
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational insights into the formation and nature of the sulfilimine bond in collagen-IV†
Anupom Roy ,
Taqred H. Alnakhli and
James W. Gauld*
,
Taqred H. Alnakhli and
James W. Gauld*
Department of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario N9B 3P4, Canada. E-mail: gauld@uwindsor.ca
First published on 22nd July 2022
Abstract
Collagen IV is essential component of basement membrane in the tissues. It provides proper cellular structure by the formation of sulfilimine bond (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) between methionine and lysine or hydroxylysine (cross-links) residues which can be formed with or without post-translational modification. The sulfilimine bond has critical roles in tissue development and human diseases. Peroxidasin, a basement membrane peroxidase, generates reactive halogen species including hypobromous (HOBr) acid and hypochlorous (HOCl) acid which help to form halosulfonium or haloamine. The sulfilamine bond can be formed either by the formation of halosulfonium or by the formation of halomine. The aim of the study is the investigation of the formation of sulfilimine bond and its nature in collagen IV using multi-scale approach that included MD, QM-cluster, systematic series of small models, and NBO analysis. These results suggest that sulfilimine bond can be formed either via brominated/chlorinated halosulfonium or haloamine pathway. The results of systematic series of small model indicate that the formation of sulfilimine complex from halosulfonium happens through the formation of positively charged halosulfonated sulfilimine complex. It also suggests that the formation of sulfilimine complex from haloamine occurs through the formation of positively charged sulfilimine complex where the S and N bond forms and halogen goes off at the same time. Furthermore, the NBO analysis suggest the S and N bond is strongly polarized toward nitrogen in both single protonated and neutral system, Nδ− ← Sδ+ and also indicate the existence of a coordinate covalent (i.e. dative) bond.
N) between methionine and lysine or hydroxylysine (cross-links) residues which can be formed with or without post-translational modification. The sulfilimine bond has critical roles in tissue development and human diseases. Peroxidasin, a basement membrane peroxidase, generates reactive halogen species including hypobromous (HOBr) acid and hypochlorous (HOCl) acid which help to form halosulfonium or haloamine. The sulfilamine bond can be formed either by the formation of halosulfonium or by the formation of halomine. The aim of the study is the investigation of the formation of sulfilimine bond and its nature in collagen IV using multi-scale approach that included MD, QM-cluster, systematic series of small models, and NBO analysis. These results suggest that sulfilimine bond can be formed either via brominated/chlorinated halosulfonium or haloamine pathway. The results of systematic series of small model indicate that the formation of sulfilimine complex from halosulfonium happens through the formation of positively charged halosulfonated sulfilimine complex. It also suggests that the formation of sulfilimine complex from haloamine occurs through the formation of positively charged sulfilimine complex where the S and N bond forms and halogen goes off at the same time. Furthermore, the NBO analysis suggest the S and N bond is strongly polarized toward nitrogen in both single protonated and neutral system, Nδ− ← Sδ+ and also indicate the existence of a coordinate covalent (i.e. dative) bond.
1 Introduction
The basement membrane is a thin, highly specialized extracellular matrix that is essential for the proper structure and function of tissues.1 In particular, it provides structural support for epithelial cells and plays important roles in, for example, intracellular signal transduction and cell behavior regulation.1,2 Its importance is further underscored by the fact that its improper functioning, due to for instance genetic mutations, can result in a number of severe or even fatal diseases or conditions in organisms.3 However, despite its physiological importance much remains unclear or even unknown including its structure.For instance, the most abundant molecular components of basement membranes are the collagen proteins of which there are least 28 sub-types.2,4 In particular, type IV Collagen (Collagen IV), discovered in 1966,5 is the most abundant molecular species of the basement membrane and is a critical component of the lamina densa,2 a sub-layer of the basement membrane.6 In particular, the collagen IV forms an essential and complex network that provides a supportive scaffold for other components of the basement membranes.7 Improper structures and/or defects in collagen IV are associated with several diseases including rheumatological and dermatological diseases.8,9 Furthermore, it is believed that the potentially fatal Goodpasture syndrome, a rare autoimmune disease, is thought to result from antibodies attacking certain alpha subunits within collagen IV in the basement membranes of the renal glomeruli and pulmonary alveoli.9 Another well-known collagen IV related pathology is the genetic disorder Alport's syndrome, caused by mutations within collagen IV, which is associated with proteinuria, progressive renal failure, ocular abnormalities, and hearing loss.9 In addition, it has been reported that collagen IV also has critical roles in wound healing10 and embryogenesis.11
While the exact structure of collagen IV remains unclear, it is known to be composed of six highly homologous polypeptide chains; α1(IV) to α6(IV), with each comprising of an (i) amino-terminal; (ii) triple-helical, and (iii) carboxy-terminal domain. Three α-chains bind together to form a triple helix, a protomer, which then aggregate end to end to form a hexamer and ultimately collagen fibrils.9,12,13 It is known that collagen IV can undergo several post-translational modifications and that protomers covalently cross-link through a variety of enzymatic and non-enzymatic processes. While these are often to help provide the structural integrity of the collagen IV network, some cross-links have also been associated with aging and disease.14 However, in 2009 it was discovered that covalent cross-links could also form between an hydroxylysine (Hyl) or lysine (Lys) residue in one chain with a Methionine (Met) residue in another.15 It is noted that this also indicates that post-translational modifications are not essential for the formation of these cross-links.15 More specifically, this cross-linking involved the side-chain amine of Hyl/Lys and the sulfur of Met and led to the formation of a sulfilimine (SN) crosslink. Furthermore, such inter-residue bonds are rare in biochemistry; so far the only known natural occurrence is in collagen IV.15 Furthermore, it is held that the sulfilimine bond of collagen IV was essential for evolution of tissues and eumetazoa.16
Recently, experimental studies have suggested that the sulfilimine bond formation in collagen IV may be induced by the hypohalous acids, hypobromous (HOBr) and hypochlorous (HOCl) acid, though their exact roles remain unclear.1,17 Furthermore, the efficiency of HOBr facilitated sulfilimine bond formation is 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold greater than for HOCl.1 These acids are produced by peroxidasin, a multifunctional extracellular matrix-associated member of the heme peroxidase family.18 Based on experimental observations it has been proposed that hypohalous acid (HOX; X = Br, Cl) mediated sulfilimine bond formation can proceed via either a halosulfonium or halomine intermediate as shown in Fig. 1.15,19 More specifically, the HOX acid may react with the side chain sulfur centre of Met93 to form a halosulfonium cation. This then reacts with the side chain amine nitrogen of Hyl, and with loss of HX forms a neutral sulfilimine crosslink between Met93 and Hyl211 (or Lys211). Alternatively, if the side chain amine of Hyl211 (or Lys211) is neutral, the HOX may react with to form a haloamine. This then reacts with the sulfur of Met93 to form the neutral sulfilimine crosslink. Of these two possible reaction pathways, the halosulfonium pathway is the major pathway for the formation of sulfilimine bond.1 While the neutral sulfilimine bond is represented as a covalent double bond, a previous computational study concluded that it is more accurately described as a polarized coordinate-covalent (dative) single bond; Nδ− ← Sδ+.20 Meanwhile, another computational study concluded that the sulfilimine bond is protonated at pH 7.21
000-fold greater than for HOCl.1 These acids are produced by peroxidasin, a multifunctional extracellular matrix-associated member of the heme peroxidase family.18 Based on experimental observations it has been proposed that hypohalous acid (HOX; X = Br, Cl) mediated sulfilimine bond formation can proceed via either a halosulfonium or halomine intermediate as shown in Fig. 1.15,19 More specifically, the HOX acid may react with the side chain sulfur centre of Met93 to form a halosulfonium cation. This then reacts with the side chain amine nitrogen of Hyl, and with loss of HX forms a neutral sulfilimine crosslink between Met93 and Hyl211 (or Lys211). Alternatively, if the side chain amine of Hyl211 (or Lys211) is neutral, the HOX may react with to form a haloamine. This then reacts with the sulfur of Met93 to form the neutral sulfilimine crosslink. Of these two possible reaction pathways, the halosulfonium pathway is the major pathway for the formation of sulfilimine bond.1 While the neutral sulfilimine bond is represented as a covalent double bond, a previous computational study concluded that it is more accurately described as a polarized coordinate-covalent (dative) single bond; Nδ− ← Sδ+.20 Meanwhile, another computational study concluded that the sulfilimine bond is protonated at pH 7.21
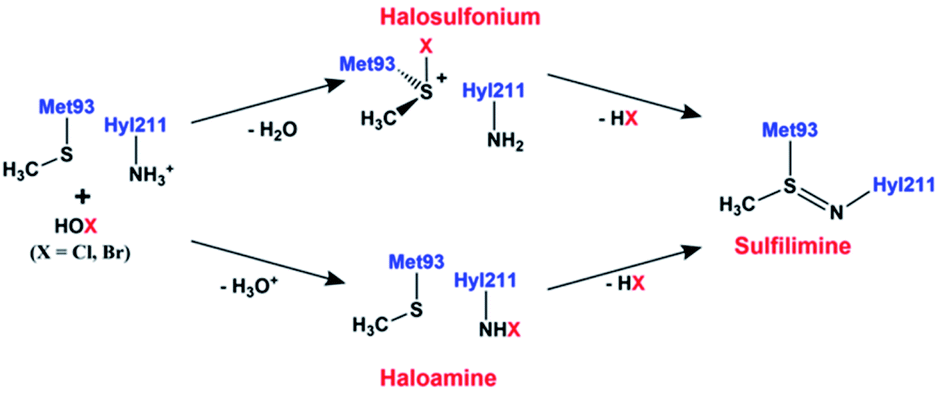 | ||
| Fig. 1 The proposed15,19 mechanism for the formation of the sulfilimine bond in collagen IV. | ||
In this present study we have applied computational chemistry methods to examine the proposed mechanisms by which the sulfilimine bond formation in collagen IV is mediated by the hypohalous acids HOX (X = Cl and Br). More specifically, using molecular dynamics (MD) simulations, quantum mechanical (QM)-cluster methods to examine possible mechanisms for formation of possible sulfilimine crosslinks. In addition, we have applied Natural Bond Orbital (NBO) analyses to gain further insights into the nature of possible intermediates, as well as the nature of the product sulfilimine in the basement membrane.
2 Computational methods
2.1 Molecular dynamic simulations (MD)
A suitable X-ray crystal structure of collagen IV, containing the sulfilimine Met–Lys crosslink, was obtained from the Protein Data Bank (PDB ID: 1M3D).22 While the structure has 6 similar chains, only chains B and D were used in the subsequent MD simulations as these two are involved in the crosslink. The structure was prepared for MD simulations using the Molecular Operator Environment (MOE).23 All parameterization, solvation, minimization, heating, equilibration and MD production runs were performed using AmberTools 16 and the AMBER 16 program.24 First, the prepared initial structure was solvated using TIP3P water model in which the structure was surrounded by a water layer of 10 Å deep with periodic boundary conditioned enabled. Then, the solvation was followed by minimization using the molecular mechanics (MM) force field AMBER ff14SB.25 The minimization was performed in two steps: (1) first hydrogen atoms on all heavy atoms were minimized, and then (2) the protein backbone and all heavy atoms using SHAKE algorithm. The resulting minimized system was gradually heated to 300 K using Anderson-like temperature coupling scheme (ntt = 2) at a constant pressure with periodic boundary conditions (ntp = 1) with a time step of 2 fs for 100 ps. Then, the heated system was submitted for equilibration and production run using NVT ensemble with constant temperature (300 K) and constant volume (ntb = 1) with a time step of 2 fs for 50 ns that includes 100 ps equilibration. The Particle Mesh Ewald (PME) method26 was used for calculating coulombic interactions, with cutoffs for nonbonded long-range interactions set to 8 Å (the default). The generated conformations from the MD simulation were analyzed based on backbone atoms' root mean square deviations (RMSD) using AmberTools 16.24 Then, the obtained RMSD values were clustered, and the structure with the shortest Met93S⋯NLys/Hyl211 distance was chosen for the QM-cluster calculations. All parameterization, solvation, minimization, heating, equilibration and MD production runs were performed using AmberTools 16 and the AMBER 16 program.24 First, the prepared initial structure was solvated using TIP3P water model in which the structure was surrounded by a water layer of 10 Å deep with periodic boundary conditioned enabled. Then, the solvation was followed by minimization using the molecular mechanics (MM) force field AMBER ff14SB.25 The minimization was performed in two steps: (1) hydrogen atoms on all heavy atoms were minimized, and then (2) the protein backbone and all heavy atoms using the SHAKE algorithm. The resulting minimized system was gradually heated to 300 K using Anderson-like temperature coupling scheme (ntt = 2) at a constant pressure with periodic boundary conditions (ntp = 1) with a time step of 2 fs for 100 ps. The system was then submitted for equilibration and production using NVT ensemble with constant temperature (300 K) and volume (ntb = 1) and a time step of 2 fs for 50 ns that includes 100 ps equilibration. The Particle Mesh Ewald method26 was used for calculating coulombic interactions, with cutoffs for nonbonded long-range interactions set to 8 Å (the default). The generated conformations from the MD simulation were analyzed based on backbone atoms' root mean square deviations (RMSD) using AmberTools 16.24 The obtained RMSD values were clustered, and the structure with the shortest Met93S⋯NLys/Hyl211 distance was chosen for the QM-cluster calculations.2.2 Quantum Mechanics (QM)-cluster and systematic series of small model studies
The Gaussian 09 and Gaussian 16 programs were used for all QM-cluster and small model calculations.27,28 QM-cluster optimization and harmonic frequency calculations were performed using M06-2X/6-31G(d,p) level of theory while the surrounding protein and solvent environment was modelled using the IEF-PCM method (ε = 78.35 for water or 4.0 for protein). The M06-2X functional was chosen because of its purported greater reliability and accuracy for main group thermochemistry and description of non-covalent long-range interactions.29,30 The results were compared where possible with those obtained at the QCISD/6-31G(d,p) or/6-311+G(2df,p) levels of theory (see text). The QM-cluster models for the formation of the halosulfonium and haloamine intermediates consisted of 30 and 33 atoms in total, respectively. More specifically, the clusters included truncated sidechains of Met93, Lys211/Hyl211, and Glu214 (that was observed in the MD simulations to be spatially close), and 1 or more water molecules. It should be noted that Met93, Lys211/Hyl211, and Glu214 were modelled by dimethylsulfide (S(CH3)2), methylamine (CH3NH2), and ethanoate/ethanoic acid (CH3COO−/CH3COOH), respectively. Single point energies were obtained at M06-2X/6-311+G(2df,p) level of theory, on the above geometries, with the solvent environment modelled using the IEF-PCM method. Gibb's Free Energy corrections were obtained at the M06-2X/6-31G(d,p) level of theory.2.3 Natural Bond Orbital (NBO) analysis
The natural bond orbital (NBO) analysis was performed using the NBO program (NBO Pro 7) with the method of Weinhold and coworkers,31 and Gaussian 09 program.27 The NBO analyses were performed at the M06-2X/6-311G+(2df,p)//M06-2X/6-31G(d,p) level of theory.3 Results and discussion
3.1 Molecular Dynamics (MD) results
As mentioned in the previous Computational methods section, a 50 ns MD simulation was first performed. This assisted examination of, for instance, key distances and interactions between residues, especially those involving the sidechains of Met93 and Lys211/Hyl211 in adjacent strands, as well as water availability around these side chains. It is noted that the side chain of Lys211/Hyl211 was protonated in the MD simulations.The distances between the side chain S of Met93 (SMet93) and side chain N of Lys211/Hyl211 (NLys211/Hyl211), in the absence of the hypohalous acids HOBr or HOCl, are shown in Fig. 2. Over the course of the MD simulation the average Lys211/Hyl211NH⋯SMet93 distance was 6.4 Å. However, during the MD simulation the distance varied and notably, shortened to less than 4.0 Å, with the shortest distance of approximately being just 3.13 Å. In these short Lys211/Hyl211NH⋯SMet93 distanced conformations, the protonated side-chain amine of Lys211/Hyl211 forms a weak electrostatic interaction with the side-chain S of Met93. In addition, water molecules were also observed in the immediate vicinity of the Met93 and Lys211/Hyl211 sidechains. Thus, these distances indicate the possibility for weak interactions and reasonable positioning of the two mechanistically key functional groups, as well as the availability of waters, for the required subsequent reactions.
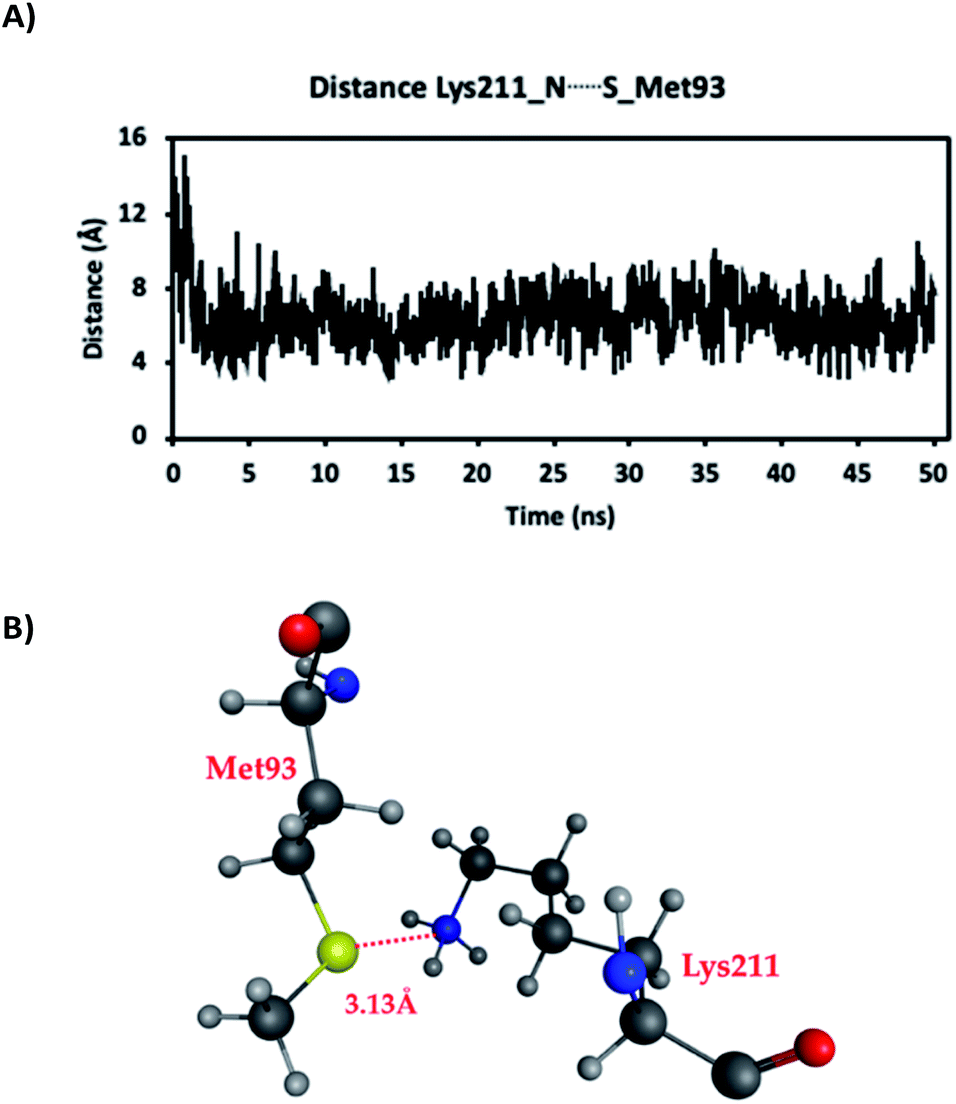 | ||
| Fig. 2 (A) The distances between SMet93 and NLys211/Hyl211 during the 50 ns MD simulations, and (B) a representative structure of the conformations with the shortest Met93S⋯NLys211/Hyl211 distance. For clarity, all other residues and water molecules are omitted (atom colours: O = red; C = dark grey; N = blue; S = yellow; H = light grey). | ||
To explore possible and proposed sulfilimine bond formation reactions involving HOBr and HOCl, as noted in the computational methods section, a suitable representative structure was then selected and hypobromous acid (HOBr) and hypochlorous acid (HOCl) manually inserted and positioned in proximity to the sidechains of Met93 and Lys211/Hyl211. It is noted that except for the addition of HOBr or HOCl, and when needed a proton in the form of H3O+, no additional changes were made.
3.2 QM-cluster studies on sulfilimine bond formation via the proposed halosulfonium or haloamine
Synthetically, the formation of sulfilimines can be achieved via reaction of a haloamine with a sulfide32,33 and several possible mechanisms, based at least in part on the results of kinetic studies, have been proposed. These include nucleophilic attack of the sulfide on the haloamine nitrogen, halogenation of the sulfide by the haloamine, or oxidative addition of the haloamine to the sulfide.32 It is noted that other possible synthetic mechanisms via other intermediates or related species (e.g., sulfoxides) have also been suggested. However, in the mechanisms proposed17 for collagen IV, it has been suggested that hypohalous acids produced by peroxidasin, HOCl and HOBr, could react with the sidechains of methionine (Met93) or the lysine/hydroxylysine (Lys211/Hyl211) residues to give the corresponding halosulfonium or haloamine derivatives, respectively. Subsequently, the halosulfonium could react with the sidechain amine of lysine/hydroxylysine (Lys211/Hyl211), or conversely the haloamine could react with the sidechain sulfide of Met93 to give a neutral sulfilimine crosslink. Thus, the reaction of HOCl and HOBr with the Met93 sulfur or Lys211/Hyl211 sidechain nitrogen was considered.Gibbs free energies were obtained for the formation of the halosulfonium or haloamine, using the chemical models described in the Computational methods section, are provided in the Tables 1 and 2, and the energy diagrams are shown in the ESI (Fig. S2–S9†). Herein, only key features of the PESs obtained are described. It is noted that for formation of the halosulfonium and haloamine the sidechain of Glu214 was neutral (i.e., CH3COOH). In both cases this resulted from addition of a H3O+ which was needed to neutralize the hydroxide (HO−) formed during the reactions.
| Reactions | Reactants | ΔG‡ (kJ mol−1) | ΔG0 (kJ mol−1) |
|---|---|---|---|
| Halosulfonium by HOBr | S(CH3)2 + CH3NH2 + CH3COOH + H2O + HOBr | 3.0 | −3.6 |
| Halosulfonium by HOCl | S(CH3)2 + CH3NH2 + CH3COOH + H2O + HOCl | 41.1 | 0.0 |
| Haloamine by HOBr | S(CH3)2 + CH3NH3 + CH3COO− + 2H2O + HOBr | 52.0 | −51.2 |
| Haloamine by HOCl | S(CH3)2 + CH3NH3 + CH3COO− + 2H2O + HOCl | 29.8 | −95.5 |
| Reactions | Reactants | ΔG‡ (kJ mol−1) | ΔG0 (kJ mol−1) |
|---|---|---|---|
| Sulfilimine via bromo halosulfonium | BrS(CH3)2 + CH3NH2 + H2O | 118.2 | −28.0 |
| Sulfilimine via chloro halosulfonium | ClS(CH3)2 + CH3NH2 + 3H2O | 15.9 | −29.9 |
| Sulfilimine via bromo haloamine | S(CH3)2 + CH3N(H)Br + H3O+ | 108.9 | −96.8 |
| Sulfilimine via chloro haloamine | S(CH3)2 + CH3N(H)Cl + H3O+ | 62.4 | −163.0 |
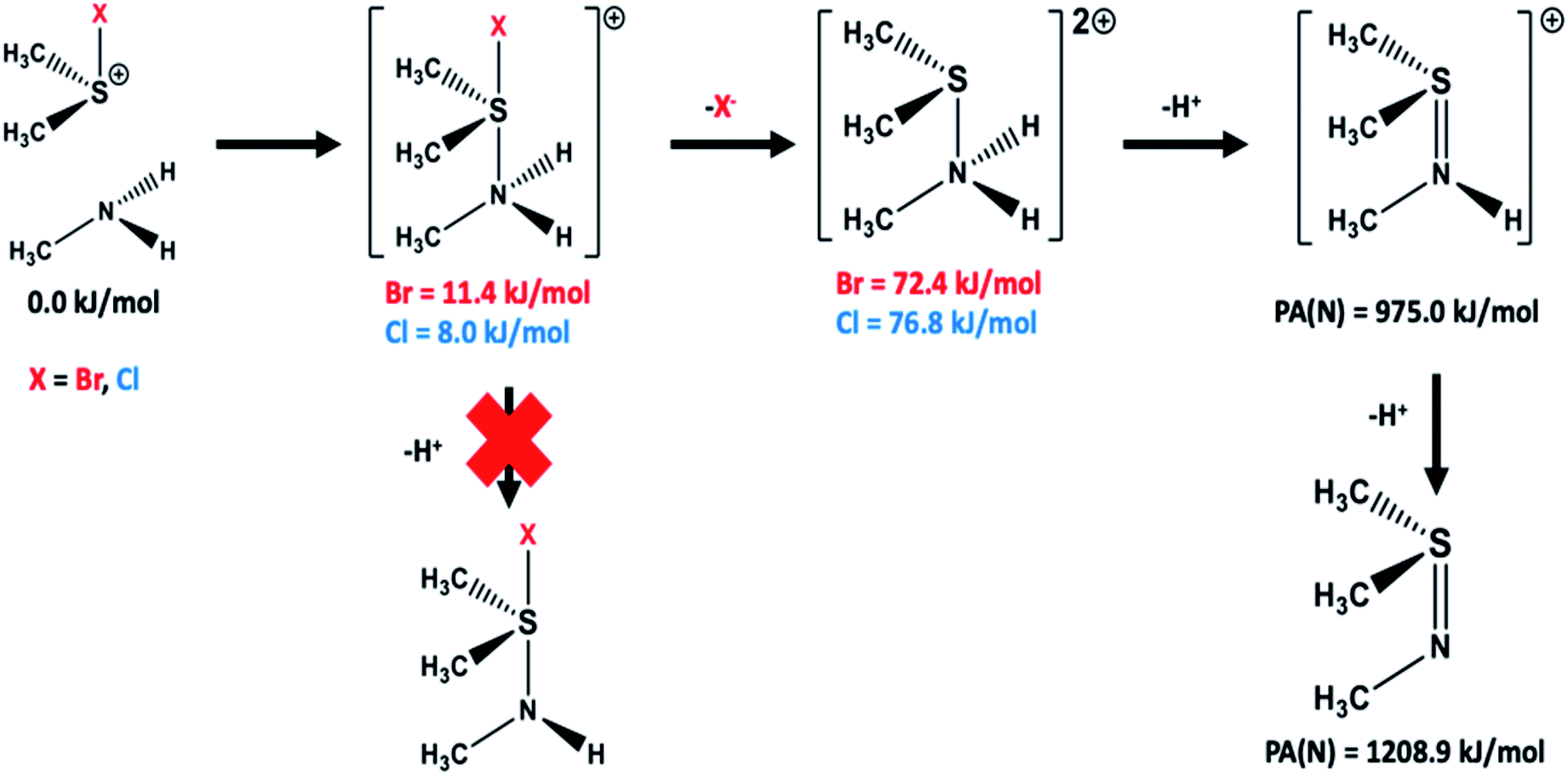 | ||
| Fig. 3 Schematic illustration of the reaction pathways examined for formation of the sulfilimine crosslink by reaction of the Met93 derived halosulfonium cation ((CH3)2SX+; X = Br, Cl) with the sidechain amine of Lys211/Hyl211 (CH3NH2). Gibb's free energies relative to the initial reactants are shown in red (bromo derived species) or blue (chloro derived species), while proton affinities of the sulfilimine nitrogen (PA(N)) in the neutral and single protonated species are also given. All energies are in kJ mol−1 and were obtained at the IEFPCM(water)-M06-2X/6-311+G(2df,p)//IEFPCM(water)-M06-2X/6-31G(d,p) with Gibb's Free Energy Corrections (see Computational methods). | ||
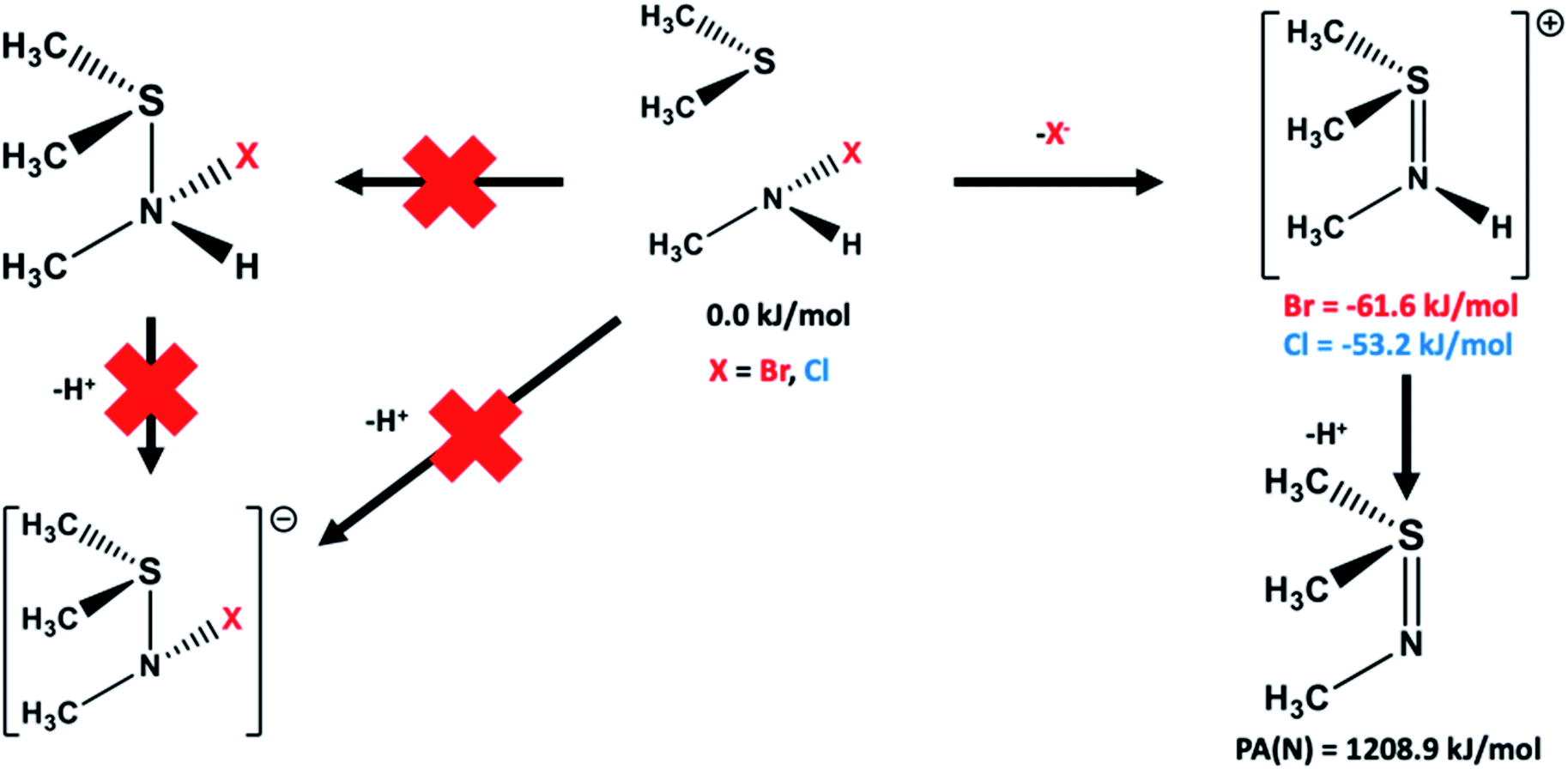 | ||
| Fig. 4 Schematic illustration of the reaction pathways examined for formation of the sulfilimine crosslink by reaction of the Lys211/Hyl211 derived haloamine's (CH3NHX; X = Br, Cl) with the sidechain sulfide of Met93 ((CH3)2S). Gibb's free energies relative to the initial reactants are shown in red (bromo derived species) or blue (chloro derived species), while proton affinities of the sulfilimine nitrogen (PA(N)) in the neutral and single protonated species are also given. All energies are in kJ mol−1 and were obtained at the IEFPCM(water)-M06-2X/6-311+G(2df,p)//IEFPCM(water)-M06-2X/6-31G(d,p) with Gibb's Free Energy Corrections (see Computational methods). | ||
Building upon these results, we also examined the possible stepwise formation of the protonated sulfilimine product. It was found that it was possible to form an initial complex between the halosulfonium and amine, with the nitrogen complexed to the sulfur trans to the halo (Fig. 3). The resulting cationic bromo- and chloro-containing complexes were calculated to be only slightly higher in free energy than the initial reactants by 11.4 and 8.0 kJ mol−1, respectively. Subsequent loss of a proton, that is formation of a neutral halo-containing species did not produce a stable species. Rather, loss of a halide to formally give the double protonated sulfilimine resulted in a species lying only 72.4 and 76.8 kJ mol−1 higher in free energy than the initial bromo- and chloro-containing reactants, respectively (see Fig. 3). Subsequent loss of a proton to the aqueous solution to give the single protonated sulfilimine species could then occur.
Sulfilimine bond formation via the haloamine intermediate was also examined. Again, it was found that the single protonated sulfilimine bond containing species described above, could be formed in one step from the initial reactants. More specifically, the sulfur of Met93 and sidechain haloamine nitrogen of Lys211/Hyl211 react directly to form an S–N interaction. Importantly, this bond formation occurs concomitantly with loss of the halide as X− from the haloamine component. Within the present models used this also required the presence of a hydronium ion (H3O+) which gives a proton to the leaving halide group to form HX (X = Br, Cl). The calculated reaction barrier heights for formation of the single protonated sulfilimine product (see Fig. S8 and S9†) were 108.9 and 62.4 kJ mol−1 for the reactions involving the bromo- and chloro haloamine's, respectively. While these are both higher than those calculated using the corresponding halosulfonium cation, it is important to note that both barriers obtained using the present models are predicted to be thermodynamically feasible. Furthermore, both reactions are calculated to be exothermic with the single protonated sulfilimine species lying lower in energy than the bromo- and chloro-halomine containing reactants by 96.8 and 163.0 kJ mol−1, respectively (see Fig. S8 and S9†).
Possible alternative pathways by which a bromo- or chloro-haloamine may react with a sulfide to form a sulfilimine species were also examined and are illustrated schematically in Fig. 4. In particular, the possibility of the haloamine and sulfide forming a neutral complex with an S⋯N interaction, or alternatively reacting with concomitant loss of a proton to form an S–N interaction containing anionic species were also considered. Neither alternate complex and pathway, at the present level of theory and with the chemical models used, was found to be feasible.
3.3 What is the likely protonated state of the sulfilimine bond-containing product?
In the feasible pathways for sulfilimine bond formation via either a halosulfonium reacting with a neutral alkyl amine, or a haloamine reacting with a disulfide, the resultant product was the protonated sulfilimine product (see Fig. 3 and 4). However, the sulfilimine crosslink in collagen IV is commonly represented and/or assumed to be neutral.15,20 Thus, we further examined whether the single protonated sulfilimine may lose a proton to the aqueous environment. It is noted that other studies have suggested that at least some sulfilimines could potentially be classed as superbases with calculated proton affinities of over 1000 kJ mol−1.34 In this present study, at the IEFPCM(water)-M06-2X/6-311+G(2df,p)//IEFPCM(water)-M06-2X/6-31G(d,p) corrected for Gibb's Free Energy, the proton affinity of the nitrogen centre in the sulfilimine bond is 1208.9 kJ mol−1. This is in general agreement with calculated values for other sulfilimines. Importantly, it is significantly higher than the proton affinity of water calculated at the same level of theory (989.1 kJ mol−1). Meanwhile, the proton affinity of the nitrogen centre in the single protonated sulfilimine bonded species is calculated to be slightly lower than that of water at 975.0 kJ mol−1. Thus, the present results indicate that under expected biological conditions the sulfilimine crosslink is most likely single protonated, in agreement with a previous computational study,21 but has the potential to vary its protonation state; but it is unlikely neutral.The protonation state of the sulfilimine bond's nitrogen centre significantly influences the length of the sulfilimine bond. For the current chemical models, the length of the sulfilimine bond in the optimized structures of the neutral, single- and double protonated nitrogen species are 1.606, 1.661, and 1.788 Å, respectively. That is, while the above proton affinities indicate that the single protonated species is more common in vivo, the neutral sulfilimine has a markedly shorter and stronger bond. We also optimized the structures of all three sulfilimine species at the considerably higher QCISD/6-311+G(2df,p) level of theory and determined the proton affinity of the nitrogen centre in the neutral and single protonated species (Table S1†). The optimized sulfilimine bond lengths obtained for the neutral and single- and double protonated nitrogen species of 1.599, 1.650, and 1.778 Å, respectively, in agreement with the trends observed when using the above M06-2X DFT-based approaches. Similar trends in the calculated proton affinities and comparison with that of water were also observed. In particular, at the IEFPCM(water)-QCISD/6-311+G(2df,p) level corrected for Gibbs Free Energy, the proton affinities of water is calculated to be 993.8 kJ mol−1, while that of the neutral sulfilimine is much higher at 1218.4 kJ mol−1, and that of the single protonated sulfilimine is slightly lower than that of H2O by 7.6 kJ mol−1 at 986.2 kJ mol−1.
3.4 Natural Bond Orbital (NBO) analyses
To gain further insights into the nature of the sulfilimine bond in the neutral, single- and double protonated species, they were examined using NBO. Pichierri20 has previously used NBO to characterize the nature of a neutral sulfilimine bond as proposed for collagen IV. For completeness, we considered all possible conformers and isomers of the neutral, single- and double protonated sulfilimine chemical species (Fig. 5). Specifically, there were two conformers for each species as the methyl group on the nitrogen can be on the same or opposite side as the sulfur's methyl groups. It is noted that thermodynamically the difference in conformers was not significant within the computational models used. In addition, there are also two possible isomers for each single protonated conformer Key NBO results are summarized in Table 1.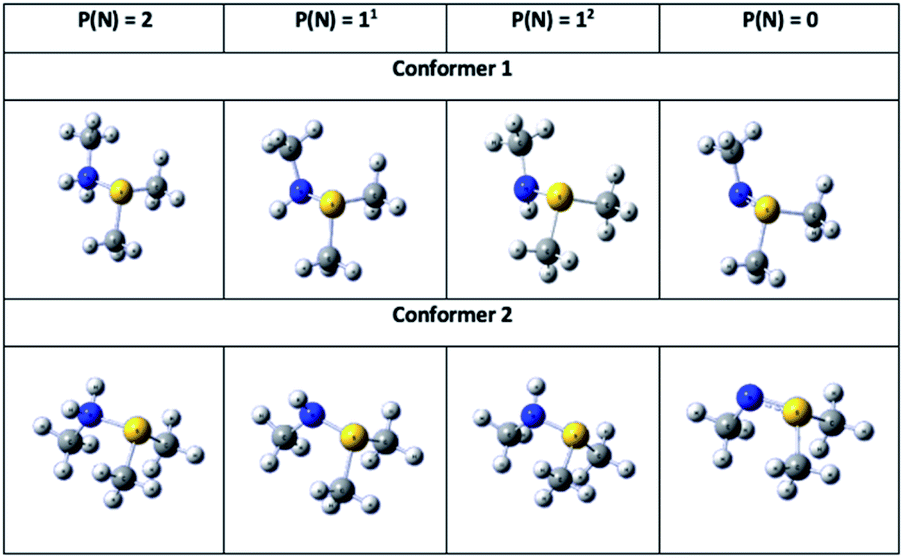 | ||
| Fig. 5 Illustration of the conformers and isomers of the neutral, and single- and double-protonated sulfilimine species considered herein for NBO analysis. P(N) indicates the protonation state of N in sulfilimine complex while 11 and 12 denote isomer 1 and 2 of the single-protonated sulfilimine species, respectively. (Atom colours: N = blue; S = yellow; C = grey; H = white). | ||
The NBO method analyzes the wavefunction in terms of localized electron pairs (i.e., natural orbitals) and provides Lewis-type descriptions for molecules in terms of two-electron two-center (σ, π) bonds and lone-pairs (n). The σNS (orbital hybridization) NBO of the conformers 1 and 2 are shown in Table 3.
| P(N) | σNS |
|---|---|
| Conformer 1 | |
| 2 | σNS = 0.8169(sp3.90d0.01)N + 0.5768(sp8.90d0.13)S |
| 11 | σNS = 0.7720(sp4.39d0.03)N + 0.6356(sp6.03d0.07)S |
| 12 | σNS = 0.7814(sp3.30d0.02)N + 0.6240(sp4.89d0.07)S |
| 0 | σNS = 0.7513(sp4.04d0.04)N + 0.6599(sp3.62d0.04)S |
|
|
| Conformer 2 | |
| 2 | σNS = 0.8166(sp3.94d0.01)N + 0.5772(sp8.91d0.13) |
| 11 | σNS = 0.7822(sp3.23d0.02)N + 0.6230(sp4.80d0.07)S |
| 12 | σNS = 0.7822(sp3.23d0.02)N + 0.6230(sp4.80d0.07)S |
| 0 | σNS = 0.7559(sp3.48d0.04)N + 0.6547(sp3.11d0.04)S |
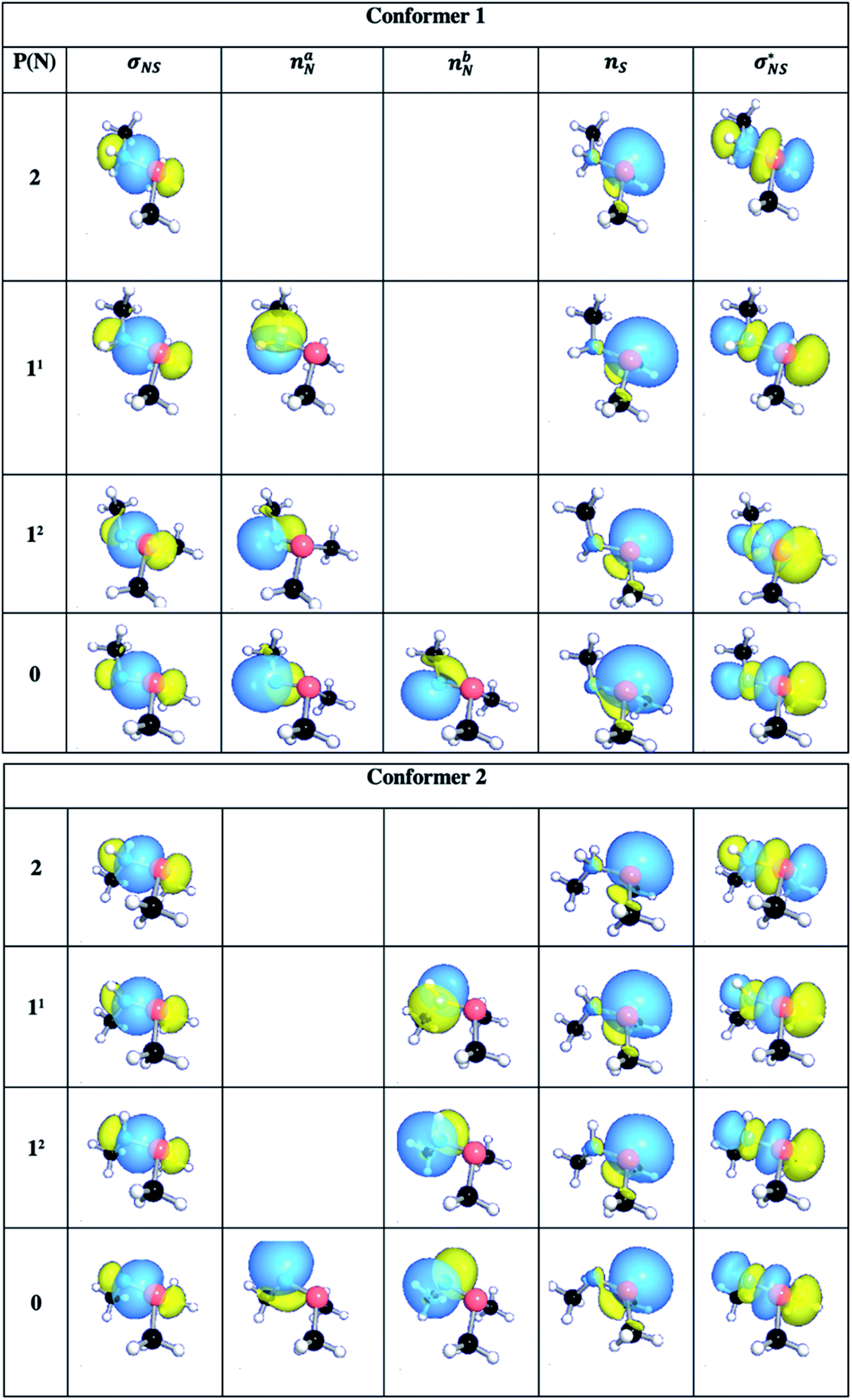 | ||
| Fig. 6 Natural bond orbitals obtained for conformers 1 and 2 showing 2e–2e σ-type covalent bond between N and S as well as the lone-pairs on sulfur (nS) and nitrogen (naN and nbN). The σNS and σ*NS represent bonding and antibonding orbitals, respectively. P(N) indicates the protonation state of N in sulfilimine complex while 11 and 12 denote isomer 1 and 2 of the single-protonated sulfilimine species, respectively. (Atom colours: N = blue; S = rose; C = black; H = white). | ||
The NBO analyses also provided descriptions of the electron lone pairs on nitrogen and sulfur, and additional bonding and antibonding orbitals which are illustrated in Fig. S6.† Notably for both conformers there is a lone pair on sulfur (nS), which remained unprotonated in all possible species considered herein. Meanwhile, the nitrogen centre in both neutral conformers has two lone pairs (naN and nbN) which are the sites of the first and second protonations (described above).
NBO analyses also provide values for the stabilization energy (E) associated with interaction(s) between filled and empty NBOs.31 For both double protonated conformers the highest stabilization energies of 4.78 and 6.19 kcal mol−1 were obtained for the CH(9) → 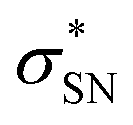 of conformer 1 and CH(12) →
of conformer 1 and CH(12) → 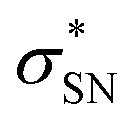 of conformer 2 interactions, respectively. For single protonated conformers and isomers, for conformer 1 stabilization energies of 5.95 and 9.48 kcal mol−1 were obtained for the naN →
of conformer 2 interactions, respectively. For single protonated conformers and isomers, for conformer 1 stabilization energies of 5.95 and 9.48 kcal mol−1 were obtained for the naN → 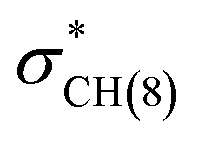 and naN →
and naN → 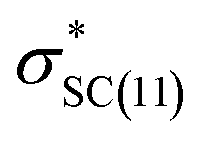 interactions in isomers 1 and 2, respectively. In contrast, for conformer 2 much closer stabilization energies of 9.35 and 9.48 kcal mol−1 were obtained for the naN →
interactions in isomers 1 and 2, respectively. In contrast, for conformer 2 much closer stabilization energies of 9.35 and 9.48 kcal mol−1 were obtained for the naN → 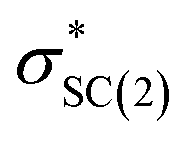 and naN →
and naN → 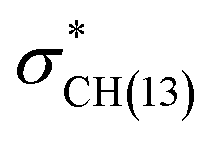 interactions in isomers 1 and 2, respectively. However, for the neutral (unprotonated) sulfilimine conformers 1 and 2 differences were observed between the origins and size of the largest stabilizing interaction(s). For conformer 1, it arose from the interaction of a lone pair on the nitrogen (naN) and an antibonding σ S–C orbital (
interactions in isomers 1 and 2, respectively. However, for the neutral (unprotonated) sulfilimine conformers 1 and 2 differences were observed between the origins and size of the largest stabilizing interaction(s). For conformer 1, it arose from the interaction of a lone pair on the nitrogen (naN) and an antibonding σ S–C orbital (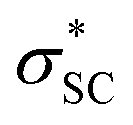 ), naN →
), naN → 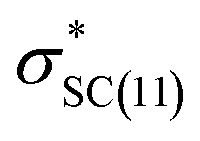 , with a value of 23.25 kcal mol−1. In contrast, for unprotonated conformer 2 the largest combined stabilization energy of 109.82 kcal mol−1 arose from interactions of both S–C bonding orbitals (SC) and the
, with a value of 23.25 kcal mol−1. In contrast, for unprotonated conformer 2 the largest combined stabilization energy of 109.82 kcal mol−1 arose from interactions of both S–C bonding orbitals (SC) and the 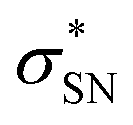 orbital; SC(2) →
orbital; SC(2) → 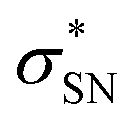 , SC(7) →
, SC(7) → 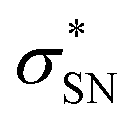 .
.
Interestingly, the NBO analyses also indicate that in both double protonated conformers 1 and 2, the S⋯N bond is strongly polarized towards S (i.e., Nδ− → Sδ+). However, upon loss of a proton to give the single protonated conformers the S⋯N bond is strongly polarized towards N (i.e., Nδ− ← Sδ+). This same polarization is observed in the neutral sulfilimine conformers as previously described by Pichierri.20 Indeed, these NBO results on all the neutral and protonated conformers support the previous conclusion of Pichierri20 that the S⋯N bond in collagen IV should be described as a coordinate covalent Nδ− ← Sδ+ bond.
The charges obtained from the NBO analyses for the sulfur and nitrogen centre in each sulfilimine species (i.e., conformers 1 and 2, and for all the protonation states) are given in Table 2. For both conformers, as the protonation state is systematically decreased from double to single the neutral, a corresponding decrease in the positive charge of the sulfur atom with a simultaneous increase in the negative charge on the nitrogen is observed. In the neutral system the positive and negative charge on the sulfur and nitrogen, are closest to equal in absolute value.
The dipole moments of the unprotonated (neutral), and single and double protonated sulfilimine species were also examined. The dipole moments in the double protonated species are in a distinctly different orientation to those of the single protonated and neutral sulfilimine species. Furthermore, in the neutral species the magnitude of the dipole moment is markedly higher than that of the double and single protonated species due to the changes of natural charges in S and N. This phenomenon has previously35 been said to be a characteristic of dative bonding such as suggested for the sulfilimine bond in collagen IV (Table 4).
| P(N) | Atoms & charges |
|---|---|
| Conformer 1 | |
| 2 | S = 1.118, N = −0.705 |
| 11 | S = 1.065, N = −0.831 |
| 12 | S = 1.096, N = −0.840 |
| 0 | S = 1.048, N = −1.056 |
|
|
| Conformer 2 | |
| 2 | S = 1.119, N = −0.703 |
| 11 | S = 1.100, N = −0.840 |
| 12 | S = 1.100, N = −0.840 |
| 0 | S = 1.060, N = −1.026 |
4 Conclusions
We have performed a multi-scale computational investigation, using MD simulations and Quantum Mechanical (QM)-cluster methods to examine proposed mechanisms for formation of the sulfilimine bond in collagen IV between Met93 and Lys/Hyl211 residues. In addition, we have used NBO analysis to examine the nature of the possible neutral or protonated (single or double) sulfilimine bond-containing products formed.4.1 Formation of a sulfilimine product
The present results suggest that formation of the sulfilimine crosslink via the proposed halosulfonium or haloamine intermediate is thermodynamically feasible. In reaction of a halosulfonium with a neutral alkyl amine, the first step involves formation of a complex in which the sulfur of the cationic halosulfonium (Met93-X+; X = Cl, Br) interacts with the lone pair on the amine''s nitrogen (Lys/Hyl211). The halogen is then lost as a halide to give a double protonated sulfilimine species. In contrast, the first step in the formation of an S⋯N bond via reaction of a haloamine with the Met93 sulfur requires concomitant loss of the halogen as a halide. This directly gives the most likely product, the single protonated sulfilimine species.4.2 Protonation of the sulfilimine bond
Proton affinities of the nitrogen (PAN) in the neutral and single protonated sulfilimine species were calculated. In the neutral sulfilimine bond PAN is >1200 kJ mol−1, which is markedly higher than that of H2O (∼990 kJ mol−1). In contrast, PAN in the single protonated sulfilimine bond is slightly lower (<15 kJ mol−1) than that calculated for H2O (Table S1†). Thus, single protonated is the most likely state of the sulfilimine crosslink in vivo, but conditions may be able to influence its protonation state (e.g., single or double).4.3 Nature of the sulfilimine crosslink
NBO analyses indicate that, as suggested previously for the neutral sulfilmine bond,20 the S and N bond is strongly polarized toward nitrogen in the single protonated and neutral systems, Nδ− ← Sδ+, and that it is also well describe as a coordinate covalent (i.e., dative) bond.Author contributions
Anupom Roy: computational calculations and writing, Taqred H. Alnakhli: computational calculations of bromo halosulfonium and bromo haloamine formation, James W. Gauld: supervision, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Science and Engineering Research Council of Canada (NSERC) for financial support. We thank Compute Canada for computational resources.Notes and references
- A. S. McCall, C. F. Cummings, G. Bhave, R. Vanacore, A. Page-McCaw and B. G. Hudson, Bromine Is an Essential Trace Element for Assembly of Collagen IV Scaffolds in Tissue Development and Architecture, Cell, 2014, 157(6), 1380–1392 CrossRef CAS PubMed.
- R. Kalluri, Basement membranes: structure, assembly and role in tumour angiogenesis, Nat. Rev. Cancer, 2003, 3(6), 422–433 CrossRef CAS PubMed.
- M. A. Morrissey and D. R. Sherwood, An active role for basement membrane assembly and modification in tissue sculpting, J. Cell Sci., 2015, 128(9), 1661–1668 CAS.
- J. Heino, The collagen family members as cell adhesion proteins, Bioessays, 2007, 29(10), 1001–1010 CrossRef CAS PubMed.
- N. A. Kefalides, A collagen of unusual composition and a glycoprotein isolated from canine glomerular basement membrane, Biochem. Biophys. Res. Commun., 1966, 22(1), 26–32 CrossRef CAS PubMed.
- M. Paulsson, Basement-Membrane Proteins - Structure, Assembly, and Cellular Interactions, Crit. Rev. Biochem. Mol. Biol., 1992, 27(1–2), 93–127 CAS.
- Y. Wu and G. Ge, Complexity of type IV collagens: from network assembly to function, Biol. Chem., 2019, 565–574 CrossRef PubMed.
- R. E. Petty, D. W. Hunt and A. M. Rosenberg, Antibodies to type IV collagen in rheumatic diseases, J. Rheumatol., 1986, 13(2), 246–253 CAS.
- A. M. Abreu-Velez and M. S. Howard, Collagen IV in Normal Skin and in Pathological Processes, N. Am. J. Med. Sci., 2012, 4(1), 1–8 CrossRef PubMed.
- L. E. Tracy, R. A. Minasian and E. J. Caterson, Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound, Adv. Wound Care, 2016, 5(3), 119–136 CrossRef PubMed.
- X. Bai, D. J. Dilworth, Y. C. Weng and D. B. Gould, Developmental distribution of collagen IV isoforms and relevance to ocular diseases, Matrix Biol., 2009, 28(4), 194–201 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Collagen structure and stability, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- J. Khoshnoodi, V. Pedchenko and B. G. Hudson, Mammalian collagen IV, Microsc. Res. Tech., 2008, 71(5), 357–370 CrossRef CAS PubMed.
- J. Gaar, R. Naffa and M. Brimble, Enzymatic and non-enzymatic crosslinks found in collagen and elastin and their chemical synthesis, Org. Chem. Front., 2020, 7(18), 2789–2814 RSC.
- R. Vanacore, A. J. Ham, M. Voehler, C. R. Sanders, T. P. Conrads, T. D. Veenstra, K. B. Sharpless, P. E. Dawson and B. G. Hudson, A sulfilimine bond identified in collagen IV, Science, 2009, 325(5945), 1230–1234 CrossRef CAS PubMed.
- A. L. Fidler, R. M. Vanacore, S. V. Chetyrkin, V. K. Pedchenko, G. Bhave, V. P. Yin, C. L. Stothers, K. L. Rose, W. H. McDonald and T. A. Clark, A unique covalent bond in basement membrane is a primordial innovation for tissue evolution, Proc. Natl. Acad. Sci., 2014, 111(1), 331–336 CrossRef CAS PubMed.
- G. Bhave, C. F. Cummings, R. M. Vanacore, C. Kumagai-Cresse, I. A. Ero-Tolliver, M. Rafi, J. S. Kang, V. Pedchenko, L. I. Fessler, J. H. Fessler and B. G. Hudson, Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis, Nat. Chem. Biol., 2012, 8(9), 784–790 CrossRef CAS PubMed.
- G. Sirokmany, H. A. Kovacs, E. Lazar, K. Konya, A. Donko, B. Enyedi, H. Grasberger and M. Geiszt, Peroxidasin-mediated crosslinking of collagen IV is independent of NADPH oxidases, Redox Biol., 2018, 16, 314–321 CrossRef CAS PubMed.
- G. E. Ronsein, C. C. Winterbourn, P. Di Mascio and A. J. Kettle, Cross-linking methionine and amine residues with reactive halogen species, Free Radical Biol. Med., 2014, 70, 278–287 CrossRef CAS PubMed.
- F. Pichierri, Theoretical characterization of the sulfilimine bond: Double or single?, Chem. Phys. Lett., 2010, 487(4–6), 315–319 CrossRef CAS.
- M. Ončák, K. Berka and P. Slavíček, Novel covalent bond in proteins: calculations on model systems question the bond stability, ChemPhysChem, 2011, 12(17), 3449–3457 CrossRef PubMed.
- M. Sundaramoorthy, M. Meiyappan, P. Todd and B. G. Hudson, Crystal structure of NC1 domains. Structural basis for type IV collagen assembly in basement membranes, J. Biol. Chem., 2002, 277(34), 31142–31153 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), Molecular Operating Environment, 2020.09, Chemical Computing Group ULC, 1010 Sherbooke St, West, Suite #910, Montreal, QC, Canada, H3A 2R7, p. 2021 Search PubMed.
- D. Cerutti, T. Cheatham III, T. Darden, R. Duke, T. Giese, H. Gohlke, A. Goetz, N. Homeyer, S. Izadi and P. Janowski, Amber 2016, University of California, San Francisco 2016 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N log (N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- M. Walker, A. J. A. Harvey, A. Sen and C. E. H. Dessent, Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions, J. Phys. Chem. A, 2013, 117(47), 12590–12600 CrossRef CAS PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, Assessment of the Performance of the M05-2X and M06-2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules, J. Chem. Theory Comput., 2008, 4(12), 1996–2000 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint, Chem. Rev., 1988, 88(6), 899–926 CrossRef CAS.
- T. L. Gilchrist and C. J. Moody, The chemistry of sulfilimines, Chem. Rev., 1977, 77(3), 409–435 CrossRef CAS.
- K. Tsujihara, N. Furukawa, K. Oae and S. Oae, Sulfilimine. I. synthesis and formation mechanism, Bull. Chem. Soc. Jpn., 1969, 42(9), 2631–2635 CrossRef CAS.
- E. D. Raczyńska, J.-F. Gal, P.-C. Maria and M. Szeląg, Proton Transfer Chemistry in the Gas Phase. Is a Spontaneous' Neutralization'Reaction a Myth or a Reality?, Croat. Chem. Acta, 2009, 82(1), 87–103 Search PubMed.
- F. A. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective, Cambridge University Press, New York, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02105f |
| This journal is © The Royal Society of Chemistry 2022 |