
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of A-site composition in the photostability of tin–lead perovskite solar cells†
Luis
Huerta Hernandez
 ,
Md Azimul
Haque
,
Anirudh
Sharma
,
Luis
Lanzetta
,
Jules
Bertrandie
,
Aren
Yazmaciyan
,
Joel
Troughton
and
Derya
Baran
*
,
Md Azimul
Haque
,
Anirudh
Sharma
,
Luis
Lanzetta
,
Jules
Bertrandie
,
Aren
Yazmaciyan
,
Joel
Troughton
and
Derya
Baran
*
Physical Sciences and Engineering Division (PSE), KAUST Solar Center (KSC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: derya.baran@kaust.edu.sa
First published on 5th September 2022
Abstract
The efficiency of tin–lead perovskite solar cells (TLPSC) has been consistently increasing. However, their photostability continues to be a persistent challenge. Besides the oxidation of tin (Sn), the presence of methylammonium (MA) has been correlated with poor device photostability, which still remains unclear. In this work, we unravel the influence of the A-site cation choice, MA vs. cesium (Cs), on the photostability and performance of TLPSCs where Cs0.25−xMAxFA0.75Sn0.5Pb0.5I3 (FA: formamidinium; x = 0.0–0.25) is used as a photoactive layer. We observe that the addition of MA (i.e. increasing x) improves the device efficiency while reducing the photostability. This reduction is attributed to light-induced interfacial barriers, surface recombination, and ion diffusion. We found that MA volatilizes from the surface of MA-rich devices during illumination, leading to an irreversible performance loss after light exposure. We further demonstrate that MA content rather than the choice of the hole-transport layer (PEDOT:PSS vs. NiOx) dominates the device photostability. Under continuous 1 sun illumination, devices without MA and with the use of NiOx as a transport layer are able to retain 80% of their efficiency for 690 h, while MA-rich cells retain 80% of their efficiency only for the first 3 h of operation. Our work provides insights into the development of photostable TLPSC.
Introduction
In the last decade, tin–lead perovskite solar cells (TLPSCs) have witnessed a continuous and rapid development with state-of-the-art devices achieving efficiencies above 20%.1–6 In contrast to pure lead (Pb) or tin (Sn) perovskites, their combination results in a narrower bandgap that is defined according to the Sn:Pb ratio. For bandgaps between 1.2 and 1.3 eV, the common Sn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb ratio is found to be around 0.4:0.6 and 0.7:0.3.7,8 This is advantageous as TLPSCs possess a higher Shockley–Queisser limit than Pb based perovskites. They are promising for bottom cells in all-perovskite tandem solar cells, where efficiencies above 30% are predicted.9–12 Additionally, the lower content of Pb decreases the environmental toxicity of perovskite solar cells (PSCs).13 The Sn–Pb systems are more stable when compared to pure Sn perovskites because of the reduced Sn content and the stabilizing effect of Pb substitution in the lattice.14
:Pb ratio is found to be around 0.4:0.6 and 0.7:0.3.7,8 This is advantageous as TLPSCs possess a higher Shockley–Queisser limit than Pb based perovskites. They are promising for bottom cells in all-perovskite tandem solar cells, where efficiencies above 30% are predicted.9–12 Additionally, the lower content of Pb decreases the environmental toxicity of perovskite solar cells (PSCs).13 The Sn–Pb systems are more stable when compared to pure Sn perovskites because of the reduced Sn content and the stabilizing effect of Pb substitution in the lattice.14
The long-term stability of TLPSCs under operating conditions has been one of the major bottlenecks for further development of this technology. The main contributors include the oxidation of Sn, unstable hole-transport layer (HTL), and the presence of methylammonium (MA) content in the perovskite. The most typical explanation for the limited stability is the easy oxidation of Sn2+ to Sn4+. Initially, the Sn oxidation results in a self-doping effect that increases the electrical conductivity, reduces the diffusion length, and favors non-radiative recombination.15–17 On a longer time scale, this can trigger the formation of different byproducts that hinder device efficiency.18–20 The most common strategy to overcome this limitation consists of employing different additives or dopants, which help preventing the oxidation of Sn2+ or reduce the Sn4+ already present in the system.9,21,22 Likewise, the selection of a stable HTL has become increasingly more important. The usual architecture for TLPSCs is the p–i–n configuration, where PEDOT:PSS is the most common HTL so far. However, it has been shown that under operating conditions, the interface between PEDOT:PSS and the perovskite presents signs of degradation.23 Different approaches to minimize this issue include the removal of the HTL23–25 or the use of other HTLs such as NiOx or PTAA.26–29
Another critical aspect of the device stability is the selection of the A-site cation, namely Cs, MA, and formamidinium (FA). Devices based on the typical MAFA double cation or the CsMAFA triple cation compositions have consistently retained >95% of their original PCE after 1000 hours under dark and inert conditions.4,30–34 However, upon light illumination, efficiency losses of around 10% already exist after 450 h under 1 sun.6 In contrast, MA-free devices based on the CsFA mixture maintained their initial efficiency even after 1000 h of operation under 1 sun at 85 °C.23 Further work has also shown that CsFA devices remain stable for 4000 h at 85 °C.28 Hence, MA plays a critical role in the photostability or the thermal stability, which has also been observed for Pb and Sn-only compositions.35,36 Although this is commonly attributed to the highly volatile nature of MA,37,38 this issue has not been thoroughly studied in TLPSCs. Therefore, a more in-depth understanding of the role of cation compositions in performance and photodegradation is needed to enable the rational design of stable TLPSCs.
In this work, we investigate the role of MA in the efficiency and photostability of TLPSCs. To explore the effect of MA, we choose the perovskite composition Cs0.25−xMAxFA0.75Sn0.5Pb0.5I3 where the content of MA is given by x = 0, 0.10, 0.20, and 0.25. The addition of MA improves the overall performance of the cells; however, it results in a sharp and permanent decrease in the efficiency during the first hour of 1 sun illumination. To understand this rapid and irreversible decay in performance, we carried out light-intensity dependent measurements and impedance spectroscopy. We found that at higher contents of MA (x = 0.25), the devices suffered from increased surface recombination, and high mobile ion density after light exposure. Moreover, X-ray and UV photoemission experiments present evidence of MA volatilization at the surface of the x = 0.25 films after continuous illumination, which is correlated to the creation of extraction barriers at the perovskite and electron transport layer (ETL) interface. To further understand what is the limiting factor in the photostability of TLPSCs, we tested devices with and without MA by using two different hole transport layers (HTLs), PEDOT:PSS and NiOx. Devices with only MA (x = 0.25) exhibited a performance drop higher than 30% after 15 hours under illumination, regardless of the HTL, whereas MA-free devices (x = 0) retained 80% of the original PCE after 550 h and 690 h for PEDOT:PSS and NiOx, respectively. In comparison to the surface oxidation of Sn or HTL selection, our results confirm that MA content is the most critical factor for device photostability in TLPSCs.
Experimental section
Materials
CsI (dry beads 99.999%), PbI2 (dry beads, 99.999%), SnI2 (99.99%), SnF2, bathocuproine (BCP), DMSO, and DMF (both anhydrous) were purchased from Sigma Aldrich. MAI and FAI were provided by Greatcell and TCI, respectively. C60 was supplied by nanoC. NiOx nanoparticle (7 nm) solution in ethanol was provided by Avantama. PEDOT:PSS AI 4083 was obtained from Heraeus.Device fabrication
Pre-patterned ITO/glass substrates (Xinyan Technology, 10/15 Ω sq−1, CN) were sequentially sonicated in a Hellmanex aqueous solution, deionized water, acetone, and isopropanol for 10 min each. Substrates were dried with N2 and treated with UV ozone for 15 min. The NiOx nanoparticle solution was spin coated at a speed of 4000 rpm for 30 s, followed by annealing at 300 °C for 30 min; the thickness of the NiOx measured by atomic force microscopy was ∼5 nm. For the devices that utilized PEDOT:PSS as the HTL, a solution of PEDOT and isopropanol (1:1) was coated on the clean ITO at 4000 rpm for 30 s and then annealed at 150 °C for 10 min. After HTL deposition, the samples were transferred to a N2 glovebox. The perovskite precursor solution was prepared by dissolving the precursor salts of CsI, MAI, FAI, PbI2, SnI2 and SnF2 (10 mol% with respect to SnI2) in DMF:DMSO (3:1) according to the formula Cs0.25−xMAxFA0.75Sn0.5Pb0.5I3; the intended concentration was 1.6 M, and the solution was stirred for 1 h at room temperature. A two-step spin-coating procedure of 1000 rpm for 10 s and 4000 rpm for 40 s was used for the deposition of the perovskite films. Chlorobenzene (100 μl) is added at the 20th s of the 2nd step. After this, the samples were annealed at 100 °C for 10 min. Subsequently, C60 (30 nm) and BCP (8 nm) were thermally evaporated at a rate of 0.1 A s−1 under high vacuum by using an Angstrom Engineering system. Finally, 100 nm of Ag was thermally evaporated to obtain devices with an area of 0.1 cm2.
Device characterization
A LED solar simulator (WaveLabs Sinus-70, Greater than AAA rated) calibrated to ±5% of AM1.5G between 350 and 1100 nm was used to obtain current–voltage curves and for the light-soaking experiments. For light intensity dependent measurements, several neutral-density filters from Thorlabs were used to attenuate the simulated sunlight. To record the J–V response, a Keithley 2400-series source-measure unit was used with a scan speed of 0.15 V s−1. For the short-term stability measurements, a J–V curve was recorded each 10 min while devices were left under illumination (AM1.5G) and open-circuit conditions. For the entire duration of the measurement, devices where in a N2 glovebox (<0.1 ppm). For the long-term photostability measurements, solar cells were placed inside an environmental chamber and purged with a constant flow of nitrogen. This chamber was placed under a bank of metal halide lamps (Osram Powerstar HQI, 900 W) with a total intensity of approximately 1 sun. A thermoelectric chiller was used to cool the cells to a temperature of 40 °C throughout the light soaking experiment. Each solar cell was connected to an individual source-measure unit (based on the open-source “μSMU” design available at https://certification.oshwa.org/sa000002.html) calibrated against a Keithley 2400. Current–voltage curves were measured at 10 minute intervals from Jsc to Voc, with each device held at its maximum power point in-between measurements. Impedance spectroscopy measurements were performed with a Fluxim Paios characterization suite. The impedance signal was recorded in the 0.1 Hz to 10 MHz frequency range with a 10 mV amplitude voltage inside a N2 glovebox in the dark.Film characterization
SEM pictures were taken with a Zeiss Auriga microscope. UV-Vis measurements were carried out with a Perkin Lambda 850 UV/Vis spectrophotometer. Ultra-violet and X-ray photoelectron spectroscopy (UPS/XPS) measurements were performed in an ultrahigh vacuum chamber (∼10−10 mbar) built by Scienta Omicron, using a hemispherical electron analyzer (Sphera II EAC 125 7-channeltron). The analyzer was calibrated with the Fermi edge of polycrystalline silver cleaned using argon sputtering. UPS measurements were performed using a He I line with an excitation energy of 21.22 eV and a pass energy of 5 eV. The work function of the samples was determined from the secondary electron cut-off of the UPS spectrum, as the difference between the excitation energy and the length of the spectrum.39 All XPS measurements were performed using a monochromatized AlK (excitation energy of 1486.6 eV) operated at 15 kV. XPS high-resolution spectra were recorded at a pass energy of 10 eV, respectively, and referenced to the carbon 1s peak at 284.8 eV. The samples for XPS and UPS measurements were prepared by depositing the respective perovskite solution on ITO/NiOx substrates under equal conditions as explained in the device fabrication section. Then, fresh samples were kept in the dark, while light-soaked samples were under AM 1.5G (WaveLabs Sinus-70) for 6 hours; during this process, the samples were kept under N2. The X-ray diffraction (XRD) experiment was performed with a Bruker D8 advanced diffractometer.Results and discussion
PSCs with the device architecture ITO/NiOx(5 nm)/Cs0.25−xMAxFA0.75Sn0.5Pb0.5I3 (x = 0, 0.10, 0.20, and 0.25) (700 nm)/C60 (30 nm)/BCP (8 nm)/Ag (100 nm) were fabricated (Fig. 1a). The films of each composition presented a pin-hole free morphology and a similar grain size of ∼500 nm (Fig. S1†). The PCE increases with the increase in MA content, resulting in enhancement of the average PCE from 14.80 (x = 0) to 16.76 (x = 0.25) (Fig. 1b, Table 1). The improvement is due to enhanced fill factor (FF) and open-circuit voltage (Voc) at higher MA content (Fig. S2†). The short-circuit current density (Jsc) data are in agreement with the values obtained from external quantum efficiency (EQE) measurements (Fig. S3†). Moreover, the forward and reverse scans present a visible increase in hysteresis at higher x values (Fig. S4†). In the Pb-based PSC, the use of MA to boost the FF and Voc has been rationalized due to longer carrier diffusion lengths at higher quantities of MA.40,41 A longer diffusion length allows for a more effective charge extraction and sufficient light-harvesting. In Sn–Pb perovskites, longer diffusion lengths are observed for MAFA compared to CsFA-based compositions,1,10 suggesting that the inclusion of MA in TLPSCs follows a similar trend to a Pb-based PSC. Additionally, ultraviolet photoemission spectroscopy (UPS) measurements (discussed later in the text) showed a favorable energy alignment between NiOx and the perovskite ionization energies (IEs) when x = 0.25 but not at x = 0 (Fig. S5†). The IE mismatch observed for x = 0 results in formation of an energetic barrier that impedes efficient hole extraction, causing the observed lower Voc. | ||
| Fig. 1 (a) p–i–n device architecture of TLPSCs. (b) PCEs of corresponding devices at different MA contents represented as X. (c) Normalised PCEs of devices as a function of time at AM1.5G in a N2 environment; devices were left under open circuit conditions for the whole duration of the measurement (the PCE was obtained from Fig. S6†). (d) Normalised performance gain/loss of devices measured before and 24 h after light stability tests; devices were left in the dark during the recovery time. | ||
| x | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|
| 0 | 32.53 (32.71) | 0.66 (0.68) | 0.68 (0.69) | 14.80 (15.51) |
| 0.1 | 31.25 (31.81) | 0.73 (0.73) | 0.70 (0.71) | 15.88 (16.49) |
| 0.2 | 30.98 (30.78) | 0.76 (0.78) | 0.70 (0.72) | 16.47 (17.34) |
| 0.25 | 29.76 (31.19) | 0.79 (0.79) | 0.71 (0.72) | 16.77 (18.01) |
The photostability of the devices was tested by recording J–V curves for each perovskite composition every 10 min for 1 hour under AM1.5G (Fig. S6†), leaving the devices under open-circuit conditions. The normalized PCEs of representative devices are presented in Fig. 1c. For the devices with higher content of MA (x = 0.25 and x = 0.20), we observed a PCE loss of 60% during the first 10 min of operation. After this sudden drop, the efficiency remained relatively stable. The PCE drop was less severe for x = 0.10; however, the stabilization of the PCE was not observed throughout the measurement duration. In contrast, the devices without MA (x = 0) did not present significant PCE loss and remained stable during the complete duration of the measurement. The performance loss in MA-based devices was previously shown to be reversible, and this was attributed to thermally generated carriers caused during illumination.12 An alternative explanation for the performance drop was given due to mobile ions screening the built-in potential during the early stages of illumination.42 To verify the possible degradation pathway of the cells, we measured the recovery of our devices after 24 h after the light stability test. During recovery, the devices were stored in the dark in a N2 filled glovebox. The devices without MA (x = 0) exhibited a 10% efficiency improvement after the recovery time (Fig. 1d). For the intermediate compositions (x = 0.10 and x = 0.20), a nearly full recovery of their PCE is observed. In comparison, the devices with only MA (x = 0.25) lost around 25% of their initial efficiency. This irreversible performance loss indicates that thermally generated carriers are not responsible for the device degradation; instead, our findings align better with the hypothesis of light-induced ionic motion, which can further incite irreversible chemical reactions.37,43 To ensure this performance loss was due to light-soaking and not intrinsic instability of the device, we measured the shelf stability of only MA (x = 0.25) devices and found that 90% of their initial PCE was retained for at least 100 h (Fig. S7†). These results suggest that the inclusion of MA reduces the photostability and leads to an irreversible loss of performance after 1 h under illumination. To better understand the mechanisms leading to significant differences in the photostability of the devices, we further analyze the principal recombination mechanisms, the charge transport behavior and the surface chemistry of fresh and light-soaked samples with x = 0 and x = 0.25 compositions, henceforth denoted as CsFA (x = 0.0) and MAFA (x = 0.25).
Light-intensity dependent measurements can provide information regarding the recombination kinetics in a solar cell. The relation between Voc and light intensity I is given by 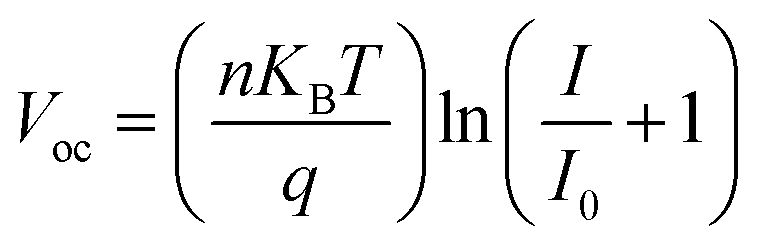 , where KB is the Boltzmann constant, T is the temperature, q is the elementary charge, and n is the ideality factor. A value of n near 1 indicates band-to-band recombination or surface recombination, while an n of around 2 indicates that recombination is considered trap-assisted.44 In the case of CsFA devices, they presented a minor increase in n from 1.25 to 1.29 after light soaking, which suggests that the recombination kinetics were not significantly influenced after illumination (Fig. 2a). For the MAFA devices, the fresh and light-soaked samples exhibited a change of n from 1.38 to 1.07, respectively (Fig. 2b). We attribute this variation to increased surface recombination for the light-soaked samples.44 Similarly, we studied the changes of Jsc against different light intensities. Here, Jsc and I are related through the power law given by Jsc ∝ Iα, where values of α less than 1 are related to space charge effects or bimolecular recombination.45 As expected, CsFA devices did not present significant changes in α between fresh and light-soaked samples (Fig. 2c). In comparison, the MAFA devices showed a reduction in α from 0.87 to 0.80 after light soaking, indicating that photocurrent in MAFA cells is limited by carrier imbalance or interfacial barriers (Fig. 2d).46
, where KB is the Boltzmann constant, T is the temperature, q is the elementary charge, and n is the ideality factor. A value of n near 1 indicates band-to-band recombination or surface recombination, while an n of around 2 indicates that recombination is considered trap-assisted.44 In the case of CsFA devices, they presented a minor increase in n from 1.25 to 1.29 after light soaking, which suggests that the recombination kinetics were not significantly influenced after illumination (Fig. 2a). For the MAFA devices, the fresh and light-soaked samples exhibited a change of n from 1.38 to 1.07, respectively (Fig. 2b). We attribute this variation to increased surface recombination for the light-soaked samples.44 Similarly, we studied the changes of Jsc against different light intensities. Here, Jsc and I are related through the power law given by Jsc ∝ Iα, where values of α less than 1 are related to space charge effects or bimolecular recombination.45 As expected, CsFA devices did not present significant changes in α between fresh and light-soaked samples (Fig. 2c). In comparison, the MAFA devices showed a reduction in α from 0.87 to 0.80 after light soaking, indicating that photocurrent in MAFA cells is limited by carrier imbalance or interfacial barriers (Fig. 2d).46
 | ||
| Fig. 2 (a and b) Light intensity-dependent Voc measurements for CsFA and MAFA devices, (c and d) light intensity-dependent Jsc measurements for CsFA and MAFA devices, and (e and f) Nyquist plots derived from impedance spectroscopy measurements of CsFA and MAFA devices; measurements were taken in a N2 atmosphere under dark conditions. | ||
To understand the charge transport behavior before and after light soaking the devices, we conducted impedance spectroscopy (IS) measurements. IS allows to individually study the electronic and ionic contribution to the charge transport. Several studies demonstrate that ion conduction dominates at low frequencies, and the electronic contribution ensues at high frequencies.47–49 The Nyquist plots from CsFA and MAFA reveal that ion diffusion has a significant contribution to charge transport (Fig. 2e and f). This phenomenon is characterized by a linear increase in the mid-frequency region and a line bending in the low and high-frequency regions.47 We observed that ion diffusion in MAFA devices occurs regardless of being fresh or stressed by light. The ion motion could explain the steep drop in performance observed during our stability measurements of the MAFA device, as well as the increased surface recombination and the creation of interfacial barriers found in our light-intensity dependent measurements.42 CsFA cells initially exhibited an ionic diffusion behavior; however, the light soaked devices presented a semicircle feature at mid and high frequencies, resulting in an improved efficiency after recovery.50
To study photoinduced changes in the electronic properties of perovskites, we performed UPS measurements. The IE of CsFA fresh samples was found to be 5.25 eV, with a minor change of 0.03 eV after light soaking, agreeing well with the observed photostable behavior. In comparison, light soaking resulted in a shift of 0.1 eV in the IE of MAFA samples, increasing from −5.54 to −5.64 eV, suggesting a possible change in surface composition. Using the optical bandgaps, the conduction band (CB) of the CsFA and MAFA was estimated to be −4.03 and −4.32 eV, respectively. Due to negligible changes in the optical bandgap after light soaking the MAFA films (Fig. S8†), the estimated CB was found to be lower (relative to the vacuum level) after the light soaking from −4.32 to −4.42 eV. Since the LUMO of C60 is known to be 4.3 eV,51 the change in the CB of MAFA after light-soaking would result in an energetic barrier for electron transfer between MAFA and the C60 (Fig. 3a). No such energetic offset was found in the case of CsFA devices upon light soaking (Fig. 3b). The extraction barrier found in the MAFA/C60 interface justifies the reduced photostability observed as compared to the CsFA cells.
 | ||
| Fig. 3 (a) Band diagram of fresh and light soaked MAFA films highlighting the energy barrier between light soaked MAFA and C60. (b) Band diagram of fresh and light soaked CsFA films. (c) XPS spectra showing the C 1s peaks of MAFA fresh and light soaked films for 6 h. (d) XPS spectra showing the N 1s peaks of MAFA fresh and light-soaked films for 6 h. | ||
X-ray photoelectron spectroscopy (XPS) was performed to elucidate the irreversible performance loss of the MAFA devices and to check prolonged photo-induced changes in the chemical composition at the perovskite surface as possible degradation pathways. Noteworthily, no significant changes were detected for the amount of Sn4+ upon light soaking, indicating that the observed degradation is not due to the Sn oxidation at the surface. No further variations were observed in I and Pb peaks upon photodegradation (Fig. S9†). However, the C 1s peak was found to have four components at 284.8 eV, 286.3 eV, 288.2 eV and 289 eV attributed to (i) the adventitious carbon contamination, (ii) MA+/C–O, (iii) FA+, and (iv) PbCO3,52 respectively (Fig. 3c). From all the components, only MA+/C–O showed a significant drop of around 13%, accompanied by a chemical shift of 0.1 eV towards higher binding energy. This suggest that the degradation of MAFA samples results in the loss of total MA+, potentially caused by the volatilization of MA from the MAFA film. The N 1s peak was found to be asymmetric, where the peaks at 400.6 eV and 402.2 eV are attributed to the nitrogen in FA+ (96%) and MA+ (4%), respectively (Fig. 3d) though no chemical shift in the N1s peak was observed upon photo-degradation; the amount of N1s associated with MA+ slightly reduced to ∼3%, in agreement with the changes observed for the C 1s peak attributed to MA+. The C 1s spectra of CsFA films presented a reduction in the area ∼4% for the C–O component at 285.5 eV. In contrast to MAFA films, the N 1s spectra only reveal a single component at 400.6 eV attributed to the FA+ (Fig. S10†); no significant peak shifts were detected after light soaking. Similarly, the spectra of Pb, Sn, and I of CsFA films did not exhibit noteworthy changes upon light soaking (Fig. S11†). Considering the above findings, we conclude that irreversible loss of performance during the initial hours of operation in TLPSCs is mainly caused by the volatilization of MA at the perovskite surface. Furthermore, the X-ray diffraction (XRD) patterns of fresh and light soaked MAFA films reveal the presence of PbI2 (Fig. S12†). We did not find any correlation between peak intensity from the PbI2 signal and light soaking time, indicating that PbI2 is not formed due to illumination. In previous studies, presence of PbI2 has been associated with accelerated perovskite degradation under illumination.53 In the case of fresh and light soaked CsFA films, no PbI2 signal was detected. Hence, the PbI2 found in MAFA samples could result in another degradation pathway that is not present in the CsFA composition.
To explore the device degradation on a longer timescale, we tracked the photovoltaic performance of unencapsulated devices at the maximum power point (MPP) (details are provided in the ESI†). As a HTL, NiOx has been considered a promising material since it has shown improved device stability compared to other polymeric HTLs.26,54,55 In the particular case of TLPSCs, there is an absence of in-depth photostability studies to offer insights on the performance of NiOx HTLs. To compare these results to state-of-the-art devices, we fabricated MAFA and CsFA cells with PEDOT:PSS as an alternative HTL. The average efficiencies obtained with PEDOT are 15.13% and 17.57% for CsFA and MAFA devices, respectively (Fig. S13†). Regardless of the HTL, the MAFA devices presented a fast decay in efficiency during the initial hours of the test. After 200 h, MAFA–PEDOT:PSS retained a slightly higher PCE of 65% of its initial value, compared to the 50% retained with the MAFA–NiOx cells. We note that MAFA–NiOx degrades faster than MAFA–PEDOT:PSS, which could be attributed to a MA deprotonation by NiOx.55 In both cases, significant losses were recorded for the Jsc and FF, indicating the formation of energetic barriers that inhibit the extraction of the photo-generated carriers.52 CsFA–PEDOT:PSS cells retained a higher PCE than CsFA–NiOx devices during the first 450 hours. However, a faster decay for the CsFA–PEDOT:PSS cells was recorded after this point. CsFA–NiOx devices retained 80% of their original PCE after 690 hours, whereas the PCE of CsFA–PEDOT:PSS cells dropped to 60% (Fig. 4). In both systems, the principal losses were observed for the Jsc, probably related to the Sn oxidation.20 The lower stability of PEDOT:PSS compared to NiOx has been attributed to higher hydrophobicity and ITO etching due to the PEDOT:PSS acidity.54–57 Comparing these results to the ones from Fig. 1d, the efficiency losses from MAFA devices held at the MPP were lower, while they remained stable for a considerably longer time. However, the trend of a faster efficiency decay for MAFA compared to CsFA remained the same. Overall, these results suggest that the A-site composition is more critical than the HTL during the early stages under operating conditions. In particular, the MA content in TLPSCs appears to be the bottleneck for photostable devices.
 | ||
| Fig. 4 Photovoltaic performance at 1 sun (metal halide lamp) as a function of time of unencapsulated MAFA and CsFA devices with NiOx and PEDOT:PSS as the HTL. J–Vs were recorded each 10 min while devices were held at MPP under continuous N2 flow. | ||
Conclusion
In this work, we investigated the influence of A-site on the performance and photostability of TLPSCs. We found that the addition of MA improves the PCE while significantly compromising the device stability under working conditions. Our results indicate that light-induced interfacial barriers, surface recombination, and ion diffusion are responsible for the poor photostability of MA-rich devices during initial operational conditions. Additionally, we observed that MAFA devices present an irreversible loss of performance after illumination. We attribute this performance drop to the creation of an extraction barrier at the perovskite/ETL interface caused by the permanent volatilization of MA at the perovskite surface. Lastly, we found that the surface oxidation of Sn and the choice of the HTL have secondary roles in the short-term photostability of the device. However, the content of MA becomes a more critical factor towards the development of photostable TLPSCs. Since MA-based devices show high performance but inferior photostability, strategies focusing on reducing MA content or reducing ion migration will be beneficial for device photostability.Conflicts of interest
There are no conflicts of interests.Acknowledgements
This publication is based upon the work supported by King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No: OSR-2019-CARF/CCF-3079 and Award No. OSR-CRG2018-3737.References
- J. Tong, Z. Song, D. H. Kim, X. Chen, C. Chen and A. F. Palmstrom, et al., Carrier lifetimes of >1 μs in Sn-Pb perovskites enable efficient all-perovskite tandem solar cells, Science, 2019, 364(6439), 475–479 CrossRef CAS PubMed.
- R. Lin, K. Xiao, Z. Qin, Q. Han, C. Zhang and M. Wei, et al., Monolithic all-perovskite tandem solar cells with 24.8% efficiency exploiting comproportionation to suppress Sn(II) oxidation in precursor ink, Nat. Energy, 2019, 4(10), 864–873 CrossRef CAS.
- K. Xiao, R. Lin, Q. Han, Y. Hou, Z. Qin and H. T. Nguyen, et al., All-perovskite tandem solar cells with 24.2% certified efficiency and area over 1 cm2 using surface-anchoring zwitterionic antioxidant, Nat. Energy, 2020, 5(11), 870–880 CrossRef CAS.
- G. Kapil, T. Bessho, T. Maekawa, A. K. Baranwal, Y. Zhang and M. A. Kamarudin, et al., Tin-Lead Perovskite Fabricated via Ethylenediamine Interlayer Guides to the Solar Cell Efficiency of 21.74, Adv. Energy Mater., 2021, 11(25), 2101069 CrossRef CAS.
- Z. Yang, Z. Yu, H. Wei, X. Xiao, Z. Ni and B. Chen, et al., Enhancing electron diffusion length in narrow-bandgap perovskites for efficient monolithic perovskite tandem solar cells, Nat. Commun., 2019, 10(1), 4498 CrossRef.
- C. Li, Z. Song, C. Chen, C. Xiao, B. Subedi and S. P. Harvey, et al., Low-bandgap mixed tin–lead iodide perovskites with reduced methylammonium for simultaneous enhancement of solar cell efficiency and stability, Nat. Energy, 2020, 5(10), 768–776 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting tin and lead iodide perovskites with organic cations: Phase transitions, high mobilities, and near-infrared photoluminescent properties, Inorg. Chem., 2013, 52(15), 9019–9038 CrossRef CAS.
- A. Goyal, S. McKechnie, D. Pashov, W. Tumas, M. van Schilfgaarde and V. Stevanović, Origin of Pronounced Nonlinear Band Gap Behavior in Lead–Tin Hybrid Perovskite Alloys, Chem. Mater., 2018, 30(11), 3920–3928 CrossRef CAS.
- R. M. I. Bandara, S. M. Silva, C. C. L. Underwood, K. D. G. I. Jayawardena, R. A. Sporea and S. R. P. Silva, Progress of Pb-Sn Mixed Perovskites for Photovoltaics: A Review, Energy Environ. Mater., 2021, 5(2), 370–400 CrossRef.
- G. E. Eperon, T. Leijtens, K. A. Bush, R. Prasanna, T. Green and J. T.-W. Wang, et al., Perovskite-perovskite tandem photovoltaics with optimized band gaps, Science, 2016, 354(6314), 861–865 CrossRef CAS PubMed.
- S. Gu, R. Lin, Q. Han, Y. Gao, H. Tan and J. Zhu, Tin and Mixed Lead–Tin Halide Perovskite Solar Cells: Progress and their Application in Tandem Solar Cells, Adv. Mater., 2020, 32(27), 1907392 CrossRef CAS PubMed.
- T. Leijtens, R. Prasanna, K. A. Bush, G. E. Eperon, J. A. Raiford and A. Gold-Parker, et al., Tin–lead halide perovskites with improved thermal and air stability for efficient all-perovskite tandem solar cells, Sustainable Energy Fuels, 2018, 2(11), 2450–2459 RSC.
- J. Li, H.-L. Cao, W.-B. Jiao, Q. Wang, M. Wei and I. Cantone, et al., Biological impact of lead from halide perovskites reveals the risk of introducing a safe threshold, Nat. Commun., 2020, 11(1), 310 CrossRef CAS PubMed.
- T. Leijtens, R. Prasanna, A. Gold-Parker, M. F. Toney and M. D. McGehee, Mechanism of Tin Oxidation and Stabilization by Lead Substitution in Tin Halide Perovskites, ACS Energy Lett., 2017, 2(9), 2159–2165 CrossRef CAS.
- M. A. Haque, L. H. Hernandez, B. Davaasuren, D. R. Villalva, J. Troughton and D. Baran, Tuning the Thermoelectric Performance of Hybrid Tin Perovskites by Air Treatment, Adv. Energy Sustain. Res., 2020, 1(2), 2000033 CrossRef.
- K. J. Savill, A. M. Ulatowski, M. D. Farrar, M. B. Johnston, H. J. Snaith and L. M. Herz, Impact of Tin Fluoride Additive on the Properties of Mixed Tin-Lead Iodide Perovskite Semiconductors, Adv. Funct. Mater., 2020, 30(52), 2005594 CrossRef CAS.
- N. K. Noel, S. D. Stranks, A. Abate, C. Wehrenfennig, S. Guarnera and A.-A. Haghighirad, et al., Lead-free organic–inorganic tin halide perovskites for photovoltaic applications, Energy Environ. Sci., 2014, 7(9), 3061–3068 RSC.
- T. Leijtens, K. Bush, R. Cheacharoen, R. Beal, A. Bowring and M. D. McGehee, Towards enabling stable lead halide perovskite solar cells; interplay between structural, environmental, and thermal stability, J. Mater. Chem. A, 2017, 5(23), 11483–11500 RSC.
- L. Lanzetta, N. Aristidou and S. A. Haque, Stability of Lead and Tin Halide Perovskites: The Link between Defects and Degradation, J. Phys. Chem. Lett., 2020, 11(2), 574–585 CrossRef CAS PubMed.
- L. Lanzetta, T. Webb, N. Zibouche, X. Liang, D. Ding and G. Min, et al., Degradation mechanism of hybrid tin-based perovskite solar cells and the critical role of tin (IV) iodide, Nat. Commun., 2021, 12(1), 2853 CrossRef CAS PubMed.
- M. A. Haque, D. Rosas Villalva, L. H. Hernandez, R. Tounesi, S. Jang and D. Baran, Role of Dopants in Organic and Halide Perovskite Energy Conversion Devices, Chem. Mater., 2021, 33(21), 8147–8172 CrossRef CAS.
- J. Sanchez-Diaz, R. S. Sánchez, S. Masi, M. Kreĉmarová, A. O. Alvarez and E. M. Barea, et al., Tin perovskite solar cells with >1300 h of operational stability in N2 through a synergistic chemical engineering approach, Joule, 2022, 6(4), 861–883 CrossRef CAS PubMed.
- R. Prasanna, T. Leijtens, S. P. Dunfield, J. A. Raiford, E. J. Wolf and S. A. Swifter, et al., Design of low bandgap tin–lead halide perovskite solar cells to achieve thermal, atmospheric and operational stability, Nat. Energy, 2019, 4(11), 939–947 CrossRef CAS.
- S. Moghadamzadeh, I. M. Hossain, M. Loy, D. B. Ritzer, H. Hu and D. Hauschild, et al., In2O3:H-Based Hole-Transport-Layer-Free Tin/Lead Perovskite Solar Cells for Efficient Four-Terminal All-Perovskite Tandem Solar Cells, ACS Appl. Mater. Interfaces, 2021, 13(39), 46488–46498 CrossRef CAS PubMed.
- F. Lv, Y. Yao, C. Xu, D. Liu, L. Liao and G. Wang, et al., Elimination of Charge Transport Layers in High-Performance Perovskite Solar Cells by Band Bending, ACS Appl. Energy Mater., 2021, 4(2), 1294–1301 CrossRef CAS.
- X. Y. Hou, F. J. Li, X. Zhang, Y. F. Shi, Y. X. Du and J. B. Gong, et al., Reducing the Energy Loss to Achieve High Open-circuit Voltage and Efficiency by Coordinating Energy-Level Matching in Sn-Pb Binary Perovskite Solar Cells, Sol. RRL, 2021, 5(8), 2100287 CrossRef CAS.
- H. Chen, Z. Peng, K. Xu, Q. Wei, D. Yu and C. Han, et al., Band alignment towards high-efficiency NiOx-based Sn-Pb mixed perovskite solar cells, Sci. China Mater., 2021, 64(3), 537–546 CrossRef CAS.
- J. Werner, T. Moot, T. A. Gossett, I. E. Gould, A. F. Palmstrom and E. J. Wolf, et al., Improving Low-Bandgap Tin–Lead Perovskite Solar Cells via Contact Engineering and Gas Quench Processing, ACS Energy Lett., 2020, 5(4), 1215–1223 CrossRef CAS.
- D. Chi, S. Huang, M. Zhang, S. Mu, Y. Zhao and Y. Chen, et al., Composition and Interface Engineering for Efficient and Thermally Stable Pb–Sn Mixed Low-Bandgap Perovskite Solar Cells, Adv. Funct. Mater., 2018, 28(51), 1804603 CrossRef.
- G. Kapil, T. Bessho, C. H. Ng, K. Hamada, M. Pandey and M. A. Kamarudin, et al., Strain Relaxation and Light Management in Tin–Lead Perovskite Solar Cells to Achieve High Efficiencies, ACS Energy Lett., 2019, 4(8), 1991–1998 CrossRef CAS.
- Q. Han, Y. Wei, R. Lin, Z. Fang, K. Xiao and X. Luo, et al., Low-temperature processed inorganic hole transport layer for efficient and stable mixed Pb-Sn low-bandgap perovskite solar cells, Sci. Bull., 2019, 64(19), 1399–1401 CrossRef.
- N. Ghimire, R. S. Bobba, A. Gurung, K. M. Reza, M. A. R. Laskar and B. S. Lamsal, et al., Mitigating Open-Circuit Voltage Loss in Pb–Sn Low-Bandgap Perovskite Solar Cells via Additive Engineering, ACS Appl. Energy Mater., 2021, 4(2), 1731–1742 CrossRef CAS.
- Z. Chen, M. Liu, Z. Li, T. Shi, Y. Yang and H.-L. Yip, et al., Stable Sn/Pb-Based Perovskite Solar Cells with a Coherent 2D/3D Interface, iScience, 2018, 9, 337–346 CrossRef CAS PubMed.
- R. Wang, H. Gao, R. Yu, H. Jia, Z. Ma and Z. He, et al., β-Diketone Coordination Strategy for Highly Efficient and Stable Pb–Sn Mixed Perovskite Solar Cells, J. Phys. Chem. Lett., 2021, 12(49), 11772–11778 CrossRef CAS PubMed.
- A. F. Akbulatov, L. A. Frolova, N. N. Dremova, I. Zhidkov, V. M. Martynenko and S. A. Tsarev, et al., Light or Heat: What Is Killing Lead Halide Perovskites under Solar Cell Operation Conditions?, J. Phys. Chem. Lett., 2020, 11(1), 333–339 CrossRef CAS.
- A. F. Akbulatov, S. A. Tsarev, M. Elshobaki, S. Y. Luchkin, I. S. Zhidkov and E. Z. Kurmaev, et al., Comparative Intrinsic Thermal and Photochemical Stability of Sn(II) Complex Halides as Next-Generation Materials for Lead-Free Perovskite Solar Cells, J. Phys. Chem. C, 2019, 123(44), 26862–26869 CrossRef CAS.
- N. H. Nickel, F. Lang, V. V. Brus, O. Shargaieva and J. Rappich, Unraveling the Light-Induced Degradation Mechanisms of CH3NH3PbI3 Perovskite Films, Adv. Electron. Mater., 2017, 3(12), 1700158 CrossRef.
- A. Latini, G. Gigli and A. Ciccioli, A study on the nature of the thermal decomposition of methylammonium lead iodide perovskite, CH3NH3PbI3: an attempt to rationalise contradictory experimental results, Sustainable Energy Fuels, 2017, 1(6), 1351–1357 RSC.
- A. Sharma, S. Singh, X. Song, D. Rosas Villalva, J. Troughton and D. Corzo, et al., A Nonionic Alcohol Soluble Polymer Cathode Interlayer Enables Efficient Organic and Perovskite Solar Cells, Chem. Mater., 2021, 33(22), 8602–8611 CrossRef CAS PubMed.
- H. Tan, F. Che, M. Wei, Y. Zhao, M. I. Saidaminov and P. Todorović, et al., Dipolar cations confer defect tolerance in wide-bandgap metal halide perovskites, Nat. Commun., 2018, 9(1), 3100 CrossRef.
- M. I. Saidaminov, K. Williams, M. Wei, A. Johnston, R. Quintero-Bermudez and M. Vafaie, et al., Multi-cation perovskites prevent carrier reflection from grain surfaces, Nat. Mater., 2020, 19(4), 412–418 CrossRef CAS PubMed.
- J. Thiesbrummel, V. M. Le Corre, F. Peña-Camargo, L. Perdigón-Toro, F. Lang and F. Yang, et al., Universal Current Losses in Perovskite Solar Cells Due to Mobile Ions, Adv. Energy Mater., 2021, 2101447 CrossRef CAS.
- X. Tang, M. Brandl, B. May, I. Levchuk, Y. Hou and M. Richter, et al., Photoinduced degradation of methylammonium lead triiodide perovskite semiconductors, J. Mater. Chem. A, 2016, 4(41), 15896–15903 RSC.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen and J. P. Correa Baena, et al., Interpretation and evolution of open-circuit voltage, recombination, ideality factor and subgap defect states during reversible light-soaking and irreversible degradation of perovskite solar cells, Energy Environ. Sci., 2018, 11(1), 151–165 RSC.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and P. W. M. Blom, Origin of the light intensity dependence of the short-circuit current of polymer/fullerene solar cells, Appl. Phys. Lett., 2005, 87(20), 203502 CrossRef.
- T. Singh and T. Miyasaka, Stabilizing the Efficiency Beyond 20% with a Mixed Cation Perovskite Solar Cell Fabricated in Ambient Air under Controlled Humidity, Adv. Energy Mater., 2018, 8(3), 1700677 CrossRef.
- W. Peng, C. Aranda, O. M. Bakr, G. Garcia-Belmonte, J. Bisquert and A. Guerrero, Quantification of Ionic Diffusion in Lead Halide Perovskite Single Crystals, ACS Energy Lett., 2018, 3(7), 1477–1481 CrossRef CAS.
- M. Bag, L. A. Renna, R. Y. Adhikari, S. Karak, F. Liu and P. M. Lahti, et al., Kinetics of Ion Transport in Perovskite Active Layers and Its Implications for Active Layer Stability, J. Am. Chem. Soc., 2015, 137(40), 13130–13137 CrossRef CAS PubMed.
- E. von Hauff and D. Klotz, Impedance spectroscopy for perovskite solar cells: characterisation, analysis, and diagnosis, J. Mater. Chem. C, 2022, 10(2), 742–761 RSC.
- A. D. Sheikh, A. Bera, M. A. Haque, R. B. Rakhi, S. D. Gobbo and H. N. Alshareef, et al., Atmospheric effects on the photovoltaic performance of hybrid perovskite solar cells, Sol. Energy Mater. Sol. Cells, 2015, 137, 6–14 CrossRef CAS.
- G. Xu, P. Bi, S. Wang, R. Xue, J. Zhang and H. Chen, et al., Integrating Ultrathin Bulk-Heterojunction Organic Semiconductor Intermediary for High-Performance Low-Bandgap Perovskite Solar Cells with Low Energy Loss, Adv. Funct. Mater., 2018, 28(42), 1804427 CrossRef.
- L. E. Mundt, J. Tong, A. F. Palmstrom, S. P. Dunfield, K. Zhu and J. J. Berry, et al., Surface-Activated Corrosion in Tin–Lead Halide Perovskite Solar Cells, ACS Energy Lett., 2020, 5(11), 3344–3351 CrossRef CAS.
- G. Tumen-Ulzii, C. Qin, D. Klotz, M. R. Leyden, P. Wang and M. Auffray, et al., Detrimental Effect of Unreacted PbI2 on the Long-Term Stability of Perovskite Solar Cells, Adv. Mater., 2020, 32(16), 1905035 CrossRef CAS PubMed.
- J. You, L. Meng, T.-B. Song, T.-F. Guo, Y. Yang and W.-H. Chang, et al., Improved air stability of perovskite solar cells via solution-processed metal oxide transport layers, Nat. Nanotechnol., 2016, 11(1), 75–81 CrossRef CAS PubMed.
- C. C. Boyd, R. C. Shallcross, T. Moot, R. Kerner, L. Bertoluzzi and A. Onno, et al., Overcoming Redox Reactions at Perovskite-Nickel Oxide Interfaces to Boost Voltages in Perovskite Solar Cells, Joule, 2020, 4(8), 1759–1775 CrossRef CAS.
- Y. Meng, Z. Hu, N. Ai, Z. Jiang, J. Wang and J. Peng, et al., Improving the Stability of Bulk Heterojunction Solar Cells by Incorporating pH-Neutral PEDOT:PSS as the Hole Transport Layer, ACS Appl. Mater. Interfaces, 2014, 6(7), 5122–5129 CrossRef CAS PubMed.
- M. P. d. Jong, L. J. v. IJzendoorn and M. J. A. d. Voigt, Stability of the interface between indium-tin-oxide and poly(3,4-ethylenedioxythiophene)/poly(styrenesulfonate) in polymer light-emitting diodes, Appl. Phys. Lett., 2000, 77(14), 2255–2257 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2se00663d |
| This journal is © The Royal Society of Chemistry 2022 |