
Dual FLT3/haspin kinase inhibitor based on 3H-pyrazolo[4,3-f]quinoline scaffold with activities against acute myeloid leukemia†
Allison L.
Kempen
 a,
Nickolas R.
Brauer
a and
Herman O.
Sintim
*abc
a,
Nickolas R.
Brauer
a and
Herman O.
Sintim
*abc
aDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA. E-mail: hsintim@purdue.edu
bPurdue Institute for Drug Discovery, 720 Clinic Drive, West Lafayette, IN 47907, USA
cPurdue Institute for Cancer Research, 201 S. University St., West Lafayette, IN 47907, USA
First published on 14th July 2023
Abstract
The 3H-pyrazolo[4,3-f]quinoline core, a privileged fusion moiety from quinoline and indazole, facilely synthesized in a one flask multi-component Doebner–Povarov reaction, is a newly described kinase hinge binder. Previous works have demonstrated that the 3H-pyrazolo[4,3-f]quinoline moiety can be tuned, via judicious substitution patterns, to selectively inhibit cancer-associated kinases, such as FLT3 and haspin. A first generation 3H-pyrazolo[4,3-f]quinoline-based haspin inhibitor, HSD972, and FLT3 inhibitor, HSD1169, were previously disclosed as inhibitors of various cancer cell lines. Given the recent revelation that haspin is over-expressed and plays critical proliferative roles in many cancers, and compounds with dual activity against FLT3 and other important kinases are now being actively developed by many groups, we became interested in optimizing the 3H-pyrazolo[4,3-f]quinoline-based compounds to improve activity against both FLT3 and haspin. Herein, we report the discovery of new 3H-pyrazolo[4,3-f]quinoline-based dual FLT3/haspin inhibitor, HSK205. HSK205 has remarkable potencies against FLT3-driven AML cell lines, inhibiting proliferation with GI50 values between 2–25 nM. Western blot analyses of treated AML cells confirm that HSK205 inhibit the phosphorylation of both FLT3 and histone H3 (a haspin target) in cells. While multi-component reactions (MCRs) have been used to make many bioactive molecules, there are very few examples of using MCRs to make compounds that target protein kinases, which have emerged as one of the top drug candidates (especially in oncology). This work highlights our recent efforts to make ultrapotent protein kinase inhibitors using multi-component reactions (especially the Doebner–Povarov reaction).
1. Introduction
Acute myeloid leukemia (AML) is a devastating disease that affects over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people in the US each year.1 About 30% of AML patients harbor mutations in the FLT3 kinase, and for these patients, the prognosis is usually poorer than those without FLT3 mutation.2 Many FLT3 inhibitors have been developed and trialed in the clinic and a few, including midostaurin, quizartinib, and gilteritinib, have been approved in the United States or Japan. These FLT3 inhibitors have moderately improved the survival of FLT3-harboring AML patients, although resistance to the approved FLT3 inhibitors has been observed in the clinic. Specifically, FLT3-F691I is resistant to midostaurin while FLT3-F691L is resistant to gilteritinib and quizartinib.3 In addition to FLT3 resistant mutations, FLT3-independent resistance pathways, such as NRAS activation or mutations of other kinases, also emerge after prolonged FLT3i treatment.4,5 In some instances, patients who were initially FLT3-positive become FLT3-negative during treatment; yet the cancer progresses, fueled by alternative pathways.6 Thus, there is a need for newer drugs that can deal with FLT3, as well as other oncogenic pathways that may arise during treatment. Others have recently disclosed FLT3 inhibitors that have activities against other cancer related kinases, such as dual FLT3/MERTK,7 FLT3/MNK,8 FLT3/CDK,9 FLT3/TOPK,10 FLT3/PIM,11 or FLT3/AURKA inhibitors,12 and in these cases the authors have argued that the concurrent inhibition of the FLT3 and other kinase pathways confer some advantages in overcoming resistance. For example, CCT245718, a dual FLT3/Aurora A inhibitor could overcome FLT3-D835Y-mediated resistance.13 In another interesting report, it was shown that the frequency of resistance to a dual FLT3/CDK4 inhibitor (AMG 925) was less than other FLT3 inhibitors (such as quizartinib) that did not inhibit CDK4.14 Considering these prior dual inhibitors, we became interested in identifying FLT3 inhibitors that also inhibited other kinases which play important roles in cancer progression. Haspin has emerged as an interesting novel target in cancer and is the main kinase that phosphorylates histone H3 at T3.15 A recent preliminary report indicated that reduction of haspin via genetic means or inhibition by a compound reduced leukemia proliferation.16
000 people in the US each year.1 About 30% of AML patients harbor mutations in the FLT3 kinase, and for these patients, the prognosis is usually poorer than those without FLT3 mutation.2 Many FLT3 inhibitors have been developed and trialed in the clinic and a few, including midostaurin, quizartinib, and gilteritinib, have been approved in the United States or Japan. These FLT3 inhibitors have moderately improved the survival of FLT3-harboring AML patients, although resistance to the approved FLT3 inhibitors has been observed in the clinic. Specifically, FLT3-F691I is resistant to midostaurin while FLT3-F691L is resistant to gilteritinib and quizartinib.3 In addition to FLT3 resistant mutations, FLT3-independent resistance pathways, such as NRAS activation or mutations of other kinases, also emerge after prolonged FLT3i treatment.4,5 In some instances, patients who were initially FLT3-positive become FLT3-negative during treatment; yet the cancer progresses, fueled by alternative pathways.6 Thus, there is a need for newer drugs that can deal with FLT3, as well as other oncogenic pathways that may arise during treatment. Others have recently disclosed FLT3 inhibitors that have activities against other cancer related kinases, such as dual FLT3/MERTK,7 FLT3/MNK,8 FLT3/CDK,9 FLT3/TOPK,10 FLT3/PIM,11 or FLT3/AURKA inhibitors,12 and in these cases the authors have argued that the concurrent inhibition of the FLT3 and other kinase pathways confer some advantages in overcoming resistance. For example, CCT245718, a dual FLT3/Aurora A inhibitor could overcome FLT3-D835Y-mediated resistance.13 In another interesting report, it was shown that the frequency of resistance to a dual FLT3/CDK4 inhibitor (AMG 925) was less than other FLT3 inhibitors (such as quizartinib) that did not inhibit CDK4.14 Considering these prior dual inhibitors, we became interested in identifying FLT3 inhibitors that also inhibited other kinases which play important roles in cancer progression. Haspin has emerged as an interesting novel target in cancer and is the main kinase that phosphorylates histone H3 at T3.15 A recent preliminary report indicated that reduction of haspin via genetic means or inhibition by a compound reduced leukemia proliferation.16
Our laboratory has previously shown that the “fusion” of two known kinase hinge binders, indazole and quinoline, which are commonly found in FDA-approved drugs, such as axitinib and erlotinib, leads to the novel 3H-pyrazolo[4,3-f]quinoline (Fig. 1A), which binds to the hinge region of kinases.17 Remarkably, the substituent at position 7 of the 3H-pyrazolo[4,3-f]quinoline moiety dictates which kinases are potently inhibited. For example, pyrazole substitution at position 7 leads to inhibitors that bind to mutant FLT3 with picomolar Kd values,18 while phenyl boronic acid substituents lead to ultrapotent ROCK1/2 inhibitors19 (Fig. 1B). Benzo[d]thiazolyl substituted compounds like HSD992 are potent CDK inhibitors,20 while phenol containing compounds, such as HSD972, are excellent haspin inhibitors (HSD972 haspin IC50 = 14 nM).21 In addition to the 3H-pyrazolo[4,3-f]quinoline core, others have shown that analogous systems, such as pyrazolo[3,4-h]quinoline scaffold, also have anticancer activities.22
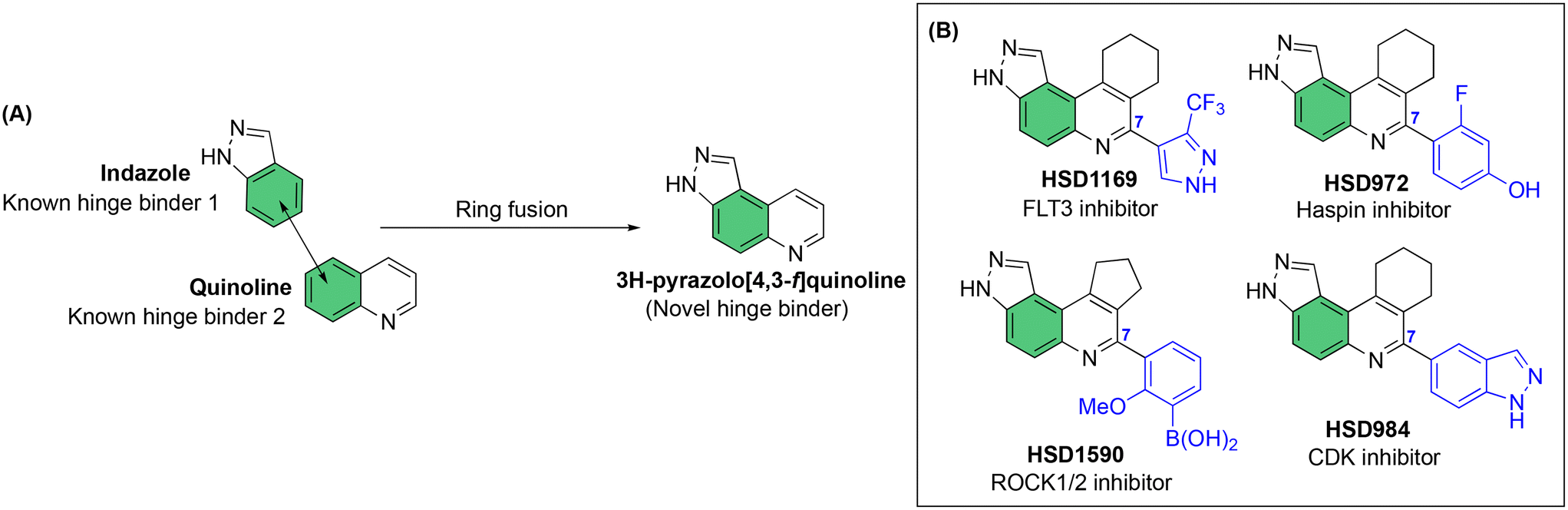 | ||
| Fig. 1 A) Fusion of indazole with quinoline to afford the novel hinge binder, 3H-pyrazolo[4,3-f]quinoline core. B) C-7 substitution of the 3H-pyrazolo[4,3-f]quinoline moiety affects kinase targeting. | ||
Considering that HSD972 and HSD1169 (see Fig. 1), both containing the 3H-pyrazolo[4,3-f]quinoline moiety inhibit haspin or FLT3 respectively, we wondered if we could find a C7 substituent that would endow dual FLT3 and haspin kinase inhibition. Indeed, we discovered that replacement of the phenol moiety at position-7 of HSD972 with benzamide led to compound HSD1217 (Fig. 2A), which inhibited haspin more potently than our previously disclosed compound, HSD972. Thus, we became interested in optimizing HSD1217.
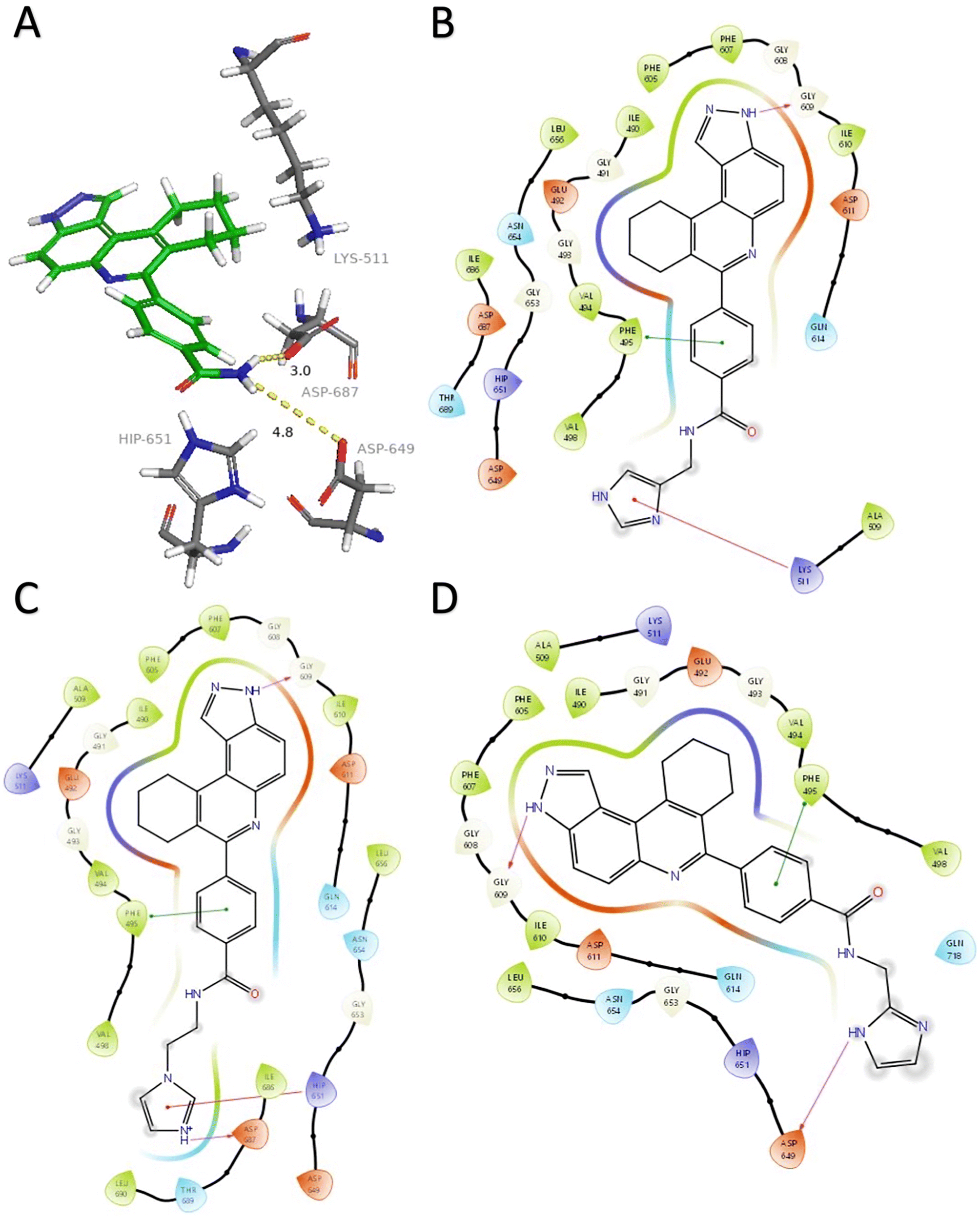 | ||
| Fig. 2 A) HSD1217 and ATP-binding residues. Docked structures showing interactions between haspin (PDB: 2WB8) and three initial compounds (B) 4, (C) 5, and (D) 6. Hydrogen bonds are represented in fuchsia, pi–cation interactions in red, and pi–pi stacking in green. Solvent exposure is represented with gray rings surrounding atoms. Docking was performed using Glide (Schrödinger) and visualization through PyMol and Maestro. | ||
Analysis of the docked structure of HSD1217 to haspin (Fig. 2A) indicated that the benzamide–NH2 moiety had a hydrogen bonding interaction with key residue D687 and was positioned towards a region harboring H651 and D649 residues, which bind to the ATP phosphate moiety. Thus, we rationalized that appending polar groups, such as imidazole, with hydrogen bonding and hydrogen donating capabilities to the benzamide that could reach the either H651 or D649 residues while maintaining hydrogen bonding with the D687 residue could provide additional interactions to increase ligand affinity for the protein. Because H651, D649, or D687 are critical for ATP binding to haspin, we speculate that inhibitors that engage these residues would have a higher chance of resisting kinase mutations that abrogate inhibitor binding.23In silico analysis of compounds 4, 5, and 6 (analogs of HSD1217), which contain the imidazole moiety, indicated that all three compounds could form key interactions with at least one of these ATP-binding residues (Fig. 2B–D). For docking analysis of 3H-pyrazolo[4,3-f]quinoline-based compounds to FLT3, the reader is referred to our previous publication, ref. 17.
With these preliminary compounds in hand, we began a structure–activity relationship (SAR) study to explore how modifications affected haspin and/or FLT3 inhibition. Herein, we disclose the identification of new imidazoyl-containing pyrazolo-based compounds that are dual FLT3/haspin inhibitors, inhibiting both kinases at ultra-low (sub-nanomolar) concentrations.
2. Results and discussion
2.1 Chemistry
Multicomponent reactions (MCRs) have often been utilized to rapidly synthesize a vast range of highly functionalized analogs in a single pot to build libraries for biological screening. In this work, compounds were synthesized using the Doebner-like MCR followed by subsequent amidation (Scheme 1). For our pilot library, we focused on using amines bearing imidazole or bioisosteres thereof as we believed that such heterocyclic amines that are found in other drugs, such as nilotinib, could facilitate ligand engagement with protein residues, vide supra. During the Doebner-like MCR, the amine and aldehyde react to form the Schiff base, which subsequently reacts with the added ketone in the presence of the acid catalyst (HCl). With the MCR product A (see Scheme 1) in hand, the final amide products (4–15) were formed via amidation, using commercially available amines, HATU and Hünig's base.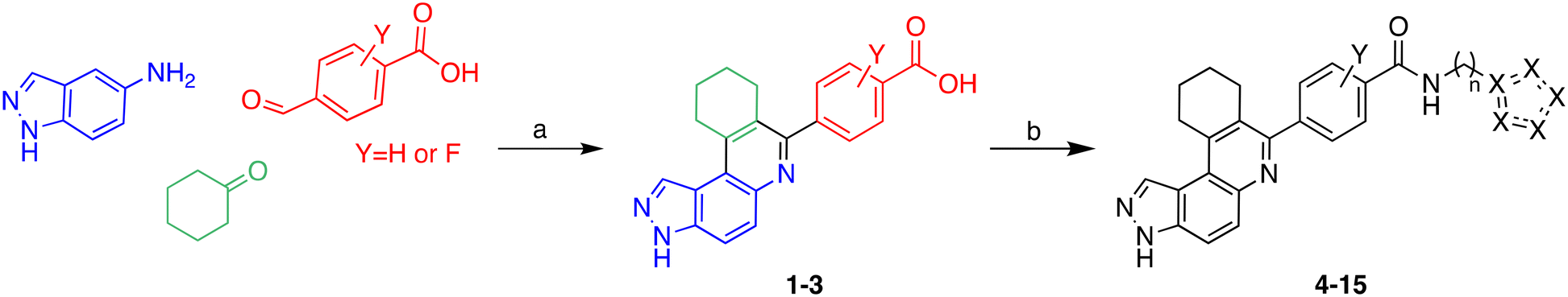 | ||
| Scheme 1 Synthesis of analogs via Doebner-like MCR followed by amidation. (a) EtOH, reflux, 2 h, then ketone (2 equiv.), cat. HCl, reflux, 12 h; (b) HATU, DIPEA, DMF, 60 °C, 12 h. | ||
2.2 Kinase assay and structure–activity relationship (SAR) study
With the amide kinase inhibitor library in hand, we began by analyzing kinase inhibition as well as AML cell line (Molm-14, which harbors FLT3-ITD, and MV4-11) growth inhibition at 50 nM compound concentration (Table 1).| Compound | Structure | Percent inhibition (%) at 50 nM inhibitor conc. | Compound | Structure | Percent inhibition (%) at 50 nM inhibitor conc. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Haspin | FLT3 | MOLM-14 | MV4-11 | Haspin | FLT3 | MOLM-14 | MV4-11 | ||||
| 4 |
|
54 ± 1 | 49 ± 6 | 67 ± 3 | 62 ± 9 | 11 |
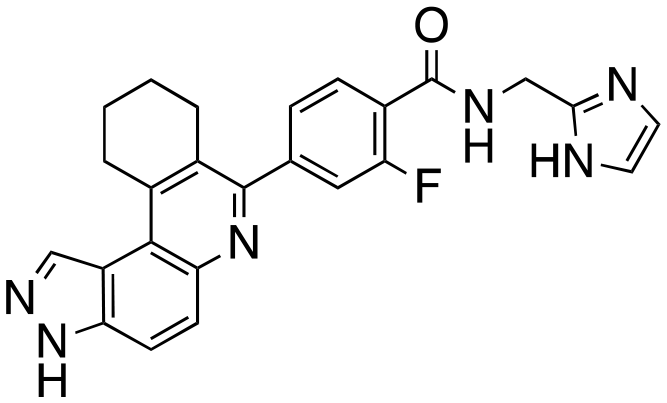
|
76 ± 4 | 85 ± 9 | 80 ± 3 | 82 ± 5 |
| 5 |
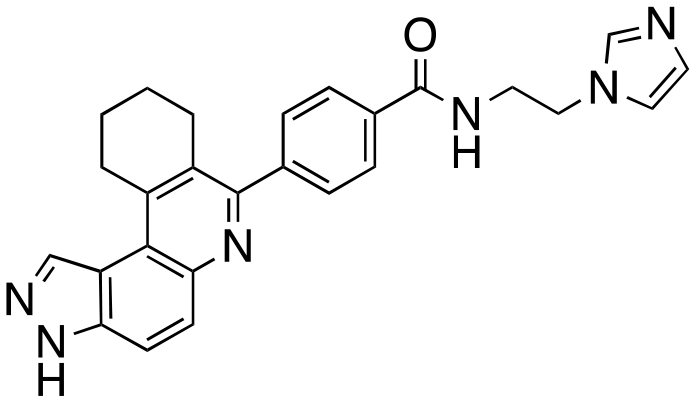
|
33 ± 4 | 31 ± 2 | 42 ± 6 | 35 ± 5 | 12 |
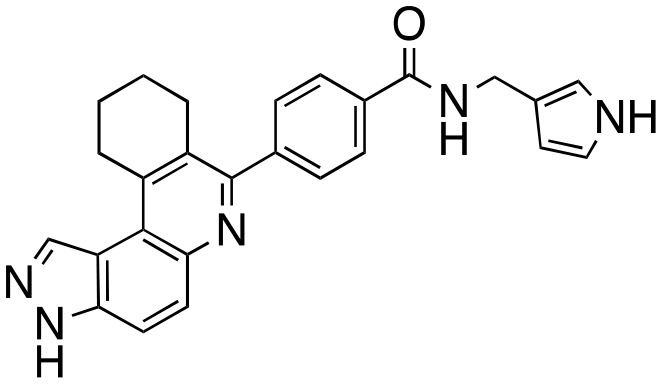
|
41 ± 8 | 46 ± 2 | 39 ± 11 | 28 ± 6 |
| 6 |
|
77 ± 3 | 76 ± 7 | 84 ± 5 | 80 ± 10 | 13 |
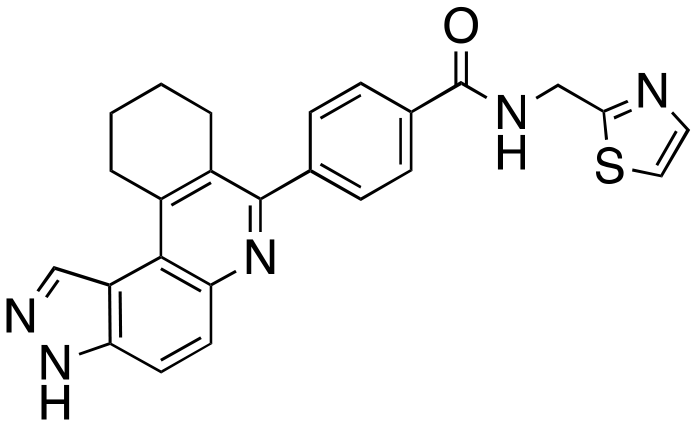
|
68 ± 6 | 53 ± 7 | 55 ± 5 | 60 ± 2 |
| 7 |
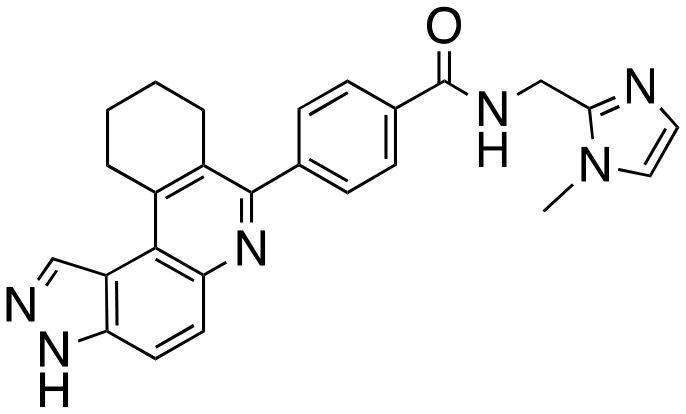
|
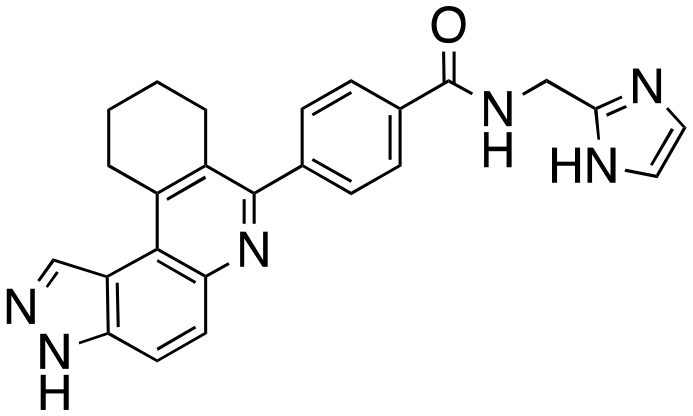
26 ± 2 | 14 ± 4 | 21 ± 7 | 25 ± 4 | 14 (HSK205) |
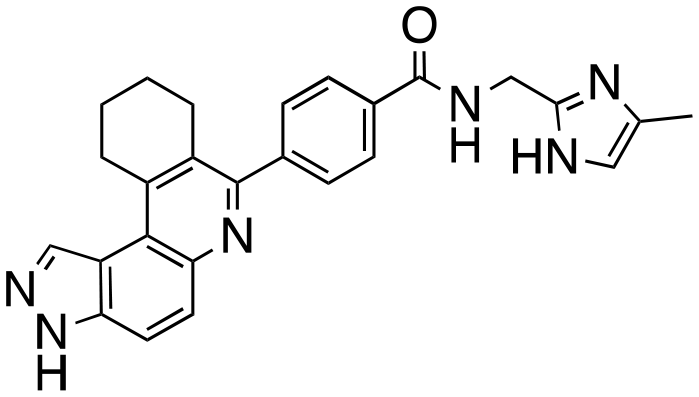
|
97 ± 2 | 94 ± 3 | 96 ± 4 | 94 ± 7 |
| 8 |
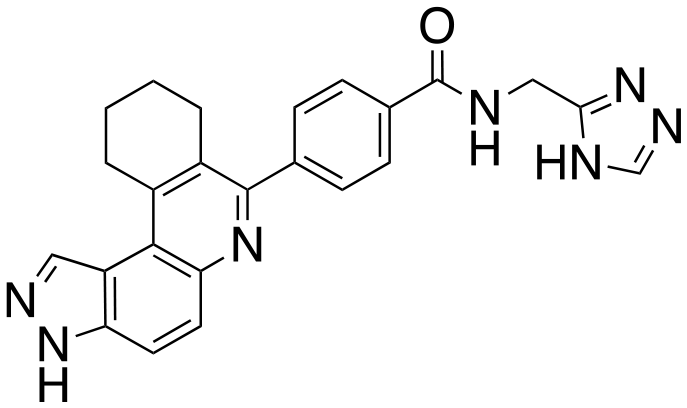
|
57 ± 8 | 49 ± 1 | 48 ± 4 | 52 ± 7 | 15 |
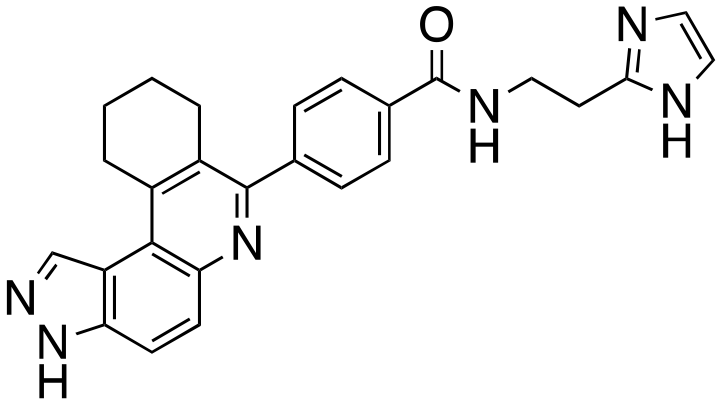
|
78 ± 3 | 71 ± 9 | 73 ± 8 | 69 ± 1 |
| 9 |
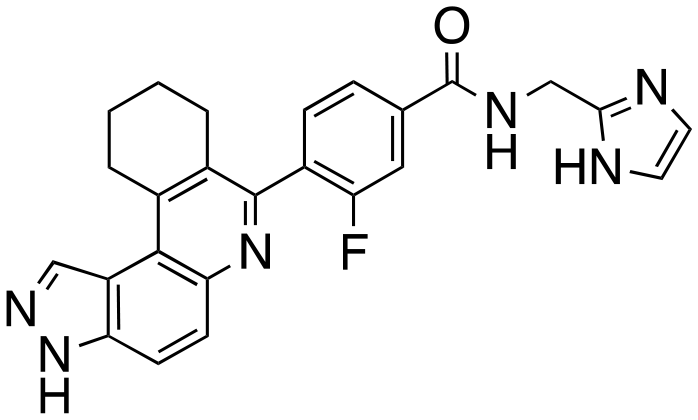
|
53 ± 2 | 88 ± 5 | 87 ± 9 | 79 ± 4 | HSD1169 |
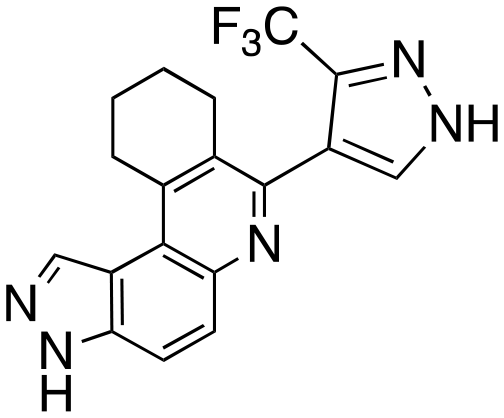
|
39 ± 10 | 81 ± 2 | 90 ± 5 | 92 ± 4 |
| 10 |
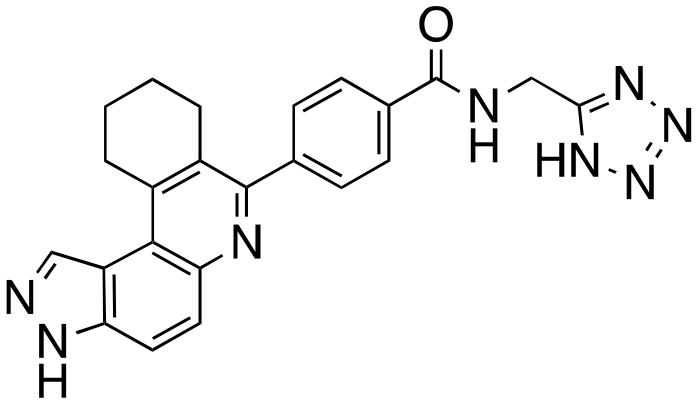
|
57 ± 2 | 51 ± 3 | 50 ± 1 | 41 ± 8 | Gilteritinib |
|
11 ± 4 | 93 ± 4 | 87 ± 6 | 91 ± 5 |
All compounds were screened in lab against haspin and FLT3 using the ADP Glo Kinase Assay (Promega, Madison, WI). Compounds were screened at a concentration of 50 nM, and HSD1169 and gilteritinib were used as positive controls. Additionally, cellular growth inhibition of the analogs was evaluated against two AML cell lines, Molm-14 (FLT3-ITD) and MV4-11.
The structure–activity relationship (SAR) study demonstrated that the imidazole ring is critical for activity against both haspin and FLT3, as even subtle modifications dramatically affected kinase inhibition. Compounds 4, 5 and 6 all contain the imidazole moiety, albeit substituted differently; their inhibition profiles against the tested kinases are however different. Compound 6, the 2-substituted imidazole, inhibited haspin and FLT3 (77% and 76%, respectively) better than 5, the 1-substituted imidazole, (33% and 31%, respectively), and 4, the 4-substituted imidazole (54% and 49%, respectively). Considering that compound 6 showed the best inhibitions of the tested kinases, we became interested in exploring how substitution patterns on 6 could potentially improve potency. Substituting the –NH on compound 6 with –NMe, as in compound 7, reduced haspin and FLT3 inhibition as well as AML growth inhibition. For example, while at 50 nM, compound 6 inhibited haspin at 77%, the –NMe analog 7 inhibited the same enzyme at 26%. For FLT3, potency dropped from 76% inhibition with compound 6 at 50 nM to only 14% inhibition with compound 7. Replacing the imidazole unit in compound 6 with other 5-membered bioisosteres (triazole (8), tetrazole (10) or thiazole (13)) afforded compounds that could inhibit both kinases and AML cell proliferation, but generally less so than compound 6. Extending the length of carbon chain between the amide and the imidazole by one unit to give compound 15 did not drastically impact inhibition of either kinase.
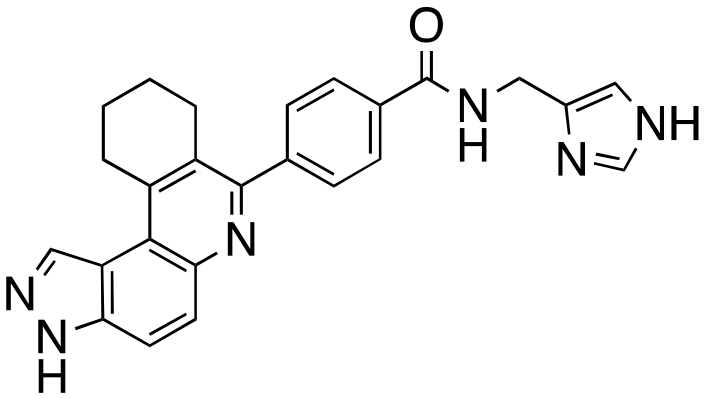
We routinely explore the effects of fluorine substitution on lead compounds as it is known that fluorine can be used to tune drug properties (both target engagement as well as general drug properties, such as metabolic stability).24 Thus, we explored fluorinating the benzamide moiety of compound 6 to afford compounds 9 and 11 but FLT3 and haspin kinase inhibitions were not significantly impacted (compare compounds 9 and 11 with 6, Table 1). Some imidazole-containing drugs, such as nilotinib, have the imidazole unit methylated at position 4. Thus, we also wanted to investigate the extent that methylation of the imidazole unit in compound 6 would affect kinase inhibition. To our delight, this significantly improved potency against haspin, FLT3, and AML cell proliferation. Compound 14 (also called HSK205) at only 50 nM inhibited haspin and FLT3 at 97% and 94%, respectively, hinting that this compound could be an ultra-potent inhibitor of both FLT3 and haspin (vide infra). Indeed, at only 50 nM, compound HSK205 almost completely inhibited (96% and 94%, respectively) the growth of Molm-14 (FLT3-ITD) and MV4-11 cells.
To validate results obtained in our laboratory, we also sent HSK205 to contract research organizations, Eurofins and Reaction Biology, to obtain binding data for haspin (Eurofins) and enzyme inhibition data for FLT3 (Reaction Biology). At 10 μM ATP, HSK205 inhibited the FLT3-ITD enzyme with an IC50 = 0.199 nM, while under similar conditions, gilteritinib had an IC50 = 0.121 nM (Fig. 3A). For the mutated FLT3 D835Y enzyme, HSK205 inhibited with an IC50 = 0.187 nM, whereas gilteritinib had an IC50 = 0.024 nM. With regards to haspin inhibition, HSK205 could bind to haspin with Kd = 0.55 nM (Kd of control haspin inhibitor CHR-6494 < 0.17 nM), Fig. 3C. This SAR study has validated earlier reports that the 3H-pyrazolo[4,3-f]quinoline moiety can target FLT3 and the moiety is highly tunable, in that substitution at the C7 position can be used to target other kinases. For example, HSD1169, also a 3H-pyrazolo[4,3-f]quinoline-containing compound and bearing a fluorinated pyrazole is a strong inhibitor of FLT3 but a weak inhibitor of haspin, while changing the C7 moiety to a benzamide leads to a dual FLT3/haspin inhibitor.
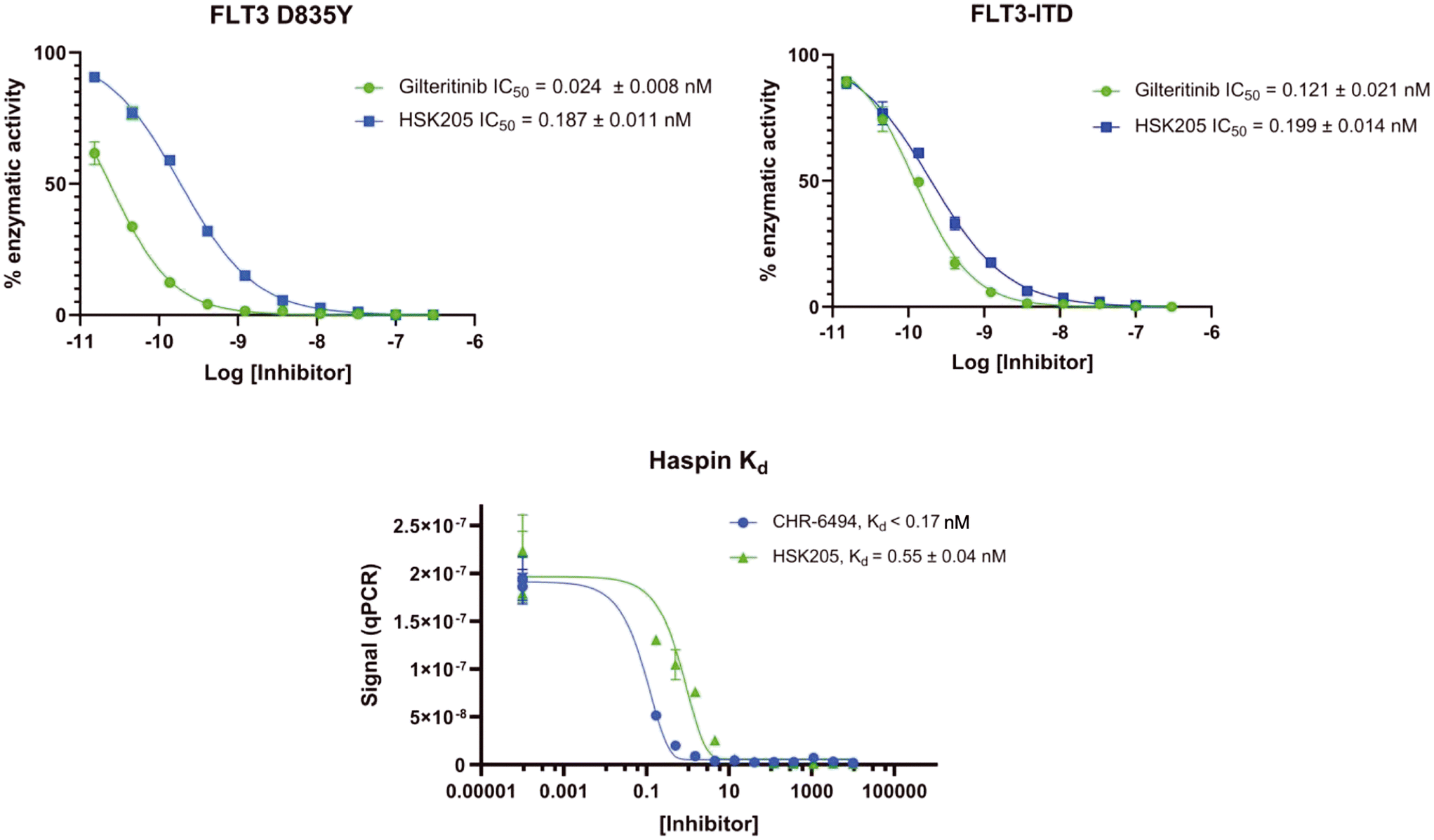 | ||
| Fig. 3 Enzyme binding and inhibition data. Compounds were tested against FLT3-ITD enzyme in 10-dose IC50 duplicate mode with a 3-fold serial dilution starting at 10 μM. Compounds were tested against FLT3 (D835Y) enzyme in 10-dose IC50 duplicate mode with a 3-fold serial dilution starting at 10 μM. Kd data for haspin kinase obtained by Eurofins. Each data point represents the mean and error bars represent the SEM of triplicates. All data was fitted to a non-linear regression equation using GraphPad Prism 9.0 software. | ||
2.3 Target engagement and in vitro biological activity
We proceeded to evaluate if HSK205 does indeed inhibit haspin in AML cells and performed immunoblotting to confirm inhibition of histone H3 (Thr3) phosphorylation by haspin (Fig. 4A). Molm-14 cells were treated with either HSK205 (5 nM or 20 nM or 100 nM) or DMSO control or gilteritinib (a negative control) at the higher 100 nM concentration, and cells were harvested after 24 hours of treatment. The samples were subsequently analyzed for the phosphorylation of histone H3 (Thr3) by haspin.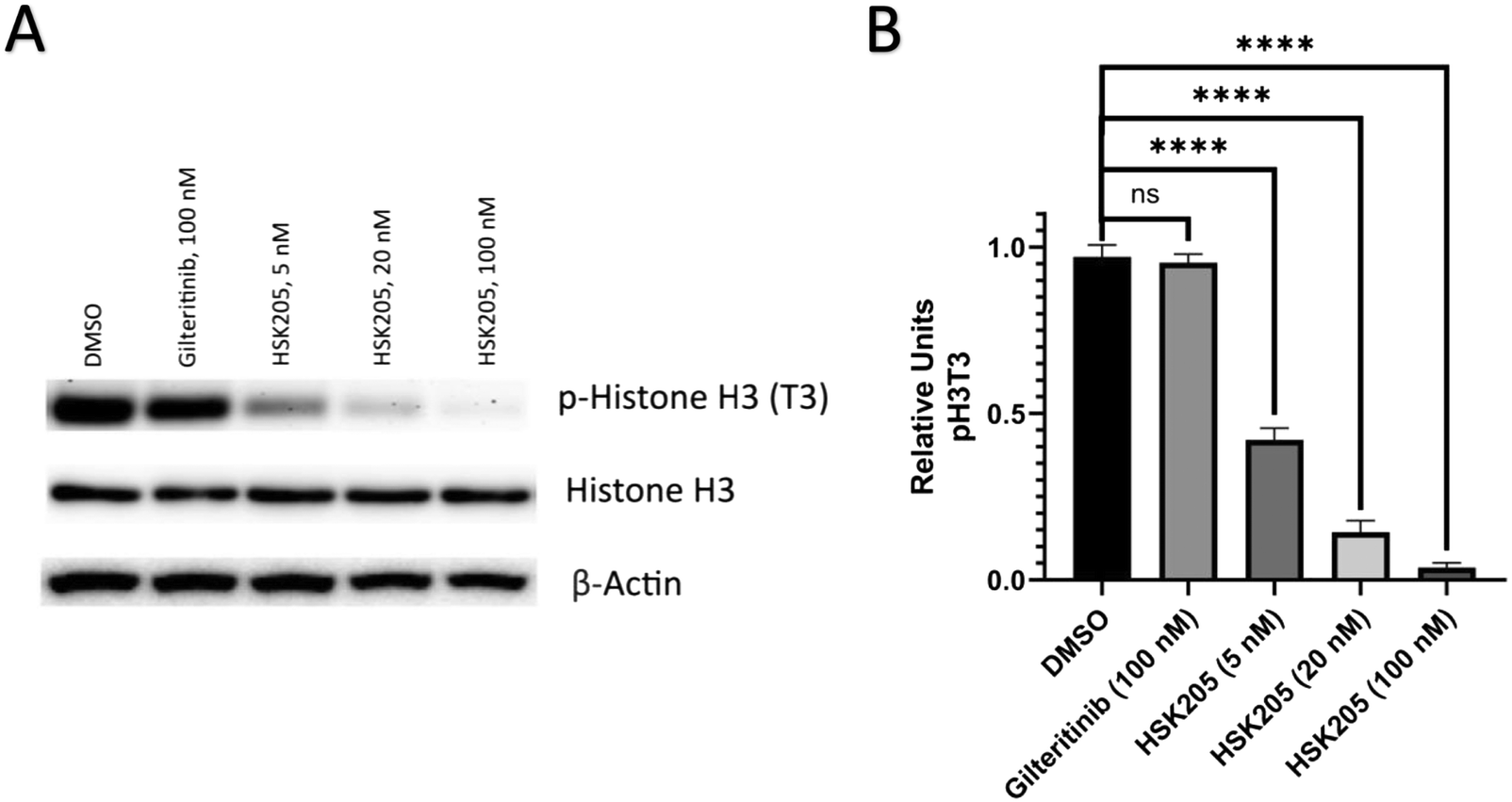 | ||
| Fig. 4 Western blot analysis. (A) Molm-14 cells were treated with HSK205 (5, 20, or 100 nM) or control for 24 hours. Gilteritinib at 100 nM was used as a positive control. (B) Bands were quantified relative actin loading control. Values are reported as means of triplicates and error bars represent standard deviation (NS: p > 0.05, *: p < 0.05, **: p < 0.01, ***p < 0.001, ****p < 0.0001). | ||
Results showed that HSK205 was effective in reducing phosphorylation of histone H3 (Thr3), with a more pronounced effect at 20 nM compared to 5 nM and 100 nM compared to 20 nM (Fig. 4B). Gilteritinib, however, did not display haspin inhibition in Molm-14 cells.
Additionally, we also evaluated FLT3 target engagement in Molm-14 cells to confirm the FLT3 inhibition seen in our preliminary kinase screen (Table 1). Cells were treated for 2 hours, harvested, and subsequently analyzed. Administration of HSK205 to Molm-14 cells led to a reduction of phosphorylated FLT3 (Fig. 5).
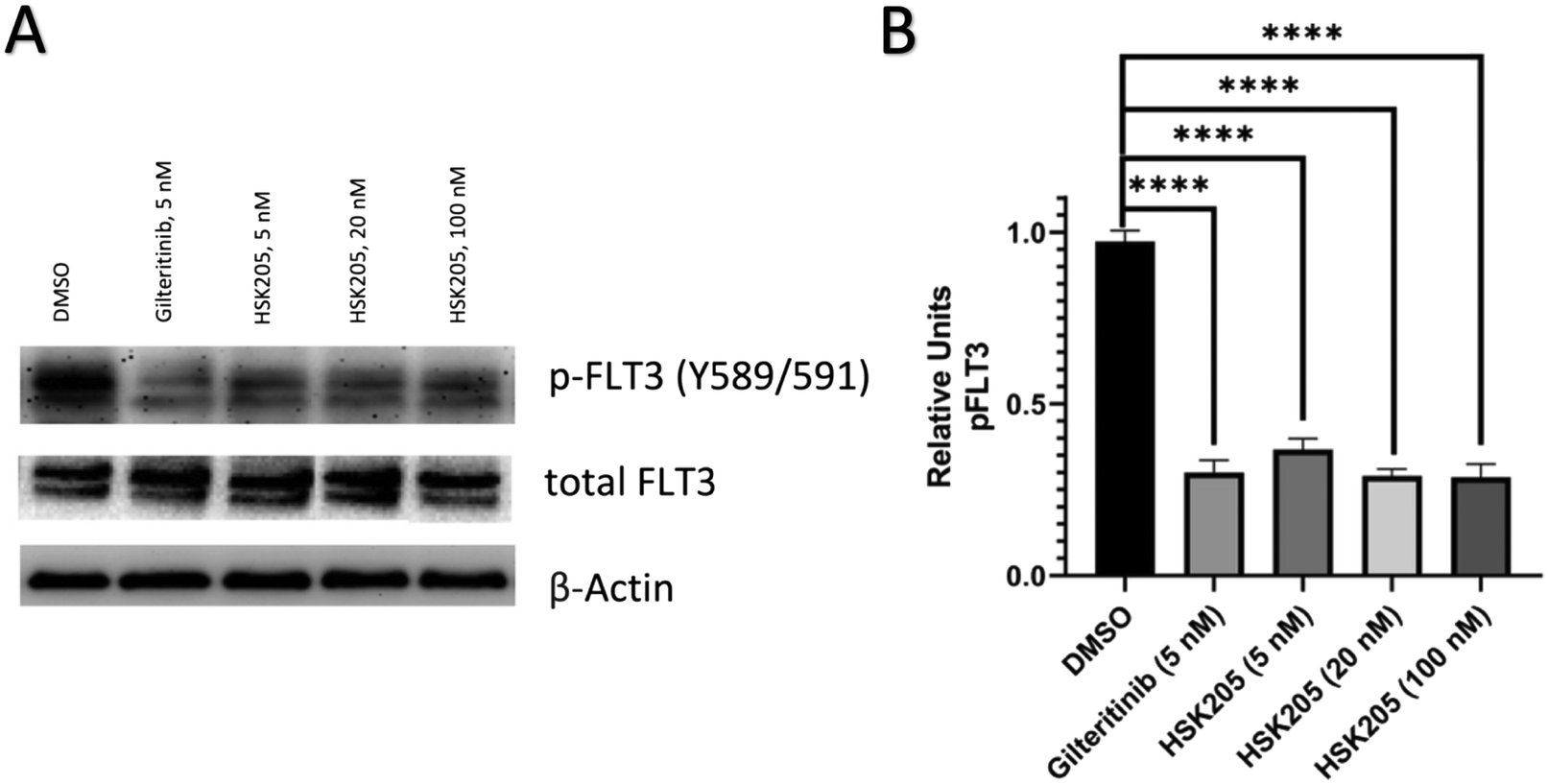 | ||
| Fig. 5 Western blot analysis. (A) Molm-14 cells were treated with HSK205 (5, 20, or 100 nM) or control for 2 hours. Gilteritinib at 5 nM was used as a positive control. (B) Bands were quantified relative actin loading control. Values are reported as means of triplicates and error bars represent standard deviation (p > 0.05: NS, p < 0.05: *, p < 0.01: **, p < 0.001: ***, p < 0.0001: ****). | ||
Many FLT3 inhibitors have been trialed in the clinic and some, such as gilteritinib, midostaurin and quizartinib, have been approved but on-target FLT3 mutations that reduce AML sensitivity to these drugs have been observed.3 Thus, we evaluated HSK205 against Molm-14 cell lines that harbor resistant mutations commonly found in the clinic, FLT3-ITD-D835Y and F691L (Fig. 6). Remarkably, we found that HSK205 showed low nanomolar GI50 values against Molm-14-ITD-F691L (2.2 nM) and Molm-14-ITD-D835Y (4.3 nM).
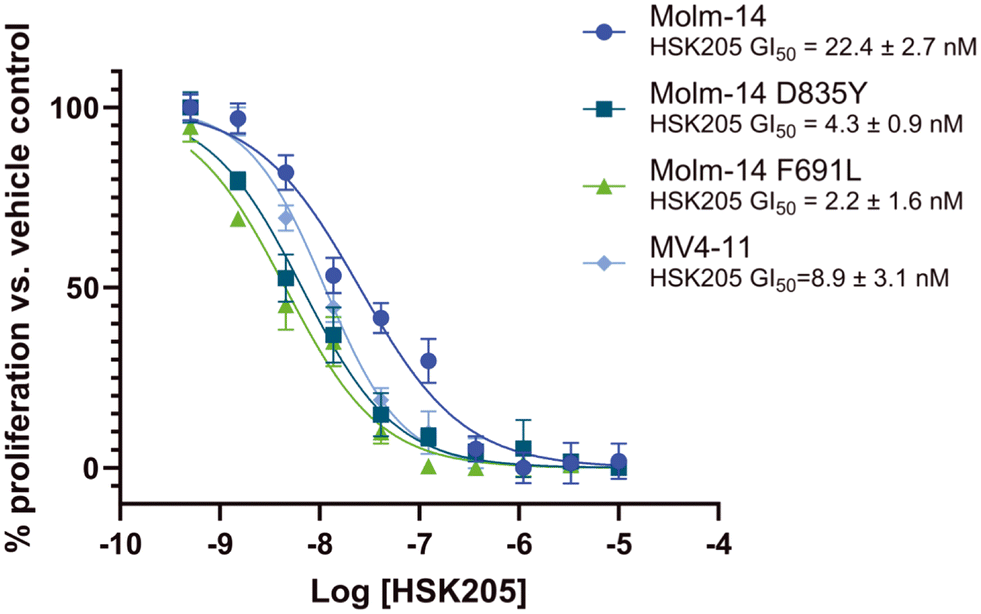 | ||
| Fig. 6 Dose response curves. FLT3-driven AML cells were treated with varying concentrations of HSK205 for 72 h. Data was fitted to a non-linear regression equation using GraphPad Prism 9.0 software. Each data point represents the mean and error bars represent the SEM of triplicates. | ||
Selectivity is important in the discovery of kinase inhibitors. It has been shown that FLT3 inhibitors, including those that have been FDA approved, commonly target additional kinases, such as KDR, PDGFR, c-KIT, and FGFR. Inhibition of these kinases can result in decreased tolerability and increased side effects. Specifically, it has been shown that co-inhibition of FLT3 and c-KIT can lead to myelosuppression and a high possibility of infection.25 KDR, FGFR and PDGFR are all important for cardio function, and it is well known that targeting these cardiovascular-related kinases can result in low tolerability and increased side effects.26–28 Additionally, our anti-target screening included CDK1, which has been demonstrated to lead to toxicity in normal proliferating cells.29 We performed an in-lab kinase screening of our top compound, HSK205, against these anti-targets and compared to known haspin inhibitor, CHR-6494, and FDA-approved FLT3 inhibitor, gilteritinib (Fig. 7). HSK205 was screened at a concentration of 100 nM, roughly 500 times its FLT3 IC50. Even at a high concentration, HSK205 showed minimal activity against these five anti-targets, which was similar to or lower than the activity of the well-tolerated gilteritinib.
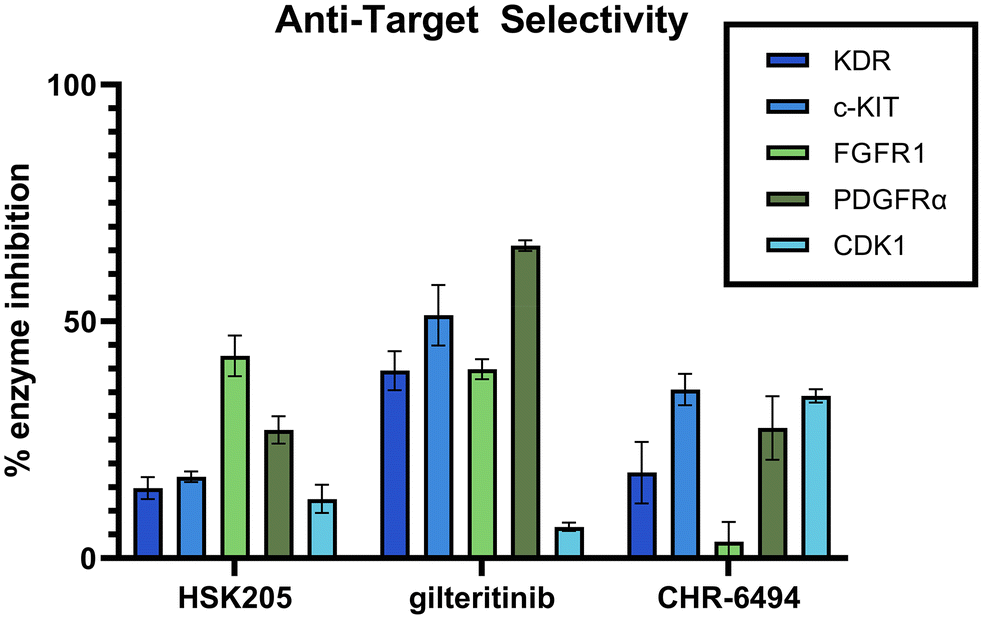 | ||
| Fig. 7 Selectivity kinase inhibition for known anti-targets. HSK205 was screened at 100 nM against various specified kinases via the ADP Glo Kinase Assay. Known FLT3 inhibitor, gilteritinib, and known haspin inhibitor, CHR-6494, were used as positive controls. Results reported are means of triplicates and error bars represent standard deviation. | ||
To move compounds forward for further development, we routinely conduct DNA intercalation assays to eliminate compounds with high affinity for DNA. Although HSK205 contains polycyclic rings, which is typically seen as features that enhance DNA intercalation and unwanted toxicity,30,31 it was determined HSK205 does not intercalate into DNA (Fig. 8). Under similar experimental conditions, mitoxantrone (a known DNA intercalator) was shown to associate with DNA (Fig. 8).
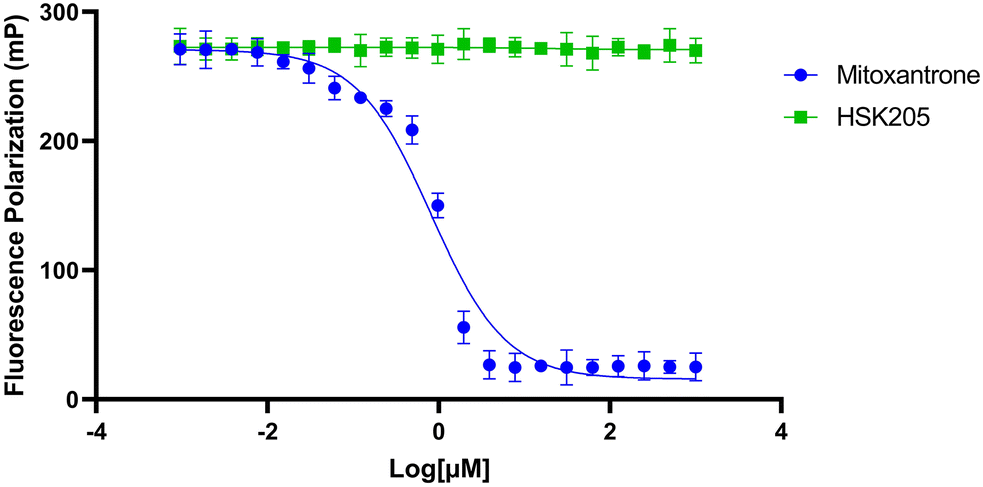 | ||
| Fig. 8 DNA intercalation assay. Mitoxantrone was used as a positive control and DMSO as a negative control. Compounds, including mitoxantrone, were tested over a range of doses from 1 mM to 10 nM. Data was fitted to a nonlinear regression curve using GraphPad Prism 9.0 software. Each data point represents the mean of triplicates and error bars represent standard deviation. | ||
3. Conclusions
In this report we demonstrate that the recently reported protein kinase hinge binder, 3H-pyrazolo[4,3-f]quinoline, which is readily synthesized via a multicomponent reaction potently inhibit FLT3 and haspin. Our lead compound, HSK205, showed remarkable potencies in vitro and in cells against both haspin and FLT3. HSK205 inhibited the proliferation of AML cell lines harboring FLT3-ITD-D835Y or F691L, which are resistant to many of the reported FLT3 inhibitors. Additionally, HSK205 showed limited activity against select anti-targets, such as c-Kit and KDR.4. Experimental section
4.1 Chemistry
Unless noted otherwise, all reagents and solvents were purchased from commercial sources and used as received. The 1H and 13C NMR spectra were obtained in DMSO-d6 as solvent using a 500 MHz spectrometer or 800 MHz spectrometer with Me4Si as an internal standard. Chemical shifts are reported in parts per million (δ) and are calibrated using residual undeuterated solvent as an internal reference. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, or combinations thereof. High resolution mass spectra (HRMS) were obtained using electron spray ionization (ESI) technique and a TOF mass analyzer. New compounds were characterized by 1H NMR, 13C NMR, and HRMS data, and purities of final compounds were reported using HPLC.4.2 Biological evaluation
4.3 Molecular docking
The crystal structure for haspin (PDB: 2WB8) was downloaded from the Protein Data Bank and edited to remove any existing bound ligand within the protein active site. Two-dimensional ligands were built in ChemDraw 21.0.0 and were transferred to three-dimensional mol2 files using OpenBabel. Docking was performed using the Glide software by Schrödinger. Waters were removed before docking into haspin. Docking trial validation for the haspin kinase was based off the previously generated docking pose of HSD972. Visualization was performed through PyMol and Maestro.Author contributions
HOS provided conceptualization. Investigation was done by ALK and NRB. ALK wrote original draft. Writing, review, and editing was performed by NRB and HOS. HOS provided funding acquisition and supervision.Conflicts of interest
HOS holds shares in KinaRx Inc., a company with interest in oncology.Acknowledgements
This study was supported by the National Institutes of Health (1R01CA267978-01) and Purdue University.Notes and references
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- M. Lin and B. Chen, Drug Des., Dev. Ther., 2018, 12, 1009–1017 CrossRef CAS PubMed.
- S. Scholl, M. Fleischmann, U. Schnetzke and F. H. Heidel, Cells, 2020, 9, 2493 CrossRef CAS PubMed.
- A. S. Alotaibi, M. Yilmaz, R. Kanagal-Shamanna, S. Loghavi, T. M. Kadia, C. D. DiNardo, G. Borthakur, M. Konopleva, S. A. Pierce, S. A. Wang, G. Tang, V. Guerra, B. Samra, N. Pemmaraju, E. Jabbour, N. J. Short, G. C. Issa, M. Ohanian, G. Garcia-Manero, K. N. Bhalla, K. P. Patel, K. Takahashi, M. Andreeff, J. E. Cortes, H. M. Kantarjian, F. Ravandi and N. Daver, Cancer Discovery, 2021, 2, 125–134 CrossRef CAS PubMed.
- P. M. Kasi, M. R. Litzow, M. M. Patnaik, S. K. Hashmi and N. Gangat, Leuk. Res. Rep., 2016, 5, 7–10 Search PubMed.
- L. K. Schmalbrock, A. Dolnik, S. Cocciardi, E. SträngStr, F. Theis, N. Jahn, E. Panina, T. J. Bïatte, J. Herzig, S. Skambraks, F. G. Rücker, V. I. Gaidzik, P. Paschka, W. Fiedler, H. R. Salih, G. Wulf, T. Schroeder, R. F. Schlenk, F. Thol, M. Heuser, R. A. Larson, A. Ganser, H. G. Stunnenberg, S. Minucci, R. M. Stone, C. D. Bloomfield and L. Bullinger, Blood, 2021, 137, 3093–3104 CrossRef CAS PubMed.
- K. A. Minson, C. C. Smith, A. B. Lee-Sherick, D. DeRyckere, E. Lasater, A. A. Hill, X. Wang, S. V. Frye, H. S. Earp, N. P. Shah and D. K. Graham, Blood, 2014, 124, 3757 CrossRef.
- S. C. Yen, L. C. Chen, H. L. Huang, W. C. HuangFu, Y. Y. Chen, T. Eight Lin, S. T. Lien, H. J. Tseng, T. Y. Sung, J. H. Hsieh, W. J. Huang, S. L. Pan and K. C. Hsu, Bioorg. Chem., 2022, 121, 105675 CrossRef CAS PubMed.
- K. C. Goh, W. C. Ong, C. Hu, A. L. Liang, W. Stunkel, Y. C. Tan, K. Sangthongpitag, S. K. Goh, Z. Q. Bonday, A. D. William, B. W. Dymock, E. Kantharaj and J. M. Wood, Blood, 2007, 110, 1593 CrossRef.
- N. Dayal, C. Opoku-Temeng, D. E. Hernandez, M. A. Sooreshjani, B. A. Carter-Cooper, R. G. Lapidus and H. O. Sintim, Future Med. Chem., 2018, 10, 823–835 CrossRef CAS PubMed.
- W. Czardybon, R. Windak, A. Gołas, M. Gałęzowski, A. Sabiniarz, I. Dolata, M. Salwińska, P. Guzik, M. Zawadzka, E. Gabor-Worwa, B. Winnik, M. Żurawska, E. Kolasińska, E. Wincza, M. Bugaj, M. Danielewicz, E. Majewska, M. Mazan, G. Dubin, M. Noyszewska-Kania, E. Jabłońska, M. Szydłowski, T. Sewastianik, B. Puła, A. Szumera-Ciećkiewicz, M. Prochorec-Sobieszek, E. Mądro, E. Lech-Marańda, K. Warzocha, J. Tamburini, P. Juszczyński and K. Brzózka, Oncotarget, 2018, 9, 16917–16931 CrossRef PubMed.
- D. Taussig, J. O'Nions, M. Jongen-Lavrencic, J. J. Janssen, D. C. de Leeuw, P. T. Tan, S. Brook, K. Stoddart, H.-T. Arkenau and G. Fisher, Blood, 2022, 140, 6222–6223 CrossRef.
- M. U. Tariq, M. Furqan, H. Parveen, R. Ullah, M. Muddassar, R. S. Z. Saleem, V. Bavetsias, S. Linardopoulos and A. Faisal, Br. J. Cancer, 2021, 125, 966–974 CrossRef CAS PubMed.
- Z. Li, X. Wang, J. Eksterowicz, M. W. Gribble Jr, G. Q. Alba, M. Ayres, T. J. Carlson, A. Chen, X. Chen, R. Cho, R. V. Connors, M. DeGraffenreid, J. T. Deignan, J. Duquette, P. Fan, B. Fisher, J. Fu, J. N. Huard, J. Kaizerman, K. S. Keegan, C. Li, K. Li, Y. Li, L. Liang, W. Liu, S. E. Lively, M.-C. Lo, J. Ma, D. L. McMinn, J. T. Mihalic, K. Modi, R. Ngo, K. Pattabiraman, D. E. Piper, C. Queva, M. L. Ragains, J. Suchomel, S. Thibault, N. Walker, X. Wang, Z. Wang, M. Wanska, P. M. Wehn, M. F. Weidner, A. J. Zhang, X. Zhao, A. Kamb, D. Wickramasinghe, K. Dai, L. R. McGee and J. C. Medina, J. Med. Chem., 2014, 57, 3430–3449 CrossRef CAS PubMed.
- N. G. Amoussou, A. Bigot, C. Roussakis and J. M. H. Robert, Drug Discovery Today, 2018, 23, 409–415 CrossRef CAS PubMed.
- M. Liu, S. A. Stoner, A. G. Davis and D.-E. Zhang, Blood, 2022, 140, 2997–2998 CrossRef.
- N. Dayal, E. Řezníčková, D. E. Hernandez, M. Peřina, S. Torregrosa-Allen, B. D. Elzey, J. Škerlová, H. Ajani, S. Djukic, V. Vojáčková, M. Lepšík, P. Řezáčová, V. Kryštof, R. Jorda and H. O. Sintim, J. Med. Chem., 2021, 64, 10981–10996 CrossRef CAS.
- N. Dayal, C. Opoku-Temeng, D. E. Hernandez, M. A. Sooreshjani, B. A. Carter-Cooper, R. G. Lapidus and H. O. Sintim, Future Med. Chem., 2018, 10, 823–835 CrossRef CAS PubMed.
- N. Dayal, C. G. Mikek, D. Hernandez, G. A. Naclerio, E. F. Yin Chu, B. A. Carter-Cooper, R. G. Lapidus and H. O. Sintim, Eur. J. Med. Chem., 2019, 180, 449–456 CrossRef CAS.
- C. Opoku-Temeng, N. Dayal, D. E. Hernandez, N. Naganna and H. O. Sintim, Chem. Commun., 2018, 54, 4521–4524 RSC.
- C. Opoku-Temeng, N. Dayal, M. Aflaki Sooreshjani and H. O. Sintim, Bioorg. Chem., 2018, 78, 418–426 CrossRef CAS.
- P. L. Nguyen, C. H. Lee, H. Lee and J. Cho, Antioxidants, 2022, 11, 117 CrossRef CAS PubMed.
- J. Eswaran, D. Patnaik, P. Filippakopoulos, F. Wang, R. L. Stein, J. W. Murray, J. M. G. Higgins and S. Knapp, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20198–20203 CrossRef CAS PubMed.
- P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS PubMed.
- A. A. Warkentin, M. S. Lopez, E. A. Lasater, K. Lin, B.-L. He, A. Y. Leung, C. C. Smith, N. P. Shah and K. M. Shokat, eLife, 2014, 3, e03445 CrossRef PubMed.
- A. Kommalapati, S. H. Tella, M. Borad, M. Javle and A. Mahipal, Cancers, 2021, 13, 2968 CrossRef CAS PubMed.
- T. Force, D. S. Krause and R. A. Van Etten, Nat. Rev. Cancer, 2007, 7, 332–344 CrossRef CAS.
- R. M. Touyz and J. Herrmann, npj Precis. Oncol., 2018, 2, 13 CrossRef PubMed.
- R. Prevo, G. Pirovano, R. Puliyadi, K. J. Herbert, G. Rodriguez-Berriguete, A. O'Docherty, W. Greaves, W. G. McKenna and G. S. Higgins, Cell Cycle, 2018, 17, 1513–1523 CrossRef CAS.
- F. P. Guengerich, Base Intercalation in DNA, in Molecular Life Sciences, ed. R. D. Wells, J. S. Bond, J. Klinman and B. S. S. Masters, Molecular Life Sciences, Springer, New York, NY, 2018, DOI:10.1007/978-1-4614-1531-2_432.
- K. Gurova, Future Oncol., 2009, 5, 1685–1704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00192j |
| This journal is © The Royal Society of Chemistry 2023 |