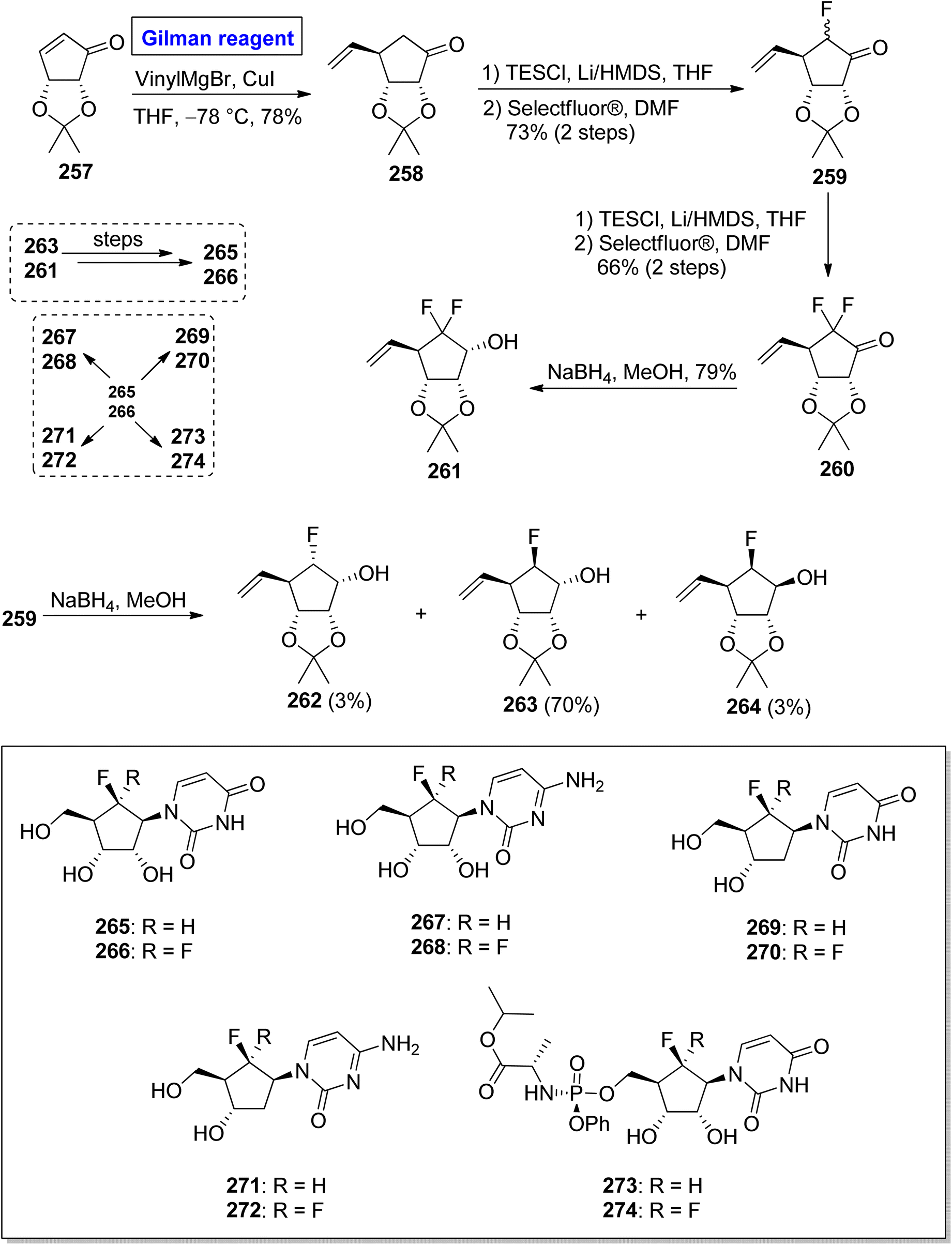
Nucleoside analogues are considered a cornerstone of the preventive strategies against cancer and various viral diseases (including COVID-19). Among these analogues, the role of carbocyclic nucleosides as naturally existing cytotoxic agents is predominant.119,120 In 2022, Benckendorff et al. presented a scalable and efficient synthesis of monofluorinated and difluorinated carbocyclic nucleoside analogues 265–274 and evaluated their cellular viability against PANC-1 and cancer cell lines U87-MG (Scheme 31).121 Their synthesis began with the readily available cyclopentenone 257, which was subjected to a conjugate addition reaction by employing the corresponding Gilman reagent. Thus, the treatment of cyclopentenone 257 with vinyl magnesium chloride and copper iodide in THF, with the adjustment of temperature at −78 °C furnished the compound 258 with a 78% yield. The compound 258 was silylated and subsequently treated with Selectfluor® in DMF to give compound 259 (with successful installation of fluorine) with a 73% yield. In order to install the second fluorine, a sequence of silyation and reaction with Selectfluor® was repeated, this time for compound 259, which resulted in compound 260 in 66% yield. In the next steps, the reduction of compound 260 by using NaBH4 in MeOH gave compound 261 with a 79% yield. The reduction of compound 259 provided a mixture of compounds 262, 263, and 264 in 3%, 70%, and 3% yield, respectively. Later on, compound 263 and compound 261 were used as precursors for the synthesis of compound 265 and compound 266, respectively, which proceeded further for the synthesis of analogues 267 to 274. All the synthesized analogues 267 to 274 were evaluated against cancer and PANC-1 cell lines. Among these, the compound 267 exhibited mild cytotoxic activity against U87-MG cells.
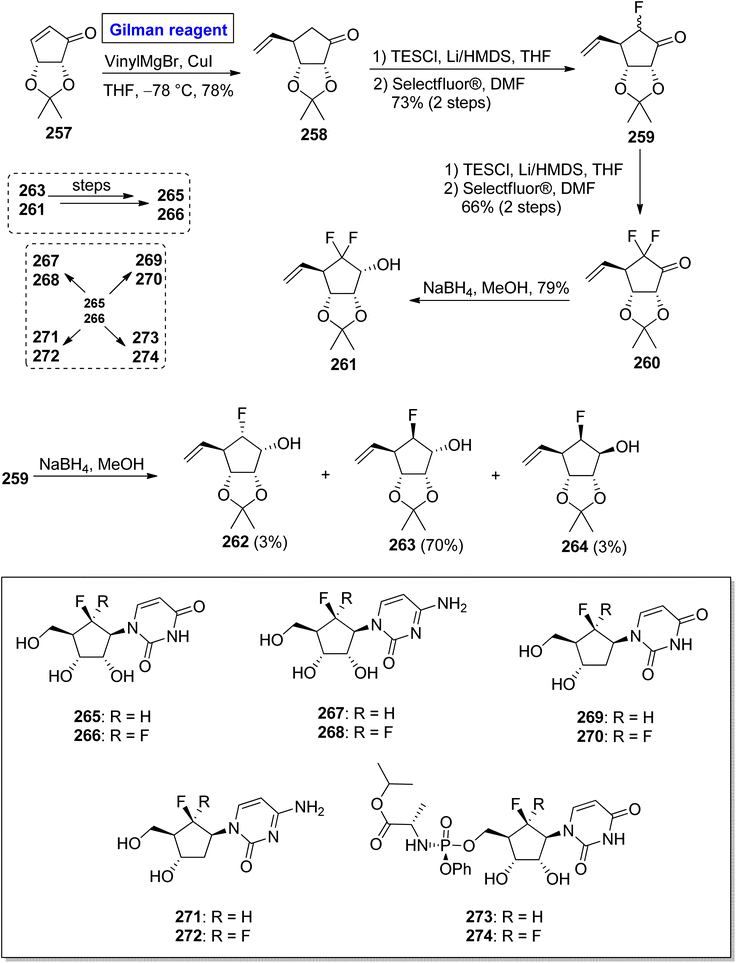 |
| | Scheme 31 Synthesis of fluorinated carbocyclic nucleosides 265–274. | |
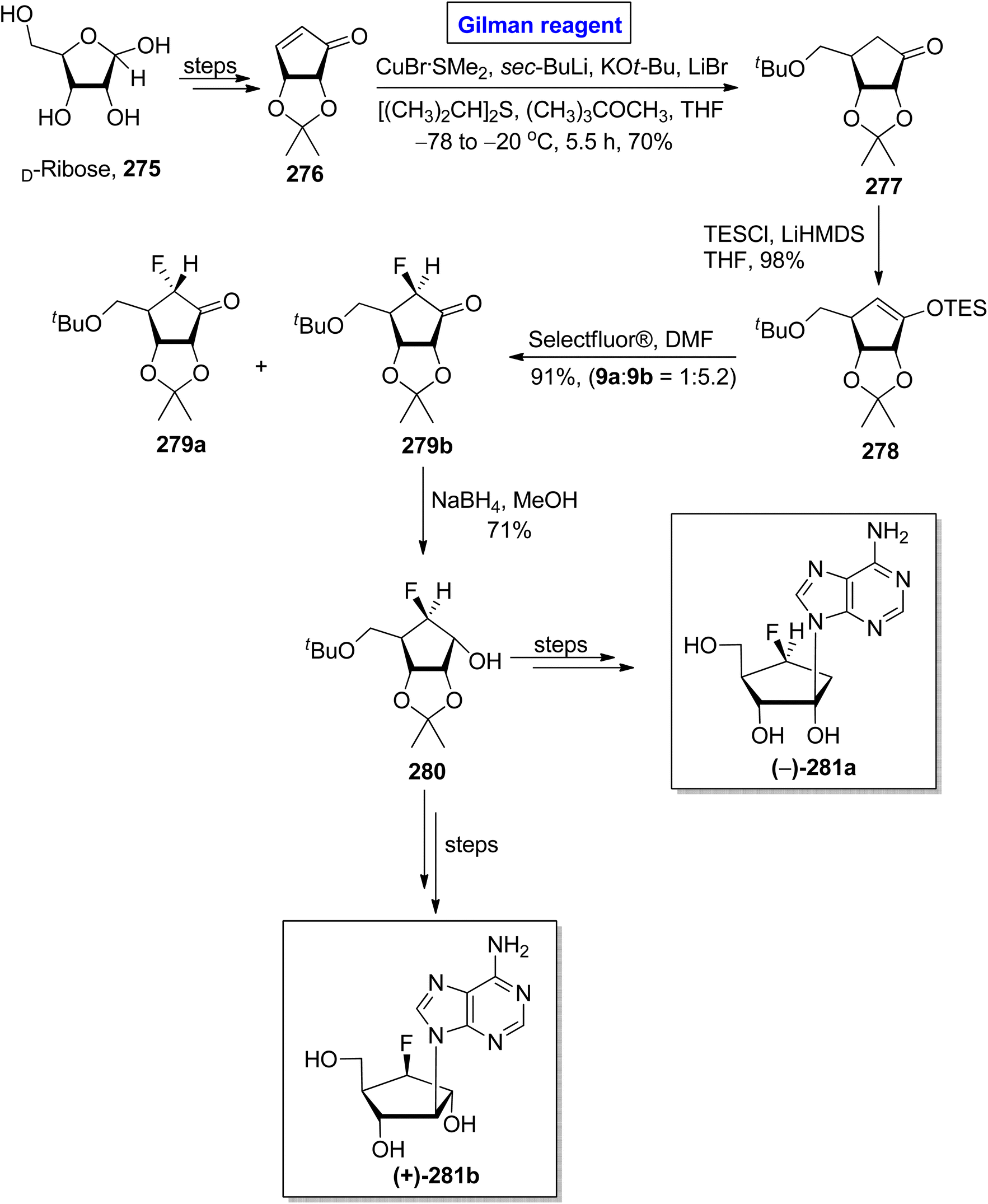
One of the carbocyclic nucleosides, (−)-aristeromycin, was isolated from a Gram-positive bacteria, Streptomyces citricolor. It is a strong anti-viral agent and adenosylhomocysteine hydrolase (AdoHcy) inhibitor.122 Its remarkable pharmacological profile prompted endeavors to synthesize this natural product as well as its analogues.123 In continuation of this effort, Kim et al. in 2017 disclosed the synthesis of (−)-6/-β-fluoro-aristeromycin 281a and (+)-5/-β-fluoro-isoaristeromycin 281b (Scheme 32).124 The key steps in their procedure entailed a regioselective conjugate addition reaction by employing Gilman reagent and electrophilic fluorination. In their methodology, compound 276 (modified from D-ribose), was treated with CuBr·SMe2, sec-BuLi, KOtBu, LiBr, [(CH3)2CH]2S, and (CH3)3COCH3 (Gilman reagent) in THF to achieve compound 277 in 70% yield. After this, the electrophilic fluorination of compound 278 (from the silylation of compound 277), by using Selectfluor® in DMF, compounds 279a and 279b were achieved in 90% yield (dr = 1:5.2). The compound 279b underwent reduction in the presence of NaBH4 in MeOH to generate the compound 280 in 71% yield. Later on, compound 280 was used as a well-suited intermediate for the synthesis of compounds 281a and 281b.
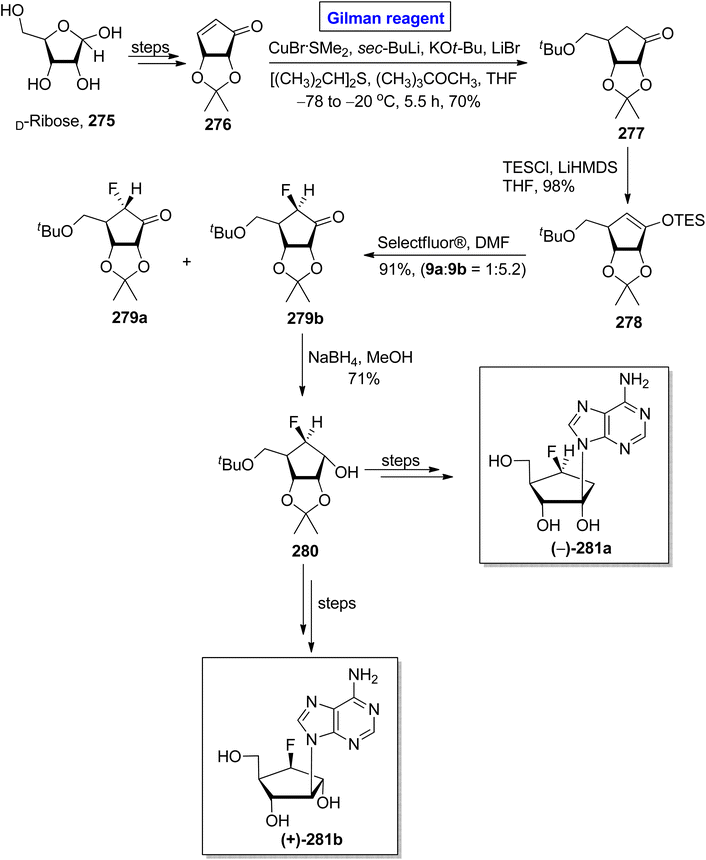 |
| | Scheme 32 Synthesis of (−)-6/-β-fluoro-aristeromycin 281a and (+)-5/-β-fluoro-isoaristeromycin 281b. | |
Synthesis of miscellaneous natural products
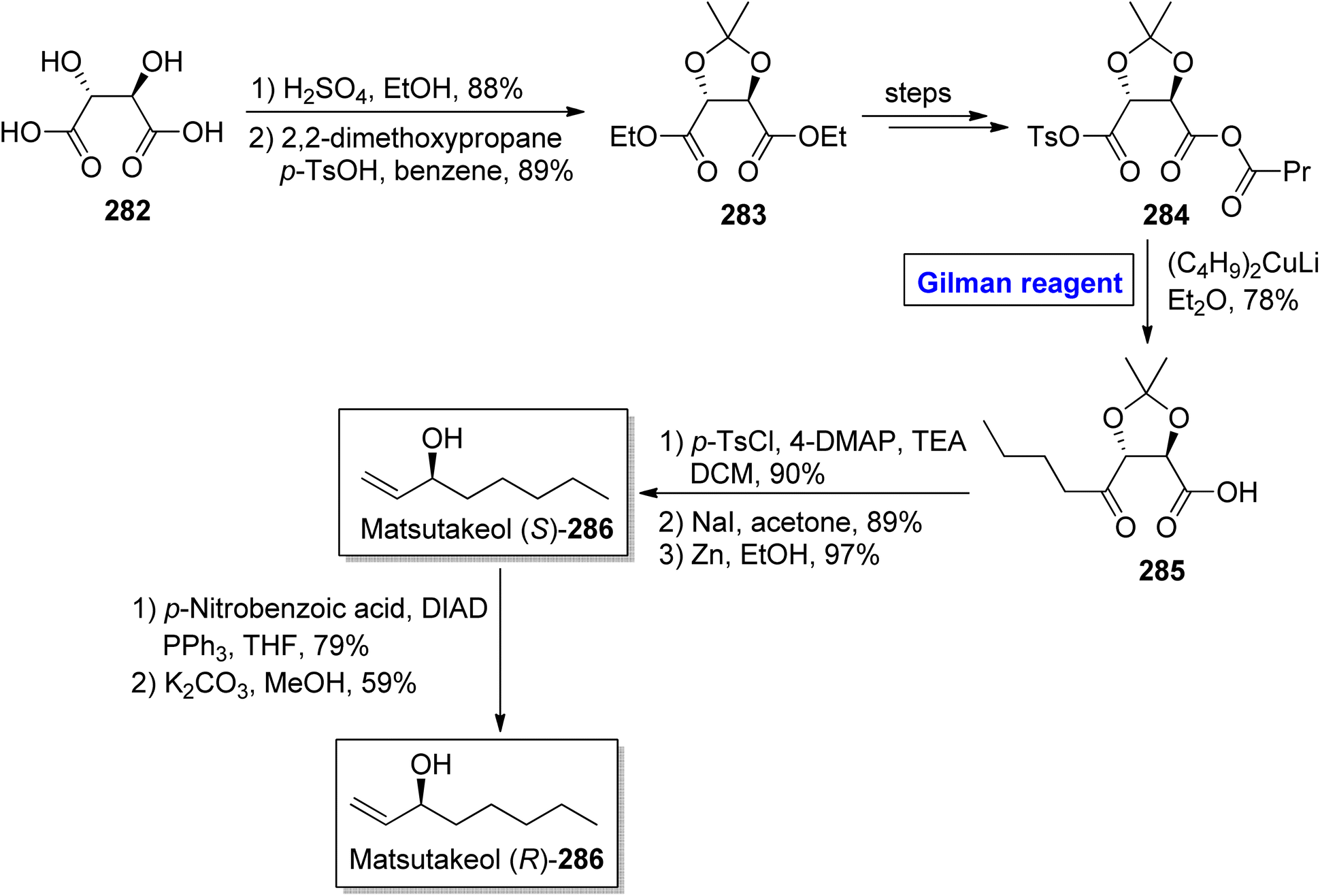
Tricholoma matsutake is an edible and expensive mushroom usually found in China, Korea, and Japan.125 It exhibits a number of health benefits, such as lowering of blood cholesterol and improvement of blood circulation. It is also an important preventive agent for cancer, diabetes, and certain heart diseases.126 Matsutakeol is an aromatic alcohol consisting of eight carbons (having a chiral carbon at the C-3 position) and a component (60 to 70%) of matsutake mushroom. In 2016, Lee et al. performed the facile and efficient synthesis of (S)-(+)-matsutakeol (S)-286 (in 32% overall yield) in just 10 steps involving the employment of Gilman reagent by using inexpensive and easily available starting material, i.e., L-tartaric acid (Scheme 33).127 Their synthesis started with the esterification of compound 282 accompanied by acetal protection to give compound 283 in 89% yield. A few steps later, the compound 284 was subjected to nucleophilic substitution reaction via treatment with (C4H9)2CuLi and Et2O (Gilman reagent) to furnish alcohol 285 in 78% yield. In the next step, the compound 285 was subjected to tosylation, iodine substitution, and acetal cleavage (in the presence of zinc in ethanol) in sequence to acquire compound (S)-286 in 97% yield. To get matsutakeol (R)-286, Mitsunobu inversion37 reaction of compound (S)-286 and further treatment with potassium carbonate in methanol provided the desired compound in 59% yield.
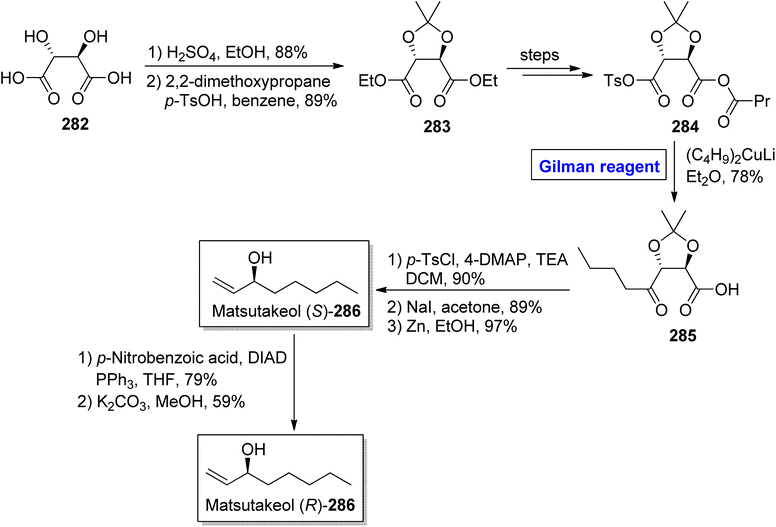 |
| | Scheme 33 Total synthesis of matsutakeol (S)-286 and (R)-286. | |
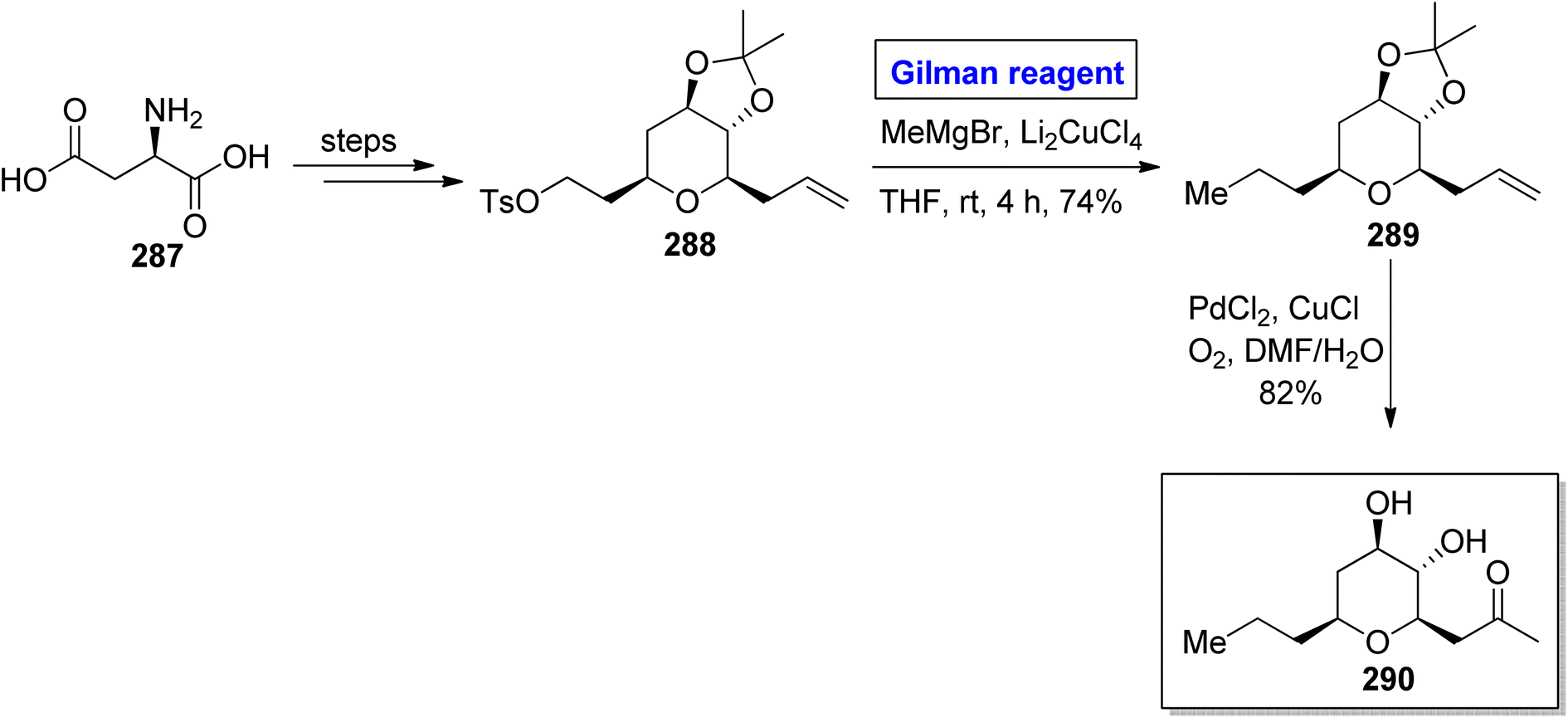
Tetrahydropyran is an intriguing heterocyclic scaffold present in many medicinally important natural products.128 One of the tetrahydropyran based natural products is phomonol 295, which was isolated from Phomopsis sp. of endophytic fungus. Its structure constitutes 2,3,4,6 tetrasubstituted pyran, having four asymmetric centers.129,130 Dada and Yaragorla reported an efficient and straightforward approach toward the total synthesis of phomonol 295 in 2022 (Scheme 34).131 Their synthesis commenced with the easily available chiral starting material, i.e., D-aspartic acid 287, which was modified in a number of steps to provide acetonide 288. In order to carry out the nucleophilic substitution reaction, a well-suited Gilman reagent was employed. For this purpose, compound 288 was treated with MeMgBr and Li2CuCl4 in THF at room temperature. After four hours, alkene 289 was obtained (in 74% yield), which was subjected to oxidation via PdCl2, CuCl in DMF/H2O to furnish compound 295 in 82% yield.
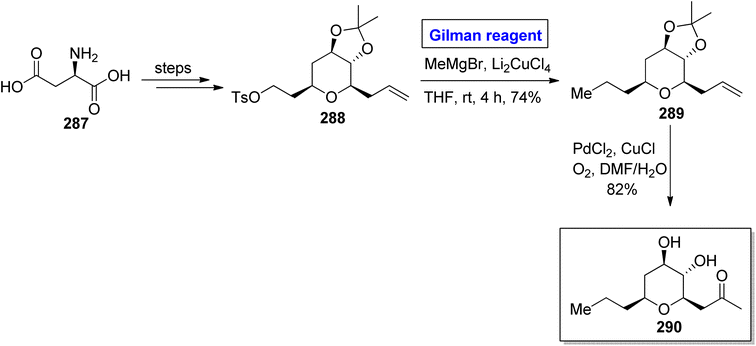 |
| | Scheme 34 Total synthesis of phomonol 290. | |
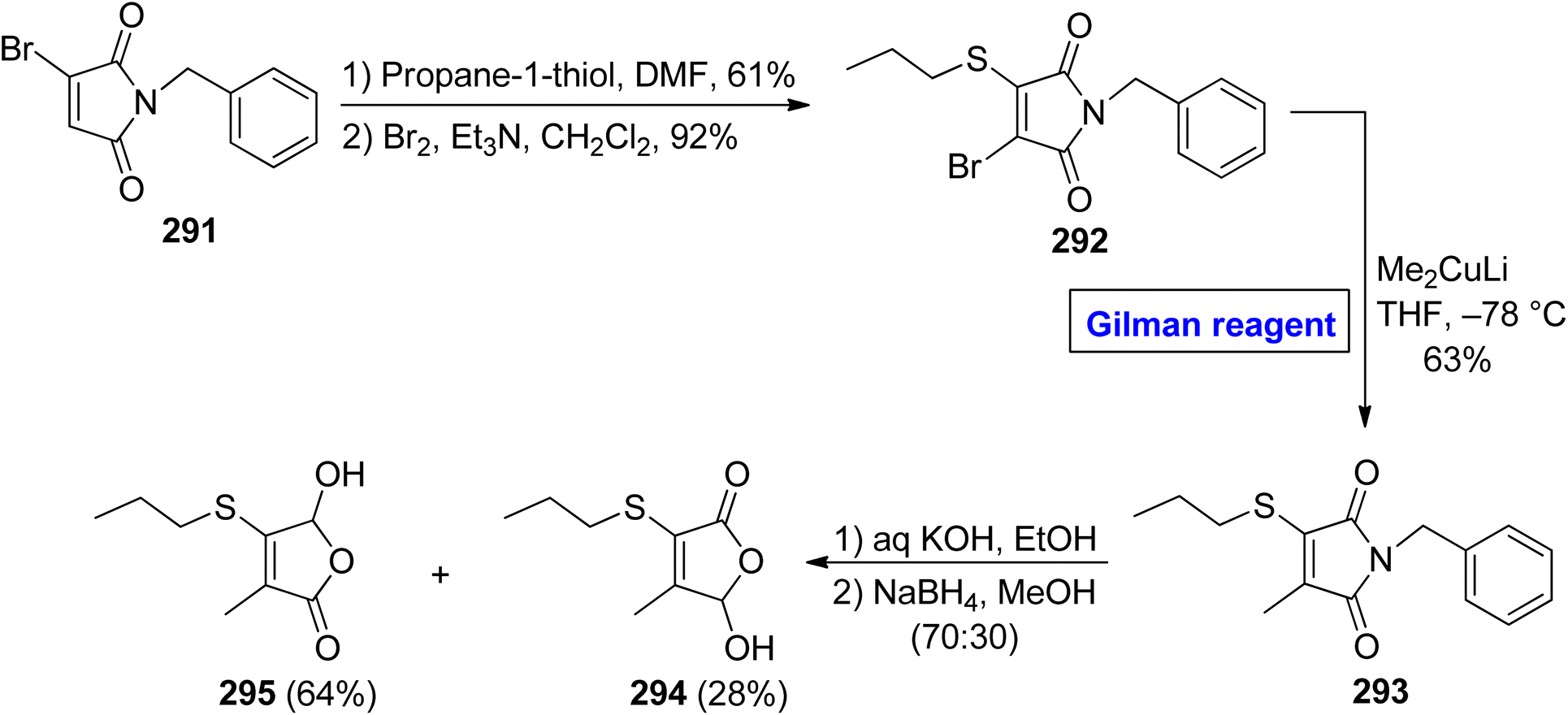
Cancer is a fatal disease and about 400000 children may develop cancer each year. With the increase in carcinogens in the environment in recent years, the demand for pharmaceutics (as effective anti-cancer agents) has also increased47,48 5-hydroxy-3-methyl-4-propylsulfanyl-5H furan-2-one 295 is an effective chemopreventive agent, which was isolated first in 2007 from Allium cepa (onions), by Parkin and Xiao.132,133 In 2011, Borikar et al. presented an efficient and facile methodology for the synthesis of this unique natural product 295 (Scheme 35).134 To accomplish this task, bromomaleimide 291 was allowed to react with propane-1-thiol (in DMF) and Br2 (in the presence of Et3N in DCM) in sequence to provide bromide 292 in 92% yield. In order to perform the nucleophilic substitution reaction, a well-suited Gilman reagent was employed by treating the bromide 292 with lithium dimethyl cuprate in THF at −78 °C to acquire compound 293 in 63% yield. The hydrolysis of thiomaleimde 293 under basic conditions, followed by treatment with NaBH4 in MeOH, resulted in a mixture of compound 295 (64%) and compound 294 (28%), which was separated by column chromatography to get our desired compound 295.
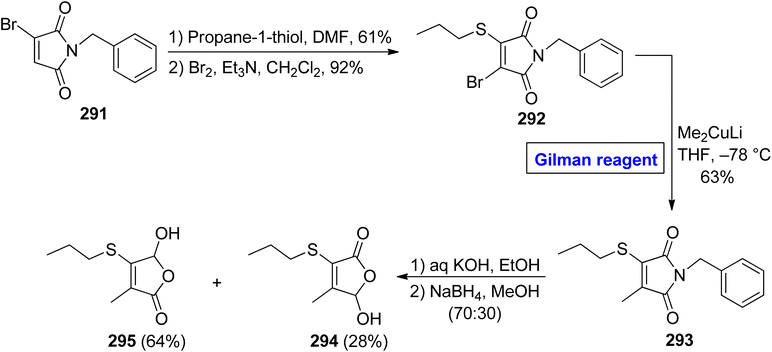 |
| | Scheme 35 Total synthesis of 5-hydroxy-3-methyl-4-propylsulfanyl-5H furan-2-one 295. | |
Conclusion
Gilman reagent is mainly based on RLi and CuX or R2CuLi with THF or Et2O as solvent. In general, the temperature range for the Gilman reaction is between −78 °C to 0 °C. The electronic and steric effects of the R group directly affect the regioselectivity of the products formed as a result of the Gilman reaction. With these salient features, the Gilman reagent seems to have opened up new routes in modern total synthesis via conjugate addition reaction, bimolecular nucleophilic substitution reaction, and ring-opening reaction of both epoxide and aziridine. This review article is an updated compilation of Gilman reagent-induced synthetic approaches for the synthesis of intriguing natural products with significant biological activities. These natural products entail alkaloids, terpenoids, polyketides, macrolides, amino acids, nucleoside analogues, and some drugs. Further, this review will urge many organic chemists to employ their efforts on the methodological studies of the Gilman reagent so that more efficient synthetic pathways can be developed for future progress in medicinally important natural products.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. Irfan extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding through the large group Research Project under grant number RGP2/276/44.
References
- G. H. Posner, Org. React., 2004, 19, 1–114 Search PubMed.
- B. H. Lipshutz, R. S. Wilhelm and D. M. Floyd, J. Am. Chem. Soc., 1981, 103, 7672–7674 CrossRef CAS.
- B. H. Lipshutz, F. Kayser and K. Siegmann, Tetrahedron Lett., 1993, 34, 6693–6696 CrossRef CAS.
- B. H. Lipshutz and B. James, J. Chem. Soc. C, 1994, 59, 7585–7587 CAS.
- E. J. Corey and G. H. Posner, J. Am. Chem. Soc., 1968, 90, 5615–5616 CrossRef CAS.
- G. M. Whitesides, W. F. Fischer Jr, J. S. Filippo Jr, R. W. Bashe and H. O. House, J. Chem. Soc. C, 1969, 91, 4871–4882 CAS.
- C. Eaborn, P. B. Hitchcock, J. D. Smith and A. C. Sullivan, J. Organomet. Chem., 1984, 263, c23–c25 CrossRef CAS.
- S. H. Bertz, G. Dabbagh, J. M. Cook and V. J. Honkan, Org. Chem., 1984, 49, 1739 CrossRef CAS.
- S. H. Bertz and G. Dabbagh, J. Am. Chem. Soc., 1988, 110, 3668–3670 CrossRef CAS.
- S. H. Bertz, A. Chopra, M. Eriksson, C. A. Ogle and P. Seagle, Eur. J. Chem., 1999, 5, 2680–2691 CrossRef CAS.
- S. H. Bertz, S. Cope, D. Dorton, M. Murphy and C. A. Ogle, Angew. Chem., Int. Ed., 2007, 46, 7082–7085 CrossRef CAS PubMed.
- E. R. Bartholomew, S. H. Bertz, S. Cope, M. Murphy and C. A. Ogle, J. Am. Chem. Soc., 2008, 130, 11244–11245 CrossRef CAS PubMed.
- S. H. Bertz, R. A. Hardin, M. D. Murphy, C. A. Ogle, J. D. Richter and A. A. Thomas, Angew. Chem., Int. Ed., 2012, 51, 2681–2685 CrossRef CAS PubMed.
- S. H. Bertz, S. K. Cope, R. A. Hardin, M. D. Murphy, C. A. Ogle, D. T. Smith, A. A. Thomas and T. N. Whaley, Organometallics, 2012, 31, 7827–7838 CrossRef CAS.
- S. H. Bertz, R. A. Hardin and C. A. Ogle, J. Am. Chem. Soc., 2013, 135, 9656–9658 CrossRef CAS PubMed.
- Y. Yamamoto, S. Yamamoto, H. Yatagai, Y. Ishihara and K. Maruyama, J. Org. Chem., 1982, 47, 119–126 CrossRef CAS.
- A. Vivien, A. Bartoli, J. L. Parrain and L. Commeiras, Tetrahedron Lett., 2020, 61, 151472 CrossRef CAS.
- F. Velazquez and H. F. Olivo, Org. Lett., 2000, 2, 1931–1933 CrossRef CAS PubMed.
- A. Bisai, S. P. West and R. Sarpong, J. Am. Chem. Soc., 2008, 130, 7222–7223 CrossRef CAS PubMed.
- S. K. Mal, G. K. Kar and J. K. Ray, Tetrahedron, 2004, 60, 2805–2811 CrossRef CAS.
- J. Vetter, Food Chem. Toxicol., 2004, 42, 1373 CrossRef CAS PubMed.
- Y. Lu, T. Yao and Z. Chen, Acta Pharm. Sin. B, 1986, 21, 829 CAS.
- S. C. Deshmukh, A. Roy and P. Talukdar, Org. Biomol. Chem., 2012, 10, 7536–7544 RSC.
- S. P. Wong, C. Y. Gan, K. H. Lim, K. N. Ting, Y. Y. Low and T. S. Kam, Org. Lett., 2015, 17, 3628 CrossRef CAS PubMed.
- S. P. Wong, K. W. Chong, K. H. Lim, S. H. Lim, Y. Y. Low and T. S. Kam, Org. Lett., 2016, 18, 1618 CrossRef CAS PubMed.
- P. Gan, J. Pitzen, P. Qu and S. A. Snyder, J. Am. Chem. Soc., 2018, 140, 919–925 CrossRef CAS PubMed.
- M. Satake, A. J. Bourdelais, R. M. Van Wagoner, D. G. Baden and J. L. C. Wright, Org. Lett., 2008, 10, 3465 CrossRef CAS PubMed.
- A. T. Hermann, S. R. Matinez and A. Zakarion, Org. Lett., 2011, 13, 3636 CrossRef PubMed.
- J. S. Yadav, N. M. Reddy, M. A. Rahman, A. R. Prasad and B. S. Reddy, Tetrahedron, 2013, 69, 8618–8625 CrossRef CAS.
- R. Akhtar, S. A. R. Naqvi, A. F. Zahoor and S. Saleem, Mol. Diversity, 2018, 22, 447–501 CrossRef CAS PubMed.
- M. Kuramoto, K. Hayashi, K. Yamaguchi, M. Yada, T. Tsuji and D. Uemura, Bull. Chem. Soc. Jpn., 1998, 71, 771–779 CrossRef CAS.
- N. Hikage, H. Furukawa, K. Takao and S. Kobayashi, Chem. Pharm. Bull., 2000, 48, 1370–1372 CrossRef CAS PubMed.
- C. B. Roa, A. S. R. Anjaneyula, N. S. Sarma, Y. Venkatateswarlu, R. M. Rosser, D. J. Faulkner, M. H. M. Chen and J. Clardy, J. Am. Chem. Soc., 1984, 106, 7983–7984 CrossRef.
- D. C. Behenna, J. L. Stockdill and B. M. Stoltz, Angew. Chem., Int. Ed., 2008, 47, 2365–2386 CrossRef CAS.
- T. Nakajima, D. Yamashita, K. Suzuki, A. Nakazaki, T. Suzuki and S. Kobayashi, Org. Lett., 2011, 13, 2980–2983 CrossRef CAS PubMed.
- J. T. Bagdanoff, D. C. Behenna, J. L. Stockdill and B. M. Stoltz, Eur. J. Org Chem., 2016, 2101–2104 CrossRef CAS PubMed.
- S. Munawar, A. F. Zahoor, S. Ali, S. Javed, M. Irfan, A. Irfan, K. K. Mojzych and M. Mojzych, Molecules, 2022, 27, 6953 CrossRef CAS PubMed.
- M. Kuramoto, H. Arimoto and D. Uemura, Mar. Drugs, 2004, 2, 39 CrossRef CAS.
- Y. Tsubosaka, T. Murata, K. Yamada, D. Uemura, M. Hori and H. Ozaki, J. Pharmacol. Sci., 2010, 113, 208 CrossRef CAS PubMed.
- C. Gignoux, A. F. Newton, A. Barthelme, W. Lewis, M. L. Alcaraz and R. A. Stockman, Org. Biomol. Chem., 2012, 10, 67–69 RSC.
- D. P. A. Gossan, A. Alabdul Magid, P. A. Kouassi-Yao, J. B. Behr, A. C. Ahibo, L. A. Djakouré, D. Harakat and N. L. Voutquenne, Phytochemistry, 2015, 109, 76 CrossRef CAS PubMed.
- M. Shibano, D. Tsukamoto and G. Kusano, Heterocycles, 2002, 57, 1539 CrossRef CAS.
- B. J. Byatt, A. Kato and S. G. Pyne, Org. Lett., 2012, 23, 4029–4033 CrossRef PubMed.
- J. Choi, N. N. Yadav and H. J. Ha, Asian J. Org. Chem., 2017, 6, 1292–1307 CrossRef CAS.
- E. Airiau, T. Spangenberg, N. Girard, B. Breit and A. Mann, Org. Lett., 2021, 12, 528–531 CrossRef PubMed.
- N. N. Yadav, Y. G. Lee, N. Srivastava and H. J. Ha, Front. Chem., 2019, 7, 460 CrossRef PubMed.
- I. Sarfraz, A. Rasul, G. Hussain, M. A. Shah, A. F. Zahoor, M. Asrar, Z. Selamoglu, X. Y. Ji, S. Adem and S. D. Sarker, BioFactors, 2020, 46, 550–562 CrossRef CAS PubMed.
- I. Shahzadi, A. F. Zahoor, B. Parveen, A. Rasul, Z. Raza, S. Ahmad, A. Irfan and G. A. El-Hiti, J. Chem., 2022, 2022, 1–8 CrossRef.
- W. Li, Y. Q. Tang, J. Sang, R. Z. Fan, G. H. Tang and S. Yin, Org. Lett., 2020, 22, 106 CrossRef CAS PubMed.
- J. Zhu, R. Wang, L. Lou, W. Li, G. Tang, X. Bu and S. Yin, J. Med. Chem., 2016, 59, 6353 CrossRef CAS PubMed.
- F. Q. Ni, S. Q. Wu, W. Li, Q. Li and S. Yin, Org. Lett., 2020, 22, 9360–9364 CrossRef CAS PubMed.
- R. Ratnayake, D. Covell, T. T. Ransom, K. R. Gustafson and J. A. Beutler, Org. Lett., 2009, 11, 57 CrossRef CAS PubMed.
- K. Molawi, N. Delpont and A. M. Echavarren, Angew. Chem., Int. Ed., 2010, 49(20), 3517–3519 CrossRef CAS PubMed.
- R. Nelson, M. Gulías, J. L. Mascareñas, F. López and F. Angew, Angew. Chem., Int. Ed., 2016, 55, 14359–14363 CrossRef CAS PubMed.
- Y. Saritas, S. H. von Reu and W. A. König, Phytochemistry, 2002, 59, 795 CrossRef CAS PubMed.
- R. Richter, S. Basar, A. Koch and W. A. König, Phytochemistry, 2005, 66, 2708 CrossRef CAS PubMed.
- D. W. Jee and H. Y. Lee, Asian J. Org. Chem., 2021, 10, 820–826 CrossRef CAS.
- M. Suchy, V. Benesova, V. Herout and F. Sorm, Tetrahedron Lett., 1959, 10, 5–9 CrossRef.
- V. Saroglou, A. Karioti, C. Demetzos, K. Dimas and H. Skaltsa, J. Nat. Prod., 2005, 68, 1404–1407 CrossRef CAS PubMed.
- K. Johrer, M. Obkircher, D. Neurelter, J. Partell, C. Zelle-Rieser, E. Maizner, J. Kern, M. Hermann, F. Hamacher, O. Markel, N. Wacht, C. Zidorn, M. Scheideler and R. Grell, J. Mol. Med., 2012, 90, 681–693 CrossRef PubMed.
- K. Kimura and T. Usuki, Tetrahedron Lett., 2022, 107, 154102 CrossRef CAS.
- D. C. Umrani, R. Seshadri, K. G. Gore and K. K. Chakravarti, Flavor Ind., 1970, 1, 623–624 CAS.
- P. Kraft and R. Cadalbert, Synthesis, 2002, 2243–2253 CrossRef CAS.
- P. Kraft and N. Denizot, Eur. J. Org Chem., 2013, 49–58 CrossRef CAS.
- M. Tsuda, K. Oguchi, R. Iwamoto, Y. Okamoto, E. Fukushi, J. Kawabata, T. Ozawa and A. Masuda, J. Nat. Prod., 2007, 70, 1661 CrossRef CAS PubMed.
- Y. Liu, J. Wang, H. Li, J. Wu, G. Feng and W. M. Dai, Synlett, 2010, 2184 CAS.
- J. Huang and J. Yang, Synlett, 2012, 23, 737–740 CrossRef CAS.
- M. Tsuda, K. Oguchi, R. Iwamoto, Y. Okamoto, J. Kobayashi, J. Kawabata, E. Fukushi, T. Ozawa and A. Masuda, Nat. Prod., 2007, 70, 1661–1663 CrossRef CAS PubMed.
- A. K. Ghosh and H. Yuan, Mar. Drugs, 2022, 20, 587 CrossRef CAS PubMed.
- P. A. Searle and T. F. Molinski, J. Am. Chem. Soc., 1995, 117, 8126 CrossRef CAS.
- A. B. Smith III, P. R. Verhoest, K. P. Minbiole and M. Schelhaas, J. Am. Chem. Soc., 2001, 123, 4834 CrossRef PubMed.
- K. B. Raju, B. N. Kumar, B. S. Kumar and K. Nagaiah, Helv. Chim. Acta, 2015, 98, 386–399 CrossRef CAS.
- J. B. MacMillan and T. F. Molinski, Org. Lett., 2002, 4, 1535 CrossRef CAS PubMed.
- D. De Joarder and M. P. Jennings, Tetrahedron Lett., 2011, 52, 5124 CAS.
- D. De Joarder and M. P. Jennings, Tetrahedron Lett., 2013, 54, 5826–5829 CrossRef CAS.
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- Y. J. Kim, K. Furihata, A. Shimazu, K. Furihata and H. J. Seto, Antibiot., 1991, 44, 1280–1282 CrossRef CAS PubMed.
- N. Terayama, E. Yasui, M. Mizukami, M. Miyashita and S. Nagumo, Org. Lett., 2014, 16, 2794–2797 CrossRef CAS PubMed.
- Y. H. Jeon, J. Y. Lee and S. Kim, Bioorg. Med. Chem., 2012, 20, 1893–1901 CrossRef CAS PubMed.
- H. Nakano, A. Sugawara, T. Hirose, H. Gouda, S. Hirono, S. Omura and T. Sunazuka, Tetrahedron, 2015, 71, 6569–6579 CrossRef CAS.
- D. Katsumi, K. Nakasone, N. Terayama, E. Yasui, M. Mizukami, M. Miyashita and S. Nagumo, J. Org. Chem., 2019, 84, 1553–1562 CrossRef CAS PubMed.
- J. Kobayashi, H. Shigemori, M. Ishibashi, T. Yamasu, H. Hirota and T. J. Sasaki, Org. Chem., 1991, 56, 5221–5224 CrossRef CAS.
- J. Kobayashi, K. Shimbo, M. Sato and M. Tsuda, J. Org. Chem., 2002, 67, 6585–6592 CrossRef CAS PubMed.
- T. Usui, S. Kazami, N. Dohmae, Y. Mashimo, H. Kondo, M. Tsuda, A. G. Terasaki, K. Ohashi, J. Kobayashi and H. Osada, Chem. Biol., 2004, 11, 1269–1277 CrossRef CAS PubMed.
- A. Hara, R. Morimoto, Y. Ishikawa and S. Nishiyama, Org. Lett., 2011, 13, 4036–4039 CrossRef CAS PubMed.
- T. Fehr, J. J. Sanglier, W. Schuler, L. Gschwind, M. Ponelle, W. Schilling and C. J. Wioland, Antibiot., 1996, 49, 230–233 CrossRef CAS PubMed.
- M. E. Maier and C. Hermann, Tetrahedron, 2000, 56, 557–561 CrossRef CAS.
- J. R. Vakiti and S. Ghosh, Tetrahedron Lett., 2014, 55, 6438–6440 CrossRef CAS.
- M. Okabe, T. Sugita, K. Kinoshita and K. J. Koyama, Nat. Prod., 2016, 79, 1208–1212 CrossRef CAS PubMed.
- H. Kumar, A. S. Reddy and R. B. V. Subba, Tetrahedron Lett., 2014, 55, 1519–1522 CrossRef CAS.
- C. W. Reed, M. G. Fulton, K. D. Nance and C. W. Lindsley, Tetrahedron Lett., 2019, 60, 743–745 CrossRef CAS.
- G. E. Chlipala, P. Huu Tri, N. Van Hung, A. Krunic, S. Hee Shim, D. D. Soejarto and J. Orjala, J. Nat. Prod., 2010, 73, 784–787 CrossRef CAS PubMed.
- R. C. Yanagita, H. Kamachi, M. Kikumori, H. Tokuda, N. Suzuki, K. Suenaga, H. Nagai and K. Irie, Bioorg. Med. Chem. Lett., 2013, 23, 4319–4323 CrossRef CAS PubMed.
- J. S. Yadav, G. Rajendar, R. S. Rao and S. Pabbaraja, J. Org. Chem., 2013, 78, 8524–8530 CrossRef CAS PubMed.
- S. Feuillastre, L. Raffier, B. Pelotier and O. Piva, Org. Biomol. Chem., 2020, 18, 1949–1956 RSC.
- C. K. Lu, H. N. Chou, C. K. Lee and T. H. Lee, Org. Lett., 2005, 7, 3893–3896 CrossRef CAS.
- C. K. Lu, Y. M. Chen and S. H. Wang, Tetrahedron Lett., 2010, 51, 6911–6914 CrossRef CAS.
- P. AnkiReddy, S. AnkiReddy and S. Gowravaram, Org. Biomol. Chem., 2018, 16, 4191–4194 RSC.
- K. Suenaga, K. Araki, T. Sengoku and D. Uemura, Org. Lett., 2001, 3, 527–529 CrossRef CAS PubMed.
- K. Araki, K. Suenaga, T. Sengoku and D. Uemura, Tetrahedron, 2002, 58, 1983–1995 CrossRef CAS.
- J. Ren, J. Wang and R. Tong, Org. Lett., 2015, 17, 744–747 CrossRef CAS PubMed.
- Y. Igarashi, L. Yu, M. Ikeda, T. Oikawa, S. Kitani, T. Nihira, B. Bayanmunkh and W. J. Panbangred, Nat. Prod., 2012, 75, 986 CrossRef CAS PubMed.
- R. García-Salcedo, R. Álvarez-Álvarez, C. Olano, L. Cañedo, A. F. Braña, C. Méndez, C. F. De-la and J. A. Salas, Mar. Drugs, 2018, 16, 259 CrossRef PubMed.
- L. Yu, T. Oikawa, S. Kitani, T. Nihira, B. Bayanmunkh, W. Panbangred and Y. J. Igarashi, Antibiot., 2014, 67, 345 CrossRef CAS PubMed.
- M. Dumpala, B. Srinivas and P. R. Krishna, Synlett, 2020, 31, 69–72 CrossRef CAS.
- J. F. Tremblay, Chem. Eng. News, 2000, 78, 14–15 Search PubMed.
- P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2012, 134, 3635–3638 CrossRef CAS PubMed.
- I. Chogii, P. Das and J. T. Njardarson, Asian J. Org. Chem., 2019, 8, 1041–1044 CrossRef CAS.
- F. Kotch and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3028–3033 CrossRef CAS PubMed.
- M. A. Cejas, W. A. Kinney, C. Chen, G. C. Leo, B. A. Tounge, J. G. Vinter, P. P. Joshi and B. E. Maryanoff, J. Am. Chem. Soc., 2007, 129, 2202 CrossRef CAS PubMed.
- S. Basu, P. S. Kandiyal, V. S. K. Neelamraju, H. Singh, R. S. Ampapathi and T. K. Chakraborty, Tetrahedron, 2014, 70, 1169–1175 CrossRef CAS.
- M. Fill and J. A. Copello, Physiol. Rev., 2002, 82, 893–922 CrossRef CAS.
- K. Shiomi, R. Matsui, A. Kakei, Y. Yamaguchi, R. Masuma, H. Hatano, N. Arai, M. Isozaki, H. Tanaka, S. Kobayashi, A. Turberg and S. Omura, J. Antibiot., 2010, 63, 77–82 CrossRef CAS PubMed.
- A. Watanabe, Y. Noguchi, T. Hirose, S. Monma, Y. Satake, T. Arai, K. Masuda, N. Murashima, K. Shiomi, S. Omura and T. Sunazuka, Tetrahedron Lett., 2020, 61, 151699 CrossRef CAS.
- Y. Sasaki, A. Niida, T. Tsuji, A. Shigenaga, N. Fujii and A. Otaka, Org. Chem., 2006, 71, 4969–4979 CrossRef CAS PubMed.
- A. Proteau-Gagne, J. F. Nadon, S. Bernard, B. Guerin, L. Gendron and Y. L. Dory, Tetrahedron Lett., 2011, 52, 6603–6605 CrossRef CAS.
- D. E. White, I. C. Stewart, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 810–811 CrossRef CAS.
- T. Kobayakawa and H. Tamamura, Tetrahedron, 2016, 72, 4968–4971 CrossRef CAS.
- A. C. Ojeda-Porras, V. Roy and L. A. Agrofoglio, Chem. Rec., 2022, 22, e202100307 CrossRef CAS PubMed.
- A. Kálmán, T. Yoritsánszky, J. Béres and G. Sági, Nucleosides, Nucleotides Nucleic Acids, 1990, 9, 235–243 CrossRef.
- C. M. Benckendorff, V. D. Slyusarchuk, N. Huang, M. A. Lima, M. Smith and G. J. Miller, Org. Biomol. Chem., 2022, 20, 9469–9489 RSC.
- E. De Clercq, D. E. Bergstrom, A. H. John and J. A. Montgomery, Antiviral Res, 1984, 4, 119 CrossRef CAS.
- E. De Clercq, Nat. Rev. Drug Discovery, 2002, 1, 13 CrossRef CAS PubMed.
- G. Kim, J. S. Yoon, D. B. Jarhad, Y. S. Shin, M. S. Majik, V. A. Mulamoottil, X. Hou, S. Qu, J. Park, H. B. Mu and L. S. Jeong, Org. Lett., 2017, 19, 5732–5735 CrossRef CAS PubMed.
- X. Yin, Q. You and Z. Jiang, Carbohydr. Polym., 2011, 86, 1358 CrossRef CAS.
- R. Zawirska-Wojtasiak, Food Chem., 2004, 86, 113 CrossRef CAS.
- W. H. Lee, I. H. Bae, B. M. Kim and Y. B. Seu, Bull. Korean Chem. Soc., 2016, 37, 1910–1911 CrossRef CAS.
- Y. Y. Li, M. Z. Wang, Y. J. Huang and Y. M. Shen, Mycology, 2010, 1, 254–261 CrossRef CAS.
- P. R. Krishna and S. Prabhakar, Tetrahedron Lett., 2013, 54, 3788–3790 CrossRef.
- B. V. S. Reddy, L. Srinivas, P. S. Reddy, B. P. Reddy, A. R. Prasad and J. S. Yadav, Helv. Chim. Acta, 2014, 97, 1326–1332 CrossRef.
- R. Dada and S. Yaragorla, Synth. Commun., 2022, 52, 445–450 CrossRef CAS.
- H. Xiao and K. L. Parkin, Phytochemistry, 2007, 68, 1059 CrossRef CAS PubMed.
- L. Gomez-Paloma, M. C. Monti, S. Terracciano, A. Casapullo and R. Riccio, Curr. Org. Chem., 2005, 9, 1419 CrossRef CAS.
- S. P. Borikar, V. Paul, V. G. Puranik, V. T. Sathe, S. Lagunas-Rivera and M. Ordónez, Synthesis, 2011, 1595–1598 CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Usman Nazeerb,
Muhammad Athar Saeeda,
Asim Manshaa,
Ahmad Irfanc and
Muhammad Umair Tariq*d
*a,
Usman Nazeerb,
Muhammad Athar Saeeda,
Asim Manshaa,
Ahmad Irfanc and
Muhammad Umair Tariq*d