 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic templated length-controlled oligomerization
Bartosz
Lewandowski
 *,
Rebecca J. B.
Schäfer
,
Etienne
Cotter
,
Dora
Harangozo
and
Helma
Wennemers
*
*,
Rebecca J. B.
Schäfer
,
Etienne
Cotter
,
Dora
Harangozo
and
Helma
Wennemers
*
Laboratory of Organic Chemistry, D-CHAB, ETH Zurich, Vladimir-Prelog-Weg 3, 8093 Zurich, Switzerland. E-mail: Bartosz.Lewandowski@org.chem.ethz.ch; Helma.Wennemers@org.chem.ethz.ch
First published on 30th January 2023
Abstract
Templated synthesis is an intriguing strategy for the length-controlled synthesis of oligomers. Traditionally, such reactions require stoichiometric amounts of the template with respect to the product. Recently we reported catalytic macrocyclic templates that promote oligomerization of a small molecule substrate with a remarkable degree of length control. Herein we present our efforts toward creating linear templates for catalytic length-controlled oligomer synthesis.
Introduction
Nature uses templated synthesis to produce oligomers from monomer building blocks.1 For example, genetic information is processed by transcription of DNA into RNA and subsequent translation into peptides and proteins. Scientists have engineered the natural DNA-based oligomerization machinery to allow for the preparation of complementary DNA or RNA strands2–4 as well as the sequence- and length-controlled synthesis of non-natural oligomers.5–10 Templated oligomerization has also been achieved with non-DNA-based synthetic templates11 and enabled access to macrocycles,12,13 cages14 and even topologically more complex products.15,16 Templating has also allowed the preparation of linear oligomers with length control.17–19 A common limitation of templated oligomerization is the tight binding between the template and the oligomer. The newly formed oligomer is therefore only accessible in stoichiometric amounts relative to the template, and the complex between the template and the synthetic oligomer needs to be disassembled in a subsequent release step.20 Recently, we reported the first catalytic length-controlled oligomerization.21 Macrocyclic templates T1–T3 catalyzed the formation of oligomers through aldol-type reactions of bifunctional substrate 1 bearing aldehyde and malonic acid half thioester moieties (Fig. 1b). The templates consist of two rigid oligoproline moieties that are functionalized with thiourea and amine catalytic sites positioned at defined distances at both sides of the macrocyclic cavity (Fig. 1a). Only 10 mol% of the template sufficed to achieve full conversion of the substrate, predominantly to a single oligomer. The length of the oligomer correlated with the size of the macrocyclic catalysts (Fig. 1c). Thus, the number of monomer molecules incorporated into the oligomer is determined by the dimensions of the macrocycle and the number of its catalytic sites. | ||
| Fig. 1 (a) Catalytic macrocyclic templates T1–T3; (b) catalytic length-controlled oligomerization of malonic acid half thioester (MAHT)-aldehyde bifunctional substrate 1; and (c) correlation between the number of catalytic sites on the template with the length of the formed oligomer. | ||
Building on these findings, we are intrigued whether catalytic length-controlled oligomerization can also be achieved with linear templates. Such linear templates would be less complex and thus easier to access and modify, e.g., by extension of the length, than macrocyclic templates. Herein we present our investigations towards the development of linear templates for catalytic length-controlled oligomer synthesis.
Results and discussion
General design of a catalytic linear oligomerization template
We envisioned that a linear template for catalytic length-controlled oligomerization needs to contain a recognition site (Fig. 2, dark green) for an “initiator” building block and activation sites (Fig. 2, dark blue) that allow for the activation of the initiator and incoming “propagator” building blocks. The initiator should contain a binding site (Fig. 2, green) that binds to the recognition site of the template. Additionally, the initiator needs a functional group (Fig. 2, blue) that can be activated by the activation site on the template for reaction with the reactive site of the propagator. The propagator should contain the reactive site (Fig. 2, brown) as well as the same activatable functional group as the initiator. Upon binding of the initiator to the template (step 1), its other functional group will become activated by the activating site of the template and react with the reactive site on the propagator (step 2). This reaction will bring the activatable functionality of the propagator into the vicinity of the next activation site on the template. The template-bound propagator will then become activated, and react with the next propagator molecule (step 3). These propagating reactions will continue until the last activation site at the end of the template is reached. Consequently, the length of the formed oligomer will correlate with the number of activation sites on the template.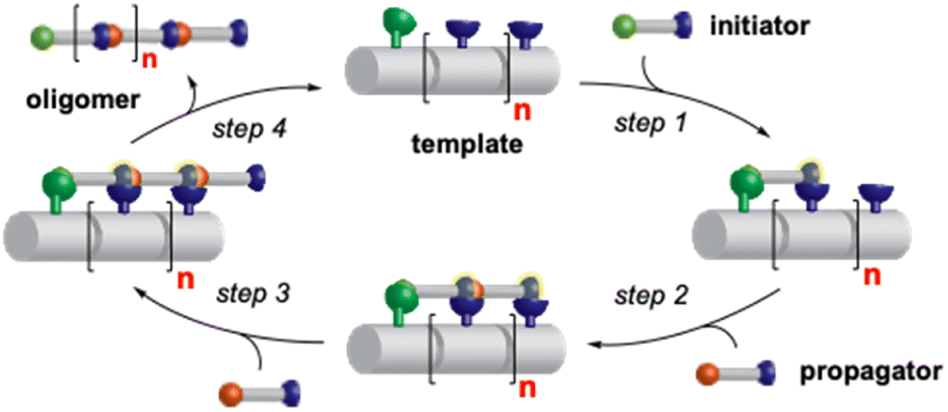 | ||
| Fig. 2 Concept of catalytic length-controlled oligomerization with a linear synthetic template. | ||
For length-controlled oligomerization, the “initiator” and the growing oligomer should only react with the “propagator” when bound and activated by the template. For the controlled activation of the reactants, the distances between the recognition and activation sites on the template need to match those of the functional groups of the initiator and the propagator. For catalytic turn-over, the template needs to bind less tightly to the oligomer than to the monomeric building blocks. These prerequisites are likely best fulfilled by rigid templates that can be functionalized with recognition and catalytic sites at geometrically well-defined positions. Ideal templates are composed of modular components that allow for facile length and functional group variations. Oligoprolines fulfil these requirements as they already adopt, at a chain-length of six residues, a polyproline II (PPII) helix, in which every third residue is located at the same face of the helix at a distance of ∼9 Å (Fig. 3).22,23 These peptides are accessible with different lengths by standard solid-phase peptide synthesis (SPPS), are soluble in aqueous as well as organic solvents, and can be derivatized if γ-azidoproline (Azp) residues are used as building blocks.23 Thus, oligoprolines allow for positioning recognition and activation sites on the same side of the helical scaffold.
 | ||
| Fig. 3 Model of a PPII-helical oligoproline 12-mer (left) and general structure of oligoprolines with (2S,4S)-Azp residues in every third position (right). | ||
Design of specific initiator, propagator, and template
Building on our catalytic length-controlled oligomerization with macrocyclic templates21 and expertise with conjugate addition reactions using nitroolefins,24–26 we started to explore addition reactions between β-ketoesters and functionalized nitroolefins with p-(carboxy)-nitrostyrene (I) as initiator and p-(β-ketoester)-nitrostyrene (P) as propagator (Fig. 4a). We started with the β-ketoester as a donor moiety en route to β-ketoacids since the latter can undergo non-productive decarboxylation and require careful control of the reaction conditions.24,25 As linear catalytic templates, we envisioned oligoproline 6-mer TL1 and 9-mer TL2, each functionalized with a guanidinium moiety at Cγ of the second Pro residue for coordination with the carboxylic acid of the initiator and thiourea moieties for coordination and activation of the nitroolefin (Fig. 4b). Within propagator P, the distance between the nitro group and the β-ketoester is approximately 9 Å, thus, matching the distance between functional groups installed at every third residue of the oligoproline template. | ||
| Fig. 4 (a) Structures and calculated lowest energy structures of the initiator I, propagator P, and oligomers; (b) templates TL1 and TL2; and (c) plausible binding and activation of initiator I by template TL1. | ||
Catalytic turnover requires (1) binding of the initiator to the template and (2) a lower binding affinity of the oligomer to the template compared to the initiator and propagator. The carboxylic acid moiety of the initiator I should ensure binding to the templates TL1 and TL2. Product release is more challenging but should be feasible since the conformation of the oligomer differs significantly from that of the initiator and propagator. The monomeric building blocks are predominantly flat with sp2-hybridized carbons, whereas the oligomer contains sp3-hybridized carbons and, therefore, tetrahedral moieties. It is noteworthy that the propagator is predominantly planar due to enolization (Fig. 4). Since the geometry of the templates is tailored to accommodate that of the initiator, the oligomer should have a lower binding affinity to the template compared to the monomers.
Synthesis of initiator, propagator, and template
Templates TL1 and TL2 were prepared by SPPS using established procedures.21 Initiator I was prepared from 4-carboxybenzaldehyde via a Henry reaction followed by dehydration. Propagator P was also readily available from the same starting material in 5 steps.Conformation and binding affinity studies
Circular dichroism (CD) spectra of templates TL1 and TL2 show the typical signature of PPII helicity; thus, the templates adopt the expected helical structure. The binding of the initiator to the template is a prerequisite for templated oligomerization. Since the binding affinity between a carboxylic acid and a guanidinium group depends on the pH and the solvent,27 we determined the binding affinity of the complex between I and TL1 in different environments. Isothermal titration calorimetry (ITC) studies in methanol revealed an association constant Ka = 8201 ± 600 M−1. In our previous studies with the macrocyclic template, the interaction between the template and the substrate was significantly weaker (Ka ∼ 20 M−1), which agrees with other interactions observed in organocatalytic reactions.21,28 Thus, the observed association constant is rather high and likely too strong to allow for catalytic turnover in the templated oligomerization reaction. In a methanol/water mixture, the binding is, as expected, weaker, Ka = 1187 ± 36 M−1, as revealed by titration monitored with 1H NMR spectroscopy (CD3OD/D2O = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). (Note, we used NMR spectroscopic monitoring in this case since large solvent-mixing heats can override the effects arising from host–guest binding.)
:1). (Note, we used NMR spectroscopic monitoring in this case since large solvent-mixing heats can override the effects arising from host–guest binding.)
Templated oligomerization
Upon mixing I and P in a 1:3 ratio and adding 0.1 equivalents of TL1 (10 mol%) in methanol (Fig. 5a), hardly any product formation was observed after 48 hours (Fig. 5b, top). In contrast, in a mixture of methanol and water (CD3OD/D2O 9:1) the expected dimer OL1 formed in approximately 50% yield (Fig. 5b, bottom). This experiment showed that template TL1 served as a catalyst and corroborated the basic design of the catalytic oligomerization. Over the course of the reaction, OL1 precipitated, indicating poor solubility.
 | ||
| Fig. 5 (a) Catalytic templated oligomerization with TL1; (b) 1H NMR spectra (selected region) of the reaction in CD3OD (top) and CD3OD/D2O 9:1 (bottom) recorded after 24 h – signals corresponding to the product are labelled with a green dot; and (c) LC-MS analysis of the precipitate formed in the reaction in CD3OD/D2O 9:1 (inset: ESI− spectrum of the peak at 3.67 min). | ||
Next, we performed the oligomerization reaction under the same conditions using template TL2 in catalytic amounts. Conversion of the initiator and the propagator took place (∼50%) along with the formation of a precipitate. LC-MS analysis of the precipitate revealed that it consisted exclusively of dimer OL1. Trimer OL2 was not detected in the precipitate or the reaction mixture. This finding suggests that the initially formed dimer precipitates before a second template-controlled reaction with another molecule of the propagator P to form trimer OL2 can take place. Thus, the results are not conclusive as to whether the current template design allows for length-controlled oligomerization.†
Conclusions
We proposed here linear templates for catalytic length-controlled syntheses of oligomers. The templates are based on rigid oligoproline scaffolds and contain one binding site and one or more activation sites located at precise distances along the scaffold. The templates are designed to bind bifunctional substrates, an initiator and propagators, and control the length of the arising oligomer by the length of the template and the number of activation sites thereon. The work provided insights into how molecular recognition and catalysis can be harnessed to achieve length-controlled assembly of oligomers from monomeric building blocks.A catalytically active template for dimer formation has been established, but poor solubility of the dimer has prevented elongation to longer oligomers. Future studies will therefore focus on fine-tuning the structure of the template and the substrates, the solvent, and other reaction parameters, such as the concentration and the temperature. These will include the use of weaker coordinating groups on the template for binding to the initiator (e.g., amino instead of guanidinium groups), and variations of the substrates (e.g., use of MAHTs instead of β-ketoesters, and use of aldehyde instead of nitro groups). Analysis of the thermodynamic parameters will provide insight into the roles of enthalpy and entropy in the oligomerization reaction.
Materials and instruments
Solvents and reagents were of the highest commercially available grade and used without further purification. They were purchased from Sigma Aldrich, Fisher Scientific, Fluka, Bachem, BioMatrix, Biotage, IRIS Biotech, Protein Technologies and Acros Organics. Solvents used for HPLC were HPLC-grade quality. Water used for HPLC purifications and Staudinger reactions was filtered in a Milli-Q system (Millipore).Solid-phase peptide synthesis (SPPS)
SPPS was performed using the Fmoc-strategy and Rink amide-PS as resin. Amino acids and coupling reagents were purchased from Bachem, IRIS Biotech and Protein Technologies. For automated peptide synthesis, a SYRO Robot peptide synthesizer (Biotage, Sweden) controlled by the SyroXP peptide software version 2.0.126 (MultiSynTech GmbH, Germany) was used.Preparative reverse phase high-pressure liquid chromatography (RP-HPLC)
Purifications of the oligoprolines were carried out on a Semiprep UltiMate 3000 chromatography system (Dionex, USA), using a Reprosil gold C18 (150 × 16 mm) column with a flow of 6 mL min−1. The system was controlled using the Chromeleon software version 6.80. Two different solvents were used. Solvent A was HPLC-grade ACN and solvent B was 1000:10:1 H2O:ACN:TFA.
Preparative medium-pressure liquid chromatography (MPLC)
Purifications of the building blocks and bifunctional substrates were carried out on a CombiFlash EZ Prep flash chromatography system (Teledyne ISCO). Two different solvent systems were used: for the building blocks for peptide functionalization, solvent A was HPLC-grade CH2Cl2 without a stabilizer and solvent B was HPLC-grade methanol. For the bifunctional substrates, solvent A was HPLC-grade hexane and solvent B was HPLC-grade EtOAc.Analytical reverse phase high-pressure liquid chromatography (RP-HPLC)
Reverse phase HPLC (RP-HPLC) analysis of the oligoprolines was performed on a Dionex UHPLC, UltiMate 3000. A Reprosil gold 120 C18 (150 × 4 mm, 5 μm) analytical column with a flow of 1.0 mL min−1 was used. The analyses were performed using a two-solvent system. Solvent A was HPLC-grade acetonitrile and solvent B was a mixture of 1% acetonitrile and 0.1% TFA in Milli-Q pure water.Lyophilization
An Alpha 2-4 LD plus lyophilizer (Christ, Germany) was used.Thin-layer chromatography (TLC)
TLC was conducted on aluminum sheets coated with silica gel 60 F254 (Merck) using UV fluorescence (254 and 366 nm). Analytical grade solvents were used.Liquid chromatography-mass spectrometry (LC-MS)
Analytical reverse phase HPLC was performed on an Agilent 1260 Infinity II Prime LC System. An Agilent Eclipse Plus C18 (150 × 4.6 mm, 5 μm) column with a flow of 0.6 mL min−1 was used. Two different solvents were used. Solvent A was a mixture of 1% formic acid in Milli-Q pure water, and solvent B was a mixture of 1% formic acid in acetonitrile. The gradient used for the HPLC analyses of the templates was 95% A to 5% A over 11 minutes. The mass analysis was performed on a maXis ESI-QTOF spectrometer (Bruker, USA). The obtained mass values are listed as the ratio of atom mass per charge (m/z).Nuclear magnetic resonance (NMR) spectroscopy
1D and 2D NMR spectra were recorded on 400 and 500 MHz Ultrashield spectrometers (Bruker, USA). 1H-NMR chemical shifts (δH) are quoted in parts per million (ppm) downfield from TMS and coupling constants (J) are quoted in Hertz (Hz). Abbreviations for NMR data are s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet).MALDI-TOF (matrix-assisted laser desorption/ionization–time-of-flight) mass spectrometry
MALDI-TOF analyses were carried out using a Bruker SolariX 94 spectrometer (Bruker, USA). α-Cyano-4-hydroxycinnamic acid (10 mg mL−1 in ACN/H2O 1:1 and 1 μL TFA) was used as the matrix. The obtained mass values are listed as the ratio of atom mass per charge (m/z).
High-resolution mass spectrometry (HR-MS)
HR-MS analyses were performed by the Molecular and Biomolecular Analysis Service of ETH Zurich (MOBIAS). High-resolution electrospray ionization (HR-ESI) spectra were measured on a Bruker maXis spectrometer.Circular dichroism spectroscopy (CD)
CD spectroscopic analyses were carried out with a Chirascan™ Plus (Applied Photophysics Ltd, Leatherhead, UK). Quartz cuvettes with a path length of 1.0 mm (Hellma 110-QS) were used. The samples were prepared at 200 μM concentration in MeOH as a solvent.Isothermal titration calorimetry (ITC)
ITC measurements were carried out on a MicroCal PEAQ-ITC (Malvern Panalytical Ltd, Malvern, UK). The raw calorimetry data were analyzed using the analysis software provided by Malvern Panalytical Ltd.Synthesis
Linear templates TL1 and TL2
The peptides were prepared following previously reported procedures21 using Fmoc-Pro-OH, Fmoc-Azp-OH (Azp)29 and Fmoc-di-(Boc)-Gup-OH (Gup)30,31 as individual amino-acid building blocks.6-mer template (TL1)
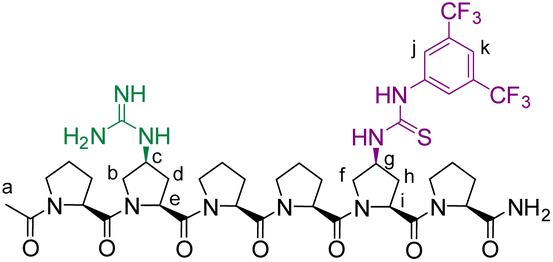
HR-MALDI-MS: m/z: 985.3927 [M + H]+, calculated for C42H55F6N12O7S+: 985.3936.

9-mer template (TL2)


Initiator I
4-Carboxybenzaldehyde (0.3 g, 2.0 mmol, 1 equiv.) and NH4OAc (0.385 g, 5 mmol, 2.5 equiv.) were dissolved in AcOH (8 mL). CH3NO2 (4 mL) was then added. The reaction mixture was stirred under reflux for 1 h. The reaction mixture was poured into a beaker containing water (15 mL). The aqueous mixture was extracted with EtOAc (3 × 15 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by recrystallization from MeOH. 4-(E)-nitrostyrene-benzoic acid I3 was obtained as yellow powder (0.34 g, 1.76 mmol, 88%).

Propagator P (Fig. 6)
4-Formyl benzoic acid (500 mg, 3.3 mmol, 1 equiv.) and NH4Cl (1 g, 18.7 mmol, 5.7 equiv.) were suspended in dry MeOH (10 mL) in a 20 mL microwave vial. The vial was sealed and the mixture was heated to reflux for 36 h, upon which the solution became clear. The solvent was then removed under reduced pressure and the solid residue was crystallized from hexane to yield 2 as glassy crystals (463 mg, 2.4 mmol, 71% yield).

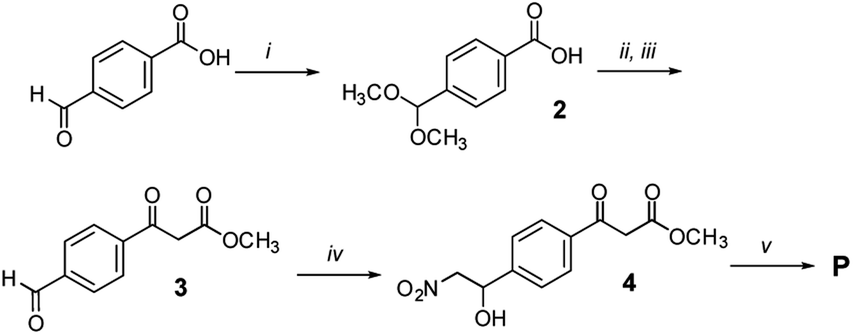 | ||
| Fig. 6 Synthesis of propagator P: (i) CH3OH, NH4Cl, reflux, o/n, 75%; (ii) Meldrum's acid, EDC × HCl, DMAP, CH2Cl2, r. t., 48 h, 70%; (iii) 1 M HCl, THF, 40 °C, 2 h, 95%; (iv) CH3NO2, NaOHaq., CH3OH, r. t., 65%; (v) Ac2O, pyridine, CH2Cl2, 0 °C, 2 h, 75%. | ||
Methyl 3-(4-formylphenyl)-3-oxopropanoate (3)
Compound 2 (450 mg, 2.3 mmol, 1 equiv.) was dissolved in dry CH2Cl2 (3 mL). Meldrum's acid (397 mg, 3.45 mmol, 1.5 equiv.) followed by DMAP (220 mg, 2.75 mmol, 1.2 equiv.) and EDC × HCl (660 mg, 3.45 mmol, 1.5 equiv.) were added. After 22 hours, the reaction was quenched by addition of water (5 mL) and diluted with CH2Cl2 (7 mL). The layers were separated. The aqueous layer was extracted with CH2Cl2 (6 × 5 mL). The combined organic extracts were washed with 1 N HCl aqueous solution (2 × 5 mL) and brine (2 × 5 mL), dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The crude product (515 mg, 1.6 mmol, 70% yield) was immediately used in the next step without purification. It was dissolved in dry MeOH (10 mL) and then refluxed under an Ar atmosphere for 2 h. Afterwards MeOH was removed under reduced pressure and the crude residue was dissolved in a THF
 :H2O mixture (4:1, 5 mL). 2 N aqueous HCl solution was added (1 mL) and the mixture was stirred on a rotary evaporator bath for 45 minutes at 40 °C. Then it was cooled down to room temperature and CH2Cl2 (10 mL) and H2O (10 mL) were added. The layers were separated and the aqueous one was extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were washed with brine (5 mL), dried over MgSO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by MPLC (hexane:EtOAc, gradient 0% to 20% EtOAc in 15 min). Compound 3 was obtained as a colourless oil (313 mg, 1.52 mmol, 95% yield).
:H2O mixture (4:1, 5 mL). 2 N aqueous HCl solution was added (1 mL) and the mixture was stirred on a rotary evaporator bath for 45 minutes at 40 °C. Then it was cooled down to room temperature and CH2Cl2 (10 mL) and H2O (10 mL) were added. The layers were separated and the aqueous one was extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were washed with brine (5 mL), dried over MgSO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by MPLC (hexane:EtOAc, gradient 0% to 20% EtOAc in 15 min). Compound 3 was obtained as a colourless oil (313 mg, 1.52 mmol, 95% yield).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 mixture of the keto and enol forms).
δ 12.47 (s, 0.33H, Hj′), 10.12 (s, 0.67H, Ha), 10.06 (s, 0.33H, Ha′), 8.10 (d, J = 8.2 Hz, 1.3H, Hc), 8.04–7.97 (m, 1.3H, Hd), 7.94 (d, J = 0.9 Hz, 1.3H, Hc′ + Hd′), 5.77 (s, 0.33H, Hg′), 4.05 (s, 1.3H, Hg), 3.83 (s, 1H, Hi′), 3.77 (s, 2H, Hi).
:1 mixture of the keto and enol forms).
δ 12.47 (s, 0.33H, Hj′), 10.12 (s, 0.67H, Ha), 10.06 (s, 0.33H, Ha′), 8.10 (d, J = 8.2 Hz, 1.3H, Hc), 8.04–7.97 (m, 1.3H, Hd), 7.94 (d, J = 0.9 Hz, 1.3H, Hc′ + Hd′), 5.77 (s, 0.33H, Hg′), 4.05 (s, 1.3H, Hg), 3.83 (s, 1H, Hi′), 3.77 (s, 2H, Hi).
Methyl 3-(4-(1-hydroxy-2-nitroethyl)phenyl)-3-oxopropanoate (4)
Nitromethane (27 μL, 0.4 mmol, 1.5 equiv.) was dissolved in MeOH (0.5 mL). 10 M aqueous NaOH (45 μL, 0.34 mmol, 1.3 equiv.) was added to the solution. The mixture was cooled to 0 °C using an ice bath. Then a solution of compound 3 (70 mg, 0.26 mmol, 1 equiv.) in MeOH (0.5 mL) was added dropwise. The reaction was stirred at 0 °C for 30 minutes and then allowed to warm to room temperature. Subsequently it was stirred at room temperature for 2 h. A few drops of AcOH, followed by H2O (5 mL) and CH2Cl2 (5 mL) were added and the layers separated. The aqueous layer was extracted with CH2Cl2 (4 × 5 mL). The combined organic extracts were washed with brine (5 mL), dried over MgSO4, filtered and the solvents evaporated under reduced pressure. The crude mixture was separated by MPLC (hexane
 :ethyl acetate, gradient 0% to 40% over 15 minutes) to yield 4 as a pale yellow solid (53 mg, 0.17 mmol, 65%).
:1 mixture of the keto and enol forms).
δ 12.46 (s, 0.11H, Hk′), 8.02–7.84 (m, 1.78H, Hf), 7.79 (d, J = 8.2 Hz, 0.22H, Hf′), 7.59–7.48 (m, 1.78H, Hd), 7.46 (d, J = 8.3 Hz, 0.22H, Hd′), 5.67 (s, 0.11H, Hh′), 5.58–5.49 (m, 1H, Ha + Ha′), 4.74–4.35 (m, 2H, Hb + Hb′), 3.98 (s, 1.8H, Hh), 3.80 (s, 0.33H, Hj′), 3.74 (s, 2.67H, Hj).
:ethyl acetate, gradient 0% to 40% over 15 minutes) to yield 4 as a pale yellow solid (53 mg, 0.17 mmol, 65%).
:1 mixture of the keto and enol forms).
δ 12.46 (s, 0.11H, Hk′), 8.02–7.84 (m, 1.78H, Hf), 7.79 (d, J = 8.2 Hz, 0.22H, Hf′), 7.59–7.48 (m, 1.78H, Hd), 7.46 (d, J = 8.3 Hz, 0.22H, Hd′), 5.67 (s, 0.11H, Hh′), 5.58–5.49 (m, 1H, Ha + Ha′), 4.74–4.35 (m, 2H, Hb + Hb′), 3.98 (s, 1.8H, Hh), 3.80 (s, 0.33H, Hj′), 3.74 (s, 2.67H, Hj).
Methyl (E)-3-(4-(2-nitrovinyl)phenyl)-3-oxopropanoate (P)
Compound 4 (40 mg, 0.15 mmol, 1 equiv.) was dissolved in CH2Cl2 (1 mL). The solution was cooled down to 0 °C. Subsequently, dry pyridine (12 μL, 0.15 mmol, 1 equiv.) and acetic anhydride (16.5 μL, 0.15 mmol, 1 equiv.) were added. The mixture was allowed to warm to room temperature and was then stirred for 4 hours. Next, CH2Cl2 (2 mL) and 0.1 M aqueous HCl (1 mL) were added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 2 mL). The combined organic layers were dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The crude mixture was separated by MPLC (hexane
 :ethyl acetate, gradient 10% to 20% over 20 minutes) to yield P as a yellow solid (28 mg, 0.113 mmol, 75%).
:2 mixture of the keto and enol forms).
δ 12.50 (s, 0.4H, Hk), 8.07–8.01 (m, 2.2H, Ha + Ha′ + Hd), 7.88 (d, J = 8.5 Hz, 0.8H, Hd′), 7.72–7.65 (m, 1.8H, Hb + He), 7.64–7.58 (m, 1.2H, Hb′ + He′), 5.76 (s, 0.4H, Hh′), 4.05 (s, 1.2H, Hh), 3.85 (s, 1.2H, Hj′), 3.79 (s, 1.8H, Hj).
:ethyl acetate, gradient 10% to 20% over 20 minutes) to yield P as a yellow solid (28 mg, 0.113 mmol, 75%).
:2 mixture of the keto and enol forms).
δ 12.50 (s, 0.4H, Hk), 8.07–8.01 (m, 2.2H, Ha + Ha′ + Hd), 7.88 (d, J = 8.5 Hz, 0.8H, Hd′), 7.72–7.65 (m, 1.8H, Hb + He), 7.64–7.58 (m, 1.2H, Hb′ + He′), 5.76 (s, 0.4H, Hh′), 4.05 (s, 1.2H, Hh), 3.85 (s, 1.2H, Hj′), 3.79 (s, 1.8H, Hj).
Binding affinity measurements
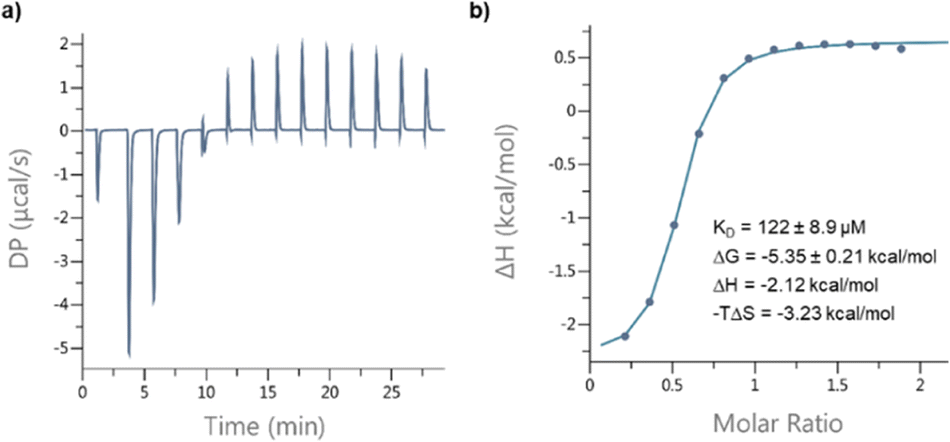 | ||
| Fig. 7 ITC analysis of the interaction between I and TL1 in CH3OH: (a) raw data; (b) fitted curve. | ||
:D2O = 9:1, at room temperature. The concentrations of the host (I) and the guest (TL1) solutions were 6 mM and 300 mM, respectively (Fig. 8). The data were fitted to a single-site non-competitive binding equation using Prism ver.9.2.0 (GraphPad) (Fig. 9).
 | ||
| Fig. 8 Stacked 1H NMR spectra of TL1 (CD3OD:D2O = 9:1) upon titration of increasing portions of I. | ||
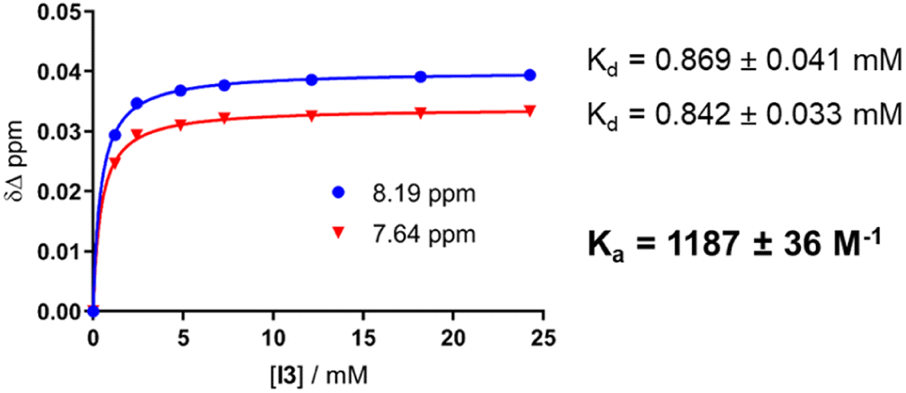 | ||
| Fig. 9 Titration curve correlating the changes in the chemical shifts of aromatic protons in TL1 with the changes in the concentration of I during the titration (left); Kd values obtained for both fits and the average Ka value (right). | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement (No 328103) and grant agreement 862081 and support by ETH Research Grant ETH-35 17-2.Notes and references
- J. M. Berg and J. L. Tymoczko, Biochemistry, W. H. Freeman, New York, 2006 Search PubMed.
- U. Gubler and B. J. Hoffman, Gene, 1983, 25, 263–269 CrossRef CAS PubMed.
- J. Tian, H. Gong, N. Sheng, X. Zhou, E. Gulari, X. Gao and G. Church, Nature, 2004, 432, 1050–1054 CrossRef CAS PubMed.
- N. K. Vaish, A. W. Fraley, J. W. Szostak and L. W. McLaughlin, Nucleic Acids Res., 2000, 28, 3316–3322 CrossRef CAS PubMed.
- Z. J. Gartner, B. N. Tse, R. Grubina, J. B. Doyon, T. M. Snyder and D. R. Liu, Science, 2004, 305, 1601–1605 CrossRef CAS PubMed.
- S. Liao and N. C. Seeman, Science, 2004, 306, 2072–2074 CrossRef CAS PubMed.
- K. Josephson, M. C. T. Hartman and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 11727–11735 CrossRef CAS PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73–77 CrossRef CAS PubMed.
- J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282–292 CrossRef CAS PubMed.
- W. Meng, R. A. Muscat, M. L. McKee, P. J. Milnes, A. H. El-Sagheer, J. Bath, B. G. Davis, T. Brown, R. K. O'Reilly and A. J. Turberfield, Nat. Chem., 2016, 8, 542–548 CrossRef CAS PubMed.
- F. Diederich and P. J. Stang, Templated Organic Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfay, T. D. W. Claridge, A. Saywell, M. O Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. Anderson, Nature, 2011, 469, 72–75 CrossRef PubMed.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- J. Cremers, R. Haver, M. Rickhaus, J. Q. Gong, L. Favereau, M. D. Peeks, T. D. W. Claridge, L. M. Herz and H. L Anderson, J. Am. Chem. Soc., 2018, 140, 5352–5355 CrossRef CAS PubMed.
- Y. Segawa, M. Kuwayama, Y. Hijikata, M. Fushimi, T. Nishihara, J. Pirillo, J. Shirasaki, N. Kubota and K. Itami, Science, 2019, 365, 272–276 CrossRef CAS PubMed.
- Z. Ashbridge, E. Kreidt, L. Pirvu, F. Schaufelberger, J. H. Stenlid, F. Abild-Petersen and D. A. Leigh, Science, 2022, 375, 1035–1041 CrossRef CAS PubMed.
- K. S. Feldman and Y. B. J. Lee, J. Am. Chem. Soc., 1987, 109, 5850–5851 CrossRef.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- D. Núñez-Villanueva, M. Ciaccia, G. Iadevaia, E. Sanna and C. A. Hunter, Chem. Sci., 2019, 10, 5258–5266 RSC.
- X. Li and D. R. Liu, Angew. Chem., Int. Ed., 2004, 43, 4848–4870 CrossRef CAS PubMed.
- B. Lewandowski, D. Schmid, R. Borrmann, D. Zetschok, M. Schnurr and H. Wennemers, Nat. Synth., 2023, 2, 331–337 CrossRef.
- P. Wilhelm, B. Lewandowski, N. Trapp and H. Wennemers, J. Am. Chem. Soc., 2014, 136, 15829–15832 CrossRef CAS PubMed.
- S. Dobitz, M. R. Aronoff and H. Wennemers, Acc. Chem. Res., 2017, 50, 2420–2428 CrossRef CAS PubMed.
- J. Saadi and H. Wennemers, Nat. Chem., 2016, 8, 276–280 CrossRef CAS PubMed.
- J. Lubkoll and H. Wennemers, Angew. Chem., Int. Ed., 2007, 46, 6841–6844 CrossRef CAS PubMed.
- T. Schnitzer, A. Budinská and H. Wennemers, Nat. Catal., 2020, 3, 143–147 CrossRef CAS.
- B. Linton and A. D. Hamilton, Tetrahedron, 1999, 55, 6027–6038 CrossRef CAS.
- C. E. Grünenfelder, J. K. Kisunzu and H. Wennemers, Angew. Chem., Int. Ed., 2016, 55, 8571–8574 CrossRef PubMed.
- H. Wennemers, J. Kisunzu, B. Lewandowski and U. Lewandowska, (2S,4S)-(9H-Fluoren-9-ylmethoxycarbonyl)-4-azidoproline, e-EROS Encycl. Reagents Org. Synth., 2016 DOI:10.1002/047084289X.rn01954.
- Y. A. Nagel, P. S. Raschle and H. Wennemers, Angew. Chem., Int. Ed., 2017, 56, 122–126 CrossRef CAS PubMed.
- M. Li, R. Puschmann, A. Herdlitschka, D. Fiedler and H. Wennemers, Angew. Chem., Int. Ed., 2020, 59, 15586–15589 CrossRef CAS PubMed.
Footnote |
| † Aside from precipitation, the oligomerization could also stop due to a non-favorable binding geometry between the dimer and the template. |
| This journal is © The Royal Society of Chemistry 2023 |