
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron clusters (ferrabisdicarbollides) shaping the future as radiosensitizers for multimodal (chemo/radio/PBFR) therapy of glioblastoma†
Miquel
Nuez-Martínez
a,
María
Queralt-Martín
 b,
Amanda
Muñoz-Juan
a,
Vicente M.
Aguilella
b,
Anna
Laromaine
a,
Francesc
Teixidor
a,
Clara
Viñas
*a,
Catarina G.
Pinto
c,
Teresa
Pinheiro
d,
Joana F.
Guerreiro
c,
Filipa
Mendes
c,
Catarina
Roma-Rodrigues
ef,
Pedro V.
Baptista
ef,
Alexandra R.
Fernandes
ef,
Srecko
Valic
g and
Fernanda
Marques
*c
b,
Amanda
Muñoz-Juan
a,
Vicente M.
Aguilella
b,
Anna
Laromaine
a,
Francesc
Teixidor
a,
Clara
Viñas
*a,
Catarina G.
Pinto
c,
Teresa
Pinheiro
d,
Joana F.
Guerreiro
c,
Filipa
Mendes
c,
Catarina
Roma-Rodrigues
ef,
Pedro V.
Baptista
ef,
Alexandra R.
Fernandes
ef,
Srecko
Valic
g and
Fernanda
Marques
*c
aInstitut de Ciència de Materials de Barcelona, ICMAB-CSIC, Campus Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain. E-mail: clara@icmab.es
bLaboratory of Molecular Biophysics, Department of Physics, Universitat Jaume I, 12071 Castelló, Spain
cCentro de Ciências e Tecnologias Nucleares and Departamento de Engenharia e Ciências Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10, 2695-066 Bobadela LRS, Portugal. E-mail: fmarujo@ctn.tecnico.ulisboa.pt
diBB – Instituto de Bioengenharia e Biociências, Departamento de Engenharia e Ciências Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal
eUCIBIO – Applied Molecular Biosciences Unit, Department of Life Sciences, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal
fAssociate Laboratory i4HB – Institute for Health and Bioeconomy, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal
gRuđer Bošković Institute, Bijenička 54, HR-10000 Zagreb, Croatia
First published on 24th October 2022
Abstract
Glioblastoma multiforme (GBM) is the most common and fatal primary brain tumor, and is highly resistant to conventional radiotherapy and chemotherapy. Therefore, the development of multidrug resistance and tumor recurrence are frequent. Given the poor survival with the current treatments, new therapeutic strategies are urgently needed. Radiotherapy (RT) is a common cancer treatment modality for GBM. However, there is still a need to improve RT efficiency, while reducing the severe side effects. Radiosensitizers can enhance the killing effect on tumor cells with less side effects on healthy tissues. Herein, we present our pioneering study on the highly stable and amphiphilic metallacarboranes, ferrabis(dicarbollides) ([o-FESAN]− and [8,8′-I2-o-FESAN]−), as potential radiosensitizers for GBM radiotherapy. We propose radiation methodologies that utilize secondary radiation emissions from iodine and iron, using ferrabis(dicarbollides) as iodine/iron donors, aiming to achieve a greater therapeutic effect than that of a conventional radiotherapy. As a proof-of-concept, we show that using 2D and 3D models of U87 cells, the cellular viability and survival were reduced using this treatment approach. We also tested for the first time the proton boron fusion reaction (PBFR) with ferrabis(dicarbollides), taking advantage of their high boron (11B) content. The results from the cellular damage response obtained suggest that proton boron fusion radiation therapy, when combined with boron-rich compounds, is a promising modality to fight against resistant tumors. Although these results are encouraging, more developments are needed to further explore ferrabis(dicarbollides) as radiosensitizers towards a positive impact on the therapeutic strategies for GBM.
1. Introduction
Cancer is a multifactorial disorder comprising a set of diseases arising from the uncontrolled growth and proliferation of abnormal cells because of the combined genetic and environmental factors. The core hallmarks of cancer include the abnormal cells’ ability to sustain proliferative signaling, evade growth suppressors, induce angiogenesis, enable replicative immortality, resist cell death, and spread to other tissues and organs (metastasis).1 Despite recent advances in therapeutics and diagnostics, cancer is still one of the leading causes of death worldwide, and given the ever-increasing number of new cases, clinical management of the disease continues to be a challenge in the 21st century.2Currently, surgery, chemotherapy and radiotherapy are the most widely used cancer treatment modalities.3,4 Their selection and application in therapeutic approaches depend on the cancer type, location and stage of progression, in addition to the inherent complexity of tumor heterogeneity.5
Chemotherapy in cancer treatment is usually employed by administering cytostatic and/or cytotoxic drugs that halt tumor progression by suppressing the cells’ ability to divide and induce apoptosis. However, most of the chemotherapeutic agents also target healthy cells, which results in severe dose-limiting side effects.6 Moreover, patients exposed to prolonged chemotherapy regimens may acquire multidrug resistance (MDR) leading to treatment failure and relapse in most of the cases.7
Radiation therapy (RT) is an important cancer treatment modality, and approximately 50% of cancer patients receive radiation therapy during the course of their illness. The main goal of radiation therapy is to maximize the radiation dose to kill cancer cells, while sparing normal cells and tissues adjacent to or in the radiation path.8 In the clinical setting, one of the most common approaches is to deliver high energy/high frequency electromagnetic radiation (X-rays and γ-rays) to tumor sites – external beam radiation. These electromagnetic X-rays and gamma rays are low-LET (Linear Energy Transfer) radiation sources composed of massless photons. Nevertheless, some of these therapeutic approaches still deliver radiation to normal tissues, thus increasing the risk of inducing additional cancers.9
Moreover, tumors resistant to radiation cannot be controlled even with high doses of radiation. Some new irradiation modalities are under development and include, for instance, particle beam therapy based on high-LET radiation10 using particles with substantial mass and charge (alpha-particles, electrons, protons, and carbon ions), boron neutron capture therapy (BNCT)11–14 using neutral particles such as neutrons and photodynamic therapy (PDT) – a light based technology that has also shown promising potential.15 However, except PDT, these newer modalities can only typically be performed in larger medical facilities and, to date, have shown no evidence of clinical superiority compared to X-rays.16,17 Although great success has been achieved with radiotherapy, further exploration of irradiation technologies is necessary to overcome the presented challenges.
A fundamental factor that needs to be considered in radiotherapy is the distribution of energy through the cells, which is of paramount importance when considering the biological damage done by a fixed dose of radiation. This distribution is influenced by the charge and mass of the particles that compose the radiation. One approach with the potential to enhance the success of radiation therapy is the use of radiosensitizers, whose purpose is to increase the radiation dose at tumor sites selectively, and, ultimately, to reduce the effective radiation dose and enhance the efficacy of treatment through a synergistic cell-killing effect when combined with radiation.18,19 The main mechanisms include radiation-induced repair inhibition of DNA damage, increasing the degree of DNA damage, and disturbing the cell cycle and organelle function to improve cytotoxicity among others.
For a low radiation dose, the probability of the photoelectric effect and its concomitant Auger cascades or Compton scattering becomes significantly higher when a radiosensitizer is introduced within a targeted cell.19
Enriching tumors with high atomic number (Z) materials is an upcoming strategy to improve the effects of high-energy photons via amplifying of primary (electronic) processes.20,21 As the photoelectric effect is dependent on the atomic number of the material, high Z materials have been used as radiosensitizers, taking advantage of their high attenuation capability for X-rays or gamma radiation. For instance, the use of gold (Au, Z = 79) nanomaterials as radiosensitizers has been greatly explored due to their ability to effectively absorb X-ray energy and interact with radiation in tumor cells, emitting photoelectrons, Auger electrons, and other secondary electrons and Compton scattering. These secondary electrons not only interact with DNA directly, but also react with water to increase the production of reactive oxygen species (ROS) and further increase the sensitivity of tumor cells to radiation.22–25
Iodine compounds were introduced as contrast agents for mammalian cell cultures; however, their application as radiosensitizers and therapeutic efficacy in radiotherapy have not been extensively studied. The use of iodine in radiation therapy has shown to enhance the cell killing effect of X-rays and cause chromosomal aberrations in irradiated cells.26 Iodine possess four radioisotopes, [123I]−, [124I]−, [125I]− and [131I]−,27–31 that have been applied in medicine and biology in positron emission tomography (PET) or single photon emission computed tomography (SPECT) imaging and radiotherapy agents.32,33 More recently, photon activation using iodine from iododeoxyuridine (IdUrd) and monochromatic photons above the K absorption edge of iodine (33.2 keV) as the activating agent exhibited a significant radiosensitization effect on glioblastoma. Nonetheless, due to their short circulation half-life and the inability to pass the blood brain barrier (BBB), iodinated compounds’ clinical application has been limited.34–36
Metallacarboranes [M(C2B9H11)2]− are unique 3D aromatics not only theoretically but also because of their advantages of high stability,37 and water-soluble38 and redox-active nature,39 in which a central metal ion, commonly Co or Fe, is the common vertex of two joined by one vertex icosahedron.40–44 These compounds share the versatility of the carboranes while the metal center introduces additional properties in redox potentials and the overall charge of the molecule. Characteristic properties of the small anionic metallabis(dicarbollide) molecules are their chemical and thermal stability (withstanding strong acid, moderate base, high temperatures and intense radiation),45,46 as well as biological stability (neither degradation nor chemical modification compounds were identified after cells’ uptake),47,48 their solubility in both polar and nonpolar solvents,37 and their amphiphilicity.49–52 Besides these, the 3D cylindrical shape of the small anionic metallabis(dicarbollides) molecules with a size of 1.1 × 0.6 nm, producing strong dihydrogen B–H⋯H–N bond interactions with the amine groups of amino acids,53–55 proteins,56,57 lipids,58 DNA48 and glycolipids,59 has gained significant interest in the medicinal chemistry field. For therapeutic applications, metallacarboranes can act as multimodal anticancer agents by enabling a simultaneous approach to chemotherapy, radiotherapy and imaging, through the incorporation of multifunctional hybrid molecules (which introduce the concept of small molecules for multitherapeutic use). Their iodinated derivatives can have additional benefits as radiosensitizers, enhancing the local dose inside tumors upon exposure to electromagnetic radiation.60,61
The present study aims to explore the biological potential of [o-FESAN]− and [8,8′-I2-o-FESAN]− (Scheme 1) as radiosensitizers in a glioblastoma cell model, since an efficient intracellular uptake of these compounds is a crucial factor affecting radiation therapy efficacy.61Scheme 1 shows the formulae of the two small anionic ferrabis(dicarbollide) molecules with their vertices numbered.
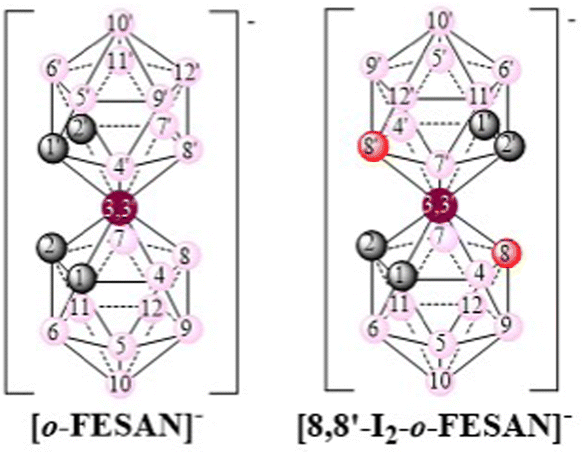 | ||
| Scheme 1 Schematic representation of the small anionic ferrabis(dicarbollide) clusters with their vertices numbered. Circles in grey correspond to Ccluster–H vertices while the pink and the red ones to B–H and B–I vertices, respectively. | ||
As metallabis(dicarbollide) and its diiodinated derivative cross the cell membrane,47 studies on the translocation of [o-FESAN]− and [8,8′-I2-o-FESAN]− anions through synthetic lipid membranes which could reveal important properties at the interface of biological and synthetic membranes are reported, as well as their in vivo evaluation using the small organism C. elegans. The radiobiological effects induced by electromagnetic radiation (γ-rays and X-rays) in glioblastoma cells treated with Na[o-FESAN] and Na[8,8′-I2-o-FESAN] are also reported. A preliminary study on the biological effects generated by a proton–boron nuclear fusion reaction (PBFR, see Scheme 2)62 with ferrabis(dicarbollides) in the U87 glioblastoma cells is reported. We have tested for the first time this reaction using metallacarboranes to enhance proton irradiation effects through the generation of short range (∼30 μm) high-LET alpha particles thus evaluating compounds’ clinical relevance for this treatment modality.
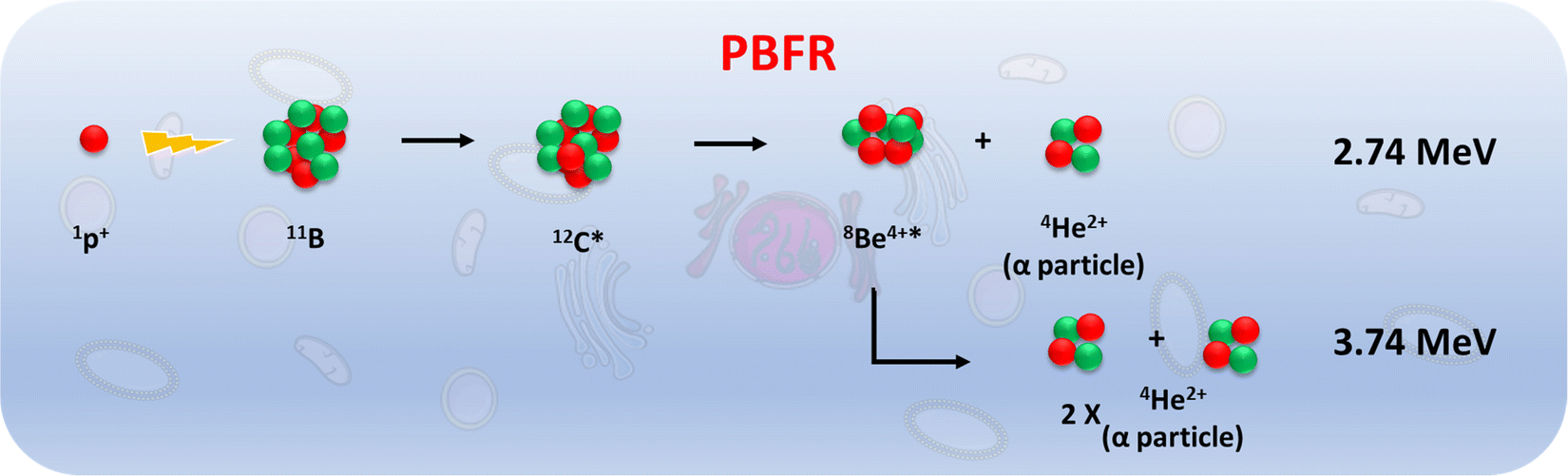 | ||
| Scheme 2 Representation of the reaction between an energetic proton and 11B resulting in the generation of excited carbon 12C, which splits into a 2.74 MeV α particle and a 8Be. Then, beryllium divides into two α particles. Consequently, a total of three α particles are generated by the proton boron reaction. | ||
2. Materials and methods
2.1. Chemistry
The sodium salts of ferrabis(dicarbollide) Na[o-FESAN] and Na[8,8′-I2-o-FESAN] were synthesized from Cs[o-FESAN]42 and Cs[8,8′-I2-o-FESAN]63 by using a cationic exchanging resin as reported in the literature64 (see ESI,† Scheme S1). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). Hexadecane and pentane were purchased from Merck KGaA (Darmstadt, Germany).2.2. Instrumentation
FTIR spectra were obtained using a Shimadzu FTIR-8300 spectrophotometer. The 1H and 1H{11B} NMR (400.13 MHz) and 11B and 11B{1H} NMR (128.37 MHz) spectra were recorded on a Bruker ARX 400 instrument equipped with appropriate decoupling accessories. All NMR spectra were performed in d6-acetone at 22 °C. The 11B and 11B{1H} NMR chemical shift values were referenced to external BF3·OEt2, while the 1H and 1H{11B} NMR chemical shift values were referenced to SiMe4. Chemical shifts are reported in units of parts per million downfield from reference. UV-Visible spectra (see ESI,† Fig. S1) were recorded on a double beam PharmaSpec UV-1700 series spectrophotometer from Shimadzu Corporation, operating from 200 nm to 800 nm in 1.0 cm quartz cells using 1 cm cuvettes with 0.08 mM of each ferrabis(dicarbollide), Na[o-FESAN] or Na[8,8′-I2-o-FESAN] samples in water and n-octanol. Thermogravimetric analyses and differential Scanning Calorimetry (TGA/DSC) were performed on a Netzsch STA 449 thermal analyzer at a heating rate of 10 °C min−1 in an Ar atmosphere. EPR measurements were performed on a Bruker Elexsys 580 at 10 K. Spectral parameters were: field sweep 800 mT, central field 405 mT, modulation amplitude 0.1 mT, modulation frequency 100 KHz, time constant 1.28 ms and attenuation 30 dB.2.3. EPR studies
EPR measurements on the Na[o-FESAN] and Na[8,8′-I2-o-FESAN] salts at 80 K and 10 K were carried out in the solid state (see Fig. S2, ESI†). The EPR spectra at 10 K of the dried U87 glioblastoma cells after uptaking either Na[o-FESAN] or Na[8,8′-I2-o-FESAN] were also obtained and compared with the EPR spectra of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] salts, respectively, which were taken as references (Fig. 1).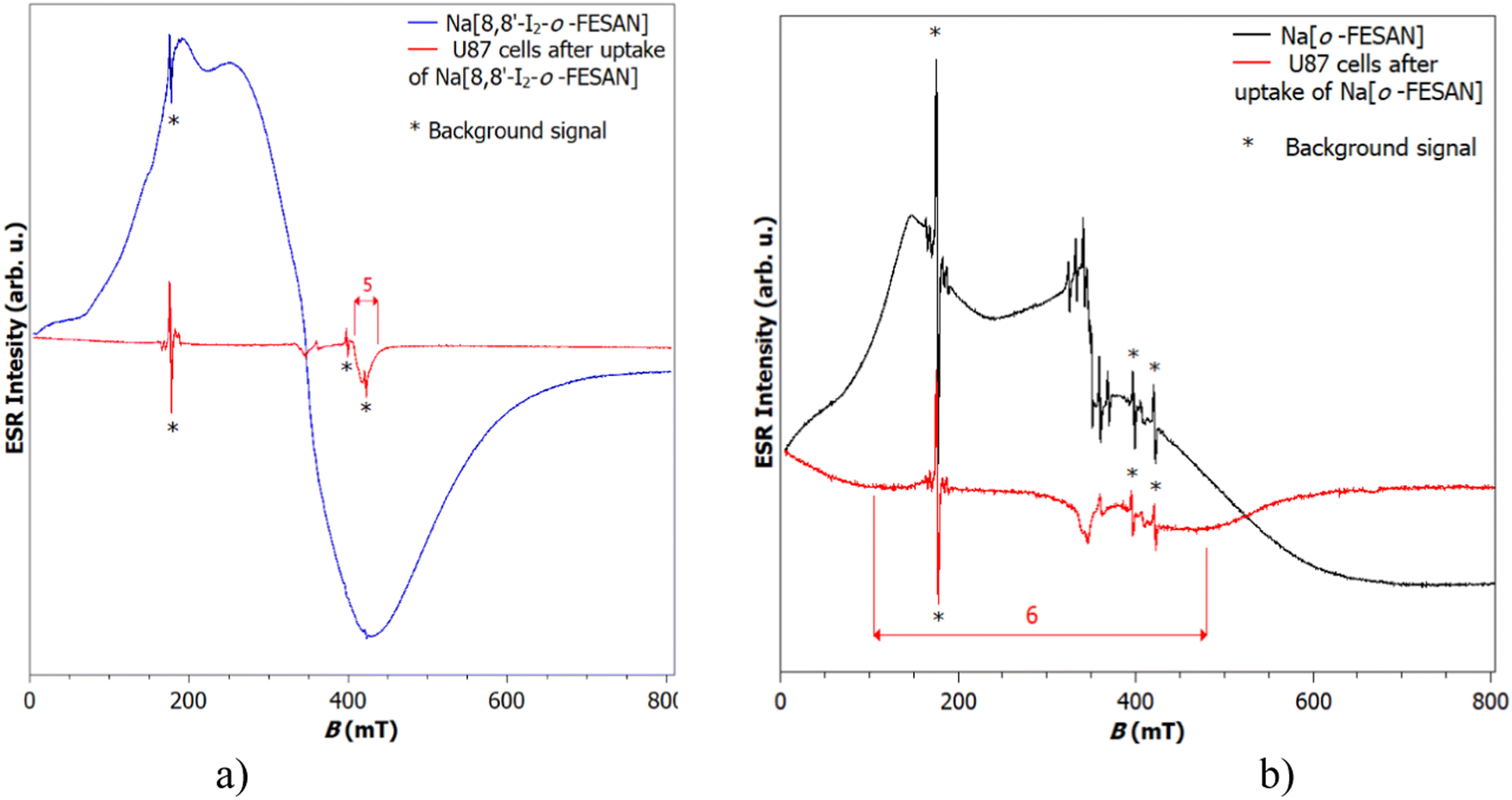 | ||
| Fig. 1 (a) Spectra of Na[8,8′-I2-o-FESAN] in powder, U87 cells after uptake of Na[8,8′-I2-o-FESAN]. The g factor indicated by number 5 is 1.7. (b) Spectra of Na[o-FESAN] in powder and U87 cells after uptake of Na[o-FESAN]. The g factor indicated by number 6 is approximately 2. | ||
The spectrum of Na[o-FESAN] consists of a broad line superposed to the cavity signal. According to the value of the g factor for broad peak 1, which is 4.7, it can be assumed that this broad line originates from Fe(III).65 This is additionally confirmed by a line of lower intensity positioned at a g value of 2.0, peak 4, on which six narrow lines are superimposed.65 The Na[8,8′-I2-o-FESAN] powder sample shows a broad signal of much stronger intensity at a g value of approximately 2.0. A shoulder at g = 4.7 is noticeable on that signal too. Both signals originate from Fe(III).65,66
Fig. 1(a), which shows the spectra of Na[8,8′-I2-o-FESAN] in powder and U87 cells after the uptake of Na[8,8′-I2-o-FESAN], diplays that, after Na[8,8′-I2-o-FESAN] uptake, U87 cells contain a clear line of weak intensity at g = 1.7, while Fig. 1(b) shows a very broad signal (indicated as 6) placed at a g value of about 2 in the spectrum of dried U87 cells after the uptake of Na[o-FESAN]; this signal undoubtedly confirms the presence of iron in the sample.
2.4. Planar bilayer electrophysiology measurements
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (w/w). Lipid was added on ∼100 μm diameter orifices in the 15 μm-thick Teflon partition that separated two identical chambers (Fig. 2). The orifices were pre-treated with a 1% solution of hexadecane in pentane. Aqueous solutions consisted of 1.6 mL of 150 mM NaCl at pH 6 and all measurements were performed at room temperature (23 ± 1 °C). Planar membrane formation was tracked by applying a periodic triangular voltage wave with an external wave generator and visualizing the squared output signal with an oscilloscope. Voltage was applied through Ag/AgCl electrodes assembled in 2 M KCl, and 1.5% agarose bridges within standard 250 μL pipette tips. The potential was defined as positive when it was higher on the feeding compartment, whereas the stripping compartment was set to ground. An Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) in the voltage-clamp mode was used to measure the current registered upon the addition of 50 μM compound at the feeding compartment of the chamber. Compound stock concentration was 10 mM in water. A constant stirring of at least 1 minute was applied after compound addition to assure homogeneous mixing. Current was filtered using an integrated low pass 8-pole Bessel filter at 1 kHz, digitized using a DigiData 1440A (Molecular Devices, Sunnyvale, CA) with a sampling frequency of 2 kHz, and analyzed using pClamp 10.7 software (Molecular Devices, Sunnyvale, CA). The chamber and the head stage were isolated from external noise sources with a double metal screen (Amuneal Manufacturing Corp., Philadelphia, PA). Current after compound addition was tracked during the entire experimental time to assure membrane integrity.
:3 (w/w). Lipid was added on ∼100 μm diameter orifices in the 15 μm-thick Teflon partition that separated two identical chambers (Fig. 2). The orifices were pre-treated with a 1% solution of hexadecane in pentane. Aqueous solutions consisted of 1.6 mL of 150 mM NaCl at pH 6 and all measurements were performed at room temperature (23 ± 1 °C). Planar membrane formation was tracked by applying a periodic triangular voltage wave with an external wave generator and visualizing the squared output signal with an oscilloscope. Voltage was applied through Ag/AgCl electrodes assembled in 2 M KCl, and 1.5% agarose bridges within standard 250 μL pipette tips. The potential was defined as positive when it was higher on the feeding compartment, whereas the stripping compartment was set to ground. An Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) in the voltage-clamp mode was used to measure the current registered upon the addition of 50 μM compound at the feeding compartment of the chamber. Compound stock concentration was 10 mM in water. A constant stirring of at least 1 minute was applied after compound addition to assure homogeneous mixing. Current was filtered using an integrated low pass 8-pole Bessel filter at 1 kHz, digitized using a DigiData 1440A (Molecular Devices, Sunnyvale, CA) with a sampling frequency of 2 kHz, and analyzed using pClamp 10.7 software (Molecular Devices, Sunnyvale, CA). The chamber and the head stage were isolated from external noise sources with a double metal screen (Amuneal Manufacturing Corp., Philadelphia, PA). Current after compound addition was tracked during the entire experimental time to assure membrane integrity.
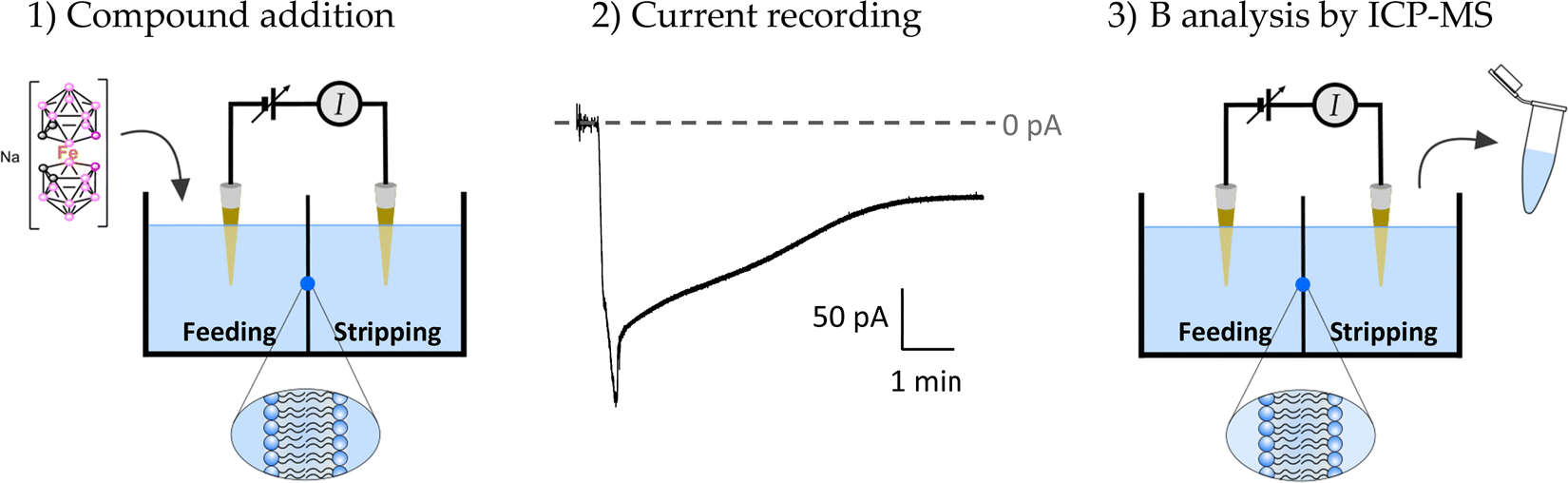 | ||
| Fig. 2 Experimental method for the recording of compound-induced current in planar bilayers and determination of compound concentration in terms of the amount of boron by ICP-MS. | ||
2.5. Cells, culture media and compound solutions
U87 glioblastoma (GBM) cells were obtained from ATCC (American Type Culture Collection). For the biological experiments, cells were cultured in DMEM with the GlutaMAX™ medium supplemented with 10% FBS and maintained at 37 °C in a 5% CO2 humidified atmosphere. Fresh stock solutions (1 mM) of the ferrabis(dicarbollide) (Na[o-FESAN] and Na[8,8′-I2-o-FESAN]) compounds were prepared in DMEM media. Serial dilutions from the stock were prepared in DMEM with Glutamax supplemented with 10% fetal bovine serum (FBS).2.6. Blood–brain barrier (BBB) translocation studies
bEnd.3 murine brain endothelioma cells (ATTCC-CRL2299) were grown in DMEM supplemented with 10% FBS (all from Invitrogen) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C (Heraeus, Germany), with the medium being changed every other day. To allow the formation of a stable in vitro BBB model, 5000 cells per well were seeded in fibronectin-coated tissue culture PET inserts (pore size of 1 μm) for 24-well plates (BD falcon) and grown for 9 days with media being changed every 2 days, as previously described.69For the evaluation of the integrity of the BBB model, fluorescein isothiocyanate-dextran with a MW of 4 (FD4 from Sigma Aldrich) was used as a fluorescent probe, as previously described.68 Only cells present in the inserts with values of integrity higher than 90% were used for subsequent studies. For the translocation assay, inserts were incubated with 100 μM of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] in the transport medium (DMEM fluorobrite + 10% FBS) in the apical side for 5 h. After incubation, samples were collected and analyzed by ICP-MS for quantification of the B and Fe contents. Control inserts with only transport media or DMSO control were also submitted to the same procedure. After the translocation assay, inserts were washed (apical and basal sides) twice with PBS and once with the transport medium and a post-translocation integrity assay was performed as described above. For ICP-MS determination of B and Fe contents on the apical and basal fraction, each sample was incubated for 24 h with a mixture of HNO3 and HCl (1:3), diluted with ultra-pure water and analyzed on a Thermo-X series spectrometer.
2.7. Viability assays with 2D cells
U87 cells were seeded in 96-well plates (1 × 104 cells/200 μL medium) and incubated at 37 °C for 24 h to adhere. Then, the medium was discarded, and cells were incubated with the corresponding sodium salt of the ferrabis(dicarbollide) compounds at serial concentrations in the range of 1–500 μM for 6, 24, 48 and 72 hours. After incubation, the viability was determined using the MTT assay. Two independent assays were performed, using at least 6 replicates per condition in each assay. For the MTT assay, the culture medium was removed and 200 μL of MTT solution (0.5 mg mL−1) were added to each well. After incubating for 3 h at 37 °C, the MTT solution was discarded and 200 μL of DMSO were added to dissolve the formazan crystals. The absorbance was measured at 570 nm using an ELISA plate reader. Absorbance readings from treated samples were normalized to controls and curve fitting was performed using Graph Pad Prism 5 software to obtain the IC50 values.2.8. Cellular uptake by PIXE
The concentrations of Fe in pellets of U87 cells, controls and samples incubated with the compounds (50 μM, 24 h) were determined by the particle-induced X-ray emission (PIXE) technique using the CTN/IST Van de Graaf accelerator. The cell pellets were obtained by centrifugation after washing the cells with PBS to remove the medium and freeze-dried until use. The detailed procedure encompassing acid digestion and analysis was previously described.70,71 The elemental concentrations of Fe were obtained in μg g−1 dry weight and converted to ng 10−6 cells.2.9. 3D spheroid cultures
U87 spheroids were prepared in Nunclon™ Sphera™ ultra-low attachment 96U-well plates. Briefly, cells from 80–90% confluent monolayer cultures were tripsinized and seeded at 1000 cells per well. The plate was then centrifuged at 1500 rpm for 5 minutes and incubated at 37 °C in a humidified atmosphere of 5% CO2. Spheroid growth was monitored daily using a Primovert Inverted ZEISS Microscope (objective 4×) with an integrated HDcam camera, and the images were analysed using SpheroidSizer, a free high-throughput MATLAB-based image analysissoftware program.72 When the spheroids reached diameters in the range of 350 μm (typically at day 3), viability assays were performed.2.10. Viability assays with 3D spheroids
U87 cells were cultured as spheroids for 3 days. Then, 100 μL of culture medium were removed from each well and 100 μL of each compound at selected concentrations were added. As a control, spheroids were incubated with 100 μL of medium supplemented with 10% FBS. After 72 hours of incubation, viability was determined using the acid phosphatase (APH) assay. Three independent assays were performed, using at least 4 spheroids per condition in each assay. In parallel, U87 cells in the monolayer incubated under the same conditions were also tested in a 96-well plate (three independent assays with at least 4 wells per condition). For the APH assay, 180 μL of culture medium was removed from each well and the spheroids were washed twice with 180 μL PBS. Then 100 μL of PBS was removed from each well and 100 μL of acid phosphatase buffer (0.1 M sodium acetate, 0.1% (v/v) Triton X-100, and 2 mg mL−1p-nitrophenylphosphate pH 4.8) was added to the wells. As a negative control, 100 μL of PBS and 100 μL of acid phosphatase buffer were added to empty wells. After 90 minutes at 37 °C in a humidified atmosphere of 5% CO2, 10 μL of 1 M NaOH was added to each well to stop the reaction, and the absorbance was measured at 405 nm using a microplate reader.2.11. In vivo studies in C. elegans
C. elegans Bristol strain N2 and Escherichia coli OP50 were obtained from the Caenorhabditis Genetics Center (Minnesota, USA). Nematodes were maintained using standard procedures73 and exposure towards materials was performed following previous protocols.74The survival rate of synchronized L4 nematodes was evaluated after 24 h exposure to Na[o-FESAN] and Na[8,8′-I2-o-FESAN] at concentrations ranging from 0–50 μM in the M9 buffer supplemented with 4% of dead OP50 in a final volume of 100 μL. After exposure, worms’ survival was computed by tapping the plate and counting the moving alive worms.73
Each well contained 15 ± 3 L4 worms (number of total worms (n) = 450, number of independent experiments (N) = 3). LD50 was calculated using GraphPad Prism 9 and fitting the curve to a four-parameter dose-response curve.
2.12. Irradiation studies
For the irradiation studies, U87 cells (104 cells per well) were seeded into 96 well plates and previously incubated with the compounds at selected concentrations for 24 h.For the X-ray irradiation studies, cells were irradiated using a Philips MCN 165 X-ray tube and an YXLON 9421 high-voltage generator at 1.5 Gy radiation dose, 15 min@20 mA. ISO beam quality N150 was used (Emax = 150 keV), filtered with 4 mm Al + 1 mm Cu. The source distance to the cell plate was 0.8 m.
A 2.0 MeV proton beam was extracted from the vacuum chamber to air using an exit nozzle with a 6.3 μm thick Mylar window and focused to tens of micrometer dimensions.
The focused beam with an average flux of 2.32 × 108 protons cm−2 s−1 was scanned over a monolayer of U87 cells, which were seeded in 96-well plates (104 cells per well) and incubated with the compounds for 24 h. The plate was positioned perpendicular to the beam path and for each well an area of 0.11 cm2, representing 34% of the total area of the well (0.32 cm2) was irradiated. The culture medium was partially removed from each well immediately before irradiation to guarantee that the irradiation area would have an equivalent cell thickness of 30 μm. The proton energy was tuned to reach the main resonance energy of the proton–boron nuclear fusion reaction (675 keV) in the cell layer.62,76 This was achieved by using an appropriate combination of attenuators (an air path of 1.34 cm and a 12.6 μm thick Mylar foil) so that the transmitted proton energy after crossing a 30 μm cell layer was 477 ± 28 keV, resulting in an average LET of 26.4 ± 0.9 keV in the cell layer. An estimated average dose of 9 Gy was delivered in each well. The stopping power and the ion range calculations were performed using the SRIM software code.77
2.13. Apoptosis analysis
U87 cells in the medium were seeded in 24-well plates (2 × 105 cells/500 μL medium) and incubated at 37 °C for 24 h to adhere. Then, the medium was discarded, and cells were incubated with Na[o-FESAN] and Na[8,8′-I2-o-FESAN] at 200 and 100 μM, respectively, for 6 h. After this, cells were irradiated with the 60Co γ-ray source at 2 Gy total dose. After treatment, cells were fixed with 4% (w/v) formaldehyde for 15 minutes, washed 3 times with PBS and then stained for 15 minutes with 7.5 μg mL−1 Hoechst 33258 in PBS (ThermoFisher Scientific, maximum excitation wavelength: 352 nm, maximum emission wavelength: 461 nm). After being washed 3 times with PBS, cells were observed using a Ti-U Eclipse inverted fluorescence microscope and respective software (Nikon, Tokyo, Japan) using a DAPI fluorescence filter cube (Nikon). At least 5 images with circa 30 nuclei were acquired using the NIS Elements Basic software (Nikon).782.14. Cell cycle evaluation
The cell cycle evaluation followed previously described procedures with a few modifications.79 Briefly, U87 cells were seeded in 24-well plates (2 × 105 cells/500 μL medium) and incubated at 37 °C for 24 h to adhere. Then, the cells were synchronized in the early S phase with a thymidine double block (2 mM per well). After this, the medium was discarded and cells were incubated with the compounds at 200 μM (Na[o-FESAN]) and 100 μM (Na[8,8′-I2-o-FESAN]) for 6 h. After this, cells were irradiated with the 60Co γ-ray source at 2 Gy total dose and the medium was changed by a fresh one. After 24 h incubation, cells were detached using Tryple Express (Thermo Fisher Scientific), washed 3 times with PBS and fixed overnight in 80% (v/v) cold ethanol. After centrifugation at 1500 × g for 10 minutes, cells were incubated for 30 minutes with 50 μg mL−1 RNAse A in PBS (NZYtech), followed by incubation with 25 μg mL−1 propidium iodide. Cell fluorescence was acquired using an Attune acoustic focusing cytometer (Thermo Fisher Scientific) with a BL2 filter and the results were processed with respective software (Attune Cytometric Software 2.1).2.15. γ-H2AX assay
U87 cells were seeded in 6-well plates containing a coverslip for each well, at a density of 2 × 105 cells/500 μL medium and, after overnight attachment, were incubated with 200 μM Na[o-FESAN] or 100 μM Na[8,8′-I2-o-FESAN] for 6 h before being irradiated as described above for the apoptotic and cell cycle assays. After 30 minutes of incubation in a fresh medium, the detection of DNA double stranded breaks was performed using the y-H2AX assay as previously described.80 Foci analysis was made using a custom ImageJ macro, with images obtained from two independent experiments.2.16. Colony formation assay
Analysis of colony formation was done after irradiation. U87 cells were seeded in 96-well plates and left to adhere. After 24 h, the cells were incubated with increasing concentrations of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] for 24 h and irradiated with the different sources of radiation: gamma rays (2 Gy), X-rays (1.5 Gy) and protons (9 Gy). For comparison, non irradiated cells subjected to the same protocol were used as the control. After irradiation, the cells were dissociated with Trypsin and the number of cells was counted. Three wells per condition were used. Dissociated cells were then seeded in 6-well plates: 200 cells under control conditions, 400 cells under the conditions where cells were only exposed to radiation or incubated with the lowest concentration of both compounds, 600 cells under the conditions where cells were incubated with the intermediate concentration of the compounds, and 800 cells under the conditions where cells were incubated with the highest concentration of the compounds. After 11 days of incubation at 37 °C, cells were fixed with a solution of 3:1 methanol:acetic acid at −20 °C for 20 min, washed, and then stained with 4% Giemsa in phosphate buffer for 10 min. Colonies with more than 50 cells were counted. One assay was performed. Results are expressed as a ratio of cellular survival upon treatment when compared with untreated control.
2.17. Statistical analysis
The data presented in the paper are shown as mean values ± SD and, unless otherwise stated, at least 3 biological replicates were used. Statistical analysis was performed using the GraphPad Prism 6 software to assess if there was a significant effect of each treatment compared with the respective control samples. For this, a one-way ANOVA followed by Dunnett's test was performed with a threshold of p ≤ 0.05.3. Results
3.1. Physical and chemical properties of ferrabis(dicarbollide) molecules
As mentioned in the introduction, several reports have been published on the properties of [o-COSAN]− and [8,8′-I2-o-COSAN]− but to the best of our knowledge, seldom related to [o-FESAN]− and [8,8′-I2-o-FESAN]− (Table 1).81–90| Compound | [o-FESAN]− | [8,8′-o-I2-FESAN]− |
|---|---|---|
| Size (Å) | 1.1 × 0.663 | 1.1 × 0.845 |
| MW | 323.75 | 572.45 |
| Rotamer | Cisoid | Transoid |
| DLS | Aggregates in H2O (d = 140 nm) in the range 1 < c < 50 mM; (d = 108 nm) at c > 50 mM | Aggregates in H2O (d = 83 nm) in the range 1 < c < 80 mM; (d = 1.5 nm) at c > 80 mM |
| Solubility (mM) | 1247 | 374 |
| logS |
3.10 | 2.57 |
| Lipophilia (P) | 45.7 | 99.3 |
| logP |
1.66 | 2.00 |
| E 1/2 Fe3+/2+(in V vs. Fc+/Fc) | −1.00 | −0.86 |
| UV-visible (nm) | 271/295 | 289/343 |
The aqueous solubility (S) and lipophilicity (P) of a compound are key parameters in drug development.89 Importantly, although the sodium salts of ferrabis(dicarbollides) are soluble in water (1247 and 374 mM for Na[o-FESAN] and Na[8,8′-I2-o-FESAN], respectively), they display lipophilic character (P values are 45.7 (logP = 1.66) for Na[o-FESAN] and 99.3 (logP = 2.00) for Na[8,8′-I2-o-FESAN]).65
These results indicate that the presence of the two iodine atoms significantly affects the lipophilicity of the small ferrabis(dicarbollide) molecule making the 8,8′-di-iodinated derivative approximately 2.17-fold more lipophilic than Na[o-FESAN]; whereas it is reported that Na[8,8′-I2-o-COSAN] has a partition value (P = 151.0) approximately 3.5-fold higher than Na[o-COSAN] (P = 43.7).47 Fe makes both ferrabis(dicarbollides) less lipophilic than their related cobaltabis(dicarbollides).
Lipophilicity is a major factor influencing passive brain transfer across the BBB in either direction. It is reported that typically, for a radiotracer to be considered as a potential efficient molecular imaging probe in the living human brain with positron emission tomography (PET), its partition coefficients (logP) should range between 2.0 and 3.5.91 Consequently, the logP of Na[8,8′-I2-o-FESAN] complex is in this range (see Table 1).
The TGA/DSC studies of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] showed that only removal of water molecules coordinated to the sodium cation was observed until 550 °C, demonstrating the thermal stability of both ferrabis(dicarbollide) clusters (see ESI,† Fig. S3 and S4). The stability of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] in water as well as in DMEM (without phenol red) in the absence and in the presence of FBS was studied by using FTIR and 11B{1H} NMR spectroscopies, which are essential analytical tools for structure elucidation and chemical determination. Recently, we have reported by using Synchrotron Radiation-Fourier Transform Infrared (FTIR) that Na[o-COSAN] after its uptake by two different glioma initiating cells (mesenchymal and proneural) strongly interacts with DNA and proteins modifying their secondary structure, while the interaction with lipids is weaker, being enough to cross the biological membranes (cell and nucleous).58 This finding has been possible because Na[o-COSAN] displays a strong and characteristic ν(B–H) frequency in the infrared range 2.600–2.500 cm−1 in which no other frequencies of organic compounds appear.92 The interaction between Na[o-COSAN] and proteins can also be observed by 11B{1H}NMR spectroscopy.48 To our knowledge, neither FTIR nor 11B{1H}NMR studies related to the interaction between small anionic ferrabis(dicarbollide) molecules and biomolecules have been reported.
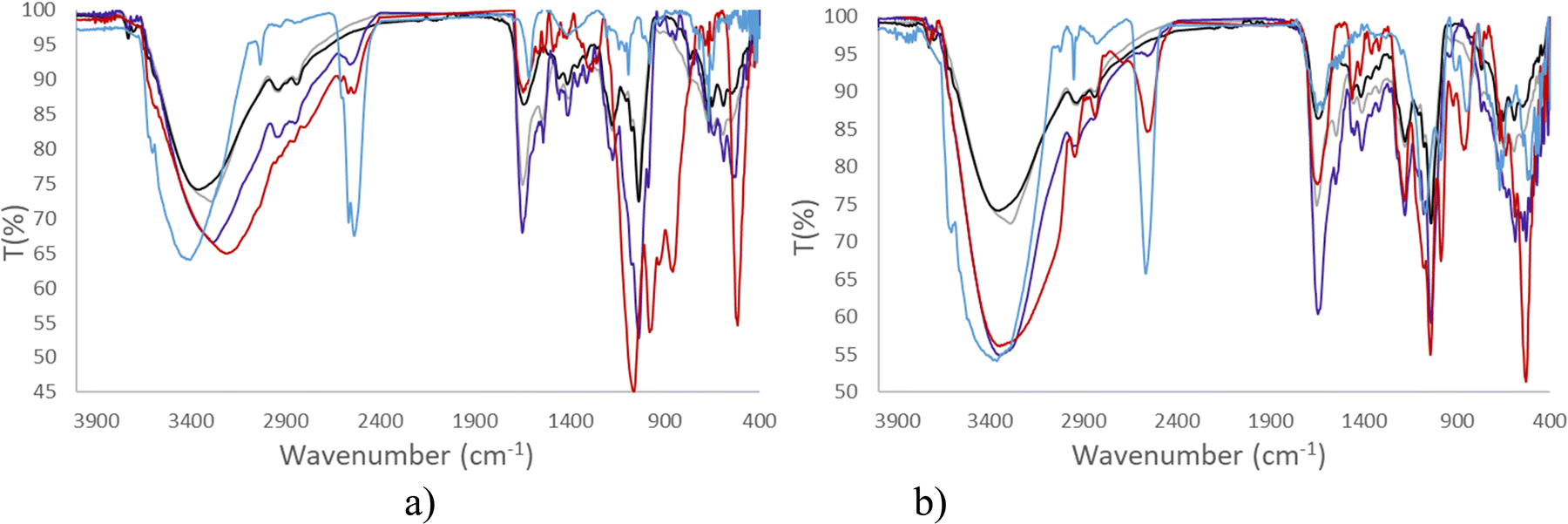
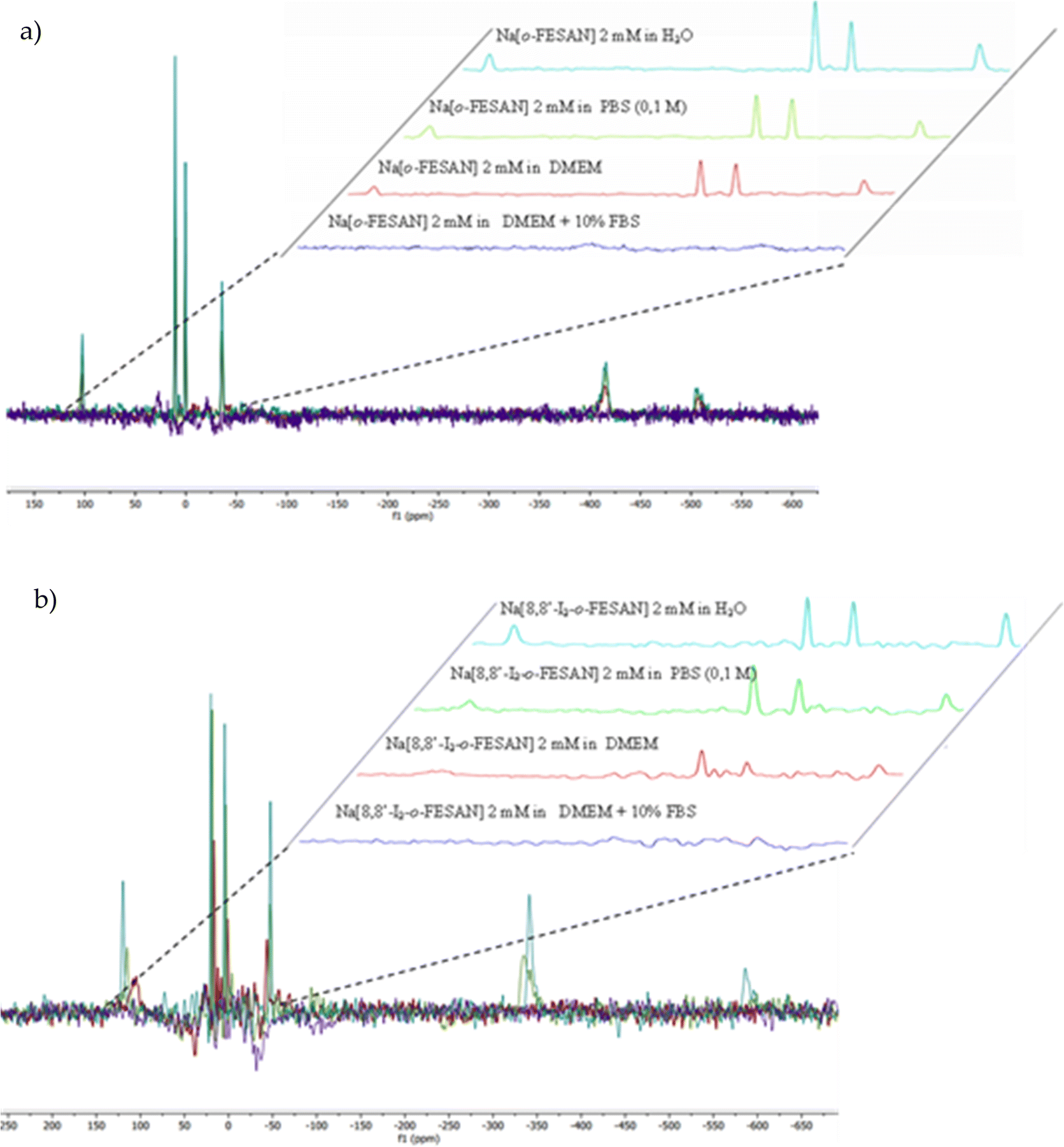
Consequently, in this article, both spectroscopies have been performed to provide information on the stability of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] in DMEM and DMEM + 10% FBS (Fig. 3 and 4).
 | ||
| Fig. 3 (a) IR spectra of 2 mM Na[o-FESAN] in aqueous solution (blue), in DMEM (without phenol red) (red), and in DMEM + 10% FBS (purple). (b) IR spectra of 2 mM Na[8,8′-I2-o-FESAN] in aqueous solution (blue), in DMEM (red), and in DMEM + 10% FBS (purple). As a control: IR spectra of DMEM (black) and DMEM + 10% FBS (grey). | ||
 | ||
| Fig. 4 (a) The 11B{1H}-NMR spectrum of 2 mM Na[o-FESAN] in aqueous solution (blue), in 0.1 M phosphate buffer (green), in DMEM (red), and in DMEM + 10% FBS (purple). (b) 11B{1H}-NMR spectrum of 2 mM Na[8,8′-I2-o-FESAN] in aqueous solution (blue), in 0.1 M phosphate buffer (green), in DMEM (red), and in DMEM + 10% FBS (purple). | ||
Fig. 3 clearly displays that the characteristic ν(B–H) frequency of Na[o-FESAN] in DMEM (red) decreases with respect to its spectrum in an aqueous solution (blue) but the frequency decreases much more if the DMEM culture medium contains 10% FBS (dark blue); to emphasize that the characteristic B–H's frequency in the infrared range 2.600–2.500 cm−1 is not present in DMEM nor in the DMEM + 10% FBS, the spectra of these culture media without ferrabis(dicarbollides) were run as references. Furthermore, the N–H stretching frequency of the culture medium shifts 170 cm−1 to lower wavenumbers. The decrease of the ν(B–H) signal intensity and the shift of the ν(N–H) to lower frequencies are probably due to the existence of the dihydrogen B–H⋯H–N bonds. It seems that most of the B–H units are within the aggregates, preventing them from interacting with the IR radiation.
Furthermore, Fig. 4 clearly shows that the resonances of the 11B{1H} NMR spectrum corresponding to a 2 mM aqueous solution of Na[o-FESAN] (green) decrease in the 11B{1H} NMR spectrum in culture medium at the same concentration (red) and disappear in DMEM containing 10% FBS (light blue). Fig. 3(b) and 4(b) show the same studies for Na[8,8′-I2-o-FESAN].
To discern if the ferrabis(dicarbollide) interaction with proteins affects the relaxation times, the 11B{1H}-NMR spectra of 2 mM Na[o-FESAN] in different solvents (aqueous solution, in 0.1 M phosphate buffer, in DMEM and in DMEM + 10% FBS) were obtained with relaxation time parameters of 2.62 s and 0.015 s (see the ESI†). No differences were observed confirming that the ferrabis(dicarbollide)–protein aggregate formation does not affect the relaxation time.
To visualize the ferrabis(dicarbollide)–protein aggregate formation, DLS measurements of a DMEM + 10% FBS solution with increasing amounts of Na[o-FESAN] were obtained. As shown in Fig. 5, the size is maintained between 0 and 0.5 mM but, at the Na[o-FESAN] concentration higher than 0.5 mM, the FBS size grows consistently.
 | ||
| Fig. 5 Graphical representation of the hydrodynamic diameter (nm) of the DMEM + 10% FBS culture medium measured by DLS versus increasing concentrations of Na[o-FESAN] in water in the concentration range from 0 to 9 mM. | ||
The latter results are analogous to the studies performed with Na[o-COSAN] in DMEM that were related with the Na[o-COSAN] aggregate formation in the presence of protein in DMEM and DMEM + 10% FBS.48 The observed signals’ decrease in the 11B{1H} NMR spectrum of the Na[o-COSAN] in DMEM without FBS is probably due to the B–H⋯H–N hydrogen bond between Na[o-COSAN] and the L-glutamine (3.4 mM) present in the medium. Interactions between [o-COSAN]− and several aminoacids (histidine, arginine and tryptophan),53 as well as bioactive molecules (isoniazid, pyrazinamide, dopamine, nicotinic acid, nicotinamide, histamine and metformin) were previously observed.54 Both spectroscopy studies support that the ferrabis(dicarbollides) interact also with amino acids and FBS protein as they do with DNA.48
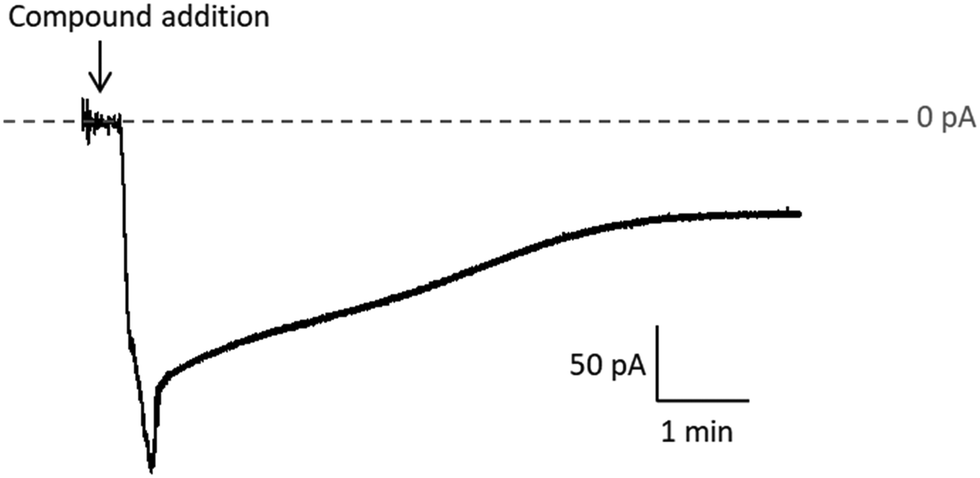 | ||
| Fig. 6 Sample current recording after Na[o-FESAN] addition to the feeding side of the chamber. The stripping side was electrically grounded. Negative current implies that the [o-FESAN]− anion crosses the membrane. | ||
Quantitative estimation of the amount of [o-FESAN]− and [8,8′-I2-o-FESAN]− that crossed the membrane was made by extracting the stripping solution from the stripping side 30, 60, 90 and 120 min after compound addition to the feeding side (independent experiments) and analysing the boron content of the sample by ICP-MS (see Fig. 2). Only experiments in which the lipid membrane integrity was preserved were considered. Interestingly, the success rate of experiments with [8,8′-I2-o-FESAN]− (41%) was half of that with [o-FESAN]− (85%). This result (48 vs 100) is consistent with the higher lipophilicity of [8,8′-I2-o-FESAN]− over [o-FESAN]− (45% vs 100%, respectively). The concentration of the compound in the feeding side (50 μM) was chosen not too high to avoid lipid membrane disruption but high enough to match the lowest detection limit of the ICP-MS equipment (nanomolar range).
The rate of translocation of [o-FESAN]− and [8,8′-I2-o-FESAN]− across the DOPC:DOPE membranes is somewhat similar: 2.6 ± 2.8 and 2.5 ± 1.8 nM min−1, respectively. For both compounds, experiments show a high variability. It is known65 that at the concentration used here both compounds form large aggregates (140 nm and 83 nm in diameter for Na[o-FESAN] and Na[8,8′-I2-o-FESAN], respectively) that exceed by far the thickness of the lipid bilayers (∼4–5 nm). The high variability of the translocation rate could be related to the bilayer formation technique. As is known,67 only the central part of the membrane assembled in the orifice is actually a bilayer. The size of the central annulus where the thickness is the smallest is not known and this may change from one experiment to another. Given the fact that Na[o-FESAN] and Na[8,8′-I2-o-FESAN] partition into the lipid membrane, the average effective membrane thickness might change their translocation rate, thus explaining its variability between independent experiments.
We also observed that there is no correlation between the measured ionic current in the steady state and the compound rate of translocation across the membrane (measured from ICP-MS analysis of the stripping side). Presently, there is no clear explanation of this fact but the formation of aggregates of different sizes could be a reason why both magnitudes seem to be not correlated.
3.2. Biological studies
| Incubation time (h) | U87 cells | |
|---|---|---|
| Na[o-FESAN] (μM) | Na[8,8′-I2-o-FESAN] (μM) | |
| 6 | 265 ± 118 | 106 ± 28 |
| 24 | 245 ± 92 | 32.1 ± 9.5 |
| 48 | 90.8 ± 22 | 7.7 ± 1.8 |
| 72 | 51.2 ± 12 | 6.0 ± 1.2 |
| 24 h, 50 μM | ng Fe/106 cells |
|---|---|
| Control | 81.0 ± 2.8 |
| Na[o-FESAN] | 548 ± 16 |
| Na[8,8-I2-o-FESAN] | 349 ± 9.4 |
PIXE results showed that the Fe uptake in U87 treated cells was 4- to 6-fold higher than that in the control (non-treated cells). However, as can be observed, the cellular uptake is not related to the cytotoxic activity, since the uptake of a more active compound was less by the cells.
In Fig. 7, the % of B and Fe detected in the apical and basal fractions of cells incubated with 100 μM of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] is presented.
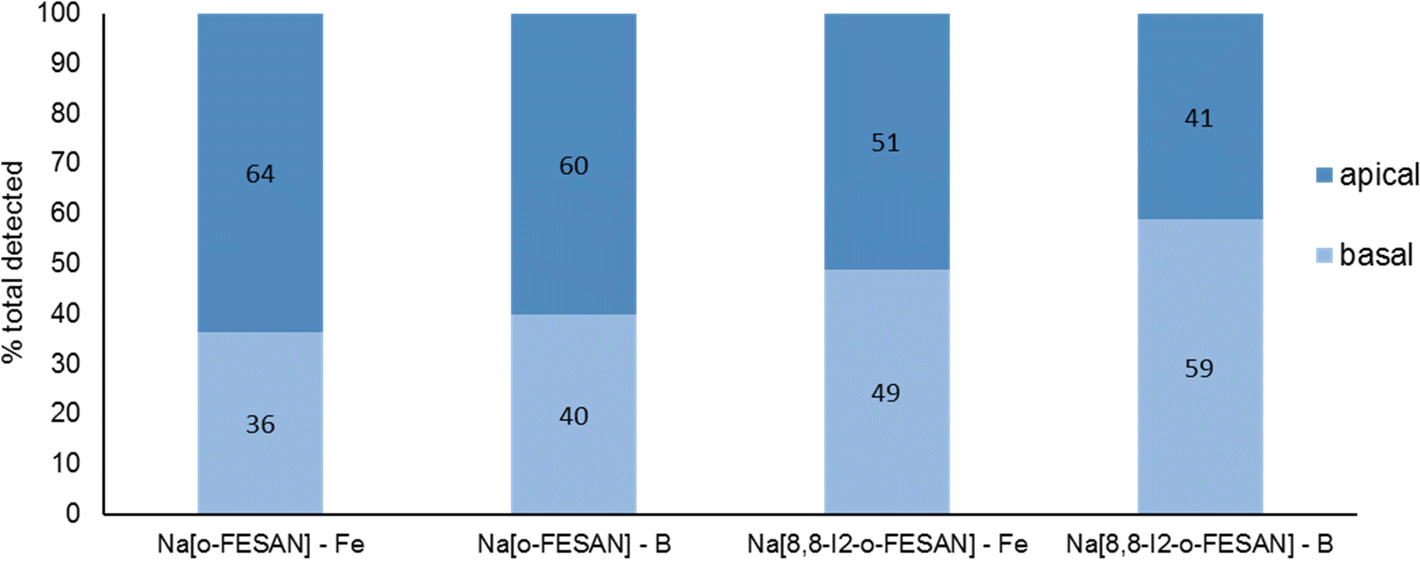 | ||
| Fig. 7 BBB translocation determined by ICP-MS determination of B and Fe and in the apical and basal media of the BBB cell model incubated with 100 μM Na[o-FESAN] and Na[8,8′-I2-o-FESAN] for 5 h. | ||
The ICP-MS determination of the content of both elements in the 2 fractions for each compound shows similar values (Na[o-FESAN]-B was compared with Na[o-FESAN]-Fe and Na[8,8′-I2-o-FESAN]-B with Na[8,8′-I2-o-FESAN]-Fe), which indicates that the metallacarboranes are able to cross the BBB model intact, i.e., without disruption of the 3D cluster structure. Furthermore, the ratio of the total mass detected of B vs. Fe is also in accordance with an intact cluster (the B/Fe ratio detected experimentally: 3.3 for Na[o-FESAN] and 2.9 for Na[8,8′-I2-o-FESAN] vs. those predicted: 3.5 and 3.1, respectively).
The translocation results show that both ferrabis(dicarbollides) are able to translocate the in vitro BBB cell model, although to a different extent, with Na[8,8′-I2-o-FESAN] presenting a higher translocation value (∼50%) than Na[o-FESAN] (∼40%), which could be correlated with its higher lipophilicity.
A post-translocation integrity assay was also performed and no decrease in the integrity of the cell model was detected, indicating the non-toxic character of both ferrabis(dicarbollides) at the time and concentration tested in the BBB model.
After 72 h of exposure to the compounds (6th day of spheroid culture), the viability of the U87 spheroids was assessed by the acid phosphatase (APH) assay. A physical characterization of the spheroids upon incubation with the compounds, which included the measurement of the diameter, area, and circularity, was performed daily, from day 3 to day 6 of culture.
Illustrative microscope images of the spheroids exposed to the compounds tested at concentrations equivalent to the IC50 and higher than the IC50 are shown in Fig. 8.
 | ||
| Fig. 8 Representative images of the U87 spheroids after 72 h (day 6 of culture) of exposure to the different complexes at the IC50 concentration and above IC50 concentration. Controls consist of spheroids incubated only with medium. Scale bars correspond to 500 μm. | ||
In a qualitative analysis, the administration of the compounds did not seem to significantly affect the shape and integrity of the spheroids. Quantitatively, concerning the growth and size of the spheroids, Na[o-FESAN] affected the growth of the U87 spheroids with a significant decrease in the spheroids’ area on day 3 of treatment (Fig. 8 panels A and B) for IC50 and higher than IC50 concentrations tested.
Overall, the circularity of the spheroids was not severely affected by the administration of the ferrabis(dicarbolide) compounds, maintaining, in most cases, a regular shape, which is reflected by circularity values very close to 1.
The viability of the spheroids after incubation with the ferrabis(dicarbollides), Na[o-FESAN] and Na[8,8′-I2-o-FESAN], for 72 h was determined using the APH assay and the test was performed in parallel also for monolayer-cultured cells. The viability results of the spheroids for the 2 compounds with the IC50 and above IC50 concentrations tested are presented in Fig. 9 panels C and D. In general, these results reflect the growth behaviour observed for the spheroids.
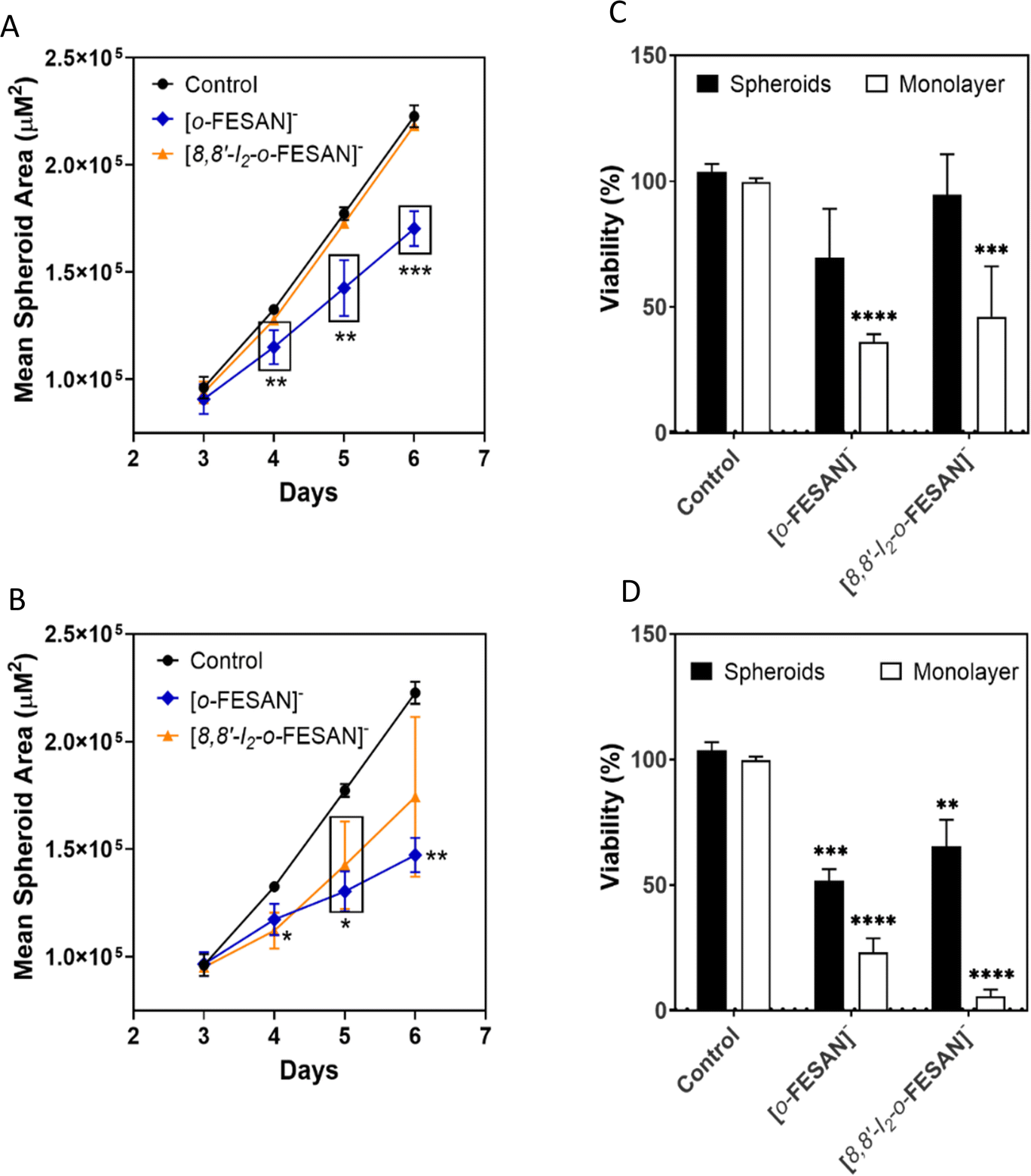 | ||
| Fig. 9 Effect of exposure to Na[o-FESANE] and Na[8,8′-I2-o-FESANE] on U87 spheroids at concentrations corresponding to the IC50 (top panels) and above the IC50 (bottom panels). (A) and (B) U87 spheroid growth, represented by the mean spheroids area (in μm2) as a function of the number of days in culture, and (C) and (D) cellular viability (%) at 72 h, assessed by the APH assay, in parallel to monolayer cultured cells. Controls consist of spheroids or monolayer cultured cells incubated only with medium. Data are presented as the average ±SD of 3 independent assays for spheroids and one for monolayer cells. Statistical significance was calculated using one-way ANOVA, followed by Dunnett's test comparing treated spheroids/cells with control spheroids/cells (*p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001). | ||
There was an obvious decrease in the viability of the spheroids treated with all the compounds. However, using the concentration corresponding to the IC50 previously determined in monolayer-cultured cells by the MTT assay, only incubation with Na[o-FESAN] led to a statistically significant decrease in the spheroids’ viability (Fig. 8 panel C). When using a higher concentration (Fig. 9 panel D), all compounds were found to be able to significantly reduce U87 spheroids’ viability.
The same assay was performed in parallel for monolayer-cultured cells (Fig. 9 panels C and D). When compared with 3D spheroids, U87 cells grown as monolayer cultures were more sensitive to the compounds tested. This is not surprising as 3D culture systems are frequently more refractory to anti-cancer treatments due to limited drug penetration and activation of several resistance mechanisms.94 In fact, spheroid models can mimic the metabolic and proliferative gradients of in vivo tumors, with consequent changes in the cellular phenotype and status, exhibiting multicellular chemoresistance.
To screen the toxicity of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] in nematodes, synchronized L4 C. elegans were exposed to both small molecules at concentrations ranging from 0–50 μM for 24 h (Fig. 10 panel A). Toxicity was determined by the survival of the worms (existence of movement) and the mean lethal dose 50% (LD50). The obtained LD50 values for Na[o-FESAN] and Na[8,8′-I2-o-FESAN] were 5.8 ± 0.6 μM and 6.0 ± 0.6 μM (Fig. 10 panel B). Na[o-FESAN] LD50 was lower than the IC50 found in U87 cells after 72 h of treatment, whereas Na[8,8′-I2-o-FESAN] LD50 was close to the described IC50 for the same compound in U87 cells after 72 h. In comparison with other metallacarboranes, LD50 values of Na[o-FESAN] and Na[8,8′-I2-o-FESAN] are slightly lower than those found for Na[o-COSAN] and Na[8,8′-I2-o-COSAN], 8.6 and 9.7 μM respectively.95
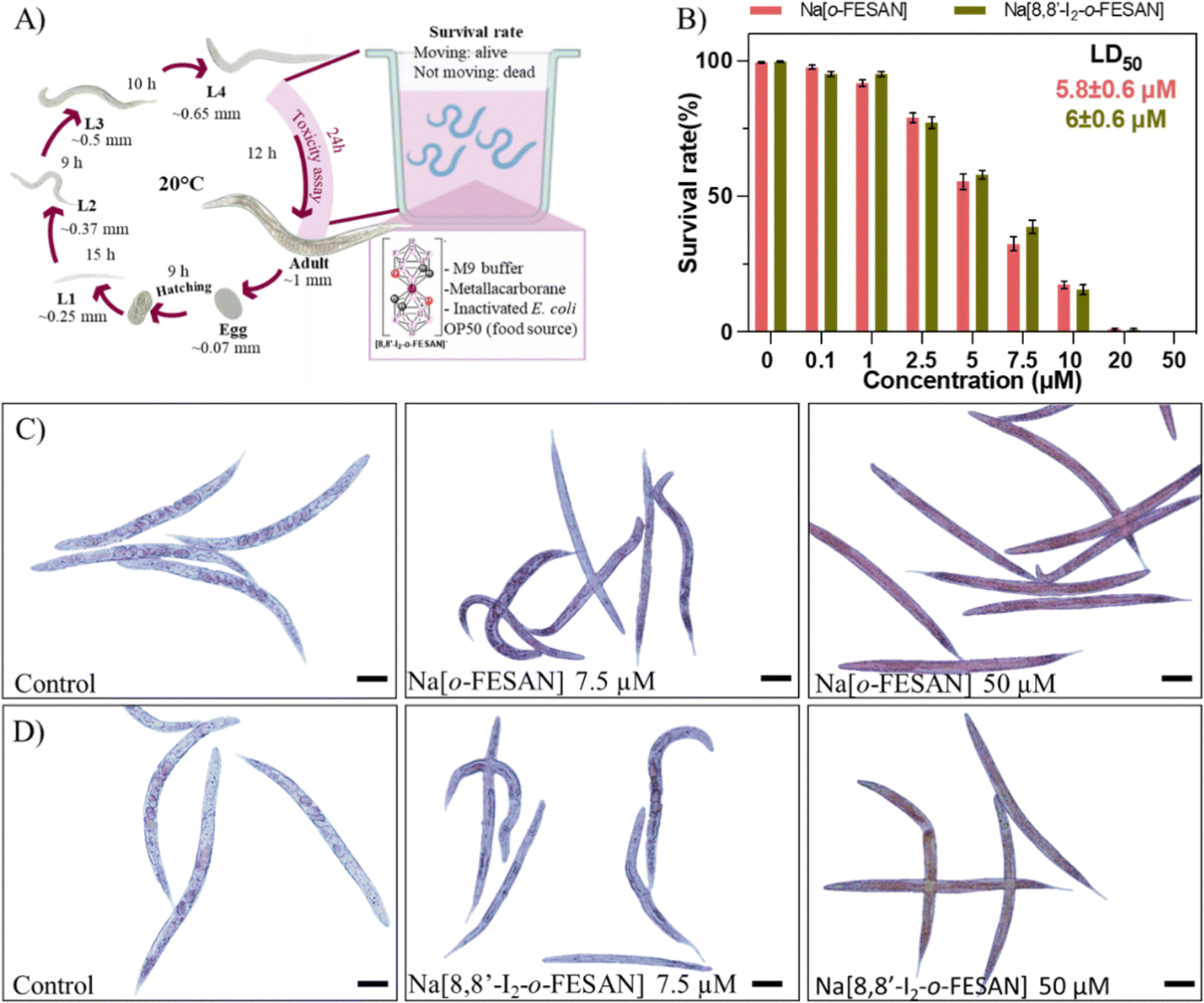 | ||
| Fig. 10 In vivo toxicological evaluation of Na[o-FESAN] and Na[8,8′-I2-o-FESAN]. (A) Schematic representation of C. elegans life cycle and exposure conditions from the L4 stage to adulthood. (B) Survival rate of worms at concentrations up to 50 μM Na[o-FESAN] (red) and Na[8,8′-I2-o-FESAN] (green). Microscopy image of Na[o-FESAN]-treated worms (C) and Na[8,8′-I2-o-FESAN]-treated worms (Na[8,8′-I2-o-FESAN]). Bar size 100 μm. | ||
Both ferrabis(dicarbollides) arrested the worm's development in the L4 stage, the initial stage in the toxicological evaluation, whereas control worms continue their growth till adulthood (Fig. 10 panels C and D). Moreover, worms exposed to the highest concentration (50 μM) were colored by the characteristic colour of each compound, maroon for Na[o-FESAN] and dark green for Na[8,8′-I2-o-FESAN], indicating the availability to cross C. elegans biological membranes in vivo.
The difference found between the IC50 in cells and C. elegans could be due to the differences between the models. In in vitro experiments, cells are independent of each other and they are not forming organs or other higher structures. These molecules have been shown to have a cytostatic effect on cells, stopping their growth. After replacing the media with compounds with fresh media, cells can recover from this state. However, in animals, cells depend on each other to maintain the well-being of the whole organism, much more in simple animals such as C. elegans, so it is understandable to have a higher impact on them and therefore have a lower IC50.
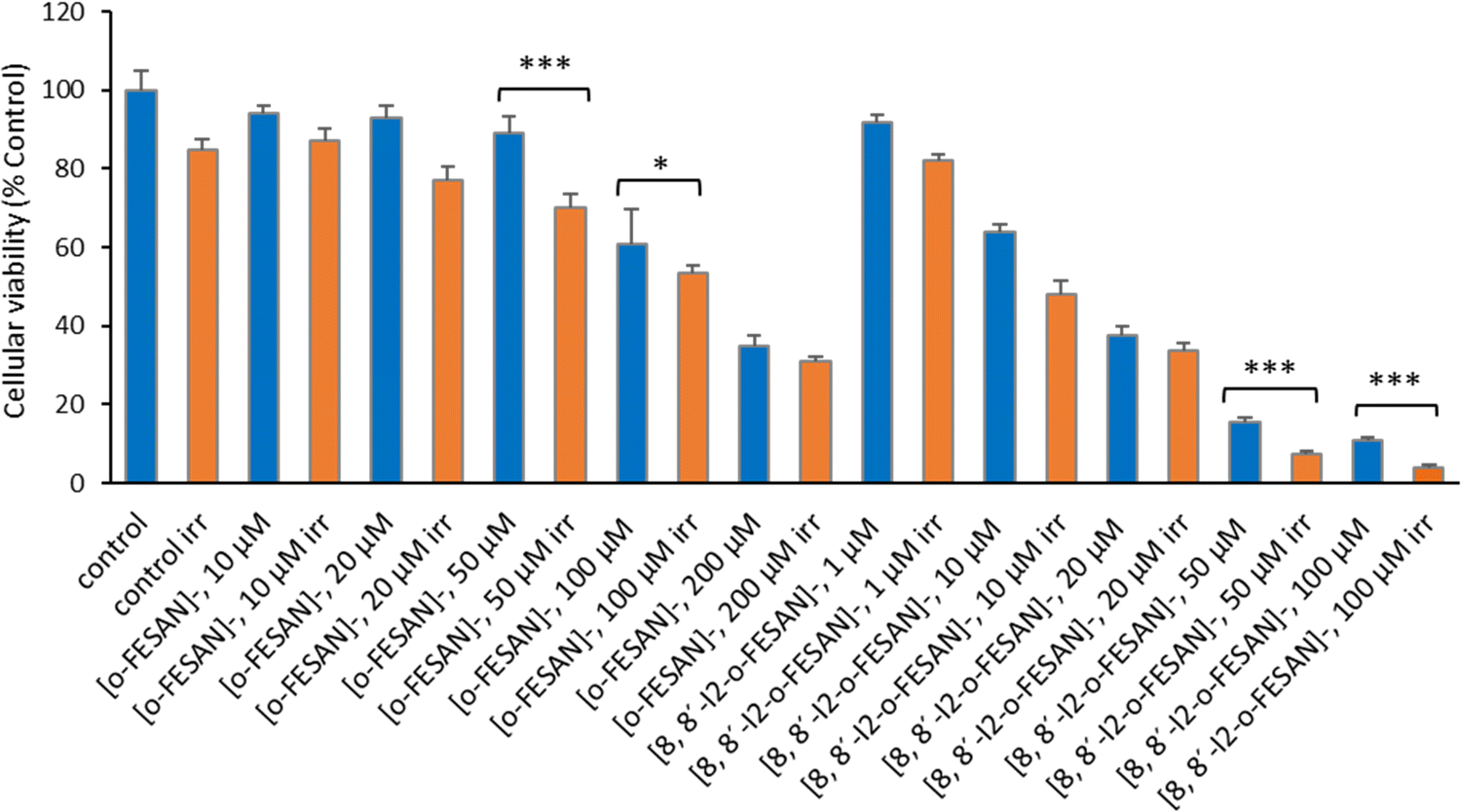 | ||
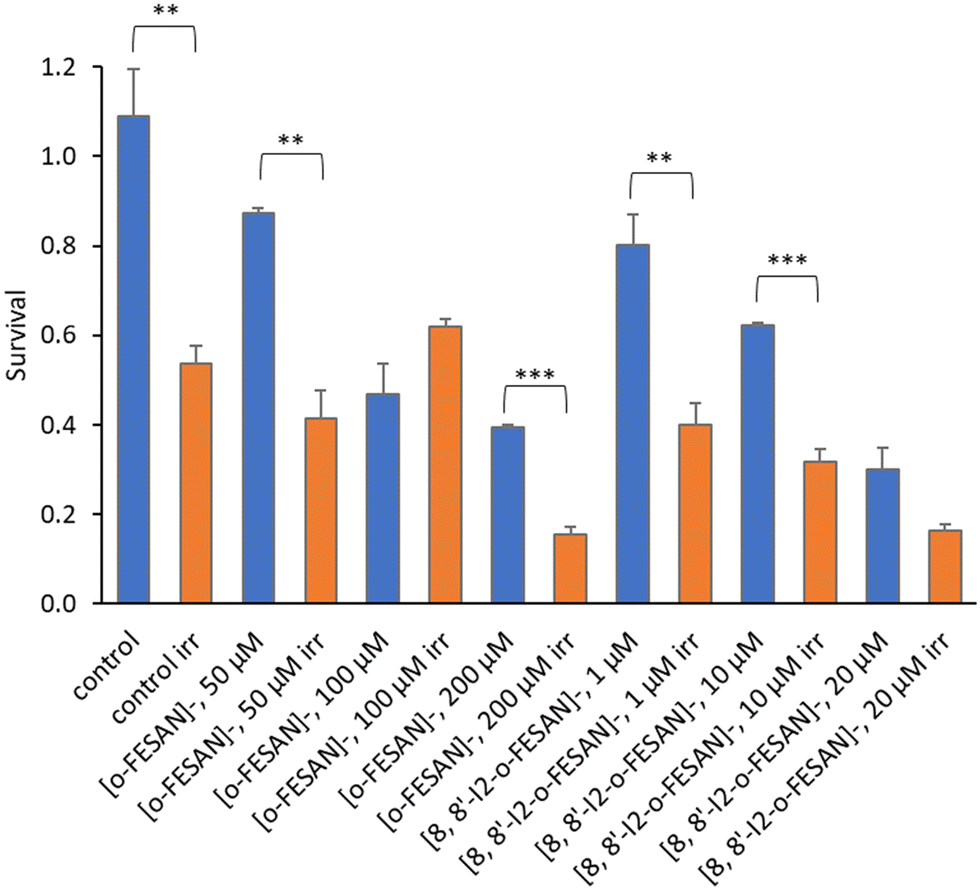
| Fig. 11 Cellular viability 72 h after irradiation with γ-rays (2 Gy) using a 60Co source. U87 cells were previously incubated with the compounds for 24 h at concentrations in the ranges of 10–200 μM ([o-FESAN]−) and 1–100 μM ([8,8′-I2-o-FESAN]−). Results are mean ± SD of two independent experiments done with at least six replicates per condition. *p < 0.05; ***p ≤ 0.0005. | ||
 | ||
| Fig. 12 Clonogenic survival fraction eleven days after irradiation with γ-rays (2 Gy) using the 60Co source. U87 Cells were previously incubated with the compounds for 24 h at concentrations in the ranges of 50–200 μM ([o-FESAN]−) and 1–20 μM ([8,8′-I2-o-FESAN]−). Results are mean ± SEM of a single experiment done with at least 3 replicates per condition. *p Value < 0.05; ***p value < 0.0005. | ||
Exposure of U87 cells to any of the two compounds under study led only to a slight increase in the foci number, which was not statistically significant, indicating that the compounds by themselves do not have the ability to induce considerable DNA damage (Fig. 13 panel A). On the contrary, irradiation of control cells led to a significant increase in foci number. However, this increase was much more marked when cells had been previously exposed to 200 μM of Na[o-FESAN] or 100 μM Na[8,8′-I2-o-FESAN] for 6 hours as shown in Fig. 13 panel B. More importantly, there was a significant increase in the damage induced by irradiation combined with incubation with Na[8,8′-I2-o-FESAN], when compared to the damage induced only by irradiation in control cells, which indicates that Na[8,8′-I2-o-FESAN] might in fact exert a potential radiosensitizing effect in this GBM cell line.
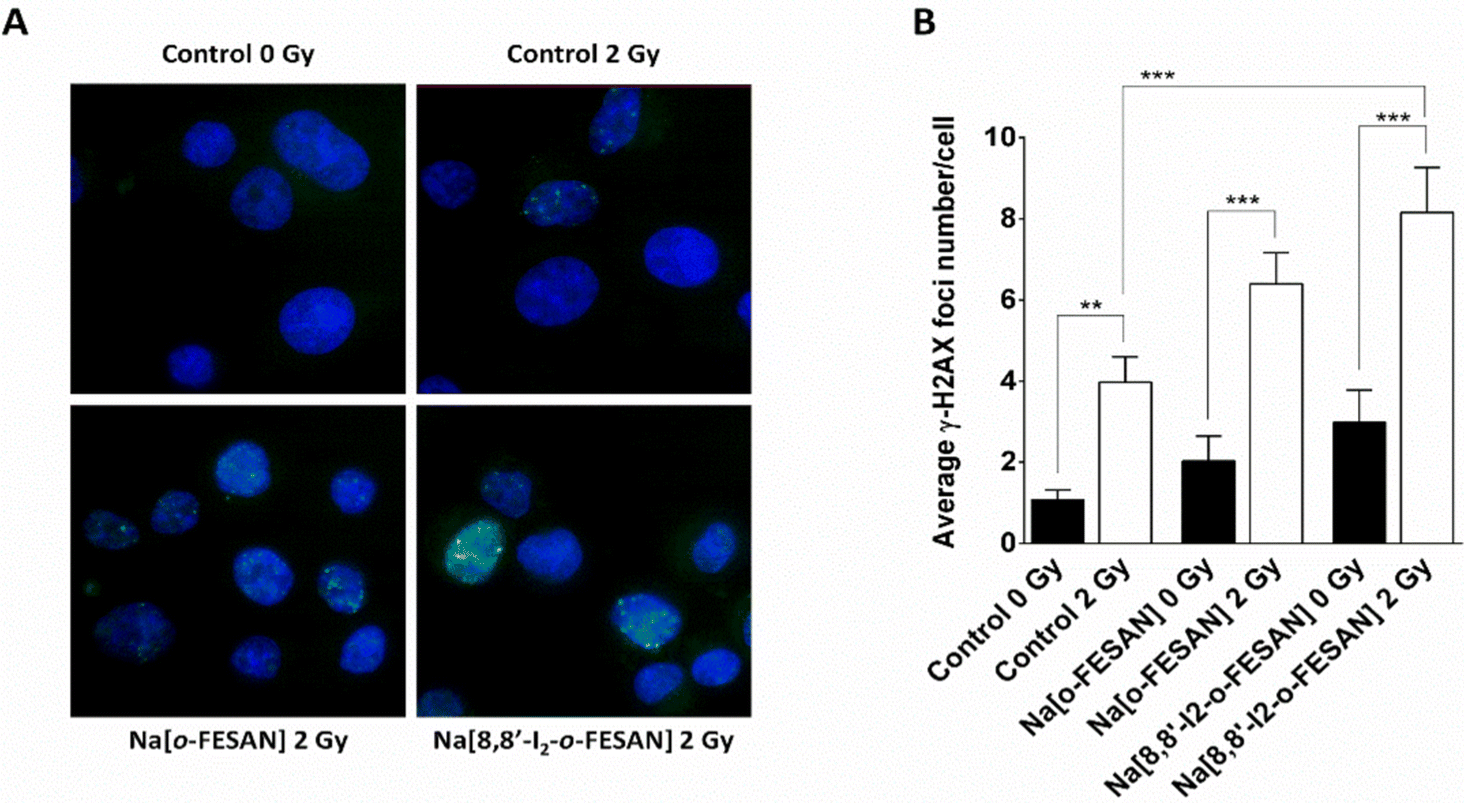 | ||
| Fig. 13 γ-H2AX foci induction ½ h post-irradiation. (A) Representative images and (B) quantification of DNA damage by 200 μM Na[o-FESAN] or 100 μM Na[8,8′-I2-o-FESAN] on U87 cells upon irradiation with 2 Gy of γ-rays. The results are shown as the mean ± SEM of two independent experiments. Statistical significance was calculated using one-way ANOVA, followed by Tukey's test (**p ≤ 0.01 and ***p ≤ 0.001). | ||
To further understand other cellular effects of the post-irradiation of cells after exposition to Na[o-FESAN] or Na[8,8′-I2-o-FESAN], apoptotic cells were visualized by fluorescence microscopy using the nucleic acid dye Hoechst 33258 (Fig. 13 panel A). As observed in Fig. 14 panel A, apoptotic events, including chromatin condensation and nucleus fragmentation were evident for [o-FESAN]− and [8,8′-I2-o-FESAN]−.
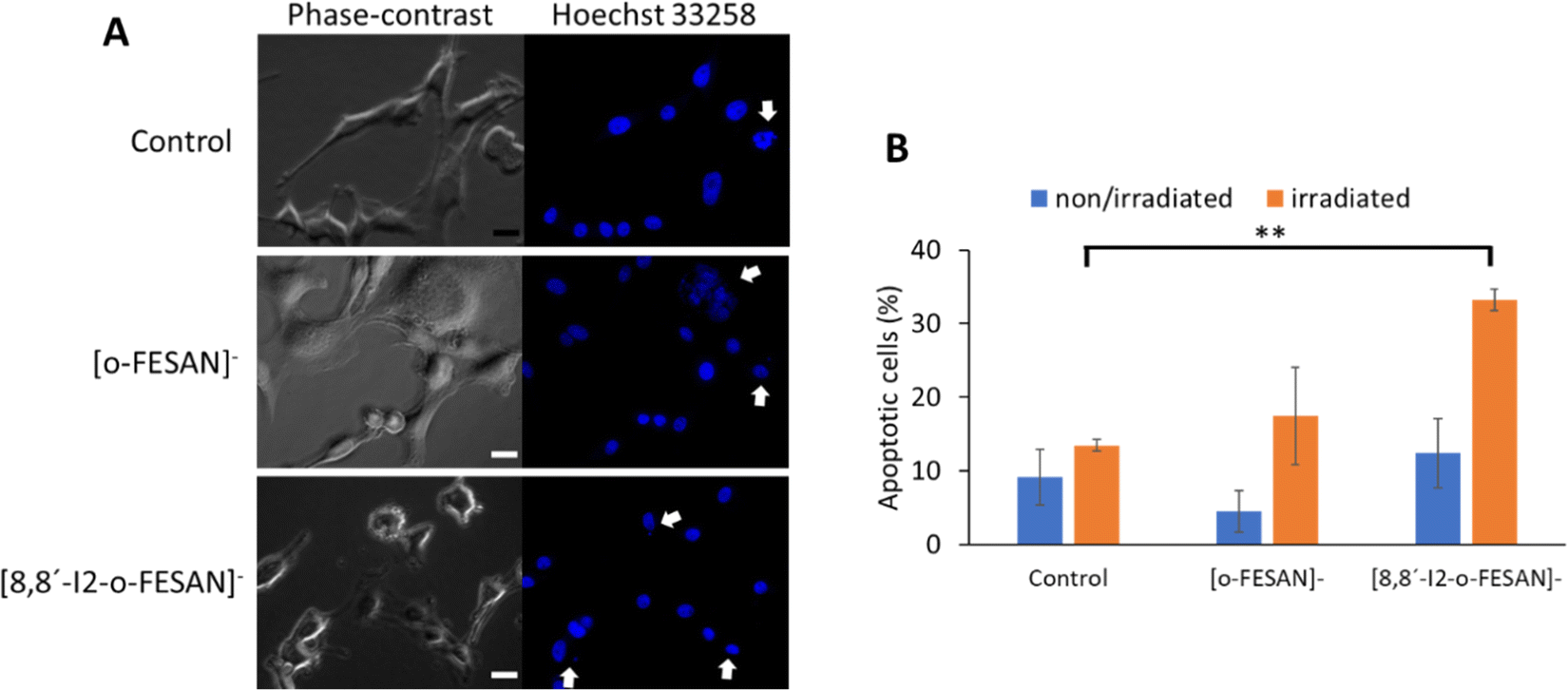 | ||
| Fig. 14 Morphological changes in the nuclei of the cells exposed to 100 μM [o-FESAN]− and 100 μM [8,8′-I2-o-FESAN]− with and without irradiation. (A) Nuclei of the cells stained with Hoechst 33258 for the visualization of apoptotic events, including chromatin condensation and nucleus fragmentation (white arrows) after exposure to control, [o-FESAN]− and [8,8′-I2-o-FESAN]− and with irradiation. Images were obtained using a Nikon TiU eclipse and the respective software. Scale bar: 20 μm. (B) % of the apoptotic cells after exposure to [o-FESAN]− and [8,8′-I2-o-FESAN]− and with and without irradiation. Six random fields with at least 10 nuclei were selected for analysis. **p Value < 0.005 relative to the respective control (control irradiated). | ||
While in non-irradiated cells there is no statistical significant difference between control cells and cells exposed to Na[o-FESAN] or Na[8,8′-I2-o-FESAN], irradiation induces an increased apoptotic events that is 3× higher in cells exposed previously to Na[o-FESAN] compared to control (Fig. 14 panel B). Moreover, despite the slight increase in the % of apoptotic cells observed after irradiation of cells previously exposed to Na[o-FESAN], this increase is not statistically significant demonstrating the potential of Na[8,8′-I2-o-FESAN] compounds as radiosensitizers.
Even if no statistically significant difference is observed between non-irradiated Na[8,8′-I2-o-FESAN]-exposed compared to control cells, there is an increase of the number of apoptotic cells in non-irradiated Na[8,8′-I2-o-FESAN] exposed cells compared to Na[o-FESAN] exposed cells (Fig. 14(B)).
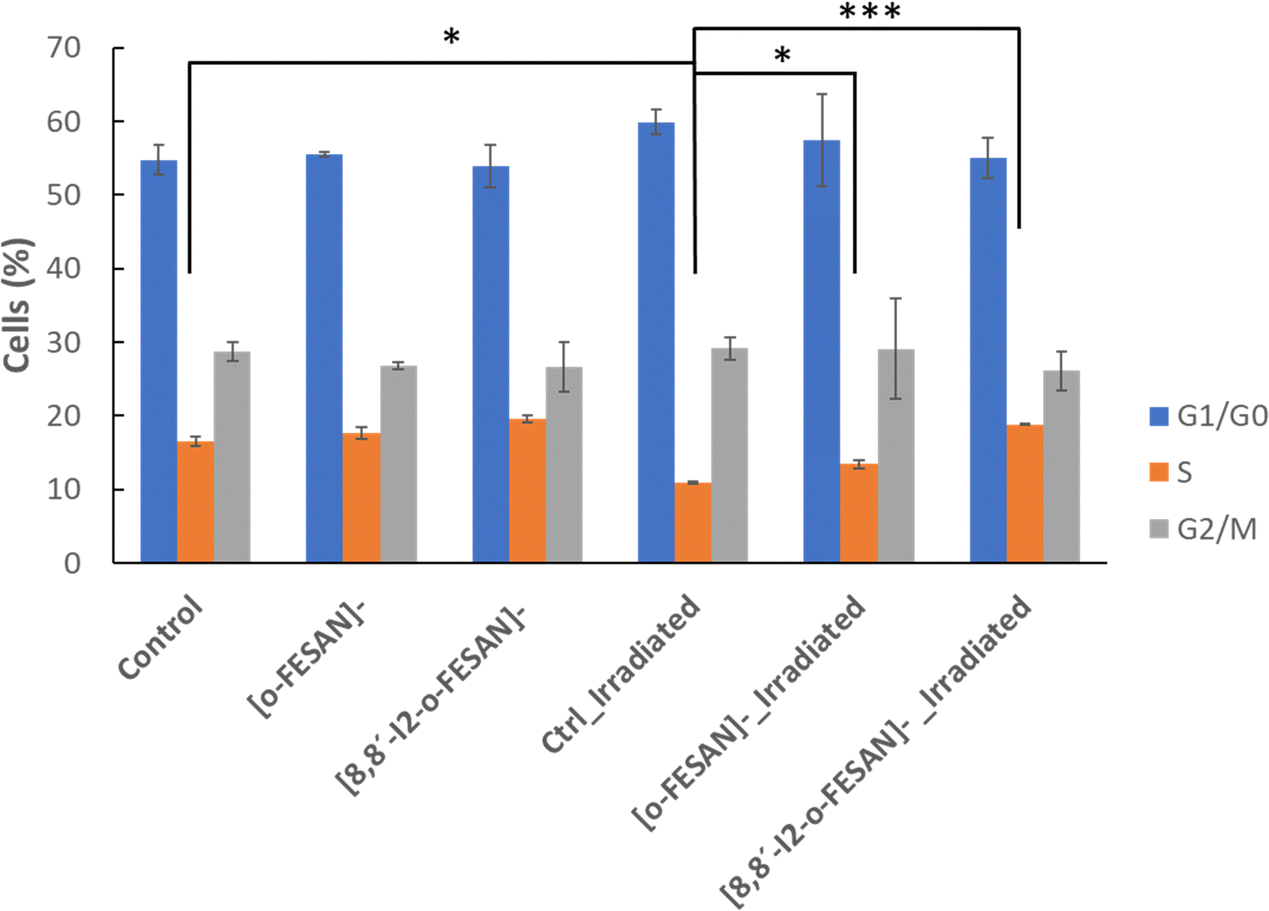
Considering these results, cell cycle effect was evaluated by flow cytometry using propidium iodide for cell staining (Fig. 14). As observed in Fig. 15, in non-irradiated cells no effect in cell cycle progression is observed after 24 h, with a similar number of cells in all phases in control, [o-FESAN]− and [8,8′-I2-o-FESAN]−. After irradiation, there is a slight decrease of control cells in the S phase and an increased number of cells in the G0/G1 phase. This trend is also observed in the [o-FESAN]− treated cells, despite not as evident as in control cells (Fig. 15). This indicates that irradiation with γ-rays is retaining cells in the G0/G1 phase to avoid progression to the S phase.
 | ||
| Fig. 15 Cell cycle evaluation of cells exposed to [o-FESAN]− and [8,8′-I2-o-FESAN]− with or without irradiation. After two periods of synchronization at the G1/S phase with thymidine, cells were exposed to [o-FESAN]− and [8,8′-I2-o-FESAN]− and irradiated. The fluorescence levels of propidium iodide were determined by flow cytometry. The represented results are the mean ± SD of two experiments. *p Value < 0.05. **p Value < 0.005. | ||
 | ||
| Fig. 16 Cellular viability 72 h after irradiation with X-rays (1.5 Gy) using a Philips MCN 165 X-ray tube and an YXLON 9421 high-voltage generator. U87 cells were previously incubated with the compounds for 24 h at concentrations in the ranges of 10–200 μM ([o-FESAN]−) and 1–100 μM ([8,8′-I2-o-FESAN]−). Results are mean ± SD of two independent experiments done with at least six replicates per condition. *p < 0.05; **p value ≤ 0.005; and ***p ≤ 0.0005. | ||
Interestingly, when it comes to [8,8′-I2-o-FESAN]− exposed cells no difference in the number of cells in the different cell cycle phases is observed with and without irradiation (Fig. 15). However, a statistically significant difference on the number of cells in the S phase compared to control cells or cells exposed to [o-FESAN]− is observed (Fig. 15).
One reason for this difference could be that the high level of γ-H2AX foci induction in cells exposed to [8,8′-I2-o-FESAN]− could induce higher levels of damage repair in cells not entering immediately into apoptosis that could, after further irradiation, survive without the need to delay the cell cycle (Fig. 13 and 15). Those cells that did not activate the repair mechanisms would be more prompt to die through apoptosis (Fig. 14 and 15).
The X-ray absorption edge is intrinsic for each atom. X-rays with higher energy than the K-edge of iodine (33.2 keV) are required for fluorescence X-ray emission. The irradiation was generated using two-layer metal filters containing Cu and Al. X-rays less than 31 KeV will be attenuated through Cu filter (1 mm thick); Al filter (4 mm thick) will absorb Cu fluorescent X-ray including Kα and Kβ line (approximately 8 keV). Fluorescent X-rays including the Kα line (28.5 keV) and L-band (3.78–5.18 keV) energy bands are released from iodine. Therefore radiation damage is sustained only locally.98
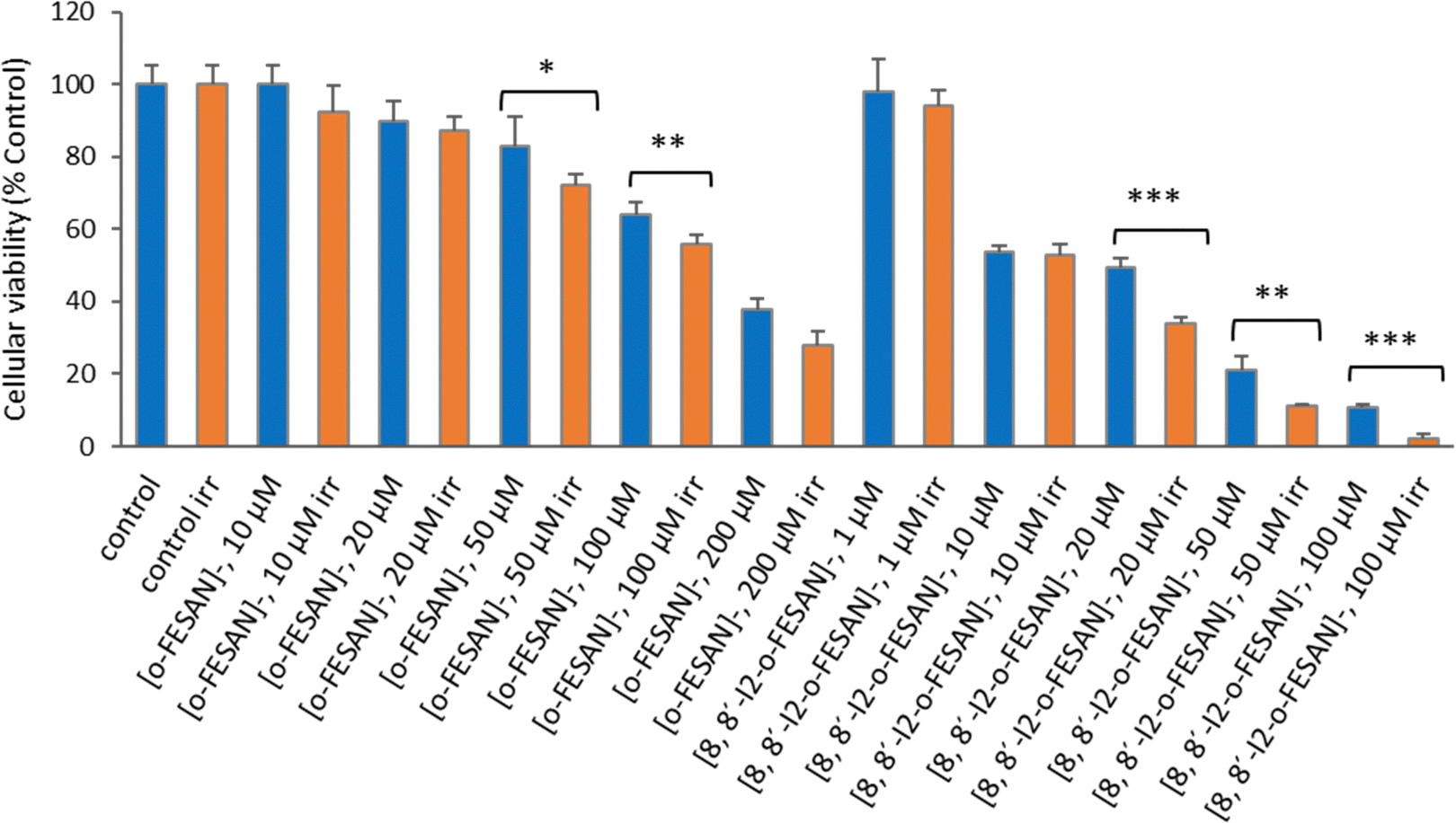
As shown in Fig. 15 a dose dependent effect was observed for the cellular viability comparable to that obtained for γ-rays. The viability loss in comparison to control conditions (without compounds and not irradiated) was more evident at higher concentrations for both compounds. Concerning survival, cells presented a significantly lower survival in comparison to control conditions at concentrations equivalent to compounds’ IC50 values (Fig. 17). With the exception of cells incubated with the lowest concentration of [o-FESAN]−, not exposed to radiation, under all conditions cells had a significantly lower survival in comparison to control conditions (without compounds and not irradiated). The cells that were incubated with [o-FESAN]− (50 and 200 μM) and [8,8′-I2-o-FESAN]− (1 and 10 μM) had a considerably lower survival when compared to the same cells not irradiated but incubated with the compounds. The MTT and the clonogenic assays showed a similar trend in particular for [8,8′-I2-o-FESAN]−. However, with X-rays a considerable lower survival was observed with [8,8′-I2-o-FESAN]− treated cells in comparison to γ-rays. Although the atomic number of iron (Fe, Z = 26) is relatively low, a dose dependent X-ray absorption was also observed for [o-FESAN]−,which caused the radiosensitizing effect observed in the cells.
 | ||
| Fig. 17 Clonogenic survival fraction eleven days after irradiation with X-rays (1.5 Gy) using a Philips MCN 165 X-ray tube and an YXLON 9421 high-voltage generator. U87 Cells were previously incubated with the compounds for 24 h at concentrations in the ranges of 50–200 μM ([o-FESAN]−) and 1–20 μM ([8,8′-I2-o-FESAN]−). Results are mean ± SEM of a single experiment done with at least 3 replicates per condition. **p Value < 0.005; ***p value < 0.0005. | ||
Results from Fig. 16 and 11 (cellular viability after irradiation with γ-rays) showed that for both [o-FESAN]− and [8,8′-I2-o-FESAN]− a dose dependent effect was observed and this effect of viability loss was significant at 50 and 100 μM, although this trend was not observed at the highest concentration of [o-FESAN]− (200 μM). Both experiments were intended to check the radiosensitizing effect of iron expecting an enhanced effect of viability loss by iodine which has a high atomic number, in particular using X-rays. The fact that [8,8′-I2-o-FESAN]− is much more cytotoxic could explain that the radiosensitizing effect induced by iron is somehow hindered.
The presence of B in the target molecule may increase the effect of protons on cell death, due to the p + 11B → 3α nuclear fusion reaction, with a resonance at 675 keV and a high cross section (∼1 barn).76 The emitted α-particles have a broad spectrum with a predominant energy of 4 MeV, whose range in water (≈25 μm) is of the order of a cell dimension. Due to these characteristics, the reaction has great potential in the context of therapeutic applications.
Fig. 18 shows the impact in the viability of U87 glioblastoma cells treated with [o-FESAN]− and [8,8′-I2-o-FESAN]− of proton irradiation, as a function of the deposited dose.
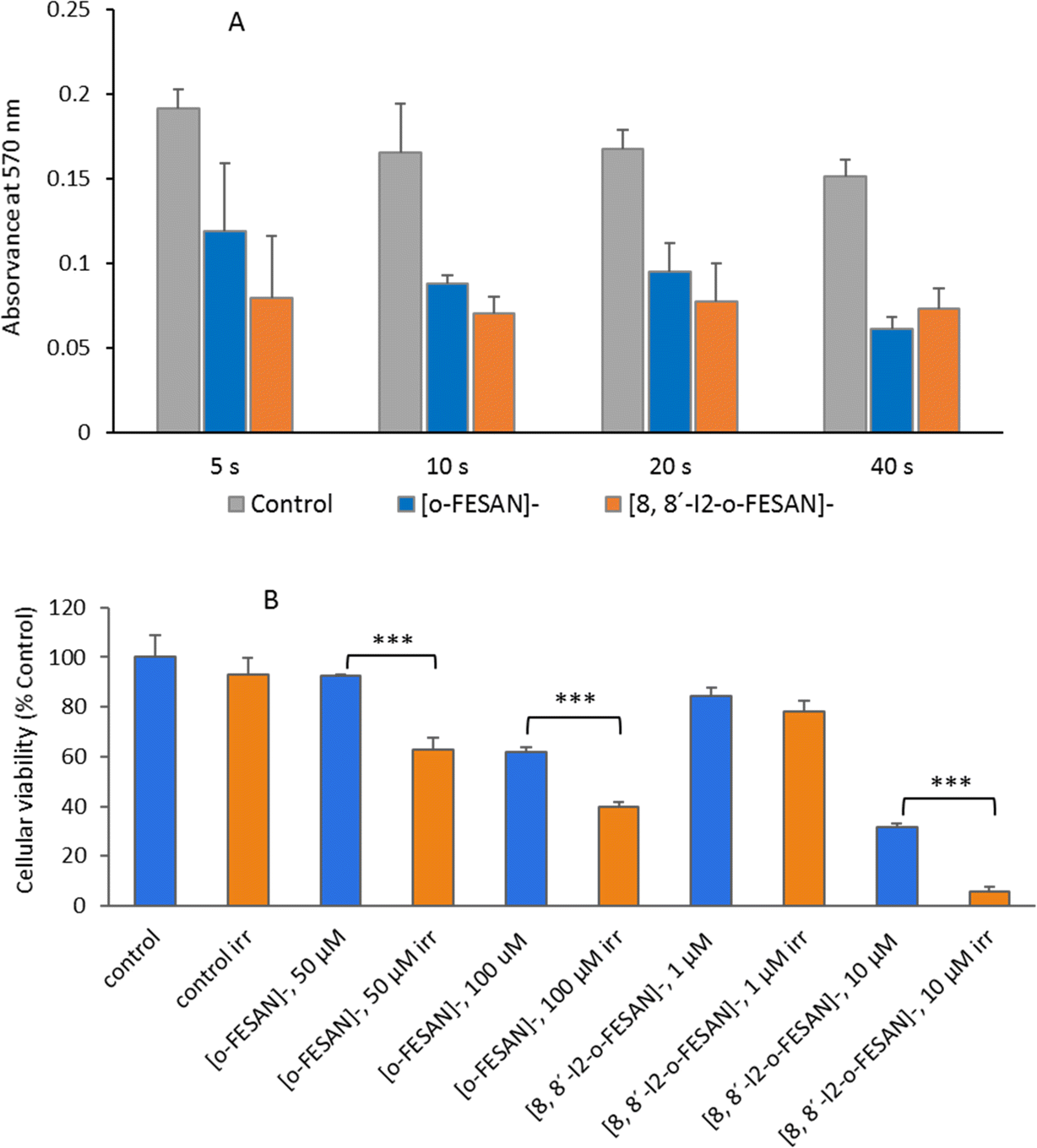 | ||
| Fig. 18 Cellular viability of U87 cells relative to control (non-irradiated cells) after proton irradiation. (A) Selection of irradiation time after treatment of cells with compounds at their IC50 values for 24 h. (B) After treatment with [o-FESAN]− (50 and 100 μM) and [8,8′-I2-o-FESAN]− (1 and 10 μM) and proton irradiation for 10 s with a dose of 9 Gy. Results are mean ± SD of two independent experiments done with at least 3 replicates per condition. ***p Value ≤ 0.0005. | ||
In non-irradiated U87 cells, the treatment with [o-FESAN]− and [8,8′-I2-o-FESAN]− caused a small decrease in viability relative to controls as measured by the MTT assay. However, after proton irradiation U87 cells exposed to [o-FESAN]− and [8,8′-I2-o-FESAN]− showed a 50% increase in lethality contrasting with a 16% increase in irradiated controls as shown in Fig. 18. It should be considered that only 34% of the cells were directly irradiated. Therefore, the high lethality rate observed suggests that α-particles generated in the fusion reaction may have direct consequences in irradiated cells and in those contiguous to the irradiated area, such as, more complex lesions at the DNA level, and indirect effects by radiolysis. Such an expressive lethality may be the enhancement effect due to α-particles but these effects should be further evaluated. The magnitude of lethality achieved evidenced the potential of both compounds to increase the cellular killing following proton irradiation.
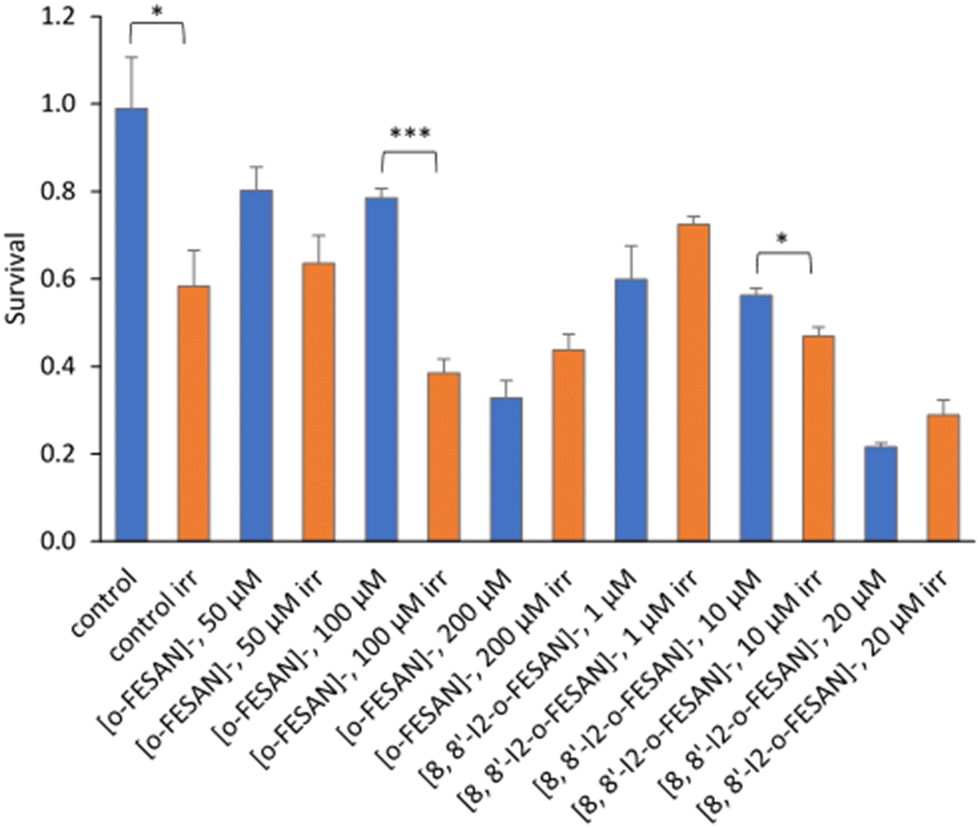
All cells incubated with [8,8′-I2-o-FESAN]− (irradiated and not irradiated) had a significantly reduced survival in comparison to control conditions (Fig. 19). Cells incubated with the highest concentration of [o-FESAN]− (100 μM) also had a significant decrease in survival in relation to control conditions.
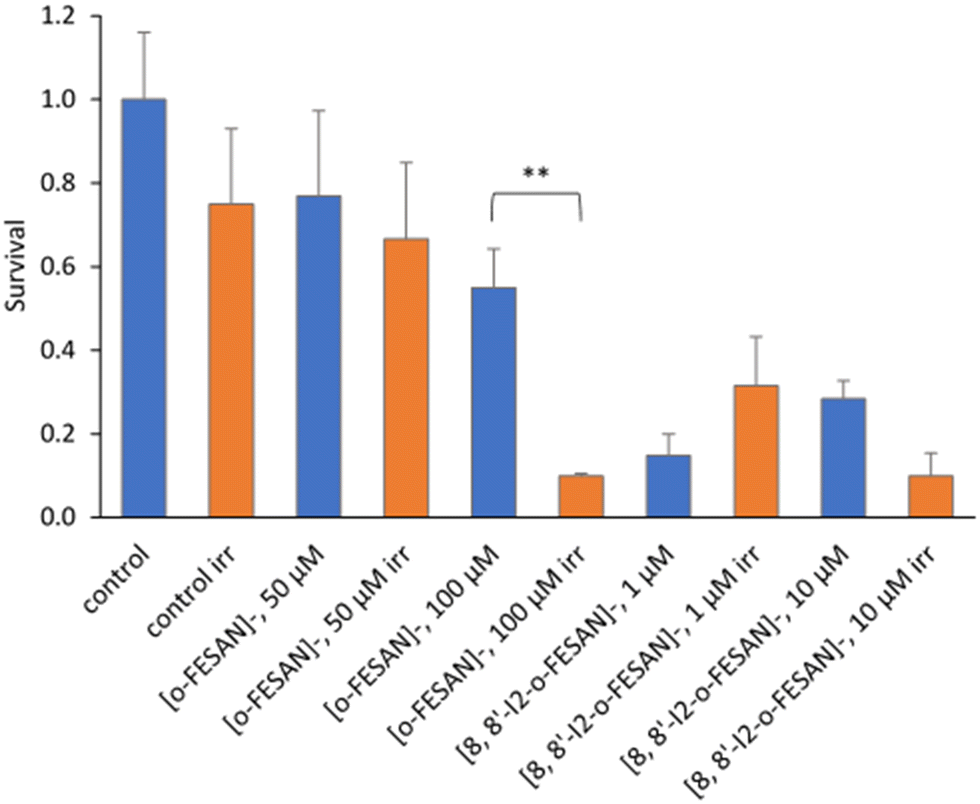 | ||
| Fig. 19 Clonogenic survival fraction eleven days after proton irradiation for 10 s at a dose of 0.98 kGy. U87 Cells were previously incubated with the compounds for 24 h at concentrations in the ranges of 50–200 μM ([o-FESAN]−) and 1–20 μM ([8,8′-I2-o-FESAN]−). Results are mean ± SEM of a single experiment done with at least 3 replicates per condition. **p Value < 0.005. | ||
The first experimental proof of PBCT using a boronated compound (BSH) and DU145 as a prostate cancer cell model was reported by Cirrone et al.99 The BSH concentration was selected based on the literature results on the use of BSH for BNCT. Authors found that the yield of chromosome aberrations is similar for cells irradiated with X-rays and proton only, but in the presence of BSH the number of aberrations per cell increase (0.08 vs. 0.18), when irradiated at 4 Gy. From our studies, we cannot directly compare these results with those herein presented. The main differences are: (i) a different cell model; (ii) the use of metallacarboranes having two boron clusters instead of one as in BSH; (iii) lower concentrations used compared to that used for BSH; (iv) different experimental set-up and different irradiation conditions; (v) different cell volume and mass as a cell monolayer was irradiated in our study; and (vi) a theoretical calculation of the dose was done; therefore, ∼9 Gy in 30 μm water equivalent showed to cause a significant loss of cellular viability and survival. Nevertheless, the results herein presented constitute the first experimental evidence of a significant enhancement of proton effectiveness by the damage caused to U87 cells, in the presence of boron-rich metallacarboranes, through irradiation with proton beams. This effect was well above the cytotoxic effect of metallacarboranes against U87 cells. Although preliminar at the moment, our results represent an important finding for further development by both in vitro and in vivo studies, particularly in the treatment of glioblastoma.
4. Conclusions
Radiotherapy (RT) is a current standard-of-care treatment for glioblastoma multiform (GBM). Since GBM is a radioresistant cancer, most patients experience tumour recurrence and disease progression. Therefore, it is mandatory to find new alternatives to overcome radiation resistance. The development of multifunctional materials with additive therapeutic effects and better safety is an urgent need and a challenge in cancer therapy research. Increasing the effectiveness of radiotherapy using high-Z materials has recently attracted much attention. The present study proposes ferrabis(dicarbollide) small anions, [o-FESAN]− and [8,8′-I2-o-FESAN]−, as prospective radiosensitizers in GBM. As is well known the radiosensitizing effect depends on the cellular uptake, and the favorable uptake of these molecules encouraged us to study their potential for multimodal radiotherapies (Mössbauer effect, γ-rays, X-rays, BNCT and PBFT) as an “all in one” approach. Moreover, the chemical and the biological stabilities of these ferrabis(dicarbollides) in physiological media are also key parameters to be considered for prospective clinical applications. FTIR and 11B{1H}-NMR spectroscopies of both ferrabis(dicarbollides) in cellular media (with and without FBS) confirmed the binding affinity of ferrabis(dicarbollides) to amino acids and proteins. The study on the translocation of the small [o-FESAN]− and [8,8′-I2-o-FESAN]− anions through a DOPC:DOPE synthetic lipid membrane, which include lipids with negative intrinsic curvature to better resembling the cells membrane, indicated that the translocation rate of both anions at 50 μM is similar. These studies support that the translocation process of ferrabis(dicarbollides) is not simply related to their lipophilic character.The viability of the ferrabis(dicarbollides) was evaluated in U87 cells, monolayer and spheroids, after treatment with both ferrabis(dicarbollides). Using U87 monolayer cells, the iodinated compound was revealed to be much more cytotoxic even at shorter incubation times. When compared with monolayer cells, U87 cells grown as spheroids were less sensitive to the compounds tested, as expected due to limited drug penetration and activation of several resistance mechanisms. However, after 72 hours U87 spheroids incubated with Na[o-FESAN] at the IC50 concentration led to a statistically significant decrease in the spheroids’ viability.
In vivo tests with both ferrabis(dicarbollide) small anions were performed in L4 C. elegans nematodes showing LD50 values similar for both Na[o-FESAN] and Na[8,8′-I2-o-FESAN] after 24 h of incubation. These anionic small molecules, whose interaction with amino acids and proteins has been demonstrated herein, could interact with the amine groups in the plethora of biomolecules of the C. elegans organism and probably contribute to their toxicity.
The development of multifunctional materials with additive therapeutic effects and better safety is an urgent need and a challenge in cancer therapy research. The radiobiological effects of radiosensitization using the ferrabis(dicarbollides) [o-FESAN]− and [8,8′-I2-o-FESAN]− were studied using electromagnetic radiation, γ-ray and X-ray. The γ-ray irradiation promoted apoptosis in [o-FESAN]− and [8,8′-I2-o-FESAN]− treated cells, with the latter inducing a 3-fold increase in irradiated vs. non-irradiated cells. Regarding the cytostatic potential of the treatment, the cell cycle analysis revealed that γ-rays retain cells in the G0/G1 phase, which is mainly observed in controls and [o-FESAN]− treated cells and to a smaller extent in [8,8′-I2-o-FESAN]− treated cells. Together with DNA damage analysis, these results suggest that γ-ray irradiation in [8,8′-I2-o-FESAN]− induces more DNA damage, without the possibility for cells to recover, triggering apoptosis. Exploring the ability of iodine-mediated X-ray radiation enhancement to induce DNA damage, we anticipate that [8,8′-I2-o-FESAN]− has potential to be further explored in this domain. Although the atomic number of iron (Fe, Z = 26) is relatively low, both iodine and iron can also be proposed in combined radiation therapies, as radiosensitizers. We also tested for the first time the proton boron fusion reaction (PBFR) with U87 cells exposed to [o-FESAN]− and [8,8′-I2-o-FESAN]−, taking advantage of their high boron (11B) content. The results obtained for the cellular damage suggest that proton boron fusion radiation therapy, when combined with boron-rich compounds, is a promising modality to fight against resistant tumors. Although encouraging, more developments are needed to further explore ferrabis(dicarbollides) as radiosensitizers towards a positive impact on the therapeutic strategies of GBM.
To sum up, our results of the in vitro, in vivo, and irradiation studies strongly suggest that these small anions are prospective candidates to be considered as future radiosensitizers in GBM through multimodal radiotherapies. More research is being conducted in our laboratories to enlighten on the irradiation mechanism when the cell pre-targeting with boron-containing compounds happens successfully. We hope that the continued developments in this area of metal-based radiosensitizers in cancer radiotherapy will have a positive impact on the therapeutic strategies for glioblastoma treatment. Furthermore, the strength of PBFR is that 11B is the key nucleus in this radiation whereas in BNCT it is 10B. In BNCT an isotopic enrichment in 10B of the boron-containing drug is required whereas in PBFR it appears be not to necessary. Also, in BNCT each B results in one α-particle while in PBFR each nucleus of 11B results in three α-particles. A simplistic calculation indicates that the use of PBFR would require 1/12th of isotopically natural molecules with respect to BNCT. Furthermore, in an ideal situation, BNCT can be used synchronously on the existing 10B and Mössbauer on 57Fe, resulting in several therapies in one compound.
Author contributions
Conceptualization: F. M. M. (Fernanda Marujo Marques) and C. V. (Clara Viñas); methodology: M. N.-M., M. Q.-M., A. M.-J., V. M. A., A. L., C. V., C. G. P., T. P., J. F. G., F. M., C. R.-R., A. R. F., P. V. B., and F. M. M.; formal analysis: M. N.-M., M. Q.-M., C. V., T. P., and S. V.; writing—original draft preparation: C. V., and F. M. M.; and writing—review and editing: all the authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors received support from the Spanish Ministerio de Economía y Competitividad (PID2019-106832RB-100), the Generalitat de Catalunya (2017SGR1720), FCT - Fundação para a Ciência e a Tecnologia, in the scope of the project UID/Multi/04349/2019 and the projects LISBOA-01-0247-FEDER-045904 and UTAP-EXPL/FMT/0020/2021 of Centro de Ciências e Tecnologias Nucleares/IST, PTDC/BTM-TEC/29256/2017, UIDP/04565/2020 of iBB/IST, UIDP/04378/2020 and UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences – UCIBIO and the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy – i4HB. The Metrology Laboratory of Ionizing Radiation team of Centro Tecnológico e Nuclear, Instituto Superior Técnico (CTN/IST) is acknowledged for their support in the irradiation setups. Miquel Nuez-Martínez is enrolled in the PhD program of the UAB. MQM and VMA acknowledge financial support by the Spanish Government MCIN/AEI/10.13039/501100011033 (project 2019-108434GB-I00 to VMA and project IJC2018-035283-I to MQM), and Universitat Jaume I (project UJI-B2018-53 to V. M. A. and project UJI-A2020-21 to MQM). SV thanks Croatian Science Foundation (project IP-2018-01-3168). Catarina I.G. Pinto is enrolled in the PhD scholarship 689 DFA/BD/07119/2020.References
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- E. Hosseinzadeh, N. Banaee and H. A. Nedaie, Curr. Cancer Ther. Rev., 2017, 13, 17–27 Search PubMed.
- E. Bidram, Y. Esmaeili, H. Ranji-Burachaloo, N. Al-Zaubai, A. Zarrabi, A. Stewart and D. E. Dunstan, J. Drug Delivery Sci. Technol., 2019, 54, 101350 CrossRef CAS.
- R. Gyanchandani, N. Priedigkeit, A. Basudan and A. V. Lee, Intratumor heterogeneity: Biological and clinical implications, Elsevier Inc., 2018; ISBN 9780128117859 Search PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Cancers, 2014, 6, 1769–1792 CrossRef PubMed.
- R. Baskar, K. A. Lee, R. Yeo and K. W. Yeoh, Int. J. Med. Sci., 2012, 9, 193–199 CrossRef PubMed.
- N. Hunter and C. R. Muirhead, J. Radiol. Prot., 2009, 29, 5–21 CrossRef PubMed.
- G. L. Jiang, Front. Med. China, 2012, 6, 165–172 CrossRef.
- R. F. Barth, Z. Zhang and T. Liu, Cancer Commun., 2018, 38, 36 CrossRef PubMed.
- W. Sauerwein, A. Wittig, R. Mos and Y. Nakagawa, Neutron Capture Therapy. Principles and Applications, Springer-Verlag, Berlin Heidelberg, Germany, 2012 DOI:10.1007/978-3-642-31334-9.
- A. E. Schwint, A. Monti Hughes, M. A. Garabalino, E. C. C. Pozzi, E. M. Heber and V. A. Trivillin, Optimizing the therapeutic efficacy of boron neutron capture therapy (BNCT) for different pathologies: Research in animal models employing different boron compounds and administration strategies (Chapter 3.6), in Boron-Based Compounds. Potential and Emerging applications in Medicine, ed. E. Hey-Hawkins and C. Viñas, John Wiley & Sons Ltd, Chichester, UK, 2018 Search PubMed.
- S. Imamichi and M. Masutani, Cancer Sci., 2018, 109, 753 Search PubMed.
- (a) J. Xu, J. Gao and Q. Wei, J. Nanomater., 2016, ID8507924, DOI:10.1155/2016/8507924; (b) A.-G. Niculesco and A. M. Grumezescu, Appl. Sci., 2021, 11, 3626 CrossRef.
- T. D. Malouff, D. S. Seneviratne, D. K. Ebner, W. C. Stross, M. R. Waddle, D. M. Trifiletti and S. Krishnan, Front. Oncol., 2021, 11, 1–11 Search PubMed.
- F. Tabbakh and N. S. Hosmane, Sci. Rep., 2020, 10, 1–12 CrossRef.
- D. Kwatra, A. Venugopal and S. Anant, Transl. Cancer Res., 2013, 2, 330–342 CAS.
- L. Gong, Y. Zhang, C. Liu, M. Zhang and S. Han, Int. J. Nanomed., 2021, 16, 1083–1102 CrossRef PubMed.
- S. Lacombe, E. Porcel and E. Scifoni, Cancer Nanotechnol., 2017, 8, 9 CrossRef.
- J. Choi, G. Kim, G. Cho, S. Bin and H. J. Im, J. Nanobiotechnol., 2020, 18, 1–23 CrossRef.
- N. Goswami, Z. Luo, X. Yuan, D. T. Leong and J. Xie, Mater. Horiz., 2017, 4, 817–831 RSC.
- M. Norouzi, Pharmacol. Res., 2020, 156, 104753 CrossRef CAS PubMed.
- S. Penninckx, A. C. Heuskin, C. Michiels and S. Lucas, Cancers, 2020, 12, 1–361 CrossRef.
- F. Silva, A. Paulo, A. Pallier, S. Même, É. Tóth, L. Gano, F. Marques, C. F. G. C. Geraldes, M. M. C. A. Castro, A. M. Cardoso, A. S. Jurado, P. López-Larrubia, S. Lacerda and M. P. Cabral Campello, Materials, 2020, 13, 1–17 Search PubMed.
- H. Marsudaira, A. Ueno and I. Furuono, Radiol. Sci., 1980, 84, 144–148 Search PubMed.
- L. S. Freudenberg, W. Jentzen, A. Stahl, A. Bockisch and A. S. J. Rosenbaum-Krumme, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38, 48–56 CrossRef.
- M. W. Barentsz, M. A. A. J. van den Bosch, W. B. Veldhuis, P. J. van Diest, R. M. Pijnappel, A. J. Witkamp and H. M. Verkooijen, Br. J. Surg., 2013, 100(5), 582–588 CrossRef CAS PubMed.
- N. W. P. Rutjes, B. A. Binnington, C. R. Smith, M. D. Maloney and C. A. Lingwood, Kidney Int., 2002, 62(3), 832–845 CrossRef CAS.
- L. Andreana, G. Isgro, L. Marelli, N. Davies, D. Yu, S. Navalkissoor and A. K. Burroughs, Cancer Treat. Rev., 2012, 38(6), 641–649 CrossRef CAS PubMed.
- G. Bleeker, G. A. M. Tytgat, J. A. Adam, H. N. Carona, L. C. M. Kremer, L. Hooft and E. C. van Dalen, Cochrane Database Syst. Rev., 2015, 9, CD009263 Search PubMed.
- M. Henze, A. Mohammed, H. P. Schlemmer, K. K. Herfarth, S. Hoffner, S. Haufe, W. Mier, M. Eisenhut, J. Debus and U. Haberkorn, J. Nucl. Med., 2004, 45(4), 579–586 Search PubMed.
- K. B. Gona, A. Zaulet, V. Gómez, F. Teixidor, J. Llop and C. Viñas, Chem. Commun., 2014, 50, 11415–11417 RSC.
- A. B. H. Laster, W. C. Thomlinson and R. G. Fairchild, Radiat. Res., 1993, 133, 219–224 CrossRef PubMed.
- M. Tamura, H. Ito and H. Matsui, Sci. Rep., 2017, 7, 1–7 CrossRef CAS.
- Y. Xie, Y. Han, X. Zhang, H. Ma, L. Li, R. Yu and H. Liu, Front. Oncol., 2021, 11, 1–14 Search PubMed.
- J. Poater, C. Vinas, I. Bennour, S. E. Gordils, M. Sola and F. Teixidor, J. Am. Chem. Soc., 2020, 142(20), 9396–9407 CrossRef CAS PubMed.
- J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS.
- R. N. Grimes, Carboranes, Elsevier Inc., New York, 3rd edn, 2016 Search PubMed.
- M. F. Hawthorne, D. C. Young and P. A. Wegner, J. Am. Chem. Soc., 1965, 87, 1818–1819 CrossRef CAS.
- M. F. Hawthorne and T. D. Andrews, J. Chem. Soc., Chem. Commun., 1965, 443–444 RSC.
- M. F. Hawthorne, D. C. Young, T. D. Andrews, D. V. Howe, R. L. Pilling, A. D. Pitts, M. Reintjes, L. F. Warren Jr and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 879–896 CrossRef CAS.
- C. Viñas, J. Pedrajas, J. Bertran, F. Teixidor, R. Kivekäs and R. Sillanpää, Inorg. Chem., 1997, 36, 2482–2486 CrossRef PubMed.
- I. Bennour, A. Cioran, F. Teixidor and C. Viñas, Green Chem., 2019, 21, 1925–1928 RSC.
- J. Rais and P. Selucky, Nucleonics, 1992, 1, 17 Search PubMed.
- S. D. Reilly, C. F. V. Mason and P. H. Smith, Cobalt(III) Dicarbollide: A Potential 137Cs and 90Sr Waste Extraction Agent, Report LA-11695, Los Alamos National Laboratory, Los Alamos, NM, 1990 DOI:10.2172/7079517.
- M. Tarrés, E. Canetta, E. Paul, J. Forbes, K. Azzouni, C. Viñas, F. Teixidor and A. J. Harwood, Sci. Rep., 2015, 5, 7804 CrossRef PubMed.
- I. Fuentes, T. García-Mendiola, S. Sato, M. Pita, H. Nakamura, E. Lorenzo, F. Teixidor, F. Marques and C. Viñas, Chem. – Eur. J., 2018, 24, 17239–17254 CrossRef CAS PubMed.
- A. M. A. Abdelgawwad, J. A. M. Xavier, D. Roca-Sanjuán, C. Viñas, F. Teixidor and A. Francés-Monerris, Angew. Chem., Int. Ed., 2021, 60, 25753–25757 CrossRef CAS PubMed.
- P. Matejícek, P. Cígler, K. Procházka and V. Král, Langmuir, 2006, 22, 575–581 CrossRef PubMed.
- P. Bauduin, S. Prevost, P. Farràs, F. Teixidor, O. Diat and T. Zemb, Angew. Chem., Int. Ed., 2011, 50, 5298–5300 CrossRef CAS PubMed.
- D. C. Malaspina, C. Viñas, F. Teixidor and J. Faraudo, Angew. Chem., Int. Ed., 2020, 59, 3088–3092 CrossRef CAS PubMed.
- A.-I. Stoica, C. Viñas and F. Teixidor, Chem. Commun., 2009, 4988–4990 RSC.
- A.-I. Stoica, C. Viñas and F. Teixidor, Chem. Commun., 2008, 6492–6494 RSC.
- A.-I. Stoica, C. Kleber, C. Viñas and F. Teixidor, Electrochim. Acta, 2013, 113, 94–98 CrossRef CAS.
- I. Fuentes, J. Pujols, C. Viñas, S. Ventura and F. Teixidor, Chem. – Eur. J., 2019, 25, 12820–12829 CrossRef CAS PubMed.
- T. M. Goszczynski, K. Fink, K. Kowalski, Z. J. Lesnikowski and J. Boratynski, Sci. Rep., 2017, 7, 9800 CrossRef PubMed.
- M. Nuez-Martínez, L. Pedrosa, I. Martinez-Rovira, I. Yousef, D. Diao, F. Teixidor, E. Stanzani, F. Martínez-Soler, A. Tortosa, À. Sierra, J. J. Gonzalez and C. Viñas, Int. J. Mol. Sci., 2021, 22, DOI:10.3390/ijms22189937.
- T. Merhi, A. Jonchère, L. Girard, O. Diat, M. Nuez, C. Viñas and P. Bauduin, Chem. – Eur. J., 2020, 26, 13935–13947 CrossRef CAS PubMed.
- P. Farràs, E. J. Juárez-Pérez, M. Lepšík, R. Luque, R. Núñez and F. Teixidor, Chem. Soc. Rev., 2012, 41, 3445–3463 RSC.
- T. Peters, C. Grunewald, M. Blaickner, M. Ziegner, C. Schütz, D. Iffland, G. Hampel, T. Nawroth and P. Langguth, Radiat. Oncol., 2015, 10, 1–13 CrossRef CAS PubMed.
- D. K. Yoon, N. Naganawa, M. Kimura, M. G. Choi, M. S. Kim, Y. J. Kim, M. W. M. Law, S. K. Djeng, H. B. Shin, B. Y. Choe and T. S. Suh, Appl. Phys. Lett., 2019, 115, 223701 CrossRef.
- I. Rojo, F. Teixidor, C. Viñas, R. Kivekäs and R. Sillanpää, Clusters, Chem. – Eur. J., 2003, 9, 4311–4323 CrossRef CAS PubMed.
- I. Bennour, M. N. Ramos, M. Nuez-Martínez, J. A. M. Xavier, A. B. Buades, R. Sillanpää, F. Teixidor, D. Choquesillo-Lazarte, I. Romero, M. Martinez-Medina and C. Viñas, Dalton Trans., 2022, 51, 7188–7209 RSC.
- M. Filek, M. Łabanowska, M. Kurdziel and A. Sieprawska, Toxins, 2017, 9, 178 CrossRef PubMed.
- H. Zhang, C. J. Carrell, D. Huang, V. Sled, T. Ohnishi, J. L. Smith and W. A. Cramer, J. Biol. Chem., 1996, 271(49), 31360–31366 CrossRef CAS PubMed.
- M. Montal and P. Mueller, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 3561–3566 CrossRef CAS PubMed.
- S. M. Bezrukov and I. Vodyanoy, Biophys. J., 1993, 64, 16–25 CrossRef CAS PubMed.
- B. Woods, R. D. M. Silva, C. Schmidt, D. Wragg, M. Cavaco, V. Neves, V. F. C. Ferreira, L. Gano, T. S. Morais, F. Mendes, J. D. G. Correia and A. Casini, Bioconjugate Chem., 2021, 32, 1399–1408 CrossRef CAS PubMed.
- A. B. Buades, L. C. J. Pereira, B. J. C. Vieira, A. C. Cerdeira, J. C. Waerenborgh, T. Pinheiro, A. P. A. Matos, C. G. Pinto, J. F. Guerreiro, F. Mendes, S. Valic, F. Teixidor, C. Viñas and F. Marques, Inorg. Chem. Front., 2022, 9, 1490–1503 RSC.
- M. A. Barreiros, T. Pinheiro, M. F. Araújo, M. M. Costa, M. Palha and R. C. da Silva, Spectrochim. Acta, Part B, 2001, 56, 2095–2106 CrossRef.
- W. Chen, C. Wong, E. Vosburgh, A. J. Levine, D. J. Foran and E. Y. Xu, J. Visualized Exp., 2014, 89, 51639 Search PubMed.
- S. Brenner, Genetics, 1974, 77, 71–94 CrossRef CAS PubMed.
- L. Gonzalez-Moragas, P. Berto, C. Vilches, R. Quidant, A. Kolovou, R. Santarella-Mellwig, Y. Schwab, S. Stürzenbaum, A. Roig and A. Laromaine, Acta Biomater., 2017, 53, 598–609 CrossRef CAS PubMed.
- E. Alves, K. Lorenz, N. Catarino, M. Peres, M. Dias, R. Mateus, L. C. Alves, V. Corregidor, N. P. Barradas, M. Fonseca, J. Cruz and A. Jesus, Eur. Phys. J. Plus, 2021, 136, 684 CrossRef.
- M. C. Spraker, M. W. Ahmed, M. A. Blackston, N. Brown, R. H. France, S. S. Henshaw, B. A. Perdue, R. M. Prior, P.-N. Seo, S. Stave and H. R. Weller, J. Fusion Energy, 2012, 31, 357–367 CrossRef CAS.
- http://www.srim.org/ (accessed 11/3/2022).
- L. R. Raposo, A. Silva, D. Silva, C. Roma-Rodrigues, M. Espadinha, P. V. Baptista, M. M. M. Santos and A. R. Fernandes, Bioorg. Med. Chem., 2021, 30, 115880 CrossRef CAS PubMed.
- T. S. Morais, Y. Jousseaume, M. F. M. Piedade, C. Roma-Rodrigues, A. R. Fernandes, F. Marques, M. J. Villa De Brito and M. Helena Garcia, Dalton Trans., 2018, 47, 7819–7829 RSC.
- E. Pereira, L. do Quental, E. Palma, M. C. Oliveira, F. Mendes, P. Raposinho, I. Correia, J. Lavrado, S. Di Maria, A. Belchior, P. Vaz, I. Santos and A. Paulo, Sci. Rep., 2017, 7, 42544 CrossRef CAS PubMed.
- J. D. McKinney, F. S. McQuillan, H. Chen, T. A. Hamor, C. J. Jones, M. Slaski, G. H. Cross and C. J. Harding, J. Organomet. Chem., 1997, 547, 253–262 CrossRef CAS.
- O. N. Kazheva, G. G. Alexandrov, A. V. Kravchenko, V. A. Starodub, I. A. Lobanova, I. B. Sivaev, V. I. Bregadze, L. V. Titov, L. I. Buravov and O. A. Dyachenko, J. Organomet. Chem., 2009, 694, 2336–2342 CrossRef CAS.
- O. N. Kazheva, G. G. Alexandrov, A. V. Kravchenko, I. B. Sivaev, I. D. Kosenko, I. A. Lobanova, M. Kajňaková, L. I. Buravov, V. I. Bregadze, A. Feher, V. A. Starodub and O. A. Dyachenko, Inorg. Chem. Commun., 2012, 15, 106–108 CrossRef CAS.
- J. M. Forward, D. M. P. Mingos and A. V. Powell, J. Organomet. Chem., 1994, 465, 251–258 CrossRef CAS.
- J. M. Forward, D. M. P. Mingos, T. E. Müller, D. J. Williams and Y.-K. Yan, J. Organomet. Chem., 1994, 467, 207–216 CrossRef CAS.
- R. J. Wiersema and M. F. Hawthorne, J. Am. Chem. Soc., 1974, 96, 761–770 CrossRef CAS.
- K. Bednarska-Szczepaniak, K. Dziedzic-Kocurek, E. Przelazly, J. Stanek and Z. J. Lesnikowski, Chem. Commun., 2021, 58(3), 391–394 RSC.
- T. Garcia-Mendiola, V. Bayon-Pizarro, A. Zaulet, I. Fuentes, F. Pariente, F. Teixidor, C. Viñas and E. Lorenzo, Chem. Sci., 2016, 7, 5786–5797 RSC.
- I. Grabowska, A. Stachyra, A. Gora-Sochacka, A. Sirko, A. B. Olejniczak, Z. J. Lesnikowski, J. Radecki and H. Radecka, Biosens. Bioelectron., 2014, 51, 170–176 CrossRef CAS PubMed.
- The compound's solubility (S) is the concentration (mol L−1) of its saturated aqueous solution, while their lipophilicity is given by its n-octanol/water Partition Coefficient (P), which is defined as the ratio of the amount of compound present in the organic phase (n-octanol) to the amount present in the aqueous phase. For convenience, the logS and logP are used.
- V. W. Pike, Trends Pharmacol. Sci., 2009, 30, 431–440 CrossRef CAS PubMed.
- L. A. Leites, Chem. Rev., 1992, 92, 279–323 CrossRef CAS.
- C. Verdiá-Báguena, A. Alcaraz, V. M. Aguilella, A. M. Cioran, S. Tachikawa, H. Nakamura, F. Teixidor and C. Viñas, Chem. Commun., 2014, 50, 6700–6703 RSC.
- F. Mittler, P. Obeïd, A. V. Rulina, V. Haguet, X. Gidrol and M. Y. Balakirev, Front. Oncol., 2017, 7, 293 CrossRef PubMed.
- M. Nuez-Martinez, C. I. G. Pinto, J. F. Guerreiro, F. Mendes, F. Marques, A. Muñoz-Juan, J. A. M. Xavier, A. Laromaine, V. Bitonto, N. Protti, S. Geninatti Crich, F. Teixidor and C. Viñas, Cancers, 2021, 13, 6367 CrossRef CAS PubMed.
- K. Buch, T. Peters, T. Nawroth, M. Sänger, H. Schmidberger and P. Langguth, Radiat. Oncol., 2012, 7, 1 CrossRef PubMed.
- L. J. Kuo and L.-X. Yang, In Vivo, 2008, 22, 305–309 CAS.
- M. Tamura, H. Ito and H. Matsui, Sci. Rep., 2017, 7, 43667 CrossRef PubMed.
- G. A. P. Cirrone, L. Manti, D. Margarone, G. Petringa, L. Giuffrida, A. Minopoli, A. Picciotto, G. Russo, F. Cammarata, P. Pisciotta, F. M. Perozziello, F. Romano, V. Marchese, G. Milluzzo, V. Scuderi, G. Cuttone and G. Korn, Sci. Rep., 2018, 8, 1141 CrossRef CAS PubMed.
- P. Bláha, C. Feoli, S. Agosteo, M. Calvaruso, F. P. Cammarata, R. Catalano, M. Ciocca, G. A. P. Cirrone, V. Conte, G. Cuttone, A. Facoetti, G. I. Forte, L. Giuffrida, G. Magro, D. Margarone, L. Minafra, G. Petringa, G. Pucci, V. Ricciardi, E. Rosa, G. Russo and L. Manti, Front. Oncol., 2021, 11, 682647 CrossRef PubMed.
- T. Mitin and A. L. Zietman, J. Clin. Oncol., 2014, 32, 2855–2863 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme S1 and Fig. S1–S4. See DOI: https://doi.org/10.1039/d2tb01818g |
| This journal is © The Royal Society of Chemistry 2022 |