
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNHC induced radical formation via homolytic cleavage of B–B bonds and its role in organic reactions†
Laura
Kuehn
a,
Ludwig
Zapf
a,
Luis
Werner
a,
Martin
Stang‡
 a,
Sabrina
Würtemberger-Pietsch
a,
Ivo
Krummenacher
a,
Holger
Braunschweig
a,
Emmanuel
Lacôte
*b,
Todd B.
Marder
*a and
Udo
Radius
*a
a,
Sabrina
Würtemberger-Pietsch
a,
Ivo
Krummenacher
a,
Holger
Braunschweig
a,
Emmanuel
Lacôte
*b,
Todd B.
Marder
*a and
Udo
Radius
*a
aInstitute for Inorganic Chemistry, Institute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: todd.marder@uni-wuerzburg.de; u.radius@uni-wuerzburg.de
bUniv Lyon, Université Claude Bernard Lyon 1, CNRS, CNES, ArianeGroup, LHCEP, Bât. Raulin, 2 rue Victor Grignard, F-69622 Villeurbanne, France. E-mail: emmanuel.lacote@univ-lyon1.fr
First published on 7th June 2022
Abstract
New borylation methodologies have been reported recently, wherein diboron(4) compounds apparently participate in free radical couplings via the homolytic cleavage of the B–B bond. We report herein that bis-NHC adducts of the type (NHC)2·B2(OR)4, which are thermally unstable and undergo intramolecular ring expansion reactions (RER), are sources of boryl radicals of the type NHC–BR2˙, exemplified by Me2ImMe·Bneop˙ 1a (Me2ImMe = 1,3,4,5-tetramethyl-imidazolin-2-ylidene, neop = neopentylglycolato), which are formed by homolytic B–B bond cleavage. Attempts to apply the boryl moiety 1a in a metal-free borylation reaction by suppressing the RER failed. However, based on these findings, a protocol was developed using Me2ImMe·B2pin23 for the transition metal- and additive-free boryl transfer to substituted aryl iodides and bromides giving aryl boronate esters in good yields. Analysis of the side products and further studies concerning the reaction mechanism revealed that radicals are likely involved. An aryl radical was trapped by TEMPO, an EPR resonance, which was suggestive of a boron-based radical, was detected in situ, and running the reaction in styrene led to the formation of polystyrene. The isolation of a boronium cation side product, [(Me2ImMe)2·Bpin]+I−7, demonstrated the fate of the second boryl moiety of B2pin2. Interestingly, Me2ImMe NHC reacts with aryl iodides and bromides generating radicals. A mechanism for the boryl radical transfer from Me2ImMe·B2pin23 to aryl iodides and bromides is proposed based on these experimental observations.
Introduction
Diboron(4) compounds are extremely useful reagents for the synthesis of organoboronates, which are valuable building blocks in organic synthesis.1 In addition to their use in Suzuki–Miyaura crossing–coupling reactions2 the C–B bond can easily be converted into numerous functional groups.1 While early catalytic borylation reactions typically employed precious metal catalysts, first row “Earth abundant” transition metals have become of increasing interest, due to their low cost and low toxicity.3,4 Recently, transition metal-free methods which activate the B–B bond of diboron(4) compounds have attracted much attention,5 having the advantage of avoiding metal contamination in the final organic product. sp2–sp3 Lewis base adducts of diboron(4) compounds show a higher reactivity of the B–B bond than their respective sp2–sp2 precursors, due to the increased bond length and polarity. Thus, they are used in various borylation reactions or for the diboration of unsaturated substrates, in which a nucleophilic boryl moiety is generated when one of the two boron atoms is coordinated to a strong Lewis base. In 2009, Hoveyda et al. proposed that an NHC sp2–sp3 diboron(4) adduct is responsible for the metal-free β-borylation of α,β-unsaturated carbonyl compounds.6Unlike heterolysis, induced by anionic nucleophiles,5b,e homolytic cleavage of the B–B bond is challenging due to the relatively high bond dissociation energies (BDE) of the diboron(4) compounds. For example, Wang et al. reported a metalfree borylation process for the direct conversion of aryl amines to aryl pinacol boronates. A radical mechanism involving single electron transfer between an aryl diazonium ion formed and a tetra-coordinated boron complex has been proposed for this reaction. Recently, new borylation methodologies have been reported, wherein the boron reagent apparently participates in free radical coupling via the homolytic cleavage of the B–B bond.5d,8 To date, there are mainly two ways to realize this homolytic cleavage, i.e., para-substituted pyridine-induced or by irradiation with light. Initially, the groups of Suginome and Ohmura reported the transition metal-free diboration of 1,4-dihydropyrazine derivatives and 4,4′-bipyridine, respectively, simply by reaction with B2pin2 at elevated temperatures.9,10 While they initially proposed a mechanism proceeding via a nucleophilic pathway,9a their more recent reports9b,10 confirm the role of radicals and the homolytic cleavage of the B–B bond in these processes.
In 2016, Li et al. reported the homolytic cleavage of the B–B bond of B2pin2via the coordination of 4-cyanopyridine to both boron atoms, generating pyridine-stabilized boryl radicals, which were employed for the synthesis of various organoboronates.11 Hydroboration of alkenes using B2pin2 and catalytic amounts of 4-cyanopyridine was demonstrated by the group of Cai, who also proposed that the reaction proceeded via the formation of a 4-cyanopyridine-stabilized boryl radical.12 Jiao et al. described the borylation of various aryl halides using potassium methoxide in the presence of catalytic amounts of 4-phenylpyridine, proposing the in situ generation of super electron donors (SED) based on boryl-pyridine species.13 In contrast to the initial assumption of a homolytic B–B bond cleavage,13a DFT studies demonstrate that the SEDs are generated by heterolytic cleavage of the B–B bond in B2pin2via coordination of the two boron atoms by pyridine and methoxide, respectively.13b In 2017, the groups of Pinet and Pucheault reported the stoichiometric borylation of various aryl iodides, mediated by an in situ formed fluoride sp2–sp3 diboron adduct from B2pin2 and CsF. This anionic adduct, previously structurally characterized by our group,5b was proposed to reduce the aryl iodide via a single-electron-transfer process (SET), forming FBpin5e and a boryl radical, which is stabilized by pyridine.14 Furthermore, a decarboxylative borylation of aryl and alkenyl carboxylic acids via a radical coupling mechanism was reported by Fu and co-workers.15 An in situ formed three-component bis-adduct between B2pin2, the carboxylic acid substrate, and a pyridine derivative is assumed to undergo homolytic B–B bond cleavage by an intramolecular SET, generating a carboxylate radical and a pyridine-stabilized boryl radical.15 Ogawa et al. reported the diboration of terminal alkynes with B2pin2 under irradiation by a high pressure mercury lamp in the presence of catalytic amounts of diphenyl disulfide and triphenylphosphine giving 1,2-diborylalkenes.16 The mechanism is not yet clear, but boron-centered radicals are proposed to be formed from B2pin2 and PPh3 under irradiation.16b In 2018, Studer and co-workers introduced the radical borylation of alkyl and aryl iodides initiated by a photoinduced C–I bond homolysis.17a The alkyl/aryl radical formed presumably adds to B2cat2 and, with coordination of DMF (dimethylformamide), the weakened B–B bond homolyzes giving the alkyl/aryl boronic ester and a DMF-stabilized boryl radical.17 Just recently, the photocatalytic, regioselective radical borylation of α,β-unsaturated esters and related compounds has been reported,18 and we reported the transition metal-free borylation of primary and secondary alkyl sulfones to yield alkylboronic esters.19 For the latter, the reaction of alkyl sulfones with bis(neopentyl glycolato) diboron(4) (B2neop2) and a stoichiometric amount of base as promotor afforded alkylboronic esters with a wide functional group tolerance. Radical clock, radical trap experiments, and EPR studies revealed that the borylation process involves radical intermediates.19
Typical methods for increasing the lifetime of radical species involve delocalization of the unpaired electron and/or the use of sterically hindered groups to prevent dimerization.8c,20 With respect to boron-centered radicals, well known anionic species of the type BR3˙− are generated by occupying the vacant p-orbital of an electron deficient borane with a single electron.21 For example, our group recently reported a structurally characterized example, in which such a radical anion is stabilized by an ortho-carborane moiety.21i In contrast, neutral analogs result from coordination of a Lewis base (L) to the boron center giving an L–BR2˙ adduct. In recent years, the electrophilicity of N-heterocyclic carbenes (NHCs) has been employed for stabilizing such boron centered radicals (Scheme 1).22 Offering an empty π*-orbital, the NHC is able to engage in π-backbonding interactions and thus delocalize the unpaired electron, which lowers the bond dissociation energies of the corresponding boranes.20,22,23
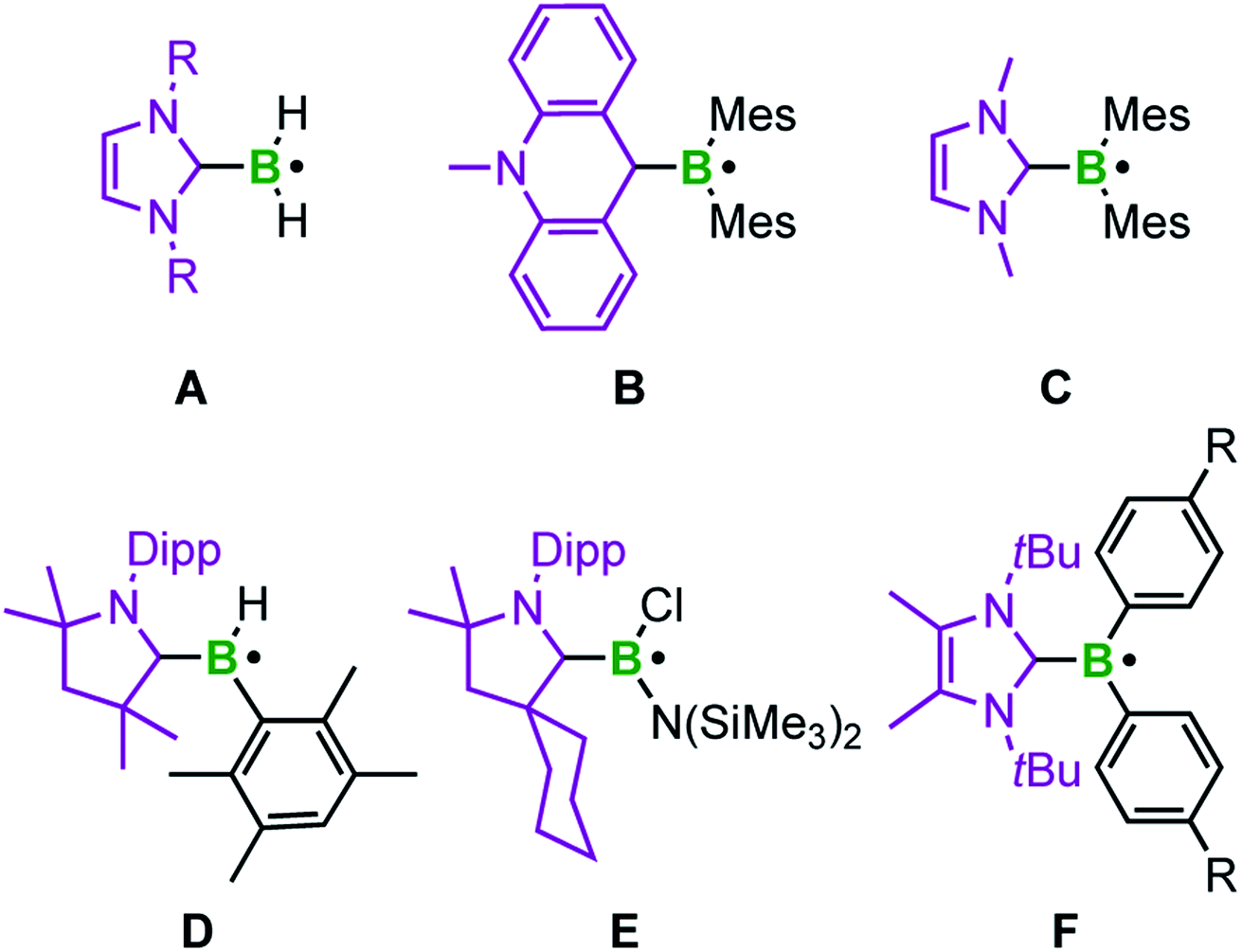 | ||
| Scheme 1 Selected neutral boryl radicals stabilized by carbenes. | ||
The groups of Curran, Lacôte, and Lalevée have extensively studied NHC-stabilized radicals of the type NHC–BH2˙ A, which are formed from the borane adduct NHC·BH3 by homolytic B–H bond cleavage. Radical formation from NHC·BH3 is facilitated by the relatively small homolytic B–H bond dissociation energy (80–88 kcal mol−1)24 in the precursor induced by the increased spin delocalization in the resulting ligated radical.25 This bond cleavage can be realized in the presence of radical initiators such as DTBP (di-tert-butylperoxide) or azobisisobutyronitrile (AIBN), triggered by light or heat. However, these radicals are transient and cannot be isolated. EPR studies and corroborating theoretical data reveal that radicals of type A are planar with the spin density delocalized over the NHC ligand and the boron center.24 This class of compounds has been applied as efficient co-initiators in radical photopolymerization reactions as well as radical reductions of halides and xanthates.26 The same groups generated the corresponding aryl boron radicals by analogously abstracting hydrogen atoms from NHC·BH2Ar compounds via homolytic B–H bond cleavage.26h The groups of Curran, Taniguchi, and Wang achieved various borylative radical cyclization reactions and the borylation of polyfluoroarenes, as well as the radical hydroboration of alkenes, also utilizing NHC·BH3 with in situ formation of the NHC-boryl radical A (Scheme 1) by hydrogen atom abstraction using di-tert-butyl hyponitrile and AIBN, respectively.27,28
The acridinyl radical B, reported by Gabbaï et al. in 2007, can be considered as the first structurally characterized carbene-stabilized neutral boryl radical (Scheme 1).29 However, in this example, the unpaired electron is largely delocalized over the acridinyl fragment with very little contribution from the boron atom, as indicated by EPR spectroscopy. The same group observed, but could not isolate, radical C by introducing a nucleophilic NHC (Scheme 1), via reversible reduction of an NHC-stabilized borenium cation.30 Radical C reveals a higher boron radical character and significant localization of the unpaired electron in a (B–CNHC)π-orbital.30 In 2014, Braunschweig and co-workers isolated the cAACMe-stabilized (cAAC = cyclic alkyl amino carbene) boryl radical D by reduction of the corresponding haloborane adduct.31 EPR and DFT data indicate that the spin density is delocalized over the strong π-accepting cAAC ligand. In the same year, the similar cAACCy-stabilized aminoboryl radical E, generated analogously by reduction of the corresponding adduct, was introduced by the groups of Bertrand and Stephan.32 Recently, Tamm et al. reported the NHC-stabilized boryl radical F, prepared by reduction of the corresponding borenium cation. Radical F was structurally confirmed and characterized by EPR spectroscopy, revealing that the unpaired electron is almost exclusively localized on the B(Ar)2 moiety.33 In general, NHCs, and especially cAACs, provide significant stabilization of boron-based radicals, with NHCs being at a unique cross-road between stabilization and reactivity, which has led to the strong focus discussed above.
For many years, our groups have been intensively studying neutral pyridine34 and NHC6c,35 as well as anionic5b adducts5a,36 of diboron(4) compounds.1b We have shown that, depending on the stoichiometry and the steric demand of the NHC and the diboron(4) reagent, mono- or bis-NHC adducts are accessible.35 In the current paper we focus on how NHC complexation of diboron(4) derivatives can be harnessed for radical reactions, beyond ring expansion reactions (RER).
Results and discussion
The RER of bis-NHC adducts typically proceeds via the insertion of one B(OR)2 moiety into the C–N bond of one NHC.35 A second exo-B(OR)2 moiety binds to the former carbene–carbon atom. In the case of B2neop2 (neop = neopentylglycolato), the endocyclic Bneop ring opens and one oxygen atom binds to the exocyclic boron atom forming an 8-membered heterocyclic ring with the second NHC coordinated to the endocyclic boron atom (Scheme 2).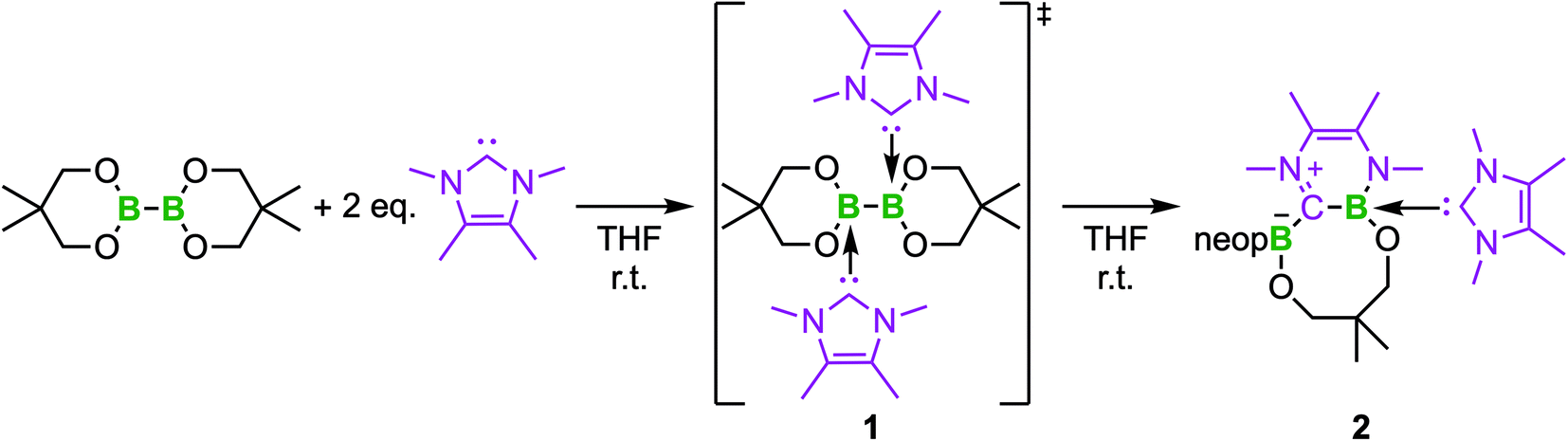 | ||
| Scheme 2 Reaction of B2neop2 with two equivalents of Me2ImMe at room temperature forming the ring expanded product RER-B2neop2·(Me2ImMe)22 with the bis-NHC adduct (Me2ImMe)2·B2neop21 as an intermediate. | ||
For B2neop2 this RER occurs at room temperature, whereas for B2cat2 (cat = catecholato) elevated temperatures of 70 °C are required.35a For small NHCs (e.g. Me2ImMe or iPr2Im) and the small diboron(4) reagent B2eg2 (eg = ethyleneglycolato), RERs are observed at temperatures as low as −40 °C.35c Furthermore, we have shown that the reactivity at diboron(4) compounds also depends on the NHC. For example, the reaction of B2cat2 with Dipp2Im (unsaturated NHC backbone) and Dipp2Sim (saturated NHC backbone) leads, in both cases, to the mono-NHC adduct. However, whereas Dipp2Im·B2cat2 is stable with regard to RER, the adduct with the backbone-saturated NHC Dipp2Sim·B2cat2 is thermally unstable generating a ring-expansion product.35c
Based on the above findings, we were interested to obtain further insight into the subsequent reactivity of bis-NHC adducts of diboron(4) compounds as well as the mechanism of the RERs. As the RER is very fast, and we could not exclude a mechanism involving radicals, we began our studies by monitoring the RER of B2neop2 with two equivalents of Me2ImMe in THF by EPR spectroscopy (Fig. 1). Beginning at −80 °C, we slowly raised the temperature, as our previous studies have shown that, at room temperature, the ring-expanded product RER-(Me2ImMe)2·B2neop22 is quantitatively generated within minutes.
 | ||
| Fig. 1 Simulated (red) and experimental (black) EPR spectrum of the RER of B2neop2 with two equivalents Me2ImMe in THF. | ||
At 290 K, a weak multiline EPR signal was detected with an isotropic g-value of 2.003 (Fig. 1). With the anticipated formation of a radical of the type NHC-BR2˙ (Me2ImMe-Bneop˙) 1a, presumably generated by homolytic cleavage of the B–B bond in B2neop2, spectral fitting (Fig. 1, red) suggests a delocalized spin distribution over the NHC fragment with hyperfine couplings to boron (a(11B) = 8.7 MHz), to nitrogen (a(14N) = 9.5 MHz), and to the methyl protons of the NHC (a(1H) = 6.9 and 7.9 MHz). Bis-NHC adducts, such as (Me2ImMe)2·B2neop21, with both boron atoms coordinated by an NHC, reveal an increased bond length and therefore a weakened boron–boron bond. X-ray diffraction of single crystals of (Me2ImMe)2·B2neop21, obtained from a saturated solution of B2neop2 and two equivalents of Me2ImMe in toluene at −30 °C (Fig. 2a) revealed, that both boron atoms of 1 are sp3-hybridized and tetrahedrally coordinated with a B–B bond distance of 1.770(2) Å, significantly elongated compared to that observed for B2neop2 (1.712(3) Å).35c According to DFT calculations (M06-2x//def2-TZVPP(B)/def2-TZVP), the dissociation energies of the B–B bonds in the diboron(4) compounds drop significantly upon NHC coordination to boron, from 408.9 kJ mol−1 for B2neop2 to 254.7 kJ mol−1 for (Me2Im)·B2neop2 to 105.5 kJ mol−1 for (Me2Im)2·B2neop2 (Fig. 2c). The reason for this weakening lies in the stabilization of the resulting boryl radicals by delocalization on the NHC. The decrasing B–B bond strengths can be demonstrated by the calculated Wiberg bond indices for the B–B bonds, which decrease going from B2neop2 (1.05) to (Me2Im)·B2neop2 (0.97) to (Me2Im)2·B2neop2 (0.85, Fig. 3c). Our EPR data for 1a are in accordance with those previously reported for NHC–BH2˙ radicals.20,22,24 The structure of NHC–BR2˙ radical Me2Im–Bneop˙ and its spin density distribution were computed by DFT calculations (Fig. 2b). The NHC-boryl radical is nearly planar with the spin density delocalized between the boron atom and the NHC ring.
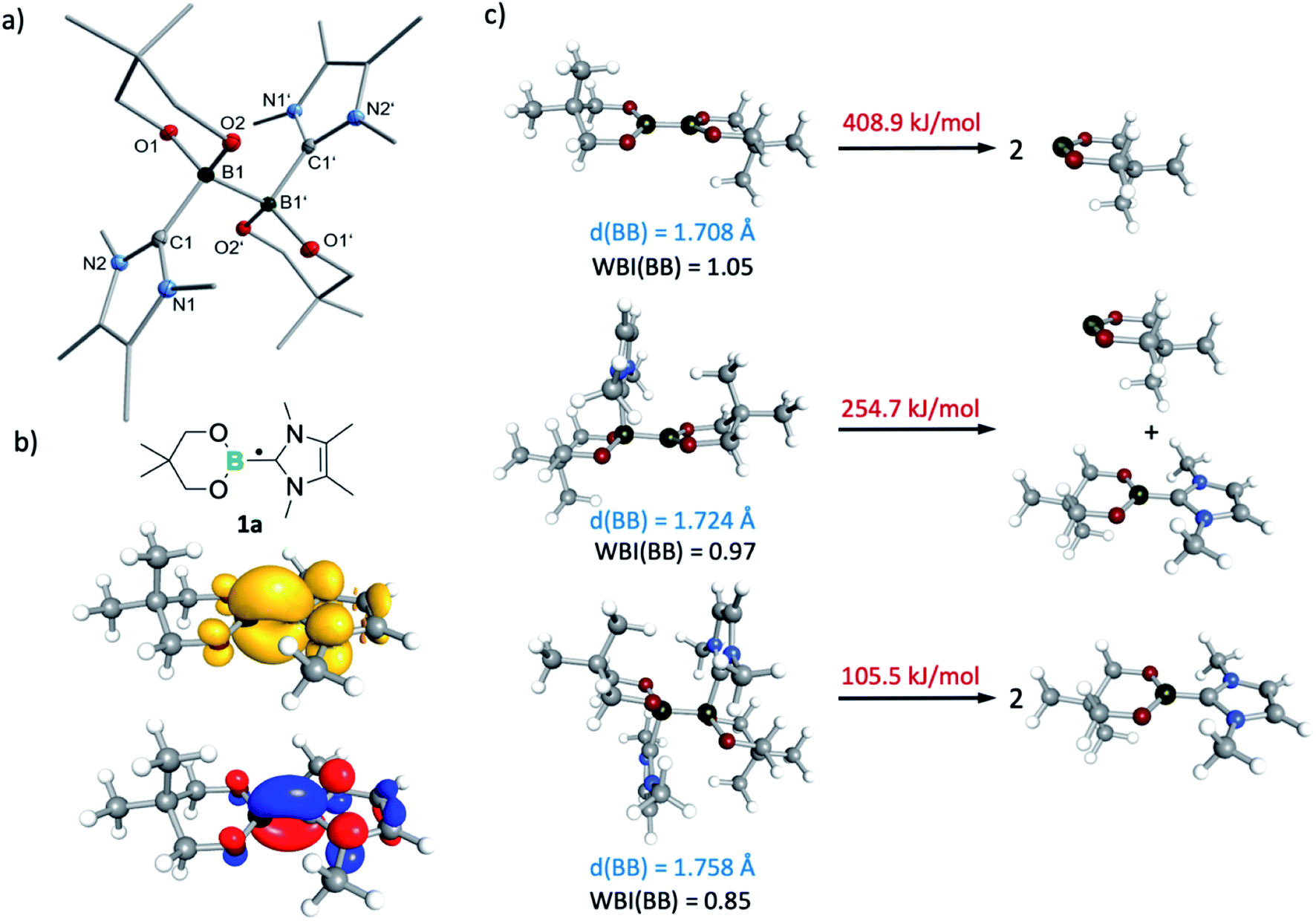 | ||
| Fig. 2 (a) Molecular structure of (Me2ImMe)2·B2neop21. Hydrogen atoms are omitted for clarity and the thermal ellipsoids are drawn at 50% probability. Selected bond lengths [Å] and angles [°]: B1–B1′ 1.770(2), B1–C1 1.662(2), B1–O1 1.482(1), B1–O2 1.486(1), C1–B1–O1 109.82(8), C1–B1–O2 110.13(8). (b) Schematic structure of the NHC–BR2˙ radical 1a (top) and the DFT-calculated (M06-2x//def2-TZVPP(B)/def2-TZVP) spin density (middle) and SOMO (bottom) for the (Me2Im)·Bneop˙ radical. (c) DFT computed (M06-2x//def2-TZVPP(B)/def2-TZVP) B–B bond lengths (blue), B–B Wiberg bond indices (black) and B–B dissociation energies (red) for B2neop2, (Me2Im)·B2neop2 and (Me2Im)2·B2neop2. | ||
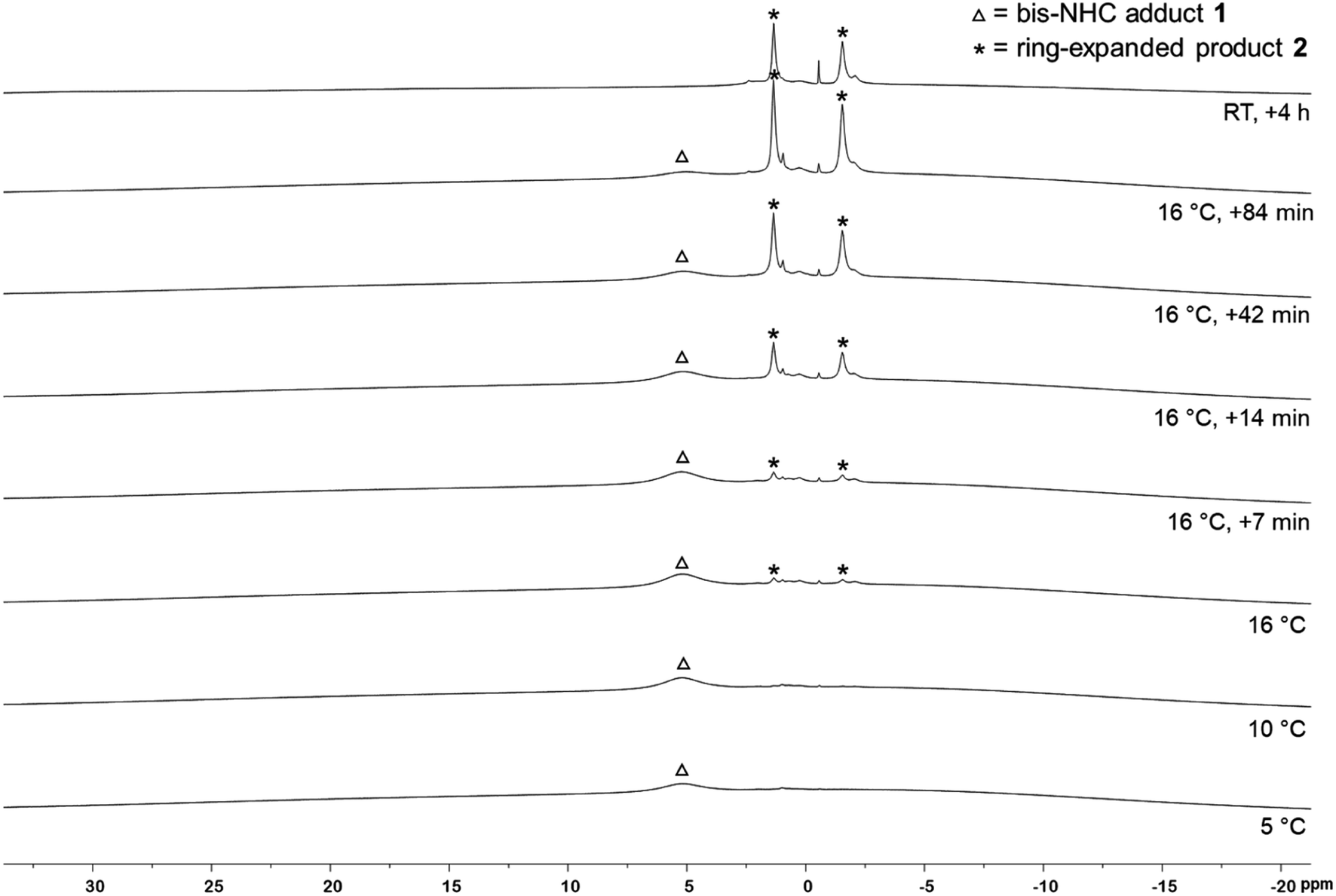 | ||
| Fig. 3 11B VT-NMR spectra of the RER of B2neop2 with two equivalents Me2ImMe in d8-THF (160 MHz). Triangle: bis-NHC adduct (Me2ImMe)2·B2neop21. Asterisk: ring-expanded product RER-(Me2ImMe)2·B2neop22. | ||
To verify that the EPR signal detected arose from a species preceding the RER of 1, we monitored the reaction by VT 1H and 11B{1H} NMR spectroscopy (Fig. 3). While the 1H NMR spectra show broad signals as expected, the presence of the bis-NHC adduct (Me2ImMe)2·B2neop21 at low temperatures and the onset of the ring expansion reaction at 16 °C were easy followed by 11B{1H} NMR (Fig. 3). Below 5 °C, no signal was detected, due to the poor solubility of the starting materials. Upon warming to 5 °C, a broad signal for the bis-NHC adduct 1 was detected at 5.12 ppm, which is significantly shifted upfield from that of free B2neop2 (28.4 ppm),37 as a result of rehybridization at boron from sp2 to sp3.
At 16 °C, the signals for the ring-expanded product RER-(Me2ImMe)2·B2neop22 began to arise at 1.83 and −1.35 ppm. The reaction was complete within 90 min. This observation, i.e., that the RER began at the same temperature (16 °C) according to NMR spectroscopy as the EPR resonance was observed at (16.9 °C), supports the assumption that the EPR signal detected is derived from a species which proceeds the RER. Another NMR spectrum recorded after 4 h at room temperature revealed that 2 had already started to decompose, which is in accordance with our previous observations.35c
Following studies by Stephan et al. on the reactivity of a doubly base-stabilized diboron(4) compound, which undergoes homolytic cleavage of the weakened B–B bond,38 we sought to provide experimental evidence for our in situ observed NHC–BR2˙ moiety 1a. Therefore, we tried to trap 1a at temperatures below the onset of the RER, as well as during the RER process, with common radical scavengers including TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl), N-tert-butyl-α-phenylnitrone, diphenyl disulfide, and sulfur. However, these attempts were unsuccessful as the intramolecular RER predominated. Based on the work of Curran, Lacôte, and Lalevée on radical polymerization reactions,26 we then employed radical 1a as an initiator for the polymerization of styrene. Therefore, 5.0 wt% of the diboron(4) compound and two equivalents of the NHC were combined in precooled (0 °C) styrene, which was used as the solvent. The reaction mixture was allowed to warm slowly to room temperature and was stirred for 16 h. Upon adding an excess of methanol, a colorless precipitate formed, which was identified as polystyrene by size exclusion chromatography (SEC) analysis (Fig. 4). The number-average molecular weight (Mn) of the polystyrene formed was determined to 8540 Da, with a dispersity (Đ) of 1.74. The analogous reaction using only the diboron(4) compound (without the NHC) did not lead to the formation of polystyrene. Thus, we confirmed experimentally the formation of radical 1a as it successfully initiated the polymerization of styrene giving polystyrene with 90% conversion.
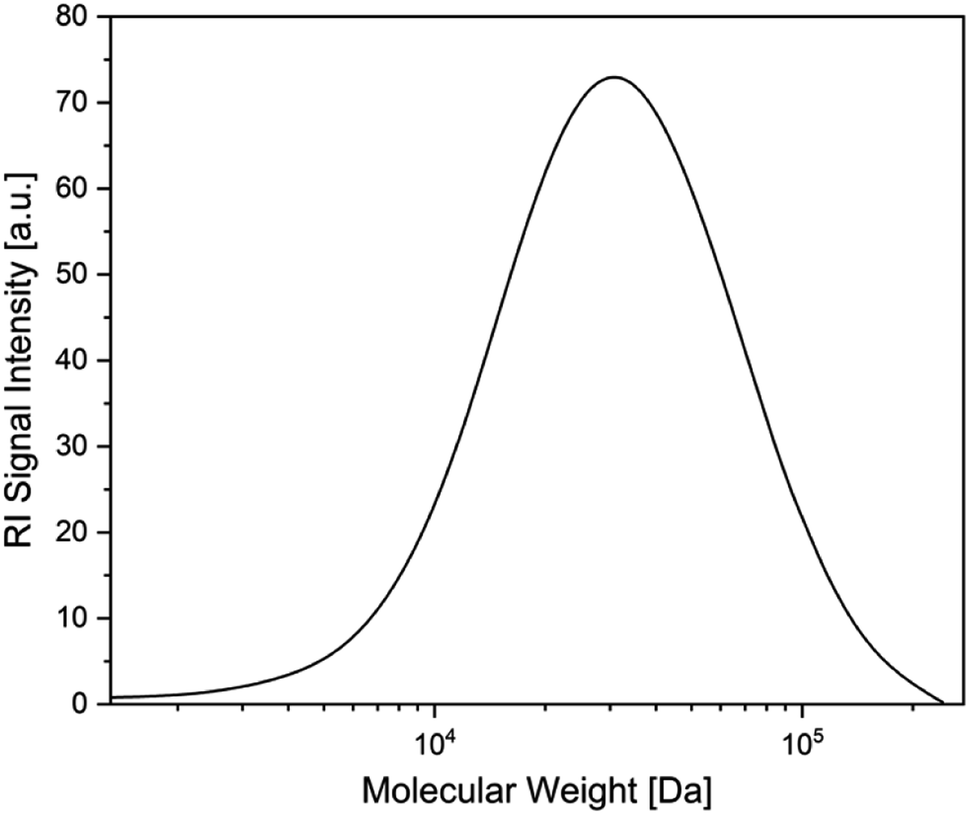 | ||
| Fig. 4 Size exclusion chromatography (SEC) analysis of polystyrene. | ||
Attempts to apply the boryl radical 1a in metal-free borylation reactions of aryl iodides failed. The ring-expanded product 2 and the unreacted substrate employed were always obtained. Given that the NHC-boryl radical arising from the homolytic scission of 1a is able to add to a styrene monomer before leading to 2, it is likely that an sp2 boron is required for the metal-free borylation to proceed.
To probe this issue, we thus investigated diboron(4) compounds with only one boron atom coordinated by an NHC (sp2–sp3 adducts). The B–B bond in these compounds is less activated, making them stable towards RER (with the exception of Dipp2Sim·B2cat2 at elevated temperatures35c). As the corresponding mono-NHC adduct Me2ImMe·B2neop2, however, is not stable and begins to decompose during its preparation,39 we synthesized the mono-NHC adduct Me2ImMe·B2pin23 in 73% yield using B2pin2 as the diboron(4) compound. Compound 3 was characterized by NMR spectroscopy, HRMS, and elemental analysis.
To study a possible transfer of one boryl moiety of a mono-NHC adduct to aryl iodides, adduct 3 was treated stoichiometrically with 4-iodotoluene 4I (Scheme 3) in C6D6 and the reaction was monitored via1H NMR spectroscopy. At 80 °C, the first new signals appeared within 1 h, whereas full conversion of 4I was achieved after the addition of 2.5 equivalents of 3. The new signals match those reported for the desired C–I bond borylated product 4a,40 which was additionally verified by GC/MS and HRMS. Interestingly, the C–C coupling product of the aryl halide with the solvent C6D6 was detected as a byproduct 4b in 25% yield (Scheme 3). Compound 4b, as well as the analogous coupling product 4b-H, which was obtained by running the reaction in non-deuterated benzene, were isolated and characterized via NMR spectroscopy, GC/MS, and HRMS.
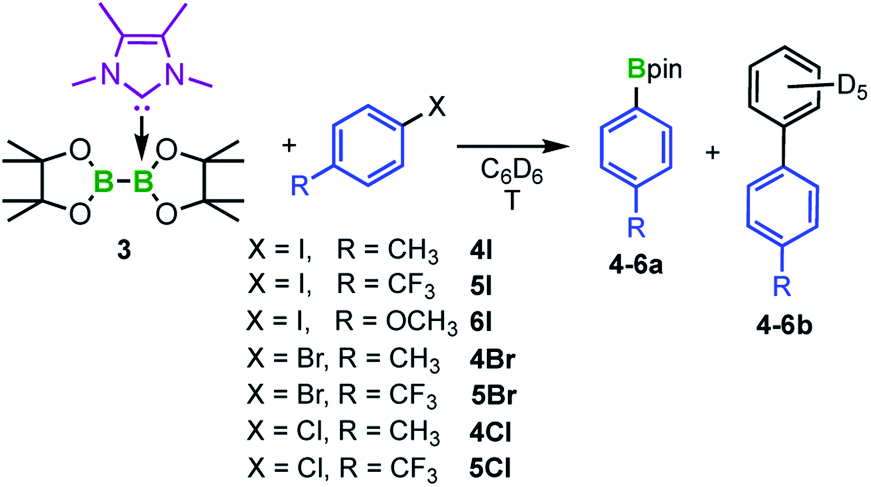 | ||
| Scheme 3 Reaction of the mono-NHC adduct Me2ImMe·B2pin23 with aryl halides (X = I, Br, Cl) forming the corresponding C–X bond borylation products 4–6a and the C–C coupling products 4–6b between the substrate and the solvent C6D6. | ||
Further reactivity studies were conducted for the reaction of 3 with different para-substituted aryl iodides, bromides, and chlorides (Scheme 3). Full conversion was achieved for the aryl iodides 4I–6I and aryl bromides 4Br and 5Br. The ratio of the borylated arene and the C–C coupling side product strongly depends on the reaction temperature (Table 1). Lower temperatures (50 °C) diminish the formation of the byproduct from 25% to 7% (cf.Table 1, entries 1 and 2), but also decrease the reaction rate. For example, in the case of 4-iodo-benzotrifluoride 5I, starting material was no longer detectable after 1.5 h at 80 °C, whereas at a reaction temperature of 50 °C, full conversion was achieved only after 16 h (Fig. 5). For 5I and 5Br, the borylated arene was obtained in solution in the form of its Me2ImMe adduct (4-F3C–C6H4-Bpin·Me2ImMe5a-ADD), possibly due to the more Lewis acidic boron atom resulting from the electron-withdrawing CF3-substituent. This observation explains why an additional 0.5 equivalents of Me2ImMe·B2pin23 are required for full conversion of 5I and 5Br. For comparison, 5a-ADD was independently synthesized and isolated (for details see ESI†).
| Entry | R | T [°C] | 4–6a [%] | 4–6b [%] |
|---|---|---|---|---|
| a Reaction conditions: 1.0 equivalent of the aryl halide, 2.5 equivalents of 3, C6D6; yields were determined via quantitative 1H NMR spectroscopy, as well as GC/MS using biphenyl as an internal standard. Isolated yields in parentheses. b 3.0 equivalents of 3. c In solution, the boron atom is coordinated by Me2ImMe forming the NHC adduct 5a-ADD. | ||||
| 1 | CH34I | 80 | 75 | 25 |
| 2 | CH34I | 50 | 93 (85) | 7 |
| 3 | CF35Ib | 80 | 44c | 56 |
| 4 | CF35Ib | 50 | 55c (44) | 45 |
| 5 | OCH36I | 80 | 80 | 20 |
| 6 | OCH36I | 50 | 91(82) | 9 |
| 7 | CH34Br | 80 | 73 | 27 |
| 8 | CH34Br | 50 | 92 (86) | 8 |
| 9 | CF35Brb | 80 | 28c | 72 |
| 10 | CF35Brb | 50 | 70c | 30 |
| 11 | CH34Cl | 80 | Trace | Trace |
| 12 | CF35Cl | 80 | 20 | 12 |
 | ||
| Fig. 5 In situ 1H NMR spectra (aromatic region) of the reaction of the mono-NHC adduct Me2ImMe·B2pin23 and 4-iodo-benzotrifluoride 5I in C6D6 at 50 °C (400 MHz). Diamond: 4-iodobenzotrifluoride 5. Triangle: aryl boronic ester 5a (* in solution, the boron atom is coordinated by Me2ImMe forming the NHC adduct 5a-ADD). Square: C–C coupling product 5b. | ||
For substrate 5I, the reaction also proceeds slowly at room temperature, with 84% conversion after one week, and a 5a/b ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The absence of light did not have any impact on the reaction outcome. For aryl chlorides, no reaction occurred at temperatures of 50 °C. The reaction of the aryl chloride 4Cl led to traces of 4a at 80 °C, whereas for the more activated aryl chloride 5Cl, 20% of the aryl boronic ester 5a and 12% of the coupling product 5b were observed at 80 °C (Table 1, entries 11–12). The aryl boronic esters were isolated and characterized by NMR spectroscopy, GC/MS, and HRMS.
:1. The absence of light did not have any impact on the reaction outcome. For aryl chlorides, no reaction occurred at temperatures of 50 °C. The reaction of the aryl chloride 4Cl led to traces of 4a at 80 °C, whereas for the more activated aryl chloride 5Cl, 20% of the aryl boronic ester 5a and 12% of the coupling product 5b were observed at 80 °C (Table 1, entries 11–12). The aryl boronic esters were isolated and characterized by NMR spectroscopy, GC/MS, and HRMS.
Attempts to suppress the C–C coupling side product by performing the reaction in MTBE (methyl tert-butyl ether) instead of benzene, not providing any aromatic C–H bonds in the solvent, resulted in hydrodehalogenation of the aryl halide.
We then analyzed the reaction mixture of the completed reaction of 3 with 5I by NMR spectroscopy and GC/MS. Besides the aryl boronic ester 5a and the C–C coupling product 5b, of 5I with C6D6, residual B2pin2 was detected in solution. The precipitate that formed during the reaction was redissolved in chloroform and analyzed by GC/MS and NMR spectroscopy. While the GC/MS only showed traces of 5a and B2pin2, 1H NMR spectroscopy additionally revealed the formation of two other species (see ESI, Fig. S1†), possibly of an ionic nature, as they were not detectable by GC/MS. In the corresponding 11B{1H} NMR spectrum, besides B2pin2, only one significant signal was observed with a chemical shift of 1.77 ppm which, together with HRMS, and X-ray diffraction analysis (Fig. 6) was assigned to the boronium cation [(Me2ImMe)2·Bpin]+7 with iodide as the counterion. This compound might demonstrate the fate of the second boryl moiety in our boryl transfer reaction based on the mono-NHC adduct 3 (see below). The amount of 7 formed is, according to the amount of isolated residue and estimation of the ratio of 7 to the unknown species, vide infra, in the 1H NMR spectrum, appropriate for it being formed stoichiometrically during the generation of the borylated arene. Thus, the formation of 7 as only a minor side product can be excluded.
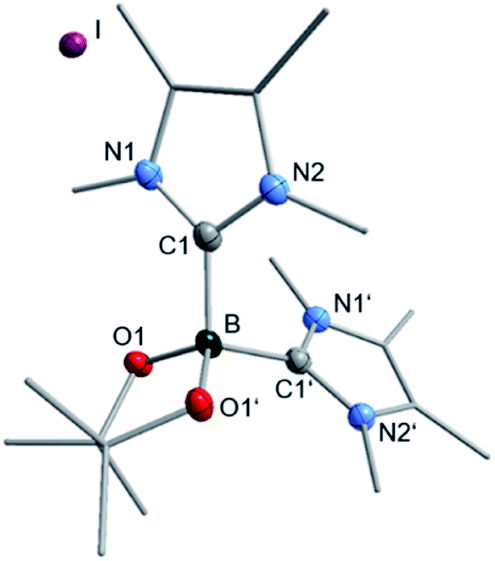 | ||
| Fig. 6 Molecular structure of [(Me2ImMe)2·Bpin]+I−7. Hydrogen atoms are omitted for clarity and thermal ellipsoids are drawn at 50% probability. Selected bond lengths [Å] and angles [°]: B–C1 1.663(4), B–O1 1.471(3), C1–B–O1 110.80(12), C1–B–O1′ 111.23(12), C1–B1–C1′ 106.9(3). | ||
The boron atom in [(Me2ImMe)2·Bpin]+I−7 is four-coordinate, bound to the pinacolato unit and the two NHCs as σ-donating ligands, with a B–C1 bond distance of 1.663(4) Å. The data obtained match those previously reported for boronium ions.41
As, for the bis-NHC adduct (Me2ImMe)2·B2neop21, we observed homolytic bond cleavage forming radical 1a, we then started preliminary mechanistic investigations to determine how this boryl transfer from the mono-NHC adduct 3 to the aryl halide proceeds, and whether boryl radicals might be involved.
The C–C coupling with the solvent, as well as the observed hydrodehalogenation, are preliminary indications that aryl radicals are formed from the aryl halides.42 The cyclic voltammograms (CV) of 4-iodotoluene 4I, and Me2ImMe were measured in THF (for details see ESI, Fig. S4†). The redox potentials are given versus Ag/Ag+ instead of Fc/Fc+, as the free NHC reacts with Fc+ and THF resulting in the imidazolium salt.43 For 4I, an irreversible reduction was observed at Ec = −2.63 V, showing a high electrochemical stability with respect to the acceptance of an electron. An irreversible oxidation for Me2ImMe was detected at Ea = −1.17 V and its reduction potential is Ec = −2.99 V. Thus, the formation of an aryl radical by reduction of the aryl iodide 4 by electron transfer from the free NHC can be excluded by comparing their respective potentials. However, it is conceivable that some NHCs can dimerize, leading to a tetra-amino alkene. These molecules are super electron donors, capable of generating radicals from aryl halides.44 Therefore, free NHCs are potentially conducive to the formation of radicals in the presence of aryl halides. The feasibility of this type of radical generation was corroborated by the fact that 1:1 mixture of 4-iodotoluene or 4-bromotoluene, respectively, and the NHC (5.0 wt% each) lead to polymerization of styrene (see ESI†).
Moreover, the reaction of 4I with stoichiometric amounts of the free NHC in the absence of B2pin2 in benzene at 80 °C also generated the C–C coupling product 4b-H in 26% isolated yield (Scheme 4). The precipitate that was formed during the reaction was identified as the imidazolium salt [4-CH3-Ph-Me2ImMe]+I−8, characterized by GC/MS (detected as m/z = 200 [M-CH3I]+), HRMS and NMR spectroscopy. Besides the imidazolium salt, another, thus far unidentified, species is formed as a side product (Scheme 4). Compound 8 was not detected in the residue of the reaction mixture of the analogous reaction in the presence of B2pin2.
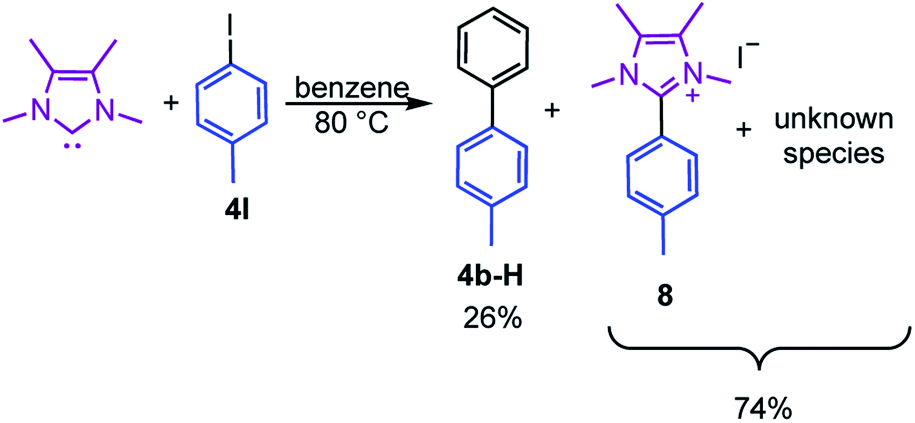 | ||
| Scheme 4 Reaction of the free NHC Me2ImMe with 4-iodotoluene 4I at 80 °C forming the C–C coupling product with the solvent 4b-H, the imidazolium salt [4-CH3-Ph-Me2ImMe]+I−8, and an unknown compound. | ||
The signals of the second species detected in the 1H NMR spectrum (Fig. 7) at a chemical shift of 2.24 and 3.86 ppm (indicated by squares), match those set of signals found in the 1H NMR spectrum recorded of the residue formed during the boryl transfer reaction (2.24 and 3.88 ppm; cf. Fig. S1†). Thus, as this compound is also generated in the absence of boron and does not show any resonances in the aromatic region, it presumably must be formed from Me2ImMe. The most obvious assumption is that the signals arise from the imidazolium salt [Me2ImMeH]+I−, generated by protonation of the free NHC during the coupling of 4I with benzene, as two singlets are detected in the expected region for the CH3 protons of the backbone (2.24 ppm) and the NCH3 protons (3.86 ppm) in a 1:1 ratio. However, no signal is detected for the CH proton at the former carbene carbon atom, and comparison of the signals with those of previously prepared [Me2ImMeH]+I− (recorded in CDCl3) also disproves this assumption (2.20 and 3.81 ppm). The possibility of residual free NHC in the residue reacting with CDCl3 forming the analogous imidazolium salt [Me2ImMeD]+Cl− was also excluded by a control experiment (2.14 and 3.78 ppm).
 | ||
| Fig. 7 1H NMR spectrum recorded in CDCl3 (400 MHz) of the residue formed in the reaction of stoichiometric amounts of the free NHC with 4-iodotoluene 4I showing the imidazolium salt [4-CH3-C6H4–Me2ImMe]+I−8, and an unknown compound (square). | ||
When the reaction shown in Scheme 4 was performed in the presence of stoichiometric amounts of the radical scavenger TEMPO, trace amounts of the coupling product aryl-TEMPO were detected by GC/MS, which provides further evidence for the existence of aryl radicals. The same coupling product between the aryl radical and TEMPO was observed in larger amounts by running our standard boryl transfer reaction using Me2ImMe·B2pin23 and 4-iodobenzotrifluoride 5I in the presence of TEMPO (for details see ESI, Fig. S5†); however, there was no evidence for trapping of a boryl moiety.
The CV of the mono-NHC adduct 3 reveals an irreversible oxidation at Ea = −1.17 V, and a second one at Ea = −0.08 V. The former potential is identical with that observed for the free NHC. DFT calculations by the groups of Marder and Lin6c showed that during the exchange of the NHC between the two boron atoms in a mono-NHC adduct, no transition state could be located for an intramolecular process. Instead, a dissociation–reassociation mechanism was suggested by their theoretical studies. Thus, it is suggested that some Me2ImMe can dissociate from B2pin2 in 3, which explains the CV. In all cases, it is unlikely that 3 serves as a single electron transfer reagent.
We next ran a reaction with only B2pin2 and 4-iodotoluene (1:1 ratio) 4I under our standard conditions. As expected, after 16 h at 80 °C, only starting materials were detected, which excludes the possibility of radicals generated by the diboron(4) compound and traces of oxygen.45 When 0.5 equivalents of NHC were added, ca. 12% conversion was obtained. Upon adding another 1.5 equivalents of B2pin2 to the reaction mixture to accommodate the need for 2.5 equivalents in our standard reaction, no further reaction progress was observed. The subsequent addition of another 0.5 equivalents of NHC resulted in 30% conversion, revealing that the NHC does not serve as a catalyst and is required in stoichiometric amounts, therefore doing more than just initiating the reaction. The need for 2.5 equivalents of B2pin2 is also required as, with smaller amounts, more of the C–C coupling side product 4b is generated. In general, the reaction also proceeds when B2pin2 and Me2ImMe are added separately to the reaction mixture but shows slightly increased conversion when the pre-prepared mono-NHC adduct Me2ImMe·B2pin23 is employed.
We then attempted the polymerization of styrene (as the solvent) at 80 °C using Me2ImMe·B2pin23 and 4-iodobenzotrifluoride 5I to see whether polystyrene is formed, which would provide further experimental proof for the presence of radicals over the course of our reaction. GPC analysis (for details see ESI, Fig. S6†) confirmed the formation of polystyrene; however, with a higher number-average molecular weight (Mn) of 25870 Da and a Đ of 4.15 than that observed in the lower temperature reaction, vide supra, initiated by 1a. The higher dispersity shows that there are likely many more initiation and termination steps at play. Therefore, the aryl radical is likely not the only radical species present, also providing circumstantial evidence for the presence of NHC-boryl radicals. The longer polymer chain indicates that there are likely less radicals generated which lead to fruitful initiations.
Our previous observations with 4-iodobenzotrifluoride 5I revealed that the boryl transfer proceeds slowly at room temperature (which is not the case for the other aryl iodides), thus prompting us to monitor the reaction by EPR spectroscopy. An EPR signal was detected; however, due to the very slow reaction progress at room temperature, it was very weak and ill-defined. Thus, the determination of hyperfine couplings and, hence, insight into the nature of the radical was precluded. Nonetheless, the isotropic g-value of 2.006, together with the broad linewidth, suggest the presence of a boron-based radical (for details see ESI, Fig. S7†).
All of the data presented lead us to propose a mechanism for the metal-free borylation process (Scheme 5).
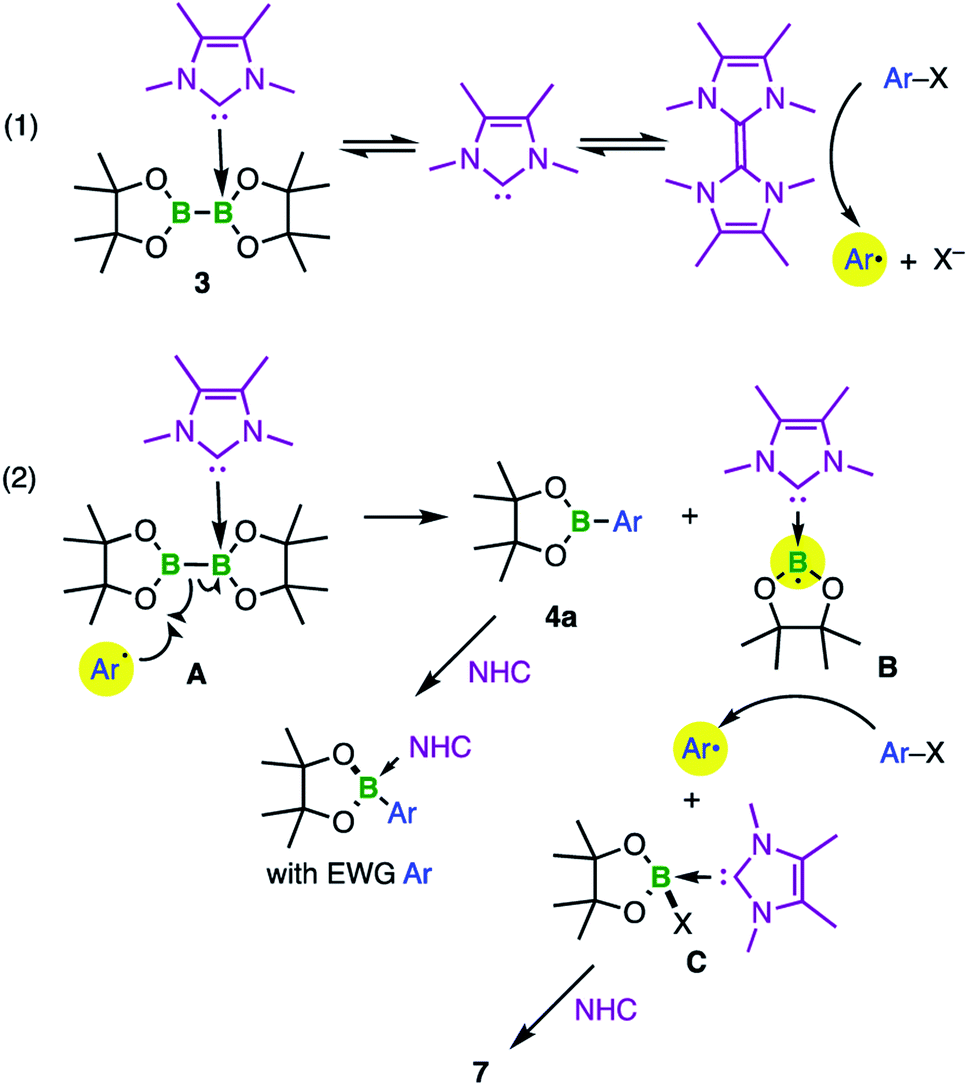 | ||
| Scheme 5 Proposed mechanism for the borylation of aryl halides. | ||
A small fraction of mono-adduct 3 is likely able to release the NHC, which ends up dimerizing. As soon as this happens, SET can occur with the aryl halide, which starts the radical chain reaction (initiation step, (1) in Scheme 5). Once the aryl radical is formed it can add to the sp2 boron of 3 (which explains why 1 does not lead to borylation of aryl halides). The B–B bond undergoes homolytic scission, presumably in a single elementary step A (homolytic substitution at boron), or via some associative mechanism (radical addition followed by radical release). In both cases, this delivers the borylated product as well as an NHC-boryl radical (B), which is stabilized by partial delocalization of the spin. B can then proceed to abstract the halogen atom from aryl halide 4, thus closing the radical chain. Alternatively, the aryl radicals can also react with the solvent, leading to the biaryls observed. We did not try to optimize this further as our focus was on the fate of the diboron compound, but the insights obtained may prove useful to develop an alternative to the pyridine-based metal-free borylations.
After termination, the desired arylboronate product can intercept the NHC, provided the Lewis acidity of the product is high enough (as is the case for the reactions of Table 1, entries 9–10). Finally, the boron byproduct C can also react with the NHC, which leads to boronium ion 7 (Fig. 6). The higher Lewis basicity of the carbenes compared with pyridines prevents the catalytic turnover of the Lewis base, which allowed us to isolate the key byproduct 7.
Conclusions
Former studies by the groups of Marder and Radius have shown that bis-NHC adducts of diboron(4) compounds are thermally unstable, undergoing ring expansion reactions (RER) via the insertion of one B(OR)2 moiety into the C–N bond of one NHC with the second B(OR)2 moiety bound exo to the former carbene–carbon atom.35 Based on these findings, the reaction of B2neop2 with two equivalents of Me2ImMe was followed by 11B NMR spectroscopy revealing that, at low temperatures, the bis-NHC adduct (Me2ImMe)2·B2neop21 is formed which, at 16 °C, starts undergoing a ring expansion reaction. Monitoring the same reaction by EPR spectroscopy provided evidence for the RER proceeding via the formation of a boryl radical of the type NHC–BR2˙ 1a, presumably formed by homolytic B–B bond cleavage. Radical 1a was successfully applied as an initiator for the radical polymerization of styrene. Further investigations with respect to applying the boryl moiety 1a in a metal-free borylation reaction by suppressing the RER failed. Therefore, the mono-NHC adduct Me2ImMe·B2pin23 was synthesized. Because it is a mono-adduct, its B–B bond is less activated and therefore stable towards RER. The stoichiometric reaction of 3 with different substituted aryl iodides and bromides in benzene, at elevated temperatures, led to boryl transfer and gave the desired aryl boronic esters in good yields. Interestingly, varying amounts of a C–C coupling product between the aryl halide and the solvent (benzene), depending on the reaction temperature, were detected as a side product. Further studies concerning the mechanism of this boryl transfer reaction revealed that radicals are likely involved, as an aryl radical was trapped by TEMPO, and running the boryl transfer reaction in styrene led to its polymerization. Monitoring the reaction by EPR spectroscopy revealed a signal (though very weak and ill-defined), which is suggestive of a boron-based radical. Furthermore, the boronium cation [(Me2ImMe)2·Bpin]+I−7 was formed stoichiometrically during the boryl transfer reaction, possibly demonstrating the fate of the second boryl moiety. NHCs are beneficial when strong complexation (and simultaneous homolytic cleavage of the B–B bond) is needed, as is the case for initiation of radical polymerizations, or stabilization of crucial intermediates in the metal-free borylation. Further work will focus on the development of safe, low-temperature initiators for radical polymerizations (in contrast to, e.g., azo initiators which decompose at room temperature, and are intrinsically dangerous) as the bis-adducts could be assembled within the polymerization vessel only when needed.Data availability
Additional data and spectra, and crystallographic data, NMR spectra, and Cartesian coordinates for calculations can be found in the ESI.†Author contributions
All authors contributed to research, writing, and editing of the article. E. L., T. B. M. and U. R. designed the project; L. K., L. Z., L. W., M. S. and S. W.-P. designed and performed the experiments; L. K., L. W. and U. R. performed the XRD studies, U. R. designed and performed the computational studies; H. B. and I. K. designed and performed the EPR studies; L. Z. performed CV studies; L. K., E. L., T. B. M. and U. R. wrote the manuscript with input from all the other co-authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by funds from the Julius-Maximilians-Universität Würzburg and the Deutsche Forschungsgemeinschaft (DFG). We thank AllyChem Co. Ltd. for a generous gift of diboron(4) reagents and Johanna Lutz for the GPC measurements. E. L. thanks the Alexander von Humboldt Foundation for a Bessel Award.References
- (a) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011 CrossRef; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- (a) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS PubMed; (b) S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799–1803 CrossRef CAS PubMed; (c) S. K. Bose, A. Deißenberger, A. Eichhorn, P. G. Steel, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2015, 54, 11843–11847 CrossRef CAS PubMed; (d) J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS PubMed; (e) S. K. Bose, S. Brand, H. O. Omoregie, M. Haehnel, J. Maier, G. Bringmann and T. B. Marder, ACS Catal., 2016, 6, 8332–8335 CrossRef CAS; (f) Y.-M. Tian, X.-N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS PubMed; (g) L. Kuehn, M. Huang, U. Radius and T. B. Marder, Org. Biomol. Chem., 2019, 17, 6601–6606 RSC; (h) L. Kuehn, D. G. Jammal, K. Lubitz, T. B. Marder and U. Radius, Chem.–Eur. J., 2019, 25, 9514–9521 CrossRef CAS PubMed; (i) Y.-M. Tian, X.-N. Guo, Z. Wu, A. Friedrich, S. A. Westcott, H. Braunschweig, U. Radius and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 13136–13144 CrossRef CAS PubMed; (j) Y.-M. Tian, X.-N. Guo, I. Krummenacher, Z. Wu, J. Nitsch, H. Braunschweig, U. Radius and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18231–18242 CrossRef CAS PubMed; (k) M. Huang, Z. Wu, J. Krebs, A. Friedrich, X. Luo, S. A. Westcott, U. Radius and T. B. Marder, Chem.–Eur. J., 2021, 27, 8149–8158 CrossRef CAS PubMed.
- (a) V. Lillo, A. Bonet and E. Fernández, Dalton Trans., 2009, 2899–2908 RSC; (b) K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Tetrahedron, 2015, 71, 2183–2197 CrossRef CAS; (c) V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef CAS; (d) K. Kubota, H. Iwamoto and H. Ito, Org. Biomol. Chem., 2017, 15, 285–300 RSC; (e) H. Yoshida, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (f) J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed; (g) D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (h) S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562–4565 CrossRef CAS PubMed; (i) Y.-M. Tian, X.-N. Guo, H. Braunschweig, U. Radius and T. B. Marder, Chem. Rev., 2021, 121, 3561–3597 CrossRef CAS PubMed; (j) S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed; (k) J. Hu, M. Ferger, Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC; (l) K. Muthuvel and T. Gandhi, ChemCatChem, 2022, e202101579 CAS.
- (a) R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC; (b) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed; (c) S. Würtemberger-Pietsch, U. Radius and T. B. Marder, Dalton Trans., 2016, 45, 5880–5895 RSC; (d) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC; (e) L. Kuehn, M. Stang, S. Würtemberger-Pietsch, A. Friedrich, H. Schneider, U. Radius and T. B. Marder, Faraday Discuss., 2019, 220, 350–363 RSC; (f) M. Huang, J. Hu, S. Shi, A. Friedrich, J. Krebs, S. A. Westcott, U. Radius and T. B. Marder, Chem.–Eur. J., 2022, 28, e202200480 CAS.
- (a) K.-s. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253–7255 CrossRef CAS PubMed; (b) K.-s. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2010, 132, 12766 CrossRef CAS; (c) C. Kleeberg, A. G. Crawford, A. S. Batsanov, P. Hodgkinson, D. C. Apperley, M. S. Cheung, Z. Lin and T. B. Marder, J. Org. Chem., 2012, 77, 785–789 CrossRef CAS PubMed.
- (a) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846–1849 CrossRef CAS PubMed; (b) D. Qiu, L. Jin, Z. Zheng, H. Meng, F. Mo, X. Wang, Y. Zhang and J. Wang, J. Org. Chem., 2013, 78, 1923–1933 CrossRef CAS PubMed.
- (a) G. Yan, D. Huang and X. Wu, Adv. Synth. Catal., 2018, 360, 1039 CrossRef CAS; (b) F. W. Friese and A. Studer, Chem. Sci., 2019, 10, 8503–8518 RSC; (c) T. Taniguchi, Eur. J. Org. Chem., 2019, 6308–6319 CrossRef CAS; (d) L. Zheng, L. Cai, K. Tao, Z. Xie, Y.-L. Lai and W. Guo, Asian J. Org. Chem., 2021, 10, 711–748 CrossRef CAS; (e) L. Qiang, Z. Lei and M. Fanyang, Acta Chim. Sin., 2020, 78, 1297–1308 CrossRef.
- (a) K. Oshima, T. Ohmura and M. Suginome, Chem. Commun., 2012, 48, 8571–8573 RSC; (b) T. Ohmura, Y. Morimasa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 2852–2855 CrossRef CAS PubMed.
- T. Ohmura, Y. Morimasa, T. Ichino, Y. Miyake, Y. Murata, M. Suginome, K. Tajima, T. Taketsugu and S. Maeda, Bull. Chem. Soc. Jpn., 2021, 94, 1894–1902 CrossRef CAS.
- (a) G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985–5989 CrossRef CAS PubMed; (b) G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904–3910 CrossRef CAS PubMed; (c) J. Cao, G. Wang, L. Gao, X. Cheng and S. Li, Chem. Sci., 2018, 9, 3664–3671 RSC.
- R. Xu, G.-p. Lu and C. Cai, New J. Chem., 2018, 42, 16456–16459 RSC.
- (a) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS PubMed; (b) L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC; (c) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef PubMed.
- S. Pinet, V. Liautard, M. Debiais and M. Pucheault, Synthesis, 2017, 49, 4759–4768 CrossRef CAS.
- W.-M. Cheng, R. Shang, B. Zhao, W.-L. Xing and Y. Fu, Org. Lett., 2017, 19, 4291–4294 CrossRef CAS PubMed.
- (a) A. Yoshimura, Y. Takamachi, L.-B. Han and A. Ogawa, Chem.–Eur. J., 2015, 21, 13930–13933 CrossRef CAS PubMed; (b) A. Yoshimura, Y. Takamachi, K. Mihara, T. Saeki, S.-i. Kawaguchi, L.-B. Han, A. Nomoto and A. Ogawa, Tetrahedron, 2016, 72, 7832–7838 CrossRef CAS.
- (a) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed; (b) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS PubMed.
- G. Li, G. Huang, R. Sun, D. P. Curran and W. Dai, Org. Lett., 2021, 23, 4353–4357 CrossRef CAS PubMed.
- (a) M. Huang, J. Hu, M. Tang, S. A. Westcott, U. Radius and T. B. Marder, Chem. Commun., 2022, 58, 395–398 RSC; (b) M. Huang, J. Hu, I. Krummenacher, A. Friedrich, H. Braunschweig, S. A. Westcott, U. Radius and T. B. Marder, Chem.–Eur. J., 2022, 28, e202103866 CAS.
- (a) P. P. Power, Chem. Rev., 2003, 103, 789–810 CrossRef CAS PubMed; (b) Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS; (c) T. Taniguchi, Chem. Soc. Rev., 2021, 8995–9021 RSC.
- (a) E. Krause and H. Polack, Ber. Dtsch. Chem. Ges., 1926, 59, 777–785 CrossRef; (b) T. L. Chu and T. J. Weismann, J. Am. Chem. Soc., 1956, 78, 23–26 CrossRef CAS; (c) W. Kaim and A. Schulz, Angew. Chem., Int. Ed., 1984, 23, 615–616 CrossRef; (d) A. Schulz and W. Kaim, Chem. Ber., 1989, 122, 1863–1868 CrossRef CAS; (e) L. Ji, R. M. Edkins, A. Lorbach, I. Krummenacher, C. Brückner, A. Eichhorn, H. Braunschweig, B. Engels, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2015, 137, 6750–6753 CrossRef CAS PubMed; (f) Y. Zheng, J. Xiong, Y. Sun, X. Pan and J. Wu, Angew. Chem., Int. Ed., 2015, 54, 12933–12936 CrossRef CAS PubMed; (g) F. Rauch, S. Fuchs, A. Friedrich, D. Sieh, I. Krummenacher, H. Braunschweig, M. Finze and T. B. Marder, Chem.–Eur. J., 2020, 26, 12794–12808 CrossRef CAS PubMed; (h) X. Jia, J. Nitsch, Z. Wu, A. Friedrich, J. Krebs, I. Krummenacher, F. Fantuzzi, H. Braunschweig, M. Moos, C. Lambert, B. Engels and T. B. Marder, Chem. Sci., 2021, 12, 11864–11872 RSC; (i) J. Krebs, M. Haehnel, I. Krummenacher, A. Friedrich, H. Braunschweig, M. Finze, L. Ji and T. B. Marder, Chem.–Eur. J., 2021, 27, 8159–8167 CrossRef CAS PubMed.
- (a) S.-H. Ueng, M. Makhlouf Brahmi, É. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS PubMed; (b) S. Telitel, A.-L. Vallet, S. Schweizer, B. Delpech, N. Blanchard, F. Morlet-Savary, B. Graff, D. P. Curran, M. Robert, E. Lacôte and J. Lalevée, J. Am. Chem. Soc., 2013, 135, 16938–16947 CrossRef CAS PubMed.
- (a) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (b) S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC.
- (a) S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS PubMed; (b) J. C. Walton, M. M. Brahmi, L. Fensterbank, E. Lacôte, M. Malacria, Q. Chu, S.-H. Ueng, A. Solovyev and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 2350–2358 CrossRef CAS PubMed.
- (a) P. R. Rablen and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4648–4653 CrossRef CAS; (b) P. R. Rablen, J. Am. Chem. Soc., 1997, 119, 8350–8360 CrossRef CAS.
- (a) D. P. Curran, A. Solovyev, M. Makhlouf Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed; (b) M.-A. Tehfe, M. M. Brahmi, J.-P. Fouassier, D. P. Curran, M. Malacria, L. Fensterbank, E. Lacôte and J. Lalevée, Macromolecules, 2010, 43, 2261–2267 CrossRef CAS; (c) S.-H. Ueng, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, Org. Lett., 2010, 12, 3002–3005 CrossRef CAS PubMed; (d) M.-A. Tehfe, J. Monot, M. M. Brahmi, H. Bonin-Dubarle, D. P. Curran, M. Malacria, L. Fensterbank, E. Lacôte, J. Lalevée and J.-P. Fouassier, Polym. Chem., 2011, 2, 625–631 RSC; (e) M.-A. Tehfe, J. Monot, M. Malacria, L. Fensterbank, J.-P. Fouassier, D. P. Curran, E. Lacôte and J. Lalevée, ACS Macro Lett., 2012, 1, 92–95 CrossRef CAS PubMed; (f) J. Lalevée, S. Telitel, M. A. Tehfe, J. P. Fouassier, D. P. Curran and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 5958–5961 CrossRef; (g) S. Telitel, S. Schweizer, F. Morlet-Savary, B. Graff, T. Tschamber, N. Blanchard, J. P. Fouassier, M. Lelli, E. Lacôte and J. Lalevée, Macromolecules, 2013, 46, 43–48 CrossRef CAS; (h) J. C. Walton, M. M. Brahmi, J. Monot, L. Fensterbank, M. Malacria, D. P. Curran and E. Lacôte, J. Am. Chem. Soc., 2011, 133, 10312–10321 CrossRef CAS.
- (a) T. Watanabe, D. Hirose, D. P. Curran and T. Taniguchi, Chem.–Eur. J., 2017, 23, 5404–5409 CrossRef CAS PubMed; (b) S.-C. Ren, F.-L. Zhang, J. Qi, Y.-S. Huang, A.-Q. Xu, H.-Y. Yan and Y.-F. Wang, J. Am. Chem. Soc., 2017, 139, 6050–6053 CrossRef CAS PubMed; (c) J. Qi, F.-L. Zhang, Y.-S. Huang, A.-Q. Xu, S.-C. Ren, Z.-Y. Yi and Y.-F. Wang, Org. Lett., 2018, 20, 2360–2364 CrossRef CAS PubMed; (d) J.-K. Jin, F.-L. Zhang, Q. Zhao, J.-A. Lu and Y.-F. Wang, Org. Lett., 2018, 20, 7558–7562 CrossRef CAS PubMed; (e) K. Takahashi, M. Shimoi, T. Watanabe, K. Maeda, S. J. Geib, D. P. Curran and T. Taniguchi, Org. Lett., 2020, 22, 2054–2059 CrossRef CAS PubMed; (f) Y.-S. Huang, J. Wang, W.-X. Zheng, F.-L. Zhang, Y.-J. Yu, M. Zheng, X. Zhou and Y.-F. Wang, Chem. Commun., 2019, 55, 11904–11907 RSC.
- For other examples, see ref. 7, 19, and 22.
- C.-W. Chiu and F. P. Gabbaï, Angew. Chem., Int. Ed., 2007, 46, 1723–1725 CrossRef CAS PubMed.
- T. Matsumoto and F. P. Gabbaï, Organometallics, 2009, 28, 4252–4253 CrossRef CAS.
- P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed.
- M. F. Silva Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135–1140 CrossRef CAS PubMed.
- (a) P. Nguyen, C. Dai, N. J. Taylor, W. P. Power, T. B. Marder, N. L. Pickett and N. C. Norman, Inorg. Chem., 1995, 34, 4290–4291 CrossRef CAS; (b) W. Clegg, C. Dai, F. J. Lawlor, T. B. Marder, P. Nguyen, N. C. Norman, N. L. Pickett, W. P. Power and A. J. Scott, J. Chem. Soc., Dalton Trans., 1997, 839–846 RSC.
- (a) S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, U. Radius and T. B. Marder, Chem.–Eur. J., 2015, 21, 9018–9021 CrossRef CAS PubMed; (b) S. Würtemberger-Pietsch, H. Schneider, T. B. Marder and U. Radius, Chem.–Eur. J., 2016, 22, 13032–13036 CrossRef PubMed; (c) M. Eck, S. Würtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. S. D. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius and T. B. Marder, Dalton Trans., 2017, 46, 3661–3680 RSC; (d) A. F. Eichhorn, S. Fuchs, M. Flock, T. B. Marder and U. Radius, Angew. Chem., Int. Ed., 2017, 56, 10209–10213 CrossRef CAS PubMed.
- M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. L. Santos, J. Org. Chem., 2011, 76, 3997–4007 CrossRef CAS PubMed.
- F. J. Lawlor, N. C. Norman, N. L. Pickett, E. G. Robins, P. Nguyen, G. Lesley, T. B. Marder, J. A. Ashmore and J. C. Green, Inorg. Chem., 1998, 37, 5282–5288 CrossRef CAS.
- L. L. Cao and D. W. Stephan, Organometallics, 2017, 36, 3163–3170 CrossRef CAS.
- S. Würtemberger-Pietsch, PhD thesis, Julius-Maximilians-Universität Würzburg, 2016.
- (a) M. Murata, T. Oyama, S. Watanabe and Y. Masuda, J. Org. Chem., 2000, 65, 164–168 CrossRef CAS PubMed; (b) A. Wolan and M. Zaidlewicz, Org. Biomol. Chem., 2003, 1, 3274–3276 RSC.
- (a) D. J. Brauer, H. Bürger, G. Pawelke, W. Weuter and J. Wilke, J. Organomet. Chem., 1987, 329, 293–304 CrossRef CAS; (b) W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed; (c) O. J. Metters, A. M. Chapman, A. P. M. Robertson, C. H. Woodall, P. J. Gates, D. F. Wass and I. Manners, Chem. Commun., 2014, 50, 12146–12149 RSC.
- (a) S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed; (b) G. Deng, K. Ueda, S. Yanagisawa, K. Itami and C.-J. Li, Chem.–Eur. J., 2009, 15, 333–337 CrossRef CAS PubMed.
- T. Ramnial, I. McKenzie, B. Gorodetsky, E. M. W. Tsang and J. A. C. Clyburne, Chem. Commun., 2004, 1054–1055 RSC.
- (a) J. A. Murphy, T. A. Khan, S.-z. Zhou, D. W. Thomson and M. Mahesh, Angew. Chem., Int. Ed., 2005, 44, 1356–1360 CrossRef CAS PubMed; (b) J. A. Murphy, S.-z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mahesh, S. R. Park, T. Tuttle and L. E. A. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178–5183 CrossRef CAS PubMed.
- C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415–3434 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed descriptions of the experimental procedures, product characterization data, NMR spectra, screening data, and data related to the computational studies. Crystal data collection and processing parameters. CCDC 2162556 (1) and CCDC 2162557 (7). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02096c |
| ‡ Present Address: Department of Chemistry, University of California, Irvine, CA 92697, USA |
| This journal is © The Royal Society of Chemistry 2022 |