DOI:
10.1039/D2MA00322H
(Paper)
Mater. Adv., 2022,
3, 5857-5870
Paramagnetic ultrasmall Ho2O3 and Tm2O3 nanoparticles: characterization of r2 values and in vivo T2 MR images at a 3.0 T MR field
Received
21st March 2022
, Accepted 20th May 2022
First published on 23rd May 2022
Abstract
Paramagnetic ultrasmall Ho2O3 and Tm2O3 nanoparticles (davg = ∼2.1 nm) grafted with various hydrophilic and biocompatible ligands such as poly(ethylene glycol) diacid (Mn = 250 and 600 amu) and polyacrylic acid (Mw = 1800 amu) were synthesized via a one-pot polyol method. Appreciable transverse (r2) and negligible longitudinal (r1) water proton spin relaxivity values were observed for all nanoparticle samples. The r2 values increased with increasing nanoparticle magnetic moment and decreased with increasing ligand size. Owing to the aforementioned r1 and r2 values, the nanoparticle samples exhibited appreciable negative contrast enhancements in in vivo T2 magnetic resonance (MR) images at a 3.0 T MR field after intravenous injection, demonstrating their potential as efficient T2 MRI contrast agents.
Introduction
Nowadays, magnetic resonance imaging (MRI) is the most commonly used technique in diagnosing diseases.1–4 MRI contrast agents are commonly intravenously injected to improve the sensitivity and resolution in MR images via contrast enhancements.5–13 MRI contrast agents are classified into T1 and T2 MRI contrast agents.10,11T1 MRI contrast agents significantly reduce longitudinal (T1) water proton spin relaxation times in the tissue, making MR images brighter (positive contrast),10,11 whereas T2 MRI contrast agents significantly reduce transverse (T2) water proton spin relaxation times in the tissue, making MR images darker (negative contrast).10,11 At present, molecular Gd-chelates as T1 MRI contrast agents have gained wide market applications because of their good contrasts and rapid excretion via the renal system within a few hours after intravenous injection.14–17 Conversely, most of the clinically approved dextran and carbohydrate-coated superparamagnetic iron oxide (Fe3O4) nanoparticles (SPIONs) as T2 MRI contrast agents18–25 are withdrawn from the market due to their lack of clinical users.18 They had been developed for liver, spleen, and lymph node imaging18–25 and are excretable via the hepatobiliary system due to their nanosizes (>3 nm).26 They have shown drawbacks, such as side effects (i.e., back pains) and less efficiency than Gd-chelates.18 Therefore, it is challenging to develop a new class of T2 MRI contrast agents made of ultrasmall nanoparticles (< 3 nm), which are excretable via the renal system27–29 like molecular agents.
The ability of nanoparticles to induce T1 and T2 water proton spin relaxations highly depends on the electron magnetic moments (j = ![[small script l]](https://www.rsc.org/images/entities/i_char_e146.gif) + s) of metal ion consisting nanoparticles5,6 where j represents the total electron magnetic moment,
+ s) of metal ion consisting nanoparticles5,6 where j represents the total electron magnetic moment, ![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) represents the orbital component, and s represents the spin component. According to the inner and outer sphere models,5,6 nanoparticles can significantly induce both T1 and T2 water proton spin relaxations if = 0, whereas they can exclusively induce only T2 water proton spin relaxations with negligible induction of T1 water proton spin relaxations if ≠ 0, corresponding to efficient T2 MRI contrast agents. Former examples of nanoparticle contrast agents are those consisting of Fe3+ (s = 5/2), Mn2+ (s = 5/2), and Gd3+ (s = 7/2). Gd2O3 nanoparticles have the highest T1 induction and their T2/T1 induction ratio is closest to one,30,31 making them the most powerful T1 MRI contrast agents among the nanoparticle contrast agents. Later examples of nanoparticle contrast agents include Ln2O3 nanoparticles (Ln = Dy, Ho, and Tb)32–37 and CoO nanoparticles38 because of the nonzero of 4f-electrons in Ln3+ and 3d-electrons in Co2+. The SPIONs can significantly induce T2 water proton spin relaxations with appreciable induction of T1 water proton spin relaxations18–25 because they are composed of the following two types of metal ions: Fe2+ ( = 2, s = 2, j = 4) and Fe3+ (j = s = 5/2), corresponding to an intermediate example between the former and latter examples. Notably, magnetic moments of Ln2O3 nanoparticles are nearly particle size-independent because of the compact 4f-electrons in Ln3+, which are nearly unaffected by surface-coating ligands as can be noticed from their small energy splitting (∼100 cm−1) by external factors.39 In contrast, 3d-transition metal oxide nanoparticles have size-dependent magnetic moments and relaxivities40,41 because of diffuse 3d-electrons, which are significantly affected by external factors as can be noticed from their large energy splitting by ligands (∼10
represents the orbital component, and s represents the spin component. According to the inner and outer sphere models,5,6 nanoparticles can significantly induce both T1 and T2 water proton spin relaxations if = 0, whereas they can exclusively induce only T2 water proton spin relaxations with negligible induction of T1 water proton spin relaxations if ≠ 0, corresponding to efficient T2 MRI contrast agents. Former examples of nanoparticle contrast agents are those consisting of Fe3+ (s = 5/2), Mn2+ (s = 5/2), and Gd3+ (s = 7/2). Gd2O3 nanoparticles have the highest T1 induction and their T2/T1 induction ratio is closest to one,30,31 making them the most powerful T1 MRI contrast agents among the nanoparticle contrast agents. Later examples of nanoparticle contrast agents include Ln2O3 nanoparticles (Ln = Dy, Ho, and Tb)32–37 and CoO nanoparticles38 because of the nonzero of 4f-electrons in Ln3+ and 3d-electrons in Co2+. The SPIONs can significantly induce T2 water proton spin relaxations with appreciable induction of T1 water proton spin relaxations18–25 because they are composed of the following two types of metal ions: Fe2+ ( = 2, s = 2, j = 4) and Fe3+ (j = s = 5/2), corresponding to an intermediate example between the former and latter examples. Notably, magnetic moments of Ln2O3 nanoparticles are nearly particle size-independent because of the compact 4f-electrons in Ln3+, which are nearly unaffected by surface-coating ligands as can be noticed from their small energy splitting (∼100 cm−1) by external factors.39 In contrast, 3d-transition metal oxide nanoparticles have size-dependent magnetic moments and relaxivities40,41 because of diffuse 3d-electrons, which are significantly affected by external factors as can be noticed from their large energy splitting by ligands (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1).42 This implies that ultrasmall Ln2O3 nanoparticles made of Ln3+ with high j-values and with ≠ 0 can have appreciable magnetic moments at room temperature close to their bulk values, allowing them to have appreciable transverse (r2) and negligible longitudinal (r1) water proton spin relaxivities. This will make them work as a new class of efficient T2 MRI contrast agents, which had been recently demonstrated in ultrasmall Ln2O3 nanoparticles (Ln = Dy, Ho, and Tb), where moderate negative contrasts in in vivo T2 MR images were observed.32,33,35,37
000 cm−1).42 This implies that ultrasmall Ln2O3 nanoparticles made of Ln3+ with high j-values and with ≠ 0 can have appreciable magnetic moments at room temperature close to their bulk values, allowing them to have appreciable transverse (r2) and negligible longitudinal (r1) water proton spin relaxivities. This will make them work as a new class of efficient T2 MRI contrast agents, which had been recently demonstrated in ultrasmall Ln2O3 nanoparticles (Ln = Dy, Ho, and Tb), where moderate negative contrasts in in vivo T2 MR images were observed.32,33,35,37
In addition to the previously described nanoparticle magnetic moments, the T2 contrast in MR images is sensitive to physical factors arising from ligands such as ligand size and hydrophilicity43–46 because these factors can influence the strength of magnetic dipole-dipole interactions between the nanoparticles and water proton spins (because r2 ∝ 1/L3 in which L is the distance between the nanoparticles and water proton spins)5,47 and the amount of water molecules interacting with the nanoparticles, whereas in metal ion-chelates, the T1 contrast in MR images is sensitive to the hydration number which is determined by the types of chelates.5–7,48 Considering the aforementioned ligand physical factors and that ligand coating is essential to make the nanoparticles colloidally stable and biocompatible for in vivo applications, appropriate ligands should be chosen for nanoparticle coating to obtain high r2 values.
Here, we synthesized ultrasmall Ho2O3 and Tm2O3 nanoparticles grafted with various hydrophilic and biocompatible ligands, namely, poly(ethylene glycol) diacid (PEGD) (Mn = 250 and 600 amu) and polyacrylic acid (PAA) (Mw = 1800 amu), and characterized them using various experimental techniques. We explored their potential as efficient T2 MRI contrast agents by measuring r1 and r2 values and in vivo T2 MR images at a 3.0 T MR field.
Results and discussion
Particle diameters
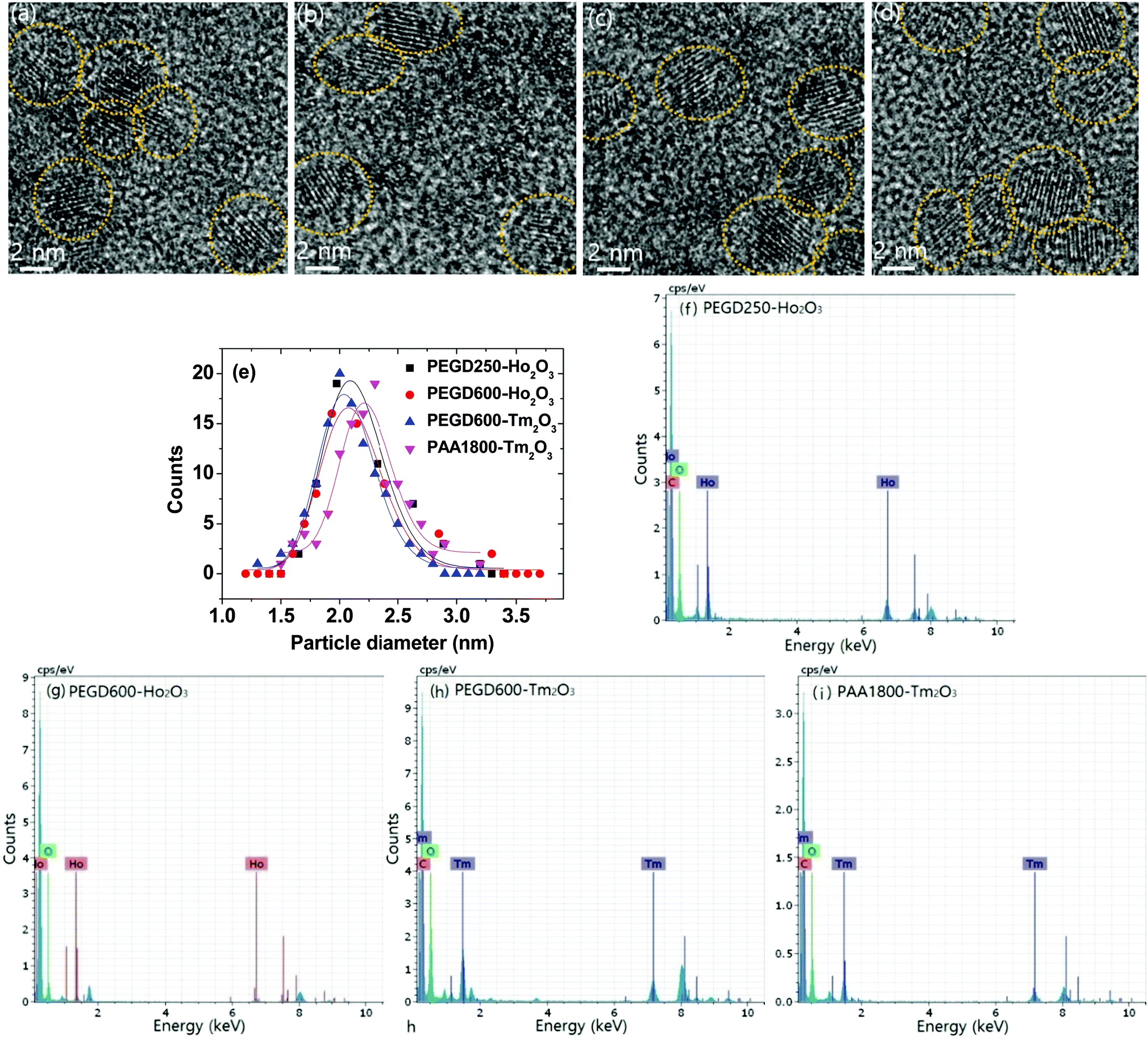
Various hydrophilic and biocompatible ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles were synthesized via a one-pot polyol method. The particle diameters were determined by obtaining high-resolution transmission electron microscope (HRTEM) images (Fig. 1a–d). The particle diameter ranged from 1.0 to 3.0 nm and the average particle diameter (davg) (Table 1) was estimated to be 2.1 nm for both PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles, and 2.1 and 2.2 nm for PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles, respectively, from log-normal function fits to the observed particle diameter distributions (Fig. 1e). The ligand-coated nanoparticles were also confirmed by energy-dispersive X-ray spectroscopy (EDS), where elements such as C, O, Ho, and Tm were strongly detected (Fig. 1f–i). The physicochemical properties of the previously studied PAA1800-coated ultrasmall Ho2O3 nanoparticles (davg = 1.7 nm)35 were added to Table 1 for comparison.
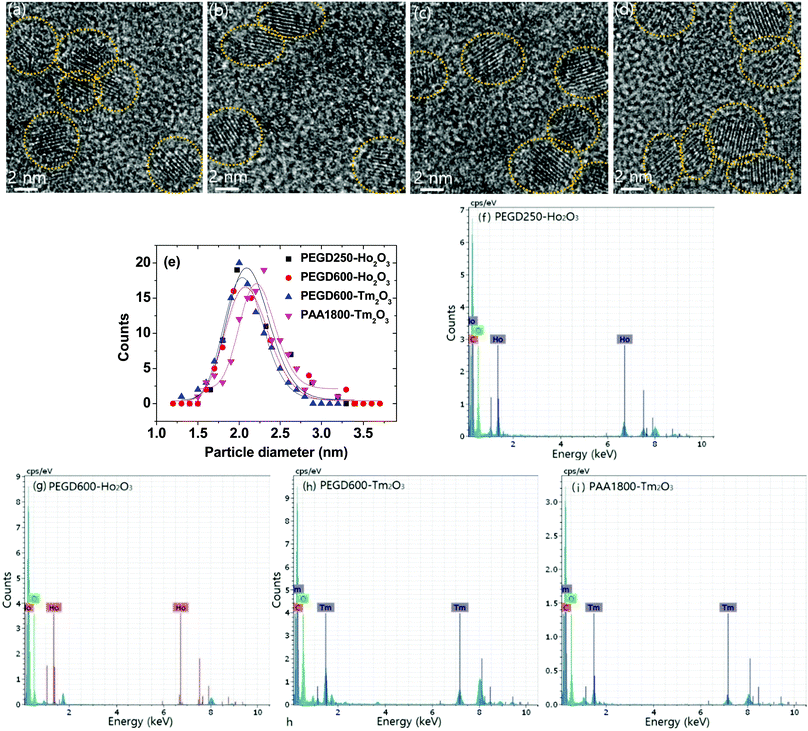 |
| | Fig. 1 HRTEM images of (a) PEGD250- and (b) PEGD600-coated ultrasmall Ho2O3 nanoparticles and (c) PEGD600- and (d) PAA1800-coated ultrasmall Tm2O3 nanoparticles. (e) Log-normal function fits to the observed particle diameter distributions to obtain davgs. EDS spectra of (f) PEGD250- and (g) PEGD600-coated ultrasmall Ho2O3 nanoparticles and (h) PEGD600- and (i) PAA1800-coated ultrasmall Tm2O3 nanoparticles. | |
Table 1 Summary of the observed physicochemical properties of various hydrophilic and biocompatible ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles
| Nanoparticle |
Surface-coating ligand |
d
avg (nm) |
a
avg (nm) |
ζ (mV) |
pHa |
Surface-coating amount |
|
P
(wt%) |
σ
(nm−2) |
N
NP
|
|
pH of nanoparticle suspension samples in aqueous media.
Average amount of ligands coating a nanoparticle (in wt%).
Grafting density, i.e., average number of ligands coating a nanoparticle unit surface area.
Average number of ligands coating a nanoparticle.
Data from ref. 35.
|
| Ho2O3 |
PEGD250 |
2.1 |
8.7 |
10.8 |
∼6.5 |
43.2 |
5.7 |
79 |
| Ho2O3 |
PEGD600 |
2.1 |
13.5 |
14.9 |
∼6.7 |
51.6 |
3.5 |
49 |
| Ho2O3e |
PAA1800 |
1.7 |
12.7 |
−32.9 |
∼9.0 |
45.5 |
0.85 |
7 |
| Tm2O3 |
PEGD600 |
2.1 |
12.0 |
14.7 |
∼6.7 |
59.5 |
4.6 |
64 |
| Tm2O3 |
PAA1800 |
2.2 |
20.6 |
−21.4 |
∼9.0 |
48.4 |
1.1 |
16 |
Hydrodynamic diameters and zeta potentials
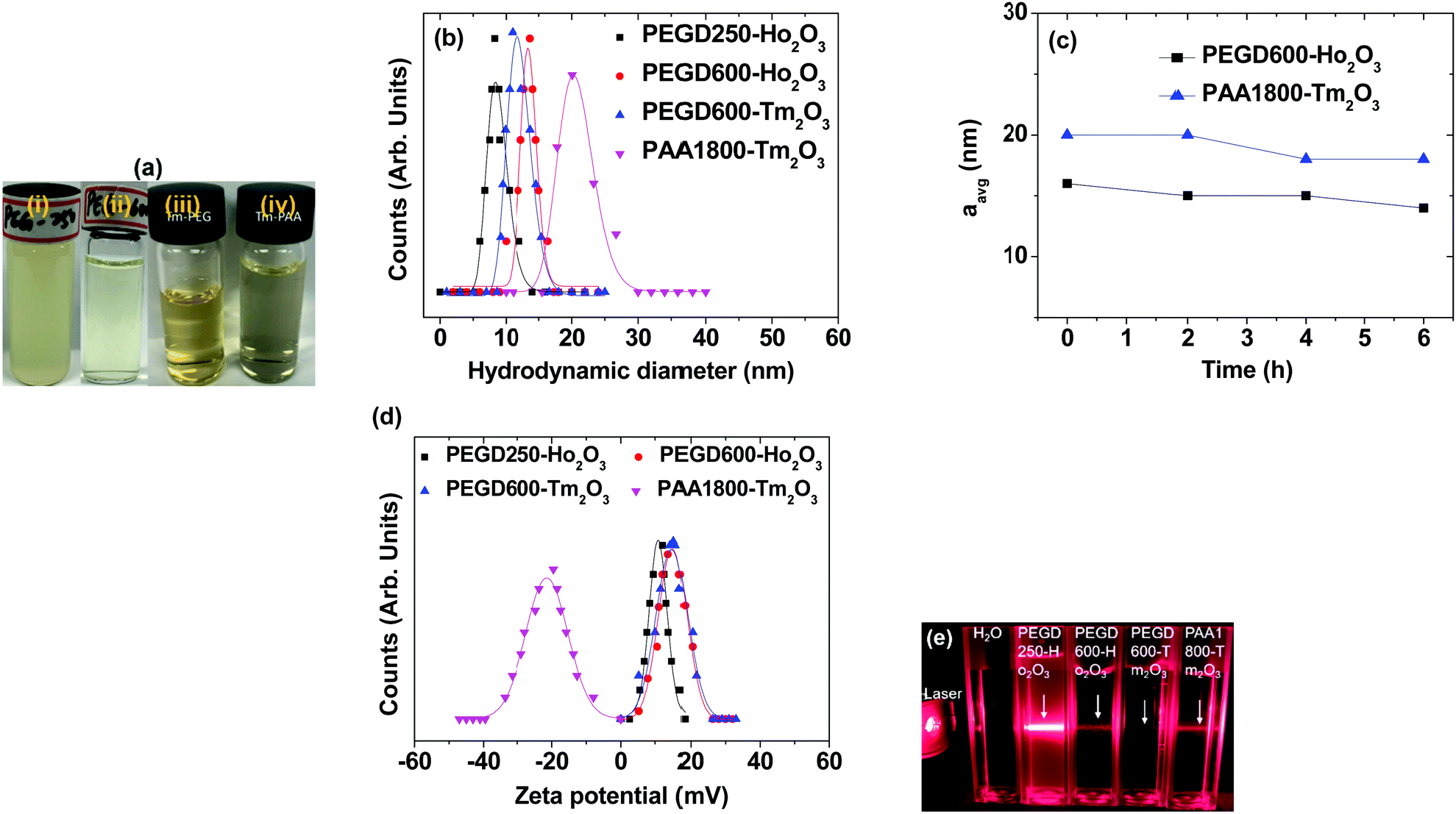
The nanoparticle suspension samples in aqueous media are presented in Fig. 2a. Except for the PEGD250-coated ultrasmall Ho2O3 nanoparticles, all samples exhibited excellent colloidal stability: they did not settle down to the beaker bottom until > 1 year after synthesis, whereas the PEGD250-coated ultrasmall Ho2O3 nanoparticles partially precipitated in a week but were redispersed via shaking. This is likely because PEGD250 is considerably short enough to attract a sufficient amount of water molecules to stabilize the nanoparticle colloids whereas PEGD600 is long enough (approximately four times longer than PEGD250) and each PAA1800 possesses abundant COO− groups (approximately 25 COO− groups per monomer) to attract a sufficient amount of water molecules to stabilize the nanoparticle colloids. The average hydrodynamic diameter (aavg) was estimated to be 8.7 and 13.5 nm for the PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles, respectively, and 12.0 and 20.6 nm for the PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles, respectively, from a log-normal function fits to the observed dynamic light scattering (DLS) patterns (Fig. 2b and Table 1). Notably, the aavg values increased with increasing ligand size, which is likely attributable to an increase in ligand-coating layer thickness and hydration spheres due to the increase in the amount of water molecules attracted by ligands around the nanoparticles with increasing ligand size. The hydrodynamic diameters measured at different times for PEGD600-coated ultrasmall Ho2O3 nanoparticles and PAA1800-coated ultrasmall Tm2O3 nanoparticles (Fig. 2c) showed nearly constant aavg values over time, indicating negligible aggregation between nanoparticles with time, as consistent with their observed good colloidal stability.
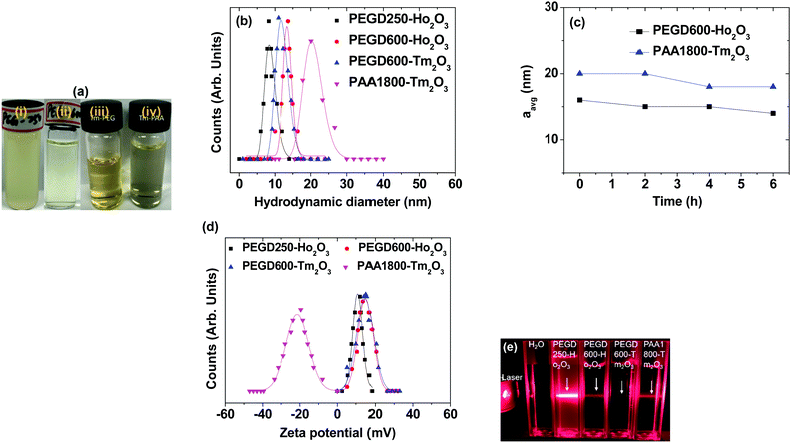 |
| | Fig. 2 (a) Photographs of the nanoparticle suspension samples in aqueous media: (i) PEGD250- and (ii) PEGD600-coated ultrasmall Ho2O3 nanoparticles, (iii) PEGD600- and (iv) PAA1800-coated ultrasmall Tm2O3 nanoparticles. (b) Plots of log-normal function fits to the observed DLS patterns. (c) Plots of aavg values as a function of time (h). (d) Plots of zeta potentials. (e) Tyndall effects confirming nanoparticle colloidal dispersions in aqueous media: arrows indicate laser light scattering by the nanoparticle colloids. | |
The zeta potential of the nanoparticle suspension samples in aqueous media was measured to be 10.4 and 14.5 mV for the PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles, respectively, and 12.7 and −20.2 mV for the PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles, respectively (Fig. 2d and Table 1). The positive zeta potential of the PEGD250- and PEGD600-coated nanoparticles in slightly acidic suspension media (pH = 6.5–6.7) is due to partially protonated oxygens and carboxyl groups of PEGD, thus providing positive values and consistent with previous observations in PEGD600-coated Fe3O4 nanoparticles,49 whereas the negative zeta potential of the PAA1800-coated nanoparticles is due to numerous negative COO− groups of PAA1800 in basic suspension sample (pH = ∼9.0) and consistent with previous observations for nanoparticles grafted with numerous COO− group containing polymers.35,50,51 The nanoparticle colloidal dispersions in aqueous media were confirmed via Tyndall effects, where laser light scattering was observed only for the nanoparticle suspension samples due to collisions between the nanoparticle colloids and laser light whereas it was not observed for triple-distilled water (Fig. 2e).
Crystal structure
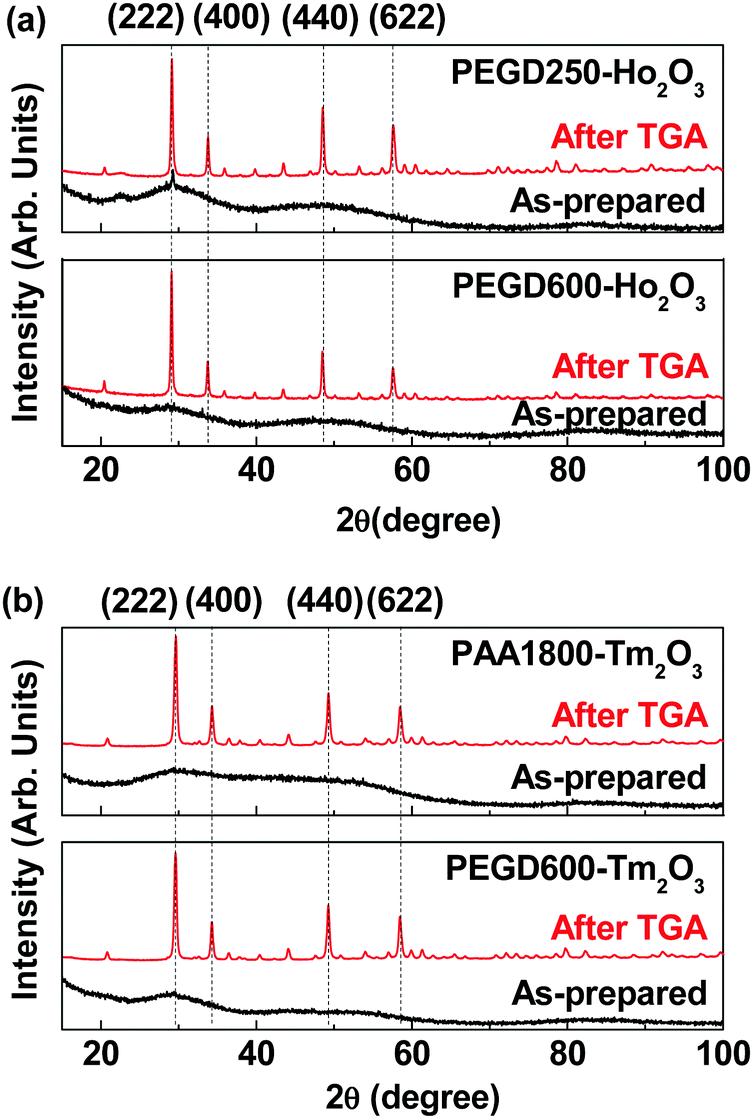
As shown in Fig. 3, X-ray diffraction (XRD) patterns of the as-synthesized nanoparticles showed broad peaks, indicating the amorphous feature of the nanoparticles.52 However, the XRD patterns after thermogravimetric analysis (TGA) displayed sharp peaks, indicating crystallization of the nanoparticles after TGA due to heating up to 900 °C. All peaks after TGA could be assigned with (hkl) Miller indices according to body-centered cubic Ho2O3 and Tm2O3.53,54 The estimated lattice constants after TGA were 10.609 and 10.482 Å for Ho2O3 and Tm2O3 nanoparticles, respectively, which are in good agreement with the reported values of 10.6186 and 10.49 Å,53,54 respectively.
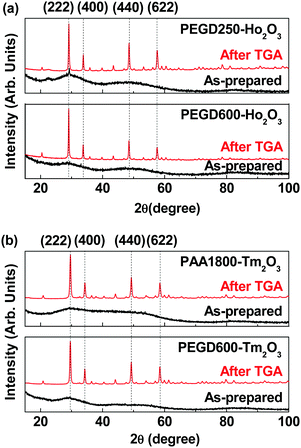 |
| | Fig. 3 XRD patterns of the nanoparticle powder samples before (i.e., as-prepared) and after TGA: (a) PEGD250- (top) and PEGD600-coated ultrasmall Ho2O3 nanoparticles (bottom) and (b) PAA1800- (top) and PEGD600-coated ultrasmall Tm2O3 nanoparticles (bottom). All peaks after TGA could be assigned with (hkl) Miller indices of body-centered cubic Ho2O3 and Tm2O3.53,54 | |
Surface-coating results
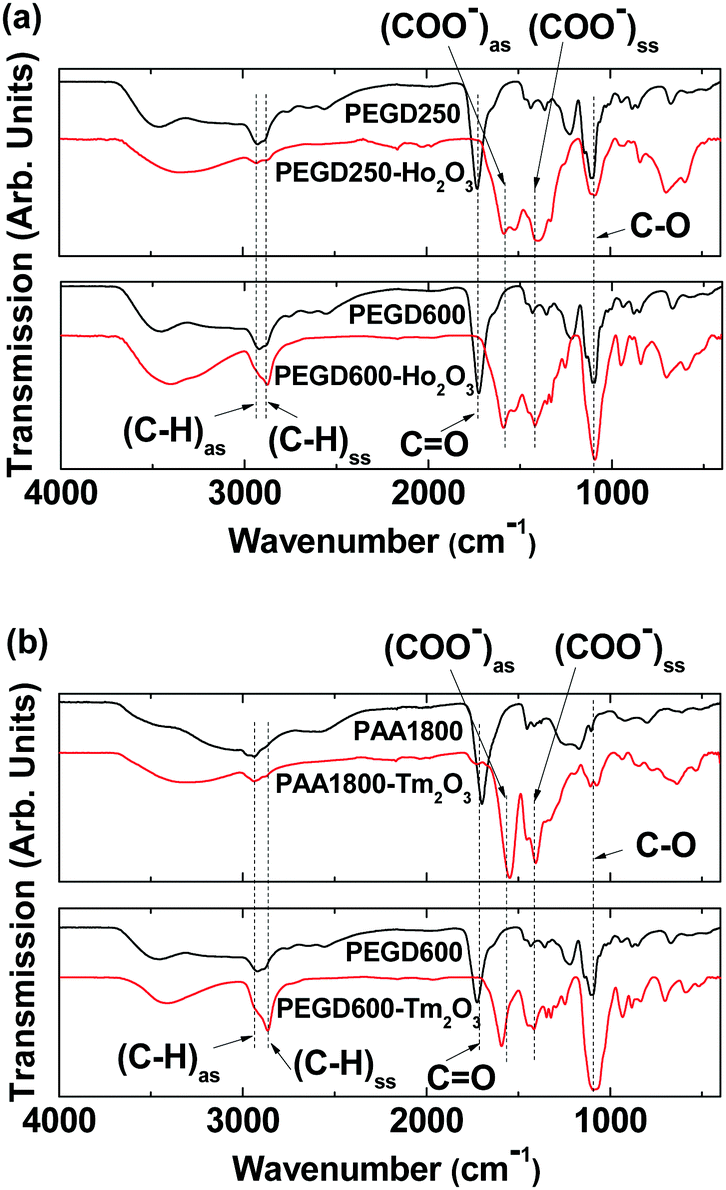
The surface coating of the nanoparticles was investigated by recording Fourier transform-infrared (FT-IR) absorption spectra of the ligand-coated nanoparticles as well as the ligands for reference. As shown in Fig. 4a and b and Table 2, characteristic IR absorption bands of the ligands such as C–H antisymmetric and symmetric stretching vibrations at 2922–2937 and 2868–2876 cm−1, respectively, COO− antisymmetric and symmetric stretching vibrations at 1547–1593 and 1398–1433 cm−1, respectively, and C–O stretching vibrations at 1087–1099 cm−1 were observed in the FT-IR absorption spectra of the PEGD250-, PEGD600-, and PAA1800-coated nanoparticles, confirming the successful ligand coating of the nanoparticles. The splitting of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of PEGD250 at 1723 cm−1 and PEGD600 at 1721 cm−1 and PAA1800 at 1700 cm−1 into the aforementioned COO− symmetric and antisymmetric stretching vibrations in the samples indicate the bridge bonding of the COO− groups of the ligands to Ho3+ and Tm3+ of the nanoparticles.55,56 This bridge bonding was strong, as confirmed from large red-shifts of the COO− antisymmetric and symmetric stretching frequencies by ∼130 and ∼300 cm−1 from the CO stretching frequencies, respectively (Table 2). This corresponds to hard-acid (COO− groups of the ligands) and hard-base (Ho3+ and Tm3+ of the nanoparticles) types of bonding.57–59 The observed absorption frequencies are consistent with the literature.56,60,61
O stretching vibrations of PEGD250 at 1723 cm−1 and PEGD600 at 1721 cm−1 and PAA1800 at 1700 cm−1 into the aforementioned COO− symmetric and antisymmetric stretching vibrations in the samples indicate the bridge bonding of the COO− groups of the ligands to Ho3+ and Tm3+ of the nanoparticles.55,56 This bridge bonding was strong, as confirmed from large red-shifts of the COO− antisymmetric and symmetric stretching frequencies by ∼130 and ∼300 cm−1 from the CO stretching frequencies, respectively (Table 2). This corresponds to hard-acid (COO− groups of the ligands) and hard-base (Ho3+ and Tm3+ of the nanoparticles) types of bonding.57–59 The observed absorption frequencies are consistent with the literature.56,60,61
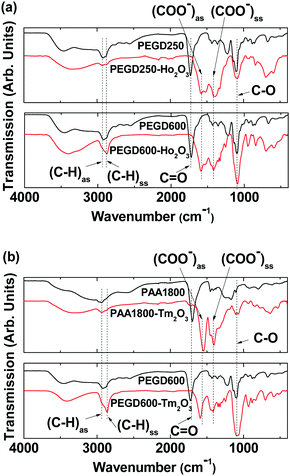 |
| | Fig. 4 FT-IR absorption spectra of (a) PEGD250 and PEGD250-coated ultrasmall Ho2O3 nanoparticles (top), and PEGD600 and PEGD600-coated ultrasmall Ho2O3 nanoparticles (bottom), and (b) PAA1800 and PAA1800-coated ultrasmall Tm2O3 nanoparticles (top), and PEGD600 and PEGD600-coated ultrasmall Tm2O3 nanoparticles (bottom). | |
Table 2 Observed FT-IR absorption frequencies in cm−1
|
|
(C–H)as |
(C–H)ss |
CO |
(COO−)asa |
(COO−)ssa |
C–O |
|
The numbers in parentheses correspond to the red shifts from the CO stretching frequencies of the ligands.
|
| PEGD250 |
2926 |
2885 |
1723 |
— |
— |
1101 |
| PEGD600 |
2923 |
2888 |
1721 |
— |
— |
1099 |
| PAA1800 |
2978 |
2937 |
1700 |
— |
— |
1101 |
| PEGD250-Ho2O3 |
2926 |
2874 |
— |
1578 (145) |
1398 (325) |
1096 |
| PEGD600-Ho2O3 |
2922 |
2876 |
— |
1593 (128) |
1418 (303) |
1093 |
| PEGD600-Tm2O3 |
2926 |
2868 |
— |
1589 (132) |
1433 (288) |
1987 |
| PAA1800-Tm2O3 |
2937 |
2868 |
— |
1547 (153) |
1402 (298) |
1099 |
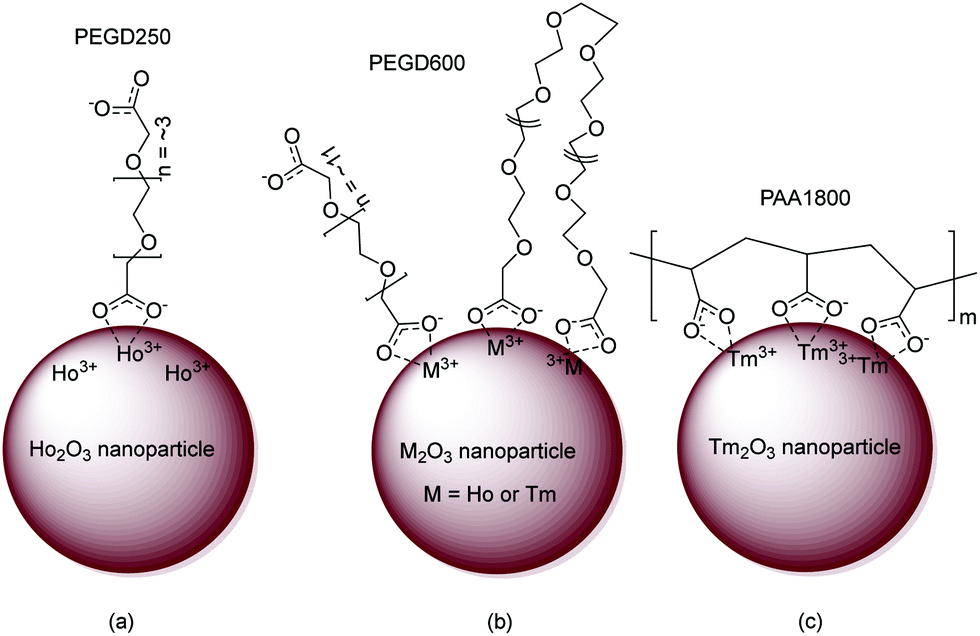
Based on FT-IR absorption spectral results, the surface-coating structures of PEGD250, PEGD600, and PAA1800 on the nanoparticle surfaces are schematically proposed in Fig. 5a–c, respectively. As shown in Fig. 5a, one of the two COO− groups of PEGD250 is likely bonded to Ho3+ of the ultrasmall Ho2O3 nanoparticles because of the short length of PEGD250. PEGD600 is likely bonded to the nanoparticles via its one or two COO− groups because of its long and flexible length (Fig. 5b). Each PAA1800 possesses approximately 25 COO− groups and thus can allow multiple bonding interactions among its many COO− groups and Tm3+ of the nanoparticles (Fig. 5c).
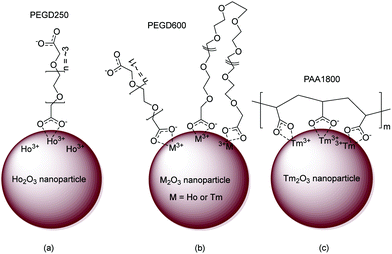 |
| | Fig. 5 Proposed ligand-coating structures of (a) PEGD250, (b) PEGD600, and (c) PAA1800 via the bridge bonding between the COO− groups of the ligands and Ho3+ or Tm3+ of the nanoparticles. | |
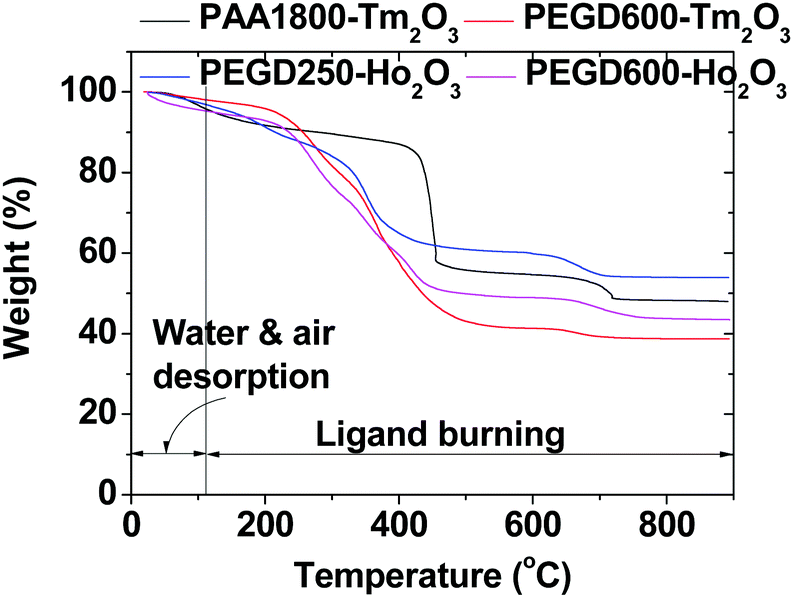
The amount (P) of ligand-coating of the nanoparticles in wt% was estimated from the mass loss in the TGA curve after considering an initial mass drop between room temperature and ∼105 °C due to water and air desorption (Fig. 6). The residual mass in the TGA curve corresponded to the net mass of the Ho2O3 or Tm2O3 nanoparticles without ligands. Grafting density (σ),62 corresponding to the average number of ligands coating a nanoparticle unit surface area, was estimated using the bulk density of Ho2O3 (8.41 g cm−3) or Tm2O3 (8.6 g cm−3),63davg estimated from HRTEM imaging, and the above P value obtained from the TGA curve. The average number (NNP) of ligands coating the nanoparticle was then estimated by multiplying σ with the nanoparticle surface area (=πd2avg). As provided in Table 1, the σ and NNP values decreased with increasing ligand size likely because a larger ligand generally occupied a larger space due to its steric effects.
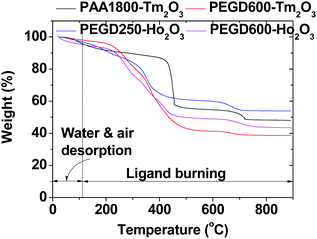 |
| | Fig. 6 TGA curves of the PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles, and PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles. | |
Magnetic properties
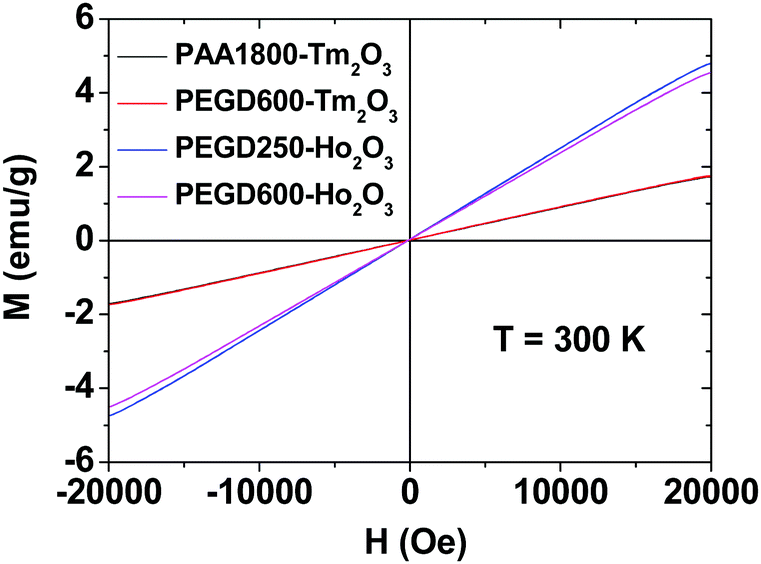
The magnetic properties of the PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles, and PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles were investigated by measuring magnetization (M) versus applied field (H) (i.e., M–H) curves at 300 K using a vibrating sample magnetometer (VSM) (Fig. 7). The mass-corrected net M values of the nanoparticles without ligands were used in the plots, which were estimated using their net masses that were extracted from their TGA curves shown in Fig. 6. All nanoparticle samples showed paramagnetism with no hysteresis, zero coercivity, and zero remanence in the M–H curves, which is similar to that of their corresponding bulk Ho2O3 and Tm2O3.64,65 From the mass-corrected M–H curves, the net M values of the ultrasmall Ho2O3 and Tm2O3 nanoparticles without ligands at 2.0 T and 300 K were estimated to be 4.64 and 1.73 emu g−1 (Table 3), respectively. The bigger net M value of the ultrasmall Ho2O3 nanoparticles compared with that of ultrasmall Tm2O3 nanoparticles is due to a higher magnetic moment of Ho3+ (5I5, 10.6 μB) compared with that of Tm3+ (3H6, 7.56 μB),66 where μB is the Bohr magneton.
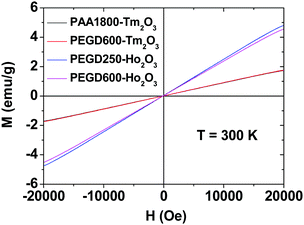 |
| | Fig. 7 Mass-corrected M–H curves of the PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles, and PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles at 300 K. The net M values of the ultrasmall Ho2O3 and Tm2O3 nanoparticles without ligands were used in the plots, which were estimated using the net masses of the nanoparticles extracted from the TGA curves. | |
Table 3 Magnetic properties and water proton spin relaxivities
| Nanoparticle |
Surface-coating ligand |
Magnetic properties at 300 K |
Water proton spin relaxivities (22 °C, 3.0 T) |
| Magnetism |
Net M (emu g−1) at 2 T |
r
1 (s−1 mM−1) |
r
2 (s−1 mM−1) |
|
Data from ref. 35 and net M was obtained at 1.8 T.
|
| Ho2O3 |
PEGD250 |
Paramagnetism |
4.76 |
Average = 4.64 |
0.14 |
30.39 |
| Ho2O3 |
PEGD600 |
Paramagnetism |
4.52 |
0.17 |
11.33 |
| Ho2O3a |
PAA1800 |
Paramagnetism |
4.1 |
|
0.13 |
1.44 |
| Tm2O3 |
PEGD600 |
Paramagnetism |
1.74 |
Average = 1.73 |
0.11 |
5.79 |
| Tm2O3 |
PAA1800 |
Paramagnetism |
1.72 |
0.10 |
1.03 |
r
1 and r2 values: nanoparticle magnetic moment and ligand-size effects on r2 values
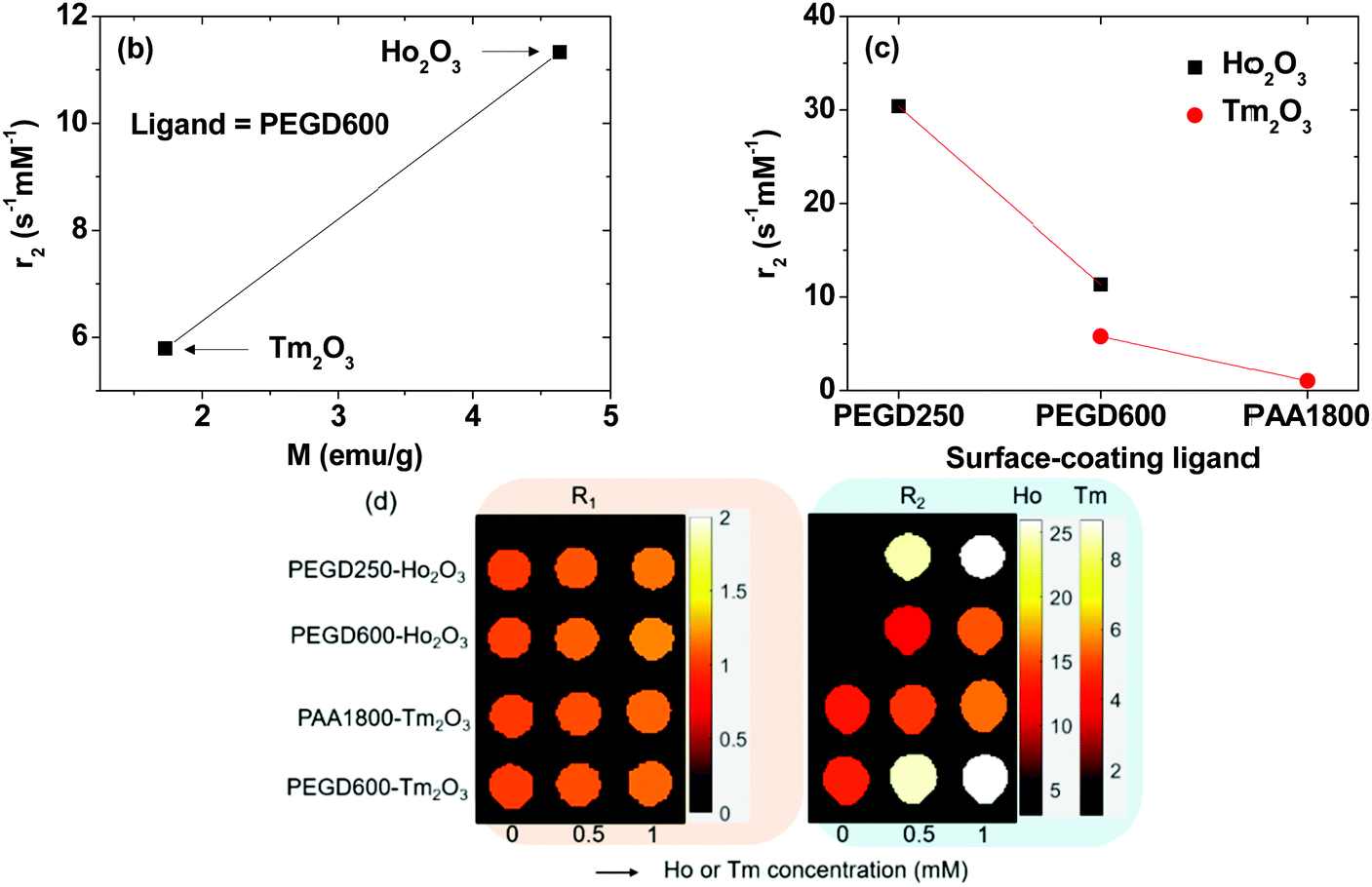
r
1 and r2 values were estimated from the slopes of 1/T1 and 1/T2 plots as a function of Ho or Tm concentration, respectively (Fig. 8a and Table 3). r1 Values were negligible for all nanoparticle samples (<0.2 s−1 mM−1), whereas r2 values were appreciable with a magnitude that depended on the nanoparticle species and surface-coating ligands. This implies that the ultrasmall Ho2O3 and Tm2O3 nanoparticles can exclusively induce only T2 water proton spin relaxations with negligible induction of T1 water proton spin relaxations. This is due to the contribution of 4f-electron orbital motions in Ho3+ and Tm3+ of the nanoparticles to the nanoparticle magnetic moments. According to the inner sphere model, only the magnetic moment from electron spin motion can significantly contribute to the r1 value,5,6 which is not the case for Ho3+ and Tm3+. However, the r2 value is proportional to the square of nanoparticle magnetic moment according to the outer sphere model,5,47 and thus, was appreciable because nanoparticle magnetic moments of the Ho2O3 and Tm2O3 nanoparticles at room temperature were appreciable (Table 3).
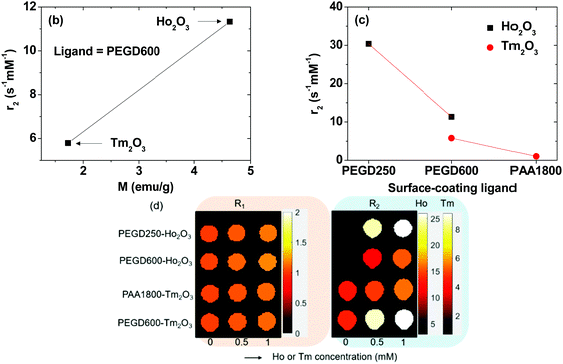 |
| | Fig. 8 (a) Plots of 1/T1 and 1/T2 of the nanoparticle suspension samples in aqueous media as a function of Ho or Tm concentration. The slopes correspond to the r1 and r2 values, respectively. Plots of the r2 values as a function of (b) nanoparticle magnetic moment (M) (using the average M in Table 3) and (c) ligand-size (PEGD250 < PEGD600 < PAA1800). (d) Dose-dependent R1 and R2 map images of the nanoparticle suspension samples in aqueous media. | |
Given that T2 water proton spin relaxation is induced by the magnetic dipole-dipole interactions between the nanoparticles and water proton spins, the r2 value is proportional to MNP2/L3 in which MNP is the nanoparticle magnetic moment (unit: emu/nanoparticle) and L is the distance between the nanoparticle and water proton spin.5,47MNP ∝ davg3M for paramagnetic nanoparticles35 and davg values are nearly the same for all nanoparticle samples for the present study (see Table 1) and thus, MNP1 (Ho2O3 nanoparticle) > MNP2 (Tm2O3 nanoparticle) and L1 (PAA1800) > L2 (PEGD600) > L3 (PEGD250) if L is assumed to be proportional to the ligand size. This explains the observed increase in r2 value with increasing M (Fig. 8b) and a decrease in r2 value with increasing ligand size (Fig. 8c). Overall, MNP12/L33 > MNP12/L23 > MNP22/L23 > MNP22/L13 explains the observed r2 values such that r2 (PEGD250-coated Ho2O3 nanoparticle) > r2 (PEGD600-coated Ho2O3 nanoparticle) > r2 (PEGD600-coated Tm2O3 nanoparticle) > r2 (PAA1800-coated Tm2O3 nanoparticle). This simple model equation also explains that r2 (PEGD600-coated Ho2O3 nanoparticle) > r2 (PAA1800-coated Ho2O3 nanoparticle; Table 3)35 > r2 (PAA1800-coated Tm2O3 nanoparticle).
As shown in the R1 and R2 map images (Fig. 8d), dose-dependent contrast enhancements in the R1 map images were negligible for all nanoparticle samples whereas R2 map images exhibited appreciable dose-dependent contrast enhancements for all nanoparticle samples, supporting in vitro that the ultrasmall Ho2O3 and Tm2O3 nanoparticles may act as efficient T2 MRI contrast agents.
In vitro cellular cytotoxicity
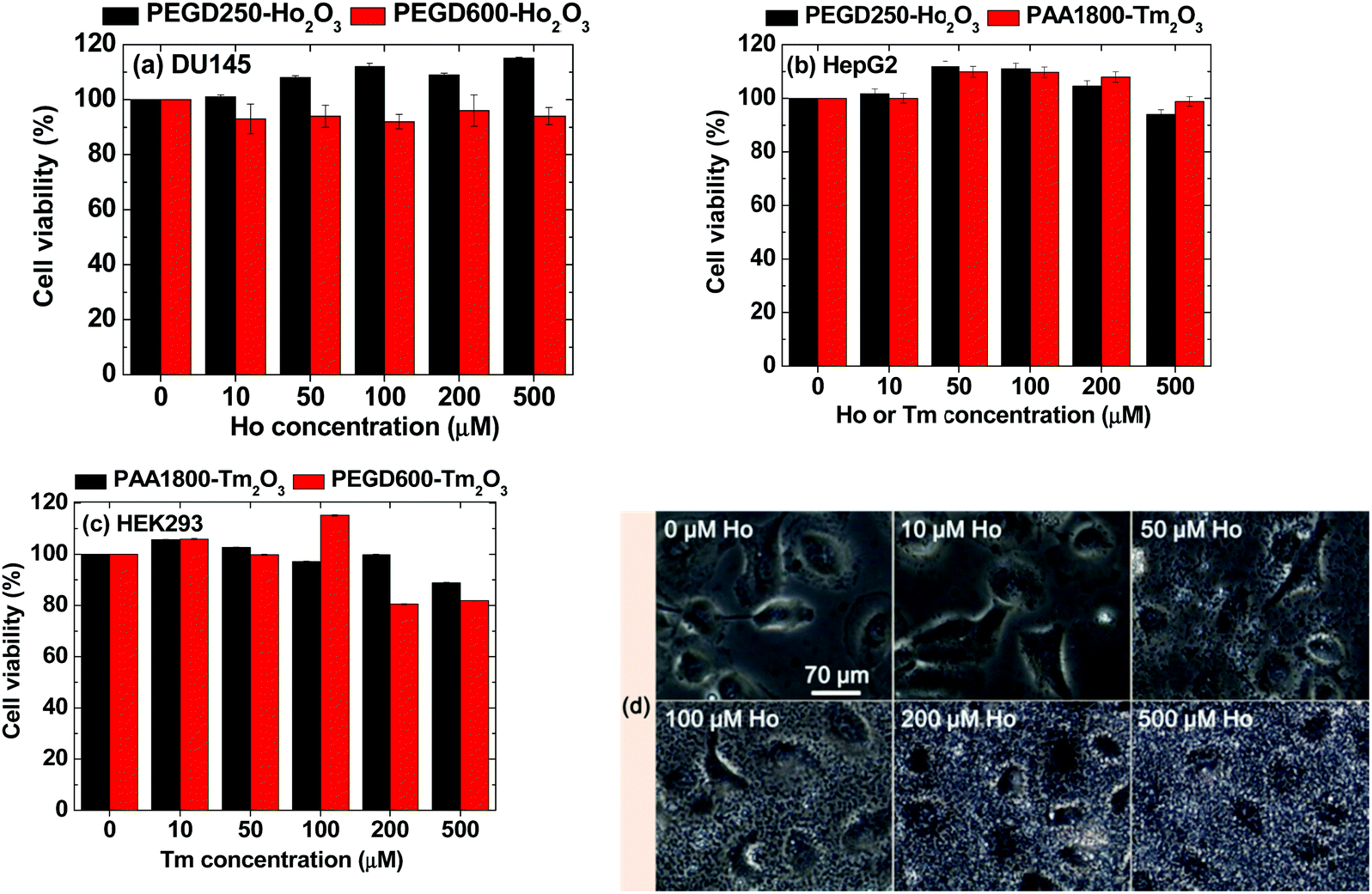
The cellular cytotoxicity of the PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles and PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles was investigated by measuring in vitro cell viabilities in various types of cell lines, such as human prostate cancer (DU145), human embryonic kidney 293 (HEK293), and human liver cancer (HepG2) cell lines 48 h after incubation. As shown in Fig. 9a–c, all samples exhibited considerably low cellular cytotoxicities of up to 500 μM Ho and Tm in various cell lines. Dose-dependent cell morphologies were investigated by measuring optical microscope images of control and treated DU145 cells with PEGD250-coated ultrasmall Ho2O3 nanoparticles at various Ho concentrations (Fig. 9d). As shown in Fig. 9, the nanoparticles were not localized in the cells but scattered all over the place and heavily covered the cells with the degree of cell coverage which increased with increasing nanoparticle concentration. In addition, the cell morphologies of the treated cells with the nanoparticles were similar to those of the control cells, likely due to the very low cytotoxicities of the nanoparticles.
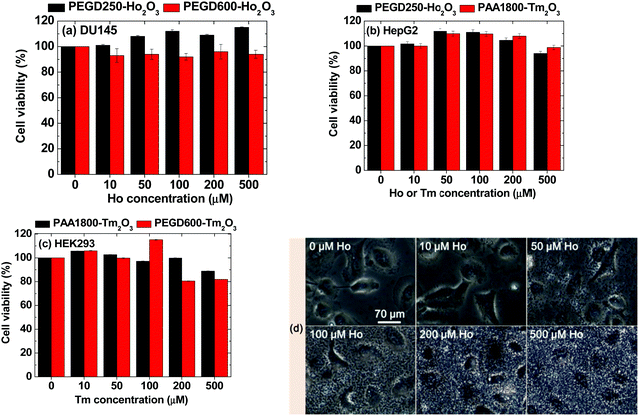 |
| | Fig. 9
In vitro cell viabilities of (a) PEGD250- and PEGD600-coated ultrasmall Ho2O3 nanoparticles in DU145 cell lines, (b) PEGD250-coated ultrasmall Ho2O3 nanoparticles and PAA1800-coated ultrasmall Tm2O3 nanoparticles in HepG2 cell lines, and (c) PEGD600- and PAA1800-coated ultrasmall Tm2O3 nanoparticles in HEK293 cell lines 48 h after incubation as a function of Ho or Tm concentration. (d) Optical microscope images of control and treated DU145 cells with PEGD250-coated ultrasmall Ho2O3 nanoparticles at various Ho concentrations: the same scale bar applies to all images. | |
Hemolysis assay results
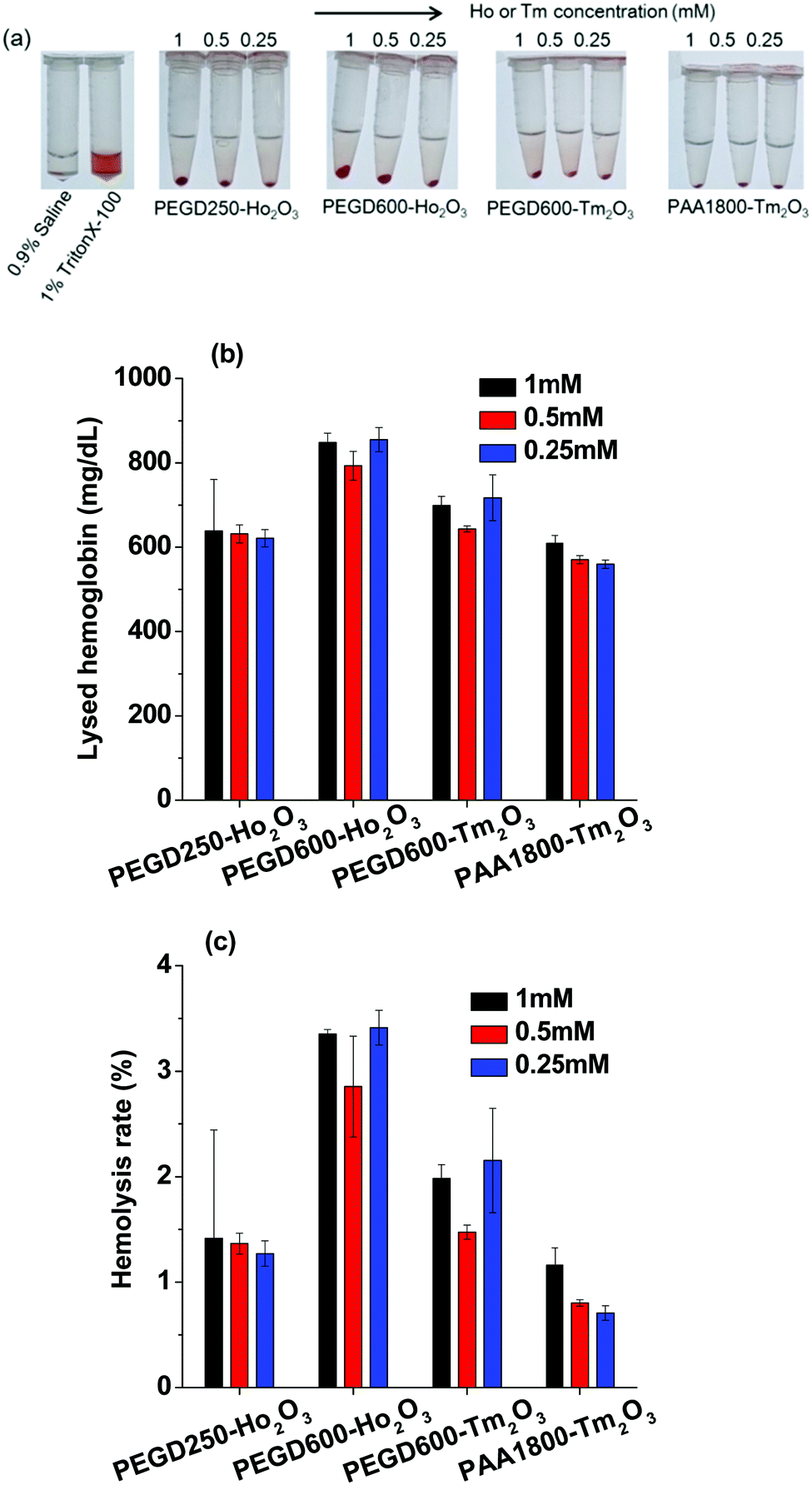
To investigate the hemolytic effects of the nanoparticle samples, the hemolysis assay was performed for all nanoparticle samples and the results are shown in Fig. 10a–c. Photographs of the lysed assay results are shown in Fig. 10a and the estimated lysed hemoglobin concentrations in mg dL−1 are plotted in Fig. 10b. The hemolysis rates of the nanoparticle samples are plotted in Fig. 10c. As shown in Fig. 10a–c, only the PEGD600-coated Ho2O3 nanoparticles exhibited slight hemolytic properties for the tested concentration range (2.85 ± 0.48% to 3.41 ± 0.16%). However, the other nanoparticle samples exhibited small hemolysis rates which were less than 2% suitable for in vivo applications.
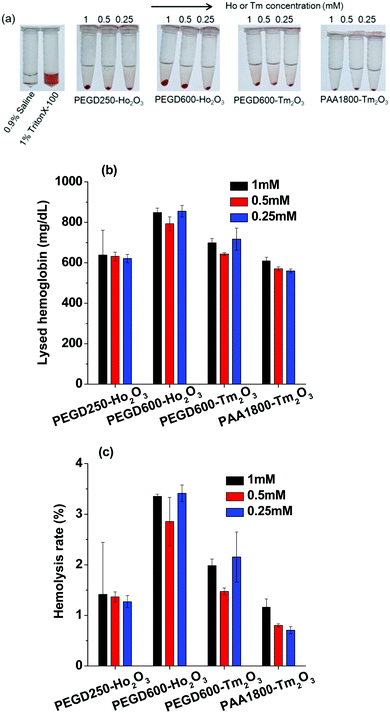 |
| | Fig. 10 Hemolysis assay results: (a) photographs of lysed blood samples, (b) lysed hemoglobin concentration in mg dL−1, and (c) hemolysis rate in %. | |
In vivo T
2 MR images at a 3.0 T MR field
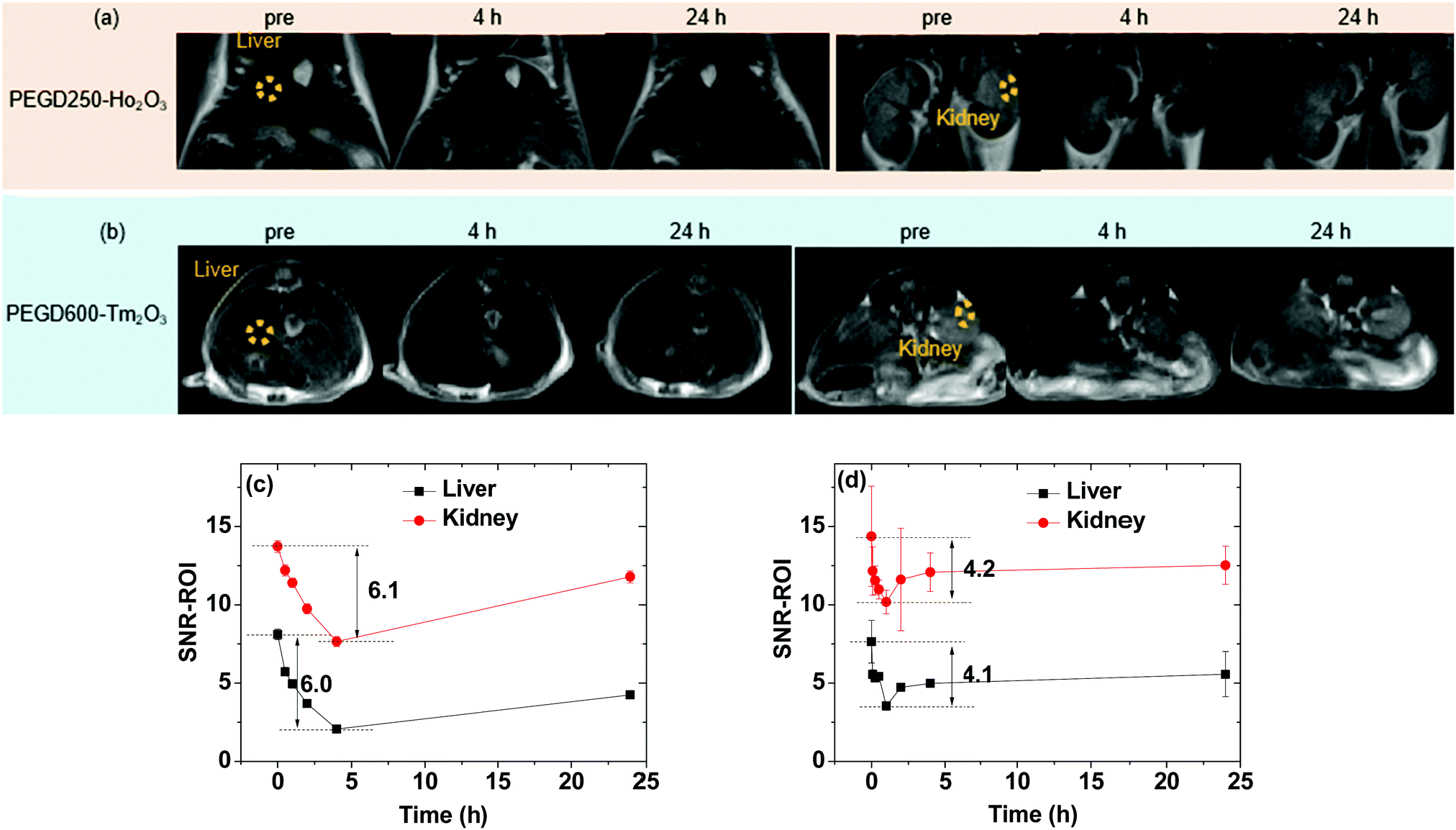
To demonstrate the effectiveness of the ultrasmall Ho2O3 and Tm2O3 nanoparticles as efficient T2 MRI contrast agents in vivo, the PEGD250-coated ultrasmall Ho2O3 nanoparticles and PEGD600-coated ultrasmall Tm2O3 nanoparticles were used for T2 MR image measurements. These nanoparticles were chosen because they possess higher r2 values compared with the same kind of nanoparticles grafted with different ligands. In vivo T2 MR images were obtained before (labelled as “pre”) and after intravenous injection of the aqueous nanoparticle suspension samples into mice tails at a 3.0 T MR field. As shown in Fig. 11a and b, negative contrast enhancements (i.e., darker images) after injection were clearly observed in the liver and kidneys for both nanoparticle samples.
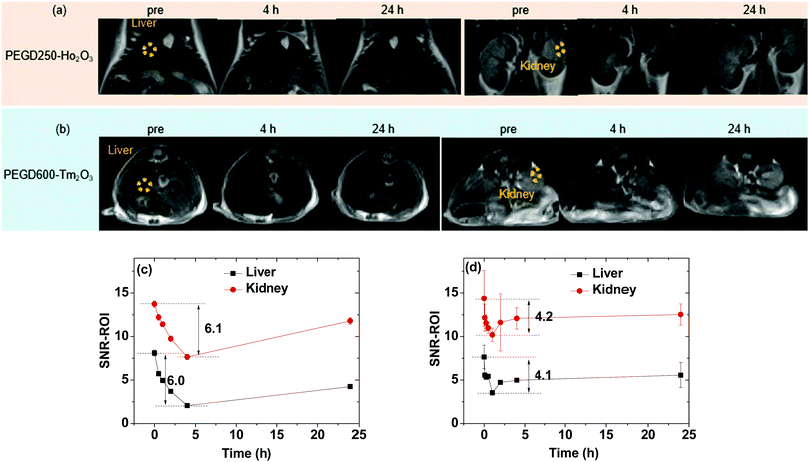 |
| | Fig. 11
In vivo T
2 MR images in the liver and kidneys of mice at a 3.0 T MR field before (labeled as “pre”) and after intravenous injection of the aqueous suspension samples of the (a) PEGD250-coated ultrasmall Ho2O3 nanoparticles and (b) PEGD600-coated ultrasmall Tm2O3 nanoparticles into mice tails (two mice were used for each sample). Dotted circles in the “pre” T2 MR images indicate regions-of-interest (ROIs). Plots of signal-to-noise ratios (SNRs) of ROIs in the T2 MR images before and after intravenous injection of the aqueous suspension samples of the (c) PEGD250-coated ultrasmall Ho2O3 nanoparticles and (d) PEG600-coated ultrasmall Tm2O3 nanoparticles. | |
To quantitatively investigate how the negative contrast enhancement changes with time, the signal-to-noise ratios (SNRs) of regions-of-interest (ROIs) in the liver and kidneys (labeled as dotted circles in “pre” T2 MR images) were plotted as a function of time. As shown in Fig. 11c and d, the negative contrast enhancements initially increased (or SNR-ROI decreased) after injection due to the accumulation of the nanoparticles in the liver and kidneys and then, decreased (or SNR-ROI increased) with time due to the excretion of the nanoparticles from the liver and kidneys because of their ultrasmall particle diameters. Notably, the PEGD250-coated ultrasmall Ho2O3 nanoparticles exhibited higher negative contrast enhancements (maximum = ∼6.0 in Fig. 11c) compared with those (maximum = ∼4.1 in Fig. 11d) obtained with the PEGD600-coated ultrasmall Tm2O3 nanoparticles because of the higher r2 value (i.e., 30.39 s−1 mM−1) of the former nanoparticles than that of the latter nanoparticles (i.e., 5.79 s−1 mM−1). Therefore, it is expected that the other untested nanoparticles will also provide negative contrast enhancements with magnitudes that are proportional to their r2 values. These results prove that the ultrasmall Ho2O3 and Tm2O3 nanoparticles should act as a new class of efficient T2 MRI contrast agents.
As shown in Fig. 11c and d, the excretion of the PEGD250-coated ultrasmall Ho2O3 nanoparticles was slightly longer compared with that of the PEGD600-coated ultrasmall Tm2O3 nanoparticles. This was likely related to the surface-coating ligands of the PEGD250-coated ultrasmall Ho2O3 nanoparticles, which resulted in less colloidal stability. Thus, the possible aggregation and interaction of the PEGD250-coated ultrasmall Ho2O3 nanoparticles with biological molecules inside the body of the mice would delay their excretion. It is worth noting that the contrast enhancements will be even higher at higher MR fields because of the unsaturated nanoparticle magnetic moments as can be seen in the M–H curves (Fig. 7) and because the r2 value is proportional to the square of nanoparticle magnetic moment which will increase with increasing MR field.
In vivo biodistribution results of the injected nanoparticles
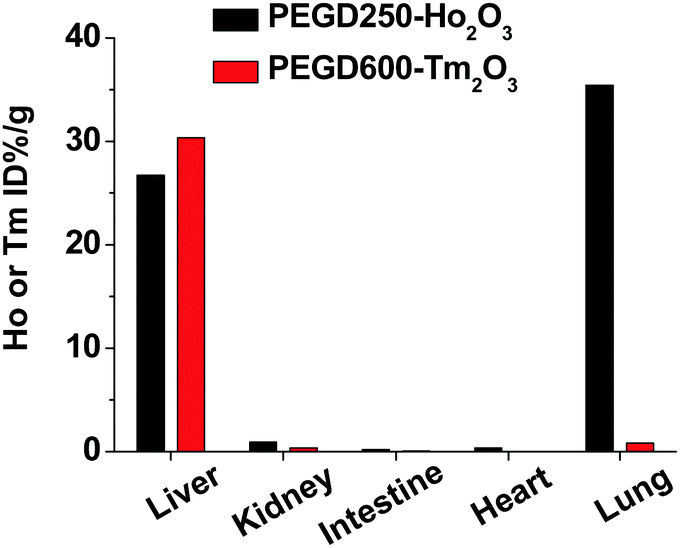
The in vivo biodistributions of the PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3 nanoparticles which were used for in vivo MRI experiments were assessed by measuring the Ho or Tm concentration for the lung, heart, liver, intestine, and kidney using ICP-AES. As shown in Fig. 12, nanoparticles were highly accumulated in the liver with 26.7 ± 0.007% and 30.4 ± 0.012% for the PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3, respectively. From the results, both nanoparticles can be expected to have a long circulation through the gastrointestinal route promising long-term diagnosis for any liver abnormality. Moreover, the PEGD250-coated Ho2O3 nanoparticles showed large lung accumulation (35.4 ± 0.002%), possibly due to their adsorption on red blood cells,67 which may be related to their observed lower colloidal stability compared with other nanoparticle samples.
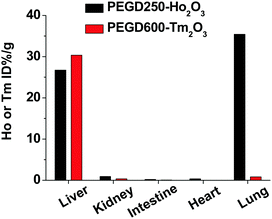 |
| | Fig. 12
In vivo biodistribution results of the PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3 nanoparticles 12 h after intravenous injection into mice tails (the number of mice used, n = 3). | |
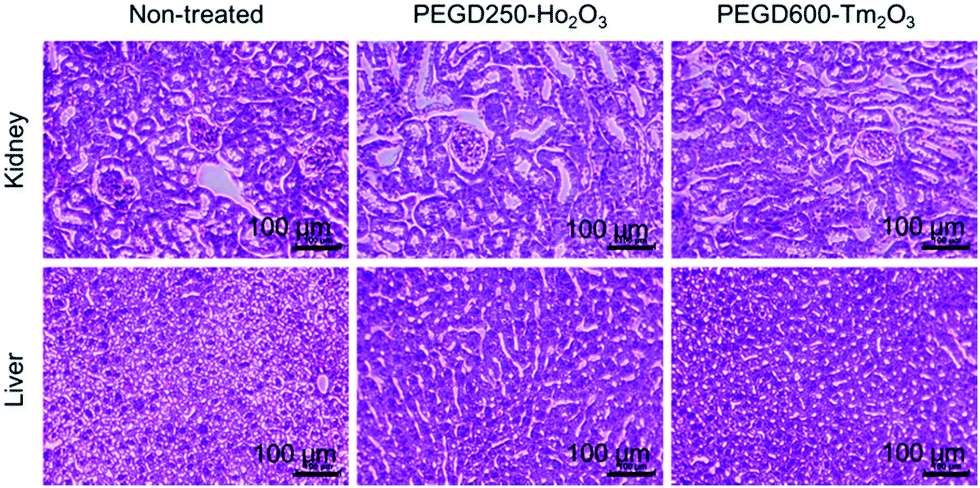
Histological analysis results
To investigate the in vivo toxicity of the PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3 nanoparticles which were used for the in vivo MRI experiments, histological changes were assessed for the two major organs, i.e., kidney and liver, which are responsible for excretion and detoxification. As shown in Fig. 13, both nanoparticle samples did not show any morphological changes for the kidney and liver, similar to the untreated mice trends, indicating negligible in vivo toxicity.
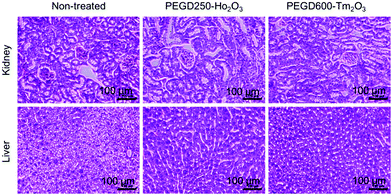 |
| | Fig. 13 Optical microscope images of the liver and kidney after H&E staining for the PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3 nanoparticles 24 h after intravenous injection into mice tails (the number of mice used, n = 3). | |
Experimental
Materials
Chemicals including Ho(NO3)3·5H2O (99.9%), Tm(NO3)3·5H2O (99.9%), NaOH (>99.9%), triethylene glycol (TEG, 99%), PEGD (99%, Mn = 250 amu, PEGD250), PEGD (99%, Mn = 600 amu, PEGD600), and PAA (analytical standard grade, Mw = 1800 amu, PAA1800) were purchased from Sigma-Aldrich (Burlington, MA, USA) and used as-received. Ethanol (99.5%) was purchased from Duksan (Ansan, South Korea) and used as-received for the initial washing of the nanoparticles. Triple-distilled water was used for the final washing of the nanoparticles and preparation of the nanoparticle suspension samples (∼20 mM Ho or Tm).
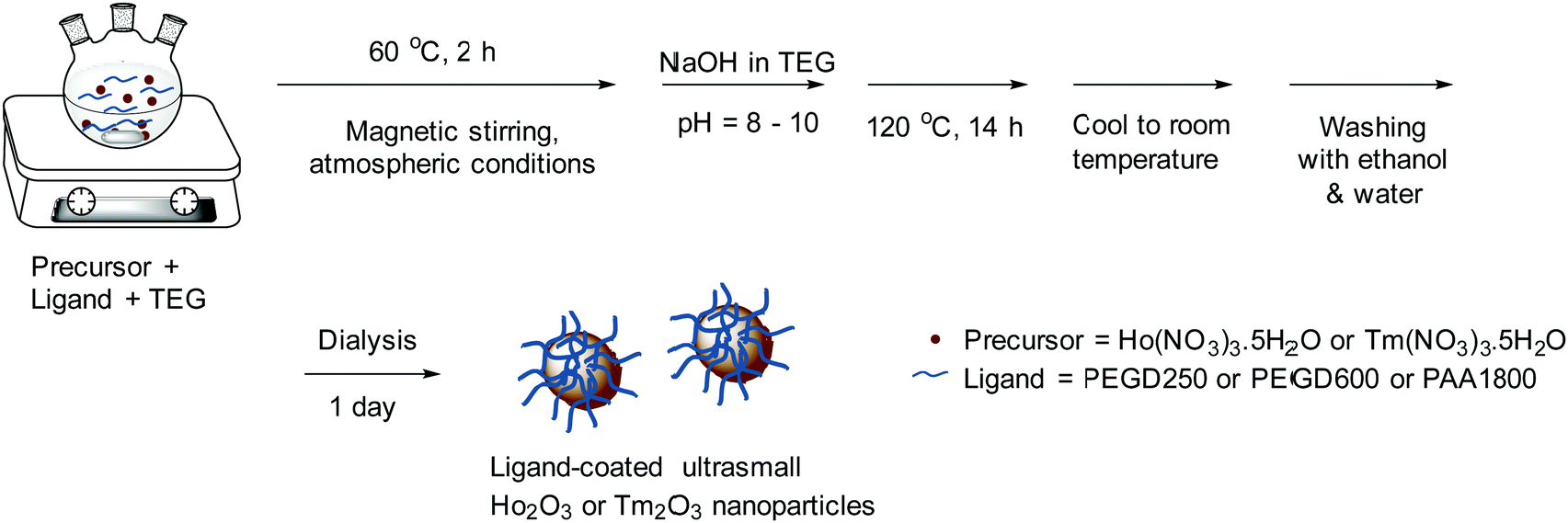
Synthesis of various ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles
The one-pot polyol synthesis of various hydrophilic and biocompatible ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles is shown in Fig. 14. In a three-necked round bottom flask, 2.0 mmol of Ho(NO3)3·5H2O or Tm(NO3)3·5H2O and ligand (3.0 mmol of PEGD250 or 2.0 mmol of PEGD600 or 1.0 mmol PAA1800) (Table 4) were dissolved in 20 mL of TEG with magnetic stirring at 60 °C for 2 h under atmospheric conditions. An NaOH solution prepared in TEG by dissolving 7.0 mmol of NaOH in 15 mL of TEG with magnetic stirring at 80 °C was added to the above precursor solution until the solution pH reached 8–10. The reaction solution was homogenized with magnetic stirring at 120 °C for 14 h before cooling to room temperature. To remove unreacted precursors, Na+, OH−, ligand, and TEG from the product solution, the solution was transferred to a 500 mL beaker and 400 mL of ethanol was added with magnetic stirring for 10 min. The solution was placed in a refrigerator until the nanoparticles settled down to the beaker bottom. The top transparent solution was decanted and the remaining product solution was washed thrice with ethanol using the same process. To remove ethanol from the nanoparticles, the product solution was diluted with 400 mL of triple-distilled water and then rotary evaporated to ∼40 mL three times. To further purify the product solution, it was dialyzed against 1.0 L of triple-distilled water using a dialysis tube (MWCO = 1000 amu for the PEGD250- and PEGD 600-coated nanoparticles, and 2000 amu for the PAA1800-coated nanoparticles) for a day with magnetic stirring.
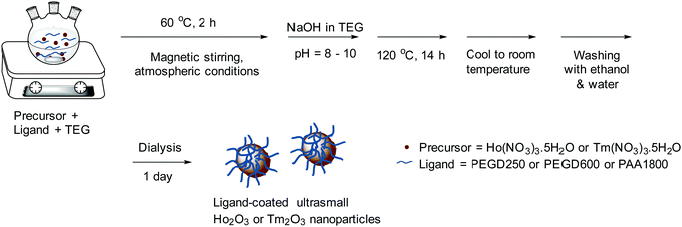 |
| | Fig. 14 One-pot polyol synthesis of various hydrophilic and biocompatible ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles (ligand = PEGD250, PEGD600, and PAA1800). | |
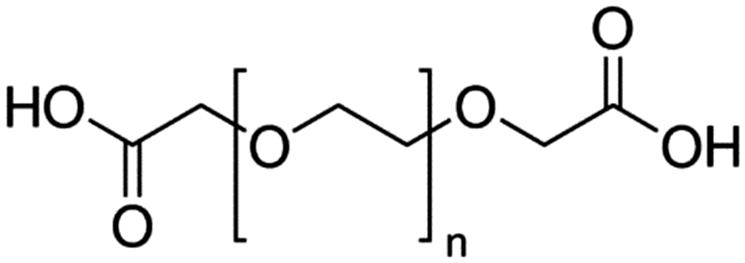
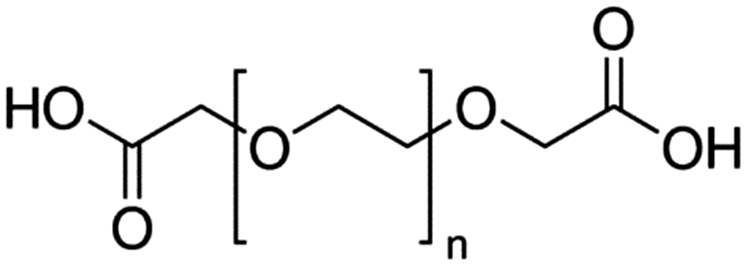
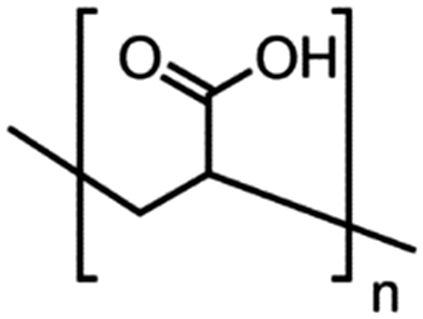
Table 4 Physical data of various hydrophilic and biocompatible ligands used for the surface coating
| Ligand |
Molecular weight (amu) |
Structure and size (n) |
| PEGD250 |
M
n = 250 |
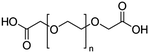
|
n = ∼3 |
| PEGD600 |
M
n = 600 |
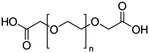
|
n = ∼11 |
| PAA1800 |
M
w = 1800 |
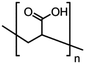
|
n = ∼25 |
Physicochemical property characterizations
The particle diameters of various hydrophilic and biocompatible ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles were measured using an HRTEM (Titan G2 ChemiSTEM CS Probe, 200 kV; FEI, Hillsboro, Oregon, USA). For measurements, a drop of the diluted nanoparticle suspension sample in ethanol was placed onto a carbon film supported by a 200-mesh copper grid (Pelco No. 160, Ted Pella Inc., Redding, CA, USA) using a micropipette (2–20 μL, Eppendorf, Hamburg, Germany) and allowed to dry in air at room temperature. The copper grid with the nanoparticles was subsequently placed inside the HRTEM vacuum chamber for measurements. An EDS instrument (Quantax Nano, Bruker, Berlin, Germany) installed inside the HRTEM was used to qualitatively identify elements (C, O, Ho, Tm) in the nanoparticle samples. The Ho and Tm concentrations of the aqueous nanoparticle suspension samples were determined using an inductively coupled plasma atomic emission spectrometer (ICP-AES) (IRIS/AP, Thermo Jarrell Ash Co., Waltham, MA, USA). A DLS particle size analyzer (Zetasizer Nano ZS, Malvern, Malvern, UK) was used to measure the hydrodynamic diameters and zeta potentials of the nanoparticle suspensions in aqueous media. A multi-purpose XRD instrument (X’PERT PRO MRD, Philips, The Netherlands) with unfiltered CuKa (λ = 0.154184 nm) radiation was used to characterize the crystal structures of the nanoparticle powder samples. The scanning step and scan range in 2θ were 0.033° and 15–100°, respectively. The attachment of the hydrophilic ligands to the nanoparticles was probed by recording FT-IR absorption spectra (Galaxy 7020A, Mattson Instrument Inc., Madison, WI, USA) using the powder samples pelletized with KBr. The scan range was 400–4000 cm−1. A TGA instrument (SDT-Q600, TA Instrument, New Castle, DE, USA) was used to estimate the ligand surface-coating amounts by recording TGA curves between room temperature and 900 °C under an air flow. The average amount of surface-coating ligands in wt% was estimated from the mass loss after considering the initial mass drop due to water and air desorption between room temperature and ∼105 °C. The net amount of nanoparticles without ligands in the samples was estimated from the remaining mass. A VSM (7407-S, Lake Shore Cryotronics Inc., Westerville, OH, USA) was used to record the M–H curves (−2.0 T ≤ H ≤ 2.0 T) at 300 K using 20–30 mg powder samples. The net M values of the nanoparticles without ligands were estimated using the net masses of the nanoparticles extracted from the TGA curves.
r
1 and r2 relaxivity and R1 and R2 map image measurements
T
1 and T2 water proton spin relaxation times and R1 and R2 water proton spin relaxation map images were measured using a 3.0 T MRI scanner (Magnetom Trio Tim, Siemens, Munich, Bayern, Germany). Aqueous dilute nanoparticle suspension samples (1.0, 0.5, 0.25, 0.125, and 0.0625 mM Ho or Tm) were prepared via dilution of the original concentrated nanoparticle suspension samples (∼20 mM Ho or Tm) with triple-distilled water. These dilute solutions were used to measure T1 and T2 relaxation times and R1 and R2 map images. Next, r1 and r2 water proton spin relaxivities of the nanoparticle suspension samples were estimated from the slopes of 1/T1 and 1/T2 plots versus the Ho or Tm concentration, respectively. T1 relaxation time measurements were conducted using an inversion recovery method. In this method, the inversion time (TI) was varied, and the MR images were acquired at 35 different TI values in the range of 50–1750 ms. T1 Relaxation times were obtained from nonlinear least-square fits to the measured signal intensities at various TI values. For the measurements of T2 relaxation times, the Carr–Purcell–Meiboom–Gill pulse sequence was used for multiple spin-echo measurements, and 34 images were acquired at 34 different echo time (TE) values in the range of 10–1900 ms. T2 relaxation times were obtained from nonlinear least-square fits to the mean pixel values of the multiple spin-echo measurements at various TE values.
In vitro cellular cytotoxicity measurements
The in vitro cellular cytotoxicity of the nanoparticles was measured using a CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA). The intracellular adenosine triphosphate was quantified using a Victor 3 luminometer (PerkinElmer, Waltham, MA, USA). The human prostate cancer (DU145), human embryonic kidney 293 (HEK293), and human liver cancer (HepG2) cell lines were used. The cells were seeded into a separate 24-well cell culture plate and incubated for 24 h (5 × 104 cell density, 500 μL cells per well, 5% CO2, and 37 °C). Four test solutions (10, 50, 100, 200, and 500 μM Ho or Tm) were prepared via dilution of the original concentrated nanoparticle suspension samples with a sterile phosphate-buffered saline solution, and 2.0 mL aliquots were used to treat the cells, which were subsequently incubated for 48 h. Cell viabilities were measured thrice to obtain average cell viabilities, which were then normalized with respect to those of untreated control cells (0.0 mM Ho or Tm).
Hemolysis assay
Mice blood (balb/c, 19–20 g, male, 6 weeks old) was collected and immediately mixed with heparinized saline (Sigma-Aldrich, catalog no. H3393-50KU, 20 units per ml) to prevent coagulation. Nanoparticle samples (1, 0.5, and 0.25 mM Ho or Tm, 490 μL) were mixed with the heparinized blood (10 μL) and incubated at 37 °C for 1 h. 1% TritonX-100 and saline (0.9% NaCl) were used as a positive and negative control, respectively. The incubated blood samples were centrifuged at 10000 rpm for 5 min to remove intact erythrocytes and the supernatants of each sample were obtained. The lysed hemoglobin in the supernatants was quantified with a hemoglobin assay kit (Sigma-Aldrich, catalog no. MAK115) according to the manufacturer's instructions. The hemolysis rate was estimated as follows:
| Hemolysis rate (%) = [(HNPs − H1% tritonX-100)/(Hsaline − H1% tritonX-100)] × 100, |
where H is the amount of lysed hemoglobin. The experiments were performed thrice.
In vivo T
2 MR image measurements
In vivo animal imaging experiments were conducted in accordance with the rules and regulations and permission of the animal research committee of the Korea Institute of Radiological and Medical Sciences (IACC number = 2021–0078). A 3.0 T MRI scanner (Magnetom Trio Tim, Siemens, Munich, Bayern, Germany) was used to obtain in vivo T2 MR images. Two balb/c male mice weighing 25–27g were used for each aqueous nanoparticle suspension sample. The mice were anesthetized using 1.5% isoflurane in oxygen. Measurements were made before and after injecting the nanoparticle suspension sample into mice's tail veins. The injection dose was approximately 0.087–0.1 mmol Ho or Tm kg−1. The fast spin-echo sequence was used to obtain T2 MR images. The typical parameters for coronal (or axial) image measurements were as follows: H = 3.0 T, echo time (TE) = 37 (36) ms, repetition time (TR) = 1620 (1629) ms, echo train length = 13 (13) mm, pixel bandwidth = 197 (197) mm, flip angle = 120 (120) degree, width = 60 (60) mm, height = 60 (30) mm, number of acquisitions (NEX) = 3 (4), slice thickness = 1.0 (1.2) mm, and slice gap = 1.1 (3.0) mm, where the numbers in parentheses are the parameters used for axial image measurements.
In vivo biodistribution study
PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3 nanoparticles which were used for in vivo MRI experiments were injected into normal balb/c mice tail veins with a 0.1 mmol Ho or Tm kg−1 dosage (19–20 g, 6 weeks old, male, n = 3). To obtain organ samples (i.e., lung, heart, liver, intestine, and kidney), the mice were anesthetized and exsanguinated 12 h after injection. The extracted organs were digested with 65% nitric acid and 30% hydrogen peroxide at 180 °C for 2 h. The digested samples were diluted with 3% nitric acid to a defined weight to measure Ho or Tm concentrations using an ICP-AES. Then, the Ho or Tm concentration was converted into the injected dose per gram of organ (ID%/g) with normalization to a 20 g mouse using the formula: ID%/g = (weight of Ho or Tm in the organ/weight of organ) × 100 × (weight of mouse/20).
Histological analysis
PEGD250-coated Ho2O3 and PEGD600-coated Tm2O3 nanoparticles which were used for in vivo MRI experiments were injected into normal balb/c mice tail veins with a 0.1 mmol Ho or Tm kg−1 dosage (19–20 g, 6 weeks old, male, n = 3). The mice were anesthetized using 1.5% isoflurane in oxygen and exsanguinated to obtain the kidney and the liver 24 h after injection. The organ samples were fixed in 4% paraformaldehyde for 72 h and treated with ethanol (concentration gradient 50, 70, 95, 100%), xylene (Junsei Chemical, Japan), and paraffin for 30 min. The organs were sectioned into 5 μm thickness and then treated with xylene for 1 h and ethanol (concentration gradient 100, 95, 70, and 50%) for 10 min at 65 °C. Hematoxylin and eosin (H&E) staining (BBC Biochemical, Mount Vernon, WA, USA) was performed according to the manufacturer's instructions and the stained samples were observed using a microscope (ECLIPSE Ti, Nikon, Tokyo, Japan) to assess acute in vivo toxicity.
Conclusions
In summary, various hydrophilic and biocompatible ligand-coated ultrasmall Ho2O3 and Tm2O3 nanoparticles were synthesized via a one-pot polyol method (ligand = PEGD250, PEGD600, and PAA1800), and their r1 and r2 values and in vivo T2 MR images at a 3.0 T MR field were measured to investigate their potential as a new class of efficient T2 MRI contrast agents. The results are summarized below:
(1) The average particle diameters were approximately 2.1 nm for all nanoparticle samples.
(2) The negligible r1 (<0.2 s−1 mM−1) and appreciable r2 values were observed for all nanoparticle samples, owing to 4f-electron orbital motion contributions of Ho3+ and Tm3+ to nanoparticle magnetic moments. The r2 value increased with increasing nanoparticle magnetic moments [from 1.73 (Tm2O3) to 4.64 (Ho2O3) emu g−1 at 2.0 T and 300 K] and decreased with increasing ligand-size (PEGD250 < PEGD600 < PAA1800). These two factors explained the observed r2 values such that 30.39 s−1 mM−1 (PEGD250-coated ultrasmall Ho2O3 nanoparticles) >11.33 s−1 mM−1 (PEGD600-coated ultrasmall Ho2O3 nanoparticles) >5.79 s−1 mM−1 (PEGD600-coated ultrasmall Tm2O3 nanoparticles) >1.03 s−1 mM−1 (PAA1800-coated ultrasmall Tm2O3 nanoparticles).
(3) Owing to the above r1 and r2 values, appreciable negative contrast enhancements were observed in in vivo T2 MR images at a 3.0 T MR field, which demonstrated the potential of the ultrasmall Ho2O3 and Tm2O3 nanoparticles as a new class of efficient T2 MRI contrast agents.
Author contributions
S. L. synthesized and characterized the samples and wrote the draft manuscript; T. T., H. Y., S. L. H., M. Y. A, A. K. A. A. S., D. Z., and Y. L. assisted with the synthesis and characterization of the samples; S. K. and J. A. P. obtained in vivo MR images; A. B. performed the hemolytic assay, H&E staining and biodistribution; S. K. measured relaxivities and map images; S. H. Y, D. W. H, and K.-S. C. measured cellular cytotoxicities; S.-W. N. assisted with the project; Y. C. and G. H. L. led the project; and G. H. L. wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Basic Science Research Program of the National Research Foundation (NRF) funded by the Ministry of Education, Science, and Technology (No. 2016R1D1A3B01007622) and the Korean government (Ministry of Science, and Information and Communications Technology: MSIT) (No. 2021R1A4A1029433). This work was also supported by a grant of the Korea Institute of Radiological and Medical Sciences (KIRAMS) funded by MSIT (No. 50461-2022).
Notes and references
- G. L.-G. Menezes, F. M. Knuttel, B. L. Stehouwer, R. M. Pijnappel and M. A.-A. J. van den Bosch, World J. Clin. Oncol., 2014, 5, 61–70 CrossRef PubMed.
- F. Zugni, A. R. Padhani, D.-M. Koh, P. E. Summers, M. Bellomi and G. Petralia, Cancer Imag., 2020, 20, 34 CrossRef PubMed.
- E. Bejer-Oleńska, M. Thoene, A. Włodarczyk and J. Wojtkiewicz, Biomed Res. Int., 2018, 2715831 Search PubMed.
- M. Haris, S. K. Yadav, A. Rizwan, A. Singh, E. Wang, H. Hariharan, R. Reddy and F. M. Marincola, J. Transl. Med., 2015, 13, 313 CrossRef PubMed.
- R. B. Lauffer, Chem. Rev., 1987, 87, 901–927 CrossRef CAS.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- G.-P. Yan, L. Robinson and P. Hogg, Radiography, 2007, 13, e5–e19 CrossRef.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- C. F.-C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS PubMed.
- Y.-D. Xiao, R. Paudel, J. Liu, C. Ma, Z.-S. Zhang and S.-K. Zhou, Int. J. Mol. Med., 2016, 38, 1319–1326 CrossRef CAS PubMed.
- G. J. Strijkers, W. J.-M. Mulder, G. A.-F. van Tilborg and K. Nicolay, Anti-Cancer Agents Med. Chem., 2007, 7, 291–305 CrossRef CAS PubMed.
- E. Peng, F. Wang and J. M. Xue, J. Mater. Chem. B, 2015, 3, 2241–2276 RSC.
- C. Do, J. DeAguero, D. Brearley, X. Trejo, T. Howard, G. P. Escobar and B. Wagner, Kidney360, 2020, 1, 561–568 CrossRef PubMed.
- C.-T. Yang and K.-H. Chuang, Med. Chem. Commun., 2012, 3, 552–565 RSC.
- Z. Zhou and Z.-R. Lu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 1–18 CrossRef CAS.
- H. Li and T. J. Meade, J. Am. Chem. Soc., 2019, 141, 17025–17041 CrossRef CAS PubMed.
- Y.-X. J. Wang, World J. Gastroenterol., 2015, 21, 13400–13402 CrossRef CAS PubMed.
- Y.-X. J. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- Y.-W. Li, Z.-G. Chen, J.-C. Wang and Z.-M. Zhang, World J. Gastroenterol., 2015, 21, 4334–4344 CrossRef PubMed.
- Z. Shen, A. Wu and X. Chen, Mol. Pharmaceutics, 2017, 14, 1352–1364 CrossRef CAS PubMed.
- I. Fernández-Barahona, M. Munoz-Hernando, J. Ruiz-Cabello, F. Herranz and J. Pellico, Inorganics, 2020, 8, 28 CrossRef.
- J. S. Weinstein, C. G. Varallyay, E. Dosa, S. Gahramanov, B. Hamilton, W. D. Rooney, L. L. Muldoon and E. A. Neuwelt, J. Cereb. Blood Flow Metab., 2010, 30, 15–35 CrossRef CAS PubMed.
- R. Qiao, C. Yang and M. Gao, J. Mater. Chem., 2009, 19, 6274–6293 RSC.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lan, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- W. Poon, Y.-N. Zhang, B. Ouyang, B. R. Kingston, J. L.-Y. Wu, S. Wilhelm and W. C.-W. Chan, ACS Nano, 2019, 13, 5786–5798 CrossRef CAS PubMed; Y.-N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C.-W. Chan, J. Controlled Release, 2016, 240, 332–348 CrossRef PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 CrossRef CAS PubMed.
- M. J. Baek, J. Y. Park, W. Xu, K. Kattel, H. G. Kim, E. J. Lee, A. K. Patel, J. J. Lee, Y. Chang, T. J. Kim, J. E. Bae, K. S. Chae and G. H. Lee, ACS Appl. Mater. Interfaces, 2010, 2, 2949–2955 CrossRef CAS PubMed.
- J. Y. Park, M. J. Baek, E. S. Choi, S. T. Woo, J. H. Kim, T. J. Kim, J. C. Jung, K. S. Chae, Y. Chang and G. H. Lee, ACS Nano, 2009, 3, 3663–3669 CrossRef CAS PubMed.
- K. Kattel, J. Y. Park, W. Xu, H. G. Kim, E. J. Lee, B. A. Bony, W. C. Heo, J. J. Lee, S. Jin, J. S. Baeck, Y. Chang, T. J. Kim, J. E. Bae, K. S. Chae and G. H. Lee, ACS Appl. Mater. Interfaces, 2011, 3, 3325–3334 CrossRef CAS PubMed.
- K. Kattel, J. Y. Park, W. Xu, H. G. Kim, E. J. Lee, B. A. Bony, W. C. Heo, S. Jin, J. S. Baeck, Y. Chang, T. J. Kim, J. E. Bae, K. S. Chae and G. H. Lee, Biomaterials, 2012, 33, 3254–3261 CrossRef CAS PubMed.
- S. Marasini, H. Yue, S. L. Ho, K.-H. Jung, J. A. Park, H. Cha, A. Ghazanfari, M. Y. Ahmad, S. Liu, Y. J. Jang, X. Miao, K.-S. Chae, Y. Chang and G. H. Lee, Eur. J. Inorg. Chem., 2019, 3832–3839 CrossRef CAS.
- S. Marasini, H. Yue, S. L. Ho, J. A. Park, S. Kim, K.-H. Jung, H. Cha, S. Liu, T. Tegafaw, M. Y. Ahmad, A. Ghazanfari, K.-S. Chae, Y. Chang and G. H. Lee, Nanomaterials, 2021, 11, 1355 CrossRef CAS PubMed.
- M. Norek, E. Kampert, U. Zeitler and J. A. Peters, J. Am. Chem. Soc., 2008, 130, 5335–5340 CrossRef CAS PubMed.
- H. Yue, J. A. Park, S. L. Ho, M. Y. Ahmad, H. Cha, S. Liu, T. Tegafaw, S. Marasini, A. Ghazanfari, S. Kim, K. S. Chae, Y. Chang and G. H. Lee, Pharmaceuticals, 2020, 13, 312 CrossRef CAS PubMed.
- Y. Kimura, T. Kurimoto, Y. Imai, H. Sugii, A. Toshimitsu, T. Matsuda, H. Imai, H. Yamada and T. Kondo, JSM Biotechnol. Bioeng., 2014, 2, 1043 Search PubMed.
-
F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, A Wiley-Interscience Publication, New York, USA, 4th edn, 1980, p. 984 Search PubMed.
- S. M. Dadfar, D. Camozzi, M. Darguzyte, K. Roemhild, P. Varvarà, J. Metselaar, S. Banala, M. Straub, N. Güvener, U. Engelmann, I. Slabu, M. Buhl, J. van Leusen, P. Kögerler, B. Hermanns-Sachweh, V. Schulz, F. Kiessling and T. Lammers, J. Nanobiotechnol., 2020, 18, 22 CrossRef CAS PubMed.
- D. Bonvin, D. T.-L. Alexander, A. Millán, R. Piñol, B. Sanz, G. F. Goya, A. Martínez, J. A.-A. Bastiaansen, M. Stuber, K. J. Schenk, H. Hofmann and M. M. Ebersold, Nanomaterials, 2017, 7, 225 CrossRef PubMed.
-
F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, A Wiley-Interscience Publication, New York, USA, 4th edn, 1980, p. 646 Search PubMed.
- C. R. Kim, J. S. Baeck, Y. Chang, J. E. Bae, K. S. Chae and G. H. Lee, Phys. Chem. Chem. Phys., 2014, 16, 19866–19873 RSC.
- T. Tegafaw, W. Xu, S. H. Lee, K. S. Chae, H. Cha, Y. Chang and G. H. Lee, AIP Adv., 2016, 6, 065114 CrossRef.
- X. Miao, W. Xu, H. Cha, Y. Chang, I. T. Oh, K. S. Chae, T. Tegafaw, S. L. Ho, S. J. Kim and G. H. Lee, Appl. Surf. Sci., 2019, 477, 111–115 CrossRef CAS.
- A. Stepanov, V. Burilov, M. Pinus, A. Mustafina, M. H. Rümmeli, R. G. Mendez, R. Amirov, S. Lukashenko, E. Zvereva, S. Katsuba, J. Elistratova, I. Nizameev, M. Kadirov and R. Zairov, Colloids Surf., A, 2014, 443, 450–458 CrossRef CAS.
- A. Roch, R. N. Muller and P. Gillis, J. Chem. Phys., 1999, 110, 5403–5411 CrossRef CAS.
- R. Zairov, S. Pizzanelli, A. P. Dovzhenko, I. Nizameev, A. Orekhov, N. Arkharova, S. N. Podyachev, S. Sudakova, A. R. Mustafina and L. Calucci, J. Phys. Chem. C, 2020, 124, 4320–4329 CrossRef CAS.
- E. Illés, E. Tombácz, M. Szekeres, I. Y. Tóth, Á. Szabó and B. Iván, J. Magn. Magn. Mater., 2015, 380, 132–139 CrossRef.
- Y. J. Jang, S. Liu, H. Yue, J. A. Park, H. Cha, S. L. Ho, S. Marasini, A. Ghazanfari, M. Y. Ahmad, X. Miao, T. Tegafaw, K.-S. Chae, Y. Chang and G. H. Lee, Diagnostics, 2021, 11, 2 CrossRef CAS PubMed.
- S. Marasini, H. Yue, S. L. Ho, J. A. Park, S. Kim, J. U. Yang, H. Cha, S. Liu, T. Tegafaw, M. Y. Ahmad, A. K.-A. A. Saidi, D. Zhao, Y. Liu, K.-S. Chae, Y. Chang and G. H. Lee, Appl. Sci., 2021, 11, 2596 CrossRef CAS.
- F. Söderlind, H. Pedersen, R. M. Petoral, P. O. Käll and K. Uvdal, J. Colloid Interface Sci., 2005, 288, 140–148 CrossRef PubMed.
- A. Boutahar, R. Moubah, E. K. Hill, H. Lassri and E. Lorenzo, Sci. Rep., 2017, 7, 13904 CrossRef CAS PubMed.
- A. Sidorowicz, A. Wajler, H. Weglarz, K. Jach, K. Orliński and A. Olszyna, Int. J. Appl. Ceram. Technol., 2016, 13, 302–307 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- H. Hu, J. Saniger, J. Garcia-Alejandre and V. M. Castaño, Mater. Lett., 1991, 12, 281–285 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581–587 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643–648 CrossRef CAS.
- F. Askari, M. Zandi, P. Shokrolahi, M. H. Tabatabaei and E. Hajirasoliha, Prog. Biomater., 2019, 8, 169–183 CrossRef CAS PubMed.
- M. Imani, S. Sharifi, H. Mirzadeh and F. Ziaee, Iran. Polym. J., 2007, 16, 13–20 CAS.
- M. K. Corbierre, N. S. Cameron and R. B. Lennox, Langmuir, 2004, 20, 2867–2873 CrossRef CAS PubMed.
-
CRC Handbook of Chemistry and Physics, Internet Version, ed., D. R. Lide, 2005, CRC Press, Boca Raton, FL, USA, pp. 4–60 (Ho2O3) and pp. 4–90 (Tm2O3) Search PubMed.
- W. C. Koehler, E. O. Wollan and M. K. Wilkinson, Phys. Rev., 1958, 110, 37–40 CrossRef CAS.
- H. B. Lal, V. Pratap and A. Kumar, Pramana, 1978, 10, 409–412 CrossRef CAS.
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth Heinemann, Oxford, UK, 2nd edn, 1997, p. 1243 Search PubMed.
- A. C. Anseimo, V. Gupta, B. J. Zern, D. Pan, M. Zakrewsky, V. Muzykantov and S. Mitragotri, ACS Nano, 2013, 7, 11129–11137 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Huan
Yue
a,
Son Long
Ho
a,
Huan
Yue
a,
Son Long
Ho