DOI:
10.1039/D3RA05700C
(Paper)
RSC Adv., 2023,
13, 34157-34166
A novel LC-MS/MS method combined with derivatization for simultaneous quantification of vitamin D metabolites in human serum with diabetes as well as hyperlipidemia
Received
21st August 2023
, Accepted 6th November 2023
First published on 22nd November 2023
Abstract
Vitamin D plays an important role in calcium homeostasis. Recent studies indicate that vitamin D deficiency has become a major public health problem. In order to define vitamin D status, many analytical methods were used to quantify 25-hydroxyvitamin D (25OHD), as circulating 25OHD is regarded as the best indicator to evaluate vitamin D status. The current LC-MS/MS technology is internationally recognized as the “gold standard” for the detection of vitamin D and its metabolites. The impediment to the analysis of vitamin D metabolites is the low level of 25OHD and 1,25(OH)2D. Therefore, it is challenging to achieve the desired sensitivity and accuracy in the determination of trace vitamin D compounds in biological liquids. Here, a method based on liquid–liquid extraction in combination with derivatization, followed by liquid chromatography-electrospray/tandem mass spectrometry was developed for determination of the vitamin D metabolites, including 25-hydroxyvitamin D2, 25-hydroxyvitamin D3, 1α,25-dihydroxyvitamin D2 and 1α,25-dihydroxyvitamin D3. The method was simple and rapid, and it was validated with good linearity (R2 > 0.998), excellent recovery (average value with 81.66–110.31%) and high precision of intra-day and inter-day (0.06–6.38% and 0.20–6.82%). The values of limit of detection (LOD) and limit of quantitation (LOQ) were as low as 0.3 ng mL−1 and 1.0 ng mL−1, respectively. Finally, the developed method was successfully applied to determination of the vitamin D metabolites from the human serum samples of healthy subjects and patients with diabetes as well as hyperlipidemia.
1. Introduction
Vitamin D plays a key role in bone health and calcium and phosphorus metabolism.1–3 In addition, it has a variety of biological functions, including inducing cell differentiation, inhibiting cell growth, immune regulation, etc.4,5 There are two main forms of vitamin D, including ergocalciferol (vitamin D2) and cholecalciferol (vitamin D3). Vitamin D2 and vitamin D3 are obtained from different sources. Vitamin D3 is mainly formed by ultraviolet B irradiation of 7-dehydrocholesterol under the skin but can also be obtained from dietary supplements, whereas vitamin D2 can be obtained from dietary supplements.6,7
Due to the essential role of vitamin D in human health, several studies have reported that vitamin D deficiency is related to many diseases, including cancer, cardiovascular disease, autoimmune disease, schizophrenia, depression, etc.8,9 Especially, a growing number of studies have also found a strong link between vitamin D deficiency and diabetes and hyperlipidemia.10–14 Vitamin D has no biological activity and needs to be converted into bioactive metabolites through two steps of hydroxylation.15,16 The biotransformation pathway of vitamin D and the related metabolites is demonstrated in Fig. 1.
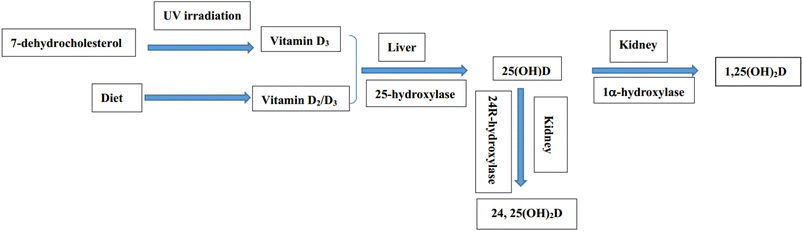 |
| | Fig. 1 Schematic diagram of the biotransformation pathway of vitamin D and the related metabolites. | |
Quantification of 25OHD in human serum is the best index to evaluate vitamin D status.17 Various methods are used for the quantification of circulating 25OHD, including radioimmunoassay (RIA), enzyme-linked immunosorbent assay (ELISA) and high-performance liquid chromatography. Radioimmunoassay (RIA) and enzyme-linked immunosorbent assay (ELISA) are unable to separate 25OHD2 and 25OHD3.17 And it's reported that an optical biosensor is used to determinate vitamin D3, which is unable to analyze multiple analytes simultaneously.18 This indicates the need for selective analytical methods for vitamin D2 and vitamin D3 forms.19 The emergence of liquid chromatography-tandem mass spectrometry (LC-MS/MS) is based on the latest development of HPLC technology. Watson et al.20 first described LC-MS/MS method to measure vitamin D2, vitamin D3 and their respective monohydroxy and dihydroxy metabolites. This method has been greatly developed in clinical laboratories in the past 10–15 years, especially for quantitative analysis of low molecular weight analytes, such as vitamins, hormones and steroids. In recent years, owing to inherent specificity and sensitivity, LC-MS/MS has become a popular method and become the gold standard for the detection of vitamin D and its metabolites.19 This technique also has ability to simultaneously measure 25OHD2 and 25OHD3 and can separate them.15
Vitamin D metabolites have the low ionization efficiency due to the lack of easily charged groups in electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) sources. Derivatization techniques have been developed to improve the detection response of low ionized compounds.7,21 However, the conjugated diene groups of vitamin D metabolites make it a specific target for Diels–Alder derivatization. 4-Phenyl-1,2,4-triazolin-3,5-dione (PTAD) reagent has been widely used in the synthesis of vitamin D compounds with s-cis-diene group. Derivatization reagents introduce polar groups, so the sensitivity of derivatization reagents is usually 10 times higher than that of non-derivatized compounds.19,22
In addition, the components of biological samples are too complex and contain more endogenous interference. When using this technology to analyze samples, the samples will pollute the instrument and the chromatographic column. If the sample extraction is not pure, it will produce a relatively high matrix effect, so the sample pretreatment technology process has attracted more and more attention of researchers.23 Generally, the pretreatment methods of samples include protein precipitation,24 liquid phase extraction,25 solid phase extraction7 and supported liquid–liquid extraction.26 Protein precipitation can release vitamin D metabolites which are closely bound with protein. Solid phase extraction (SPE) used to extract vitamin D metabolites from complex samples is labor-intensive and time-consuming, and resulting in low recovery with 59.1 and 60.2% for vitamin D2 and vitamin D3.27 While liquid–liquid extraction (LLE) is a classical and widely used technique in the analysis of vitamin D and its metabolites. The LLE method is simple and time-saving. The commonly used liquid–liquid extraction solvents are methyl tert-butyl ether, hexane, ether, ethyl acetate, chloroform or the mixture of non-polar solvents. In vitamin D analysis, LLE includes the addition and mixing of solvents with relatively small polarity and incompatible with water to extract hydrophobic vitamin D compounds. According to the subsequent analytical techniques, the organic layer is separated, the solvent is removed, and the sample is recombined in a suitable solvent.25
In this study, a highly sensitive method based on LLE and derivatization combined with LC-MS/MS was developed to achieve simultaneous analysis of the vitamin D metabolites. Initially, the vitamin D metabolites in biological sample were captured using LLE method. Then, a derivatization with high efficiency was carried out by introducing the derivative reagent of PTAD via Diels–Alder reaction. Next, a LC-MS/MS approach was used to analyze the derivatives of the vitamin D metabolites. The linearity, limit of detection (LOD), limit of quantitation (LOQ), matrix effects, recovery and precision were further investigated. Finally, the established method was used to the analysis of human serum vitamin D metabolites from healthy persons, diabetes and hyperlipidemia patients to prove its practicability and applications.
2. Materials and methods
2.1 Chemicals and reagents
25-Hydroxyvitamin D2 (25OHD2), 25-hydroxyvitamin D3 (25OHD3), 1,25-dihydroxyvitamin D2 [1,25(OH)2D2], 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] and internal standard [26,26,26,27,27,27-2H6]-25-hydroxy vitamin D3 (25OHD3-d6) were purchased from Zhen Zhun company (Shanghai, China). The purity of the above standards is more than 98%. 4-Phenyl-1,2,4-triazole-3,5-dione (PTAD) was obtained from French Latin company (Shanghai, China). Acetonitrile (ACN, LC grade), methanol (MeOH, LC grade), methyl tert-butyl ether (MTBE) and ethyl acetate were purchased from J&K Scientific Ltd (Beijing, China). Formic acid (FA, analytic grade, 88%) was acquired form Kernel (Tianjin, China). Nitrogen (N2) was purchased from Da Te Gas Co, Ltd (Dalian, China). Ammonium hydroxide solution was purchased from Sigma-Aldrich (St. Louis, MO, USA).
2.2 Stock solutions preparation
Vitamin D metabolites were obtained as standard solutions in ethanol and they were diluted with methanol. 1 mg internal standard (25OHD3-d6) was dissolved in 1 mL methanol in the original vial and then transferred into an amber glass HPLC vial. The internal standard work solution containing 100 ng mL−1 25OHD3-d6 in methanol was used for the calibration study. The vitamin D metabolites work solutions with concentrations of 1–100 ng mL−1 were prepared for standard curves. 10 mg PTAD was dissolved in 1 mL ethyl acetate with a concentration of 10 mg mL−1. All the solutions were stored in amber vials at −20 °C in the darkness until use.
2.3 Sample collection
In this study, the human serum samples from healthy people and patients with diabetes and hyperlipidemia were collected from Dalian Friendship People's Hospital. Before laboratory study, written informed consent was signed by all volunteers, and the research protocol (no. 2021(002)) was approved by the ethics committee of Dalian Medical University. The human serum samples without hemolysis, jaundice and cyclosis were frozen at −80 °C before use.
2.4 Sample pretreatment
The human serum was thawed, and 100 μL aliquots of human serum in 1.5 mL EP tubes were spiked with 50 μL internal standard (25OHD3-d6) solution with a concentration of 200 ng mL−1 in methanol and allowed to equilibrate for 3 min at room temperature. The sample was added with 400 μL methanol to precipitate proteins, and then vortexed on a vortex mixer for 1 min, followed by centrifugation at 3500×g for 10 min. The supernatant was transferred into 1.5 mL EP tube and spiked with 400 μL MTBE and vortexed using a vortex mixer for 1 min, then centrifuged at 3500×g for 10 min, and subsequently the upper organic layer was transferred into 1.5 mL EP tube. The residue solution was added 200 μL MTBE and mixed using a vortex mixer for 1 min, and then centrifuged at 3500×g for 10 min. Finally, two extraction solutions were merged, and were dried by N2 gas stream. A 50 μL aliquot of 0.4 mg mL−1 PTAD in ethyl acetate was added to above dried sample with vortexing for 1 min, and then allowed to react at 60 °C for 10 min. In order to terminate the reaction, 50 μL ethanol was added to the mixture. The above mixed solution was evaporated to complete dryness under N2 gas stream. The dry residue was reconstituted with methanol–water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) containing 0.1% FA, and centrifuged for 10 min at 10000×g before transferring the supernatant into UHPLC vials. An aliquot (10 μL) was injected into the LC-MS/MS system for analysis. AA schematic diagram of the vitamin D metabolites analysis process of human serum samples was displayed in Fig. 2.
:1, v/v) containing 0.1% FA, and centrifuged for 10 min at 10000×g before transferring the supernatant into UHPLC vials. An aliquot (10 μL) was injected into the LC-MS/MS system for analysis. AA schematic diagram of the vitamin D metabolites analysis process of human serum samples was displayed in Fig. 2.
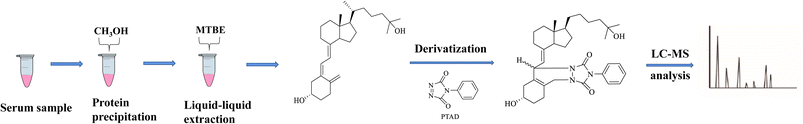 |
| | Fig. 2 Schematic diagram of the vitamin D analysis process of human serum samples. | |
2.5 Optimization of solvents to precipitate protein
To investigate protein precipitation efficiency, ACN and CH3OH were commonly used as protein precipitation solvents. For each protein precipitation, 400 μL of individual solvent was added to 100 μL of human serum spiked with mixed standard solution at the concentration of 500 ng mL−1. And the above mixture was vortexed for 1 min. The remaining steps were the same as that described in Section 2.4. According to the signal response intensity of the vitamin D metabolites, the best protein precipitation solvent was selected.
2.6 Optimization of extraction conditions
In order to extract as much vitamin D metabolites as possible, common extraction solvents, including methyl tert-butyl ether (MTBE) and chloroform-methanol (CHCl3–CH3OH, v/v = 2:1) were examined. For each extraction, 400 μL of individual solvent was added to the remaining solution after protein precipitation, and the mixed solution was vortexed for 1 min, followed by centrifugation at 3500×g for 10 min. The remaining steps were the same as Section 2.4. In addition, the extraction times was also investigated. Six replicated samples were prepared, each three was in a group. Experimental steps of the first group were carried out as the same as Section 2.4, while the experimental steps of the second group were different from those of the first group only in the extraction times. The second group was extracted with MTBE after protein precipitation and then dried with nitrogen streams. The other steps were consistent with those described in Section 2.4.
2.7 Optimization of derivatization conditions
The molar weight of the derivatization reagent has a key effect on the derivative reaction. In order to make the derivatization reagent to react with vitamin D metabolites completely in human serum samples, the ratio of the derivative substance to the standard substance with a certain molar amount was investigated to select the optimal molar amount of the derivatization reagent. 18 EP tubes divided into six groups were prepared, 100 μL 100 ng mL−1 vitamin D metabolites mixed standard solution was added to each EP tube, then the PTAD at six different concentrations were added. The molar ratios of the derivatization reagent to the standard were 2, 20, 200, 2000, 9000, and 12000. The mixtures were vortexed for 1 min, and then incubated in a water bath at 60 °C for 10 min. The excess PTAD was reacted with 50 μL ethanol. Then the mixtures were vortexed for 1 min and centrifuged at 10000×g for 10 min. Subsequently, 200 μL of supernatant was transferred to an HPLC vial and analyzed using LC-MS/MS. Finally, the corresponding signal intensities were compared. Moreover, for the sake of improving derivatization efficiency, the derivatization reaction time was optimized. 1.5 mL 100 ng mL−1 vitamin D metabolites mixed standard solutions were prepared, 100 μL 0.4 mg mL−1 PTAD was added into each tube. The above mixture was vortexed for 1 min, and then incubated in a water bath at 60 °C for 5 min, 10 min, 20 min, 40 min, and 60 min, respectively. 50 μL ethanol was added to terminate the reaction. After vortexed for 1 min and centrifuged at 10000×g for 10 min, 200 μL of the supernatant was transferred into an HPLC vial and analyzed by LC-MS/MS. The corresponding signal intensities were compared.
2.8 Optimization of redissolved solvents
Prior to LC-MS/MS analysis, it is essential to redissolve the dried samples. According to the polarity of vitamin D, methanol, ethanol and acetonitrile were selected as the redissolving solvent. The dried sample was redissolved with methanol, ethanol and acetonitrile, respectively. A certain proportion of water was added to improve the ionization strength. After the sample was dried in nitrogen streams, the sample was dissolved with the above-mentioned proportion of solvent, the mixed solution was vortexed for 1 min, followed by centrifugation at 3500×g for 10 min. Then the samples were analyzed by LC-MS/MS. The corresponding signal intensities were compared, and the group of redissolved solvents with better effect was selected.
2.9 LC-MS/MS analysis
The LC-MS analysis was performed with an Agilent 1290 UHPLC system, coupled with an API 3200 MS/MS system (Applied Biosystems SCIEX, USA). An ACQUITY UPLC separation module was used for separation. The UPLC was carried out on a CSH C18 column (2.1 mm × 100 mm, 1.7 μm, Waters Corporation, USA). The column temperature was kept at 50 °C. Samples were eluted at a flow rate of 0.3 mL min−1 using binary phases. Phase A was water (0.1% formic acid and 10 mM ammonium acetate) and phase B was methanol (0.1% formic acid and 10 mM ammonium acetate). The linear gradient conditions were as follows: 0 min, 98% B; 1 min, 98% B; 2 min, 65% B; 4–7 min, 100% B; 10 min, 2% B; 12 min, 2% B. The total chromatographic run time was 12 min. The injection volume was 10 μL.
Mass spectrometer conditions were set as follows: CUR at 25 psi, GS1 at 45 psi, GS2 at 45 psi, IS at 5500 V, and TEM at 500 °C. The mass spectrometer was auto-equilibrated for 3 min. According to the structural characteristics of vitamin D metabolites, positive ion mode was used. The mass spectrometer was performed in the multiple reaction monitoring (MRM) mode. Diluted standards were used to optimize delustering potential (DP), entrance potential (EP), collision potential (CEP), Collision chamber outlet voltage (cell exit potential, CXP) and collision energies (CE). The acquisition parameters for vitamin D analytes were listed in Table 1.
Table 1 MRM parameters of the derivatives of the four vitamin D metabolites and internal standarda
| Analytes |
Precursor/product (Da) |
DP (V) |
EP (V) |
CEP (V) |
CE (V) |
CXP (V) |
Ion mode |
Retention time (RT) |
| DP: delustering potential; EP: entrance potential, the inlet voltage that can direct and focus ions through the high-voltage Q0 region; CEP: collision entrance potential; CE: collision energy; CXP: cell exit potential. |
| 25(OH)D2 |
570.1/298.0 |
60 |
6 |
10 |
30 |
6 |
+ |
6.44 |
| 25(OH)D3 |
558.5/298.1 |
60 |
5 |
8 |
25 |
12 |
+ |
6.40 |
| 1,25(OH)2D2 |
586.3/314.2 |
60 |
5 |
7 |
28 |
6 |
+ |
6.23 |
| 1,25(OH)2D3 |
574.2/314.2 |
60 |
6 |
12 |
22 |
13 |
+ |
6.18 |
| 25(OH)D3-d6 |
564.2/298.2 |
60 |
5 |
8 |
23 |
13 |
+ |
6.38 |
2.10 Method validation
In order to check the reliability of the established method, the linearity, limit of detection (LOD), limit of quantitation (LOQ), matrix effect, recovery and intra-day/inter-day precision were investigated. For the purpose of analysis of extraction recovery and matrix effect, three groups of samples were prepared. The vitamin D metabolites were added to human serum samples to form three spiked samples at low concentrations of 1, 10, 5, 5 ng mL−1, medium concentrations of 5, 50, 10, 10 ng mL−1, and high concentration of 20, 100, 20, 20 ng mL−1 for 25OHD2, 25OHD3, 1,25(OH)2D2 and 1,25(OH)2D3, respectively. Similarly, the standard samples were prepared at the same concentrations as above and blank samples were individually prepared. A certain concentration of deuterium isotope internal standard was added to each group of samples to minimize the matrix effect.
The matrix effect was calculated by the following formula:
| | |
R (%) = 100 × (S3 − S2)/S1
| (1) |
The formula for calculating the recovery is as follows:
| | |
R (%) = 100 × (S3 − S2)/S0
| (2) |
where
S0 referred to the relative peak area of vitamin D metabolites under the corresponding concentration standard curve,
S1,
S2 and
S3 represented the peak area of vitamin D metabolites in standard solutions, blank human serum samples and spiked human serum samples
via the same pretreatment conditions, respectively.
Linear regression of the standard concentrations on the theoretical peak area ratios of the target vitamin D metabolites and its corresponding internal standard was used to evaluate linearity. The intra-day precision was estimated by measuring five times in a single day, while the inter-day precision was estimated by measuring five times for 3 consecutive days.
3. Results and discussion
3.1 Optimization of deproteinized solvent
Protein precipitation is a common method used to release vitamin D metabolites from human serum biding proteins. In this study, methanol and acetonitrile were selected as two commonly used protein precipitation reagents for protein precipitation28 and the protein precipitation effects of the two groups were compared. The results were shown in Fig. 3a. The corresponding intensity of each vitamin D metabolite was 100% when deproteinized with methanol, while the corresponding intensity of each vitamin D metabolite when deproteinized with acetonitrile was a percentage of the corresponding strength of methanol. The results showed that the intensity of vitamin D metabolites was higher than that of acetonitrile when using methanol for protein precipitation detection, showing a higher extraction efficiency with methanol. Because of the hydroxyl group, 25OHD and 1,25(OH)2D have good solubility in methanol. Finally, methanol was selected to precipitate protein for detection of vitamin D metabolites in human serum.
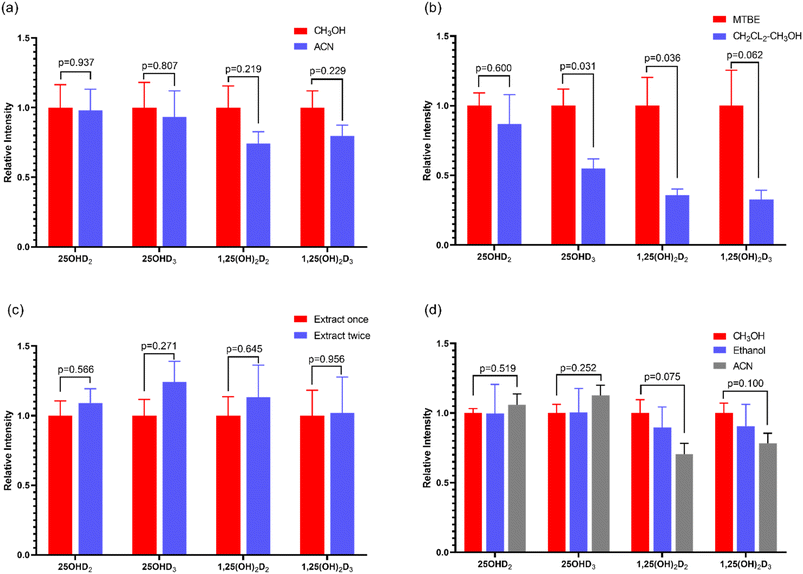 |
| | Fig. 3 MS intensity of vitamin D metabolites with protein precipitation by CH3OH and ACN (a), extracted by MTBE and CH2Cl2–CH3OH (b), extracted by MTBE for once and twice (c), and redissolved in CH3OH, ethanol and ACN (d). | |
3.2 Evaluation of the extraction approach
Through the investigation of the signal intensity of the compounds tested, the extraction effects of two vitamin D metabolites with different extraction solvents were compared. The results were displayed in Fig. 3b. The signal intensity of extraction with MTBE method was significantly higher than that of CH2Cl2–CH3OH (2:1, v/v). In addition, the extraction times were also optimized. Only one extraction may not be complete, so MTBE was used for the secondary extraction for vitamin D metabolites. Results in Fig. 3c, 25OHD2 and 25OHD3 showed obvious difference, that was, the efficiency of the twice extraction was higher than that of one extraction. However, there was no difference between the effect of 1,25 (OH)2D2 and 1,25 (OH)2D3 with extraction once or twice, which may be due to the very low content of 1,25 (OH)2D in human body, only in the range of pg mL−1. As a result, MTBE was selected as the extraction solvent of vitamin D metabolites in human serum. And it was used to extract twice.
3.3 Evaluation of derivatization approach
Some studies have shown that PTAD-derivatization might improve the ESI-MS/MS response of vitamin D metabolites, thus the quite low abundant analytes in biological fluids was allowed to be detected.29 In our study, the influence of the molar amount of PTAD on the signal intensity of vitamin D metabolites was investigated, and the optimal molar amount of PTAD was selected. The results were shown in Fig. 4a–d, the ratio of the molar amount of PTAD to that of the standard is 9000 As the molar amount of PTAD increases, the signal response intensity does not change much, which was almost the same as reported in many studies. Moreover, some studies have clarified that if the molar amount of PTAD is too high, it will inhibit the rate of derivatization reaction. The derivatization time from 5 to 60 min was also examined, as is shown in Fig. 4e–h. It is indicated in previous studies that derivatization time was 30–60 min.7 There are even 24 hours long for the derivatization reaction in some studies. Besides, the derivatization temperature has a key effect on the derivatization between vitamin D metabolites and PTAD. It can be seen from Fig. 4i–l, the derivatization reaction was more completed with the increase of temperature from 25 °C to 70 °C. When the derivatization temperature was at 60 °C, almost all vitamin D metabolites were derivatized. Further increase of temperature would be unfavorable to the stability of vitamin D metabolites. Thus, the derivatization for the vitamin D metabolites was carried out at 60 °C. It is indicated in this study that the optimum conditions for derivatization were PTAD at 0.4 mg mL−1 in ethyl acetate and an incubation time of 10 min at 60 °C. In our study, the completion time of the derivatization reaction is shorter, saving a lot of pretreatment time. This also shows that the rate of the derivatization reaction is fast and the conditions of the derivatization reaction are mild.
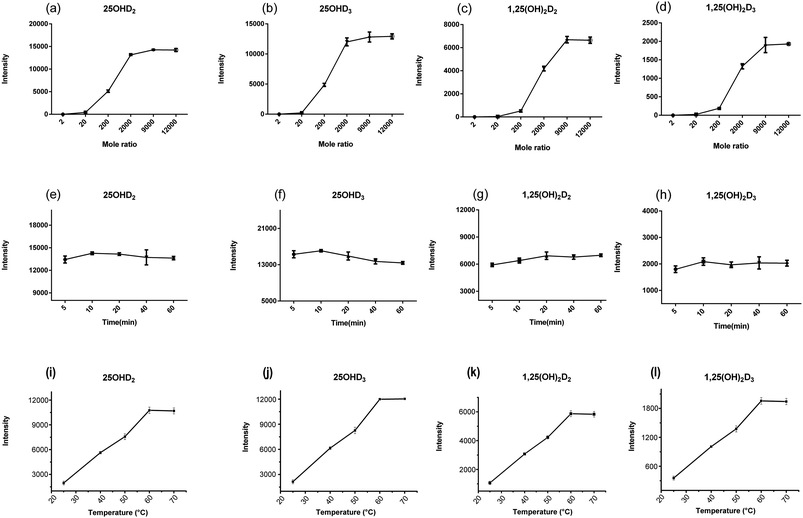 |
| | Fig. 4 The optimization of the amount of derivative reagent (a–d), derivatization time (e–h) and derivatization temperature (i–l) for the vitamin D metabolites. | |
After derivatization, the sensitivity of the vitamin D metabolites was increased by 10 to 66 times. The mass chromatogram response of the metabolites before and after derivatization with the derivatization reagent PTAD was shown in Fig. 5a and b, respectively.
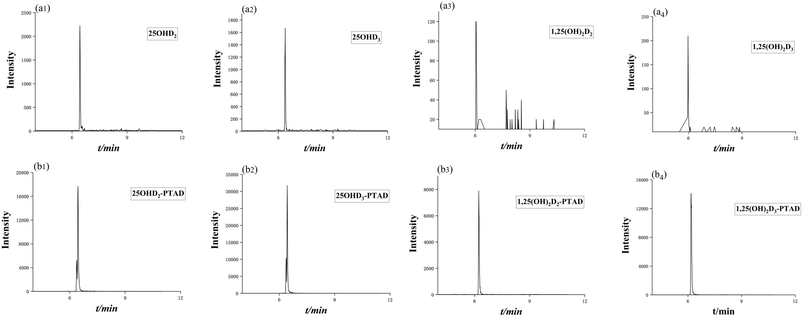 |
| | Fig. 5 Mass chromatogram response of vitamin D metabolites before (a) and after derivatization (b). | |
3.4 Optimization of redissolving solvents
In order to dissolve the detected vitamin D as much as possible, the solvent with good solubility was selected for redissolution. In this study, methanol, ethanol and acetonitrile were selected as the solvents. According to the signal response intensity of the sample after redissolution, the optimal solvent was selected. The results were shown in Fig. 3d. For 25OHD2 and 25OHD3, the solubilization effect of acetonitrile was better than that of methanol and ethanol. The properties of methanol and ethanol were similar, and the solubility of vitamin D metabolites in these two solvents was similar. However, for 1,25(OH)2D2 and 1,25(OH)2D3, methanol is more soluble than 1,25(OH)2D3. Due to the low content of 1,25(OH)2D2 and 1,25(OH)2D3 in vivo, it is difficult to be detected. Therefore, methanol was selected as the redissolving solvent. When the ratio of methanol to water was 9:1, the response intensity of vitamin D was higher.
3.5 Validation of the method
3.5.1 Linear range, LOD and LOQ. The linearity of vitamin D metabolites was generated by plotting the peak area ratio (analyte/internal standard) versus six levels of concentrations. Co-mingled calibration standards were prepared at different levels in methanol. Internal standard solution was prepared in methanol at a concentration of 100 ng mL−1. The calibration curve was composed of the ratio of the chromatographic peak area of the substance measured and the chromatographic peak area of the internal standard substance. As summarized in Table 2, good linearities were acquired over wide concentration ranges with 1–100 ng mL−1 for 25OHD2 and 25OHD3, 5–100 ng mL−1 for 1,25(OH)2D2 and 1,25(OH)2D3, and the correlation coefficients (R2) were more than 0.998. The limits of detection (LOD) were 0.3, 0.3, 1.5 and 1.5 ng mL−1 for 25OHD2, 25OHD3, 1,25(OH)2D2 and 1,25(OH)2D3, respectively. The limits of quantification (LOQ) were 1.0, 1.0, 5.0 and 5.0 ng mL−1 for 25OHD2, 25OHD3, 1,25(OH)2D2 and 1,25(OH)2D3, respectively.
Table 2 The linear range, R2, LOD and LOQ for the derivatives of the four vitamin D metabolites
| Compounds |
Linear equation |
Linear range (ng mL−1) |
R2 |
LOD (ng mL−1) |
LOQ (ng mL−1) |
| 25(OH)D2 |
Y = 0.0076x + 0.0022 |
1–100 |
0.9999 |
0.3 |
1.0 |
| 25(OH)D3 |
Y = 0.0073x + 0.0181 |
1–100 |
0.9996 |
0.3 |
1.0 |
| 1,25(OH)2D2 |
Y = 0.0039x − 0.0008 |
5–100 |
0.9984 |
1.5 |
5.0 |
| 1,25(OH)2D3 |
Y = 0.0014x − 0.0013 |
5–100 |
0.9995 |
1.5 |
5.0 |
3.5.2 The matrix effects, recovery and precision. To evaluate the reliability of the developed method, the matrix effects, recovery and intra-day/inter-day precision were further investigated. As listed in Table 3, the matrix effects of the derivatives of vitamin D metabolites at low, medium, high levels were 67.55–91.41%, 73.25–111.13% and 64.68–95.88%, respectively. And the coefficient of variations (CVs) of all the analytes at three concentration levels were in the range of 1.52–11.01%. It indicated that the matrix effect produced by endogenous matrix was acceptable. The recovery is mainly to determine whether the established sample treatment method could completely extract the components to be analyzed in the sample. The average recoveries of the derivatives of vitamin D metabolites were 81.66–110.31%. And the CVs of all the analytes at three levels were in the range of 0.91–13.95%. The relative standard deviations (RSDs) of intra-day precision were 2.92–11% and the RSDs of inter-day precision were 0.20–6.82%. These results are satisfying and acceptable, suggesting the method has a good accuracy.
Table 3 Matrix effect, recovery, intra- and inter-day precision of analytes in human serum
| Spiking analytes |
Concentration (ng mL−1) |
Matrix effect (n = 3) |
Recovery (n = 3) |
Intra-day (n = 5) |
Inter-day (n = 15) |
| Mean ± SD |
CV (%) |
Mean ± SD |
CV (%) |
RSD (%) |
RSD (%) |
| 25OHD2 |
1 |
67.55 ± 3.94 |
3.82 |
124.11 ± 4.31 |
3.65 |
10.74 |
0.20 |
| 5 |
111.13 ± 9.73 |
8.53 |
105.63 ± 13.48 |
12.10 |
6.15 |
0.20 |
| 20 |
91.16 ± 1.25 |
1.52 |
101.19 ± 4.56 |
5.63 |
2.92 |
3.39 |
| 25OHD3 |
10 |
75.61 ± 8.52 |
10.22 |
96.78 ± 4.44 |
4.10 |
6.10 |
0.40 |
| 50 |
101.95 ± 3.53 |
3.30 |
100.43 ± 2.18 |
1.96 |
4.51 |
6.82 |
| 100 |
95.88 ± 6.91 |
5.48 |
103.65 ± 4.07 |
5.49 |
3.78 |
2.80 |
| 1,25(OH)2D2 |
5 |
91.41 ± 2.40 |
2.16 |
106.13 ± 0.97 |
0.91 |
11.01 |
0.96 |
| 10 |
73.25 ± 6.12 |
7.18 |
125.24 ± 1.90 |
1.49 |
11.22 |
2.61 |
| 20 |
72.57 ± 4.75 |
5.99 |
85.69 ± 11.36 |
11.34 |
5.26 |
5.26 |
| 1,25(OH)2D3 |
5 |
89.19 ± 6.40 |
6.43 |
76.12 ± 3.84 |
6.24 |
11.46 |
2.18 |
| 10 |
76.02 ± 3.30 |
3.02 |
96.84 ± 9.74 |
13.95 |
10.37 |
2.58 |
| 20 |
64.68 ± 7.44 |
11.01 |
72.03 ± 5.35 |
8.32 |
7.00 |
2.61 |
3.5.3 Comparison with previously reported methods. The results obtained from the developed method using LC-MS/MS combined with derivatization was compared with other previously reported methods18,30–32 as presented in Table 4. The developed method has high sensitivity. And compared with other reported method with SPE or LLE combined with SPE,32,33 only LLE pretreatment method used in this study is generally relatively simple to operate and requires more common equipment and reagents. Besides, compared with the reported method with 0.5–1 h for derivatization time,7 only 10 min for derivatization time was needed in this work, saving pretreatment time, which is very conducive to high throughput analysis.
Table 4 Comparison of the developed method versus some previously reported methods for determination of vitamin D2 and vitamin D3
| Analyte |
Methods |
Linearity |
Limit of detection |
References |
| Vitamin D2 [25(OH)D2] |
LLE + derivatization + LC-MS/MS |
1.5–48 nmol L−1 |
0.3 nmol L−1 |
Ref. 30 |
| HPLC-MS/MS |
3.9–183.6 nmol L−1 |
3.9 nmol L−1 |
Ref. 31 |
| LLE + derivatization + LC-MS/MS |
1–100 ng mL−1 |
0.3 ng mL−1 |
This work |
| Vitamin D3 [25(OH)D3] |
Spectrofluorometric using Tb3+-ACV |
1.0–320 nmol L−1 |
1.19 nmol L−1 |
Ref. 18 |
| LLE + derivatization + LC-MS/MS |
7.8–250 nmol L−1 |
1.5 nmol L−1 |
Ref. 30 |
| HPLC-MS/MS |
4.0–265.3 nmol L−1 |
4.0 nmol L−1 |
Ref. 31 |
| SPE + LC-MS/MS |
1–60 ng mL−1 |
1 ng mL−1 |
Ref. 32 |
| LLE + derivatization + LC-MS/MS |
1–100 ng mL−1 |
0.3 ng mL−1 |
This work |
3.6 Determination of vitamin D metabolites in human serum samples from healthy subjects and patients with diabetes and hyperlipidemia
As an endogenous substance, vitamin D metabolites may have significant variation and disorder in healthy people and some metabolic diseases. The developed derivatization and LC-MS/MS method was applied to analyze the vitamin D metabolites in human serum samples of healthy people (n = 10), diabetes (n = 10) and hyperlipidemia (n = 10) patients. Among all the vitamin D metabolites, 25OHD2 and 25OHD3 could be detected by using the established method. The MRM chromatograms of the vitamin D metabolites from the standard mixture and the human serum sample were shown in Fig. 6a and b. As a result, the concentrations of 25OHD2 in the human serum samples from 10 healthy people, 10 diabetes and 10 hyperlipidemia patients were 1.00–7.80 ng mL−1, 1.10–4.04 ng mL−1 and 1.00–2.32 ng mL−1, respectively. And the concentrations of 25OHD3 in the above three groups were 19.66–52.30 ng mL−1, 9.86–20.83 ng mL−1 and 10.00–41.00 ng mL−1, respectively. The contents of vitamin D metabolites in human serum determined in this study were in accordance with the order of magnitude of the contents in the literatures.23,24 Univariate analysis showed that total contents of 25OHD2 and 25OHD3 had significant difference (p < 0.01) between normal subjects and patients with diabetes as well as hyperlipidemia (Fig. 7). Most experts believed that the concentration of 25OHD below 20 ng mL−1 (50 nmol L−1) were signs of vitamin D deficiency, while the concentration with 21–29 ng mL−1 (51–74 nmol L−1) were considered to be signs of vitamin D insufficiency.28 Most studies in the literature showed that the content of 25OHD in patients with diabetes mellitus was deficient.34 Vitamin D insufficiency may contribute to the pathogenesis of diabetes, and epidemiological evidence suggests that it is associated with insulin resistance.35 The content of 25OHD measured in this study was in the range of deficiency in patients of diabetes, which is in line with expectations.34 The content of 25OHD measured in patients with hyperlipidemia in this study was also in the range of deficiency. It was reported that vitamin D deficiency is associated with hyperlipidemia, and elevated human serum 25OHD levels were associated with decreased human serum TC and TG levels, especially in patients with hyperlipidemia who were vitamin D deficiency.14 Therefore, increasing the concentration of circulating 25OHD may be used as an adjunct therapy for hyperlipidemia.14 It has a certain reference value for clinical diagnosis and treatment.
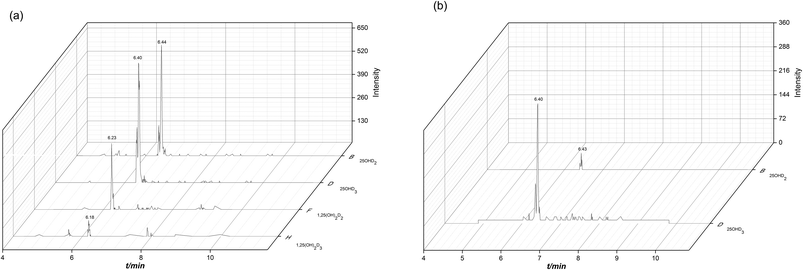 |
| | Fig. 6 MRM chromatograms of the derivatives of the vitamin D metabolites obtained from standard mixture (a) and the human serum sample (b). | |
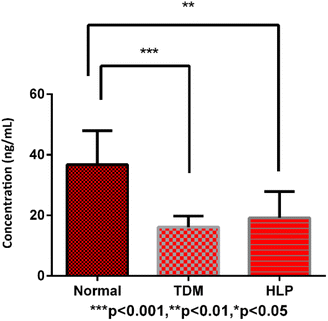 |
| | Fig. 7 Univariate analysis of the vitamin D metabolites from normal subjects, diabetes (TDM) and hyperlipidemia (HLP) patients. | |
4. Conclusions
In this study, a novel method based on liquid–liquid extraction, derivatization and LC-MS/MS was established to simultaneously analyze the vitamin D metabolites. PTAD was used to derivatize the vitamin D metabolites. The sample pretreatment process of the method was simple and rapid, and the derivatization reaction conditions were mild. Moreover, the method with good linearity, precision, recovery and sensitivity was reliable to analyze the vitamin D metabolites in real samples. The method showed good practicability in studying the vitamin D metabolites in relation to metabolic diseases. The results indicated that there were significant differences in human serum 25OHD levels between healthy individuals, diabetes and hyperlipidemia patients. Consequently, the developed method was useful to determine the vitamin D metabolites, and it's promising to apply it to the research of other vitamin D metabolites-related diseases.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the financial support of the foundations from Natural Science Foundation of China (No. 22104008) and Natural Science Foundation of Liaoning (No. 20170540270, No. LZ2020043, No. 2021-BS-217, No. 2022-BS-241). All the authors have declared no financial/commercial conflicts of interest.
Notes and references
- D. A. Volmer, L. R. Mendes and C. S. Stokes, Analysis of vitamin D metabolic markers by mass spectrometry: current techniques, limitations of the “gold standard” method, and anticipated future directions, Mass Spectrom. Rev., 2015, 34(1), 2–23 CrossRef CAS PubMed.
- E. van Etten, C. Gysemans, D. D. Branisteanu, A. Verstuyf, R. Bouillon and L. Overbergh, et al., Novel insights in the immune function of the vitamin D system: synergism with interferon-beta, J. Steroid Biochem. Mol. Biol., 2007, 103, 546–551 CrossRef CAS PubMed.
- S. Samuel and M. D. Sitrin, Vitamin D's role in cell proliferation and differentiation, Nutr. Rev., 2008, 66(10 Suppl 2), S116–S124 CrossRef PubMed.
- H. E. Harrison, Vitamin D, Prog. Clin. Biol. Res., 1981, 61, 95–108 CAS.
- Á. Gil, J. Plaza-Diaz and M. D. Mesa, Vitamin D: Classic and Novel Actions, Ann. Nutr. Metab., 2018, 72(2), 87–95 CrossRef PubMed.
- S. Yu, W. Zhou, D. Wang, Y. Yin, Q. Cheng and S. Xie, et al., Rapid liquid chromatography-tandem mass spectrometry method for determination of 24,25(OH)(2)D and 25OHD with efficient separation of 3-epi analogs, J. Steroid Biochem. Mol. Biol., 2019, 187, 146–151 CrossRef CAS PubMed.
- C. J. Hedman, D. A. Wiebe, S. Dey, J. Plath, J. W. Kemnitz and T. E. Ziegler, Development of a sensitive LC/MS/MS method for vitamin D metabolites: 1,25 Dihydroxyvitamin D2&3 measurement using a novel derivatization agent, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 953–954, 62–67 CrossRef CAS PubMed.
- H. Lyu, S. Wang, Y. Jin, R. Shen, J. Chen and C. Zhu, et al., Simultaneous determination of VD2, VD3, 25(OH) D2, and 25(OH) D3 in human plasma using electrospray LC-MS/MS as well as its application to evaluate VD plasma levels in depressive, schizophrenic patients and healthy individuals, Biomed. Chromatogr., 2020, 34(11), e4932 CrossRef CAS PubMed.
- A. Hossein-nezhad and M. F. Holick, Vitamin D for health: a global perspective, Mayo Clin. Proc., 2013, 88(7), 720–755 CrossRef CAS PubMed.
- V. Hirani, R. G. Cumming and D. G. Couteur, et al., Low levels of 25-hydroxy vitamin D and active 1,25-dihydroxy vitamin D independently associated with type 2 diabetes mellitus in older Australian men: the concord health and ageing in men project, J. Am. Geriatr. Soc., 2014, 62, 1741–1747 CrossRef PubMed.
- Y. Song, L. Wang and A. G. Pittas, et al., Blood 25-hydroxy vitamin D levels and incident type 2 diabetes: a meta-analysis of prospective studies, Diabetes Care, 2013, 36, 1422–1428 CrossRef CAS PubMed.
- P. Lucato, M. Solmi and S. Maggi, et al., Low vitamin D levels increase the risk of type 2 diabetes in older adults: a systematic review and meta-analysis, Maturitas, 2017, 100, 8–15 CrossRef CAS PubMed.
- C. Lu, Y. Yin and Y. Cui, et al., 1,25(OH)2D3 improves blood lipid metabolism, liver function, and atherosclerosis by constraining the TGF-beta/Smad signaling pathway in rats with hyperlipidemia, Cell Cycle, 2019, 18(22), 3111–3124 CrossRef CAS PubMed.
- Y. L. Cheng, T. I. Lee and Y. M. Chien, et al., Vitamin D level regulates serum lipids discrepantly in adults with and without dyslipidemia, Endocr. Connect., 2023, 12(9), e230013 CAS.
- S. W. Zhang, W. Jian, S. Sullivan, B. Sankaran, R. W. Edom and N. Weng, et al., Development and validation of an LC-MS/MS based method for quantification of 25 hydroxyvitamin D2 and 25 hydroxyvitamin D3 in human serum and plasma, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 961, 62–70 CrossRef CAS PubMed.
- F. G. Strathmann, T. J. Laha and A. N. Hoofnagle, Quantification of 1α,25-dihydroxy vitamin D by immunoextraction and liquid chromatography-tandem mass spectrometry, Clin. Chem., 2011, 57(9), 1279–1285 CrossRef CAS PubMed.
- C. T. Sempos, E. Lindhout and N. Heureux, et al., Towards harmonization of directly measured free 25-hydroxyvitamin D using an enzyme-linked immunosorbent assay, Anal. Bioanal. Chem., 2022, 414(27), 7793–7803 CrossRef CAS PubMed.
- S. Alharthi, M. S. Attia and M. N. Abou-Omar, et al., pH assists simultaneous determination of folic acid and vitamin
D(3) in biological fluids using a novel Tb(3+)-acyclovir optical biosensor, RSC Adv., 2021, 11(34), 20865–20873 RSC.
- P. A. Aronov, L. M. Hall, K. Dettmer, C. B. Stephensen and B. D. Hammock, Metabolic profiling of major vitamin D metabolites using Diels-Alder derivatization and ultra-performance liquid chromatography-tandem mass spectrometry, Anal. Bioanal. Chem., 2008, 391(5), 1917–1930 CrossRef CAS PubMed.
- D. Watson, K. D. Setchell and R. Ross, Analysis of vitamin D and its metabolites using thermospray liquid chromatography/mass spectrometry, Biomed. Chromatogr., 1991, 5(4), 153–160 CrossRef CAS PubMed.
- T. Higashi, K. Shimada and T. Toyo'oka, Advances in determination of vitamin D related compounds in biological samples using liquid chromatography-mass spectrometry: a review, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878(20), 1654–1661 CrossRef CAS PubMed.
- N. S. Abu Kassim, F. P. Gomes, P. N. Shaw and A. K. Hewavitharana, Simultaneous quantitative analysis of nine vitamin D compounds in human blood using LC-MS/MS, Bioanalysis, 2016, 8(5), 397–411 CrossRef CAS PubMed.
- M. J. Müller and D. A. Volmer, Mass spectrometric profiling of vitamin D metabolites beyond 25-hydroxyvitamin D, Clin. Chem., 2015, 61(8), 1033–1048 CrossRef PubMed.
- I. Shah, R. James, J. Barker, A. Petroczi and D. P. Naughton, Misleading measures in Vitamin D analysis: a novel LC-MS/MS assay to account for epimers and isobars, Nutr. J., 2011, 10, 46 CrossRef CAS PubMed.
- F. P. Gomes, P. N. Shaw, K. Whitfield, P. Koorts and A. K. Hewavitharana, Recent trends in the determination of vitamin D, Bioanalysis, 2013, 5(24), 3063–3078 CrossRef CAS PubMed.
- C. Bylda, R. Thiele, U. Kobold and D. A. Volmer, Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS, Analyst, 2014, 139(10), 2265–2276 RSC.
- A. Mena-Bravo, C. Ferreiro-Vera and F. Priego-Capote, et al., Quantitative analytical method to evaluate the metabolism of vitamin D, Clin. Chim. Acta, 2015, 442, 6–12 CrossRef CAS PubMed.
- M. F. Holick, N. C. Binkley, H. A. Bischoff-Ferrari, C. M. Gordon, D. A. Hanley and R. P. Heaney, et al., Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline, J. Clin. Endocrinol. Metab., 2011, 96(7), 1911–1930 CrossRef CAS PubMed.
- A. Alexandridou, P. Schorr and D. A. Volmer, Comparing derivatization reagents for quantitative LC-MS/MS analysis of a variety of vitamin D metabolites, Anal. Bioanal. Chem., 2023, 415(19), 4689–4701 CrossRef CAS PubMed.
- S. Zelzer, A. Meinitzer and D. Enko, et al., Simultaneous determination of 24,25- and 25,26-dihydroxyvitamin D3 in serum samples with liquid-chromatography mass spectrometry - A useful tool for the assessment of vitamin D metabolism, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, 1158, 122394 CrossRef CAS PubMed.
- S. Baecher, A. Leinenbach and J. A. Wright, et al., Simultaneous quantification of four vitamin D metabolites in human serum using high performance liquid chromatography tandem mass spectrometry for vitamin D profiling, Clin. Biochem., 2012, 45(16–17), 1491–1496 CrossRef CAS PubMed.
- S. Xu, R. Ni and L. Lv, et al., Simultaneous determination of vitamin D metabolites 25(OH)D(3) and 1alpha,25(OH)(2)D(3) in human plasma using liquid chromatography tandem mass spectrometry, J. Mass Spectrom. Adv. Clin. Lab, 2022, 24, 65–79 CrossRef CAS PubMed.
- L. Usoltseva, V. Ioutsi and Y. Panov, et al., Serum Vitamin D Metabolites by HPLC-MS/MS Combined with Differential Ion Mobility Spectrometry: Aspects of Sample Preparation without Derivatization, Int. J. Mol. Sci., 2023, 24, 8111 CrossRef CAS PubMed.
- A. G. Pittas and B. Dawson-Hughes, et al., Vitamin D Supplementation and Prevention of Type 2 Diabetes, N. Engl. J. Med., 2019, 381(6), 520–530 CrossRef CAS PubMed.
- S. Devaraj, G. Jialal, T. Cook, D. Siegel and I. Jialal, Low vitamin D levels in northern American adults with the metabolic syndrome, Horm. Metab. Res., 2011, 43(1), 72–74 CrossRef CAS PubMed.
Footnote |
| † Co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Qian Qin†a,
Fasheng Lia,
Ying Fua and
Na Liu*a
ab,
Qian Qin†a,
Fasheng Lia,
Ying Fua and
Na Liu*a






