
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancement of thermally activated delayed fluorescence properties by substitution of ancillary halogen in a multiple resonance-like diplatinum(II) complex†
Piotr
Pander
 *ab,
Andrey V.
Zaytsev
c,
Amit
Sil
d,
J. A. Gareth
Williams
*d,
Valery N.
Kozhevnikov
*c and
Fernando B.
Dias
a
*ab,
Andrey V.
Zaytsev
c,
Amit
Sil
d,
J. A. Gareth
Williams
*d,
Valery N.
Kozhevnikov
*c and
Fernando B.
Dias
a
aDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: piotr.h.pander@durham.ac.uk
bCentre for Organic and Nanohybrid Electronics, Silesian University of Technology, Konarskiego 22B, 44-100 Gliwice, Poland
cDepartment of Applied Sciences, Northumbria University, Ellison Building, Newcastle upon Tyne, NE1 8ST, UK. E-mail: valery.kozhevnikov@northumbria.ac.uk
dDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 14th January 2022
Abstract
We present an in-depth investigation of the influence of chloro-to-iodo exchange on the thermally activated delayed fluorescence (TADF) of a dinuclear platinum(II) complex featuring monodentate halide ancillary ligands. The complexes are constructed using a ditopic bis-N^C^N-chelating ligand of the form (N^C^N–N^C^N)Pt2X2. The initially formed chloro complex (X = Cl) is readily transformed into the iodo analogue (X = I). The change is found to increase the radiative decay rate constant kr by around 3-fold to 3–4 × 105 s−1. This remarkably high value is comparable with the state-of-the-art iridium(III) organometallic phosphors. The improved luminescence properties of the iodo compound are shown to be due to a smaller singlet–triplet energy gap ΔEST compared to the chloro analogue. Iodide reduces the HOMO–LUMO overlap in a multiple resonance-like orbital structure of the complex. Iodination is therefore the first practical strategy shown to improve the TADF properties of diplatinum(II) complexes. The analogous monoplatinum(II) phosphorescent complex is studied in parallel in order to shed light more generally on the effect of iodo ligands on the triplet and singlet states and on spin–orbit coupling (SOC) in platinum(II) complexes.
Introduction
Highly luminescent iridium(III) and platinum(II) organometallic compounds have become widespread in OLEDs in recent years due to their high stability and short phosphorescence lifetimes.1–5 The triplet radiative rate constants (kTr) of such complexes are generally four to five orders of magnitude higher than those of metal-free organic phosphors.6 Efforts towards further accelerating their radiative decay, to reduce the influence of luminescence quenching processes in devices, have mostly focused on strategies that increase the metal character of the excited states to enhance spin–orbit coupling (SOC).1,7 Recently, it has been found that the use of dinuclear structures – featuring two metal ions – may lead to larger SOC and accelerated triplet radiative rate constants relative to structurally comparable mononuclear complexes.8–10 Meanwhile, the decay rates of organometallic complexes may also be increased if they exhibit thermally activated delayed fluorescence (TADF).11,12 This phenomenon relies on the up-conversion of triplets into singlets (T1 → S1) and thus benefits from the high singlet radiative rates (S1 → S0) rather than relying on the promotion of the triplet radiative rate constant (T1 → S0).1 It results in radiative decay rates being less dependent on SOC than those of conventional phosphorescent complexes. TADF shows particular promise for promoting kr in red and near infrared (NIR) organometallic emitters, where SOC is invariably diminished due to increased ligand character to the excited state upon extending the ligand conjugation, for example.13,14So far, TADF properties have been found in a plethora of metal-free compounds15,16 as well as in organometallic complexes of Cu(I),17–19 Ag(I),20 Au(I)21 and Pd(II),11,22,23 but lesser known examples involving other metal ions such as W(VI)24 or Sn(IV)25 also exist. The latest discovery of TADF properties in dinuclear platinum(II)26 and of somewhat similar behaviour in di- and mononuclear iridium(III)27 complexes has demonstrated that large radiative rates can be obtained without the need for strong SOC from the metal. Further research has indicated that dinuclear complexes of platinum(II) are more likely to demonstrate TADF than their mononuclear counterparts, as the former have smaller ΔEST and larger S1 → S0 radiative rates – factors crucial for efficient delayed fluorescence.28 There persists a need, however, to better understand all the effects that lead to efficient TADF in this newly emerged group of emitters. Having recently studied the first example of a Pt(II) complex 1 involving a ditopic bis-N^C^N-chelating ligand that demonstrates TADF under ambient conditions, we sought possible ways to reduce ΔEST and thus further promote the radiative decay. In this contribution, we show how such an effect can be readily achieved through metathesis of the halide co-ligands from chloro to iodo (compound 3) and we also probe the effect of iodo ligands more generally by studying the mononuclear complex 4 (Chart 1).
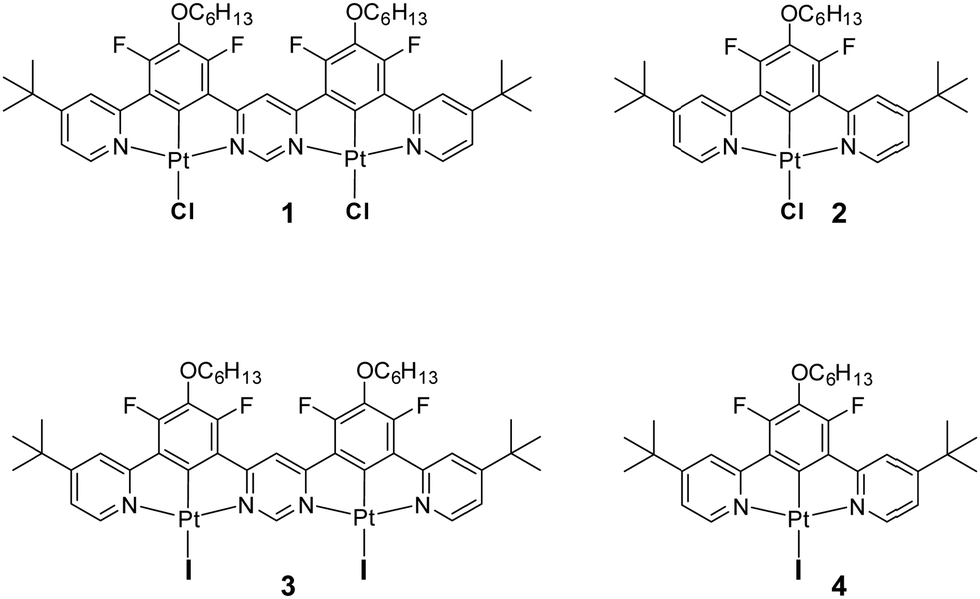 | ||
| Chart 1 Top: Structure of the dinuclear Pt(II) complex 1 recently discovered to show TADF28 and of its mononuclear analogue 2, which feature monodentate chloro ligands. Bottom: The structures of the iodo derivatives 3 and 4 prepared and studied in the current work, being the analogues of 1 and 2 respectively. | ||
Synthesis
The rationale behind the use of ditopic, bis-N^C^N-coordinating ligands has been discussed in our earlier work.28 Briefly, N^C^N-coordinating ligands – especially those based on 2,6-dipyridylbenzenes – have been shown to form cyclometallated complexes with metal ions such as Pt(II) (e.g., 2) and Ir(III).27 The short M–C bonds coupled with the strong σ donation properties of the metallated ring ensure signficant metal character to the lowest-energy excited states, and hence efficient spin–orbit coupling.29 The synergy between such σ donation and π* acceptor character of the heterocycles meanwhile ensures that potentially deactivating d–d states are kept high in energy. Finally, the rigidity associated with such structures helps to minimise non-radiative decay pathways.30 The coupling of two such N^C^N units together through a mutually common pyrimidine ring offers an attractive route to dinuclear complexes where these desirable features are not only retained, but are also accompanied by significant lowering of the emissive excited state energy and enhancement of the radiative rate constant.31The synthesis of the derivative incorporating fluoro, hexoxy, and tert-butyl substituents (enhancing solubility of the ensuing complex) has been described in our earlier work, together with its metallation with two Pt(II) ions. In the present work, the resulting dinuclear complex 1 featuring monodentate chloro ligands has been transformed to the diiodo derivative 3 upon treatment with an excess of sodium iodide in acetone under reflux (see Section S2, ESI†). The mononuclear model 4 was similarly obtained from 2. The identity and purity of the two new complexes were confirmed by 1H, 13C, 19F NMR spectroscopy and by elemental analysis. Crystals of 3 suitable for X-ray diffraction analysis were obtained from several separate crystallisations. Although the data could not be refined to a level sufficient for in-depth consideration of bond lengths and angles, the identity of the complex is unequivocally confirmed (see Section S3, ESI†).
Results and discussion
Solution state photophysics
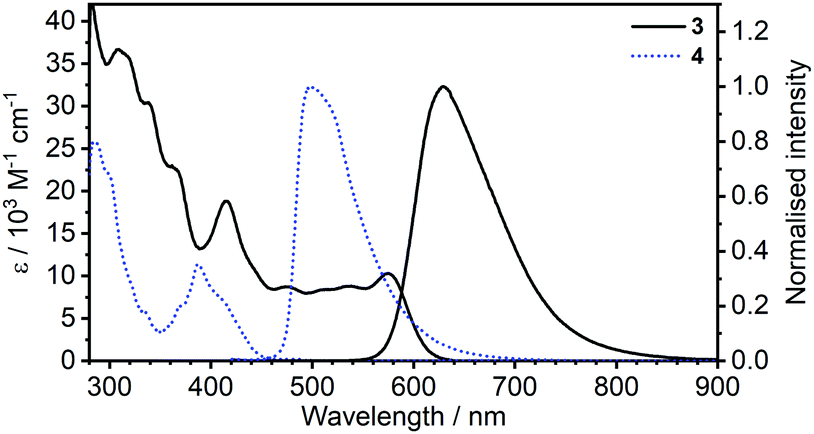 | ||
| Fig. 1 Comparison of absorption and photoluminescence spectra of complexes 3 and 4 in toluene, c = 10−5 M for absorption and c = 5 × 10−7 M for emission spectra. | ||
| Complex | Solvent | λ abs /nm (ε/M−1 cm−1) | λ em /nm | Φ PL | τ /μs | k r /105 s−1 | k nr /105 s−1 |
|---|---|---|---|---|---|---|---|
| a Absorption maxima and molar absorption coefficients. b Emission maxima. c Photoluminescence quantum yield recorded against rhodamine 6G (ΦPL = 0.9133) or coumarin 153 (ΦPL = 0.5333) in air-equilibrated absolute ethanol solutions. d Photoluminescence lifetime at room temperature. e Observed radiative rate constant, kr = ΦPL/τ. f Observed non-radiative rate constant, knr = (1 − ΦPL)/τ. | |||||||
| 3 | Toluene | 575 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300), 536 (8800), 509 (8400), 475 (8700), 443 (10800), 415 (18800), 368sh (22300), 339 (30400), 318sh (35800), 309 (36700) 300), 536 (8800), 509 (8400), 475 (8700), 443 (10800), 415 (18800), 368sh (22300), 339 (30400), 318sh (35800), 309 (36700) |
628 | 0.43 | 1.0 | 4.3 | 5.6 |
| Chlorobenzene | 571 (10100), 538 (8200), 511 (7900), 473sh (7500), 411 (16800), 363sh (23400), 337 (27000), 311 (31300) |
633 | 0.57 | 1.7 | 3.3 | 2.5 | |
| CH2Cl2 | 555 (9800), 528 (8700), 503 (8200), 463 (7300), 439sh (8700), 402 (16500), 374sh (22000), 353 (26400), 332 (27800), 309 (32900) |
635 | 0.27 | 1.5 | 1.8 | 5.0 | |
| 4 | Toluene | 478 (200), 411sh (7300), 388 (11400), 370sh (6400), 336sh (5700), 300sh (22000), 285 (25700) |
498 | 0.82 | 5.1 | 1.6 | 0.4 |
| Chlorobenzene | 479 (200), 412sh (5900), 387 (10600), 368sh (6000), 336 (5700), 299sh (21500) |
510 | 0.83 | 5.6 | 1.5 | 0.3 | |
| CH2Cl2 | 475 (200), 404sh (6300), 381 (11300), 361sh (7000), 333 (7500), 288 (29600), 259 (35100) |
511 | 0.83 | 6.0 | 1.4 | 0.3 | |
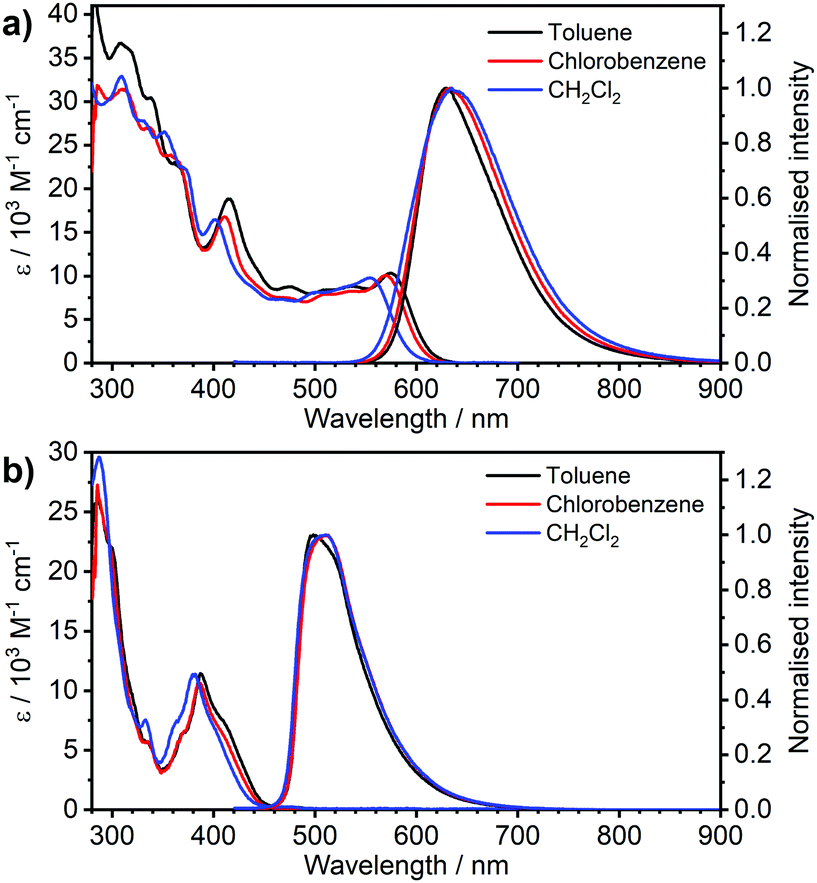 | ||
| Fig. 2 Absorption and photoluminescence spectra of complexes (a) 3 and (b) 4 in the three solvents indicated in the figure legends, c ≈ 10−5 M for absorption and c = 5 × 10−7 M for emission spectra. | ||
Most strikingly however, the photoluminescence and absorption spectra of 3 clearly overlap, indicating that the emissive state might be associated with a radiative transition possessing large oscillator strength (ε ∼ 104 M−1 cm−1), such as of singlet and not triplet character.11,23,26,28 On the other hand, 4 presents a typical picture of a phosphorescent transition metal complex with a relatively large apparent Stokes shift and no overlap of the emission spectrum with transitions of high oscillator strength. As this behaviour of complex 3 is in common with other recently discovered diplatinum(II) complexes26,28 that demonstrate TADF properties, we have studied the temperature-dependent behaviour of its photoluminescence in toluene solution (Fig. 3 and 4). The photoluminescence intensity of 3 increases by ∼25% upon decreasing the temperature from 300 to 230 K, which is attributed to the suppression of non-radiative decay pathways affecting the triplet state. At temperatures below 230 K we note a strong decline in photoluminescence intensity and an associated change in the spectrum, which becomes more clearly visible at temperatures close to 160 K. Further analysis allows the emission to be deconvoluted into the sum of two separate emission spectra: energetically higher, attributed to TADF (λem = 627 nm), and lower, attributed to phosphorescence (λem = 671 nm) (Fig. S5.4, ESI†). Having made this analysis we observe a significant decline in the TADF intensity and a small associated increase in phosphorescence intensity upon cooling from 230 K to 160 K. The ratio of TADF to phosphorescence shows a trend consistent with the models used in early studies of the phenomenon32 which allows the activation energy for the process to be estimated as Ea = 116 ± 2 meV. We also note the TADF-to-phosphorescence ratio does not follow the trend in the temperature range above 230 K – where non-radiative processes strongly affect the T1 state. Although the photoluminescence spectrum of 3 at 300 K is clearly dominated by TADF, phosphorescence still contributes to ∼7% of the total photoluminescence intensity (Fig. S5.4, ESI†).
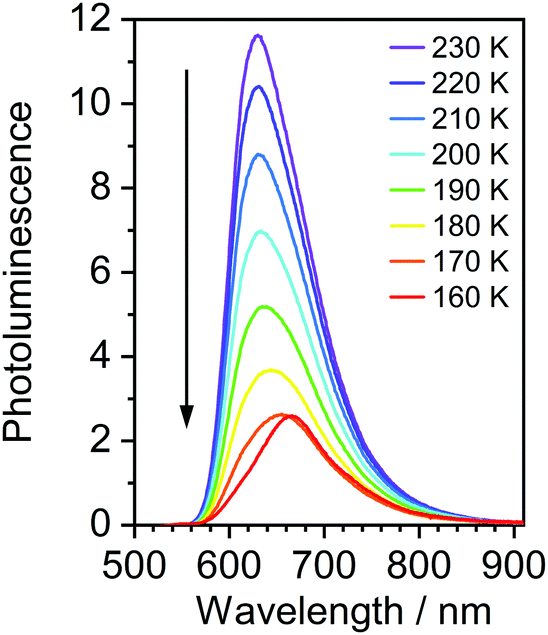 | ||
| Fig. 3 Photoluminescence spectra of 3 in toluene at various temperatures from 230 to 160 K, c = 10−5 M. | ||
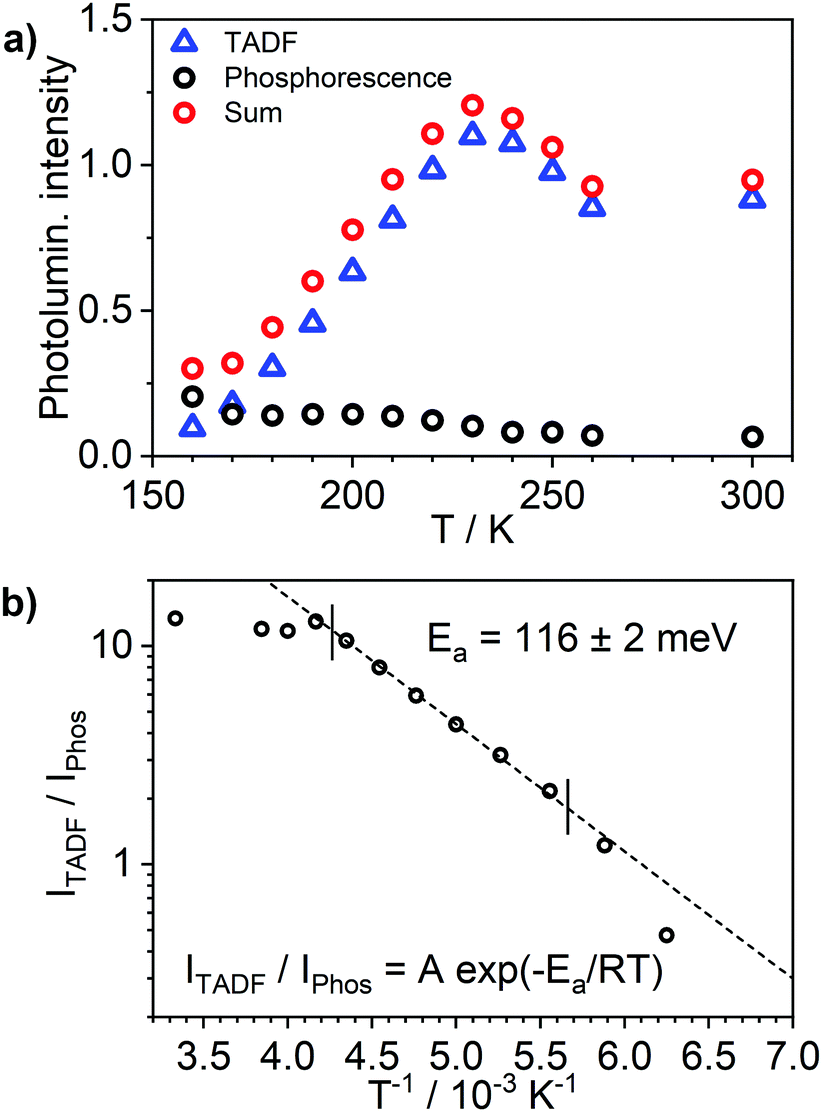 | ||
| Fig. 4 Photoluminescence of complex 3 in toluene solution (c = 10−5 M) as a function of temperature: (a) intensity of TADF, intensity of phosphorescence and their sum (total intensity); (b) ratio of TADF to phosphorescence. | ||
The photoluminescence decay of 3 was studied as a function of temperature in toluene (Fig. 5). At each temperature, the decay lifetime recorded at 630 nm is identical with the values registered at 650 and 700 nm (Fig. S5.6 and S5.7, ESI†) (note phosphorescence maximum at λem = 671 nm), which suggests that the two emissive states involved in the luminescent process, S1 and T1, remain in equilibrium.1,34 This behaviour of the photoluminescence decay,34 together with the other experimental features presented in this work, provide an unequivocal confirmation for the TADF mechanism being at work. The dependence of the observed lifetime with temperature is well fitted using the model described by Eqn 11,35 in Fig. 5, Fig. S5.6 and S5.7 (ESI†), where τobs(T) is the observed emission lifetime (s); Ea is the activation energy of the reverse intersystem crossing process in J mol−1; τPH is the phosphorescence lifetime (s); kSr is the radiative rate constant of the singlet state (s−1); R is the universal gas constant, 8.314 J mol−1 K−1; and T is the temperature in K. The model yields Ea = 101 ± 4 meV which is close to the value obtained from the energy difference between S1 and T1 measured from the onset of the deconvoluted steady state emission spectra.
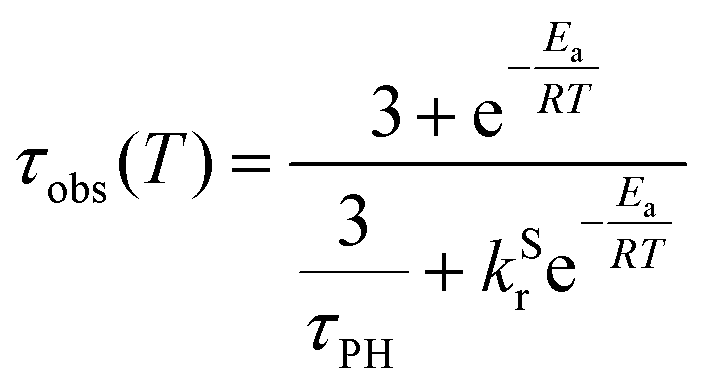 | (1) |
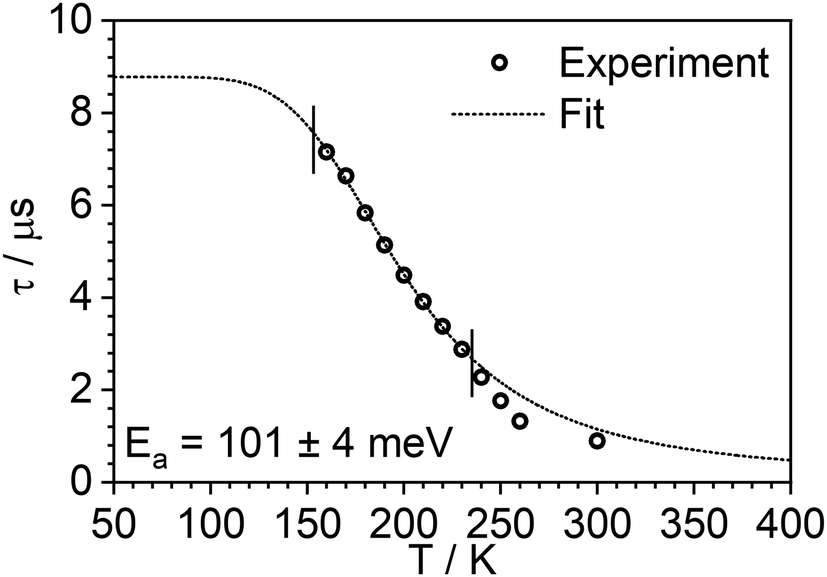 | ||
| Fig. 5 Photoluminescence decay lifetime as a function of temperature in 3 at 635 nm and the best fit according to Eqn (1). Note that experimental data points at higher temperature are excluded from the fit, as they are found from the steady-state experiments to be affected by changes in non-radiative decay. | ||
With the experimental picture presented by complex 3, the energetically higher state is associated with a transition demonstrating faster radiative decay to the ground state than the lower-energy luminescent state. Thus the former is attributed to a singlet and the latter to a triplet state. Such attribution of the emissive state multiplicity justifies the use of the term delayed fluorescence in the description of the experimental findings.
Solid state photophysics
Solid films are a convenient medium for studying luminescent complexes at cryogenic temperatures as they do not undergo phase transitions (freezing/melting) in the 300–80 K temperature window. Therefore, to further confirm the TADF properties of complex 3, we have studied the photoluminescence decay, as well as the steady-state and time-resolved photoluminescence spectra as a function of temperature in polystyrene films (Fig. 6 and Fig. S5.9–S5.12, ESI†). A similar analysis was made for the phosphorescent complex 4 and is presented in the ESI† (Fig. S5.13 and S5.14).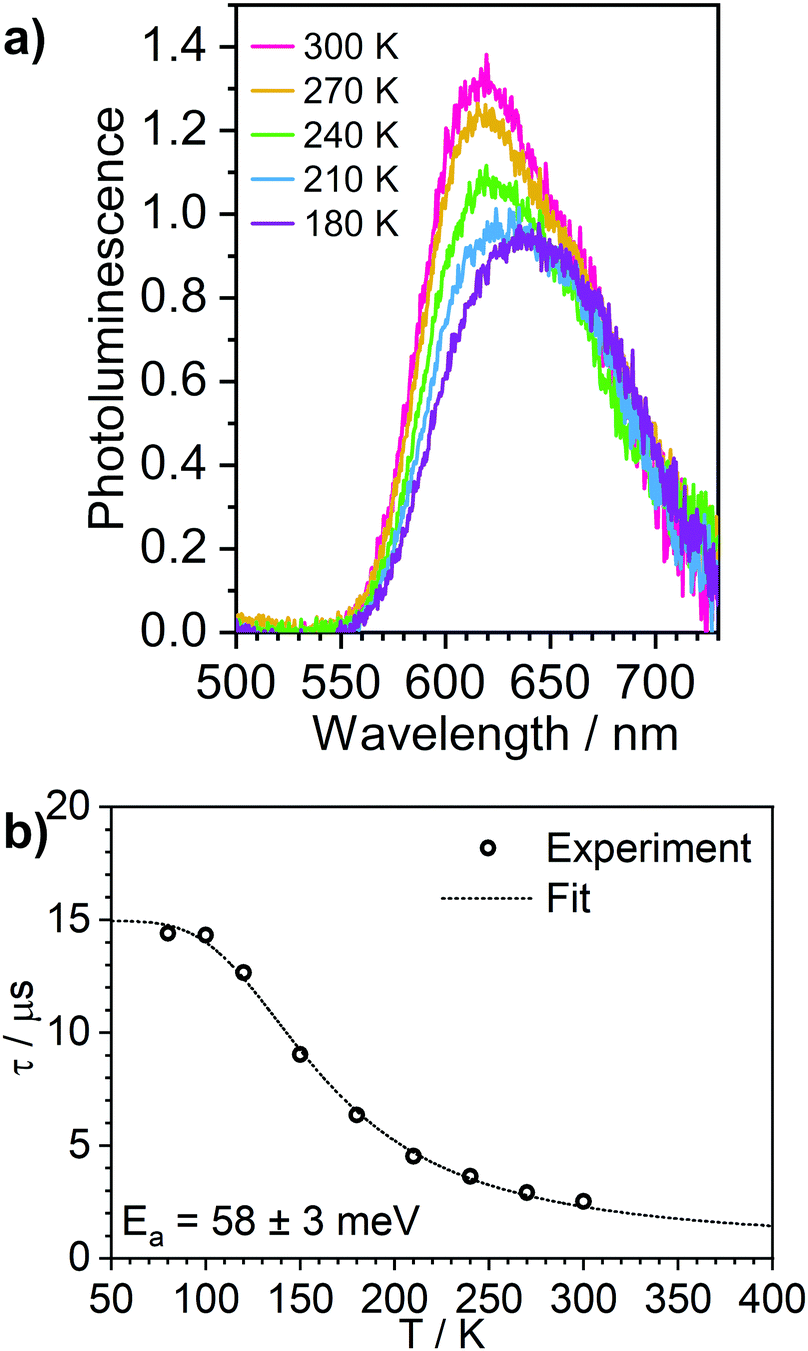 | ||
| Fig. 6 Photoluminescence of 3 in polystyrene matrix at 0.1% (w/w). (a) steady state spectra recorded at temperatures from 300 to 180 K. (b) photoluminescence decay lifetime at temperatures from 300 to 80 K, showing the experimental data points and the best fit according to Eqn (1). | ||
The photoluminescence behaviour of 3 in polystyrene matrix is in agreement with that in toluene solution. In this case, however, the Ea = 58 ± 3 meV (obtained by fitting photoluminescence lifetime as a function of temperature with Eqn (1)) is significantly smaller than in toluene. This also leads to the phosphorescence and TADF having very similar spectra so that the change in contribution of both components observed in function of temperature is less striking than in toluene solution. The radiative rate of the singlet state, kSr = 1 × 107 s−1, obtained from the fit is nearly an order of magnitude smaller than that reported in 1.28
Explaining the role of the ancillary halogen ligand using DFT and TDDFT calculations
Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations at the B3LYP/def2-TZVP/ZORA level of theory were employed in order to explain the divergent behaviour between complexes 1 and 2, and their iodo-analogues 3 and 4. First of all, we recognise that iodine, being a heavier halogen than chlorine, should introduce additional SOC to the complex. It is widely recognised that heavier halogens, Br and I, in metal-free organic molecules are often implicated in the room temperature phosphorescence (RTP) properties of such compounds and numerous examples are known.36–41 Heavy halogens, however, induce relatively long-lived RTP in comparison with typical platinum(II) or iridium(III) organometallic compounds, which can be attributed to differences in electron structure favouring the mixing of p orbitals into the excited state in halogens rather than d orbitals as with transition metals. Interestingly, efficient emission from a di-iridium(III) complex has been interpreted recently in terms of the strong SOC of T1 with singlets, associated with the change from chloride to iodide co-ligands.42 Such an effect is favoured by increasing halide to heterocycle charge-transfer character, as observed previously for Re(N^N)CO3X complexes (where X = halide), for example.43,44We first simulate the electronic properties of the phosphorescent monoplatinum(II) analogues 2 and 4 as a control group for the TADF complexes 1 and 3. Given the good correlation between simulated and experimental properties obtained earlier28 for 1 and 3, we again follow the approach of Younker and Dobbs45 to study molecules at their ground state geometry. The analysis of excited states is performed at two levels of theory: routine time-dependent density functional theory (TDDFT) and TDDFT including spin–orbit coupling (SOC-TDDFT). We will refer to the former as TDDFT or zero-order states (Sn, Tn), and to the latter as SOC-TDDFT states (Γn), where Γ0 denotes ground state. The single point energy calculations of TDDFT and SOC-TDDFT states have been performed using the ground state (S0) geometry optimised at the B3LYP/def2-TZVP level of theory. Further details are given in Section 1. General of the ESI.†
We have not observed any significant differences in luminescent properties between 2 and 4 (Table 2), nor in the calculations (Table S4.1, ESI†). We note however that complex 4 displays slightly lower experimental energy gap than 3, while the triplet energy from the phosphorescence spectrum remains virtually the same. At first, we consider the effect of the ancillary halogen on the molecular orbital structure. To minimise the number of orbitals in consideration, we chose HOMO, HOMO−1, LUMO, and LUMO+1, being the most relevant orbitals to the two lowest singlet and triplet excitations (Fig. 7, Tables S4.3 and S4.5, ESI†). Iodine as a stronger electron donor than chlorine affects mostly HOMO and HOMO−1, reducing the relative contribution of the π-conjugated N^C^N ligand to the molecular orbital. As a consequence, HOMO and HOMO−1 are localised mostly at the central atom and the ancillary iodide (Pt–I axis). Apart from that, the respective orbital contours in both 2 and 4 are nearly identical in each case. HOMO and HOMO−1 are visibly destabilised in 4 while LUMOs are stabilised, which leads to a narrower HOMO–LUMO gap in 4 than in 2 (Fig. S4.1 and S4.2, ESI†). However, SOC-dependent properties, such as zero-field splitting (ZFS) or the reciprocal triplet radiative rate kT−1r, are comparable, and the latter is in good agreement with experimental figures. Based on these results, we conclude that iodine per se does not introduce any significant SOC into the complex due to it being a heavier atom than chlorine. It appears that the HOMO-LUMO overlap in 4 is smaller than in 2 leading to the ΔEST in the former being nearly half the value of the latter (Table S4.1, ESI†).
| 1 | 3 | 2 | 4 | ||
|---|---|---|---|---|---|
| a Absorption spectrum onset. b Emission maxima. c Photoluminescence quantum yield. d Photoluminescence lifetime at room temperature. e Observed radiative rate constant, kr = ΦPL/τ. f Observed non-radiative rate constant, knr = (1 − ΦPL)/τ. g Photoluminescence lifetime at 80 K. h Singlet radiative decay rate. i Activation energy of the TADF process. j Singlet energy recorded from fluorescence (TADF) spectrum onset at room temperature. k Triplet energy recorded from phosphorescence spectrum onset at 80 K. l Singlet–triplet energy splitting. Figures for complexes 1 and 2 are reproduced from earlier work.28 Some of the values for complexes 3 and 4 are reproduced from Table 1. | |||||
| Chlorobenzene | λ abs /nm (onset) | 574 | 607 | 431 | 443 |
| λ em /nm | 578sh, 635 | 633 | 514 | 510 | |
| Φ PL | 0.51 | 0.57 | 0.85 | 0.83 | |
| τ /μs | 5.0 | 1.7 | 5.7 | 5.6 | |
| k r /105 s−1 | 1.0 | 3.3 | 1.5 | 1.5 | |
| k nr /105 s−1 | 1.0 | 2.5 | 0.3 | 0.3 | |
| Polystyrene | λ em /nm | 579sh, 640 | 618 | 498 | 502 |
| τ /μs | 5.3 | 2.5 | 4.1 | 4.1 | |
| τ /μs (80 K) | 12.3 | 14.3 | 4.1 | 4.1 | |
| k Sr /s | 9 × 107 | 1 × 107 | — | — | |
| E a /eV | 0.159 ± 0.020 | 0.058 ± 0.003 | — | — | |
| S1j/eV | 2.30 | 2.20 | — | — | |
| T1k/eV (80 K) | 2.10 | 2.14 | 2.68 | 2.67 | |
| ΔESTl/eV | 0.20 | 0.06 | — | — | |
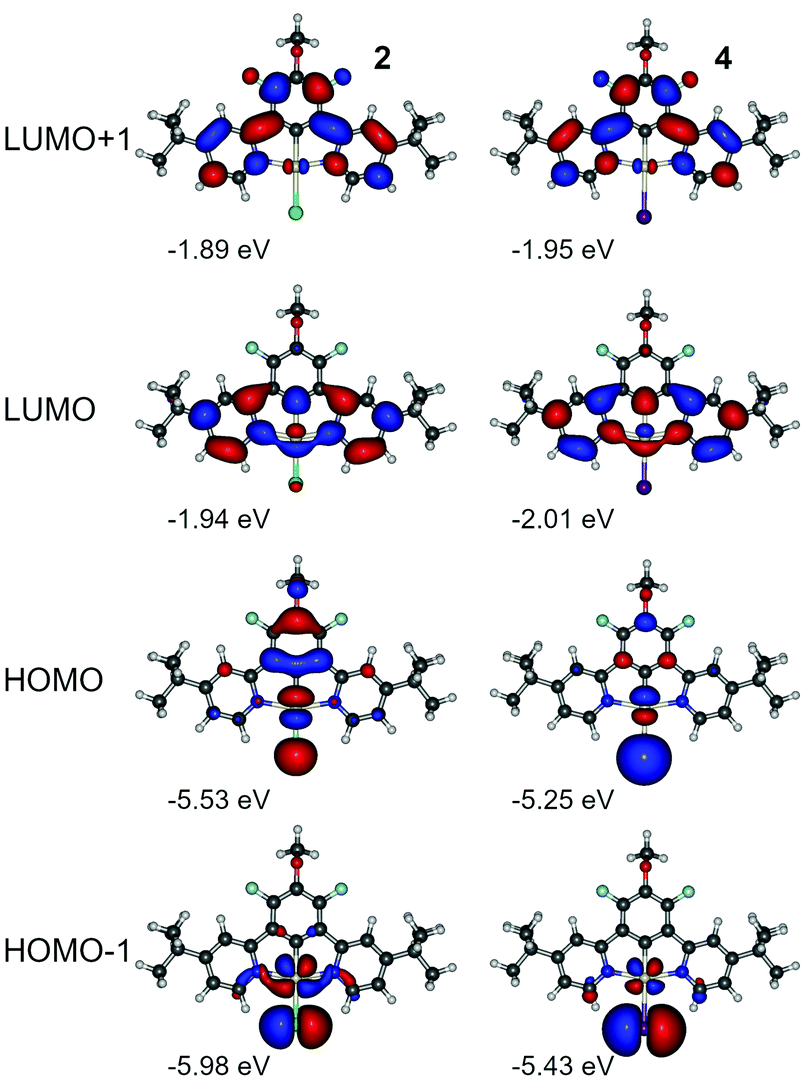 | ||
| Fig. 7 Molecular orbital contour plots at the B3LYP/dev2-TZVP/ZORA level for monoplatinum(II) complexes 2 and 4. | ||
Having the above in mind, we now analyse the properties of 1 and 3. In this case we look into HOMO to HOMO-2 and LUMO as being most relevant to the lowest three singlet and triplet excitations (Fig. 8, Fig. S4.1, S4.2 and Tables S4.2 and S4.4, ESI†). We first note the orbital geometry is divergent between 1 and 3 with the HOMO in 3 being of similar geometry to the HOMO−2 in 1. At the same time HOMO−1 and HOMO−2 in 3 resemble HOMO and HOMO−1 in 1, respectively. The HOMOs in 3 exhibit a decreased contribution of the π-conjugated ligand to the molecular orbital than in complex 1 due to the presence of the ancillary iodide, which is similar to behaviour of complexes 2 and 4. The LUMO remains overall very similar in both dinuclear complexes and is located mostly on the electron-deficient pyrimidine linker. The HOMOs in complex 3 are significantly destabilised in respect to analogue 1 – consistently with behaviour of 2 and 4. Destabilisation of HOMO together with stabilisation of LUMO in 3 leads to a narrower energy gap than in the chloro analogue 1. The iodo-complex 3 shows a significantly smaller ΔEST than its chloro analogue (Table S4.1, ESI†). The ancillary iodide contributes to further reduction in HOMO–LUMO overlap, resulting in near orthogonality of the frontier molecular orbitals (MOs) and leading to reduction of S1 and Sn energy without affecting the energy of the T1 state. The frontier MO pattern of 3 resembles that presented by B–N multiple resonance TADF emitters,46–48 yielding an excited state presenting a combination of small orbital overlap and negligible solvatochromic effects. We note that simulated ZFS is larger in 3 than in 1 and so is the kT−1r. According to the simulation (Table S4.1, ESI†), ZFS in 3 is larger than in its monoplatinum(II) analogue 4 while the opposite can be said about 1 and 2. So far, simulation appears to be consistent with the experimental data (Table 2).
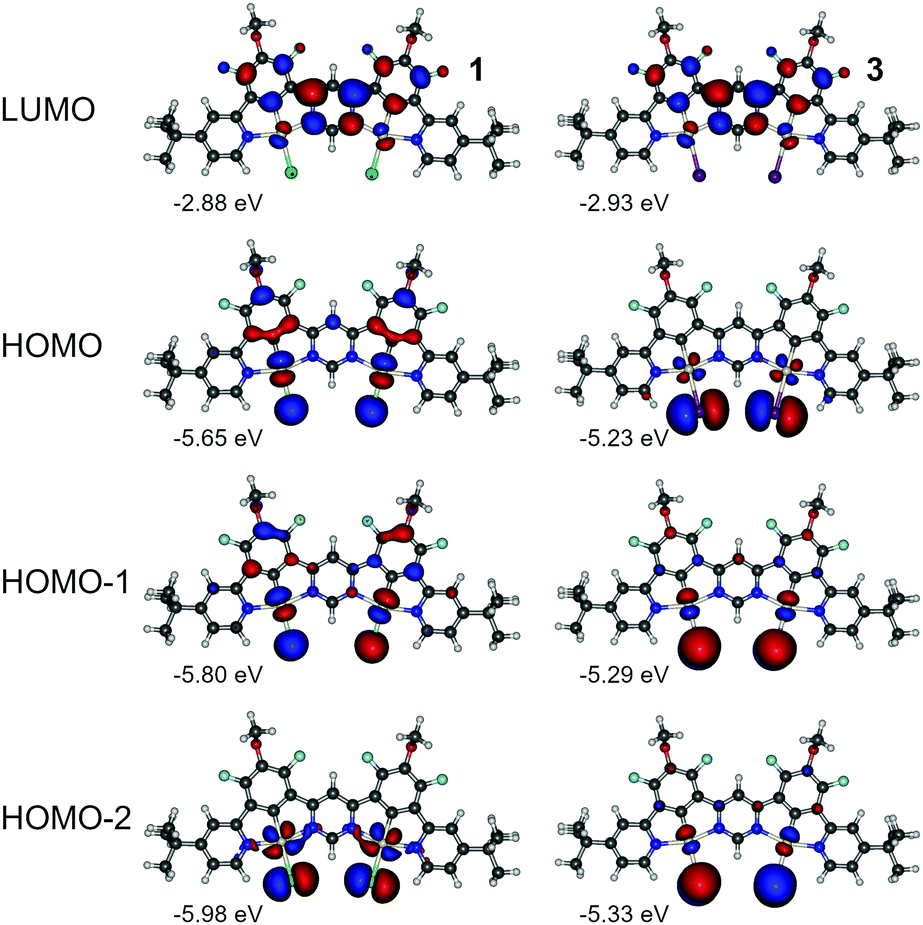 | ||
| Fig. 8 Molecular orbital contour plots at the B3LYP/dev2-TZVP/ZORA level for diplatinum(II) complexes 1 and 3. | ||
In all three complexes, 1, 2, 4, we find that both the S1 and T1 have a dominant HOMO → LUMO character while in 3, the T1 is dominated by the HOMO−1 → LUMO transition involving d orbitals that are different from those of the S1 (HOMO → LUMO). Thus, the S1–T1 SOC matrix element (SOCME) in 3, 2276 cm−1, is at least 100-fold larger than in the other three complexes. A large S1–T1 SOCME and small ΔEST should lead to a very high kTr. However, the oscillator strength f(T1 → S0) scales with that of the strongly coupled S1 with an extremely small f(S1 → S0) = 6 × 10−5, thus this mixing does not provide a significant promotion of the radiative rate of T1.49–51 The small S1 → S0 oscillator strength in 3 is a result of the near orthogonality between HOMO and LUMO in 3. The value of f(S1 → S0) in 3 suggests a natural S1 decay lifetime of ∼100 μs which is not only slower than the calculated natural phosphorescence decay rate of ∼60 μs but would render the molecule non-emissive through the S1 due to incurring non-radiative processes. This contradiction demonstrates that a simple TDDFT calculation does not give a full picture of molecule 3 and SOC has to be taken into account.
SOC causes mixing between singlet and triplet states which often leads to equivocal spin character to the excited states in organometallic complexes. We identified that S1 contributes in 45% into triplet state Γ3 which, however, retains small f(Γ3 → Γ0) = 1.6 × 10−5 due to the low f(S1 → S0) as discussed in the previous paragraph. In this case, the lowest SOC-TDDFT state with singlet-like properties, such as large oscillator strength f, is Γ4 which comprises 25% S2, 59% T2, and 12% T3, among other states. Γ4 is a singlet state with a strong contamination by triplet states, due to SOC. The oscillator strength f(Γ4 → Γ0) = 0.018 gives kS−1r= 560 ns, which is of the same order of magnitude as the experimentally derived value in polystyrene kS−1r = 100 ns (Table 2). Further to that, the ΔEST calculated as Γ1–Γ4 = 0.072 eV is also close to the experimental figure 0.058 eV in the same host. This demonstrates that the model used in this work correctly describes the excited state properties of complex 3.
The theoretical picture of excited states in organometallic compounds, especially those with strong SOC, is consistent with a loss of unequivocal state multiplicity.52 For example, complex 1 undergoes radiative decay through an excited state with ∼70% S1 character, whereas in 3 it is only 25% S2. In this case the luminescence arising from the energetically upper state, attributed to the S1 in metal-free systems, cannot always be identified as conventional fluorescence. However, due to the similar experimental picture of organometallic emitters to classical TADF molecules, we consider the term to be also applicable to the former and thus the unequivocal attribution of state multiplicity, i.e. S1 and T1, to still be helpful in the description of experimental results. In this case, the emissive states with substantially larger transition oscillator strength f(Γn → Γ0) than the lowest emissive triplet states Γ1–3 and clear admixture of singlets can be treated as singlets despite being strongly contaminated by triplet state admixtures.
The conclusions presented in this section strengthen the view that the profound luminescence properties of many dinuclear platinum(II) complexes do not necessarily come from the increased metal contribution to the excited state. Instead, it appears that the two metals benefit the complex by provision of additional singlet states involving metal d orbitals that are different from those of T1, while also bringing them closer together.8,9,42 The ancillary iodide acts in a similar way in complex 3, by destabilising HOMOs and reducing energy difference between HOMO and HOMO−1 etc. In extreme situations, this also leads to re-ordering of orbital patterns, which promotes even stronger SOC.
OLED devices
Complex 3 shows a lesser tendency to form NIR-emitting excimers than its chloro analogue 1. It is therefore more amenable to application in an OLED, taking advantage of its efficient monomolecular luminescence through TADF. The complex is highly soluble in toluene, in contrast to 1 which only shows negligible solubility in this solvent. This has allowed more complex OLED structures to be explored, incorporating high molecular weight poly(N-vinylcarbazole) (PVKH) as the hole transport and electron blocking layer (Devices 1 and 2 in Table 3). The device structures used are shown in the ESI† (Table S7.1). The electroluminescence spectrum of Devices 1 and 2 with 5% emitter doping is dominated by the red (CIE 0.62, 0.36) monomolecular emission of 3 (λEL = 612 nm) with a shoulder of a NIR-emitting excimer at 800–900 nm (Fig. 9). The luminescence at λEL = 612 nm shows a more narrowband (FWHM = 89 nm) distribution than that of typical charge-transfer red TADF emitters,16 thanks to the multiple resonance-like orbital pattern in the complex 3. The shoulder at 450–550 nm in the electroluminescence spectrum of Device 1 is attributed to the exciplex formed at the interface between PVKH and PO-T2T53 {2,4,6-tris[3-(diphenylphosphinyl)phenyl]-1,3,5-triazine} from the mCP:PO-T2T54 host {mCP – 1,3-bis(carbazol-9-yl)benzene}. Device 2, which uses a different electron transport material in the blend namely PBD {2-(4-biphenyl)-5-(4-tert-butylphenyl)-1,3,4-oxadiazole}, does not show any electroluminescence in this region. The OLED produced with higher, 33% emitter load (Device 3) demonstrates strong contribution from an NIR excimer (λEL = 782 nm), although the higher energy electroluminescence band at λEL = 612 nm does remain significant. We have also fabricated a non-doped device with the complex used neat as the emitting layer (Fig. S7.2, ESI†), but the electroluminescence was too weak to be fully characterised. This OLED shows excimer electroluminescence peaking at 824 nm, a longer wavelength than the value of 805 nm reported for the chloro analogue 1. Interestingly, a small contribution from the monomolecular emission can still be noticed in the electroluminescence spectrum of the non-doped device.| Device | x,a % | λ el,b nm | % λ > 700,c nm | Φ PL | EQEmax,e % | Max. luminance, cd m−2 | Max. radiosity, mW cm−2 |
|---|---|---|---|---|---|---|---|
| a Weight doping concentration of 3 in the emissive layer. b Electroluminescence maxima. c Percent of spectral power at wavelengths above 700 nm. d Photoluminescence quantum yield of the emissive layer in nitrogen. e Device maximum external quantum efficiency. | |||||||
| Dev 1 | 5 | 612 | 22 | 0.13 ± 0.04 | 3.11 | 1820 | 2.94 |
| Dev 2 | 5 | 612 | 21 | 0.11 ± 0.05 | 2.29 | 1840 | 2.86 |
| Dev 3 | 33 | 782 | 69 | 0.04 ± 0.02 | 0.36 | 160 | 0.82 |
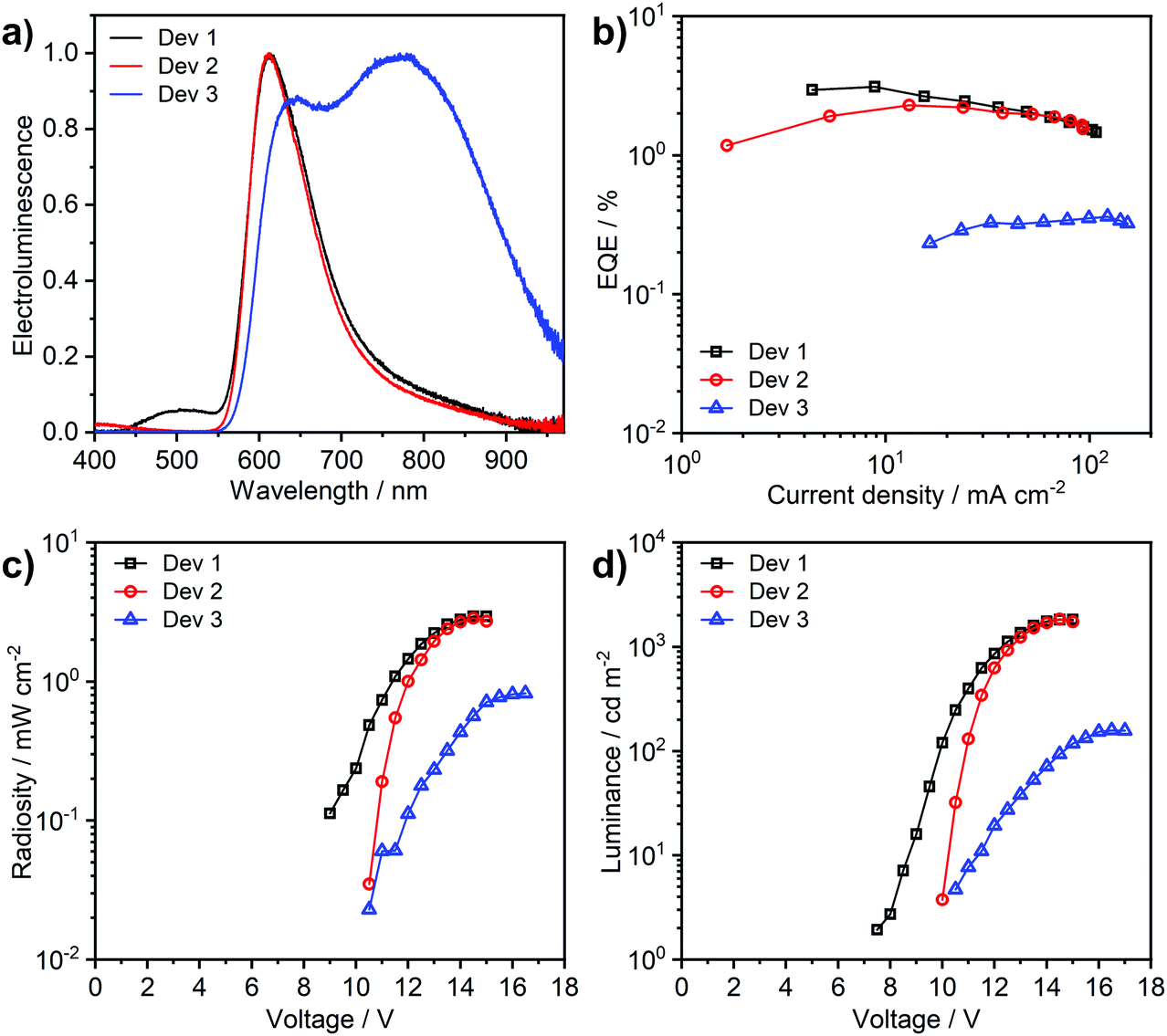 | ||
| Fig. 9 Characteristics of devices 1–3: (a) electroluminescence spectra; (b) external quantum efficiency (EQE) vs. current density; (c) radiosity–voltage characteristics; (d) luminance–voltage characteristics. | ||
The device external quantum efficiency agrees with the ΦPL in film in each case (Table 3). We note that the long-wavelength excimer formed from 3 has very low emission efficiency and acts as a luminescence quencher in solid film, significantly reducing the ΦPL from the high yields reported in solution. Therefore, there is still a need for further improvement of complexes such as 3 towards reducing intermolecular interactions in solid film, if highly-efficient multiple resonace-like TADF OLEDs are to be achieved.
Conclusions
We report a relatively simple practical strategy for enhancing the TADF properties of diplatinum(II) complexes via substitution of the ancillary ligand. The newly reported, delayed fluorescence di-Pt(II) complex 3 shows a very small Ea = 58 ± 3 meV in polystyrene and a large radiative decay rate in toluene, kr = 4.3 × 105 s−1, comparable with those of state-of-the-art iridium(III) complexes.55 Thanks to the TADF mechanism, a simple substitution of the ancillary ligand with iodide results in a radiative decay rate ∼3-fold larger than in the chloro analogue 1. We identify that, although the ancillary iodide does improve luminescent characteristics of complex 3 thanks to the TADF mechanism, it also leads to somehow greater SOC in the dinuclear complex. However, the role of iodine does not relate to its intrinsic SOC constant, but rather to its role in destabilising the highest occupied molecular orbitals in the molecule. Such a situation leads to a better mixing of singlet character into the triplet states as there is a large number of Sn states energetically close to the T1 in complex 3.To demonstrate potential applicability of this group of complexes in OLEDs, we have produced solution-processed devices demonstrating relatively narrow-band red electroluminescence (λEL = 612 nm, FWHM = 89 nm), with EQE of 3.11%. Some improvements are clearly necessary in order to prevent the occurrence of luminescence-quenching through aggregation and/or excimer formation in the solid state, in order to achieve highly efficient OLEDs.
Author contributions
P. P. – conceptualization, formal analysis, investigation, visualization, writing – original draft, writing – review and editing; A. V. Z. – investigation; A. S. – investigation; J. A. G. W. – conceptualization, funding acquisition, project administration, supervision, writing – review and editing; V. N. K. – conceptualization, funding acquisition, project administration, writing – original draft, writing – review and editing; F. B. D. – funding acquisition, project administration, resources, validation, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC (grant refs EP/S012788/1 and EP/S01280X) for support of this work. We are grateful to Dr Dmitry Yufit at Durham Chemistry for determining the crystal structure of complex 3 and for his continued assistance with crystallography. The authors thank Dr Gleb Baryshnikov for fruitful discussions. This work made use of the facilities of the Hamilton HPC Service of Durham University.Notes and references
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley, 2008 Search PubMed.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- Y. J. Cho, K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4050–4055 CrossRef CAS PubMed.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- A. Bossi, A. F. Rausch, M. J. Leitl, R. Czerwieniec, M. T. Whited, P. I. Djurovich, H. Yersin and M. E. Thompson, Inorg. Chem., 2013, 52, 12403–12415 CrossRef CAS PubMed.
- M. Z. Shafikov, P. Pander, A. V. Zaytsev, R. Daniels, R. Martinscroft, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, J. Mater. Chem. C, 2021, 9, 127–135 RSC.
- M. Z. Shafikov, R. Daniels, P. Pander, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, ACS Appl. Mater. Interfaces, 2019, 11, 8182–8193 CrossRef CAS PubMed.
- R. E. Daniels, S. Culham, M. Hunter, M. C. Durrant, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Dalton Trans., 2016, 45, 6949–6962 RSC.
- Z. Q. Zhu, C. Do Park, K. Klimes and J. Li, Adv. Opt. Mater., 2019, 7, 1801518 CrossRef.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS PubMed.
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- A. S. Romanov, L. Yang, S. T. E. Jones, D. Di, O. J. Morley, B. H. Drummond, A. P. M. Reponen, M. Linnolahti, D. Credgington and M. Bochmann, Chem. Mater., 2019, 31, 3613–3623 CrossRef CAS.
- R. Czerwieniec, J. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed.
- R. Czerwieniec and H. Yersin, Inorg. Chem., 2015, 54, 4322–4327 CrossRef CAS PubMed.
- A. S. Romanov, S. T. E. Jones, L. Yang, P. J. Conaghan, D. Di, M. Linnolahti, D. Credgington and M. Bochmann, Adv. Opt. Mater., 2018, 6, 1801347 CrossRef.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. A. Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- G. Li, Q. Chen, J. Zheng, Q. Wang, F. Zhan, W. Lou, Y. F. Yang and Y. She, Inorg. Chem., 2019, 58, 14349–14360 CrossRef CAS PubMed.
- P. W. Zach, S. A. Freunberger, I. Klimant and S. M. Borisov, ACS Appl. Mater. Interfaces, 2017, 9, 38008–38023 CrossRef CAS PubMed.
- K. Chan, T. Lam, D. Yu, L. Du, D. L. Phillips, C. Kwong, G. S. M. Tong, G. Cheng and C. Che, Angew. Chem., 2019, 131, 15038–15042 CrossRef.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- P. Pander, R. Daniels, A. V. Zaytsev, A. Horn, A. Sil, T. J. Penfold, J. A. G. Williams, V. N. Kozhevnikov and F. B. Dias, Chem. Sci., 2021, 12, 6172–6180 RSC.
- M. Z. Shafikov, R. Martinscroft, C. Hodgson, A. Hayer, A. Auch and V. N. Kozhevnikov, Inorg. Chem., 2021, 60, 1780–1789 CrossRef CAS PubMed.
- P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, P.-H. Lanoe, V. N. Kozhevnikov and F. B. Dias, J. Mater. Chem. C, 2021, 9, 10276–10287 RSC.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed.
- P.-H. Lanoë, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831–6834 RSC.
- C. A. Parker and C. G. Hatchard, Trans. Faraday Soc, 1961, 57, 1894 RSC.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- The IUPAC Compendium of Chemical Terminology, ed. V. Gold, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2019 Search PubMed.
- J. R. Kirchhoff, R. E. Gamache, M. W. Blaskie, A. A. Del Paggio, R. K. Lengel and D. R. McMillin, Inorg. Chem., 1983, 22, 2380–2384 CrossRef CAS.
- Y. Liu, G. Zhan, Z.-W. Liu, Z.-Q. Bian and C.-H. Huang, Chinese Chem. Lett., 2016, 27, 1231–1240 CrossRef CAS.
- J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447 RSC.
- H. Chen, L. Xu, X. Ma and H. Tian, Polym. Chem., 2016, 7, 3989–3992 RSC.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- Z. Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed.
- M. Shimizu, in Principles and Applications of Aggregation-Induced Emission, ed. Y. Tang and B. Z. Tang, Springer International Publishing, Cham, 2019, pp. 43–76 Search PubMed.
- M. Z. Shafikov, A. V. Zaytsev and V. N. Kozhevnikov, Inorg. Chem., 2021, 60, 642–650 CrossRef CAS PubMed.
- B. D. Rossenaar, D. J. Stufkens and A. Vlček, Inorg. Chem., 1996, 35, 2902–2909 CrossRef CAS.
- A. Cannizzo, A. M. Blanco-Rodríguez, A. El Nahhas, J. Šebera, S. Záliš, A. Vlček and M. Chergui, J. Am. Chem. Soc., 2008, 130, 8967–8974 CrossRef CAS PubMed.
- J. M. Younker and K. D. Dobbs, J. Phys. Chem. C, 2013, 117, 25714–25723 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53 CrossRef CAS.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- K. Nozaki, J. Chinese Chem. Soc., 2006, 53, 101–112 CrossRef CAS.
- K. Mori, T. P. M. Goumans, E. van Lenthe and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14523–14530 RSC.
- G. A. Crosby, K. W. Hipps and W. H. Elfring, J. Am. Chem. Soc., 1974, 96, 629–630 CrossRef CAS.
- P. Pander, S. Gogoc, M. Colella, P. Data and F. B. Dias, ACS Appl. Mater. Interfaces, 2018, 10, 28796–28802 CrossRef CAS PubMed.
- M. T. Walden, P. Pander, D. S. Yufit, F. B. Dias and J. A. G. Williams, J. Mater. Chem. C, 2019, 7, 6592–6606 RSC.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and characterisation of new materials; X-ray diffraction and crystal data; further information on the equipment and methods for theory, photophysical characterisation, electrochemistry, and OLED devices. Crystallographic data for complex 3. CCDC 2121802. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc05026e |
| This journal is © The Royal Society of Chemistry 2022 |