DOI:
10.1039/D2SC01232D
(Edge Article)
Chem. Sci., 2022,
13, 5707-5717
Heme compound II models in chemoselectivity and disproportionation reactions†
Received
1st March 2022
, Accepted 11th April 2022
First published on 12th April 2022
Abstract
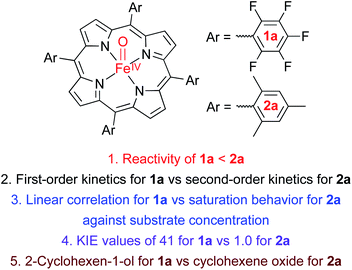
Heme compound II models bearing electron-deficient and -rich porphyrins, [FeIV(O)(TPFPP)(Cl)]− (1a) and [FeIV(O)(TMP)(Cl)]− (2a), respectively, are synthesized, spectroscopically characterized, and investigated in chemoselectivity and disproportionation reactions using cyclohexene as a mechanistic probe. Interestingly, cyclohexene oxidation by 1a occurs at the allylic C–H bonds with a high kinetic isotope effect (KIE) of 41, yielding 2-cyclohexen-1-ol product; this chemoselectivity is the same as that of nonheme iron(IV)-oxo intermediates. In contrast, as observed in heme compound I models, 2a yields cyclohexene oxide product with a KIE of 1, demonstrating a preference for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C epoxidation. The latter result is interpreted as 2a disproportionating to form [FeIV(O)(TMP+˙)]+ (2b) and FeIII(OH)(TMP), and 2b becoming the active oxidant to conduct the cyclohexene epoxidation. In contrast to 2a, 1a does not disproportionate under the present reaction conditions. DFT calculations confirm that compound II models prefer C–H bond hydroxylation and that disproportionation of compound II models is controlled thermodynamically by the porphyrin ligands. Other aspects, such as acid and base effects on the disproportionation of compound II models, have been discussed as well.
C epoxidation. The latter result is interpreted as 2a disproportionating to form [FeIV(O)(TMP+˙)]+ (2b) and FeIII(OH)(TMP), and 2b becoming the active oxidant to conduct the cyclohexene epoxidation. In contrast to 2a, 1a does not disproportionate under the present reaction conditions. DFT calculations confirm that compound II models prefer C–H bond hydroxylation and that disproportionation of compound II models is controlled thermodynamically by the porphyrin ligands. Other aspects, such as acid and base effects on the disproportionation of compound II models, have been discussed as well.
Introduction
Heme enzymes, such as cytochromes P450 (CYP 450), utilize high-valent iron(IV)-oxo porphyrin π-cation radical intermediates, referred to as compound I (Cpd I) and two oxidizing equivalents above the resting ferric state, to achieve highly efficient and selective oxidation reactions, such as in alkane hydroxylation and olefin epoxidation.1 In biomimetic studies, a number of synthetic Cpd I models have been synthesized and characterized with various spectroscopic techniques, and their reactivities have been investigated intensively in various oxidation reactions.2 In contrast, high-valent iron(IV)-oxo porphyrin intermediates, referred to as compound II (Cpd II) and one oxidizing equivalent above the resting ferric state, have been investigated less intensively in enzymatic and biomimetic reactions, since they are not the key active oxidants in enzymatic reactions and their reactivities are low compared to the strong oxidizing power of Cpd I intermediates.3 Therefore, the chemistry of Cpd II models still remains elusive in biomimetic oxidation reactions, although an elegant study on the acid-catalyzed disproportionation reaction of Cpd II models to Cpd I and iron(III) porphyrin complexes has been reported by Fujii and co-workers very recently.4
Iron(IV)-oxo intermediates have also been trapped and characterized in nonheme iron enzymes.5 In biomimetic studies, since the first crystal structure of a synthetic nonheme iron(IV)-oxo complex, [(TMC)FeIV(O)(MeCN)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane), was reported in 2003,6 a large number (>80) of nonheme iron(IV)-oxo complexes have been synthesized and characterized spectroscopically and/or structurally and their reactivities have been extensively investigated in various oxidation reactions, such as hydrogen atom transfer (HAT) and oxygen atom transfer (OAT) reactions;7 some of the nonheme iron(IV)-oxo complexes have shown reactivities similar to Cpd I models in the oxidation of organic substrates with a strong oxidizing power.8 However, in a couple of reactions, the reactivities and mechanisms of the nonheme iron(IV)-oxo complexes were shown to be different from those of Cpd I models.9,10a One example is the different chemoselectivities observed in the oxidation of cyclohexene by Cpd I models and nonheme iron(IV)-oxo complexes; Cpd I models afford cyclohexene oxide as the product,11 whereas nonheme iron(IV)-oxo complexes prefer allylic C–H bond hydroxylation to CC bond epoxidation (see Scheme 1B).10a The distinct chemoselectivity shown by nonheme metal(IV)-oxo complexes was interpreted with density functional theory (DFT) calculations.10
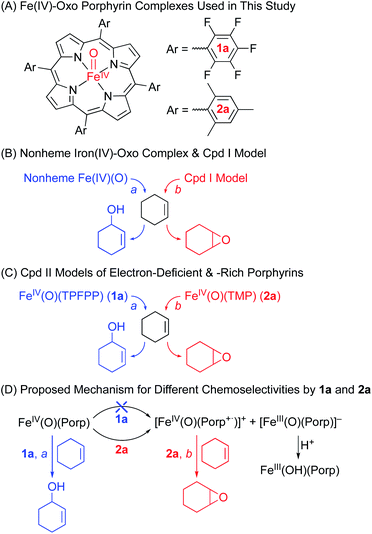 |
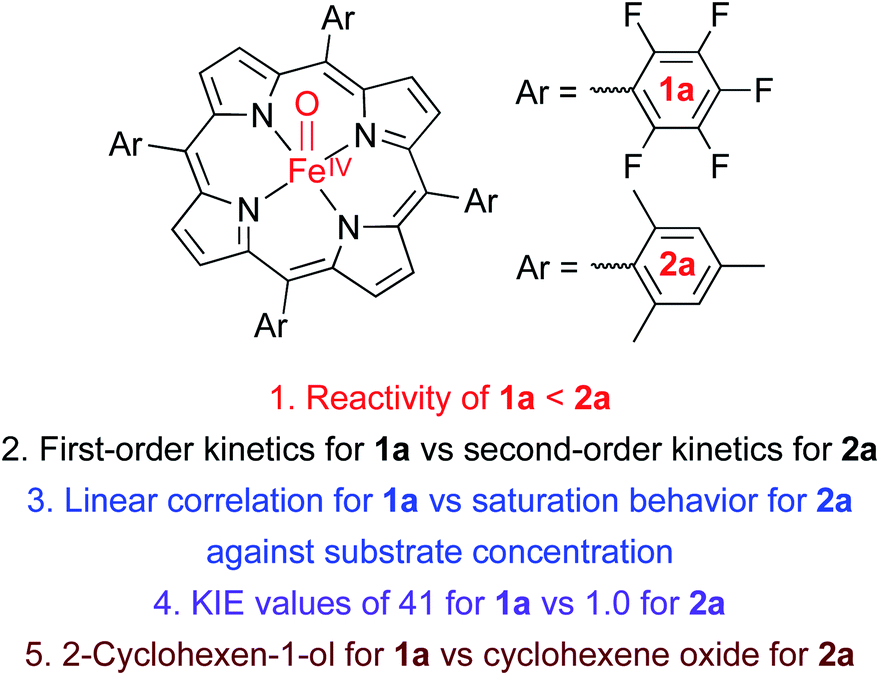
| | Scheme 1 (A) Structures of Cpd II models, 1a and 2a. (B) Chemoselectivities of nonheme iron(IV)-oxo and Cpd I complexes. (C) Chemoselectivities of Cpd II models with electron-deficient and -rich porphyrins. (D) Proposed mechanism for the different chemoselectivities of 1a and 2a. | |
As mentioned above, the chemistry of Cpd II models, iron(IV)-oxo porphyrin complexes, has been explored less intensively in oxidation reactions, compared to the well-studied Cpd I models and nonheme iron(IV)-oxo complexes. For example, while the electronic effect of porphyrin ligands, such as electron-rich versus electron-deficient porphyrins, has been well addressed in Cpd I models,11b,12,13 the porphyrin ligand effect(s) of Cpd II models in oxidation reactions has rarely been discussed. Also, the effect(s) of heme and nonheme ligands on the reactivities of iron(IV)-oxo complexes in oxidation reactions has rarely been compared. We therefore decided to investigate the reactivities of Cpd II models bearing electron-rich and -deficient porphyrins in the oxidation of cyclohexene; cyclohexene was chosen as a mechanistic probe to compare the chemoselectivity of Cpd II models to those of nonheme iron(IV)-oxo complexes (i.e., the effect of heme vs. nonheme ligands on the chemoselectivity of iron(IV)-oxo species) and Cpd I models (i.e., the chemoselectivity of Cpd I vs. Cpd II in heme systems) (Scheme 1B and C).10,11
We now report that the chemoselectivity of Cpd II models in the oxidation of cyclohexene varies depending on the electron-richness of the porphyrin ligands (see Scheme 1A for structures); that is, a Cpd II model with an electron-deficient porphyrin ligand, [FeIV(O)(TPFPP)(Cl)]− (1a, TPFPP = meso-tetrakis(pentafluorophenyl)porphyrinato dianion), affords the allylic oxidation product, as observed in the reactions of nonheme iron(IV)-oxo complexes (see Scheme 1B and C, reaction pathway a),10 whereas a Cpd II model with an electron-rich porphyrin ligand, [FeIV(O)(TMP)(Cl)]− (2a, TMP = meso-tetrakis(2,4,6-trimethylphenyl)porphyrinato dianion), yields the cyclohexene oxide product, as observed in the Cpd I model reactions (see Scheme 1B and C, reaction pathway b).11 We also report that the preference for CC bond epoxidation by 2a results from the disproportionation of 2a that forms Cpd I, [FeIV(O)(TMP+˙)]+ (2b), as the active oxidant that effects the epoxidation of cyclohexene to give the corresponding epoxide product selectively (see Scheme 1D, reaction pathway b). In contrast to 2a, the disproportionation of 1a does not occur under the reaction conditions and 1a abstracts a hydrogen atom (H-atom) from the allylic C–H bonds to give the allylic oxidation product (see Scheme 1D, reaction pathway a). The different chemoselectivities shown by 1a and 2a are then interpreted with DFT calculations. Other mechanistic aspects, such as the effects of acid and base on the disproportionation of Cpd II models (1a and 2a) and the reactivities of Cpd I models (i.e., 1b and 2b) in oxidation reactions, have been discussed as well.
Results and discussion
Synthesis of [FeIV(O)(TPFPP)(Cl)]− (1a)
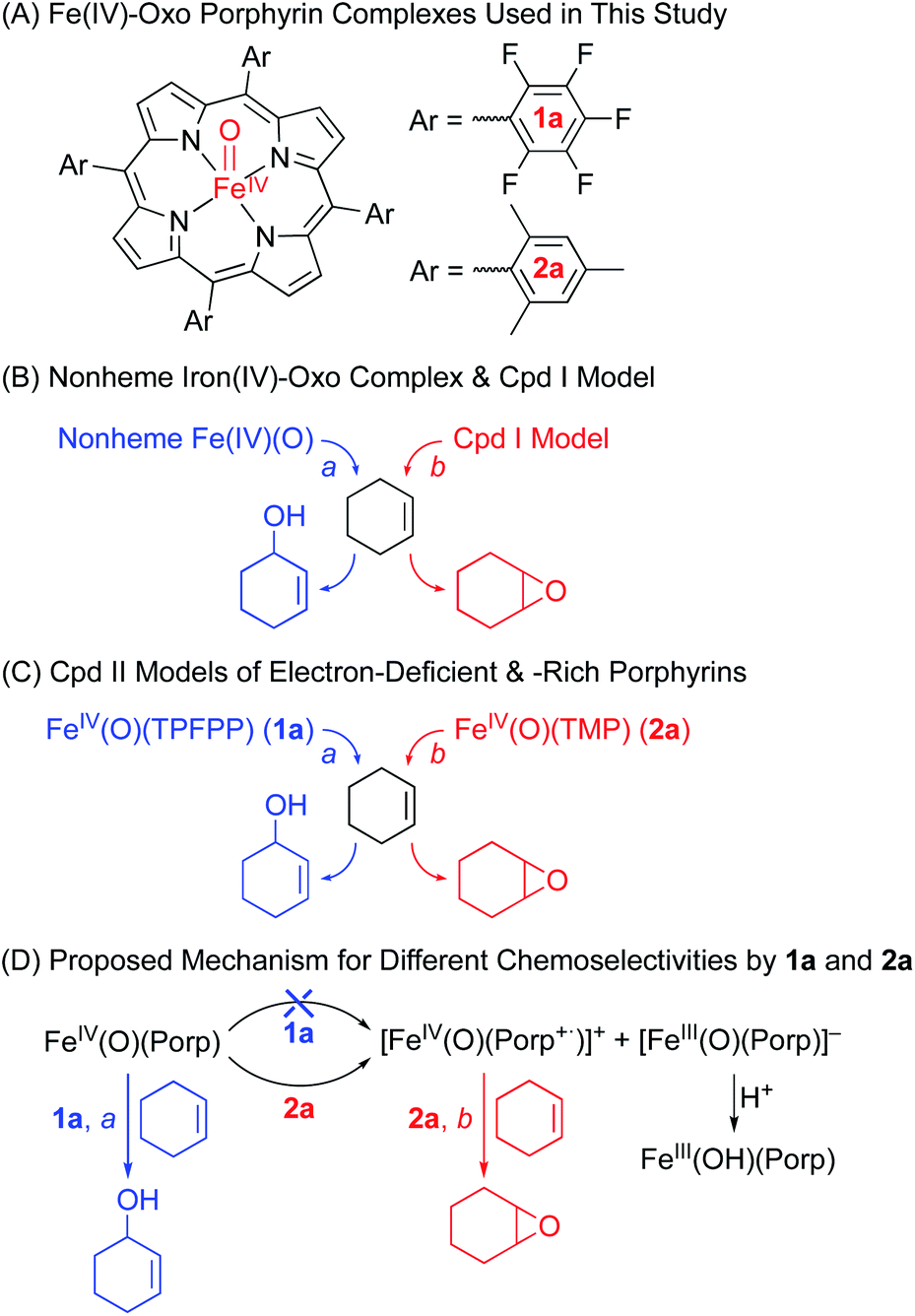
The iron(IV)-oxo porphyrin complex, [FeIV(O)(TPFPP)(Cl)]− (1a), was synthesized by reacting FeIII(TPFPP)(Cl) (1) with 4.0 equiv. of meta-chloroperbenzoic acid (m-CPBA) in acetonitrile (MeCN, 2 mL) containing H2O (15 μL) at 283 K; the colour of the reaction solution changed from brown to red and 1a was formed within 2 min (see Fig. 1a for the electronic absorption spectral change in the Q-band region; also see Fig. S1† for the full range of spectra including the Soret band). 1a was also synthesized by reacting 1 with 4.0 equiv. of iodosylbenzene (PhIO, dissolved in 50 μL of MeOH) (Fig. S2†). 1a was stable at 233 K, allowing us to characterize it using various spectroscopic techniques, such as cold spray time-of-flight mass spectrometry (CSI-MS), electron paramagnetic resonance (EPR), resonance Raman (rRaman), and X-ray absorption spectroscopy/extended X-ray absorption fine structure (XAS/EXAFS).
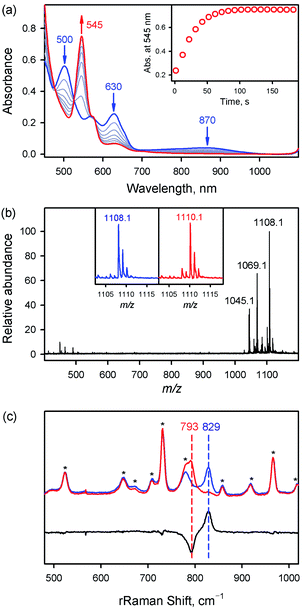 |
| | Fig. 1 (a) UV-vis spectra of FeIII(TPFPP)(Cl) (1, blue line) and [FeIV(O)(TPFPP)(Cl)]− (1a, red line); 1a was synthesized by reacting 1 (0.50 mM) with 4.0 equiv. of m-CPBA (2.0 mM) in MeCN at 283 K. Inset shows the time trace monitored at 545 nm due to 1a. (b) CSI-MS spectrum of 1a. The peaks at m/z = 1045.1, 1069.1, and 1108.1 correspond to [FeIV(O)(TPFPP)(H)]+ (calcd m/z = 1045.0), [FeIII(TPFPP)(MeCN)]+ (calcd m/z = 1069.0), and [FeIV(O)-(TPFPP)(MeCN)(Na)]+ (calcd m/z = 1108.0), respectively. Insets show the observed isotope distribution patterns for 1a-16O (blue line) and 1a-18O (red line); 1a-16O and 1a-18O were prepared by reacting 1 (0.10 mM) with PhI16O (0.40 mM) and PhI18O (0.40 mM), respectively, in MeCN at 233 K. (c) Resonance Raman spectra of 1a-16O (blue line) and 1a-18O (red line) obtained upon excitation at 405 nm. The black line is the difference spectrum of 1a-16O and 1a-18O; 1a-16O and 1a-18O were prepared by reacting 1 (1.0 mM) with PhI16O (4.0 mM) and PhI18O (4.0 mM), respectively, in MeCN at 233 K. The peaks marked with an asterisk originated from the solvent. | |
First, the CSI-MS of 1a in positive mode exhibits a prominent ion peak at a mass-to-charge ratio (m/z) of 1108.1 with mass and isotope distribution patterns corresponding to [FeIV(16O)(TPFPP)(MeCN)(Na)]+ (calcd m/z = 1108.0) (Fig. 1b). When 1a was generated using PhI18O (prepared in 50 μL of MeOH containing 15 μL of H218O), a two-mass unit shift from 1108.1 {[FeIV(16O)(TPFPP)(MeCN)(Na)]+} to 1110.1 {[FeIV(18O)(TPFPP)(MeCN)(Na)]+} was observed (Fig. 1b, inset). This CSI-MS result indicates that 1a contains one oxygen atom. The X-band EPR spectrum of 1a was silent, suggesting an Fe(IV) oxidation state (Fig. S3†). The rRaman spectrum of 1a, measured upon 405 nm excitation at 233 K, displayed one isotopically sensitive band at 829 cm−1, which shifted to 793 cm−1 upon 18O-substitution (Fig. 1c). The observed isotopic shift of −36 cm−1 with 18O-substitution is in good agreement with the calculated value for a diatomic Fe–O oscillator (−37 cm−1). The rRaman data indicate that 1a possesses an FeO unit, as reported in Cpd I, Cpd II, and nonheme iron(IV)-oxo species.14,15
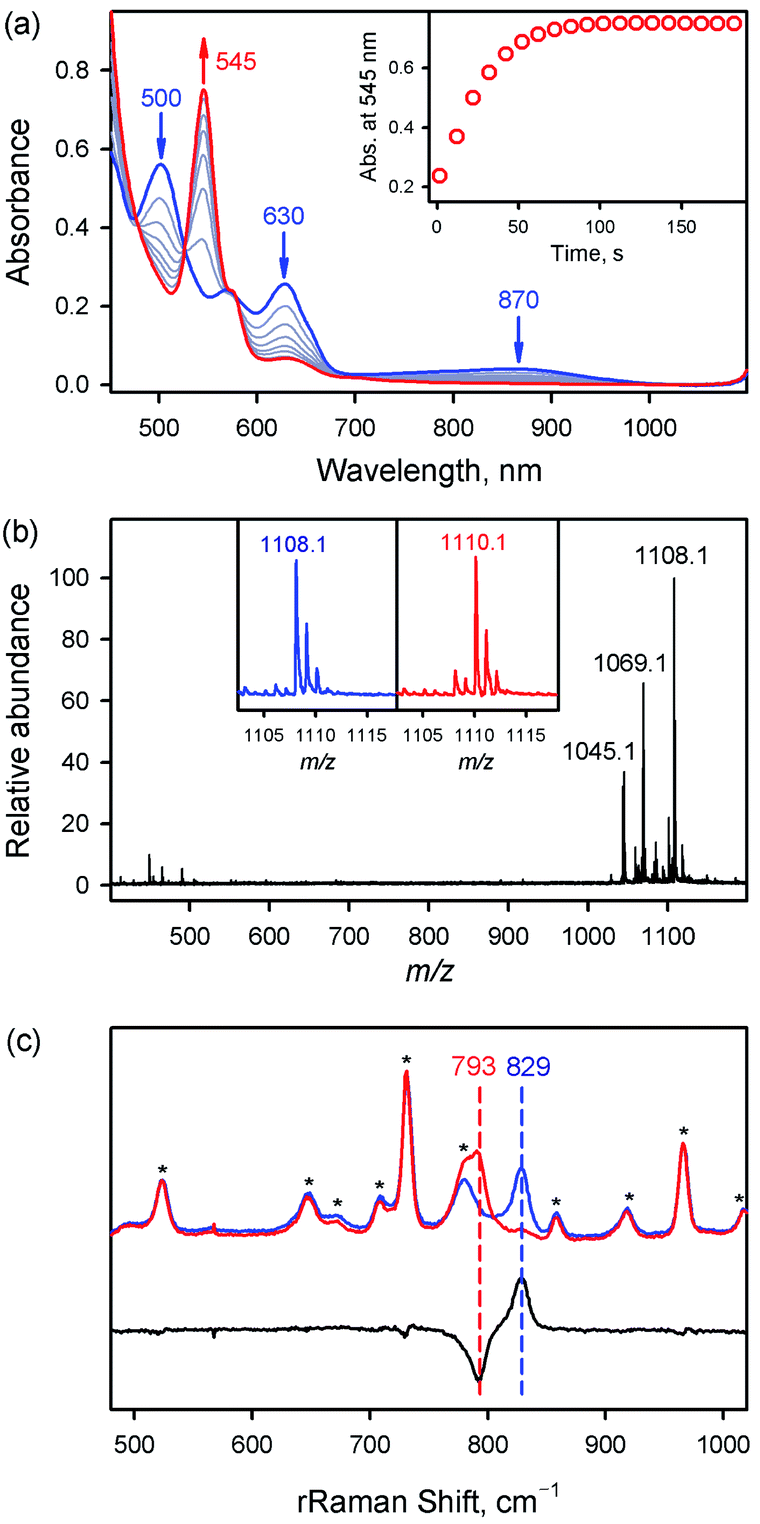
Fe K-edge XAS and EXAFS data for 1 and 1a are presented in Fig. 2 (see also Table S1 and Fig. S4†). The rising-edge energy position at half maxima shifts from 7123.6 eV to 7124.3 eV, indicating an increase in oxidation state on going from 1 (FeIII) to 1a (FeIV). The pre-edge region, which represents the electric dipole forbidden, quadrupole allowed 1s → 3d transition, shows a dramatic increase in intensity in 1a relative to 1.16 This is characteristic of a short bond (i.e., FeO) along the molecular z-axis, which enhances Fe 3dz2–4pz mixing, leading to the enhancement of dipole allowed character into the pre-edge feature. In contrast, the longer Fe–Cl bond in 1 disallows strong mixing and the pre-edge intensity remains modest. A qualitative comparison of the EXAFS data (Fig. 2b) shows a significant phase shift, indicating a large structural change between 1 and 1a. FEFF fits to the data for 1a are consistent with 1 Fe–O at 1.65 Å, 4 Fe–N at 2.00 Å, and 1 Fe–Cl at 2.24 Å. The second and third shells were fitted with single and multiple scattering contributions from the TPFPP ligand. Therefore, the data are consistent with the formation of an Fe(IV)O species with a short 1.65 Å Fe–O bond distance.
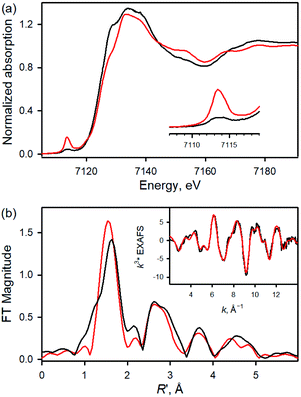 |
| | Fig. 2 (a) Normalized Fe K-edge XAS data for FeIII(TPFPP)(Cl) (1, black line) and [FeIV(O)(TPFPP)(Cl)]− (1a, red line). Inset shows the expanded pre-edge region. (b) Comparison of the nonphase-shift-corrected Fourier transform (FT) data for 1 (black line) and 1a (red line). Inset shows the EXAFS data for 1a (black line) and fit (red line). | |
Synthesis of [FeIV(O)(TMP)(Cl)]− (2a)
When 3.0 equiv. of PhIO, dissolved in 50 μL of MeOH, was added to a solution of FeIII(TMP)(Cl) (2) in butyronitrile at 253 K, the formation of an iron(IV)-oxo porphyrin complex, [FeIV(O)(TMP)(Cl)]− (2a), was observed within 10 min (see Fig. S5a† for the electronic absorption spectral change in the Q-band region; also see Fig. S5b† for the full range of spectra including the Soret band). The X-band EPR spectrum of 2a was silent, suggesting an Fe(IV) oxidation state (Fig. S6†). The rRaman spectrum of 2a, measured upon 442 nm excitation at 218 K, displayed one isotopically sensitive band at 828 cm−1, which shifted to 791 cm−1 upon 18O-substitution (Fig. S7†). The observed isotopic shift of −37 cm−1 with 18O-substitution is in good agreement with the calculated value for a diatomic Fe–O oscillator (−37 cm−1). The rRaman data indicate that 2a possesses an FeO unit.14,15 Based on the spectroscopic characterization presented above, we have confirmed that iron(IV)-oxo complexes bearing electron-deficient and -rich porphyrin ligands, 1a and 2a, were successfully synthesized for further reactivity studies.
Cyclohexene oxidation by 1a and 2a
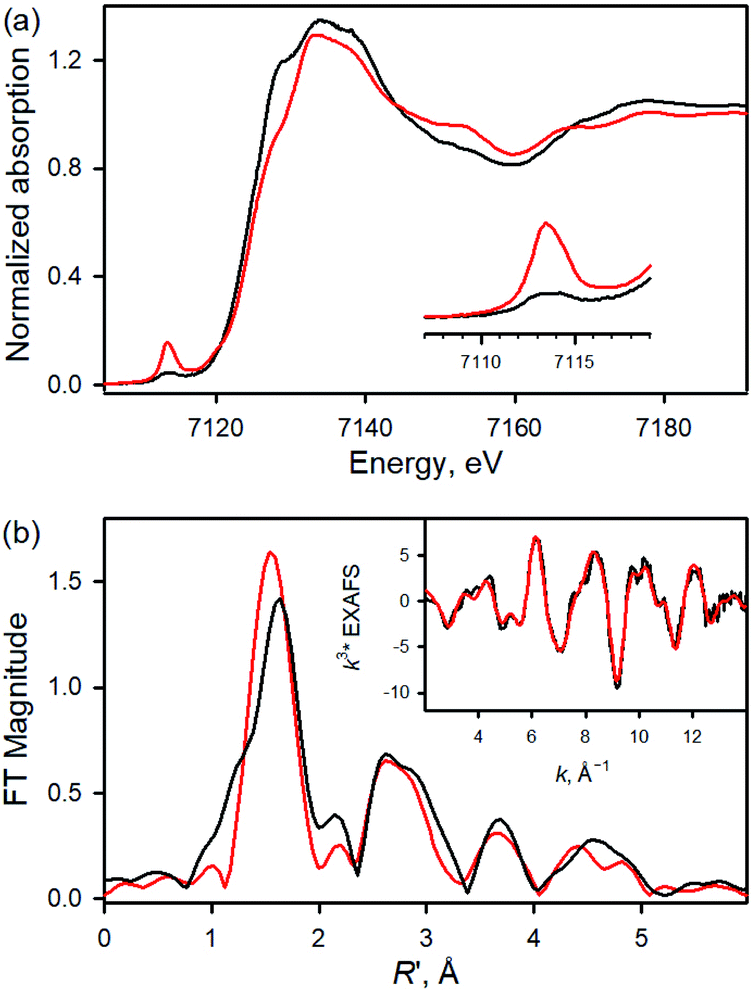
We then investigated the reactivities of 1a and 2a in cyclohexene oxidation. Since 1a reacted slowly with cyclohexene in MeCN at 253 K, the reactivity of 1a with cyclohexene was investigated at 283 K. Upon addition of cyclohexene to the reaction solution of 1a, the electronic absorption band at 545 nm corresponding to 1a disappeared with the concomitant formation of bands at 500 and 630 nm corresponding to the starting [FeIII(TPFPP)]+ complex with clear isosbestic points at 470, 523, and 564 nm (Fig. 3a). The decay rate of 1a obeyed first-order kinetics (Fig. 3a, insets) and increased linearly with an increase in the concentration of cyclohexene, affording a second-order rate constant of k2(H) = 6.1 × 10−3 M−1 s−1 at 283 K (Fig. 3b, blue line). The second-order rate constant determined in the oxidation of deuterated cyclohexene (cyclohexene-d10) by 1a was k2(D) = 1.5 × 10−4 M−1 s−1 (Fig. 3b, red line), giving a large kinetic isotope effect (KIE) value of 41. Product analysis of the reaction solution revealed the formation of 2-cyclohexen-1-ol as a major product in the oxidation of cyclohexene by 1a (85% yield based on the amount of 1a used). Also, the oxygen atom in the 2-cyclohexen-1-ol product was found to derive from the oxidant by carrying out 18O-labeling experiments with 1a-18O; the 18O-percent in the 2-cyclohexen-1-ol product was ∼75% (Fig. S8†). The formation of 2-cyclohexen-1-ol with the large KIE value of 41 demonstrates that the allylic C–H bond oxidation pathway is preferred over the CC bond epoxidation pathway in the oxidation of cyclohexene by 1a (Scheme 1C, pathway a); the preference for C–H bond hydroxylation in cyclohexene oxidation was observed in nonheme iron(IV)-oxo and other nonheme metal(IV)-oxo complexes.10 In addition, the decay product of 1a was found to be [FeIII(TPFPP)]+ by UV-vis and EPR spectroscopies (Fig. 3a and S9†); it is noted that nonheme iron(IV)-oxo species are converted to iron(III) species in C–H bond hydroxylation reactions.9,10
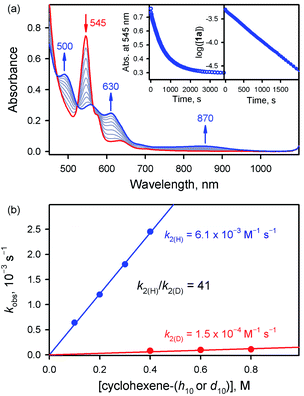 |
| | Fig. 3 (a) UV-vis absorption spectral changes observed in the reaction of 1a (0.50 mM) and cyclohexene (100 mM) in MeCN at 283 K. Insets show the time profile of the absorbance change at 545 nm (left panel) and the first-order plot of log([1a]) vs. time (right panel). (b) Plots of the pseudo-first-order rate constants (kobs) against the concentration of cyclohexene-h10 (blue circles) and cyclohexene-d10 (red circles) to determine k2 values in MeCN at 283 K. | |
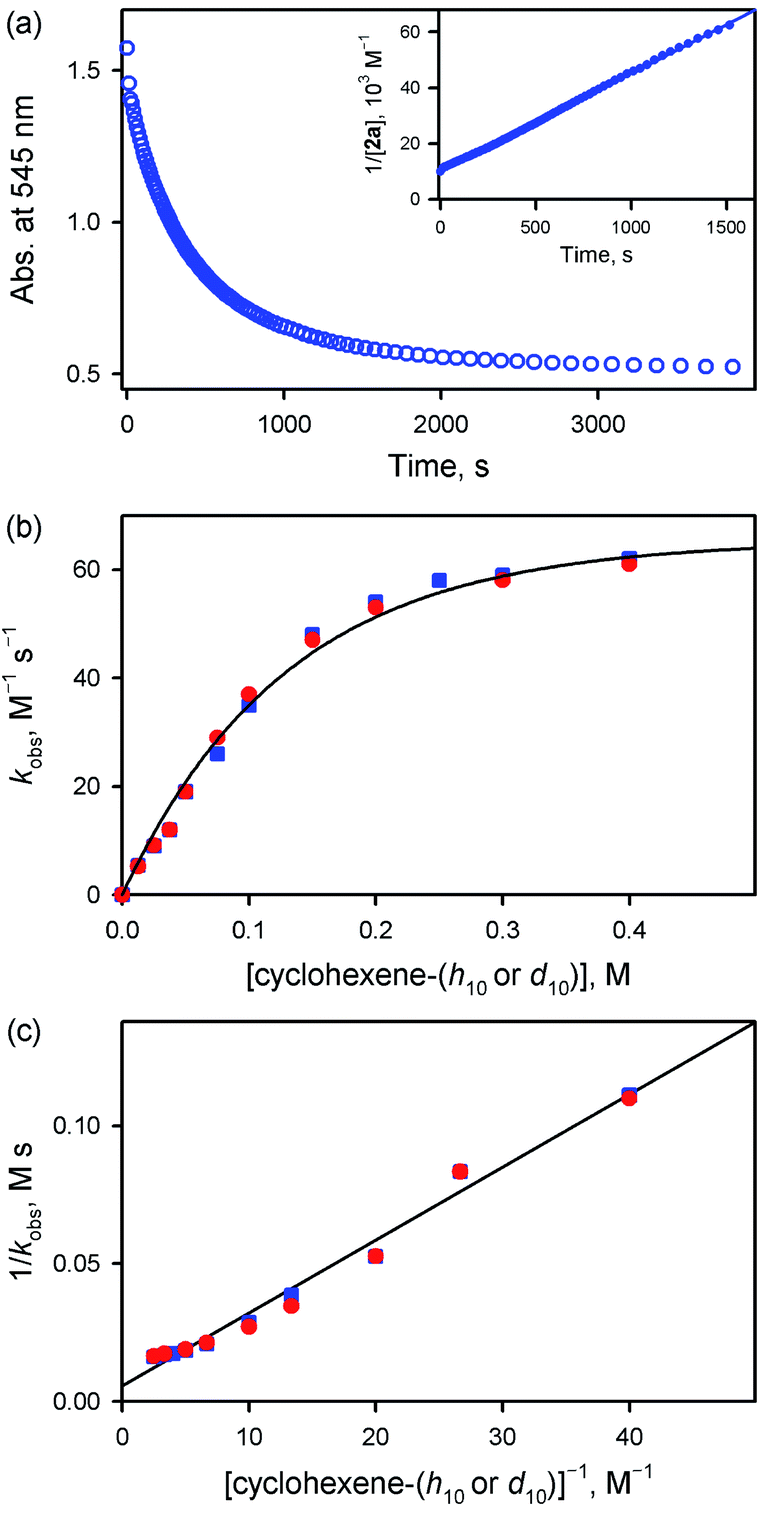
Interestingly, the reactivity of 2a bearing an electron-rich porphyrin ligand was greater than that of 1a bearing an electron-deficient porphyrin ligand (Scheme 2, reaction 1); therefore, the reaction of 2a and cyclohexene was carried out at 253 K, which is lower than the reaction temperature for 1a (e.g., 283 K). Upon addition of cyclohexene to the reaction solution of 2a, the electronic absorption band at 545 nm corresponding to 2a disappeared with the concurrent formation of bands at 505 and 575 nm corresponding to the starting [FeIII(TMP)]+ complex with clear isosbestic points at 465, 527, 585, and 710 nm (Fig. S10†). Surprisingly, the reaction was found to obey second-order kinetics, as shown in Fig. 4a (Scheme 2, reaction 2); it is noted that the reaction of 1a and cyclohexene followed first-order kinetics (Fig. 3a). The observed rate constant (kobs) of 2a increased with an increase in the concentration of cyclohexene, but a saturation plot was obtained at high concentrations of cyclohexene (Fig. 4b); the observation of saturation behavior suggests that there is a relatively fast equilibrium that precedes the oxygen atom transfer reaction (vide infra).17 The latter result is also distinct from the observation of good linear correlation between the rate constant and the cyclohexene concentration in the reaction of 1a and cyclohexene (Fig. 3b) (Scheme 2, reaction 3). When cyclohexene-h10 was replaced by cyclohexene-d10 in the oxidation reaction by 2a, the reaction exhibited second-order kinetics, saturation behavior, and a KIE value of 1.0 (Fig. 4b and c). It is of interest to note that the KIEs determined in the reactions of 1a and 2a are very different: 41 for 1a and 1.0 for 2a (Scheme 2, reaction 4) (vide infra).18
 |
| | Scheme 2 Observed reactivities of 1a and 2a. | |
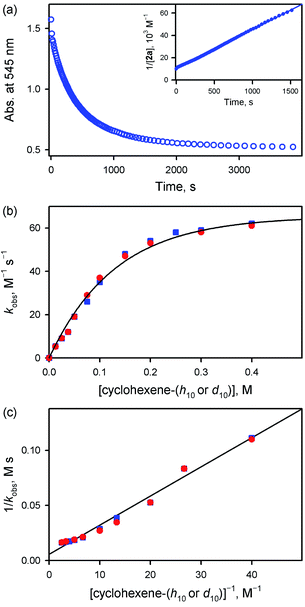 |
| | Fig. 4 (a) Time profile of the absorbance change at 545 nm due to the decay of 2a (0.10 mM) in the reaction of 2a with cyclohexene (100 mM) in butyronitrile at 253 K. Inset shows the second-order plot of 1/[2a] vs. time. (b) Plots of the observed second-order rate constants (kobs) against concentrations of cyclohexene-h10 (blue squares) and cyclohexene-d10 (red circles) obtained in the reactions of 2a (0.10 mM) with cyclohexene-h10 and cyclohexene-d10 in butyronitrile at 253 K, respectively. (c) Plots of kobs−1vs. [cyclohexene-h10]−1 (blue squares) and [cyclohexene-d10]−1 (red circles) obtained in the reactions of 2a (0.10 mM) with cyclohexene-h10 and cyclohexene-d10 in butyronitrile at 253 K, respectively. | |
Product analysis of the reaction solution of 2a and cyclohexene revealed the formation of cyclohexene oxide as the sole product (∼50% based on the concentration of 2a) (Scheme 1C, pathway b), which is very different from the 2-cyclohexen-1-ol produced in the reaction of 1a and cyclohexene (Scheme 1C, pathway a) (see also Scheme 2, reaction 5); it is noted that the reactions of Cpd I models and cyclohexene afford the cyclohexene oxide product (Scheme 1B, pathway b).11 The source of oxygen atoms in the cyclohexene oxide product was found to derive from the oxidant by carrying out 18O-labeling experiments with 2a-18O; the 18O-percent in the cyclohexene oxide product was >85% (Fig. S11†). In addition to the UV-vis spectrum of the reaction solution (Fig. S10†), the decay product of 2a was confirmed to be [FeIII(TMP)]+ by EPR spectroscopy (Fig. S12†). As a conclusion, we have shown that the reaction of 1a and cyclohexene prefers allylic C–H bond oxidation to give 2-cyclohexen-1-ol (Scheme 1C, pathway a), whereas the reaction of 2a and cyclohexene favors CC bond epoxidation and cyclohexene oxide is the product (Scheme 1C, pathway b). Further, it is notable that the reaction of 1a resembles the mononuclear nonheme iron(IV)-oxo reaction (i.e., C–H bond hydroxylation; Scheme 1B, pathway a),10 whereas the reaction of 2a follows the Cpd I reaction (i.e., CC bond epoxidation; Scheme 1B, pathway b).11
Disproportionation of Cpd II to Cpd I and [FeIII(Porp)]+
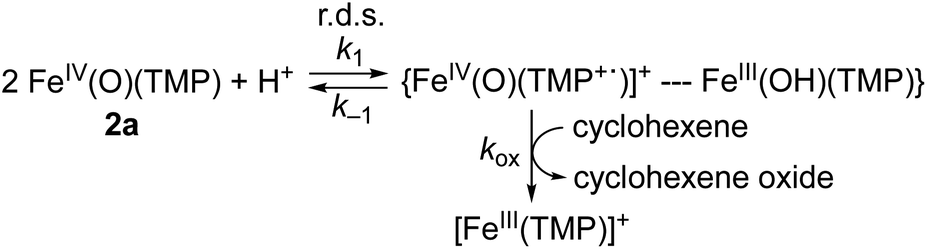
Why are the reactions of 1a and 2a very different in the oxidation of cyclohexene, such as the preference for C–H bond hydroxylation with a large KIE value vs. CC bond epoxidation with a KIE value of 1.0, first-order kinetics vs. second-order kinetics, and no saturation behavior vs. saturation behavior (see Scheme 2)? These differences are rationalized by disproportionation of 2a (Scheme 1D, reaction pathway of 2a), but not 1a (Scheme 1D, reaction pathway of 1a). That is, in the reaction of 2a, the second-order kinetics together with the saturation plot of kobsvs. concentration of cyclohexene and the absence of a kinetic isotope effect can be well explained by the disproportionation of [FeIV(O)(TMP)(Cl)]− (2a) to afford [FeIV(O)(TMP+˙)]+ (2b), which is the active oxidant that epoxidizes cyclohexene to give the corresponding epoxide product (Scheme 3). According to Scheme 3, the rate of disappearance of 2a is given by eqn (1),| | | d[2a]/dt = −k1[2a]2 + k−1[C] | (1) |
where C is a pair of [FeIV(O)(TMP+˙)]+ and FeIII(OH)(TMP), as shown in Scheme 3. The formation rate of C is given by eqn (2), where [S] is the substrate concentration, [cyclohexene]. Eqn (3) is derived from eqn (1) and (2).| | | d[C]/dt = k1[2a]2 − k−1[C] − kox[C][S] | (2) |
| | | d([2a] + [C])/dt = −kox[C][S] | (3) |
 |
| | Scheme 3 Disproportionation reaction of 2a. | |
Since no [FeIV(O)(TMP+˙)]+ was observed, [C] ≪ [2a]. Under such conditions, eqn (3) is rewritten as eqn (4).
By applying the steady-state approximation (d[C]/dt ≈ 0), eqn (5) is obtained from eqn (2). Thus, eqn (6) is obtained from eqn (4) and (5), which fits the second-order kinetics for the disappearance of 2a. The observed second-order constant (kobs) is given by eqn (7), which can be converted to a linear correlation between kobs−1 and [S]−1 [eqn (8)].
| | | [C] = k1[2a]2/(k−1 + kox[S]) | (5) |
| | | d[2a]/dt = −k1kox[2a]2[S]/(k−1 + kox[S]) | (6) |
| | | kobs = k1kox[S]/(k−1 + kox[S]) | (7) |
| | | kobs−1 = (k−1/k1kox)[S]−1 + (k1)−1 | (8) |
Such a linear correlation is confirmed in a plot of kobs−1vs. [cyclohexene]−1 in Fig. 4c. The intercept corresponds to the disproportionation rate constant (k1−1) and the slope corresponds to k−1/(k1kox). As expected, no KIE was observed for k1 (Fig. 4c and Table 1). The k1 and koxK1 (K1 = k1/k−1) values are summarized in Table 1.
Table 1 Rate constants of the oxidation of cyclohexene-h10 and cyclohexene-d10 (see the column of koxK1) and the disproportionation (see the column of k1) of 2a in butyronitrile at 253 K
| Substrate |
k
ox
K
1, M−2 s−1 |
k
1, M−1 s−1 |
| Cyclohexene-h10 |
3.8 × 102 |
1.9 × 102 |
| Cyclohexene-d10 |
3.8 × 102 |
1.9 × 102 |
We therefore conclude that the disproportionation of 1a bearing an electron-deficient porphyrin ligand does not occur under the reaction conditions, whereas 2a bearing an electron-rich porphyrin ligand is disproportionated to form an iron(IV)-oxo porphyrin π-cation radical species, [FeIV(O)(TMP+˙)]+ (2b), as an active oxidant. This is the reason why 1a and 2a exhibited different reactivities in the oxidation of cyclohexene, as summarized in Scheme 2; that is, 1a abstracts a hydrogen atom from allylic C–H bonds, whereas 2a is disproportionated to 2b that epoxidizes cyclohexene to give cyclohexene oxide selectively (Scheme 1D).
Although the disproportionation reactions of high-valent metal-oxo (and metal-imido) complexes in the presence of protons have been well addressed previously,19–21 we report an example showing that the disproportionation reaction can be modulated by the electron-richness of porphyrin ligands and that the different reactivities of iron(IV)-oxo porphyrin complexes, 1a and 2a result from the disproportionation reaction by 2a, but not by 1a. Further, the present study demonstrates that iron(IV)-oxo porphyrins prefer C–H bond hydroxylation to CC bond epoxidation, as reported in nonheme iron(IV)-oxo (and other metal(IV)-oxo) systems.10
Cyclohexene oxidation by 1b and 2b
In order to prove the possibility of the disproportionation reaction of 2a, but not 1a, and the formation of iron(IV)-oxo porphyrin π-cation radical species as an active oxidant in the cyclohexene oxidation by 2a, but not by 1a (Schemes 1D and 3), we synthesized iron(IV)-oxo porphyrin π-cation radical species independently, [FeIV(O)(TPFPP+˙)]+ (1b) and [FeIV(O)(TMP+˙)]+ (2b), investigated their reactivities with cyclohexene, and compared the reactivities of 1b and 2b to those of 1a and 2a. First, the addition of 4.0 equiv. of m-CPBA to [FeIII(TPFPP)](OTf) (OTf = CF3SO3−) and [FeIII(TMP)]Cl led to the formation of [FeIV(O)(TPFPP+˙)]+ (1b) and [FeIV(O)(TMP+˙)]+ (2b), respectively (Fig. S13†).3c,11–13,22 Then, the reactivities of 1b and 2b were investigated in cyclohexene oxidation. Upon the addition of cyclohexene-h10 to a CH2Cl2 solution of 1b at 213 K, the absorption band at 660 nm due to 1b disappeared and absorption bands corresponding to [FeIII(TPFPP)]+ appeared concurrently (Fig. S14†). The second-order rate constant, k2(H), was determined to be 1.4 × 104 M−1 s−1 at 213 K (Fig. S15†). Similarly, the second-order rate constant in the oxidation of cyclohexene-d10 by 1b was determined to be k2(D) = 1.4 × 104 M−1 s−1 (Fig. S15†), giving a KIE value of 1.0. Organic product analysis revealed that cyclohexene oxide, but no allylic oxidation products, was formed as a product. The observations of the KIE of 1.0 and the cyclohexene oxide product in the reaction of 1b and cyclohexene were different from the results obtained in the reaction of 1a and cyclohexene, such as the KIE value of 41 and the formation of the allylic oxidation product in the latter reaction. Based on the reactivity comparison of 1a and the in situ generated iron(IV)-oxo porphyrin π-cation radical species, [FeIV(O)(TPFPP+˙)]+ (1b), we conclude that the disproportionation of 1a does not occur to form 1b (vide infra) and that 1a, but not 1b, is the active oxidant that oxidizes cyclohexene to give the 2-cyclohexen-1-ol product (Scheme 1D, pathway a).
Similarly, upon the addition of cyclohexene-h10 to a butyronitrile solution of [FeIV(O)(TMP+˙)]+ (2b), the absorption band at 666 nm corresponding to 2b disappeared with the concurrent formation of an absorption band at 505 nm corresponding to [FeIII(TMP)]+ (Fig. S16†). From the slope of a linear correlation of the pseudo-first-order rate constants vs. concentration of cyclohexene, the second-order rate constant was determined to be k2(H) = 21 M−1 s−1 in butyronitrile at 253 K (Fig. S17†). Also, the second-order rate constant in the oxidation of deuterated cyclohexene (cyclohexene-d10) by 2b was determined to be k2(D) = 21 M−1 s−1 (Fig. S17†), giving a KIE value of 1.0. Organic product analysis revealed the formation of cyclohexene oxide without the allylic oxidation product (e.g., 2-cyclohexen-1-ol). Thus, the reactivity of 2b was found to be the same as that of 2a in cyclohexene oxidation, such as the KIE of 1.0 and the cyclohexene oxide product. Conclusively, these results confirm that the active oxidant in the reaction of 2a and cyclohexene is not 2a but 2b, which is formed via a disproportionation reaction of 2a (Scheme 1D, pathway b and Scheme 3).
Effects of acid and base on the reactions of 1a and 2a
To investigate further this unusual behavior of Cpd II models in the oxidation of cyclohexene, such as the preference for C–H bond hydroxylation vs. CC bond epoxidation by 1a and 2a, respectively, we investigated the effects of acid and base on the reactions of 1a and 2a with cyclohexene. According to Scheme 3, the disproportionation step should be affected by the proton concentration and the rate should increase with an increase in the concentration of protons.4,19 This was confirmed by showing that the kobs values of 2a increased with an increase in the concentration of acetic acid (AcOH) (Fig. S18†). In contrast, such an acid concentration effect was not observed in the reaction of 1a and cyclohexene upon the addition of AcOH up to 100 mM (Fig. S19†).
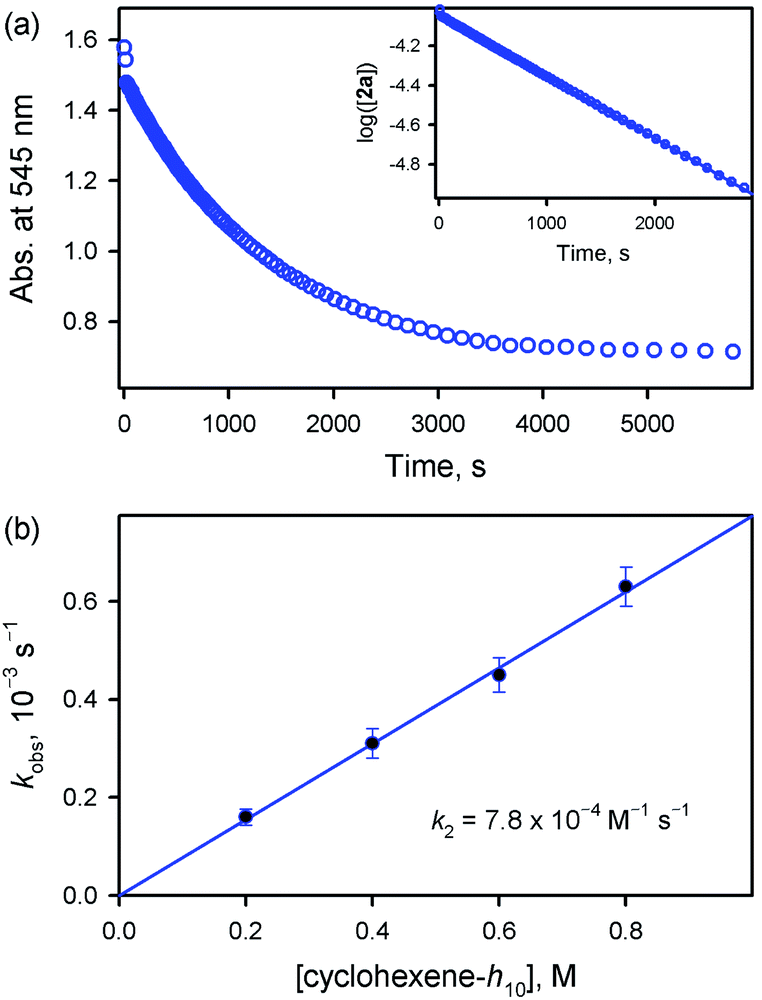
On the other hand, the addition of a base, such as tetramethylammonium hydroxide (TMAH), to the solution of 2a reduced the reaction rate of 2a (Fig. 5). In addition, interestingly, the decay of 2a came to obey first-order kinetics (Fig. 5a). Moreover, the pseudo-first-order rate constants increased linearly with an increase in cyclohexene concentration, affording the second-order rate constant of k2 = 7.8 × 10−4 M−1 s−1 in the presence of 0.25 mM TMAH (Fig. 5b). Also, product analysis of the reaction solution revealed the formation of 2-cyclohexen-1-ol product. These results, such as the first-order kinetics, the linear correlation against substrate concentration, and the formation of 2-cyclohexen-1-ol product, are in sharp contrast to those results obtained in the reactions of 2a and cyclohexene carried out in the absence of a base, such as the second-order kinetics, the saturation behavior with substrate concentration, and the formation of cyclohexene oxide. This base effect is rationalized by the change in the mechanism in the presence of a base, such as the equilibrium shift towards the inhibition of the disproportionation of 2a in the presence of a base (Scheme 3). Thus, our conclusion is that the active oxidant is 2a (e.g., [FeIV(O)(TMP)]), not 2b (e.g., [FeIV(O)(TMP+˙)]+), in the presence of a base, showing reactivity patterns similar to those observed in the reactions of 1a with cyclohexene (see Scheme 2).
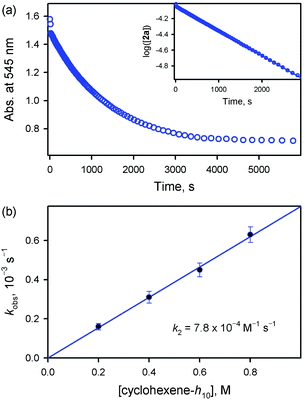 |
| | Fig. 5 (a) Time profile of the absorbance change at 545 nm due to 1a observed in the reaction of 2a (0.10 mM) with cyclohexene (400 mM) in the presence of TMAH (0.25 mM) in butyronitrile at 253 K. Inset shows the first-order plot of log([2a]) vs. time. (b) Plot of pseudo-first-order rate constant (kobs) against the concentration of cyclohexene-h10 to determine the k2 value for the reaction of 2a and cyclohexene in the presence of TMAH (0.25 mM) in butyronitrile at 253 K. | |
Density functional theory (DFT) calculations
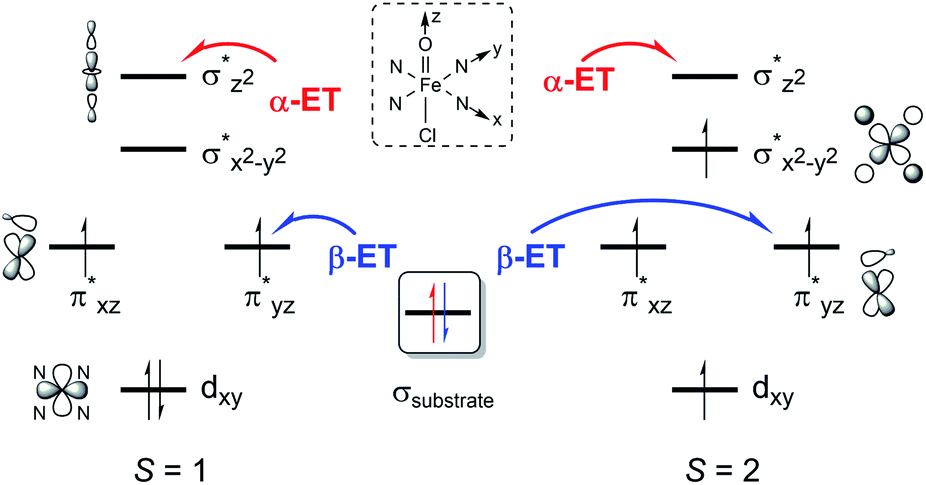
To explain the experimentally observed different reactivities of 1a and 2a, systematic DFT calculations on the reactions of 1a and 2a with cyclohexene were investigated. The electronic structures of 1a and 2a were calculated at the B3LYP/Def2-TZVPP//B3LYP/Def2-SVP level. Both species have a triplet S = 1 ground spin state (Table S2†), which is experimentally supported.22 Interestingly, we find that changing the substituents on the porphyrin ring does not affect the Fe–O bond distance (1.63 Å in both 1a and 2a) or Fe–N bond distance (2.03 Å in both 1a and 2a) for the triplet species, even though a slight difference is found for the Fe–Cl distance, of 2.45 and 2.52 Å in 1a and 2a, respectively (Table S3†). The same Fe–O bond distance in these two species is consistent with the almost equal stretching vibration frequencies of the Fe–O bond obtained from the rRaman experiment, of 829 and 828 cm−1 for 1a and 2a, respectively (vide supra). For both 1a and 2a, the lowest excited state is a singlet state, which is higher in energy than the triplet ground state by ca. 9 kcal mol−1. The second excited state is a quintet state, which lies ca. 13 kcal mol−1 higher in energy than the triplet ground state. Thus, these two excited states have been considered in the following reactivity study; the septet state with an electronic configuration of either dxy1, π*xz1, π*yz1, σ*x2−y21, σ*z21, a2u1 or dxy1, π*xz1, π*yz1, σ*x2−y21, σ*z20, a2u1, e2g1 was too high in energy to be considered feasible. Given the difference in the oxidation reactivities determined by the experiments, the apparent lack of differences between triplet 1a and 2a was puzzling. To further explain the reactivity differences between these two FeIVO species, we investigated their oxidation reactions using the DFT approach. In the present work, we studied two possible electron transfer pathways, a σ pathway and a π pathway (Fig. 6), for both hydroxylation and epoxidation reactions on both triplet and quintet state surfaces. In the case of the σ pathway, an α-electron is transferred from the substrate bonding orbital to the σ*z2 orbital of the catalyst. In the case of the π pathway, a β-electron is transferred from the substrate to the π*yz orbital of the catalyst (the y-direction here is understood as the direction forming an O–H bond). For the same reaction on the singlet state surface, only a π pathway with an α-electron being transferred from the substrate to the π*yz orbital of the catalyst was studied (Fig. S20†).
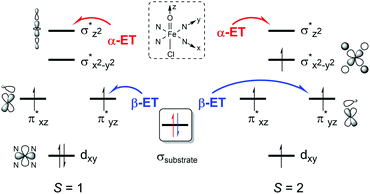 |
| | Fig. 6 Two possible electron transfer pathways (α-ET vs. β-ET) on both triplet (S = 1) and quintet (S = 2) spin state surfaces during cyclohexene oxidation by 1a and 2a. | |
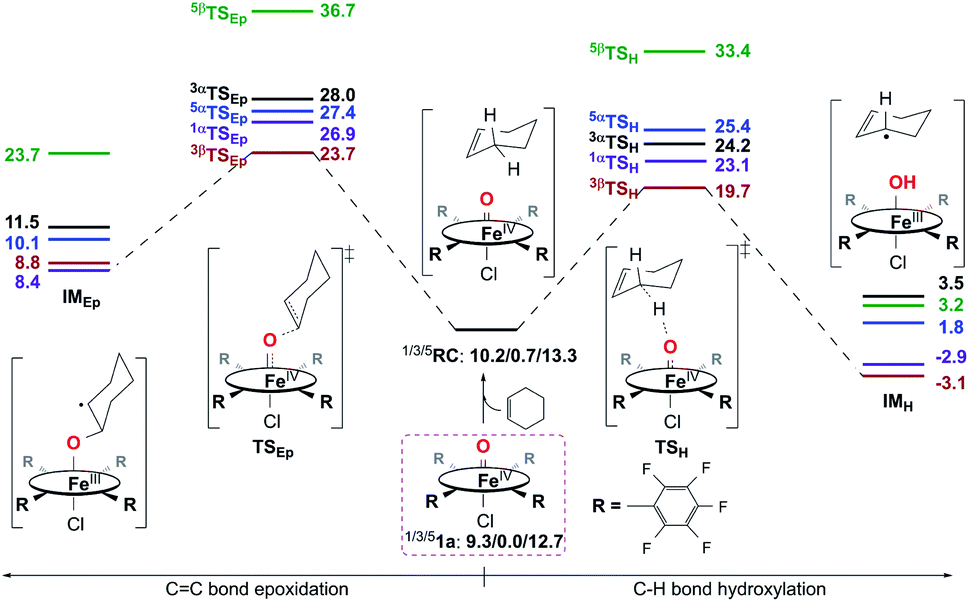
Reactivity study for 1a.
Calculation results reveal that both the C–H bond hydroxylation and CC bond epoxidation reactions mediated by 1a should occur mainly on the S = 1 ground spin state surface. Therein, the C–H bond hydroxylation has a lowest energy barrier of 19.7 kcal mol−1 (Fig. 7), which is very close to the experimental value of 20.2 kcal mol−1 obtained from k2(H) of 6.1 × 10−3 M−1 s−1 at 283 K using the Eyring equation. In contrast, the CC bond epoxidation affords a lowest energy barrier of 23.7 kcal mol−1, which indicates that this is an impossible reaction route. These computational results are in accordance with the experimental findings discussed above, in which the hydroxylation product was found to be the sole product. After inspecting the spin density populations along the reaction coordinate on the triplet state surface (Table S4†), it is observed that a β-electron has been transferred from the substrate to the catalyst during the C–H bond reaction, affording the lowest transition state, 3βTSH. After the initial 3βTSH, an intermediate (3βIMH) with a doublet Fe(III)–OH ferromagnetically coupled to a doublet cyclohexenyl radical was obtained. Compared to other possible reaction routes, this one is both kinetically and thermodynamically favorable (Fig. 7).
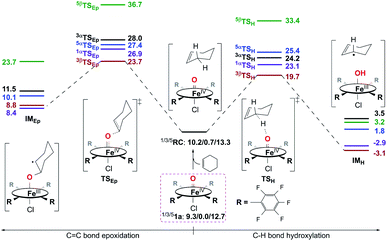 |
| | Fig. 7 Energy profiles (in kcal mol−1) calculated at the B3LYP/Def2-TZVPP//Def2-SVP level for the oxidation of cyclohexene by 1a. α/β in the superscript represents an α/β-electron transferring from substrate to catalyst during the first step of the reaction. RC is a reactant complex, TS is a transition state, and IM is an intermediate species. | |
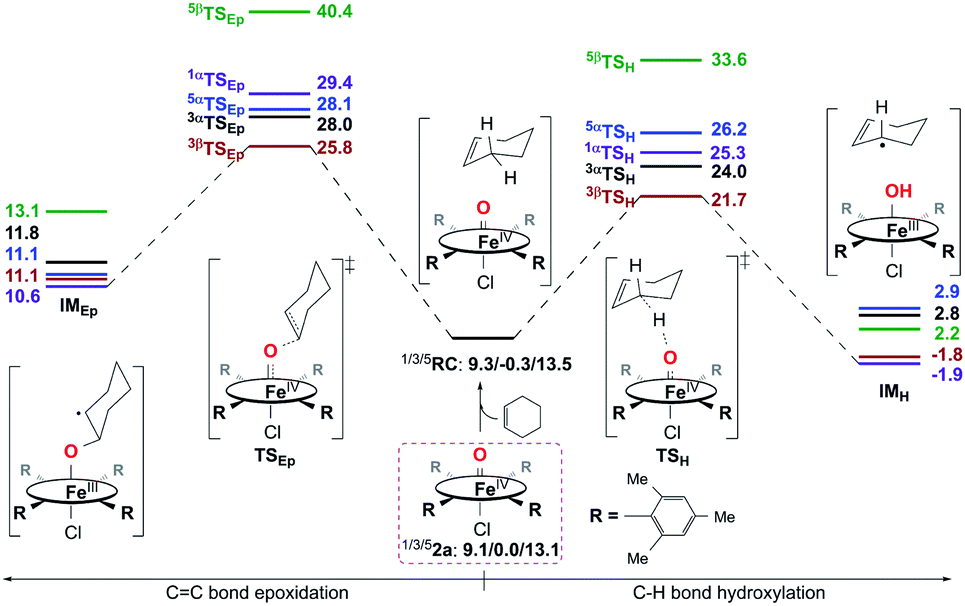
Reactivity study for 2a.
Then, the same reactivity studies were carried out for 2a. Even though neither C–H bond hydroxylation nor CC bond epoxidation of cyclohexene by 2a can take place easily as a consequence of the very high reaction energy barriers (Fig. 8), C–H bond hydroxylation with a smaller energy barrier of 21.7 kcal mol−1 is still preferred to CC bond epoxidation with a higher energy barrier of 25.8 kcal mol−1. This is also supported by the experiment performed in the presence of a base, in which 2a was considered to react with the substrate directly (vide supra). Thus, the reactions of both 1a and 2a with cyclohexene should lead to the hydroxylation product being obtained as the sole product, which is similar to the previous findings for nonheme iron(IV)-oxo cases.10d,e In contrast, without adding a base, an epoxide product was observed as the sole product instead in the reaction of 2a and cyclohexene, which is different from that with a base. Combined with the experimental findings shown above, 2a can be excluded as the real intermediate that completes the substrate oxidation in the absence of a base.
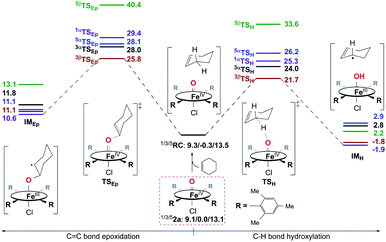 |
| | Fig. 8 Energy profiles (in kcal mol−1) calculated at the B3LYP/Def2-TZVPP//Def2-SVP level for the oxidation of cyclohexene by 2a. α/β in the superscript represents an α/β-electron transferring from substrate to catalyst during the first step of the reaction. RC is a reactant complex, TS is a transition state, and IM is an intermediate species. | |
Disproportionation reaction.
According to the current experimental results and previous reports,4,19–21 an FeIVO porphyrin π-cation radical species generated from the disproportionation of an FeIVO intermediate is believed to be the real oxidant to complete the substrate oxidation in the TMP (i.e., 2a) case. The thermodynamics of this disproportionation process were therefore studied for both 1a and 2a cases (Table 2). Three cases were studied: disproportionation of (i) two FeIVO intermediates (1a or 2a), forming FeIIIO and 1b or 2b, (ii) protonated 1a or 2a with 1a or 2a forming FeIIIOH and 1b or 2b and (iii) two protonated 1a or 2a forming FeIIIOH2 and 1b or 2b. Case (i) was found to be thermodynamically impossible, while case (iii), which would fit a scenario where the proton availability is abundant, would have resulted in a non-rate-limiting activation barrier for the disproportionation, which should then have led to reaction rates comparable to pure 1b and 2b (Table 2). Therefore, case (ii), which fits a scenario where limited protons are available, gives the best agreement with the current experimental results. It supports the disproportionation reaction in the case of 2a, whereas endothermicity makes disproportionation less likely in the case of 1a. This can be attributed to the higher electron-donating ability of the substituent in TMP, which should make the disproportionation reaction thermodynamically more favourable.19a The estimated upper energy barrier for the disproportionation reaction of 2a is the same as the hydroxylation barrier of cyclohexene. Furthermore, the need for protons to facilitate the disproportionation reaction of the Fe(IV)(O) species has also been previously documented.4,19a,20
Table 2 Thermodynamics of the disproportionation reactions of 1a and 2a
| Reaction |
ΔE (kcal mol−1) |
|
1a
|
2a
|
| FeIV(O)(L) + FeIV(O)(L) → FeIV(O)(L+˙) + FeIII(O)(L) |
55.5 |
51.5 |
| FeIV(OH)(L) + FeIV(O)(L) → FeIV(O)(L+˙) + FeIII(OH)(L) |
7.18 |
−1.87 |
| FeIV(OH)(L) + FeIV(OH)(L) → FeIV(O)(L+˙) + FeIII(OH2)(L) |
−1.04 |
−12.0 |
Reactivity study for 1b.
The reaction of 1b with cyclohexene afforded a lowest energy barrier of 8.4 kcal mol−1 (Table S5†), which reproduces the experimental value of 8.3 kcal mol−1, obtained from k2 of 1.4 × 104 M−1 s−1 at 213 K (Table 1) using the Eyring equation. This reaction is the epoxidation on the doublet spin state surface, which is also in agreement with the experimental KIE result of 1.0 as well as the epoxide product distribution.
Reactivity study for 2b.
A theoretical study investigating the reaction of 2b with cyclohexene has been done previously, using a slightly different basis set.11c According to those results, the epoxidation pathway should be preferred under the present experimental conditions, which is in good agreement with the current experimental findings. This result therefore confirms that a disproportionation reaction in the 2a case must occur; otherwise, a C–H bond hydroxylation product should have been seen.
Comparison between 1b and 2b.
To make a comparison between 1b and 2b, single-point calculations using the same method as in the 1b case were carried out on the previously optimized geometries in the reaction of 2b and cyclohexene (Fig. 9).11c The lowest energy barrier obtained was 12.9 kcal mol−1 (Fig. 9 and Table S5†), which is close to the experimental value of 13.6 kcal mol−1 obtained from kox of 21 M−1 s−1 at 253 K (Table 1) using the Eyring equation. The epoxidation reaction route is preferred in both 1b and 2b cases, which is also similar to previous findings for nonheme iron(V)-oxo cases.10d,e1b presents a higher oxidation capability than 2b (Table S5†),12d which is consistent with the experiments. Furthermore, the good agreement between theory and experiment in turn demonstrates the accuracy and reliability of the DFT results discussed above.
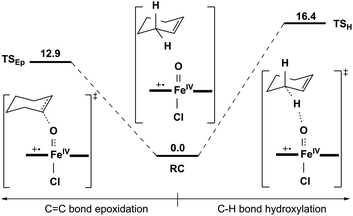 |
| | Fig. 9 Energy profiles (in kcal mol−1) calculated at the B3LYP/Def2-TZVPP//LACVP level of two lowest transition states for the oxidation of cyclohexene by 2b. RC is the reactant complex. TS is the transition state. | |
Conclusion
In summary, the present study provides valuable mechanistic insights into the chemical properties of heme Cpd II models, such as the chemoselectivity of iron(IV)-oxo porphyrin complexes, which varies depending on the electron-richness of porphyrin ligands. That is, in the oxidation of cyclohexene, a high-valent iron(IV)-oxo complex bearing an electron-deficient porphyrin ligand, [FeIV(O)(TPFPP)(Cl)]− (1a), prefers allylic C–H bond hydroxylation with a high KIE of 41, yielding 2-cyclohexen-1-ol as the product, whereas a high-valent iron(IV)-oxo complex bearing an electron-rich porphyrin ligand, [FeIV(O)(TMP)(Cl)]− (2a), favors CC bond epoxidation to afford the cyclohexene oxide product with a KIE of 1. The latter result is rationalized with the disproportionation of 2a, resulting in the generation of [FeIV(O)(TMP+˙)]+ (2b) as the active oxidant that effects the cyclohexene epoxidation. In the presence of a base, this disproportionation reaction is prohibited and the cyclohexene oxidation by 2a occurs via C–H bond hydroxylation to yield 2-cyclohexen-1-ol as the product. Thus, based on experimental and theoretical investigations, we conclude that the chemoselectivity of heme Cpd II models is the same as that of nonheme iron(IV)-oxo species and C–H bond hydroxylation is preferred to the CC bond epoxidation pathway in the oxidation of cyclohexene.
We have also shown that the disproportionation of heme Cpd II models depends on the electron-richness of the iron(IV)-oxo porphyrin complexes. That is, an electron-deficient iron(IV)-oxo porphyrin complex is difficult to disproportionate, whereas an electron-rich iron(IV)-oxo porphyrin complex readily disproportionates to form Cpd I as an active oxidant. The disproportionation reaction is shown to be affected by the presence of acid or base. Thus, we conclude that care should be taken in investigating the chemical properties of heme Cpd II models because of the possible involvement of the disproportionation of Cpd II to Cpd I, and Cpd I becoming the actual intermediate for the oxidation of organic substrates.
Data availability
All data (experimental and theoretical details) required to understand and verify the research in this article have been provided as the ESI† in the online version of the article at the publisher’s website.
Author contributions
Wonwoo Nam conceived and designed the experiments; Ranjana Gupta, Mi Sook Seo, and Ritimukta Sarangi performed the experiments; Xiao-Xi Li and Youngseob Lee performed the theoretical calculation (DFT); Yong-Min Lee, Ranjana Gupta, Xiao-Xi Li, Mi Sook Seo, Sachiko Yanagisawa, Minoru Kubo, Ritimukta Sarangi, and Kyung-Bin Cho analysed the data; Wonwoo Nam, Shunichi Fukuzumi, Kyung-Bin Cho, Ritimukta Sarangi, and Yong-Min Lee co-wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the NRF of Korea through CRI (NRF-2021R1A3B1076539 to W. N.) and Basic Science Research Program (NRF-2020R1I1A1A01074630 to Y.-M. L., NRF-2019R1I1A1A01055822 to M. S. S. and NRF-2021R1A2C1012851 to K.-B. C.). The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894).
Notes and references
-
(a) K. D. Dubey and S. Shaik, Acc. Chem. Res., 2019, 52, 389–399 CrossRef PubMed;
(b) P. C. E. Moody and E. L. Raven, Acc. Chem. Res., 2018, 51, 427–435 CrossRef CAS PubMed;
(c) F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS PubMed;
(d) C. M. Krest, E. L. Onderko, T. H. Yosca, J. C. Calixto, R. F. Karp, J. Livada, J. Rittle and M. T. Green, J. Biol. Chem., 2013, 288, 17074–17081 CrossRef CAS PubMed;
(e) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
-
(a) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed;
(b) J. J. D. Sacramento and D. P. Goldberg, Acc. Chem. Res., 2018, 51, 2641–2652 CrossRef CAS PubMed;
(c) X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed;
(d) R. A. Baglia, J. P. T. Zaragoza and D. P. Goldberg, Chem. Rev., 2017, 117, 13320–13352 CrossRef CAS PubMed.
-
(a) A. P. Ledray, C. M. Krest, T. H. Yosca, K. Mittra and M. T. Green, J. Am. Chem. Soc., 2020, 142, 20419–20425 CrossRef CAS PubMed;
(b) K. Mittra and M. T. Green, J. Am. Chem. Soc., 2019, 141, 5504–5510 CrossRef CAS PubMed;
(c) Y. J. Jeong, Y. Kang, A.-R. Han, Y.-M. Lee, H. Kotani, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2008, 47, 7321–7324 CrossRef CAS PubMed;
(d) R. van Eldik, Coord. Chem. Rev., 2007, 251, 1649–1662 CrossRef CAS;
(e) W. Nam, S.-E. Park, I. K. Lim, M. H. Lim, J. Hong and J. Kim, J. Am. Chem. Soc., 2003, 125, 14674–14675 CrossRef CAS PubMed.
- K. Nishikawa, Y. Honda and H. Fujii, J. Am. Chem. Soc., 2020, 142, 4980–4984 CrossRef CAS PubMed.
-
(a) S. Kal and L. Que Jr, J. Biol. Inorg. Chem., 2017, 22, 339–365 CrossRef CAS PubMed;
(b) E. I. Solomon, S. Goudarzi and K. D. Sutherlin, Biochemistry, 2016, 55, 6363–6374 CrossRef CAS PubMed;
(c) E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed;
(d) C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger Jr, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que Jr, Science, 2003, 299, 1037–1039 CrossRef CAS PubMed.
-
(a) V. A. Larson, B. Battistella, K. Ray, N. Lehnert and W. Nam, Nat. Rev. Chem., 2020, 4, 404–419 CrossRef CAS;
(b) X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS PubMed;
(c) S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407–2414 CrossRef CAS PubMed;
(d) W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS PubMed;
(e) M. Puri and L. Que Jr, Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed;
(f) W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed.
-
(a) M. S. Seo, N. H. Kim, K.-B. Cho, J. E. So, S. K. Park, M. Clémancey, R. Garcia-Serres, J.-M. Latour, S. Shaik and W. Nam, Chem. Sci., 2011, 2, 1039–1045 RSC;
(b) A. N. Biswas, M. Puri, K. K. Meier, W. N. Oloo, G. T. Rohde, E. L. Bominaar, E. Münck and L. Que Jr, J. Am. Chem. Soc., 2015, 137, 2428–2431 CrossRef CAS PubMed;
(c) I. Monte-Pérez, X. Engelmann, Y.-M. Lee, M. Yoo, E. Kumaran, E. R. Farquhar, E. Bill, J. England, W. Nam, M. Swart and K. Ray, Angew. Chem., Int. Ed., 2017, 56, 14384–14388 CrossRef PubMed.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
-
(a) Y. H. Kwon, B. K. Mai, Y.-M. Lee, S. N. Dhuri, D. Mandal, K.-B. Cho, Y. Kim, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2015, 6, 1472–1476 CrossRef CAS PubMed;
(b) S. N. Dhuri, K.-B. Cho, Y.-M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623–8632 CrossRef CAS PubMed;
(c) S. Kim, K.-B. Cho, Y.-M. Lee, J. Chen, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 10654–10663 CrossRef CAS PubMed;
(d) W. N. Oloo, Y. Feng, S. Iyer, S. Parmelee, G. Xue and L. Que Jr, New J. Chem., 2013, 37, 3411–3415 RSC;
(e) V. Dantignana, J. Serrano-Plana, A. Draksharapu, C. Magallón, S. Banerjee, R. Fan, I. Gamba, Y. Guo, L. Que Jr, M. Costas and A. Company, J. Am. Chem. Soc., 2019, 141, 15078–15091 CrossRef CAS PubMed;
(f) X.-X. Li, K.-B. Cho and W. Nam, Bull. Korean Chem. Soc., 2021, 42, 1506–1512 CrossRef CAS.
-
(a) W. J. Song, Y. O. Ryu, R. Song and W. Nam, J. Biol. Inorg. Chem., 2005, 10, 294–304 CrossRef CAS PubMed;
(b) A.-R. Han, Y. J. Jeong, Y. Kang, J. Y. Lee, M. S. Seo and W. Nam, Chem. Commun., 2008, 1076–1078 RSC;
(c) R. Gupta, X.-X. Li, K.-B. Cho, M. Guo, Y.-M. Lee, Y. Wang, S. Fukuzumi and W. Nam, J. Phys. Chem. Lett., 2017, 8, 1557–1561 CrossRef CAS PubMed.
-
(a) H. Fujii, J. Am. Chem. Soc., 1993, 115, 4641–4648 CrossRef CAS;
(b) H. Fujii, Coord. Chem. Rev., 2002, 226, 51–60 CrossRef CAS;
(c) M. Asaka and H. Fujii, J. Am. Chem. Soc., 2016, 138, 8048–8051 CrossRef CAS PubMed;
(d) Y. Ishimizu, Z. Ma, M. Hada and H. Fujii, J. Biol. Inorg. Chem., 2019, 24, 483–494 CrossRef CAS PubMed;
(e) N. Fukui, K. Ueno, M. Hada and H. Fujii, Inorg. Chem., 2021, 60, 3207–3217 CrossRef CAS PubMed;
(f) Y. Suzuki, M. Hada and H. Fujii, J. Inorg. Biochem., 2021, 223, 111542 CrossRef CAS PubMed;
(g) K. Ueno, Y. Ishimizu and H. Fujii, Inorg. Chem., 2021, 60, 9243–9247 CrossRef CAS PubMed;
(h) Y. Ishimizu, Z. Ma, M. Hada and H. Fujii, Inorg. Chem., 2021, 60, 17687–17698 CrossRef CAS PubMed.
-
(a) Y. M. Goh and W. Nam, Inorg. Chem., 1999, 38, 914–920 CrossRef CAS PubMed;
(b) D. Kumar, L. Tahsini, S. P. de Visser, H. Y. Kang, S. J. Kim and W. Nam, J. Phys. Chem. A, 2009, 113, 11713–11722 CrossRef CAS PubMed.
-
(a) T. Kitagawa and Y. Mizutani, Coord. Chem. Rev., 1994, 135/136, 685–735 CrossRef CAS;
(b) K. Nakamoto, Coord. Chem. Rev., 2002, 226, 153–165 CrossRef CAS;
(c) J. Terner, V. Palaniappan, A. Gold, R. Weiss, M. M. Fitzgerald, A. M. Sullivan and C. M. Hosten, J. Inorg. Biochem., 2006, 100, 480–501 CrossRef CAS PubMed.
-
(a) C. V. Sastri, M. J. Park, T. Ohta, T. A. Jackson, A. Stubna, M. S. Seo, J. Lee, J. Kim, T. Kitagawa, E. Münck, L. Que Jr and W. Nam, J. Am. Chem. Soc., 2005, 127, 12494–12495 CrossRef CAS PubMed;
(b) Y.-M. Lee, S. N. Dhuri, S. C. Sawant, J. Cho, M. Kubo, T. Ogura, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1803–1806 CrossRef CAS PubMed;
(c) S. Hong, H. So, H. Yoon, K.-B. Cho, Y.-M. Lee, S. Fukuzumi and W. Nam, Dalton Trans., 2013, 42, 7842–7845 RSC.
-
(a) R. Sarangi, Coord. Chem. Rev., 2013, 257, 459–472 CrossRef CAS PubMed;
(b) T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
-
(a) T. Kojima, Y. Hirai, T. Ishizuka, Y. Shiota, K. Yoshizawa, K. Ikemura, T. Ogura and S. Fukuzumi, Angew. Chem., Int. Ed., 2010, 49, 8449–8453 CrossRef CAS PubMed;
(b) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed;
(c) I. Garcia-Bosch, A. Company, C. W. Cady, S. Styring, W. R. Browne, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2011, 50, 5648–5653 CrossRef CAS PubMed;
(d) J. Park, Y.-M. Lee and W. Nam, J. Am. Chem. Soc., 2013, 135, 5052–5061 CrossRef CAS PubMed;
(e) T. Ishizuka, S. Ohzu, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Chem. Sci., 2014, 5, 1429–1436 RSC;
(f) L. Zhang, Y.-M. Lee, M. Guo, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2020, 142, 19879–19884 CrossRef CAS PubMed.
-
(a) S. Fukuzumi, Y.-M. Lee and W. Nam, Bull. Korean Chem. Soc., 2021, 42, 1558–1568 CrossRef CAS;
(b) P. T. Truong, S. G. Miller, E. J. M. S. Maria and M. A. Bowring, Chem.–Eur. J., 2021, 27, 14800–14815 CrossRef CAS PubMed;
(c) S. Fukuzumi, Y.-M. Lee and W. Nam, Bull. Korean Chem. Soc., 2020, 41, 1217–1232 CrossRef CAS.
-
(a) Z. Pan and M. Newcomb, Inorg. Chem. Commun., 2011, 14, 968–970 CrossRef CAS PubMed;
(b) R. Zhang and M. Newcomb, Acc. Chem. Res., 2008, 41, 468–477 CrossRef CAS PubMed;
(c) Z. Pan and M. Newcomb, Inorg. Chem., 2007, 46, 6767–6774 CrossRef CAS PubMed;
(d) R. Zhang, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 2005, 127(18), 6573–6582 CrossRef CAS PubMed.
-
(a) X. Lu, Y.-M. Lee, M. S. Seo and W. Nam, Chem. Commun., 2020, 56, 11207–11210 RSC;
(b) S. Fukuzumi, N. Fujioka, H. Kotani, K. Okhubo, Y.-M. Lee and W. Nam, J. Am. Chem. Soc., 2009, 131, 17127–17134 CrossRef CAS PubMed.
-
(a) M. Wolak and R. van Eldik, Chem.–Eur. J., 2007, 13, 4873–4883 CrossRef CAS PubMed;
(b) J. T. Groves, Z. Gross and M. K. Stern, Inorg. Chem., 1994, 33, 5065–5072 CrossRef CAS.
- J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S19 for spectroscopic characterization and kinetic data, Tables S1–S5 and Fig. S20 for EXAFS and DFT calculation results, and calculated Cartesian coordinates. See https://doi.org/10.1039/d2sc01232d |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2022 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xiao-Xi
Li‡
a,
Xiao-Xi
Li‡