
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVOC emissions by fresh and old asphalt pavements at service temperatures: impacts on urban air quality†
J.
Lasne
 *a,
A.
Lostier
a,
M. N.
Romanias
a,
S.
Vassaux
b,
D.
Lesueur
b,
V.
Gaudion
a,
M.
Jamar
a,
R. G.
Derwent
c,
S.
Dusanter
a and
T.
Salameh
*a
*a,
A.
Lostier
a,
M. N.
Romanias
a,
S.
Vassaux
b,
D.
Lesueur
b,
V.
Gaudion
a,
M.
Jamar
a,
R. G.
Derwent
c,
S.
Dusanter
a and
T.
Salameh
*a
aIMT Nord Europe, Institut Mines-Télécom, Univ. Lille, Centre for Energy and Environment, F-59000 Lille, France. E-mail: jerome.lasne@imt-nord-europe.fr; therese.salameh@imt-nord-europe.fr
bIMT Nord Europe, Institut Mines-Télécom, Univ. Lille, Centre for Materials and Processes, F-59000 Lille, France
crdscientific, Newbury, Berkshire, RG14 6LH, UK
First published on 18th September 2023
Abstract
Outdoor air pollution is a major cause of chronic illness and of mortality, with an estimated 4.5 million deaths every year. Its effects are amplified in urban areas where the global population concentrates at an increasing pace. Asphalt-covered surfaces dominate urban areas. Although pollutant emissions from asphalt have been investigated at temperatures above 120 °C during laying, data on emissions at service temperatures are lacking. In the present work, we characterize and quantify in the laboratory volatile organic compound (VOC) emissions by fresh and old asphalt mixtures with gas chromatography – mass spectrometry/flame ionization detector (GC-MS/FID) and proton transfer reaction – time of flight mass spectrometry (PTR-ToFMS) under simulated atmospheric conditions at service temperatures (23–60 °C). The impact of asphalt aging on VOC emissions is assessed. We show that asphalt pavements contribute significantly to urban pollution and therefore need to be included to emission inventories and taken into account by air quality models. VOC emissions from these extended surfaces are shown to be important contributors to ozone and secondary organic aerosol formed in urban areas. This study reveals a large background source of VOCs, precursors of ozone and particulate matter in cities. Specifically, we estimate that in Paris (France), asphalt pavements at service temperatures are responsible for the emission of 0.148 Gg NMVOC per year, corresponding to 21.3% of emissions from road transportation, and 2.9% of total NMVOC emissions in 2019. The corresponding mass of formed Secondary Organic Aerosol (SOA) is estimated as 1.60–4.12 tonnes per year, accounting for 1.5–3.8% of PM1 emitted by road transportation. It suggests that “net zero emissions” targets, such as the zero pollution action plan of the European Green Deal, cannot be met until this new challenge from asphalt pavements is acknowledged and tackled. Reducing emissions from asphalt pavements during service is a major challenge for sustainable cities and needs to be addressed by the scientific community, policy makers and industrial partners.
Environmental significanceThe world's urban population grows at an increasing pace. Urban atmospheric pollution is a prime concern for health and environmental sustainability. Inventories of pollution sources are necessary to account for observations, and to set relevant mitigation targets. Our study provides for the first time the speciation and quantification of volatile organic compound (VOC) emissions from an uncharacterized and widespread urban surface, asphalt pavements, at service temperatures. We determine ozone and secondary organic aerosol (SOA) formation potentials in Paris, France. Our work shows that asphalt pavements significantly contribute to VOC emissions and SOA formation, and might become a dominant urban pollution source in the future. It has strong implications for urban atmospheric chemistry, with the characterization of an extended pollution source. |
1. Introduction
The global death toll of ambient air pollution is estimated at 8.8 million deaths per year, with an average life expectancy loss of 3 years.1 Recent estimates suggest that outdoor air pollution accounts for 4.5 million deaths every year.2 Outdoor air pollution is exacerbated in cities, where many pollution sources are concentrated together in the same area. Moreover, the proportion of the global population living in urban areas is growing, from around 50% today to a projected 70% by 2050.3 Air quality, especially in urban areas, and its impact on health are therefore major challenges faced by our society, with their importance expected to grow in a future threatened by climate change.Air pollution can affect human health and the environment in several ways. Greenhouse gases, such as methane and carbon dioxide increase Earth's surface temperature. Oxidants, such as ozone (O3), and their precursors, nitrogen oxides (NOx) and volatile organic compounds (VOCs), are irritant and toxic species, causing airway inflammation and increased respiratory symptoms in people suffering from asthma. Additionally, secondary organic aerosols (SOA) represent up to 90% of organic aerosols which contribute to about 20 to 90% of the particulate matter (PM) mass in ambient air.4 SOA form by condensation of photo-oxidized atmospheric VOCs, and increase cardio-respiratory disease mortality.5 VOCs therefore play a central role in air pollution as precursors of ozone and SOA.
The main anthropogenic VOC sources have been identified as road transport, solvent use, extraction and distribution of fossil fuels, production and combustion processes, agriculture, and wastes.6 Consequently, policies have been implemented at national and international levels to reduce VOC emissions from these sources. As a result of the decrease of VOC emissions from road transport, the relative contribution of non-combustion sources has increased, and now represents a major contribution to SOA and ozone precursors.7 Emerging sources, such as volatile chemical products (VCP) and as-yet unidentified sources, have not been fully assessed at present, and are therefore missing from inventories.8 Unknown sources represent a major knowledge gap in urban atmospheric chemistry models.8 New undocumented sources of VCPs could represent 50% of VOC emissions in industrialized cities;9 these sources are ubiquitous, and scale with population density,10 pointing to urban environments. Field measurements in urban environments show that the oxygenated VOCs (OVOC) to non-methane VOCs (NMVOC) flux ratio is two to four times higher than current inventories suggest.11 One or several major OVOC sources are therefore missing in urban environments. These missing sources need to be identified and quantified to make emission inventories and hence air quality models more accurate, helping the assessment of the polluting character of urban materials and assisting policy makers.
Paved surfaces represent about 40% of urban areas.12 The role of asphalt pavement in the urban heat island (UHI) phenomenon and as an impervious surface hindering rainfall infiltration is well-established13 and technical solutions are being developed in order to minimize the UHI.14 Numerous laboratory studies have investigated the impact on the physical properties, chemical composition and chemical structure of the oxidation of hot asphalt mixtures and their aging at deposition temperature (i.e., T > 120–140 °C).15–17 These studies highlight the strong influence of UV radiation, humidity and temperature on asphalt binder aging.17 These factors likely also influence the chemistry occurring at the surface of asphalt mixtures, and hence, emissions by asphalt mixtures are also expected to be sensitive to these parameters.
Temperatures lower than 70 °C are relevant for road surfaces during most of their lifetime under typical weather conditions experienced at mid- and low-latitude regions, where asphalt surfaces can reach 60 °C under sunlight during a hot day.18–21 In the context of global warming associated with climate change, asphalt temperatures as high as 50–60 °C will become common in summer-times in the future.
The emission of atmospheric pollutants by hot asphalt mixtures at deposition temperatures above 120 °C is well constrained at least regarding polycyclic aromatic hydrocarbons (PAHs),22–25 and volatile organic compounds.26–28 It is therefore striking that VOC emissions by asphalt mixtures at service temperatures, i.e. below 70 °C, had never been investigated before the year 2020.29 Emissions from asphalt surfaces may account for part of the gap identified in VOC inventories of urban areas, due to the large surface they provide for atmospheric exchange of trace gases and the petroleum-based nature of the materials constituting the binder of asphalt mixtures. Their contribution to urban air quality needs to be assessed as it may contribute to the background levels of VOCs and of other key pollutants considered in atmospheric models.
The pioneering study of Khare et al. has shown that bituminous materials could emit VOCs at ambient temperatures, not only at deposition temperatures, making them important contributors to SOA formation.29 Despite the importance of their results, some key features of their work suggest that their results are not representative of the real-world environment.
• Firstly, asphalt samples studied in Khare et al., have been collected prior to, or shortly after (28 hours), application.29 They are therefore hardly relevant to real asphalt-covered surfaces because roads, for example, are usually resurfaced every 10 to 50 years. Investigations need to be directed to old asphalt mixtures, which are globally much more representative of paved surfaces.
• Secondly, temperatures investigated by Khare et al., range from 40 °C to 200 °C, with 20 °C steps.29 Relevant asphalt temperatures rarely go above 60–70 °C and are most of the time lower than 40 °C.29 In-use asphalt materials are addressed by two temperatures (40 °C and 60 °C) in Khare et al.;29 in the present work, we extend the temperature range investigated towards lower and more relevant temperatures.
The aim of our work is to investigate how and to what extent asphalt-covered surfaces contribute to urban VOC emissions. From real-world samples, we quantify the emissions of 65 VOCs, from fresh and old asphalt mixtures under temperature conditions relevant for spring/summer/autumn at mid-latitudes (23–60 °C). Our results will be key for modelers to improve atmospheric simulations of O3 and SOA concentrations in urban environments and reveal a major but previously overlooked pollutant source in urban areas. This source needs to be considered in environmental policies and its identification should drive research on innovative zero-emission road-paving materials. Asphalt-covered surfaces may represent one of the major urban sources of anthropogenic SOA in the future, precluding reaching the net zero emissions goal set to tackle climate change.
2. Materials and methods
2.1. Asphalt mixture samples
| Sample reference | STA3 | STA4 | STA5 | LTA2 | LTA3 | LTA4 |
|---|---|---|---|---|---|---|
| a The geometric emitting area represents the area of the top and side surfaces of the sample which are in contact with air in these experiments and can therefore contribute to the emissions. | ||||||
| Mass (g) | 326.0 | 357.8 | 373.7 | 743.2 | 729.4 | 425.6 |
| Geometric emitting area Sasphalt, top and sides (cm2) | 128.3 | 137.4 | 175.0 | 266.7 | 102.6 | 63.5 |
Before their use in experiments, STA samples were shaped into disks of roughly 13 cm diameter and 1.5–1.7 cm thickness, pictured on Fig. S1 of ESI.† STA asphalt mixtures were not subjected to any treatment prior to their use in the atmospheric chamber. After collection, LTA asphalt mixture samples were cut into rectangular fragments with a thickness of 1.3–1.7 cm ensuring that a large majority of the emissions come from the oxidized surface layer. LTA asphalt samples are pictured on Fig. S1 of ESI.† These samples were flushed with dry air and gently washed with water to remove dust before use. The geometric surface of the samples was calculated based on their measured dimensions. The total STA asphalt surface responsible for the measured emissions range between 128.3 and 175 cm2; the surface of LTA asphalt samples range from 63.5 and 266.7 cm2, as shown in Table 1. Note that only results obtained with samples STA3 and LTA2 are presented here; other samples have been used to test repeatability of the results, as described in Section 3 of ESI.†
The extracted binders were also characterized by simulated distillation to assess their volatile fractions. Details of the method and results are available in Section 1.3 of ESI.† The chromatograms obtained by the simulated distillation process (see Fig. S3 of ESI†) demonstrated the evolution of the mass of evaporated product as a function of retention time in the column. Briefly, the light (carbon number < C35) and heavy (carbon number > C35) fractions were not the same in fresh and old bitumens. This observation may account for part of the differences observed between the emissions from the different types of asphalt mixtures.
2.2. Experimental setup
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 617, ensuring that the flow was turbulent; hence, emissions of the asphalt mixtures were well mixed in the chamber, and the air sample composition was not biased by a concentration gradient effect. Moreover, this value is similar to the critical Re (≈11000) required for reduced-scale and full-scale experiments in street canyons with a high-to-width ≤ 1.0 to match.31 Our “reduced-scale” experiments are therefore relevant to full-scale systems with this geometry. All the experiments presented here were performed in the dark.
617, ensuring that the flow was turbulent; hence, emissions of the asphalt mixtures were well mixed in the chamber, and the air sample composition was not biased by a concentration gradient effect. Moreover, this value is similar to the critical Re (≈11000) required for reduced-scale and full-scale experiments in street canyons with a high-to-width ≤ 1.0 to match.31 Our “reduced-scale” experiments are therefore relevant to full-scale systems with this geometry. All the experiments presented here were performed in the dark.
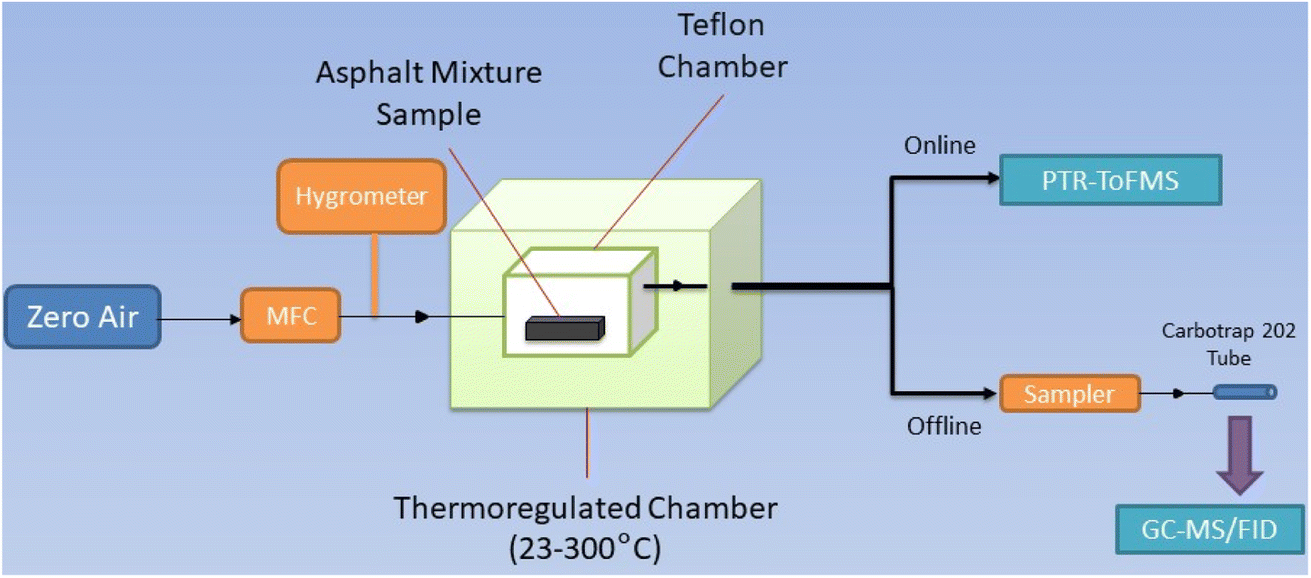 | ||
| Fig. 1 Schematic representation of the experimental setup. The dry zero air flow rate is controlled with a MFC and flowed in the Teflon chamber sitting in a thermoregulated chamber. The gas injected in the chamber carries the emissions of the asphalt sample out of the chamber where they are analyzed online by PTR-ToFMS and offline by GC-MS/FID after a sample is taken in a Carbotrap 202 tube. | ||
Emissions of the asphalt mixture samples were mixed with the air flow and exit the chamber through a Silcosteel tube. Silcosteel is used to prevent VOCs adsorption in the lines. The air flow was then directed downstream of the chamber for analysis with online and offline techniques described in the next section.
2.2.2.1. Real-time online analysis: PTR-ToFMS. PTR-ToFMS is widely used for real-time online characterization and quantification of VOCs in air.34 In our setup, a PTR-ToFMS (series 2, Kore Technology) was directly connected to the exit line of the chamber with a Silcosteel tube.
In the PTR-ToFMS instrument, H3O+ reagent ions transfer a proton to the trace species sampled and mass spectrometry is used to detect the reaction products at mass M + 1, where M is the mass of the parent molecule. This technique causes low fragmentation of the parent ions, has a high sensitivity (detection limit < 10 ppt, ppt = part per trillion), and a high time resolution (≈100 ms), so that it can be used for online monitoring of concentrations. H3O+ reagent ions do not react with major species in ambient air (N2, O2, CO2, Ar, etc.), giving this technique a decisive advantage for the study of trace species, such as VOCs. Nonetheless, molecular species with a proton affinity lower than that of H2O (691 kJ mol−1), such as most alkanes, remain neutral and therefore are not detected with this technique.35 Identification of unknown species at elevated masses was difficult because all isobaric species were detected at the same exact mass. The measured mass resolution M/δM was in the 3700–5500 range. During the experiments presented in this paper, conducted under dry conditions (RH < 0.1%) in the air flow, concentrations of the major impurities were respectively lower than 2.6% (O2+), 1.3% (NO+), and 2.3% (NO2+). Overall, impurities in the PTR-ToFMS systematically account for less than 10% of the reactant ion concentration, meaning than more than 90% of ionization is achieved by H3O+.
The sampling flow rate of the instrument was 180 sccm. The voltage applied to the drift tube was 400 V, and the pressure 1.39 mbar, leading to an electric field to gas density ratio (E/N) of 136 Townsend (1 Td = 10−17 V cm−2). The number of counts given by the mass spectrometer at all masses recorded (15–225 Da) was accumulated over 2 minutes, then normalized by the H3O+ signal measured at mass 21, corresponding to H318O+, and multiplied by 500 to account for the isotopic abundance of 18O with respect to 16O. The result was then multiplied by 150000 to account for instrumental sensitivity to H3O+. The blank spectrum of the experimental chamber on the day of the experiment was subtracted from all acquisitions. Finally, the signal recorded for individual masses was divided by the sensitivity of the instrument determined by the calibration to obtain a concentration in ppb (part per billion in volume). For an unknown compound, X, absent from the calibration mixture, the sensitivity of the instrument, SX, was determined based on the sensitivity factors of the nearest species A and B present in the calibration gas, SA and SB, and considering a proton transfer reaction constant kX of 3 × 10−9 cm3 s−1, according to (1):
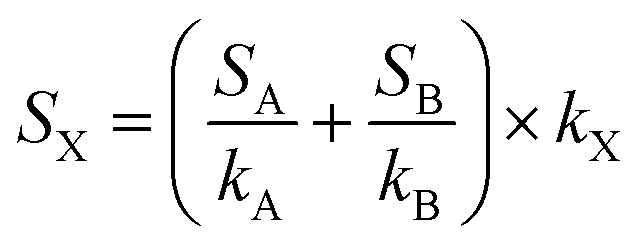 | (1) |
Factors influencing the response of the PTR-ToFMS, like isotopic abundances (75.76% 35Cl and 24.24% 37Cl in chlorobenzene and dichlorobenzene), or fragmentation probability upon reaction with H3O+ (e.g., 50% for α-pinene), were accounted for to determine the sensitivity of the instrument.
The response of the PTR-ToFMS was calibrated following the ACTRIS guidelines.33 Calibration was conducted once to twice a week with a custom-made standard (Messer: methanol 0.95 ppm, acetonitrile 1.24 ppm, acetaldehyde 1.19 ppm, acetone 1.20 ppm, isoprene 1.14 ppm, crotonaldehyde 1.18 ppm, butanone 1.17 ppm, benzene 1.15 ppm, toluene 1.19 ppm, o-xylene 1.22 ppm, chlorobenzene 1.18 ppm, α-pinene 1.19 ppm, and 1,2-dichlorobenzene 1.40 ppm). The gas standard was subjected to several dilution ratios, allowing determination of the sensitivity factors for the compounds of the calibration using the gradient of the response vs. concentration plots.
The acquisition software of the PTR-ToFMS did not allow deconvolution of the peaks observed at the same unit mass. We have therefore gathered the peaks for each unit mass and integrated them to obtain the total VOC concentrations at each unit mass.
During the experiments, after the temperature of the thermoregulated chamber was raised, 30 to 60 minutes were left for the sample to thermalize at the temperature of the chamber, as confirmed by direct measurements of the surface temperature of the asphalt mixture with an infrared thermometer. After one hour spent at a fixed temperature, VOC concentrations were stable as observed by PTR-ToFMS. A typical signal recorded at M + 1 = 71 Da during a step-by-step annealing of the STA3 asphalt mixture is shown on Fig. 2. The signal was considered constant in the second hour of each temperature step. Hence, emissions were determined from data recorded during the second hour of each temperature step. This raises the question of the exhaustion of the emissions from each sample by the temperature cycles in the experimental chamber. Firstly, note that old asphalt pavements have been exposed to the atmosphere for years and yet VOC emissions were still detectable in the laboratory. Hence, these samples were not exhausted and their emissions were at steady-state. Regarding fresh asphalt mixtures, their emissions were monitored over longer timescales and were found constant within experimental uncertainty.
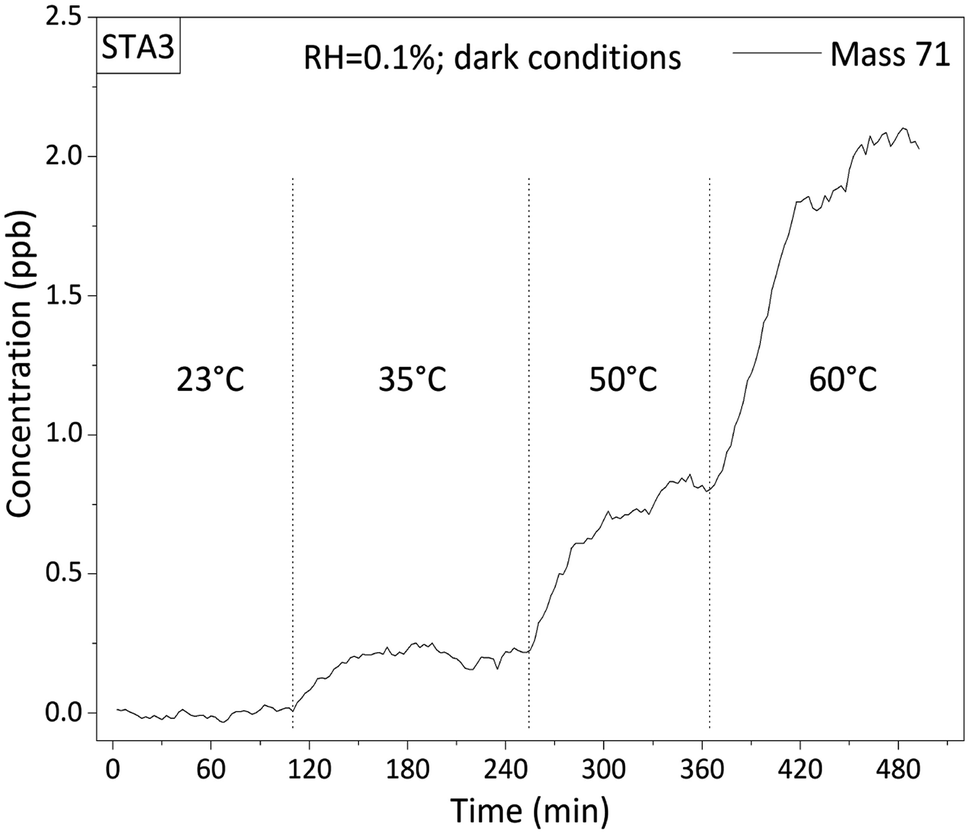 | ||
| Fig. 2 Concentration profile determined with the PTR-ToFMS signal recorded at mass 71 during a typical experiment: sample STA3 was annealed from 23 °C to 35 °C, 50 °C, and 60 °C, under dry (RH = 0.1%) and dark conditions. | ||
2.2.2.2. Offline analysis: GC-MS/FID. GC-MS/FID was used offline to characterize and quantify VOCs. Air samples were taken with a SYPAC V2 sampler (Tera Environment),36,37 connected with Silcosteel tubing to the chamber. The sampler is based on a pump and MFCs controlled with a software to set the flow, time and duration of sampling. Any potential leak where the sorbent tubes are connected to the sampler, is assessed, as well as any possible contamination. After these regular checks, the sampling can be performed through Carbotrap 202 tubes (Sigma-Aldrich) at 150 sccm for 1 hour. Carbotrap 202 tubes are composed of stainless steel, filled with multibed Carbopack B and C and were used to trap C5 to C20 species in air.32,38 These species were expected to be widely present in asphalt binder and emitted by asphalt mixtures. We assume that loss of low-volatility species is minimum in our experimental setup based on the chamber design, the Teflon walls of the chamber, the regular checks of the chamber's blank and the analytical systems, and the Silcosteel tubing connecting the chamber to the instruments.
As discussed in Section 2.2.2.1, real-time monitoring with PTR-ToFMS showed that emissions were constant in the second hour following a temperature increase; therefore, samples were taken for GC-MS/FID analysis during the second hour of each temperature step. After exposure to the asphalt mixture emissions, the Carbotrap tubes were analyzed with a 7890A GC system (Agilent Technologies equipped with an Agilent 5975C Inert MSD mass spectrometer and FID detector). Species adsorbed in the Carbotrap 202 tube were thermally desorbed with a Gerstel TDS-G thermodesorber from room temperature to 250 °C, with a 50 °C per minute ramp. The desorbed species were separated in a CP-Sil 8Cb column (standard polysiloxane, Agilent; 60 m length, 0.32 mm inner diameter, 1 μm film thickness) under a helium flux (4 mL min−1). The flux at the end of the column was split in two; one part was sent to the mass spectrometer, where the molecular components were characterized, while the other part was sent to the FID, where they are quantified.
The calibration process of the GC-MS/FID is described in Section 2 of ESI.† Briefly, Carbotrap 202 tubes were doped with 1 μL of solutions containing known concentrations of toluene, octanal, decanal, dodecane, tridecane, tetradecane, pentadecane, 2-ethyl-1-hexanol, and 1-octene (see Table S2 of ESI†). The response of unexposed Carbotrap tubes and of the empty experimental chamber under all experimental conditions investigated were measured and subtracted from the experimental measurements. The main contaminants observed in the empty experimental chamber and tubes were decanoic, dodecanoic, tridecanoic, and tetradecanoic acids.
2.2.2.3. Experimental protocol. After the asphalt mixtures were laid down in the atmospheric simulation chamber, the chamber was sealed. At least two hours were left for the chamber to be evacuated (air exchange rate n = 3 h−1). Every experiment started with the PTR-ToFMS recording the spectrum of zero air for 20 minutes. Then, the PTR-ToFMS instrument was connected to the exit port of the atmospheric chamber. The concentrations were recorded at the lowest temperature investigated (23 °C) to start and two 1 hour samplings were conducted successively. The temperature of the chamber was then raised to 35 °C. Once the temperature of the chamber was stable, we waited 30 minutes for the temperature of the asphalt sample to stabilize at the value set for the chamber. Two 1 hour samplings were then recorded. The same protocol was repeated at 50 and 60 °C.
2.3. Determination of the emission factors (EF)
Emission Factors (EFs, in μg m−2 h−1) were obtained by converting the measured concentrations, C (ppb, part per billion), of a species of molar mass M (g mol−1), into mass emissions per unit area of asphalt mixture and per unit of time. EFs were normalized by the area (Sasphalt, in m2) and temperature (T, in °C) of the sample and account was taken for the air flow rate in the Teflon chamber, Qe (in m3 h−1), following eqn (2), commonly used in calculations of emissions from materials.39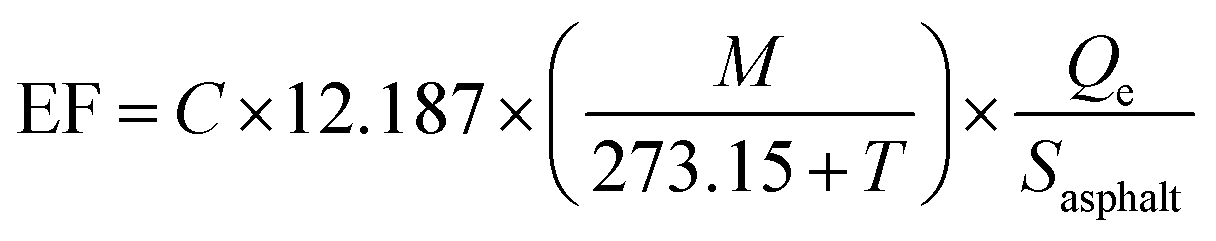 | (2) |
Normalization of EFs in eqn (2) by the surface of each sample investigated, together with the presence of a turbulent flow limiting the depth of the layer where diffusion in the gas phase drives VOC emissions, allowed comparison of experiments involving different samples and conducted under different environmental conditions (T, RH, UV, etc.).
It is important to stress that VOC concentrations determined in the atmospheric simulation chamber may not be directly applicable as VOC emission source terms in atmospheric models. These source terms can be provided by laboratory studies in the form of VOC emission factors (EF) if account is taken of the different environmental parameters dictating outdoor emissions and those in simulation chambers. As discussed in Section 2.2.1, the Reynolds number of the air flow in our experimental chamber required matching with full scale street canyons, with high-to-width ratios below or equal to unity.
3. Results and discussion
In this paper, we present VOC emissions of STA2 and LTA3 samples at 23 °C, 35 °C, 50 °C and 60 °C under dry (RH = 0.1%) and dark conditions. A discussion of the repeatability of the VOC emission factors measured for different STA and LTA samples is given in Section 3 of the ESI;† briefly, the results presented below are conservative lower values of the EFs measured on three samples of each type.3.1. Emission factors derived from PTR-ToFMS measurements
PTR-ToFMS was used to monitor online and in real time VOC emissions of STA and LTA asphalt mixtures. It is complementary with GC-MS/FID which cannot track the concentrations online in real time, but provided a better identification of the molecular species detected. When a STA or LTA asphalt mixture was inserted in the atmospheric chamber, concentrations were measured with the PTR-ToFMS, and then converted into EFs using eqn (2). Peaks observed on mass spectra at M + 1 were tentatively assigned using molecular formulae of VOCs (of molar mass M) observed by other groups;35,40–43 a comprehensive assignment of PTR-ToFMS masses can be found in Yañez-Serrano et al.44 102 masses have thus been followed (see Table S3 in ESI†), from mass 15 to 225. VOC emission factors were calculated based on 78 masses, after inorganic ions, reagent ions, fragments (M + 1 < 40 Da), and M + 1 > 200 Da have been excluded (lines shaded in orange in Table S3†). In the latter case (M + 1 > 200 Da), the uncertainty on the determination of the sensitivity of the instrument was high, hence the calculation of the emission factors was not reliable and has been excluded.The total EF of VOCs detected with PTR-ToFMS, emitted by STA and LTA asphalt mixtures annealed from 23 °C to 35 °C, 50 °C, and 60 °C, under dry (RH = 0.1%) and dark conditions, are displayed in Table 2. These values show that fresh and old asphalt mixtures emit more VOCs when the temperature increases, confirming the results of Khare et al.29 VOC EFs of three different STA and LTA samples have been monitored at 50 °C with PTR-ToFMS to estimate the variability of our measurements (see Section 3 of ESI† for a discussion); they vary by a factor of roughly 3. The values displayed here for STA3 and LTA2 asphalt mixtures represent conservative lower values of VOC emissions by the three samples of each type.
| T (°C) | Total VOC EFPTR STA (μg m−2 h−1) | Total VOC EFPTR LTA (μg m−2 h−1) |
|---|---|---|
| a Values of the uncertainty (±1σ) are calculated as shown in Section 4 of ESI. | ||
| 23 | 2 ± 2 | 223 ± 49 |
| 35 | 64 ± 29 | 314 ± 66 |
| 50 | 413 ± 110 | 466 ± 81 |
| 60 | 1033 ± 194 | 989 ± 134 |
The evolution of EFPTR values for total VOCs as a function of temperature in the 23–60 °C range can be accurately fitted with simple exponential functions given in eqn (3), for both STA and LTA asphalt:
| EFPTR(T) = EF(0 °C) + A × exp(αT) | (3) |
The data and fits are plotted in Fig. 3. The adjusted R2 is > 0.95 for both fits of STA and LTA total VOC EF, showing the excellent agreement of the fits with the data. The growth coefficient, α, is similar for both types of asphalt mixtures: α = 0.095 ± 0.007 K−1 for STA, α = 0.105 ± 0.029 K−1 for LTA, meaning that the total VOC EFs of STA and LTA specimens grow roughly by a factor of 2.7 for every 10 °C-increase of the samples' temperature. This is a much faster increase of VOC emissions with temperature than observed by Khare et al.:29 in their work, the total VOC EF doubles between 40 °C and 60 °C, whereas our experiments suggest that it would be multiplied by more than 7 over the same temperature range. An explanation for this is that we also measure the VOC fraction < C10 that Khare et al. considered negligible based on their experiments.29 Most of the emissions they observe come from VOCs > C10, and from sulfur-bearing species, the former being mostly undetected in our work. Moreover, Khare et al.29 used asphalt samples “collected during paving operations”, i.e. what we define as STA asphalt mixtures. LTA asphalt produces stronger total VOC emissions than STA asphalt, except at 50 °C and 60 °C, where they are similar. Atmospheric oxidation of asphalt binders, and deposition of VOCs by various sources in the urban environment may account for the larger VOC emissions seen for old asphalt pavements. Generally, in the real world, road pavements have aged and have been weathered and are not freshly deposited, therefore bearing more similarities with LTA than with STA asphalt. Estimates of emissions based on the VOC emissions from STA asphalt mixtures therefore largely underestimate emissions from road asphalt at relevant atmospheric temperatures of 23 °C and 35 °C.
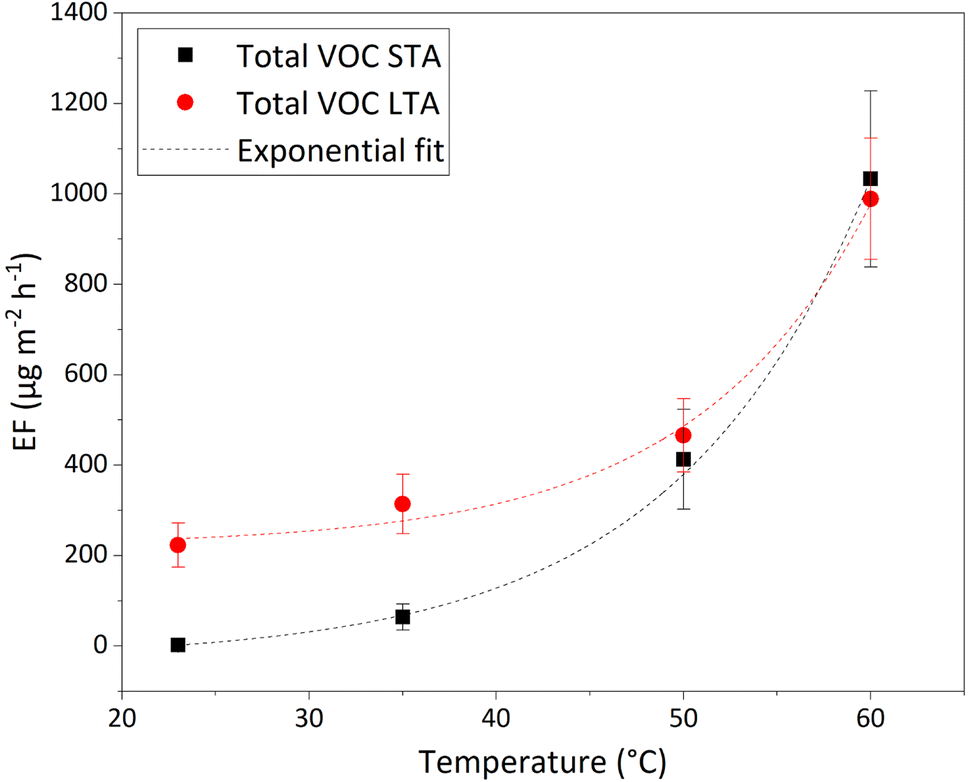 | ||
| Fig. 3 Evolution of the total EF of VOCs measured by PTR-ToFMS, as a function of temperature, under dry (RH = 0.1%) and dark conditions, for STA3 (black squares) and LTA2 (red circles) asphalt mixtures. Fits of the data are shown with dashed lines. | ||
Protonated VOCs corresponding to the 13 compounds used for calibration of the PTR-ToFMS have been tracked. Their EFs are displayed in Table 3 for STA and LTA asphalt mixtures in the 23–60 °C range. Although these compounds contribute to emissions at their respective mass, EFs measured at a specific mass generally cannot be uniquely assigned to the corresponding calibration species because of the contributions from other species (or fragments). This, however, does not hold for M + 1 = 33, and 42 which, to the best of our knowledge, can be assigned solely to methanol and acetonitrile, respectively. When a signal was detected above the limit of detection (LOD), masses in Table 3 behaved like the total VOC emissions, i.e., they increase with temperature for both STA and LTA asphalt mixtures. The only exception is mass 59, which decreased slightly between 23 °C and 35 °C for LTA asphalt, but this decrease was small compared to the increase observed for other masses and was therefore not considered significant.
| STA3 EF (μg m−2 h−1) | LTA2 EF (μg m−2 h−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| T (°C) | 23 | 35 | 50 | 60 | 23 | 35 | 50 | 60 |
| Mass detected with PTR-ToFMS M + 1 (Da) | ||||||||
| 33 | <13.5 | <6.7 | ||||||
| 42 | <1.8 | <0.8 | ||||||
| 45 | <12.3 | <2.3 | 4.05 ± 1.25 | 18.31 ± 2.02 | ||||
| 59 | <3.1 | 3.43 ± 0.42 | 12.80 ± 1.34 | 2.93 ± 0.38 | 1.97 ± 0.27 | 3.59 ± 0.39 | 9.80 ± 0.91 | |
| 69 | <2.5 | <1.1 | ||||||
| 71 | <1.6 | 3.03 ± 0.73 | 9.09 ± 1.29 | 22.52 ± 2.37 | 7.29 ± 0.94 | 11.51 ± 1.31 | 15.09 ± 1.43 | 30.05 ± 2.33 |
| 73 | <2.5 | 5.33 ± 1.19 | <1.1 | 2.63 ± 0.60 | ||||
| 79 | <22.5 | <9.4 | ||||||
| 93 | <1.5 | <0.6 | 0.92 ± 0.49 | |||||
| 107 | <2.1 | <0.9 | 1.36 ± 0.65 | |||||
| 113 | <4.4 | 6.18 ± 1.98 | 14.19 ± 3.37 | <2.6 | 2.64 ± 0.75 | |||
| 137 | <1.7 | 8.01 ± 3.18 | 15.77 ± 4.70 | 3.86 ± 1.69 | 4.31 ± 1.77 | 6.11 ± 1.95 | 13.72 ± 3.09 | |
| 147 | 0.69 ± 0.88 | 0.51 ± 0.55 | 2.88 ± 1.70 | 4.29 ± 2.30 | <2.1 | 2.35 ± 1.22 | 8.54 ± 2.62 | |
The 20 most important contributors to the total emissions shown in Table S3 of ESI† (>75% overall, and up to 99% in individual experiments) have been tracked. It is important to note that Table S3† provides only tentative assignments for masses observed by PTR-ToFMS in terms of molecular formulae, without giving a quantitative proportion of the species and fragments, as reported in the literature.
These 20 masses (M + 1) are, in Da units: 57, 71, 85, 111, 125, 127, 129, 139, 141, 143, 151, 155, 157, 166, 169, 178, 179, 182, 183, 195, and the sum of the 58 other masses. Their individual EFs are >22 μg m−2 h−1 for STA and LTA asphalt mixtures at 60 °C. The EF of each mass increased with temperature for both fresh and old asphalt mixtures, confirming the general trend observed on Fig. 3 with the total EF of all VOCs.
Fig. 4 presents the relative proportion of the emissions of STA asphalt mixtures, after total emissions were normalized to 100% at each temperature. This figure shows that the contribution of “other” masses dominates the emissions from STA asphalt at all temperatures, except at 35 °C, and represent 100% of emissions at 23 °C. At 35 °C, twelve of the main masses contribute to the emissions; all masses contributed to emissions at 50 °C and 60 °C, except mass 157 which was not detected at 50 °C. Emissions were dominated by masses 183, 155, 179, and 169. The other major masses measured were lower than the four dominant masses by a factor of 2 or more at 60 °C.
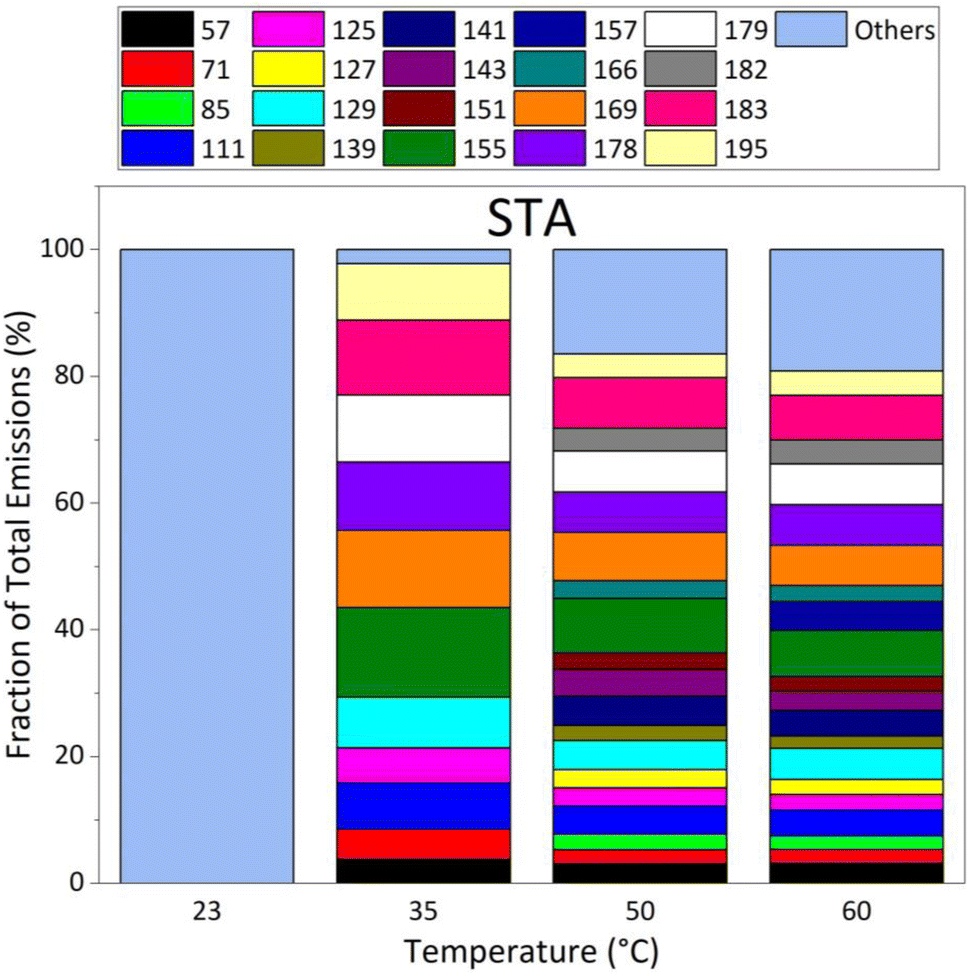 | ||
| Fig. 4 Evolution of VOC emission composition in % for the 20 masses exhibiting the strongest contributions (EF > 22 μg m−2 h−1 at 60 °C) as a function of temperature, as measured by PTR-ToFMS under dry (RH = 0.1%) and dark conditions, for the STA3 fresh asphalt sample. | ||
Fig. 5 focuses on emissions by LTA (old) asphalt mixtures. It shows that the relative contribution of “other” masses dominates LTA asphalt emissions at all temperatures. Besides “other” masses, LTA asphalt emissions are dominated by masses 169, 155, and 183, masses also observed to dominate emissions of STA asphalt mixtures. However, at 60 °C, emissions at mass 179 are a factor of three lower than those at masses 169, 155 and 183. Overall, the composition of the emissions of LTA asphalt remains roughly stable between 23 °C and 60 °C, with all masses contributing at all temperatures investigated. Masses 166, 178, 182 and 195 have increasing relative contributions with growing temperature.
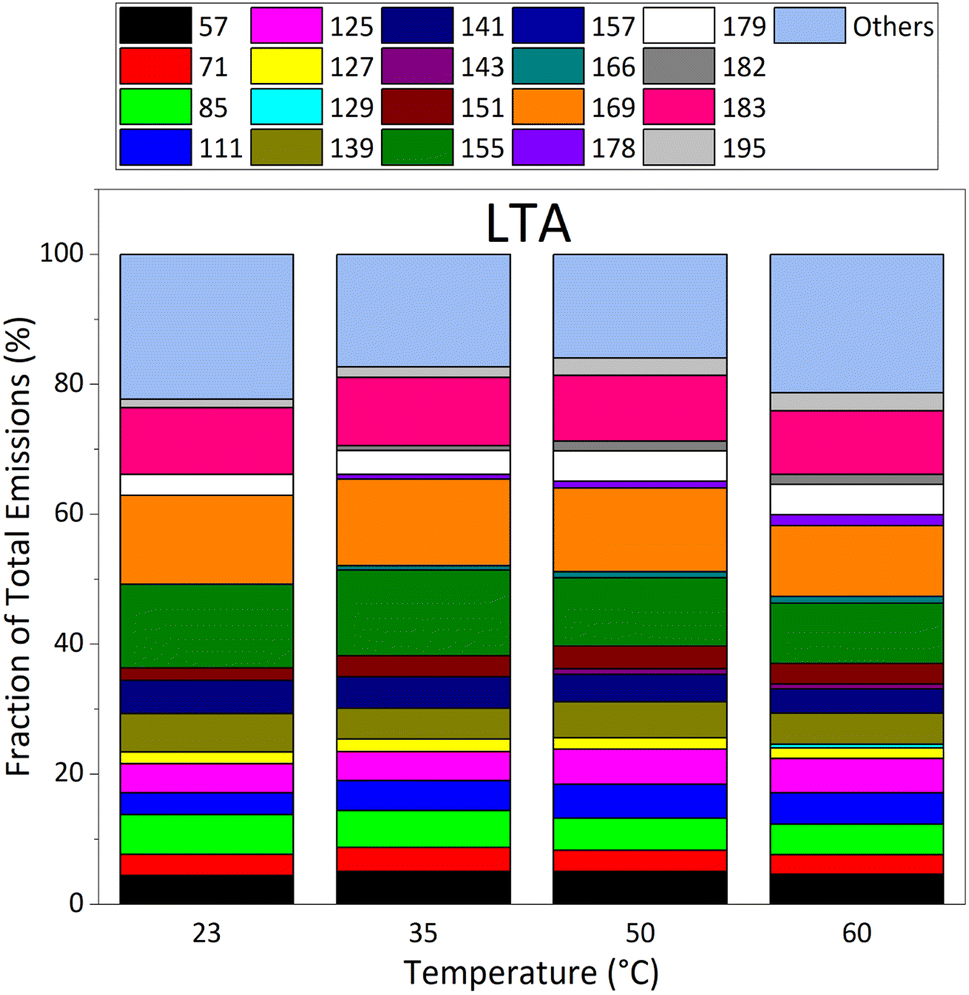 | ||
| Fig. 5 Evolution of VOC emission composition in % for the 20 masses exhibiting the strongest contributions (EF > 22 μg m−2 h−1 at 60 °C) as a function of temperature, as measured by PTR-ToFMS under dry (RH = 0.1%) and dark conditions, for the LTA2 old asphalt sample. | ||
Besides dominant masses, emissions of other individual masses were generally similar in STA and LTA asphalt mixtures' emissions at 50 °C and 60 °C. The larger individual emissions of LTA asphalt at 23 °C and 35 °C pointed again to old asphalt mixtures being stronger emitters of individual VOCs at service temperatures than STA asphalt mixtures. We also note that potential contributors to masses 155, 169, 179, and 183 (see Table S3 in ESI†) more often bear a heteroatom (N or S) than contributors to other major masses. It suggests important emissions of nitrogen- and sulfur-bearing species, besides the importance of oxygenated compounds described here that may follow atmospheric aging of old asphalt pavements. The emission patterns of STA and LTA asphalts were therefore different qualitatively, especially at 23 °C and 35 °C.
These results suggest that old asphalt pavements emit more VOCs than fresh ones in the temperature range investigated. It seems in contradiction with the results of simulated distillation (Section 1.3 of ESI†), that show that fresh binders contain a higher fraction of low carbon, and hence high-volatility, compounds. However, since the materials, in particular the binders, may not be the same in STA and LTA mixtures, this hypothesis needs to be further investigated by comparing emissions from asphalt mixtures from the same material, fresh and after aging on site for several years. At all events, given that asphalt layers can remain in service more than 20 years, emissions from old asphalt mixtures should be more representative of the global contribution to atmospheric chemistry. In addition, it is unclear at this point whether the emitted VOCs come from the binder and its oxidation products, or if they accumulate at the surface after being released by traffic-related sources. Clearly, more work is needed to fully understand the origin of VOCs emitted by asphalt mixtures.
3.2. Emission factors derived from GC-MS/FID measurements
GC-MS/FID was used offline to identify and quantify emitted VOCs, with 65 compounds being identified; they are listed in Table S4 of ESI.† They consist of 25 alkanes, from octane (C8H18) to 2,6,10,14-tetramethylhexadecane (C20H42); 7 alkenes, from 2-ethyl-1-hexene (C8H16) to 1-tetradecene (C14H28); 12 aromatics, from o/p-xylene (C8H10) to x,y,z-trimethylnaphatalene (C13H14); 6 alcohols, from 2-ethyl-1-hexanol (C8H18O) to 4-methyl-1-(1-methylethyl)-3-cyclohexene-1-ol (C10H18O); 13 carbonyls, divided into 9 aldehydes from pentanal (C5H10O) to tetradecanal (C14H28O), and 4 ketones, from 3-heptanone (C7H14O) to 2-dodecanone (C12H24O); and 2 other compounds, dibenzofuran (C12H8O) and 1,1′-oxybis-octane (C16H34O). We note that emissions of toluene and 1-octene were below detection limits at all temperatures investigated. Few species with a heteroatom (besides oxygen) were detected, due to the selective Carbotrap surface preferentially trapping hydrocarbons. EFs of individual species are given in Table S4†, for STA and LTA asphalt mixtures, at 23 °C, 35 °C, 50 °C, and 60 °C, under dry (RH = 0.1%) and dark conditions. The trend for total VOC emissions, shown in Table 4, is for EFs to increase with temperature, both for STA and LTA asphalts. The numbers are broadly consistent with the values found with PTR-ToFMS (Table 2); the differences observed can be accounted for by the inherent limitations and selectivity of both techniques.| T (°C) | Total VOC EFGC STA (μg m−2 h−1) | Total VOC EFGC LTA (μg m−2 h−1) |
|---|---|---|
| 23 | 3 ± 1 | 53 ± 18 |
| 35 | 14 ± 4 | 51 ± 18 |
| 50 | 109 ± 34 | 129 ± 44 |
| 60 | 222 ± 70 | 186 ± 63 |
VOC EFs for three different STA and LTA samples have been monitored with GC-MS/FID at 50 °C to estimate the repeatability of our measurements (see Section 3 of ESI† for a discussion). The values are displayed here for STA3 and LTA2 asphalt mixtures, and represent conservative lower values of VOC emissions among the three samples of each type tested.
The total VOC EFs obtained with GC-MS/FID shown in Table 4 were fitted to eqn (3), as performed for the PTR-ToFMS data. The good agreement of the fits with the data was confirmed by the values of the adjusted R2: it is ≈0.91 for STA, and >0.74 for LTA. The growth coefficient, α, is also similar within error for both types of asphalt: α = 0.107 ± 0.019 K−1 for STA, α = 0.086 ± 0.062 K−1 for LTA. These values are in line with those obtained with PTR-ToFMS data, which confirms the agreement of the measurements of total VOCs with both techniques.
A discussion of VOC emissions by chemical family (i.e., alkanes, aromatics, alkenes, carbonyls, alcohols, and other species) can be found in Section 8 of ESI.† In the following, we focus on emissions of individual VOCs. The major contributors are defined, as for PTR-ToFMS results, as species whose added contributions represent more than 75% of the total EF measured by GC-MS/FID at all temperatures.
VOC emissions from STA (fresh) asphalt mixtures are shown on Fig. 6; it displays the fractions of the total emissions at each temperature investigated. The major compounds in emissions from STA asphalt mixtures are, from the most to the least abundant at 60 °C: 2-ethyl-1-hexanol, acenaphthene, x,y-dimethylnaphthalene, nonanal, decanal, 5,6-dipropyldecane, tetradecane, hexanal, pentadecane, x-methylnaphthalene, 2-ethylhexanal, hexadecane, x,y,z-trimethylnaphthalene, octanal, tridecane, tridecanal, 2-dodecanone, 1-ethylnaphthalene, and undecane. These compounds accounted for more than 75% of VOC emissions by STA asphalt mixtures, whereas they only represented 17.6–35.1% of old (LTA) asphalt emissions, depending on temperature.
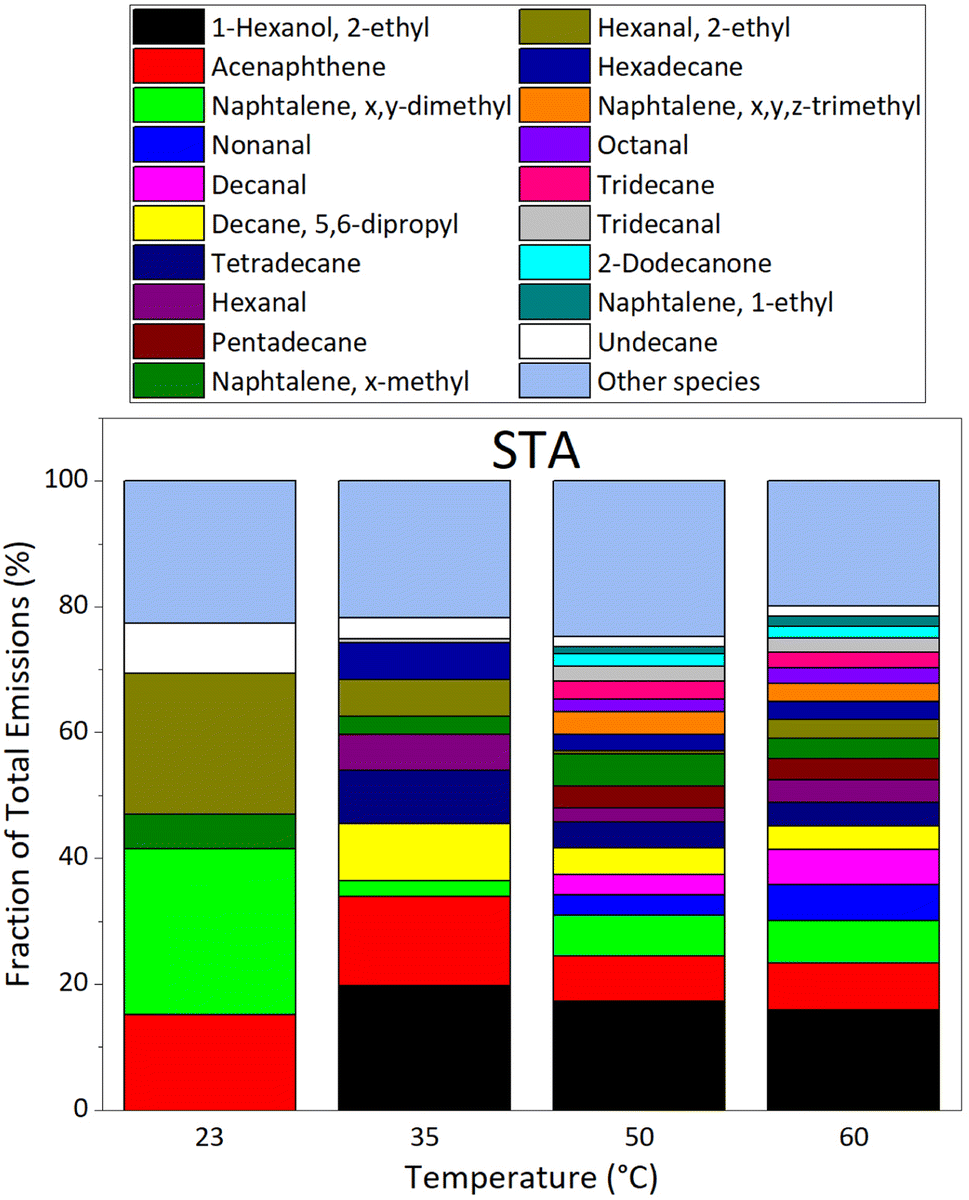 | ||
| Fig. 6 Evolution of VOC emission composition in % for the species accounting for more than 75% of VOC emissions (EF > 3.49 μg m−2 h−1 for STA asphalt mixtures) as a function of temperature, as measured by GC-MS/FID under dry (RH = 0.1%) and dark conditions, for the STA3 fresh asphalt sample. | ||
At temperatures of 40–60 °C, Khare et al. observed dominant emissions from STA asphalt of C10–C32 alkanes, followed by x,y-dimethylnaphthalene.29 This is in agreement with our observations; 5,6-dipropyldecane, tetradecane, pentadecane, hexadecane, tridecane, undecane, and x-methyldodecane, are C10–C32 alkanes, and the sum of their EFs is higher than that of x,y-dimethylnaphthalene at all temperatures investigated. Khare et al. measured EF(x,y-dimethylnaphthalene) ≈ 0.08 μg min−1 kg−1 at 40 °C, and ≈0.2 μg min−1 kg−1 at 60 °C.29 Our EF values for STA asphalt give EF(x,y-dimethylnaphthalene) ≈ 2.2 × 10−4 μg min−1 kg−1 at 35 °C, and ≈0.01 μg min−1 kg−1 at 60 °C; this is a factor of 360 lower than Khare et al. at 35 °C, and a factor of 20 lower at 60 °C. Total n-alkanes emissions may also be compared. Khare et al., measure the C10–C32 alkane fraction: EF(C10–C32 alkanes) ≈ 3 μg min−1 kg−1 at 40 °C, and ≈10 μg min−1 kg−1 at 60 °C.29 Our measurements of the C8–C16 alkane fraction give: EF(C8–C16 alkane) ≈ 0.003 μg min−1 kg−1 at 35 °C, and ≈0.04 μg min−1 kg−1 at 60 °C. These values differ by a factor of 1000 at 35 °C, and of 250 at 60 °C, with those of Khare et al. for C10–C32 alkanes.29 We note however that the alkane fractions measured in emissions are not the same in our work and in Khare et al.29 Although we anticipate a lower emission of higher mass alkanes due to their lower volatility, these species generally contribute more to mass emissions as the number of carbon atoms increases (see Table S4†). Extrapolation of this trend suggests that the C16–C32 alkane fraction, measured by Khare et al.,29 but not in the present work, could explain part of the difference with our results. Moreover, we note that the asphalt samples used in our work are different from those used by Khare et al.,29 who studied fresh asphalt mixtures shortly after deposition, whereas our fresh samples were studied a longer time after deposition. The kinetics of VOC emissions in Khare et al., suggest that they measure peak values, whereas we measure steady-state emissions that take place after longer times.29 Also, the custom-made adsorbent tubes46 used in Khare et al. to sample VOCs in air allow detection of many species, including sulfur-bearing compounds, at sub-ppt levels, and may perform differently than our Carbotrap tubes that target specifically C5 to C20 species in air, leading to quantitatively different results. Also, the bitumens used in the asphalt mixtures probably have different compositions, from which different emission profiles certainly arise. Lastly, emissions were recorded in experimental setups exhibiting different characteristics, especially in terms of air exchange rate and charge ratio of the samples; it certainly creates another difference in the emission factors measured.
VOC emissions of LTA (old) asphalt mixtures are shown on Fig. 7; the top panel reports EF values, whereas the bottom panel displays emissions as fractions of the total emissions at each temperature investigated. The major compounds in emissions of STA asphalt mixtures were, from the most to the least abundant at 60 °C: dodecane, undecane, x-methyldecane, decane, tridecane, x-methylundecane, x,y-dimethylnaphthalene, 2,6-dimethylundecane, benzaldehyde, nonane, x-methyldodecane, decanal, x,y,z-trimethylnaphthalene, x-methylnaphthalene, and 2-methylnonane. These compounds accounted for more than 75% of VOC emissions by LTA asphalt mixtures, whereas they only represent 13.6–57.9% of fresh (STA) asphalts emissions, depending on temperature.
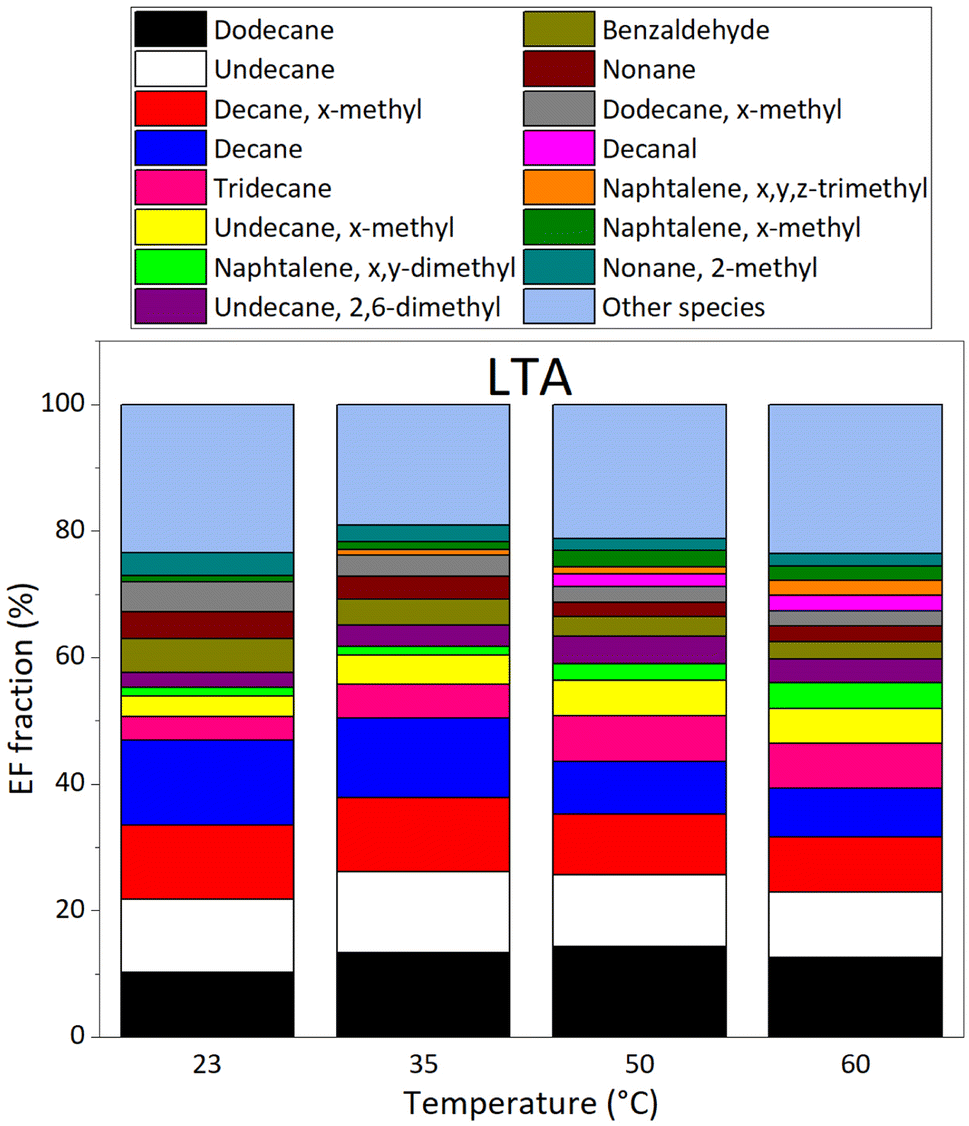 | ||
| Fig. 7 Evolution of VOC emission composition in % for the species accounting for more than 75% of VOC emissions (EF > 3.63 μg m−2 h−1 for LTA asphalt mixtures) as a function of temperature, as measured by GC-MS/FID under dry (RH = 0.1%) and dark conditions, for the LTA2 old asphalt sample. | ||
To the best of our knowledge, VOC emissions by old asphalt mixtures have never been studied before; this precludes comparison of our results with relevant data on LTA asphalt mixtures. At all temperatures, LTA asphalt emits mostly alkanes (75–80%), in contrast with emissions of STA asphalt, to which alkanes contribute for ≈30% (see Section 8 of ESI†). Methyl-, dimethyl-, and trimethyl-naphthalene are major contributors both in old and fresh asphalt mixtures emissions. Nonetheless, their relative contributions follow opposite trends with temperature. Taking the emissions from old asphalt, the contribution of these species to total VOC emissions represent 2.4% at 23 °C, growing continuously to reach 8.8% at 60 °C. Whereas in the emissions from fresh asphalt, their contribution decreases from 31.9% at 23 °C, to 12.9% at 60 °C. The total EF of methylated naphthalenes was higher in LTA asphalts' emissions than in STA's at 23 °C and 35 °C, by a factor of roughly two; it is the opposite at 50 °C and 60 °C, with emissions of methylated naphthalenes in STA asphalt being stronger by a factor of two. Emissions of fresh asphalt are also abundant in carbonyl compounds (14–28%), especially aldehydes, which are less abundant in old asphalt emissions (5–7%).
3.3. Complementarity of GC-MS/FID and PTR-ToFMS results
Several compounds detected with GC-MS/FID have M + 1 values corresponding to major masses observed with PTR-ToFMS. However, several of them are alkanes whose proton affinities (PA) are lower than that of water, precluding their detection with PTR-ToFMS. Four of these species have PAs higher than PA(H2O): acenaphthene,47 menthol,48x-methylnaphthalene47,49 and x-methyl(1,1′)biphenyl.47 Therefore, these compounds are likely major contributors to the PTR-ToFMS signal at their respective mass. Note that x-methylnaphthalene corresponds to a major mass in PTR-ToFMS spectra and is identified as a major emitter by GC-MS/FID for both STA and LTA asphalt mixtures; this is also true for acenaphthene in STA asphalt. Therefore, these two compounds can be confidently identified as major contributors both to PTR-ToFMS and GC-MS/FID data.We focus here on the quantitative comparison of PTR-ToFMS and GC-MS/FID techniques, having already discussed the qualitative aspects. We have seen that the quantitative trends drawn by both techniques are consistent although these techniques do not give access to the same information, and do not monitor the same VOC species.
We have developed two comparative methods to get the most exhaustive view of VOC EFs of asphalt mixtures. In a first method (I), we add the total EF of VOCs identified with GC-MS/FID, to the EFs determined with PTR-ToFMS from which we subtract the masses of compounds observed with GC-MS/FID, to prevent double-counting. The result is shown on Fig. 8 (left column) for STA (A) and LTA (B) asphalt mixtures. In a second method (II), we add the total EF determined with PTR-ToFMS to the EFs of alkanes determined with GC-MS/FID, this type of compounds not being detected with PTR-ToFMS. Adding EFs in such manner leads to Fig. 8 (right column) for STA (C) and LTA (D) asphalts.
 | ||
| Fig. 8 EFs determined in this work by PTR-ToFMS and GC-MS/FID for STA (A and C) and LTA (B and D) asphalt mixtures. In the left column (A and B), the total EF determined by GC-MS/FID is added to the EF determined with PTR-ToFMS, from which masses of the species detected with GC-MS/FID are subtracted (method I). In the right column (C and D), EFs of alkanes determined with GC-MS/FID are added to the total EF determined with PTR-ToFMS (method II). | ||
Comparison of A with C (STA) on the one hand, and of B with D (LTA) on the other hand, shows that both methods agree on the trend of increasing VOC emissions with temperature. However, method (II) systematically leads to higher values than method (I). For STA asphalt, the difference is, at 50 °C and 60 °C, close to 25%; total EFs are too small at 23 °C and 35 °C for the relative difference to be statistically significant. For LTA asphalt, the difference varies between 31.3% at 50 °C and 42.3%; averaged over all temperatures investigated, it is 36.4%. It means that masses subtracted from the PTR-ToFMS data in method (I) actually include many species that are not seen with GC-MS/FID. Method (II) therefore quantifies more realistically VOC emissions than method (I). At all events, it points to species missing from urban emission inventories and advocates for a search for these species with adapted and selective techniques. Sulfur-bearing species are important contributors to fresh asphalt emissions and may contribute to bridge this gap.29
4. Implications for urban atmospheric chemistry
We set out to evaluate the impact of VOC emissions from asphalt mixtures in Paris, France. Old asphalt mixtures are assumed to be the most representative asphalt surfaces, because asphalt-covered surfaces are renewed after years to decades of service. The city of Paris covers an area of 105.4 km2. Based on the fraction of paved surfaces (roads, parking lots, sidewalks) determined in Sacramento, USA12 we estimate that 40% of Paris downtown area, i.e., 42.16 km2, is covered with asphalt pavements. Emissions determined in this work are compared with the EMEP (European Monitoring and Evaluation Program) emission inventory [https://www.ceip.at/the-emep-grid/gridded-emissions]. EMEP emissions are determined with a 0.1° spatial resolution. The 0.1° × 0.1° minimum area resolved represents 82.14 km2 at Paris latitude, close to the actual area of Paris (105.4 km2). In the following, emissions for the city of Paris are therefore taken as EMEP emissions of a 0.1° × 0.1° area at longitude 2.35°E, and latitude 48.85°N, the available coordinates nearest to the actual position of Paris. Since the grid cell is smaller than Paris and its center (2.35°E; 48.85°N) is close to the center of Paris, we consider that the emissions in this grid cell are representative of those of Paris itself.Climate models suggest that average temperatures may increase by, on average, +2 °C by 2050, and by +3 °C by 2100, relative to temperatures in 1850–1900.50 Heatwaves will become stronger and more frequent, making temperatures of asphalt pavements in cities higher over extended periods. Based on this, we put forward a scenario for temperatures experienced by asphalt mixtures in Paris in the future. The temperatures over a year are approximated with those studied in this work (23 °C, 35 °C, 50 °C, 60 °C) for convenience. We note that a quantitative extrapolation of total VOC emissions is possible based on the fits to our data shown on Fig. 3. However, in our opinion, it is unlikely that the composition of the emissions remains the same at different temperatures, based on our results in the 23–60 °C range. Hence, dedicated experiments at lower and higher temperatures would be more reliable but in the calculations below, we stay with the temperatures investigated in this work (23–60 °C). Asphalt temperatures of 60 °C (or higher) may be experienced during 4 months in summer (June to September), for 12 hours per day. During two months (April and May), asphalt temperature could reach 50 °C for half of the day. During another two months (March and October), it could reach 35 °C during 12 hours per day. During the coldest months of the year (November to February), we use the lowest temperature studied in this work, 23 °C. Nighttime temperatures are also approximated with 23 °C throughout the year. Results based on this scenario are given below.
Lastly, we point out that the experiments presented in this paper are conducted under dry and dark conditions to focus on the effect of temperature on VOC emissions. However, realistic conditions of relative humidity and solar irradiation increase the VOC emission of asphalt-covered surfaces and modify them qualitatively. The quantitative values determined here and used for atmospheric implications, are therefore lower limits to real-world VOC emissions and hence to ozone and SOA formation. Actual values for these parameters are certainly much higher than calculated here.
4.1. Total VOC emissions in Paris
Firstly, we compare EMEP VOC emissions in Paris, with emissions of a source missing from inventories, asphalt-covered surfaces. Based on the annual asphalt temperature scenario described above and using the total VOC EF determined in Section 3.3 with method (II) (see Fig. 8), we estimate VOC emissions from asphalt-covered surfaces for the city of Paris over a year as 0.148 Gg. The total yearly NMVOC EMEP emissions of Paris for 2019 are 5.058 Gg, with the main contributions from solvents (3.906 Gg), road transportation (0.694 Gg), fugitive emissions (0.129 Gg), and industry (0.099 Gg). Our work shows that, for Paris in 2019, emissions from asphalt-covered surfaces represent 21.3% of NMVOC emissions from road transportation, and 2.9% of total NMVOC emissions. These values demonstrate that asphalt surfaces will represent a significant fraction of future total NMVOC emissions. This source needs consideration in the context of reducing urban pollution, especially from the road transportation sector through extending the use of electric vehicles.Moreover, these VOC emissions, missing from current inventories, could significantly participate to the missing OH reactivity in urban areas, which can reach 75% in Paris (54% on average over the entire MEGAPOLI campaign)51 and 50% in Los Angeles.52 The extent of this contribution from asphalt-covered surfaces needs to be evaluated with model simulations of VOC concentrations, but is beyond the scope of this paper.
4.2. Ozone formation potential
The ozone formation potential (OFP) is a useful concept to gain an understanding of how much ozone may be formed from different molecular species and from different emission sources. The Maximum Incremental Reactivity (MIR) scale was developed to estimate the maximum amount of ozone (Δ[O3]) that may be formed by an increment of VOC concentration (Δ[VOC]) in high-NOx urban environments,53 as the ratio Δ[O3]/Δ[VOC]. It is based on “model calculations of effects of additions of the VOCs on ozone formation in one-day box model scenarios representing conditions where ambient ozone is most sensitive to changes in VOC emissions”.54 MIR values therefore depend on the location and atmospheric parameters for which they are calculated, and on models determining the profile concentration of VOCs.55 The MIR scale has been applied mostly to US urban conditions.With a different approach, the POCP (Photochemical Ozone Creation Potential) scale was developed56 to assess the impact of VOCs on ozone formation over longer timescales (up to 5 days) using a photochemical trajectory model. The POCPi index is defined, here for VOCi, as the ratio of the additional mass of ozone formed in the presence of a fixed amount of VOC, Δ[O3]with VOCi, to the additional mass of ozone formed in the presence of the same amount of ethene, and multiplied by 100 (POCPethene is set as 100), as shown in eqn (4). The POCP scale is mostly applied to north-west European conditions and is therefore relevant to this work. Note that MIR and POCP scales are in very good general agreement.57–59
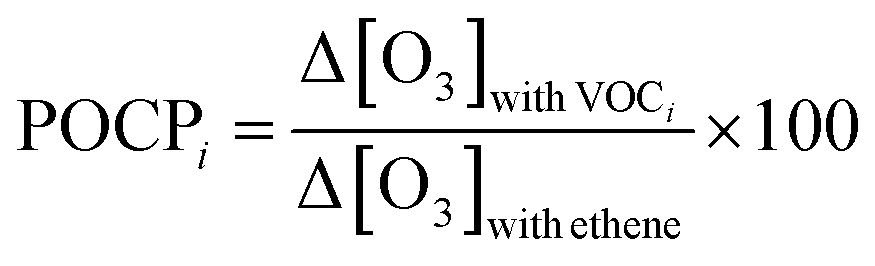 | (4) |
To allow comparison with other sources, the POCP index of asphalt-covered surfaces is estimated by weighting the POCPi values of individual compounds, with their relative contributions to the total VOC emissions as measured with GC-MS/FID in the present work and summing over all detected species for which POCP values are available, as shown with eqn (5).
 | (5) |
Our estimation of the POCP index of asphalt-covered surfaces was based only on the data of the current study, and particularly the GC-MS/FID results from which the speciation of the emitted compounds is known. We were able to extract or infer POCP values for 58 compounds59,60 out of the 65 species identified with GC-MS/FID (see Table S5 in ESI†). It also means that compounds detected with PTR-ToFMS, but not identified with GC-MS/FID, are not taken into account in these values, making the latter lower estimates. The values obtained for STA and LTA asphalt mixtures between 23 °C and 60 °C and for the temperature scenario defined above, are shown in Table 5. Another approach to estimate the POCP index of asphalt pavements would be to combine the GC-MS/FID results of the current study with those reported by Khare et al.29 However, the samples considered in the study of Khare et al.,29 were taken from North American asphalt pavements, and are therefore of a different nature to those used in the current study (the samples are relevant to Europe, particularly to France), which is likely to result in different emission profiles. In addition, Khare et al.,29 did not provide a detailed speciation analysis of the emissions from the asphalt pavements in their study, which precludes assigning POCP coefficients specific to each chemical compound, as opposed to what is made possible by our GC-MS/FID results. In conclusion, the estimation of the POCP index of asphalt pavements is based on the results of the current study and should be considered as a lower limit, because other species which were not identified and quantified with our sampling method could also contribute to ozone formation.
| T (°C) | POCP index STA (fresh) asphalt | POCP index LTA (old) asphalt |
|---|---|---|
| 23 | 62.0 | 38.9 |
| 35 | 64.3 | 39.1 |
| 50 | 60.7 | 40.9 |
| 60 | 53.6 | 42.7 |
| Annual temperature scenario (see text) | 60.7 | 39.7 |
The POCP index of LTA asphalt mixtures increases gradually with temperature. Therefore, with increasing surface temperatures, emissions from asphalt mixtures participate more in ozone formation in urban environments. This suggests that asphalt emissions may contribute to the increasing trend of urban ozone concentration with air temperature.61 The POCP index of fresh asphalt mixtures is higher than that of old asphalt at all temperatures investigated; it increases between 23 °C and 35 °C, but decreases above. POCP indexes determined for a realistic annual asphalt temperature scenario are mostly relevant for LTA asphalt because STA asphalt mixtures would age over a year, shifting from STA to LTA surfaces, as time passes.
To the best of our knowledge, POCP indexes have not been evaluated for VOC emitting sectors in Paris, nor in France. Therefore, we compare our results to the most comprehensive POCP inventory made for a neighboring country, the United Kingdom. The POCP indexes are:6 Road transport – exhaust: 69; agriculture: 56; road transport – petrol evaporation: 50; combustion: 48; wastes: 47; off-road: 46; solvent: 44. LTA asphalt mixtures have a POCP index of 39.7 for our annual temperature scenario, a value lower than the other ozone precursor sources listed above. The ozone concentration eventually formed by VOC emissions of asphalt-covered surfaces remains to be precisely estimated by air quality models.
4.3. Secondary organic aerosol formation potential
The Secondary Organic Aerosol formation Potential (SOAP) concept is analogous to the POCP concept and allows the assessment of the propensities of VOCs to form secondary organic aerosol (SOA).62 The SOAP index is defined as the ratio of the additional mass of SOA formed in the presence of a fixed amount of VOC, Δ[SOA]with VOCi, to the additional mass of SOA formed in the presence of the same amount of toluene and multiplied by 100 (SOAPtoluene is set as 100), as shown in eqn (6).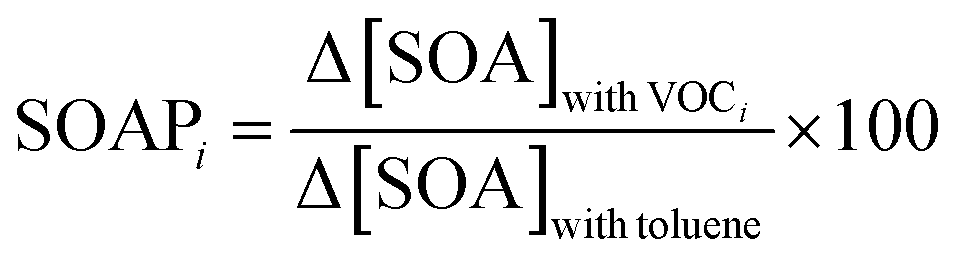 | (6) |
Similarly to the calculation of the POCP index, only the data of the current study were used to estimate the SOAP indices of asphalt pavements. In particular, the SOAP indices of 32 VOCs detected in this work in emissions of asphalt mixtures have been retrieved from the literature,62,63 or are presented for the first time in the present work (see Table S5 in ESI†). This set of SOAP indices gives no value for 12 of the 28 major compounds identified in emissions of STA and LTA asphalt mixtures, for which estimates have been attempted when possible; nonetheless, it allows estimation of a conservative lower value of potential SOA formation. To determine the total mass of SOA potentially formed, each VOC's EF determined in this work with GC-MS/FID is multiplied with its SOAP index divided by 100 (so that it is normalized to 1 for toluene), and multiplied with the SOA yield of toluene, Ytoluene(SOA). Contributions of each VOC are then summed to get the total EF(SOA), as described in eqn (7).
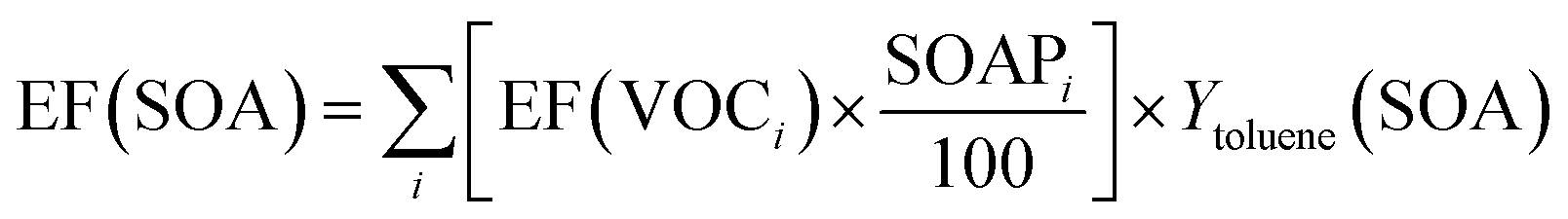 | (7) |
Values of Ytoluene(SOA) are in the 0.15–0.25 range;64 we choose the average value of 0.2 for our calculations. The resulting EF(SOA) value is multiplied by the asphalt-covered area in Paris (42.16 km2) to give the annual potential production of SOA for the city of Paris. It leads to a potential SOA mass formed by VOC emissions of asphalt mixtures in Paris over a year of 1.04 tonnes at 23 °C, 0.93 tonnes at 35 °C, 2.47 tonnes at 50 °C, and 3.76 tonnes at 60 °C, where the temperature is that of the surface of the asphalt mixture. Our annual temperature scenario elaborated for Paris gives a value of 1.60 tonnes of SOA emitted for a year.
These values are conservative lower values based only on VOCs identified with GC-MS/FID, and do not include emissions measured by PTR-ToFMS. Here, we set out to determine values of SOA formed by VOCs emitted by old asphalt mixtures by including, when possible, measurements with PTR-ToFMS. In Table S6†, we tentatively assign the major masses measured in emissions by old asphalt mixtures with PTR-ToFMS. To do so, we limit our investigation to VOCs with C, H and O atoms, which have been identified in previous works (listed in Table S3 of ESI†) and whose SOAP indices have been calculated. Then, we added the EFs measured for masses (M + 1) 57, 71, 85, and 111; mass 129 may be attributed to naphthalene, in line with the identification of several substituted naphthalenes with GC-MS/FID. However, in our setup, naphthalene may be co-eluted with decanal. For old asphalt mixtures, emission factors at mass 129 are similar to those of decanal/naphthalene measured with GC-MS/FID at all temperatures investigated. Hence, we did not count mass 129 in our calculation, but used the SOAP value of naphthalene (=106) for the EF of decanal/naphthalene measured with GC-MS/FID. This leads to potential SOA mass formed by VOC emissions of asphalt mixtures in Paris over a year of 2.17 tonnes at 23 °C, 3.12 tonnes at 35 °C, 6.31 tonnes at 50 °C, and 11.3 tonnes at 60 °C. Our annual temperature scenario gives a value of 4.12 tonnes of SOA emitted for a year in Paris.
Particulate matter can form following oxidation of VOCs in the gas-phase. The PM1 fraction is the most hazardous for human health, because it can penetrate the respiratory track down to pulmonary alveoli, due to its small size. In urban environments, PM1 is the most abundant, typically representing 60–90% of the mass of PM2.5 aerosols.65,66 Consequently, we multiply emissions of anthropogenic PM2.5 by the average value of 0.75 to estimate PM1 emissions. According to the EMEP inventory for Paris in 2019, annual PM2.5 emissions represent 670 tonnes; hence, annual PM1 emissions represent 502.5 tonnes. The major contributing sectors to PM1 emissions are: waste with 160.5 tonnes, industry with 127.5 tonnes, and road transportation with 108 tonnes. SOA formed by VOCs emitted by asphalt-covered surfaces therefore represent 1.5–3.8% of PM1 emitted by road transportation, and 0.32–0.82% of total PM1 emissions. This suggests that the contribution of asphalt-covered surfaces to SOA-forming VOC emissions in urban environments is modest but should be taken into account in inventories and in models as a source of fine atmospheric particles. Note that, as mentioned previously, these values correspond to a lower limit, for three major reasons: (i) we do not detect all VOCs or SVOCs, but only a fraction of them; (ii) the SOAP values are not available for many measured compounds; and (iii), VOC emissions of asphalt pavements are significantly higher under humid and light-irradiation conditions, i.e., closer to environmental conditions prevailing in the atmosphere (this work will be the subject of another publication). An estimate of SOA formation by chemistry transport models is necessary to get a clearer view of the contribution of VOC emissions by asphalt pavements to urban particulate matter concentrations.
Interestingly, the SOA mass concentration bias in models with respect to observations can be as high as −75%, as deduced from observations in California in 2010.67 The correlation between the SOA concentration bias and temperature could link the former to missing IVOC and SVOC emitters, such as asphalt-covered surfaces.67 Our results support this, the SOA potentially formed by VOCs emitted by LTA asphalt mixtures increasing with temperature between 23 °C and 60 °C. Our results also suggest that during summertime or heatwaves, the contribution of asphalt pavements to the total SOA formation will become more important and may therefore contribute to bridge the gap between models and observations of SOA in urban environments.
The European Green Deal targets to reduce the number of premature deaths caused by air pollution in 2030 by more than 55% with respect to 2005 levels, through a reduction in PM2.5 emissions.68 Moreover, the United Nations advocate “net zero emissions” by 2050. Even if the current decrease in PM1 emissions holds, they would be precluded from reaching zero by asphalt-covered surfaces which are expected to play a more dominant role as source of VOCs, and therefore of SOA in the future. For example, if the target of the European Green Deal is met by 2030, assuming constant VOC emissions by asphalt mixtures in Paris and that the reduction in premature deaths is directly proportional to PM emissions, this would suggest annual total PM1 emissions of 226 tonnes per year in Paris, of which 49 tonnes would be caused by road transportation. Asphalt pavements would then represent 3.3–8.4% of road transportation PM1 emissions, and 0.7–1.8% of total PM1 emissions.
Therefore, for net zero anthropogenic PM1 emissions to be reached, new strategies need to be elaborated and new inventories of pollution sources need to be completed. Previously overlooked VOC sources, like asphalt-covered surfaces, could represent in the future one of the main SOA precursors in urban environments. More work is therefore urgently needed to assess the environmental impact of urban surfaces and to find alternative and innovative materials with lower emission levels.
5. Conclusion
This work provides a comprehensive laboratory study of VOC emissions by fresh (STA) and old (LTA) asphalt mixtures, under dark and dry conditions, at relevant atmospheric temperatures. Two complementary techniques have been used, PTR-ToFMS and GC-MS/FID. PTR-ToFMS allows real-time quantification of the emissions by mass; GC-MS/FID allows identification and quantification of specific VOCs. This work has revealed some salient features of the emissions of asphalt-covered surfaces, which we summarize below:• Under dry and dark conditions, VOC emissions of STA and LTA asphalt mixtures exhibit an exponential increase with temperature in the 23–60 °C range;
• Old asphalt is a stronger VOC emitter than fresh asphalt at 23 °C and 35 °C. The difference could be due to atmospheric aging, and oxidation of surface organics leading to more volatile species. VOC emissions of both types of surface are similar at 50 °C and 60 °C;
• STA and LTA asphalt emissions are mostly composed of alkanes and aromatics at all temperatures. Old asphalt emissions are dominated by alkanes (>75%). Carbonyl compounds and alcohols represent a non-negligible fraction of emissions, especially for fresh asphalt (20–45%). Substituted naphthalenes are important contributors to the emissions: they account for 5–32% of total emissions of fresh asphalt and for 2–9% of total emissions of old asphalt;
• The nature of dominant compounds in old asphalt emissions are different from fresh asphalt, suggesting an impact of aging and of atmospheric oxidation on asphalt mixture emissions;
• The atmospheric implications of this work have been assessed for the city of Paris, France. Yearly VOC emissions from asphalt-covered surfaces are equivalent to 21.3% of NMVOC emissions from road transportation and to 2.9% of total NMVOC emissions;
• The ozone formation potential determined through the POCP index of fresh asphalt mixtures, is higher than that of petrol evaporation, but lower than that of motor vehicle exhausts. The POCP index of old asphalt mixtures is lower than inventoried sources but is not negligible. Models are needed to evaluate the impact of asphalt-covered surfaces on O3 concentrations and on the missing OH reactivity in cities;
• The estimated amount of SOA formed by asphalt-covered surfaces in Paris represents 1.5% to 3.8% of the estimated PM1 mass emitted by road transportation and 0.32% to 0.82% of the total PM1 emissions from inventoried sources. These numbers will likely increase significantly after studies are conducted under relevant conditions of relative humidity and solar irradiation;
• Lastly, SOA formed from the VOCs emitted asphalt-covered surfaces will preclude PM from reaching net zero emissions, by providing a non-negligible urban background source.
Most importantly, the results of this work should be considered lower limits, and future works should focus on VOC emissions in a humid atmosphere and under UV-light, to evaluate how environmental conditions impact emissions from asphalt mixtures. VOC emissions from asphalt-covered surfaces need to be included in inventories of urban pollutants because they represent a major missing source, mostly due to the large area covered with asphalt products in cities. This work calls for more studies of the emissions from asphalt mixtures at service temperatures, a study area that has been overlooked so far.
Author contributions
Jérôme Lasne: investigation, formal analysis, writing – original draft, visualization; Anaïs Lostier: investigation, formal analysis, writing – original draft; Manolis Romanias: conceptualization, methodology, resources, writing – review and editing, supervision, funding acquisition; Sabine Vassaux: investigation, writing – review and editing; Didier Lesueur: writing – review and editing, funding acquisition; Vincent Gaudion: methodology, investigation, technical support; Marina Jamar: methodology, investigation, formal analysis, technical support; Richard G. Derwent: resources, writing – review and editing; Sébastien Dusanter: formal analysis, resources, writing – review and editing; Thérèse Salameh: conceptualization, methodology, resources, writing – review and editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
JL acknowledges funding from IMT Nord Europe through a post-doctoral fellowship in the frame of the IRAPAQ project. TS acknowledges support from “Institut National des Sciences de l’Univers” of CNRS, and the LEFE (Les Enveloppes Fluides et l’Environnement) program through funding of the EmAPI project. The authors acknowledge Dr Florent Caron and Prof. Frédéric Thévenet (IMT Nord Europe) for insightful discussions on atmospheric simulation chambers, and Dr Joël de Brito (IMT Nord Europe) for interesting discussions about SOA formation. We are grateful to Mr. Jean-Etienne Régniez, Mr. Benjamin Choisel, and Mr. Benoit Varoqui (Mairie de Douai), for their assistance in the identification and characterization of the asphalt mixture samples, and to Mr. Alexis Guilloteau (IMT Nord Europe) for granting access to the data of the weather station. The authors acknowledge ECCAD for archiving and distribution of the EMEP data.References
- J. Lelieveld, A. Pozzer, U. Pöschl, M. Fnais, A. Haines and T. Münzel, Loss of life expectancy from air pollution compared to other risk factors: a worldwide perspective, Cardiovasc. Res., 2020, 116, 1910–1917 CrossRef CAS PubMed.
- H. E. Institute, State of Global Air 2020, 2020 Search PubMed.
- U. Nations, World Urbanization Prospects: the 2018 Revision (ST/ESA/SER.A/420), Department of Economic and Social Affairs, Population Division, 2019 Search PubMed.
- G. M. Lanzafame, D. Srivastava, O. Favez, B. A. M. Bandowe, P. Shahpoury, G. Lammel, N. Bonnaire, L. Y. Alleman, F. Couvidat, B. Bessagnet and A. Albinet, One-year measurements of secondary organic aerosol (SOA) markers in the Paris region (France): Concentrations, gas/particle partitioning and SOA source apportionment, Sci. Total Environ., 2021, 757, 143921 CrossRef CAS PubMed.
- H. O. T. Pye, C. K. Ward-Caviness, B. N. Murphy, K. W. Appel and K. M. Seltzer, Secondary organic aerosol association with cardiorespiratory disease mortality in the United States, Nat. Commun., 2021, 12, 7215 CrossRef CAS PubMed.
- R. G. Derwent, M. E. Jenkin, N. R. Passant and M. J. Pilling, Photochemical ozone creation potentials (POCPs) for different emission sources of organic compounds under European conditions estimated with a Master Chemical Mechanism, Atmos. Environ., 2007, 41, 2570–2579 CrossRef CAS.
- P. Khare and D. R. Gentner, Considering the future of anthropogenic gas-phase organic compound emissions and the increasing influence of non-combustion sources on urban air quality, Atmos. Chem. Phys., 2018, 18, 5391–5413 CrossRef CAS.
- S. Zhu, M. M. Kinnon, B. P. Shaffer, G. S. Samuelsen, J. Brouwer and D. Dabdub, An uncertainty for clean air: Air quality modeling implications of underestimating VOC emissions in urban inventories, Atmos. Environ., 2019, 211, 256–267 CrossRef CAS.
- B. C. McDonald, J. A. de Gouw, J. B. Gilman, S. H. Jathar, A. Akherati, C. D. Cappa, J. L. Jimenez, J. Lee-Taylor, P. L. Hayes, S. A. McKeen, Y. Y. Cui, S.-W. Kim, D. R. Gentner, G. Isaacman-VanWertz, A. H. Goldstein, R. A. Harley, G. J. Frost, J. M. Roberts, T. B. Ryerson and M. Trainer, Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Sci, 2018, 359, 760–764 CrossRef CAS PubMed.
- M. M. Coggon, G. I. Gkatzelis, B. C. McDonald, J. B. Gilman, R. H. Schwantes, N. Abuhassan, K. C. Aikin, M. F. Arend, T. A. Berkoff, S. S. Brown, T. L. Campos, R. R. Dickerson, G. Gronoff, J. F. Hurley, G. Isaacman-VanWertz, A. R. Koss, M. Li, S. A. McKeen, F. Moshary, J. Peischl, V. Pospisilova, X. Ren, A. Wilson, Y. Wu, M. Trainer and C. Warneke, Volatile chemical product emissions enhance ozone and modulate urban chemistry, Proc. Natl. Acad. Sci. U. S. A., 2021, 118 Search PubMed.
- T. Karl, M. Striednig, M. Graus, A. Hammerle and G. Wohlfahrt, Urban flux measurements reveal a large pool of oxygenated volatile organic compound emissions, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1186–1191 CrossRef CAS PubMed.
- H. Akbari, L. Shea Rose and H. Taha, Analyzing the land cover of an urban environment using high-resolution orthophotos, Landsc. Urban. Plan., 2003, 63, 1–14 CrossRef.
- D. Li, W. Liao, A. J. Rigden, X. Liu, D. Wang, S. Malyshev and E. Shevliakova, Urban heat island: Aerodynamics or imperviousness?, Sci. Adv., 2019, 5, eaau4299 CrossRef PubMed.
- A. Mohajerani, J. Bakaric and T. Jeffrey-Bailey, The urban heat island effect, its causes, and mitigation, with reference to the thermal properties of asphalt concrete, J. Environ. Manage., 2017, 197, 522–538 CrossRef PubMed.
- D. Lesueur, The colloidal structure of bitumen: consequences on the rheology and on the mechanisms of bitumen modification, Adv. Colloid Interface Sci., 2009, 145, 42–82 CrossRef CAS PubMed.
- B. Hofko, L. Porot, A. Falchetto Cannone, L. Poulikakos, L. Huber, X. Lu, K. Mollenhauer and H. Grothe, FTIR spectral analysis of bituminous binders: reproducibility and impact of ageing temperature, Mater. Struct., 2018, 51, 1–16 CrossRef CAS.
- R. E. Villegas-Villegas, A. Baldi-Sevilla, J. P. Aguiar-Moya and L. Loria-Salazar, Analysis of Asphalt Oxidation by Means of Accelerated Testing and Environmental Conditions, J. Transp. Res. Rec., 2018, 2672, 244–255 CrossRef.
- E. Toraldo, E. Mariani, S. Alberti and M. Crispino, Experimental investigation into the thermal behavior of wearing courses for road pavements due to environmental conditions, Constr. Build. Mater., 2015, 98, 846–852 CrossRef.
- H. Higashiyama, M. Sano, F. Nakanishi, O. Takahashi and S. Tsukuma, Field measurements of road surface temperature of several asphalt pavements with temperature rise reducing function, Case Stud. Constr. Mater., 2016, 4, 73–80 Search PubMed.
- N. Solatifar, M. Abbasghorbani, A. Kavussi, H. Sivilevičius and Management, Prediction of depth temperature of asphalt layers in hot climate area, J. Civ. Eng. Manag., 2018, 24, 516–525 CrossRef.
- S. Parison, M. Hendel, A. Grados and L. Royon, Analysis of the heat budget of standard, cool and watered pavements under lab heat-wave conditions, Energy Build., 2020, 228, 110455 CrossRef.
- P. T. Nilsson, U. Bergendorf, H. Tinnerberg, E. Nordin, M. Gustavsson, B. Strandberg, M. Albin and A. Gudmundsson, Emissions into the Air from Bitumen and Rubber Bitumen-Implications for Asphalt Workers' Exposure, Ann. Work Expo. Health, 2018, 62, 828–839 CrossRef CAS PubMed.
- J. M. Cavallari, L. V. Osborn, J. E. Snawder, A. J. Kriech, L. D. Olsen, R. F. Herrick and M. D. McClean, Predictors of airborne exposures to polycyclic aromatic compounds and total organic matter among hot-mix asphalt paving workers and influence of work conditions and practices, Ann. Occup. Hyg., 2012, 56, 138–147 CrossRef CAS PubMed.
- J. M. Cavallari, L. M. Zwack, C. R. Lange, R. F. Herrick and M. D. McClean, Temperature-dependent emission concentrations of polycyclic aromatic hydrocarbons in paving and built-up roofing asphalts, Ann. Occup. Hyg., 2012, 56, 148–160 CrossRef CAS PubMed.
- A. Preiss, W. Koch, H. Kock, M. Elend, M. Raabe and G. Pohlmann, Collection, validation and generation of bitumen fumes for inhalation studies in rats Part 1: Workplace samples and validation criteria, Ann. Occup. Hyg., 2006, 50, 789–804 CAS.
- J. Espinoza, C. Medina, A. Calabi-Floody, E. Sánchez-Alonso, G. Valdés and A. Quiroz, Evaluation of Reductions in Fume Emissions (VOCs and SVOCs) from Warm Mix Asphalt Incorporating Natural Zeolite and Reclaimed Asphalt Pavement for Sustainable Pavements, Sustainability, 2020, 12, 9546 CrossRef.
- J. B. Borinelli, J. Blom, M. Portillo-Estrada, P. Kara De Maeijer, W. Van den Bergh and C. Vuye, VOC Emission Analysis of Bitumen Using Proton-Transfer Reaction Time-Of-Flight Mass Spectrometry, Materials, 2020, 13, 3659 CrossRef CAS PubMed.
- E. Gasthauer, M. Mazé, J. P. Marchand and J. Amouroux, Characterization of asphalt fume composition by GC/MS and effect of temperature, Fuel, 2008, 87, 1428–1434 CrossRef CAS.
- P. Khare, J. Machesky, R. Soto, M. He, A. A. Presto and D. R. Gentner, Asphalt-related emissions are a major missing nontraditional source of secondary organic aerosol precursors, Sci. Adv., 2020, 6, eabb9785 CrossRef CAS PubMed.
- J. Lasne, A. Lostier, T. Salameh, E. Athanasopoulou, D. Karagiannis, A. Kakouri, S. Vassaux, D. Lesueur and M. N. Romanias, NOx emissions by real-world fresh and old asphalt mixtures: Impact of temperature, relative humidity, and UV-irradiation, Urban Clim., 2023, 49, 101457 CrossRef.
- L. W. Chew, A. A. Aliabadi and L. K. Norford, Flows across high aspect ratio street canyons: Reynolds number independence revisited, Environ. Fluid Mech., 2018, 18, 1275–1291 CrossRef.
- S. Reimann, R. Wegener, A. Claude and S. Sauvage, Updated Measurement Guideline for NOx and VOCs. ACTRIS Deliverable D3.17, 2018 Search PubMed.
- ACTRIS, Measurement Guideline for VOC Analysis by PTRMS. ACTRIS Deliverable D3.20, 2019 Search PubMed.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 2017, 117, 13187–13229 CrossRef CAS PubMed.
- R. Holzinger, A. Kasper-Giebl, M. Staudinger, G. Schauer and T. Röckmann, Analysis of the chemical composition of organic aerosol at the Mt. Sonnblick observatory using a novel high mass resolution thermal-desorption proton-transfer-reaction mass-spectrometer (hr-TD-PTR-MS), Atmos. Chem. Phys., 2010, 10, 10111–10128 CrossRef CAS.
- A. Detournay, S. Sauvage, N. Locoge, V. Gaudion, T. Leonardis, I. Fronval, P. Kaluzny and J.-C. Galloo, Development of a sampling method for the simultaneous monitoring of straight-chain alkanes, straight-chain saturated carbonyl compounds and monoterpenes in remote areas, J. Environ. Monit., 2011, 13, 983–990 RSC.
- T. Salameh, S. Sauvage, C. Afif, A. Borbon, T. Léonardis, J. Brioude, A. Waked and N. Locoge, Exploring the seasonal NMHC distribution in an urban area of the Middle East during ECOCEM campaigns: very high loadings dominated by local emissions and dynamics, Environ. Chem., 2015, 12, 316–328 CrossRef CAS.
- E. Gallego, F. J. Roca, J. F. Perales and X. Guardino, Comparative study of the adsorption performance of a multi-sorbent bed (Carbotrap, Carbopack X, Carboxen 569) and a Tenax TA adsorbent tube for the analysis of volatile organic compounds (VOCs), Talanta, 2010, 81, 916–924 CrossRef CAS PubMed.
- F. Caron, R. Guichard, L. Robert, M. Verriele and F. Thevenet, Behaviour of individual VOCs in indoor environments: How ventilation affects emission from materials, Atmos. Environ., 2020, 243, 117713 CrossRef CAS.
- K. Sekimoto and A. R. Koss, Modern mass spectrometry in atmospheric sciences: Measurement of volatile organic compounds in the troposphere using proton-transfer-reaction mass spectrometry, J. Mass Spectrom., 2021, 56, e4619 CrossRef CAS PubMed.
- Z. Deuscher, I. Andriot, E. Sémon, M. Repoux, S. Preys, J. M. Roger, R. Boulanger, H. Labouré and J. L. Le Quéré, Volatile compounds profiling by using proton transfer reaction-time of flight-mass spectrometry (PTR-ToF-MS). The case study of dark chocolates organoleptic differences, J. Mass Spectrom., 2019, 54, 92–119 CrossRef CAS PubMed.
- E. A. Bruns, J. G. Slowik, I. El Haddad, D. Kilic, F. Klein, J. Dommen, B. Temime-Roussel, N. Marchand, U. Baltensperger and A. S. H. Prévôt, Characterization of gas-phase organics using proton transfer reaction time-of-flight mass spectrometry: fresh and aged residential wood combustion emissions, Atmos. Chem. Phys., 2017, 17, 705–720 CrossRef CAS.
- B. Loubet, P. Buysse, L. Gonzaga-Gomez, F. Lafouge, R. Ciuraru, C. Decuq, J. Kammer, S. Bsaibes, C. Boissard, B. Durand, J. C. Gueudet, O. Fanucci, O. Zurfluh, L. Abis, N. Zannoni, F. Truong, D. Baisnée, R. Sarda-Estève, M. Staudt and V. Gros, Volatile organic compound fluxes over a winter wheat field by PTR-Qi-TOF-MS and eddy covariance, Atmos. Chem. Phys., 2022, 22, 2817–2842 CrossRef CAS.
- A. M. Yáñez-Serrano, I. Filella, J. Llusià, A. Gargallo-Garriga, V. Granda, E. Bourtsoukidis, J. Williams, R. Seco, L. Cappellin, C. Werner, J. de Gouw and J. Peñuelas, GLOVOCS - Master compound assignment guide for proton transfer reaction mass spectrometry users, Atmos. Environ., 2021, 244, 117929 CrossRef.
- R. F. Hansen, S. M. Griffith, S. Dusanter, P. S. Rickly, P. S. Stevens, S. B. Bertman, M. A. Carroll, M. H. Erickson, J. H. Flynn, N. Grossberg, B. T. Jobson, B. L. Lefer and H. W. Wallace, Measurements of total hydroxyl radical reactivity during CABINEX 2009 – Part 1: field measurements, Atmos. Chem. Phys., 2014, 14, 2923–2937 CrossRef.
- R. Sheu, A. Marcotte, P. Khare, S. Charan, J. C. Ditto and D. R. Gentner, Advances in offline approaches for chemically speciated measurements of trace gas-phase organic compounds via adsorbent tubes in an integrated sampling-to-analysis system, J. Chromatogr. A, 2018, 1575, 80–90 CrossRef CAS PubMed.
- E. P. L. Hunter and S. G. Lias, Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- M. Malásková, B. Henderson, P. D. Chellayah, V. Ruzsanyi, P. Mochalski, S. M. Cristescu and C. A. Mayhew, Proton transfer reaction time-of-flight mass spectrometric measurements of volatile compounds contained in peppermint oil capsules of relevance to real-time pharmacokinetic breath studies, J. Breath Res., 2019, 13, 046009 CrossRef PubMed.
- J. Zhao and R. Zhang, Proton transfer reaction rate constants between hydronium ion (H3O+) and volatile organic compounds, Atmos. Environ., 2004, 38, 2177–2185 CrossRef CAS.
- C. Intergovernmental Panel on Climate, in Climate Change 2021 – the Physical Science Basis: Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2023, pp. 3–32, DOI:10.1017/9781009157896.001.
- C. Dolgorouky, V. Gros, R. Sarda-Esteve, V. Sinha, J. Williams, N. Marchand, S. Sauvage, L. Poulain, J. Sciare and B. Bonsang, Total OH reactivity measurements in Paris during the 2010 MEGAPOLI winter campaign, Atmos. Chem. Phys., 2012, 12, 9593–9612 CrossRef CAS.
- S. M. Griffith, R. F. Hansen, S. Dusanter, V. Michoud, J. B. Gilman, W. C. Kuster, P. R. Veres, M. Graus, J. A. de Gouw, J. Roberts, C. Young, R. Washenfelder, S. S. Brown, R. Thalman, E. Waxman, R. Volkamer, C. Tsai, J. Stutz, J. H. Flynn, N. Grossberg, B. Lefer, S. L. Alvarez, B. Rappenglueck, L. H. Mielke, H. D. Osthoff and P. S. Stevens, Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets, J. Geophys. Res. Atmos., 2016, 121, 4211–4232 CrossRef CAS.
- W. P. L. Carter, Development of Ozone Reactivity Scales for Volatile Organic Compounds, J. Air Waste Manag. Assoc., 1994, 44, 881–899 CrossRef CAS.
- W. P. Carter, Updated maximum incremental reactivity scale and hydrocarbon bin reactivities for regulatory applications, College of Engineering Center for Environmental Research and Technology, University of California, Riverside, CA 92521, 2010 Search PubMed.
- M. A. Venecek, W. P. L. Carter and M. J. Kleeman, Updating the SAPRC Maximum Incremental Reactivity (MIR) scale for the United States from 1988 to 2010, J. Air Waste Manag. Assoc., 2018, 68, 1301–1316 CrossRef CAS PubMed.
- R. G. Derwent and M. E. Jenkin, Hydrocarbons and the long-range transport of ozone and pan across Europe, Atmos. Environ., Part A, 1991, 25, 1661–1678 CrossRef.
- R. G. Derwent, M. E. Jenkin, M. J. Pilling, W. P. Carter and A. Kaduwela, Reactivity scales as comparative tools for chemical mechanisms, J. Air Waste Manag. Assoc., 2010, 60, 914–924 CrossRef CAS PubMed.
- A. Mellouki, T. J. Wallington and J. Chen, Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate, Chem. Rev., 2015, 115, 3984–4014 CrossRef CAS PubMed.
- M. E. Jenkin, R. G. Derwent and T. J. Wallington, Photochemical ozone creation potentials for volatile organic compounds: Rationalization and estimation, Atmos. Environ., 2017, 163, 128–137 CrossRef CAS.
- N. R. Passant, Speciation of UK emissions of non-methane volatile organic compounds, 2002 Search PubMed.
- J. Coates, K. A. Mar, N. Ojha and T. M. Butler, The influence of temperature on ozone production under varying NOx conditions – a modelling study, Atmos. Chem. Phys., 2016, 16, 11601–11615 CrossRef CAS.
- R. G. Derwent, M. E. Jenkin, S. R. Utembe, D. E. Shallcross, T. P. Murrells and N. R. Passant, Secondary organic aerosol formation from a large number of reactive man-made organic compounds, Sci. Total Environ., 2010, 408, 3374–3381 CrossRef CAS PubMed.
- J. B. Gilman, B. M. Lerner, W. C. Kuster, P. D. Goldan, C. Warneke, P. R. Veres, J. M. Roberts, J. A. de Gouw, I. R. Burling and R. J. Yokelson, Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 2015, 15, 13915–13938 CrossRef CAS.
- P. Simonen, E. Saukko, P. Karjalainen, H. Timonen, M. Bloss, P. Aakko-Saksa, T. Rönkkö, J. Keskinen and M. Dal Maso, A new oxidation flow reactor for measuring secondary aerosol formation of rapidly changing emission sources, Atmos. Meas. Tech., 2017, 10, 1519–1537 CrossRef CAS.
- R. Vecchi, G. Marcazzan, G. Valli, M. Ceriani and C. Antoniazzi, The role of atmospheric dispersion in the seasonal variation of PM1 and PM2.5 concentration and composition in the urban area of Milan (Italy), Atmos. Environ., 2004, 38, 4437–4446 CrossRef CAS.
- Y. Zhang, J. Lang, S. Cheng, S. Li, Y. Zhou, D. Chen, H. Zhang and H. Wang, Chemical composition and sources of PM1 and PM2.5 in Beijing in autumn, Sci. Total Environ., 2018, 630, 72–82 CrossRef CAS PubMed.
- E. A. Pennington, K. M. Seltzer, B. N. Murphy, M. Qin, J. H. Seinfeld and H. O. T. Pye, Modeling secondary organic aerosol formation from volatile chemical products, Atmos. Chem. Phys., 2021, 21, 18247–18261 CrossRef CAS PubMed.
- E. Commission, Communication from the commission to the European parliament the council, the European economic and social committee and the committee of the region, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00034f |
| This journal is © The Royal Society of Chemistry 2023 |