
Three-dimensional CoOOH nanoframes confining high-density Mo single atoms for large-current-density oxygen evolution†
Lei
Tang‡
ab,
Liang
Yu‡
ab,
Chao
Ma
c,
Yao
Song
ab,
Yunchuan
Tu
ab,
Yunlong
Zhang
ab,
Xin
Bo
ab and
Dehui
Deng
 *ab
*ab
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Zhongshan Road 457, Dalian 116023, China. E-mail: dhdeng@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cCollege of Materials Science and Engineering, Hunan University, Changsha 410082, China
First published on 28th February 2022
Abstract
Layered transition-metal oxyhydroxides (MOOHs) emerge as promising noble-metal-free electrocatalysts for the oxygen evolution reaction (OER), yet are subject to a limited number of active sites at edges with an inactive basal plane. Herein, we report that a large number of in-plane active sites can be generated by confining high density of 16 wt% molybdenum single atoms in the basal-plane lattice of CoOOH (Mo-CoOOH). By constructing robust three-dimensional (3D) nanoframes to prevent layer-stacking and maximize exposure of active basal planes, the catalyst achieves an unprecedented OER activity at a large current density of 2000 mA cm−2, exhibiting the lowest overpotential of 400 mV among all previously reported catalysts with a high durability of over 120 hours. Multiple spectrometry characterization studies and first-principles calculations reveal that lattice-confined Mo atoms can bond moderately with OER intermediates, thereby serving as active sites for the reaction. This strategy provides a new path to design high-performance MOOH electrocatalysts with rich in-plane active sites.
Introduction
A critical bottleneck in application of water electrolysis and metal-air batteries as promising clean energy conversion and storage technologies,1–4 is the sluggish oxygen evolution reaction (OER) on the anode,5–12 which typically requires high-cost and scarce noble metal-based electrocatalysts such as iridium and ruthenium oxides.13,14 In the past decade, a wide range of earth-abundant transition-metal compounds including oxide perovskites,15 metal oxyhydroxides,16 amorphous metal oxyhydroxides,17 and molecular complexes,18 have been developed as candidate OER electrocatalysts. Among these materials, layered transition-metal oxyhydroxides (MOOHs, e.g. CoOOH, NiOOH, and FeOOH) are promising alternatives to noble metal-based electrocatalysts for the OER because of their low cost, well-defined atomic structure, and high activity at edge sites.19–27 Nevertheless, their electrocatalytic performances are severely restricted owing to the limited number of active sites at the edges and that their pristine basal plane is inactive for the OER. Two-dimensional (2D) MOOH layers with wide-open basal planes,28–30 on the one hand, provide opportunities for engineering the in-plane activity via tuning local compositions and structures. On the other hand, however, they tend to stack together to reduce surface energies, which could decrease the specific surface area and lead to burial of the basal planes. These attributes pose significant challenges in the development of MOOH-based high-performance electrocatalysts for the OER.In this work, we report that via confining high density of up to 16 wt% single molybdenum atoms in the basal plane lattice of ultrathin CoOOH nanosheets (Mo-CoOOH) and simultaneously fabricating robust 3D nanoframes of the nanosheets, a superior large-current-density OER activity is achieved over the catalyst. The lattice-confined high-density Mo atoms effectively activate the basal plane of Mo-CoOOH, leading to the formation of abundant in-plane active sites. The nanoframe architecture promotes the dispersion of the Mo-CoOOH nanosheets and prohibits the stacking of layers, thereby facilitating the exposure of the activated basal planes as well as the mass transfer of the reaction process. At an industry-level large current density of 2000 mA cm−2, the catalyst exhibits an unprecedented OER activity with an overpotential of only 400 mV, which, to the best of our knowledge, is the lowest among all previously reported catalysts, and can be well maintained for over 120 hours. Multiple spectrometry characterization studies and first-principles calculations reveal that the large number of Mo atoms confined in the basal plane lattice serve as highly efficient active centers for the OER by bonding moderately with the OER intermediates. This work presents a promising strategy for creating abundant in-plane active sites in MOOH electrocatalysts by confining high density of heteroatoms in the lattice combined with building 3D nanoframes, toward large-scale and high-performance OER applications.
Results and discussion
3D nanoframes of ultrathin Mo-CoOOH nanosheets with high Mo-loadings are synthesized using zeolitic imidazolate framework-67 (ZIF-67) templates as illustrated in Fig. 1a. The initial ZIF-67 templates, which are prepared on the basis of a previously reported method,31 are cubic-shaped with an average edge length of 400 nm as shown in the transmission electron microscope (TEM) images (Fig. 1b, ESI Fig. 1†). The as-synthesized ZIF-67 cubes are dispersed in an ethanol/water solution of Na2MoO4 with vigorous stirring for 2 hours at 82 °C, during which they transform into a cubic hollow nanoframe consisting of interconnected nanosheets, as shown in the TEM characterization for the evolution of structures as time goes on (Fig. 1b). During the process, the nanosheets are preferentially grown along the surface of the ZIF-67 template, thereby forming a cubic nanoframe for supporting the spatially separated nanosheets (Fig. 1b). The obtained material is further characterized by TEM, scanning electron microscopy (SEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) (Fig. 1c, Supplementary Fig. 2–4), which confirm the formation of the nanoframes as the dominant product. The X-ray diffraction (XRD) pattern of the material (ESI Fig. 5a†) showing sharp peaks corresponding to the 003 and 006 planes of oxyhydroxides reveals the formation of a layered oxyhydroxide structure,32,33 the thicknesses of which is around 1–2 nm as measured from the high-resolution (HR) TEM images (ESI Fig. 6†). Since isolated 2D nanosheets are prone to stack together to reduce surface energies based on the van der Waals interactions between layers, building of nanoframes to anchor the Mo-CoOOH nanosheets is advantageous for both preventing layer-stacking and promoting exposure of the basal planes. Thus, ZIF-67, serving not only as the Co source but also the nanoframe template, plays a key role in implementing the featured structure. The use of water as a weak corrosive agent to slowly destruct the crystal structure of ZIF-67 and dissolve Co2+ is critical for the crystallization of the ultrathin Mo-CoOOH nanosheets along the template surface and the nanoframe afterwards, because in the absence of the H2O solvent, the characteristic ultrathin nanosheets cannot even be formed with other conditions unchanged (ESI Fig. 7†). In addition, an appropriate ratio of Mo and Co precursors is also important to obtain the nanoframe structure, since an excessive amount of Mo leads to stacking of the layer structure (ESI Fig. 23†).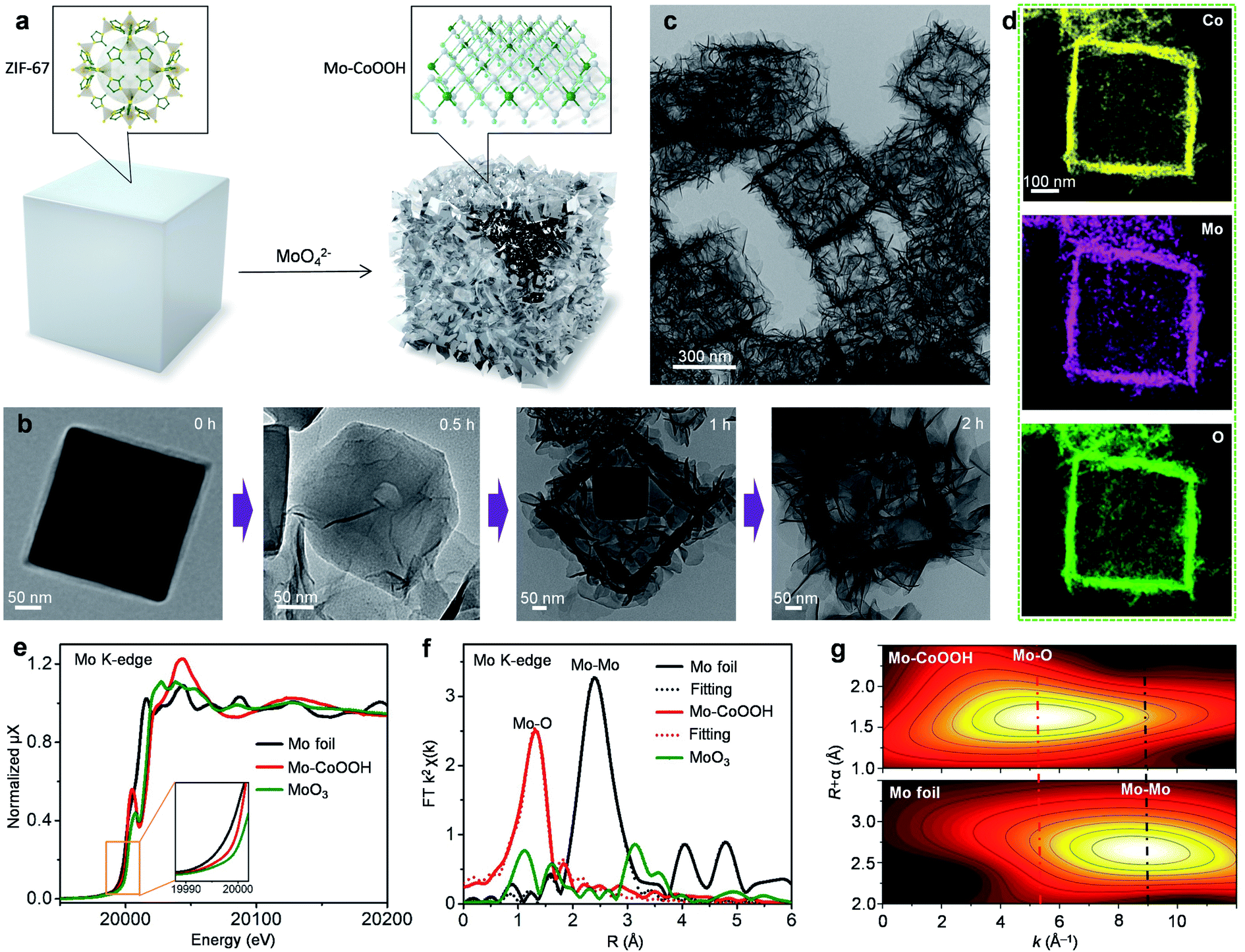 | ||
Fig. 1 Synthesis and characterization of the Mo-CoOOH 3D nanoframes. (a) Schematic illustration of the preparation process for the Mo-CoOOH nanoframes. (b) TEM images of the intermediate products collected at different times during the synthetic process. (c and d) TEM image and EDS mapping of the Mo-CoOOH nanoframes. (e) Mo K-edge XAFS spectra of Mo-CoOOH, Mo foil and MoO3. (f) k2-weighted Fourier transformation of the Mo K-edge EXAFS spectra. (g) Wavelet transformation of the k2-weighted Mo K-edge EXAFS signals of Mo-CoOOH and Mo foil. The maxima at around 5.3 and 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Å−1 (dashed lines) are related with the Mo–O and Mo–Mo contributions, respectively. Å−1 (dashed lines) are related with the Mo–O and Mo–Mo contributions, respectively. | ||
Synthesis of Mo-CoOOH involves a long-term oxidation-reduction process, in which Co2+ in ZIF-67 is oxidized to Co3+ in CoOOH and Mo6+ in MoO42− is reduced to low-valence Mo with an octahedral structure similar to that of CoOOH, so that Mo can be incorporated into the CoOOH lattice. This is reflected by the Mo K-edge X-ray absorption fine structure (XAFS) spectra, which exhibit that the absorption edge of Mo-CoOOH is between those of MoO3 and Mo foil references (Fig. 1e), indicating that Mo in Mo-CoOOH possesses a lower oxidation state than MoO3. Both Fourier and wavelet transformations of the extended XAFS (EXAFS) spectra show only Mo–O coordination peaks in Mo-CoOOH (Fig. 1f, g). A least-squares EXAFS curve fit is performed to simulate the quantitative structural parameters of Mo atoms in Mo-CoOOH, where the Mo–O coordination number is about 5.8 and close to the standard coordination number of 6 in CoOOH (Fig. 1f and ESI Table 1†). These results indicate that the Mo atoms are confined into the CoOOH lattice. The results of X-ray absorption near-edge structure (XANES) at the Co K-edge show that the absorption edge of the Mo-CoOOH sample shifts to a higher energy level compared with that of the CoO reference, being similar to that of the CoOOH reference (ESI Fig. 5b†), suggesting that the Co oxidation state in Mo-CoOOH is higher than that of CoO and close to that of CoOOH. The k2-weighted Fourier-transformation of the Co K-edge EXAFS spectra shows that the coordination environment of Co in Mo-CoOOH remains similar to that in the pure CoOOH (ESI Fig. 5c†). The elongated first-shell Co–O bond and second-shell Co-Co or Co-Mo bond in Mo-CoOOH may be attributed to that high-valence Mo atoms can provide more electrons to form stronger bonds with O than the Co atom, which in turn weakens the Co–O bond and thereby leads to elongated coordinating bonds for Co. The results of the Co 2p XPS spectra show that the Co valence in Mo-CoOOH is +3 and similar to that in CoOOH (ESI Fig. 5d†).34 The blue-shift in the Co 2p XPS of Mo-CoOOH may be due to the higher electronegativity of Mo atoms attracting outer-shell electrons from the Co atoms.
The physicochemical properties and distribution of the Co and Mo atoms in the ultrathin Mo-CoOOH nanosheets are investigated using a series of electronic and structural characterization studies. No Mo-oxide cluster or nanoparticle is observed in the HRTEM and STEM images of the nanosheets. In addition, X-ray energy-dispersive spectroscopy (EDS) for elemental mapping indicates that the Mo and Co elements are homogeneously distributed in the nanoframe (Fig. 1d). These results suggest that the Mo atoms are atomically dispersed in the Mo-CoOOH nanosheets. Abreaction-corrected HAADF-STEM characterization is further conducted to study the atomic distribution of Mo in the Mo-CoOOH nanosheets (Fig. 2a and b), which show a high density of evenly distributed bright spots in the nanosheet lattice with a d-spacing of 2.6 Å corresponding to that of CoOOH. The atomically-resolved brighter and less brighter spots indicating the Mo and Co atoms, respectively, according to their atomic numbers,3,34–37 are verified by the simulated HAADF images based on DFT-optimized structures, in which both the contrast and intensity profile analysis match well with those in the experimental images (Fig. 2b–d, ESI Fig. 8†). Since additional anions such as OH− are present between the layers of CoOOH, we used inductively coupled plasma atomic emission spectroscopy (ICP-AES) to obtain a Mo/Co atomic ratio of 1/3 corresponding to a high Mo loading of 16 wt% in Mo-CoOOH. These results demonstrate that high density of atomically dispersed Mo atoms is successfully confined in the CoOOH lattice replacing the Co atoms.
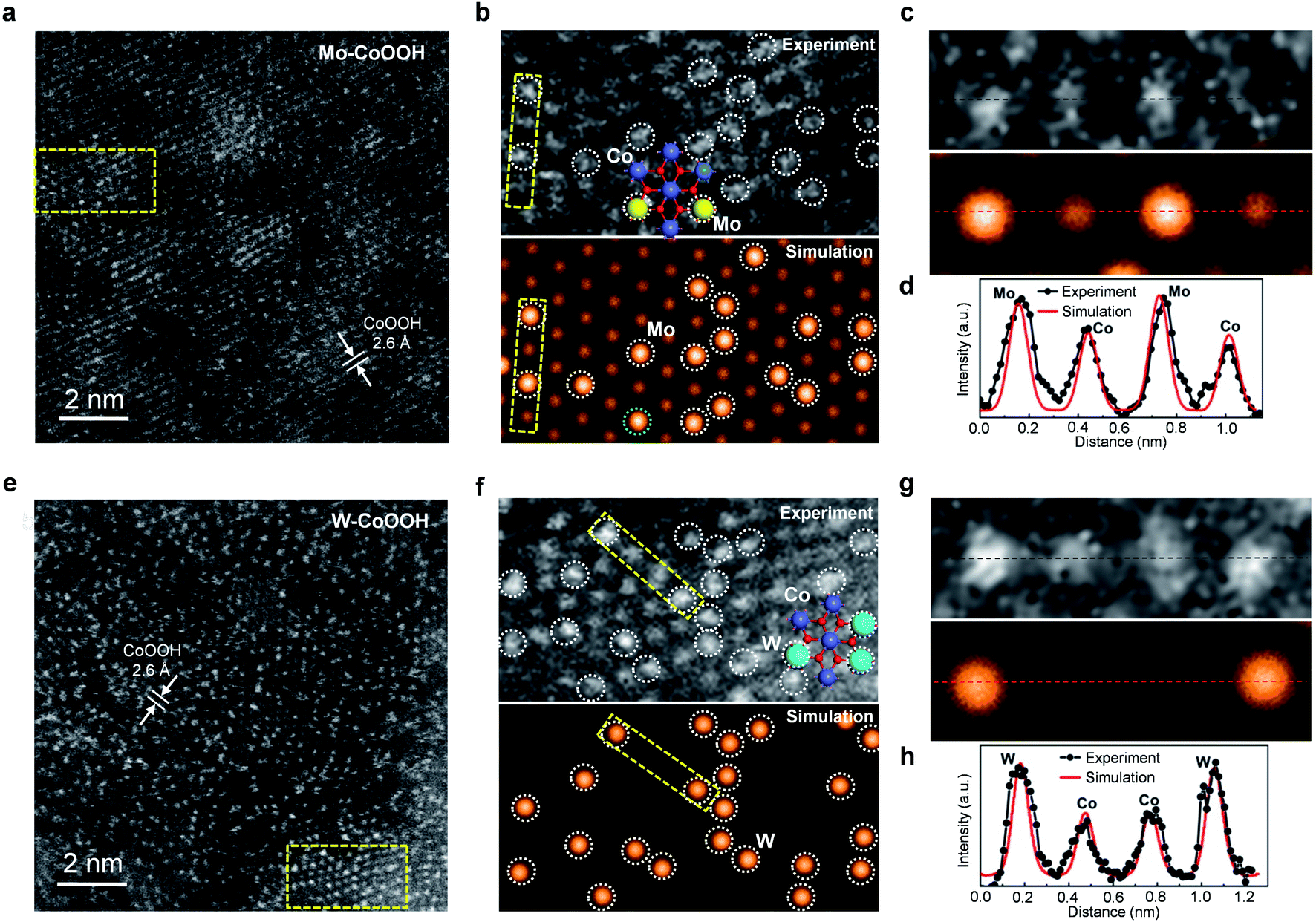 | ||
| Fig. 2 Atomic-resolution STEM imaging of Mo-CoOOH and W-CoOOH. (a and e) Atomic-resolution HAADF-STEM image of Mo-CoOOH and W-CoOOH, respectively. (b and f) Enlarged dashed rectangular regions in (a) and (e), respectively, and the corresponding simulated HAADF images based on DFT-optimized structures. The doted circles denote the lattice-confined Mo and W atoms. (c and g) Magnified views of the confined Mo and W atoms in the dashed regions in (b) and (f), respectively. (d and h) Experimental and simulated HAADF intensity profiles along the lines in (c) and (g), respectively. | ||
This strategy can be applied to synthesizing 3D nanoframes of ultrathin CoOOH layers confining other metallic heteroatoms with a high density by simply adjusting the corresponding metal precursors. For instance, 3D nanoframes of W-CoOOH are synthesized by using ZIF-67 and Na2WO4 (ESI Fig. 9†). The morphology, electronic structure, and atomic composition of the as-synthesized W-CoOOH are investigated using the same characterization techniques as those in the case of Mo-CoOOH. The TEM images show the formation of nanoframes, which is similar to that of Mo-CoOOH. EXAFS analysis combined with EDS elemental mapping and abreaction-corrected HAADF-STEM characterization verify that high-density atomically dispersed W atoms are confined in the lattice of ultrathin CoOOH layers (Fig. 2e–h, ESI Fig. 9†). These results signify the generality of this strategy in building nanoframes of ultrathin oxyhydroxide nanosheets confining high-density of heteroatoms in the basal plane lattice.
Linear sweep voltammetry (LSV) curves of different catalysts, including Mo-CoOOH, W-CoOOH, pure CoOOH, and commercial IrO2, are measured to investigate their OER performance in 1 M KOH solution at 25 °C (Fig. 3a, ESI Fig. 10†). Compared with pure CoOOH, Mo-CoOOH and W-CoOOH show remarkably improved activity with lower onset potentials (defined as the potential to reach a current density of 1 mA cm−2) of 1.38 V and 1.49 V versus the reversible hydrogen electrode (RHE), respectively, than the 1.55 V on pure CoOOH (Fig. 3b). At a current density of 10 mA cm−2, Mo-CoOOH and W-CoOOH present much lower OER overpotentials of 249 and 330 mV, respectively, compared with the 410 mV for the pure CoOOH. Tafel plots are further analysed to investigate the OER reaction kinetics over these catalysts. The Tafel slopes of the linear portion at a low potential fitted to the Tafel equation are 60.5 and 91.1 mV dec−1 for Mo-CoOOH and W-CoOOH, respectively, which are lower than the 120.4 mV dec−1 for CoOOH (Fig. 3c), indicating faster OER kinetics over Mo-CoOOH and W-CoOOH. Moreover, the OER activity of Mo-CoOOH is even higher than that of the commercial IrO2 catalyst with an onset potential of 1.42 V, an overpotential of 290 mV at 10 mA cm−2 and a Tafel slope of 61.3 mV dec−1. These results indicate that confining high density of Mo or W atoms in the CoOOH lattice prominently enhances the electrocatalytic activity, and confinement of Mo atoms can induce a superior OER activity outperforming most of the previously reported OER catalysts including the commercial precious IrO2 (ESI Table 2†). The activity of the Mo-CoOOH and W-CoOOH catalysts can be well-maintained for over 36 hours with only a slight increase in the potential and little change in the atomic structures and morphologies, showing the high stability of the material (Fig. 3d, ESI Fig. 11 and 12†).
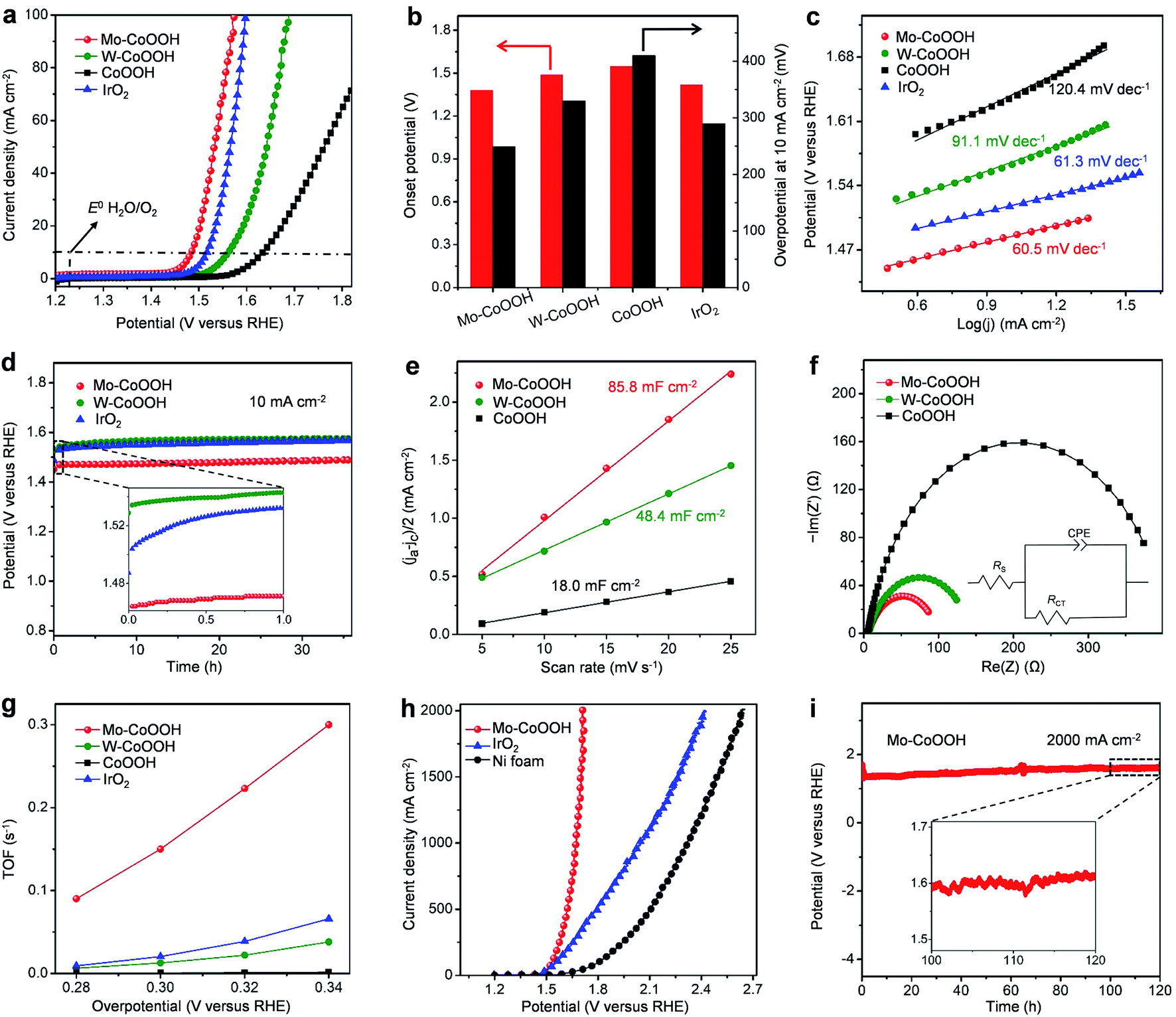 | ||
| Fig. 3 Electrocatalytic OER measurements. All the OER measurements were conducted in 1 M KOH solution at 25 °C. (a) The OER polarization curves of different catalysts loaded on the glassy carbon electrode after iR correction (i, current; R, resistance). (b) Comparison of onset potentials at 1 mA cm−2 and overpotentials at 10 mA cm−2 over different catalysts. (c) Tafel plots for different catalysts. (d) Chronopotentiometric curves obtained with Mo-CoOOH, W-CoOOH and IrO2 on a carbon fiber paper electrode with constant current densities of 10 mA cm−2 after iR correction. (e) Plot for the extraction of the double-layer capacitance for Mo-CoOOH, W-CoOOH and CoOOH. (f) Electrochemical impedance spectroscopy (EIS) Nyquist plots of Mo-CoOOH, W-CoOOH and CoOOH. The inset shows the equivalent circuit model. (g) OER turnover frequencies (TOFs) over Mo-CoOOH, W-CoOOH, pure CoOOH, and IrO2 at different overpotentials. (h) OER polarization curves for Mo-CoOOH under large current densities in comparison with IrO2 and Ni foam with iR corrections. Mo-CoOOH and IrO2 are drop-cast on Ni foam for measurements. (i) The chronopotentiometric measurements for the long-term stability test of Mo-CoOOH loaded on Ni foam after iR correction. The inset shows details of current fluctuations caused by oxygen gas bubble release. | ||
To further investigate the electrocatalytic properties of the catalysts, the electrochemical active surface areas (ECSA) of Mo-CoOOH, W-CoOOH, and pure CoOOH are measured, which are 85.8, 48.4, and 18.0 mF cm−2, respectively (Fig. 3e, ESI Fig. 13†). However, Mo-CoOOH, W-CoOOH, and CoOOH possess similar Brunauer–Emmett–Teller (BET) specific surface areas of 207.8, 198.3, and 204.9 m2 g−1, respectively, which are measured by using N2 adsorption/desorption isotherms (ESI Fig. 14†). Therefore, the significant increase in the ECSA of Mo-CoOOH and W-CoOOH indicates that confining high-density of Mo and W atoms increases markedly the number of active sites. In addition, electrochemical impedance spectroscopy (EIS) is conducted to characterize the charge-transfer kinetics at the electrode/electrolyte interface. The obtained charge transfer resistances (RCT) of Mo-CoOOH and W-CoOOH are 93 and 140 Ω, respectively, which are obviously lower than that of CoOOH (402 Ω) (Fig. 3f), thus suggesting that the electron-transfer process is effectively promoted by introducing the Mo and W atoms into the lattice. These results indicate that confining high density of Mo atoms or W atoms in the basal-plane lattice of CoOOH enhances the electrocatalytic OER activity by inducing abundant active sites and improving the electron conductivity. The intrinsic activities of Mo-CoOOH and W-CoOOH are further analysed by calculating the turnover frequencies (TOFs) based on the Mo or W content, which is remarkably larger than that of pure CoOOH calculated based on the Co content (Fig. 3g). The TOF over Mo-CoOOH is even much higher than that over the commercial precious IrO2 catalyst (Fig. 3g), thus indicating an excellent intrinsic OER activity of Mo-CoOOH.
The large-current-density OER performance of Mo-CoOOH is further compared with that of the commercial IrO2 by coating the catalysts on Ni foam electrodes. At an industry-level large current density of 2000 mA cm−2, the working voltage of Mo-CoOOH is significantly lower than that of IrO2 by 702 mV (Fig. 3h, ESI Fig. 15†), and can be stably maintained for over 120 hours (Fig. 3i). SEM, TEM, HAADF-STEM and EDS mapping characterization of the used Mo-CoOOH show that the atomic structure and morphology of the catalyst are barely changed after the large-current-density stability test (ESI Fig. 16–18†), indicating the robustness of the catalyst. The OER overpotential at 2000 mA cm−2 after the iR correction is 400 mV and is superior to all previously reported values under similar measurement conditions (ESI Table 3†). To further explore the potential of the Mo-CoOOH catalyst in practical industry applications, we build an alkaline anion exchange membrane (AEM) water electrolyser to assess its catalytic performance as an anode catalyst (Fig. 4a and b). Commercial 20% Pt/C is used as the cathode. Under various cell potentials, the current densities of the cell with the Mo-CoOOH anode are always higher than those of the cell with the commercial precious IrO2 anode (Fig. 4c). Our device needs a cell voltage of 2.08 V to reach a large current density of 1000mAcm−2, and the performance can be stably maintained for 100 hours (Fig. 4d). These results render Mo-CoOOH a promising electrocatalyst for large-scale industrial OER application.
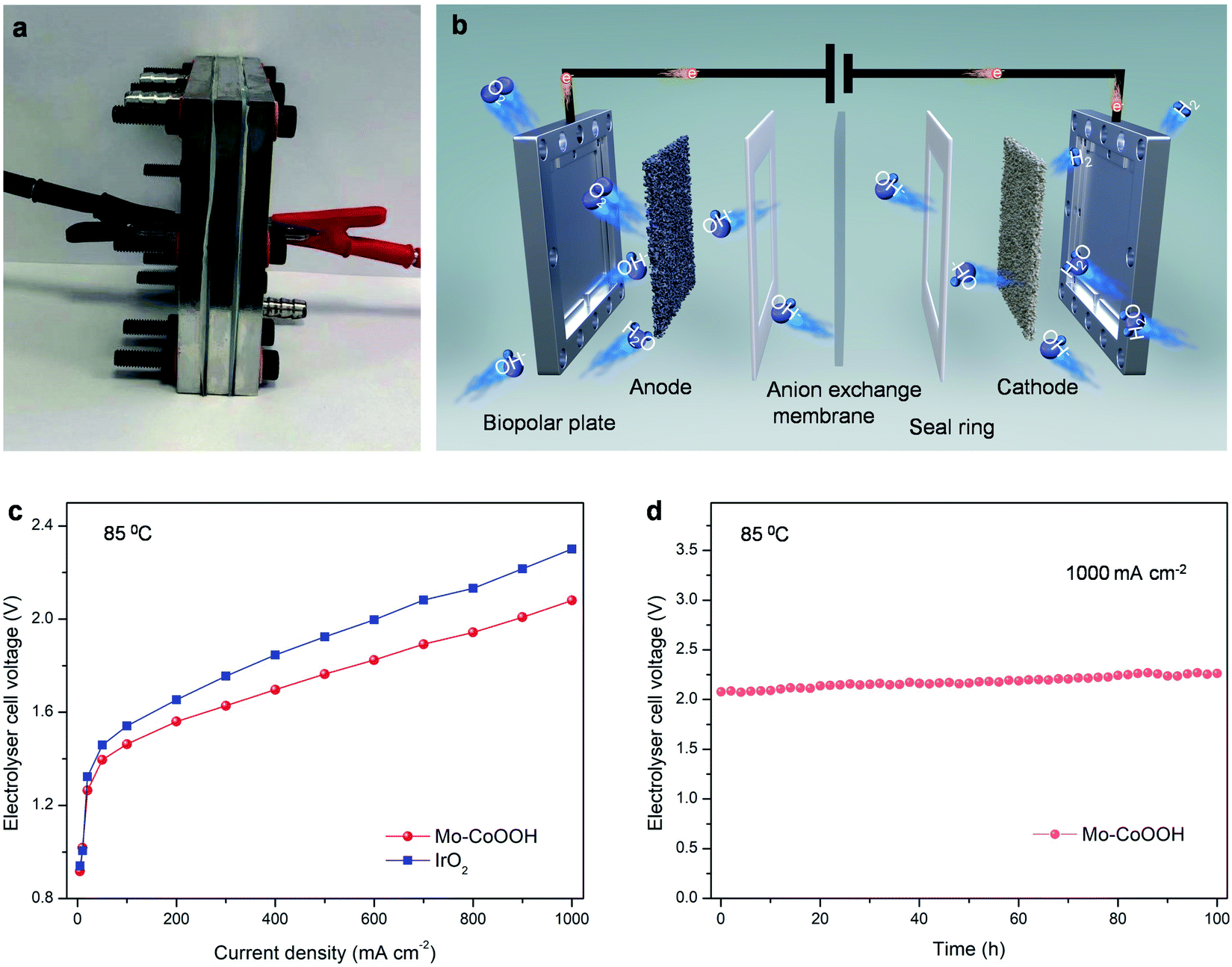 | ||
| Fig. 4 Performance of Mo-CoOOH catalysts in practical electrolyser systems. (a) Photograph of an alkaline anion exchange membrane electrolyzer device. (b) Schematic illustration of the electrolyser structure. (c) Polarization curves for water electrolysis using Mo-CoOOH or commercial precious IrO2 as the anode catalyst and 20% Pt/C as the cathode catalyst. (d) Durability of the electrolyser at a constant current density of 1000mAcm−2 for 100h at 85°C. | ||
To deeply understand the effect of confining Mo or W in the basal plane lattice on improving the OER activity of CoOOH, we performed DFT studies of the active sites and reaction mechanisms on the layered Mo-CoOOH, W-CoOOH, and pure CoOOH catalysts (ESI Fig. 19†). Since the OER reaction conditions are highly oxidative, electrochemical oxidation of the catalysts is first investigated by calculating their differential dehydrogenation free energy, which shows that the equilibrium potentials for full dehydrogenation of Mo-CoOOH, W-CoOOH and pure CoOOH are all below 1.5 V (ESI Fig. 20†). Thus, under a working potential above 1.5 V, they tend to be oxidized to Mo-CoOO, W-CoOO and CoOO, which were thereby employed as the catalyst structures to study the reaction mechanisms under practical reaction conditions (Fig. 5a). The difference in the Mo–O, W–O and Co–O bond lengths in the optimized Mo-CoOO and W-CoOO structures is only around 0.05 Å, indicating no obvious lattice distortion after confining the Mo and W atoms. The following electrochemical reaction steps are calculated for the OER process:
| * + OH− → *OH + e−, | (i) |
| *OH + OH− → *O + H2O (l) + e−, | (ii) |
| *O + OH− → *OOH + e−, | (iii) |
| *OOH + OH− → *OO + H2O (l) + e−, | (iv) |
| *OO → * + O2 (g), | (v) |
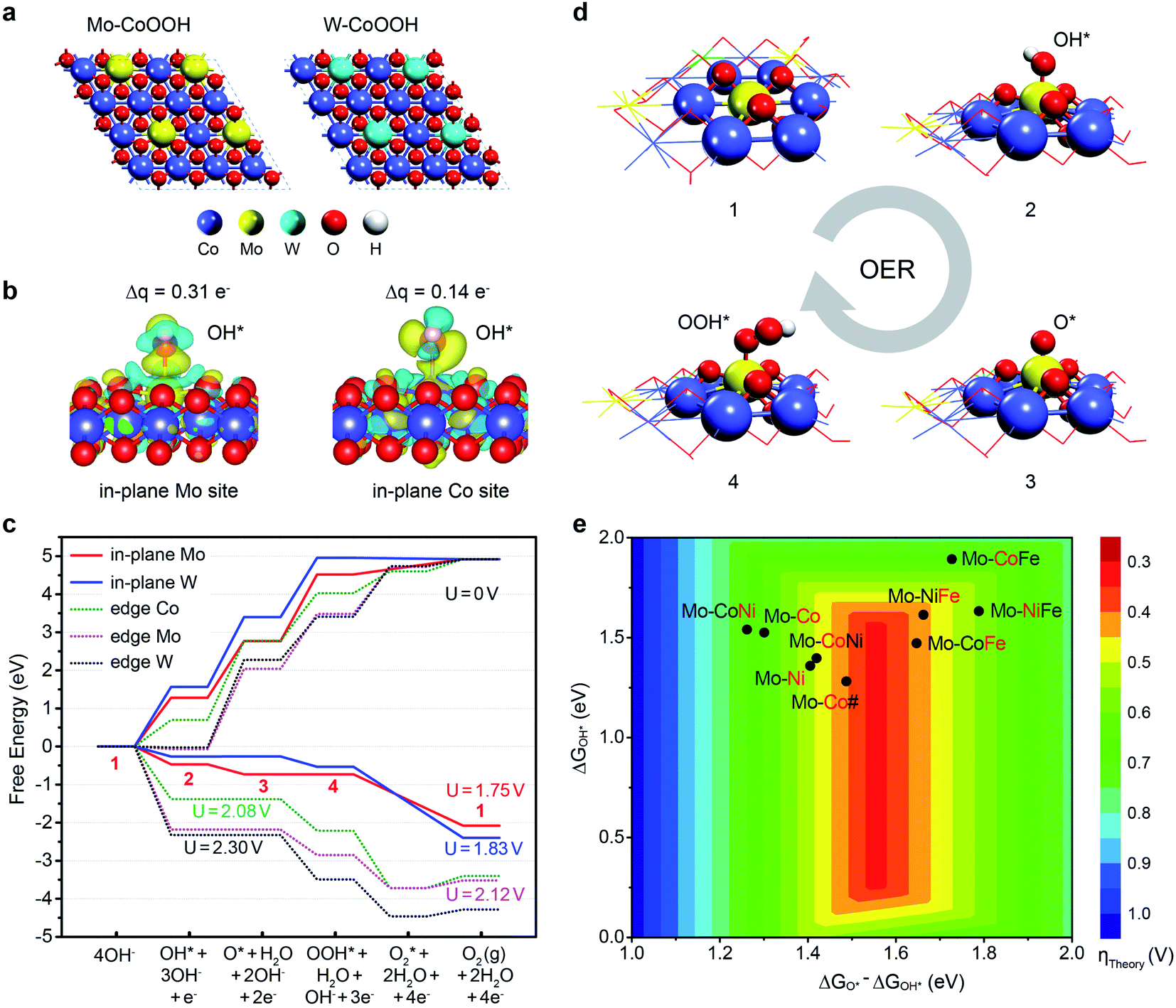 | ||
| Fig. 5 DFT insight into the OER activity of MOOH with lattice-confined Mo or W. (a) Models for simulating the basal planes of the fully oxidized Mo-CoOOH and W-CoOOH, i.e. Mo-CoOO and W-CoOO. (b) Differential charge density and Bader charge analysis of OH* adsorbed on Mo and constrained to the Co sites in the basal plane. (c) OER free energy diagram on the Mo and W active sites in the basal plane and at the edges at 0 V and their corresponding limiting potentials. (d) Atomic structures of the OER intermediates on the in-plane Mo site. The numbers represent the states in correspondence to those in (c). (e) OER activity map for 2D transition-metal (Co, Ni, and Fe) oxyhydroxides confining Mo atoms based on the linear relation between the reaction free energies of the elementary steps (ii) and (iii) (ESI Fig. 22†). Mo-CoNi denotes the catalyst of CoNiOOH confining Mo, and so on for the naming of other catalysts. The red symbols denote the elements that are substituted by Mo. Mo-Co# denotes the catalyst with a Co/Mo atomic ratio of 3/1. | ||
To further verify the electrocatalytic mechanism experimentally, operando Raman spectroscopy and operando electrochemical attenuated total reflection infrared (ATR-IR) characterization were performed for the OER over the Mo-CoOOH and pure CoOOH catalysts in the potential range from the open-circuit voltage (OCV) to 1.60 V with an interval of 0.05 V (Fig. 6). In the operando Raman spectra of the pure CoOOH catalyst, the peaks at 481 cm−1 and 690 cm−1 are assigned to the depolarized Eg mode (bending) and the polarized A1g mode (stretching) of Co–O, respectively.33 In comparison, a pair of new peaks appear at 500 cm−1 and 590 cm−1 for the Mo-CoOOH catalyst, which are attributed to the bending and stretching vibrations of Mo–O, respectively. The red-shifted Co–O peak at 475 cm−1 relative to the 481 cm−1 in CoOOH should be due to the elongated Co–O bond in Mo-CoOOH as indicated in the EXAFS analysis (ESI Fig. 5c†). Furthermore, the Raman peaks corresponding to the stretching vibrations of Mo–O at 590 cm−1 shift to lower frequencies with the increase of the applied potential from 1.25 to 1.60 V, which is correlated to the elongation of the metal–O bonds,38 thus reflecting the formation of reaction intermediates on the Mo site with longer Mo–O bonds than the lattice Mo–O during the reaction process. The results of operando electrochemical ATR-IR show that at applied potentials, except for the rise of two peaks at 1165 and 1186 cm−1 for both samples, a new peak appears at 1218 cm−1 for Mo-CoOOH, which could be attributed to the O–O bond vibration in OOH* species in situ formed on the Mo site.39 With the increase of potential from 1.25 to 1.60 V, the blue shift of the new absorption peak is observed, which may be due to the weakened Mo–OOH* interaction at higher potentials, which in turn leads to stronger O–OH* interactions. These results provide evidence for the proposed reaction mechanism that the Mo atoms are the active sites for the OER over the Mo-CoOOH catalyst.
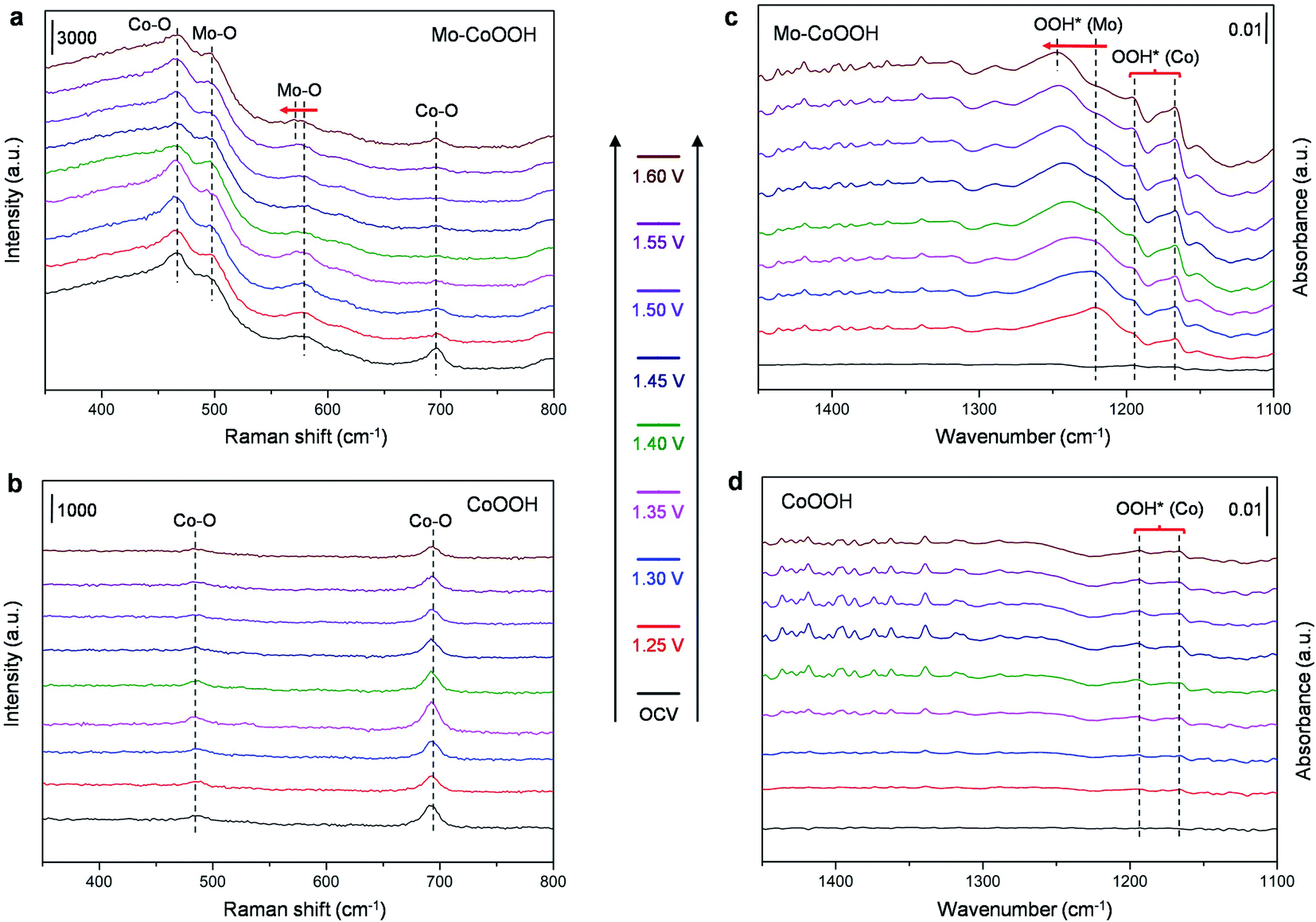 | ||
| Fig. 6 Operando Raman and ATR-IR spectroscopy characterization. (a and b) Operando Raman spectra of the Mo-CoOOH catalyst and the CoOOH catalyst under different applied potentials, respectively. (c and d) Operando ATR-IR spectra of the Mo-CoOOH catalyst and the CoOOH catalyst under different applied potentials, respectively. | ||
In summary, we report a strategy for introducing OER active sites in the basal plane of ultrathin CoOOH nanosheets by confining high density of single Mo atoms in the lattice, in combination with simultaneous fabrication of anti-stacking 3D nanoframes of the nanosheets. The catalysts reach 2000 mA cm−2 current density at an overpotential of 400 mV, surpassing those of all previously reported catalysts and even the commercial precious IrO2 catalysts. Detailed structural characterization and density-functional theory calculations reveal that the lattice-confined Mo sites in the 2D basal-plane are active sites for the OER, which bond moderately with the reaction intermediates and thereby enhance the activity. This strategy can be applied to confining different heteroatoms in the basal plane lattice of transition metal oxyhydroxides to trigger abundant active sites and design highly active catalysts for electrochemical energy conversion devices.
Author contributions
D. D. conceived the study. L. T. performed the synthetic experiments and characterization of the catalysts. C. M. and Y. Z. performed the aberration-corrected scanning transmission electron microscopy characterization. L. T., Y. S., Y. T. and X. B. performed the electrochemical tests. L. Y. performed the DFT calculations. L. T., L. Y. and D. D. co-wrote the paper. All authors discussed the results and contributed to the interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21988101, 21890753, and 21872140), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB36030200), the Key Research Program of Frontier Sciences of the Chinese Academy of Sciences (No. QYZDB-SSW-JSC020), and the DNL Cooperation Fund, CAS (No. DNL201918). We thank the staff at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facilities (SSRF) for assistance with the XAFS measurements.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spori, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Perez-Ramirez, B. R. Cuenya, R. Schlogl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- J. Deng, H. Li, J. Xiao, Y. Tu, D. Deng, H. Yang, H. Tian, J. Li, P. Ren and X. Bao, Energy Environ. Sci., 2015, 8, 1594–1601 RSC.
- Q. Xu, J. Zhang, H. Zhang, L. Zhang, L. Chen, Y. Hu, H. Jiang and C. Li, Energy Environ. Sci., 2021, 14, 5228–5259 RSC.
- E. Detsi, J. B. Cook, B. K. Lesel, C. L. Turner, Y.-L. Liang, S. Robbennolt and S. H. Tolbert, Energy Environ. Sci., 2016, 9, 540–549 RSC.
- A. Grimaud, A. Demortiere, M. Saubanere, W. Dachraoui, M. Duchamp, M.-L. Doublet and J.-M. Tarascon, Nat. Energy, 2017, 2, 16189 CrossRef CAS.
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Joule, 2020, 4, 2549–2561 CrossRef CAS.
- J. W. D. Ng, M. Garcia-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 16053–16061 CrossRef CAS.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
- W. Li, S. Watzele, H. A. El-Sayed, Y. Liang, G. Kieslich, A. S. Bandarenka, K. Rodewald, B. Rieger and R. A. Fischer, J. Am. Chem. Soc., 2019, 141, 5926–5933 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schaublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. Garcia-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS.
- M. Martin-Sabi, J. Soriano-Lopez, R. S. Winter, J.-J. Chen, L. Vila-Nadal, D.-L. Long, J. Ramon Galan-Mascaros and L. Cronin, Nat. Catal., 2018, 1, 208–213 CrossRef CAS.
- A. M. Ullman, C. N. Brodsky, N. Li, S.-L. Zheng and D. G. Nocera, J. Am. Chem. Soc., 2016, 138, 4229–4236 CrossRef CAS PubMed.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- H. Xiao, H. Shin and W. A. Goddard III, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 5872–5877 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- K. Zhu, X. Zhu and W. Yang, Angew. Chem., Int. Ed., 2019, 58, 1252–1265 CrossRef CAS PubMed.
- L. Tang, X. Meng, D. Deng and X. Bao, Adv. Mater., 2019, 31, 1901996 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- H. Y. Jin, C. X. Guo, X. Liu, J. L. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- H. Hu, B. Y. Guan and X. W. Lou, Chem, 2016, 1, 102–113 CAS.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- S. Ding, M. J. Hulsey, J. Perez-Ramirez and N. Yang, Joule, 2019, 3, 2897–2929 CrossRef CAS.
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- S. Lee, L. Bai and X. Hu, Angew. Chem., Int. Ed., 2020, 59, 8072–8077 CrossRef CAS PubMed.
- S. Nayak, I. J. McPherson and K. A. Vincent, Angew. Chem., Int. Ed., 2018, 57, 12855–12858 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details are provided. See DOI: 10.1039/d1ta09729f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |