
Hybrid palladium nanoparticles and nickel single atom catalysts for efficient electrocatalytic ethanol oxidation†
Si
Li
a,
Anxiang
Guan
a,
Huining
Wang
a,
Yaqin
Yan
a,
Haoliang
Huang
b,
Chao
Jing
b,
Lijuan
Zhang
*a,
Linjuan
Zhang
*b and
Gengfeng
Zheng
 *a
*a
aLaboratory of Advanced Materials, Department of Chemistry, Shanghai Key Laboratory of Molecular Catalysis & Innovative Materials, Faculty of Chemistry and Materials Science, Fudan University, Shanghai 200438, China. E-mail: zhanglijuan@fudan.edu.cn; gfzheng@fudan.edu.cn
bKey Laboratory of Interfacial Physics and Technology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China. E-mail: zhanglinjuan@sinap.ac.cn
First published on 15th November 2021
Abstract
The electrocatalytic ethanol oxidation reaction (EOR) is a critical component of direct ethanol fuel cells, while its reactivity, stability, and selectivity toward C1 products are severely hindered by the poisoning effect of CO species on the surface of noble-metal catalysts. In this work, we developed a hybrid material of palladium nanoparticles and nickel single-atom catalysts (Pd NPs@Ni SAC) for efficient EOR. The Pd nanoparticles catalyze the breaking of carbon–carbon bonds of ethanol, while the adsorbed CO intermediates are removed by the Ni single-atom catalyst. The Pd NPs@Ni SAC structure exhibits outstanding EOR performances, including high mass activity, good selectivities of C1 products, and excellent electrocatalytic stability. Our work suggests an attractive perspective of utilizing single-atom electrocatalysts for ethanol oxidation.
Introduction
Direct ethanol fuel cells (DEFCs) are of great potential for energy storage and conversion, as ethanol possesses the characteristics of low cost, low toxicity, and high volumetric energy density.1,2 The ethanol oxidation reaction (EOR) can involve the C2 pathway of the incomplete oxidation process by transferring 2 or 4 electrons to generate acetaldehyde or acetic acid,3 or the C1 pathway of full oxidation by transferring 12 electrons to form carbon dioxide (CO2).4 The C1 pathway toward CO2 has the potential of realizing low overpotentials and large energy densities, while the selectivity of the C1 pathway remains low even for the best-reported noble-metal-based catalysts such as Pt or Pd.5 Moreover, the CO intermediates generated during the EOR easily poison the noble metal catalyst surface, resulting in poor electrochemical activity and durability.6,7Two strategies have generally been applied to promote the selectivity and durability of the C1 pathway on Pt- or Pd-based catalysts. One is to reduce the adsorption affinity of CO intermediates on the Pt or Pd surface by forming alloys with other metals, such as Au,8 Ag,9 Cu,10 Sn,11 and so on. Alternatively, it is also possible to eliminate the adsorbed CO intermediates by introducing hydroxide or oxide components, such as Bi(OH)3,12 Ni(OH)2,13 SnO2,6,14 and CuOx.15 An optimal catalyst to promote the C1 pathway should enable efficient breaking of C–C bonds and removal of CO intermediates.16,17 Recently, single-atom catalysts (SACs) have received extensive attention in catalysis due to their unique reaction sites and high reactivity.18–22 Among them, Ni SACs have been reported to provide abundantly adsorbed OH (OHad) species.23,24 The OHad species have been suggested to facilitate oxidative removal of CO species on noble metal (Pd or Pt) sites through the Langmuir–Hinshelwood mechanism and improve the EOR stability.12,13
Herein, we developed a hybrid material composed of Pd nanoparticles and Ni SACs (denoted as Pd NPs@Ni SAC), where Pd nanoparticles were uniformly dispersed on the carbon framework-supported Ni SACs (Fig. 1). The Pd nanoparticles served as the main EOR catalytic sites to break the C–C bonds of ethanol and promote the C1 oxidation pathway. Meanwhile, the produced CO species adsorbed on the Pd surface were electrochemically oxidized by Ni SACs in the alkaline electrolyte. The Pd NPs@Ni SAC exhibited an outstanding EOR performance with a peak mass activity of 1093 mA mgPd−1, good selectivity of the C1 pathway (28%), and excellent electrocatalytic stability, substantially exceeding those of Pd nanoparticles with Cu SACs or pure carbon substrates, thus suggesting the unique structures of Pd NPs@Ni SAC for efficient EOR activity.
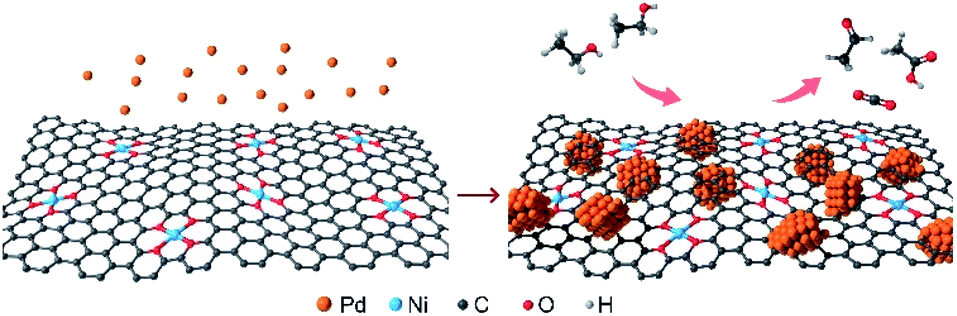 | ||
| Fig. 1 Schematic illustration of the preparation process of Pd NPs@Ni SAC (left panel), and the electrocatalytic oxidation of ethanol on Pd NPs@Ni SAC (right panel). | ||
Experimental section
Synthesis of the Ni SAC, Cu SAC, and carbon substrate
In a typical synthesis, 300 mg of sucrose and 12 mg of nickel(II) chloride hexahydrate were dispersed in 10 mL of deionized (DI) water, and 11 μL of sulfuric acid (H2SO4) was injected into the solution. Next, 3 g of sodium chloride (NaCl) was added with stirring. The mixture was transferred to a plastic container and frozen rapidly in liquid nitrogen. After being dried in a freeze dryer overnight, the obtained sample was annealed at 100 °C for 5 h to ensure the complete removal of any residual water. Afterwards, the sample was transferred into a quartz tube and annealed at 700 °C for 120 min with a heating rate of 5 °C min−1 under an Ar atmosphere. Finally, the Ni SAC was obtained by centrifugation and washed with DI water several times after annealing. The Cu SAC was also synthesized under similar conditions, except for replacing nickel(II) chloride hexahydrate with 11 mg of copper(II) chloride dihydrate. In comparison, the synthesis of the carbon substrate did not require any metal salts.Synthesis of Pd NPs@Ni SAC, Pd NPs@Cu SAC and Pd NPs@C
In brief, 15 mg of Ni SAC and 25 mg of palladium(II) chloride were dispersed in 50 mL of DI water, and sonicated until evenly dispersed. Then, 17 mg of sodium borohydride (NaBH4) was dissolved in 20 mL of DI water, and the solution was added dropwise to the above mixture with stirring. After stirring for 30 min, the Pd NPs@Ni SAC was collected by centrifugation and washed with DI water to remove excess NaBH4. The Pd NPs@Cu SAC and Pd NPs@C were synthesized by the same method by replacing Ni SAC with Cu SAC and the carbon substrate, respectively.Results and discussion
The Pd NPs@Ni SAC and Pd NPs@Cu SAC were synthesized through a two-step method. First, Ni SAC or Cu SAC was prepared by freeze-drying followed by thermal annealing. Next, Pd nanoparticles were formed on Ni SAC or Cu SAC by in situ reduction of palladium salt with sodium borohydride (Experimental section). The X-ray diffraction (XRD) patterns of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C (i.e., without Ni or Cu content) showed the same diffraction patterns with diffraction peaks at 40.1°, 46.7°, 68.1°, 82.1°, and 86.6° (Fig. 2a), corresponding to the (111), (200), (220), (311), and (222) crystal planes of Pd (PDF 46-1043). In comparison, when no Pd nanoparticles were loaded, the XRD patterns of the Ni SAC, Cu SAC, and carbon substrate only showed a broad peak centered at 26.4° (Fig. S1†), assigned to graphitic carbon.25 Meanwhile, no diffraction peaks of Ni or Cu metal were observed, suggesting that the Ni or Cu atoms were uniformly dispersed in the form of single atom metal centers on the carbon substrate.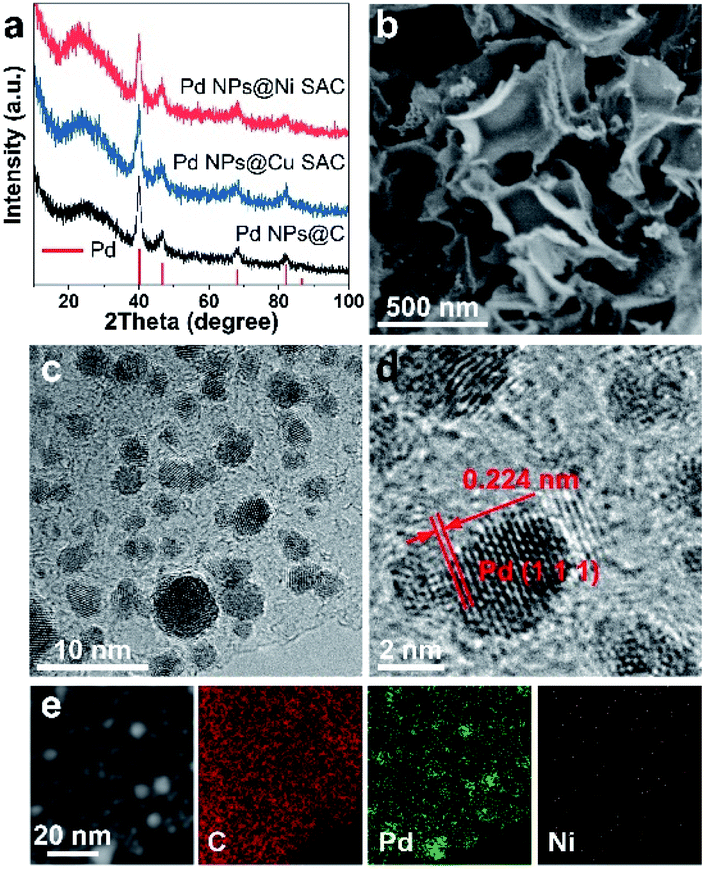 | ||
| Fig. 2 Structural characterization. (a) XRD patterns of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C. (b) SEM images, (c and d) TEM images, and (e) STEM images and corresponding elemental maps of Pd NPs@Ni SAC. | ||
Scanning electron microscopy (SEM) of all the samples showed that the carbon substrates were constructed from 2D ultrathin graphene-like nanosheets to form interconnected frameworks (Fig. 2b and S2†). Transmission electron microscopy (TEM) showed that Pd nanoparticles with an average size of 2 to 5 nm were evenly dispersed on the carbon substrate (Fig. 2c and S3†). High-resolution TEM images of the Pd nanoparticles on Ni SAC showed lattice fringes of the (111) planes with a distance of 0.224 nm (Fig. 2d), indicating the high crystallinity. The scanning transmission electron microscopy (STEM) image and corresponding elemental maps showed the uniform and isolated distribution of Ni or Cu elements in the Pd NPs@Ni SAC, Ni SAC, and Pd NPs@Cu SAC, respectively (Fig. 2e and S4†). The percentages of Pd, Ni, and Cu contents in these samples were quantified by inductively coupled plasma measurements (Table S1†).
X-ray photoelectron spectroscopy (XPS) was conducted to probe the chemical compositions of all samples and the chemical states of metal elements. The overall survey scans confirmed the existence of Pd, Ni, Cu, O, and C elements (Fig. S5, Tables S2 and S3†). The Pd 3d XPS spectra of the Pd NPs@Ni SAC exhibited a 3d5/2 peak at 334.9 eV and 3d3/2 peak at 340.2 eV (Fig. 3a).26,27 The Pd 3d5/2 XPS spectra was split into two peaks at 334.9 and 340.2 eV corresponding to the metallic Pd species, and the Pd 3d3/2 peaks at 336.6 and 341.9 eV were ascribed to the Pd(II) species. The different chemical states of Pd in the Pd NPs@Ni SAC suggested the existence of an oxide layer on the surface of Pd nanoparticles.28 Moreover, the Ni 2p XPS spectra of Pd NPs@Ni SAC were deconvoluted into three peaks at 855.8, 859.6, and 862.5 eV, respectively (Fig. 3b).23 The peaks at 855.8 and 859.6 eV were attributed to the Ni species higher than Ni(II).29,30 Due to the higher valence state, the metal atoms centered in the Ni SAC preferred to form the Ni–OH and Ni–O bonds. The C 1s, Pd 3d, Ni 2p, Cu 2p, and O 1s XPS spectra of other samples are provided in Fig. S6–S8.† From XPS measurements, the contents of Pd in Pd NPs@Ni SAC, Pd NPs@Cu SAC and Pd NPs@C were 25.2, 26.2, and 27.5 wt%, respectively. The contents were utilized to normalize the current density, as they indicated the element percentages within a few nanometers of the catalyst surface where the electrocatalysis occurred.
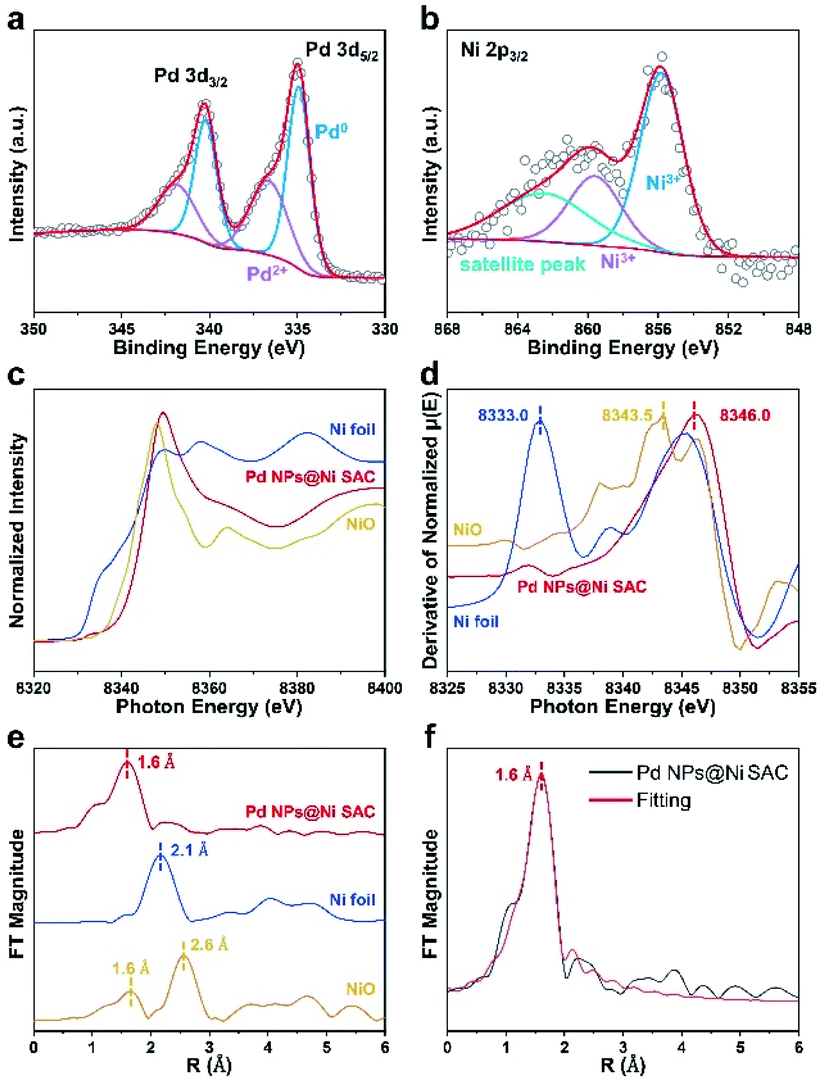 | ||
| Fig. 3 XPS and synchrotron radiation X-ray absorption fine structure spectra of Pd NPs@Ni SAC. (a) Pd 3d and (b) Ni 2p XPS spectra of Pd NPs@Ni SAC. (c) XANES spectra, (d) first derivatives of the XANES region, and (e) Fourier-transform of Ni K-edge EXAFS spectra of the NiO, Pd NPs@Ni SAC, and Ni foil. (f) Ni K-edge EXAFS fitting results of the Pd NPs@Ni SAC. | ||
The synchrotron radiation X-ray absorption near-edge structure (XANES) spectroscopy was conducted to further investigate the atomic bonding in the Ni SAC structures. The energy of the absorption edges became higher from Ni foil and NiO, to Pd NPs@Ni SAC (Fig. 3c). Meanwhile, the characteristic peaks of Ni foil, NiO, and Pd NPs@Ni SAC in the first derivative spectra were positioned at 8333.0, 8343.5, and 8346.0 eV, respectively (Fig. 3d), implying that the valence of Ni species in Pd NPs@Ni SAC was higher than that of Ni(II), consistent with the XPS analysis results.31,32 As elucidated by the Fourier-transform (FT) k2-weighted extended X-ray absorption fine structure (FT-EXAFS) spectroscopy of Ni foil, NiO and Pd NPs@Ni SAC (Fig. 3e), there was only one peak at ∼1.6 Å in Pd NPs@Ni SAC that was attributed to the scattering of the Ni–O interaction. Compared with the peaks at ∼1.6 and 2.6 Å observed in NiO, there was only one peak at ∼2.1 Å in Ni foil, confirming that the Ni atoms in Pd NPs@Ni SAC were atomically dispersed by forming Ni–O coordination. In addition, the EXAFS fitting curves showed that the Ni atoms in Pd NPs@Ni SAC were coordinated with O atoms with a bond length of 1.6 Å (Fig. 3f). The structural parameters of Pd NPs@Ni SAC obtained from EXAFS fittings are given in Table S4.†
The electrocatalytic EOR performances of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C were evaluated in the three-electrode system with 1 M KOH and 1 M ethanol as the electrolyte (Methods section in the ESI†). All the applied potentials reported in this work were converted into the reversible hydrogen electrode (RHE) scale. Cyclic voltammetry (CV) measurements of the Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C samples were first conducted between 0.04 and 1.19 V (Fig. 4a). In all the curves, the peaks in the forward scan from 0.84 to 1.04 V were attributed to the oxidation of ethanol to several intermediate products, while the peaks in the backward scan from 0.84 to 0.64 V were assigned to the oxidation of the intermediate products.33 The maximum current density of Pd NPs@Ni SAC reached 1093 mA mgPd−1 at 0.92 V, suggesting its high EOR reactivity, while the peak current densities of Pd NPs@Cu SAC and Pd NPs@C were 965 and 787 mA mgPd−1, respectively.
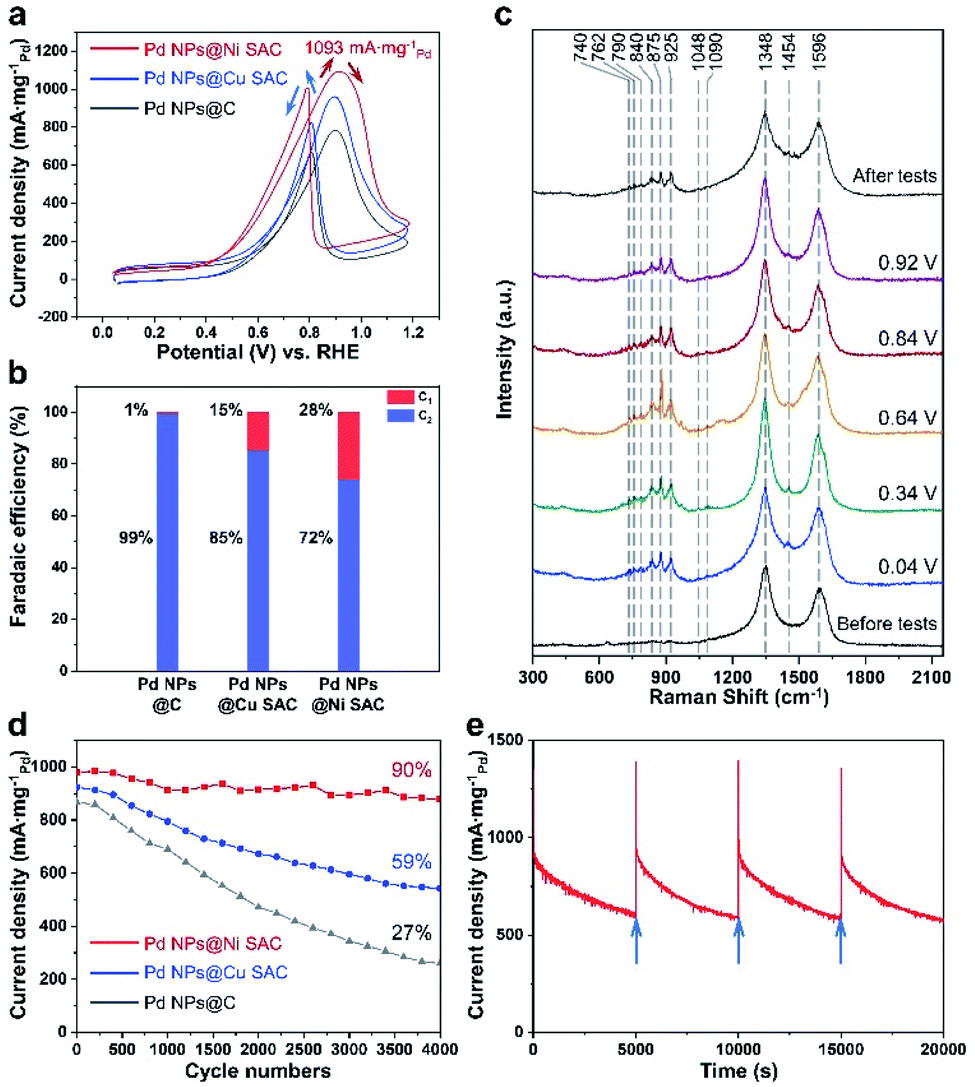 | ||
| Fig. 4 Electrocatalytic EOR measurements. (a) CV curves of Pd NPs@Ni SAC, Pd NPs@Cu SAC and Pd NPs@C at 50 mV s−1 in 1 M KOH with 1 M ethanol. (b) Faradaic efficiencies of C1 and C2 products. (c) Electrochemical Raman spectra of Pd NPs@Ni SAC. (d) Cycling stability of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C. Data were constructed by periodically extracting the anodic current at 0.92 V during continuous cycling. (e) Long-term durability of Pd NPs@Ni SAC. The arrows indicate when the catalyst was reactivated and electrolyte was replaced. | ||
The liquid products after the chronoamperometric EOR tests were detected by 1H NMR to study the different selectivities of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C (Fig. S9†). The representative data of nuclear magnetic resonance spectra are shown in Fig. S10.† During the chronoamperometric tests, the initial current density (at time = 0 s) reached higher than 1000 mA mgPd−1, and then became stable with the progress of the EOR. The calculated faradaic efficiencies (FE) of C2 and C1 products are shown in Fig. 4b. At the applied potential of 0.92 V, the C1 product selectivity on the Pd NPs@Ni SAC reached 28%, higher than those of the Pd NPs@Cu SAC (15%) and Pd NPs@C (1%), suggesting the better anti-toxicity of the Pd NPs@Ni SAC.
Electrochemical Raman spectroscopy was conducted to further investigate the EOR process. The characteristic bands of ethanol, acetaldehyde, acetic acid, and carbon were observed in the Raman spectra under different applied voltages (Fig. 4c and S11†). Specifically, bands at 1348 and 1596 cm−1 were related to the D and G bands of carbon,34 which were also observed in all the spectra with applied potentials. During the electrochemical tests, several new peaks were observed at a potential as low as 0.04 V. For the products of the EOR, the bands located at 740, 762, 790, and 840 cm−1 were attributed to the C–H vibrations of acetaldehyde, while the peak at 925 cm−1 was assigned to the CH3 band of acetic acid.35 Moreover, the peaks at 875 and 1454 cm−1 were assigned to the characteristic νs(C–C–O) and δ(C–O–H) bands of ethanol.36 The νa(C–C–O) and γ(C–O–H) bands were also found at 1048 and 1090 cm−1.37
The polarization curves and the Levich plot (Fig. S12†) of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C with a rotating disk electrode at 100 mV s−1 in 1 M KOH indicated that Pd NPs@Ni SAC had a higher electron transfer number for the EOR, consistent with the C1 selectivity under the test conditions. Furthermore, the CO stripping and the electrochemically active surface area (ECSA) measurements of Pd NPs@Ni SAC, Pd NPs@Cu SAC, and Pd NPs@C were displayed (Fig. S13†). The potential of scavenging CO was more negative on Pd NPs@Ni SAC than the other two samples, also suggesting the inhibition of CO poisoning on Pd NPs@Ni SAC.13 From the CV curves, the ECSA was calculated to be 85 m2 g−1 for Pd NPs@Ni SAC. In addition, the electrochemical impedance spectroscopy (EIS, Fig. S14†) measurements suggested that the similar electrochemical impedance would not be the main factor to affect its EOR performance.
The durability of Pd NPs@Ni SAC was investigated by conducting cycling stability and long-term chronoamperometric measurements. After 4000 cycles of cyclic voltammetry measurements, the retention was 90% of the initial current density on Pd NPs@Ni SAC (Fig. 4d), while the retention for Pd NPs@Cu SAC and Pd NPs@C was 59% and 27%, respectively. Furthermore, Pd NPs@Ni SAC exhibited an outstanding electrocatalytic durability by chronoamperometric tests, with periodic reactivation by performing several CV cycles (Fig. 4e). After about 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, the current was still maintained at 50% of the initial value. Compared to previous literature studies including the Pd-based and other noble metal-based catalysts,13,38–43 our Pd NPs@Ni SAC exhibited not only a high current density, but also a much higher electrochemical stability (Fig. S15†).
000 s, the current was still maintained at 50% of the initial value. Compared to previous literature studies including the Pd-based and other noble metal-based catalysts,13,38–43 our Pd NPs@Ni SAC exhibited not only a high current density, but also a much higher electrochemical stability (Fig. S15†).
Conclusions
In summary, we successfully developed a hybrid material of Pd nanoparticles and Ni SACs, proving to be an efficient EOR catalyst. The Pd nanoparticles served as active sites for breaking the C–C bonds, and the Ni single atom catalyst functioned as a substrate to promote elimination of CO intermediates. The Pd NPs@Ni SAC presented an outstanding EOR performance, including improved selectivity of the C1 pathway, excellent activity, and one of the highest electrochemical stabilities. Our work suggests an attractive route of combining noble-metal nanoparticles and single atom catalysts toward efficient electrochemical ethanol oxidation.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflicting interest.Acknowledgements
We thank the following funding agencies for supporting this work: the National Key Research and Development Program of China (2018YFA0209401 and 2017YFA0206901), the National Science Foundation of China (22025502, 21975051, and 21773036), the Science and Technology Commission of Shanghai Municipality (21DZ1206800 and 19XD1420400), the Shanghai Municipal Education Commission (2019-01-07-00-07-E00045), the “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences (XDA2100000), the K. C. Wong Education Foundation (GJTD-2018-10), the Youth Innovation Promotion Association, Chinese Academy of Science (Y201842), the Instrument and Equipment Development Program Chinese Academy of Science (YJKYYQ20180066), and the DNL Cooperation Fund, CAS (DNL202008). We acknowledge the support from the group of BL14W1 beamline in Shanghai Synchrotron Radiation Facility (SSRF) for the XAFS measurements.References
- J. Bai, D. Liu, J. Yang and Y. Chen, ChemSusChem, 2019, 12, 2117–2132 CrossRef CAS.
- Z. Zhang, Q. Wu, K. Mao, Y. Chen, L. Du, Y. Bu, O. Zhuo, L. Yang, X. Wang and Z. Hu, ACS Catal., 2018, 8, 8477–8483 CrossRef CAS.
- I. Kim, O. H. Han, S. A. Chae, Y. Paik, S. H. Kwon, K. S. Lee, Y. E. Sung and H. Kim, Angew. Chem., Int. Ed., 2011, 50, 2270–2274 CrossRef CAS PubMed.
- K. L. Hohn and Y. C. Lin, ChemSusChem, 2009, 2, 927–940 CrossRef CAS PubMed.
- L. X. Dai, X. Y. Wang, S. S. Yang, T. Zhang, P. J. Ren, J. Y. Ye, B. Nan, X. D. Wen, Z. Y. Zhou, R. Si, C. H. Yan and Y. W. Zhang, J. Mater. Chem. A, 2018, 6, 11270–11280 RSC.
- A. Kowal, M. Li, M. Shao, K. Sasaki, M. B. Vukmirovic, J. Zhang, N. S. Marinkovic, P. Liu, A. I. Frenkel and R. R. Adzic, Nat. Mater., 2009, 8, 325–330 CrossRef CAS PubMed.
- W. Du, G. Yang, E. Wong, N. A. Deskins, A. I. Frenkel, D. Su and X. Teng, J. Am. Chem. Soc., 2014, 136, 10862–10865 CrossRef CAS.
- H. Lin, M. Muzzio, K. Wei, P. Zhang, J. Li, N. Li, Z. Yin, D. Su and S. Sun, ACS Appl. Energy Mater., 2019, 2, 8701–8706 CrossRef CAS.
- W. Huang, X. Kang, C. Xu, J. Zhou, J. Deng, Y. Li and S. Cheng, Adv. Mater., 2018, 30, 1706962 CrossRef PubMed.
- J. Xue, G. T. Han, W. N. Ye, Y. T. Sang, H. L. Li, P. Z. Guo and X. S. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 34497–34505 CrossRef CAS.
- X. Yu, J. Liu, J. Li, Z. Luo, Y. Zuo, C. Xing, J. Llorca, D. Nasiou, J. Arbiol, K. Pan, T. Kleinhanns, Y. Xie and A. Cabot, Nano Energy, 2020, 77, 105116 CrossRef CAS.
- X. Yuan, Y. Zhang, M. Cao, T. Zhou, X. Jiang, J. Chen, F. Lyu, Y. Xu, J. Luo, Q. Zhang and Y. Yin, Nano Lett., 2019, 19, 4752–4759 CrossRef CAS PubMed.
- W. Huang, X. Y. Ma, H. Wang, R. Feng, J. Zhou, P. N. Duchesne, P. Zhang, F. Chen, N. Han, F. Zhao, J. Zhou, W. B. Cai and Y. Li, Adv. Mater., 2017, 29, 1703057 CrossRef PubMed.
- S. Bai, Y. Xu, K. Cao and X. Huang, Adv. Mater., 2021, 33, 2005767 CrossRef CAS.
- Z. Chen, Y. Liu, C. Liu, J. Zhang, Y. Chen, W. Hu and Y. Deng, Small, 2020, 16, 1904964 CrossRef CAS.
- Y. Wang, S. Z. Zou and W. B. Cai, Catalysts, 2015, 5, 1507–1534 CrossRef CAS.
- H. A. Asiri and A. B. Anderson, J. Electrochem. Soc., 2014, 162, F115–F122 CrossRef.
- L. L. Li, X. Chang, X. Y. Lin, Z. J. Zhao and J. L. Gong, Chem. Soc. Rev., 2020, 49, 8156–8178 RSC.
- W. H. Lee, Y. J. Ko, J. Y. Kim, B. K. Min, Y. J. Hwang and H. S. Oh, Chem. Commun., 2020, 56, 12687–12697 RSC.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X. A. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- Y. S. Wu, Z. Jiang, X. Lu, Y. Y. Liang and H. L. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- Y. J. Chen, S. F. Ji, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- H. L. Fei, J. C. Dong, Y. X. Feng, C. S. Allen, C. Z. Wan, B. Volosskiy, M. F. Li, Z. P. Zhao, Y. L. Wang, H. T. Sun, P. F. An, W. X. Chen, Z. Y. Guo, C. Lee, D. L. Chen, I. Shakir, M. J. Liu, T. D. Hu, Y. D. Li, A. I. Kirkland, X. F. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- Y. Li, Z. S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS.
- H. B. Yang, S. F. Hung, S. Liu, K. D. Yuan, S. Miao, L. P. Zhang, X. Huang, H. Y. Wang, W. Z. Cai, R. Chen, J. J. Gao, X. F. Yang, W. Chen, Y. Q. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- X. L. Xing, Y. F. Zhao, H. Li, C. T. Wang, Q. X. Li and W. B. Cai, J. Electrochem. Soc., 2018, 165, J3259–J3265 CrossRef CAS.
- R. K. Singh, R. Rahul and M. Neergat, Phys. Chem. Chem. Phys., 2013, 15, 13044–13051 RSC.
- K. Fujiwara, U. Müller and S. E. Pratsinis, ACS Catal., 2016, 6, 1887–1893 CrossRef CAS.
- C. C. Yan, H. B. Li, Y. F. Ye, H. H. Wu, F. Cai, R. Si, J. P. Xiao, S. Miao, S. H. Xie, F. Yang, Y. S. Li, G. X. Wang and X. H. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS.
- Y. Q. Xu, W. F. Zhang, Y. G. Li, P. F. Lu, Y. Wang and Z. S. Wu, Front. Mater., 2019, 6, 271 CrossRef.
- M. Zhou, Y. Jiang, G. Wang, W. Wu, W. Chen, P. Yu, Y. Lin, J. Mao and L. Mao, Nat. Commun., 2020, 11, 3188 CrossRef CAS.
- C. W. Xu, H. Wang, P. K. Shen and S. P. Jiang, Adv. Mater., 2007, 19, 4256–4259 CrossRef CAS.
- R. Vie, E. Drahi, O. Baudino, S. Blayac and S. Berthon-Fabry, Flex. Print. Electron., 2016, 1, 015003 CrossRef.
- A. Gómez-Monsiváis, I. Velázquez-Hernández, L. Álvarez-Contreras, M. Guerra-Balcázar, L. Arriaga, N. Arjona and J. Ledesma-García, Energies, 2017, 10, 290 CrossRef.
- S. C. S. Lai and M. T. M. Koper, Phys. Chem. Chem. Phys., 2009, 11, 10446–10456 RSC.
- R. F. B. De Souza, E. T. Neto, M. L. Calegaro, E. A. Santos, H. S. Martinho and M. C. dos Santos, Electrocatalysis, 2011, 2, 28–34 CrossRef.
- F. Lv, W. Zhang, M. Sun, F. Lin, T. Wu, P. Zhou, W. Yang, P. Gao, B. Huang and S. Guo, Adv. Energy Mater., 2021, 11, 2100187 CrossRef CAS.
- X. Yang, Z. Liang, S. Chen, M. Ma, Q. Wang, X. Tong, Q. Zhang, J. Ye, L. Gu and N. Yang, Small, 2020, 16, 2004727 CrossRef CAS.
- Q. Gao, T. Mou, S. Liu, G. Johnson, X. Han, Z. Yan, M. Ji, Q. He, S. Zhang, H. Xin and H. Zhu, J. Mater. Chem. A, 2020, 8, 20931–20938 RSC.
- S. He, Y. Liu, H. Li, Q. Wu, D. Ma, D. Gao, J. Bi, Y. Yang and C. Cui, ACS Appl. Mater. Interfaces, 2021, 13, 13311–13318 CrossRef CAS.
- C. V. S. Almeida, K. I. B. Eguiluz and G. R. Salazar-Banda, J. Electroanal. Chem., 2020, 878, 114683 CrossRef CAS.
- M. D. Obradovic, Z. M. Stancic, U. C. Lacnjevac, V. V. Radmilovic, A. Gavrilovic-Wohlmuther, V. R. Radmilovic and S. L. Gojkvic, Appl. Catal., B, 2016, 189, 110–118 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08518b |
| This journal is © The Royal Society of Chemistry 2022 |