
Electrocatalytic amino acid synthesis from biomass-derivable keto acids over ball milled carbon nanotubes†
Yiying
Xiao
 ab,
Chia Wei
Lim
b,
Jinquan
Chang
ab,
Qixin
Yuan
b,
Lei
Wang
*b and
Ning
Yan
*ab
ab,
Chia Wei
Lim
b,
Jinquan
Chang
ab,
Qixin
Yuan
b,
Lei
Wang
*b and
Ning
Yan
*ab
aJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China. E-mail: ning.yan@nus.edu.sg
bDepartment of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585, Singapore. E-mail: wanglei8@nus.edu.sg
First published on 18th March 2023
Abstract
Electrocatalytic reductive amination (ERA) offers an attractive way to synthesise organonitrogen chemicals from renewable feedstocks. Here, we report carbon nanotubes (CNTs) as an effective catalyst for the ERA of biomass-derivable α-keto acids into amino acids using NH3 as the nitrogen source. Through a facile ball milling (BM) treatment, the intrinsic defects in the CNTs were increased while the electrocatalytic activity of CNTs converting 2-ketoglutaric acid into glutamic acid was enhanced by approximately seven times. A high faradaic efficiency (FE) of ∼90% with a corresponding glutamic acid formation rate of up to 180.9 mmol g−1cat h−1 was achieved, and ∼60% molar yield of glutamic acid was obtained after 8 h of electrolysis. Electrokinetic analyses indicated that the BM–CNT catalysed ERA exhibits a first-order dependence on the substrate and NH3, with a rate-determining step (RDS) involving the first electron transfer. Following this protocol, a number of amino acids were prepared with moderate to high FEs and formation rates. Significantly, we synthesised long carbon chain amino acids, which are typically obtained in lower yields using the existing methods.
Introduction
Amino acids are essential compounds used in a myriad of industrial applications, including human food, animal feed, pharmaceutical products, bio-based polymer synthesis and so on.1–3 Currently, amino acid production mainly relies on microbial fermentation processes, which suffer from a long culturing time, complicated and energy intensive separation/purification, and the need for single-use enzymes that can be costly.4,5 Chemical methods such as the Strecker reaction,6 the Gabriel synthesis,7 and the Miller experiment8 may offer higher space–time productivities, but they usually employ toxic feedstocks or harsh reaction conditions, hindering practical applications.The construction of the C–N bond through thermocatalytic reductive amination provides amino acids from keto acids and a nitrogen source under comparatively mild conditions.9,10 This pathway involves a two-step procedure as illustrated in Scheme 1: a carbonyl compound and a nitrogen source (e.g., NH3) first condense to form an imine intermediate which is subsequently reduced to afford an amine product, while the direct reduction of the carbonyl compound is usually the competing side reaction. The selective reduction of imines can be achieved by using reducing agents such as formates,11,12 silanes,13 and hydrides,14–16 which generate organic waste and require solvent clean-up. Molecular H2 is also commonly used for reductive amination in the presence of Ni-based catalysts17,18 or noble metal-based catalysts, including Pt,19 Pd,20 Ru,21,22 Rh,23 and Ir.24 Although more environmentally friendly than other reductants, H2 still has several disadvantages of (1) needing to be generated from other energy sources; (2) requiring relatively severe reaction conditions (e.g., 10 bar H2, 80 to 150 °C), and (3) being able to hydrogenate other functional groups such as carbonyl groups or aromatic rings depending on the property of the catalysts.
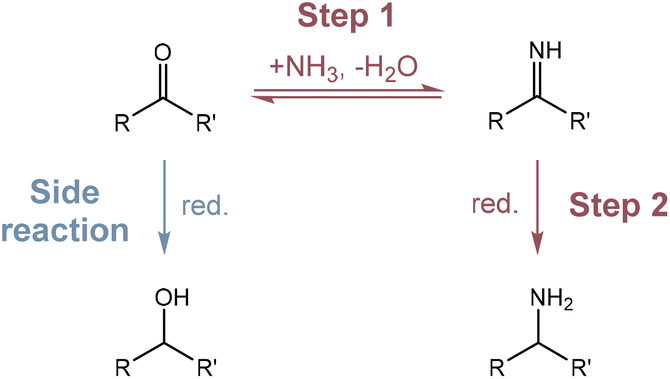 | ||
| Scheme 1 The reaction pathway of reductive amination. Step 1: condensation of a carbonyl compound and a nitrogen source to form an imine intermediate. Step 2: reduction of the imine intermediate to afford an amine product. Side reaction: reduction of the carbonyl compound to form a hydroxyl compound. | ||
In recent years, the electrochemical valorisation of biomass derivatives to value-added chemicals has emerged as a new frontier.25–31 As an example, electrocatalytic reductive amination (ERA), occurring via the pathway shown in Scheme 1, has several advantages. Similar to other well studied electrocatalytic reactions including CO2/CO reduction,32–36 N2 reduction37–40 and O2 reduction41–43 reactions, it proceeds under environmentally benign conditions, using electrons as reductants and water as the hydrogen source. Additionally, the selectivity of ERA can be facilely controlled by altering the applied potentials. As an early example, Meisters et al. prepared several amino acids from the corresponding α-keto acids in aqueous NH3/NH4Cl solution, with yields ranging from 24% to 88% after long-time constant potential electrolysis.44,45 Recently, Choi et al. investigated the ERA of 5-hydroxymethylfurfural (5-HMF) with methylamine, achieving high faradaic efficiency (FE) and selectivity by using a high surface area Ag electrode.46 In 2021, Palkovits and co-workers discovered that Cu was a highly selective cathode material to convert levulinic acid to 1,5-dimethyl-2-pyrrolidone in 78% yield through ERA and dehydration.47 These works often relied on toxic and/or expensive metals as the main component in the catalyst. For metal oxide based catalyst, Yamauchi et al. synthesised seven amino acids with FEs of 77–99% using TiO2, but hydroxylamine (NH2OH) was used as the nitrogen source, which is more poisonous and costly than NH3.48,49 Interestingly, Sun et al. demonstrated that ERA using the same catalyst could be combined with alcohol oxidation to obtain amino acids directly from alcohols.50
Carbon materials have been recognised as prospective catalysts in diverse electrochemical processes including O2 reduction,51–53 CO2 reduction,54–57 and organic electrosynthetic reactions.58,59 The electrocatalytic reactivity of carbon materials is tuneable by defect engineering, heteroatomic doping, and surface functional group modification. Herein, we have extended the electrocatalytic application of carbon nanotubes to convert biomass-derivable α-keto acids into the corresponding amino acids with moderate to high formation rates and FEs (Fig. 1). Ball milling (BM) treatment has been identified as an effective strategy to introduce intrinsic defects into CNTs to substantially boost the activity of ERA. We also systematically investigated the influences of applied potentials and substrate/ammonia concentrations on the FE and formation rate of glutamic acid, as well as catalyst recycling.
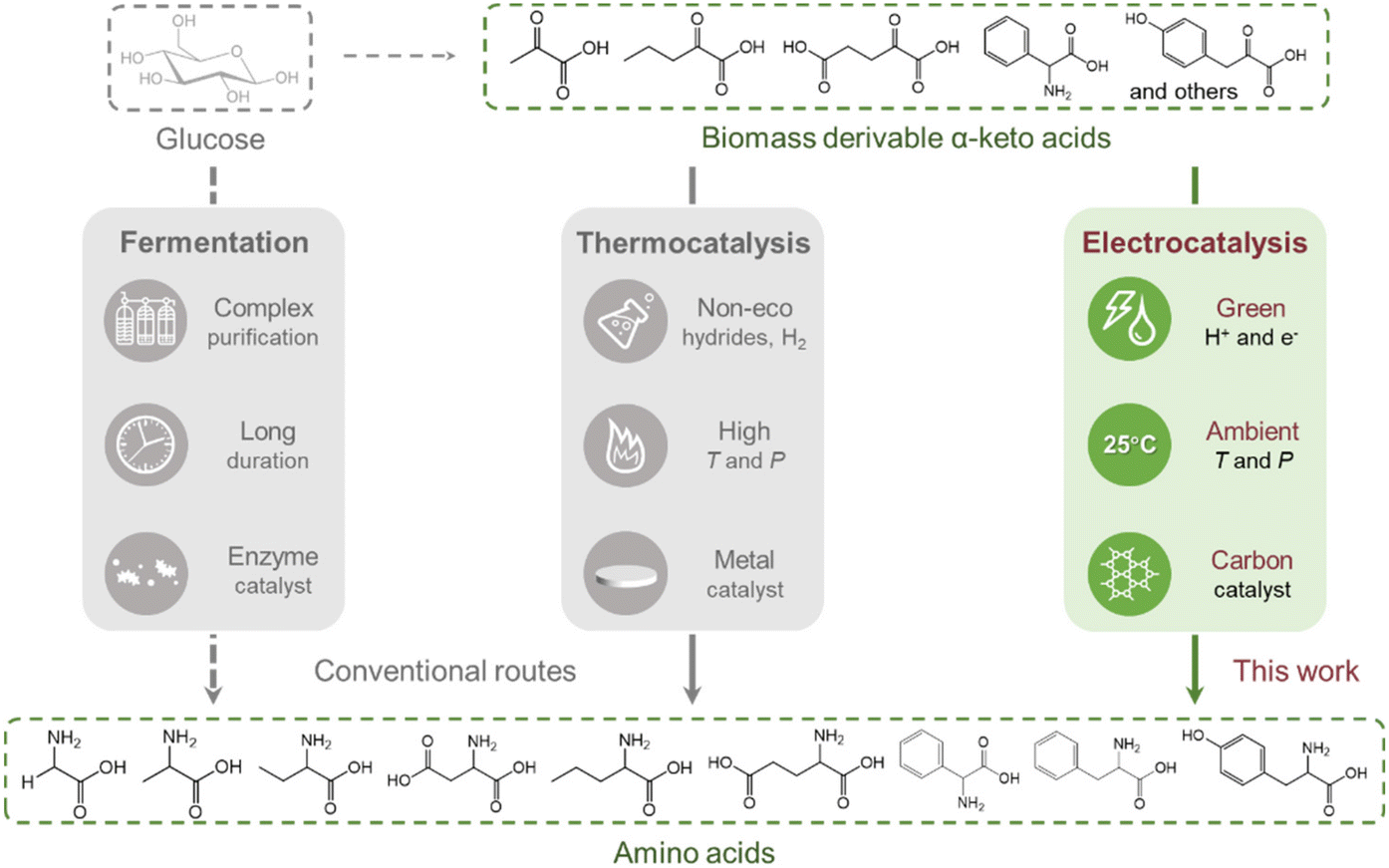 | ||
| Fig. 1 Comparison of the amino acid synthesis protocols via bio-, thermal-, and electrocatalytic routes. | ||
Materials and methods
Chemicals and materials
Multi-walled carbon nanotubes (bulk CNTs, MA-EN-CO-0R0113) were purchased from Canrd Co., Ltd. N-doped multi-walled carbon nanotubes (N–CNTs, XFM63) and amino-modified multi-walled carbon nanotubes (CNT–NH2, XFM62) were purchased from Nanjing XFNANO Materials Tech Co., Ltd. The specifications of the CNT materials are provided (Table S3†). Carbon paper (TGP-H-060) was purchased from Suzhou Sinero Tech Co., Ltd. The Ag/AgCl electrode (3 M KCl) and graphite rod were purchased from Shanghai Jingchong Electronic Technology Development Pte. Ltd. The Nafion 117 membrane (N117, DuPont) was purchased from Xianfeng Instrument Tech Co., Ltd. Nitrogen gas (N2, 99.9995%) was supplied by Air Liquide. Sodium chloride (NaCl, ≥99.0%), silver nitrate (AgNO3, ≥99.0%), Nafion® perfluorinated resin solution (5 wt% in 45% water balanced with lower aliphatic alcohols), ammonium sulfate ((NH4)2SO4, ≥99.0%), ammonia solution (∼25 wt% NH3 in water), pyruvic acid (98%), glyoxylic acid solution (50 wt% in water), 2-ketobutyric acid (97%), oxaloacetic acid (97%), 2-oxovaleric acid (98%), 2-ketoglutaric acid (99.0–101.0%), phenylglyoxylic acid (97%), sodium phenylpyruvate, 4-hydroxyphenyl pyruvic acid (98%), L-glutamic acid (≥99.0%) and 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (97%) were supplied by Sigma-Aldrich. Nitric acid (HNO3, 65%) and absolute ethanol (>99.7%) were supplied by VWR Chemicals. Acetone (HPLC grade) was purchased from Fisher Chemical. Deuterium oxide (D2O, D, 99.9%) was purchased from Cambridge Isotope Laboratories. Except for bulk CNTs, N–CNTs and CNT–NH2, commercially available chemicals were used as received without purification. All aqueous solutions were prepared using ultra-pure water (Milli-Q®, conductivity of 0.056 μS cm−1).Catalyst preparation
Bulk CNTs, N–CNTs and CNT–NH2 were separately sonicated in ultra-pure water and acetone (three times in each solvent) to remove impurities. After the removal of the supernatant by centrifugation, the precipitate was dried at 80 °C overnight. In a typical fabrication procedure of ball-milled CNTs (BM–CNTs), 0.1 g of bulk CNT powder together with 0.5 g of NaCl and 10 g of zirconium oxide (ZrO2) balls was introduced to a zirconium oxide ball-mill chamber (45 mL). Ball milling (Pulverisette 7, Fritsch) was carried out at 300 rpm for 1, 2, 3, 4 and 5 hours. The ball milling process was paused for 10 min after every 20 min of grinding to avoid overheating. After ball milling, the powder mixture was separated from the balls using a steel mesh. It was washed with ultra-pure water and vacuum-filtered until there was no precipitate in the filtrate when AgNO3 was added. Then, the collected powder was oven-dried at 80 °C overnight, resulting in the BMX h–CNT (where X represents the number of hours of the ball milling process).Electrochemical measurements
Carbon paper cut into 1.5 × 3 cm was pre-treated by immersing it in 1 M nitric acid and sonicating for 30 min. Following this step, the carbon paper was washed thoroughly with ultra-pure water and absolute ethanol, and then dried at 80 °C overnight. To prepare the catalyst ink, 4.5 mg of the prepared catalyst was dispersed in a mixture of 1.2 mL of absolute ethanol and 10 μL of Nafion® perfluorinated resin solution and then sonicated for 1.5 hours to form a homogeneous suspension. The working electrode was prepared by dropping the catalyst ink onto both sides of a pre-treated carbon paper.All the electrochemical measurements were performed with an electrochemical workstation (CHI 760E, CH Instruments Ins), except for the electrochemical impedance spectroscopy (EIS) tests, which were carried out using a Gamry Reference 3000 potentiostat. The measurements were carried out in a two-compartment (H-type) electrochemical cell with a three-electrode configuration at room temperature under stirring, except for the cyclic voltammetry (CV) tests which were conducted without stirring. A graphite rod and an Ag/AgCl electrode (3 M KCl) were used as the counter and reference electrodes, respectively. NH3/(NH4)2SO4 buffers ([NH3]/[NH4+] = 1, pH 10) with and without organic substrates were used as the cathodic and anodic electrolyte solutions, respectively. The two compartments (50 mL each), each containing 40 mL of electrolytes, were sealed with Teflon caps and separated with a piece of Nafion 117 membrane (Fig. S18†). In all cases, the cathodic electrolyte solution was purged with purified nitrogen gas for 30 min at the start of each experiment to remove atmospheric air. During the measurements, nitrogen gas was continuously bubbled into the cathodic electrolyte. Every measurement, except for the stability test, was carried out on a freshly prepared working electrode. The potentials applied against the Ag/AgCl reference electrode were converted to the iR-corrected reversible hydrogen electrode (RHE) scale on the basis of the following equation:
| ERHE (V) = EAg/AgCl (V) + 0.210 V + 0.059 V × pH−iR |
The electrosynthesis of amino acids was carried out at a constant potential and investigated systematically using different working electrodes, potentials, substrate concentrations, ammonia concentrations and substrates. The linear sweep voltammetry (LSV) measurements were conducted from −0.6 V to −1.45 V versus Ag/AgCl at a scan rate of 20 mV s−1 with and without substrates with different working electrodes. The CV measurements were performed from 0.2 V to −1.35 V versus Ag/AgCl at scan rates ranging from 10 to 200 mV s−1 under various conditions. The potentiostatic EIS measurements were performed at frequencies ranging from 106 to 0.1 Hz with an RMS amplitude of 10 mV at −1.1 V versus Ag/AgCl.
Product identification and quantification
The reaction solution collected from the cathodic compartment was analysed by 1H nuclear magnetic resonance (NMR) spectroscopy using a Bruker Ascend™ 400 (400 MHz) at room temperature with water suppression. The anodic compartment was also analysed, confirming that the Nafion 117 membrane was in a good condition. Typically, 300 μL of the electrolyte was mixed with 300 μL of deuterium oxide containing 20 mM 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt, which was used as an internal standard. The amino acids and corresponding hydroxy acids were identified from the signals at approximately 3.4–3.8 ppm and 4.0–4.2 ppm assignable to the proton on the α-carbon of the amino acids and hydroxy acids, respectively (Fig. S11,† all presented data are the average results of 8 scans). For quantification, the concentration of each compound could be calculated from the equation of the calibration curve (Fig. S12† as an example) since the concentration of amino acids or hydroxy acids exhibits a linear relationship with the integral area of the characteristic peak. More specifically, the integral value of the peak corresponding to the product was obtained assuming that the integral value of the internal standard peak was 1.0000.The faradaic efficiency (FE) for each compound was calculated as follows:
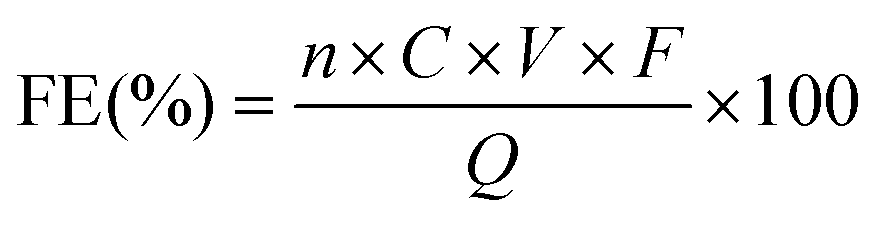
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and Q represents the total charge passed during the electrolysis.
485 C mol−1), and Q represents the total charge passed during the electrolysis.
Material characterisation
The defective level of the CNT materials was determined using a Raman microscope (HORIBA Jobin Yvon) with a 633 nm excitation laser. The chemical environments of the CNT materials were identified by X-ray photoelectron spectroscopy (XPS, Kratos AXIS UltraDLD, Kratos Analytical Ltd) with a mono Al Kα X-ray source. All XPS data were calibrated to C 1s (C–C bond) at 284.80 eV and analysed using the Vision Processing and XPSPEAK Version 4.1 software. X-ray diffraction (XRD) analysis was performed using a diffractometer (Bruker D8 Advance), where 2θ ranged from 10° to 80° at a scan speed of 5° min−1 with a step size of 0.02°, to study the crystal structure of the CNT materials. The surface morphology and microstructure of the CNT materials were characterised using a field emission scanning electron microscope (FESEM, JEOL JSM-7610F) operating at 5 kV and a field emission transmission electron microscope (FETEM, JEOL JEM-2100F) operating at 200 kV. The specific surface areas of different CNT materials were measured by nitrogen adsorption/desorption based on the Brunauer–Emmett–Teller (BET, NOVAtouch NT 4LX-1) method. About 200 mg of samples were degassed in nitrogen at 150 °C for 6 hours before analysis. The contact angles for carbon paper and BM3 h–CNT covered carbon paper were measured using a Krüss G10 contact angle measurement system.Results and discussion
Characterisation of the electrocatalysts
The surface morphology of CNTs was characterised before and after the ball milling treatment by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). As illustrated in the TEM image (Fig. 2a), most of the bulk CNTs were curved, entangled and twisted, together with the end tips almost closed. After 3 h of ball milling (sample denoted as BM3 h–CNT), a large portion of CNTs was cut shorter with open ends, while plenty of individual CNTs were separated from the agglomerates (Fig. 2b). It could also be observed that the destroyed walls were kinked and discontinuous with a multilayer polyaromatic carbon structure present in certain parts (Fig. 2b, arrow and Fig. S1 and S3†), implying structural defects and graphitic disorder in the CNT sample. Consistent with the TEM results, the SEM images show that the agglomeration of CNTs was markedly reduced after ball milling, while amorphous carbon was sporadically observed on the surface of CNTs (Fig. S2†).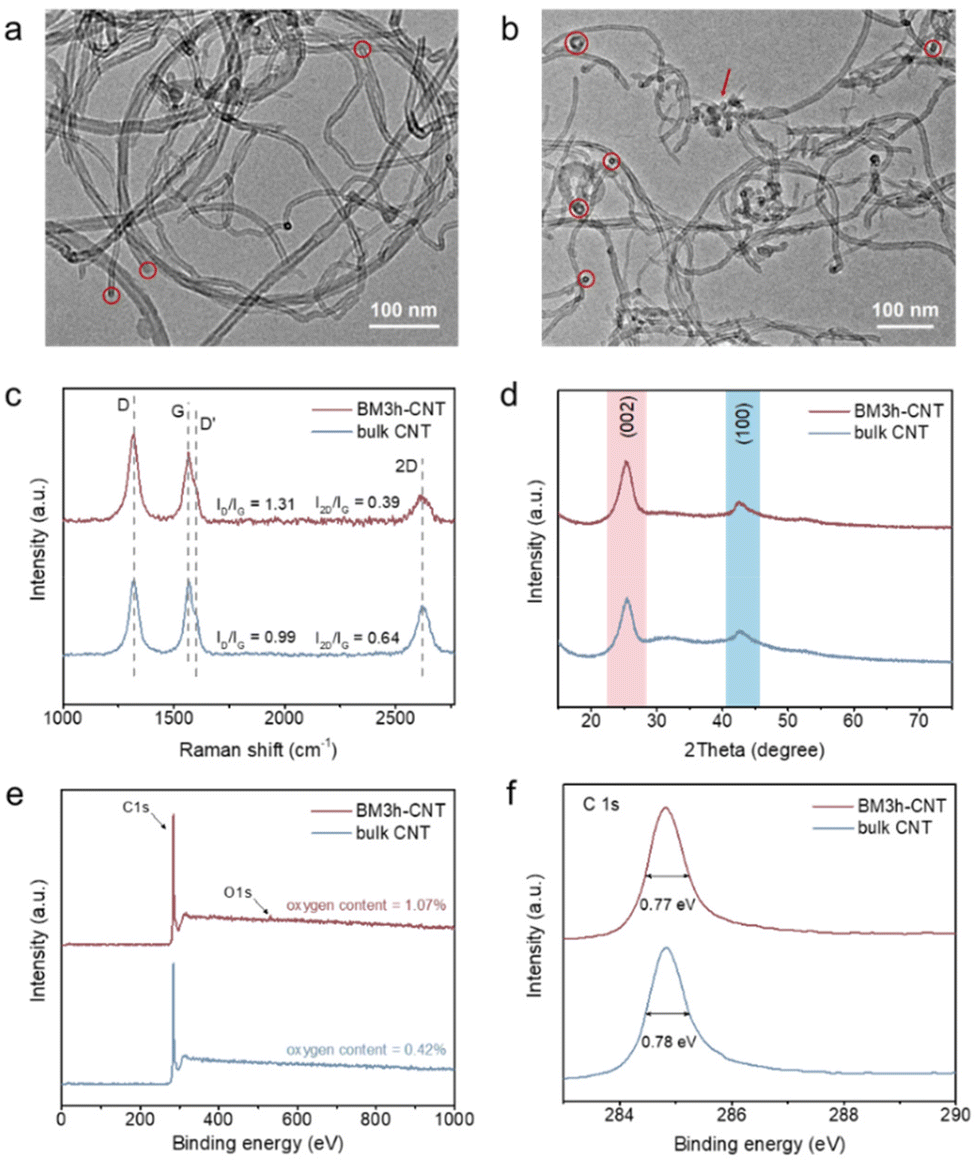 | ||
| Fig. 2 Characterisation of pristine and ball milled CNT. TEM images of (a) bulk CNT and (b) BM3 h–CNT. The red circles and arrows highlight the end tips and amorphous structure of the CNTs, respectively. (c) Raman spectra of bulk CNT and BM3 h–CNT. The values of ID/IG and I2D/IG are averages calculated based on three independent measurements. (d) XRD patterns of bulk CNT and BM3 h–CNT. The peaks at around 25.4° and 42.6° are attributed to the (002) graphite plane reflection and the (100) in-plane reflection, respectively. (e) XPS survey spectra of bulk CNT and BM3 h–CNT. (f) XPS high resolution C 1s spectra of bulk CNT and BM3 h–CNT. | ||
In the Raman spectra, four characteristic peaks were identified at around 1321 cm−1, 1602 cm−1, 1566 cm−1 and 2620 cm−1, which correspond to the disordered D and D′ bands, graphitic G band and graphene-layer-sensitive 2D band, respectively (Fig. 2c). The intensity ratio of the D band to the G band (ID/IG), reflecting the defective level of carbon materials,60 increased from 0.99 in bulk CNTs to 1.31 in BM3 h–CNTs, demonstrating the existence of a more defective structure and disordered phase in the latter. Additionally, the declining 2D/G intensity ratios (I2D/IG) from 0.64 to 0.39 after ball milling suggests that there were less graphene layers in BM3 h–CNTs compared with those in bulk CNTs,61 which is consistent with the observation in the high resolution TEM images (Fig. S3†).
The XRD patterns of the samples exhibit two characteristic peaks of CNT materials. There were no observable changes in the XRD patterns after ball milling (Fig. 2d and Fig. S4†), indicating that the ball milling treatment has no significant effects on the crystal structure of the CNTs. As displayed in the XPS survey spectra (Fig. 2e), only C and a small amount of O (oxygen contents of 0.42% and 1.07% in bulk CNTs and BM3 h–CNTs, respectively) were detected, demonstrating that the ball milling treatment does not introduce non-oxygen heteroatom elements into CNTs. The high-resolution C 1s spectra of BM3 h–CNTs and untreated CNTs are nearly identical, with similar Full Width at Half Maximum (FWHM) values of 0.77 eV (Fig. 2f). The XPS spectra of CNTs after different durations of ball milling treatment are provided in Fig. S5.†
Electrocatalytic reductive amination of 2-ketoglutaric acid to glutamic acid
We initially examined the characteristics of electrocatalytic reductive amination by linear sweep voltammetry (LSV) using 2-ketoglutaric acid as a model compound. The polarisation curve in the absence of 2-ketoglutaric acid was ascribed to the hydrogen evolution reaction (HER) (Fig. 3a, grey line). Upon the addition of 2-ketoglutaric acid, there was a dramatic shift in the onset potential in the positive direction together with a steep increase in the current density, indicating that the electrocatalytic reductive amination is favoured over HER on CNTs. Compared to bulk CNTs (blue line), BM3 h–CNTs exhibited substantially enhanced electrocatalytic performance reflected by the much higher cathodic current density (violet line). It is also worth emphasising that two irreversible reduction peaks, around −0.2 V vs. reversible hydrogen electrode (RHE) and −0.5 V vs. RHE, were observed in the cyclic voltammetry (CV) curves when 2-ketoglutaric acid was added (Fig. 3b), attributable to the reduction of the 2-iminoglutaric acid intermediate (forming the target compound, glutamic acid) and 2-ketoglutaric acid (forming the side product, 2-hydroxyglutaric acid), respectively.44,62–64 This suggests that the selectivity towards glutamic acid or 2-hydroxyglutaric acid can be effectively tuned by altering the applied potentials when ball milled CNTs are used as the electrode.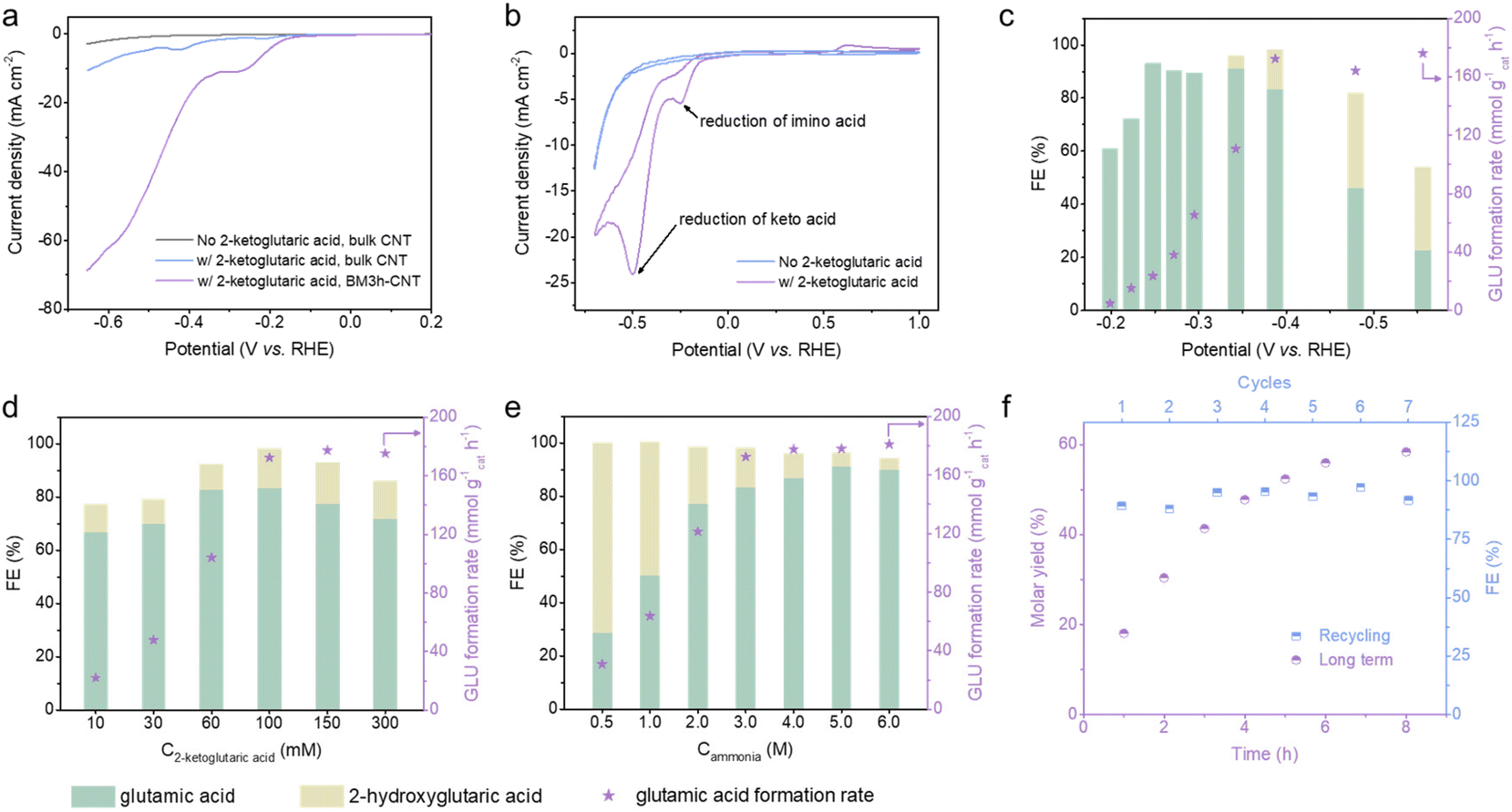 | ||
| Fig. 3 Electrocatalytic reductive amination of biomass-derivable 2-ketoglutaric acid to glutamic acid. (a) LSV curves of bulk CNTs without (grey line) and with (blue line) 100 mM 2-ketoglutaric acid, and BM3 h–CNTs with 100 mM 2-ketoglutaric acid (violet line) in a 3 M NH3/NH4+ buffer (pH 10) at a scan rate of 20 mV s−1 at room temperature. (b) CV curves of BM3 h–CNT without (blue line) and with (violet line) 100 mM 2-ketoglutaric acid in a 3 M NH3/NH4+ buffer (pH 10) at a scan rate of 10 mV s−1 at room temperature. The FE and GLU formation rate in the BM3 h–CNT catalysed room-temperature constant potential electrolysis of 2-ketoglutaric acid for 1 h at different (c) applied potentials (100 mM 2-ketoglutaric acid, 3 M NH3, pH 10), (d) 2-ketoglutaric acid concentrations (−0.39 V vs. RHE, 3 M NH3, pH 10) and (e) NH3 concentrations (−0.39 V vs. RHE, 100 mM 2-ketoglutaric acid, pH 10). (f) Extended duration room-temperature electrolysis (violet dots) and recycling test (blue dots) over BM3 h–CNTs with 100 mM 2-ketoglutaric acid, 6 M NH3 (pH 10) at −0.39 V vs. RHE. For recycling experiments, the reaction time was 1 h. | ||
Next, the effects of different applied potentials, substrate/ammonia concentrations and pH on the FE and glutamic acid formation rate (GLU formation rate) were investigated (Fig. 3c–e, Fig. S8 and S9†). As the applied potential varied from −0.2 to −0.6 V vs. RHE, the formation rate of glutamic acid first increased from 4.6 to 172.4 mmol g−1cat h−1 and began to level off when the potential was more negative than −0.39 V vs. RHE (Fig. 3c). In the range of −0.2 to −0.3 V vs. RHE, only glutamic acid was detectable, with the FE showing an upward trend as the potential became more negative and reaching its highest value (93.1%) at −0.25 V vs. RHE. In agreement with earlier CV analyses, the FE towards glutamic acid declined sharply at potentials more negative than −0.39 V vs. RHE, while the FE for 2-hydroxyglutaric acid displayed a significant increase as the keto acid reduction became more favourable.
The impact of reactant concentrations (C2-ketoglutaric acid and Cammonia) on glutamic acid synthesis was investigated at −0.39 V vs. RHE (Fig. 3d and e). As the concentration of 2-ketoglutaric acid increased, the glutamic acid formation rate showed a remarkable increase before increasing at a slower pace. The plateau of the glutamic acid formation rate can be rationalised by the possibility that the active sites on the cathode were saturated by the reactants. There was a moderate decline of FE for glutamic acid when the concentration of 2-ketoglutaric acid was higher than 100 mM (Fig. 3d). Possible reasons for the drop in FE include the formation of dimers or oligomers which also participate in reduction reactions (Scheme S1a,† electrons were utilised, but the products were not quantified) and/or the condensation between the substrate and product (Scheme S1b†), based on NMR analysis (Fig. S10†).44,46 Due to the shifted ketone–imine equilibrium to the imino acid side, the FE for glutamic acid showed a sharp increase from 28.8% to 91.2% when the ammonia concentration increased from 0.5 M to 5 M (Fig. 3e). The production of glutamic acid was found to be favoured at high pH, which similarly promotes the shifting of the ketone–imine equilibrium to produce more imino acid (Fig. S8†). Altogether, a high FE of ∼90% and a corresponding formation rate of 180.9 mmol g−1cat h−1 for glutamic acid synthesis were achieved under the optimum reaction conditions: 100 mM 2-ketoglutaric acid, 6 M ammonia and pH 10 at an applied potential of −0.39 V vs. RHE.
An extended duration electrosynthesis was conducted on BM3 h–CNTs under the optimised conditions; nearly 60% molar yield of glutamic acid was obtained after 8 h of electrolysis (Fig. 3f, violet dots). In the recycling experiments, no obvious performance loss was observed even after seven repetitions (Fig. 3f, blue dots), highlighting the durability of the catalyst. Overall, the BM3 h–CNT catalyst exhibited a high current density, FE, GLU formation rate and yield achieved at a relatively low overpotential in a relatively short reaction time, which are comparable or superior to the those of catalysts reported elsewhere44,45,48,49,62 (Table S1†).
Correlation of CNT structure and electrocatalytic reductive amination reactivity
A series of CNTs prepared by varying the ball milling duration and two types of N configurated CNTs (N–CNT and CNT–NH2) were comparatively studied. As displayed in Fig. 4a, the specific surface area (Table S2†) normalised GLU formation rate (denoted as the GLU formation rateBET surface area) increased substantially with increasing milling time between 0 and 3 h, followed by a downward trend when the milling time exceeded 3 h. Decreased electrical conductivity (Fig. 4d) and clear structural deterioration of the CNTs (Fig. S1 and S2†) were observed in the samples after long time ball milling treatment, which are likely to be the reasons for their lower activity. The FE towards glutamic acid increased after the ball milling treatment and remained constant at around 85% with prolonging milling time. No obvious relationships between the catalytic performance and the CNT morphology (Fig. S1 and S2†), crystallinity (Fig. S4†) and functionality (Fig. S5†) were observed. On the other hand, the effect of the defective degree of the CNTs was much more profound on the reaction. In accordance with the trend of the electrocatalytic activity, the D band intensity and the ID/IG ratio of the BM–CNTs increased progressively with increasing milling time (Fig. 4b); the GLU formation rateBET surface area was linearly correlated with the ID/IG ratio of the CNTs (R2 = 0.999, Fig. 4c), implying that the intrinsic carbon defects in CNTs potentially serve as the active sites and contribute to the high activity in ERA.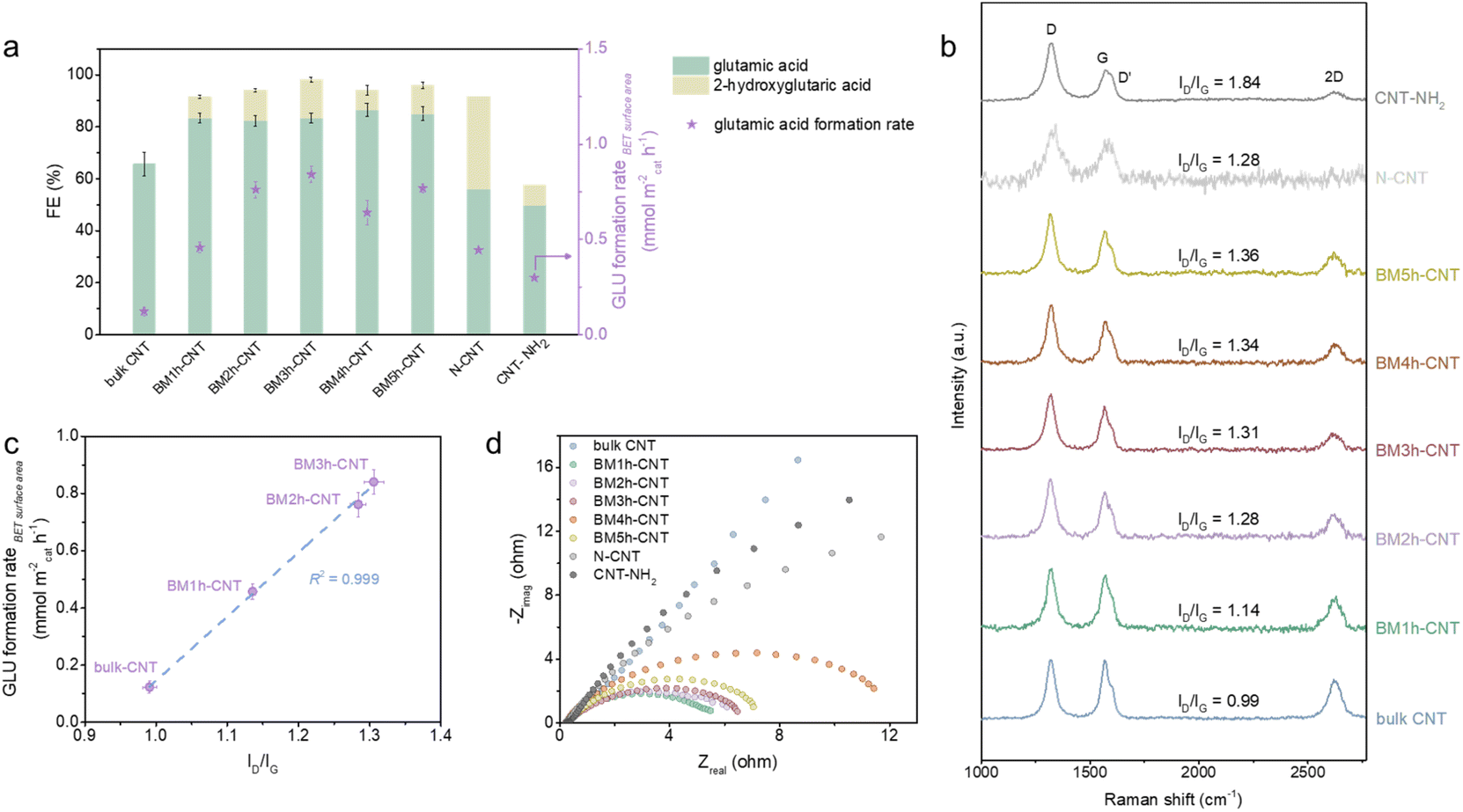 | ||
| Fig. 4 Correlation of glutamic acid synthesis activity and CNT structure. (a) FE and GLU formation rateBET surface area of room-temperature electrolysis of 100 mM 2-ketoglutaric acid and 3 M NH3 (pH 10) at −0.39 V vs. RHE for 1 h with different CNT materials. (b) Raman spectra of various CNT materials. (c) Correlation of CNT defective level ID/IG and surface area normalised glutamic acid formation rate. (d) EIectrochemical Impedance Spectra (EIS) Nyquist plots of various CNT materials at −0.29 V vs. RHE in 100 mM 2-ketoglutaric acid and 3 M NH3/NH4+ buffer (pH 10) solutions. The error bars denote the standard deviation of data from three independent experiments. | ||
Despite having high ID/IG ratios (Fig. 4b), interestingly, the N configurated CNTs exhibited relatively low GLU FE and formation rateBET surface area compared with BM–CNTs (Fig. 4a). N–CNTs even displayed a relatively higher FE towards the by-product, 2-hydroxyglutaric acid, compared with BM–CNTs and CNT–NH2. According to the characterisation results (Fig. S6 and S7†), we speculate that the causes for the inferior performance of the N configurated CNTs towards ERA are: (1) the low charge transfer efficiency of N–CNTs and CNT–NH2 (Fig. 4d) owing to the graphene lattice distortion by N heteroatoms and (2) the high amounts of pyridinic N and pyrrolic N in N–CNTs (Fig. S6f†) that may be favourable for the reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond65–69 rather than the CN double bond. As the high defective degrees for N–CNTs and CNT–NH2 are due to the incorporation of extrinsic N defects,70 these control samples further highlight the important role of the intrinsic defects in CNTs for the reductive amination.
O double bond65–69 rather than the CN double bond. As the high defective degrees for N–CNTs and CNT–NH2 are due to the incorporation of extrinsic N defects,70 these control samples further highlight the important role of the intrinsic defects in CNTs for the reductive amination.
Investigation of glutamic acid synthesis mechanism
The formation of glutamic acid followed first order dependence with respect to both 2-ketoglutaric acid and ammonia at low concentrations, but approached zeroth order dependence at 2-ketoglutaric acid concentrations above 100 mM and ammonia concentrations above 3 M (Fig. 5a and b). As displayed in Fig. 5c, the Tafel slope of BM3 h–CNTs in the region of glutamic acid production was 113.2 mV dec−1, implying that the rate determining step (RDS) for the overall reaction is the first electron transfer process. Kinetic modelling was also conducted to verify the observed reaction orders and our proposed RDS, showing a good agreement between the model predictions and the experimental results (Fig. S13†). In the literature, there are mainly two possible mechanisms for ERA in aqueous electrolytes. The first one, as previously found on mercury, is characterised by an initial direct electron addition to the imino acid44 (Scheme S2a†). The second mechanism involves the initial formation of a hydride on the cathode surface, followed by the addition of a hydrogen atom to the imino acid, which often happens on metals with low HER overpotentials, such as platinum and palladium45 (Scheme S2b†). In our system, the CV curve of 2-ketoglutaric acid displayed in Fig. 3b shows a peak at around −0.2 V vs. RHE, attributable to the electron transfer to the imino acid.44,62–64 Furthermore, the observed Tafel slope of 113.2 mV dec−1 rules out a pathway through hydride formation as its RDS may involve the adsorption of the imino acid on the cathode surface,45 resulting in a theoretical Tafel slope of 60 mV dec−1. Thus, the ERA on BM3 h–CNTs is likely to occur through the direct electron transfer pathway.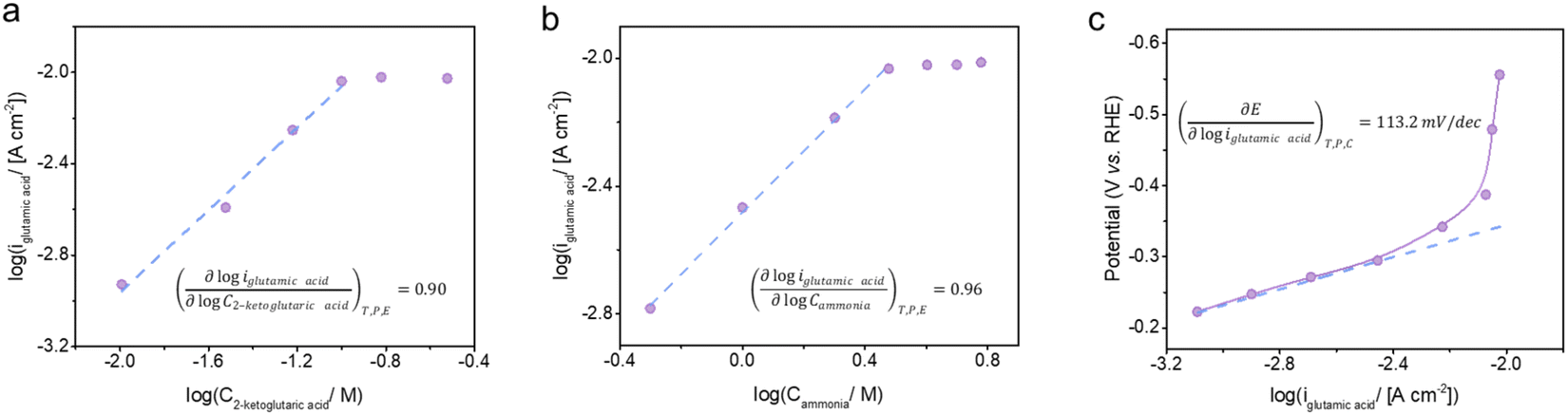 | ||
| Fig. 5 Kinetic analyses of BM3 h–CNT catalysed electro-reductive amination of 2-ketoglutaric acid to glutamic acid. The dependence of the glutamic acid partial current density on the concentration of (a) 2-ketoglutaric acid (3 M ammonia) and (b) ammonia (100 mM 2-ketoglutaric acid) at −0.39 V vs. RHE, pH 10. (c) Tafel plot of glutamic acid synthesis with 100 mM 2-ketoglutaric acid and 3 M ammonia at pH 10. | ||
Taken together, the electrokinetic study suggests the following BM3 h–CNT catalysed ERA mechanism (Scheme 2): firstly, a nucleophilic attack by ammonia on a keto acid A results in a 2-amino-2-hydroxy acid B, followed by the elimination of a water molecule to afford an imino acid intermediate C. Subsequently, the in situ formed imino acid directly accepts an electron at the electrode surface to form a radical anion D, which extracts a proton from water to generate a radical E. E then undergoes a fast proton-coupled electron transfer to form an amino acid F at the cathode surface.
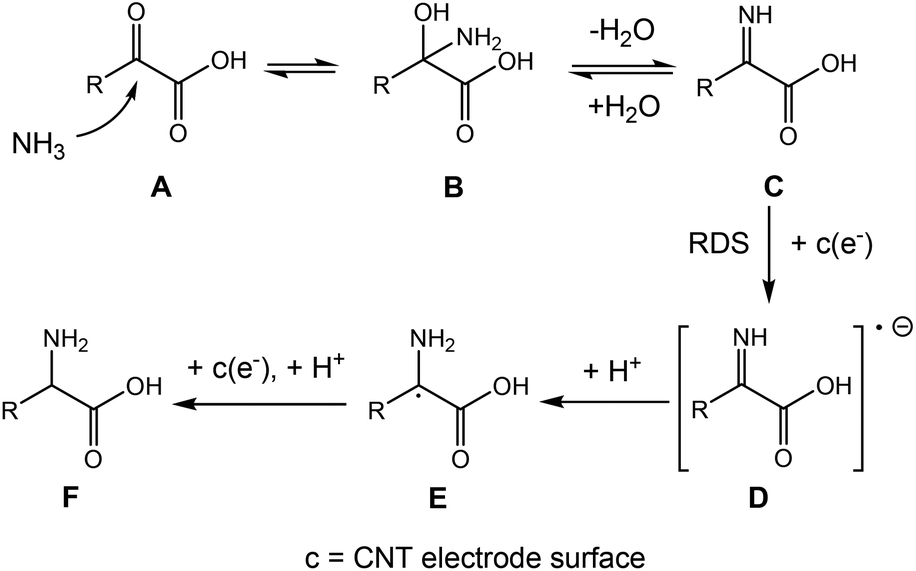 | ||
| Scheme 2 Proposed mechanism for the BM3 h–CNT catalysed ERA. | ||
Electrocatalytic synthesis of other amino acids
We further extended the substrate scope to verify the applicability of our method and investigate the influence of the keto acid structure on the reaction. A series of aliphatic substrates with increasing carbon chain lengths (Table 1, entries 1–6) and aromatic substrates (Table 1, entries 7–9) were used to produce the corresponding amino acids. Interestingly, in contrast to literature reports showing that the low solubility of organic compounds or steric hindrance of substrates caused by long carbon chains would decrease the formation rate,23,44,45,47,48,64 the substitution of hydrophobic functional groups or elongation of the carbon chain was observed to promote the formation rate in our case. The highest formation rate of 166.9 mmol g−1cat h−1 was achieved with norvaline. This suggests that higher hydrophobicity of a keto acid accelerated the formation of the corresponding amino acid, presumably due to the enhancement of the adsorption of the organic species on the hydrophobic BM3 h–CNT (Fig. S14†). To validate this hypothesis, the CV analyses of keto acids were carried out under the reaction conditions, showing that keto acids with larger adsorption peak areas in the CV curves exhibited higher amino acid formation rates (Fig. S15 and S16†). In addition, as shown in the LSV curves (Fig. S17†), the trend of the current densities for different substrates is roughly consistent with the trend of the corresponding formation rates.| Entry | Substrate | Product | FE (%) | Formation rate (mmol g−1 h−1) | |
|---|---|---|---|---|---|
| Structure | Name | ||||
| Reaction conditions: 4.5 mg of the catalyst, 60 mM substrate, 6 M ammonia, pH 10, −0.39 V vs. RHE, room temperature, 1 h reaction time.a Due to the solubility limit of the substrate, the initial concentration of the substrate was adjusted to 20 mM. | |||||
| 1 |
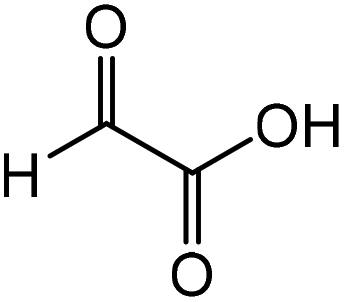
|
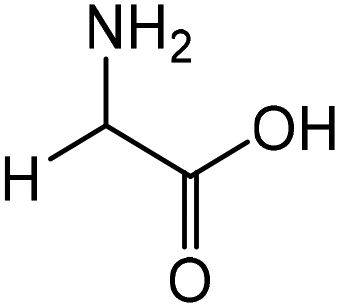
|
Glycine | 87.4 | 16.6 |
| 2 |
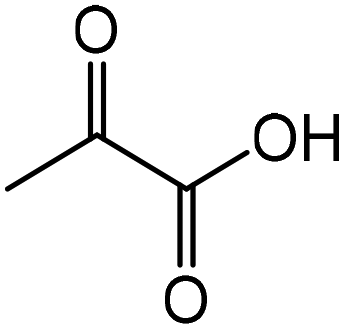
|
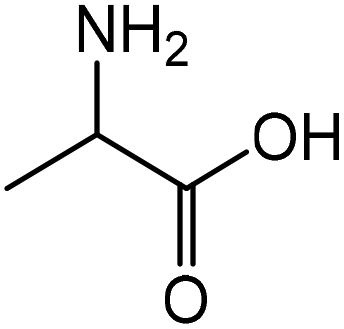
|
Alanine | 45.0 | 43.3 |
| 3 |
|
|
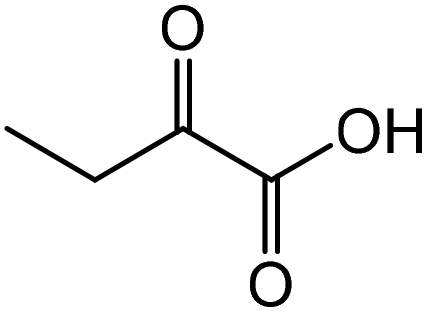
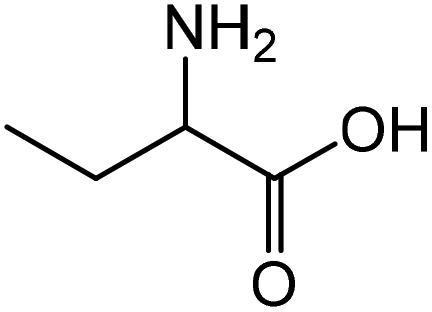
2-Aminobutyric acid | 59.2 | 64.0 |
| 4 |
|
|
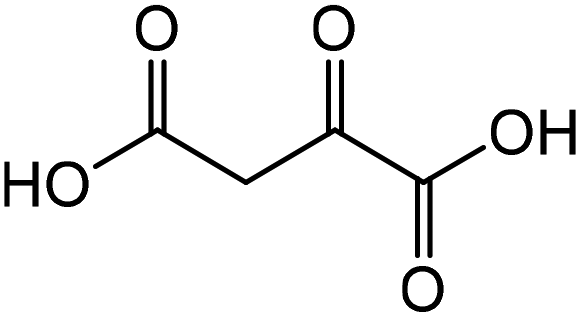
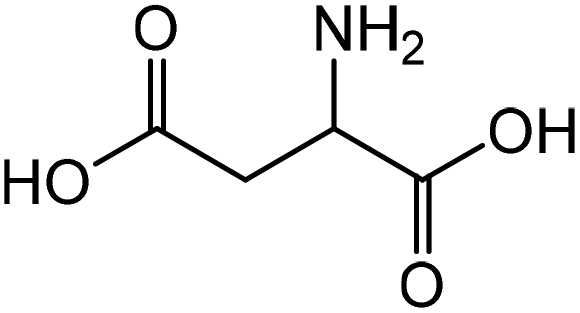
Aspartic acid | 46.9 | 14.8 |
| 5 |
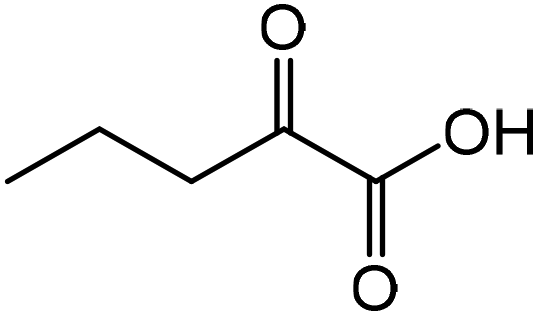
|
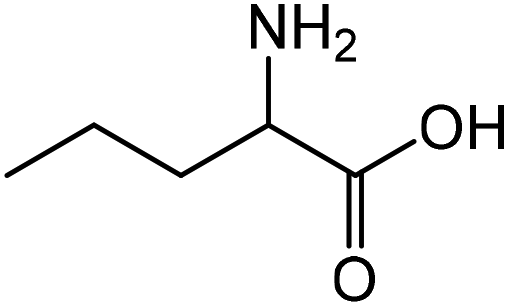
|
Norvaline | 91.3 | 166.9 |
| 6 |
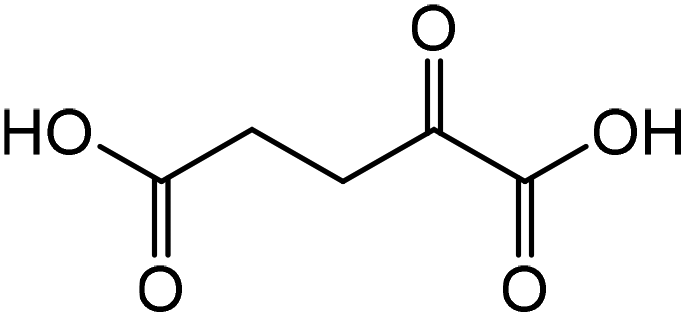
|
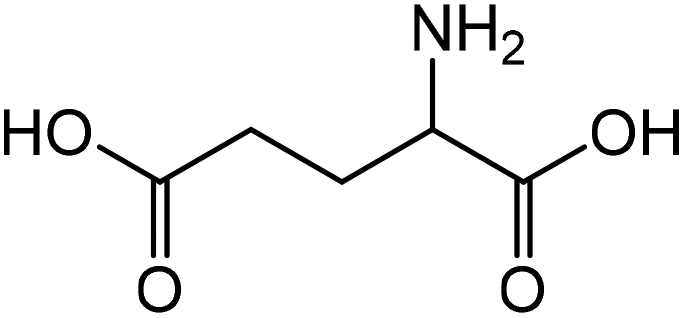
|
Glutamic acid | 90.0 | 123.7 |
| 7 |
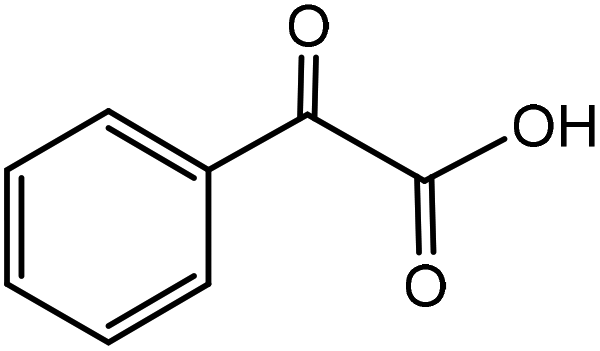
|
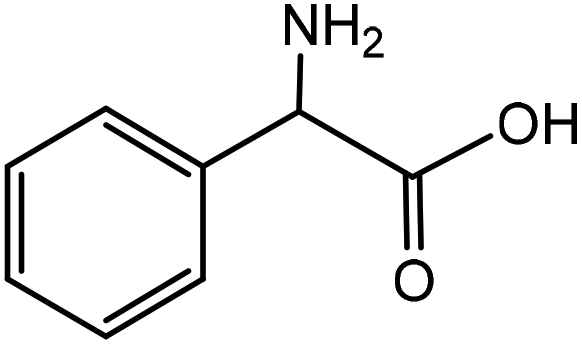
|
Phenylglycinea | 31.4 | 46.8 |
| 8 |
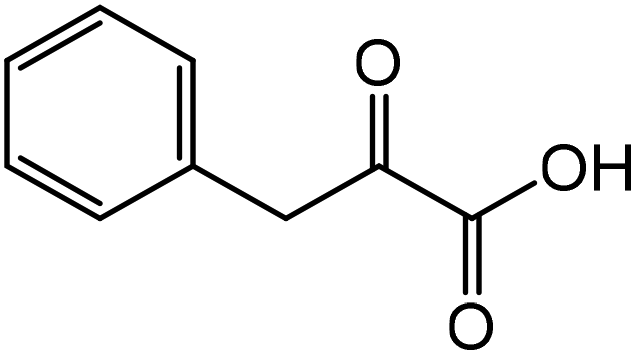
|
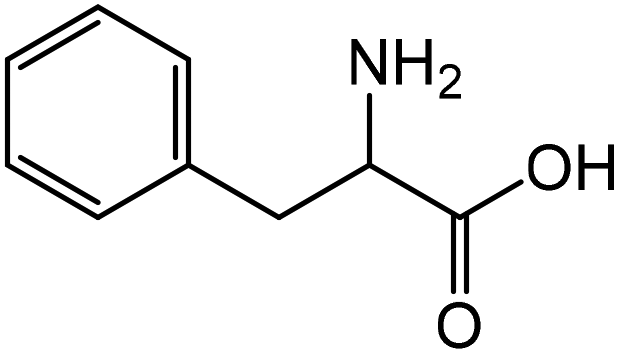
|
Phenylalaninea | 92.8 | 45.9 |
| 9 |
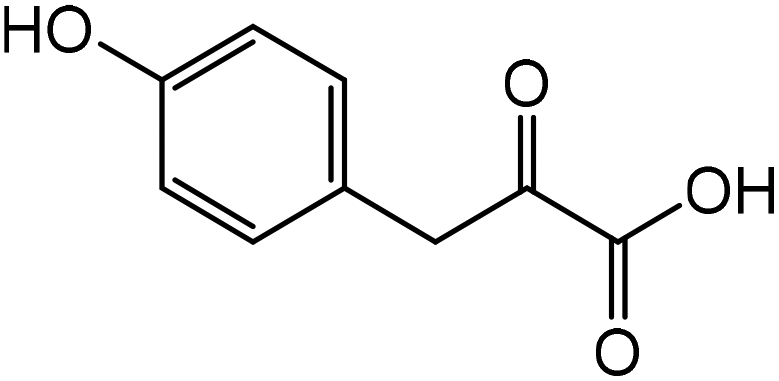
|
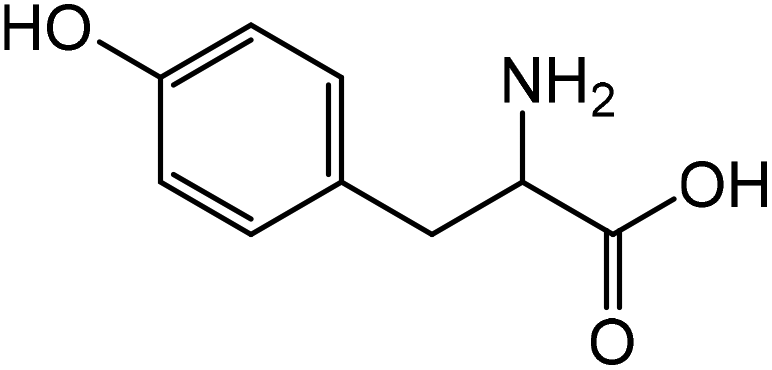
|
Tyrosinea | 72.9 | 21.1 |
Conclusions
We have developed an electrosynthetic route to produce amino acids from α-keto acids with moderate to high FEs and formation rates using ball milled CNTs as the catalyst. Under the optimum reaction conditions (−0.39 V vs. RHE, 100 mM substrate, 6 M NH3, pH 10), a formation rate of 180.9 mmol g−1cat h−1 and a FE of ∼90% were attained for glutamic acid synthesis. The synthetic methodology has been extended to the synthesis of eight other amino acids. In particular, we successfully produced long carbon chain amino acids, whose syntheses are conventionally difficult due to their high steric hindrance. Investigation on the structure–activity relationship of CNTs suggests that the high-density intrinsic defects in the BM–CNTs are likely to be the origin of their superior electrocatalytic activity. Electrokinetic studies imply that the pathway of the ERA involves the direct reduction of in situ formed imino acids. Further efforts are needed to elucidate the type of defects and their specific ERA activities.Author contributions
N. Y., L. W., and Y. X. designed the study. Y. X. performed most of the experiments. N. Y., L. W., Y. X., C. W. L., J. C., and Q. Y. were involved in data collection and discussion. N. Y., L. W., Y. X., and C. W. L. wrote and edited the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Research Foundation Singapore NRF Investigatorship (Award No. NRF-NRFI07–2021–0006). We express our gratitude to Dr Zeliang Yuan for performing XPS measurements. We appreciate Zhicheng Liu for the help with TEM imaging. We thank Yanqiu Lu for conducting contact angle measurements.References
-
A. Karau and I. Grayson, in Biotechnology of Food and Feed Additives, ed. H. Zorn and P. Czermak, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 189–228, DOI:10.1007/10_2014_269
.
- N. Tonouchi and H. Ito, Adv. Biochem. Eng./Biotechnol., 2017, 159, 3–14 CrossRef PubMed
- Global Amino Acids Market Size & Share Report, 2022–2030 .
- M. D'Este, M. Alvarado-Morales and I. Angelidaki, Biotechnol. Adv., 2018, 36, 14–25 CrossRef PubMed
-
Y.-X. Huo, in Engineering Microbial Metabolism for Chemical Synthesis, WORLD SCIENTIFIC (EUROPE), 2017, pp. 69–95, DOI:10.1142/9781786344304_000310.1142/9781786344304_0003
- A. Strecker, Justus Liebigs Ann. Chem., 1850, 75, 27–45 CrossRef
- M. S. Gibson and R. W. Bradshaw, Angew. Chem., Int. Ed. Engl., 1968, 7, 919–930 CrossRef CAS
- T. Parker Eric, J. Cleaves Henderson, P. Dworkin Jason, P. Glavin Daniel, M. Callahan, A. Aubrey, A. Lazcano and L. Bada Jeffrey, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5526–5531 CrossRef CAS PubMed
-
J. Clayden, N. Greeves and S. G. Warren, Organische Chemie, Springer, 2013, vol. 2 Search PubMed
- K. Harada and K. Matsumoto, J. Org. Chem., 1967, 32, 1794–1800 CrossRef CAS
- R. Kadyrov and T. H. Riermeier, Angew. Chem., Int. Ed., 2003, 42, 5472–5474 CrossRef CAS PubMed
- S. Ogo, K. Uehara, T. Abura and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 3020–3021 CrossRef CAS PubMed
- O.-Y. Lee, K.-L. Law, C.-Y. Ho and D. Yang, J. Org. Chem., 2008, 73, 8829–8837 CrossRef CAS PubMed
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed
- R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897–2904 CrossRef CAS
- R. J. Mattson, K. M. Pham, D. J. Leuck and K. A. Cowen, J. Org. Chem., 1990, 55, 2552–2554 CrossRef CAS
- G. Chieffi, M. Braun and D. Esposito, ChemSusChem, 2015, 8, 3590–3594 CrossRef CAS PubMed
- G. Hahn, P. Kunnas, N. de Jonge and R. Kempe, Nat. Catal., 2019, 2, 71–77 CrossRef CAS
- Y. Nakamura, K. Kon, A. S. Touchy, K.-I. Shimizu and W. Ueda, ChemCatChem, 2015, 7, 921–924 CrossRef CAS
- X. Jv, S. Sun, Q. Zhang, M. Du, L. Wang and B. Wang, ACS Sustainable Chem. Eng., 2020, 8, 1618–1626 CrossRef CAS
- G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., 2017, 129, 3096–3100 CrossRef
- S. Song, Y. Wang and N. Yan, Mol. Catal., 2018, 454, 87–93 CrossRef CAS
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC
- Y. Chi, Y. G. Zhou and X. Zhang, J. Org. Chem., 2003, 68, 4120–4122 CrossRef CAS PubMed
- H. Duan and F. Wang, Chem. Catal., 2022, 2, 641–643 CrossRef CAS
- H. Zhou, Z. Li, L. Ma and H. Duan, ChemComm, 2022, 58, 897–907 RSC
- K. Li and Y. Sun, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS PubMed
- F. J. Holzhäuser, J. B. Mensah and R. Palkovits, Green Chem., 2020, 22, 286–301 RSC
- X. Liu, D. C. Y. Leong and Y. Sun, Green Chem., 2020, 22, 6531–6539 RSC
- X. Shang, Y. Yang and Y. Sun, Green Chem., 2020, 22, 5395–5401 RSC
- M. T. Bender, X. Yuan, M. K. Goetz and K.-S. Choi, ACS Catal., 2022, 12, 12349–12368 CrossRef CAS
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS
- Z. Xu, M. Sun, Z. Zhang, Y. Xie, H. Hou, X. Ji, T. Liu, B. Huang and Y. Wang, ChemCatChem, 2022, 14, e202200052 CAS
- Y. Yang, M. Z. Ertem and L. Duan, Chem. Sci., 2021, 12, 4779–4788 RSC
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed
- H. Zou, L. J. Arachchige, W. Rong, C. Tang, R. Wang, S. Tan, H. Chen, D. He, J. Hu, E. Hu, C. Sun and L. Duan, Adv. Funct. Mater., 2022, 32, 2200333 CrossRef CAS
- Y. Ma, T. Yang, H. Zou, W. Zang, Z. Kou, L. Mao, Y. Feng, L. Shen, S. J. Pennycook, L. Duan, X. Li and J. Wang, Adv. Mater., 2020, 32, 2002177 CrossRef CAS PubMed
- Y. J. Jang and K.-S. Choi, J. Mater. Chem. A, 2020, 8, 13842–13851 RSC
- H. Zou, W. Rong, B. Long, Y. Ji and L. Duan, ACS Catal., 2019, 9, 10649–10655 CrossRef CAS
- W. Sui, W. Li, Z. Zhang, W. Wu, Z. Xu and Y. Wang, J. Power Sources, 2023, 556, 232438 CrossRef CAS
- X. Min, Y. Chen and M. W. Kanan, Phys. Chem. Chem. Phys., 2014, 16, 13601–13604 RSC
- T. Sönmez, K. S. Belthle, A. Iemhoff, J. Uecker, J. Artz, T. Bisswanger, C. Stampfer, H. H. Hamzah, S. A. Nicolae, M.-M. Titirici and R. Palkovits, Catal. Sci. Technol., 2021, 11, 6191–6204 RSC
- E. Jeffery and A. Meisters, Aust. J. Chem., 1978, 31, 73–78 CrossRef
- E. Jeffery, O. Johansen and A. Meisters, Aust. J. Chem., 1978, 31, 79–84 CrossRef
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC
- S. D. Mürtz, N. Kurig, F. J. Holzhäuser and R. Palkovits, Green Chem., 2021, 23, 8428–8433 RSC
- T. Fukushima and M. Yamauchi, ChemComm, 2019, 55, 14721–14724 RSC
- T. Fukushima and M. Yamauchi, J. Appl. Electrochem., 2021, 51, 99–106 CrossRef CAS
- K. Yan, M. L. Huddleston, B. A. Gerdes and Y. Sun, Green Chem., 2022, 24, 5320–5325 RSC
- Z. Liu, Z. Zhao, Y. Wang, S. Dou, D. Yan, D. Liu, Z. Xia and S. Wang, Adv. Mater., 2017, 29, 1606207 CrossRef PubMed
- S. Wang, E. Iyyamperumal, A. Roy, Y. Xue, D. Yu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756–11760 CrossRef CAS PubMed
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J.-B. Baek and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. A. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819 CrossRef
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang and H. Yu, Angew. Chem., 2017, 129, 15813–15817 CrossRef
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed
- G. Gan, S. Fan, X. Li, J. Wang, C. Bai, X. Guo, M. Tade and S. Liu, ACS Catal., 2021, 11, 14284–14292 CrossRef CAS
- X. Zhang, H. Jing, S. Chen, B. Liu, L. Yu, J. Xiao and D. Deng, Chem. Catal., 2022, 2, 1–2 CrossRef
- M. J. Matthews, M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus and M. Endo, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, R6585–R6588 CrossRef CAS
- L. G. Bulusheva, A. V. Okotrub, I. A. Kinloch, I. P. Asanov, A. G. Kurenya, A. G. Kudashov, X. Chen and H. Song, Phys. Status Solidi B, 2008, 245, 1971–1974 CrossRef CAS
- A. Anne, S. Daninos and J. Moiroux, New J. Chem., 1994, 18, 1169–1174 CAS
- H. Lund, Acta Chem. Scand., 1959, 13, 249–267 CrossRef CAS
- T. Pienemann and H. J. Schäfer, Synthesis, 1987, 1005–1007 CrossRef CAS
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai, B. I. Yakobson, J. Lou and P. M. Ajayan, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh and R. Vajtai, Nat. Commun., 2016, 7, 1–6 Search PubMed
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson, J. Lou and P. M. Ajayan, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed
- R. M. Yadav, Z. Li, T. Zhang, O. Sahin, S. Roy, G. Gao, H. Guo, R. Vajtai, L. Wang and P. M. Ajayan, Adv. Mater., 2022, 34, 2105690 CrossRef CAS PubMed
- W. Tian, C. Wang, R. Chen, Z. Cai, D. Zhou, Y. Hao, Y. Chang, N. Han, Y. Li, J. Liu, F. Wang, W. Liu, H. Duan and X. Sun, RSC Adv., 2018, 8, 26004–26010 RSC
- B. J. Matsoso, K. Ranganathan, B. K. Mutuma, T. Lerotholi, G. Jones and N. J. Coville, RSC Adv., 2016, 6, 106914–106920 RSC
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, SEM images, XRD patterns, XPS spectra, NMR spectra, contact angles, pH effects, formation rates of 2-hydroxyglutaric acid, CV curves, LSV curves, kinetic modelling, electrochemical setup, schemes, a table of comparison of the electrocatalytic amino acid synthesis from keto acids in this work with those in the literature, a table of the BET surface area of various CNTs, and a table of the specifications of the CNT materials. See DOI: https://doi.org/10.1039/d3gc00265a |
| This journal is © The Royal Society of Chemistry 2023 |