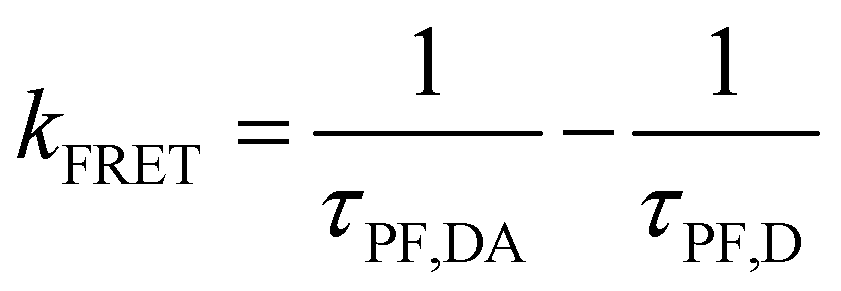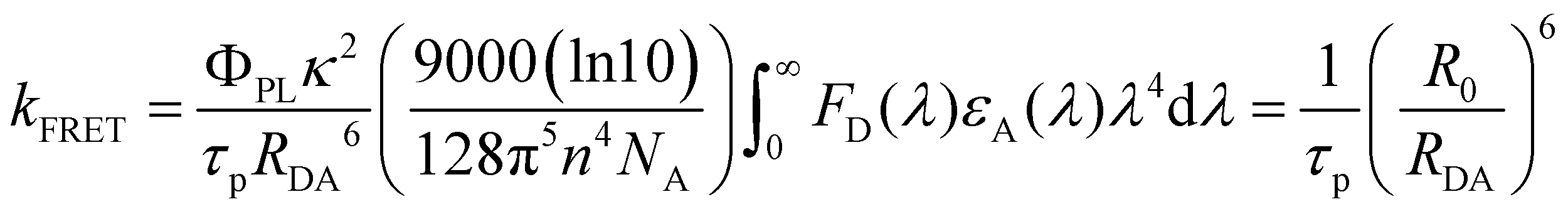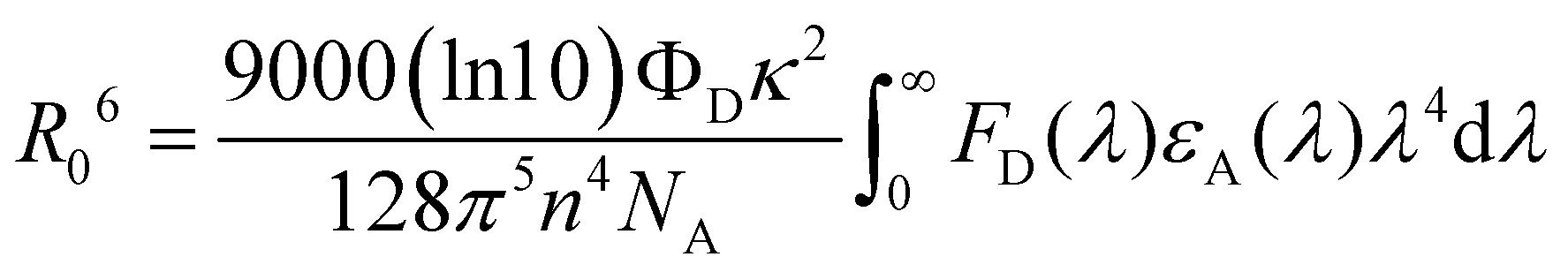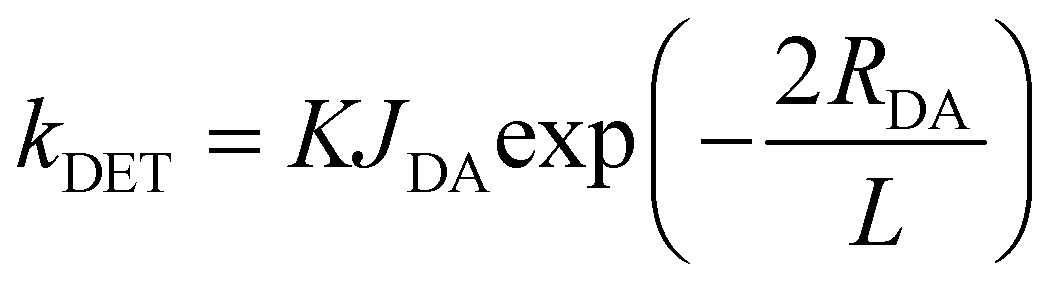DOI:
10.1039/D1TC05470H
(Paper)
J. Mater. Chem. C, 2022,
10, 4637-4645
Manipulating Förster and Dexter interactions between a thermally activated delayed fluorescence host and a phosphorescent dopant for highly efficient solution-processed red and white OLEDs†
Received
13th November 2021
, Accepted 2nd January 2022
First published on 4th January 2022
Abstract
Solution-processed phosphorescent organic light-emitting diodes (PhOLEDs) have been known as promising alternatives to their vacuum deposited counterparts due to their excellent cost-effectiveness. However, the solution-processed devices still suffer from low efficiencies especially for the saturated red emission. Herein, we designed and demonstrated very efficient solution-processed red PhOLEDs by introducing a thermally activated delayed fluorescence (TADF) host with and without inert peripheral units, i.e., 4CzDMAC-DPS and DMAC-DPS respectively, to manipulate Förster and Dexter interactions. Compared with the steric hindrance on both the host and guest, Förster and Dexter energy transfers were simultaneously promoted when removing the hindrance units on the TADF host. This is mainly attributed to the larger radius of Förster energy transfer and closer intermolecular distance between DMAC-DPS and the guest, compared with those of the 4CzDMAC-DPS and guest system. As a result, the state-of-the-art solution-processed saturated red PhOLEDs achieved a high maximum external quantum efficiency (EQE) of 22.2% with an emission peak beyond 610 nm. In contrast, the TADF host with the additional hindrance units played an important role in achieving a low efficiency roll-off (< 10% at 1000 cd m−2) without sacrificing the EQE. Moreover, ternary-blended solution-processed PhOLEDs exhibited easy color tuning from red to white, which was dominated by the energy transfer radius mediated by the hindrance effect of the TADF compound.
Introduction
Due to the capability of harvesting all the electrically generated excitons, phosphorescent organic light-emitting diodes (PhOLEDs) have received intensive interest in solid-state lighting and full-color displays.1,2 Different from fluorescent OLEDs, the internal quantum efficiency of PhOLEDs can theoretically reach 100%.3 Generally, phosphorescent dopants are dispersed into the host matrix to diminish the concentration quenching effect. In a host–guest system, the device performances are mainly determined by the non-radiative resonance energy transfer, including Förster resonance energy transfer (FRET) and Dexter energy transfer (DET). FRET is a Coulombic interaction having a long-range (up to 10 nm) dipole–dipole interaction, while DET is a short-range (typically 1.5–2 nm) electron-exchange interaction originating from intermolecular orbital overlap. Generally, FRET is predominant if the transitions from the host to guest are allowed. However, in the heavily doped phosphorescent emitting layer, DET tends to emerge and FRET is imperceptible. Given the nature of the phosphorescent emission from the radiative decay of the triplet excitons, the short-range DET between the host and the guest is dominant in PhOLEDs. Therefore, the optimization of the doping concentration to maximize the electroluminescence (EL) efficiency is crucial.3 Unfortunately, at high doping concentrations, the triplet accumulation caused annihilation will inevitably take place and thus lead to a severe efficiency roll-off.
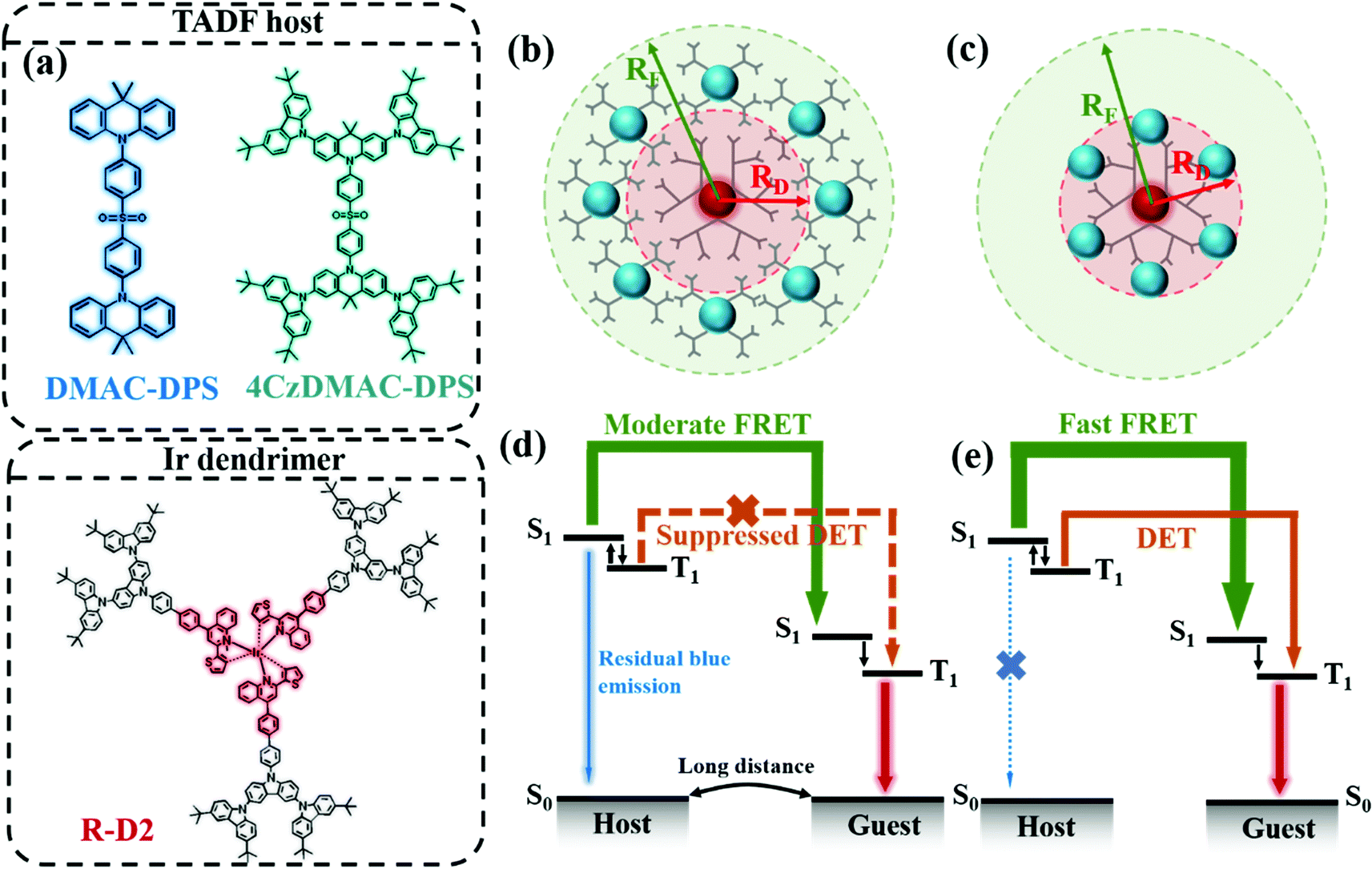
The emerging thermally activated delayed fluorescence (TADF) materials are promising hosts and sensitizers for PhOLEDs.4–13 The TADF host can up-convert the excitons from the triplet state (T1) to the singlet state (S1) via reverse intersystem crossing (RISC) and then resonantly transfer the excitons to either S1 or T1 of the phosphorescent emitter respectively via FRET or DET. Considering the distinct radii of FRET (RF) and DET (RD) (see Fig. 1b and c), it would be feasible to manipulate the Coulombic interaction and intermolecular orbital overlap by changing the doping concentration and/or introducing the bulky substituents of the host and/or the guest. Since the Duan group firstly proposed the idea of the TADF materials as the host for a phosphorescent emitter,11 many researchers followed the strategy to simultaneously improve the device efficiency and stability in both monochromatic and white PhOLEDs.4–10 For example, Liao et al. developed two TADF hosts with the titled spiro geometry for the red phosphorescence emitter Ir(MDQ)2(acac), rendering an external quantum efficiency (EQE) of 31.2% and a low efficiency roll-off of 1.8% at a brightness of 1000 cd m−2.4 Wang et al. synthesized a sky-blue TADF molecule PHCz2BP and constructed a double-layer emitting system and demonstrated white OLEDs with an EQE of 25.1% at 1000 cd m−2.10 However, all these PhOLEDs were fabricated using the vacuum-deposited technique, which is expensive and complicated. Alternatively, solution-processing techniques, including spin-coating, inkjet printing and transfer printing, are advantageous for low cost and large-area mass production.12–15 For multilayered device fabrication, the issue of orthogonal solvents remains a great challenge.13,16 Therefore, the EL performances of most solution-processed OLEDs are inferior,17–19 lagging far behind those of their vacuum-deposited counterparts.4–10 It is urgent to develop highly efficient solution-processed emitting layers with a simple structure.
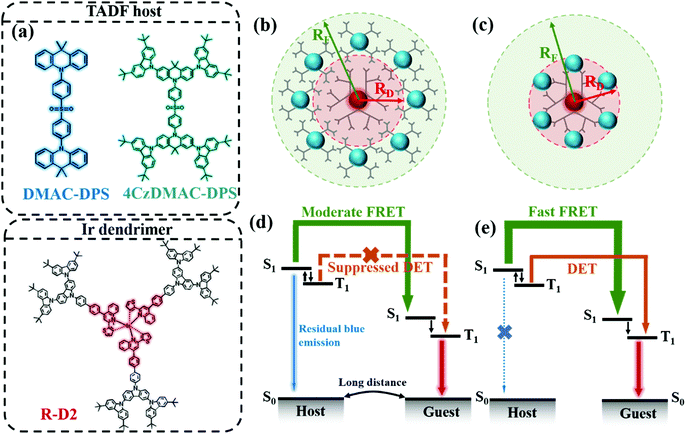 |
| | Fig. 1 (a) The molecular structures of DMAC-DPS, 4CzDMAC-DPS and R-D2. The simplified schematic radii of FRET and DET depending on the intermolecular distance: (b) both the host and guest possessing high steric hindrance and (c) only the guest equipped with the inert substituents. The blue and red spheres denote the TADF and phosphorescent cores, respectively. The corresponding energy transfer processes from the blue TADF emitter to phosphorescent dopants are respectively illustrated in (d) long and (e) short intermolecular distance. | |
Compared with small molecules20,21 and polymers,22 the emissive dendrimers encapsulated with the TADF23–25 or phosphorescent12,26–28 cores are more favorable for solution-processed OLEDs attributed to both the merits of the well-defined molecular structures of small molecules and the outstanding solution processability of polymers. The carbazole-based substituents are typical hole-transporting groups which have been widely adopted as the hosting dendrons around the emitting core of the dendrimer to suppress concentration quenching. More importantly, the peripheral carbazole substituents can modulate the FRET and DET processes, both of which are highly sensitive to the intermolecular distance.29
Herein, we adopted the TADF hosts with and without the inert steric hindrances and a phosphorescent dendrimer emitter to manipulate the multiple energy-funneling paths. In this contribution, we selected the unencapsulated blue TADF emitter, i.e., 10,10′-(4,4′-sulfonylbis(4,1-phenylene))bis(9,9-dimethyl-9,10-dihydroacridine) (DMAC-DPS), with a high photoluminescence quantum yield (PLQY) of 88% in neat films,30 and the analogue 4CzDMAC-DPS encapsulated by carbazole substituents as the hosts, respectively.23 The iridium dendrimer R-D2 with the core tris(2-thienyl-4-phenyl)iridium [Ir(Th-PQ)3] was selected as the guest.26,31 The molecular structures are depicted in Fig. 1a. The host–guest system provides a good platform for manipulating FRET and DET, schematically illustrated in Fig. 1b and c. Because of the inefficient energy transfer between the host and the guest both with large steric hindrance, the emission of the host 4CzDMAC-DPS is hardly quenched completely even at a high doping concentration of 10 wt% under either photo-excitation or electrical excitation. Nevertheless, the profoundly shortened intermolecular distance between DMAC-DPS and the guest efficiently promote DET and FRET (see Fig. 1d and e). Eventually, the optimal distance endowed the solution-processed red PhOLED with a considerably high maximum EQE of 22.2% with an emission peak beyond 610 nm. In contrast, owing to the faster exciton up-conversion from T1 to S1 in 4CzDMAC-DPS, the emitting layer 4CzDMAC-DPS:R-D2 achieved a much lower efficiency roll-off of 9.0% without sacrificing the EQE at high brightness. Furthermore, the white emission could be realized by mediating the FRET and DET processes. For instance, at a low doping concentration of 1 wt%, the white OLEDs rendered a maximum EQE of 13.2%, excellent Commission Internationale de I’Eclairage (CIE) coordinates of (0.36, 0.39), a high color rendering index (CRI) of 80, and ultra-stable spectra with ΔCIE(x, y) of (0.001, 0.002) over a wide range of luminance.
Results and discussion
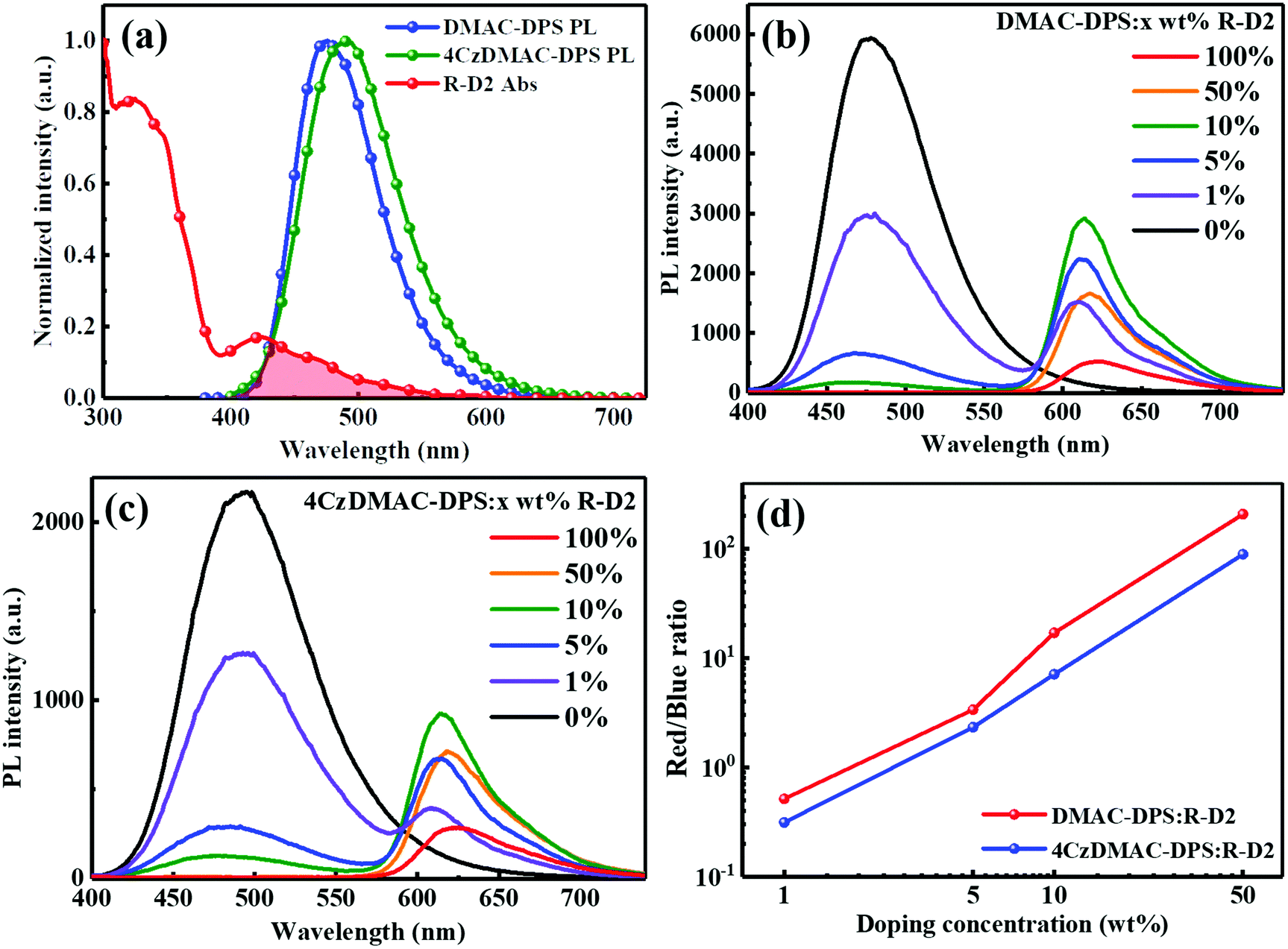
To exemplify the feasible paths of energy transfer, firstly, we measured the absorption of the iridium dendrimer R-D2 and the photoluminescence (PL) spectra of the two TADF hosts (Fig. 2a). The spectral overlap implies the potentially efficient energy transfer from TADF hosts to R-D2. The electron-donating property of carbazole dendrons endows 4CzDMAC-DPS with strong charge transfer features and thus bathochromic-shift emission, compared with DMAC-DPS.23 The steady-state PL spectra of the doped films at various concentrations were recorded and analyzed under the same test conditions (see Fig. 2b–d). As the concentration increases, the PL intensities of TADF hosts gradually decrease. However, the residual blue emission at a high doping concentration of 10 wt% suggests that the energy transfer efficiencies have been somewhat limited, which is partially due to the introduction of inert substituents on the phosphorescent dendrimer. Meanwhile, the PL intensities of R-D2 are dramatically promoted at low doping concentrations (1–10 wt%). Ascribed to the quenching effect of the iridium core, the PL quenching took place at higher doping concentrations. To quantitatively compare the efficiencies of energy transfer between TADF hosts and R-D2, the ratios of red/blue PL intensities from the emission peak in different host–guest systems were estimated and are shown in Fig. 2d. Obviously, the ratios of red/blue components in DMAC-DPS:R-D2 are much higher than those of 4CzDMAC-DPS:R-D2, respectively, at the corresponding concentration. This implies the more efficient energy transfer from DMAC-DPS to R-D2.
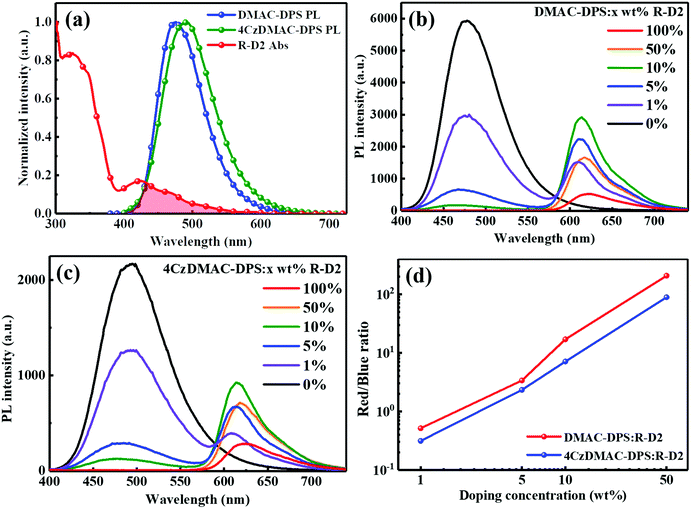 |
| | Fig. 2 (a) The overlapping of the absorption of the R-D2 film and the PL spectra of DMAC-DPS and 4CzDMAC-DPS. PL spectra of DMAC-DPS:R-D2 (b) and 4CzDMAC-DPS:R-D2 (c) in films at different dopant concentrations. (d) The ratios of red/blue components estimated from the emission peak of the emitting layer. | |
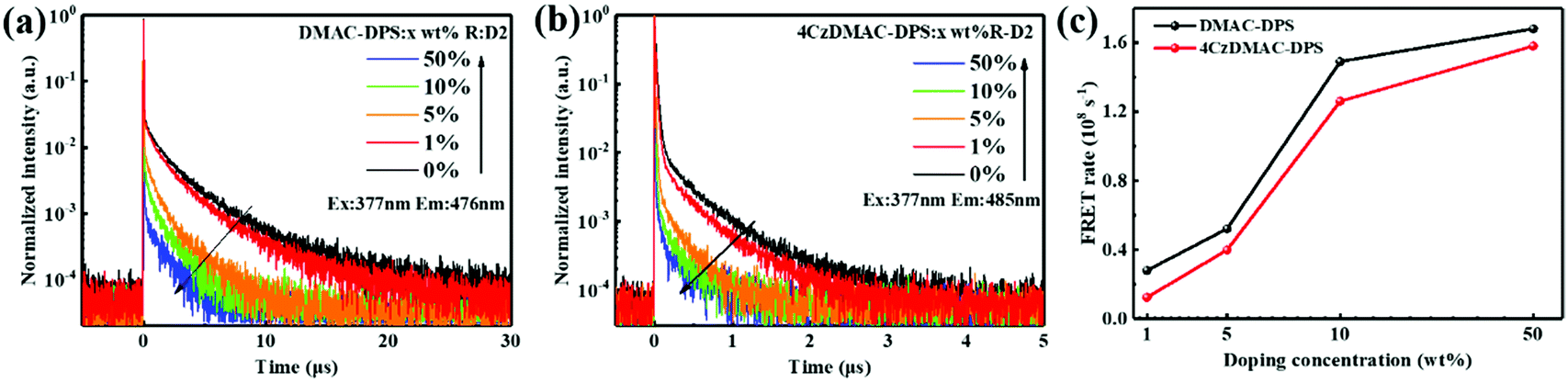
In addition to the steady-state PL spectra, the time-resolved PL spectra of the hosts in the doped films were also measured (see Fig. 3a, b and Table S1, ESI†). Both the prompt and delayed lifetimes are gradually reduced, accompanied by the increasing doping concentration. The prompt lifetime (τPF) of the TADF host is derived from the sum of the rate constants of all decay processes in which singlet excitons are involved. When the FRET process is introduced, the τPF can be written as below.32
| | 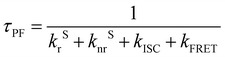 | (1) |
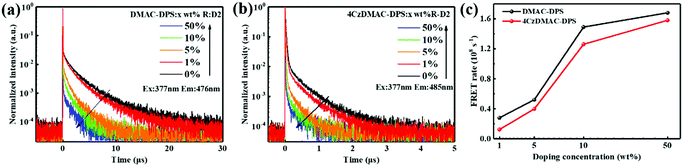 |
| | Fig. 3 Transient PL decays of DMAC-DPS:R-D2 (a) and 4CzDMAC-DPS:R-D2 (b) observed at 476 and 485 nm, respectively. (c) The estimated dopant-concentration-dependent FRET rate constants respectively from two TADF hosts to R-D2. | |
Therefore, the FRET process will introduce an extra relaxation channel of the host singlet excitons, leading to a shorter lifetime. Following eqn (1), the FRET rate can be calculated as below.32
| | 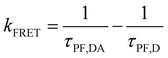 | (2) |
where τ
PF,DA and τ
PF,D are the prompt lifetime constants of the TADF films with and without R-D2, respectively. As shown in
Fig. 3c and Table S2 (ESI
†), the
kFRETs from DMAC-DPS and 4CzDMAC-DPS to R-D2 can be calculated to be 0.28 × 10
8–1.68 × 10
8 s
−1 and 0.12 × 10
8–1.58 × 10
8 s
−1, respectively, when increasing the doping concentration from 1 wt% to 50 wt%. This is in line with the steady PL analysis presented in
Fig. 2,
i.e., the faster FRET process from DMAC-DPS to R-D2 allows the more efficient quenching of blue emission and thus enhancement of red emission. Referring to the FRET theory, the
kFRET can be expressed as follows.
33| | 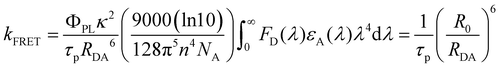 | (3) |
where
ΦPL is the PLQY of the donor,
i.e., TADF hosts in this study.
κ2 is a factor describing the relative orientation of the transition dipoles of the donor and the acceptor, assumed to be 2/3.
τp is the fluorescence lifetime of the donor.
RDA is the intermolecular distance between the donor and the acceptor.
n is the refractive index of the medium, assumed to be 1.7 here.
34NA is Avogadro's number.
FD(
λ) is the normalized emission spectra of the donor and
εA(
λ) is the molar extinction coefficient of the acceptor. The FRET radius (
R0) is defined as the intermolecular distance at which
kFRET is equal to the decay rate constant (
τp−1), and thus
R0 can be written as follows:
| | 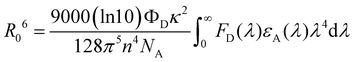 | (4) |
Using
eqn (4), the
R0s for DMAC-DPS:R-D2 and 4CzDMAC-DPS:R-D2 were calculated to be 8.3 and 7.9 nm, respectively, which are much larger than those of TADF sensitized PhOLEDs reported in previous literature.
34–36 Considering the similar spectral overlap integrals between DMAC-DPS (5.1 × 10
16 nm
−4 M
−1 cm
−1) and 4CzDMAC-DPS (5.2 × 10
16 nm
−4 M
−1 cm
−1), the main reason for the radius difference is that the PLQY of DMAC-DPS (88%) is higher than that of 4CzDMAC-DPS (68%).
23 Although the relatively larger
R0 for DMAC-DPS:R-D2 indicates a more efficient energy transfer process which is consistent with the photophysical analysis mentioned above, k
FRET indeed depends on both
R0 and
RDA based on
eqn (3). Therefore, we estimated
RDA as the center-to-center distance between the TADF hosts and the R-D2 core,
i.e., the sum of the van der Waals (VDW) radius of two molecules.
37 We calculated the VDW radius of the compounds using the Multiwfn program (Fig. S1, ESI
†).
38 As expected, by virtue of the inert peripheral units, the
RDA of 4CzDMAC-DPS:R-D2 (4.6 nm) is larger than that of DMAC-DPS:R-D2 (3.9 nm). Owing to the almost identical prompt fluorescence lifetimes, the larger R
0 and more intimate interaction for DMAC-DPS:R-D2 concurrently account for a faster and more efficient FRET process.
To guarantee complete energy transfer, the doping concentration should be at least 5 wt%, at which the short-range DET channel from TADF triplets to R-D2 triplets would be activated regardless of the peripheral dendrons on R-D2. The DET rate constant (kDET) can be expressed as follows:39
| | 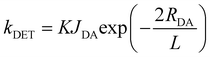 | (5) |
where
K is a parameter relative to the specific orbital interactions,
JDA is the spectral overlap integral, and
L is the VDW radius between the donor and the acceptor. Therefore,
kDET is exponentially dependent on the intermolecular distance, indicating that
kDET will drop dramatically once the distance increases slightly. Obviously, the much shorter
RDA for DMAC-DPS:R-D2 endows it with a relatively higher
kDET with respect to 4CzDMAC-DPS:R-D2. Now, it is confirmed that both the FRET and DET can efficiently take place in the DMAC-DPS:R-D2 blend system. Regarding 4CzDMAC-DPS:R-D2, these two processes are relatively suppressed. Furthermore, longer lifetimes of R-D2 doped in DMAC-DPS (
τPh) at low doping concentrations (1–10 wt%) were calculated, in contrast to those at high doping concentrations (50–100 wt%), as shown in Fig. S2 and Table S3 (ESI
†). The photophysical data of TADF materials and the calculated energy transfer radii between TADF hosts and R-D2 are summarized in
Table 1.
Table 1 The photophysical properties of TADF hosts and TADF:R-D2
| TADF host |
λ
em
[nm] |
HOMO/LUMOb [eV] |
ΔESTc [eV] |
τ
p/τdd [ns μs−] |
SOCe (S1,T1) |
k
RISC
[106 s−1] |
R
0
[nm] |
R
DA
[nm] |
|
Emission peak measured in neat film.
HOMO: highest occupied molecular orbital and LUMO: lowest unoccupied molecular orbital. The values were obtained from the literature.23,30
Singlet–triplet energy gap.
prompt and delayed fluorescence lifetimes measured in neat film.
Spin–orbital coupling constant.
Rate constant of reverse intersystem crossing.
FRET radius.
Estimated intermolecular distance between the host and R-D2.
|
| DMAC-DPS |
477 |
−5.33/−2.32 |
0.09 |
25.4/3.3 |
0.067 |
0.8 |
8.3 |
3.9 |
| 4CzDMAC-DPS |
492 |
−5.24/−2.31 |
0.09 |
26.4/0.5 |
0.105 |
2.6 |
7.9 |
4.6 |
To reveal the energy transfer mechanisms in the EL process, which may be different from PL cases, we further designed solution-processed OLEDs with the structures of indium tin oxide (ITO)/modified poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (m-PEDOT:PSS)40 (45 nm)/DMAC-DPS or 4CzDMAC-DPS:R-D2 [(100 − x):x, wt/wt] (50 nm)/bis[2-(diphenylphosphino)-phenyl] ether oxide (DPEPO) (10 nm)/1,3,5-tri(m-pyrid-3-ylphenyl)benzene (TmPyPB) (50 nm)/8-hydroxyquinolatolithium (Liq) (1 nm)/Al (100 nm), where x = 100 represent neat R-D2 for the emitting layer (Device A). Devices B1–B4 (x = 50, 10, 5 and 1) denote the ones with DMAC-DPS as the host. Similarly, devices C1–C4 (x = 50, 10, 5 and 1) refer to the one hosted by 4CzDMAC-DPS. The energy level alignments of the devices are shown in Fig. 4a. The corresponding EL performances are summarized in Table 2.
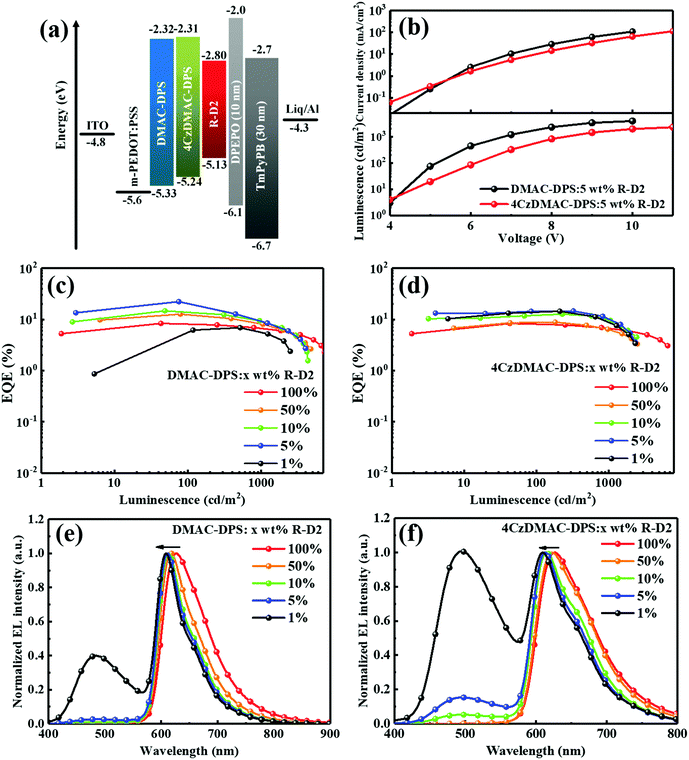 |
| | Fig. 4 (a) Energy level alignment of the devices. (b) Current density and luminescence versus driving voltage curves of Devices B3 and C3 (x = 5). EQE versus luminescence curves of the devices with DMAC-DPS (c) and 4CzDMAC-DPS (d), respectively. The corresponding normalized EL spectra are shown in (e) and (f), respectively. | |
Table 2 Summary of the EL data for the binary devices
|
|
Device |
DCa [wt%] |
V10b |
EQEc [%] |
Roll-offd (%) |
CIEe (x, y) |
η
eue
(%) |
| Maximum |
1000 cd m−2 |
|
Doping concentration.
Operational voltage at a luminance of 10 cd m−2.
External quantum efficiency.
Efficiency roll-off at a luminance of 1000 cd m−2.
Commission Internationale de I’Eclairage coordinates.
Exciton utilization efficiency assuming the ideal charge balance (γ = 1) and an out-coupling efficiency of 0.3.44
|
| R-D2 |
A |
100 |
4.5 |
8.3 |
6.6 |
26.5 |
(0.67, 0.33) |
65.9 |
| DMAC-DPS:R-D2 |
B1 |
50 |
4.2 |
12.6 |
8.1 |
35.7 |
(0.66, 0.34) |
87.5 |
|
|
B2 |
10 |
4.5 |
14.7 |
9.1 |
38.1 |
(0.64, 0.35) |
68.1 |
|
|
B3 |
5 |
4.4 |
22.2 |
9.1 |
59.0 |
(0.62, 0.35) |
99.7 |
|
|
B4 |
1 |
4.2 |
6.9 |
5.6 |
18.8 |
(0.42, 0.32) |
32.6 |
| 4CzDAMC-DPS:R-D2 |
C1 |
50 |
5.2 |
8.8 |
6.3 |
28.4 |
(0.67, 0.33) |
49.0 |
|
|
C2 |
10 |
4.7 |
12.7 |
9.7 |
23.6 |
(0.62, 0.35) |
53.3 |
|
|
C3 |
5 |
4.6 |
14.6 |
10.1 |
28.8 |
(0.55, 0.36) |
51.0 |
|
|
C4 |
1 |
4.3 |
13.2 |
8.8 |
33.3 |
(0.36, 0.39) |
58.0 |
For a doped OLED, excitons can be formed on the host and then transferred to the guests (Langevin recombination), while excitons can also be directly formed in the guests by trapping, i.e., trap-assisted recombination.41 Forrest et al. proved that the inert steric hindrance on the dopant is helpful for impeding charge trapping.42 Experimentally, as shown in Fig. S3 (ESI†), we compared the current densities of red devices (Devices B3 and C3) with a doping concentration of 5 wt% and white devices (Devices B4 and C4) with a doping concentration of only 1 wt%. The current densities are very similar, especially at high driving voltages, indicating that the guest R-D2 has limited impacts on the carrier transport at low doping concentrations. Generally, the EL spectral shift with increased operational voltages is closely related to the charge trapping effect on the guest.43 As can be seen in Fig. S4 (ESI†), the EL spectra of the devices are merely influenced by the driving voltage. Therefore, at low doping concentrations, the energy transfer is supposed to be dominant.
Fig. 4b–f compare the EQE and the normalized EL spectra of the devices. The current density–voltage (J–V) and luminance–voltage (L–V) characteristics shown in Fig. 4b and Fig. S5 (ESI†) indicate that all the DMAC-DPS-based devices exhibit better charge transport and higher luminance, compared with the 4CzDMAC-DPS-based devices at the same driving voltages. The lower electrical conductivity of 4CzDMAC-DPS might be attributed to the suppressed intermolecular π–π stacking with the introduction of bulky substituents with the reduced charge carrier mobility.44 Benefiting from the better bipolar transport ability of DMAC-DPS and the efficient FRET and DET paths as discussed above, the red devices based on DMAC-DPS exhibited a maximum EQE (Device B3) of 22.2% which is almost 2.7-fold enhanced compared with that of Device A based on the neat R-D2 (8.3%). This EQE is one of the highest values among the solution-processed red PhOLEDs (see Table 3). Note that the EQEs were gradually reduced with the increasing doping concentrations from 5 wt% to 100 wt% (neat R-D2), due to the concentration quenching nature of R-D2. The intensive DMAC-DPS emission (see Fig. 4e) could only be observed in Device B4 with 1 wt% R-D2, indicating the more efficient energy transfer between the host and the guest at higher doping concentration under EL excitation. The estimated exciton utilization efficiency (ηeue) was obtained according to eqn (S1) (ESI†) and is summarized in Table 2. Device B3 achieved the highest ηeue of 99.7%, further verifying the efficient and complete exciton harvesting by energy transfer processes.
Table 3 Summary of EL performances of solution-processed red PhOLEDs
| Device |
λ
max
[nm] |
EQEmaxb [%] |
EQE1000c [%] |
CEmaxd [cd A−1] |
CE1000e [cd A−1] |
CIE (x, y) |
|
Peak wavelength.
Maximum external quantum efficiency.
External quantum efficiency at 1000 cd m−2.
Maximum current efficiency.
Current efficiency at 1000 cd m−2.
|
| B3 |
610 |
22.2 |
9.1 |
30.2 |
12.5 |
(0.62, 0.35) |
| C7 |
610 |
15.6 |
14.2 |
24.5 |
22.4 |
(0.59, 0.37) |
| Ref. 45 |
602 |
19.3 |
— |
32.4 |
— |
(0.64, 0.36) |
| Ref. 46 |
605 |
17.2 |
— |
32.2 |
— |
— |
| Ref. 47 |
640 |
25.8 |
∼10.3 |
12.8 |
∼6.5 |
(0.68, 0.30) |
| Ref. 48 |
— |
11.1 |
— |
19.9 |
— |
(0.60, 0.39) |
| Ref. 49 |
636 |
11.1 |
9.0 |
8.7 |
7.0 |
(0.67, 0.33) |
| Ref. 31 |
608 |
∼21 |
— |
∼26 |
— |
(0.64, 0.33) |
As shown in Fig. 4d, the devices based on 4CzDMAC-DPS exhibit the same trend of the concentration-dependent EQE, which was gradually reduced from 14.6% (Device C3) to 8.8% (Device C1). As anticipated, even at a doping concentration of up to 10 wt%, the residual emission of 4CzDMAC-DPS was easily detectable (see Fig. 4f). However, the efficiency roll-off of Devices C1–C4 is relatively smaller than those of the corresponding Devices B1–B4, which is attributed to the faster RISC process of 4CzDMAC-DPS (2.6 × 106 s−1) than that of DMAC-DPS (0.8 × 106 s−1) and thus the reduced triplet exciton accumulation on host under high current density.50 Considering the same ΔEST (0.09 eV), the faster RISC could be ascribed to the higher spin–orbital coupling (SOC) of 4CzDMAC-DPS (0.105) assisted by the carbazole dendron, compared with that of DMAC-DPS (0.067).
When fixing the doping concentration at 1 wt%, the single emitting layer (EML) white OLEDs exhibited high color stability and a wide spectrum covering from 400 to 800 nm. The variations of the CIE coordinates are only (0.030, 0.031) and (0.001, 0.002) for Devices B4 and C4, respectively. Typically, WOLEDs with the single EML suffer from poor color stability, partially due to the inefficient triplet energy transfer from the blue host to orange/red dopants and consequently exciton annihilation processes.51–54 In our white devices, the short-range Dexter interaction was supposed to be blocked in the presence of the large steric hindrance, especially at low doping concentration. In addition to the stable EL spectra, Device C4 exhibits an outstanding color rendering index (CRI) of 80 and a maximum EQE 13.2%, which are merely reported in the complementary white OLEDs.
Although the optimized red Devices B3 and C3 have achieved the state-of-the-art EL performances among the solution-processed OLEDs, the residual blue emission still affected the color purity (Fig. 4e–f). To address this issue, a dual-host engineering strategy was employed to broaden the exciton recombination zone by introducing a common host 1,3-di(9H-carbazol-9-yl)benzene (mCP) with a high triplet energy of 2.9 eV. The ternary-blended EMLs were composed of mCP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMAC-DPS:R-D2 or mCP:4CzDMAC-DPS:R-D2 with weight ratios of 40:55:5, 60:35:5 and 80:15:5 respectively for Devices B5–B7 and C5–C7. The corresponding EL performances are summarized in Table 4. As predicted, the blue emission was gradually reduced with the decreasing concentration of the TADF host (see Fig. 5a and b), and thus the CIEx values were gradually increased from 0.62 to 0.64 and 0.55 to 0.59 for Devices B3–B7 and C3–C7, respectively. The ternary-blended Device B5 with DMAC-DPS exhibited not only higher color purity but also a considerably high EQE of 20% (see Fig. S6a (ESI†) and Table 4), which is comparable to that of the binary-blended Device B3. Furthermore, the introduction of mCP can further increase the intermolecular distance and thus effectively separate the TADF molecules, blocking the triple–triplet and triplet–polaron interaction induced exciton quenching. Accordingly, the efficiencies at 1000 cd m−2 were gradually increased upon reducing the concentration of the TADF component from 95 wt% to 15 wt%. For the mCP:4CzDMAC-DPS:R-D2 system, the triplet excitons on the host could be completely isolated owing to the large intermolecular distance among 4CzDMAC-DPS molecules. Therefore, Device C7 exhibited a considerably low efficiency roll-off of 9.0% with a high EQE of 14.2% at 1000 cd m−2 (see Fig. S6b, ESI†), which are the best values among those of the reported solution-processed OLEDs (Table 3).
:DMAC-DPS:R-D2 or mCP:4CzDMAC-DPS:R-D2 with weight ratios of 40:55:5, 60:35:5 and 80:15:5 respectively for Devices B5–B7 and C5–C7. The corresponding EL performances are summarized in Table 4. As predicted, the blue emission was gradually reduced with the decreasing concentration of the TADF host (see Fig. 5a and b), and thus the CIEx values were gradually increased from 0.62 to 0.64 and 0.55 to 0.59 for Devices B3–B7 and C3–C7, respectively. The ternary-blended Device B5 with DMAC-DPS exhibited not only higher color purity but also a considerably high EQE of 20% (see Fig. S6a (ESI†) and Table 4), which is comparable to that of the binary-blended Device B3. Furthermore, the introduction of mCP can further increase the intermolecular distance and thus effectively separate the TADF molecules, blocking the triple–triplet and triplet–polaron interaction induced exciton quenching. Accordingly, the efficiencies at 1000 cd m−2 were gradually increased upon reducing the concentration of the TADF component from 95 wt% to 15 wt%. For the mCP:4CzDMAC-DPS:R-D2 system, the triplet excitons on the host could be completely isolated owing to the large intermolecular distance among 4CzDMAC-DPS molecules. Therefore, Device C7 exhibited a considerably low efficiency roll-off of 9.0% with a high EQE of 14.2% at 1000 cd m−2 (see Fig. S6b, ESI†), which are the best values among those of the reported solution-processed OLEDs (Table 3).
Table 4 Summary of the EL data for the ternary red OLEDs
| Device |
TADF concentration [wt%] |
V
10
|
EQE [%] |
Roll-off (%) |
CIE (x, y) |
| Maximum |
At 1000 cd m−2 |
|
Operational voltage at a luminance of 10 cd m−2.
|
| B3 |
95 |
4.4 |
22.2 |
9.1 |
59.0 |
(0.62, 0.35) |
| B5 |
55 |
4.7 |
20.0 |
10.1 |
49.5 |
(0.63, 0.35) |
| B6 |
35 |
4.9 |
14.6 |
12.0 |
17.8 |
(0.63, 0.35) |
| B7 |
15 |
5.4 |
12.7 |
12.6 |
0.8 |
(0.64, 0.35) |
| C3 |
95 |
4.6 |
14.6 |
10.1 |
28.8 |
(0.55, 0.36) |
| C5 |
55 |
3.6 |
11.8 |
9.7 |
17.8 |
(0.57, 0.36) |
| C6 |
35 |
4.0 |
13.1 |
12.3 |
6.1 |
(0.57, 0.36) |
| C7 |
15 |
4.3 |
15.6 |
14.2 |
9.0 |
(0.59, 0.37) |
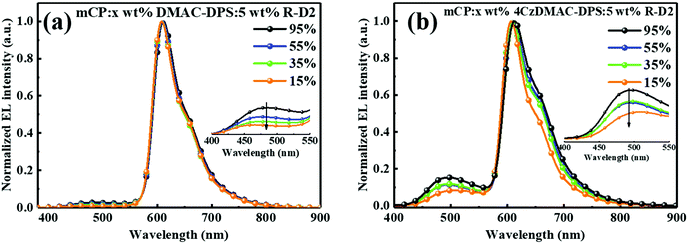 |
| | Fig. 5 Normalized EL spectra of (a) mCP:DMAC-DPS:R-D2 and (b) mCP:4CzDMAC-DPS:R-D2 doping systems. Inset: enlarged EL spectra from 400–550 nm. | |
Conclusions
In conclusion, we have demonstrated the state-of-the-art solution-processed red and white OLEDs by manipulating the intermolecular distance with the sterically shielded red phosphorescent dopant and the blue TADF host. Compared with the larger steric hindrance system with the dendron units both in the host and guest (4CzDMAC-DPS:R-D2), both DET and FRET were promoted by adopting the host DMAC-DPS without any peripherals and thus short intermolecular distance. Consequently, the red PhOLEDs achieved a considerably high maximum external quantum efficiency (EQE) of 22.2%. For 4CzDMAC-DPS:R-D2, the relatively faster reverse intersystem crossing rate in the host suppressed the triplet accumulation, allowing lower efficiency roll-off, i.e., a saturated red device with an efficiency roll-off down to 6.1% at 1000 cd m−2. Furthermore, a high-performance complementary white OLED with a maximum EQE of 13.2% was achieved, accompanied by ultra-stable CIE coordinates of (0.36, 0.39) over a wide range of luminance and high color rendering index (CRI) of 80. These results verify the feasibility of manipulating the energy transfer process in the simplified solution-process devices by changing the intermolecular distance between the host and the emissive cores.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the funding support from the National Natural Science Foundation of China (No. 62175189 and 51873159). This work was partially supported by the Open Fund of Guangdong Provincial Key Laboratory of Luminescence from Molecular Aggregates, South China University of Technology (No. 2019B030301003), and the Exchange Program for Academic Collaboration of Senior Talents between China and Canada, Australia, New Zealand, and Latin America (2021-109).
References
- X. Yang, G. Zhou and W. Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC
 .
.
- H.-T. Mao, G.-F. Li, G.-G. Shan, X.-L. Wang and Z.-M. Su, Coord. Chem. Rev., 2020, 413, 213283 CrossRef CAS .
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC .
- Y.-K. Wang, S.-H. Li, S.-F. Wu, C.-C. Huang, S. Kumar, Z.-Q. Jiang, M.-K. Fung and L.-S. Liao, Adv. Funct. Mater., 2018, 28, 1706228, DOI:10.1002/adfm.201706228 .
- Y. Jiang, K. F. Li, K. Gao, H. Lin, H. L. Tam, Y. Y. Liu, Y. Shu, K. L. Wong, W. Y. Lai and K. W. Cheah, Angew. Chem., Int. Ed., 2021, 60, 10007–10015 CrossRef CAS PubMed .
- C. Zang, S. Liu, M. Xu, R. Wang, C. Cao, Z. Zhu, J. Zhang, H. Wang, L. Zhang and W. Xie, Light: Sci. Appl., 2021, 10, 1–10 CrossRef PubMed .
- J. Xu, H. Liu, J. Li, Z. Zhao and B. Z. Tang, Adv. Opt. Mater., 2021, 9, 2001840 CrossRef CAS .
- Z. Wang, X.-L. Li, Z. Ma, X. Cai, C. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1706922 CrossRef .
- Q. Wang, Y. X. Zhang, Y. Yuan, Y. Hu, Q. S. Tian, Z. Q. Jiang and L. S. Liao, ACS Appl. Mater. Interfaces, 2018, 11, 2197–2204 CrossRef PubMed .
- J. Liang, C. Li, X. Zhuang, K. Ye, Y. Liu and Y. Wang, Adv. Funct. Mater., 2018, 28, 1707002 CrossRef .
- D. Zhang, L. Duan, D. Zhang, J. Qiao, G. Dong, L. Wang and Y. Qiu, Org. Electron., 2013, 14, 260–266 CrossRef CAS .
- Y. Tang, G. Xie, X. Yin, Y. Gao, J. Ding and C. Yang, J. Phys. Chem. Lett., 2020, 11, 5255–5262 CrossRef CAS PubMed .
- Y. Tang, Y. Gao, G. Xie and C. Yang, Nanoscale Horiz., 2020, 5, 144–149 RSC .
- Y. Zou, S. Gong, G. Xie and C. Yang, Adv. Opt. Mater., 2018, 6, 1800568 CrossRef .
- J. Park, H. Yoon, G. Kim, B. Lee, S. Lee, S. Jeong, T. Kim, J. Seo, S. Chung and Y. Hong, Adv. Funct. Mater., 2019, 29, 1902412 CrossRef .
- S. Wang, H. Zhang, B. Zhang, Z. Xie and W.-Y. Wong, Mater. Sci. Eng., R, 2020, 140, 100547 CrossRef .
- X. Ban, F. Chen, Y. Liu, J. Pan, A. Zhu, W. Jiang and Y. Sun, Chem. Sci., 2019, 10, 3054–3064 RSC .
- L. Tu, Y. Xie, Z. Li and B. Tang, SmartMat, 2021, 2, 326–346 CrossRef .
- D. Sun, Z. Ren and S. Yan, J. Mater. Chem. C, 2018, 6, 4800–4806 RSC .
- K. Wu, Z. Wang, L. Zhan, C. Zhong, S. Gong, G. Xie and C. Yang, J. Phys. Chem. Lett., 2018, 9, 1547–1553 CrossRef CAS PubMed .
- W. Zeng, T. Zhou, W. Ning, C. Zhong, J. He, S. Gong, G. Xie and C. Yang, Adv. Mater., 2019, 31, 1901404 CrossRef PubMed .
- G. Xie, J. Luo, M. Huang, T. Chen, K. Wu, S. Gong and C. Yang, Adv. Mater., 2017, 29, 1604223 CrossRef PubMed .
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442–2446 RSC .
- Y. Li, G. Xie, S. Gong, K. Wu and C. Yang, Chem. Sci., 2016, 7, 5441–5447 RSC .
- Y. Li, T. Chen, M. Huang, Y. Gu, S. Gong, G. Xie and C. Yang, J. Mater. Chem. C, 2017, 5, 3480–3487 RSC .
- L. Zhao, S. Wang, J. Lü, J. Ding and L. Wang, J. Mater. Chem. C, 2017, 5, 9753–9760 RSC .
- L. Liu, R. Dong, D. Ye, Y. Lu, P. Xia, L. Deng, Y. Duan, K. Cao and S. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 12268–12277 CrossRef CAS PubMed .
- S. Chen, Q. Zhang, W. Shang, L. Liu, H. Yu, S. Zhang, L. Deng, M. Wang, M. Wang and X. Li, Sci. Rep., 2018, 8, 1–9 Search PubMed .
- H. Dai, Q. Shen, J. Shao, W. Wang, F. Gao and X. Dong, The Innovation, 2021, 2, 100082 CrossRef PubMed .
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS .
- T. Giridhar, T. H. Han, W. Cho, C. Saravanan, T. W. Lee and S. H. Jin, Chem. – Eur. J., 2014, 20, 8260–8264 CrossRef CAS PubMed .
- D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050–5055 CrossRef CAS PubMed .
-
J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science & Business Media, 2013 Search PubMed .
- P. Wei, D. Zhang and L. Duan, Adv. Funct. Mater., 2020, 30, 1907083 CrossRef CAS .
- D. Zhang, L. Duan, D. Zhang and Y. Qiu, J. Mater. Chem. C, 2014, 2, 8983–8989 RSC .
- S. Hu, J. Zeng, X. Zhu, J. Guo, S. Chen, Z. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 27134–27144 CrossRef CAS PubMed .
- K. Sudha, S. Sundharamurthi, S. Karthikaikumar, K. Abinaya and P. Kalimuthu, J. Phys. Chem. C, 2017, 121, 5941–5948 CrossRef CAS .
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed .
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS .
- Y. Xiang, G. Xie, Q. Li, L. Xue, Q. Xu, J. Zhu, Y. Tang, S. Gong, X. Yin and C. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 29105–29112 CrossRef CAS PubMed .
- J.-H. Lee, S. Lee, S.-J. Yoo, K.-H. Kim and J.-J. Kim, Adv. Funct. Mater., 2014, 24, 4681–4688 CrossRef CAS .
- M. Baldo, M. Thompson and S. Forrest, Nature, 2000, 403, 750–753 CrossRef CAS PubMed .
- Z. Wu and D. Ma, Mater. Sci. Eng., R, 2016, 107, 1–42 CrossRef .
- D. Zhang, L. Duan, Y. Li, H. Li, Z. Bin, D. Zhang, J. Qiao, G. Dong, L. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS .
- X. Liu, B. Yao, Z. Zhang, X. Zhao, B. Zhang, W.-Y. Wong, Y. Cheng and Z. Xie, J. Mater. Chem. C, 2016, 4, 5787–5794 RSC .
- J. Wang, X. Zhang, L. Fan, X. Zhang, Y. Qin, R. Li, Y. Chen, W.-Y. Lai, X. Zhang and W. Huang, J. Mater. Chem. C, 2020, 8, 9909–9915 RSC .
- Y. Sun, X. Yang, Z. Feng, B. Liu, D. Zhong, J. Zhang, G. Zhou and Z. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 26152–26164 CrossRef CAS PubMed .
- X. Zhang, X. Guo, Y. Chen, J. Wang, Z. Lei, W. Lai, Q. Fan and W. Huang, J. Lumin., 2015, 161, 300–305 CrossRef CAS .
- L. Chen, S. Wang, Z. Yan, J. Ding and L. Wang, J. Mater. Chem. C, 2017, 5, 5749–5756 RSC .
- H. Wang, L. Meng, X. Shen, X. Wei, X. Zheng, X. Lv, Y. Yi, Y. Wang and P. Wang, Adv. Mater., 2015, 27, 4041–4047 CrossRef CAS PubMed .
- C. Cao, W. C. Chen, J. X. Chen, L. Yang, X. Z. Wang, H. Yang, B. Huang, Z. L. Zhu, Q. X. Tong and C. S. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 11691–11698 CrossRef CAS PubMed .
- Z. Chen, X.-K. Liu, C.-J. Zheng, J. Ye, X.-Y. Li, F. Li, X.-M. Ou and X.-H. Zhang, J. Mater. Chem. C, 2015, 3, 4283–4289 RSC .
- J. Ye, C. J. Zheng, X. M. Ou, X. H. Zhang, M. K. Fung and C. S. Lee, Adv. Mater., 2012, 24, 3410–3414 CrossRef CAS PubMed .
- X. K. Liu, Z. Chen, J. Qing, W. J. Zhang, B. Wu, H. L. Tam, F. Zhu, X. H. Zhang and C. S. Lee, Adv. Mater., 2015, 27, 7079–7085 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05470h |
|
| This journal is © The Royal Society of Chemistry 2022 |

 *d,
Maolin
Yu
a,
Hengjia
Liu
a,
Yuhan
Gao
a,
Guohua
Xie
*d,
Maolin
Yu
a,
Hengjia
Liu
a,
Yuhan
Gao
a,
Guohua
Xie