
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing integrated CO2 electrochemical conversion with amine-based CO2 capture: a review
Mengran
Li
 *a,
Kailun
Yang
a,
Maryam
Abdinejad
a,
Chuan
Zhao
b and
Thomas
Burdyny
*a
*a,
Kailun
Yang
a,
Maryam
Abdinejad
a,
Chuan
Zhao
b and
Thomas
Burdyny
*a
aMaterials for Energy Conversion and Storage (MECS), Department of Chemical Engineering, the Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: m.li-8@tudelft.nl; t.e.burdyny@tudelft.nl
bSchool of Chemistry, The University of New South Wales, Sydney, 2052, New South Wales, Australia
First published on 3rd August 2022
Abstract
Carbon dioxide (CO2) electrolysis is a promising route to utilise captured CO2 as a building block to produce valuable feedstocks and fuels such as carbon monoxide and ethylene. Very recently, CO2 electrolysis has been proposed as an alternative process to replace the amine recovery unit of the commercially available amine-based CO2 capture process. This process would replace the most energy-intensive unit operation in amine scrubbing while providing a route for CO2 conversion. The key enabler for such process integration is to develop an efficient integrated electrolyser that can convert CO2 and recover the amine simultaneously. Herein, this review provides an overview of the fundamentals and recent progress in advancing integrated CO2 conversion in amine-based capture media. This review first discusses the mechanisms for both CO2 absorption in the capture medium and electrochemical conversion of the absorbed CO2. We then summarise recent advances in improving the efficiency of integrated electrolysis via innovating electrodes, tailoring the local reaction environment, optimising operation conditions (e.g., temperatures and pressures), and modifying cell configurations. This review is concluded with future research directions for understanding and developing integrated CO2 electrolysers.
 Mengran Li | Dr Li obtained his PhD degree at the University of Queensland (UQ), Australia in 2016, and then started his postdoc position at UQ on the development of viable CO2 electrolysis technology for steel mills. His research in the field of CO2 electrochemical reduction includes material and system development and experiment testing, with a goal of understanding electrode interfacial interactions and developing industrially relevant electrochemical processes via material innovation and engineering. He will soon join the University of Melbourne as a Lecturer to continue his research and passion for material engineering and process innovation. |
 Kailun Yang | Dr Yang completed her PhD degree at TU Delft in 2022. After graduation, she continued her research as a postdoctoral fellow in the same group advancing electrochemical CO2 reduction to value-added products. Her main research interests lie in integrating renewable electricity into the energy transition process. Her research is mainly focused on improving the efficiency of electrochemical CO2 reduction by optimizing multiphase transport in different CO2 electrolyzer configurations. She is also interested in building and analyzing Multiphysics models to understand and predict the transport phenomena, and their relationship with the relevant reaction environment and performance. |
 Maryam Abdinejad | Dr Abdinejad received her first and second MSc degrees in Pharmaceutical Science at the University of Greenwich/England and Molecular Science at Ryerson University/Canada, respectively. She obtained her Ph.D. from the University of Toronto/Canada and joined Professor Tom Burdyny's research group at the Delft University of Technology (TU Delft)/Netherlands in 2021 as a postdoctoral researcher. Her current research focus is on the rational design and development of novel high-performance homogeneous and heterogeneous catalysts for the electrochemical reduction of CO2 to value-added materials. |
 Chuan Zhao | Professor Chuan Zhao is a full Professor at the School of Chemistry at the University of New South Wales (UNSW), Sydney. He also holds a Professorial Future Fellowship from Australian Research Council. He is interested in discovering novel electrochemical methodologies and nanomaterials for energy applications including water splitting, hydrogen fuel cells, CO2 & N2 reduction, batteries, and sensors. |
 Thomas Burdyny | Professor Burdyny completed his PhD from the University of Toronto (2017) followed by a brief postdoc at the Delft University of Technology. In 2019 he began his independent career at TU Delft, where his group focuses on applied and process integration aspects of electrochemical technologies, namely CO2 reduction. Tom has been the recipient of the prestigious VENI personal grant (2019) from the Dutch government to pursue scale-up research of electrochemical technologies, as well as acting as the ethylene work package leader in the Horizon 2020 EU project SELECTCO2. His work takes a combined modelling and experimental approach. |
Introduction
Carbon dioxide (CO2) electrolysis powered by renewable or low-carbon electricity is one of the promising routes to convert CO2 into valuable chemicals, such as carbon monoxide (CO), formic acid, ethylene, and ethanol.1–4 Recent years have seen a remarkable advancement of this process in achieving industrially relevant current densities (up to over 1 A cm−1) and high product faradaic efficiency by applying gas-diffusion electrodes.5–7 However, the state-of-the-art CO2 electrolysis usually uses pure CO2 as the feed, while CO2 is diluted in most industrial sources (<20% for flue gases from blast furnaces and post-combustion power plants8–10).5,11–21 When implemented in practice, CO2 electrolysis requires costly upstream CO2 capture processes22,23 to concentrate CO2, and an energy-intensive product separation process24,25 to recycle CO2 and concentrate product streams. In addition, gaseous CO2 reacts with hydroxide ions generated within CO2 electroreduction systems to form (bi)carbonate.6,26–29 The additional recovery of CO2 from the (bi)carbonate is another energy-intensive process that usually demands >230 kJ mol−1CO2.30 In a zero-gap membrane electrode assembly configuration, the (bi)carbonates are also prone to precipitate at electrode pores, block CO2 diffusion to the catalytically active surface, and severely degrade the overall cell performance.7,31,32 These technical issues constitute the significant challenges faced by the practical deployment of gas-fed CO2 electrochemical conversion at a large scale.An emerging strategy to address these challenges is to intensify the CO2 capture and electrochemical conversion processes.33–35 A schematic illustration is shown in Fig. 1a and b. The CO2 electrolysis process can potentially replace the energy-intensive stripper in the capture step. Sullivan et al.33 recently defined such a coupled process as Type-III fully integrated processes, including direct electroreduction of CO2 in amine-based, (bi)carbonate, and ionic liquids. As shown in Fig. 1c, the integrated electrolyser consists of an anode for water oxidation, an ion-conductive membrane, and a cathode that should be able to convert CO2 to products and recover the capture medium simultaneously.36,37 In the integrated conversion step, taking amine-based capture media as an example, the absorbed CO2 in the liquid capture media becomes the primary CO2 source for the CO2 conversion instead of the gaseous CO2. In this case, the coupled processes have the potential to prevent unwanted carbonation issues and achieve a concentrated product stream without significant downstream product separation if the conversion product is a gas product such as CO. In addition, such an intensification also has a promise of lowering the overall cost of the CO2 capture by displacement of the regeneration unit and CO2 compression.
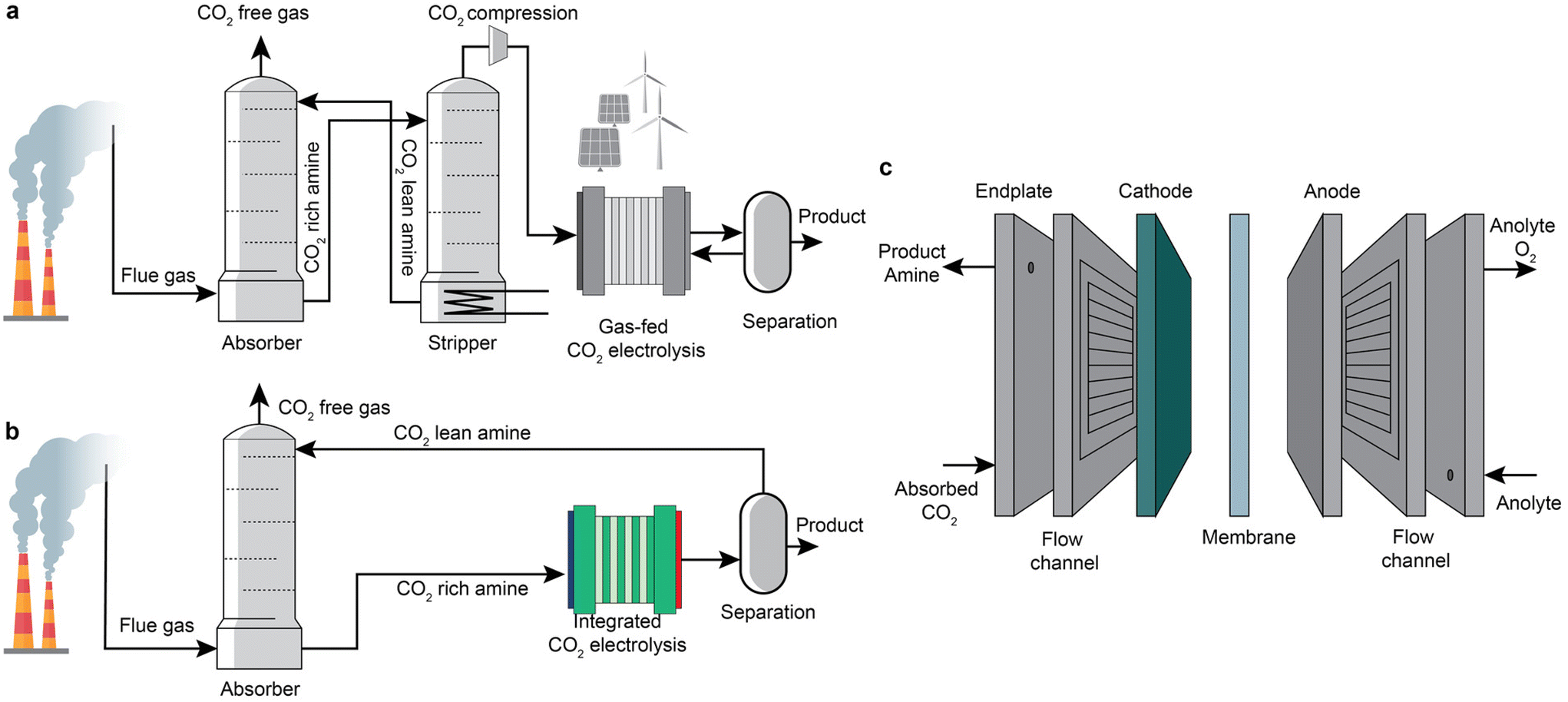 | ||
| Fig. 1 Schematic illustration of (a) sequential CO2 capture and electrolysis based on gas-fed electrolysers, (b) an integrated route based on integrated electrolysers, and (c) cell configuration of a single cell of the integrated electrolysers. | ||
Our recent energy analyses36 over sequential and coupled routes revealed that about 42% of overall energy could be saved for the integrated route if the integrated electrolyser can be operated at the same energy efficiency (∼1000 kJ molCO2 converted−1) as the state-of-the-art gas-fed electrolyser (3 V and 90% CO faradaic efficiency). The development of the integrated electrolyser is the crucial step enabling a more efficient coupled process and further cost reduction for the CO2 capture and utilisation. As amine scrubbing is the most commercially available CO2 capture process for industrial exhaust, the scope of this review will focus on the development of integrated CO2 electrolysis with amine-based CO2 capture.
The development of the integrated CO2 conversion is still at the early stage. Most studies still apply similar techniques and strategies implemented in gas-fed electroreduction to advance integrated CO2 electroreduction. Most of the reported activity and product selectivity for CO2 electroreduction in the amine-based capture medium is also inferior to the performance of gas-fed CO2 conversion. As a result, the overall energy efficiency of the integrated electrolyser is much lower than the gas-fed electrolyser36 and makes the overall process intensification less economically attractive than the sequential route.
This review aims to provide an overview of the most recent advances in improving CO2 electroreduction, mainly in amine-based capture media, focusing on the unique features of CO2 electroreduction in the capture media compared to the gas-fed electroreduction. This review starts with the discussion of catalytically active species for CO2 absorption in amines and potential catalytically active species available for CO2 reductions in the capture media. In the following section, we summarise current strategies to advance integrated electrolysers via the development of electrodes, capture media, operating conditions (e.g., temperature and pressure), and cell configuration. The review concludes with challenges and an outlook for developing efficient integrated CO2 electrolysers. Through this review, we anticipate providing new insights that can benefit the understanding and development of the integrated CO2 electrolysers.
CO2 speciation in amine-based capture media
Understanding the speciation of the CO2-rich capture medium is vital for understanding the catalytically active species for direct CO2 electro-conversion in the capture medium. The mechanisms for CO2 absorption in the amine-based capture medium have been widely studied.38–40 This section discusses the evolution of the primary CO2 species in the amine capture medium.CO2 loading effects
A typical amine scrubbing process (as shown in Fig. 1a) absorbs CO2 from the post-combustion process using an aqueous alkanolamine solution, such as primary, secondary, and tertiary amines.41 The benchmark amine that has been widely studied for CO2 capture is monoethanolamine (MEA). In the absorption unit, CO2 can react with primary amines to form carbamate (RNHCO2−) via the zwitterion mechanism at the first absorption stage (eqn (1)) and bicarbonate (HCO3−) ions at the second absorption stage (eqn (2) and (3)).42 For tertiary amines, such as methyldiethanolamine (MDEA)43,44 and 1-cyclohexylpiperidine (CHP) amine,45 CO2-amine reaction can only produce HCO3− as the primary product.40 (eqn (4))| 2RNH2 + CO2 ↔ RNHCO2− + RNH3+ | (1) |
| RHN2 + CO2 + H2O ↔ HCO3− + RNH3+ | (2) |
| RNHCO2− + H+ + H2O ↔ HCO3− + RNH3+ | (3) |
| R1R2R3N + CO2 + H2O ↔ R1R2R3NH+ + HCO3− | (4) |
The product concentrations in CO2-rich primary amines vary significantly with CO2 loading. Aqueous MEA capture medium is a typical example,46 as shown in Fig. 2. The MEA carbamate and protonated MEAs are the dominant products of CO2 absorption when the CO2 loading is below 0.4–0.5 molCO2/molamine. (Fig. 2a) When the CO2 loading increases further beyond 0.5 molCO2/molamine, carbamate ions start to undergo hydrolysis to form bicarbonate ions (eqn (3)). At this stage, bicarbonate and freely dissolved CO2 become the primary CO2 species in the capture medium. Such speciation transformation with CO2 loading usually takes place for primary (e.g., MEA) and secondary amines such as diethanolamine (DEA),46 as shown in Fig. 2b. In contrast, only bicarbonate concentration rises with the CO2 loading in MDEA aqueous solution in tertiary amines (see Fig. 2c).44
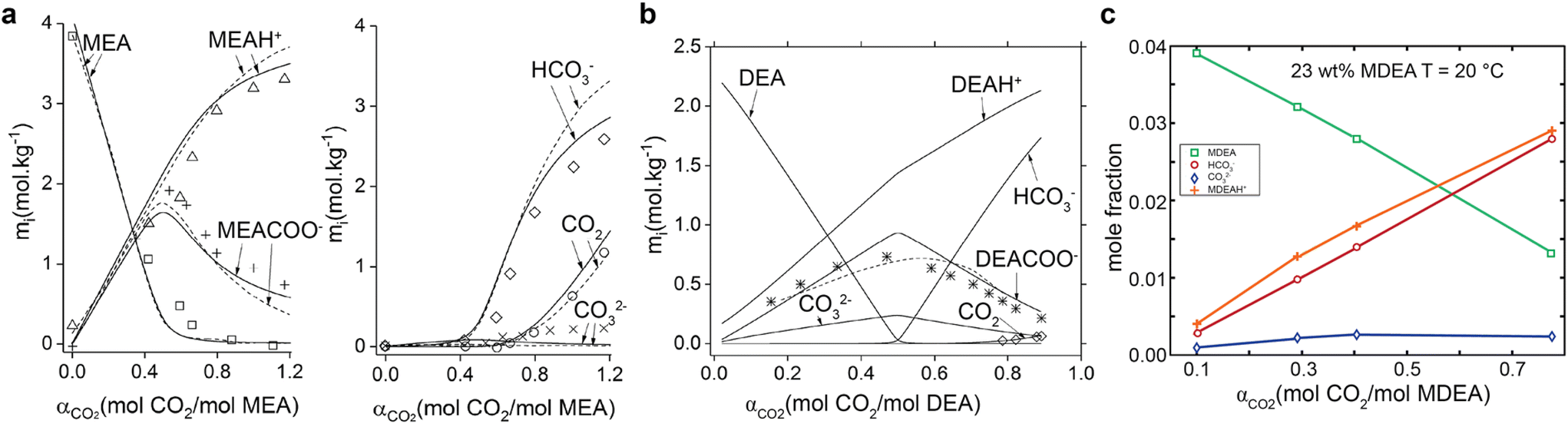 | ||
| Fig. 2 Evolution of chemical species in the monoethanolamine-CO2–H2O system in (a) 20 wt% MEA at 40 °C, (b) 20 wt% DEA at 25 °C with symbols for experimental results and lines for calculated values (reproduced with permission,46 Copyright 2018, American Chemical Society), (c) 23 wt% MDEA aqueous solution at 20 °C. Reproduced with permission.44 Copyright 2005, American Chemical Society. | ||
In a typical CO2 capture process using 20–30 wt% (equivalent to 3–5 M) aqueous MEA solution, the CO2 loading is at 0.2–0.35 molCO2/molamine for the CO2-lean stream, and at 0.4–0.5 molCO2/molamine for CO2-rich stream.47,48 These loadings indicate that: the carbamate is the dominant species at an estimated molar concentration of 1.7–2.5 M in the CO2-rich MEA solutions. For secondary amines such as 40 wt% DEA, the typical CO2 loadings are at similar levels to the MEA case, so the dominant products are carbamate ions. For tertiary amines such as MDEA solutions, the bicarbonate concentration is equivalent to CO2 loading due to the dominant bicarbonate formation.
Temperature effects
Temperature serves a critical role in determining the CO2-loading and speciation by affecting the solubility of CO2 in amines and the equilibrium constants for multiple homogenous reactions. CO2 solubility in the capture medium decreases with increasing temperatures, evident from the Henry constant relations with temperature shown in Fig. 3a.49 Consequently, as implied in Fig. 3b, the CO2 equilibrium partial pressure is higher for the CO2-loaded MEA system at elevated temperatures.50Fig. 3b also shows that a high CO2 loading lowers the heat of CO2 absorption (which is an exothermic process), meaning that more CO2 can be liberated from high-loading amines than low-loading ones.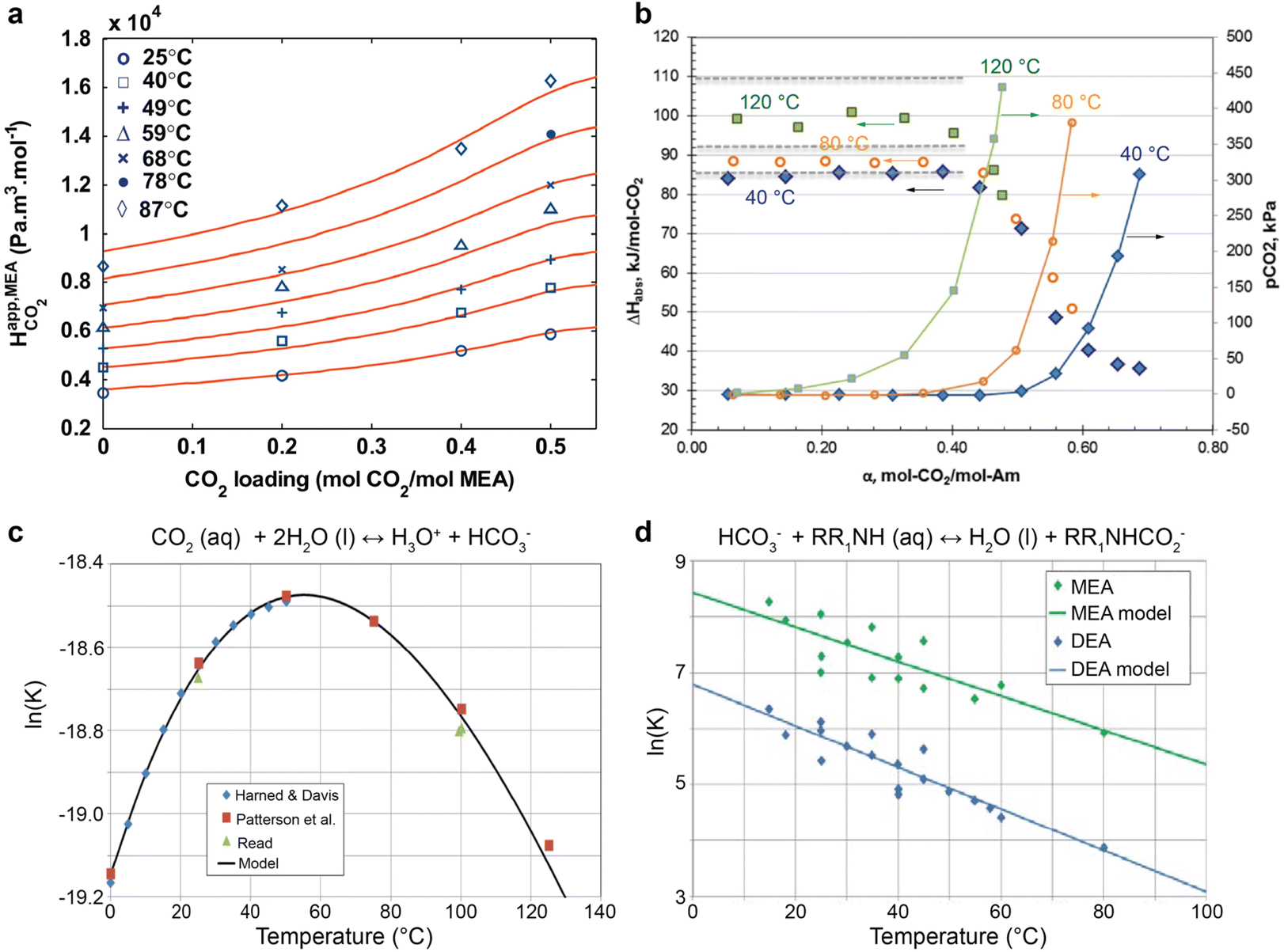 | ||
| Fig. 3 The temperature effects on (a) Henry constant of CO2, reproduced with permission,49 copyright 2011, Elsevier. (b) CO2 partial pressure and heat of CO2 absorption for 30 wt% MEA aqueous solutions, reproduced with permission,50 copyright 2014, Elsevier, (c) bicarbonate formation, and (d) carbamate formation for MEA and DEA aqueous solution. Reproduced with permission,51 copyright 2017, Elsevier. | ||
The equilibrium constants (K) for the equilibrium reactions are governed by the enthalpy of the reaction (ΔH) and temperatures (T), as expressed in eqn (5). Fig. 3c suggested that the formation of bicarbonate ions from water and solvated CO2 is promoted with increasing temperatures below 60 °C and is suppressed at a higher temperature. In Fig. 3d, the bicarbonate is prone to react with amines to form carbamate at lower temperatures for both MEA and DEA aqueous solutions.51 The temperature governs the contents of the CO2-related species in CO2-rich amines and should profoundly impact the local speciation for the industrially relevant integrated electrolysers.
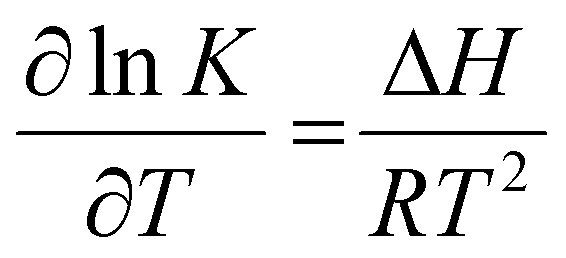 | (5) |
pH effects
Recent studies on gas-fed CO2 electrolysis have shown that a more alkaline local reaction environment (pH > 10, see Fig. 4a)52,53 close to the electrode surface than the electrolyte bulk due to the generation of the hydroxide ions from electroreduction reactions for both CO2 and water (i.e., hydrogen evolution reaction). The pH values in the capture medium usually change with the CO2 loading. As an acidic gas, CO2 usually acidifies the alkaline amine capture medium.54 (Fig. 4b) A further reduction of the pH value by the absorbed CO2 due to such acidification causes the second stage of the hydrolysis of the carbamate to bicarbonate, as described in eqn (3).55 Additionally, a low pH value could reduce the energy required to recover CO2 from the diluted amine solutions.56 The pH can also swing back to alkaline conditions when the solvents are heated to separate CO2 from the liquid. For high-rate CO2 conversion in the amine capture medium, understanding the role of pH in regulating the homogenous amine reactions is important to study the reaction pathways for CO2 delivery to the catalytically active sites and amine recovery.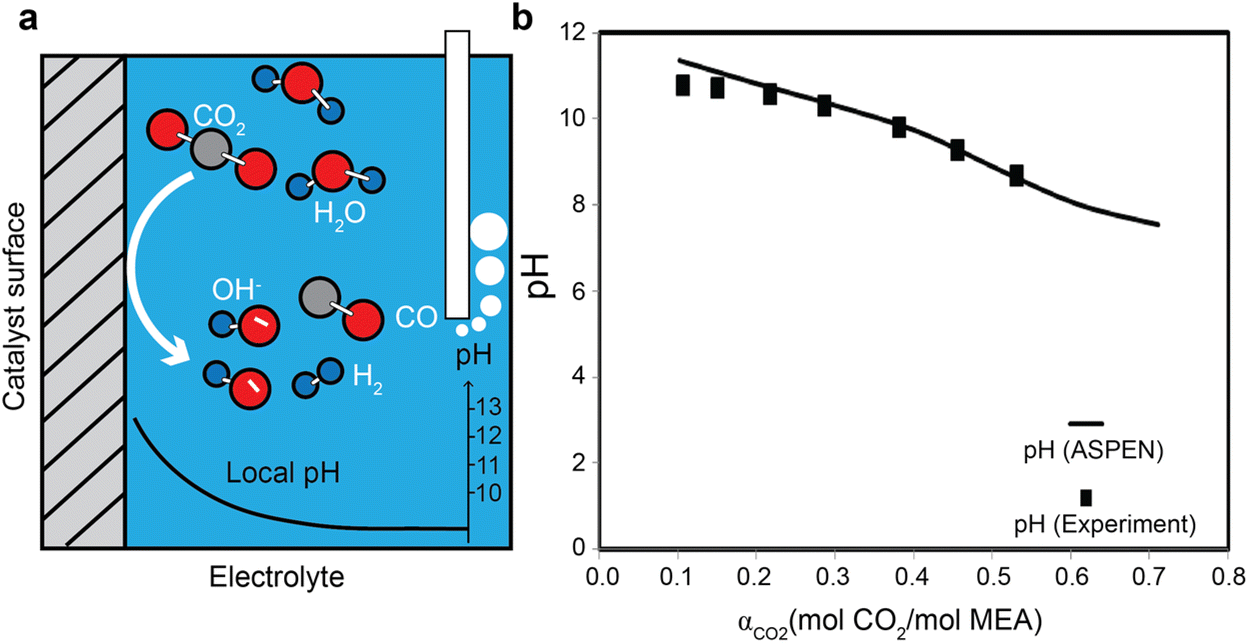 | ||
| Fig. 4 (a) A schematic illustration of local pH difference for gas-fed CO2 electroreduction close to the catalyst surface. (b) The pH evolution as a function of CO2 loading in 30 wt% MEA aqueous solution at 21 °C. Reproduced with permission,54 copyright 2012, American Chemical Society. | ||
The pH value of an amine solution is strongly correlated with the speciation. The pH usually swings between 8 and 10 for CO2 absorption and desorption cycle. In the MEA aqueous solution, for example, the bicarbonate formation and protonation of MEA take place at pH = ∼6, CO2 transformation to bicarbonate at pH = ∼7, the carbonation from bicarbonate ions at pH = ∼10.3, and deprotonation of MEAH+ to MEA at a pH = ∼10.54
These homogenous reactions could contribute to the recovery of the amines in the integrated electrolysers. By incorporating hydroxide ions into the system from electrochemical reactions, for example, one could expect the formation of free MEA and carbonates at the end equivalent points.54 If added with protons from anode reactions and water electrochemical dissociation, the amine solution will end up with the formation of protonated amines and the evolution of gaseous CO2.54 Under electrochemical reduction conditions, it is likely that the solvent close to the catalyst surface experiences a much higher local pH than the bulk solution so that locally free amine tends to be regenerated and CO2 species tend to form carbonate ions.
The recovery of amines and conversion of the absorbed CO2 into valuable products take place either via electrochemical reactions or homogenous equilibrium reactions induced by the pH changes. Because the electrolyser is energy efficient when converting CO2 to valuable product selectively, the recovery of the amines is desired to proceed with the reduction of absorbed CO2 (i.e., free dissolved CO2, carbamate, and bicarbonate ions) and neutralisation of protonated amines inside the cathode channel, rather than electroreduction of protonated amines to hydrogen and amines. The locally produced carbonate ions, as a result of the raised local pH, should be reversed back to catalytically active species in the liquid bulk via reactions such as reacting with protons from the membrane to form CO2 or carbamate.55 In this case, we could envision that the recovery and recycling of the capture medium from the electrolyser requires dedicated control of the ion transport within the cell.
Catalytically active species for CO2 electroreduction
As discussed in the section above, the carbamate, bicarbonate, and free dissolved CO2 could contribute to the direct CO2 electro-conversion in amine-based capture media because they are the primary species for the absorbed CO2. Although there are limited direct findings for the underlying mechanisms for CO2 conversion in amine capture media, we could explore the potential mechanisms by bringing the knowledge and mechanistic insights from the studies on gas-fed CO2 electroreduction and direct bicarbonate reductions.57 For gas-fed CO2 electroreduction, freely dissolved CO2 is widely deemed as the primary catalytically active reactant, no matter in a planar electrode (where CO2 diffuses to the catalyst surface via the electrolyte bulk) or in gas-diffusion electrodes (where CO2 diffuses to the catalyst surface via a hydrophobic porous matrix). In the direct bicarbonate reduction, where concentrated bicarbonate serves as the feed for CO2 conversion, the evolved CO2 molecules from acidification of bicarbonate are also recently unveiled as the actual primary active species for CO2 conversion.58,59 This section discusses recent reports focusing on the proposed mechanisms and active species for the integrated CO2 conversion.Free dissolved CO2 as the active reactant
Unlike gas-fed CO2 conversion and direct bicarbonate reduction, the CO2-rich primary or secondary amines with a CO2 loading below 0.5 molCO2/ molamine has a negligible concentration of free dissolved CO2 due to the carbamate formation. Such a low CO2 concentration is usually considered the main cause for the observed lower conversion rates than in gas-fed scenarios. Chen et al.60 claimed that the free dissolved CO2 is the primary active reactant for MEA solution because they found that hydrogen evolution reaction dominates over smooth indium (In) and silver (Ag) foils for 30 wt% MEA aqueous solution with a CO2 loading of 0.3, 0.4, and 0.48 molCO2/molMEA in the presence of 0.1 wt% cetrimonium bromide (CTAB) surfactant. Such an independency between product faradaic efficiency and CO2 loading indicates that the free dissolved CO2 should serve as the primary active species, rather than the carbamate ions that should increase their concentrations with the CO2 loading (see Fig. 2a). Similarly, Gallent et al.61 also proposed that CO2 needs to be liberated from amine solutions first before being reduced to different products. They confirmed their theory by demonstrating an enhancement of CO2 conversion faradaic efficiency and rates over lead (Pb) or gold (Au) catalyst in CO2-rich 1 M 2-amino-2-methyl-1-propanol (AMP) in propylene carbonate (PC) solutions. A similar mechanism was also proposed by Ahmad et al.,62 who argued that the CO2 liberated from AMP contributes to the CO2 conversion.In another report by Diaz et al.,45 CHP was employed as the switchable polarity capture media for CO2 conversion. This compound is a tertiary amine that is insoluble in water but can become soluble after reaction with CO2 to produce bicarbonates and protonated amine. In this case, the bicarbonate becomes the primary CO2 source. The authors used a proton-exchange membrane to promote protons produced from the anode chamber to release CO2 from bicarbonate ions for the reaction. As a result, they detected a small amount of CO2 in the product stream (see Fig. 5c), which experimentally confirms that CO2 is released from the bicarbonate and contributes to CO production. In addition, Gallent et al.61 reported a much more reduction in CO2 loading (or more CO2 liberated) at higher temperatures (75 °C vs. 15 °C) in 0.7 M tetraethylammonium chloride (TEACl) in PC solutions with 1 M AMP. (Fig. 5e) However, the CO2 molecules converted are only up to 30% of the liberated CO2. (see Fig. 5d and f) This comparison points to the essential role of released CO2 as the active species for CO2 conversion.
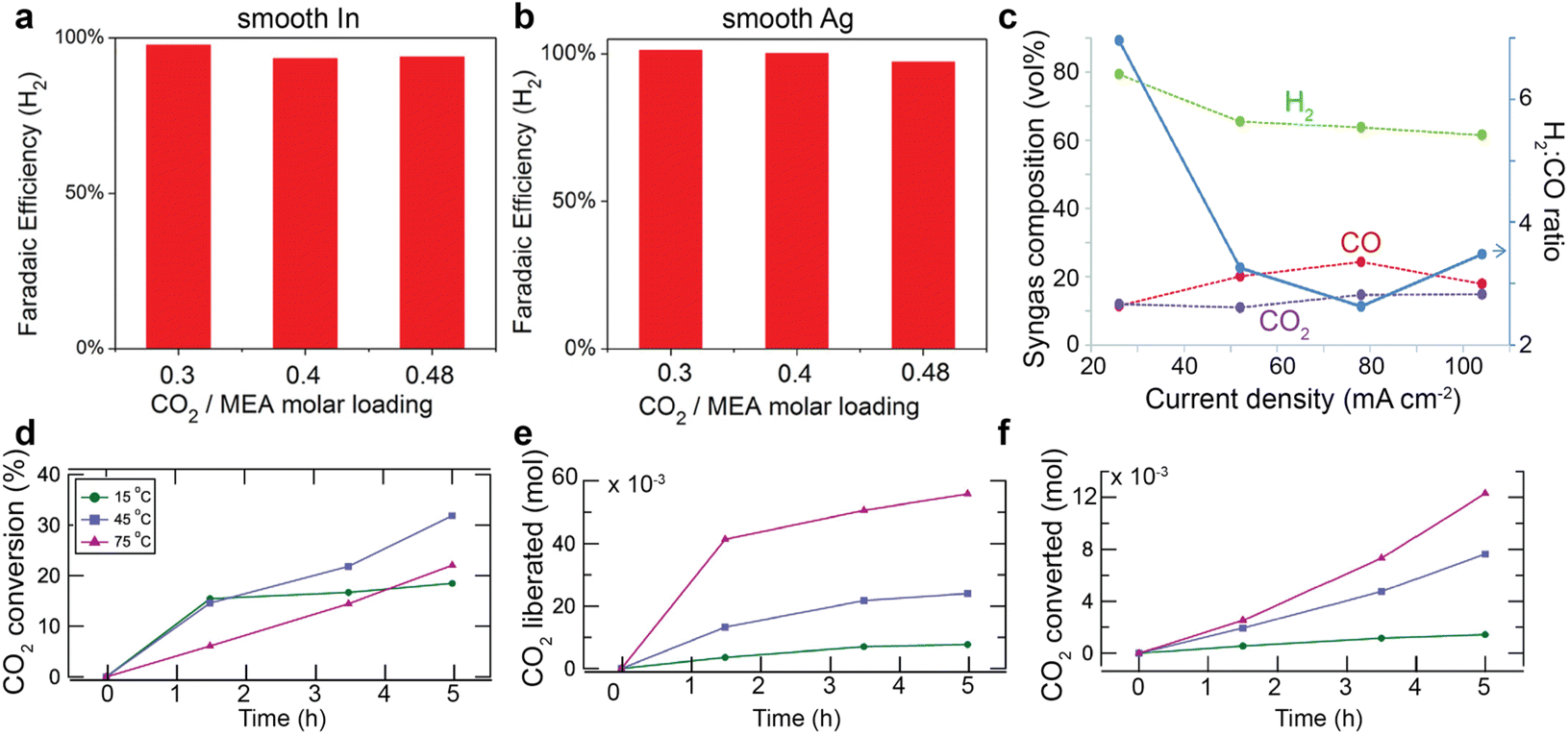 | ||
| Fig. 5 Faradaic efficiency of H2 over (a) smooth In and (b) smooth Ag surface in 30 wt% MEA aqueous solution loaded with 0.3, 0.4, 0.48 molCO2/molamine in the presence of 0.1 wt% CTAB. Reproduced with permission,60 copyright 2017, John Wiley and Sons. (c) Volumetric fractions of H2, CO, and CO2 as a function of current densities at 20 psig backpressure at Ag catalyst deposited on a reticulated vitreous carbon in CO2 loaded CHP aqueous solution. Reproduced with permission,45 copyright 2018, Royal Society of Chemistry. (d) The CO2 conversion rate, the amount of (e) liberated CO2 and (f) converted CO2 as a function of time and temperatures. Reproduced with permission,61 copyright 2021, American Chemical Society. | ||
Because bicarbonate is the dominant product in the CO2-loaded tertiary amine-based capture medium, the primary active reactant for conversion should be the free dissolved CO2 from the acidification of bicarbonate. Therefore, as reported in recent work, CO2 conversion in tertiary amine is anticipated to follow similar mechanisms for direct bicarbonate reduction.57 Similar to direct bicarbonate reduction, CO2 conversion in a bicarbonate-dominated CO2-rich capture medium requires a supply of protons to produce CO2 from bicarbonate ions. The proton flux is usually current-dependent and supplied from either anode reaction via a proton-exchange membrane63,64 or the bipolar membrane29,65,66 under a reversed bias. Protons could also cause an acidic local reaction environment close to the catalyst surface and contribute to the unwanted hydrogen evolution reaction.67,68 Our recent two-phase one-dimensional model for direct bicarbonate reduction unveiled that the rate of CO evolution can be limited more by the formation and mass transfer of CO2. Under high current densities, protons tend to either directly get reduced to hydrogen or react with the hydroxyls produced, so CO2 regeneration and reduction pathways are limited.68
Carbamate as the active reactant
An alternative explanation for the observed low CO2 conversion efficiency is the limited carbamate availability close to the catalyst surface due to the repulsion between the negatively charged carbamate anions and the negatively charged cathode. Lee et al.37 explained the observed low CO faradaic efficiency from 2 M MEA aqueous electrolyte by the inefficient charge transfer between the cathode surface and the carbamate molecule. From the results of the electrochemical impedance analyses and in situ Raman spectroscopy, the authors proposed an interfacial electron transfer process, where the electron transfer must reach the carbamate ions through large MEAH+ cations that are packed at the electrochemical double layer. The authors also showed that incorporating alkali cations such as K+ helps achieve a more compact double layer with reduced availability of MEAH+ and an improved CO faradaic efficiency.Although the CO2 species such as carbamate, bicarbonate, and free dissolved CO2 should be the source for CO2 reduction in the amine capture media, understanding the underlying mechanisms for CO2 conversion remains unclear and needs further research efforts to detangle the complex electroreduction that involve multiple species, transport, and homogenous reactions. Generally, the reported performance for CO2 electroreduction in amines is inferior to gas-fed conversion, which is usually explained by the low availability of reactants (either free dissolved CO2 or negatively charged carbamate anions) close to the electrode surface. As compared to primary and secondary amines, it is more straightforward to understand the mechanisms for CO2 reduction in tertiary amines by sourcing the knowledge and insights from the field of direct bicarbonate electro-reduction.
Strategies in achieving selective CO2 conversion
Recent findings have shown that the catalysts that are selective for gas-fed CO electroreduction are also efficient for catalysis of CO2 conversion in the amine capture media.33,37,45,60,62,69,70 However, the performance of the integrated electrolyser is usually inferior to the state-of-the-art gas-fed electrolyser in terms of product faradaic efficiency, current densities, and cell potentials. Fig. 6a compares the energy performance of these two electrolysers to convert CO2 to CO.36 More importantly, recent studies demonstrate the potential to improve the faradaic efficiency and current densities for direct CO2 conversion in an amine-based capture medium by tailoring the electrode structures, local reaction environment, and operating conditions such as temperatures and pressures, and cell design. A summary of achievements in recent studies is listed in Table 1.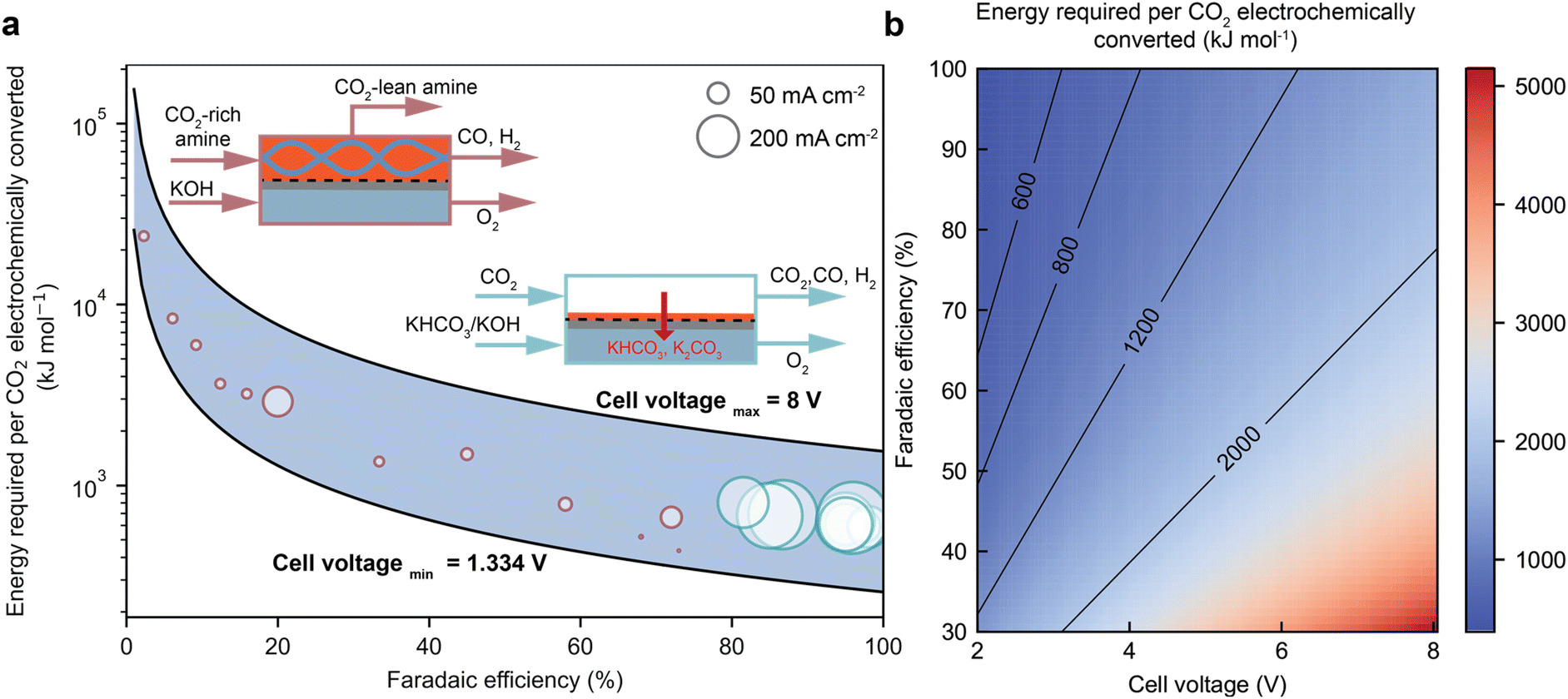 | ||
| Fig. 6 (a) A comparison of the energy consumption, CO faradaic efficiency, and cell potentials over integrated and gas-fed CO2 electrolysis. (b) The role of CO faradaic efficiency and cell voltage in determining the overall energy consumption of CO2 electrolysis to produce CO.36 | ||
| Cathode | Cathode Potential (V) vs. RHE | Product | Peak FEproduct (%) | Current densities at peak FEproduct (mA cm−2) | Solvents and conditions | Ref. |
|---|---|---|---|---|---|---|
| a Current efficiency calculated from (jCO2 − jAr)/jCO2, where j is the current density, and the current densities were collected in the presence of the subscript gases. | ||||||
| Ag/carbon-black on 300 nm Ag film on ePTFE | −0.8 | CO | 72 | 50 | 30 wt% MEA aqueous solution mixed with 2M KCl at 60 °C | 37 |
| Ag/carbon-black on 300 nm Ag film on ePTFE | −1.2 | CO | 20 | 100 | ||
| Smooth Ag | −0.8 | CO | 12.4 | — | 30 wt% MEA aqueous solution at 22 °C | 60 |
| Smooth Bi | −0.8 | Formate | 35.7 | — | ||
| Porous Ag | −0.8 | CO | 39.1 | — | ||
| Smooth Ag | −0.8 | CO | 33.4 | — | 30 wt% MEA aqueous solution with 0.1 wt% CTAB loaded with CO2 at 22° | |
| Smooth In | −0.8 | Formate | 45.4 | — | ||
| Smooth Sn | −0.8 | Formate | 19 | — | ||
| Porous In | −0.8 | Formate | 54.5 | — | ||
| Porous Pb | −0.8 | Formate | 60.8 | — | ||
| Porous Ag | −0.8 | CO | 38.2 | — | ||
| Cu | −0.78 | CO | 45 | 18.4 | 0.1 mM ethylenediamine carbamate in 0.1 M NaClO4 saturated with CO2 | 71 |
| Smooth Au foil | −1.9 vs. Ag|AgCl | CO | ∼45 | ∼15 | 1 M 2-amino-2-methyl-1-propanol (AMP) and propylene carbonate (PC) solution | 61 |
| Pb electrode | −2.5 vs. Ag|AgCl | Formate | ∼40 | ∼28 | 2 M AMP in PC solution at 75 °C | |
| HCl treated Ag foil | −0.91–−1.01 | CO | 91 ± 7 | ∼10 | 0.25–1 M AMP aqueous solution at room temperature with 0.3 mM CTAB saturated with CO2 | 62 |
| Ag foil | −0.91 | CO | 72 ± 8 | ∼11 | 1 M AMP aqueous solution | |
| Au/MgAl-LDHs | −0.4 | CO | ∼68 | ∼1.1 | 1.0 M alcohol amine solution (n(ethanolamine): n(diethanolamine) = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) :3) |
72 |
| Cu/MgAl-LDHs | −0.25 | CO | ∼73 | ∼0.5 | ||
| Ag | −1.1 | CO | 71 | 15 | [MEAHCl][MDEA], where MEAHCl is ethanolamine hydrochloride, and MDEA is methyl diethanolamine | 73 |
| Au nano dendrites | −1.0 | Formate | 60.3a | ∼46 | 0.05 M MEA aqueous solution | 74 |
| Ag microparticles mixed with Nafion and PTFE deposited on a reticulated vitreous carbon | — | CO | ∼30 | 78 | 1-Cyclohexylpiperidine aqueous solution with 0.2 M K2SO4 loaded with CO2 with back pressure of 20 psig | 45 |
| Ni-N–C single-atom-catalyst | −0.6 | CO | 63.2 | ∼4 | CO2-rich 5 M MEA solution | 75 |
A desired integrated CO2 electroreduction should achieve a high current density with a high product faradaic efficiency at a low overpotential (i.e., the excess of potential vs. the thermodynamic potential to drive the electrochemical reaction). Ideally, the amine as the electrolyte should exhibit a high ionic conductivity to minimise ohmic losses and reduce cell voltage. Fig. 6b from our recent energy analyses for CO2-to-CO highlight the importance of product faradaic efficiency and cell potentials for the overall energy efficiency, where product faradaic efficiency serves a more critical role than cell potential.36 Therefore, this section will review recent advances in improving the faradaic efficiency and current densities via electrode innovation and modification of the local reaction environment.
Ionic conductivity in amine solutions
The ionic conduction is essential for a complete electrochemical process. The ionic conductivity of the electrolyte (or capture medium) predetermines the ohmic loss, and thus has an impact on the overall cell potential.1 However, amine capture media is poorly conductive as there are typically no added supporting ions, with ions only provided via CO2 absorption. Taking MEA aqueous solution as an example,76 as shown in Fig. 7a and b, CO2 absorption causes an increase in ionic conductivity of the capture medium because the CO2 reactions increase the availability of ions (such as carbamate anions, bicarbonate anions, and MEAH+) as charge carriers for ion conduction. Fig. 7b shows that 3–4 M of MEA could be an optimal concentration range for the highest ionic conduction. However, even the highest conductivity (∼40 mS cm−1) for CO2-saturated MEA solution is not comparable to the ionic conductivity of 1 M KOH (215 mS cm−1).77 Such a trend remains valid for the CHP-CO2–H2O system shown in Fig. 7c. The ionic conductivity peaks at the CHP-H2CO3 concentration of ∼1.2 M and then drops with a further increase in the concentration. In contrast, in the system added with 0.2 M K2SO4, the overall ionic conductivity nearly doubles the ionic conductivity for bare CHP-H2CO3 at diluted conditions but also decreases with the concentration of CHP-H2CO3.45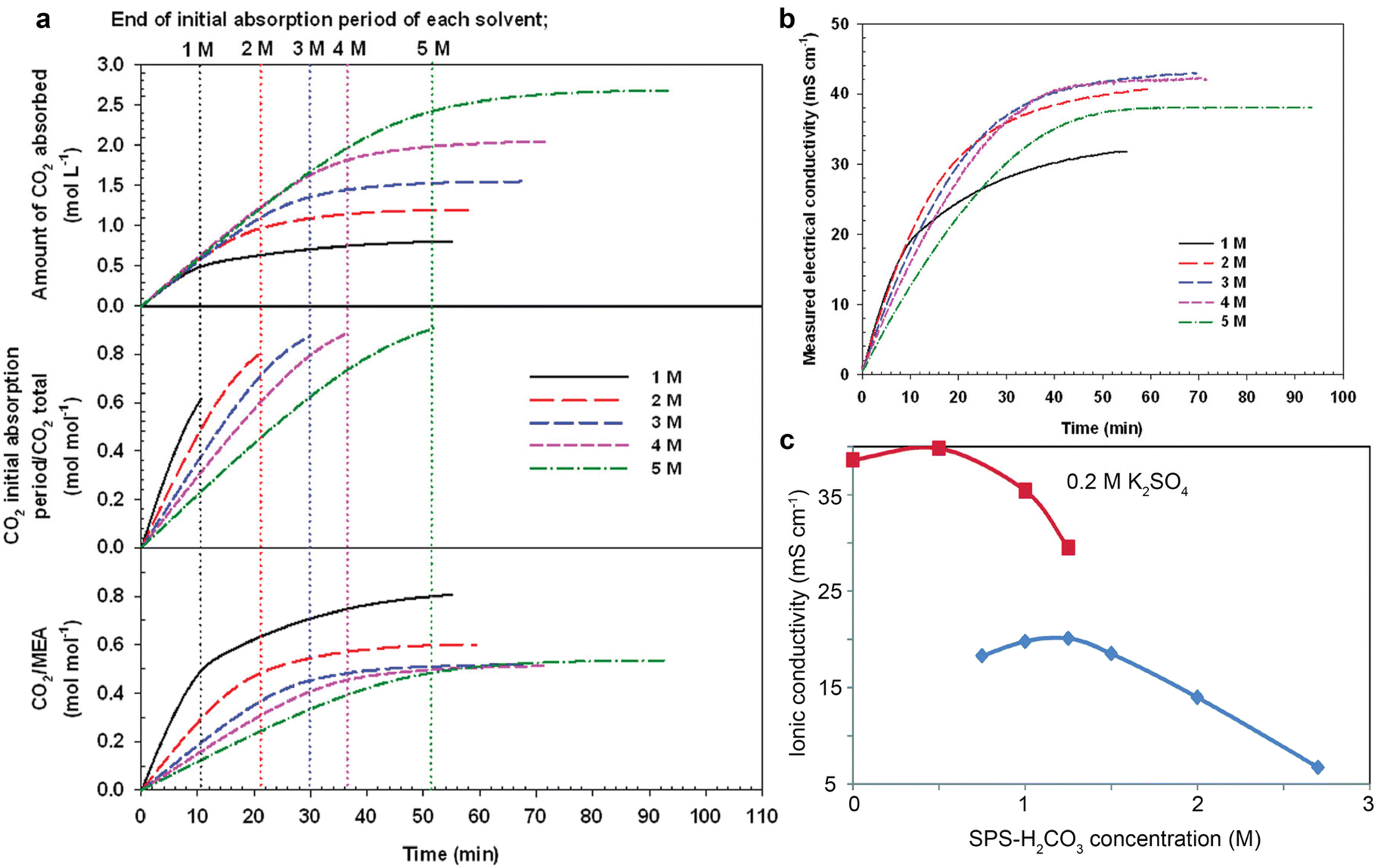 | ||
| Fig. 7 (a) The CO2 absorption amount, molar ratio, and CO2/MEA ratio as a function of time at different MEA concentrations. (b) The electrical conductivity of the MEA aqueous solutions with different concentrations as a function of CO2 absorption durations. Reproduced with permission,76 copyright 2016, American Chemical Society. (c) The ionic conductivity of CO2 loaded CHP aqueous solution with and without 0.2 M K2SO4 as a function of amine concentration. Reproduced with permission,45 copyright 2017, Royal Society of Chemistry. | ||
Electrode materials and structures
The electrode for the integrated CO2 conversion is desired to provide a high density of the electrochemically active sites with high catalytic activity. A few studies performed similar techniques used for gas-fed CO2 electroreduction to deposit catalyst nanoparticles (e.g., Ag) onto a substrate such as reticulated vitreous carbon foam or gas-diffusion layer (made from carbon or polytetrafluoroethylene (PTFE)) via coating (e.g., spray coating) and sputter deposition techniques, which are commonly used for electrode preparation for gas-fed CO2 electroreduction. (see Fig. 8a) For example, Lee et al. prepared their Ag electrodes by first sputtering ∼300 nm Ag film on the PTFE porous membrane and then spraying Ag nanoparticles and carbon black on top of the sputtered Ag film. Such an electrode can achieve 50 mA cm−2 with a CO faradaic efficiency of about 72% at −0.8 V vs. reversible hydrogen electrode (RHE). In another recent report, Ahmad et al.73 boosted the CO faradaic efficiency of CO up to ∼91% in 0.25–1 M CO2-saturated AMP aqueous solution with 0.3 mM CTAB by treating Ag foils with HCl. More recently, Kim et al.75 showed that applying nickel (Ni) single-atom catalyst as shown in Fig. 8b on a gas-diffusion electrode can achieve 64.9% CO faradaic efficiency from 5 M MEA aqueous solution at 50 mA cm−2 in a membrane electrode assembly. | ||
| Fig. 8 (a) A scanning electron micrograph of the cross-section (left panel) and illustration (right panel) for a gas-diffusion electrode. Reproduced with permission,78 copyright 2020, American Chemical Society. (b) A high-angle annular dark-field transmission electron micrograph of the Ni-nitrogen–carbon single-atom catalyst. Reproduced with permission,75 CC BY 4.0 License. (c) A scanning electron micrograph of the porous silver metal electrode for direct bicarbonate reduction. Reproduced with permission,79 copyright 2022, Royal Society of Chemistry. | ||
The requirement for the electrode structure for integrated electrolysis should be different from the gas-diffusion electrode used for gas-fed CO2 reduction due to the absence of CO2 supply in the integrated electrolysers.80–82 Therefore, a highly hydrophilic electrode surface should help improve its contact with solvent and minimise the contact with the gas product. Most metallic electrodes are hydrophilic, and their hydrophilicity is anticipated to increase if the electrode surface is roughened according to the Wenzel equation83 and under electric potential due to the electrowetting phenomenon.80 Compared to the development of the gas-diffusion electrodes, the requirement for the electrode development is less stringent on maintaining a stable wetting condition and gas–liquid interfaces within the electrode.75,80,84,85 In the field of direct bicarbonate reduction, for example, when the Berlinguette group directly incorporated Ag foam instead of carbon-based Ag gas-diffusion electrodes, the bicarbonate electrolyser can achieve a further improvement of the performance.79,86
As bicarbonate is the main source of CO2 reduction in tertiary amines, we could also apply the advances in the development of direct bicarbonate reduction to improve the performance further. Although direct bicarbonate reduction is outside the scope of this review, we reckon it is worth mentioning an example reported by Zhang et al.79 They demonstrated that the gas diffusion layer without a microporous layer and PTFE showed the best performance during direct bicarbonate reduction.78 Furthermore, the same group developed new electrodes to enhance CO2 electroreduction to CO in a 3 M bicarbonate aqueous solution. They showed that a porous Ag electrode (see Fig. 8c) is superior to the Ag-based gas-diffusion electrodes in evolving CO from bicarbonate solutions, especially at current densities >100 mA cm−2.63 These findings confirm that a hydrophilic porous electrode is likely applicable to CO2 conversion in the tertiary amines than electrode structures used in gas-fed electrolysis.
A few studies reported that applying porous electrodes can improve product faradaic efficiency. For example, Chen et al.60 prepared a few porous electrode structures based on metals, such as Ag, zinc (Zn) and Indium (In), using the hydrogen-bubble templated electrodeposition technique. The microstructures of some porous electrodes are shown in Fig. 9a. The authors observed an enhancement of formate production over porous In and CO production over Zn and Ag porous electrodes in a CO2-saturated 30 wt% MEA aqueous solution. Therefore, the porous microstructure could be beneficial for CO2 conversion. In another example, Hossain et al.74 prepared nano dendrites based on Cu, gold (Au), and Ag by growing the metals on a glassy carbon via a galvanic replacement reaction in the mixed solution of metal precursors and Zn dust. (see Fig. 9b) From a more significant increment of current densities in argon- versus CO2-saturated 0.05 M MEA aqueous solutions, the authors concluded that the nanostructured catalyst could improve CO2 conversion current densities and charge-transfer efficiency.
 | ||
| Fig. 9 Scanning electron micrographs of (a) porous Zn, In, and Ag electrodes prepared by hydrogen-bubble-templated electrodeposition technique, reproduced with permission,60 copyright 2017, John Wiley and Sons. (b) Cu, Ag, and Au nano dendrites on glassy carbon prepared via galvanic replacement reaction. Reproduced with permission,74 copyright 2020, John Wiley and Sons. | ||
Local reaction environment
In addition to the electrode, a local reaction environment close to the catalyst surface is essential to provide sufficient reactants and suppress unwanted side reactions such as hydrogen evolution reactions. As mentioned earlier, the local chemical speciation could vary vastly with the amines,61,71,75 CO2 loading,60 local pH, proton supply,45,75 alkali cations,37,69,75 temperatures,37,61 and electrochemical operating conditions.62 An ideal local reaction environment should maintain sufficient catalytically active species, minimise surface coverage of protonated amines and protons that leads to hydrogen formation, facilitate fast ion conduction and charge transfer, and provide a benign environment for amine recovery.A straightforward strategy to modify the local reaction environment is to change the types and concentrations of the amines, which predetermine the CO2 species and their concentration in the solvent bulk and thus in the local reaction environment. In the previous sections, we have discussed some examples showing that the product Faraday efficiencies are different in MEA solutions and CHP solutions. In addition, Abdinejad et al.71 reported different product distribution over Cu electrode in 0.1 M NaClO4 solution containing carbamate from MEA, ethylenediamine (EDA), and decylamine (DCA). As shown in Fig. 10a. The EDA-containing electrolyte achieved the highest CO faradaic efficiency among the tested amines, likely due to its two primary amines in one EDA molecule. Another report by Gallent et al.61 shows that 2 M AMP in PC solution is the optimal concentration to convert CO2 to formate at the highest faradaic efficiency and reaction rates, but leads to the lowest overall current densities over lead (Pb) electrode. (Fig. 10b) Similarly, Ahmad et al.62 also report that the optimal AMP concentration is within 0.25–1 M for CO production over HCl-treated Ag electrode.
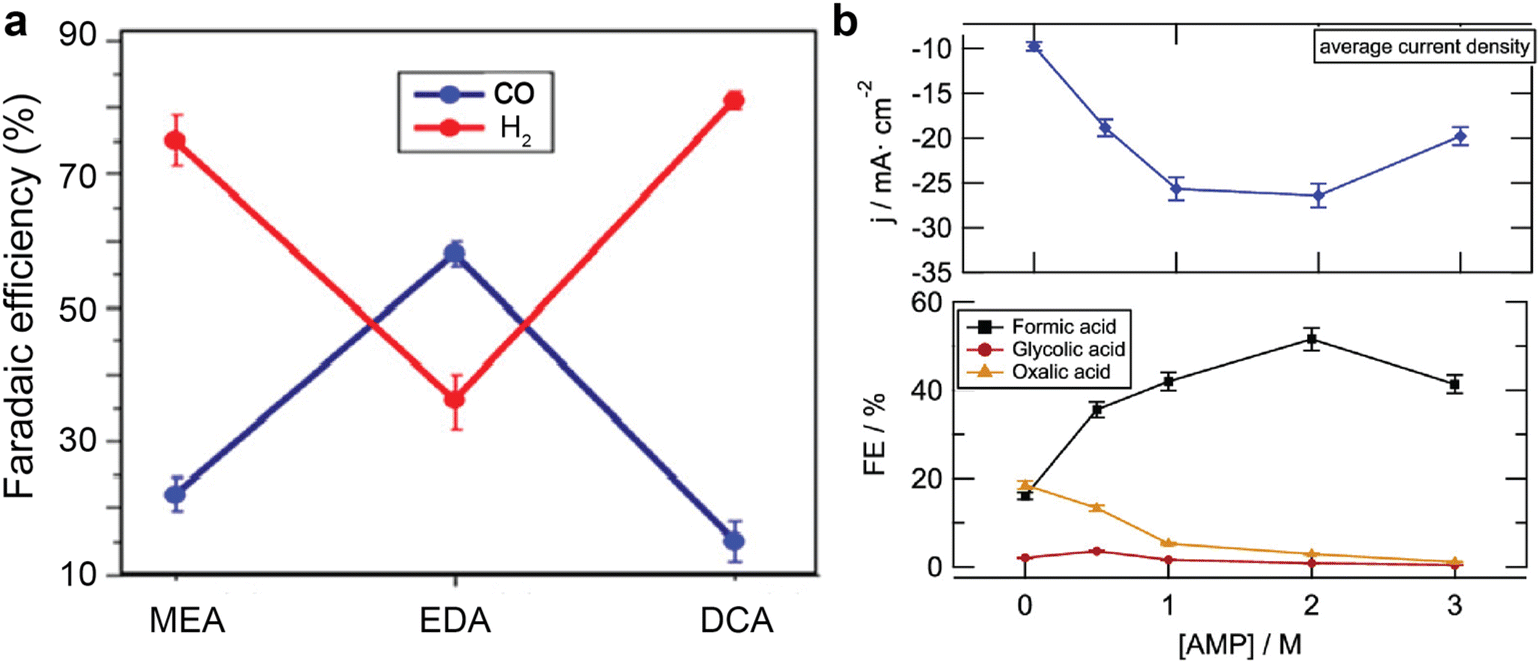 | ||
| Fig. 10 (a) Faradaic efficiency of CO and H2 over Cu catalyst in 0.1 M NaClO4 solution with MEA, ethylenediamine (EDA), and decylamine (DCA) at −0.78 V vs. RHE. Reproduced with permission,71 copyright 2020, American Chemical Society. (b) The change of current densities and product faradaic efficiency over Pb electrode as a function of 2-amino-2-methyl-1-propanol (AMP) in propylene carbonate solutions at −2.5 V vs. Ag|AgCl at 75 °C. Reproduced with permission,61 copyright 2021, American Chemical Society. | ||
A few studies37,62,69 drew consistent conclusions that the addition of alkali salts can improve product selectivity mainly due to the presence of large alkali cations (e.g., K+ or Cs+), while anions show negligible effects on the CO2 conversion. In brief, large alkali cations are reported to promote CO2 conversions in amines via multiple benefits, such as (i) enhancing charge transfer from the electrode to carbamate,37 (ii) facilitating fast ion pairing with carbamate,69 (iii) destabilising the formation of carbamate hence facilitating carbon–oxygen bond cleavage,69 (iv) suppressing hydrogen evolution reaction, (v) strengthening local electrified field87 (for free dissolved CO2), and (vi) stabilising key reaction intermediates.88,89
When CO2 serves as the active species for CO2 conversion, according to recent reports,88,90–92 cations play a crucial role in activating CO2 electroreduction in the CO2-gas-fed system, rather than modifying local electric field or buffering local pH. The findings from Monteiro's report88 show that weakly hydrated cations (e.g., Cs+ and K+) can be concentrated at the catalyst surface and tend to stabilise the negatively charged intermediates (e.g., *CO2−) via a local electric field effect, including medium-range electric field-dipole interaction and short-range electrostatic interaction. In the case of the integrated electrolysis, Lee et al.37 observed an improvement of CO faradaic efficiency in MEA solutions with Cs+ cations as compared to K+ cations. An earlier report also unveiled that the large alkali cations weaken carbamate formation and promote CO2 reduction current densities for CO2-loaded 0.1 M 2-ethoxyethylamine (EEA) in dimethyl sulfoxide (DMSO).69 In their CO2-EEA-DMSO system, the primary product is carbamic acid if there are no salts added. In contrast, the primary product distribution shifts towards a more significant proportion of carbamate ions with alkali cations following the order K+ < Na+ < Li+ (see Fig. 11a). Density functional theory (DFT) calculations suggest that the large alkali cations weaken the C–N bond and increase O–C–O bond angles due to low inductive effects between –COO− and the soft Lewis acids. Despite the low availability of carbamate ions, K+ cations achieve the highest current densities among other small cations. Their molecular dynamics (MD) simulation results, as shown in Fig. 11b, point out that the improvement could be correlated with an easier desolvation and more rapid pairing kinetics with carbamate over K+ than over Li+.
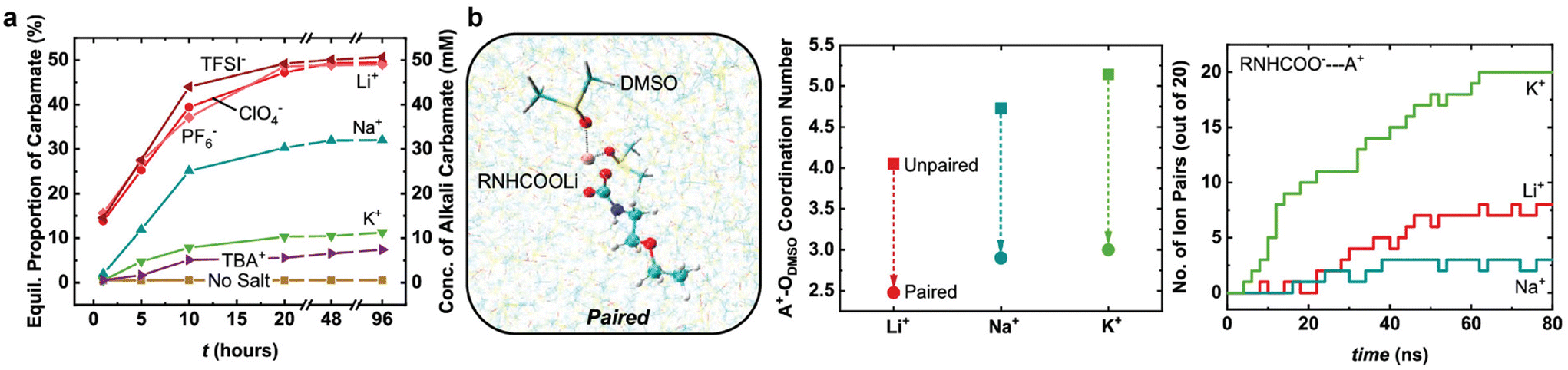 | ||
| Fig. 11 (a) Comparison of equilibrium population and concentration of alkali and tetrabutylammonium (TBA+) cations in CO2 -loaded 0.1 M EEA in DMSO. (b) A Li-carbamate pairing configuration solvated in DMSO (left) and molecular dynamics simulations results in alkali cations coordination numbers (middle) and the number of ion pairs formed against computation time (right). Reproduced with permission,69 copyright 2019, American Chemical Society. | ||
Interestingly, Kim et al.75 report that the effects of cations vary with the catalyst types: Ni single-atom catalyst is less sensitive to cation effects than a metallic catalyst. The authors attributed such trend to the high potential of zero charges of single-atom catalysts that can maintain a high surface charge density no matter the size of the alkali cations. In addition, Ni single-atom catalyst is considered a unique catalyst that has a weak binding with protons and poor kinetics to evolve hydrogen during gas-fed CO2 electrolysis.70,93 The suppression of hydrogen evolution reaction for Ni single-atom catalyst could contribute to the observed insensitivity of the catalyst to alkali cations.
Further, the side hydrogen evolution reaction during CO2 electroreduction in amines can be suppressed by incorporating surfactant in the solvents. The results shown in Fig. 12, as reported by Chen et al.,60 suggest the cation surfactant (i.e., CTAB) can boost the CO2 conversion to formate and CO, while anion surfactant (i.e., sodium dodecyl sulfate) only improve the selectivity towards formate. In contrast, there is no noticeable improvement when Triton surfactant is present in the solution. The authors also observed a lower current density in the presence of CTAB surfactant, which is an indication that the CTAB enhance the CO2 conversion selectivity mainly via the suppression of hydrogen evolution reaction. Therefore, the beneficial effect of these additives is different from the cation effect. Including cation mainly promotes CO2 conversion, while including these additives could alter proton availability and proton reduction activity.
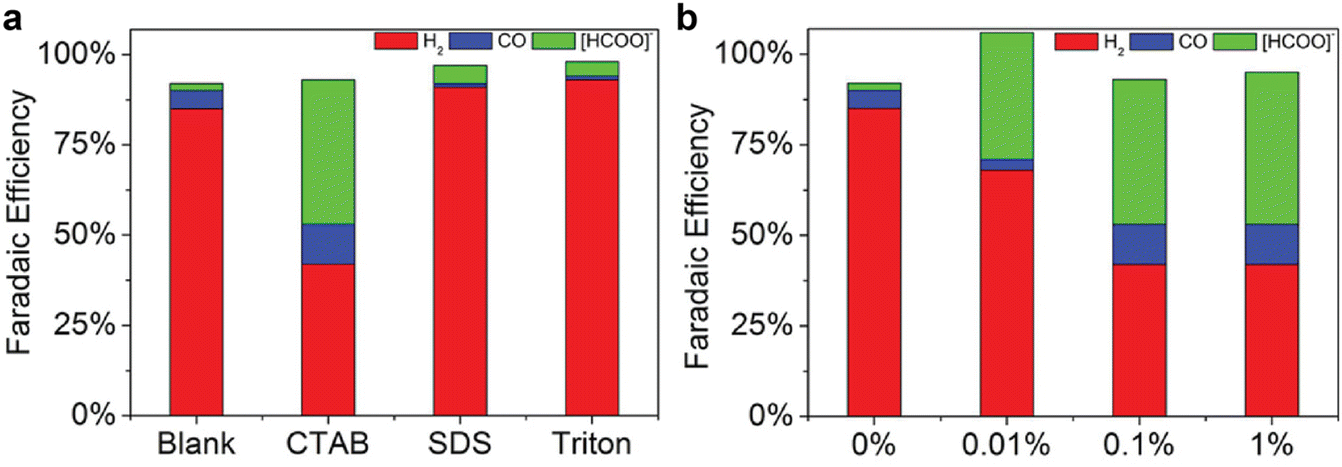 | ||
| Fig. 12 Product distributions of CO2 reduction over indium catalyst at −0.8 V vs. RHE in CO2-saturated 30 wt% MEA aqueous solution with (a) 0.1 wt% of CTAB, sodium dodecyl sulfate (SDS), and Triton surfactants, and (b) with different concentrations of CTAB surfactant. Reproduced with permission,60 copyright 2017, John Wiley and Sons. | ||
As shown in Fig. 12b, additionally, the content of the CTAB surfactant also has a profound impact on the product distribution. A high content (>0.1 wt%) of CTAB is helpful in promoting CO, and a low content (0.01 wt%) is sufficient to improve the formate production. If combined with the porous indium electrode, one could achieve a formate faradaic efficiency of 45.4% and a CO of 17.0% in 30 wt% MEA aqueous solution. Similar beneficial effects of the CTAB are also reported for quaternary ammonium compounds for gas-fed CO2 electroreduction,94–97 highlighting the similarity of mechanisms between integrated and gas-fed electrolysis. The observed faradaic-efficiency overshoot in Fig. 12b could be a result of the mismatch of the effluent flow rate, current densities, and gas concentration while calculating the faradaic efficiency. This issue takes place when the reaction system does not reach a steady state, which is sometimes challenging to achieve experimentally and adds complexity to the measurement of the performance for CO2 electrolysis.
Temperatures and pressures
As mentioned previously, temperature is an important factor in determining the chemical speciation in the capture medium. An elevated temperature normally shifts the reaction equilibrium towards the evolution of CO2 from the capture medium (see Fig. 3a and b) but decreases CO2 solubility in water. On the electrochemical reaction side, due to the Joule heating, the local temperature at the catalytic interface is generally higher than the solvent bulk, especially when the electrolyser is at high current densities.98 High operating temperatures could also accelerate the mass transport and reaction kinetics for both CO2 conversion and side hydrogen evolution reaction. Tailoring the operating temperature could be an effective strategy to enhance the current densities and product selectivity.By lifting the operating temperature up to 60 °C, Lee et al.37 demonstrated the feasibility of achieving 50 mA cm−2 with 72% CO faradaic efficiency over Ag gas-diffusion electrode in a flow cell system fed with 2 M MEA solution. Fig. 5f also shows another impact of temperature on CO2 conversion. Elevated temperatures (e.g., 75 °C) can enhance the rate of CO2 conversion over Pb for the CO2-AMP-PC system but may leave too much liberated CO2 unreacted before leaving the system.61 Therefore, a moderate temperature is beneficial to achieve a relatively high conversion rate and CO2 conversion efficiency. Similarly, Kim et al.75 also observed that the CO partial current densities reach peak values when the temperature is moderate (40 °C vs. 60 °C) in 5 M MEA aqueous solutions.
The pressure is another important factor that affects the dissolved CO2 in the capture medium. High pressure at cathode chamber increases CO2 solubility in the solvents and enhances the concentration of the free dissolved CO2 in the solvent. Diaz et al.45 reported a promoted CO faradaic efficiency (2.9%) when there is a back pressure of 20 psig compared to the faradaic efficiency (1.1%) without backpressure in CHP. Further increase in back pressure to 40 psig can improve CO faradaic efficiency by 20% at about 104 mA cm−2. These results highlight the importance of reporting the backpressure at the cathode side for CO2 conversion in the capture medium. Increasing the pressure of the CO2-loaded liquid is also effective in improving CO selectivity from bicarbonate reduction.79
Cell configuration
The cell configuration design also matters to achieve an optimal CO2 conversion rate from amines. As the active species, products, and recovered amines need to transport between solvent bulk and the electrode surface. Such configuration is similar to the scenario in CO2 transport within an H-cell configuration, where there is a thick hydrodynamic layer about 40–120 μm in thickness.99,100 Such a thick hydrodynamic boundary layer could slow down the local mass transfer of reactants, products, and ions. The flow cell configuration reported by Lee et al.37 partially contributed to the enhancement of CO2 conversion rate in amines. Flow cells allow the solvent to continuously flow in and out within the cell and can help reduce the reduce the thickness of the stagnant film. In addition, membrane electrode assembly is another promising configuration (see Fig. 1c), where there is no gap between electrodes (both cathode and anode) and membrane. Such configuration can further reduce ohmic loss arising from electrolyte ion conduction. Kim et al.75 employed such cell configuration and significantly improved the rates of CO2 conversion. Similarly, Zhang et al.79 demonstrated that a membrane electrode assembly with porous flow-through the electrode could also boost the overall CO2 conversion selectivity at industrially relevant rates from concentrated bicarbonate solutions. If the primary reactant is bicarbonate ions, the cell should allow sufficient CO2 liberation from bicarbonate by supplying protons and accelerating concentrated free dissolved CO2 locally at the catalyst surface.68 Employment of proton-conducting membrane45 or bipolar membrane75 could ensure the proton supply to the solvents. Modelling studies67,68 have shown that by adjusting the thickness, porosity and permeability of the catalyst layer, together with the thickness and ion transport activity of the cation membrane or bipolar membrane, bicarbonate electrolysers can reach higher activity and selectivity under lower cell voltages.Conclusions and outlook
In conclusion, integrating CO2 capture based on amine scrubbing and electrochemical conversion has the potential to reduce the overall capital and operational cost further. The critical enabler is the development of energy-efficient integrated CO2 electroreduction that can convert the absorbed CO2 to valuable products and regenerate amines simultaneously. However, CO2-amine solution could be a complex system involving multiple homogeneous equilibrium reactions, which are determined by factors such as CO2 loading, temperatures, pressures and pH. The catalytic interface for CO2 conversion usually shows a much higher pH and temperature than the solvent bulk during high-rate electrolysis, which could affect the chemical speciation. Although the catalytically active species for CO2 electroreduction is under debate, these species should certainly come from carbamate, bicarbonate, and free dissolved CO2. Free dissolved CO2 molecules are the active species for the latter two. All these species are found limited close to the catalyst surface, particularly at high current densities, mainly due to either limited liberation of CO2, slow transport of CO2, or electrostatic repulsion. Recent studies also demonstrated exciting improvements in CO2 conversion and suppression of side reactions by innovating electrode structures and catalysts, optimising the local reaction environment (amine types, amine content, alkali cations), increasing pressures and temperatures, and designing cell configurations.Further research efforts are demanded to address challenges in advancing integrated electrolysis. First, it remains unclear about the dominant active reactant for CO2 conversion in the capture media, but the understanding of the active species is essential for the future rational design of the catalytic interfaces and systems. It is possible to take advantage of operando or in situ spectroscopy technology, such as attenuated total reflectance – surface-enhanced infrared absorption spectroscopy (ATR – SEIRAS), Raman spectroscopy, and mass spectroscopy, to probe the local reaction environment and products under electrochemical conditions. In addition, multiscale theoretical calculations such as DFT, MD, and multiphysics modelling and simulation could help provide insights into the reaction mechanisms and local reaction environment. The new knowledge generated from these investigations can also guide us in designing the next generation of integrated electrolysis through the choice of the capture medium, modulation of ion transport, electrocatalyst and electrode structure design, process optimisation and cell configuration innovation.
The electrocatalysts and electrode structures in recent reports are similar to the electrodes used in gas-fed CO2 reduction. However, the electrode is desired to be more hydrophilic for the integrated electrolysis to increase electrochemically active surface area and minimise gas-electrode contact, which is different from the gas-fed reaction that requires a stable combination of the hydrophobic and hydrophilic surface. Therefore, further electrode development could explore the hydrophilic porous electrode, such as porous metallic films, allowing the solvent to flow within the electrode structure.
Furthermore, the cell configuration is another key contributor to the overall performance of the integrated electrolysers because it determines the conditions of the reaction zones for homogeneous reactions, such as the liberation of CO2 through bicarbonate acidification and ion transportation. Additional research is urgently needed to understand the compatibility between the CO2-rich amines and ion-exchange membranes. More modelling work could also help understand the role of cell dimensions in determining flow dynamics, CO2 liberation, and ion conduction.
Finally, both CO2 capture and electrolysis are fast-growing fields. It is essential to keep evaluating the technical and economic viability of both sequential and integrated pathways. The advancement of materials, processes, and techniques in both fields could contribute to the improvement of the integrated electrolysis and further cost reduction for CO2 capture and utilisation.
Conflicts of interest
We declare no conflicts of interest.Acknowledgements
C. Z. acknowledges the support by Australian Research Council (FT170100224).References
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed.
- F. P. G. d. Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. d. Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- B. Endrődi, A. Samu, E. Kecsenovity, T. Halmágyi, D. Sebők and C. Janáky, Nat. Energy, 2021, 6, 439–448 CrossRef PubMed.
- P. Grammelis, N. Margaritis and E. Karampinis, in Fuel Flexible Energy Generation, ed. J. Oakey, Woodhead Publishing, Boston, 2016, pp. 29–58, DOI:10.1016/B978-1-78242-378-2.00002-X.
- J.-M. G. Amann and C. Bouallou, Energy Procedia, 2009, 1, 909–916 CrossRef CAS.
- X. Wang and C. Song, Front. Energy Res., 2020, 8 DOI:10.3389/fenrg.2020.560849.
- C. J. o. C. Engineering, M. Abdinejad, C. Dao, B. Deng, F. Dinic, O. Voznyy, X.-a. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 9549–9557 CrossRef.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X.-a. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- M. Abdinejad, K. Tang, C. Dao, S. Saedy and T. Burdyny, J. Mater. Chem. A, 2022, 10, 7626–7636 RSC.
- M. Abdinejad, C. Dao, X.-A. Zhang and H. B. Kraatz, J. Energy Chem., 2021, 58, 162–169 CrossRef.
- M. Li, X. Tian, S. Garg, T. E. Rufford, P. Zhao, Y. Wu, A. J. Yago, L. Ge, V. Rudolph and G. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 22760–22770 CrossRef CAS PubMed.
- M. Li, M. N. Idros, Y. Wu, S. Garg, S. Gao, R. Lin, H. Rabiee, Z. Li, L. Ge, T. E. Rufford, Z. Zhu, L. Li and G. Wang, React. Chem. Eng., 2021, 6, 345–352 RSC.
- M. Sassenburg, R. de Rooij, N. T. Nesbitt, R. Kas, S. Chandrashekar, N. J. Firet, K. Yang, K. Liu, M. A. Blommaert, M. Kolen, D. Ripepi, W. A. Smith and T. Burdyny, ACS Appl. Energy Mater., 2022, 5, 5983–5994 CrossRef CAS PubMed.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS PubMed.
- M. Abdinejad, I. S. d. Silva and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 9791–9797 RSC.
- H. Liu, S. Cao, L. Chen, K. Zhao, C. Wang, M. Li, S. Shen, W. Wang and L. Ge, Chem. Eng. J., 2022, 433, 133594 CrossRef CAS.
- M. Abdinejad, E. Irtem, A. Farzi, M. Sassenburg, S. Subramanian, H. P. I. v. Montfort, D. Ripepi, M. Li, J. Middelkoop, A. Seifitokaldani and T. Burdyny, ACS Catal., 2022, 12, 7862–7876 CrossRef CAS PubMed.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 CrossRef CAS.
- D. Cebrucean, V. Cebrucean and I. Ionel, Energy Procedia, 2014, 63, 18–26 CrossRef CAS.
- T. Alerte, J. P. Edwards, C. M. Gabardo, C. P. O'Brien, A. Gaona, J. Wicks, A. Obradović, A. Sarkar, S. A. Jaffer, H. L. MacLean, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 4405–4412 CrossRef CAS.
- J. B. Greenblatt, D. J. Miller, J. W. Ager, F. A. Houle and I. D. Sharp, Joule, 2018, 2, 381–420 CrossRef CAS.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O'Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Nat. Catal., 2022, 5, 288–299 CrossRef CAS.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- L. Ge, H. Rabiee, M. Li, S. Subramanian, Y. Zheng, J. H. Lee, T. Burdyny and H. Wang, Chem, 2022, 8, 663–692 CAS.
- S. Garg, M. Li, T. Hussain, M. N. Idros, Y. Wu, X. S. Zhao, G. G. X. Wang and T. E. Rufford, ACS Appl. Mater. Interfaces, 2022 DOI:10.1021/acsami.2c05918.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- R. Sharifian, R. M. Wagterveld, I. A. Digdaya, C. Xiang and D. A. Vermaas, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- O. Gutiérrez-Sánchez, B. Bohlen, N. Daems, M. Bulut, D. Pant and T. Breugelmans, ChemElectroChem, 2022, 9, e202101540 CrossRef.
- M. Li, E. Irtem, H. P. I. v. Montfort and T. Burdyny, ChemRxiv, 2021 DOI:10.26434/chemrxiv-2021-33k4d.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. S. Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- H. Yamada, Polym. J., 2021, 53, 93–102 CrossRef.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- 1930.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- P. W. J. Derks, P. J. G. Huttenhuis, C. van Aken, J.-H. Marsman and G. F. Versteeg, Energy Procedia, 2011, 4, 599–605 CrossRef CAS.
- J. P. Jakobsen, J. Krane and H. F. Svendsen, Ind. Eng. Chem. Res., 2005, 44, 9894–9903 CrossRef.
- L. A. Diaz, N. Gao, B. Adhikari, T. E. Lister, E. J. Dufek and A. D. Wilson, Green Chem., 2018, 20, 620–626 RSC.
- M. C. Simoes, K. J. Hughes, D. B. Ingham, L. Ma and M. Pourkashanian, Ind. Eng. Chem. Res., 2018, 57, 2346–2352 CrossRef CAS.
- Z. Liang, K. Fu, R. Idem and P. Tontiwachwuthikul, Chin. J. Chem. Eng., 2016, 24, 278–288 CrossRef CAS.
- E. Gjernes, S. Pedersen, T. Cents, G. Watson, B. F. Fostås, M. I. Shah, G. Lombardo, C. Desvignes, N. E. Flø, A. K. Morken, T. de Cazenove, L. Faramarzi and E. S. Hamborg, Energy Procedia, 2017, 114, 1146–1157 CrossRef CAS.
- U. E. Aronu, S. Gondal, E. T. Hessen, T. Haug-Warberg, A. Hartono, K. A. Hoff and H. F. Svendsen, Chem. Eng. Sci., 2011, 66, 6393–6406 CrossRef CAS.
- I. Kim, K. A. Hoff and T. Mejdell, Energy Procedia, 2014, 63, 1446–1455 CrossRef CAS.
- K. G. Joback, J. R. Heberle and A. S. Bhown, Energy Procedia, 2017, 114, 1689–1708 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. d. Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- K. Yang, R. Kas and W. A. Smith, J. Am. Chem. Soc., 2019, 141, 15891–15900 CrossRef CAS PubMed.
- N. S. Matin, J. E. Remias, J. K. Neathery and K. Liu, Ind. Eng. Chem. Res., 2012, 51, 6613–6618 CrossRef CAS.
- B. Lv, B. Guo, Z. Zhou and G. Jing, Environ. Sci. Technol., 2015, 49, 10728–10735 CrossRef CAS PubMed.
- M. Du, AIP Conf. Proc., 2017, 1864, 020091 CrossRef.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- W. Deng, T. Yuan, S. Chen, H. Li, C. Hu, H. Dong, B. Wu, T. Wang, J. Li, G. A. Ozin and J. Gong, Fundam. Res., 2021, 1, 432–438 CrossRef CAS.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS PubMed.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2021, 60, 4269–4278 CrossRef.
- N. Ahmad, Y. Chen, X. Wang, P. Sun, Y. Bao and X. Xu, Renewable Energy, 2022, 189, 444–453 CrossRef CAS.
- Z. Zhang, E. W. Lees, S. Ren, A. Huang and C. P. Berlingette, ChemRxiv, 2021 DOI:10.26434/chemrxiv.13665074.v1.
- C. P. O'Brien, R. K. Miao, S. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- E. W. Lees, J. C. Bui, D. Song, A. Z. Weber and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 834–842 CrossRef CAS.
- R. Kas, K. Yang, G. P. Yewale, A. Crow, T. Burdyny and W. A. Smith, Ind. Eng. Chem. Res., 2022, 61, 10461–10473 CrossRef CAS.
- A. Khurram, L. Yan, Y. Yin, L. Zhao and B. M. Gallant, J. Phys. Chem. C, 2019, 123, 18222–18231 CrossRef CAS.
- M. Li, S. Garg, X. Chang, L. Ge, L. Li, M. Konarova, T. E. Rufford, V. Rudolph and G. Wang, Small Methods, 2020, 4, 2000033 CrossRef CAS.
- M. Abdinejad, Z. Mirza, X.-a. Zhang and H.-B. Kraatz, ACS Sustainable Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- L. Li, J. Yang, L. Li, Y. Huang and J. Zhao, Electrochim. Acta, 2022, 402, 139523 CrossRef CAS.
- N. Ahmad, X. Wang, P. Sun, Y. Chen, F. Rehman, J. Xu and X. Xu, Renewable Energy, 2021, 177, 23–33 CrossRef CAS.
- M. N. Hossain, S. Ahmad, I. S. da Silva and H.-B. Kraatz, Chem. – Eur. J., 2021, 27, 1346–1355 CrossRef CAS PubMed.
- J. H. Kim, H. Jang, W. Choi, H. Yun, E. C. Lee, D. Kim, J. W. Kim, S. Y. Lee and Y. J. Hwang, Res. Sq., 2022 DOI:10.21203/rs.3.rs-1310811/v1.
- S.-J. Han and J.-H. Wee, J. Chem. Eng. Data, 2016, 61, 712–720 CrossRef CAS.
- R. J. Gilliam, J. W. Graydon, D. W. Kirk and S. J. Thorpe, Int. J. Hydrogen Energy, 2007, 32, 359–364 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. L. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- Y. Wu, S. Garg, M. Li, M. N. Idros, Z. Li, R. Lin, J. Chen, G. Wang and T. E. Rufford, J. Power Sources, 2022, 522, 230998 CrossRef CAS.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988–994 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- A. G. Fink, E. W. Lees, Z. Zhang, S. Ren, R. S. Delima and C. P. Berlinguette, ChemElectroChem, 2021, 8, 2094–2100 CrossRef CAS.
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- D. Kim, W. Choi, H. W. Lee, S. Y. Lee, Y. Choi, D. K. Lee, W. Kim, J. Na, U. Lee, Y. J. Hwang and D. H. Won, ACS Energy Lett., 2021, 6, 3488–3495 CrossRef CAS.
- S.-F. Zhao, M. Horne, A. M. Bond and J. Zhang, J. Phys. Chem. C, 2016, 120, 23989–24001 CrossRef CAS.
- S. Garg, M. Li, T. E. Rufford, L. Ge, V. Rudolph, R. Knibbe, M. Konarova and G. G. X. Wang, ChemSusChem, 2020, 13, 304–311 CrossRef CAS PubMed.
- S. Garg, M. Li, Y. Wu, M. N. Idros, H. Wang, A. J. Yago, L. Ge, G. G. X. Wang and T. E. Rufford, ChemSusChem, 2021, 14, 2601–2611 CrossRef CAS PubMed.
- S. Banerjee, X. Han and V. S. Thoi, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- H. P. I. v. Montfort and T. Burdyny, ACS Energy Lett., 2022, 7, 2410–2419 CrossRef.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L.-T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, ACS Catal., 2018, 8, 6560–6570 CrossRef CAS.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984 RSC.
| This journal is © The Royal Society of Chemistry 2022 |