
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclic polymers: synthesis, characteristics, and emerging applications
Chaojian
Chen
 *ab and
Tanja
Weil
*a
*ab and
Tanja
Weil
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: chenc@northwestern.edu; weil@mpip-mainz.mpg.de
bDepartment of Chemistry and International Institute for Nanotechnology, Northwestern University, Evanston, Illinois 60208, USA
First published on 2nd August 2022
Abstract
Cyclic polymers with a ring-like topology and no chain ends are a unique class of macromolecules. In the past several decades, significant advances have been made to prepare these fascinating polymers, which allow for the exploration of their topological effects and potential applications in various fields. In this Review, we first describe representative synthetic strategies for making cyclic polymers and their derivative topological polymers with more complex structures. Second, the unique physical properties and self-assembly behavior of cyclic polymers are discussed by comparing them with their linear analogues. Special attention is paid to highlight how polymeric rings can assemble into hierarchical macromolecular architectures. Subsequently, representative applications of cyclic polymers in different fields such as drug and gene delivery and surface functionalization are presented. Last, we envision the following key challenges and opportunities for cyclic polymers that may attract future attention: large-scale synthesis, efficient purification, programmable folding and assembly, and expansion of applications.
 Chaojian Chen | Chaojian Chen is a Walter Benjamin Fellow and an International Institute for Nanotechnology Fellow at Northwestern University, United States. He received his bachelor's and master's degrees from Sichuan University and Zhejiang University, respectively. After working as a Research Assistant at the Hong Kong Polytechnic University for two years, he started his doctoral journey in Prof. Tanja Weil's group at Ulm University, Germany. In 2016, he moved with the group to the Max Planck Institute for Polymer Research in Mainz and completed his doctoral training in 2020. His research interests include bioinspired polymer architectures, programmable supramolecular assembly, and Nature-derived functional materials. |
 Tanja Weil | Tanja Weil completed her PhD at the Max Planck Institute for Polymer Research (MPIP) in Mainz, for which was awarded the Otto-Hahn Medal. After several years in industry, she returned to academia by accepting an associate professor position at the National University of Singapore. In 2010, she joined the University of Ulm as Director of the Institute of Organic Chemistry III. Since 2017, she has been Director at the MPIP, heading the department “Synthesis of Macromolecules”. She has received an ERC Synergy Grant, the Science Prize of the City of Ulm as well as the Netherlands Supramolecular Chemistry Scholar Award. |
1. Introduction
Synthetic cyclic polymers are a relatively uncommon class of macromolecules with a ring-like architecture. Several decades before the intentional synthesis of ring polymers, different types of cyclic biomacromolecules had already been identified in nature. The existence of circular deoxyribonucleic acids (DNA) was hypothesized by Jacob and Wollman in 1958,1 and first verified by Freifelder and coworkers who directly observed the cyclic DNA structure from coliphage φX174 using electron microscopy in 1964.2 In addition, various cyclic peptides have been isolated from different plants, bacteria, fungi, and animals, exhibiting enhanced stability against heating and proteolytic degradation compared to their linear analogues.3,4 Similarly, cyclic architectures have also been discovered in the ether lipids of archaea that provide them with exceptional stability under extreme heat and pH.5 Inspired by the early studies of cyclic biopolymers, enormous efforts have been devoted to preparing synthetic cyclic polymers and exploring their unique topology-related properties. Since the 1980s, significant advances have been made in establishing reliable methods for synthesizing well-defined cyclic polymers.6In contrast to linear and branched polymers, cyclic polymers have no chain ends and therefore feature a range of unexpected physical properties. For example, folding flexible polymer chains into the ring shape results in a reduced hydrodynamic size, improved stability, and a slower degradation profile compared with their linear counterparts.7,8 In addition, clear differences in crystallization and glass transition behaviors as well as rheological properties were observed for cyclic polymers compared to linear structures.9–11 Since cyclization of polymer chains alters their mode of movement, cyclic polymers also show distinct self-assembly behaviors both in solution and in thin films.12,13 Importantly, the unique properties arising from their cyclic topology render ring polymers promising candidates for some emerging applications, such as drug delivery, surface modification, and hierarchical assembly.14,15
This Review aims to introduce recent developments on the synthesis, inherent properties, and applications of cyclic polymers. We first summarize typical strategies for preparing cyclic polymers and their derivative topological polymers with more complex architectures. Second, the unique physical properties of cyclic polymers as well as their self-assembly behaviors in solution and on surfaces are discussed. Particularly, we review very recent efforts in the construction of hierarchical ordered nanostructures by folding synthetic polymers into the cyclic topology. Third, some emerging applications of cyclic polymers for creating functional surfaces and for biomedical applications are presented. In the last section, we discuss the key challenges and provide our own perspectives on the future development of this field. Our intention is to highlight the most exciting advances in the past two decades, when large-scale synthesis of high-purity cyclic polymers with good control over molecular weight and distribution became possible. For a more comprehensive overview of the historical development of cyclic polymers, readers are referred to other excellent reviews.7,12,16,17 It should be noted that some macrocycles, cyclic compounds containing a cyclic framework of at least 12 atoms but having low relative molecular mass,18,19 are not classified as “polymers” and are therefore not covered in this Review.
2. Synthesis
In the past half century, polymer chemists have devoted great efforts into the synthesis of cyclic polymers, establishing a variety of effective strategies. Among them, two of the most popular and successful routes are the ring-closure (Fig. 1A) and ring-expansion (Fig. 1B) strategies. In addition, some novel methods for polymer cyclization have also emerged in the past decade (Fig. 1C and D). These different approaches have their own advantages and limitations, as well as scope of applicability. For example, ring-closure strategies are compatible with various polymerization techniques and backbones, but they are only suitable for cyclizing polymers of relatively low molecular weights (<20 kDa).12 In contrast, the ring-expansion strategy can be used for preparing much larger cyclic polymers (up to 1200 kDa). However, it is challenging to control the molecular weight and distribution of the obtained samples, and the applicable backbones are limited. These strategies are discussed below with specific examples. Furthermore, we also introduce the synthesis of more complex polymer architectures derived from cyclic polymers.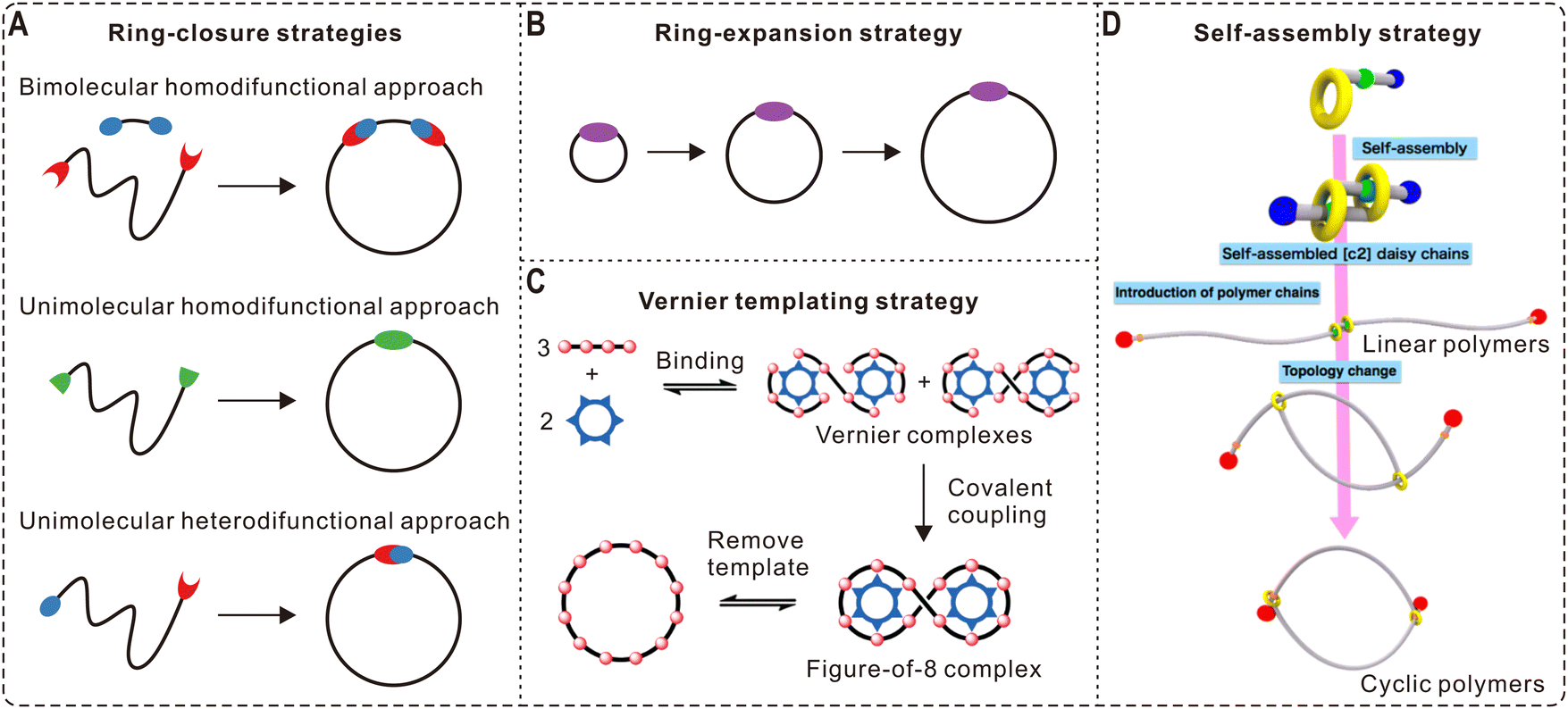 | ||
| Fig. 1 Synthesis strategies for cyclic polymers. (A) Ring-closure strategies. (B) Ring-expansion strategy. (C) Vernier templating for the synthesis of a 12-porphyrin macrocycle using a six-site template. Copyright 2011. Reproduced with permission from Springer Nature.20 (D) Synthesis of cyclic polymers via the introduction of two polymer chains to a self-assembled [c2] daisy-chain structure followed by a topology change. Copyright 2017. Reproduced with permission from the American Chemical Society.21 | ||
2.1 Ring-closure strategy
As the most straightforward method for polymer cyclization, the ring-closure strategy can be further divided into three categories: bimolecular homodifunctional approach, unimolecular homodifunctional approach, and unimolecular heterodifunctional approach (Fig. 1A). The bimolecular homodifunctional ring-closure refers to the coupling between a homotelechelic polymer and a difunctional linker, and therefore equimolar amounts of reagents are important to obtain pure products. In addition, the strategy contains two steps: (1) the intermolecular reaction between a functional terminal end group of the polymer and a complementary group originating from the small linker; (2) the intramolecular coupling between the remaining two functional groups from the polymer and the linker. Because the second step involves the competition between forming cyclic polymers and linear byproducts, this approach should be conducted under high dilution conditions, which unfortunately will slow down the first step. Due to this contradiction, early studies using this approach showed limited success for making cyclic polymers of high purity, and tedious separation procedures were required.22To address this problem, Tezuka and coworkers pioneered an electrostatic self-assembly and covalent fixation (ESA-CF) technique, which has been recognized as a great breakthrough in this field.23 Specifically, the group synthesized bifunctional poly(tetrahydrofuran) (PTHF) precursors with pyrrolidinium salt as terminal groups and bifunctional carboxylate anions. Based on their electrostatic self-assembly at low concentrations, a unique ionic complex with a balanced charge of cations and anions was formed, which could be further covalently linked through the ring-opening reaction of the pyrrolidinium salt groups. After heating at 66 °C for 3 h, cyclic PTHF with a number average molecular weight (Mn) ∼4300 g mol−1 was obtained with good yields of about 74%. The ESA-CF technique is also suitable for making various topological polymers such as multicyclic polymers, which is introduced in more detail in Section 2.4. In 2017, Zhang and coworkers employed a self-accelerating double strain-promoted azide–alkyne click reaction for cyclizing homodifunctional polymers.24 In contrast to traditional bimolecular ring-closure methods, this new approach does not require exact stoichiometric quantities of difunctional polymers and small linkers. Importantly, the intermolecular coupling reaction can be accelerated by using excess amounts of small linkers because their side reactions can be counteracted by the self-accelerated ring-closing reaction.
To avoid contamination and limitations due to stoichiometric aspects in the bimolecular ring-closure, a more practical solution is the unimolecular ring-closure of difunctional polymers (Fig. 1A). Under highly diluted conditions, the oligomerization is largely suppressed, while the intramolecular coupling is unaffected, favoring the formation of cyclic polymers. The unimolecular approach includes the cyclization of homo- and heterodifunctional polymers, and highly efficient coupling reactions are crucial for both to reach satisfactory cyclization yields. The advantage of the unimolecular homodifunctional approach is that it is easier to introduce two identical and self-complementary end groups in the same linear polymer. In this regard, Wegner et al. reported the oxidative coupling of linear polymer precursors with two diacetylene ends, and cycloalkanes up to C288H576 were obtained after hydrogenation.25 In 2011, Schappacher and Deffieux designed and synthesized homodifunctional polystyrene (PS) chains with iron(III) porphyrin groups incorporated on both ends, which could undergo fast cyclization through the formation of a stable diiron(III)-μ-oxobis(porphyrin) dimer under mild basic conditions.26 In addition, this process is fully reversible after treating the cyclic PS with dilute hydrochloric acid. Similarly, Yamamoto and coworkers also reported the light-triggered cyclization of homotelechelic polymers with anthryl terminal groups.27 This process was successful for both hydrophilic poly(ethylene oxide) (PEO) and hydrophobic PTHF telechelics, and it can also be reversed upon heat treatment at 150 °C. By introducing naphthalene or anthracene groups onto both ends of PEO chains, Chen et al. demonstrated an unimolecular cyclization method assisted by host–guest complexation.28 In the presence of cucurbit[8]uril, the π–π stacking of two functional ends can be stabilized and then covalently fixed under UV irradiation. However, a non-negligible fraction of linear impurities was observed from gel permeation chromatography (GPC) characterization. Recently, Honda et al. synthesized cyclic poly(dimethylsiloxane)s of different sizes with photo-responsive hexaarylbiimidazole linkers in the backbone, and reversible linear-cyclic topology switching can be realized by photostimulation.29
Because the number of efficient homocoupling reactions is relatively limited, the unimolecular heterodifunctional approach has become a more popular method for polymer ring-closure since the 1990s.30 By combining atom transfer radical polymerization (ATRP) and click chemistry, Laurent and Grayson reported the first example of using copper-catalyzed azide–alkyne cycloaddition (CuAAC) for polymer cyclization in 2006, which is an important milestone in the development of cyclic polymers.31 Specifically, linear PS precursors were synthesized by conventional ATRP using propargyl 2-bromoisobutyrate as an initiator, and the bromine end group was then reacted with sodium azide to obtain heterodifunctional polymers. The intramolecular cyclization reaction was conducted by slowly adding the linear polymer solution into a large amount of catalyst solution using a syringe pump. Intensive characterization by 1H nuclear magnetic resonance (NMR) spectroscopy, matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry, Fourier transform infrared (FT-IR) spectroscopy, and GPC confirms the nearly quantitative preparation of cyclic polymers with narrow dispersities. Owing to the excellent control over molecular weight and structure by ATRP, this technique has been extended to various polymer backbones and architectures such as block copolymers.30,32–34 For example, Eugene and Grayson reported the synthesis of cyclic poly(methyl acrylate)-b-PS by combining two steps of consecutive ATRP reactions and click cyclization.35 However, CuAAC-based click cyclization still needs to be conducted at highly dilute conditions (<0.1 g L−1) and therefore is time-, solvent-, and catalyst-consuming, making it difficult to scale up. To address these issues, Monteiro and coworkers performed a systematic study under the guidance of the Jacobson–Stockmayer theory,36 showing that the percentage of monocyclic products is only dependent on polymer concentration and the cyclization conditions can be optimized.37 Particularly, they reported the successful synthesis of cyclic PS51 at a high concentration (∼9.5 g L−1) and a low temperature (25 °C) in less than 9 min. In addition, Liu and Chen et al. also reported the facile synthesis of cyclic block copolymers via selective click coupling in micellar media at an even higher concentration (10 g L−1).33 This was realized by controlling the accessibility between reactive alkynyl and azide end groups within self-assembled aggregates, and intramolecular cyclization occurred exclusively for unimers exchanged from the micelles. Recently, Kim and coworkers synthesized large poly(rac-lactide) and its block copolymers via an iterative convergent approach, which were then employed as precursors for preparing cyclic polymers.34 Based on CuAAC, pure discrete cyclic poly(rac-lactide)s with high molecular weights (up to 37 kDa) and no linear contaminants were obtained after separation by preparative GPC. In addition to click cyclization, Harada et al. prepared cyclic PEO based on the host–guest complexation between cyclodextrin and azobenzene.38 Very recently, Wang and coworkers designed a cobalt salen pentenoate catalyst that can be used for one-pot ring-opening copolymerization of epoxides/anhydrides/CO2 and intramolecular cyclization, generating well-defined cyclic polyesters and CO2-based polycarbonates.39
2.2 Ring-expansion strategy
For both bimolecular and unimolecular ring-closure methods, they normally require high dilution conditions and share one common problem: the meeting chance for chain ends becomes smaller as the polymer chain length increases due to entropic penalty.36 As another popular method for creating cyclic polymers, the ring-expansion strategy refers to the continued insertion of monomers into a weak bond of a cyclic initiator or growing chain (Fig. 1B). Compared to the ring-closure strategy, this method eliminates the entropic penalty as well as the need for high dilution conditions and linear precursors, providing access to large quantities of high-molecular-weight cyclic polymers. The feasibility of this strategy was established by Kricheldorf and Lee in 1995, when they polymerized different lactone monomers using cyclic dibutyltin initiators.40In 2002, Grubbs et al. pioneered the synthesis of cyclic polyoctenamers with the Mn up to 1200 kDa and polydispersity indices (Đ) ∼ 2.0 via ring-expansion metathesis polymerization (REMP) by using a cyclic ruthenium-based catalyst.41 Since then, REMP catalyzed by ruthenium-, tungsten-, or molybdenum-based compounds has been successfully developed for the synthesis of various cyclic polymers, showing the great power of the ring-expansion strategy.42 By reacting a tungsten alkylidyne precatalyst with CO2 to produce a tungsten–oxo alkylidene catalyst, Veige and coworkers reported the stereoselective REMP of norbornene, yielding cyclic polynorbornenes with high molecular weights (up to 578 kDa), narrow distributions (Đ < 1.3), and high stereocontrol (≥97% cis-selectivity).43 When the precatalyst was treated with excess tert-butylacetylene, the obtained tungsten catalyst showed excellent activity (9 × 106 g mol−1 h−1) and complete conversion in the polymerization of phenylacetylene to obtain cyclic poly(phenylacetylene)s.44 Importantly, this new catalyst is also applicable for other mono- and disubstituted functional acetylenes, providing access to a novel class of cyclic polymers. Very recently, the same group synthesized cyclic polyacetylene with >99% trans double bonds at −94 °C based on this catalyst.45 The cyclic topology was directly proved by atomic force microscopy (AFM) imaging of cyclic bottlebrush polymers derived from the cyclic polyacetylene. In 2021, the Veige group also reported a double tethered metallacyclobutane catalyst from commercially available molybdenum alkylidene for the REMP of norbornene, obtaining cyclic polynorbornenes with Mn up to 82.6 kDa in high yields (≥91%).46 In addition, Golder and coworkers developed a stable tethered ruthenium–benzylidene complex that can initiate the polymerization of two substituted norbornene-imide monomers to form functionalized cyclic polynorbornenes with controlled molecular weights.47
Zwitterionic ring-opening polymerization (ZROP) of strained heterocyclic monomers, also called zwitterionic ring expansion polymerization (ZREP), represents another important class of ring-expansion methods. During the ZROP process, zwitterionic propagating species bearing two oppositely charged end groups keep the cyclic topology because of the electrostatic interaction.16 One famous example is the N-heterocyclic carbene (NHC)-mediated nucleophilic ZROP of lactone monomers pioneered by Waymouth and coworkers.48 In the absence of alcohol initiators, cyclic poly(lactide)s with Mn = 7–26 kDa and narrow distributions (Đ = 1.15–1.35) were obtained in short time (5–900 s) at room temperature. The nucleophilic ZROP has also been employed for preparing cyclic polymers with different catalysts (e.g., pyridines, amidines) and monomers (e.g., β-lactones, ε-caprolactone, δ-valerolactone, N-butyl N-carboxyanhydride).42 For instance, the Zhang group pioneered the synthesis of cyclic poly(α-peptoid)s via ZROP of N-substituted N-carboxylanhydrides mediated by NHC49–51 or 1,8-diazabicycloundec-7-ene (DBU).52 Very recently, Bonduelle and Verhaeghe et al. reported the lithium bis(trimethylsilyl)amide-catalyzed ZROP of N-alkylated-N-carboxyanhydrides in dimethylformamide, providing a facile route to cyclic polypeptoids with interesting antibacterial properties.53 In addition, ZROP reactions based on different mechanisms include the electrophilic ZROP and the Lewis pair-mediated ZROP. For example, Grayson and Barroso-Bujans et al. reported the electrophilic ZREP of functional epoxides monosubstituted by different groups into cyclic polyethers using tris-(pentafluorophenyl)borane as a catalyst and investigated the reaction mechanism by MALDI-ToF mass spectrometry and computational tools.54 This field has been summarized in other excellent reviews16,42,55 and therefore is not discussed further.
Not only REMP and ZROP, but also recent developments in other ring-expansion polymerizations have further enriched the toolbox of cyclic polymer synthesis. Otsuka and Aoki et al. prepared various macrocyclic monomers containing bis(hindered amino)disulfide units, which allowed the effective synthesis of cyclic polymers via heat-induced radical ring-expansion polymerization.56 Importantly, this simple procedure does not require catalysts and initiators and can be conducted under concentrated conditions. Ouchi and coworkers designed a cyclic molecule via the reaction between divinyl ether and dicarboxylic acid, which can initiate the ring-expansion cationic polymerization of vinyl ether, generating tadpole and figure-eight polymers after post-polymerization modification.57 Moore and coworkers reported the anionic ring-opening polymerization of cyclic disulfide using aryl thiol initiators, achieving large cyclic polymers with an Mn up to 630 kDa.58
2.3 Other synthetic strategies
While most of the cyclic polymers reported in the literature are prepared by either ring-closure or ring-expansion strategies, researchers have been trying to develop new methods for making polymer rings. For example, Anderson and coworkers demonstrated the formation of Vernier complexes between a simple six-site template and a building block with four binding sites (Fig. 1C). After covalent coupling and template removal, a monodisperse 12-porphyrin nanoring was obtained, showing the power of Vernier templating for preparing large cyclic polymers with small templates.20 Through a rational extension of this strategy, larger porphyrin nanorings including 24-mer, 30-mer, 40-mer, and 50-mer with a diameter of 21 nm (750 C–C bonds) have also been synthesized by the same group.59,60 In 2017, Takata and Aoki et al. reported the gram-scale preparation of cyclic poly(ε-caprolactone) (PCL) via an elegant self-assembly strategy that involves growing two polymer chains from a self-assembled [c2] daisy-chain initiator and a linear-to-cyclic topology transformation (Fig. 1D).21 Based on ring-opening metathesis polymerization, Xie et al. developed a blocking-cyclization technique by alternatively feeding di- and monofunctional monomers, generating mono- and multicyclic polymers with controlled size.612.4 Topological polymers derived from cyclic polymers
With the significant progress in ring polymer synthesis and coupling chemistry, other mono- and multicyclic macromolecules derived from cyclic polymers are now available, largely broadening the library of polymer architectures in addition to conventional linear and branched structures. Representative monocyclic topologies include tadpole-shaped, α-shaped, and cyclic brush polymers (Fig. 2A). By introducing functional monomers at desired times during the ATRP of styrene, Lutz and coworkers demonstrated the controlled folding of PS chains into different topologies such as the tadpole-shape and α-shape.62 The folding was realized by CuAAC in dilute concentrations, which are typical conditions for ring-closing of linear polymer chains.31 Monocyclic architectures can also be obtained by grafting linear chains from/to a cyclic backbone. For example, Sumerlin and Veige et al. grafted PS and poly(butyl acrylate) chains from a cyclic polyphenylacetylene initiator via ATRP, forming large cyclic brush polymers (up to 8.76 MDa) whose circular shape can be directly detected by AFM.63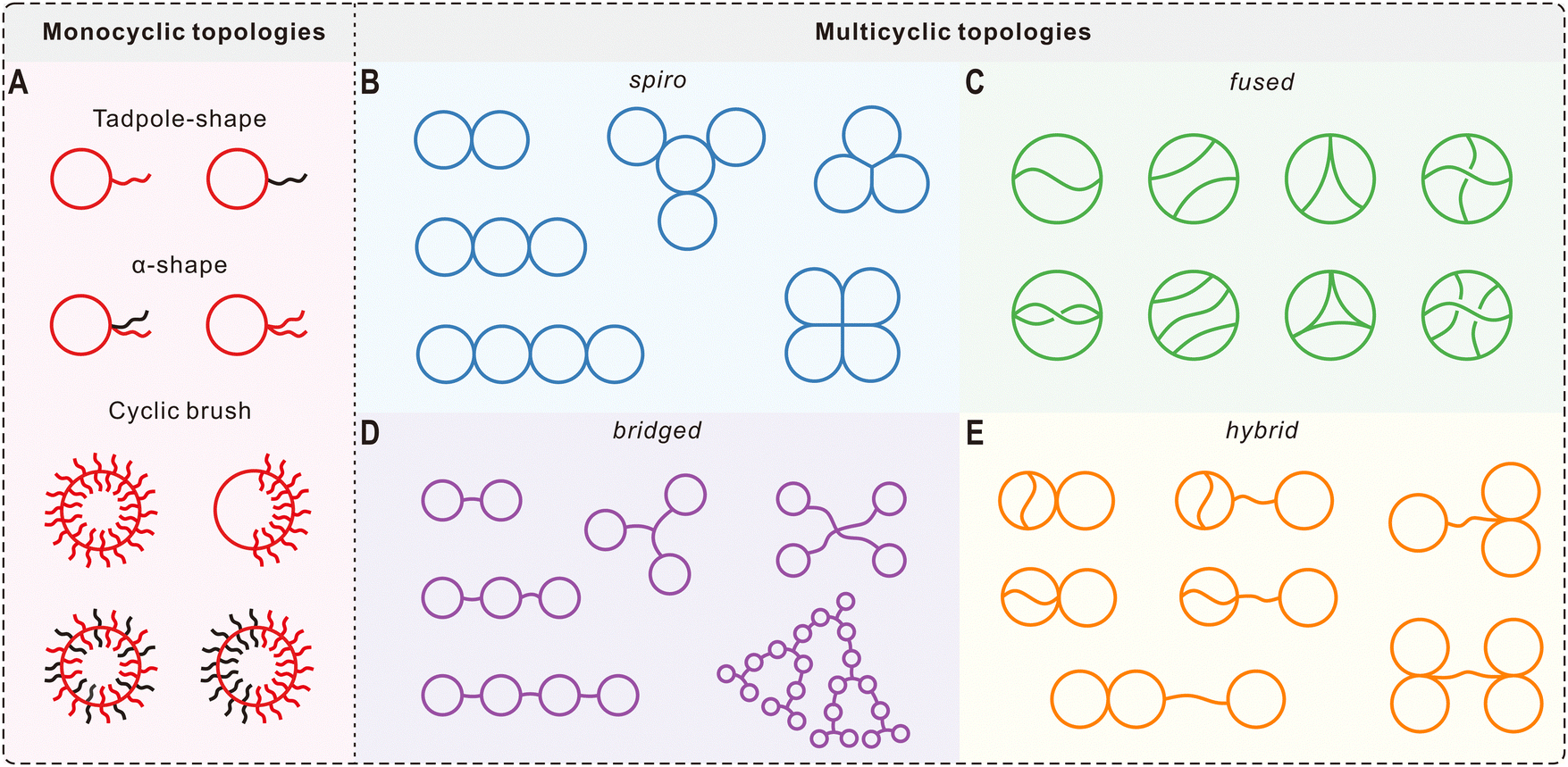 | ||
| Fig. 2 Selected macromolecular architectures derived from cyclic polymers. (A) Monocyclic topologies. (B–E) Spiro-, fused-, bridged-, and their hybrid-forms of multicyclic topologies. | ||
For more complex multicyclic polymers, they can be divided into four subclasses: spiro-, fused-, bridged-, and their hybrid-forms (Fig. 2B–E). These complex macromolecular architectures can be produced by controlled folding of linear, branched, and/or cyclic precursors with determined number of functional groups at prescribed positions using highly efficient coupling reactions. Summarized in an account published in 2017,64 most of the multicyclic polymers illustrated in Fig. 2 have been constructed by the Tezuka group using the ESA-CF technique as mentioned in Section 2.1.23 Perhaps the most representative example is the folding of a dendritic polymer precursor containing six cyclic ammonium salt terminals into a triply fused tetracyclic K3,3 graph, which was achieved through ESA-CF with two trifunctional carboxylate counteranions followed by separation with recycling GPC.65 Recently, Tezuka et al. also reported the programmed folding of telechelic PTHFs with four cyclic ammonium salt functional groups by ESA-CF into three distinct dicyclic topologies: 8 (spiro, Fig. 2B), θ (fused, Fig. 2C), and manacle (bridged, Fig. 2D).66 Other synthetic methods have also been developed by different groups to obtain interesting multicyclic architectures. In 2019, Hong et al. constructed hyperbranched multicyclic PS based on the self-accelerating click reaction between a difunctional linker and a cyclic polymer possessing three azides.67 A similar click reaction was applied by Zhang et al. for the synthesis of cage-shaped PCLs with four or six arms, which can be classified as fused topologies (Fig. 2C).68 Recently, a number of complex multicyclic polythioethers with cyclic units in the linear backbone and hyperbranched chain, as well as brush polymers with cyclic side chains, have been produced by combining rhodanine-based Knoevenagel reaction and anionic ring-opening polymerization.69 These synthetic developments provide exciting opportunities to study the structure-property relationships of macromolecules, leading to better understanding on the biological properties of topologically unique biomolecules and new concepts in designing novel polymeric materials.
3. Characteristics
3.1 Physical properties
Compared with their linear counterparts, cyclic polymers with constrained conformations possess many unique physical properties. For example, early studies on cyclic polymers have shown that they have smaller hydrodynamic volumes.70,71 This feature can be used to determine the purity of cyclic polymers and even to separate them from their linear analogues by preparative GPC.72 Like cyclic peptides and lipids in nature, cyclization of synthetic polymers can afford them with enhanced stability. In this regard, Hoskins and Grayson synthesized cyclic PCL via ring-opening polymerization and CuAAC click coupling.8 Compared with its linear precursor, the cyclic polymer showed a significant lag in the molecular weight loss during its acid-catalyzed degradation.The lack of terminal groups and reduced hydrodynamic size significantly affect thermal properties of cyclic polymers both in solution and in bulk, such as the diffusion mode, entanglement, glass transition temperature (Tg), and crystallization behavior. For example, cyclic polymers are widely accepted to have higher Tg than their linear counterparts and the difference is more obvious for low molecular weight polymers.10 Using a hemiacetal ester-based cyclic initiator, Sawamoto and Ouchi et al. synthesized two cyclic poly(vinyl ether)s via ring-expansion cationic polymerization.73 For cyclic poly(vinyl ether)s with bulky tricyclic alkane pendant groups [poly(TCDVE)s, Fig. 3A], they have higher Tg values with respect to their linear analogues of similar molecular weights (Fig. 3B). Interestingly, cyclic polymers showed a much weaker molecular weight dependence for the Tg than their linear counterparts, which was attributed to the relatively smaller free volume of cyclic polymers (Fig. 3C). To study the topological effect on thermo-sensitivity, cyclic and linear poly(dodecyl vinyl ether)s [poly(DDVE)s, Fig. 3D] were also prepared.73 During cooling, the ethyl acetate solution of linear poly(DDVE) suddenly turned from transparent to opaque at ∼42 °C (Fig. 3E), showing almost same temperatures at the solution transmittance of 50% (cloud point, CP) and 99% (upper critical solution temperature, UCST). However, the solution of cyclic poly(DDVE) started to get cloudy at a higher temperature of 50 °C and recorded a CP of 46 °C, exhibiting a markedly duller transition. By plotting the subtraction from UCST and CP (UCST-CP) against the logarithm of polymer concentration, the difference between linear and cyclic polymers was found to be more pronounced when the concentration was decreased (Fig. 3F). O’Reilly and Dove et al. also reported significant effects of cyclization on solution conformation and CP for cyclic graft copolymers with a polycarbonate backbone and poly(N-acryloylmorpholine) side chains.74 Similar to Tg, increased temperatures of crystallization (Tc) and melting (Tm) have also been observed for different cyclic polymer systems experimentally9,41,75 and by molecular dynamic simulations.76 For instance, Müller and Coulembier et al. demonstrated that cyclic polylactide chains can nucleate and crystallize much faster than their linear analogues and the Tm values were 5 °C higher.9
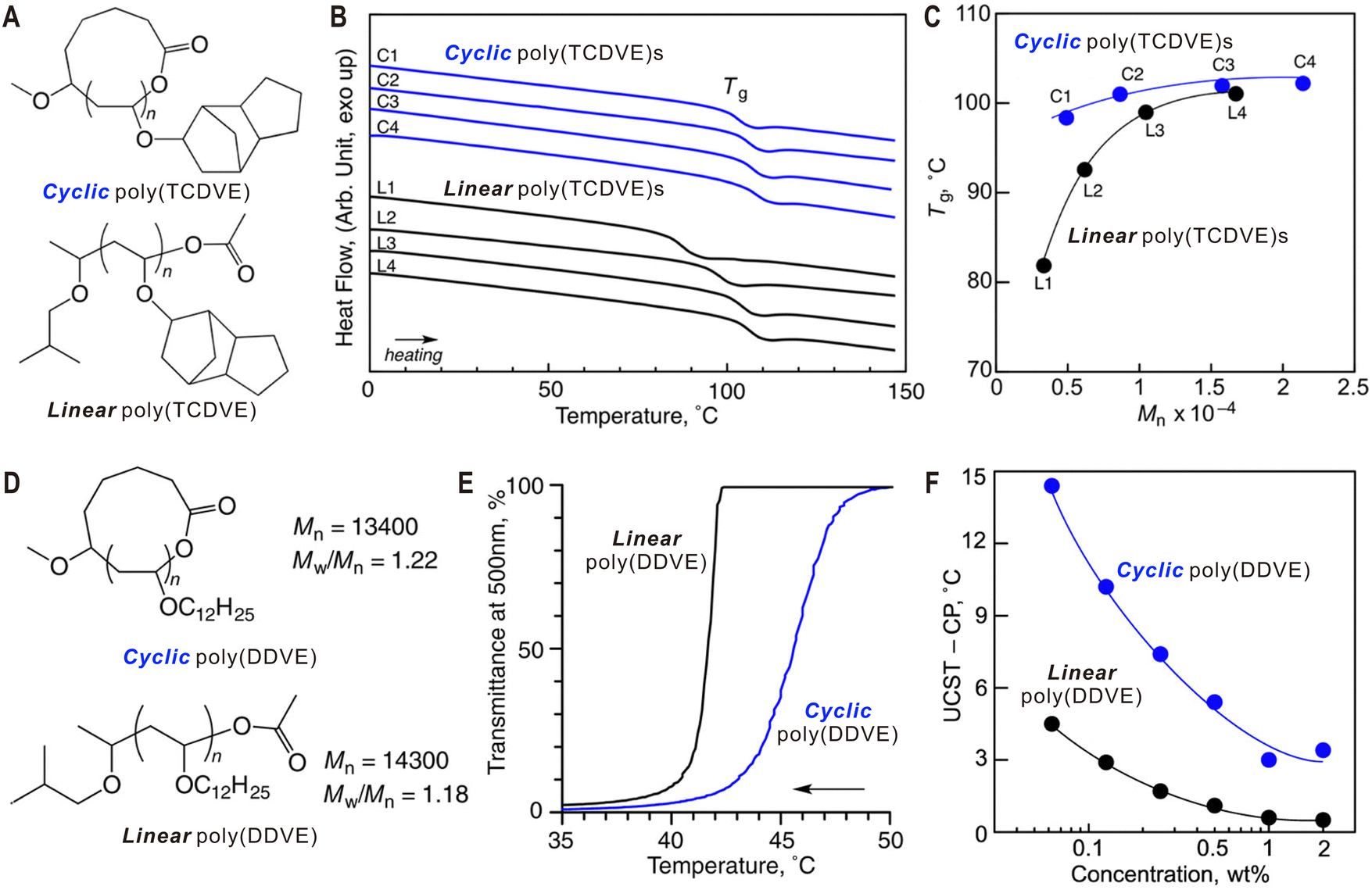 | ||
| Fig. 3 Thermal properties of linear and cyclic poly(vinyl ether)s. (A) Molecular structures of cyclic and linear poly(TCDVE)s. (B) DSC curves of cyclic and linear poly(TCDVE)s with different molecular weights. (C) The molecular weight dependence of Tg for cyclic and linear poly(TCDVE)s. (D) Molecular structures of cyclic and linear poly(DDVE)s. (E) Transmittance changes for 1 wt% ethyl acetate solutions of cyclic and linear poly(DDVE)s upon cooling. (F) Comparison of the UCST-CP for cyclic and linear poly(DDVE)s at different concentrations. Copyright 2017. Reproduced with permission from the American Chemical Society.73 | ||
The rheological properties of cyclic polymers have attracted extensive interests.11,77 However, early studies on cyclic PS samples generated contradictory results, which were later attributed to sample contamination by linear byproducts.78 Recently, Chang et al. prepared high purity cyclic PS samples (Mn = 16–370 kDa) via a ring-closure reaction and fractionation by liquid chromatography. The intrinsic viscosities of cyclic samples were found to be clearly lower than that of linear ones (0.63 ≥ [η]cyclic/[η]linear ≥ 0.57), likely due to reduced chain entanglements.78 Interestingly, the presence of small amounts of linear contaminants in cyclic polymer melts can dramatically increase the viscosity due to the threading of linear chains through polymer rings.77 Cyclic polymers also show interesting physicochemical properties when they are grafted onto surfaces,79 which are further discussed in Section 4.2.
3.2 Self-assembly behaviors
Self-assembly of amphiphilic block copolymers is an effective approach for making a wide variety of soft nanostructures including spherical and wormlike micelles and hollow polymersomes. However, most polymeric assemblies reported in literature are constructed from linear polymers. Cyclic polymer assembly was an unexplored area for a long time due to the difficulties in preparing high purity cyclic products.13 Because significant progress has been made in the last two decades to overcome those synthetic challenges, this field is now attracting growing attention. To understand the cyclization effect on micelle formation, Borsali and coworkers compared the self-assembly behavior of linear and cyclic poly(styrene-b-isoprene) (PS290-b-PI110) copolymers.80 Dynamic light scattering (DLS) and AFM results reveal that linear PS290-b-PI110 self-assembled into spherical micelles at different concentrations. The copolymers showed drastically different self-assembly behavior after cyclization, forming sunflower-like micelles at a low concentration of 0.1 mg mL−1, which could further stack and evolve into wormlike structures over 1 μm in length when the concentration was increased to 5 mg mL−1. In another study, Grayson and coworkers demonstrated that cyclic PEO-b-PCL could self-assemble into spherical micelles whose size is greatly reduced compared to micelles based on corresponding linear copolymers.81 In addition to morphological and size changes, polymer cyclization may also have a remarkable influence on the stability of assemblies. Tezuka and Yamamoto et al. compared the self-assembly behavior of a linear triblock copolymer poly(butylacrylate)-b-poly(ethylene oxide)-b-poly(butyl acrylate) and its cyclized product.82 Although both copolymers self-assembled into spherical micelles with a diameter of 20 nm, the micelles from cyclic polymers showed significantly improved thermal stability. In their subsequent study, the same cyclization strategy was successfully used for enhancing the salt resistance of polymeric micelles.83Furthermore, polymer cyclization has also been developed as a novel method for making complex hierarchical assemblies. For instance, Qiao and Kamigaito et al. reported the topology-guided formation of a “polypseudorotaxane-type” supramolecular assembly via the stereospecific complexation between cyclic syndiotactic poly(methyl methacrylate) (PMMA) and linear isotactic PMMA.84 Schappacher and Deffieux demonstrated the self-assembly of supramolecular tubes from macrocyclic brush polymers with randomly distributed PS and PI side chains.85 Very recently, our group synthesized three linear poly(2-hydroxyethyl methacrylate) (l-PHEMAn-Br) of different chain lengths (number of repeating units 10 < n < 25), whose bromine ends were then converted to azide groups by reacting with sodium azide (Fig. 4A). After cyclization via the highly efficient CuAAC click reaction, we compared the self-assembly behaviors of the obtained cyclic homopolymers (f-PHEMAn) and their linear counterparts.32 Interestingly, the folded cyclic PHEMA polymers could form stable assemblies in water while their linear analogues generated some gel-like precipitates. Further investigation of the self-assembly process by DLS, 1H NMR spectroscopy, and transmission electron microscopy (TEM) suggested the formation of wormlike structures from cyclic PHEMA, which was also confirmed by molecular dynamics simulations (Fig. 4B). Encouraged by these results, we further designed and synthesized macrocyclic brush polymers with amphiphilic polystyrene-b-poly(acrylic acid) (PS-b-PAA) side chains (Fig. 4C). AFM, TEM, and cryo-TEM images in Fig. 4D reveal that they self-assembled into stable wormlike and higher-ordered network structures. Importantly, the weight fraction of PAA and the polymer concentration were found to have profound impact on the self-assembly, and the architectural outcome can be manipulated by tuning the polymer composition, proportion, and block length.32 The self-assembly pathway from the simple block copolymer to hierarchical network is a vivid model to mimic the increasing complexity in the folding of linear polypeptides into 3D protein structures (Fig. 4E). Therefore, this work not only establishes polymer cyclization as a novel approach for fabricating wormlike assemblies which may find broad applications, but also points out the profound influence of polymer folding in macromolecular self-assembly and provides an effective avenue for constructing hierarchical bioinspired structures with significantly increased complexity.86
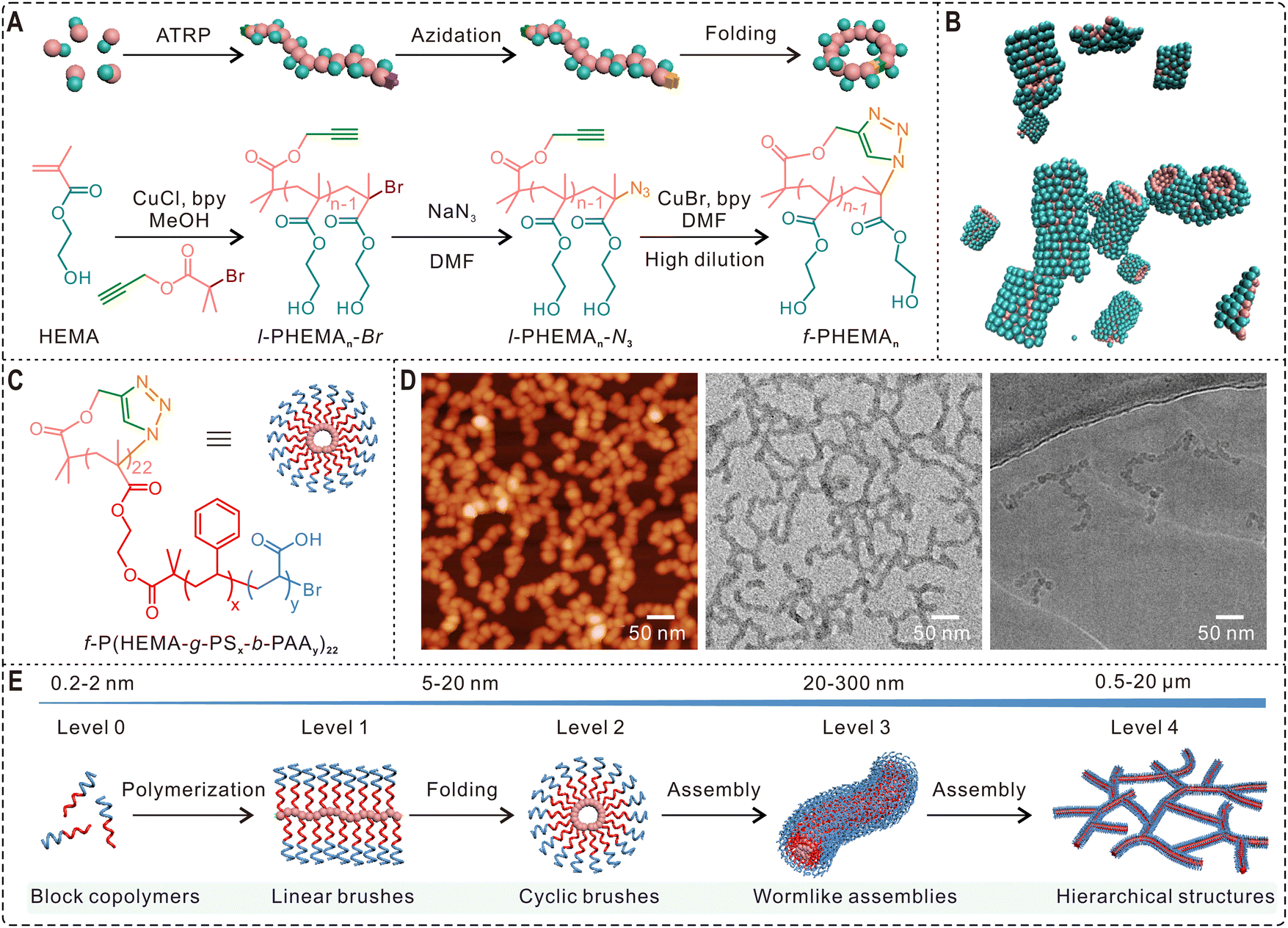 | ||
| Fig. 4 Hierarchical nanostructures self-assembled from cyclic polymers. (A) Scheme of the synthesis of l-PHEMAn-Br and its azidation and folding into the cyclic topology. (B) Molecular dynamic simulation snapshot showing the self-assembly of f-PHEMA15 into wormlike structures. (C) Molecular structure and scheme of cyclic brush polymer f-P(HEMA-g-PSx-b-PAAy)22. (D) AFM (left), TEM (middle), and cryo-TEM (right) images showing the self-assembly of cyclic brush polymers into 1D wormlike assemblies and hierarchical structures at 0.4 mg mL−1 in water. (E) Schematic of the formation of hierarchical structures by synthetic polymers via biomimetic folding and self-assembly. Copyright 2021 The Authors. Reproduced with permission.32 Published by Springer Nature. | ||
Cyclic polymers exhibit interesting and unique assembly behaviors not only in solution but also on surfaces. For example, Hawker and coworkers compared the thin film self-assembly of cyclic block copolymer PS-b-PEO with its linear counterpart and found that the domain spacing was reduced by ∼33% after polymer cyclization.87 This reveals that macromolecular topology can serve as an effective tool for achieving smaller feature sizes in block copolymer lithography, which could be potentially important for writing smaller structures for applications i.e. in semiconductor technology. Inspired by the folded tertiary structures of biopolymers, Anderson and Beton et al. demonstrated that flexible and monodisperse cyclic porphyrin polymers (Mn = 30–60 kDa) can form two-dimensional nested structures on a gold surface, with one cyclic polymer folded and captured inside a second cyclic polymer that adopts a near-circular conformation, which was observed by scanning tunneling microscopy.60
4. Applications
4.1 Drug and gene delivery
In terms of applications, cyclic polymers were first explored in the construction of drug and gene carriers, because polymer cyclization could provide opportunities to tune the stability, degradation behavior, biodistribution and pharmacokinetics of these polymer additives.15 In a pioneering study, Szoka et al. synthesized linear and cyclic brush polymers by grafting different lengths of polyethylene glycol (PEG) side chains to PCL-based backbones, and compared their pharmacokinetics and biodistribution in vivo.88 Longer blood circulation times were observed for cyclic brush polymers of high molecular weight (50 and 90 kDa) with respect to their linear counterparts of similar mass, which can be attributed to the faster reptation of linear polymers through the nanopores in the kidneys. However, cyclic polymer with a mass (∼32 kDa) below the renal filtration threshold (30–40 kDa) exhibited faster elimination because its hydrodynamic size is smaller than that of the corresponding linear polymer. In addition, both 50 kDa and 90 kDa cyclic polymers showed tissue accumulation advantages compared to their linear analogues. Because this work did not take the degradability of PCL into consideration, the same group further studied the topological effect on pharmacokinetics using more stable PEG-grafted poly(acrylic acid) brush polymers, and confirmed the above findings.89 In these two examples, the cyclic brush polymers used were supposed to adopt loosely curled conformations instead of ring-like morphologies because their relatively low side chain densities. To explore the influence of a ring-like morphology on the resulting biological properties of synthetic polymers, Jiang and Wu et al. compared the in vitro and in vivo behaviors of linear and cyclic cylindrical polymer brushes (CPBs) with high side chains densities.90 Due to their reduced flexibility, cyclic CPBs with a ring-like shape exhibited higher non-specific protein adsorption than the linear CPBs. This further led to shorter blood circulation, higher liver and spleen uptake, and thus lower tumor accumulation.Owning to their distinctive biological properties, cyclic polymers have been used as different formulations for drug delivery.15 For example, the Pun group synthesized polymer–drug conjugates by incorporating doxorubicin (Dox) into folate-functionalized sunflower polymers through pH-sensitive hydrazone bonds.91 This platform demonstrated folate receptor-mediated internalization and pH-triggered Dox release in cancer cells (Fig. 5A). Liu et al. reported polymeric micelles based on tadpole-shaped diblock copolymers with a cyclic poly(N-isopropylacrylamide) (PNIPAM) block and a linear PCL block (Fig. 5B). Compared with their linear analogues, the obtained micelles with a cyclic PNIPAM shell exhibited improved drug loading and releasing capacity.92 Similarly, Wei et al. prepared micelles with a cyclic PCL core and a linear PEG shell, which showed increased stability and drug loading capacity, as well as slower degradation (Fig. 5C).93 More recently, the same group reported micelles based on dumbbell-shaped copolymers by linking two PCL cycles with a linear PEG chain for drug delivery.94
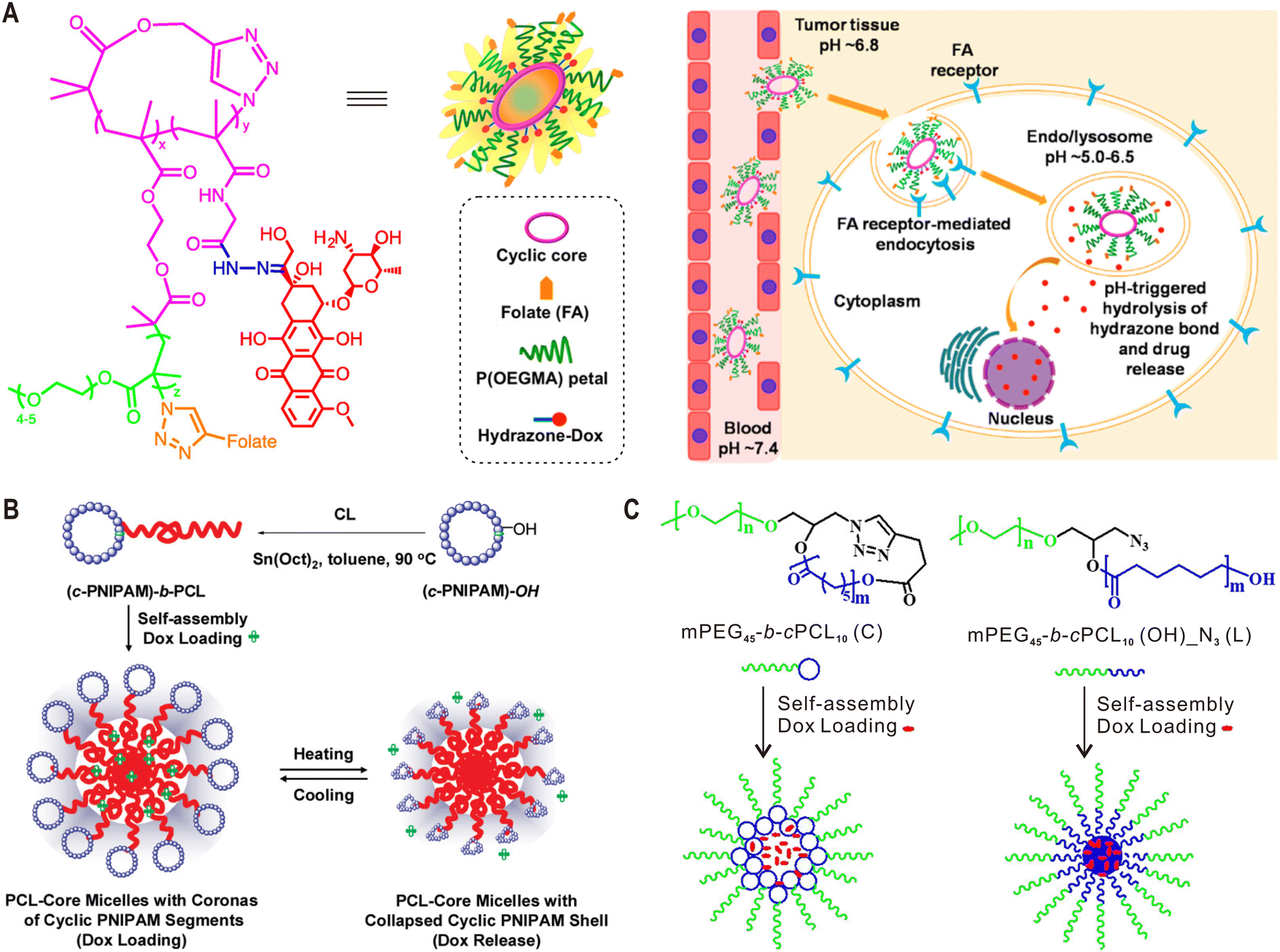 | ||
| Fig. 5 Cyclic polymers for drug delivery. (A) Chemical structure of folate-functionalized sunflower polymer–Dox conjugate and schematic for its folate receptor-mediated endocytosis and pH-triggered drug release. (B) Self-assembly of tadpole-shaped linear-cyclic diblock copolymers into micelles with a cyclic PNIPAM shell for temperature-controlled drug delivery. (C) Self-assembly of tadpole-shaped linear-cyclic diblock copolymers into micelles with a cyclic PCL core for controlled drug delivery. Copyright 2016, 2011 and 2019. Reproduced with permission from the American Chemical Society.91–93 | ||
Gene transfection is another promising area of application for cyclic polymers. In this regard, Pun et al. reported that cyclic poly[(2-dimethylamino)ethyl methacrylate]s (PDMAEMAs) could form more compact complexes with plasmid DNA than their linear analogues.95 While similar gene transfection efficiencies to mammalian cells were achieved for both linear and cyclic polymers, the latter displayed lower cytotoxicity. The same group then prepared cyclic brush polymers with PDMAEMA side chains as gene transfer vehicles, which showed higher efficiencies than the linear brush analogues in both in vitro and in vivo studies.96 Grayson et al. synthesized cyclic poly(ethylene imine) (PEI) and investigated its gene delivery activity by comparing to linear PEI.97 For cyclic polymers with four different molecular weights and N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratios, higher transfection efficiencies were recorded when compared to the corresponding linear PEIs. In addition, cyclic PEIs also showed lower toxicity than the 25 kDa branched PEI, a gold standard in gene delivery. Recently, Zhou et al. also developed cyclic poly(β-amino ester)s, which exhibited superior gene transfer performance than their linear counterparts.98
:P ratios, higher transfection efficiencies were recorded when compared to the corresponding linear PEIs. In addition, cyclic PEIs also showed lower toxicity than the 25 kDa branched PEI, a gold standard in gene delivery. Recently, Zhou et al. also developed cyclic poly(β-amino ester)s, which exhibited superior gene transfer performance than their linear counterparts.98
4.2 Surface functionalization
When linear polymers are grafted onto a surface, the chain ends in the outer layer determine many physicochemical properties of the formed polymer brushes such as antifouling and frictional properties. Cyclic polymers have no chain ends and it will undoubtedly have a significant impact on these properties. However, the interfacial topological effects of cyclic macromolecules have only been explored recently.79,99 In 2016, Benetti and coworkers grafted linear and cyclic poly(2-ethyl-2-oxazoline)s (PEOXAs) chains on titanium oxide surfaces and found that cyclic PEOXAs could be adsorbed faster and form denser brushes than their linear anologues.99 Further measurements by colloidal-probe AFM indicate an increased steric stabilization provided by the denser cyclic brushes. Importantly, this topological characteristic improved its resistance against different proteins. In addition, due to the absence of chain ends and reduced interdigitation, these cyclic brushes exhibited increased lubricity when two polymer brush-modified surfaces were sheared against each other.79The same strategy was soon applied for the surface functionalization of superparamagnetic Fe3O4 nanoparticles (NPs).100 In comparison to planar surfaces, the high curvature of NPs further amplifies the topological effect of cyclic polymers, allowing the formation of highly dense and compact PEOXA brush shells on nanoparticle surface (Fig. 6A). After storage in ultra-pure water or in phosphate buffer saline for two months, the colloidal stability of cyclic brush-coated Fe3O4 NPs was found to be much better than those coated with linear PEOXAs of equivalent chain lengths. Moreover, while linear PEOXA-coated NPs could not be redispersed after heating to a temperature above the lower critical solution temperature (LCST) of PEOXA, the aggregation process for cyclic PEOXA-functionalized NPs was fully reversible (Fig. 6B). More importantly, the nonspecific interactions between bovine serum albumin (BSA) and linear brush-coated NPs can be prevented by using cyclic PEOXA brushes (Fig. 6C). In a more detailed study, Reimbult and Benetti et al. demonstrated that cyclic PEOXA brushes could efficiently suppress the formation of a protein corona of the blood plasma protein human serum albumin (HSA).101 Very recently, Yamamoto and coworkers also reported the surface modification of gold NPs with cyclic PEG through physisorption.102 The coated NPs exhibited better dispersion stability during freezing, lyophilization, and heating than corresponding linear PEG system based on the classic gold-thiol chemisorption.
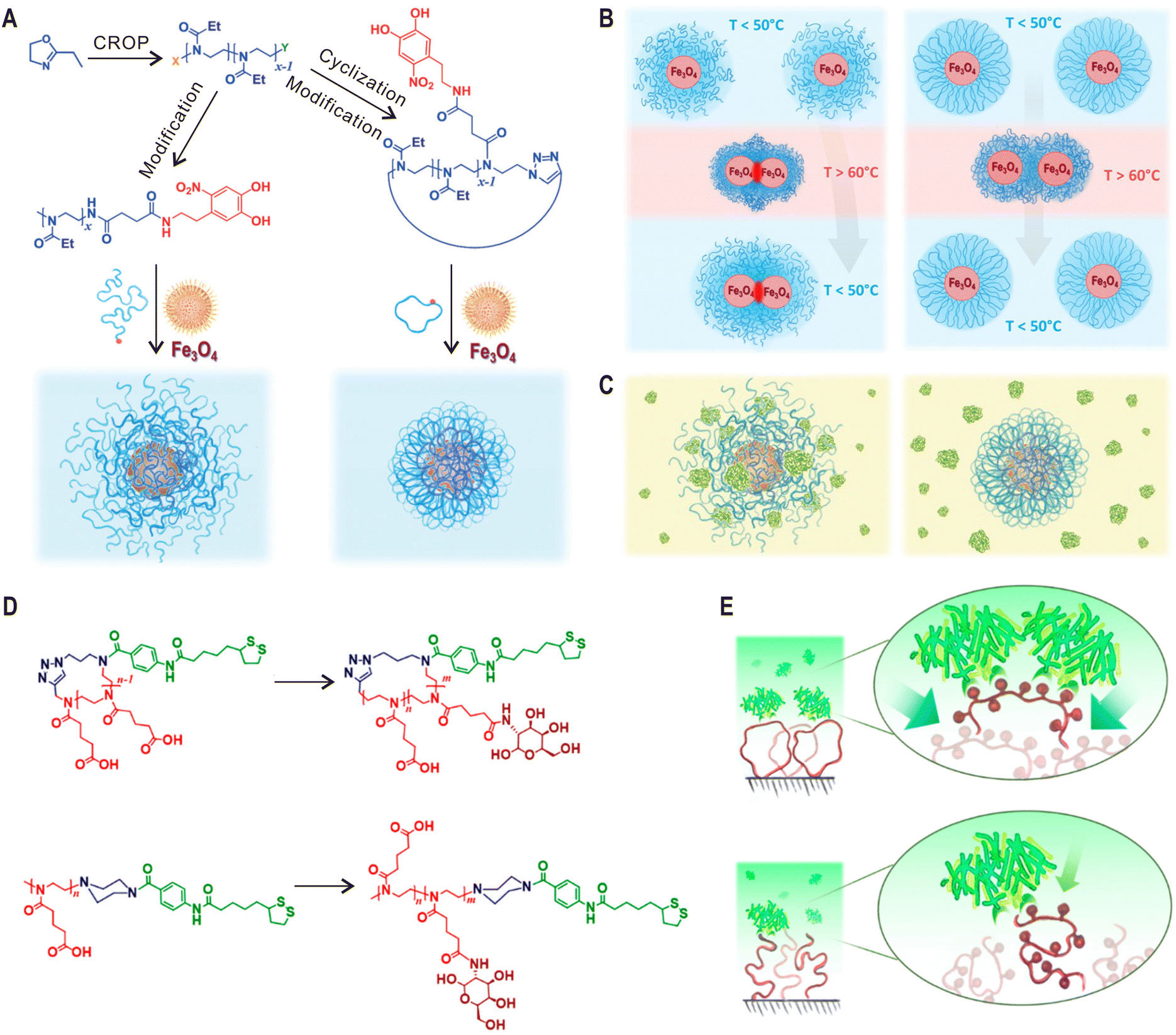 | ||
| Fig. 6 Cyclic polymers for surface functionalization. (A) Synthesis of linear and cyclic PEOXAs by cationic ring opening polymerization (CROP) and post-modification for the surface functionalization of Fe3O4 NPs. (B) Temperature-induced transitions for suspensions of NPs coated with linear (left) and cyclic (right) PEOXAs. (C) Schematics showing the interaction of BSA with linear (left) and cyclic (right) PEOXA brush-coated NPs. (D) Synthesis of cyclic and linear glycopolymers by introducing galactose moieties into to corresponding PCPOXA chains. (E) Functional surfaces modified with cyclic glycopolymers reveal more efficient presentation of functional groups and increased lectin-binding ability. Copyright 2017. Reproduced with permission from John Wiley and Sons Inc.100 Copyright 2020. Reproduced with permission from the American Chemical Society.103 | ||
In the above examples, chemically inert polymers with the cyclic topology were employed for surface modification. However, the topological effects on interfacial physicochemical properties are different if functional groups that interact with surrounding environments are introduced to the polymer backbone. For instance, Benetti and coworkers modified gold surfaces with linear and cyclic poly(2-carboxypropyl-2-oxazoline) (PCPOXA) brushes containing multiple carboxyl groups.103 By comparing their swelling and nanomechanical properties at different pH, amplified response toward pH variation was observed for the cyclic brush-modified surfaces. Furthermore, galactose moieties were introduced to PCPOXA chains, generating cyclic and linear glycopolymers (Fig. 6D). Compared with the linear brush system, the surfaces functionalized with cyclic glycopolymers exhibited 45% higher lectin-binding ability due to their expanded architecture and enhanced exposure of galactose functions (Fig. 6E).103
The above pioneering studies opened a new area by applying cyclic polymers to manipulate the physicochemical properties of various surfaces for practical applications. To protect cartilage from proteolytic enzymes and improve its lubrication properties, cyclic poly(2-methyl-2-oxazoline) (PMOXA) side chains and hydroxyl benzaldehyde (HBA) groups were grafted onto a poly(glutamic acid) (PGA) backbone.104 The obtained PGA-PMOXA-HBA copolymers were then attached to damaged cartilage tissues via the formation of Schiff bases between HBA and the amino groups from the cartilage surface, forming denser and more lubricious cyclic brush films in comparison to corresponding copolymers with linear PMOXA side chains. In addition, the more compact cyclic PMOXA chains and the intrinsic absence of chain ends also brought improved biopassive and tribological properties, showing great potential of cyclic polymers as synthetic lubricants. Wu and coworkers reported that cyclic poly(ionic liquids) grafted on surfaces can provide better antibacterial properties against Gram-negative Escherichia coli than corresponding linear polymers, as a consequence of notably different surface morphologies and higher positive charge density derived from the cyclic polymer architecture.105 In their following study, tadpole-shaped poly(N-hydroxyethylacrylamide) was used for constructing smoother and more compact surface coatings, which exhibited enhanced antifouling properties.106
4.3 Other applications
Apart from therapeutic application and surface functionalization, cyclic polymers have also been explored for other material uses.14 Because of their unique shapes, cyclic polymers grafted with functional side chains can serve as ideal templates for synthesizing donut-shaped nanoparticles. By introducing different diblock copolymers to cyclic poly(norbornene imide) backbones, Zhang and Wu et al. synthesized two cyclic bottlebrush polymers with different functional core domains, which were successfully employed for the templated preparation of donut-shaped nanoparticles with different internal structures including organo-silica cross-linked hybrids and gold nanoclusters.107Polymer rings lack chain ends, which makes them unique building blocks for constructing polymer networks. Tew and coworkers synthesized cyclic poly(5-hydroxy-1-cyclooctene) (PACOE), whose internal double bonds were crosslinked via thiol–ene chemistry to form networks, being the first reported example of gels based on cyclic polymers.108 Interestingly, the gel fraction, swelling ratio, and modulus of the obtained gels increased simultaneously at higher initial polymer concentrations. This is distinctly different from that of conventional gels based on linear PACOE, and it can be ascribed to the presence of cyclic PACOE backbones, which serve as a secondary topological cross-link.108 Recently, Benetti and Zenobi-Wong et al. prepared hydrogels from cyclic poly(2-oxazoline)s, which can incorporate more solvent while demonstrating improved mechanical properties compared with their linear counterparts, showing promise for biological applications such as 3D cell culture and tissue engineering.109
5. Outlook
In summary, we have reviewed recent advances in the synthesis, the resulting properties, self-assembly, and most prominent applications of cyclic polymers. This research field was initiated by scientific curiosity and it has meanwhile evolved from controlled synthesis to advanced materials for biomedical applications and surface engineering.110 Represented by click chemistry-based ring-closure strategies and Grubbs catalyst-mediated ring-expansion methods, substantial progress has been made to synthesize these sophisticated macromolecules in the past two decades, which allowed for the intensive exploration of various topological effects in comparison to their linear analogues. Although major efforts in this area are still devoted to the synthesis of various polymeric rings with controlled structures, an increasing number of studies have been published describing their unique properties and applications. Therefore, we believe that cyclic polymers will play even more important roles in both fundamental research and for practical applications in the future. To fully exploit the great potential of these fascinating macromolecules, several major challenges and future opportunities in this field have been summarized below from our own perspective.First, the large-scale synthesis of cyclic polymers is the basis for their practical application. Most ring-closure methods require high dilution conditions and thus are difficult to scale up. As another mainstream approach, the ring-expansion synthesis holds an obvious advantage in terms of scalability but is only suitable for limited number of special monomers. In addition, these reactions require strict conditions and the design of unique catalysts, which could be limiting for commercialization strategies. Therefore, it will be necessary to revisit different cyclization mechanisms and develop more generalized methods. In this regard, Zhang and coworkers reported the synthesis of 1 g cyclic PS in 3 h by combining the continuous-flow technique with a light-induced Diels–Alder ring-closing reaction.111 It should be mentioned that a large quantity of solvent (17.3 L) was consumed during the process. However, this limitation was later resolved by Barner-Kowollik and Junkers et al., whom employed a looped flow reactor that can reduce the required solvent volume by a factor of 43.112 Based on the Glaser coupling reaction, Shen and Wang also designed a micromixer system for the high-efficiency cyclization of linear PEO.113 In addition, the Zhang group developed the large-scale bimolecular ring-closing reaction between sym-dibenzo-1,5-cyclooctadiene-3,7-diyne (DIBOD) and linear polymer precursors with 2,6-diisopropylaryl-azide terminals connected by para-ether bonds.114 Compared with conventional linear precursors with benzyl azide ends, the reaction rate constants were 12 times larger, which enabled the production of high-purity (>95%) cyclic poly(lactic acid) and cyclic PCL at high productivities of 1.93 g L−1/12 h and 1.5 g L−1/12 h, respectively. Although this modified click reaction can increase the synthetic scale, its cost is another issue that needs careful consideration because DIBOD may involve expensive synthesis.
Second, the efficient purification of cyclic polymers is of paramount importance for studying their topological effects. It is well-known that even a tiny fraction of linear impurities may largely alter their rheological properties and self-assembly behaviors. Moreover, the rapid purification can also help solve the scalability issue. In this regard, there are two major challenges: (1) quantification of the purity of cyclic products; and (2) elimination of impurities. To address the first challenge, results generated by some conventional characterization techniques such as NMR, GPC, and Fourier-transform infrared spectroscopy are often unsatisfactory due to the small structural differences between cyclic and linear samples.16 However, mass spectrometry (especially MALDI-ToF) has proven to be a powerful tool for many polymer systems.54,115 For example, Memboeuf and Gerbaux et al. reported a tandem mass spectrometry method to detect the unreacted linear residual (<5%) in a cyclic poly(L-lactide) sample synthesized by CuAAC cyclization.116 To overcome the second challenge, some commonly-used technologies include preparative GPC34,72 and high performance liquid chromatography.117 In addition, the application of functional resins that can interact with the end groups of linear impurities is another effective strategy for the purification of cyclic polymer samples.115,118 However, these methods also have limitations such as low separation efficiency or they are only suitable for polymers with specific backbones or functional end groups. Therefore, additional efforts should be devoted to developing new materials and techniques for the efficient separation of different cyclic polymers. It is great to see that some progress has been made very recently. Uemura and Hosono et al. demonstrated that metal-organic frameworks (MOFs) with tunable pore structures can isolate cyclic and linear PEGs.119 After passing through an MOF-based liquid chromatography system, gram-scale cyclic PEGs with an ∼100% cyclic purity were obtained in high yield (83%) due to the selective intercalation of linear chains into MOF nanopores. Similarly, Barroso-Bujans et al. also observed that cyclic PEG chains have slower melt intercalation rates in graphite oxide (GO) than their linear counterparts.120 Especially when the pristine GO was pillared with 1 wt% of 1,6-hexanediamine, the topological effect can be dramatically amplified, showing great potential of GO-based materials for the purification of cyclic polymers.
Third, the programmable folding and assembly of cyclic polymers can provide valuable insights to help understand the complex structure of biomolecules and design new materials. It is well known that biomolecules such as peptides, proteins and DNA have precise 3D folded structures, which are crucial for performing their unique biofunctions. As introduced in Section 3.2, our recent attempts have shown that even simple cyclic homopolymers and cyclic brush polymers can assemble into controlled wormlike and hierarchical structures.32 Therefore, the fabrication of complex multicyclic polymers and hierarchical assemblies via controlled folding and assembly can not only serve as simple yet effective means to mimic those 3D bio-structures, but also cumulate important experience for making the next generation of bioinspired soft materials with increased complexity.
Finally, the expansion of applications for cyclic polymers will undoubtedly be the focus of future research. Remarkable synthetic advances in the past 20 years have made it possible to synthesize a variety of cyclic polymers with well-defined structures. Past studies on their applications in the biomedical field and surface functionalization already support the importance of topology for different uses. We believe that the application of cyclic polymers is still in its infancy and cyclic polymers offer numerous opportunities in various fields including nanomedicine, surface engineering, and marine biofouling.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Max Planck-Bristol Centre for Minimal Biology. C. J. Chen is grateful for a Walter Benjamin Fellowship funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 453265186. Open Access funding provided by the Max Planck Society.References
- F. Jacob and E. L. Wollman, C. R. Hebd. Seances Acad. Sci., 1958, 247, 154–156 CAS
.
- D. Freifelder, R. L. Sinsheimer and A. K. Kleinschmidt, Science, 1964, 146, 254–255 CrossRef CAS PubMed
- A. C. Conibear and D. J. Craik, Angew. Chem., Int. Ed., 2014, 53, 10612–10623 CrossRef CAS PubMed
- D. J. Craik, Science, 2006, 311, 1563–1564 CrossRef PubMed
- G. D. Sprott, J. Bioenerg. Biomembr., 1992, 24, 555–566 CrossRef CAS PubMed
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC
- H. R. Kricheldorf, J. Polym. Sci., Polym. Chem., 2010, 48, 251–284 CrossRef CAS
- J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406–6413 CrossRef CAS
- N. Zaldua, R. Lienard, T. Josse, M. Zubitur and A. Mugica,
et al.
, Macromolecules, 2018, 51, 1718–1732 CrossRef CAS
- M. D. Hossain, D. R. Lu, Z. F. Jia and M. J. Monteiro, ACS Macro Lett., 2014, 3, 1254–1257 CrossRef CAS PubMed
- M. Kapnistos, M. Lang, D. Vlassopoulos, W. Pyckhout-Hintzen and D. Richter,
et al.
, Nat. Mater., 2008, 7, 997–1002 CrossRef CAS PubMed
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed
- R. J. Williams, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2015, 6, 2998–3008 RSC
- M. Romio, L. Trachsel, G. Morgese, S. N. Ramakrishna and N. D. Spencer,
et al.
, ACS Macro Lett., 2020, 9, 1024–1033 CrossRef CAS PubMed
- B. Golba, E. M. Benetti and B. G. De Geest, Biomaterials, 2021, 267, 120468 CrossRef CAS PubMed
- R. Lienard, J. De Winter and O. Coulembier, J. Polym. Sci., 2020, 58, 1481–1502 CrossRef CAS
- D. Richter, S. Goossen and A. Wischnewski, Soft Matter, 2015, 11, 8535–8549 RSC
- R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray and T. D. W. Claridge,
et al.
, Nature, 2011, 469, 72–75 CrossRef PubMed
- D. Aoki, G. Aibara, S. Uchida and T. Takata, J. Am. Chem. Soc., 2017, 139, 6791–6794 CrossRef CAS PubMed
- D. Geiser and H. Hocker, Macromolecules, 1980, 13, 653–656 CrossRef CAS
- H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka and A. Uchibori,
et al.
, J. Am. Chem. Soc., 2000, 122, 9592–9599 CrossRef CAS
- P. Sun, J. Q. Chen, J. A. Liu and K. Zhang, Macromolecules, 2017, 50, 1463–1472 CrossRef CAS
- K. S. Lee and G. Wegner, Makromol. Chem., Rapid Commun., 1985, 6, 203–208 CrossRef CAS
- M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2011, 133, 1630–1633 CrossRef CAS PubMed
- T. Yamamoto, S. Yagyu and Y. Tezuka, J. Am. Chem. Soc., 2016, 138, 3904–3911 CrossRef CAS PubMed
- Z. W. Ji, Y. P. Li, Y. Ding, G. S. Chen and M. Jiang, Polym. Chem., 2015, 6, 6880–6884 RSC
- S. Honda, M. Oka, H. Takagi and T. Toyota, Angew. Chem., Int. Ed., 2019, 58, 144–148 CrossRef CAS PubMed
- T. Josse, J. De Winter, P. Gerbaux and O. Coulembier, Angew. Chem., Int. Ed., 2016, 55, 13944–13958 CrossRef CAS PubMed
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed
- C. J. Chen, M. K. Singh, K. Wunderlich, S. Harvey and C. J. Whitfield,
et al.
, Nat. Commun., 2021, 12, 3959 CrossRef CAS PubMed
- Z. S. Ge, Y. M. Zhou, J. Xu, H. W. Liu and D. Y. Chen,
et al.
, J. Am. Chem. Soc., 2009, 131, 1628–1629 CrossRef CAS PubMed
- M. B. Koo, S. W. Lee, J. M. Lee and K. T. Kim, J. Am. Chem. Soc., 2020, 142, 14028–14032 CrossRef CAS PubMed
- D. M. Eugene and S. M. Grayson, Macromolecules, 2008, 41, 5082–5084 CrossRef CAS
- H. Jacobson and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1600–1606 CrossRef CAS
- D. E. Lonsdale, C. A. Bell and M. J. Monteiro, Macromolecules, 2010, 43, 3331–3339 CrossRef CAS
- Y. Inoue, P. Kuad, Y. Okumura, Y. Takashima and H. Yamaguchi,
et al.
, J. Am. Chem. Soc., 2007, 129, 6396–6397 CrossRef CAS PubMed
- Y. J. Zhao, S. S. Zhu, C. Liao, Y. Wang and J. W. Y. Lam,
et al.
, Angew. Chem., Int. Ed., 2021, 60, 16974–16979 CrossRef CAS PubMed
- H. R. Kricheldorf and S. R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS
- C. W. Bielawski, D. Benitez and R. H. Grubbs, Science, 2002, 297, 2041–2044 CrossRef CAS PubMed
- Y. A. Chang and R. M. Waymouth, J. Polym. Sci., Polym. Chem., 2017, 55, 2892–2902 CrossRef CAS
- S. A. Gonsales, T. Kubo, M. K. Flint, K. A. Abboud and B. S. Sumerlin,
et al.
, J. Am. Chem. Soc., 2016, 138, 4996–4999 CrossRef CAS PubMed
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Nat. Chem., 2016, 8, 791–796 CrossRef CAS PubMed
- Z. H. Miao, S. A. Gonsales, C. Ehm, F. Mentink-Vigier and C. R. Bowers,
et al.
, Nat. Chem., 2021, 13, 792–799 CrossRef CAS PubMed
- U. Mandal, I. Ghiviriga, K. A. Abboud, D. W. Lester and A. S. Veige, J. Am. Chem. Soc., 2021, 143, 17276–17283 CrossRef CAS PubMed
- T. W. Wang, P. R. Huang, J. L. Chow, W. Kaminsky and M. R. Golder, J. Am. Chem. Soc., 2021, 143, 7314–7319 CrossRef CAS PubMed
- D. A. Culkin, W. H. Jeong, S. Csihony, E. D. Gomez and N. R. Balsara,
et al.
, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS PubMed
- L. Guo and D. H. Zhang, J. Am. Chem. Soc., 2009, 131, 18072–18074 CrossRef CAS PubMed
- L. Guo, S. H. Lahasky, K. Ghale and D. H. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS PubMed
- N. S. Jiang, J. X. Chen, T. Y. Yu, A. Chao and L. Y. Kang,
et al.
, Macromolecules, 2020, 53, 7601–7612 CrossRef CAS PubMed
- A. Li, L. Lu, X. Li, L. L. He and C. Do,
et al.
, Macromolecules, 2016, 49, 1163–1171 CrossRef CAS
- P. Salas-Ambrosio, A. Tronnet, M. Since, S. Bourgeade-Delmas and J. L. Stigliani,
et al.
, J. Am. Chem. Soc., 2021, 143, 3697–3702 CrossRef CAS PubMed
- F. M. Haque, C. M. Schexnayder, J. M. Matxain, F. Barroso-Bujans and S. M. Grayson, Macromolecules, 2019, 52, 6369–6381 CrossRef CAS
- H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed
- R. Takashima, D. Aoki and H. Otsuka, Macromolecules, 2020, 53, 4670–4677 CrossRef CAS
- N. Kusuyama, Y. Daito, H. Kubota, Y. Kametani and M. Ouchi, Polym. Chem., 2021, 12, 2532–2541 RSC
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075–17080 CrossRef CAS PubMed
- D. V. Kondratuk, L. M. A. Perdigao, M. C. O. Sullivan, S. Svatek and G. Smith,
et al.
, Angew. Chem., Int. Ed., 2012, 51, 6696–6699 CrossRef CAS PubMed
- D. V. Kondratuk, L. A. Perdigao, A. M. S. Esmail, J. N. O'Shea and P. H. Beton,
et al.
, Nat. Chem., 2015, 7, 317–322 CrossRef CAS PubMed
- J. Chen, H. F. Li, H. C. Zhang, X. J. Liao and H. J. Han,
et al.
, Nat. Commun., 2018, 9, 5310 CrossRef CAS PubMed
- B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J. F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS PubMed
- D. Pal, Z. H. Miao, J. B. Garrison, A. S. Veige and B. S. Sumerlin, Macromolecules, 2020, 53, 9717–9724 CrossRef CAS
- Y. Tezuka, Acc. Chem. Res., 2017, 50, 2661–2672 CrossRef CAS PubMed
- T. Suzuki, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2014, 136, 10148–10155 CrossRef CAS PubMed
- K. Kyoda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2019, 141, 7526–7536 CrossRef CAS PubMed
- C. Liu, Y. Y. Fei, H. L. Zhang, C. Y. Pan and C. Y. Hong, Macromolecules, 2019, 52, 176–184 CrossRef CAS
- Y. X. Zhang, Y. Wu, Y. L. Zhao, L. C. Zhang and K. Zhang, Macromolecules, 2021, 54, 6901–6910 CrossRef CAS
- Z. Zhang, X. Nie, F. Wang, G. Chen and W. Q. Huang,
et al.
, Nat. Commun., 2020, 11, 3654 CrossRef CAS PubMed
- M. Duval, P. Lutz and C. Strazielle, Makromol. Chem., Rapid Commun., 1985, 6, 71–76 CrossRef CAS
- G. Hadziioannou, P. M. Cotts, G. Tenbrinke, C. C. Han and P. Lutz,
et al.
, Macromolecules, 1987, 20, 493–497 CrossRef CAS
- K. Dodgson, D. Sympson and J. A. Semlyen, Polymer, 1978, 19, 1285–1289 CrossRef CAS
- H. Kammiyada, M. Ouchi and M. Sawamoto, Macromolecules, 2017, 50, 841–848 CrossRef CAS
- R. J. Williams, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Macromolecules, 2016, 49, 2802–2813 CrossRef CAS PubMed
- M. E. Cordova, A. T. Lorenzo, A. J. Muller, J. N. Hoskins and S. M. Grayson, Macromolecules, 2011, 44, 1742–1746 CrossRef CAS
- H. Y. Xiao, C. F. Luo, D. D. Yan and J. U. Sommer, Macromolecules, 2017, 50, 9796–9806 CrossRef CAS
- J. D. Halverson, G. S. Grest, A. Y. Grosberg and K. Kremer, Phys. Rev. Lett., 2012, 108, 038301 CrossRef PubMed
- Y. Jeong, Y. Jin, T. Chang, F. Uhlik and J. Roovers, Macromolecules, 2017, 50, 7770–7776 CrossRef CAS
- M. Divandari, G. Morgese, L. Trachsel, M. Romio and E. S. Dehghani,
et al.
, Macromolecules, 2017, 50, 7760–7769 CrossRef CAS
- E. Minatti, P. Viville, R. Borsali, M. Schappacher and A. Deffieux,
et al.
, Macromolecules, 2003, 36, 4125–4133 CrossRef CAS
- B. Y. Zhang, H. Zhang, Y. J. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845–848 CrossRef CAS PubMed
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251–10253 CrossRef CAS PubMed
- S. Honda, T. Yamamoto and Y. Tezuka, Nat. Commun., 2013, 4, 1574 CrossRef PubMed
- J. M. Ren, K. Satoh, T. K. Goh, A. Blencowe and K. Nagai,
et al.
, Angew. Chem., Int. Ed., 2014, 53, 459–464 CrossRef CAS PubMed
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512–1515 CrossRef CAS PubMed
-
C. J. Chen, Unfolding of Natural Macromolecules and Folding of Synthetic Polymers: Bioinspired Strategies for Constructing Precision Nanomaterials, PhD thesis, Universität Ulm, Ulm, 2020 Search PubMed
- J. E. Poelma, K. Ono, D. Miyajima, T. Aida and K. Satoh,
et al.
, ACS Nano, 2012, 6, 10845–10854 CrossRef CAS PubMed
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox and J. M. J. Frechet,
et al.
, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed
- B. Chen, K. Jerger, J. M. J. Frechet and F. C. Szoka, J. Controlled Release, 2009, 140, 203–209 CrossRef CAS PubMed
- C. F. Yin, R. N. Wang, Y. Sun, S. Li and X. K. Zhang,
et al.
, Nano Today, 2021, 41, 101293 CrossRef CAS
- C. E. Wang, H. Wei, N. Tan, A. J. Boydston and S. H. Pun, Biomacromolecules, 2016, 17, 69–75 CrossRef CAS PubMed
- X. J. Wan, T. Liu and S. Y. Liu, Biomacromolecules, 2011, 12, 1146–1154 CrossRef CAS PubMed
- G. Y. Kang, L. Sun, Y. P. Liu, C. Meng and W. Ma,
et al.
, Langmuir, 2019, 35, 12509–12517 CrossRef CAS PubMed
- G. Y. Kang, Y. P. Liu, L. X. Li, L. Sun and W. Ma,
et al.
, Chem. Commun., 2020, 56, 3003–3006 RSC
- H. Wei, D. S. H. Chu, J. Zhao, J. A. Pahang and S. H. Pun, ACS Macro Lett., 2013, 2, 1047–1050 CrossRef CAS PubMed
- Y. L. Cheng, H. Wei, J. K. Y. Tan, D. J. Peeler and D. O. Maris,
et al.
, Small, 2016, 12, 2750–2758 CrossRef CAS PubMed
- M. A. Cortez, W. T. Godbey, Y. L. Fang, M. E. Payne and B. J. Cafferty,
et al.
, J. Am. Chem. Soc., 2015, 137, 6541–6549 CrossRef CAS PubMed
- C. F. Wang, X. B. Huang, L. T. Sun, Q. X. Li and Z. L. Li,
et al.
, Chem. Commun., 2022, 58, 2136–2139 RSC
- G. Morgese, L. Trachsel, M. Romio, M. Divandari and S. N. Ramakrishna,
et al.
, Angew. Chem., Int. Ed., 2016, 55, 15583–15588 CrossRef CAS PubMed
- G. Morgese, B. S. Shaghasemi, V. Causin, M. Zenobi-Wong and S. N. Ramakrishna,
et al.
, Angew. Chem., Int. Ed., 2017, 56, 4507–4511 CrossRef CAS PubMed
- M. Schroffenegger, N. S. Leitner, G. Morgese, S. N. Ramakrishna and M. Willinger,
et al.
, ACS Nano, 2020, 14, 12708–12718 CrossRef CAS PubMed
- Y. B. Wang, J. E. Q. Quinsaat, T. Ono, M. Maeki and M. Tokeshi,
et al.
, Nat. Commun., 2020, 11, 6089 CrossRef CAS PubMed
- L. Trachsel, M. Romio, B. Grob, M. Zenobi-Wong and N. D. Spencer,
et al.
, ACS Nano, 2020, 14, 10054–10067 CrossRef CAS PubMed
- G. Morgese, E. Cavalli, J. G. Rosenboom, M. Zenobi-Wong and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 1621–1626 CrossRef CAS PubMed
- W. Y. Liu, Y. S. Dong, S. J. Liu, T. Wei and Z. Q. Wu,
et al.
, Macromol. Rapid Commun., 2019, 40, 1900379 CrossRef CAS PubMed
- Y. P. Cao, S. J. Liu, Z. Q. Wu and H. Chen, J. Mater. Chem. B, 2021, 9, 2877–2884 RSC
- L. F. Xiao, L. Qu, W. Zhu, Y. Wu and Z. P. Liu,
et al.
, Macromolecules, 2017, 50, 6762–6770 CrossRef CAS
- K. Zhang, M. A. Lackey, J. Cui and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 4140–4148 CrossRef CAS PubMed
- L. Trachsel, M. Romio, M. Zenobi-Wong and E. M. Benetti, Macromol. Rapid Commun., 2021, 42, 2000658 CrossRef CAS PubMed
- B. Verbraeken and R. Hoogenboom, Angew. Chem., Int. Ed., 2017, 56, 7034–7036 CrossRef CAS PubMed
- P. Sun, J. A. Liu, Z. B. Zhang and K. Zhang, Polym. Chem., 2016, 7, 2239–2244 RSC
- E. Baeten, M. Rubens, K. N. R. Wuest, C. Barner-Kowollik and T. Junkers, React. Chem. Eng., 2017, 2, 826–829 RSC
- H. Y. Shen and G. W. Wang, Polym. Chem., 2017, 8, 5554–5560 RSC
- L. C. Zhang, Y. Wu, S. M. Li, Y. X. Zhang and K. Zhang, Macromolecules, 2020, 53, 8621–8630 CrossRef CAS
- F. M. Haque, A. Alegria, S. M. Grayson and F. Barroso-Bujans, Macromolecules, 2017, 50, 1870–1881 CrossRef CAS
- T. Josse, J. De Winter, P. Dubois, O. Coulembier and P. Gerbaux,
et al.
, Polym. Chem., 2015, 6, 64–69 RSC
- H. C. Lee, H. Lee, W. Lee, T. H. Chang and J. Roovers, Macromolecules, 2000, 33, 8119–8121 CrossRef CAS
- C. H. Hovelmarin, S. Goossen and J. Angaier, Macromolecules, 2017, 50, 4169–4179 CrossRef
- T. Sawayama, Y. B. Wang, T. Watanabe, M. Takayanagi and T. Yamamoto,
et al.
, Angew. Chem., Int. Ed., 2021, 60, 11830–11834 CrossRef CAS PubMed
- F. Barroso-Bujans, J. Allgaier and A. Alegria, Macromolecules, 2020, 53, 406–416 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2022 |