
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic reactions in a Co12 cuboctahedral cage arising from guest encapsulation and cage-based redox activation†
Xuejian
Zhang
,
Burin
Sudittapong
 and
Michael D.
Ward
*
and
Michael D.
Ward
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: m.d.ward@warwick.ac.uk
First published on 9th January 2023
Abstract
A Co12 coordination cage with a cuboctahedral architecture, and incorporating a mixture of tritopic (face-capping) and ditopic (edge-bridging) ligands, shows strong guest binding of large aromatic fluorophores (fluorescein and its derivatives 6-carboxyfluorescein and Eosin-Y) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding constants in water in the range logK = 6.7–7.9; its large central cavity (>1000 Å3) facilitates binding of much larger guests than was possible with the smaller Co8 cage that we have reported previously. Guest binding is accompanied by catalysed reactions of bound guests because the high positive charge on the cage surface (24+) also attracts anions, allowing the organic guests and anionic reaction partners to be co-located, resulting for example in cage-catalysed hydrolysis of phosphate esters (the insecticides Me-paraoxon and Et-paraoxon) and conversion of diacetyl fluorescein to fluorescein. In addition, we demonstrate a new type of cage-based catalysis which relies on the redox activity of the Co(II)/Co(III) couple in the cage to activate the peroxymonosulfate (PMS) anion by converting it to the highly reactive SO4˙− radical ion, which bleaches cavity-bound fluorescein by complete oxidation. This is an example of an ‘advanced oxidation process’ in which the host cage not only brings the fluorescein and the PMS together via orthogonal hydrophobic and electrostatic interactions, but also provides redox activation of the PMS via a Co(II)/Co(III) couple, with the cage taking an active role in the catalytic process rather than acting simply as a passive reaction vessel.
:1 binding constants in water in the range logK = 6.7–7.9; its large central cavity (>1000 Å3) facilitates binding of much larger guests than was possible with the smaller Co8 cage that we have reported previously. Guest binding is accompanied by catalysed reactions of bound guests because the high positive charge on the cage surface (24+) also attracts anions, allowing the organic guests and anionic reaction partners to be co-located, resulting for example in cage-catalysed hydrolysis of phosphate esters (the insecticides Me-paraoxon and Et-paraoxon) and conversion of diacetyl fluorescein to fluorescein. In addition, we demonstrate a new type of cage-based catalysis which relies on the redox activity of the Co(II)/Co(III) couple in the cage to activate the peroxymonosulfate (PMS) anion by converting it to the highly reactive SO4˙− radical ion, which bleaches cavity-bound fluorescein by complete oxidation. This is an example of an ‘advanced oxidation process’ in which the host cage not only brings the fluorescein and the PMS together via orthogonal hydrophobic and electrostatic interactions, but also provides redox activation of the PMS via a Co(II)/Co(III) couple, with the cage taking an active role in the catalytic process rather than acting simply as a passive reaction vessel.
Introduction
The enduring interest in the chemistry of self-assembled coordination cages1 is due in large part to their ability to act as catalysts for chemical transformations of guests bound in their central cavity.2 Binding of a potential substrate inside a cage cavity can be considered as an approximate model for binding of substrates at enzyme active sites, and the mechanisms by which reactions are accelerated have some obvious similarities between the two cases. Effects responsible for catalysed reactions of substrates bound in cages include conformational changes which bring a flexible substrate close to the conformation of a reaction transition state;3 electronic effects associated with the high charges which may be carried by multinuclear metal/ligand assemblies;4 and high local concentrations when two reaction partners are co-located in or around the cage.5,6Our recent work on cage-based catalysis6 has focussed on our octanuclear cubic [M8L12]16+ system in which a ditopic bis(pyrazolyl-pyridine) bridging ligand spans each of the twelve edges of the assembly of eight M(II) ions which lie at the vertices.7 The cage has a cavity size (ca. 400 Å3) which is ideal for accommodating a wide range of small organic guests; in its water-soluble forms, the hydrophobic effect ensures strong binding of such guests, with binding constants of up to 106 M−1 for optimally-sized guests;6a,b and the high positive charge allows the cage to accumulate anions around its surface very effectively.6,8 This last effect is the basis for its catalytic activity as the cage uses two orthogonal8c interactions to bring two reaction partners into close proximity, viz. (i) an organic substrate which binds in the cage cavity via the hydrophobic effect; and (ii) accumulation of anions around the cage surface via electrostatic/ion-pairing effects. The result is a high local concentration of ions around a cage-bound guest, leading to catalysed bimolecular reactions such as the Kemp elimination (reaction of benzisoxazole with hydroxide,6c or with a basic phenolate anion6d), phosphate ester hydrolysis,6e or an aldol condensation.6f
Whilst this [Co8L12]16+ cubic cage (hereafter abbreviated as Co8) has provided many examples of cage-based catalysis6 it is of course size- and shape-limited in the guests that it can bind. We were therefore interested to see if we could extend cage-based catalysis to larger members of our coordination cage family which would be able to accommodate a wider range of guests in the central cavity, whilst maintaining the key characteristics that make catalysis possible (a hydrophobic interior cavity surface for organic guest binding; and a high positive charge which attracts anions to the cage surface). This is non-trivial as it requires solubility in water, requiring in some cases significant changes to ligand syntheses to introduce external solubilising substituents such as HO groups7b or PEG chains.7c Additionally, some of the larger cages that we have prepared exist in solution in slow equilibria between larger cages and smaller assemblies which prevents meaningful analysis of host/guest chemistry.9
In this paper we introduce the cuboctahedral cage Co12 (Fig. 1) as a catalyst for reactions between hydrophobic substrates and anions in water, using a range of ester hydrolysis reactions as illustrations. Significantly, we also report extension of the range of cage-catalysed reactions to include some that require not just co-location of reaction partners around the cage, but redox activation of one of the reagents by the Co(II) ions in the cage. This is an additional degree of sophistication in the cage-based catalysed reactions, making the cage not just an inert container that holds components in close proximity to one another, but also an active participant in electron-transfer steps required for the catalytic cycle in a way that has been demonstrated in recent examples of photo-redox catalysis.10 This additional redox-based participation of the cage in catalytic processes will substantially extend the scope of reaction types that can be catalysed.
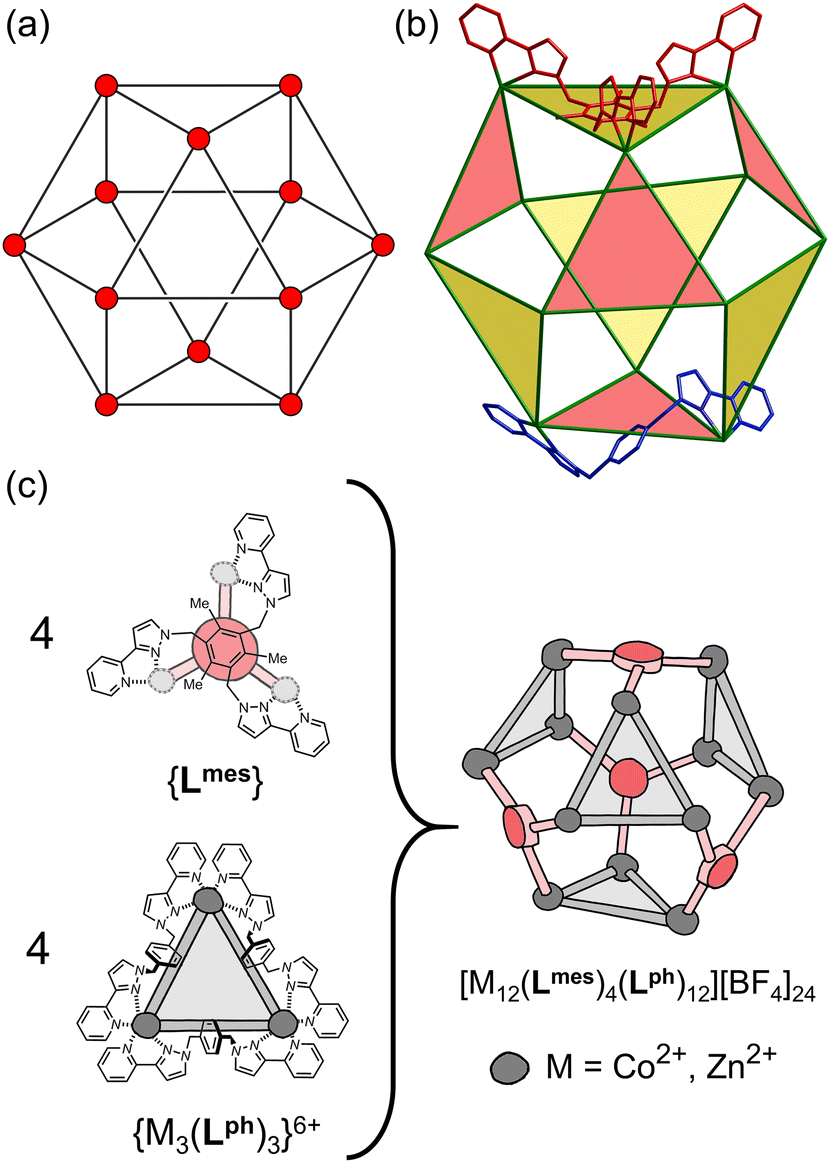 | ||
| Fig. 1 (a) An idealised cuboctahedron; (b) partial crystal structure of [Cu12(Lph)12(Lmes)4](BF4)24 (taken from ref. 11a) emphasising the cuboctahedral M12 core with the four triangular faces capped by tritopic ligands Lmes coloured yellow, and the alternate four triangular faces – which are Cu3(μ-Lph)3 cyclic helicates – coloured pink; (c) additional sketch emphasising the disposition of the four M3(μ-Lph)3 cyclic helicate faces in these structures (grey triangles) and the four Lmes ligands which connect them (red). Figure reproduced with modifications from ref. 11b. | ||
Results and discussion
The Co12 cage and its water solubilisation
As the candidate for extending our cage-based catalysis studies we have chosen the M12 cuboctahedral cage shown in Fig. 1.11 This is based on a mixture of two different types of ligand (edge-bridging, ditopic, Lph; and face-capping, tritopic, Lmes). A combination of four equivalents of Lmes and twelve equivalents of Lph combine with twelve M(II) ions to form a cuboctahedral array [M12(Lph)12(Lmes)4]24+ in which four of the eight M3 triangular faces are capped by an Lmes ligand, and the other four are edge-bridged, forming a set of (chiral) M3(Lph)3 cyclic helicates.11a Interestingly the four triangular cyclic helicate faces can vary their sense of helicity between ‘anticlockwise’ (A) and ‘clockwise’ (C) independently of one another, which means that the [M12(Lph)12(Lmes)4]24+ cage can exist as a mixture of three different diastereoisomers: (i) all four cyclic helicate faces homochiral (enantiomeric pair AAAA/CCCC, T symmetry); (ii) three cyclic helicate faces of one chirality and one of the other (enantiomeric pair AAAC/CCCA, C3 symmetry); and finally (iii) two cyclic helicates of each chirality (AACC, S4 symmetry, which has no enantiomer).11b The result is a set of three M12 cages of essentially the same size and with very similar arrangement of metal ions at the vertices, differing only in the chiralities of the cyclic helicate faces. Examples of all three diastereoisomers of this type of cuboctahedral cage have been individually structurally characterised in previous work.11b Separation of the three diastereoisomers in significant quantities beyond picking individual crystals was not possible given their essentially identical size/shape and identical charge. Importantly for the purposes of this work, the cavity volumes of the three diastereoisomers are very similar to one another (1036–1121 Å3)11b and substantially larger than the cavity of the M8L12 cubic cage (409 Å3)6 so we expect fundamentally different host/guest chemistry. For the purposes of this work, therefore, the set of three similar diastereoisomers of the [Co12(Lph)12(Lmes)4]24+ cage is considered as a single species.Initially therefore we combined Co(BF4)2, Lph and Lmes in a 12:12:4 ratio in the previously-reported way11 to obtain solid [Co12(Lph)12(Lmes)4](BF4)24 (hereafter abbreviated as Co12). High-resolution ES mass spectrometry confirmed formation of the cage with a characteristic sequence of signals associated with loss of different numbers of anions giving the species {[Co12(Lph)12(Lmes)4](BF4)24−n}n+ (n = 4–9, Fig. S1–S4;† with the correct fractional isotope spacings and isotopic patters in the high-resolution signals). In the 1H NMR spectrum in CD3CN (Fig. S5†), the paramagnetism associated with use of high-spin Co(II) ions acts as a convenient shift reagent, spreading out the signals over a 200 ppm chemical shift range.7a The spectrum is clearly highly complex due to the mixture of three diastereoisomers with different symmetries and cannot be assigned in detail: but we can identify in some parts of the spectrum a characteristic set of eight signals (subsets of 1, 3 and 4 signals with the same intensity within each subset)‡ which arise from one chemical type of proton (e.g. a pyridyl H6 from Lmes) in the three diastereoisomers, as described earlier.11b In the 1H NMR spectrum for Co12 in MeCN we can identify exactly this pattern between −10 and −23 ppm (Fig. S5,† top panel) which, together with the mass spectrum, confirms formation of Co12 with all three diastereoisomers present.
The complex formed beautiful crystals on recrystallisation from a variety of solvents which however failed to diffract X-rays significantly, even using synchrotron radiation, which is likely a consequence of co-crystallisation of the three diastereoisomers which will lead to extensive disorder in the organic ligand positions and (to a lesser extent) in the metal atom positions. Indeed the NMR spectrum of redissolved apparently ‘single’ crystals matched that of the bulk as-prepared material, showing the mixture of diastereoisomers to be present in the crystals. We note however that several other crystal structures of this M12 cage family have been previously reported.11
For control experiment purposes we also prepared the isostructural Zn12 cage in exactly the same way; this complex too was characterised by high-resolution ES mass spectrometry (Fig. S7–S10†), showing the sequence {[Zn12(Lph)12(Lmes)4](BF4)24−n}n+ (n = 4–9) with the expected isotope patterns and spacings for the individual species. The 1H NMR spectrum of Zn12 (Fig. S11†) is mostly unassignable due to the overlap of signals in the aromatic region associated with the mixture of diastereoisomers, but in the aliphatic region we can clearly see the singlets for the methyl protons of Lmes: a set of four of the same intensity for the S4 isomer and a set of three of the same intensity for the C3 isomer. We cannot see the additional signal arising from the T isomer (expected between 0 and 1 ppm)11b so we conclude that the Zn12 complex consists of the S4 and C3 isomers: in the previously-reported Cd12 species, the T isomer was by far the least abundant of the three.11b A DOSY spectrum (Fig. S12†) confirms that a single species (ignoring the isomerism) is present in solution, and from the diffusion coefficient [log(D/m2 s−1) = −9.8] we can estimate from the Stokes–Einstein equation that the radius of the complex is ca. 12 Å, which is close to the crystallographically-observed average radius of ca. 14 Å and far larger than any species such as individual ligands or low-nuclearity complexes.
Having confirmed formation of Co12 (and Zn12 for control experiments) the next problem is water-solubilisation to allow the hydrophobic effect to be exploited for guest binding. Appending hydroxymethyl or PEG substituents to the ligands is possible but laborious,7b,c and we found that ligand substituents in the obvious positions (pyridyl C4) caused steric problems which prevented cage assembly – a problem which did not arise earlier in the M8L12 cube where the pyridyl C4 substituents do not clash.7b We have also used anion metathesis, with the usual fluoroborate anions associated with the cage synthesis being replaced by chloride ions which rendered the [M8L12]16+ cage highly water soluble.6d However similar anion-exchange experiments to convert Co12 to its chloride salt did not lead to sufficiently high water solubility and also results in partial decomposition, possibly because chloride is a good competing ligand for Co(II) and the high positive charge on this cage will result in a high chloride ion concentration around the cage following anion metathesis.
We found however that we could successfully water-solubilise Co12 for catalysis in two different ways which did not require anion-exchange or complex synthetic chemistry associated with incorporation of ligand substituents. These are (i) inclusion of 1% dmso in the aqueous solvent;6g and (ii) more unusually, use of a non-ionic surfactant. Use of a small amount of dmso in the aqueous solvent (dmso/water 1:99, v/v) provided sufficient solubility for many spectroscopic and catalysis measurements with Co12 as we report below.§ The non-ionic surfactant polysorbate-20 (also known as Tween-20) also proved to be effective. Tween-20 contains a water-soluble branched poly-oxyethylene terminus functionalised with a lauryl ester chain which provides a hydrophobic terminus. As such one of the applications of Tween-20 is to render water-soluble a range of hydrophobic species, including drug molecules for formulation purposes12 and hydrophobic membrane proteins during extraction following cell lysis,13 and it turned out to be very effective at allowing Co12 to be water-solubilised to a surprisingly high (mM range) concentration. Addition of solid Co12 to commercial Tween-20, followed by overnight stirring, sonication, dilution with water, and filtration through a 2-micron filter afforded a clear aqueous solution whose concentration was established by UV/Vis spectroscopy. A dynamic light scattering experiment (Fig. S14†) confirmed formation of particles with an average size of 12 nm, associated with formation of the Tween-20 micelles which contain Co12 inside their hydrophobic interiors: we denote the Tween-encapsulated cage molecules as TW20⊂Co12. 1H NMR spectra of the resulting solutions were uninformative due to the large excess of Tween present, and the slow tumbling of Co12 molecules in the micellar environment which broadened the spectra beyond the normal effects of paramagnetism: however the UV/Vis spectrum in the presence of Tween/water was the same as in MeCN/water (10/90).
Guest binding in Co12 in water
Initial studies on guest binding focussed on the set of four of guests shown in Scheme 1: these are fluorescein (FLU), 6-carboxyfluorescein (6FLU), Eosin-Y (EY) and hydroxypyrene-tris-sulfonate (HPTS). All of these are fluorescent so we could monitor guest uptake into Co12 by following fluorescence quenching as portions of TW20⊂Co12 were added to a fixed concentration (1 μM) of the potential guest in aqueous solution. Binding is clearly strong (Fig. 2): fitting the curve for quenching of FLU (a dianion under the prevailing conditions) for example, to a 1:1 binding isotherm, afforded a logK value of 6.7. A Job plot based on the fluorescence titration data confirmed 1:1 binding, and an electrospray mass spectrum of a Co12/FLU mixture showed a set of strong signals associated with the 1:1 Co12·FLU adduct associated with varying numbers of anions (Fig. S13†). This behaviour is notably different from what we observed for the interaction of FLU with the smaller Co8 cage, which is too small to accommodate FLU inside the cavity: in that case external surface binding occurred with up to five FLU units interacting with the Co8 cage exterior according to the Job plot, and a much smaller binding constant.8b In contrast, with Co12 as the host, we are clearly seeing (a) much stronger binding of fluorescein and (b) this binding is in a 1:1 ratio as shown by both a Job plot and an electrospray mass spectrum, all of which is consistent with cavity-based binding of the single FLU guest.
 | ||
| Scheme 1 The aromatic, anionic fluorophores used in this work (shown in the forms that exist in weakly acidic solution). | ||
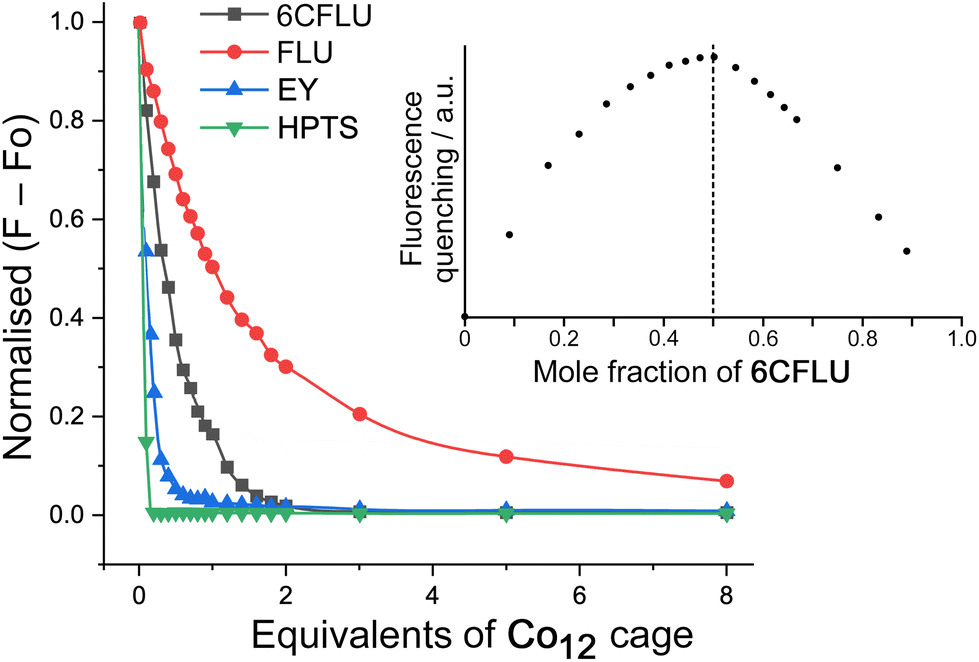 | ||
| Fig. 2 Fluorescence titrations performed by adding Co12 (up to 8 equivalents) to a range of different fluorescent dyes (1 μM in water, pH 7, no buffer) showing progressive fluorescence quenching of the dyes as they bind in the Co12 cavity; the resulting 1:1 binding constants are in the main text. A Job plot confirming 1:1 binding for guest 6CFLU is shown in the inset; exactly similar Job plots were obtained with FLU and EY. | ||
The binding constant (logK = 6.7) with FLU is already much larger than we have observed with virtually all other cage/guest complexes using Co8,6a which can be ascribed to a combination of (i) the higher hydrophobic surface area of fluorescein compared to the smaller guests that can fit inside the Co8 cage, and (ii) an electrostatic contribution associated with the 2− charge on fluorescein and the 24+ charge on Co12. We found no evidence for further association of larger numbers of FLU guests associated with external surface binding which could still in principle be possible: if it does occur, it is clearly much weaker than the cavity binding.
The related guests 6CFLU and EY bind even more strongly (logK values of 7.0 and 7.9 respectively, with 1:1 Co12:guest binding according to Job plots – see Fig. 2, inset, for an example), which we ascribe to the higher negative charge (of 6CFLU, 3−) and higher hydrophobicity (of EY) respectively, compared to FLU.8bHPTS – a trianion – binds too strongly for a binding constant to be determined under these conditions but clearly behaves differently: the very rapid quenching (≪1 equivalent of Co12 is needed to completely quench the HPTS fluorescence) implies that binding is not just cavity-based but there is also a surface interaction whereby one Co12 binds and quenches multiple equivalents of HPTS around its exterior, in a manner analogous to the Co8/FLU system.8b
Co 12 -catalysed phosphotriester hydrolysis
To examine the ability of TW20⊂Co12 to act as a catalyst using the same mechanism as we observed with the cubic Co8 cage (co-location of hydrophobic substrate and anions using orthogonal interactions)6e we examined the catalysed hydrolysis of the phosphotriesters methyl- and ethyl-paraoxon (4-O2NC6H4O–P(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OR)2, with R = Me or Et respectively), which are potent acetylcholinesterase-inhibiting insecticides, and contain 4-nitrophenolate as the leaving group from the hydrolysis reaction, allowing facile monitoring of hydrolysis reaction rates by UV/Vis spectroscopy.
O)(OR)2, with R = Me or Et respectively), which are potent acetylcholinesterase-inhibiting insecticides, and contain 4-nitrophenolate as the leaving group from the hydrolysis reaction, allowing facile monitoring of hydrolysis reaction rates by UV/Vis spectroscopy.
Under a range of catalyst and substrate concentration conditions (using fixed pH 8, borate buffer, see Fig. 3 and S16†) we obtained second-order rate constants for the cage-catalysed hydrolysis of these paraoxons of ca. 0.5 M−1 s−1 for methyl-paraoxon and 0.4 M−1 s−1 for ethyl-paraoxon which are 1–2 orders of magnitude faster than we observed using Co8 as the catalyst under similar conditions.6e Exactly similar behaviour (with slightly smaller rate accelerations) was observed using 1% dmso in water as the solvent rather than the aqueous Tween-20 surfactant (see ESI†).
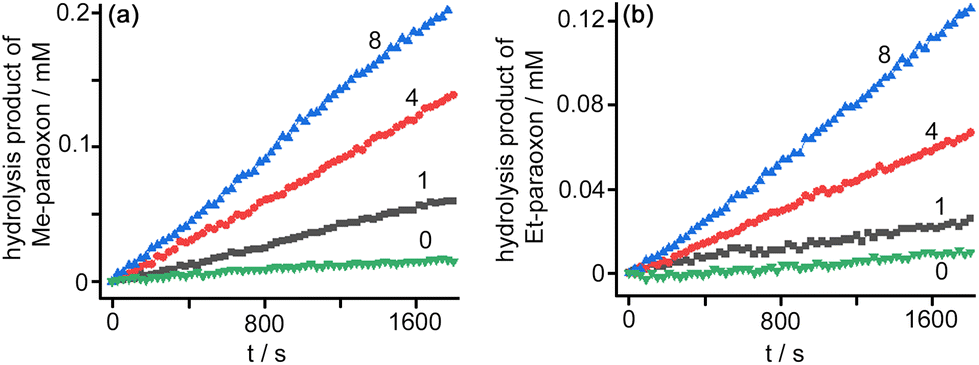 | ||
| Fig. 3 Catalysed hydrolysis (pH 8, 298 K) of the phosphate esters (a) Me-paraoxon (0.45 mM) and (b) Et-paraoxon (0.49 mM) monitored by UV/Vis absorbance of the accumulating 4-nitrophenolate ion at 400 nm. The green, black, red and blue traces correspond to addition of 0, 1, 4 and 8 mol% of Co12 (used as the TW20⊂Co12 solution). | ||
In our earlier work using Co8 we found that the cage-catalysed hydrolysis of methyl-paraoxon occurred at the external surface of the cage, as the substrate did not bind in the Co8 cavity for steric reasons. Nonetheless the hydrophobicity of the exterior surface allowed methyl-paraoxon to associate with it in aqueous solution, and the 16+ charge on the cage attracted hydroxide ions, so the cage surface still served as a medium to attract and co-locate the two reaction partners around the exterior surface, even if the resulting catalysis did not benefit from the substrate lying inside the cavity. With TW20⊂Co12 as the catalyst the much (2.5×) larger cage cavity will allow methyl- and ethyl-paraoxon to bind inside it, surrounded by hydroxide ions that are also attracted by the 24+ surface charge, resulting in the catalysed hydrolysis of both substrates that we observe at a rate higher than was previously observed for an external surface-based reaction. From these observations we can expect that the larger cavity of Co12 will facilitate catalysed reactions of hydrophobic cavity-binding guests with anions with a much wider range of substrates compared to Co8.
Co 12 -catalysed carboxylate ester hydrolysis
Another illustration of a cage-catalysed reaction involving an ester substrate and hydroxide ions as the reaction partner is provided by the hydrolysis of diacetyl fluorescein (DAF; see Scheme 2) to fluorescein which can be readily followed by monitoring the grow-in of the absorption band for the fluorescein dianion as the reaction proceeds.6g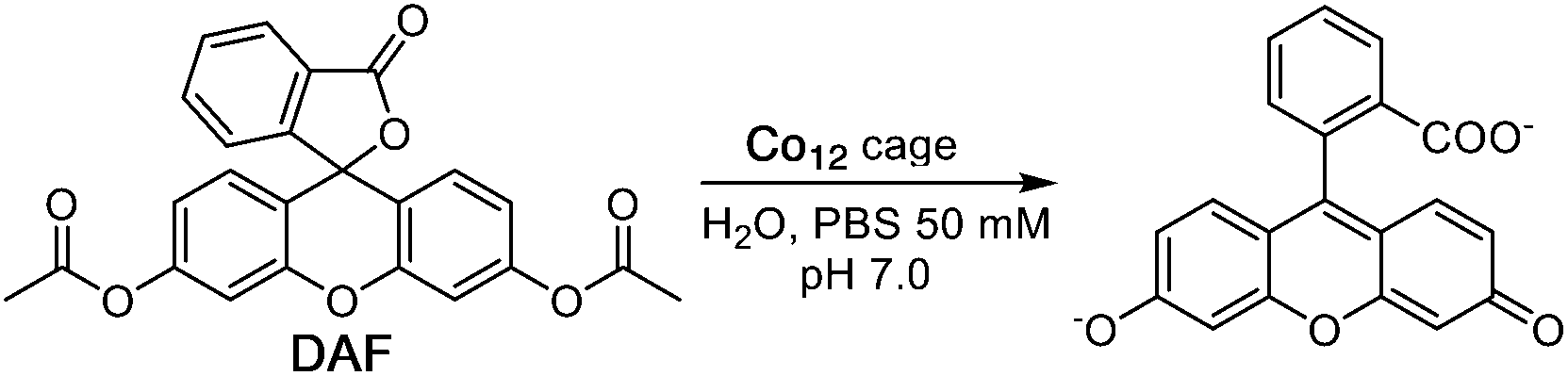 | ||
| Scheme 2 Hydrolysis of diacetyl fluorescein. | ||
Under conditions where DAF shows no significant hydrolysis over several hours (0.25 mM DAF, 50 mM PBS buffer at pH 7) the presence of Co12 results in appearance of the fluorescein absorption band at a rate which increases linearly with Co12 concentration (Fig. 4): dividing the initial rate constant by the Co12 concentration gives a second order rate constant for the cage-catalysed hydrolysis of DAF of 0.24 M−1 s−1. This is between one and two orders of magnitude greater than the catalysis of DAF hydrolysis by the smaller cage Co8 under similar conditions:6g in that case DAF cannot bind inside the cavity and the reaction occurs at the external surface of the cage with the substrate held by hydrophobic interactions in the hydroxide-rich region around the cationic surface. With Co12 in contrast the improvement in catalytic rate enhancement compared to Co8 implies that DAF is binding inside the cage cavity, surrounded by the layer of surface-bound hydroxide ions, and we have observed before how encapsulation of guests can result in much higher catalysed reaction rates.6c
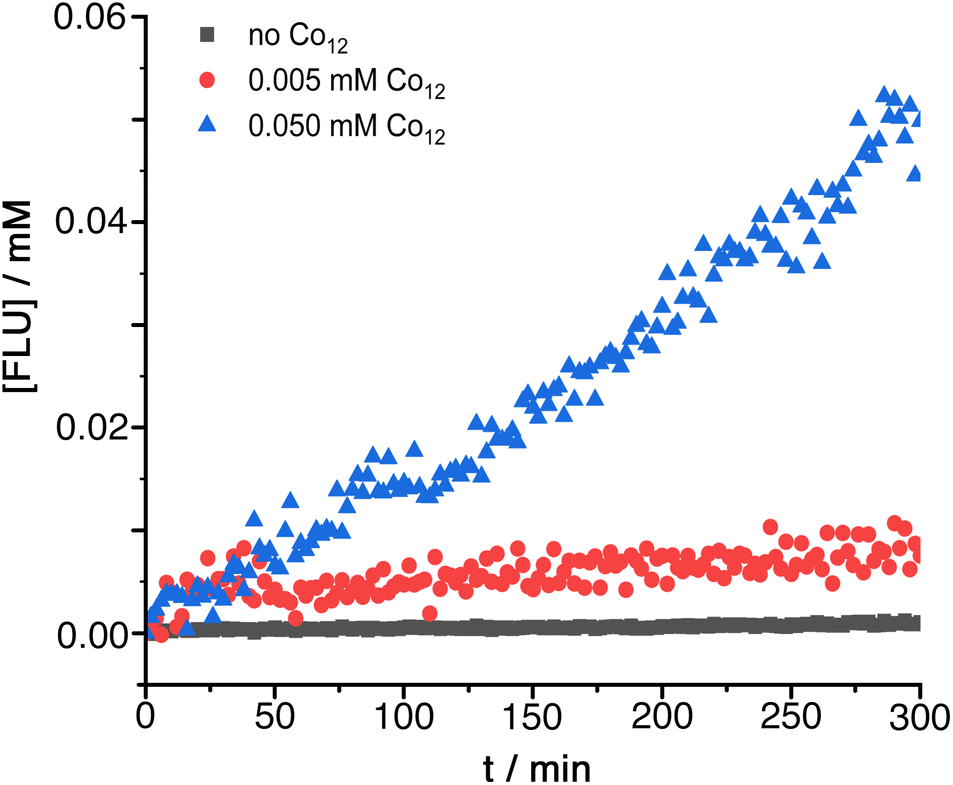 | ||
| Fig. 4 Catalysed hydrolysis of DAF by Co12 in 99:1 water/dmso at the indicated catalyst concentrations: 0.25 mM DAF, 50 mM PBS buffer, pH 7, 298 K. | ||
Co 12 -catalysed oxidation of dyes using peroxymonosulfate: an ‘advanced oxidation process’ involving cage-based redox activation
We demonstrate here a quite different example of a cage-catalysed reaction which requires not only the co-location of two reaction partners [the dye fluorescein, and the powerful anionic oxidant peroxy-monosulfate (PMS)] but also participation of the Co(II) ions in the cage in a redox cycle. PMS is of great interest as a powerful oxidant for destroying toxins and organic impurities as part of waste-water treatment where it has emerged as a desirable alternative to H2O2 and other sources of more conventional ‘reactive oxygen species’ such as 1O2 and HO˙ in so-called ‘advanced oxidation processes’.14 Importantly, despite its strong thermodynamic oxidising ability, direct reaction of PMS with the contaminants that it is intended to destroy is kinetically slow, and in practice PMS requires activation by any of a wide range of methods that cleaves the peroxide O–O bond to generate the SO4˙− sulfate radical anion which is the species that is predominantly responsible for rapid oxidation of organic contaminants in water, ultimately to innocuous CO2 and H2O.14A common method to activate PMS is a redox reaction with a low oxidation state metal ion, with Co(II) being reportedly particularly effective,14b according to eqn (1).
| Co2+ + HSO5− → Co3+ + SO4˙− + HO− | (1) |
Given that Co12 can encapsulate fluorescein in the cavity, and the high positive charge of Co12 means that it should also have the ability to accumulate anions in the same way as Co8 does (as the ester hydrolysis catalytic studies reported above confirm), we can expect that fluorescein and PMS will be co-located in and around molecules of Co12. In addition, the Co(II) ions in the cage can, if necessary, provide the necessary activation of PMS via a Co(II)/Co(III) couple, with the resulting SO4˙− again being held close to the Co12 cage (and its encapsulated substrate) by the 24+ charge on the cage.
In the absence of TW20⊂Co12, an aqueous solution of fluorescein (7.5 μM) also containing 45 equivalents of PMS showed no significant degradation of fluorescein as shown by the invariance of its UV/Vis absorption spectrum over time. In the presence of 10 mol% of Co12 however the fluorescein absorption at 490 nm was quickly bleached (Fig. 5, red trace; Fig. S17†), disappearing completely within half an hour (97% loss in 20 minutes) with a pseudo-first-order rate constant of 0.0016 s−1 under the prevailing conditions. Effects of different concentrations of Co12 are shown in Fig. S18.† Varying the concentration of added PMS (between 15 and 60 equivalents compared to Co12 catalyst) made little difference, and we speculate that this is because the high affinity of anions for the 24+ cage surface means that the surface becomes quickly saturated with the anions such that addition of more makes no difference. We observed the same non-intuitive effect with the Kemp elimination of cavity-bound benzisoxazole using the Co8 catalyst: HO− ions accumulate around the cage surface sufficiently strongly even at pH ≈ 8 that the cage surface becomes saturated, and increasing the pH makes no further difference to the local concentration of hydroxide ions around the guest.6c Consequently the reaction rate was invariant in [HO−] over a considerable pH range despite the mechanism implying that the reaction should be first order in [HO−].6c
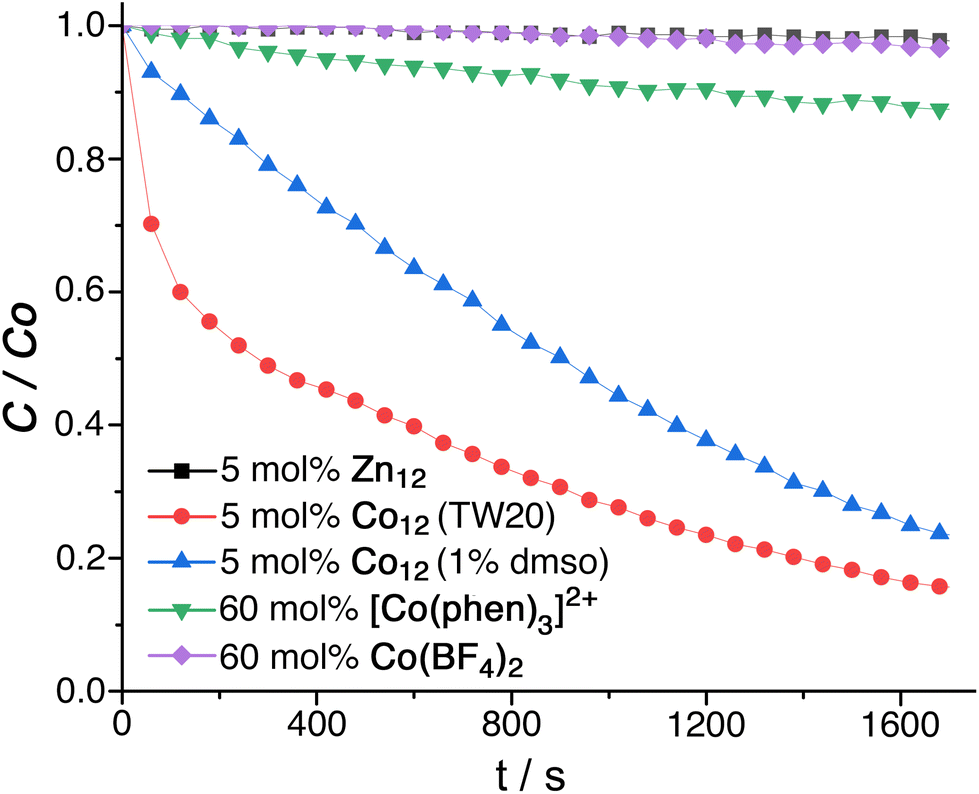 | ||
| Fig. 5 Destruction of fluorescein by oxidation with peroxo-monosulfate under different sets of conditions, showing the importance of both cage-based encapsulation (red and blue lines vs. green and purple) and the importance of having Co(II) ions in the cage superstructure as redox agents for activation of peroxymonosulfate (red vs. black lines). | ||
Similar results were obtained with the dyes rhodamine and methylene blue, with 5 mol% of TW20⊂Co12 resulting in 98% degradation of rhodamine after 20 minutes and 90% degradation of methylene blue after 3 minutes. We note that this type of PMS-based oxidative degradation has not been reported before with fluorescein, and it will be facilitated by the relative ease of oxidation of fluorescein compared to the other organic materials present in the cage superstructure.15
We observed that addition of 0.2 M t-butanol to the catalytic system – a known scavenger of hydroxyl radicals, but not of SO4˙− radicals16 – did not change the reaction rate. In contrast addition of 0.2 M MeOH, which can scavenge both hydroxyl and SO4˙− radicals,17 reduced the rate of degradation of fluorescein and resulted in only ca. 75% bleaching by the end of the experiment. This sensitivity of the reaction to MeOH, but not to t-BuOH, confirms that SO4˙− radicals (but not hydroxyl radicals) are involved, which in turn confirms that redox activation of PMS by Co(II) ions is required before the reaction can occur.
Thus, Co12 in water could in principle accelerate the destruction of fluorescein by PMS in two ways. Firstly, co-location of the two reaction partners, either inside the cage cavity or around its exterior surface, could provide the catalysis following the example provided by the Kemp elimination and other reactions catalysed by Co8.6 Secondly the presence of Co(II) ions in the cage provides redox activation of the PMS by converting it to the more reactive species SO4˙− according to eqn (1).14b Some control experiments clarify these issues.
Firstly, replacement of Co12 as catalyst by the isostructural cuboctahedral cage Zn12, whilst keeping all other conditions the same, completely stopped the degradation of fluorescein (Fig. 5, black trace). Co-location of fluorescein and PMS in and around the M12 cage cavity alone is an insufficient condition for catalysis: the redox-based activation of the PMS to generate SO4˙− is also required, as the quenching experiments with MeOH/t-BuOH showed. Use of Co(BF4)2 as activator was ineffective, with a mixture of fluorescein/PMS/Co(BF4)2 showing no degradation of fluorescein (Fig. 5, purple trace). However the Co2+/Co3+ couple in water is highly positive – around +1.8 V – meaning that aqueous Co2+ ions are poor reducing agents: a better comparison is with a Co(II) complex in an N6 coordination environment similar to that which occurs in this cage family, with a much lower redox potential, that would make it a more effective activation agent for PMS according to eqn (1).
Accordingly the final control experiment was to investigate bleaching of fluorescein in a mixture of fluorescein/PMS/[Co(phen)3]2+ (phen = 1,10-phenanthroline)18 in which the N6-coordinated Co(II) ions are present at the same total concentration as in Co12, i.e. the concentration of mononuclear [Co(phen)3]2+ is 12 times the molar concentration that was used for Co12. The [Co(phen)3]2+/3+ redox potential has been reported as +0.62 V vs. NHE (−0.02 V vs. ferrocene/ferrocenium, Fc/Fc+)18a and in a separate paper as −0.04 V vs. Fc/Fc+.18b This is much less positive than for Co(BF4)2, and is also lower than the Co2+/Co3+ couple of Co12 for which we observed an electrochemically irreversible oxidation (presumably due to a forbidden spin-state change associated with the Co2+/Co3+ interconversion) at +0.62 V vs. Fc/Fc+ in MeCN (Fig. S19†), making [Co(phen)3]2+ an even better reducing agent for activation of PMS than is Co12. Despite this, in the presence of [Co(phen)3]2+ but in the absence of Co12 (i.e. with redox activation of PMS available, but without cage-based encapsulation of components) we observed only slow degradation of fluorescein, to the extent of about 10% reduction in absorbance after half an hour (Fig. 5, green trace): this is far slower than was observed in the presence of Co12.
These experiments confirm that the effective oxidative destruction of fluorescein using Co12 as catalyst arises from a combination of three factors. These are the ability of Co12 to (i) bind fluorescein strongly in water inside its cavity; (ii) accumulate anionic reaction partners around its positively-charged surface, bringing PMS anions close to the fluorescein substrate; and (iii) provide redox activation of the PMS using the Co(II) ions in the cage superstructure which facilitate its conversion to reactive SO4˙− radicals. Removal of either the redox activation provided by the Co(II) ions in the cage (control experiment using Zn12) or the cage-based co-encapsulation of reactants (control experiment using [Co(phen)3]2+ for activation) stops the effective oxidation catalysis provided by Co12/PMS system. Finally we note that using the alternative method of water-solubilisation of Co12 (addition of 1% dmso as co-solvent) also provided a clear catalysis of the fluorescein oxidation reaction by PMS (Fig. 5, blue trace) although the effect is less strong than when using Tween-20, an observation which mirrors what we observed (above) for cage-catalysed phospho-ester hydrolysis when the water/Tween-20 reaction medium afforded slightly better catalysis than water/dmso (99:1).
Conclusions
In conclusion we have demonstrated that the Co12 cage can be solubilised in water (either using a neutral surfactant or by inclusion of 1% dmso in the solvent), permitting a range of guest binding and reactivity studies that rely on the hydrophobic effect. In aqueous solution Co12 binds a range of organic fluorophores (fluorescein and analogues) very strongly (K ≈ 107 M−1) in the central cavity. The ability of the Co12 cage to co-locate hydrophobic organic substrates and anions using two orthogonal interactions results in catalysed hydrolysis destruction of two phosphate-ester based insecticides and also of diacetyl fluorescein which incorporates two carboxylic ester groups. In both cases the second order rate constants for the catalysed reactions are significantly (1–2 orders of magnitude) larger than we previously observed using the smaller cage Co8, which is suggestive of cavity-based catalysis in the larger cavity of Co12 compared to exterior surface-based catalysis using Co8.In addition, Co12 exhibits a new type of cage-catalysed reaction, specifically the destruction of fluorescein by PMS as a model for the ‘advanced oxidation process’ reactions that are studied to remove organic pollutants from water using peroxy-monosulfate. This involves not just the ability of the Co12 cage to bring the organic substrate and the anionic reaction partners into close proximity, but also requires redox activation of PMS (initial conversion to SO4˙− radicals) using the Co(II) ions in the cage – which is therefore an active redox partner in the reaction as well as acting to co-locate the substrates in and around the cage cavity. This type of direct participation of redox-active ions in the cage superstructure in catalysed reactions, which means that the cage is no longer just acting as an inert reaction vessel, offers substantial promise for use of the cages in redox- and photo-redox catalysis processes.
Author contributions
X. Z.: cage synthesis, guest binding and catalysis studies, data analysis. B. S.: NMR and MS studies. M. D. W.: project supervision and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank (i) the University of Warwick (UK) for financial support via a Warwick/Monash Alliance accelerator grant involving X. Z.; and (ii) the Development and Promotion of Science and Technology Talents Project, Thailand, for a PhD studentship to B. S.References
- (a) T. R. Cook and P. J. Stang, Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (b) T. R. Cook, Y.-R. Zheng and P. J. Stang, Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed; (c) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (d) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Building on architectural principles for three-dimensional metallosupramolecular construction, Chem. Soc. Rev., 2013, 42, 1728 RSC; (e) D. Zhang, T. K. Ronson and J. R. Nitschke, Functional Capsules via Subcomponent Self-Assembly, Acc. Chem. Res., 2018, 51, 2423 CrossRef CAS PubMed; (f) H. Vardhan, M. Yusubov and F. Verpoort, Self-assembled metal-organic polyhedra: An overview of various applications, Coord. Chem. Rev., 2016, 306, 171 CrossRef CAS; (g) E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Design and Applications of Water-Soluble Coordination Cages, Chem. Rev., 2020, 120, 13480 CrossRef PubMed.
- (a) Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Catalytic reactions within the cavity of coordination cages, Chem. Soc. Rev., 2019, 48, 4707 RSC; (b) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Supramolecular catalysis in metal-ligand cluster hosts, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; (c) M. Yoshizawa, J. K. Klosterman and M. Fujita, Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (d) M. Otte, Size-Selective Molecular Flasks, ACS Catal., 2016, 6, 6491 CrossRef CAS; (e) C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts, Acc. Chem. Res., 2018, 51, 2447 CrossRef CAS PubMed; (f) W.-X. Gao, H.-N. Zhang and G.-X. Jin, Supramolecular catalysis based on discrete heterometallic coordination-driven metallacycles and metallacages, Coord. Chem. Rev., 2019, 386, 69 CrossRef CAS; (g) M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Advances in supramolecular host-mediated reactivity, Nat. Catal., 2020, 3, 969 CrossRef CAS.
- (a) D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, Enabling New Modes of Reactivity via Constrictive Binding in a Supramolecular-Assembly-Catalyzed Aza-Prins Cyclization, J. Am. Chem. Soc., 2015, 137, 9202 CrossRef CAS PubMed; (b) C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, Enzymelike Catalysis of the Nazarov Cyclization by Supramolecular Encapsulation, J. Am. Chem. Soc., 2010, 132, 6938 CrossRef CAS PubMed.
- (a) M. D. Pluth, R. G. Bergman and K. H. Raymond, Acid catalysis in basic solution: a supramolecular host promotes orthoformate hydrolysis, Science, 2007, 316, 85 CrossRef CAS PubMed; (b) M. D. Pluth, R. G. Bergman and K. N. Raymond, Proton-Mediated Chemistry and Catalysis in a Self-Assembled Supramolecular Host, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (c) J. Wang, T. A. Young, F. Duarte and P. J. Lusby, Synergistic Noncovalent Catalysis Facilitates Base-Free Michael Addition, J. Am. Chem. Soc., 2020, 142, 17743 CrossRef CAS PubMed; (d) C. M. Hong, M. Morimoto, E. A. Kapustin, N. Alzakhem, R. G. Bergman, K. N. Raymond and F. D. Toste, Deconvoluting the Role of Charge in a Supramolecular Catalyst, J. Am. Chem. Soc., 2018, 140, 6591 CrossRef CAS PubMed.
- (a) J. Kang and J. Rebek Jr., Acceleration of a Diels–Alder reaction by a self-assembled molecular capsule, Nature, 1997, 385, 50 CrossRef CAS PubMed; (b) M. Yoshizawa, M. Tamura and M. Fujita, Diels-alder in aqueous molecular hosts: unusual regioselectivity and efficient catalysis, Science, 2006, 312, 251 CrossRef CAS PubMed; (c) Y. Nishioka, T. Yamaguchi, M. Yoshizawa and M. Fujita, Unusual [2+4] and [2+2] Cycloadditions of Arenes in the Confined Cavity of Self-Assembled Cages, J. Am. Chem. Soc., 2007, 129, 7000 CrossRef CAS PubMed; (d) S. Horiuchi, T. Murase and M. Fujita, Diels–Alder Reactions of Inert Aromatic Compounds within a Self-Assembled Coordination Cage, Chem. – Asian J., 2011, 6, 1839 CrossRef CAS PubMed.
- (a) M. D. Ward, C. A. Hunter and N. H. Williams, Coordination Cages Based on Bis(pyrazolylpyridine) Ligands: Structures, Dynamic Behavior, Guest Binding, and Catalysis, Acc. Chem. Res., 2018, 51, 2073 CrossRef CAS PubMed; (b) M. D. Ward, C. A. Hunter and N. H. Williams, Guest Binding and Catalysis in the Cavity of a Cubic Coordination Cage, Chem. Lett., 2017, 46, 2 CrossRef CAS; (c) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Highly efficient catalysis of the Kemp elimination in the cavity of a cubic coordination cage, Nat. Chem., 2016, 8, 231 CrossRef CAS PubMed; (d) W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, Catalysis in a Cationic Coordination Cage Using a Cavity-Bound Guest and Surface-Bound Anions: Inhibition, Activation, and Autocatalysis, J. Am. Chem. Soc., 2018, 140, 2821 CrossRef CAS PubMed; (e) C. G. P. Taylor, A. J. Metherell, S. P. Argent, F. M. Ashour, N. H. Williams and M. D. Ward, Coordination-Cage-Catalysed Hydrolysis of Organophosphates: Cavity- or Surface-Based?, Chem. – Eur. J., 2020, 26, 3065 CrossRef CAS PubMed; (f) C. Mozaceanu, C. G. P. Taylor, J. R. Piper, S. P. Argent and M. D. Ward, Catalysis of an Aldol Condensation Using a Coordination Cage, Chemistry, 2020, 2, 22 CrossRef CAS; (g) A. B. Solea, B. Sudittapong, C. G. P. Taylor and M. D. Ward, Inside or outside the box? Effect of substrate location on coordination-cage based catalysis, Dalton Trans., 2022, 51, 11277 RSC.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, Octanuclear cubic coordination cages, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Quantification of solvent effects on molecular recognition in polyhedral coordination cage hosts, Chem. Sci., 2013, 4, 2744 RSC; (c) G. D. Jackson, M. B. Tipping, C. Pritchard, C. G. P. Taylor, J. R. Piper, C. Mozaceanu and M. D. Ward, A Family of Externally-Functionalised Coordination Cages, Chemistry, 2021, 3, 1203 CrossRef CAS.
- (a) M. D. Ludden, C. G. P. Taylor, M. B. Tipping, J. S. Train, N. H. Williams, J. C. Dorrat, K. L. Tuck and M. D. Ward, Interaction of anions with the surface of a coordination cage in aqueous solution probed by their effect on a cage-catalysed Kemp elimination, Chem. Sci., 2021, 12, 14781 RSC; (b) M. D. Ludden and M. D. Ward, Outside the box: quantifying interactions of anions with the exterior surface of a cationic coordination cage, Dalton Trans., 2021, 50, 2782 RSC; (c) M. D. Ludden, C. G. P. Taylor and M. D. Ward, Orthogonal binding and displacement of different guest types using a coordination cage host with cavity-based and surface-based binding sites, Chem. Sci., 2021, 12, 12640 RSC.
- (a) W. Cullen, C. A. Hunter and M. D. Ward, An Interconverting Family of Coordination Cages and a meso-Helicate; Effects of Temperature, Concentration, and Solvent on the Product Distribution of a Self-Assembly Process, Inorg. Chem., 2015, 54, 2626 CrossRef CAS PubMed; (b) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, Structures and Dynamic Behavior of Large Polyhedral Coordination Cages: An Unusual Cage-to-Cage Interconversion, J. Am. Chem. Soc., 2011, 133, 858 CrossRef CAS PubMed.
- (a) X. Jing, C. He, Y. Yang and C. Duan, A Metal–Organic Tetrahedron as a Redox Vehicle to Encapsulate Organic Dyes for Photocatalytic Proton Reduction, J. Am. Chem. Soc., 2015, 137, 3967 CrossRef CAS PubMed; (b) L. Yang, X. Jing, C. He, Z. Chang and C. Duan, Redox-Active M8L6 Cubic Hosts with Tetraphenylethylene Faces Encapsulate Organic Dyes for Light-Driven H2 Production, Chem. – Eur. J., 2016, 22, 18107 CrossRef CAS PubMed.
- (a) N. K. Al-Rasbi, I. S. Tidmarsh, S. P. Argent, H. Adams, L. P. Harding and M. D. Ward, Mixed-Ligand Molecular Paneling: Dodecanuclear Cuboctahedral Coordination Cages Based on a Combination of Edge-Bridging and Face-Capping Ligands, J. Am. Chem. Soc., 2008, 130, 11641 CrossRef CAS PubMed; (b) S. P. Argent, F. C. Jackson, H. M. Chan, S. Meyrick, C. G. P. Taylor, T. K. Ronson, J. P. Rourke and M. D. Ward, A family of diastereomeric dodecanuclear coordination cages based on inversion of chirality of individual triangular cyclic helicate faces, Chem. Sci., 2020, 11, 10167 RSC.
- (a) P. Saveyn, E. Cocquyt, W. Zhu, D. Sinnaeve, K. Haustraete, J. C. Martins and P. Van der Meeren, Solubilization of flurbiprofen within non-ionic Tween 20 surfactant micelles: a 19F and 1H NMR study, Phys. Chem. Chem. Phys., 2009, 11, 5462 RSC; (b) K. Hoppe and M. Sznitowska, The Effect of Polysorbate 20 on Solubility and Stability of Candesartan Cilexetil in Dissolution Media, AAPS PharmSciTech, 2014, 15, 1116 CrossRef CAS PubMed; (c) J. Akbari, M. Saeedi, K. Morteza-Semnani, H. R. Kelidari, F. S. Moghanlou, G. Zareh and S. Rostamkalaei, The Effect of Tween 20, 60, and 80 on Dissolution Behavior of Sprionolactone in Solid Dispersions Prepared by PEG 6000, Adv. Pharm. Bull., 2015, 5, 435 CrossRef CAS PubMed.
- (a) S. Hjertén and K.-E. Johansson, Selective solubilization with Tween 20 of membrane proteins from Acholeplasma laidlawii, Biochim. Biophys. Acta, 1972, 288, 312 CrossRef PubMed; (b) L. Liljas, P. Lundahl and S. Hjertén, The major sialoglycoprotein of the human erythrocyte membrane. Release with a non-ionic detergent and purification, Biochim. Biophys. Acta, 1974, 352, 327 CrossRef CAS PubMed; (c) S. Schuck, M. Honsho, K. Ekroos and K. Simons, Resistance of cell memebranes to different detergents, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5795 CrossRef CAS PubMed; (d) V. Kotov, K. Bartels, K. Veith, I. Josts, U. K. T. Subhramanyam, C. Günther, J. Labahn, T. C. Marlovits, I. Moraes, H. Tidow, C. Löw and M. Garcia-Alai, High-throughput stability screening for detergent-solubilized membrane proteins, Sci. Rep., 2019, 9, 10379 CrossRef PubMed.
- (a) J. Wang and S. Wang, Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants, Chem. Eng. J., 2018, 334, 1502 CrossRef CAS; (b) F. Ghanbari and M. Moradi, Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review, Chem. Eng. J., 2017, 310, 41 CrossRef CAS; (c) X. Zheng, X. Niu, D. Zhang, M. Lv, X. Ye, J. Ma, Z. Lin and M. Fu, Metal-based catalysts for persulfate and peroxymonosulfate activation in heterogeneous ways: A review, Chem. Eng. J., 2022, 429, 132323 CrossRef CAS; (d) J. Lee, U. von Gunten and J.-H. Kim, Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks, Environ. Sci. Technol., 2020, 54, 3064 CrossRef CAS PubMed.
- (a) N. L. Queiroz, J. A. M. Nascimento, M. L. Nascimento, V. B. Nascimento and S. C. B. Oliveira, Oxidation Mechanism of Fluorescein at Glassy Carbon Electrode, Electroanalysis, 2017, 29, 489 CrossRef CAS; (b) O. K. Lebedeva, V. S. Snytko, I. I. Kuznetsova, D. Y. Kultin, A. N. Zakharov and L. M. Kustov, Unusual Behavior of Fluorescein under Conditions of Electrochemical Oxidation in an Aqueous Phosphate Buffer Solution, Russ. J. Phys. Chem., 2019, 93, 168 CrossRef CAS; (c) S. Pirillo, F. S. G. Einschlag, M. L. Ferreira and E. H. Rueda, Eriochrome Blue Black R and Fluorescein degradation by hydrogen peroxide oxidation with horseradish peroxidase and hematin as biocatalysts, J. Mol. Catal. B: Enzym., 2010, 66, 63 CrossRef CAS.
- M. Piechowski, M. A. Thelen, J. Hoigné and R. E. Bühler, tert-Butanol as an OH-Scavenger in the Pulse Radiolysis of Oxygenated Aqueous Systems, Ber. Bunsenges. Phys. Chem., 1992, 96, 1448 CrossRef.
- R. Li, J. Kong, H. Liu, P. Chen, G. Liu, F. Li and W. Lv, A sulfate radical based ferrous–peroxydisulfate oxidative system for indomethacin degradation in aqueous solutions, RSC Adv., 2017, 7, 22802 RSC.
- (a) S. M. Feldt, G. Wang, G. Boschloo and A. Hagfeldt, Effects of Driving Forces for Recombination and Regeneration on the Photovoltaic Performance of Dye-Sensitized Solar Cells using Cobalt Polypyridine Redox Couples, J. Phys. Chem. C, 2011, 115, 21500 CrossRef CAS; (b) H. Ferreira, M. M. Conradie and J. Conradie, Electrochemical properties of a series of Co(II) complexes containing substituted phenanthrolines, Electrochim. Acta, 2018, 292, 489 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental and characterisation data. See DOI: https://doi.org/10.1039/d2qi02223k |
| ‡ In the T-symmetry isomer all such protons are equivalent, giving a single signal for all twelve of them. In the C3-symmetry isomer there are four inequivalent environments (with equal likelihood) for each proton, and in the S4-symmetry isomer there are three inequivalent environments (with equal likelihood) for each such proton. The result is a set of eight signals, being sets of 1 + 3 + 4 for the three diastereoisomers, and if the assembly of the four triangular helicate faces in the cuboctahedron is purely statistical with no preference for any one diastereoisomer, all eight signals should be of equal intensity (see ref. 11b). Any bias towards one diastereoisomer and away from the others will result in a very characteristic set of closely-spaced 1 + 3 + 4 signals for each chemical environment of proton. |
| § We could also achieve significant water solubility using 10% MeCN in water, a solvent mixture that allowed greater solubility than 1% dmso in water, and we used 10% MeCN in water for some of the NMR and MS experiments to confirm stability of the cages in water. Notably this gave cleaner ES mass spectra than did 1% dmso in water, as the presence of dmso results in greater fragmentation and adduct formation at the high temperatures required for solvent evaporation under MS conditions (compare Fig. S1/S3, and S7/S9†). For the catalysis experiments however we used either Tween-20 or 1% dmso in water to achieve water solubility, to maximise the amount of water present and hence the hydrophobic effect responsible for guest binding. We note that a small admixture of dmso to facilitate water solubility has not caused any stability problems with other members of this cage family based on Co(II) tris(pyrazolyl-pyridine) metal complex units, see for example ref. 6g. |
| This journal is © the Partner Organisations 2023 |