
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiviral role of nanomaterials: a material scientist's perspective
Muhammad Aanish Ali
 a,
Nagina Rehman
b,
Tae Joo Park
*c and
Muhammad Abdul Basit
*a
a,
Nagina Rehman
b,
Tae Joo Park
*c and
Muhammad Abdul Basit
*a
aDepartment of Materials Science and Engineering, Institute of Space Technology, Islamabad 44000, Pakistan. E-mail: ab_saim@hotmail.com; m.abdulbasit@mail.ist.edu.pk
bDepartment of Zoology, Government College University Allama Iqbal Road, Faisalabad 38000, Pakistan
cDepartment of Materials Science and Chemical Engineering, Hanyang University, Ansan 15588, Republic of Korea. E-mail: tjp@hanyang.ac.kr
First published on 21st December 2022
Abstract
The present world continues to face unprecedented challenges caused by the COVID-19 pandemic. Collaboration between researchers of multiple disciplines is the need of the hour. There is a need to develop antiviral agents capable of inhibiting viruses and tailoring existing antiviral drugs for efficient delivery to prevent a surge in deaths caused by viruses globally. Biocompatible systems have been designed using nanotechnological principles which showed appreciable results against a wide range of viruses. Many nanoparticles can act as antiviral therapeutic agents if synthesized by the correct approach. Moreover, nanoparticles can act as carriers of antiviral drugs while overcoming their inherent drawbacks such as low solubility, poor bioavailability, uncontrolled release, and side effects. This review highlights the potential of nanomaterials in antiviral applications by discussing various studies and their results regarding antiviral potential of nanoparticles while also suggesting future directions to researchers.
 Muhammad Aanish Ali | Engr Muhammad Aanish received his bachelor’s degree in Materials Science and Engineering from Institute of Space Technology (IST), Islamabad, Pakistan in September 2022. He is an active researcher in the fields of development of Nanomaterials for various engineering applications such as photovoltaics and biotechnological treatment of waste water. He is currently a member of research group “Nanomaterials for Advanced Energy Applications” (NAEA) headed by Dr Basit at IST. He has participated in several review/research papers during his academic career so far. His key ambition is to conduct extensive and valuable research that could provide a good value to research community. |
 Nagina Rehman | Dr Nagina Rehman graduated in Zoology from Bahauddin Zakariya University Multan, Pakistan in 2009. Then she joined University of Agriculture Faisalabad to accomplish her MPhil Zoology in 2013 with research specialization of biodiversity. Having PhD from Government College University Faisalabad Pakistan (2020), Dr Nagina chose a multidisciplinary research career in Nanotechnology for Zoological Species. She is currently working as Assistant Professor at Riphah University, Faisalabad (Campus) and motivated to investigate and research the toxicological effect/control of nanomaterials in human beings. |
 Tae Joo Park | Dr Tae Joo Park is currently serving as Professor of Materials Science & Chemical Engineering at Hanyang University (HYU) Ansan, Republic of Korea. Dr Park has PhD from Seoul National University, Korea and research experience from University of Texas Dallas, USA. He established Nanodevice Engineering Laboratory at HYU and conducted many funded research projects in parallel to the research supervision of many local/international students in the domain of energyharvesting/storage/nanoelectronic/neuromorphic devices, film/particle ALD and 2D-electron gas. Owing to expertise in research and innovation, Dr Park has won many awards and honors from various firms including Samsung (2019) and Merck (2021) etc. |
 Muhammad Abdul Basit | Dr Muhammad Abdul Basit is serving as Associate Professor at Department of Materials Science & Engineering at IST Pakistan, where he established: “Nanomaterials for Advanced Energy Applications” research group too. After having graduation from UET Lahore, Dr Basit joined BZU Multan as Lecturer and received MS-leading PhD scholarship by Higher Education Commission of Pakistan (2011–16) for his PhD from NEL-HYU, South Korea. In addition to post-doctoral research at HYU, he won intellectual capability awards from various agencies such as PCK (2012), DAAD-Germany (2017) etc. He holds reputable record for scientific publications/keynotes/book chapters as well as research in arts (Urdu Poetry/Iqbal). |
1. Introduction
The modern world faces serious challenges due to viral infections and their outbreaks in the form of epidemics and pandemics. Viruses are basically small obligate parasites and are non-living things residing outside of the host cell. These viruses interact with cells via various mechanisms such as receptor–ligand interactions. Among these, respiratory viruses pose a crucial threat to all sorts of demographics. Researchers continue to work on their treatment and suppression by utilizing nano-based options such as functional metallic nanoparticles (NPs) and nano-enabled drug delivery systems. The drug delivery systems must be capable of dealing with inherent features of viral infections like their replication dynamics, level of drug resistance, and complex lifecycle stages. Frequent drug use causes enhancement of drug resistance against viruses and has now become a crucial health issue globally for nearly all sorts of drugs that are known to be effective against one or more specific viruses. The past two decades have witnessed the emergence of old viral infections and introduction of new types of viruses such as severe acute respiratory syndrome (SARS-CoV-2) which is a form of coronavirus.1–4 The human body can contract these viruses via various routes such as ear, nose, mouth, and skin.5 Once a virus is contracted, it is difficult for the human body to develop an immune response against it. Due to larger categories of viruses than the number of treatment options available, there are still many infections for which there is either no effective treatment or treatment has not proved to be fully effective.6Besides posing threat to human health and life, the viral outbreaks have affected human life in several ways ranging from disruption of daily life activities, economic collapse of societies, shortages of food and medical supplies, and mass-scale deaths.7,8 A recent example is of COVID-19 pandemic in which the world witnessed the peak of these problems till the time when an adequate number of vaccine doses were administered globally to most of the population and the trends showed a decline in emergence of new cases and decrease in deaths due to increased immunity among people, thanks to these vaccines.
Research has previously shown that nanomaterials possess great potential in antiviral applications.9 Materials scientists and chemists have conducted extensive research to explore and synthesize newer, efficient nanomaterials which can successfully inhibit and treat a wide range of viruses. Different functional NPs and nanostructures have shown antiviral activities against a broad spectrum of viruses, each having its mechanism and viricidal activity. Every system comes with its limitations and benefits and hence it is important to employ the most suitable candidate for treatment of specific viruses.10 Fig. 1 presents an outlook on the use of nanoparticles and nano-based drug delivery systems which have proved to be effective against many viruses.
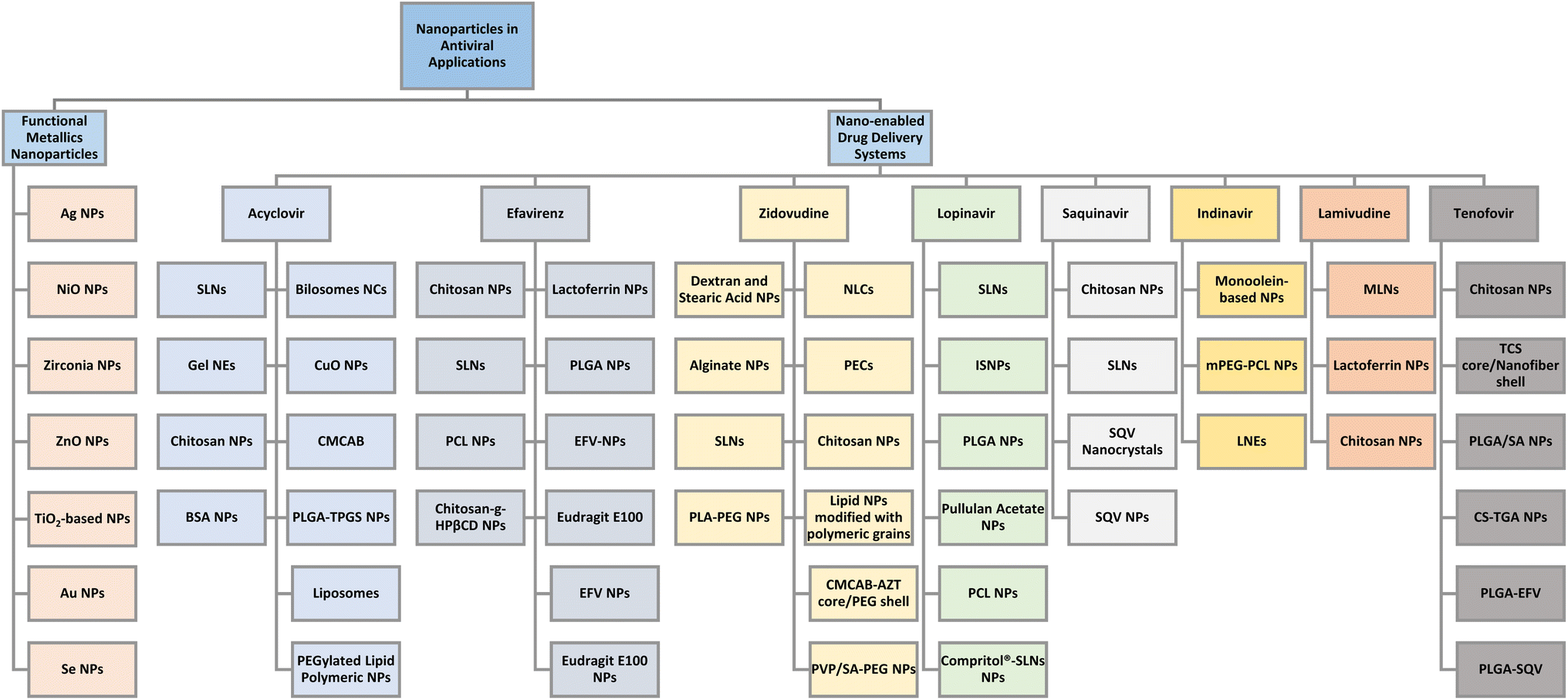 | ||
| Fig. 1 An overview of applications of nanoparticles and their combinations with a variety of antiviral drugs. | ||
Before the advent and advance of nanotechnology, antiviral drugs were extensively used for treatment of viruses. The efficiency of these drugs was limited due to their inherent low bioavailability, shorter half-life, cytotoxicity, uncontrolled release as well as serious side effects that limited their use.11,12 Material scientists have developed nanoparticles that were capable of acting as delivery vehicles for these drugs, releasing a controlled amount of drug to the targeted site for the required amount of time. They showed appreciable entrapment of drug while increasing the efficacy and enhancing the viricidal effects. The key property of nano-enabled delivery systems is that due to their extremely smaller size and high surface areas, they facilitate better digestion, absorption, and penetration of drugs.13,14 In this review, we have discussed well-known functional NPs which exhibit antiviral action, their synthesis, properties, and mechanism along with exhaustive and informative discussion of existing nano-enabled drug delivery systems which are known to have overcome the limitations of antiviral drugs such as low biocompatibility, cytotoxicity, hemocompatibility and difficulty in administration to patients, among various other issues associated with them which we have covered in this review.
2. Metal-based nanoparticles as effective antivirals
2.1 Ag-based NPs
Silver nanoparticles (AgNPs) have shown great potential in biomedical applications such as anti-bacterial and anti-cancer. However, there is only a handful of research on antiviral activity of Ag-based NPs. Results from recent studies show that AgNPs are quite effective against a wide range of viruses such as respiratory syncytial virus (RSV), enterovirus 71, herpesvirus (HSV-1 and HSV-2), poliovirus (PV), dengue virus (DENV), influenza (H1N1, H3N2), hepatitis (HSV-1, HAV-10, and CoxB4) and coronaviruses (porcine epidemic diarrhea virus (PEDV) and feline coronavirus (FCoV)).15Morris et al. reported the first in vivo study to examine the antiviral nature of AgNPs against RSV infections. A significant decrease in pro-inflammatory cytokines and chemokines was recorded in the infected mice's lungs. AgNPs were able to effectively block the entry of RSV to host cells by binding to the surface of glycoproteins and inhibiting the spread of RSV. While the development of vaccines for RSV infections remains a challenge, employing AgNPs can be a novel strategy to treat RSV-infected patients.16 While small interfering RNA (siRNA) holds promise in antiviral activity against EV71, its major limitation is its inability to cross cell membranes. The study prepared surface-decorated AgNPs using polyethyleneimine (PEI) and siRNA and monitored for their antiviral activity. The accumulation of reactive oxygen species (ROS) was effectively inhibited, and EV71 was not able to infect the host cells.17 The potential of AgNPs against PV was monitored in vitro, as the electrochemically synthesized AgNPs played a key role in the disinfection of PV, a non-enveloped virus.18
The use of AgNPs in antiviral applications is generally limited since many AgNPs are synthesized in a liquid atmosphere, a technique not easily applicable. Recent studies have suggested numerous solutions to address this limitation. Szymańska et al. reported preparation of mucoadhesive hydrogel based on tannic acid (TA)-modified AgNPs (TA-AgNPs) as an effective route for treatment of HSV-1 and HSV-2.19 For enhancing the antiviral action, the NPs were encapsulated by a hydrogel called Carbopol 974P for effective delivery to the targeted site (ex vivo vaginal mucosa). The gelation (100% crosslinking) was facilitated by an initiator, thereby providing closer contact between drug carrier and the mucosal tissue. The size of nanoparticles was found to be between 13 and 54 nm as estimated from the micrographs of transmission electron microscope (TEM) (Fig. 2(a) and (b)). The antiviral efficacy of NP-based hydrogel was evaluated in vitro. To ensure that components of hydrogel do not affect the infectivity of HSV-1 and 2, placebo hydrogel and TA-AgNPs were simultaneously applied. The scheme of virus inhibition assay is illustrated in Fig. 2c(A). Both types of HSVs were considerably inhibited after a 24 h incubation period, which indicates the potential of prepared formulation against HSV infections. However, the inhibition was roughly 20% more for HSV-2 (Fig. 2c(B)). The results shown in Fig. 2c(C) and (D) indicate that inhibition of HSV-1 and 2 is greater in cells that were exposed to hydrogels as compared to the control cells, with more inhibition in case of HSV-2. It is pertinent to mention that inhibition rate of HSV-1 infection was dependent on the concentration of the NPs in hydrogel while concentration played no role in inactivation of HSV-2. Possible mechanisms of virus suppression may be due to NP interacting with HSV envelope and blockage of virus interaction with cells due to layer of hydrogel around them. The study further explored the mode of antiviral activity of prepared formulation by employing assays of virus attachment and penetration. The variables of both assays are shown in Fig. 2d(A). After 24 h post-infection, it was observed that around 85–90% inhibition for HSV-1 attachment to the cell surface with a considerable decrease in plaque numbers (greater than 60%) was recorded for HSV-2 (Fig. 2d(B)). This time the percentage of HSV-1 inhibition after treatment with H2/NP25 (Fig. 2d(B)) was found to be greater than the inhibition of HSV-2. The penetration assay showed that HSV-2 entry to HaCaT cells was suppressed by exposure to both hydrogels. Interestingly, both formulations of hydrogel showed lower inactivation for HSV-1 in contrast to attachment assay. NPs inside the cell could have played a vital role in inhibition. Hence cell-to-cell spread assays were conducted (Fig. 2e). It can be seen that the inhibitory effect of both hydrogels on the cells infected with HSV-2 is significantly greater than that of the control and placebo (Fig. 2e(C)). In contrast, the cell-to-cell spread was inhibited for HSV-1 infected cells only by the hydrogel with 50 ppm TA-AgNPs (Fig. 2e(D)). The Carbopol 974P provided two distinct properties; transporting antiviral (TA-AgNPs) to targeted site and inherent antiviral activity. Therefore, a synergistic effect of both hydrogel and NPs played a key role in suppression of HSV infection. The authors, however, suggested the need for future in vivo studies for further exploration and innovation of this novel route (readers are suggested to consult the cited literature for information about the specific composition of the H2/NP25 Hydrogel formulation).
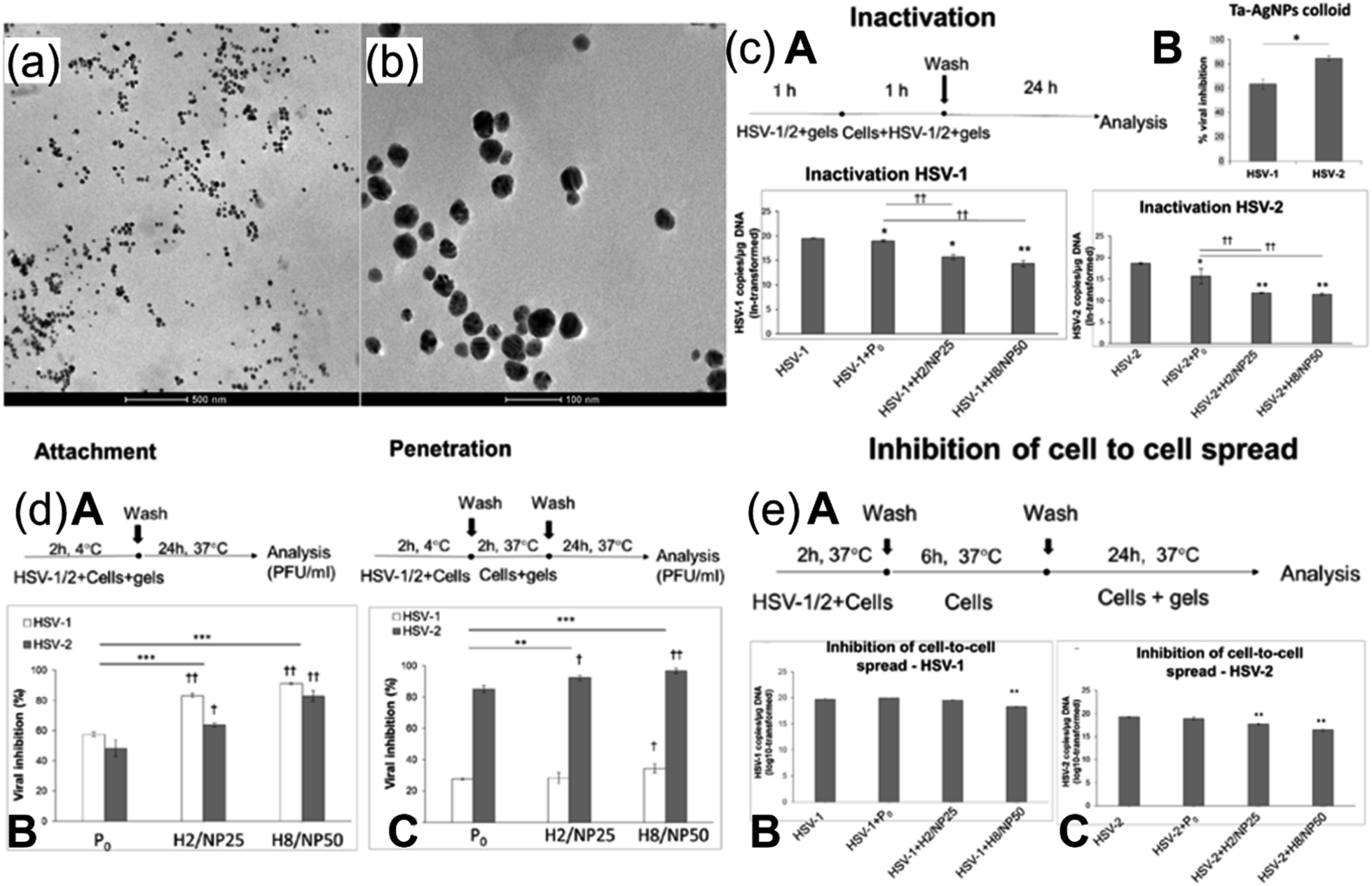 | ||
| Fig. 2 TEM images of H2/NP25 hydrogel containing 25 parts per million (ppm) TA-AgNPs. (a) Shows a scale bar of 500 nm; (b) shows a scale bar of 100 nm. (c) (A) HSV inhibition assay; (B) inhibition of HSV-1 and HSV-2 in HaCaT cells; (C) DNA titers of HSV-1; (D) of HSV-2. (d) (A) HSV attachment and penetration scheme; (B) HSV-1 and HSV-2 attachment and (C) penetration. (e) (A) Cell-to-cell inhibition assay scheme; (B) DNA titers of HSV-1 and (C) HSV-2 (reprinted with permission from ref. 19 Copyrights 2018 MDPI). | ||
Another study reported two algae, Oscillatoria sp. and Spirulina platensis, mediated by green Ag2O|AgO-NPs and Au-NPs, respectively, and evaluated their effect on HSV-1 infection.20 The spherical-shaped Ag2O|AgO-NPs with smaller size (nm) than non-spherical AuNPs showed a greater reduction rate of HSV-1. The results indicate the potential of bio-synthesized NPs as suppressing agents for HSV-1 infection. Remarkable antiviral activity of green synthesized AgNPs of two plant extracts, L. coccineus and M. lutea, was shown against HSV-1, HAV-10, and CoxB4 virus.21
Sreekanth et al. reported virucidal effects of AgNPs, 5–15 nm in size, against influenza A virus with an easy, smooth, and convenient approach used for synthesis of AgNPs i.e., ultrasonication method. Green synthesized AgNPs were studied for in vitro cytotoxic and antiviral activities and showed to possess good antiviral action against H1N1 variant of Influenza A virus.22 Another study reported AgNPs (not green synthesized) as potential antiviral agents against H1N1.23 The potential of AgNPs was shown against H3N2 variant of Influenza virus with the help of in vitro and in vivo studies.24 For in vivo studies, the survival of mice was enhanced while in vitro studies showed that AgNPs were able to protect cells by inhibiting viral infections. The mechanism of antiviral action appeared to be the destruction of virus morphology by AgNPs.
2.2 NiO nanostructures (NONS)
The leading cause of the infection in cucumber crops is cucumber mosaic virus (CMV), resulting in a significant reduction in crop yield globally, especially in Egypt. Viral diseases are not usually countered by chemical pesticides, one of the reasons being their ineffectiveness resulting from repeated use.25 Several studies have related the growth of plants with the amount of nickel (Ni) present. The need evolved to investigate the effect of Ni-based nanostructures against CMV.26 NiO was fabricated via one-pot hydrothermal synthesis and the efficacy of NONS against CMV in cucumber plants was evaluated against reduction in disease severity, assessed via immunosorbent assay. NONS particles of about 15–20 nm in size with greater exposed surface area were administered via foliar spray or soil drench to infected plants. The nanostructures were characterized by FESEM, X-ray diffraction (XRD), and Raman spectroscopy, and results are shown in Fig. 3. FE-SEM images in Fig. 3a and b show stable morphological nanostructures of NiO. Semi-spherical head ends are composed of nanotubes aligned normally within body of head. The distribution of these nanotubes across the surface of head indicates that average size of tube is in between 15 and 20 nm. Moreover, thick distribution of nanotubes resembles tubular vessels resulting from carefully designed NONS. WA-XRD was employed to determine the crystal structure of NONS. Crystal planes are indicated at well-resolved and sharp diffraction peaks of NiO and indicated accordingly in Fig. 3c. Raman spectroscopy revealed the presence of pristine NiO as appeared peaks result from presence of two TO modes and two LO modes as indicated in Fig. 3d. In vivo study concluded that plants treated with NONS showed visible reduction in CMV compared to non-treated counterparts, showing potential for NONS to be used as antiviral agent against CMV. | ||
| Fig. 3 (a) FESEM micrograph of NONS. (b) FESEM micrograph showing uniform formation of head and trunk. (c) XRD pattern of NONS. (d) Raman spectroscopy results of NONS (reprinted with permission from ref. 26 Copyrights 2019 Elsevier). | ||
2.3 Zirconia NPs
Zirconia NPs have shown promise in treatment of cancer in previous studies, but their antiviral activity was not reported previously. Huo and collaborators studied the antiviral effect of ZrO2 NPs on H5N1 variant of influenza A virus.27 The mice infected with H5N1 virus were treated with ZrO2 NPs of about 200 nm in size and administered intraperitoneally. The in vivo study concluded that survival chances of mice were increased by about 85.7%, as the ZrO2 NPs promoted the release of cytokines in mice. Thus, this novel strategy needs further exploration for future innovations.2.4 ZnO NPs
The antiviral drugs currently employed for the treatment of influenza viruses are becoming insignificant as they cause adverse side effects with drug-resistant strains.28 There is an urgent need to develop better and more efficient anti-influenza agents. A few advantages of using nanoparticles for the treatment of viral infections are that their synthesis is cheaper, they possess good antiviral efficacy, and can be tailored to improve properties via coating. ZnO-NPs have previously shown substantial antimicrobial and antibacterial activities.29 However, a handful of research has been done to evaluate the antiviral efficacy of ZnO-NPs. Ghaffari et al. were the first to examine the antiviral efficacy of ZnO-NPs and PEGylated ZnO-NPs in vitro.30 The size of NPs was reduced to a large extent as a result of massive ball milling. Scanning electron microscopy (SEM) confirmed the sizes of ZnO-NPs to be 20–50 nm and ZnO-PEG-NPs to be 16–20 nm (Fig. 4a). The SEM images show NPs to be of spherical morphology with uniform size distribution. The surface coating of ZnO-NPs was confirmed via TEM micrographs in Fig. 4b. The presence of ZnO-NPs was confirmed by XRD power diffraction pattern. When compared to the standard powder diffraction file (PDF), the peaks and their respective intensities of ZnO exhibit a similar pattern as depicted in Fig. 4c. Thermogravimetric analysis (TGA) was employed for weight loss measurements (Fig. 4f and g)). It can be seen that at a temperature of 400 °C, there is a significant decrease in weight (%) in the case of ZnO-PEG-NPs (Fig. 4g) while the weight loss is very small at the same temperature for ZnO (Fig. 4f). This gives a reasonable explanation for coating of PEG which resulted in greater weight loss. The key finding of the study was that the NPs cause antiviral action only after the virus has infected the cells. There was considerable inhibition when H1N1 virus was exposed after infection to ZnO-PEG-NPs as there was a significant reduction in virus titers in contrast to the virus control, as shown at concentrations of 75, 100, 200 μg mL−1. The maximum concentration of ZnO-NPs alone could only result in 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10 TCID50 reduction which is much lower than reductions caused due to PEGylated ZnO-NPs. At its maximum non-toxic concentration, PEG alone could reduce by 0.7log10 TCID50, which is even lower than ZnO-NPs. These results are schematically depicted in Fig. 4h. It was observed in quantitative real-time PCR tests that significant inhibition resulted by ZnO-PEG-NPs at concentrations of 75, 100, 200 μg mL−1, which are much superior to ZnO-NPs inhibition rates at respective concentrations. However, it is quite interesting to note that oseltamivir was able to fully inhibit influenza at a concentration of 75 μg mL−1. These results are summarized in Fig. 4e. While the precise antiviral mechanism of virus inhibition largely remained unexplored, PEGylating the surface of ZnO-NPs resulted in increased antiviral activity and can be employed as a novel strategy for improving the antiviral efficacy. Hence ZnO-PEG-NPs can be an effective antiviral agent against influenza viruses.
log10 TCID50 reduction which is much lower than reductions caused due to PEGylated ZnO-NPs. At its maximum non-toxic concentration, PEG alone could reduce by 0.7log10 TCID50, which is even lower than ZnO-NPs. These results are schematically depicted in Fig. 4h. It was observed in quantitative real-time PCR tests that significant inhibition resulted by ZnO-PEG-NPs at concentrations of 75, 100, 200 μg mL−1, which are much superior to ZnO-NPs inhibition rates at respective concentrations. However, it is quite interesting to note that oseltamivir was able to fully inhibit influenza at a concentration of 75 μg mL−1. These results are summarized in Fig. 4e. While the precise antiviral mechanism of virus inhibition largely remained unexplored, PEGylating the surface of ZnO-NPs resulted in increased antiviral activity and can be employed as a novel strategy for improving the antiviral efficacy. Hence ZnO-PEG-NPs can be an effective antiviral agent against influenza viruses.
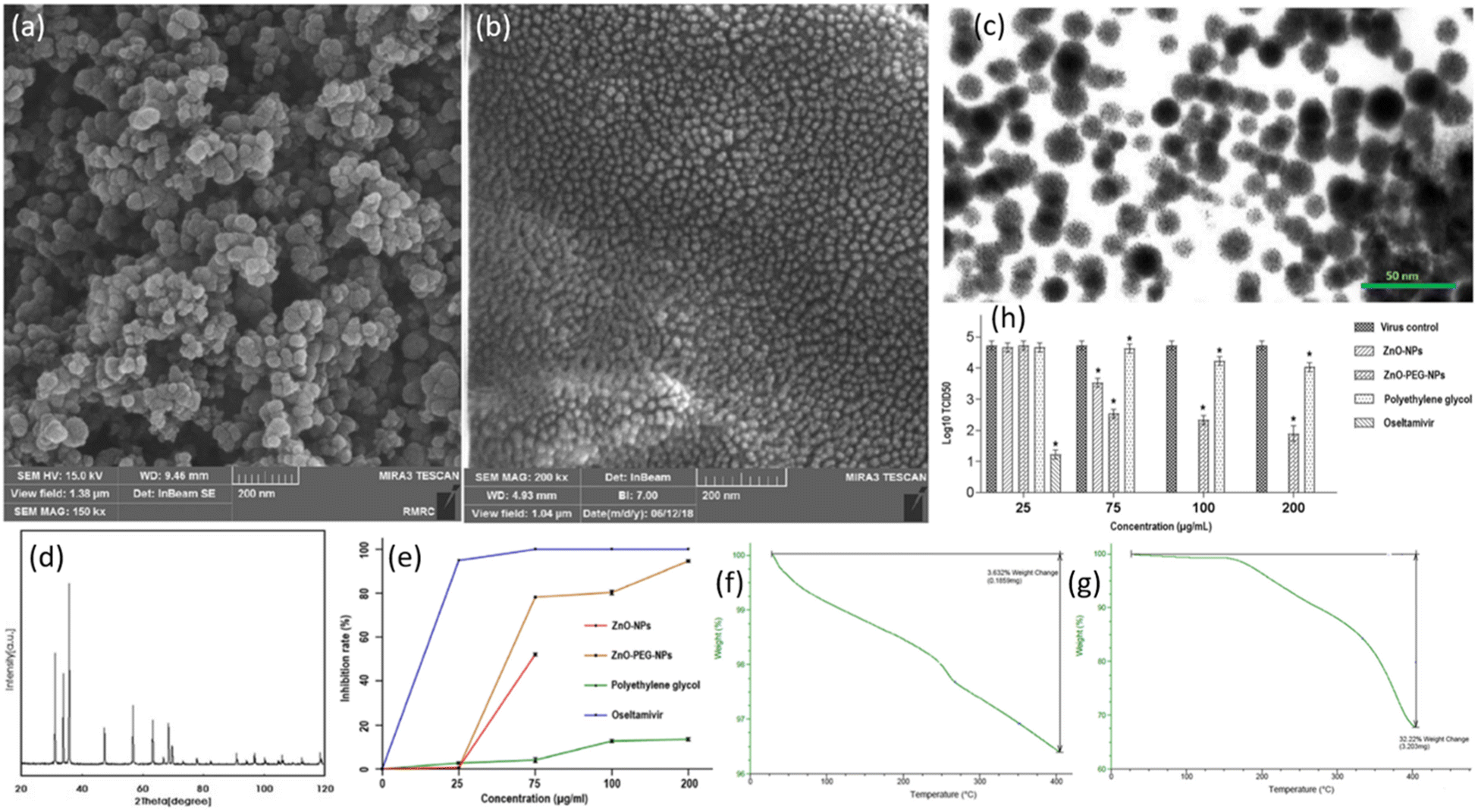 | ||
| Fig. 4 (a) FESEM micrograph of ZnO-NPs. (b) FESEM micrograph of ZnO-PEG-NPs. (c) TEM micrograph of ZnO-PEG-NPs. (d) Powder XRD results for ZnO-NPs. (e) Real-time PCR assay results for inhibition rates of four compounds against H1N1 influenza. (f) TGA results of ZnO-NPs. (g) TGA of ZnO-PEG-NPs. (h) Post-exposure antiviral activity of different compounds against H1N1 influenza virus assessed via TCID50 assay (reprinted with permission from ref. 30 Copyrights 2019 BioMed Central). | ||
2.5 TiO2-based NPs
Broad bean stain virus (BBSV) infects seeds of many food crops. Faba bean crop is significant globally, especially in Egypt. They are quite sensitive to viral diseases like BBSV.31 Recent studies have proposed nanotechnology as a novel method to control pathogens in plants.32 The excellent property profile offered by TiO2 allows it to be used in a variety of applications, including antiviral. However, the efficacy of TiO2 is negatively affected by several factors such as its inherent crystallinity, unfavorable surface-to-size area ratio, and its surface morphology.33 The material scientists have focused on developing new modes of synthesis of TiO2 that induce functionalities that can prove effective in the treatment of plant-related diseases. The mechanism of antiviral action against plant viruses can be different and varies from material to material. For instance, it can include the direct effects of NPs against the virus which can result in death of virus or may also cause the defensive system of plants to become stronger and hence increase its inherent immunity to deal with virus.34 One such study was done by Elsharkaway et al., who fabricated TiO2 nanostructures (TDNS) and examined its antiviral efficacy against BBSV.35 The approach used for synthesis was similar to that reported by Gomaa et al.36 i.e., direct mild hydrothermal synthesis, with slight changes. To determine the crystal structure of the synthesized NPs, wide-angle X-ray diffraction (WA-XRD) was utilized which shows a similar pattern as that for standard TiO2 as seen in Fig. 5a. FE-SEM micrographs confirm the successful synthesis of TiO2 structures as surfaces can be seen to be smooth from inside and rough from outside with a small thickness of walls (Fig. 5b). The further morphological examination also shows that size distribution was in the range of 3–5 μm for diameter, while 3–4 μm for length and width. Combining of a large number of TiO2 nano-sheets can form a hollow shape as hydrogen from oleic acid bond to TiO2 surface.37 This can result in favorable exposed surface area for interaction between virus and TDNS with greater antiviral activity. Experimental results after a 14 days treatment are shown in Fig. 5c. The control leaves can be seen to be smaller and deformed while the leaves treated with TDNS exhibited great reduction in symptoms of BBSV. Outstanding reduction in disease severity of faba bean plants due to TDNS can be seen in Fig. 5d. Foliar spray method appeared to be more effective in decreasing the disease severity than soil drench technique. (In foliar spray, fertilizers (in our case TiO2 nanostructures or TDNS) are sprayed on the plant leaves rather than pouring them in the soil. In contrast, soil drench technique involves pouring the chemicals mixed with water directly onto the soil). Moreover, concentration of BBSV in plants treated via foliar spray method is also lower than in soil drench. Plants treated with both approaches showed no significant difference in gene expression which was, however, much greater than control. Thus, due to TDNS inducing systematic resistance, there was a significant decrease in disease severity. Structural features of TDNS such as particle size and suitable shape were the governing factors for its good overall efficacy against BBSV. This study is only one of its kind which evaluated and suggested an eco-friendly approach to treating plants against BBSV. | ||
| Fig. 5 (a) Wide-angle (WA) XRD pattern of TDNS. (b) FESEM image of TDNS. (c) Symptoms of BBSV caused in un-treated faba plants (control), treated with TDNS via foliar spray and soil drench after 2 weeks. (d) Degree of disease severity in treated plants and un-treated plants (Different letters above columns indicate significant differences by the Steel–Dwass test for faba bean (Pd 0.05)) (reprinted with permission from ref. 35 Copyrights 2018 Society of Chemical Industry). | ||
Nakano et al. proposed use of TD thin film for photocatalytic inactivation of H1N1 Influenza. This can be attributed to the strong oxidation effect produced when TiO2 is exposed to UV light.38 The study demonstrated that TiO2 substantially increases the disinfection rates of H1N1 by a strong oxidation effect which degrades viral proteins. A slight amendment in the ISO method was made for evaluating the anti-bacterial effects of TD which can prove beneficial for enhancing the antiviral activity. There are a few studies that relate the antiviral mechanism of action of TiO2 to the best of our knowledge.
2.6 GO-Ag NPs
Nanomaterials are of key importance in treatment of viral infections. In this regard, AgNPs have shown good potential in antiviral actions against a variety of viruses, as discussed before in this review. Chen et al. reported that there have been no studies on the antiviral efficacy of nanomaterials against non-enveloped viruses.39 Their study evaluated the antiviral activity of GO sheets and GO-AgNPs against both enveloped and non-enveloped viruses. The enveloped virus chosen was Feline Coronavirus (FCoV) while the non-enveloped virus was Infectious Bursal Disease Virus (IBDV). The particle size distribution (PSD) of Ag NPs was between 5–25 nm while thickness of each GO layer was 0.355 nm and there were 2 to 5 layers in total. The virus inhibition assay reported a significant reduction of FCoV (25%) and IBDV (23%) by GO-Ag NPs than GO sheets whose inhibition was restricted to merely 16%. The virus inhibition mechanism of GO-Ag NPs is illustrated schematically in Fig. 6.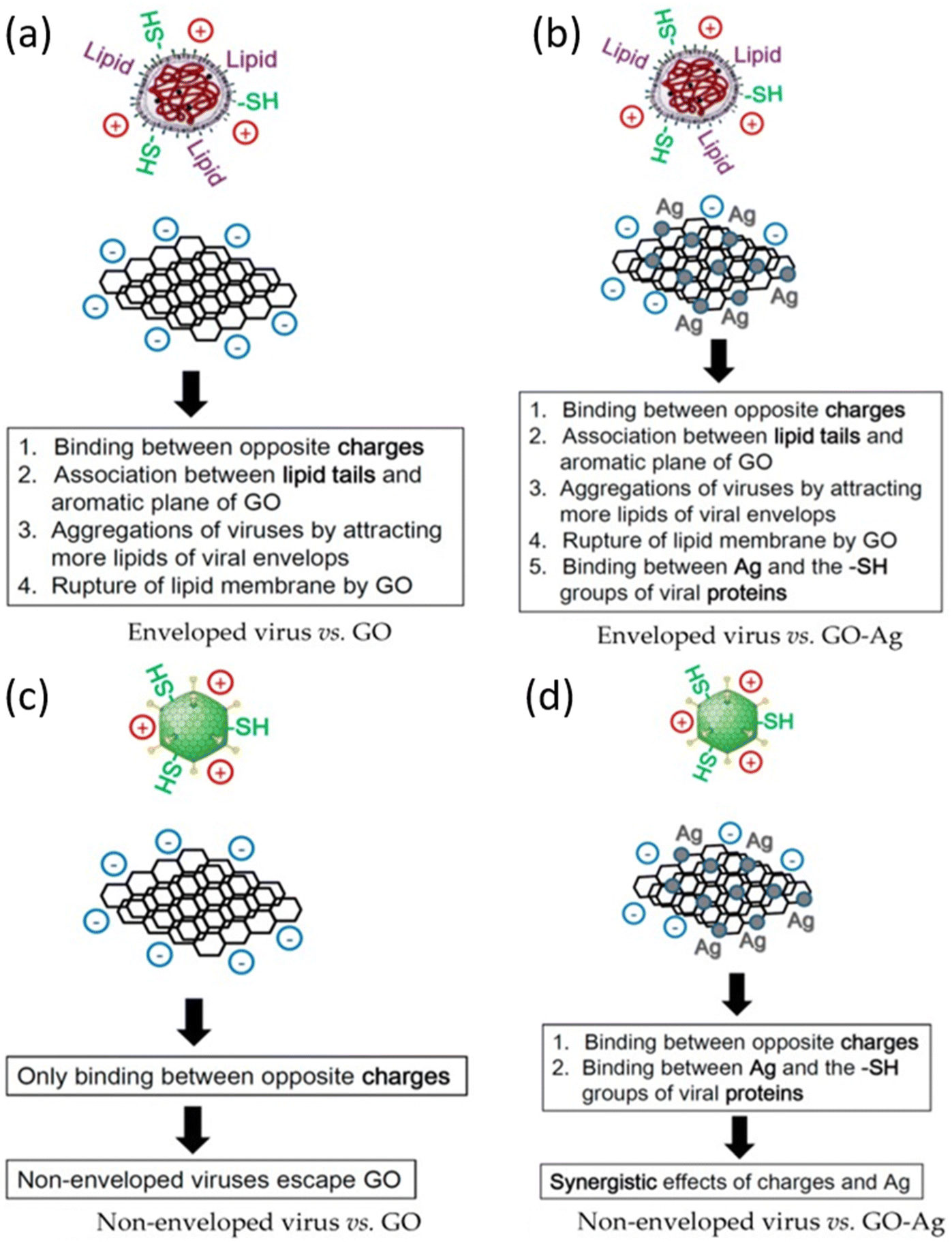 | ||
| Fig. 6 Schematic illustration of antiviral mechanism of (a) GO; (b) GO-Ag against enveloped viruses; (c) GO and (d) GO-Ag against non-enveloped viruses (reprinted with permission from ref. 39 Copyrights 2016 MDPI). | ||
2.7 Gold nanoparticles (AuNPs)
The exceptional property profile package offered by AuNPs such as exquisite quantum size effect, surface effect, and macroscopic quantum tunneling effect make them useful in nanomedical applications. Several studies have evaluated antiviral efficacy of AuNPs against a variety of viral infections. For example, Asim and his coworkers synthesized AuNPs of 7.86 ± 3.3 nm in size by sonication of gallic acid in a bath Sonicator and examined their antiviral mechanism of action.40 They reported that AuNPs prevent the attachment and penetration of HSV. The amount of virus inhibited is dependent on the time of exposure of AuNPs and hence the method employed uses highly monodispersed AuNPs. Their bio-friendly nature makes them a strong candidate for prevention of HSV. Another study reported by Gianvincenzo et al. evaluated AuNPs antiviral efficacy against HIV.41 Two compounds used Au clusters of about 1.7 nm and 2.6 nm, respectively, which accommodated about 140 sulfated ligands and were enough to perform anti-HIV action. The in vitro study revealed that AuNPs do this by binding to glycoprotein (gp120). AuNPs can be coated with sulfate-ended ligands which in turn bind the HIV, causing its inhibition. This result opens doors of opportunities to tailor those surface ligands for development of more anti-HIV systems. The potential of AuNPs is not limited till here. Papp et al. have demonstrated in previous studies about the capability of AuNPs in the treatment of Influenza A Viruses (IAVs).42 They showed that sialic-acid coated glycerol dendrons when immobilized on 14 nm AuNPs caused an appreciable anti-influenza effect. Moreover, AuNPs were non-toxic under operating conditions. The AuNPs have also been found compatible with the treatment of Dengue virus (DENV). For instance, Paul and his collaborators have reported that biocompatible AuNPs can be used to improve the delivery and stability of siRNA.43 When entered Vero cells, they decrease DENV-serotype 2 replication to a great extent by releasing infectious virions. Hence AuNPs can be used for DENV infection control in vitro.2.8 Selenium NPs
There are no effective drugs for the treatment of EV-A71. Selenium (Se) is present in our body as a nutritional element that is responsible for protection against viral infections. Recent studies have reported that SeNPs have excellent bioavailability and lower side effects due to their smaller size44,45 and hence have the potential to be used as an antiviral agent against EV71.Li et al. investigated the effect of SeNPs on the suppression of EV-A71 virus as they interfere with JNK signaling pathways.46 As seen in Fig. 7a(A), the SeNPs can pass through the cell membrane and suppress the generation of Reactive Oxide Species (ROS) by EV-A71. They interfered with and successfully inhibited JNK signaling pathways by mechanisms of caspase-8 and caspase-9-mediated apoptosis in the cells infected by the virus. They synthesized SeNPs via a simple method and were able to prepare uniform NPs of size roughly 100 nm, as confirmed by the TEM micrograph in Fig. 7a(B). Due to their smaller size, SeNPs were highly stable and able to penetrate the cells. The presence of Se and Cu was confirmed in energy dispersive X-ray spectroscopy (EDX) as they form the SeNPs and the copper grid (Fig. 7a(C)). Thiazolyl blue tetrazolium bromide (MTT) assay was employed to examine the cell viability with SeNPs. Cell viability can be seen to slightly decrease in Fig. 7a(D). The decrease can be attributed to the concentration of SeNPs. This indicates that SeNPs can inhibit the EV-A71 virus proliferation. The MTT assay also measured the antiviral activity of SeNPs (concentration maintained at 15.625 μM). The results show that cell viability was increased to 71% by SeNPs as compared to 59% when treated with EV-A71 virus (Fig. 7a(D)). This suggests good antiviral efficacy of SeNPs against EV-A71 virus.
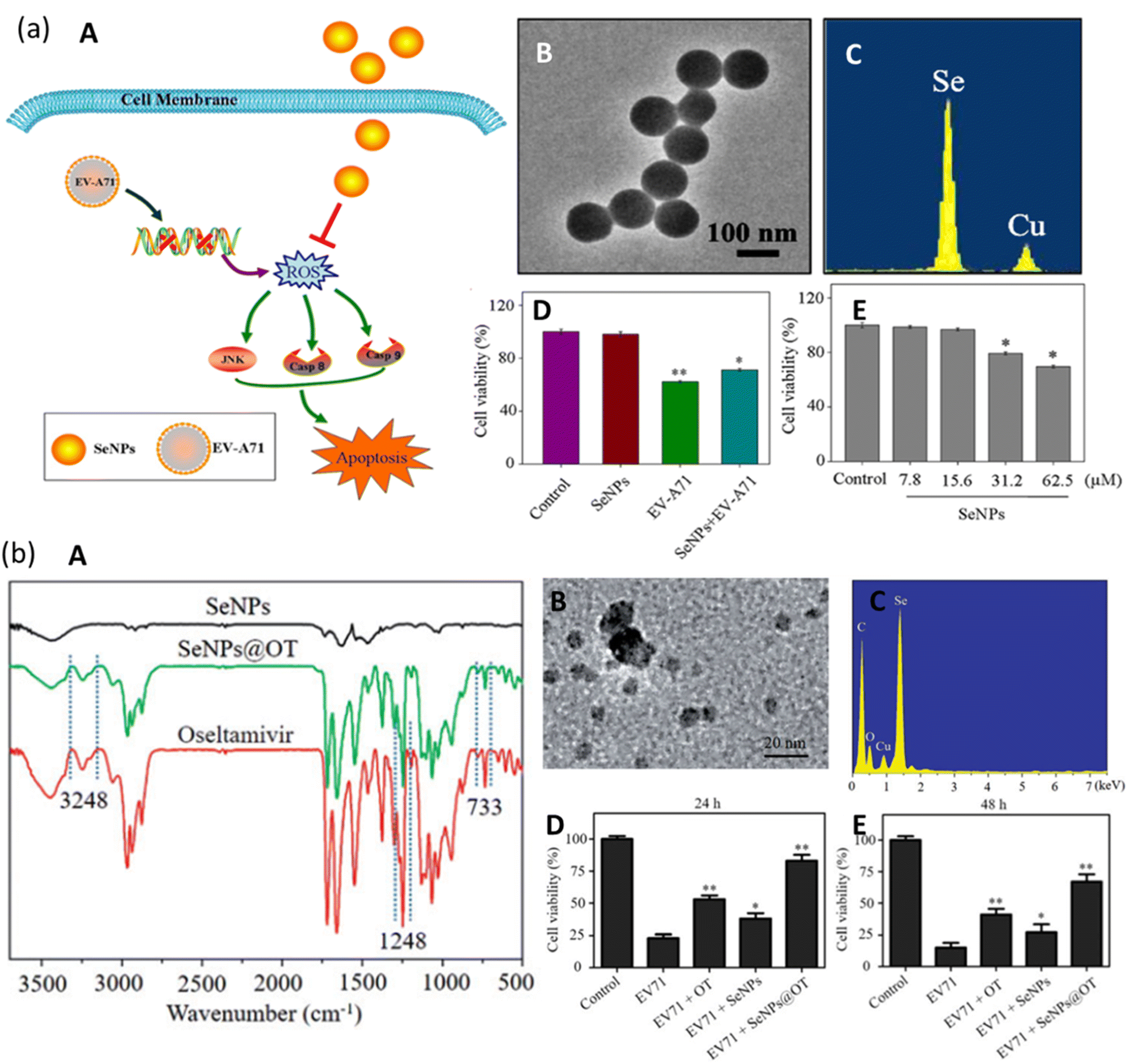 | ||
| Fig. 7 (a) Mechanism of JNK and caspase signaling pathways. Green arrows show direct stimulatory modifications while orange shows direct inhibitory modifications (A). TEM micrographs of SeNPs (B). EDX pattern of SeNPs (C). MTT assay for antiviral activity (D). MTT assay for cell viability (reprinted with permission from ref. 46 Copyrights 2019 ACS). (a) FTIR spectrum of SeNPs, SeNPs loaded with OT and of OT (A). TEM micrograph of SeNPs@OT (b). EDX pattern of SeNPs@OT (C). Stability of SeNPs assessed in aqueous solution (D) and in PBS (E) (reprinted with permission from ref. 50 Copyrights 2019 Tailor and Francis Group). | ||
For the treatment of EV-71 variant of enterovirus, oseltamivir (OT) is commonly used as an antiviral therapeutic agent. Although it has been approved for use by FDA, due to continuous usage, it has shown a decrease in antiviral efficacy.47 To overcome this problem of inherent drug resistance against the virus, nanotechnology holds promise.48 As discussed previously, Se is an important element in human body and hence its deficiency can result in increased susceptibility to virus infections.49 Zhong et al. proposed a novel nanotechnological approach for increased antiviral efficacy against EV-71.50 They fabricated a nano-sized functional antiviral system by loading OT on the surface of SeNPs and evaluated its antiviral activity. The cell model for this particular study was chosen to be a human astrocytoma cell (U251). To confirm the chemical bonding between the drug and the NPs, Fourier transform infrared spectroscopy (FTIR) was performed whose graph is depicted in Fig. 7b(A). The spectrum is same as that of oseltamivir and peaks corresponding to 3248, 1248, and 733 cm−1 can be seen for SeNPs@OT which is an indication of successful formation of a nano-sized antiviral system. The mean size of this nanosystem is confirmed by TEM micrograph in Fig. 7b(B), which confirms it to be around 10 nm. EDX results can also be seen in Fig. 7b(C), which shows characteristic peaks corresponding to Se (from NPs), O, and C (from OT) which further confirms the success in the preparation of SeNPs@OT. The cell viability of model U251 cells was examined and in Fig. 7b(D), the viability is lowest for EV-71 infected cells which increased to a greater amount with OT than SeNPs. However, when the prepared nanosystem SeNPs@OT was used, the viability of the infected cells was the highest. A similar experiment was conducted after 48 hours post infection and results can be seen to be similar as in Fig. 7b(E). Hence SeNPs loaded onto OT suppressed the cell apoptosis caused by EV-71 virus via mitochondrial pathway and reduced the generation of reactive oxygen species. Table 1 shows a summary of different metallic NPs which exhibit antiviral activity against a wide category of viruses along with their key parameters.
| Nanoparticle(s) | Virus | Classification of virus | In vitro or in vivo | Size (nm) | Synthesis method | Mechanism | Reference |
|---|---|---|---|---|---|---|---|
| Ag2O|AgO-NPs and Au-NPs | Herpesvirus | HSV-1 | In vitro | 14.42–48.97 nm [Ag2O–AgO-NPs]; 15.60–77.13 nm [Au] | Biosynthesis | — | 20 |
| Ag2S nanoclusters | Coronavirus | Porcine epidemic diarrhea virus (PEDV) | In vitro, in vivo | 3.7 nm and 5.3 nm | One-pot method | Prevents synthesis of viral negative-strand RNA and viral budding | 51 |
| AgNPs | Dengue virus | DENV | In vitro | 100 nm | Biological (seed extract) | — | 52 |
| Influenza | H3N2 | In vitro, in vivo | 9.5 nm | Oxidation-reduction method | Destruction of morphologic viral structures | 24 | |
| H1N1 | In vitro | 10 nm | — | — | 23 | ||
| H1N1 | In vitro | 5–15 nm | Ultra-sonication method | — | 22 | ||
| Enterovirus 71 | EV71 | In vitro | - | Simple method | — | 17 | |
| Respiratory syncytial virus | RSV infections | In vitro, in vivo | 8–12 nm | — | Prevents entry of viral glycoproteins into the host cell | 16 | |
| Dengue virus | DENV | In vitro | 30–70 nm | Biological (plant extract) | — | 53 | |
| Hepatitis | HSV-1, HAV-10 and CoxB4 | Used design studio | 8.91–27.89 nm | Biological (plant extracts) | — | 21 | |
| Herpesvirus | HSV-1 and HSV-2 | In vitro, in vivo, ex vivo | 13 to 54 nm | — | Affect viral attachment | 19 | |
| Poliovirus | PV | In vitro | 7.1 nm | Electrochemical method | Cytopathic effect (CPE) | 18 | |
| Au-MES NPs | Herpesvirus | HSV-1 | — | 4 nm [Au-MES NPs] | Solution-based method | Prevents virus from attaching, entering and spreading from cell to cell | 54 |
| AuNPs | Herpesvirus | HSV | — | ∼7 nm | Gallic acid in a bath sonicator | Prevents the attachment/penetration of virus | 40 |
| Human immunodeficiency virus | HIV-1 | — | 1.7 nm and 2.6 nm | — | Binding to gp120 | 41 | |
| Influenza | Influenza A viruses (IAVs) | — | 2 nm and 14 nm | — | Multivalent interaction with sialic-acid-functionalized AuNPs | 42 | |
| AuNPs interfering RNA | Dengue virus | DENV | In vitro, in vivo | 12.92–43.25 nm | Chemical | Release infectious virion | 43 |
| Carbon dots NPs | Human immunodeficiency virus | HIV-1 | — | 2 nm | Pyrolysis of citric acid | Suppressing the syncytium formation | 55 |
| Carbon quantum dots (CQDs) | Highly pathogenic coronavirus | HCoV | — | 7.6 ± 0.2 nm | Hydrothermal carbonization | — | 56 |
| Copper(I) iodide NPs | Influenza | H1N1 | — | 160 nm | — | OH− radicals are generated and viral proteins undergo degradation | 57 |
| Feline calicivirus | FCV | — | 100–400 nm | — | Cu+ ions generated followed by generation of ROS and capsid protein oxidation | 58 | |
| Copper–graphene (Cu–Gr) nanocomposite | Influenza | Influenza A viruses (IAVs) | — | - | — | Inactivate the virion particles within a half hour, preventing entry to the host cell | 59 |
| CuO NPs | Herpesvirus | HSV-1 | In vitro | 40 nm | — | Production of ROS via Cu+ OR degradation of viral genome | 60 |
| Cuprous oxide NPs | Hepatitis | HCV | In vitro | 45.4 nm | Solution phase | Inhibited entry of HCV pp | 61 |
| GO-Ag NPs | Feline coronavirus (FCoV): non-enveloped virus and infectious bursal disease virus (IBDV): Enveloped virus | FCoV and IBDV | — | 5–25 nm | Hummers' method | — | 39 |
| Gold nanorod-based HR1 peptide | Coronavirus | SARS CoV-2 | — | 18 nm (diameter) | Chemical solid phase | Increases the immune indicators and decreases the inflammation indicators | 62 |
| Iron oxide NPs | Influenza | H1N1 | In vitro | 10–15 nm | Chemical reduction and magnetic separation | — | 63 |
| NiO nanostructures (NONS) | Cucumber mosaic virus | CMV | In vivo | 15 to 20 nm | One-pot hydrothermal synthetic approach | Increase the expression of pod, pr1 and pal1 genes | 26 |
| Se@PEI@siRNA | Enterovirus 71 | EV71 | - | 80 nm | — | Chances of SK-N-SH cells for staying in sub-G1 phase are reduced | 64 |
| SeNPs | Enterovirus 71 | EV71 | In vitro | 10 nm | — | SeNPs@OT entered host cells by clathrin-associated endocytosis while suppressing EV71 proliferation | 50 |
| Enterovirus A71 | EV-A71 | — | 100 nm | Simple method | Cytopathic effect | 46 | |
| Ag NCs with SiO2 composite sputtered coating | Coronavirus | SARS CoV-2 | — | Less than 200 nm | Co-sputtering with argon at radio frequency | Coating possessed a virucidal effect | 65 |
| GO-Ag nanocomposite | Porcine epidemic diarrhea virus | PEDV | In vitro | 17 ± 3.4 nm | Self-assembly via interfacial electrostatic force | Prevent entry of PRRSV to host cells | 66 |
| TiO2 nanostructures (TDNS) | Broad bean stain virus | BBSV | — | — | Modified direct hydrothermal synthesis | Inducing systemic resistance | 35 |
| TiO2 NPs | Influenza | H1N1 | — | — | — | Strong oxidation effect | 38 |
| H3N2 | — | 4–10 nm | — | Fragmentation of viral envelope | 67 | ||
| Tungsten carbide nanoparticles (WC NPs) | Poliovirus type-1, vaccinia virus ankara, human adenovirus type 5, Murine norovirus | PV-1, MVA, HAdV-5, MNV | In vitro | 10–20 nm | Plasma atomization | — | 68 |
| Zirconia NPs | Influenza | H5N1 | In vivo | 200 nm | Two-step selective etching method | Promote the release of cytokines in mice | 27 |
| ZnO NPs and PEGylated ZnO NPs | Influenza | H1N1 | In vitro | 20–50 nm ZnO-NPs; 16–20 nm ZnO-PEG-NPs | Mechanical method | Virus inhibited once it enters the host cell | 30 |
3. Nanoparticles for enhancing the efficacy of antiviral drugs
3.1 Acyclovir
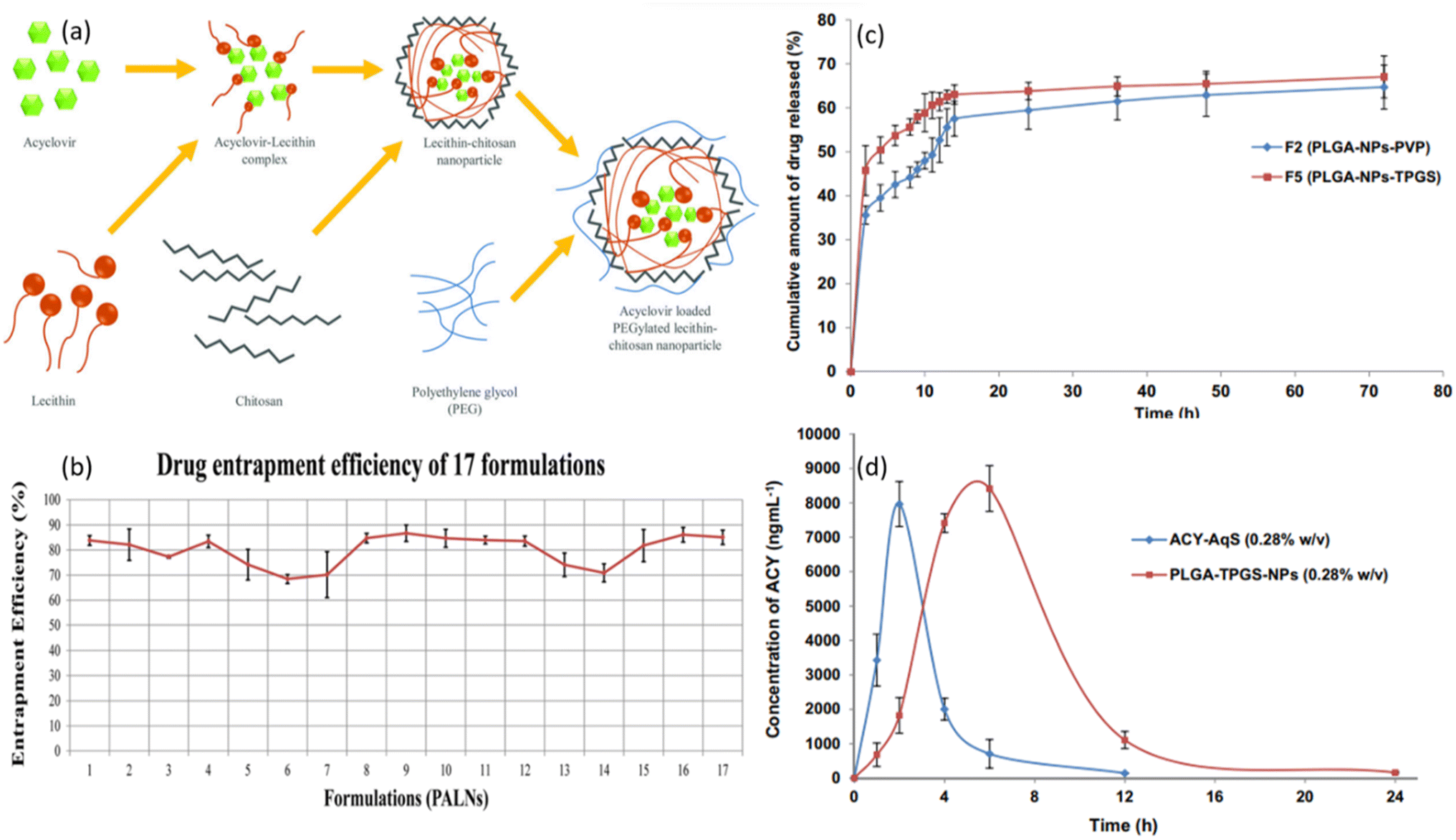 | ||
| Fig. 8 (a) Schematic illustration of the mechanism of PALNs formation. (b) Graphical illustration of entrapment efficiency of 17 tested drug formulations (reprinted with permission from ref. 69 Copyrights 2020 AAPS). (c) In vitro release profiles of PLGA-NPs in STF with pH of 7.4. (d) The concentration of ACV in aqueous humor after topical ocular administration of formulation “F5” and ACV-AqS in eyes of rabbits (reprinted with permission from ref. 81 Copyrights 2018 Elsevier). | ||
Similarly, Saifi and her collaborators employed bilosomes nanocarriers (NCs) to enhance the oral bioavailability of ACV.75 The in vitro, ex vivo, and in vivo assessments showed that a vesicle size of 121.2 ± 3.21 nm was obtained via thin film hydration method (optimized via BBD) and showed 71.87–88.67% entrapment efficiency. The biocompatible bilosomes were found to be an effective drug carrier as they enhanced gut absorption of ACV at a considerably lesser amount dose than needed for a typical tablet.
Poorly soluble drugs can be employed for ocular delivery if their resulting eye irritation is under tolerance. The approach employed by Suwannoi et al. examined the ocular delivery route of ACV-loaded with BSA NPs which were surface modified with transactivating transduction (TAT) peptide, to deal with viral-related keratitis.80 About 200 nm-sized NPs showed less cytotoxic effects on HCE-T cells and resulted in the greatest ACV permeation in NPs. In vitro study showed that a novel formulation can be used for effective trans-corneal delivery of ACV.
3.2 Efavirenz (EFV)
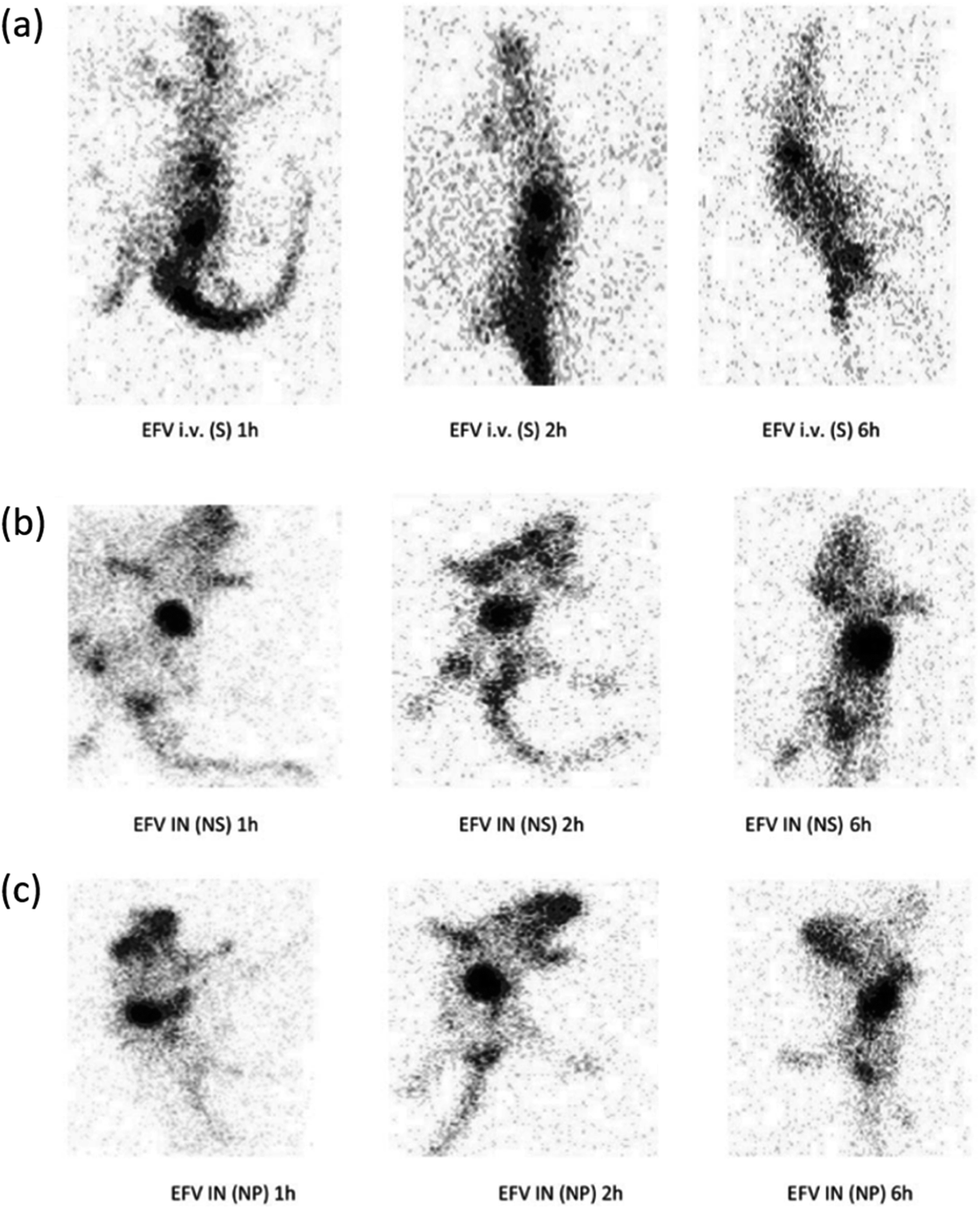 | ||
| Fig. 9 Gamma scintigraphy images of rats showing the presence of radioactivity post-administration of intravenous efavirenz solution [EFV i.v. (S)], intranasal efavirenz solution [IN EFV (S)] and intranasal efavirenz nanoparticles (IN EFV-NPs). (a) EFV-i.v. (S), (b) EFV IN (S) and (c) IN EFV-NPs (reprinted with permission from ref. 83 Copyrights 2019 Tailor and Francis Group). | ||
3.3 Zidovudine (AZT)
The ideal approach for treating many diseases by using the same delivery system is to employ multi-functional NPs. In another study, they prepared hybrid NPs of carboxy methyl cellulose–AZT core enclosed by a Compritol (Comp)–polyethylene glycol shell.90 In vitro study results showed that the system was biocompatible, cytocompatible, and showed appreciable loading coupled with controlled release of AZT drug and encapsulation efficiency of 82%. The results open doors for more opportunities for the development of LPNs as an efficient delivery vehicle of antiviral drugs.
Previous research has shown that Aloe vera can be administered to patients via oral, transdermal, and buccal drug delivery.93 However, all these delivery routes for the application of Aloe vera gel have corresponding setbacks. For instance, when administered through oral route, although it is much easier for patient to intake the drug, however, drug administration is associated with poor bioavailability coupled with low absorption of proteins.94 An alternate to oral delivery, buccal route of administration has been researched extensively.95 A major drawback of buccal administration is the need for permeation enhancers which if absent would limit membrane permeation of some compounds across cheek mucosa and therefore cause unsuccessful delivery of Aloe vera. But perhaps the most effective route for administration, according to our perspective, is applying Aloe vera drug via skin or transdermal route. It has many positive outcomes associated which include but are not limited to successful avoidance of first-pass metabolism, comparatively and adequately greater surface area for absorption, less frequent doses required and its inherent noninvasive nature.96 We suggest more research should be conducted on exploring the nanocarriers systems for achieving synergistic effects of Aloe vera gel and nanoparticles used for its transport to treat viral infections.
3.4 Lopinavir (LPN)
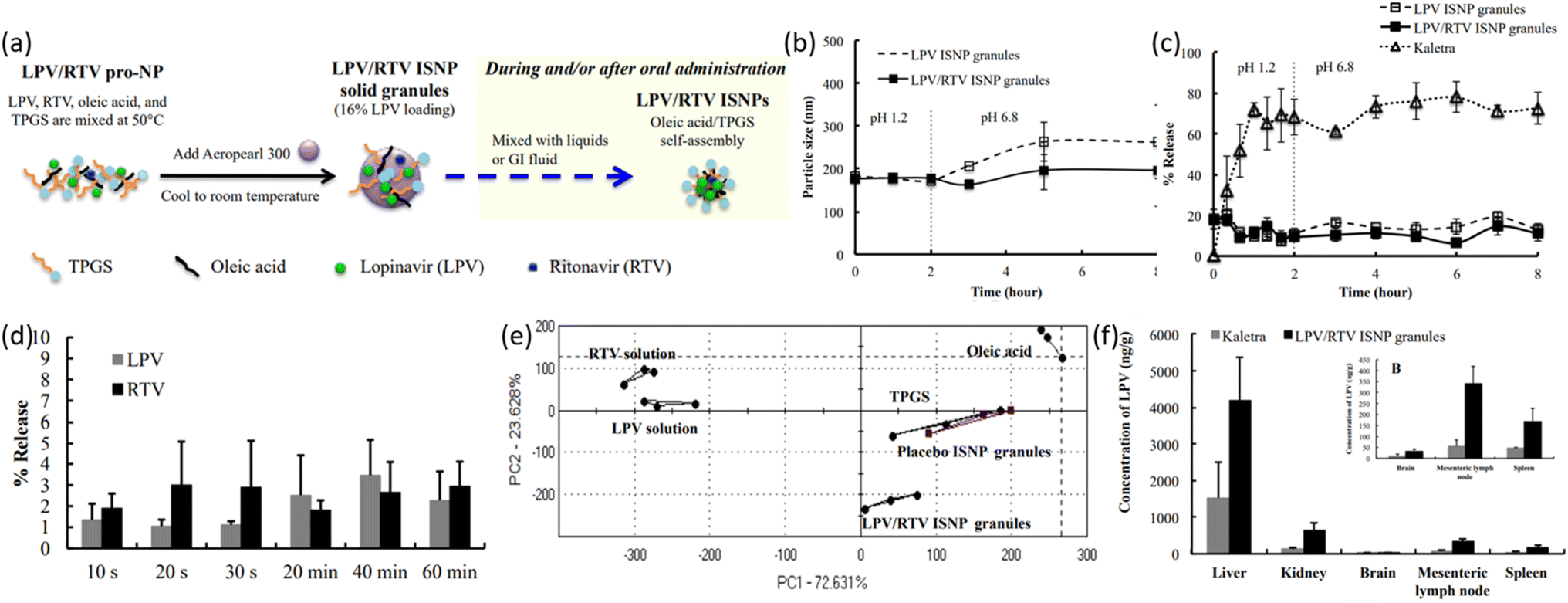 | ||
| Fig. 10 (a) Schematic illustration of LPV/RTV ISNP granules preparation and LPV/RTV ISNPs formation. (b) Physical stability of LPV/RTV ISNP granules and LPV ISNP granules in physiological conditions (simulated). (c) Dissolution profiles of LPV from LPV ISNP granules and Kaletra. (d) Taste examination of LPV/RTV ISNP granules in PBS maintained at pH 6.8 and (e) using an Astree e-tongue. (f) Distribution of tissues by LPV after oral administration to rats (reprinted with permission from ref. 100 Copyrights 2016 Elsevier). | ||
Ravi and Vats prepared SLNs-LPV formulation for enhanced HIV action by increased oral bioavailability, evaluated in vivo with a rat model.101 They employed warm oil-in-water (O/W) micro-emulsion technique and prepared NPs of 196.5 ± 3.5 nm size. Entrapment efficiency of 76.5 ± 3.5% was recorded. Another study also reported SLN-LPV formulation prepared via hot self nano-emulsification (SNE) method which achieved 180.6 ± 2.32 nm NP size and entrapment efficiency of 91.5 ± 1.3%.102 The results showed higher oral bioavailability and lymphatic drug transport. The novel synthesis method of SLN preparation was explored and can be used for the preparation of SLNs of higher fatty acids.
Another novel method for enhancing the oral delivery of LPV via Pullulan Acetate NPs was explored by Ravi et al. by employing Motozato's method and achieving a particle size of nearly 197 nm.103 The prepared formulation was quite effective in the treatment of HIV infections by using LPV as an antiviral drug and pullulan acetate NPs as nanocarriers which enhanced the oral bioavailability and hence the effectiveness of the formulation against HIV. The bioavailability of LPV from NPs was about 2× greater compared to free LPV. Higher distribution of LPV-loaded NPs to lymphoid organs was recorded with an entrapment efficiency of 75%.
3.5 Saquinavir (SQV)
Yuan et al. examined the result of employing nanocrystal suspension on the oral bioavailability of SQV.116 Nanocrystals of SQV were prepared via the anti-solvent precipitation-high pressure homogenization method. The size of nanocrystals was 205.93 ± 3.74 nm having a narrow poly-dispersity index (PDI) of 0.1. The nanocrystals were hence uniformly distributed. The zeta potential showed a high negative value which is a representation of the good stability of the prepared formulation. The rod-shaped particles were confirmed from TEM micrographs. Fig. 11a shows the results of the cellular uptake study performed on Caco-2 cells. The higher fluorescent intensity of SAQ nanocrystals indicates high drug uptake in cells. Small red dots seen in the cytoplasm of cells after 2 h could potentially be the SQV nanocrystals. In contrast, there are only a few particles observed in the cytoplasm of SQV coarse crystals after a similar 2 h period and at the same level of excitation intensity. The level of fluorescence intensity after 0.5, 1, and 2 hours of SQV nanocrystals can be seen to be significantly higher than SQV coarse crystals which are in line with increased cellular uptake of nanocrystals in Caco-2 cells, as shown in Fig. 11b. The better cellular uptake profile of SQV nanocrystals can be linked to their lower crystallinity. As shown in Fig. 11c, SQV nanocrystals and coarse powder exhibit similar degrees of crystallinity. The drug release study was performed on both coarse and nanocrystals. After a period of 2 h, roughly 20% coarse crystals were able to dissolve compared to 60% dissolution of nanocrystals. As expected, the dissolution of nanocrystals is much quicker than coarse crystals of SQV. A similar experiment was performed by monitoring the release profile of ethyl rhodamine B (RHD) from both coarse and nanocrystals. Release profiles exhibited a similar pattern as before with a greater percentage release from SQV nanocrystals. The RHD was entrapped in the crystal lattice of SQV nanocrystals and showed immediate release once nanocrystals dissolved. The effect of particle size of SQV on the drug transport across the Caco-2 cells monolayer from apical (AL) to basolateral (BL) side was assessed. As seen in Fig. 11d, the percentage of SQV in the receiving chamber (BL side) is increasing as time progresses. Yet again, a greater percentage of drug was able to pass through the monolayers when they were treated with nanocrystals, hence indicating faster drug transport for SQV nanocrystals than coarse crystals. The apparent coefficient of permeability (Papp) of SQV was determined by treating Caco-2 cells with both formulations. The greater permeability of SQV nanocrystals is seen as shown in Fig. 11e. All these results together indicate that SQV nanocrystals have enhanced oral drug absorption. A recent study by Krieser and her group aimed to improve the taste masking and stability of the SQV nanostructures developed for enhanced pediatric adherence.105 They employed interfacial polymer technique and prepared SQV NPs of 136–158 nm average diameters. The prepared formulation exhibited sustained release, a high drug loading capacity of 80%, ability to encapsulate 97% of the drug with low dynamic viscosity. The in vitro studies showed that SQV NPs showed excellent stability and controlled release properties. The dose can be given in a liquid form to the children with a taste acceptable to them. This amounts to an appreciation for designing drug delivery systems to treat children suffering from HIV.
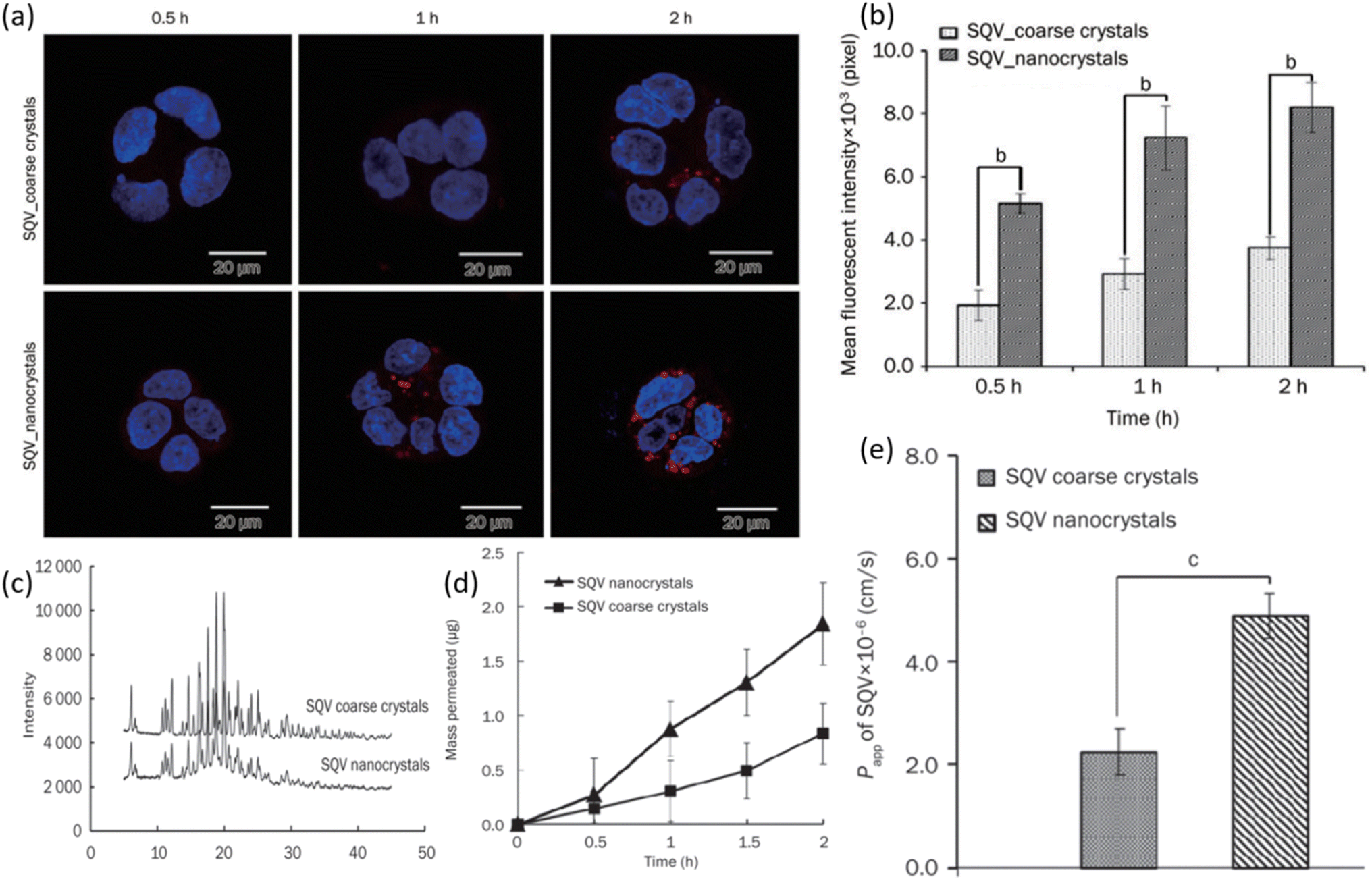 | ||
| Fig. 11 (a) Cellular uptake of SQV coarse and nanocrystals was observed via laser scanning microscope. (b) The fluorescence intensity of cells was analyzed quantitatively. (c) XRPD diffraction pattern of SQV coarse and nanocrystals. (d) Permeation profiles of SQV through a monolayer of Caco-2 cells and (f) permeability coefficient of SQV across monolayers (reprinted with permission from ref. 106 Copyrights 2015 Nature). | ||
3.6 Indinavir (IDV)
To treat children suffering from HIV, the dose being administered to them via oral route must be taste-masked and in liquid form to prevent difficulties in swallowing the tablets. A study reported the preparation of 155 ± 7 nm size NPs of formulation of Monoolein and IDV, using magnetic stirring and high-pressure homogenization method.108 The incorporation efficiency of the formulation was 96% and IDV was able to remain in the same concentration for a month while exhibiting a sustained release profile. This combination was not only biocompatible but showed no irritation with improved taste-masking and the ability to overcome the bitter taste of the drug. Thus, it can be used for pediatric HIV treatment.
3.7 Lamivudine
Glycyrrhizin LMW CS NPs can be used as an effective drug carrier system for liver targeting with decreased damage to tissues and sustained release of the drug. Glycyrrhizin has demonstrated antiviral activities against a broad spectrum of viruses.110,111 Chitosan is a non-toxic, biocompatible, and biodegradable polymer that is widely used as a carrier molecule for various vaccines, genes, protein molecules, etc.112–116 Mishra et al. investigated the controlled release of GL conjugated LMW CS-NPs in liver targeting. Model drug lamivudine was encapsulated within GL-CS-NPs and intravenously administered to a mouse to examine the targeting efficacy. Conjugation of GL was determined by FTIR spectrum as shown in Fig. 12a. The amino functionality (–NH2) of CS has caused stretching of N–H at wave numbers 3346 cm−1 and 3371 cm−1 in Fig. 12a(A) and hence this spectrum is of CS-NPs. While in Fig. 12a(B), –NH deformation of –NH–CH2 verifies the successful conjugation of GL with an amino group in NPs and hence this spectrum represents GL-CS-NPs. In vitro release profile of lamivudine from NPs was examined in PBS solution maintained at a pH of 7.4. The in vitro study showed that CS-NPs were able to release 59.2 ± 2.1% while GL-CS-NPs exhibited 42.9 ± 1.8% release of lamivudine after 72 hours. It is obvious from the graph that the release profile showed a biphasic fashion, with an initial burst followed by a sustained release. The reason for initial burst release can be the attachment of drug particles on the surface of NPs. Lower drug release from GL-CS-NPs can be attributed to the presence of structural features which could have resulted in a double barrier effect. The TEM micrographs showed a smoother surface for CS-NPs than GL-CS-NPs which can be due to the substitution of –NH2 of LMWC by GL (Fig. 12b). The fluorescence micrograph of GL-CS-NPs showed a higher accumulation of hepatocytes cells than plain dye solution and dye-loaded CS-NPs. The fluorescence image of the liver shown in Fig. 12c shows that GL-CS-NPs were able to localize preferentially in the liver and hence greater percentage of lamivudine was seen in hepatocyte tissues with GL-CS-NPs as compared to plain drug. The results of the study indicate that loading lamivudine in GL combined with LMWC-NPs improves the resistance time and quantity of drugs in the liver. This can lead to a reduction in the dosage and quantity of each dose. Moreover, hepatic targeting and controlled release can decrease the toxicity associated with lamivudine. Hence, liver targeting via controlled release of GL-CS NPs can be an effective strategy for the treatment of HIV.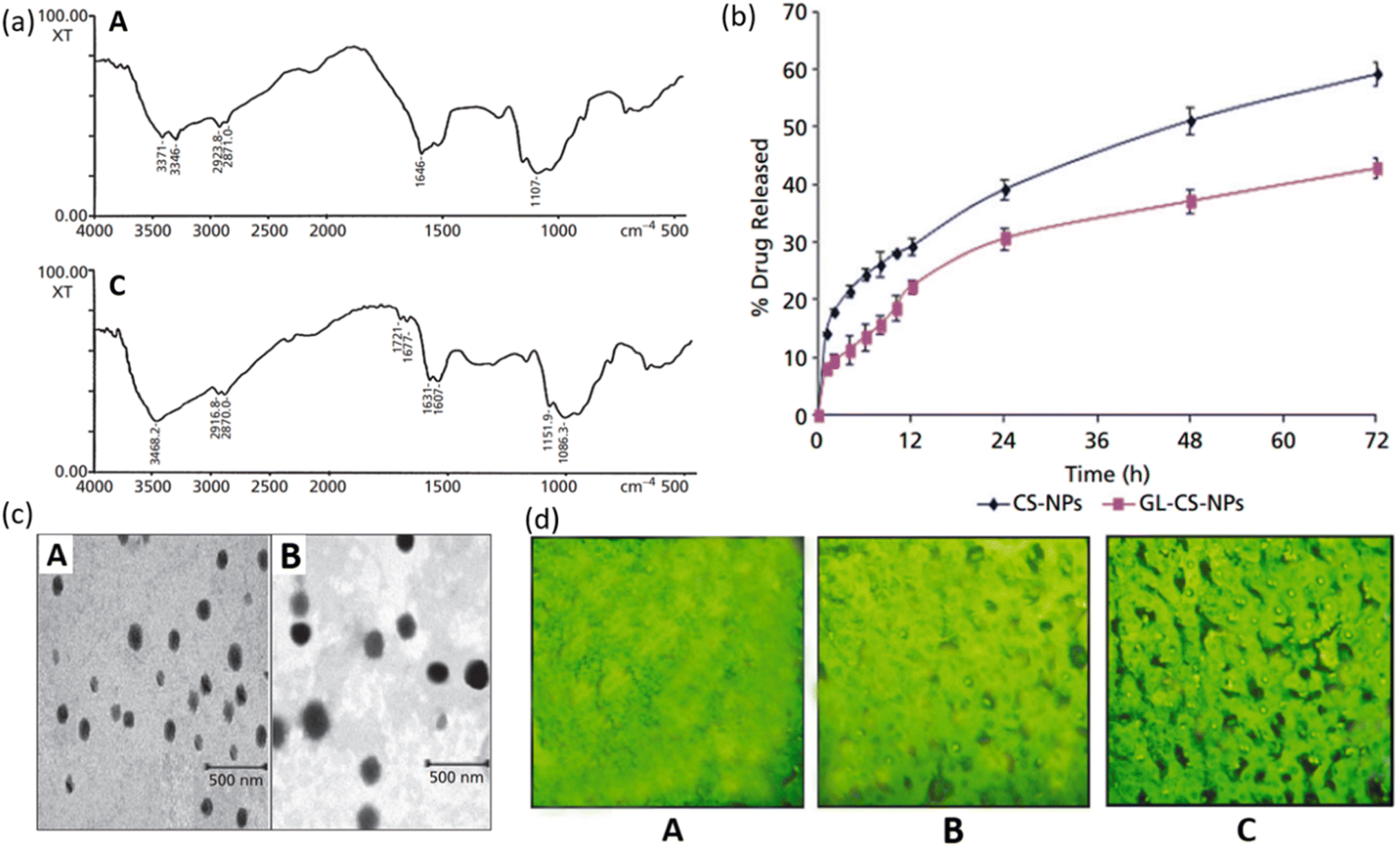 | ||
| Fig. 12 (a) FTIR spectrum of (A) CS-NPs and (B) glycyrrhizin conjugated LMW CS-NPs. (b) TEM micrographs of (A) CS-NPs and (B) glycyrrhizin conjugated LMW CS-NPs. (c) Fluorescent photograph of FITC-labelled (A) plain drug solution; (B) CS-NPs; (C) glycyrrhizin conjugated LMW CS-NPs in the liver sac (reprinted with permission from ref. 117 Copyrights 2014 Wiley). | ||
Another route employed the hot homogenization method to produce lamivudine-MLN (multiple lipid NPs) to enhance the oral administration of the formulation.118 The size reached after the combination of the drug and the NPs was about 450 nm. The simulation studies indicate that around 1.3% of MLN-lamivudine would be released in 4 hours in gastric fluid. The release profile showed sustained and controlled release for about 45 hours. The developed system can be applied as a topical drug or orally administered (after resuspension).
3.8 Tenofovir (TFV)
 | ||
| Fig. 13 (a) Characterization of films via SEM and EDS. (b) Drug release profiles for various films. (c) Analysis of H&E-stained vaginal mucosa (d) quantitative assessment of an average number of epithelial cell layers. (e) Schematic illustration of the proposed idea (reprinted with permission from ref. 120 Copyrights 2016 Elsevier). | ||
Another study examined a thermogelling system containing TFV-loaded with chitosan NPs synthesized via ionic gelation method.121 The biocompatible formulation reached a size of 545.1 ± 69.17 nm once the drug was incorporated into NPs. Gelation temperature of the gel was tolerable for the administration to women and the gelation starts once it is fully administered to the vagina. However, due to the high water solubility of TFV, the NPs showed very low encapsulation efficiency (6.8 ± 3.1%) and drug loading content of 1.86 ± 0.85%. The in vitro study showed that the initial burst release effect was reduced to 27% with the formulation. The prepared vaginal gel holds significance in ease of administration as well as effectiveness for the treatment of women suffering from HIV.
Meng et al. examined the effects of TFV loaded with CS-TGA NPs against HIV prevention.122 They used ionotropic gelation to prepare CS-TGA NPs of mean size between 240–252 nm. Greater encapsulation efficiency of 22.60% was recorded than the study quoted previously. Both NPs did not show any toxicity in 2 days. The percentage of mucoadhesion was five times greater in CS-TGA NPs than CS-NPs. This shows that the prepared NPs have the potential to increase the retention time of TFV, hence making it more effective in the treatment of HIV.
Thiolated chitosan (TCS) core/shell nanofiber (NF) can improve the loading capacity of TFV.123 As stated previously, TFV is highly water-soluble. The coaxial electrospinning technique was utilized to prepare NFs having a core of PEO and shell composed of PLA/TCS. The NFs reached a mean diameter of 99.53 nm with smooth surface morphology. They were found to be safe for topical administration. A significant increase in drug loading capacity was recorded. At concentration of 1 mg mL−1, NFs were non-cytotoxic. Their biocompatibility was proved from in vivo studies. Hence TCS core/shell NFs can be employed as a delivery vehicle of TFV. Table 2 presents a summary of various studies over the past decade showing well known antiviral drugs and their combinations with NPs along with other important parameters.
| Antiviral Drug | Nanoparticle(s) | Action against virus | Route of administration | In vitro/in vivo/ex vivo | Size (nm) | Polydispersity index [PDI] | Synthesis method | Efficiency | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Acyclovir | SLNs | [HSV]-TK | — | In vitro | 180 nm | — | Emulsification and low-temperature solidification method | 78% EE2% | 124 |
| PEGylated lipid polymeric NPs | HSV-1 and HSV-2 | Oral | In vitro, ex vivo | 187.7 ± 3.75 nm | 0.179 ± 0.03 to 0.429 ± 0.12 | Box–Behnken design (BBD) | 83.81 ± 1.93% EE2% | 69 | |
| Bilosomes NCs | HSV-1, HSV-2, and varicella-zoster (VZV) | Oral | In vitro, in vivo, ex vivo | 121.2 ± 3.21 nm | 0.261 ± 0.023 | Thin-film hydration technique. (Optimize by Box–Behnken statistical design) | 71.87–88.67% EE2% | 75 | |
| Gel nanoemulsions (NEs) | HSV | Ocular | In vitro, in vivo, ex vivo | 28 nm to 34 nm | 0.38 ± 0.04 to 0.47 ± 0.05 | Low energy method | 2.8× increase in drug permeation | 125 | |
| Bovine serum albumin (BSA) NPs | HSV | Transcorneal/Ocular | In vitro | 173.0 ± 9.5 to 204.7 ± 15.5 nm | 0.079 ± 0.023 to 0.226 ± 0.025 | — | — | 80 | |
| PLGA polymer stabilized with TPGS nanosystem | HSV | Ocular | In vitro, in vivo, ex vivo | 262.38 ± 11.85 nm | 0.255 ± 0.011 | — | 58.42 to 80.15% EE1% | 81 | |
| Carboxymethyl cellulose acetate butyrate NPs | HSV | Intravenous | In vitro | ∼125–450 nm | — | Precipitation processes (one simple and other rapid) | Drug loading efficiency of 40% | 82 | |
| Eudragit RLPO® based NPs | — | Oral | In vitro | 82 ± 3.83 nm to 532 ± 4.86 nm | 0.308 ± 0.24 to 0.716 ± 0.25 | Nanoprecipitation technique | 79.34 ± 1.64% EE2% | 126 | |
| Chitosan NPs | HSV | Topical delivery | In vitro | 240.0 ± 62.4 nm | 0.53 ± 0.12 | Using cross-linked chitosan with tripolyphosphate (TPP) | 16% EE1% | 127 | |
| Chitosan NPs | OVI | Ocular | In vitro | 200–495 nm | — | Ionic gelation technique | 56 to 80% EE1% | 128 | |
| Microemulsions (ME) | Herpes virus infections | Topical | In vitro | 6.2 ± 0.2 nm to 15.1 ± 1.5 nm | — | Pseudo ternary phase diagrams | 2× fold increase in ACV accumulation | 129 | |
| β-Cyclodextrin-poly(4-acryloylmorpholine) mono-conjugate (β-CD-PACM) | HSV-1 | Oral | In vitro | 150 nm (unloaded) and 200 nm when (loaded) | — | Solvent injection technique | 83% EE1% | 130 | |
| Liposomes | HSV | Intranasal | In vivo | 1048.1 ± 101.3 nm and 627.4 ± 36.9 nm (for two methods) | — | Drug lipid film hydration method | 43.20% | 131 | |
| Bovine serum albumin (BSA) NPs | HSV | Ocular | In vitro | ∼200 nm | — | Desolvation method | 84.59 ± 1.81 and 52.05 ± 2.03 EE2% | 132 | |
| Adefovir dipivoxil | SLNs | HBV | — | In vitro | 389.4 ± 166.5 | −0.371 | Solvent diffusion method | 15% EE2% | 133 |
| Atazanavir | Eudragit RL100 NPs (ATV NPs) | HIV | Oral | In vitro, in vivo | 465.59 nm | 0.372 | Nanoprecipitation method | 41.3 to 56.9% EE1% | 134 |
| Atazanavir and darunavir | LNPs | HIV | Subcutaneous | — | 33.6–35.6 nm | — | Sonication of hydrated lipid–drug suspension | 85.5 ± 8.2 [ATV], 85.1 ± 7.1 [RTV], and 6.1 ± 0.8% [TFV] EE2% | 135 |
| Atazanavir, efavirenz, and ritonavir | Atazanavir, efavirenz, and ritonavir NPs (nano ART) | HIV-1 | Parenteral administration | — | 300–645 nm | — | High-pressure homogenization | — | 136 |
| Azidothymidine | Galactosylated liposomes | AIDS6 | Intravenous | In vitro | 120.01 ± 2.11 nm | — | Esterification of galactose | EE2% (L1 to L4): 42.35 ± 0.38, 54.26 ± 3.25, 36.69 ± 3.10, 31.44 ± 2.22 (%) | 137 |
| Atazanavir and darunavir | Lipid polymer hybrid NPs (LPHNs) | HIV | Oral | In vitro | 50 nm | — | One-step optimized nanoprecipitation method | 62, 68.1 and 68.5% w/w EE1% | 138 |
| Dolutegravir | Chitosan-based polymeric NPs | HIV | Oral | — | 140–548 nm | — | 3-Step process demineralization, deproteinization, and deacetylation | - | 139 |
| Dolutegravir sodium | β-Cyclodextrin-based NPs | Neuro-AIDS | Intranasal | In vitro, in vivo | 72.47 ± 4.8 to 106.5 ± 5.6 nm | 0.306 ± 0.002 and 0.475 ± 0.004 | Cross-linking hydroxypropyl β-cyclodextrin (HPβCD) with diphenyl carbonate | 77 ± 3.35% EE2% | 140 |
| Efavirenz | Chitosan NPs | HIV | Oral | In vitro | ±104 nm | — | Ionotropic gelation method | 91.09% EE2% | 141 |
| Eudragit E100 | HIV/AIDS | Oral | In vitro, in vivo | 110 ± 5 nm | 0.201 ± 0.05 | Emulsion solvent evaporation method | 99% EE2% | 84 | |
| Lactoferrin NPs | HIV | Oral | In vitro, in vivo | 45 ± 60 nm | <0.341 | Sol-oil protocol | 2× times improved anti-HIV-1 action compared to free EFV | 85 | |
| SLNs | HIV | Oral | In vitro, in vivo | 168 nm | <0.220 | Hot homogenization technique followed by ultrasonication method | 60 ± 5% EE2% | 142 | |
| SLNs | HIV | Oral | In vitro | 124.5 ± 3.2 nm | 0.234 | — | 86% EE2% | 143 | |
| Poly(epsilon-caprolactone) (PCL) NPs | HIV/AIDS | Oral | In vitro, in vivo | 200–250 nm | Narrow | Double-emulsion/spray-drying method | 86–93% EE1% | 144 | |
| Nanoemulsion of EFV | HIV/AIDS | Oral | In vitro, in vivo | Less than 30 nm | — | Phase inversion composition method | 80% release within 6 hours | 145 | |
| Chitosan-g-HPβCD NPs | Neuro-AIDS | Intranasal | In vitro, in vivo | 198 ± 4.4 nm | 0.325 ± 0.004 to 0.675 ± 0.005 | Ionic gelation method | 38 ± 1.43% EE2% | 145 | |
| Efavirenz (EFV) and lopinavir/ritonavir (for boost) | PLGA NPs | HIV | Intracellular | In vitro | 138.3–55.4 nm | — | High-pressure homogenization method | >79% EE2% | 146 |
| Efavirenz and nevirapine | SLNs | HIV/AIDS | — | In vitro | 128.7 nm to 182.2 nm | — | Modified emulsion/microemulsion procedure | EFV ∼98% and NVP ∼30% EE2% | 147 |
| Elvitegravir | PLGA-EVG NPs | HIV-1 | Intraperitoneally | In vitro, in vivo | Less than 200 nm | — | Nano-precipitation technique | ∼95% loading efficiency of drug | 148 |
| PLGA-EVG NPs | HIV-1 | — | In vitro | ∼47 nm | - | Nano-precipitation technique | ∼92% EE1% | 149 | |
| Enfuvirtide and protoporphyrin IX | Nano-liposome | HIV-1 | Intravenous and intramuscular administration | In vitro | — | — | Surfactant-based nanoparticles A rapid extrusion procedure | — | 150 |
| Foscarnet | Chitosan NPs | HIV-1, herpesvirus DNA polymerase | Oral, topical | In vitro, in vivo | 292 ± 5 nm to 497 ± 13 nm | 0.26 ± 0.01 to 0.78 ± 0.21 | — | — | 151 |
| Griffithsin (GRFT) (an anti-viral lectin) | mPEG-PLGA GRFT NPs | HIV-1, HSV-2 | Topical | In vitro, in vivo | 152 to 345 nm | — | Double emulsion solvent evaporation technique | 85.6 ± 11.0 EE1% | 152 |
| Indinavir | Lipid nanoemulsion (LNE) | HIV | Intravenous | In vitro, in vivo | 200.1 ± 73.2 nm (lowest value) | 0.05 ± 0.04 | — | 98.8%, 98.9% and 99.0% EE2% | 109 |
| Monoolein-based NPs | HIV | Oral | In vitro | 155 ± 7 nm | 0.16 ± 0.03 | Magnetic stirring and high-pressure homogenization | 96% drug incorporation efficiency | 108 | |
| mPEG-PCL NPs | HIV | Oral | In vivo | 211 ± 10.12 nm (mean particle size) | 0.22 to 0.68 | Emulsification solvent evaporation method | 60%, 40% and 15% drug release percent | 107 | |
| Indinavir and lactoferrin | Nanoemulsion | HIV | Drug injection | In vitro, in vivo | 112 ± 3.5 nm | 0.20 ± 0.02 | High-speed homogenization method | - | 153 |
| Ivermectin | Ivermectin NPs | ZIKV | Oral | In vitro, in vivo | ∼65 nm | — | — | Conjugation efficiency of ∼60% for empty NPs and ∼40% for 20% IVM feed loaded NPs | 154 |
| Lamivudine | Chitosan NPs | HIV-1 | Oral | In vitro, in vivo | 120.7 ± 3.1 nm [CS-NPs]; 145.8 ± 4.2 [GL-LMWC-NPs] | 0.09 ± 0.01 [CS-NPs]; 0.11 ± 0.06 [GL-LMWC-NPs] | Depolymerization followed by ionotropic gelation method | 71.37 ± 1.19% EE1% | 117 |
| MLNs | — | Topical (semisolid) or oral (after resuspension) | In vitro | ∼450 nm | <0.3 | Hot homogenization method in conjunction with high shear and ultrasonication | — | 118 | |
| Lopinavir | SLNs | HIV/AIDS | Oral | In vivo | 196.5 ± 3.5 nm | 0.11 ± 0.01 | Warm oil-in-water (O/W) micro-emulsion technique | EE (%) 76.5 ± 3.5% | 101 |
| In situ self-assembly nanoparticles (ISNPs) | HIV | Oral | In vitro, in vivo | Less than 158 nm | – | Warn microemulsion precursors with modification | 95% EE2% | 100 | |
| PLGA NPs | HIV/AIDS | Oral | In vitro, in vivo | 142.1 ± 2.13 nm | – | Solvent diffusion (nanoprecipitation) method | 93.03 ± 1.27% EE2% | 155 | |
| Pullulan acetate NPs | HIV/AIDS | Oral | In vitro, in vivo | 197 ± 4 nm (∼197 nm) | <0.2 | Motozato's method | 75% EE2% | 103 | |
| Poly-e-caprolactone (PCL) nanoparticles (NPs) | HIV/AIDS | Oral | In vitro, in vivo, ex vivo | 195.3 ± 2.3 nm | 0.10 ± 0.01 | Oil-in-water emulsion-solvent evaporation technique | 93.9% EE2% | 156 | |
| SLNs | HIV/AIDS | Oral | In vitro, in vivo | 180.6 ± 2.32 nm | 0.133 ± 0.001 | Hot self-nano emulsification (SNE) technique | 91.5 ± 1.3% EE2% | 102 | |
| Compritol®-SLNs | HIV/AIDS | Oral | In vivo | 156 nm | – | Hot homogenization method followed by ultrasonication | 98.99% EE2% (highest) | 157 | |
| Lopinavir–ritonavir–tenofovir | Drug-combination nanoparticles (DcNPs) | HIV | Subcutaneous | In vitro | — | — | Aseptic technique | Highest drug association efficiency of 99 ± 8.2% for lopinavir, 92 ± 7.1% for ritonavir and 10 ± 0.8% for tenofovir | 158 |
| Nevirapine | Mesoporous silica nanoparticles (MSNPs) | HIV-1 | — | In vitro | 60 nm | — | Stober's method | — | 159 |
| PS80-coated PCL NPs | HIV/AIDS | Intravenous | In vitro, in vivo | 218.3 ± 7.3 nm | 0.283 ± 0.038; 0.179 ± 0.00 | Emulsion solvent evaporation technique | 50.71% EE2% (highest) | 160 | |
| Cellulose acetate butyrate (CAB) NPs | HIV/AIDS | — | In vitro | 305.76 ± 5.7 nm | 0.29 ± 0.03 | Emulsification solvent evaporation method | 75.89 ± 1.36% EE1% | 161 | |
| Nanoliposomes | HIV/AIDS | — | In vitro | 157 nm | — | Thin-film hydration | 78.14% and 76.25% EE1% | 162 | |
| Oseltamivir | SeNPs | EV71 | — | In vitro | 10 nm | — | — | — | 50 |
| Raltegravir + efavirenz | PLGA NPs | HIV | Intravaginal | In vitro | 81.8 ± 6.4 nm | — | Emulsion–solvent evaporation method | 55.5% [RAL] and 98.2% [EFV] EE1% | 163 |
| Ritonavir | SLNs | HIV-1 | Oral | In vitro | 170–250 nm | 0.2 | Solvent emulsification method and double emulsion method | 53.2% EE2% | 164 |
| SLNs | HIV/AIDS | Oral | In vitro, in vivo | Less than 300 nm | 0.361 | Solvent evaporation followed by ultrasonication | 53.20 ± 4.13 to 73.04 ± 2.85% EE1% | 165 | |
| PLGA NPs | HIV/AIDS | — | — | 42–102 nm | 0.381 | Solid-in-oil-in-water (s/o/w) solvent evaporation technique with some changes | 75% EE1% | 166 | |
| Lopinavir (LPN) NPs | HIV-1 | Oral | In vitro, in vivo | ∼320 nm | <0.2 | Antisolvent precipitation and high-pressure homogenization techniques | — | 167 | |
| Saliphenylhalamide (SaliPhe) | SiNPs | Influenza A viruses (IAVs) | Inhalation or intravenous (envisioned) | In vitro | 129 ± 10 nm | 0.112 | — | — | 168 |
| Saquinavir | SQV NPs | HIV | Oral | In vitro | 136–158 nm | — | Interfacial polymer technique | > 97% EE1% | 105 |
| SQV nanocrystals | HIV | Oral | In vivo, ex vivo | 205.93 ± 3.74 nm | 0.1 | Anti-solvent precipitation high-pressure homogenization method | — | 106 | |
| Chitosan NPs | AIDS | — | In vitro | 10–200 nm | — | Ionic gelation technique | 72% EE1% | 169 | |
| SLNs | HIV/AIDS | Intravenous | — | 120 nm to 450 nm | — | — | — | 170 | |
| SLNs | HIV | Oral | In vitro, in vivo | 215 ± 9 nm [SQSLNs]; 344 ± 16 nm [SNS] | 0.196 ± 0.019 of SNS | Hot high-pressure homogenization (HPH) method | 79.24 ± 1.53% EE2% | 104 | |
| Stavudine | Chitosan NPs | HIV | Oral | In vitro | 212 nm (PSD) | — | Ionic gelation of chitosan with tripolyphosphate anions | 85.8 ± 0.16% EE2% (highest) | 171 |
| SLNs | HIV-1/AIDS | Intravenous | In vitro, in vivo, ex-vivo | 75 ± 1.22 nm | 0.12 | Homogenization | High labeling efficiency | 172 | |
| Mannosylated liposomes | HIV | Intravenous | In vitro, in vivo | 120 ± 1.52 nm | — | Esterification of mannose | 47.2 ± 1.57% EE2% | 173 | |
| Stavudine, delavirdine, and saquinavir | SLNs | HIV/AIDS | — | — | 142–294 nm | — | Involves emulsion | — | 174 |
| Tenofovir | Thiolated chitosan (TCS) core/shell nanofiber (NF) | HIV-1 | Topical | In vitro, in vivo | 58.81 nm | - | Coaxial electrospinning technique | 95% (in 5 hours) | 123 |
| Chitosan NPs | HIV | Vaginal route | In vitro | 545.1 ± 69.17 nm | 0.663 ± 0.107 | Ionic gelation | 6.8 ± 3.1 EE1% | 121 | |
| (PLGA)/stearylamine (SA) composite NPs | HIV | Vaginal route | In vitro, in vivo, ex vivo | 127 ± 1 nm | 0.27 ± 0.01 | Double emulsion/solvent evaporation method | Drug association efficiency >50% | 120 | |
| Chitosan–thioglycolic acid-conjugated (CS–TGA) NPs | HIV/AIDS | Topical | In vitro | 240.1 nm CS NPs; 252.3 nm CS-TGA-NPs | 0.298 ± 0.002 [CS]; 0.317 ± 0.052 [CS-TGA] | Ionotropic gelation | 22.60% EE1% | 122 | |
| PLGA NPs loaded with efavirenz NPs or saquinavir NPs | HIV-1 BaL infection | Topical | In vitro | 227 ± 1.8 nm [EFV]; 189 ± 96.3 nm [SQV] | 0.05 [EFV]; 0.486 [SQV] | Emulsion or nanoprecipitation techniques | 44.5 ± 2.7 [EFV] and 48.3 ± 15.2 [SQV] | 175 | |
| Tenofovir, alafenamide and elvitegravir | TAF + EVG NPs | HIV | Subcutaneous | In vitro, in vivo | 190.2 ± 2.3 nm | 0.14 ± 0.01 | Oil-in-water emulsion solvent evaporation technique | 54.1 ± 3.6 [TAF] and 44.6 ± 2.4% [EVG] EE1% | 176 |
| Tenofovir alafenamide | Emtricitabine (FTC) loaded NPs | HIV-1 | Subcutaneous and oral | In vivo, ex vivo | 233.2 ± 12.8 nm | 0.11 ± 0.05 | Oil-in-water emulsion solvent evaporation technique | 69.2 ± 14.5% [TAF] and 65.9 ± 18.2% [FTC] EE1% | 177 |
| Tenofovir disoproxil fumarate | Chitosan NPs | HIV/AIDS | Oral | In vitro, in vivo, ex vivo | 156 ± 5 nm | 0.16 ± 0.06 | Ionic gelation technique | 48.2 ± 1% EE2% | 178 |
| Valacyclovir | PLA-PEG NPs | HSV | Oral | In vitro, in vivo | ∼30 nm | — | Nanoprecipitation | 11.4 ± 0.5 EE2% (highest) | 179 |
| SLNs | HSV | Ocular | In vitro, in vivo, ex vivo | 202.5 ± 2.56 nm | 0.252 ± 0.06 | Solvent emulsification/evaporation method | 28.01 ± 1.89 to 58.82 ± 2.45% EE2% | 180 | |
| Zidovudine | NLCs | HIV | Oral | In vitro | 100 to 300 nm | < 0.3 | Hot ultrasonication and microwave assisted method | 44 ± 3%, 22 ± 2% EE2% | 181 |
| Alginate NPs | HIV/AIDS | Intravenous | In vitro | 432 ± 11.9 nm | — | Emulsion solvent evaporation method | Loading efficacy of 29.5 ± 3.2% | 88 | |
| Lipid NPs modified with polymer gelatin | HIV/AIDS | Oral and topical | In vitro | 224 ± 31.2 nm [PLNs of SA]; 291.2 ± 38 nm [PLNs of comp] | — | — | 87.4 ± 0.58% EE1% | 89 | |
| Nanosized polyelectrolyte complexes (PECs) | HIV | — | In vitro | 100–200 nm | 0.125–0.305 | — | Drug release of 38.1% at pH 4.5 and 31.2% at pH 7.4 | 182 | |
| SLNs | HIV | Parenteral, oral, ophthalmic, and topical | — | 222–227 nm [AZT-SA], 402 nm to 434 nm [AZT-SA-AV] | 0.2 to 0.3 [AZA-SA], 0.38–0.45 [AZT-SA-AV] | Simple emulsion solvent evaporation method | 74.92 ± 1.2% EE1% | 92 | |
| Chitosan NPs | AIDS | Nasal | — | 260 ± 1.70 nm, 330 ± 12.9 [NP1, NP2]; 406 ± 14.0 and 425 ± 14.5 for AZT-loaded NP1 and NP2 | 0.247, 0.329, 0.390, 0.381 | Ionotropic gelation method | 17.58% ± 1.48 and 11.02% ± 2.05 EE2% for NP1 and NP2 | 183 | |
| Lipid-polymer hybrid NP | HIV | — | In vitro | 175 ± 2.5 nm | 0.196 | Melt emulsification-probe sonication technique | 6.5 ± 0.50 to 49.26 ± 0.75% EE2% | 184 | |
| PLA–PEG blend NPs | AIDS | Intranasal | In vivo | 328.1 ± 8.6 nm | 0.383 | Double emulsion–evaporation method | 52% EE1% | 185 | |
| SLNs | AIDS | Oral | In vitro | 621 nm | — | W/o/w double-emulsion solvent–evaporation method | 27% EE2% | 186 | |
| Dextran and stearic acid NPs | HIV/AIDS | Intravenous | In vitro, in vivo | 356 nm to 730 nm | — | Double emulsion solvent evaporation method | 93.46% EE1% | 86 | |
| PVP/SA-PEG NPs (PSNPs) | AIDS | Intravenous | In vitro | 341 ± 4.34 nm | 0.3 ± 0.04 | Emulsification-solvent evaporation method | 37.19% to 79.2% | 87 | |
| Hybrid NPs of CMC-AZT core enclosed by shell of Comp-PEG | AIDS | Oral | In vitro | 161.65 ± 44.06 nm | — | — | 82% EE1% | 90 | |
| Zidovudine + efavirenz + lamivudine | Lactoferrin NPs | HIV | Oral | In vitro, in vivo | 67 nm | — | Sol-oil protocol | 58 to 71% EE1% | 187 |
4. Conclusion
The established potential of nanotechnology in antiviral applications coupled with massive progress in designing wide categories of different nanomaterials-based antiviral drugs gives many expectations to the research community to explore their potential further to overcome their existing drawbacks. The nano-based systems can prove fruitful in overcoming the associated drawbacks with the current therapies such as low oral bioavailability, issues with hemocompatibility, chances for skin diseases due to topical exposure of drug, toxic nature of the drug, reduction in drug's effectiveness due to frequent administration, to name a few. However, as per the best of our knowledge, most of these nano-based systems designed to date are not approved for clinical use or are still under clinical trials, despite exhaustive and continued effort for more than 24 years. It is expected that in upcoming years, we shall witness more clinical approvals and applications of these nano-based systems which will definitely help overcome the health issues globally.5. Future perspectives
Tuning at the nanoscale can give us interesting properties for a range of applications. In our review, we have covered many functional metallic NPs as well as a variety of drugs that can be tailored by using NPs as their delivery vehicles. However, in order to treat patients globally in vast numbers (such as with COVID-19 pandemic recently), it is not possible to develop antiviral drugs in such a short span of time and fully overcome their existing drawbacks. As we discussed earlier, frequent drug administration is itself counterproductive since it generates an immune response from body and increases its drug resistance resulting in a decline in the drug's efficiency. Moreover, the route of administration for each drug must be carefully chosen. There are associated disadvantages with the choice of each route. For instance, oral administration may not be feasible for children, ocular delivery could prove detrimental for eyes and topical exposure of drug to skin may cause skin diseases. On contrary, vaccines have proven to be a rather better option in fighting pandemics in the past century. However, vaccines also have associated shortcomings such as insufficient cellular immunity, risk of antibodies dependent enhancements (ADE) and lack of cross-permeation. Our discussion regarding the potential of nanomaterials in antiviral applications gives useful insight that they can be a good candidate in development of novel vaccines against various viral infections. We suggest the scientific community and interested researchers to also focus on further development of these “nanovaccines”, evaluate their potential against viruses, weigh their pros and cons, and explore methods for their cost-effective and large-scale manufacturing so that they can deal with current and upcoming global health challenges posed by such highly unpredictable viral outbreaks.Author contributions
Muhammad Aanish Ali (drafting and paper writing); Dr Nagina Rehman (drafting and reviewing the paper); Dr Tae Joo Park (resources and supervision); Dr Muhammad Abdul Basit (conceptualization, resources and data analysis).Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The work is mainly done at Department of Materials Science and Engineering at Institute of Space Technology, Pakistan and supported by NEL, Hanyang University South Korea from Project No. 20010727 (Technology Innovation Program by Ministry of Trade, Industry and Energy Korea).References
- R. E. Kahn, W. Ma and J. A. Richt, in Influenza Pathogenesis and Control - Volume I, ed. R. W. Compans and M. B. A. Oldstone, Springer International Publishing, Cham, 2014, pp. 205–218 Search PubMed.
- M. Qasim, D.-J. Lim, H. Park and D. Na, J. Nanosci. Nanotechnol., 2014, 14, 7374–7387 CrossRef CAS PubMed.
- C. Wejse, C. B. Patsche, A. Kühle, F. J. V. Bamba, M. S. Mendes, G. Lemvik, V. F. Gomes and F. Rudolf, Int. J. Infect. Dis., 2015, 32, 128–134 CrossRef CAS PubMed.
- C. R. Braden, S. F. Dowell, D. B. Jernigan and J. M. Hughes, Emerging Infect. Dis., 2013, 19, 864–869 CrossRef PubMed.
- M. Breitbart and F. Rohwer, Trends Microbiol., 2005, 13, 278–284 CrossRef CAS PubMed.
- L. Chen and J. Liang, Mater. Sci. Eng. C, 2020, 112, 110924 CrossRef CAS PubMed.
- A. Rivera and I. Messaoudi, ACS Infectious Diseases, 2015, 1, 186–197 CrossRef CAS PubMed.
- C. J. Schweitzer and T. J. Liang, ACS Infectious Diseases, 2016, 1, 416–419 CrossRef PubMed.
- B. A. Aderibigbe, Molecules, 2017, 22, 1370 CrossRef PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, M. Cantisani, V. Marra and M. Galdiero, Molecules, 2011, 16, 8894–8918 CrossRef CAS PubMed.
- D. M. Coen and R. J. Whitley, Curr. Opin. Virol., 2011, 1, 545–547 CrossRef PubMed.
- F. Corsi, L. Sorrentino, S. Mazzucchelli, M. Truffi, A. Capetti, G. Rizzardini and L. Fiandra, J. Pharm. Pharmacol., 2016, 4, 328–339 Search PubMed.
- D. Lembo, M. Donalisio, A. Civra, M. Argenziano and R. Cavalli, Expert Opin. Drug Delivery, 2018, 15, 93–114 CrossRef CAS PubMed.
- L. Calderón, R. Harris, M. Cordoba-Diaz, M. Elorza, B. Elorza, J. Lenoir, E. Adriaens, J. P. Remon, A. Heras and D. Cordoba-Diaz, Eur. J. Pharm. Sci., 2013, 48, 216–222 CrossRef PubMed.
- T. Q. Huy, N. T. Hien Thanh, N. T. Thuy, P. Van Chung, P. N. Hung, A. T. Le and N. T. Hong Hanh, J. Virol. Methods, 2017, 241, 52–57 CrossRef CAS PubMed.
- D. Morris, M. Ansar, J. Speshock, T. Ivanciuc, Y. Qu, A. Casola and R. Garofalo, Viruses, 2019, 11, 732 CrossRef CAS PubMed.
- Y. Li, Z. Lin, T. Xu, C. Wang, M. Zhao, M. Xiao, H. Wang, N. Deng and B. Zhu, RSC Adv., 2017, 7, 1453–1463 RSC.
- T. Q. Huy, N. T. Hien Thanh, N. T. Thuy, P. Van Chung, P. N. Hung, A. T. Le and N. T. Hong Hanh, J. Virol. Methods, 2017, 241, 52–57 CrossRef CAS PubMed.
- E. Szymańska, P. Orłowski, K. Winnicka, E. Tomaszewska, P. Bąska, G. Celichowski, J. Grobełny, A. Basa and M. Krzyżowska, Int. J. Mol. Sci., 2018, 19, 387 CrossRef PubMed.
- M. M. El-Sheekh, M. T. Shabaan, L. Hassan and H. H. Morsi, Int. J. Environ. Health Res., 2020, 1–12 Search PubMed.
- E. G. Haggag, A. M. Elshamy, M. A. Rabeh, N. M. Gabr, M. Salem, K. A. Youssif, A. Samir, A. Bin Muhsinah, A. Alsayari and U. R. Abdelmohsen, Int. J. Nanomed., 2019, 14, 6217–6229 CrossRef CAS PubMed.
- T. V. M. Sreekanth, P. C. Nagajyothi, P. Muthuraman, G. Enkhtaivan, S. V. P. Vattikuti, C. O. Tettey, D. H. Kim, J. Shim and K. Yoo, J. Photochem. Photobiol., B, 2018, 188, 6–11 CrossRef CAS PubMed.
- D. xi Xiang, Q. Chen, L. Pang and C. long Zheng, J. Virol. Methods, 2011, 178, 137–142 CrossRef PubMed.
- D. Xiang, Y. Zheng, W. Duan, X. Li, J. Yin, S. Shigdar, M. L. O'Connor, M. Marappan, X. Zhao, Y. Miao, B. Xiang and C. Zheng, Int. J. Nanomed., 2013, 8, 4103–4114 CrossRef PubMed.
- B. Gerhardson, Trends Biotechnol., 2002, 20, 338–343 CrossRef CAS PubMed.
- A. S. Hamed Derbalah and M. M. Elsharkawy, J. Biotechnol., 2019, 306, 134–141 CrossRef CAS PubMed.
- C. Huo, J. Xiao, K. Xiao, S. Zou, M. Wang, P. Qi, T. Liu and Y. Hu, Int. J. Nanomed., 2020, 15, 661–674 CrossRef CAS PubMed.
- J. N. Makau, K. Watanabe, M. M. D. Mohammed and N. Nishida, J. Med. Food, 2018, 21, 777–784 CrossRef CAS PubMed.
- K. S. Siddiqi, A. ur Rahman, Tajuddin and A. Husen, Nanoscale Res. Lett., 2018, 13(1), 1–13 CrossRef PubMed.
- H. Ghaffari, A. Tavakoli, A. Moradi, A. Tabarraei, F. Bokharaei-Salim, M. Zahmatkeshan, M. Farahmand, D. Javanmard, S. J. Kiani, M. Esghaei, V. Pirhajati-Mahabadi, A. Ataei-Pirkooh and S. H. Monavari, J. Biomed. Sci., 2019, 26, 1–10 CrossRef CAS PubMed.
- K. W. BAILISS and S. Senananyake, Plant Pathol., 1984, 33, 185–192 CrossRef.
- Y. H. Hsueh, K. S. Lin, W. J. Ke, C. Te Hsieh, C. L. Chiang, D. Y. Tzou and S. T. Liu, PLoS One, 2015, 10, e0144306 CrossRef PubMed.
- C. Adán, J. Marugán, E. Sánchez, C. Pablos and R. Van Grieken, Electrochim. Acta, 2016, 191, 521–529 CrossRef.
- R. M. Di Piero, Q. S. de Novaes and S. F. Pascholati, Braz. Arch. Biol. Technol., 2010, 53, 269–278 CrossRef.
- M. M. Elsharkawy and A. Derbalah, Pest Manage. Sci., 2019, 75, 828–834 CrossRef CAS PubMed.
- H. Gomaa, H. Khalifa, M. M. Selim, M. A. Shenashen, S. Kawada, A. S. Alamoudi, A. M. Azzam, A. A. Alhamid and S. A. El-Safty, ACS Sustainable Chem. Eng., 2017, 5, 10826–10839 CrossRef CAS.
- C. Jia, P. Yang, H. S. Chen and J. Wang, CrystEngComm, 2015, 17, 2940–2948 RSC.
- R. Nakano, H. Ishiguro, Y. Yao, J. Kajioka, A. Fujishima, K. Sunada, M. Minoshima, K. Hashimoto and Y. Kubota, Photochem. Photobiol. Sci., 2012, 11, 1293–1298 CrossRef CAS PubMed.
- Y. N. Chen, Y. H. Hsueh, C. Te Hsieh, D. Y. Tzou and P. L. Chang, Int. J. Environ. Res. Public Health, 2016, 13, 4–6 Search PubMed.
- A. Halder, S. Das, D. Ojha, D. Chattopadhyay and A. Mukherjee, Mater. Sci. Eng. C, 2018, 89, 413–421 CrossRef CAS PubMed.
- P. Di Gianvincenzo, M. Marradi, O. M. Martínez-Ávila, L. M. Bedoya, J. Alcamí and S. Penadés, Bioorg. Med. Chem. Lett., 2010, 20, 2718–2721 CrossRef CAS PubMed.
- I. Papp, C. Sieben, K. Ludwig, M. Roskamp, C. Böttcher, S. Schlecht, A. Herrmann and R. Haag, Small, 2010, 6, 2900–2906 CrossRef CAS PubMed.
- A. M. Paul, Y. Shi, D. Acharya, J. R. Douglas, A. Cooley, J. F. Anderson, F. Huang and F. Bai, J. Gen. Virol., 2014, 95, 1712–1722 CrossRef CAS PubMed.
- Y. Li, Z. Lin, M. Guo, Y. Xia, M. Zhao, C. Wang, T. Xu, T. Chen and B. Zhu, Int. J. Nanomed., 2017, 12, 5733–5743 CrossRef CAS PubMed.
- Z. Lin, Y. Li, G. Gong, Y. Xia, C. Wang, Y. Chen, L. Hua, J. Zhong, Y. Tang, X. Liu and B. Zhu, Int. J. Nanomed., 2018, 13, 5787–5797 CrossRef CAS PubMed.
- Y. Li, T. Xu, Z. Lin, C. Wang, Y. Xia, M. Guo, M. Zhao, Y. Chen and B. Zhu, ACS Omega, 2019, 4, 6720–6725 CrossRef CAS.
- Y. Li, Z. Lin, M. Guo, M. Zhao, Y. Xia, C. Wang, T. Xu and B. Zhu, Int. J. Nanomed., 2018, 13, 2005–2016 CrossRef CAS PubMed.
- A. EA and P. EB, Int. J. Med. Inform., 2019, 125, 30–36 CrossRef PubMed.
- Z. Cheng, X. Zhi, G. Sun, W. Guo, Y. Huang, W. Sun, X. Tian, F. Zhao and K. Hu, J. Med. Virol., 2016, 88, 653–663 CrossRef CAS PubMed.
- J. Zhong, Y. Xia, L. Hua, X. Liu, M. Xiao, T. Xu, B. Zhu and H. Cao, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 3485–3491 CrossRef CAS PubMed.
- T. Du, J. Liang, N. Dong, J. Lu, Y. Fu, L. Fang, S. Xiao and H. Han, ACS Appl. Mater. Interfaces, 2018, 10, 4369–4378 CrossRef CAS PubMed.
- V. Sujitha, K. Murugan, M. Paulpandi, C. Panneerselvam, U. Suresh, M. Roni, M. Nicoletti, A. Higuchi, P. Madhiyazhagan, J. Subramaniam, D. Dinesh, C. Vadivalagan, B. Chandramohan, A. A. Alarfaj, M. A. Munusamy, D. R. Barnard and G. Benelli, Parasitol. Res., 2015, 114, 3315–3325 CrossRef PubMed.
- J. Jampílek and K. Král’ová, in Nanotheranostics: Applications and Limitations, Nature Publishing Group, 2019, pp. 137–178 Search PubMed.
- D. Baram-Pinto, S. Shukla, A. Gedanken and R. Sarid, Small, 2010, 6, 1044–1050 CrossRef CAS PubMed.
- M. Z. Fahmi, W. Sukmayani, S. Q. Khairunisa, A. M. Witaningrum, D. W. Indriati, M. Q. Y. Matondang, J. Y. Chang, T. Kotaki and M. Kameoka, RSC Adv., 2016, 6, 92996–93002 RSC.
- A. Łoczechin, K. Séron, A. Barras, E. Giovanelli, S. Belouzard, Y. T. Chen, N. Metzler-Nolte, R. Boukherroub, J. Dubuisson and S. Szunerits, ACS Appl. Mater. Interfaces, 2019, 11, 42964–42974 CrossRef PubMed.
- Y. Fujimori, T. Sato, T. Hayata, T. Nagao, M. Nakayam, T. Nakayam, R. Sugamat and K. Suzuki, Appl. Environ. Microbiol., 2012, 78, 951–955 CrossRef CAS PubMed.
- N. Shionoiri, T. Sato, Y. Fujimori, T. Nakayama, M. Nemoto, T. Matsunaga and T. Tanaka, J. Biosci. Bioeng., 2012, 113, 580–586 CrossRef CAS PubMed.
- I. Das Jana, P. Kumbhakar, S. Banerjee, C. C. Gowda, N. Kedia, S. K. Kuila, S. Banerjee, N. C. Das, A. K. Das, I. Manna, C. S. Tiwary and A. Mondal, ACS Appl. Nano Mater., 2021, 4, 352–362 CrossRef CAS.
- A. Tavakoli and M. S. Hashemzadeh, J. Virol. Methods, 2020, 275, 113688 CrossRef CAS PubMed.
- X. Hang, H. Peng, H. Song, Z. Qi, X. Miao and W. Xu, J. Virol. Methods, 2015, 222, 150–157 CrossRef CAS PubMed.
- L. Tang, Y. Jiang, M. Zhu, L. Chen, X. Zhou, C. Zhou, P. Ye, X. Chen, B. Wang, Z. Xu, Q. Zhang, X. Xu, H. Gao, X. Wu, D. Li, W. Jiang, J. Qu, C. Xiang and L. Li, Front. Med., 2020, 14, 664–673 CrossRef PubMed.
- R. Kumar, M. Nayak, G. C. Sahoo, K. Pandey, M. C. Sarkar, Y. Ansari, V. N. R. Das, R. K. Topno, Bhawna, M. Madhukar and P. Das, J. Infect. Chemother., 2019, 25, 325–329 CrossRef CAS PubMed.
- Z. Lin, Y. Li, T. Xu, M. Guo, C. Wang, M. Zhao, H. Chen, J. Kuang, W. Li, Y. Zhang, T. Lin, Y. Chen, H. Chen and B. Zhu, ACS Omega, 2020, 5, 12495–12500 CrossRef CAS PubMed.
- C. Balagna, S. Perero, E. Percivalle, E. V. Nepita and M. Ferraris, Open Ceram., 2020, 1, 100006 CrossRef CAS.
- T. Du, J. Lu, L. Liu, N. Dong, L. Fang, S. Xiao and H. Han, ACS Appl. Bio Mater., 2018, 1, 1286–1293 CrossRef CAS PubMed.
- N. A. Mazurkova, Y. E. Spitsyna, N. V. Shikina, Z. R. Ismagilov, S. N. Zagrebel’nyi and E. I. Ryabchikova, Nanotechnol. Russ., 2010, 5, 417–420 CrossRef.
- F. Pfaff, B. Glück, T. Hoyer, D. Rohländer, A. Sauerbrei and R. Zell, Lett. Appl. Microbiol., 2019, 69, 302–309 CrossRef CAS PubMed.
- S. Mahmood, K. C. Kiong, C. S. Tham, T. C. Chien, A. R. Hilles and J. R. Venugopal, AAPS PharmSciTech, 2020, 21, 1–15 CrossRef PubMed.
- S. Ilk, N. Saglam and M. Özgen, Artif. Cells, Nanomed., Biotechnol., 2017, 45, 907–916 CrossRef CAS PubMed.
- S. Raut, S. S. Bhadoriya, V. Uplanchiwar, V. Mishra, A. Gahane and S. K. Jain, Acta Pharm. Sin. B, 2012, 2, 8–15 CrossRef CAS.
- Q. Tan, W. Liu, C. Guo and G. Zhai, Int. J. Nanomed., 2011, 6, 1621 CrossRef CAS PubMed.
- J. J. F. Verhoef and T. J. Anchordoquy, Drug Delivery Transl. Res., 2013, 3, 499–503 CrossRef CAS PubMed.
- S. Mahmood, K. C. Kiong, C. S. Tham, T. C. Chien, A. R. Hilles and J. R. Venugopal, AAPS PharmSciTech, 2020, 21(7), 1–15 CrossRef PubMed.
- Z. Saifi, M. Rizwanullah, S. R. Mir and S. Amin, J. Drug Delivery Sci. Technol., 2020, 57, 101634 CrossRef.
- J. F. Fangueiro, T. Andreani, L. Fernandes, M. L. Garcia, M. A. Egea, A. M. Silva and E. B. Souto, Colloids Surf., B, 2014, 123, 452–460 CrossRef CAS PubMed.
- K. Cholkar, S. P. Patel, A. D. Vadlapudi and A. K. Mitra, J. Ocul. Pharmacol. Ther., 2013, 29, 106–123 CrossRef CAS PubMed.
- M. H. Warsi, M. Anwar, V. Garg, G. K. Jain, S. Talegaonkar, F. J. Ahmad and R. K. Khar, Colloids Surf., B, 2014, 122, 423–431 CrossRef CAS PubMed.
- U. V. Bhosale, V. Kusum Devi and N. Jain, J. Young Pharm., 2011, 3, 275–283 CrossRef CAS PubMed.
- P. Suwannoi, M. Chomnawang, A. Tunsirikongkon, A. Phongphisutthinan, C. C. Müller-Goymann and N. Sarisuta, J. Drug Delivery Sci. Technol., 2019, 52, 624–631 CrossRef CAS.
- M. Alkholief, H. Albasit, A. Alhowyan, S. Alshehri, M. Raish, M. Abul Kalam and A. Alshamsan, Saudi Pharm. J., 2019, 27, 293–302 CrossRef PubMed.
- V. B. Vedula, M. Chopra, E. Joseph and S. Mazumder, Appl. Nanosci., 2016, 6, 197–208 CrossRef CAS.
- A. Belgamwar, S. Khan and P. Yeole, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 374–386 CrossRef CAS PubMed.
- B. N. V. Hari, N. Narayanan, K. Dhevendaran and D. Ramyadevi, Mater. Sci. Eng. C, 2016, 67, 522–532 CrossRef CAS PubMed.
- P. Kumar, Y. S. Lakshmi and A. K. Kondapi, HIV Med., 2017, 18, 452–462 CrossRef CAS PubMed.
- K. S. Joshy, A. George, S. Snigdha, B. Joseph, N. Kalarikkal, L. A. Pothen and S. Thomas, Mater. Sci. Eng. C, 2018, 93, 864–872 CrossRef CAS PubMed.
- K. S. Joshy, S. Snigdha, G. Anne, K. Nandakumar, A. P. Laly and T. Sabu, Chem. Phys. Lipids, 2018, 210, 82–89 CrossRef CAS PubMed.
- K. S. Joshy, M. A. Susan, S. Snigdha, K. Nandakumar, A. P. Laly and T. Sabu, Int. J. Biol. Macromol., 2018, 107, 929–937 CrossRef CAS PubMed.
- K. S. Joshy, S. Snigdha, N. Kalarikkal, L. A. Pothen and S. Thomas, Chem. Phys. Lipids, 2017, 207, 24–37 CrossRef CAS PubMed.
- K. S. Joshy, S. Snigdha, A. George, N. Kalarikkal, L. A. Pothen and S. Thomas, Cellulose, 2017, 24, 4759–4771 CrossRef CAS.
- A. L. M. Carvalho, J. A. da Silva, A. A. M. Lira, T. M. F. Conceição, R. de S. Nunes, R. L. C. de Albuquerque Junior, V. H. V. Sarmento, L. B. Leal and D. P. de Santana, J. Pharm. Sci., 2016, 105, 2188–2193 CrossRef CAS PubMed.
- K. S. Joshy, C. P. Sharma, N. Kalarikkal, K. Sandeep, S. Thomas and L. A. Pothen, Mater. Sci. Eng. C, 2016, 66, 40–50 CrossRef CAS PubMed.
- A. Laux, C. Gouws and J. H. Hamman, Expert Opin. Drug Delivery, 2019, 16, 1283–1285 CrossRef PubMed.
- I. Gomez-Orellana, Expert Opin. Drug Delivery, 2005, 2, 419–433 CrossRef CAS PubMed.
- S. I. Pather, M. J. Rathbone and S. Senel, Expert Opin. Drug Delivery, 2008, 5, 531–542 CrossRef CAS PubMed.
- M. R. Prausnitz and R. Langer, Nat. Biotechnol., 2008, 26, 1261–1268 CrossRef CAS PubMed.
- T. B. Ernest, D. P. Elder, L. G. Martini, M. Roberts and J. L. Ford, J. Pharm. Pharmacol., 2010, 59, 1043–1055 CrossRef PubMed.
- A. Cram, J. Breitkreutz, S. Desset-Brèthes, T. Nunn and C. Tuleu, Int. J. Pharm., 2009, 365, 1–3 CrossRef CAS PubMed.
- P. N.-W. D. Information and U. 2012, WHO Drug Information International Regulatory Harmonization.
- K. Pham, D. Li, S. Guo, S. Penzak and X. Dong, J. Controlled Release, 2016, 226, 88–97 CrossRef CAS PubMed.
- P. R. Ravi and R. Vats, J. Pharm. Pharmacol., 2017, 69, 823–833 CrossRef CAS PubMed.
- J. S. Negi, P. Chattopadhyay, A. K. Sharma and V. Ram, Eur. J. Pharm. Sci., 2013, 48, 231–239 CrossRef CAS PubMed.
- P. R. Ravi, R. Vats, J. Balija, S. P. N. Adapa and N. Aditya, Carbohydr. Polym., 2014, 110, 320–328 CrossRef CAS PubMed.
- S. S. Dodiya, S. S. Chavhan, K. K. Sawant and A. G. Korde, J. Microencapsulation, 2011, 28, 515–527 CrossRef CAS PubMed.
- K. Krieser, J. Emanuelli, R. M. Daudt, S. Bilatto, J. B. Willig, S. S. Guterres, A. R. Pohlmann, A. Buffon, D. S. Correa and I. C. Külkamp-Guerreiro, Mater. Sci. Eng. C, 2020, 117, 111315 CrossRef CAS PubMed.
- Y. He, D. N. Xia, Q. X. Li, J. S. Tao, Y. Gan and C. Wang, Acta Pharmacol. Sin., 2015, 36, 1151–1160 CrossRef CAS PubMed.
- M. Kurd, S. S. Malvajerd, S. Rezaee, M. Hamidi and K. Derakhshandeh, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 2123–2133 CrossRef CAS PubMed.
- M. D. Bianchin, G. Prebianca, M. F. Immich, M. L. Teixeira, M. Colombo, L. S. Koester, B. V. de Araújo, F. Poletto and I. C. Külkamp-Guerreiro, Drug Dev. Ind. Pharm., 2021, 47, 83–91 CrossRef CAS PubMed.
- K. Prabhakar, S. M. Afzal, G. Surender and V. Kishan, Acta Pharm. Sin. B, 2013, 3, 345–353 CrossRef.
- R. Pompei, O. Flore, M. A. Marccialis, A. Pani and B. Loddo, Nature, 1979, 281, 689–690 CrossRef CAS PubMed.
- J. Cinatl, B. Morgenstern, G. Bauer, P. Chandra, H. Rabenau and H. W. Doerr, Lancet, 2003, 361, 2045–2046 CrossRef CAS PubMed.
- J. L. Chew, C. B. Wolfowicz, H. Q. Mao, K. W. Leong and K. Y. Chua, Vaccine, 2003, 21, 2720–2729 CrossRef CAS PubMed.
- Y. Wu, W. Yang, C. Wang, J. Hu and S. Fu, Int. J. Pharm., 2005, 295, 235–245 CrossRef CAS PubMed.
- D. Depan, B. Girase, J. S. Shah and R. D. K. Misra, Acta Biomater., 2011, 7, 3432–3445 CrossRef CAS PubMed.
- D. Depan, P. K. C. Venkata Surya, B. Girase and R. D. K. Misra, Acta Biomater., 2011, 7, 2163–2175 CrossRef CAS PubMed.
- Q. Yuan, S. Hein and R. D. K. Misra, Acta Biomater., 2010, 6, 2732–2739 CrossRef CAS PubMed.
- D. Mishra, N. Jain, V. Rajoriya and A. K. Jain, J. Pharm. Pharmacol., 2014, 66, 1082–1093 CrossRef CAS PubMed.
- S. M. T. Cavalcanti, C. Nunes, S. A. C. Lima, J. L. Soares-Sobrinho and S. Reis, Pharm. Res., 2017, 34, 1204–1216 CrossRef CAS PubMed.
- C. Woodsong and J. D. S. Holt, Adv. Drug Delivery Rev., 2015, 92, 146–154 CrossRef CAS PubMed.
- A. Machado, C. Cunha-Reis, F. Araújo, R. Nunes, V. Seabra, D. Ferreira, J. das Neves and B. Sarmento, Acta Biomater., 2016, 44, 332–340 CrossRef CAS PubMed.
- S. S. Timur, A. Şahin, E. Aytekin, N. Öztürk, K. H. Polat, N. Tezel, R. N. Gürsoy and S. Çalış, Pharm. Dev. Technol., 2018, 23, 301–310 CrossRef CAS PubMed.
- J. Meng, T. Zhang, V. Agrahari, M. J. Ezoulin and B. B. C. Youan, Nanomedicine, 2014, 9, 1595–1612 CrossRef CAS PubMed.
- J. Meng, V. Agrahari, M. J. Ezoulin, C. Zhang, S. S. Purohit, A. Molteni, D. Dim, N. A. Oyler and B. B. C. Youan, Mol. Pharm., 2016, 13, 4129–4140 CrossRef CAS PubMed.
- R. Parthiban, Shanmugapriya, Indhu, Sathishkumar, Surendhar and R. Prakash, Drug Invent. Today, 2020, 14, 108–111 Search PubMed.
- M. M. Mahboobian, M. Mohammadi and Z. Mansouri, J. Drug Delivery Sci. Technol., 2020, 55, 101400 CrossRef CAS.
- A. Gandhi, S. Jana and K. K. Sen, Int. J. Biol. Macromol., 2014, 67, 478–482 CrossRef CAS PubMed.
- L. Calderón, R. Harris, M. Cordoba-Diaz, M. Elorza, B. Elorza, J. Lenoir, E. Adriaens, J. P. Remon, A. Heras and D. Cordoba-Diaz, Eur. J. Pharm. Sci., 2013, 48, 216–222 CrossRef PubMed.
- N. Rajendran, R. Natrajan, R. Kumar and S. Selvaraj, Asian J. Pharm., 2010, 4, 220–226 CrossRef CAS.
- E. Peira, D. Chirio, M. E. Carlotti, R. Spagnolo and M. Trotta, J. Drug Delivery Sci. Technol., 2009, 19, 191–196 CrossRef CAS.
- R. Cavalli, M. Donalisio, A. Civra, P. Ferruti, E. Ranucci, F. Trotta and D. Lembo, J. Controlled Release, 2009, 137, 116–122 CrossRef CAS PubMed.
- I. A. Alsarra, A. Y. Hamed and F. K. Alanazi, Drug Delivery, 2008, 15, 313–321 CrossRef CAS PubMed.
- P. Suwannoi, M. Chomnawang, N. Sarisuta, S. Reichl and C. C. Müller-Goymann, J. Ocul. Pharmacol. Ther., 2017, 33, 743–752 CrossRef CAS PubMed.
- Z. Xing-Guo, M. Jing, L. Min-Wei, J. Sai-Ping, H. Fu-Qiang and D. Yong-Zhong, J. Zhejiang Univ., Sci., B, 2008, 9, 506–510 CrossRef PubMed.
- G. Singh and R. S. Pai, Drug Delivery, 2016, 23, 532–539 CrossRef CAS PubMed.
- J. Duan, J. P. Freeling, J. Koehn, C. Shu and R. J. Y. Ho, J. Pharm. Sci., 2014, 103, 2520–2529 CrossRef CAS PubMed.
- A. S. Nowacek, J. McMillan, R. Miller, A. Anderson, B. Rabinow and H. E. Gendelman, J Neuroimmune Pharmacol., 2010, 5, 592–601 CrossRef PubMed.
- M. Garg and N. K. Jain, J. Drug Targeting, 2006, 14, 1–11 CrossRef CAS PubMed.
- H. Elkateb, L. M. Tatham, H. Cauldbeck, E. Niezabitowska, A. Owen, S. Rannard and T. McDonald, Int. J. Pharm., 2020, 588, 119794 CrossRef CAS PubMed.
- K. Priya Dharshini, H. Fang, D. Ramya Devi, J. X. Yang, R. H. Luo, Y. T. Zheng, M. Brzeziński and B. N. Vedha Hari, Carbohydr. Polym., 2021, 256, 117440 CrossRef CAS PubMed.
- A. V. Belgamwar, S. A. Khan and P. G. Yeole, J. Drug Delivery Sci. Technol., 2019, 52, 1008–1020 CrossRef CAS.
- R. Rozana, Y. Yulizar, A. Saefumillah and D. O. B. Apriandanu, AIP Conf. Proc., 2020, 2242, 040004 CrossRef CAS.
- V. Makwana, R. Jain, K. Patel, M. Nivsarkar and A. Joshi, Int. J. Pharm., 2015, 495, 439–446 CrossRef CAS PubMed.
- P. K. Gaur, S. Mishra, M. Bajpai and A. Mishra, BioMed Res. Int., 2014, 2014, 363404 Search PubMed.
- L. Tshweu, L. Katata, L. Kalombo, D. A. Chiappetta, C. Hocht, A. Sosnik and H. Swai, Nanomedicine, 2014, 9, 1821–1833 CrossRef CAS PubMed.
- S. Kotta, A. W. Khan, S. H. Ansari, R. K. Sharma and J. Ali, Int. J. Pharm., 2014, 462, 129–134 CrossRef CAS PubMed.
- A. Shibata, E. McMullen, A. Pham, M. Belshan, B. Sanford, Y. Zhou, M. Goede, A. A. Date and C. J. Destache, AIDS Res. Hum. Retroviruses, 2013, 29, 746–754 CrossRef CAS PubMed.
- M. de Sousa and F. B. T. Pessine, Adv. Sci., Eng. Med., 2014, 6, 1135–1142 CrossRef CAS.
- Y. Gong, K. Zhi, P. K. B. Nagesh, N. Sinha, P. Chowdhury, H. Chen, S. Gorantla, M. M. Yallapu and S. Kumar, Viruses, 2020, 12, 564 CrossRef CAS PubMed.
- Y. Gong, P. Chowdhury, N. M. Midde, M. A. Rahman, M. M. Yallapu and S. Kumar, Biochem. Biophys. Rep., 2017, 12, 214–219 Search PubMed.
- T. N. Figueira, M. M. Domingues, F. Illien, I. Cadima-Couto, T. Todorovski, D. Andreu, S. Sagan, M. A. R. B. Castanho, A. Walrant and A. S. Veiga, ACS Infectious Diseases, 2020, 6, 224–236 CrossRef CAS PubMed.
- E. Russo, N. Gaglianone, S. Baldassari, B. Parodi, S. Cafaggi, C. Zibana, M. Donalisio, V. Cagno, D. Lembo and G. Caviglioli, Colloids Surf., B, 2014, 118, 117–125 CrossRef CAS PubMed.
- K. M. Tyo, A. B. Lasnik, L. Zhang, M. Mahmoud, A. B. Jenson, J. L. Fuqua, K. E. Palmer and J. M. Steinbach-Rankins, J. Controlled Release, 2020, 321, 84–99 CrossRef CAS PubMed.
- Z. Karami, M. R. Saghatchi Zanjani, S. Rezaee, K. Rostamizadeh and M. Hamidi, Drug Dev. Ind. Pharm., 2019, 45, 736–744 CrossRef CAS PubMed.
- B. Surnar, M. Z. Kamran, A. S. Shah, U. Basu, N. Kolishetti, S. Deo, D. T. Jayaweera, S. Daunert and S. Dhar, ACS Nano, 2019, 13, 11034–11048 CrossRef CAS PubMed.
- G. Joshi, A. Kumar and K. Sawant, Drug Delivery, 2016, 23, 3492–3504 CrossRef CAS PubMed.
- P. R. Ravi, R. Vats, V. Dalal, N. Gadekar and N. Aditya, Drug Dev. Ind. Pharm., 2015, 41, 131–140 CrossRef CAS PubMed.
- A. Alex, W. Paul, A. J. Chacko and C. P. Sharma, Ther. Delivery, 2011, 2, 25–35 CrossRef CAS PubMed.
- S. Perazzolo, L. M. Shireman, J. Koehn, L. A. McConnachie, J. C. Kraft, D. D. Shen and R. J. Y. Ho, J. Pharm. Sci., 2018, 107, 3153–3162 CrossRef CAS PubMed.
- L. Fotooh Abadi, P. Kumar, V. Gajbhiye, K. M. Paknikar and S. Kulkarni, Colloids Surf., B, 2020, 194, 111227 CrossRef CAS PubMed.
- S. Lahkar and M. K. Das, J. Nanopart. Res., 2020, 22, 109 CrossRef CAS.
- J. Varshosaz, S. Taymouri, E. Jafari, A. Jahanian-Najafabadi and A. Taheri, J. Drug Delivery Sci. Technol., 2018, 48, 9–20 CrossRef CAS.
- L. N. Ramana, S. Sethuraman, U. Ranga and U. M. Krishnan, J. Biomed. Sci., 2010, 17, 1–9 CrossRef PubMed.
- A. A. Date, A. Shibata, M. Goede, B. Sanford, K. La Bruzzo, M. Belshan and C. J. Destache, Antiviral Res., 2012, 96, 430–436 CrossRef CAS PubMed.
- F. Javan, A. Vatanara, K. Azadmanesh, M. Nabi-Meibodi and M. shakouri, J. Pharm. Pharmacol., 2017, 69, 1002–1009 CrossRef CAS PubMed.
- S. Kumar, R. Narayan, V. Ahammed, Y. Nayak, A. Naha and U. Y. Nayak, J. Drug Delivery Sci. Technol., 2018, 44, 181–189 CrossRef CAS.
- I. F. Abou-El-Naga, E. D. El Kerdany, R. F. Mady, T. I. Shalaby and E. M. Zaytoun, Parasitol. Int., 2017, 66, 735–747 CrossRef CAS PubMed.
- S. Jain, J. M. Sharma, A. K. Jain and R. R. Mahajan, Nanomedicine, 2013, 8, 1639–1655 CrossRef CAS PubMed.
- L. M. Bimbo, O. V. Denisova, E. Mäkilä, M. Kaasalainen, J. K. De Brabander, J. Hirvonen, J. Salonen, L. Kakkola, D. Kainov and H. A. Santos, ACS Nano, 2013, 7, 6884–6893 CrossRef CAS PubMed.
- L. N. Ramana, S. Sharma, S. Sethuraman, U. Ranga and U. M. Krishnan, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 476–484 CrossRef CAS PubMed.
- Y. C. Kuo and H. F. Ko, Biomaterials, 2013, 34, 4818–4830 CrossRef CAS PubMed.
- J. Adlin Jino Nesalin and A. Anton Smith, J. Pharm. Res., 2013, 6, 268–274 CAS.
- R. Shegokar and K. K. Singh, Pharmazie, 2011, 66, 264–271 CAS.
- M. Garg, A. Asthana, H. B. Agashe, G. P. Agrawal and N. K. Jain, J. Pharm. Pharmacol., 2010, 58, 605–616 CrossRef PubMed.
- Y. C. Kuo and C. Y. Chung, Colloids Surf., B, 2011, 88, 682–690 CrossRef CAS PubMed.
- T. Chaowanachan, E. Krogstad, C. Ball and K. A. Woodrow, PLoS One, 2013, 8, 61416 CrossRef PubMed.
- S. Mandal, P. K. Prathipati, G. Kang, Y. Zhou, Z. Yuan, W. Fan, Q. Li and C. J. Destache, Aids, 2017, 31, 469–476 CrossRef CAS PubMed.
- S. Mandal, G. Kang, P. K. Prathipati, Y. Zhou, W. Fan, Q. Li and C. J. Destache, J. Controlled Release, 2019, 294, 216–225 CrossRef CAS PubMed.
- J. Shailender, P. R. Ravi, M. Reddy Sirukuri, A. Dalvi and O. Keerthi Priya, Drug Dev. Ind. Pharm., 2018, 44, 1109–1119 CrossRef CAS PubMed.
- B. Gourdon, C. Chemin, A. Moreau, T. Arnauld, P. Baumy, S. Cisternino, J. M. Péan and X. Declèves, Int. J. Pharm., 2017, 529, 357–370 CrossRef CAS PubMed.
- R. Kumar and V. R. Sinha, AAPS PharmSciTech, 2017, 18, 884–894 CrossRef CAS PubMed.
- S. M. T. Cavalcanti, C. Nunes, S. A. Costa Lima, J. L. Soares-Sobrinho and S. Reis, Eur. J. Pharm. Sci., 2018, 122, 22–30 CrossRef CAS PubMed.
- J. K. Yan, Y. Y. Wang, W. Y. Qiu and J. Y. Wu, Carbohydr. Polym., 2017, 174, 209–216 CrossRef CAS PubMed.
- M. Da Silva Barbi, F. C. Carvalho, C. P. Kiill, H. Da Silva Barud, S. H. Santagneli, S. J. L. Ribeiro and M. P. D. Gremião, J. Nanosci. Nanotechnol., 2015, 15, 865–874 CrossRef PubMed.
- D. D. Kumbhar and V. B. Pokharkar, Colloids Surf., A, 2013, 436, 714–725 CrossRef CAS.
- R. M. Mainardes, N. M. Khalil and M. P. D. Gremião, Int. J. Pharm., 2010, 395, 266–271 CrossRef CAS PubMed.
- S. Singh, A. K. Dobhal, A. Jain, J. K. Pandit and S. Chakraborty, Chem. Pharm. Bull., 2010, 58, 650–655 CrossRef CAS PubMed.
- P. Kumar, Y. S. Lakshmi and A. K. Kondapi, Pharm. Res., 2017, 34, 257–268 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |