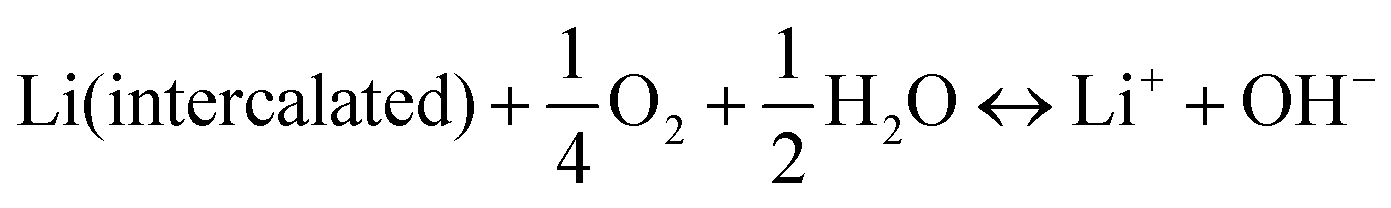DOI:
10.1039/D2EE00004K
(Review Article)
Energy Environ. Sci., 2022,
15, 1805-1839
Historical development and novel concepts on electrolytes for aqueous rechargeable batteries†
Received
2nd January 2022
, Accepted 8th April 2022
First published on 8th April 2022
Abstract
In battery systems, aqueous electrolytes are superior in ionic conductivity, interfacial wettability, safety and environmentally benign compared to organic liquids, polymers, inorganic solid-state and ionic liquid electrolytes. However, their narrow electrochemical stability window, electrode dissolution/side reactions and temperature-variation instability result in a low energy density, undesirable cyclic lifespan and limited working temperature window in aqueous rechargeable batteries. Herein, the historical development of aqueous electrolytes in various types of batteries, ranging from monovalent-ion batteries, multivalent-ion batteries, metal–air batteries, metal–chalcogen batteries, and hybrid batteries to redox flow batteries is summarized through the representative pioneering works. Meanwhile, the strategies of electrolyte additive, pH-management, salt-concentrated electrolyte, gelling, solvent hybridizing, interface tuning and beyond concentrated electrolyte to mitigate the challenges associated with aqueous electrolytes are reviewed and discussed in detail. All these new electrolyte concepts mutually stabilize aqueous rechargeable batteries and improve their corresponding energy density in a broad temperature range, ushering in a promising aqueous energy storage era.
Broader context
Batteries are key technology to mitigate carbon emissions, which are closely related to energy and environmental science. The electrolyte is the most important material in batteries, which thermodynamically and kinetically affect their overall performance. Therefore, it is necessary to understand the principles, historical development and new concepts of electrolytes for the development of enhanced batteries, particularly for next-generation batteries such as aqueous rechargeable batteries. Aqueous rechargeable batteries exhibit high safety, excellent rate capability, desirable environmentally benign nature and cost-effective manufacturing, making them suitable for stationary energy storage. Their corresponding electrolytes can be improved in terms of electrochemical stability window, interfacial stability and temperature-variation stability to potentially extend their applications to electric vehicles to achieve a carbon neutral world. In this review, not only the historical development and new concepts of electrolytes for aqueous rechargeable batteries have been comprehensively and systematically summarized, but a visionary perspective is also provided. The perspective part concentrates on both an academic roadmap for fundamental understanding (characterization and modeling) of electrolytes and the electrode/electrolyte interface and industrial routes for commercializing aqueous rechargeable batteries. This review offers readers a convenient and fast way to understand the key electrolytes for aqueous rechargeable batteries, and also promotes research on improving the relevant electrolytes.
|
1. Introduction
Climate change has attracted global attention, and hence under the UN Paris Agreement, countries are committed to restricting global warming to less than 2 °C and are actively pursuing a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C limitation.1–3 This has triggered a boom in research and practice towards carbon dioxide mitigation, ranging from carbon avoidance (photovoltaic power stations and hydrogen energy) and carbon embedding (methanol) to carbon removal (bioenergy crops and carbon capture technology).4,5 The popular strategy of using electric energy in automobiles, which are known as electric vehicles (EVs), can reduce the overall energy usage by enhancing energy efficiency, thus reducing carbon emissions, which can also be categorized into carbon avoidance.4,6,7 Transport is the second largest source of CO2 emissions, with 24% of the total emissions, where the fuel efficiency of the mode of travel can significantly influence CO2 emissions largely.8 Compared with current internal combustion engine vehicles, EVs are expected to reduce well-to-wheel emissions by 70–85% by 2030 with virtually zero tailpipe emissions and excellent energy utilization efficiency, which requires the total use of EVs in the transport sector.8 The commercialization of EVs, involving full battery electric vehicles (BEVs), hybrid electric vehicles (HEVs) and hybrid plug-in electric vehicles (HPEVs), demands mature battery technology with the properties of high energy density, high power density and long lifespan, which currently, commercialized batteries especially lithium-ion batteries (LiBs) fail to satisfy.7,9,10
°C limitation.1–3 This has triggered a boom in research and practice towards carbon dioxide mitigation, ranging from carbon avoidance (photovoltaic power stations and hydrogen energy) and carbon embedding (methanol) to carbon removal (bioenergy crops and carbon capture technology).4,5 The popular strategy of using electric energy in automobiles, which are known as electric vehicles (EVs), can reduce the overall energy usage by enhancing energy efficiency, thus reducing carbon emissions, which can also be categorized into carbon avoidance.4,6,7 Transport is the second largest source of CO2 emissions, with 24% of the total emissions, where the fuel efficiency of the mode of travel can significantly influence CO2 emissions largely.8 Compared with current internal combustion engine vehicles, EVs are expected to reduce well-to-wheel emissions by 70–85% by 2030 with virtually zero tailpipe emissions and excellent energy utilization efficiency, which requires the total use of EVs in the transport sector.8 The commercialization of EVs, involving full battery electric vehicles (BEVs), hybrid electric vehicles (HEVs) and hybrid plug-in electric vehicles (HPEVs), demands mature battery technology with the properties of high energy density, high power density and long lifespan, which currently, commercialized batteries especially lithium-ion batteries (LiBs) fail to satisfy.7,9,10
In fact, the bottleneck in promoting the use of EVs promotion is the battery requirements, given that the current batteries exhibit undesirable energy densities, low power densities and safety issues, which are caused by the relatively low capacity of their positive electrode, sluggish electrode kinetics and/or electrode/electrolyte interface kinetics, and flammable organic solvent of electrolytes, respectively.11–15 Furthermore, although the cyclic lifespan of commercial LiBs (electrode) is acceptable, the degradation of their electrolyte can shorten the lifespan of a whole cell and even induce potential safety hazards.13,14 Concerning these issues, several new concepts ranging from lithium//air batteries, lithium//sulfur batteries and flow batteries (innovations on the cathodes)16–18 to solid-state batteries, quasi-solid-state batteries and aqueous batteries (innovations in the electrolyte) have been proposed and widely researched.19–21 Meanwhile, the capacity of the cathode is influenced by the interfacial kinetics and mass transportation between the electrolyte and cathode, where the working potential of the cathode will decrease due to interfacial polarization, thereby reducing the energy density of the whole cell.22,23 Therefore, electrolytes are of paramount importance for the development of batteries with high energy density and good safety.
An ideal electrolyte material should possess properties such as good electronic insulation, high ionic conductivity, wide electrochemical stability widow (ESW), thermal stability and safety, facilitate interfacial kinetics, low chemical “cross-talk” between electrodes, be produced via simple large-scale manufacturing process, inexpensive, environmentally friendly and non-toxic;24,25 however, currently no electrolyte can meet all these criteria. The conventional electrolytes can be classified into organic liquid electrolytes, polymer electrolytes, inorganic solid-state electrolytes, ionic liquid electrolytes and aqueous electrolytes, which are semi-quantitatively compared in Fig. 1. Among them, aqueous electrolytes are impressive in terms of their ionic conductivity, environmentally benign nature and interfacial wettability.26–33
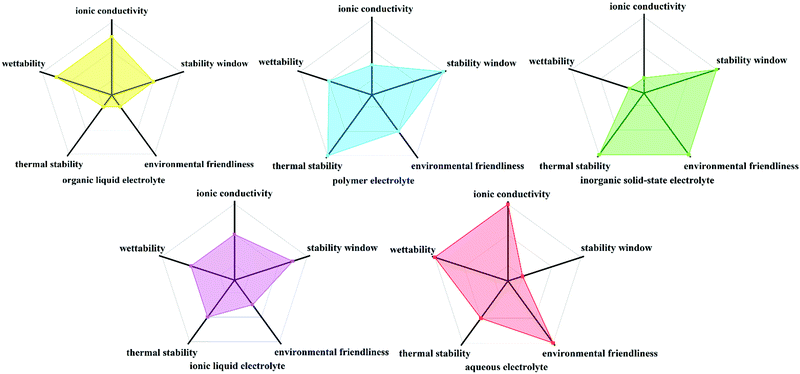 |
| | Fig. 1 Semi-quantitative comparison of organic liquid electrolyte, polymer electrolyte, inorganic solid-state electrolyte, ionic liquid electrolyte and aqueous electrolyte. | |
Exploring safe and cost-effective electrochemical energy storage systems is of great significance and importance.34,35 Based on aqueous electrolytes, aqueous rechargeable batteries (ARBs) have move to the forefront, which are destined to avoid the potential flammability and explosion of organic liquid electrolyte-based batteries and sophisticated (atmosphere controlled) manufacturing processes.36,37 Furthermore, due to the high permittivity and low viscosity of water, the ionic conductivity of aqueous electrolytes is considerably higher than that of organic liquid electrolytes, resulting in an excellent rate capability and power density in ARBs.29,36 Thus, based on the above-mentioned advantages of aqueous electrolytes, ultrafast charge–discharge energy storage systems can be established on a grid-scale with ARBs.29,38 However, there is a noticeable gap between conventional LiBs and ARBs in terms of energy density, which restricts the large-scale application and eliminates the low cost and good safety benefits of ARBs in the EV industry.39,40
The relatively low energy density of ARBs is basically derived from the low working voltages caused by their narrow ESWs, which is related to the oxygen evolution in high-potential and hydrogen evolution in low-potential aqueous electrolytes. To some extent, the narrow ESWs of aqueous electrolytes is the Achilles’ heel in improving the energy density of ARBs.36,41 In 2013, Wu et al. innovatively utilized a sandwich polymer membrane and LISICON to coat an Li metal anode. Due to the ‘‘cross-over’’ effect of the Li ion in the coated LISICON, this ARB delivered an output voltage of about 4.0 V, a big breakthrough in the theoretically ESW of water; however, it exhibited poor cyclic stability.42 Furthermore, the issue regarding the ESW can induce a short cyclic lifespan, thus increasing the cost to run energy storage stations based on ARBs.43 It is noteworthy that electrode dissolution in aqueous electrolytes can hinder the long-term charge–discharge cycling due to the strong polarity of water molecules. Theoretically, mass dissolution is a thermodynamically favorable process, as determined by the Gibbs free energy, regardless of the acid/alkaline environment of the electrolyte in real situations.44,45 Besides narrow ESWs and electrode dissolution, batteries can become unstable due to the side reactions of the electrodes with water and/or oxygen, especially in the case of aqueous rechargeable lithium-ion batteries (ARLiBs).41 Moreover, the phase transition of water with a variation in temperature can limit the application of ARBs at elevated or subzero temperatures, thereby confining the operation temperature windows of aqueous electrolytes.46 Hereon, the methods that can be employed to increase the energy density, expand the cyclic lifespan and extend the working temperature window of ARBs by tailoring aqueous electrolytes are summarized and discussed. Before that, the design concepts (principles) and historical development of aqueous electrolytes are briefly introduced and summarized in the following section.
2. Design concepts and historical development of aqueous electrolytes
2.1. Principles of battery electrolyte
The typical liquid electrolyte used in rechargeable batteries, fuel cells, and electrochemical capacitors is composed of a solvent, anion, and solvent-solvated cation. In the case of organic liquid electrolytes, the solid electrolyte interface (SEI) can function as a critical component to equip electrolytes with both electrochemical and thermal stability as well as excellent rate capability,47,48 which is absent in conventional aqueous electrolytes. Thus, a reasonable design is necessary for aqueous electrolytes, enabling good stability of the electrolyte/electrode interfaces. The design of an electrolyte is based not only on whether the reactants in both sides may be solid, liquid, or gaseous (processing consideration), but also the limiting electrochemical potentials μA and μC for the anode and cathode (their Fermi energies), respectively.49 The ESW of the electrolyte originates from the separation energy (Eg) of the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) of the electrolyte.50 Thus, to achieve thermodynamic stability in a battery, μA and μC should be located in the ESW of the electrolyte, as exhibited in the schematic illustration of the electron energies of the electrode and electrolyte in Fig. 2a. The open circuit potential (VOC) of a battery cell can be defined as follows:51where e is the magnitude of the electronic charge, while for organic liquid electrolytes, the passivating SEI film offers kinetic stability to a large VOC with no extra-large.51 This is because an anode with a μA above the LUMO can reduce the electrolyte unless a passivation layer generates a barrier to electron transfer from the anode to the electrolyte LUMO. Similarly, a cathode with a μC below the HOMO will oxidize the electrolyte unless a passivation layer blocks electron transfer from the electrolyte HOMO to the cathode.51
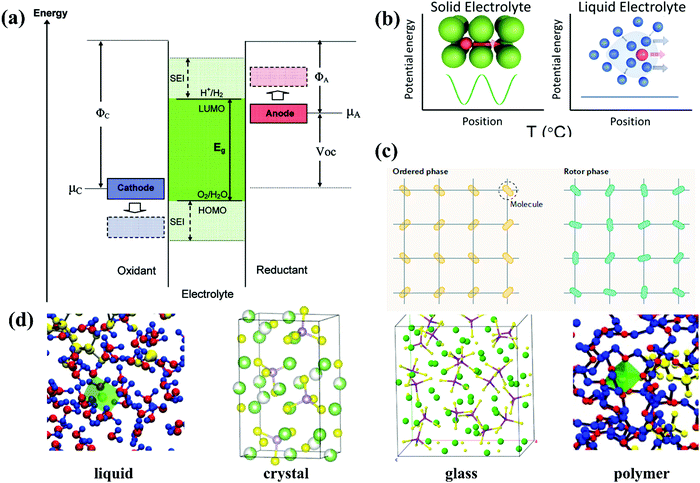 |
| | Fig. 2 (a) Schematic open-circuit energy diagram of an aqueous electrolyte.51 (b) Potential energy of migration in liquid electrolytes of a charged species in red with a solvation shell of electrolyte molecules (highlighted in blue) and an interstitial mobile ion in a crystalline solid, respectively.59 (c) Comparison of the orientations of molecules in ordered phases and rotor phases (plastic crystals).62 (d) Solvation structure of liquid, crystal, glass and polymer examples.63 | |
Experimentally, the ESW can be determined through standard potentiodynamic approaches such as linear sweep voltammetry (LSV) and cyclic voltammetry (CV), despite the various electrochemical environments of electrolytes during actual applications.52,53 The restricted reduction and oxidation potentials are assigned to a predefined value (reaching the decomposition current). The cutoff criteria may be arbitrary given that it lacks theoretical significance. In fact, the observed total current of the working electrode, iT, can be expressed as the sum of two parts, as shown in the following equation:53
where
inF represents the nonfaradaic component (capacitive, interface double layer) and
iF represents the faradaic component (impurity reactions, redox process, and decomposition of the electrolyte at the interface).
53 Ideally,
inF should be close to zero if
iT indicates the ESW of the electrolyte exactly. Thus, practically,
inF should be eliminated when predefining the current level for
iT. Normally,
inF and
iF can be distinguished by the slope of the LSV/CV curve. A lower slope in the LSV/CV curve is assigned to
inF, appearing earlier, whereas a higher slope is assigned to
iF, appearing later.
54,55 Also, non-porous electrodes such as Pt and glassy carbon can create the circumstance of
inF ≈ 0.
53
Conducting ions are the essential requirement of electrolytes, which affect how fast the energy stored in the electrodes can be delivered. In liquid electrolytes, the transport of ions is realized by the solvation and dissociation of ionic compounds and the migration of these solvated ions through the solvent.56 In terms of solvation and migration, ionic conductivity can be qualified as follows:57
| | 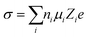 | (3) |
while for most liquid electrolytes, the conductivity results from the overall migration of both anions and cations. Also, for most batteries, only the portion of current carried by the cation is important, and thus the concept of transference number (
tcation) is introduced as follows:
57| |  | (4) |
In diluted electrolyte, the transference number of cations is normally 0.2 to 0.4, meaning that anions are much more mobile than cations due to the solvation sheath around cations.56 A transference number of less than 1 can remarkably induce concentration polarization during battery operation through the resultant overwhelming anion movement and enrichment near the electrode surface, which requires a relatively “naked” cation with a high cation current.56
Meanwhile, solvents with lower viscosity (η) can enhance the mobility of ions and ameliorate the concentration polarization according to the Stokes–Einstein relation, as follows:57
| | 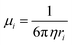 | (5) |
where
ri is the solvation radius. A lower
η leads to a higher
μi, while a lower solvation degree of cation (solvation radius) leads to a higher
μi. Besides, the dielectric constant,
ε, of the solvents is also critical, which determines the charge carrier number (
ni). With a higher
ε, ions have a higher probability of remaining free.
58 Therefore, in terms of ionic conductivity, an ideal solvent should demonstrate a high
ε and low
η, which is difficult to realize in organic solvent systems, while water can be an optimized option, thereby offering desirable ionic conductivity to high power density ARBs.
The ion conduction mechanisms between liquid and solid electrolytes are different. The potential energy profile of mobile ions in liquid electrolytes can be considered flat (Fig. 2b, right) due to the reasonably fast exchange between the solvating molecules and the solvent molecules, and the uniform surroundings. In contrast, the diffusion of mobile species in a solid-state crystalline network needs to pass through periodic bottleneck points, which define an energetic barrier that separates two local minima (typically crystallographic sites for ions) along the minimum energy pathway (Fig. 2b, left).59 Therefore, the ion conduction mechanism in liquid electrolytes is concentration diffusion, whereas that in solid especially crystal solid electrolytes is ion hopping between the ground-state stable sites and/or intermediate metastable sites of the crystalline framework. In the crystalline case, vacancies of cations or interstitials are perceived as the mobile charged species, the migration mechanisms of which can be summarized as ion migration to a neighboring vacant site, direct interstitial mechanism between sites not fully occupied and concerted or correlated interstitially (knock-on) mechanism.22 The ionic conductivity of a solid crystal electrolyte can be thermally activated, the process of which is described by a modified Arrhenius relationship, while the ion absolute mobility, μ, can be linked to the ion diffusion coefficient, D, through the Nernst–Einstein relationship.
In the case of polymer and glass, which are two solid electrolytes exhibiting no long-range structural order, their mechanisms vary accordingly. In polymer electrolytes, the relationship curve between σ and 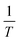 is bent and is best fitted to VTF behavior (after Vogel, Tammann, and Fulcher). To (equilibrium glass-transition temperature) is introduced for amendment, which is related to the (kinetic) measured glass-transition temperature.60 Glass is a promising electrolyte for use in solid-state batteries. Although the Arrhenius relationship and Nernst–Einstein relationship can still be applied to glass electrolytes, cation migration occurs via a mechanism that combines the concerted motion of cations with the large, quasi-permanent reorientation of anions. This effect, known as the ‘paddlewheel’ mechanism, is typically observed in high-temperature crystalline polymorphs. In contrast to the behavior of crystalline materials, the glass paddlewheel dynamics contribute to ion mobility at room temperature.61 The dynamics of ion motions in these materials are often described as being strongly coupled to the structural modes, whereas intermediate degrees of coupling are observed in organic ionic plastic crystals. Owing to the relatively weak interactions between molecules, reorientational processes occur with little hindrance. The orientations of molecules in ordered phases and rotor phases are illustrated in Fig. 2c.62 Recently, in one study, a unified framework was built for all liquid and solid (crystal, polymer and glass) electrolytes based on the concepts of solvation shell for liquids and solvation cage for solids. An LiTFSI aqueous solution, β-Li3PS4 at 537 K, amorphous Li3PS4 glass at 298 K and LiTFSI in polyethylene oxide (PEO) at 363 K were utilized as examples in that work for liquid, crystal, glass and polymer, respectively (Fig. 2d), to study the Li-ion solvation structure.63 The ultimate goal was enabling the rational design of the solvation environment to propel the migration of the working ions in a unified framework.
is bent and is best fitted to VTF behavior (after Vogel, Tammann, and Fulcher). To (equilibrium glass-transition temperature) is introduced for amendment, which is related to the (kinetic) measured glass-transition temperature.60 Glass is a promising electrolyte for use in solid-state batteries. Although the Arrhenius relationship and Nernst–Einstein relationship can still be applied to glass electrolytes, cation migration occurs via a mechanism that combines the concerted motion of cations with the large, quasi-permanent reorientation of anions. This effect, known as the ‘paddlewheel’ mechanism, is typically observed in high-temperature crystalline polymorphs. In contrast to the behavior of crystalline materials, the glass paddlewheel dynamics contribute to ion mobility at room temperature.61 The dynamics of ion motions in these materials are often described as being strongly coupled to the structural modes, whereas intermediate degrees of coupling are observed in organic ionic plastic crystals. Owing to the relatively weak interactions between molecules, reorientational processes occur with little hindrance. The orientations of molecules in ordered phases and rotor phases are illustrated in Fig. 2c.62 Recently, in one study, a unified framework was built for all liquid and solid (crystal, polymer and glass) electrolytes based on the concepts of solvation shell for liquids and solvation cage for solids. An LiTFSI aqueous solution, β-Li3PS4 at 537 K, amorphous Li3PS4 glass at 298 K and LiTFSI in polyethylene oxide (PEO) at 363 K were utilized as examples in that work for liquid, crystal, glass and polymer, respectively (Fig. 2d), to study the Li-ion solvation structure.63 The ultimate goal was enabling the rational design of the solvation environment to propel the migration of the working ions in a unified framework.
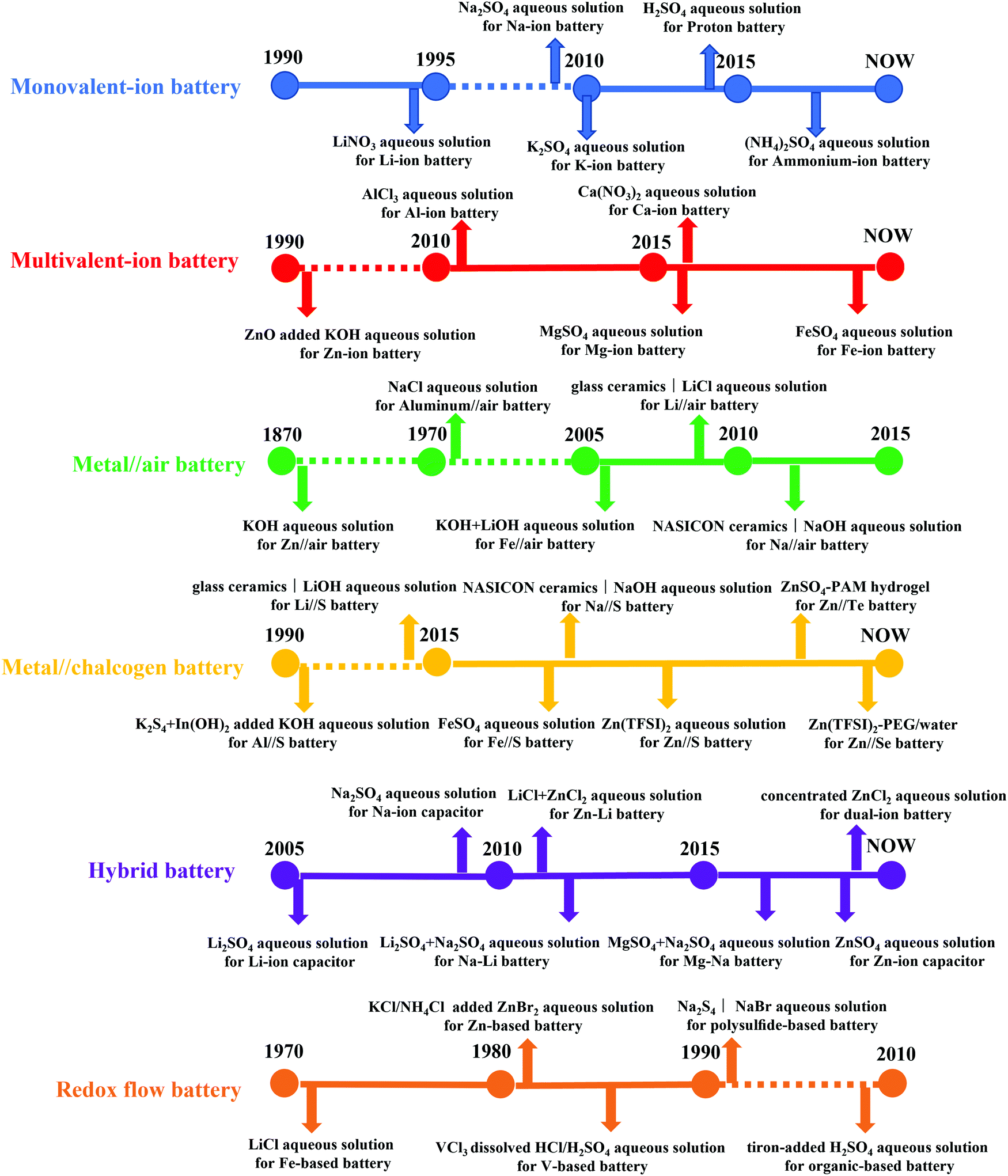
Based on this design concept, numerous aqueous electrolytes have been developed and applied in various types of ARBs. In the following sections, the historic development of aqueous electrolytes is summarized in different types of ARBs, ranging from monovalent-ion batteries, multivalent-ion batteries, metal//air batteries, metal//chalcogen batteries, and hybrid batteries to redox flow batteries.
2.2. Monovalent-ion battery
Among the monovalent-ion batteries, LiBs are the most popular and practical, while sodium-ion batteries (SiBs), potassium-ion batteries (PiBs), even proton batteries (PBs) and ammonium-ion batteries (AiBs) are starting to play an important role at this stage owing to the growing concern regarding lithium resources, manipulating the development of the corresponding aqueous electrolytes.64 In 1994, the first ARLiB was introduced by Jeff Dahn and co-workers with VO2//LiMn2O4 chemistry in 5 M LiNO3 aqueous solution, delivering an energy density of 75 W h kg−1, which was regarded as a breakthrough in energy storage systems.65 In 2009, Na2SO4 aqueous solutions were employed as electrolytes for realizing Na+ reversible intercalation/deintercalation in an NaMnO2 cathode,66 which were further developed in 2010, coupling an NaTi2(PO4)3 anode and Na4Mn9O18 cathode.67,68 Compared with lithium, sodium offers multiple advantages ranging from its wide abundance in the Earth's crust and dramatically lower cost to even distribution worldwide, and hence has attracted a great deal of attention.69,70 Meanwhile, K2SO4 aqueous solution facilitated K+ reversible intercalation/deintercalation in the K0.27MnO2·0.6H2O cathode in 2010.71 Subsequently, a Prussian blue analogue (PBA), nickel hexacyanoferrate was proposed to function as a cathode for PiB with 1 M KNO3 aqueous electrolyte. Its open-framework structure allowed the retention of 66% of the initial capacity even at a C rate of 41.7. The high power density, safety, and low-cost production method of this device make an attractive candidate for use in large-scale energy storage to support the electrical grid.72 Proton insertion/extraction involving electrochemical energy chemistries have been reported as hybrid devices,73 until the discovery of a pure proton rocking-chair capacitor with anthraquinone (AQ)//tetrachlorohydroquinone (TCHQ) redox couple and 0.5 M H2SO4 aqueous electrolyte.74 Recently, 62 wt% (9.5 m, molality, molar salt in kg solvent) H3PO4 was reported as an electrolyte for a high-rate aqueous proton battery, with Mn2O3 as the anode and CuFe-Turnbull's blue analogue (H-TBA) as the cathode. It is noteworthy that the unit of m is different from the conventional solution concentration unit of M (molarity, molar salt in liter solution) because of the evident variation in the density of solution, and thus the volume change caused by a high salt concentration.75–77 As a eutectic mixture electrolyte with a low melting point, the 9.5 m H3PO4 electrolyte enhanced the low-temperature performance of PBs even at −78 °C.78 In 2017, the first rocking-chair ammonium-ion battery was demonstrated with an ammonium Prussian white analogue cathode, organic solid as the anode and 1.0 M (NH4)2SO4 aqueous solution as the electrolyte. Although the average working voltage and energy density of that battery were ∼1.0 V and ∼ 43 W h kg−1, respectively, this performance was still encouraging, inspiring research towards the utilization of ammonia waste.79 Subsequently, Wu and co-workers further introduced sodium iron hexacyanoferrate and copper hexacyanoferrate cathodes in 1 M (NH4)2SO4 (pH ≈ 5.4) aqueous solution to enhance the rate performance and stability of ammonium-ion batteries.80,81
2.3. Multivalent-ion Battery
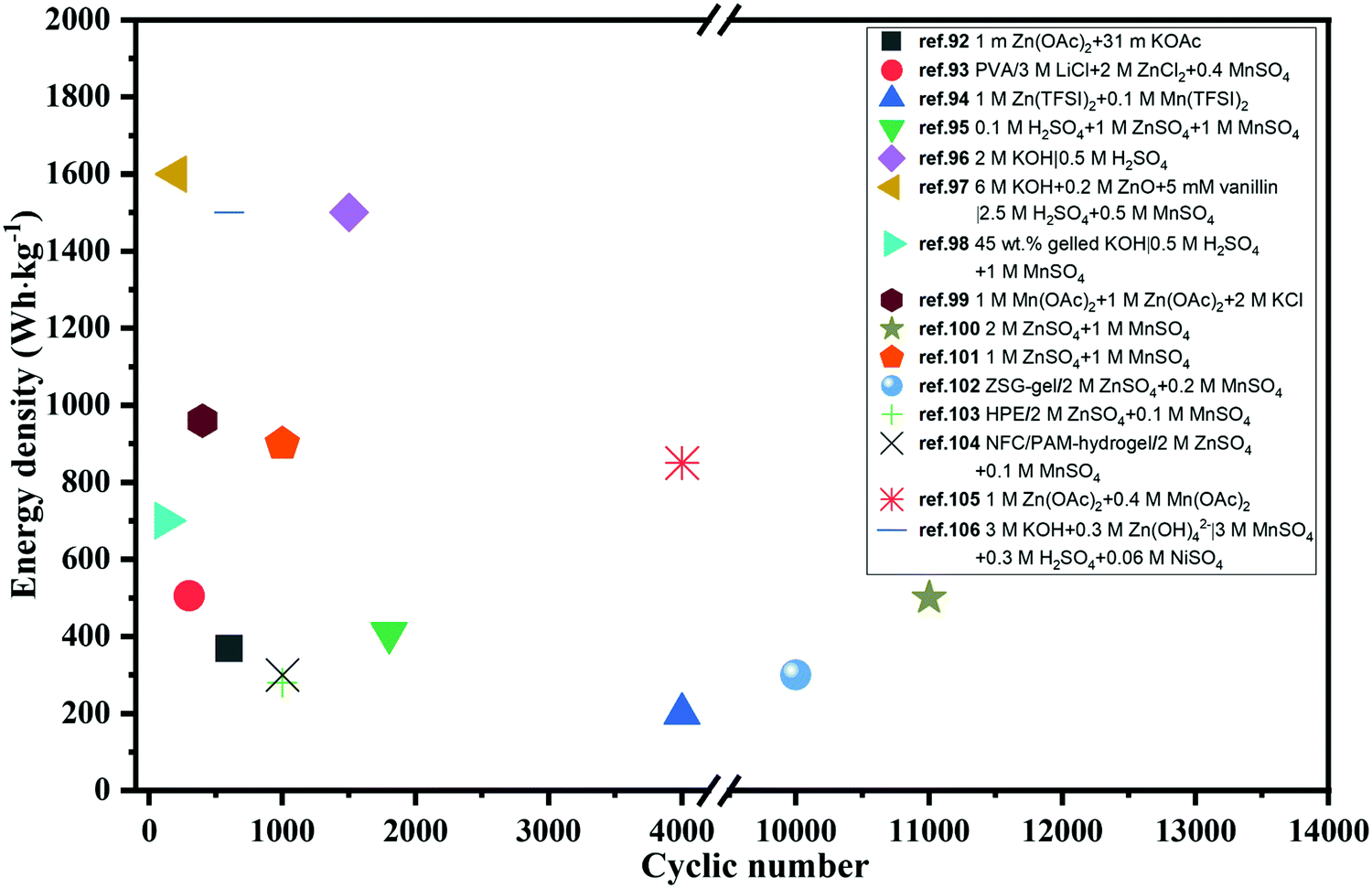
The energy density of multivalent-ion batteries (MViBs) is theoretically higher than that of the presently commercialized LiBs due to the higher capacity induced by the multivalent ion insertion in the cathode.82 Magnesium (Mg), aluminum (Al), calcium (Ca) and zinc (Zn) ion batteries dominate among the MViBs, while iron (Fe) and copper (Cu) ion batteries have also emerged recently.83–85 Similar with ARLiBs, aqueous electrolytes make MViBs safe and environmentally friendly, even perform bettering than ARLiBs because of their higher energy density, and hence water in MViBs can open doors to new opportunities.86 Among them, aqueous rechargeable zinc ion batteries (ARZiBs) can be further selected as the crown jewel due to their features of high abundance, large-scale production, nontoxicity, low redox potential (−0.76 V vs. standard hydrogen electrode (SHE)) and high overpotential for hydrogen evolution reactions (contributing to the thermodynamic stability against aqueous electrolytes).87,88 Alkaline Zn//MnO2 batteries with KOH aqueous solution served as the primary battery since the 1960s, and efforts to make them rechargeable started in the 1990s, even expanding manganese-based cathodes to vanadium-based materials and PBA.89–91 In the last three years, remarkable progress has been achieved in the case of ARZiBs, especially for aqueous Zn–MnO2 batteries through the technical routines towards aqueous electrolytes such as hydrogel, salt-concentrated, pH-adjusting and decoupling electrolytes. Some representative works are exhibited with the employed electrolytes and compared in terms of energy density and lifespan (cyclic number) in Fig. 3,92–106 where the energy density of various aqueous Zn//MnO2 batteries is unified based on the mass of MnO2, and some of them are basically estimated. There is an obvious tendency to employ aqueous Zn//MnO2 batteries as high-energy storage devices, which are almost ready for further commercialization on a large scale, while their flexible and stretchable properties enabled by hydrogel electrolytes extend their applications from electric grids to daily-use, wearable electronic devices.96,107
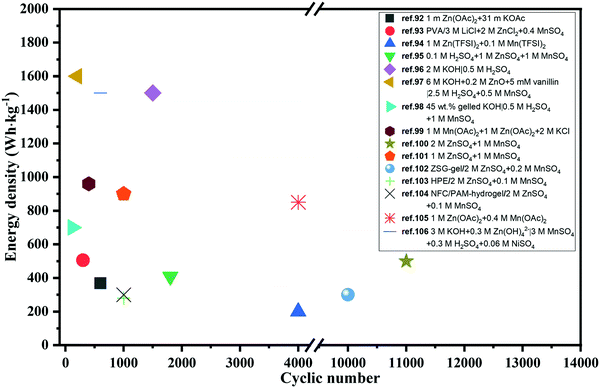 |
| | Fig. 3 Performance comparison of recently reported Zn//MnO2 batteries based on various aqueous electrolytes in terms of energy density and cyclic number. | |
Besides Zn, Al is also an abundant and cheap element, which possesses a superior volumetric capacity to that of Mg and Ca.108 As early as around 1980, an NaCl aqueous solution was used as the electrolyte for Al-alloy//air batteries.109 In 2012, Liu et al. demonstrated Al-ion insertion/extraction in anatase TiO2 electrode with an AlCl3 aqueous solution as the electrolyte;110 however, it is highly acidic in nature and enhances the dissolution of the active material and corrosion of auxiliary battery parts.108 To overcome this drawback, Al(NO3)3 and Al2(SO4)3 aqueous solutions have been developed as electrolytes for aqueous rechargeable aluminum-ion batteries (ARAiBs). However, the corresponding electrochemical activity was weakened in these electrolytes, especially for Al-ion insertion/extraction in anatase TiO2 electrodes. Therefore, mesoporous nanosized electrodes have been demonstrated to improve the electrode and interfacial kinetics, and thus enhance the discharge capacity.108 Mg is regarded another alternative to Li, but it suffers a lot from sluggish kinetics in organic liquid electrolytes. Therefore, water was introduced in a non-aqueous system to enhance the kinetics in electrolytes and cathodes, and thus the rate capability of the whole cell.111 Since 2013, numerous works focused on aqueous Mg chemistries have been reported in half-cells (mostly using Mg(NO3)2 aqueous solutions as electrolytes);112,113 however, it was only in 2017 that Chen et al. reported the fabrication of a full Mg-ion battery (MiB) based on 1 M MgSO4 aqueous solution with a polyimide//nickel hexacyanoferrate redox couple.114 In the same year, Wang et al. used a 4 m (molar salt in kg solvent) magnesium bis(trifluoromethane sulfonyl)imide (Mg(TFSI)2) super-concentrated electrolyte to expand the ESW to 2.0 V, enabling poly pyromellitic dianhydride//lithium vanadium phosphate battery chemistry with a working voltage of 1.9 V.111 Likewise, water was introduced in a Ca ion battery to improve the electrolyte and electrode/electrolyte kinetics, while a super-concentrated aqueous electrolyte (8.37 m Ca(NO3)2) was used with a CuHCF working electrode in a half-cell system, delivering a capacity of almost 70 mA h g−1.115 In 2017, the combination of 2.5 M Ca(NO3)2 aqueous electrolyte with an organic polyimide anode and CuHCF cathode as a full cell was reported, displaying 1000 cycles of charge–discharge under 400 mA g−1 current density with a cut-off voltage of 0.5–1.9 V.116 Among the metal elements used as cations (charge carriers) in MViBs, Fe element is even more abundant than Zn, Mg and Ca, which has a content of ∼46500 ppm in the Earth's crust.84 Reversible Fe plating/stripping was realized in an FeSO4 aqueous electrolyte with Fe-based PBAs, which exhibited an excellent cycling performance of 1000 cycles. The authors also showed that the Fe metal anode could pair with an LiFePO4 cathode as a hybrid cell, displaying a discharge capacity of ∼155 mA h g−1.84 In summary, ARZiBs and ARAiBs are more suitable to be industrialized than the other types of ARBs at this stage in terms of reliable performance and cost. Particularly, ARZiBs are almost ready for application in portable electronic devices and large-scale energy storage stations, especially Zn//MnO2 batteries, as shown in Fig. 3, but their energy density needs to be further improved for EV applications.
2.4. Metal//air battery
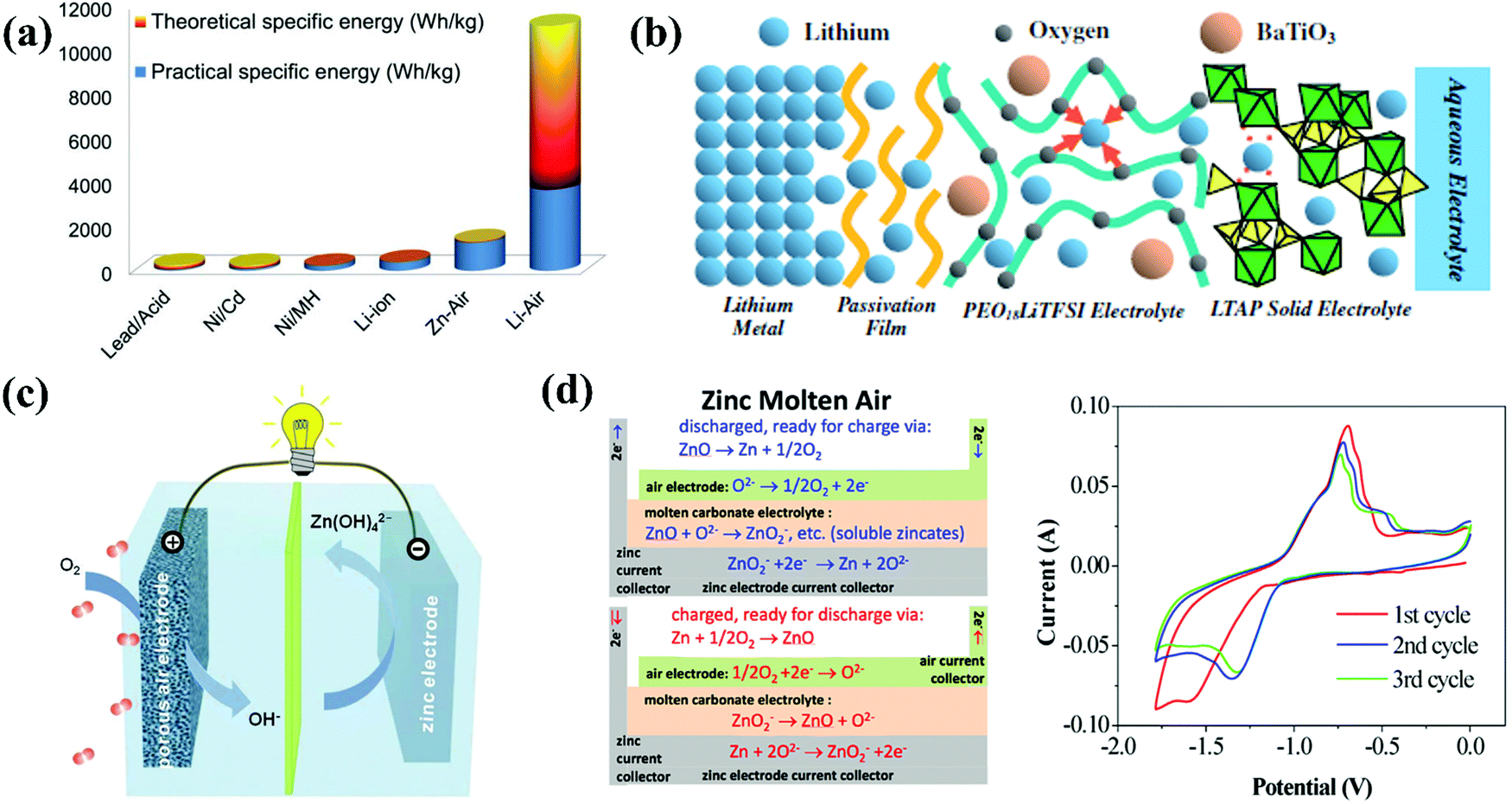
Since the 1960s and early 1970s, considerable work has been carried out on metal//air batteries (MABs) for a variety of applications.117 MABs are unique compared with other batteries owing to the fact that their electroactive material (oxygen) does not require storage. In this respect, this type of battery is similar to fuel cells and is essentially a fuel cell/battery hybrid, which principally simplifies the battery design and increases the energy density.117 Therefore, MABs have attracted much attention as a possible alternative due to their extremely high energy density compared to that of other batteries, as shown in Fig. 4a.118 The electrolytes utilized for MABs can be basically classified into aqueous electrolytes (not sensitive to moisture) and non-aqueous electrolytes (water-sensitive system with aprotic solvents). Li and Na are intrinsically suitable for non-aqueous electrolytes, while metals such as Al, Fe and Zn are appropriate for aqueous systems.118 For the first time, a water-stable Li metal anode with water-stable lithium-conducting glass ceramics (Li super ion conductor, LISICON) and a lithium-conducting polymer electrolyte was proposed as the anode for an Li//air battery (LAB) with LiCl aqueous electrolyte at the air electrode in 2008,119 possessing a similar cell configuration to that shown in Fig. 4b. Although the nonaqueous electrolyte-based LABs possessed a higher energy density/power density than that of the aqueous electrolyte-based LABs, the uncertain side reactions in the oxygen electrode and aprotic solvent-insoluble Li2O2, which usually occur in the former, can be avoided in the latter.120 In 2014, the application of a gel polymer/LISICON multilayer-coated Li metal anode was expanded to an Li//Br2 battery with 1 M LiBr2 as the electrolyte.121 Based on a similar design concept, in 2013, a mixed aqueous (NaOH)/aprotic electrolyte was realized through a separator of Na3Zr2Si2PO12 (NASICON) ceramics and applied in an Na//air battery (SAB), discharging a capacity of ∼600 mA h g−1 (energy density of ∼1500 W h kg−1) and outputting an areal powder density of 11 mW cm−2.122 Unlike LABs and SABs, Zn//air batteries (ZABs) naturally suit aqueous electrolytes, which mostly operate in alkaline media, such as KOH and NaOH, due to the higher activity of both the zinc electrode and air electrode since the first invention in 1878.123 The structures of cells based on alkaline aqueous electrolytes are much simpler than that of aqueous LABs and SABs, as shown in Fig. 4c, because no protection layers are required for Zn metal anodes. For ZABs with open structures, water loss from the aqueous electrolyte is a crucial cause of performance degradation, whereas the alkalinity of these electrolytes leads to the problem of insoluble carbonate precipitation and promotes zinc-dendrite growth and hydrogen evolution, all limiting the performance of ZABs.124 Thus, to overcome these problems through electrolytes, a molten Li0.87Na0.63K0.50CO3 eutectic electrolyte with added NaOH was prepared and employed in a ZAB, which was cycled for 110 cycles at 550 °C, having an average charging potential of ∼1.43 V and discharge potential of ∼1.04 V.125 The relevant cell configuration is shown in Fig. 4d, together with the CV curve upon Zn plating/stripping (∼1.38 V vs. Ni/Ni(OH)2 for zinc deposition and ∼0.7 V vs. Ni/Ni(OH)2 for the oxidation of Zn to Zn2+).125 Al and Fe are deemed as the first and second most abundant metallic elements in the Earth's crust. The theoretical energy density of Al//air batteries (AABs, 8140 W h kg−1) is much higher than that of ZABs (1350 W h kg−1), while that of Fe//air batteries (IABs) is 1200 W h kg−1.126,127 Considering this, AABs should be more promising; however, they suffer significantly from self-corrosion of the anode and sluggish kinetics, which demand a rational electrolyte with good stability and electrode/electrolyte interface process. Initially, NaCl aqueous solution was utilized as the electrolyte for AABs, but the energy density was restricted in the neutral condition, and then an alkaline system was introduced for not only AABs (KOH) but also IABs (KOH + LiOH).117,128
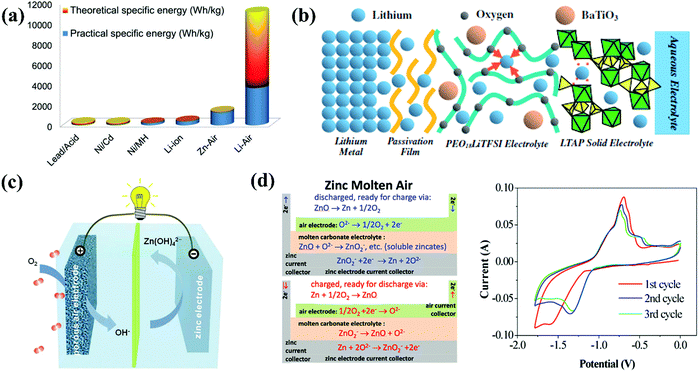 |
| | Fig. 4 (a) Theoretical and practical energy densities of various types of rechargeable batteries.118 (b) Schematic diagram of the proposed water stable lithium metal electrode with polymer buffer layer and glass ceramic separator.120 (c) Schematic principle of operation for Zn//air batteries.123 (d) Schematic representation of the molten Zn//air battery and CV curve of Zn redox reaction in that electrolyte with a sweep rate of 100 mV s−1.125 | |
2.5. Metal//chalcogen battery
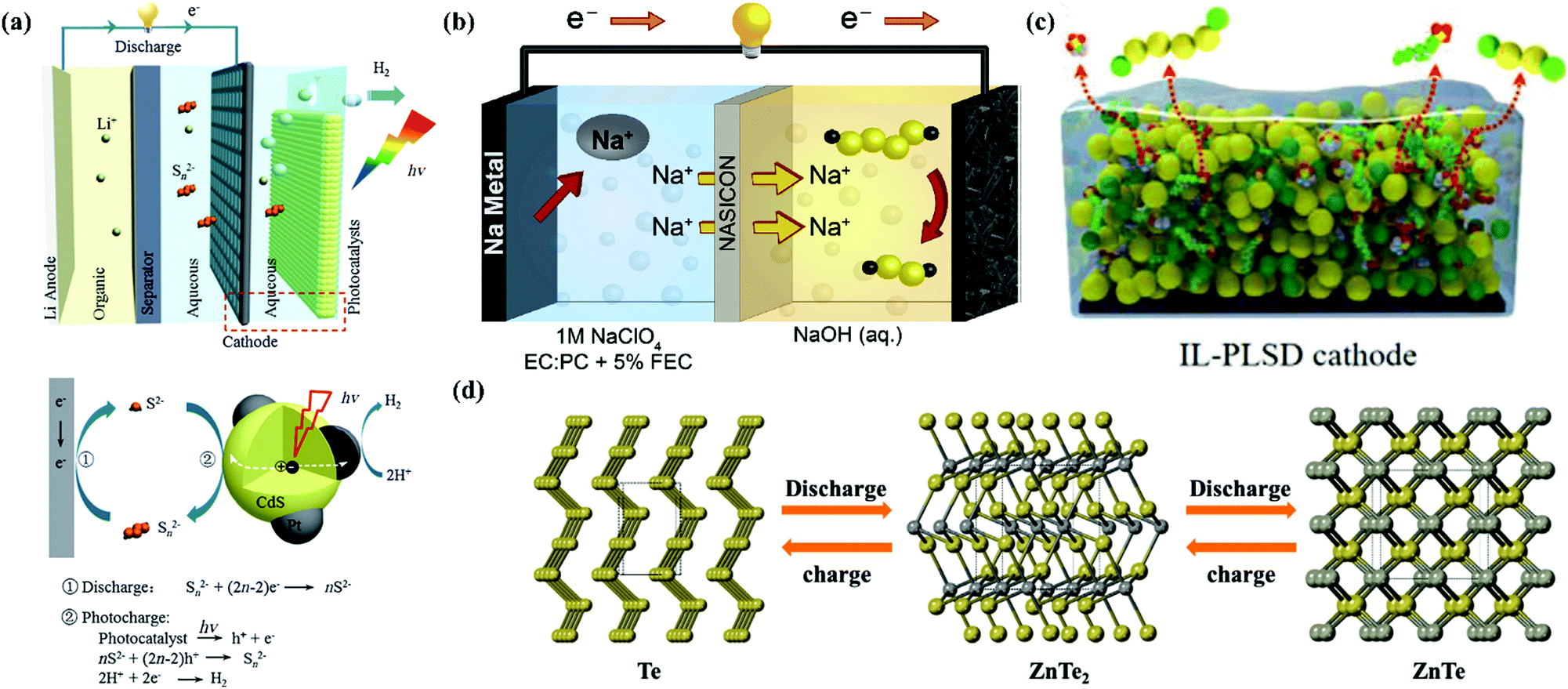
Besides MABs, metal//chalcogen batteries (MCBs) are promising alternatives because they can provide remarkable capacities, while using cheaper and more reliable electrodes of sulfur (S), selenium (Se) and tellurium (Te) than air electrodes.129,130 MCBs possess an exceptional energy density (for instance, 2600 W h kg−1 for Li//S batteries, LSBs) and may be readily sealed, alleviating electrolyte leakage/loss issues, hence making them a safe option. However, this type of secondary batteries (especially for LSBs) faces the challenges of inferior electrical conductivity of their cathodes, polysulfide dissolution and their shuttling between electrodes, and considerable volume variation during charge/discharge cycles.129 In conventional LSBs, ether solvents were mostly used in the liquid electrolytes, which can dissolve polysulfides. Traditional carbonate-based LiB electrolytes (carbonates) and ionic liquids, which are either polysulfide incompatible or sparingly polysulfide-soluble, have been adopted in some special cases. Unfortunately, the use of these flammable liquid electrolytes raises the risk of electrolyte fire.131 Therefore, some studies attempted to introduce aqueous electrolytes in metal–chalcogen chemistry.132 Aqueous Al//S batteries (ASBs) were reported as early as 1993, which used KOH aqueous solution with both K2S4 and In(OH)2 additives as the electrolyte, while the relevant working voltage was only 1.3 V; however, their cyclic performance was not demonstrated.133 In 2019, one Fe//S battery (ISB) was reported with 0.5 M FeSO4 aqueous solution as the electrolyte, although its working voltage was very low (0.6–0.8 V), its cyclic performance was improved to 200 charge/discharge cycles by changing the Fe metal anode midway, avoiding polysulfide shuttling.134 In the case of LSBs and Na//S batteries (SSBs), analogous to LABs/SABs, protection layers (membrane electrolytes) were necessary in the metal anode side when introducing aqueous electrolytes. Li et al. integrated a photocatalyst in a hybrid LSB involving 0.01 M Li2S4 and 0.2 M LiOH aqueous electrolyte (catholyte) to store solar energy directly. At the Li anode side, an organic liquid electrolyte of 1 M LiClO4 in ethylene carbonate (EC)/dimethyl carbonate (DMC) was used, coupled with an Li1.35Ti1.75Al0.25P2.7Si0.3O12 (LATP) glass ceramic separator.135 The structure of this solar energy directly stored LSB and the reaction mechanism is shown in Fig. 5a. For the Na metal anode-based LSB, 0.25 M Na2S4 + 0.1 M NaOH aqueous electrolyte (catholyte) was matched with 1 M NaClO4 in EC: propylene carbonate (PC) + 5% fluoroethylene carbonate (FEC) organic electrolyte (anolyte) with the assistance of an NASICON membrane, as shown in Fig. 5b.136 This configuration for both LSBs and SSBs not only stabilized the Li/Na metal anodes in aqueous electrolytes, but also prevented dendrite formation and polysulfide shuttling and increased the utilization of the active materials.
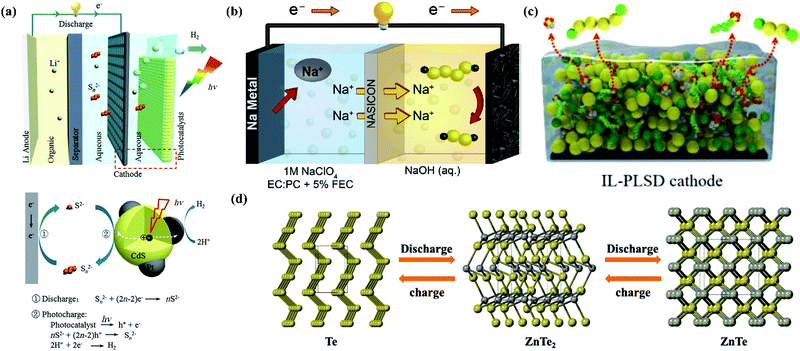 |
| | Fig. 5 (a) Battery consisting of an Li anode with organic electrolyte, Li-ion conductive LATP glass ceramic separator, and aqueous Li2Sn alkalic catholyte containing Pt/CdS photocatalyst and corresponding photocharging and discharging processes.135 (b) Schematic of a sodium–aqueous polysulfide hybrid battery with a sodium–metal anode, organic anolyte, NASICON separator, and alkaline aqueous polysulfide catholyte.136 (c) Schematic of PLSD cathodes coated with Zn2+-conducting IL.137 (d) Schematic representation of phase transformation during discharging (yellow: Te atoms and grey: Zn atoms).139 | |
Zn stands out among the aqueous systems, and recently aqueous Zn//chalcogen batteries have also become eye-catching, as reported by Zhi's group. An aqueous Zn//S battery was initially realized using a “liquid film”-comprising ionic liquid encapsulated in a copolymer (working as the cathode, as shown in Fig. 5c) and 1 M Zn(TFSI)2 aqueous electrolyte. This Zn//polysulfide system delivered an extraordinary capacity of 1148 mA h g−1 and an overwhelming energy density of 724.7 W h kg−1 (calculated based on the cathode mass, under a current density of 0.3 A g−1).137 The same aqueous solution (1 M Zn(TFSI)2) was also used as the electrolyte of a Zn//Se battery, but its performance was undesirable. Thus, a 2 M Zn(TFSI)2/polyethylene glycol (PEG)/water electrolyte was prepared, extending the ESW of the aqueous electrolyte and enhancing the performance of the Zn//Se battery.138 The overall reaction mechanism of this battery was also studied, as follows:138
which was same conversion reaction mechanism in both the organic electrolyte and aqueous electrolyte. Meanwhile, an aqueous Zn//Te battery was also reported by Zhi's group, utilizing a 1 M ZnSO
4 polyamide (PAM) hydrogel as the electrolyte, which displayed an ultra-flat discharge plateau and high volumetric capacity (2619 mA h cm
−3 under 0.05 A g
−1 current density).
139 This battery exhibited obvious flat discharge plateaus at about 0.59 and 0.48 V, originating from the reduction of Te. Furthermore, this was proved to be a two-step solid-to-solid conversion with the successive formation of ZnTe
2 and ZnTe, obeying the following two equations:
139| | | 2Te + Zn2+ + 2e− ↔ ZnTe2 (first step) | (7) |
| | | ZnTe2 + Zn2+ + 2e− ↔ 2ZnTe (second step) | (8) |
as demonstrated in
Fig. 5d. This work presented a new direction to design conversion-type aqueous MCBs with high capacity, stable output potential, excellent rate capability and long cyclic performance.
2.6. Hybrid battery
Aqueous hybrid batteries (AHBs) are new members of the ARB family, attracting great interest from researchers, which can be classified into hybrid ion batteries (HiBs), dual ion batteries (DiBs) and capacitor batteries (CBs). Besides CBs integrating battery behaviour in one side and capacitor behaviour in the other side, the mechanism of HiBs and DiBs is similar to that of single-ion ARBs, while their electrolytes are composed of various electrochemically active ions, diffusing from the electrolyte to electrodes simultaneously (for DiBs) or individually (for HiBs).140 Compared with the ARBs of single ions, AHBs exhibit several significant benefits, which can be summarized as follows: (1) enhanced rate performance by combining the ions of sluggish diffusion with that exhibiting better kinetics; (2) superior coulombic efficiency (CE) by avoiding ion trapping; (3) high flexibility and adaptability (more options for electrode materials); (4) low cost due to the less occupancy of Li/other costly elements in the whole cell; and (5) wide applications (energy storage and lithium purification).141 HiBs are the most popular among AHBs, which can be further divided into monovalent/monovalent ion batteries and monovalent/multivalent ion batteries.140 In 2013, Xia's group initially reported two monovalent/monovalent systems (Na0.22MnO2//LiMn2O4 and TiP2O7//Na0.44MnO2) based on Li2SO4 + Na2SO4 mixed electrolytes, exhibiting a specific energy of 17 W h kg−1 and 25 W h kg−1 based on the total weight of the active electrode materials, respectively.142 In 2018, a low-cost aqueous Mg–Na battery was proposed as a monovalent/multivalent system, employing Mn3O4 as the cathode of MiB, NaTi2(PO4)3 as the anode of SiB and 2 M MgSO4 + 1 M Na2SO4 aqueous solution as the electrolyte, which avoided the cathode problems of SiBs and anode problems of MiBs.143 A Pb//LiMn2O4 battery was reported in 0.5 M Li2SO4 aqueous solution as a monovalent/multivalent system, which realized the reversible dissolution/deposition of Pb ↔ PbSO4 in a neutral environment.144 Furthermore, an Mg//LiFePO4 battery was developed in a hybrid electrolyte comprised of a non-aqueous electrolyte (1 M PhMgBr and 0.1 M LiBr in tetrahydrofuran) against an Mg anode and aqueous electrolyte (0.5 M H2SO4) against an LiFePO4 cathode, while LISICON was utilized as the separator to stabilize both the non-aqueous and aqueous electrolytes.145 In the case of monovalent/multivalent systems, Zn metal anode-based HiBs have been reported intensively,140 especially that in mild aqueous electrolytes, which can overcome poor cyclic performances and the formation of Zn dendrites in alkaline conditions, as shown by the following equation:| | | Zn + 4OH− ↔ Zn(OH)42− + 2e− ↔ ZnO + 2OH− + H2O + 2e− | (9) |
In 2012, Chen et al. proposed Zn metal anode-based HiBs for the first time, which employed LiMn2O4 as the cathode and 3 M LiCl + 4 M ZnCl2 solution (pH = 4) as electrolyte.146 Afterwards, aqueous Zn–LiMn2O4 batteries were investigated frequently, while the relevant reaction mechanism was determined as follows:
| | | Negative: Zn2+ + 2e− ↔ Zn | (10) |
| | | Positive: LiMn2O4 ↔ Li1−xMn2O4 + xLi+ + xe− | (11) |
In 2018, Wang's group made a breakthrough in aqueous Zn//LiMn
2O
4 batteries
via the use of 1 m Zn(TFSI)
2 + 20 m LiTFSI highly concentrated electrolyte, which was cycled for 4000 cycles under a rate of 4C. In their work, a symmetrical Zn cell based on the highly concentrated electrolyte was cycled for 500 cycles (∼170 h) under a current density of 0.2 mA cm
−2 without any dendrite and ZnO observed.
147 The Zn-based monovalent/multivalent system was extended by Wu's group from Zn//LiMn
2O
4 to the Zn//Na
0.95MnO
2 redox couple with 0.5 M Zn(OAc)
2 + 0.5 M NaOAc electrolyte, which was charged/discharged for 1000 cycles under a rate of 4C in the voltage range of 1–2 V.
148 Meanwhile, Wu's group made a noticeable contribution to rechargeable Zn-based HiBs in an alkaline environment, reporting the fabrication of Zn//Co
3O
4 and Zn//NiO batteries in 1 M KOH + 10 mM Zn(OAc)
2 electrolyte with [Zn(OH)
4−]
2− and OH
− as the charge carriers.
149,150 These works achieved a balance between high energy and high stability in alkaline battery systems.
The ions in HiBs participating in the electrode reaction are only cations, while both cations and anions are involved in DiBs, which need to be considered as active species. The typical cathodes of DiBs are graphitic carbons, metal organic frameworks (MOFs), organic cathodes or electroactive polymers and MXenes, among which graphitic carbon cathodes are the most suitable candidates for practical applications due to their unique layered structure, environmentally friendly nature, and low cost.151,152 An aqueous DiB using 1 M (NH4)2SO4 aqueous solution as the electrolyte was reported by Zhang et al. in 2019, in which the n-type polyimide anode reacted with NH4+, while the p-type radical polymer reacted with SO42− to obtain a large cell voltage of 1.9 V.134 Alternatively, Ji's group made contributions to aqueous DiBs by using concentrated ZnCl2 aqueous solutions (30 m) with the ZnCl42−-insertion molecular solid of ferrocene as the anode and Zn2+-insertion PBA as the cathode. This electrolyte not only minimized the dissolution of the ferrocene anode, but also widened the voltage of the full cell by 0.35 V compared with dilute ZnCl2 electrolyte.153 Both cations and anions contribute to the capacity in CBs, which is similar to that in DiBs, while the cations are involved in the redox-active oxide cathode and anions are absorbed/desorbed by an inert activated carbon (AC) anode in CBs. Therefore CBs is one type of relatively new device that is intermediate in energy between batteries and supercapacitors, whilst principally offering supercapacitor-like power and cyclability values.154 The high ionic conductivity of aqueous electrolytes can induce a high electrical double layer (EDL) capacitance in the active carbon anode/electrolyte interfaces, and hence can be a good option for CBs.29 An Li2SO4 aqueous solution was assembled with an LiMn2O4 cathode and AC anode as an Li+ ion capacitor by Wang and Xia in 2005,155 and before that, an Ni(OH)2 cathode was demonstrated as a good cathode for aqueous systems with an AC anode in alkaline aqueous electrolyte (KOH).156 Brousse et al. prepared an Na+ ion capacitor based on an Na2SO4 aqueous solution with AC anode, while MnO2 was utilized as a cathode for Na ion intercalation/deintercalation in the bulk according to the following equation:157
| | | MnO2 + Na+ + e− = MnOONa | (12) |
while the surface redox could be enhanced once the cathode was nanosized as follows:
| | | (MnO2)surface + Na+ + e− = (MnOONa)surface | (13) |
Recently, a Zn-ion capacitor was innovatively proposed by Kang et al., in which AC, Zn metal and ZnSO4 aqueous solution served as the cathode, anode and electrolyte, respectively. A metric of 121 mA h g−1 capacity (84 W h kg−1 energy density), 14.9 kW kg−1 power output and excellent cycling stability (over 10000 cycles with 91% capacity retention) was exhibited.158 Numerous works towards Zn ion capacitors have been reported since then, indicating another feasible route to perfect electrochemical energy storage systems.
2.7. Redox flow battery
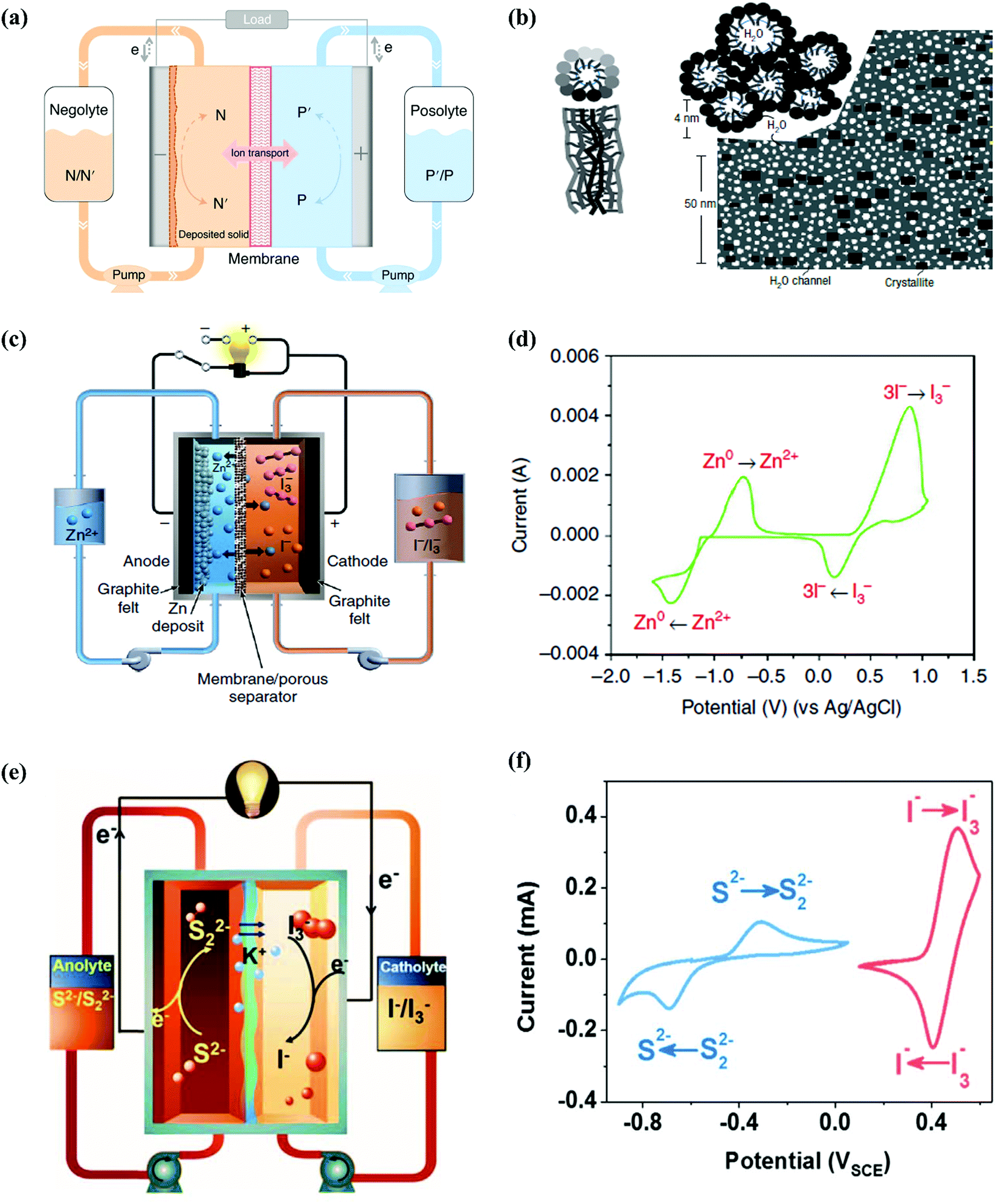
Redox flow batteries (RFBs) are novel electrochemical energy storage devices, storing electrical energy in two redox-active matters with distinct redox potentials. The redox species are usually dissolved or suspended in the electrolyte tanks (negolyte and posolyte, as shown in in Fig. 6a). Electrochemical charge-transfer reactions occur on the electrode stack, consisting of a pair of porous electrodes separated by an ion-conducting membrane. The capacity of the RFB depends on the available charges stored in the electrolyte tank, whilst the current output of the RFB is determined by the rate of the electrochemical reaction on the electrode stack. This unique configuration enables design flexibility in decoupling energy and power, which is critical for large-scale energy storage and energy distribution.159 Aqueous redox flow batteries (ARFBs) are one of the most important candidates for large-scale energy storage, which can be applied over a wide power range with long lifetimes over a flexible discharge duration. Moreover, the natural scalability induced by their unique design of decoupled power and energy is considered the most prominent advantage. Since the first ARFB was invented by NASA in the 1970s with Fe(III)/Fe(II) and Cr(III)/Cr(II) as the active species and FeCl2- and CrCl3-containing HCl solution as the electrolyte, a vast number of ARFBs have been invented, which are classified as vanadium (V)-, Zn-, Fe-, polysulfide (PS)-based (inorganic) and organic-based ARFBs.160 In 1985, a V(III)/V(II) redox couple was introduced as the first V-based ARFB with VCl3-dissolved HCl/H2SO4 solution as the electrolyte, but there was considerable room for ameliorating the reversibility of the redox reactions.161 Furthermore, V-based ARFBs face the challenges of low thermal stability, crossover of V-species, and limited kinetics, which are basically correlated with the electrolyte issues, where porous nanofiltration-enhanced Nafion membranes (perfluorinated cation exchange membranes), as shown in Fig. 6b, can be the effective solutions.162,163 Besides V-based ARFBs, Zn-based ARFBs are widely reported due to the high adaptability of Zn metal anodes towards aqueous systems, and Zn//Br2 systems were the first to be reported among Zn-based species, in which KCl/NH4Cl-containing ZnBr2 concentrated solutions served as the electrolytes.164 However, Br2 is considered a hazardous substrate and the corresponding redox potential exceeds the ESW of water. Hence, Li et al. introduced Zn//I2 species with a battery structure scheme, as shown in Fig. 6c, consisting of a Nafion membrane and ZnI2 solution as the electrolyte. The redox mechanism was revealed as follows:165
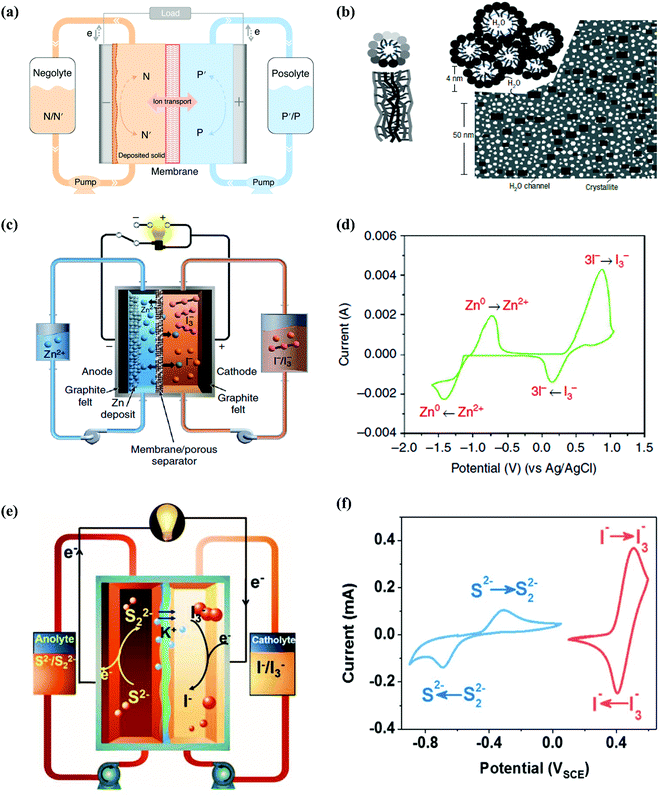 |
| | Fig. 6 (a) Schematic representation of a general RFB cell. The solid and dashed curly arrows represent the charging and discharging process, respectively. N/N′, negolyte in oxidized/reduced state; P/P′, posolyte in oxidized/reduced state.159 (b) Two views of an inverted-micelle cylinder and schematic diagram of the approximately hexagonal packing of several inverted-micelle cylinders.163 (c) Schematic representation of the proposed Zn//I2 system.165 (d) CV curves of 0.085 M ZnI2 on a glassy carbon electrode at the scan rate of 50 mV s−1.165 (e) Schematic illustration of the proposed aqueous polysulfide/iodide redox flow batteries.190 (f) CV curves of 5 mM K2S2–0.5 M KCl solution (blue) and 5 mM KI–0.5 M KCl solution (red) at 5 mV s−1 on a gold electrode.190 | |
Anode: Zn ↔ Zn2+ + 2e−
| | | E0 = −0.7626 V vs. SHE | (14) |
Cathode: I2(s) + I− ↔ I3− I3− + 2e− ↔ 3I−
| | | E0 = 0.5360 V vs. SHE | (15) |
Overall: Zn + I3−↔ Zn2+ + 3I−
with the relevant CV curve shown in
Fig. 6d. In this case, ZnI
2 electrolytes with various concentrations were investigated, and the concentration of 3.5 M was selected for further cyclic performance tests due to the optimum CE, energy efficiency (EE) and voltage efficiency (VE) dependent on that condition, whereas ethanol was further added for expanding the stable electrolyte temperature window from −20 °C to 50 °C.
165 Similar to Zn-based ARFBs, Fe-based ARFBs have been widely studied and applied. As mentioned above, the initially studied ARFB was an Fe-based battery with Fe
3+/Fe
2+ and Cr
3+/Cr
2+ as the active species and HCl solution as the electrolyte, which was limited by the sluggish reaction kinetics of Cr
3+/Cr
2+, parasitic hydrogen evolution reaction (HER), and the crossover of active materials. Thus, management of the electrolyte pH was chosen as an effective strategy to mitigate these challenges,
160 which will be discussed in detail in the next section. The first PS-based ARFB was invented by Regenesys Ltd in 1991 with NaBr on the positive side and Na
2S
4 on the negative side as the electrolyte,
166 extending the utilization of PS from MCBs to ARFBs. Analogous to Zn-based ARFBs, the Br
−/Br
3− chemistry was replaced by I
−/I
3− in the PS system, demonstrating higher solubility, faster kinetics and a lower vapour pressure.
167 This cell was comprised of KI and K
2S
2 + KOH aqueous solution and Nafion 115/117 membranes with the cell structure shown in
Fig. 6e and CV curves of the redox species shown in
Fig. 6f. Compared with inorganic-based ARFBs, organic-based ARFBs demonstrate large Earth abundance and high tunability through functionalization. In 2009, Xu
et al. innovatively reported the concept of organic-based ARFBs by adopting 1,2-dihydrobenzoquinone-3,5-disulfonic acid (BQDS) or 1,4-dihydrobenzoquinone-2-sulfonic acid (BQS) as the cathode and conventional PbSO
4 in iron-containing aqueous H
2SO
4 solution as the anolyte, which delivered an average CE of 99% and EE of 70% over 100 cycles. The obtained high performance indicated that soluble quinones are promising positive materials for ARFBs.
168 Recently, reversible ketone hydrogenation and dehydrogenation were realized in an organic-based ARFB using a design based on the molecular structure of 9-fluorenone (FL) as an example. An ARFB consisting of a ferro/ferricyanide catholyte and 1.36 M 4-carboxylic-7-sulfonate fluorenol (4C7SFL)/1 equiv. NaOH anolyte (equivalent to 2.72 M electron transfer) was subjected to current density testing and extended cycling, which delivered 1000 charge–discharge cycles at 25 °C (75% capacity retention) and 780 cycles (70% capacity retention) at 50 °C.
169
Overall, the historical development of aqueous electrolytes for ARBs in various species was briefly introduced with the timelines of representatively innovative works summarized in Fig. 7. The flourishing of aqueous electrolytes facilitated the all-around development of ARBs, while their challenges of low energy density, poor cyclic stability and narrow working temperature window were exposed during practice. From the perspective of electrolytes, the narrow ESWs, electrode dissolution/side reactions, and phase transition/instability of water at different working temperatures can account for the issues associated with ARBs. Therefore, it is critical to develop more mature aqueous electrolytes via new concepts.
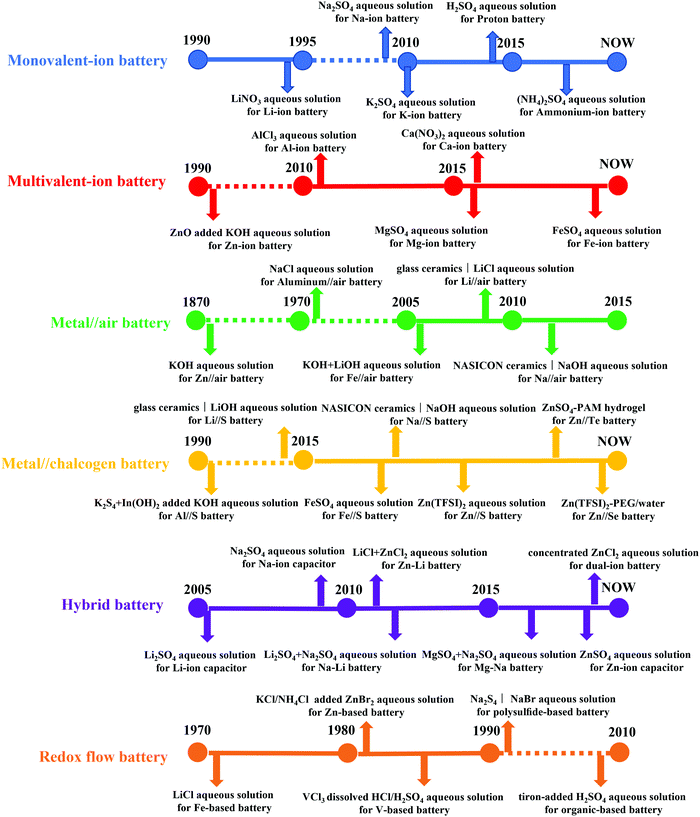 |
| | Fig. 7 Historical development of aqueous electrolytes with representative innovative works for various ARBs. | |
3. New concepts for better aqueous electrolyte
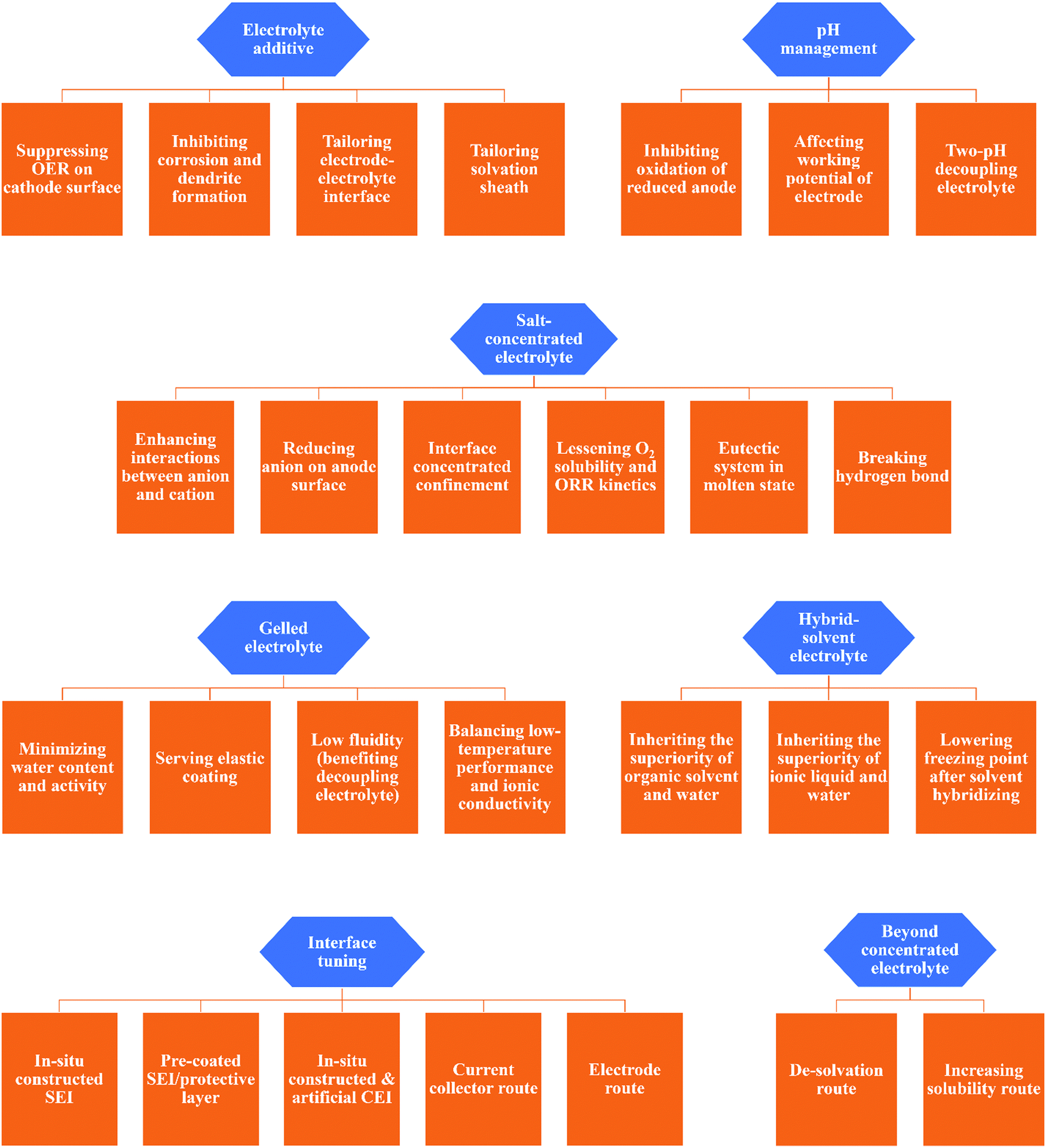
Electrolytes in battery systems have been tailored to the specific chemistry and structure of the electrodes and reactions present in the cell.48 Although the requirements vary with the electrodes, there are still some common demands towards electrolytes such as wide ESW, high ionic conductivity, desirable thermal stability and wide working temperature range, while negligible electrode dissolution/side reactions in the case of aqueous electrolytes.170 In 2013, Wu et al. published a pioneering work on ARBs with an exceptionally high working voltage above 3 V, which inspired intensive and extensive research towards better aqueous electrolytes, and consequently better ARBs.42 The recently developed concepts for better aqueous electrolytes can be categorized into electrolyte additive, salt-concentrated electrolyte, gelled electrolyte, solvent-hybrid electrolyte, electrode/current collector-electrolyte interface tuning, and beyond salt-concentrated electrolyte, which have formed a dense network with different working mechanisms and different specific routes (as shown in Fig. 8). The concepts presented in this review are the common strategies for liquid-phase electrolytes, except pH management (including two-pH decoupling electrolyte), which is a unique strategy for aqueous electrolytes.
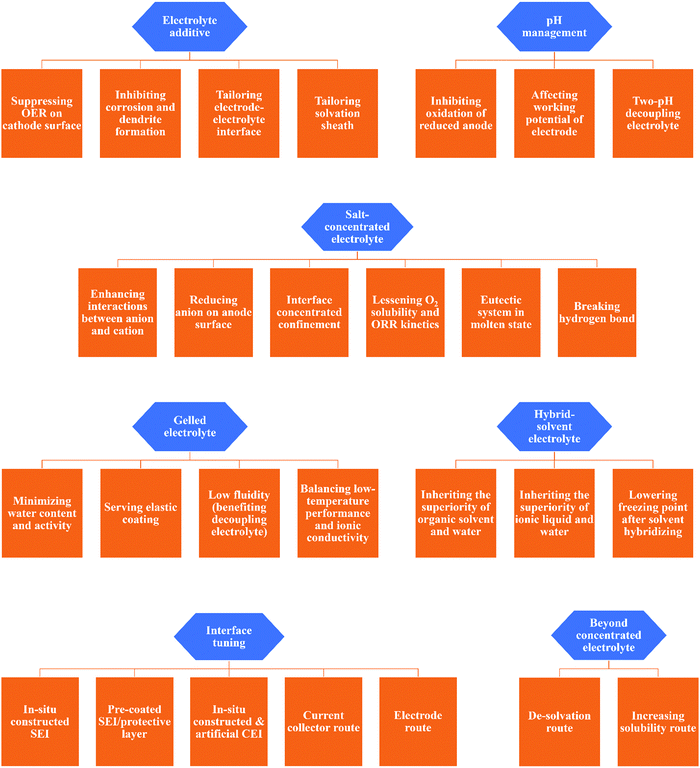 |
| | Fig. 8 Summary of specific working mechanisms and implementation paths of various strategies for mitigating aqueous electrolyte challenges. | |
3.1. Electrolyte additive
For conventional LiBs, the use of electrolyte additives is one of the most economic ways to improve their performance. Usually, the amount of additive in the electrolyte is no more than 5% either by weight or by volume, but it significantly improves the reversibility and cyclic life of LiBs.171 The strategy of electrolyte additives is also appropriate for ARBs, which was intensively reported in ARZiBs in the last five years. The additives added to aqueous electrolytes can be classified as ion, metal, organic and inorganic, offering the function of enhancing ion transportation, manipulating ESWs, and tuning electrode surfaces/interfaces.172
3.1.1. Suppressing OER on electrode surface.
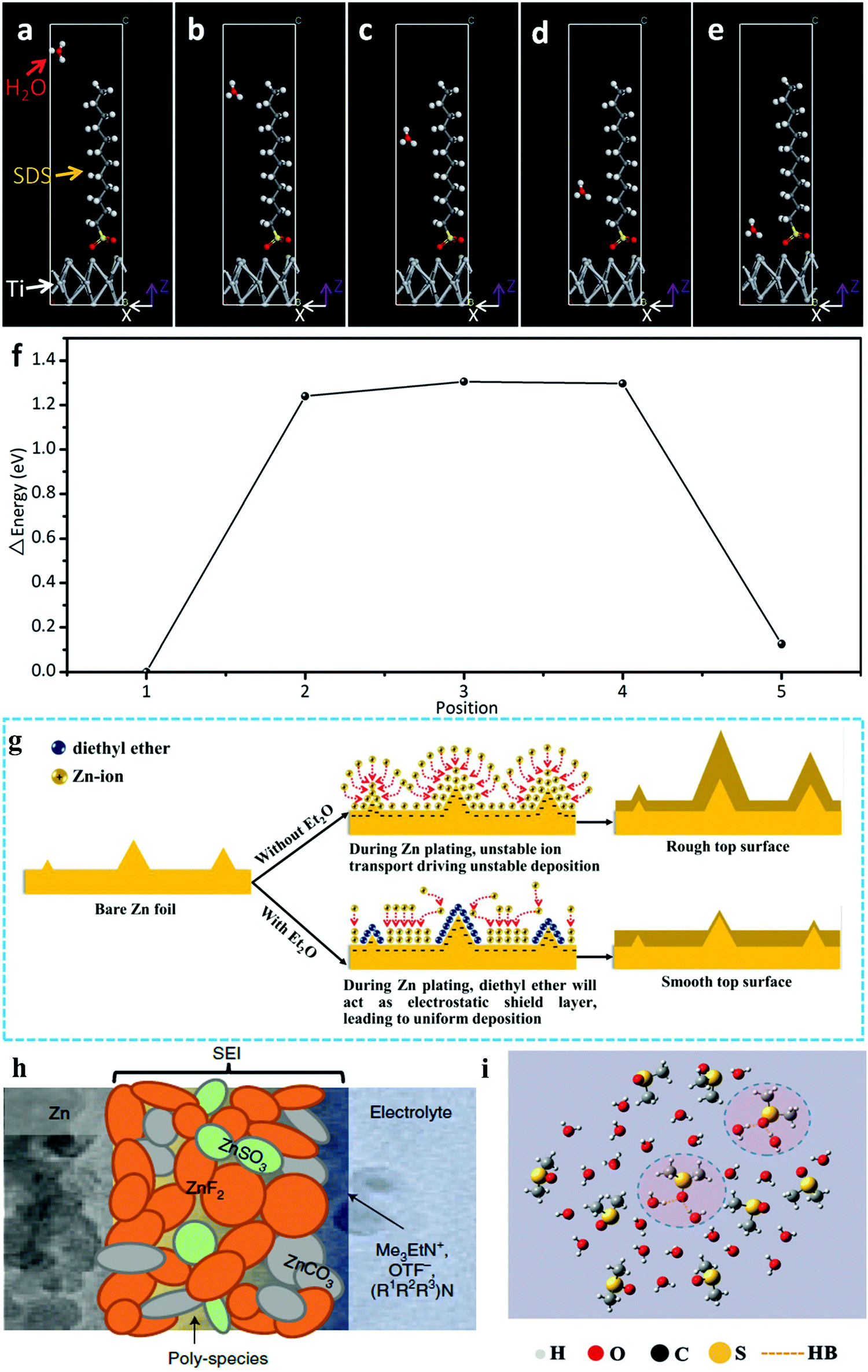
In a typical study on widening the ESWs of aqueous electrolytes, sodium dodecyl sulfate (SDS) was adding to 1 M Na2SO4 + 1 M ZnSO4 aqueous electrolyte, which equipped the HiB of Zn//Na2MnFe(CN)6 with a 2.0 V operation voltage and capacity retention of 75% over 2000 cycles. In this study, the surfactant additive could effectively suppress the oxygen evolution reaction (OER), and thus extended the ESW to 2.5 V, which was attributed to the SDS adsorption layer and theoretically confirmed by density functional theory (DFT) calculation, as shown in Fig. 9a–f.173 Beside SDS, Mn ion additive was proven to suppress the OER and broaden the ESW through the competition mechanism of the dissolution/deposition reaction of MnO2/Mn2+ and the OER in the solvent.95
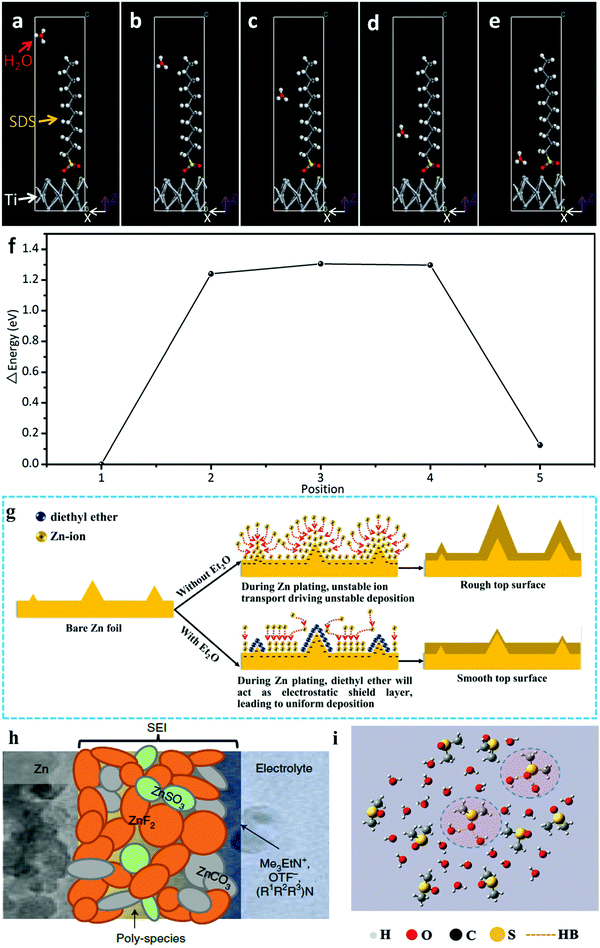 |
| | Fig. 9 (a–e) Models of density functional theory calculations, showing a water molecule passing through the SDS adsorption interlayer at different positions.173 (f) Energy change tendency at different positions.173 (g) Schematic of the morphology evolution of Zn anodes in mild aqueous electrolyte with and without Et2O additive during Zn stripping/plating.175 (h) Cartoon of proposed Zn2+-conducting SEI, characterized by small nodular particles embedded in a polymeric framework.177 (i) Local structure of the χDMSO = 0.3 system from MD simulations.46 | |
3.1.2. Inhibiting corrosion and dendrite formation in Zn metal anode.
The above-introduced two additives expanded the ESWs by suppressing the OER, while additives such as PbSO4, diethyl ether (Et2O) and sodium dodecyl benzene sulfonate (SDBS) were used for inhibiting corrosion and suppressing the formation of dendrites in the Zn metal anode, thus enhancing the cyclability. PbSO4 was added to 1 M Li2SO4 + 2 M ZnSO4 electrolyte gelled by 5% fumed silica until saturation, which provided a 20% lower corrosion current density on the Zn anode. The Zn metal anode in the tested Zn//LiMn2O4 HiB with PbSO4-containing electrolyte was determined to possess a dendrite-free morphology through scanning electron microscopy (SEM).174 In the Zn//MnO2 system, 2 vol% Et2O was added to 3 M Zn(CF3SO3)2 + 0.1 M Mn(CF3SO3)2 electrolyte, which was believed to be absorbed near the tip of the metal anode substrate surface, inducing an electrostatic shield and suppressing dendrite formation by maintaining a smooth deposition layer, as shown in Fig. 9g.175 SDBS was reported to improve the electrochemical behaviors of a Zn//LiFePO4 HiB. With the assistance of this additive, Zn ions were deposited smoothly on the surface of the Zn metal instead of participating in the vertical growth of flake-shaped Zn dendrites. Simultaneously, the Li-ion diffusion coefficient was increased from 1.78 × 10−11 to 8.22 × 10−11 cm2 s−1 by ameliorating the wettability of the LiFePO4 cathode, contributing to the high power density of the whole cell.176
3.1.3. Interfacial engineering.
In LiBs, additives are used intensively for engineering interfaces, especially engineering SEIs, which are also employed to tailor the interfaces of ARBs. Recently, Wang's and Xu's group reported the use of trimethylethyl ammonium trifluoromethanesulfonate (Me3EtNOTF) as an additive in the fluorinated interface between a Zn metal anode and aqueous zinc trifluoromethanesulfonate (Zn(OTF)2) electrolyte, which enabled the stable cycling of both a Zn//VOPO4 full cell (136 W h kg−1 and retained 88.7% of its capacity for >6000 cycles) and Zn//O2 cell (325 W h kg−1 for >300 cycles). The ZnF2-rich composition in that interface was caused by the additive, while ZnCO3, ZnSO3 and poly-species were also discovered there (Fig. 9h). DFT calculation suggested that the downstream reactions resulting from alkylammonium decomposition were responsible for the formation of ZnCO3.177 In another work, ZnF2 was added to 2 M ZnSO4 electrolyte directly for the formation of a stable F-rich interfacial layer. The F-rich interfacial layer could not only regulate the growth orientation of zinc crystals, but also serve as an inert protection layer against side reactions such as the HER. Under the protection of that interfacial layer, Zn stripping/plating was stably repeated for 600 h, and an anode-free Zn//LiMn2O4 HiB was realized.178 Besides the interfaces between the Zn metal anode and aqueous electrolytes, the additive can also function on the cathode. In 2016, Wang's group constructed a cathode electrolyte interphase (CEI) between an LiCoO2 cathode and LiTFSI aqueous electrolyte through the electrochemical oxidation of tris(trimethylsilyl) borate. The LiCoO2 cathode was stabilized at a high cut-off voltage corresponding to 0.7 e− electron charge transfer, which delivered a working voltage of 2.5 V and 120 W h kg−1 energy density.179
3.1.4. Solvation sheath tailoring.
To widen the working temperature windows of ARBs, some efforts focused on tailoring their solvation sheath, which adjusted the correlated ARBs to the frozen environments. Dimethyl sulfoxide (DMSO), a highly polar aprotic solvent, can form strong H-bonds with water molecules and significantly change the original H-bond structure of water molecules.180 According to previous studies, the HER of water molecules is a competitive process with the H-bond formation process in solution,181 and hence DMSO is an effective additive to suppress the HER. Thus, DMSO was added to an aqueous solution of 2 M NaClO4 with a molar fraction of 0.3 (2M-0.3 electrolyte). The 2M-0.3 electrolyte exhibited a sufficient ionic conductivity of 0.11 mS cm−1 at −50 °C. The Raman and Fourier transform infrared (FTIR) spectra proved that hydrogen bonds were stably formed between DMSO and water molecules, facilitating the operation of the electrolyte at ultra-low temperatures, while molecular dynamics (MD) modelling, as shown in Fig. 9i, demonstrated consistent results.46 In ARFB systems, additives are also employed to widen their working temperature windows. For example, 10 vol% ethanol served as an additive in a near-neutral 5 M ZnI2 electrolyte of Zn//I2 ARFB, enabling a stable battery performance in the temperature range of −20 to 50 °C. Nuclear magnetic resonance (NMR) study and DFT-based simulation together with flow test data illustrated that the addition of ethanol induced ligand formation between the oxygen on the hydroxyl group and the zinc ions, which not only extended the stable electrolyte temperature window but ameliorated the interface of the zinc metal anode.165 In the PB system, anions such as BF4− and SO4− were discovered to enhance the adaptability of relevant aqueous electrolytes towards frozen conditions. In one work on a PB, FTIR, Raman and NMR analyses synergistically demonstrated that the introduction of BF4− anions efficiently broke the hydrogen-bond networks of the original water molecules, resulting in an ultralow freezing point. Therefore, the alloxazine//MnO2 redox couple could run regularly even at −90 °C and displayed a high specific discharge capacity of 85 mA h g−1.182 Similarly, the hydrogen bonds between water molecules in the electrolyte could be remarkably damaged by modulating the interaction between SO42− and water molecules, lowering the freezing point of the electrolyte. Consequently, the p-chloranil-reduced graphene oxide redox couple of the proton insertion mechanism delivered an extraordinary electrochemical performance even at −70 °C.183 Basically, the electrolyte additive strategy can be a feasible route to improve the stability and widen working temperature windows of ARBs. The performance of representative works is summarized in Table S1 (ESI†). However, some additives probably fail to become a universal way to achieve better main aqueous electrolytes. Furthermore, electrolyte additives only function as minor modification to some cases rather than a fundamental transformation, while solvents and salts have a greater effect on aqueous electrolytes.
3.2. pH management and two-pH decoupling electrolyte
In aqueous electrolytes, the pH value can have a considerable impact on the ESW by thermodynamically affecting the HER and OER. Furthermore, the pH and H2O in aqueous electrolytes with or without O2 can influence the stability and working potential of the electrodes in ARLiBs, as follows:41| | 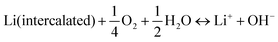 | (17) |
Based on the chemical potential of intercalated Li in the electrode (μintLI(x)) and Li in Li metal (μ0LI), the working potential of the electrode (V(x)) can be derived as follows:184| | | V(x) = 4.268 − 0.059 pH (V) | (18) |
where x for both μintLI (x) and V (x) is the amount of intercalated Li in the lithium-ion intercalated compounds. According to eqn (18), V(x) in equilibrium condition is determined to be 3.85 V at pH 7 and 3.50 V vs. Li/Li+ at pH 13, indicating that the reduction state of all usual anode materials in ARLiBs would theoretically be chemically oxidized by O2 and H2O rather than undergoing the electrochemical redox process. However, after eliminating O2 in aqueous electrolytes, the reaction becomes:185| | 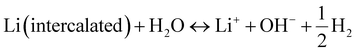 | (19) |
and the correlated V(x) can be further determined as:41| | | V(x) = 3.039 − 0.059 pH(V) | (20) |
revealing that H2O can also chemically oxidize some reduction-state anodes in ARLiBs.
3.2.1. pH management for higher stability.
Theoretically, the pH value of aqueous electrolytes can serve as a benchmark to determine whether the electrodes are stable against the corresponding aqueous electrolytes. Practically, pH adjusting can be another route to manipulate the stability of ARBs.41 Base on this idea, Cui's group used 1 M KH2PO4 (pH = 1) solution as an electrolyte for a PiB with a copper hexacyanoferrate cathode and AC/polypyrrole hybrid anode, which had zero-capacity loss after 1000 deep-discharge cycles.38 Xie et al. adjusted the pH value of 5 M LiNO3 aqueous electrolyte by controlling the concentration of LiOH, and found that when the pH was ∼8.5, the ARLiB with the CuV2O5//LiMn2O4 redox couple demonstrated the optimum cyclic performance.186 pH management of aqueous electrolytes is also a popular strategy to stabilize ARFBs. For example, Xie et al. utilized a 1.5 M HAc/NaAc buffer electrolyte to maintain the pH value of negative electrolyte in a Zn//Fe-based ARFB in the range of 2.0–6.0. The chemical reaction of Zn species with proton species was very insignificant, while the electro-reduction of protons on the negative electrode was significantly suppressed in this pH range.187 In a PS-based ARFB system, PS could be stabilized in alkaline electrolyte by avoiding the formation of H2S in neutral and acidic conditions. The equilibrium potential of polysulfide was pH-dependent for pH < 11.5, while it remained at −0.51 V vs. SHE when pH > 11.5. The reaction of PS in water was as follows:188
| S22− + 2H2O + 2e− ↔ 2HS− + 2OH− |
In a Zn//I2 ARFB, fast overcharge self-protection ability was demonstrated with a smart pH-responsive electrolyte. Once overcharged, the electrolyte pH increased, which was induced by hydrogen evolution, and consequently the irreversible formation of insulating ZnO at the anode side and soluble Zn(IO3)2 at the cathode side, which could rapidly switch off the Zn//I2 ARFB with capacity degrading to 6% of the initial capacity, thereby avoiding continuous battery damage. Noticeably, the stimulus-responsive ARFB could be switched on with nearly 100% capacity recovery by re-adjusting the electrolyte pH, delivering excellent stability.189
3.2.2. pH management for higher energy density.
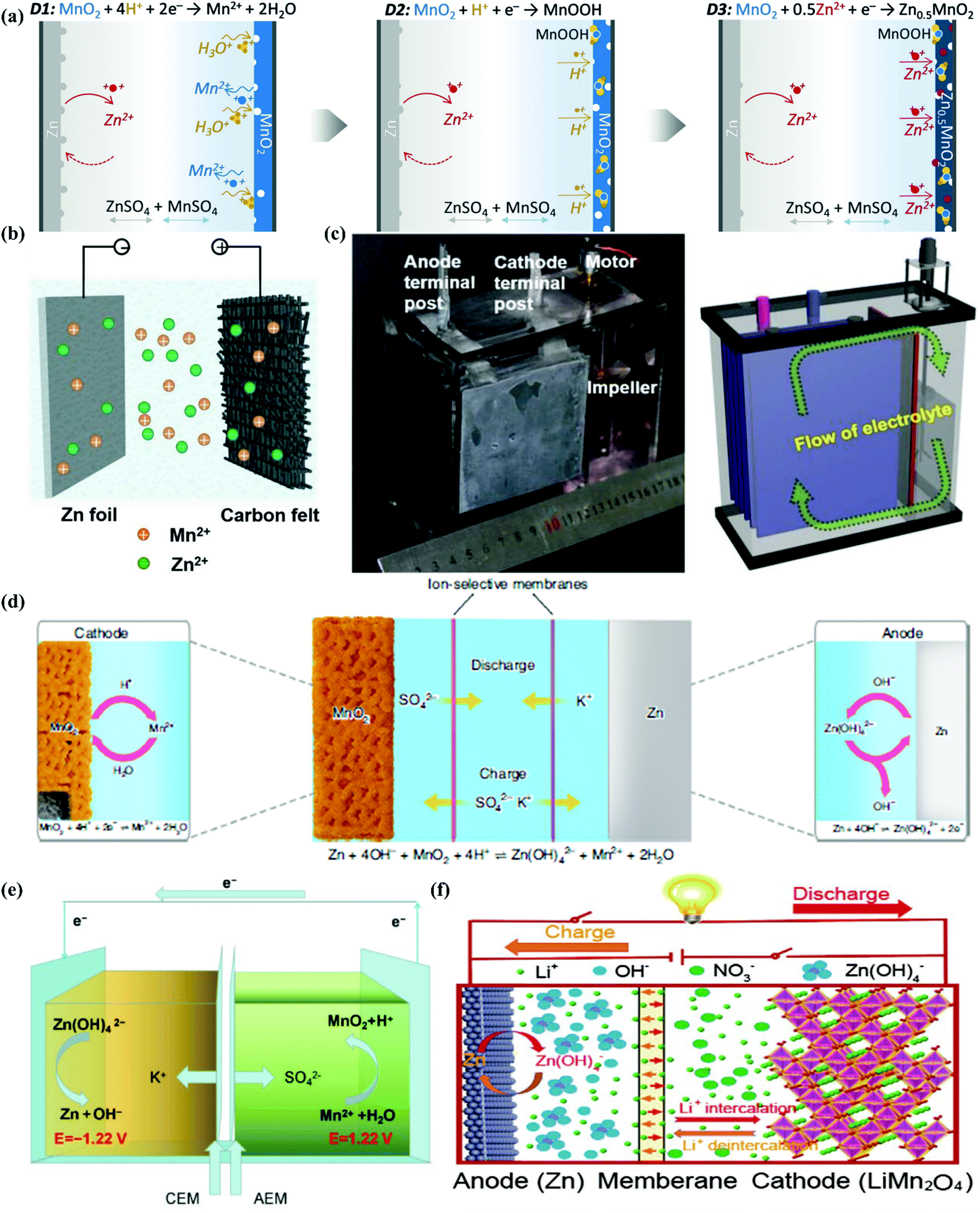
Besides stability, pH management even can improve the energy density of ARBs, especially for the Zn//MnO2 system, in which the cathode mechanism can be classified as proton-participating conversion in mild/strong alkaline environment,190,191 reversible Zn ion insertion/extraction in neutral/mild acidic aqueous electrolyte,91,192 proton and Zn ion co-insertion in mild acid condition,193 and cathode dissolution/deposition mechanism under strong acid condition.95,194 Various cathode mechanisms generate different working potentials in the cathode, among which the that of the dissolution/deposition mechanism is the highest. Therefore, the pH value of 1 M ZnSO4 + 1 M MnSO4 aqueous electrolyte was adjusted using H2SO4 to create a strong acid environment, while 0.1 M was chosen as the optimized concentration of H2SO4 for balancing between a high working voltage and wide ESW (desirable cyclic stability). The discharge profiles of the pH-adjusted electrolyte-based Zn–MnO2 battery exhibited three discharge regions including D1 (2.0–1.7 V), D2 (1.7–1.4 V), and D3 (1.4–0.8 V), which represented MnO2 dissolution, proton insertion, and Zn ion insertion, respectively (Fig. 10a). The average out-put voltage of that cell was 1.95 V, together with ∼570 mA h g−1 capacity (active matter of both cathode and anode), exhibiting ∼409 W h kg−1 energy density.95 Based on the dissolution/deposition mechanism of MnO2, a membrane-free Zn//MnO2 ARFB was proposed for large-scale energy storage with the corresponding schematic illustration and digital image, and schematic cross-section of the cell shown in Fig. 10b and c, respectively.101
 |
| | Fig. 10 (a) Schematic representation and charge storage mechanism of the aqueous Zn//MnO2 battery in H2SO4-adjusted 1 M ZnSO4 + 1 M MnSO4 electrolyte.95 (b) Configuration of a membrane-free Zn//MnO2 ARFB.101 (c) Digital image and cross-section schematic of the cell, where the electrolyte flow propelled by the impeller is indicated by a green arrow.101 (d) Schematic diagram of the cell structure and chemical reactions at the cathode and anode during the discharge and charge based on decoupled reactions in acidic and alkaline electrolytes separated by a neutral electrolyte in the central chamber.97 (e) Schematic illustration and mechanism of Zn//MnO2 ARFB battery using an acid/alkaline decoupling electrolyte.96 (f) Schematic representation of the structure and working mechanism of the assembled Zn–Li HiB with alkaline/mild decoupling electrolyte.198 | |
3.2.3. Two-pH decoupling electrolyte.
Actually, the pH of aqueous electrolytes not only affect the working mechanism of the MnO2 cathode but also the Zn metal anode. The redox potential of Zn stripping/plating is −0.76 V vs. SHE, while that for Zn/Zn(OH)42− conversion in an alkaline environment can be extended to −1.199 V vs. SHE.98 Thus, decoupling the working conditions of the MnO2 cathode and Zn anode to enable both acidic MnO2 and alkaline Zn redox reactions in a single cell can break the 2.0 V barrier of the aqueous Zn//MnO2 battery. One design (Fig. 10d) used both an alkaline (6 M KOH + 0.2 M ZnO + 5 mM vanillin) and an acidic (3 M H2SO4 + 0.1 M MnSO4) electrolyte in two chambers separated by a neutral (0.1 M K2SO4) electrolyte to avoid their neutralization. The neutral electrolyte chamber was encapsulated by two membranes (cation-exchange or bipolar membrane) with ion selectivity on each side. Basing on this design, the decoupled Zn//MnO2 battery exhibited an exceptionally high open-circuit voltage of 2.83 V and a specific energy density of 1621.7 W h kg−1 (based on the active mass of MnO2).97 The three-chamber structure was sophisticated to some extent, and hence one study attempted to construct this type of decoupled Zn//MnO2 battery into a ARFB with one anolyte of 2.4 M KOH + 0.1 M Zn(CH3COO)2 and one catholyte of 0.5 M H2SO4 + 1.0 M MnSO4, separated by a bipolar membrane (Fig. 9e).96 Recently, a polyacrylate (PAA)-based alkaline hydrogel and PAM-cellulose-based acid hydrogel were prepared and employed as a two-pH decoupling electrolyte. The commonly used cellophane worked as a separator, effectively solving the problem of electrolyte neutralization caused by diaphragm ruptures. The flexible Zn//MnO2 battery displayed a high working voltage of 2.7 V, high area capacity of 2.63 mA h cm−2, and acceptable cyclic stability of 86.7% capacity retention after 400 cycles.195
The alkaline/acid decoupling electrolytes were designed for an aqueous Zn//MnO2 battery, whereas alkaline/mild ones were proposed by Wu's group for Zn–Li HiBs. In that system, 5 M LiNO3 and 5 M LiOH + 1 M Zn(OAc)2 aqueous solutions were selected as the cathodic and anodic electrolytes, respectively, coupling the waste Nafion NR117 membrane treated with 3 wt% H2O2 and 1 M H2SO4 as the separator (Fig. 9f). Before that, the same membrane was employed in an aqueous Zn//MnO2 battery and an aqueous Li-ion capacitor with alkaline/mild electrolytes, showing its feasibility and validity as a separator.196,197 While in the Zn–Li HiB, a Zn anode in alkaline solution and LiMn2O4 cathode in mild solution together equipped the whole cell with a working voltage of above 2.3 V. This battery system delivered a steady energy density of 208 W h kg−1 (based on the total weight of active materials) at 1.69C with a high average output voltage of up to 2.31 V, and could be cycled for over 1000 cycles with an average CE of >98%.198 In summary, the pH of aqueous electrolytes can influence the redox potential and even the reaction mechanism of electrodes, thereby influencing the stability and working voltages of the whole cell, which is an important move towards overcoming the disadvantages of ARBs. The performance of representative works is summarized in Table S2 (ESI†). However, managing the pH of aqueous electrolytes may fail to shift the electrode redox potential considerably according to eqn (18) and (20). In contrast, for two-pH decoupling electrolytes, their complicated cell structures probably restrict their application and popularization in ARBs, together with the unwanted HER and OER at the electrode/electrolyte interfaces under a high working voltage. Therefore, the strategies of pH management and two-pH decoupling electrolyte need more studies and further optimization to maturely enhance the stability and working voltage/energy density of ARBs.
3.3. Salt-concentrated electrolyte
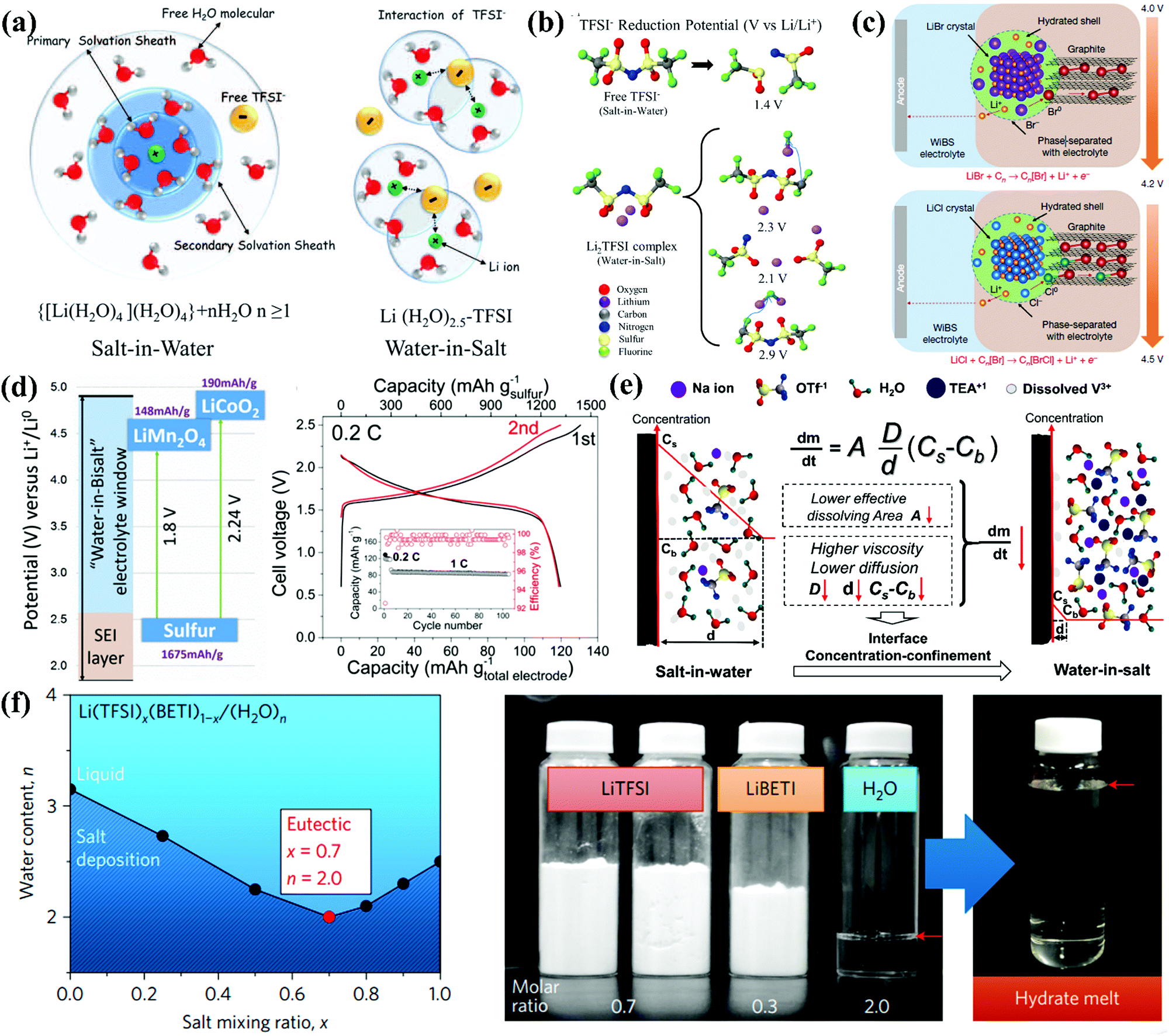
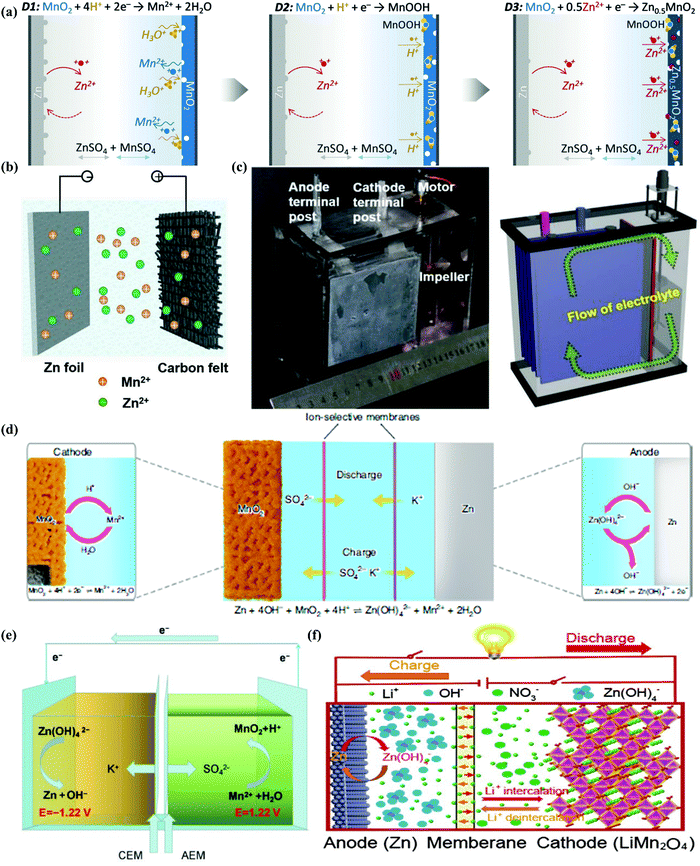
Liquid electrolytes are a combination of solvents and salts, while salt concentration is a critical parameter, impacting the ESWs, ionic conductivity, and electrode/electrolyte interfaces through solvation structures, viscosity, ionic mobility and overall electrolyte structure (morphology).56,199 Based on this, salt concentration manipulation is an easy and effective way to tailor the corresponding liquid electrolytes. Increasing the salt concentration in liquid electrolytes will result in enhanced interactions between cations and anions/solvents and a decrease in the content of free-state solvent molecules, which will demonstrate unusual physicochemical and electrochemical properties that are remarkably distinct from conventional dilute electrolytes.199
3.3.1. “Water-in-salt” electrolyte.
In 2015, one significant breakthrough was made by Wang's and Xu's group, proposing the concept of “water-in-salt” electrolyte (WiSEs), which ushered salt-concentrated electrolytes and ARBs into a new era.75 The flourished salt-concentrated electrolytes in ARBs can be categorized into WiSEs, hydrate-melt electrolytes (HMEs) and regular super-concentration electrolytes (RSCEs), which generate wide ESWs, and then high energy density. The definition of WiSEs is that salt outnumbers the solvent in a binary system in terms of both weight and volume, leading to an anion-containing Li-ion solvation sheath (Fig. 11a) and the formation of a dense interphase on the anode surface (mainly arising from anion reduction, Fig. 11b). Combined with the substantially reduced electrochemical activity of water in a WiSE of 21 m LiTFSI, an ESW of ∼3.0 V was provided, which equipped a full ARLiB (Mo6S8//LiMn2O4) with a working voltage of 2.3 V and 1000 charge/discharge cycles.75 Since that, WiSEs have demonstrated all-round versatility in raising the energy density, enhancing the stability and widening the working temperature windows of ARBs. In terms of energy density enhancement, Wang's and Xu's groups applied WiSEs to an ARLiB with halogen conversion–intercalation chemistry in graphite (Fig. 11c), delivering a capacity of 243 mA h g−1 (based on the total weight of the electrode) at an average potential of 4.2 V vs. Li/Li+. Pairing a graphite anode with an artificial SEI, a 4 V class ARLiB was created with an energy density of 460 W h kg−1.76 Furthermore, Wang's and Xu's group employed the WiSE in ZAB chemistry with a porous carbon air cathode, demonstrating an energy density of 1000 W h kg−1 (based on the mass of the cathode) in 200 cycles.147 Meanwhile, in the LAB system, WiSEs provide the necessary functionalities to support aprotic Li//O2 operations via reversible Li2O2 formation and decomposition. The lack of organic solvent molecules was a highlight there, eliminating the known reaction pathways towards by-product formation in organic electrolyte systems.200 In another theoretically high energy density system of LSB, a WiSE played a role as well, coupling a sulfur anode with solid–liquid two-phase, and LiMn2O4/LiCoO2 cathode, delivering a working voltage of 1.8/2.24 V (Fig. 11d). The capacity of the sulfur anode was tested as 1327 mA h g−1, while the energy density of the full cell was determined to be 200 W h kg−1 (total electrode mass) for >1000 cycles at ∼100% CE.201
 |
| | Fig. 11 (a) Illustration of the evolution of the Li-ion primary solvation sheath in diluted and WiSE.75 (b) Predicted reduction potentials of free and complex TFSI− from G4MP2 quantum chemistry calculations.75 (c) Schematic drawing of the conversion-intercalation mechanism occurring in (LiBr)0.5(LiCl)0.5-graphite during its oxidation in the WiSE. Two-stage reactions involved in the oxidation of Br− (∼4.0 V) and Cl− (∼4.2 V) and their subsequent intercalation into the graphitic structure.76 (d) Illustration of S//LiMn2O4 and S//HV-LiCoO2 full cell in WiSE with expanded ESW. Voltage profiles of S//HV-LiCoO2 full cell in WiSE at a rate of 0.2C. (inset) Capacity stability and CE during cycling.201 (e) Schematic illustration of vanadium dissolution according the Noyes–Whitney equation.45 (f) Liquidus line of Li(TFSI)x(BETI)1−x salt–water mixtures and stoichiometric amounts of LiTFSI, LiBETI and water used to prepare an Li(TFSI)0.7(BETI)0.3·2H2O hydrate melt. The red arrows indicate the liquid levels.208 | |
The stability endowed by WiSEs is also significant among ARBs, not only suppressing the HER/OER of water but also dissolution of the electrodes in aqueous electrolytes. In the above-described LSB system, excellent cyclic stability was induced by the solid–liquid two-phase reaction pathway, where the liquid polysulfide phase in the sulfide electrode was thermodynamically phase-separated from the WiSE.201 The phenomenon of negligible polysulfide dissolution in WiSEs can also be discovered in SiB chemistry. Yue et al. investigated concentration-controlled cathode dissolution through a susceptible Na3V2(PO4)3 cathode, whose time-, cycle-, and state-of-charge-dependent dissolubility were evaluated by multiple electrochemical and chemical methods. In that study, a novel mechanism of interface concentrated-confinement was proven with WiSEs, which enhanced the cyclic stability of the NaTi2(PO4)3//Na3V2(PO4)3 full cell. The high viscosity, low vanadium ion diffusion, low polarity of solvated water and scarce solute-water dissolving surfaces in WiSEs (9 m NaOTf + 22 m tetraethylammonium triflate, TEAOTf), remarkably decreased the thermodynamic-controlled solubility, the dissolving kinetics, and physical space local mass interfacial confinement (Fig. 11e).45 It was discovered that the WiSEs could reduce the O2 solubility in water and the kinetics of the oxygen reduction reaction (ORR) by slowing down O2 diffusion, as shown by the Koutecky–Levich equation.202 Hence, the WiSE-based cells could work with an open configuration, which exhibited superior ability to dissipate heat and pressure and eliminated the risk of explosion.202 Besides stability, WiSEs can also expand the working temperature widows of ARBs. A WiSE of 22 m KCF3SO3 (KFSI) was prepared for a PiB, matched with an Fe-substituted Mn-rich PBA K1.85Fe0.33Mn0.67[Fe(CN)6]0.98·0.77H2O (KFeMnHCF-3565) cathode and 3,4,9,10-perylenetetracarboxylic diimide (PTCDI) anode. Beside the impressive performance of 80 W h kg−1 and superior capacity retention of 85% at 20C after more than 10000 charge–discharge cycles at room temperature (RT), the full PiB was assembled in a pouch cell, demonstrating a superior performance at a wide temperature range of −20 °C to 60 °C.203 In the study reported by Reber et al., a key factor limiting the application of WiSEs at low temperatures was recognized due to their salt crystallization tendency even near RT. Thus, 10 m sodium (fluorosulfonyl)(trifluoromethylsulfonyl)imide (NaFTFSI) with asymmetric anions was introduced into 25 m NaFSI with symmetric anions. Subsequently, an SiB based on the asymmetric anion optimized WiSE in combination with an NaTi2(PO4)3 anode and Na3(VOPO4)2F cathode demonstrated excellent capacity retention and high energy density down to a temperature of −10 °C.204 However, in WiSEs, expensive organic salts were intensively used, which increased the cost of the whole cell. Some studies attempted to use cost-effective salts as substitutions, and obtained some notable achievements, especially for that based on acetates and chlorates. Ji's group applied potassium acetate (KOAc) to form a WiSE, which was capable of cycling relevant PiBs up to 11000 cycles under a current density of 1 A g−1 with an ESW of ∼3.2 V.205 Through blending with LiOAc and Zn(OAc)2, this acetate-based WiSE could be used in ARLiBs and Zn//MnO2 systems with desirable performances and potentials on a large scale.92,206 Meanwhile, Ji's group reported the preparation of ZnCl2-based WiSE (30 m ZnCl2), enabling a dendrite-free Zn metal anode with high CE and a novel DiB with high reversibility.153,207
3.3.2. Hydrate-melt electrolyte.
Among the salt-concentrated systems, another significant electrolyte reported was HMEs, which utilize a eutectic system thereof. LiTFSI and LiN(SO2C2F5)2 (LiBETI) salts were firstly selected for HMEs because these anions are weakly Lewis basic and only interact with Li+ weakly, thereby promoting the solvation of Li+ by water molecules rather than the abundant formation of ion pairs. Furthermore, the anions exerted a ‘plasticizing’ effect, avoiding the crystallization of the salts and hydrates. Fig. 11f demonstrates the miscibility limits of the blended LiTFSI–LiBETI salts in water at RT, while the eutectic composition of Li(TFSI)0.7(BETI)0.3 was determined to exhibit the highest miscibility, forming a stable, transparent liquid with an extremely low water content (H2O:Li+ = 2.0 molar ratio), equalling a water molar concentration of 10.1 mol L−1 (Fig. 10f). Because of this low water concentration, the water molecules could be isolated from each other by the bulky anions, thus suppressing the activity of water by decreasing the opportunity for the formation of clusters of free water molecules. This HME equipped Li4Ti5O12//LiNi0.5Mn1.5O4 redox couples with an upper limitation working voltage of 3.1 V and energy density of >130 W h kg−1 within 100 cycles.208 Besides, the concept of HME was applied to SiBs and PiBs, employing Na(PTFSI)0.65(TFSI)0.14(OTf)0.21·3H2O and K(PTFSI)0.12(TFSI)0.08(OTf)0.8·2H2O hydrate melts with ternary anions including TFSI−, OTf− and especially asymmetric the N(SO2CF3)(SO2C2F5)− (PTFSI−) anion, which significantly expanded the ESWs to 2.7 and 2.5 V, respectively. Na(PTFSI) and K(PTFSI) with asymmetric anions normally possess high solubility, which is attributed to their high vibrational mobility and flexibility offered by their asymmetric structure, realizing Na and K hydrate melts at super-high concentration.209 The HMEs were also appropriate in LABs, coupling LiSICON glass ceramic-protected Li metal anode and Ketjen Black (KB) cathode, which showed a low charge potential of ∼3.16 V, a high discharge capacity of 38 mA h cm−2, and a stable cyclic ability of 50 cycles with a capacity limitation of 1000 mA h g−1 under a current density of 500 mA g−1.210 After substituting into ZnCl2·2.33 H2O, the HME could work with a Zn metal anode and Pt/C cathode as a ZAB, delivering a reversible capacity of 1000 mA h g−1 (based on catalyst) over 100 cycles.211 In the WiSE system, a similar concept to HME was proposed, namely “water-in-bisalt” electrolyte (WiBSE), which could dissolve another unhydrated salt with similar chemical properties and form binary salt systems in the molten state with higher cation/water ratios. In the typical WiBSE of 21 M LiTFSI + 7 m LiOTf, a more compact SEI than that in WiSE was created, allowing a 2.5 V TiO2//LiMn2O4 full LiB with an average discharge voltage of 2.1 V and an energy density of 100 W h kg−1 (total electrode mass).77 Additionally, the acetate could replace the organic FSI− anion-based salts in WiBSEs, while the cation-size effect on ESWs in the acetate system was further investigated.212
3.3.3. Regular super-concentration electrolyte.
Normally, the cation/water ratio of WiSEs and HMEs reach above 2.0 for aqueous electrolytes with a high salt concentration but fail to approach the ratio or meet the definitions of WiSEs/HMEs, which can be categorized as RSCEs. Pan et al. investigated the thermodynamic and kinetic contributions to the ESW of an LiNO3-based RSCE (LiNO3:H2O = 1:2.5) through MD simulation and spectral analysis. The local structure of the intimate Li+–water interaction was discovered at a super-concentration, inducing (Li+(H2O)2)n polymer-like chains rather than the ubiquitous hydrogen bonding between water molecules, thereby extending the ESW to 2.55 V.213 Based on RSCEs, a universal strategy was proposed to ameliorate the specific energy, reversibility and safety of MViBs, which used Ca(NO3)2-based electrolytes as an example, driving the Ca-ion/sulfur–metal oxide full cell stably.214 A 3 M Zn(FSI)2 aqueous electrolyte was employed by Chen's group to fabricate a Zn//ZnMn2O4 battery, which exhibited excellent stability by suppressing Mn dissolution (capacity retention of 94% over 500 cycles under a high current density of 500 mA g−1).192 Subsequently, the same group optimized the concentration of ZnCl2 to 7.5 m, which achieved an ultralow solid–liquid transition temperature via the breakage of H-bonds, and thus an ultra-wide temperature window from −90 to 60 °C.215 Xue et al. introduced super-concentrated sugars into 2 M NaNO3 to reduce the free water molecules and destroy the tetrahedral structure, thus expanding the ESW to 2.812 V by breaking the hydrogen bonds. Consequently, LiBs, SiBs, PiBs and supercapacitors with excellent electrochemical performances were derived from the sugar-based electrolytes.216
Briefly, salt-concentrated electrolytes can be a universal strategy to improve the energy density and cyclic stability and widen the working temperature windows of ARBs, which have been employed intensively and extensively, as summarized in Table 1 (both units of m and M were used herein, according to the different salt concentrations of various electrolytes).45,75–77,92,147,153,192,200–222 However, their cost probably impedes their large-scale applications because of the large amount of salt in these systems, although desirable performances have been obtained. The unit price of some representative salts utilized in salt-concentrated electrolytes is compared in Fig. 12, in which the corresponding information was collected from Sigma-Aldrich in USA, Sinoreagent in China and Alfa Aesar in the UK.190–192 It can be concluded from Fig. 12 that some organic salts are exceptionally expensive but only a little better in electrolyte performance than the cost-effective salts ranging from nitrites, perchlorates, and chlorates to acetates, and hence it is important for us to strike a balance between the properties of electrolytes and their cost in real applications.
Table 1 A summary of recent representative works on salt-concentrated electrolytes for ARBs
| Electrolyte |
Electrode |
ESW (V) |
Energy density |
Cyclic stability |
Working temperature |
Ref. |
| LiNO3·2.5H2O |
AC//LiMn2O4 or Li Ni1/3Mn1/3Co1/3O2 |
2.55 |
n/a |
n/a |
RT |
213
|
| 8.37 m Ca(NO3)2–PVA |
S∣C//Ca0.4MnO2 |
2.6 |
110 W h kg−1 (total electrode) |
150 cycles (0.2C) |
RT |
214
|
| 2 M NaNO3/66.7 wt% sucrose |
AC//PBA |
2.8 |
n/a |
2000 cycles (2 A g−1) |
−50 °C to 80 °C |
216
|
| 7.5 m ZnCl2 |
Zn//polyaniline |
n/a |
n/a |
2000 cycles (0.2 A g−1) |
−90 °C to 60 °C |
215
|
| ZnCl2·0.03KBr·2H2O |
Zn//graphite |
n/a |
∼440 W h kg−1 (cathode) |
100 cycles (0.25 A g−1) |
RT |
217
|
| 30 m ZnCl2 |
Zn symmetrical cell |
2.3 |
n/a |
600 hours (∼1800 cycles, 0.2 mA cm−2) |
RT |
207
|
| 30 m ZnCl2 |
Fc/C//PBA |
2.3 |
n/a |
2000 cycles (5C) |
RT |
153
|
| ZnCl2·2.33 H2O |
Zn//Pt/C |
n/a |
n/a |
100 cycles (0.1 A g−1) |
30 °C |
211
|
| 30 m KOAc |
KTi2(PO4)3//AC |
3.2 |
n/a |
11000 cycles (1 A g−1) |
RT |
205
|
| 8 m LiOAc + 32 m KOAc |
TiO2//LiMn2O4 |
∼3.0 |
n/a |
50 cycles (0.5C) |
RT |
206
|
| 7 m LiOAc + 45 m CsOAc |
VO2//LiNi0.5Mn1.5O4 |
∼3.0 |
114.8 W h kg−1 (total electrode) |
1200 cycles (4C) |
RT |
212
|
| 1 m Zn(OAc)2 + 31 m KOAc |
Zn//MnO2 |
3.1 |
232.7 W h kg−1 (activated electrode and electrolyte) |
600 cycles (C/3) |
RT |
92
|
| 4 M KOH + 16 M KOAc |
Al alloy//MnxOy@Ag |
2.2 |
436.1 W h kg−1 (activated electrode and electrolyte) |
2300 min (∼30 cycles, 25 mA cm−2) |
RT |
218
|
| 17 m NaClO4 |
NaTi2(PO4)3//Na4Fe3(PO4)2(P2O7) |
2.7 |
36 W h kg−1 (total electrode) |
>200 cycles (1C) |
RT |
221
|
| 21 m LiTFSI |
Mo6S8//LiMn2O4 |
∼3.0 |
84 W h kg−1 (total electrode) |
1000 cycles (4.5C) |
RT |
75
|
| 21 m LiTFSI |
LICGC∣Li//Ru/TiSi2 |
∼3.0 |
n/a |
300 cycles (0.05 A g−1) |
RT |
200
|
| 9.26 m NaOTf |
NaTi2(PO4)3//Na0.66[Mn0.66Ti0.34]O2 |
2.5 |
31 W h kg−1 (total electrode) |
>1200 cycles (1C) |
RT |
219
|
| 22 m KFSI |
PTCDI//KFeMnHCF-3565 |
∼3.0 |
80 W h kg−1 (total electrode) |
10000 cycles (100C) |
−20 °C to 60 °C |
203
|
| 3 M ZnFSI |
Zn//ZnMn2O4 |
n/a |
∼202 W h kg−1 (total electrode) |
500 cycles (0.5 A g−1) |
RT |
192
|
| 21 m LiTFSI + 7 m LiOTf |
C/TiO2//LiMn2O4 |
∼3.1 |
100 W h kg−1 (total electrode) |
100 cycles (0.5C) |
RT |
77
|
| 21 m LiTFSI + 7 m LiOTf-PEO/PVA |
passivated graphite//(LiBr)0.5(LiCl)0.5-graphite |
∼3.1 |
460 W h kg−1 (total electrode) |
∼250 cycles (0.08 A g−1) |
RT |
76
|
| 21 m LiTFSI + 7 m LiOTf-PVA |
S-KB//LiMn2O4 or LiCoO2 |
∼3.1 |
200 W h kg−1 (total electrode) |
>1000 cycles (1C) |
RT |
201
|
| 21 m LiTFSI + 7 m LiOTf (open battery configuration) |
Al2O3@LiTi2(PO4)3//LiMn2O4 |
∼3.1 |
170 W h kg−1 (total electrode) |
1000 cycles (5C) |
RT |
202
|
| 42 m LiTFSI + 21 m Me3EtN·TFSI |
Li4Ti5O12//LiMn2O4 |
3.25 |
145 W h kg−1 (total electrode) |
150 cycles (0.2C) |
RT |
216
|
| 1 m Zn(TFSI)2 + 20 m LiTFSI |
Zn//LiMn2O4 |
n/a |
180 W h kg−1 (total electrode) |
4000 cycles (4C) |
RT |
147
|
| 1 m Zn(TFSI)2 + 20 m LiTFSI |
Zn//Super P |
n/a |
300 W h kg−1 (total electrode) |
200 cycles (0.05 A g−1) |
RT |
147
|
| 9 m NaOTf + 22 m TEAOTf |
NaTi2(PO4)3//Na3V2(PO4)3 |
n/a |
n/a |
100 cycles (1C) |
RT |
45
|
| 25 m NaFSI + 10 m NaFTFSI |
NaTi2(PO4)3//Na3(VOPO4)2F |
2.7 |
64 W h kg−1 (total electrode) |
500 cycles (C/5, 1C) |
−10 °C, 10 °C, 30 °C |
204
|
| Li(TFSI)0.7(BETI)0.3·2H2O |
Li4Ti5O12//LiNi0.5Mn1.5O4 |
3.8 |
>130 W h kg−1 (total electrode) |
100 cycles (6.8C) |
RT |
208
|
| Na(PTFSI)0.65(TFSI)0.14(OTf)0.21·3H2O |
NaTi2(PO4)3//Na3(VOPO4)2F |
2.7 |
77.9 W h kg−1 (total electrode) |
500 cycles (5C) |
RT |
209
|
| Li(TFSI)0.7(BETI)0.3·2H2O |
LiSICON∣Li//KB |
3.8 |
950 W h kg−1 (activated electrode and electrolyte) |
50 cycles (0.5 A g−1) |
RT |
210
|
| Zn(ClO4)2·6H2O–succinonitrile |
Zn//poly(2,3-dithiin-1,4-benzoquinone) |
2.55 |
n/a |
3500 cycles (0.3C) |
−20 °C, −10 °C, RT |
222
|
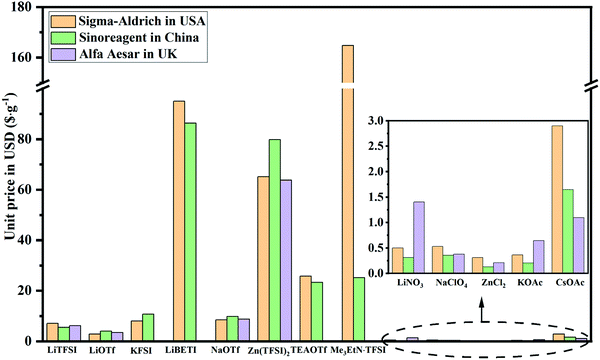 |
| | Fig. 12 Comparison of the unit price of various representative salts utilized in salt-concentrated electrolytes. | |
3.4. Gelled electrolyte
Gelled aqueous electrolytes can be deemed as hydrogel electrolytes, consisting of a crosslinked network of polymer chains with interstitial spaces filled with solvent water, which endows the aqueous electrolyte with quasi-solid state and flexible property. Hydrogel electrolytes can be fabricated and applied in flexible and stretchable ARBs not only because of their physical properties but also highly available and tunable polymer chemistries. Hydrogel-based ARBs with self-healing property, shape memory and stretchability can enable the fabrication of wearable devices for health care, implantable devices, soft electronics, and strain sensors.107,223 The frequently used polymer host materials in hydrogel electrolytes include polyvinyl alcohol (PVA), PAA, PAM, gelatin, alginate, agarose, chitosan, poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) (PEO–PPO–PEO) and poly(N-isopropylacrylamide-co-acrylamide) (PNIPAM/AM), which possess various features, functional groups, crosslinking/gelation methods (Fig. 13a), and cooperate with aqueous solutions of different cations, anions and pH values.223
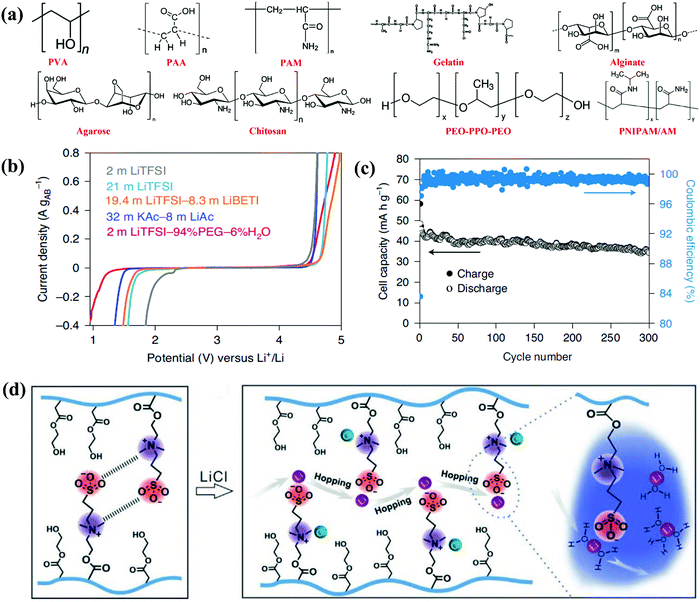 |
| | Fig. 13 (a) Molecular structure of frequently used polymer host materials for hydrogel electrolytes.223 (b) Comparison of ESWs between molecular crowding electrolyte and other salt-concentrated electrolytes.228 (c) Cyclic stability and CE of Li4Ti5O12//LiMn2O4 cell based on molecular crowding electrolyte under 1C rate.228 (d) Schematic diagram of electrostatic interaction between anionic and cationic groups without salts, and proposed Li+ migration mechanism in zwitterionic polymer hydrogel electrolyte.231 | |
3.4.1. Minimizing water content for higher stability.
The original function of hydrogel electrolytes based on their polymers was endowing ARBs with quasi-solid properties,224 while additional functions were also discovered to mitigate the challenges of ARBs. In a 5 M LiCl–PVA hydrogel electrolyte for supercapacitors, PVA could prevent the chemical dissolution of the V3O7 electrode by minimizing the water content, providing a neutral pH medium, and serving as an elastic coating.225 With further development, PVA was found to further extend the ESW of an LiClO4 aqueous solution, hence resulting in a higher energy density and cyclic stability in Zn–Li HiBs.226 By integrating PVA with the currently popular WiSE (21 m LiTFSI), a flexible and wearable symmetrical ARLiB was developed with an energy density of 141 W h kg−1 during 4000 cycles using a single LiVPO4F as both the anode and cathode. In that cell, high energy and power density and flexibility were realized mutually in the presence of PVA.227 Besides PVA, carboxymethyl cellulose (CMC) was added to an acetate-based WiSE (20 m KOAc) in a small amount (2 wt%) for gelling, which maintained a similar ESW as 32 m KOAc WiSE with less salt dissolved. The existence of CMC reduced the dissolution of Mn and Fe in the KMHCF cathode, and then enhanced the cyclic stability of this PiB to 400 cycles.212 In 2020, a significant work was reported by Lu's group using LiTFSI salt and poly(ethylene glycol) (PEG) polymer based on the common phenomenon of molecular crowding in living cells, which changed the hydrogen-bonding structure in water molecules, and thus reduced the activity of the water solvent. This molecular crowding electrolyte was optimized to 2 m LiTFSI–94% PEG–6% H2O, delivering an ESW of 3.2 V and especially excellent stability against the HER (Fig. 13b), which endowed the Li4Ti5O12//LiMn2O4 full cell with an energy density ranging from 75 to 110 W h kg−1 over 300 charge–discharge cycles (Fig. 13c).228 Another similar work was reported in 2021 as a “water-in-gel” electrolyte, which consisted of water, NaCl, ZnCl2 and sodium alginate (SA), expanding the ESW to 2.72 V. A “pass-way” for cation transportation was built within that electrolyte through coordinate bonds between the carboxylate groups of SA and Na/Zn ions. Also, due to the random coil conformation of the alginate chains, which was affected by dissolved ions, the 3D-fibrilar, porous polymer network ensured the uniform distribution and efficient transportation of Na/Zn ions in an aqueous environment. A Zn–Na hybrid battery based on this electrolyte was proposed, achieving a high extraction voltage of Na ions (2.1 V vs. Zn/Zn2+) and high capacity retention of 96.8% after 450 cycles.229
3.4.2. Lowering fluidity for higher energy density.
Besides wide ESWs, the quasi-solid state and poor fluidity of hydrogel electrolytes also contribute to the high energy density of ARBs based on their decoupling structure. As noted above, a PAA-based alkaline hydrogel and PAM-cellulose-based acid hydrogel were prepared as a two-pH decoupling electrolyte, enabling a high working voltage in a Zn//MnO2 battery (2.7 V). and thus high energy density. Compared with liquid decoupling electrolytes, hydrogel-based electrolytes simplify the three-chamber into a two-chamber structure, thus improving the relevant processibility.195 A gelled decoupling electrolyte with gelatin/ZnSO4 analyte, gelatin/CuSO4 catholyte, and anion-exchange separator was proposed, in which decoupling charge carriers of anolyte and catholyte simultaneously endowed the Zn anode and S cathode with optimal redox chemistry, as follows:230| | | Cathode: S+ 2Cu2+ + 4e− ↔ Cu2S | (22) |
| | | Anode: Zn ↔ Zn2+ + 2e− | (23) |
Derived from this, the theoretical volumetric energy density of this flexible HiB was regarded as 3868 W h L−1, while the battery delivered a stable output voltage (released 92% of its full capacity within a small voltage drop of 0.15 V) and an ultrahigh reversible capacity of 2063 mA h g−1 (based on mass of S) under a current density of 100 mA g−1.230
3.4.3. Anti-freezing for wider temperature window.
Hydrogel electrolytes also possess anti-freezing function, which guarantees the stable operation of ARBs at low temperatures. A new type of zwitterionic polymer hydrogel electrolyte was fabricated, achieving high conductivity without sacrificing the flexibility of hydrogels at subzero temperatures. The anionic and cationic counterions on the zwitterionic chains facilitated the dissociation of the LiCl salt, while the salt concentration significantly affected the conductivity and anti-freezing performance of the electrolytes. A direct hopping migration mechanism of hydrated Li+ through the channel of zwitterion groups was further proposed (Fig. 13d). The ultrahigh ionic conductivity of 12.6 mS cm−1 was achieved at −40 °C, owing to the synergistic effects including the electrostatic interactions between the zwitterionic groups and Li+ and the formation of an Li+(H2O)n hydration structure.231 Furthermore, a borax-crosslinked PVA/glycerol hydrogel electrolyte with a freezing point of below −60 °C was developed, in which glycerol could strongly interact with the PVA chains, thereby effectively prohibiting the formation of ice crystals in the whole gel network. This type of hydrogel electrolyte with 10.1 mS cm−1 at −35 °C, drove a Zn//MnO2 battery to charge/discharge for over 2000 cycles with around 90% capacity retention at that temperature.232 In summary, the performance of representative works is listed in Table S3 (ESI†). To some extent, hydrogel electrolytes are a solvent strategy to solidify/inactivate water and achieve wider ESWs, higher stability, and a wider working temperature range, which are effective and economic for better ARBs. In next section, another method for solvent manipulation, named hybrid solvent, will be reviewed.
3.5. Hybrid-solvent electrolyte
The SEIs formed in WiSEs were found to be predominantly inorganic metal fluorides, originating from the reduction of salt anions of TFSI− or OTf−. However, the formation of this anion-originating SEI faces a severe “cathodic challenge” as result of the repulsion of anions by the negatively polarized anode surfaces, making the expansion of the cathodic limits extremely difficult.233,234 Therefore, a new class of electrolytes hybridizing aqueous and non-aqueous solvents was reported, which inherited the intrinsic merits of each system and successfully resolved the conflicts among performance, cost, interfacial chemistry, ambient control, and environmental concerns.
3.5.1. Hybridization with organic (aprotic) solvent.
Dimethyl carbonate (DMC) was blended with an LiTFSI WiSE, introducing a secondary ingredient (alkylcarbonate) in the protective interphase on the anode in addition to LiF from anion reduction. This hybrid interphase, consisting of LiF and alkylcarbonate, stabilized the electrolyte down to 1.0 V vs. Li/Li+, allowing 3.2 V Li-ion cell chemistry of Li4Ti5O12 anode and LiNi0.5Mn1.5O4 cathode.233 Subsequently, the LiTFSI-DMC/water hybrid-solvent electrolyte was applied for DiBs, in which Li+ intercalation/deintercalation occurred on the Li4Ti5O12 side, while intercalation/deintercalation occurred on the graphite side.235 Furthermore, DMC was introduced in an LiTFSI + LiOTf WiBSE with both broad ESW and excellent safety, and assembled with a KS6 graphite cathode and Nb2O5 anode as DIB, which exhibited a good comprehensive performance including capacity, cycling stability, rate performance, and medium discharge voltage.236 Besides Li+-based chemistries, the hybrid solvent concept also suits SiBs. 7 m NaOTf aqueous solution was mixed with 8 m NaOTf propylene carbonate (PC) solution. Through Raman spectral characterization, intensified cation–anion association was observed in this electrolyte, which benefited the formation of an interphase at the electrode/electrolyte interface, thereby stabilizing the water molecules against the reducing surfaces of the electrodes. The Na+-based solvent-hybrid electrolyte offered an ESW of up to 2.8 V and enabled the NaTi2(PO4)3//Na3V2(PO4)3 redox pair.237
3.5.2. Hybridization with ionic liquid.
The above-mentioned DMC and PC are regular organic solvents for conventional LiBs, while some ionic liquids were also mixed with aqueous solutions in some studies.238,239 Ionic liquids exhibit unique properties such as flame-retardant ability, non-volatility, and high thermal stability, but their high viscosity may hinder their further applications. Therefore, a ‘‘water in salt/ionic liquid” electrolyte composed of a salt of LiTFSI and a molten salt of 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl) imide (EMIM-TFSI) was mixed with an extremely small amount of water (water/LiTFSI/EMIM-TFSI molar ratio of 1:1:2). In this electrolyte, the ESW was considerably widened without sacrificing the ionic conductivity, allowing accessibility to the full capacity of a commercial Nb2O5 material at a low potential window (−1.6 V vs. Ag/AgCl).239 Meanwhile, deep eutectic solvents (DESs), generally obtained by simply mixing Lewis acids and bases in a eutectic molar ratio, have emerged as attractive alternatives to ionic liquids due to their high degree of design freedom. 6 wt% water was added to a urea-based DES, endowing the Zn anode with unusual reversibility and durability. The intensified water–DES interactions significantly suppressed the water reactivity, while the merits of the aqueous system on the ionic conductivity and viscosity were conserved.238
3.5.3. Hybridization with low-freezing point solvent.
Some organic solvents possessing low freezing points can be employed to enhance the low-temperature performance of ARBs by hybridizing aqueous electrolytes. For instance, the freezing point of acetonitrile (AN) is −48 °C, together with the properties of high dielectric constant (35.9), high oxidation stability (>5 V vs. Li+/Li), and high miscibility with water, enabling an ARLiB with the Li4Ti5O12//LiMn2O4 redox pair to exhibit high capacity at both RT and 0 °C.240 In addition, ethylene glycol (EG) has been widely used as antifreeze in engine coolant owing to its high boiling point and relatively low freezing point (−12 °C). When mixed with water, its freezing point can be further reduced (even to −40 °C), while the solvation interaction of Zn2+ with H2O is partly reduced by the introduction of EG, contributing to fewer side reactions such as the HER on the Zn anode side. In a reported work by Li's group, the portion of EG in the mixture was optimized to 40% in volume, considering both ionic conductivity and ESW, which drove the Zn//PANI–V2O5 battery to more than 250 cycles under a current density of 0.2 A g−1 at −20 °C.241 Qiao's group proposed a route using low-cost antisolvents, in which, for example, methanol was added to ZnSO4 aqueous electrolyte to weaken the Zn2+ solvation sheath and minimize water activity by strengthening the interaction between the free and coordinated water molecules with antisolvents. The Zn reversibility was significantly boosted in the antisolvent electrolyte of 50% methanol in volume (anti-M-50%) at low or elevated temperatures. Based on the anti-M-50% electrolyte, the merits of the Zn//PANI coin cell/pouch cell were impressive, even repeatedly charged/discharged to 2000 cycles at −10 °C.242 Hybridizing solvents were also applied to ARFBs by Yu et al., who systematically studied a wide range of salts (LiCl, Li2SO4 and LiTFSI) and solvents (dimethoxyethane, DME; tetraethylene glycol dimethyl ether, TEGDME; acetonitrile, AN; and dimethylformamide, DMF). Finally, 1.5 M LiI in DMF/water was chosen because of its 2.8 V ESW and retained ionic conductivity at low temperatures, which enabled the successful operation of Zn//LiI ARFBs at −20 °C for 150 cycles with nearly no capacity loss.243 The merits of the works involving hybrid-solvent electrolytes are summarized (Table 2) based on representative papers in the literature.233,235–245 Also, depending on the merits, it can be concluded that the strategy of hybridizing solvents is promising if a balance among wide ESW, high ionic conductivity, low freezing point and cost can be found.
Table 2 A summary of recent representative works on hybrid-solvent electrolytes for ARBs
| Electrolyte |
Electrode |
ESW |
Energy density |
Cyclic stability |
Working temperature |
Reference |
| 14 M LiTFSI in DMC/water |
Li4Ti5O12//LiNi0.5Mn1.5O4 |
4.1 V |
165 W h kg−1 (total electrode) |
>1000 cycles (6C) |
RT |
233
|
| 14 M LiTFSI in DMC/water |
Li4Ti5O12//graphite |
4.1 V |
n/a |
50 cycles (0.2 A g−1) |
RT |
235
|
| 9.25 m LiTFSI + 3.1 LiOTf in DMC/water |
Nb2O5//graphite |
4.0 V |
n/a |
300 cycles (0.2 A g−1) |
RT |
236
|
| Li4(TEGDME)(H2O)7 |
Li4Ti5O12//LiMn2O4 |
4.2 V |
120 W h kg−1 (total electrode) |
500 cycles (3C) |
RT |
244
|
| 50 wt% Li(LiOH)PAA |
TiO2/LiTi2(PO4)3//LiMn2O4/LiNi0.5Mn1.5O4 |
2.7 V |
142.2 W h kg−1 (total electrode) |
100 cycles (0.5C) |
RT |
245
|
| Mixture of 7 m NaOTf in water and 8 m NaOTf in PC |
NaTi2(PO4)3//Na3V2(PO4)3 |
2.8 V |
45 W h kg−1 (total electrode) |
100 cycles (10C) |
RT |
237
|
| LiTFSI + Zn(TFSI)2 (Li+:Zn2+ = 20 in molar) in DES/water |
Zn//LiMn2O4 |
>2.5 V |
224.6 W h kg−1 (cathode) |
600 cycles (0.1, 0.5, 1C) |
RT |
238
|
| LiTFSI/H2O/(EMIM-TFSI)2.0 |
Nb2O5//AC |
4.7 V |
51.9 W h kg−1 (total electrode) |
3000 cycles (1 A g−1) |
RT |
239
|
| 15.3 m LiTFSI in AN/water |
Li4Ti5O12//LiMn2O4 |
4.5 V |
173 W h kg−1 (total electrode) |
1000 cycles (5C) |
RT, 0 °C |
240
|
| 2 M ZnSO4 in EG/water |
Zn//PANI/V2O5 |
2.8 V |
1700 W h kg−1 (cathode) |
>250 cycles (0.2 A g−1) |
−40 °C to RT |
241
|
| 2 M ZnSO4 in Anti-M-50% |
Zn//PANI |
∼2.5 V |
n/a |
2000 cycles (5 A g−1) |
RT, −10 °C |
242
|
| 1.5 M LiI in DMF/water |
Zn//LiI |
2.8 V |
n/a |
2000 cycles (0.2 mA cm−2) |
RT, −20 °C |
243
|
3.6. Electrode/current collector–electrolyte interface tuning
Interface tuning is a common strategy for all types of electrolytes, while the goals of tuning can be generally classified into thermodynamics (chemical and thermal stability) and kinetics (charge and mass transportation). Aqueous electrolytes possess superior interfacial kinetics and wettability, and thus it is more importance to cover the shortage in thermodynamics. The SEI can kinetically stabilize electrolytes at potentials far beyond their thermodynamic stability limits, allowing cell reactions to proceed reversibly. However, it failed to occur in aqueous electrolytes until the discovery of WiSE. The formation mechanism of an SEI in WiSE are possibly contributed by two pathways, as follows: (1) reduction of anion complexes or clusters and (2) reduction of O2 and CO2 dissolved in the electrolyte (Fig. 14a),246 implied by combining various spectroscopic, electrochemical and computational techniques.
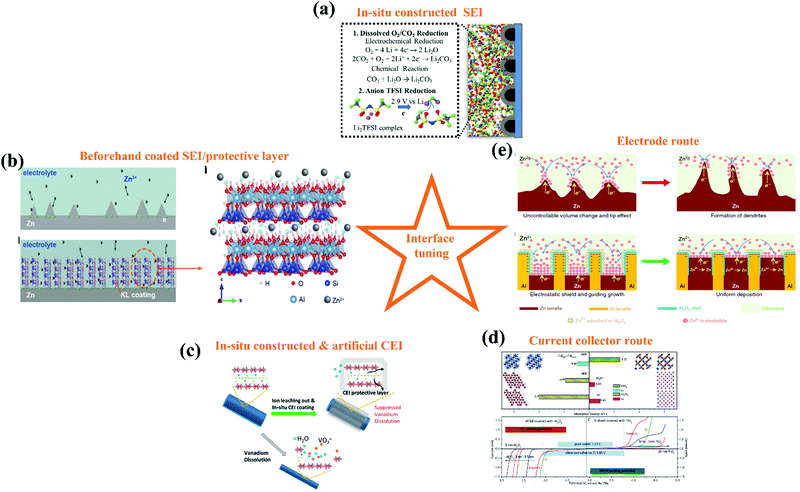 |
| | Fig. 14 (a) SEI formation mechanisms in WiSE.246 (b) Schematic diagrams of Zn deposition process on bare Zn and KL–Zn with a detailed schematic diagram of confined Zn2+ transmission in kaolin.250 (c) Illustration of the in situ CEI layer strategy design.252 (d) Stability of Al and Ti current collectors in 15 m NaClO4 aqueous electrolyte.254 (e) Schematic illustration of eutectic strategy for dendrite and crack suppression.257 | |
3.6.1. Solid electrolyte interface.
The in situ constructed-SEI in WiSEs not only enabled a series of high voltage/energy density ARLiBs with unprecedented stability, but also brought high flexibility and even “open configurations”, which have been hitherto unavailable for any LiB chemistries.75,77,202 In contrast, in Zn2+-based aqueous systems, severe the HER during Zn plating/stripping makes the in situ formation of SEI impossible. As mentioned for the electrolyte additive strategy, some additives were added to facilitate the formation of an SEI in ARZiBs. Wang at el. used 0.5 m Me3EtNOTF additive to build a fluorinated and hydrophobic interphase that conducts Zn2+, while suppressing the HER through the decomposition of alkylammonium ions.177 Meanwhile, the same group developed inorganic a ZnF2–Zn5(CO3)2(OH)6–organic bilayer SEI based on a low-concentration aqueous Zn(OTF)2–Zn(NO3)2 electrolyte, mutually considering Zn2+ diffusion (inorganic inner layer) and water inactivation (organic outer layer). A Zn5(OH)8(NO3)2·2H2O layer was initially formed on the Zn anode surface via the self-terminated chemical reaction of NO3− with Zn2+ and OH−, and then it converted into Zn2+ conducting Zn5(CO3)2(OH)6, promoting the formation of ZnF2 as the inner layer. Meanwhile, the organic-dominated outer layer was generated by the reduction of OTf−.247
Another type of interfacial engineering is pre-coating an SEI/protective layer, which is more mechanically robust than and in situ-grown SEI. In the WiSE system, a pre-coated anode-protected layer was prepared by adding 0.5 M LiTFSI and 10 wt% PEO to a mixture of 1,1,2,2-tetrafluoroethyl-2′,2′,2′-trifluoroethyl ether (HFE) and DMC (95:5 in volume), vigorously heating and stirring at 75 °C, which stabilized the graphite anode in a WiBSE and guaranteed a full cell with a working voltage of 4.0 V.248 The HFE–PEO protective gel was applied to another graphite anode in the same WiBSE, paired with the cathode of halogen conversion–intercalation chemistry, which not only provided a working voltage of more than 4.0 V but also considerably high capacity of 243 mA h g−1 (for total electrode mass).76 Besides ARLiBs, pre-coated protective layers were massively used on the surface of Zn metal anodes for the purpose of dendrite-control.249 A layer of sieve-element function (selective channel of Zn2+) and uniform-pore distribution (≈3.0 nm) of a kaolin was prepared and coated on a Zn metal anode for alleviating dendrite and corrosion issues. Owing to the selective channel of Zn2+ and narrow distribution pore diameter of kaolin (Fig. 14b), homogeneous Zn2+ migration confined by this channel was achieved with dendrite-free deposition. Moreover, the coated kaolin (KL)–Zn anode exhibited long-time stability (800 h under current density of 1.1 mA h cm−2), and accordingly suppressed side reactions at the anode surface/interface. This property was further verified by the KL-Zn//MnO2 cell with high initial specific capacity and good capacity retention and a reasonably well-preserved morphology, as characterized by SEM.250 A porous nano-CaCO3 coating was discovered to possess the ability to guide uniform and position-selected Zn stripping/plating at the nano-CaCO3-layer/Zn metal anode interfaces. This Zn-deposition-guiding ability was mainly ascribed to the porous nature of the nano-CaCO3-layer, considering the discovery of a similar functionality (even though relatively inferior) in Zn metal anodes coated with porous acetylene black or nano-SiO2 layers. The coated Zn anode was matched with a CNT–MnO2 cathode in ZnSO4 + MnSO4 electrolyte, showing a 42.7% higher discharge capacity than bare Zn metal anode (177 vs. 124 mA h g−1 at 1 A g−1) after 1000 cycles.251
3.6.2. Cathode electrolyte interface.
Compared with the SEI, the CEI has been rarely reported, but it is still worthy to be investigated and applied for improving the stability of the cathode–electrolyte interface against side reactions and cathode dissolution. One general strategy to construct a CEI for suppressing the dissolution of vanadium-based cathodes in aqueous electrolytes and beyond was proposed, which introduced Sr ions into vanadium oxide layers as a sacrifice guest, leaching from the vanadium-based cathode as an in situ CEI coating layer on the surface (Fig. 14c).252 In ARLiBs, it is generally accepted that CEIs can only be formed in super-concentrated electrolytes containing fluorine-based organic anions (TFSI−, OTf−, etc.), while in a traditional nitrite-based aqueous electrolyte (pH-adjusted saturated LiNO3), a CEI was initiatively discovered on the surface of the LiNiO2 cathode with the composition of Li2CO3 and LiOH, as demonstrated by transmission electron microscopy (TEM) and X-ray photoelectron (XPS). Under the protection of the CEI, the capacity retention of LiNO2 was significantly enhanced in comparison of that in 1 M LiNO3 + LiOH (pH = 10) aqueous electrolyte after 50 cycles.253
3.6.3. Current collector.
In the study regarding the HME of Li(TFSI)0.7(BETI)0.3·2H2O, the effect from current collector to the ESW was confirmed, which could further expand the observed ESW when changing the cathode current collector to Ti and that of the anode into Al.208 Zhu et al. reported that oxidizing an Al current collector to Al2O3 (3 nm thickness surface) for the anode and Ti current collector to TiO2 (5 nm thickness surface) for the cathode could expand the ESW of 15 M NaClO4 aqueous electrolyte to 3.5 V (Fig. 14d). This wide ESW enabled the TiS2 anode of SiBs to exhibit a low working potential (1.5 V vs. Na/Na+) for the first time. Thus, a full cell coupling a TiS2 anode with PBA cathode in 15 M NaClO4 aqueous electrolyte demonstrated a working voltage of 2.6 V and cyclic life up to 1000 times (30C rate) with a high energy density of 100 W h kg−1.254 In Zn metal anode-based ARBs, the substrates (current collectors) can also affect Zn stripping/plating. Archer's group found that graphene with a low lattice mismatch for Zn was shown to be effective in driving the deposition of Zn with a locked crystallographic orientation relation. The resultant epitaxial Zn anodes achieved exceptional reversibility in 2 M ZnSO4 aqueous electrolyte (10000 cycles) and 1000 cycles of full-cell charging/discharging together with an α-MnO2 cathode.255
3.6.4. Electrode.
In aqueous supercapacitor systems, tuning the electrode itself was also reported as a method to widen the ESW. In that work, the W atoms in the WO3 crystal were partially replaced by Mo atoms, thus introducing oxygen vacancies. The negative limitation of the ESW was tunable between −0.4 and −1.2 V vs. SHE in 1 M Li2SO4 aqueous electrolyte owing to the consolidation process-customized band gap and HER overpotentials of the Mo-substituted WO3 electrode.256 Zn alloying is another electrode route-pertaining interface, especially alloying with Al. Eutectic-composition alloying of Zn and Al as an effective strategy substantially tackled the irreversibility issues of Zn metal anodes by making use of their lamellar structure composed of alternating Zn and Al nano lamellas. The lamellar nanostructure not only promoted Zn stripping from the precursor eutectic Zn88Al12 (at%) alloys, but produced core/shell Al/Al2O3 interlamellar nanopatterns in situ to guide the subsequent growth of zinc (Fig. 14e), enabling dendrite-free Zn stripping/plating for more than 2000 h in oxygen-free aqueous electrolyte of 2 M ZnSO4.257 Zn–Al alloying was also employed in an ARAiB, which consisted of an AlxMnO2 cathode, a Zn substrate-supported Zn–Al alloy anode, and an Al(OTf)3 aqueous electrolyte. The featured alloy interface layer could effectively alleviate the passivation and suppress the dendrite growth, ensuring ultralong-term stable Al stripping/plating. The relevant architected cell exhibited a record-high discharge voltage plateau near 1.6 V and specific capacity of 460 mA h g−1 for over 80 cycles.258 Generally, interface tuning can be another universal method for ameliorating aqueous electrolyte and any other electrolytes, which is worthy to be investigated more deeply. The performance of representative works is summarized in Table S4 (ESI†).
3.7. Beyond concentrated electrolyte
The benefits of salt-concentrated electrolytes towards ARBs have been intensively claimed here, while type kind of electrolyte has to use salts with high solubility, restricting its applications in scale and scope. Therefore, the routes concerning breaking the ceiling of salt solubility, thereby introducing more cost-effective salts with relatively low solubility, and further broadening ESW emerge, which can be classified into de-solvation/salting-out and solubility enhancement.
3.7.1. De-solvation.
Zhou's group developed a new liquid electrolyte for Li metal batteries with de-solvated Li+ (“Li+ de-solvated electrolyte”), which was merely composed of inactive “frozen-like” solvent and crystal-like salt solute.259 The addition of salts to the concentrated electrolytes could effectively eliminate the abundance of free solvents in the electrolytes and make the concentrated electrolytes more aggregative than their original counterparts. Specifically, the electrolyte configuration evolved from solvent-separated ion pair (SSIP)-dominated structures to contact ion pair (CIP)-dominated structures (Fig. 15a).260 This type of beyond concentrated electrolyte was even more aggregative than the saturated state and was composed of only crowded solvent-depleted CIPs without any free solvents.260 Tao's group introduced this concept in aqueous electrolytes with inexpensive inorganic salts.55,226,261 An “oversaturated gel electrolyte” (OSGE) was simply prepared with PVA and saturated LiNO3 at 95 °C. Subsequently, the excess salt was crystallized at room temperature, which was dispersed equally by the continuous room-temperature saturated gel (RTSG), forming a heterogeneous morphology (Fig. 15b). The continuous ion pathway ensured the considerable ionic conductivity of OSGE, regardless of the existence of ion-insulated crystallized salt, while the ESW was further expanded compared with that of the RTSGE due to the further compacted CIPs (Fig. 15c). Moreover, the stability window of the OSGE was still wide enough at elevated temperatures even at 80 °C, ensuring the desirable elevated temperature performance of ARLiBs.55 Meanwhile, a perchlorate OSGE (1 m Zn(ClO4)2 + 10 m LiClO4–PVA) was also employed in Zn–Li HiBs by the same group, which showed an almost dendrite-free morphology with better cyclic stability.226 Yan et al. applied this strategy for the fabrication of electrolytes for low-temperature Zn-ion capacitors, which possessed superior ionic conductivity (even 1.3 × 10−3 S cm−1 at −60 °C) based on Zn(ClO4)2 salty ice, which is attributed to the unique 3D ionic transport channels inside this ice.262
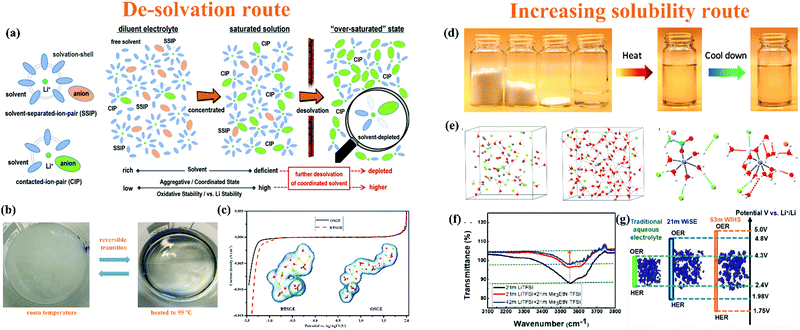 |
| | Fig. 15 (a) Design idea of further depletion of solvent molecules in the Li+ solvation sheath.260 (b) Optical images of top view of OSGE at RT and 95 °C.55 (c) Comparison of ESW and Li+ solvation sheath between OSGE and RTSGE.55 (d) WSOE45-1 prepared by stoichiometric amounts of ZnCl2, ZnBr2, Zn(OAc)2, and water.263 (e) Snapshots and extracted typical molecules conformations of MD simulation on WSOE45-1 and 5 m ZnBr0.5Cl1.5 aqueous solution.263 (f) FTIR spectra of WiSE, 42 m WiHS, and 63 m WiHS electrolytes.220 (g) MD simulation results and ESWs of traditional aqueous electrolyte, WiSE and WiHS electrolyte.220 | |
3.7.2. Increasing solubility.
Beside de-solvation/salting-out, the solubility increasing route was also attempted. A ZnCl2 + ZnBr2 + Zn(OAc)2 aqueous electrolyte with a record super-solubility of up to 75 m was reported, which broke through the physical solubility limit, owing to the formation of acetate-capped water–salt oligomers (WSOs) bridged by Br−/Cl−–H and Br−/Cl−/O–Zn2+ interactions (Fig. 15d and e). This supersoluble electrolyte drove a high-performance DiB to display a reversible capacity of 605.7 mA h g−1 (corresponding energy density of 908.5 W h kg−1).263 A 63 m “water-in-hybrid-salt” (WiHS) electrolyte (42 m LiTFSI + 21 m Me3EtN·TFSI) was prepared, which doubled the solubility of LiTFSI due to the inert salt based on the asymmetric ammonium cation of Me3EtN+. The FTIR spectrum in Fig. 15f reveals that the hydrogen bonding network of the WiHS electrolyte was severely disrupted, leading to a further broadened ESW (Fig. 15g).220 The performance of representative works is summarized in Table S5 (ESI†). Basically, the mechanism behind beyond concentrated electrolytes still needs more studies, especially more fundamental studies, although some excellent works about this electrolyte tuning strategy have been reported.
4. Future perspective and conclusions
To mitigate the challenges of narrow ESWs, electrode dissolution/side reactions and temperature-variation instability of aqueous electrolytes, various strategies including electrolyte additive, pH management, salt concentration, electrolyte gelling, hybrid solvent, interface tuning, and breaking through salt solubility limitation were reported intensively during the past five years. The relevant achievements are remarkable, promoting ARBs in energy storage systems rapidly, particularly the achievements in salt-concentrated electrolytes. The beyond concentrated electrolytes can be included in salt-concentrated electrolytes, together with “solvate ionic liquid”, “super-concentrated”, “solvent-in-salt”, and “localized concentrated” electrolytes, which are inherited from “polymer-in-salt” electrolytes.264 At present, the salt-concentrated strategy is the most effective and efficient for high-energy and high-stability ARBs, whereas the cost of the salt is a challenge for large-scale applications. In the future, the hybrid solvent strategy can be more competitive if better non-aqueous solvents are discovered, which not only can reduce the cost, but also generate more stable electrode/electrolyte interfaces, thereby stabilizing ARBs. The artificial SEI/CEI can be another promising method; however, it is technically more difficult to create than hybrid solvents, and thus may take longer to be developed.
Despite the significant progress, there are still some potential challenges for the further commercialization of ARBs. Firstly, simulation and characterization towards aqueous electrolytes need to be improved. The parameters of MD simulation for the solvation sheath and DFT calculation for the electrode–electrolyte interface should be pre-set conscientiously rather than intentionally for supporting the as-obtained experimental results. In the case of the experimental characterization of the ion solvation structure, Raman, FTIR and NMR spectroscopy are used mostly as indirect methods, while seldom works have been reported using direct methods. X-ray nanoimaging was utilized for understanding the benefits of WiSEs, which provided direct evidence for the stability of WiSEs compared with conventional aqueous electrolytes.265 However, that was micron/sub-micron characterization, failing to approach the ion solvation sheath with a sub-nanosize. In situ liquid cell TEM was reported to reveal the nanoscale mineralization pathways of hydroxyapatite, which exhibits potential to characterize liquid aqueous electrolytes.266 Secondly, the necessity of high concentration should be reconsidered.267,268 For instance, in the ZnCl2 system, the concentration of 7.5 m rather than 30 m in the saturated state was selected as the optimum concentration for modulating the electrolyte structure, and thus suppressing the freezing of water,215 implying that, in some cases, too concentrated may be as bad as too diluted. Moreover, seawater can function as a desirable electrolyte for ARZiBs with a stable interfacial process through a Zn–Mn alloy anode,269 further demonstrating that suitable concentration and rational interfacial design are more critical than pursuing even more concentrated electrolytes. Further, Tarascon et al. indicated that there was still a long way to apply WiSEs, the most popular salt-concentrated species, because of their fragile in situ-grown SEI and poor elevated-temperature performances.270 The most evident benefit induced by salt-concentrated electrolytes is their expanded ESWs, and thus higher energy density of ARBs by pairing with high-working voltage electrodes. However, the electrode kinetics and mass transportation in salt-concentrated systems are usually undesirable due to their high viscosity and sluggish interfacial process. Nanosized electrodes can be an effective way to overcome kinetic issues, which balance the wide ESWs of the electrolyte and high capacity of the electrode when utilized in salt-concentrated electrolytes.92,212,261 Besides their relatively low working voltage, ZABs and ARFBs are regarded as effective aqueous chemistries to significantly improve the energy density through their dramatically enhanced capacity, which are promising alternatives for the salt-concentrated strategy. To some extent, ZABs and ARFBs are suitable substitutes for LiBs owing to both their high energy density and excellent safety,159,160,271 which are equally important in the future. The last issue is the gap between industry and academics. In fact, reporting the performance based on a limited number of metrics does not give a realistic picture of the battery performance required for practical use. Although it is not necessary to force the standardization of the electrode and button cell design for fundamental research, it is of paramount importance to understand the influence of button cell design, fabrication, and testing protocol on the tested results, providing more reliable results as guidelines for future studies and applications.272,273 For real applications, the issue of self-discharge in ARBs should not be neglected, which is not only induced by macroscopical and microcosmic short circuit and electrode dissolution, but also direct chemical reactions (corrosion) between the electrode and electrolyte, especially for Zn foil electrodes. The reported long cyclic life of Zn metal-based batteries is normally obtained by testing a button cell immediately, neglecting the aging effects. Using Zn foil as both the working electrode and current collector will cause many problems when the battery is scaled up. Therefore, it is necessary to build up a Zn powder/current collector configuration with a longer calendar life.274
As mentioned above, ZABs and ARFBs with high capacity (high energy density) are promising for large-scale energy storage. Among the ARFBs, Zn//Br2 and Zn//I2 batteries are kinetically better than the Fe//Cr battery, while Zn corrosion is obstructive together with the corrosion of ARFBs. In the case of ZABs, besides the issues with the Zn metal anode, the sluggish kinetics of the ORR/OER at the cathode side also results in large polarization and low energy efficiency. The introduction of light has been demonstrated to boost the reaction kinetics of the ORR/OER, leading to enhanced electrochemical performances.275 Therefore, the synergic effect of electricity and light can cultivate thermodynamically and kinetically better ZABs, if the theoretical understanding of the photoelectrochemical process at oxygen cathodes is deeper, whilst relevant characterization technology becomes more advanced. Overall, aqueous energy storage systems have been extensively investigated through the development of novel electrolyte concepts, while there are still challenges to be tackled before their large-scale applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
One of the authors (S. C.) thanks China Scholarship Council for a fully funded PhD studentship to study at University of Warwick (No. 201706690053).
Notes and references
- M. C. Hänsel, M. A. Drupp, D. J. A. Johansson, F. Nesje, C. Azar, M. C. Freeman, B. Groom and T. Sterner, Nat. Clim. Change, 2020, 10, 781–789 CrossRef.
- J. H. R. Lambers, Science, 2015, 348, 501–502 CrossRef CAS PubMed.
- S. M. S. U. Eskander and S. Fankhauser, Nat. Clim. Change, 2020, 10, 750–756 CrossRef CAS.
- O. Babacan, S. De Causmaecker, A. Gambhir, M. Fajardy, A. W. Rutherford, A. Fantuzzi and J. Nelson, Nat. Energy, 2020, 5, 720–728 CrossRef CAS.
- N. Bauer, S. K. Rose, S. Fujimori, D. P. van Vuuren, J. Weyant, M. Wise, Y. Cui, V. Daioglou, M. J. Gidden, E. Kato, A. Kitous, F. Leblanc, R. Sands, F. Sano, J. Strefler, J. Tsutsui, R. Bibas, O. Fricko, T. Hasegawa, D. Klein, A. Kurosawa, S. Mima and M. Muratori, Clim. Change, 2018, 163, 1553–1568 CrossRef.
- C. E. Thomas, Int. J. Hydrogen Energy, 2009, 34, 6005–6020 CrossRef CAS.
- X. Zeng, M. Li, D. Abd El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- M. Tran, D. Banister, J. D. K. Bishop and M. D. McCulloch, Nat. Clim. Change, 2012, 2, 328–333 CrossRef.
- Y. Lu, X. Rong, Y.-S. Hu, L. Chen and H. Li, Energy Storage Mater., 2019, 23, 144–153 CrossRef.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- W. Lee, S. Muhammad, C. Sergey, H. Lee, J. Yoon, Y.-M. Kang and W.-S. Yoon, Angew. Chem., Int. Ed., 2020, 59, 2578–2605 CrossRef CAS PubMed.
- Q. Wang, L. Jiang, Y. Yu and J. Sun, Nano Energy, 2019, 55, 93–114 CrossRef CAS.
- D. Wang, J. Coignard, T. Zeng, C. Zhang and S. Saxena, J. Power Sources, 2016, 332, 193–203 CrossRef CAS.
- G.-L. Zhu, C.-Z. Zhao, J.-Q. Huang, C. He, J. Zhang, S. Chen, L. Xu, H. Yuan and Q. Zhang, Small, 2019, 15, 1805389 CrossRef PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS PubMed.
- A. Parasuraman, T. M. Lim, C. Menictas and M. Skyllas-Kazacos, Electrochim. Acta, 2013, 101, 27–40 CrossRef CAS.
- F. Zheng, M. Kotobuki, S. Song, M. O. Lai and L. Lu, J. Power Sources, 2018, 389, 198–213 CrossRef CAS.
- Q. Pang, A. Shyamsunder, B. Narayanan, C. Y. Kwok, L. A. Curtiss and L. F. Nazar, Nat. Energy, 2018, 3, 783–791 CrossRef CAS.
- D. Bin, Y. Wen, Y. Wang and Y. Xia, J. Energy Chem., 2018, 27, 1521–1535 CrossRef.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- W. Zhao, J. Yi, P. He and H. Zhou, Electrochem. Energy Rev., 2019, 2, 574–605 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- S. Xin, Y. You, S. Wang, H.-C. Gao, Y.-X. Yin and Y.-G. Guo, ACS Energy Lett., 2017, 2, 1385–1394 CrossRef CAS.
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- J. Liu, C. Xu, Z. Chen, S. Ni and Z. X. Shen, Green Energy Environ., 2018, 3, 20–41 CrossRef.
- H. Manjunatha, G. S. Suresh and T. V. Venkatesha, J. Solid State Electrochem., 2011, 15, 431–445 CrossRef CAS.
- W. Tang, Y. Zhu, Y. Hou, L. Liu, Y. Wu, K. P. Loh, H. Zhang and K. Zhu, Energy Environ. Sci., 2013, 6, 2093–2104 RSC.
- A. Gurung, J. Pokharel, A. Baniya, R. Pathak, K. Chen, B. S. Lamsal, N. Ghimire, W.-H. Zhang, Y. Zhou and Q. Qiao, Sustainable Energy Fuels, 2019, 3, 3279–3309 RSC.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Mater., 2016, 5, 139–164 CrossRef.
- A. Manuel Stephan, Eur. Polym. J., 2006, 42, 21–42 CrossRef CAS.
- Y. Wang and W.-H. Zhong, ChemElectroChem, 2015, 2, 22–36 CrossRef CAS.
- X. Yuan, F. Ma, L. Zuo, J. Wang, N. Yu, Y. Chen, Y. Zhu, Q. Huang, R. Holze, Y. Wu and T. van Ree, Electrochem. Energy Rev., 2021, 4, 1–34 CrossRef CAS.
- Z. Chang, Y. Yang, M. Li, X. Wang and Y. Wu, J. Mater. Chem. A, 2014, 2, 10739–10755 RSC.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef PubMed.
- X. Wu and X. Ji, Nat. Chem., 2019, 11, 680–681 CrossRef PubMed.
- A. Eftekhari, Adv. Energy Mater., 2018, 8, 1801156 CrossRef.
- J.-Y. Luo, W.-J. Cui, P. He and Y.-Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- X. Wang, Y. Hou, Y. Zhu, Y. Wu and R. Holze, Sci. Rep., 2013, 3, 1401 CrossRef PubMed.
- J. O. G. Posada, A. J. R. Rennie, S. P. Villar, V. L. Martins, J. Marinaccio, A. Barnes, C. F. Glover, D. A. Worsley and P. J. Hall, Renewable Sustainable Energy Rev., 2017, 68, 1174–1182 CrossRef CAS.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- J. Yue, L. Lin, L. Jiang, Q. Zhang, Y. Tong, L. Suo, Y.-S. Hu, H. Li, X. Huang and L. Chen, Adv. Energy Mater., 2020, 10, 2000665 CrossRef CAS.
- Q. Nian, J. Wang, S. Liu, T. Sun, S. Zheng, Y. Zhang, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 16994–16999 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, Chem. Rev., 2020, 120, 6783–6819 CrossRef CAS PubMed.
- J. B. Goodenough, Solid State Ionics, 1994, 69, 184–198 CrossRef CAS.
- N. Mahmood, T. Tang and Y. Hou, Adv. Energy Mater., 2016, 6, 1600374 CrossRef.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- M. Ishikawa, M. Ihara, M. Morita and Y. Matsuda, Electrochim. Acta, 1995, 40, 2217–2222 CrossRef CAS.
- K. Xu, J. Electrochem. Soc., 1999, 146, 4172 CrossRef CAS.
- S. G. Chen, R. Lan, J. Humphreys and S. W. Tao, ACS Appl. Energy Mater., 2020, 3, 2526–2536 CrossRef CAS.
- S. Chen, P. Sun, B. Sun, J. Humphreys, P. Zou, K. Xie and S. Tao, Energy Storage Mater., 2021, 37, 598–608 CrossRef.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
-
W. B. Kirby, Linden's Handbook of Batteries, McGraw-Hill Education, New York, 5th edn, 2019 Search PubMed.
- A.-K. Kontturi, K. Kontturi, L. Murtomäki and D. J. Schiffrin, J. Chem. Soc., Faraday Trans., 1994, 90, 2037–2041 RSC.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- M. A. Ratner, P. Johansson and D. F. Shriver, MRS Bull., 2000, 25, 31–37 CrossRef CAS.
- J. G. Smith and D. J. Siegel, Nat. Commun., 2020, 11, 1483 CrossRef CAS PubMed.
- Z. Zhang and L. F. Nazar, Nat. Rev. Mater., 2022 DOI:10.1038/s41578-021-00401-0.
- D. J. Siegel, L. Nazar, Y.-M. Chiang, C. Fang and N. P. Balsara, Trends Chem., 2021, 3, 807–818 CrossRef CAS.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed.
- Q. T. Qu, Y. Shi, S. Tian, Y. H. Chen, Y. P. Wu and R. Holze, J. Power Sources, 2009, 194, 1222–1225 CrossRef CAS.
- J. F. Whitacre, A. Tevar and S. Sharma, Electrochem. Commun., 2010, 12, 463–466 CrossRef CAS.
- S. I. Park, I. Gocheva, S. Okada and J.-I. Yamaki, J. Electrochem. Soc., 2011, 158, A1067 CrossRef CAS.
- D. Bin, F. Wang, A. G. Tamirat, L. Suo, Y. Wang, C. Wang and Y. Xia, Adv. Energy Mater., 2018, 8, 1703008 CrossRef.
- X. Pu, H. Wang, D. Zhao, H. Yang, X. Ai, S. Cao, Z. Chen and Y. Cao, Small, 2019, 15, 1805427 CrossRef PubMed.
- Q. Qu, L. Li, S. Tian, W. Guo, Y. Wu and R. Holze, J. Power Sources, 2010, 195, 2789–2794 CrossRef CAS.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS PubMed.
- S. Liu, G. L. Pan, N. F. Yan and X. P. Gao, Energy Environ. Sci., 2010, 3, 1732–1735 RSC.
- T. Tomai, S. Mitani, D. Komatsu, Y. Kawaguchi and I. Honma, Sci. Rep., 2014, 4, 3591 CrossRef PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lü, C.-J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS PubMed.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed.
- H. Jiang, W. Shin, L. Ma, J. J. Hong, Z. Wei, Y. Liu, S. Zhang, X. Wu, Y. Xu, Q. Guo, M. A. Subramanian, W. F. Stickle, T. Wu, J. Lu and X. Ji, Adv. Energy Mater., 2020, 10, 2000968 CrossRef CAS.
- X. Wu, Y. Qi, J. J. Hong, Z. Li, A. S. Hernandez and X. Ji, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed.
- C. Li, J. Wu, F. Ma, Y. Chen, L. Fu, Y. Zhu, Y. Zhang, P. Wang, Y. Wu and W. Huang, ACS Appl. Energy Mater., 2019, 2, 6984–6989 CrossRef CAS.
- C. Li, W. Yan, S. Liang, P. Wang, J. Wang, L. Fu, Y. Zhu, Y. Chen, Y. Wu and W. Huang, Nanoscale Horiz., 2019, 4, 991–998 RSC.
- Z. Rong, R. Malik, P. Canepa, G. Sai Gautam, M. Liu, A. Jain, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 6016–6021 CrossRef CAS.
- M. Li, J. Lu, X. Ji, Y. Li, Y. Shao, Z. Chen, C. Zhong and K. Amine, Nat. Rev. Mater., 2020, 5, 276–294 CrossRef CAS.
- X. Wu, A. Markir, Y. Xu, C. Zhang, D. P. Leonard, W. Shin and X. Ji, Adv. Funct. Mater., 2019, 29, 1900911 CrossRef.
- L. Wei, L. Zeng, M. C. Wu, H. R. Jiang and T. S. Zhao, J. Power Sources, 2019, 423, 203–210 CrossRef CAS.
- W. Manalastas Jr, S. Kumar, V. Verma, L. Zhang, D. Yuan and M. Srinivasan, ChemSusChem, 2019, 12, 379–396 CrossRef PubMed.
- X. Zeng, J. Hao, Z. Wang, J. Mao and Z. Guo, Energy Storage Mater., 2019, 20, 410–437 CrossRef.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS.
- W. J. Wruck, J. Electrochem. Soc., 1991, 138, 3560 CrossRef CAS.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- C. Xu, B. Li, H. Du and F. Kang, Angew. Chem., Int. Ed., 2012, 51, 933–935 CrossRef CAS PubMed.
- S. Chen, R. Lan, J. Humphreys and S. Tao, Energy Storage Mater., 2020, 28, 205–215 CrossRef.
- Y. Zeng, X. Zhang, Y. Meng, M. Yu, J. Yi, Y. Wu, X. Lu and Y. Tong, Adv. Mater., 2017, 29, 1700274 CrossRef PubMed.
- Y. Jin, L. Zou, L. Liu, M. H. Engelhard, R. L. Patel, Z. Nie, K. S. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31, 1900567 CrossRef PubMed.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S.-Z. Qiao, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed.
- C. Liu, X. Chi, Q. Han and Y. Liu, Adv. Energy Mater., 2020, 10, 1903589 CrossRef CAS.
- C. Zhong, B. Liu, J. Ding, X. Liu, Y. Zhong, Y. Li, C. Sun, X. Han, Y. Deng, N. Zhao and W. Hu, Nat. Energy, 2020, 5, 440–449 CrossRef CAS.
- G. G. Yadav, D. Turney, J. Huang, X. Wei and S. Banerjee, ACS Energy Lett., 2019, 4, 2144–2146 CrossRef CAS.
- C. Xie, T. Li, C. Deng, Y. Song, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 135–143 RSC.
- M. Li, Q. He, Z. Li, Q. Li, Y. Zhang, J. Meng, X. Liu, S. Li, B. Wu, L. Chen, Z. Liu, W. Luo, C. Han and L. Mai, Adv. Energy Mater., 2019, 9, 1901469 CrossRef.
- G. Li, W. Chen, H. Zhang, Y. Gong, F. Shi, J. Wang, R. Zhang, G. Chen, Y. Jin, T. Wu, Z. Tang and Y. Cui, Adv. Energy Mater., 2020, 10, 1902085 CrossRef CAS.
- F. Mo, Z. Chen, G. Liang, D. Wang, Y. Zhao, H. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2020, 10, 2000035 CrossRef CAS.
- H. Li, C. Han, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Wang, Z. Liu, Z. Tang, Y. Wang, F. Kang, B. Li and C. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- D. Wang, H. Li, Z. Liu, Z. Tang, G. Liang, F. Mo, Q. Yang, L. Ma and C. Zhi, Small, 2018, 14, 1803978 CrossRef PubMed.
- X. Zeng, J. Liu, J. Mao, J. Hao, Z. Wang, S. Zhou, C. D. Ling and Z. Guo, Adv. Energy Mater., 2020, 10, 1904163 CrossRef CAS.
- D. Chao, C. Ye, F. Xie, W. Zhou, Q. Zhang, Q. Gu, K. Davey, L. Gu and S.-Z. Qiao, Adv. Mater., 2020, 32, 2001894 CrossRef CAS PubMed.
- W.-J. Song, S. Lee, G. Song and S. Park, ACS Energy Lett., 2019, 4, 177–186 CrossRef CAS.
- V. Verma, S. Kumar, W. Manalastas Jr, R. Satish and M. Srinivasan, Adv. Sustainable Syst., 2019, 3, 1800111 CrossRef.
- S. Zečević, L. Gajić, A. R. Despić and D. M. Drazić, Electrochim. Acta, 1981, 26, 1625–1631 CrossRef.
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743–9746 RSC.
- F. Wang, X. Fan, T. Gao, W. Sun, Z. Ma, C. Yang, F. Han, K. Xu and C. Wang, ACS Cent. Sci., 2017, 3, 1121–1128 CrossRef CAS PubMed.
- Y. Zhang, K. Cheng, K. Ye, Y. Gao, W. Zhao, G. Wang and D. Cao, Electrochim. Acta, 2015, 182, 971–978 CrossRef CAS.
- Y. Mizuno, M. Okubo, E. Hosono, T. Kudo, K. Oh-ishi, A. Okazawa, N. Kojima, R. Kurono, S.-I. Nishimura and A. Yamada, J. Mater. Chem. A, 2013, 1, 13055–13059 RSC.
- L. Chen, J. L. Bao, X. Dong, D. G. Truhlar, Y. Wang, C. Wang and Y. Xia, ACS Energy Lett., 2017, 2, 1115–1121 CrossRef CAS.
- C. Lee and S.-K. Jeong, Chem. Lett., 2016, 45, 1447–1449 CrossRef CAS.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- K. F. Blurton and A. F. Sammells, J. Power Sources, 1979, 4, 263–279 CrossRef CAS.
- J.-S. Lee, S. Tai Kim, R. Cao, N.-S. Choi, M. Liu, K. T. Lee and J. Cho, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- T. Zhang, N. Imanishi, S. Hasegawa, A. Hirano, J. Xie, Y. Takeda, O. Yamamoto and N. Sammes, J. Electrochem. Soc., 2008, 155, A965 CrossRef CAS.
- T. Zhang, N. Imanishi, Y. Takeda and O. Yamamoto, Chem. Lett., 2011, 40, 668–673 CrossRef CAS.
- Z. Chang, X. Wang, Y. Yang, J. Gao, M. Li, L. Liu and Y. Wu, J. Mater. Chem. A, 2014, 2, 19444–19450 RSC.
- K. Hayashi, K. Shima and F. Sugiyama, J. Electrochem. Soc., 2013, 160, A1467–A1472 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- J. Fu, R. Liang, G. Liu, A. Yu, Z. Bai, L. Yang and Z. Chen, Adv. Mater., 2019, 31, 1805230 CrossRef PubMed.
- S. Liu, W. Han, B. Cui, X. Liu, F. Zhao, J. Stuart and S. Licht, J. Power Sources, 2017, 342, 435–441 CrossRef CAS.
- M. A. Deyab and Q. Mohsen, Renewable Sustainable Energy Rev., 2021, 139, 110729 CrossRef CAS.
- N. Chawla, Mater. Today Chem., 2019, 12, 324–331 CrossRef CAS.
- B. T. Hang, T. Watanabe, M. Egashira, I. Watanabe, S. Okada and J.-I. Yamaki, J. Power Sources, 2006, 155, 461–469 CrossRef CAS.
- Y. Boyjoo, H. Shi, Q. Tian, S. Liu, J. Liang, Z.-S. Wu, M. Jaroniec and J. Liu, Energy Environ. Sci., 2021, 14, 540–575 RSC.
- S. Rehman, M. Pope, S. Tao and E. McCalla, Energy Environ. Sci., 2022, 15, 1423–1460 RSC.
- H.-J. Peng, J.-Q. Huang and Q. Zhang, Chem. Soc. Rev., 2017, 46, 5237–5288 RSC.
- S. Yun, S. H. Park, J. S. Yeon, J. Park, M. Jana, J. Suk and H. S. Park, Adv. Funct. Mater., 2018, 28, 1707593 CrossRef.
- S. Licht and D. Peramunage, J. Electrochem. Soc., 1993, 140, L4–L6 CrossRef CAS.
- X. Wu, A. Markir, Y. Xu, E. C. Hu, K. T. Dai, C. Zhang, W. Shin, D. P. Leonard, K.-I. Kim and X. Ji, Adv. Energy Mater., 2019, 9, 1902422 CrossRef CAS.
- N. Li, Y. Wang, D. Tang and H. Zhou, Angew. Chem., Int. Ed., 2015, 54, 9271–9274 CrossRef CAS PubMed.
- M. M. Gross and A. Manthiram, Energy Storage Mater., 2019, 19, 346–351 CrossRef.
- Y. Zhao, D. Wang, X. Li, Q. Yang, Y. Guo, F. Mo, Q. Li, C. Peng, H. Li and C. Zhi, Adv. Mater., 2020, 32, 2003070 CrossRef CAS PubMed.
- Z. Chen, F. Mo, T. Wang, Q. Yang, Z. Huang, D. Wang, G. Liang, A. Chen, Q. Li, Y. Guo, X. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2021, 14, 2441–2450 RSC.
- Z. Chen, Q. Yang, F. Mo, N. Li, G. Liang, X. Li, Z. Huang, D. Wang, W. Huang, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 2001469 CrossRef CAS PubMed.
- H. Ao, Y. Zhao, J. Zhou, W. Cai, X. Zhang, Y. Zhu and Y. Qian, J. Mater. Chem. A, 2019, 7, 18708–18734 RSC.
- H.-R. Yao, Y. You, Y.-X. Yin, L.-J. Wan and Y.-G. Guo, Phys. Chem. Chem. Phys., 2016, 18, 9326–9333 RSC.
- L. Chen, Q. Gu, X. Zhou, S. Lee, Y. Xia and Z. Liu, Sci. Rep., 2013, 3, 1946 CrossRef PubMed.
- X. Cao, L. Wang, J. Chen and J. Zheng, J. Mater. Chem. A, 2018, 6, 15762–15770 RSC.
- Y. Liu, Z. Wen, X. Wu, X. Wang, Y. Wu and R. Holze, Chem. Commun., 2014, 50, 13714–13717 RSC.
- Z. Chang, Y. Yang, X. Wang, M. Li, Z. Fu, Y. Wu and R. Holze, Sci. Rep., 2015, 5, 11931 CrossRef CAS PubMed.
- J. Yan, J. Wang, H. Liu, Z. Bakenov, D. Gosselink and P. Chen, J. Power Sources, 2012, 216, 222–226 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- B. Zhang, Y. Liu, X. Wu, Y. Yang, Z. Chang, Z. Wen and Y. Wu, Chem. Commun., 2014, 50, 1209–1211 RSC.
- X. Wang, F. Wang, L. Wang, M. Li, Y. Wang, B. Chen, Y. Zhu, L. Fu, L. Zha, L. Zhang, Y. Wu and W. Huang, Adv. Mater., 2016, 28, 4904–4911 CrossRef CAS PubMed.
- X. Wang, M. Li, Y. Wang, B. Chen, Y. Zhu and Y. Wu, J. Mater. Chem. A, 2015, 3, 8280–8283 RSC.
- T. Placke, A. Heckmann, R. Schmuch, P. Meister, K. Beltrop and M. Winter, Joule, 2018, 2, 2528–2550 CrossRef CAS.
- L. Zhang, H. Wang, X. Zhang and Y. Tang, Adv. Funct. Mater., 2021, 2010958 CrossRef CAS.
- X. Wu, Y. Xu, C. Zhang, D. P. Leonard, A. Markir, J. Lu and X. Ji, J. Am. Chem. Soc., 2019, 141, 6338–6344 CrossRef CAS PubMed.
- J. Ding, W. Hu, E. Paek and D. Mitlin, Chem. Rev., 2018, 118, 6457–6498 CrossRef CAS PubMed.
- Y.-g. Wang and Y.-y. Xia, Electrochem. Commun., 2005, 7, 1138–1142 CrossRef CAS.
- J. P. Zheng, J. Electrochem. Soc., 2003, 150, A484 CrossRef CAS.
- L. Athouël, F. Moser, R. Dugas, O. Crosnier, D. Bélanger and T. Brousse, J. Phys. Chem. C, 2008, 112, 7270–7277 CrossRef.
- L. Dong, X. Ma, Y. Li, L. Zhao, W. Liu, J. Cheng, C. Xu, B. Li, Q.-H. Yang and F. Kang, Energy Storage Mater., 2018, 13, 96–102 CrossRef.
- Y. Yao, J. Lei, Y. Shi, F. Ai and Y.-C. Lu, Nat. Energy, 2021, 6, 582–588 CrossRef.
- Z. Li and Y.-C. Lu, Adv. Mater., 2020, 32, 2002132 CrossRef CAS PubMed.
- E. Sum and M. Skyllas-Kazacos, J. Power Sources, 1985, 15, 179–190 CrossRef CAS.
- D. Ma, B. Hu, W. Wu, X. Liu, J. Zai, C. Shu, T. Tadesse Tsega, L. Chen, X. Qian and T. L. Liu, Nat. Commun., 2019, 10, 3367 CrossRef PubMed.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2008, 7, 75–83 CrossRef CAS PubMed.
- P. Singh and B. Jonshagen, J. Power Sources, 1991, 35, 405–410 CrossRef CAS.
- B. Li, Z. Nie, M. Vijayakumar, G. Li, J. Liu, V. Sprenkle and W. Wang, Nat. Commun., 2015, 6, 6303 CrossRef CAS PubMed.
- A. Price, S. Bartley, S. Male and G. Cooley, Power Eng. J., 1999, 13, 122–129 CrossRef.
- Z. Li, G. Weng, Q. Zou, G. Cong and Y.-C. Lu, Nano Energy, 2016, 30, 283–292 CrossRef CAS.
-
Y. Xu, Y. Wen, J. Cheng, Y. Yanga, Z. Xie and G. Cao, Novel Organic Redox Flow Batteries Using Soluble Quinonoid Compounds As Positive Materials, World Non-Grid-Connected Wind Power and Energy Conference, Nanjing, 2009, pp. 475–478 Search PubMed.
- R. Feng, X. Zhang, V. Murugesan, A. Hollas, Y. Chen, Y. Shao, E. Walter, N. P. N. Wellala, L. Yan, K. M. Rosso and W. Wang, Science, 2021, 372, 836 CrossRef CAS PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef.
- Z. Hou, X. Zhang, X. Li, Y. Zhu, J. Liang and Y. Qian, J. Mater. Chem. A, 2017, 5, 730–738 RSC.
- T. K. A. Hoang, M. Acton, H. T. H. Chen, Y. Huang, T. N. L. Doan and P. Chen, Mater. Today Energy, 2017, 4, 34–40 CrossRef.
- W. Xu, K. Zhao, W. Huo, Y. Wang, G. Yao, X. Gu, H. Cheng, L. Mai, C. Hu and X. Wang, Nano Energy, 2019, 62, 275–281 CrossRef CAS.
- J. Hao, J. Long, B. Li, X. Li, S. Zhang, F. Yang, X. Zeng, Z. Yang, W. K. Pang and Z. Guo, Adv. Funct. Mater., 2019, 29, 1903605 CrossRef.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- Y. An, Y. Tian, K. Zhang, Y. Liu, C. Liu, S. Xiong, J. Feng and Y. Qian, Adv. Funct. Mater., 2021, 31, 2101886 CrossRef CAS.
- F. Wang, Y. Lin, L. Suo, X. Fan, T. Gao, C. Yang, F. Han, Y. Qi, K. Xu and C. Wang, Energy Environ. Sci., 2016, 9, 3666–3673 RSC.
- Q. Nian, X. Zhang, Y. Feng, S. Liu, T. Sun, S. Zheng, X. Ren, Z. Tao, D. Zhang and J. Chen, ACS Energy Lett., 2021, 6, 2174–2180 CrossRef CAS.
- J. Cui, X. Liu, Y. Xie, K. Wu, Y. Wang, Y. Liu, J. Zhang, J. Yi and Y. Xia, Mater. Today Energy, 2020, 18, 100563 CrossRef CAS.
- T. Sun, H. Du, S. Zheng, J. Shi and Z. Tao, Adv. Funct. Mater., 2021, 31, 2010127 CrossRef CAS.
- F. Yue, Z. Tie, S. Deng, S. Wang, M. Yang and Z. Niu, Angew. Chem., Int. Ed., 2021, 133, 14001–14005 CrossRef.
- W. Li, W. R. McKinnon and J. R. Dahn, J. Electrochem. Soc., 1994, 141, 2310–2316 CrossRef CAS.
- W. Li and J. R. Dahn, J. Electrochem. Soc., 1995, 142, 1742–1746 CrossRef CAS.
- L. Bai, J. Zhu, X. Zhang and Y. Xie, J. Mater. Chem., 2012, 22, 16957–16963 RSC.
- Z. Xie, Q. Su, A. Shi, B. Yang, B. Liu, J. Chen, X. Zhou, D. Cai and L. Yang, J. Energy Chem., 2016, 25, 495–499 CrossRef.
- R. Demir-Cakan, M. Morcrette, J.-B. Leriche and J.-M. Tarascon, J. Mater. Chem. A, 2014, 2, 9025–9029 RSC.
- F. Wang, J. Tseng, Z. Liu, P. Zhang, G. Wang, G. Chen, W. Wu, M. Yu, Y. Wu and X. Feng, Adv. Mater., 2020, 32, 2000287 CrossRef CAS PubMed.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Nat. Energy, 2016, 1, 16039 CrossRef CAS.
- F. Y. Cheng, J. Chen, X. L. Gou and P. W. Shen, Adv. Mater., 2005, 17, 2753–2756 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- W. Sun, F. Wang, S. Hou, C. Yang, X. Fan, Z. Ma, T. Gao, F. Han, R. Hu, M. Zhu and C. Wang, J. Am. Chem. Soc., 2017, 139, 9775–9778 CrossRef CAS PubMed.
- X. Guo, J. Zhou, C. Bai, X. Li, G. Fang and S. Liang, Mater. Today Energy, 2020, 16, 100396 CrossRef.
- L. Dai, Y. Wang, L. Sun, Y. Ding, Y. Yao, L. Yao, N. E. Drewett, W. Zhang, J. Tang and W. Zheng, Adv. Sci., 2021, 8, 2004995 CrossRef CAS PubMed.
- C. Li, W. Wu, S. Zhang, L. He, Y. Zhu, J. Wang, L. Fu, Y. Chen, Y. Wu and W. Huang, J. Mater. Chem. A, 2019, 7, 4110–4118 RSC.
- W. Fan, F. Liu, Y. Liu, Z. Wu, L. Wang, Y. Zhang, Q. Huang, L. Fu and Y. Wu, Chem. Commun., 2020, 56, 2039–2042 RSC.
- X. Yuan, X. Wu, X.-X. Zeng, F. Wang, J. Wang, Y. Zhu, L. Fu, Y. Wu and X. Duan, Adv. Energy Mater., 2020, 10, 2001583 CrossRef CAS.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- Q. Dong, X. Yao, Y. Zhao, M. Qi, X. Zhang, H. Sun, Y. He and D. Wang, Chem, 2018, 4, 1345–1358 CAS.
- C. Yang, L. Suo, O. Borodin, F. Wang, W. Sun, T. Gao, X. Fan, S. Hou, Z. Ma, K. Amine, K. Xu and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6197–6202 CrossRef CAS PubMed.
- L. Chen, L. Cao, X. Ji, S. Hou, Q. Li, J. Chen, C. Yang, N. Eidson and C. Wang, Nat. Commun., 2020, 11, 2638 CrossRef CAS PubMed.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- D. Reber, R.-S. Kühnel and C. Battaglia, ACS Mater. Lett., 2019, 1, 44–51 CrossRef CAS.
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, ACS Energy Lett., 2018, 3, 373–374 CrossRef CAS.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang, C. Fang and X. Ji, Chem. Commun., 2018, 54, 14097–14099 RSC.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- Q. Zheng, S. Miura, K. Miyazaki, S. Ko, E. Watanabe, M. Okoshi, C.-P. Chou, Y. Nishimura, H. Nakai, T. Kamiya, T. Honda, J. Akikusa, Y. Yamada and A. Yamada, Angew. Chem., Int. Ed., 2019, 58, 14202–14207 CrossRef CAS PubMed.
- S. Wu, Y. Qiao, S. Yang, J. Tang, P. He and H. Zhou, ACS Catal., 2018, 8, 1082–1089 CrossRef CAS.
- C.-Y. Chen, K. Matsumoto, K. Kubota, R. Hagiwara and Q. Xu, Adv. Energy Mater., 2019, 9, 1900196 CrossRef.
- S. Chen, R. Lan, J. Humphreys and S. Tao, Appl. Mater. Today, 2020, 20, 100728 CrossRef.
- J. Zheng, G. Tan, P. Shan, T. Liu, J. Hu, Y. Feng, L. Yang, M. Zhang, Z. Chen, Y. Lin, J. Lu, J. C. Neuefeind, Y. Ren, K. Amine, L.-W. Wang, K. Xu and F. Pan, Chem, 2018, 4, 2872–2882 CAS.
- X. Tang, D. Zhou, B. Zhang, S. Wang, P. Li, H. Liu, X. Guo, P. Jaumaux, X. Gao, Y. Fu, C. Wang, C. Wang and G. Wang, Nat. Commun., 2021, 12, 2857 CrossRef CAS PubMed.
- Q. Zhang, Y. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed.
- H. Bi, X. Wang, H. Liu, Y. He, W. Wang, W. Deng, X. Ma, Y. Wang, W. Rao, Y. Chai, H. Ma, R. Li, J. Chen, Y. Wang and M. Xue, Adv. Mater., 2020, 32, 2000074 CrossRef CAS PubMed.
- H. Liu, C.-Y. Chen, H. Yang, Y. Wang, L. Zou, Y.-S. Wei, J. Jiang, J. Guo, W. Shi, Q. Xu and P. Cheng, Adv. Mater., 2020, 32, 2004553 CrossRef CAS PubMed.
- S. Wu, S. Hu, Q. Zhang, D. Sun, P. Wu, Y. Tang and H. Wang, Energy Storage Mater., 2020, 31, 310–317 CrossRef.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef.
- L. Chen, J. Zhang, Q. Li, J. Vatamanu, X. Ji, T. P. Pollard, C. Cui, S. Hou, J. Chen, C. Yang, L. Ma, M. S. Ding, M. Garaga, S. Greenbaum, H.-S. Lee, O. Borodin, K. Xu and C. Wang, ACS Energy Lett., 2020, 5, 968–974 CrossRef CAS.
- M. H. Lee, S. J. Kim, D. Chang, J. Kim, S. Moon, K. Oh, K.-Y. Park, W. M. Seong, H. Park, G. Kwon, B. Lee and K. Kang, Mater. Today, 2019, 29, 26–36 CrossRef CAS.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- Z. Wang, H. Li, Z. Tang, Z. Liu, Z. Ruan, L. Ma, Q. Yang, D. Wang and C. Zhi, Adv. Funct. Mater., 2018, 28, 1804560 CrossRef.
- Z. Liu, H. Li, M. Zhu, Y. Huang, Z. Tang, Z. Pei, Z. Wang, Z. Shi, J. Liu, Y. Huang and C. Zhi, Nano Energy, 2018, 44, 164–173 CrossRef CAS.
- G. Wang, X. Lu, Y. Ling, T. Zhai, H. Wang, Y. Tong and Y. Li, ACS Nano, 2012, 6, 10296–10302 CrossRef CAS PubMed.
- S. Chen, R. Lan, J. Humphreys and S. Tao, ACS Appl. Energy Mater., 2020, 3, 2526–2536 CrossRef CAS.
- C. Yang, X. Ji, X. Fan, T. Gao, L. Suo, F. Wang, W. Sun, J. Chen, L. Chen, F. Han, L. Miao, K. Xu, K. Gerasopoulos and C. Wang, Adv. Mater., 2017, 29, 1701972 CrossRef PubMed.
- J. Xie, Z. Liang and Y.-C. Lu, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS PubMed.
- W. Pan, Y. Wang, X. Zhao, Y. Zhao, X. Liu, J. Xuan, H. Wang and D. Y. C. Leung, Adv. Funct. Mater., 2021, 31, 2008783 CrossRef CAS.
- C. Dai, X. Jin, H. Ma, L. Hu, G. Sun, H. Chen, Q. Yang, M. Xu, Q. Liu, Y. Xiao, X. Zhang, H. Yang, Q. Guo, Z. Zhang and L. Qu, Adv. Energy Mater., 2021, 11, 2003982 CrossRef CAS.
- J. Yang, Z. Xu, J. Wang, L. Gai, X. Ji, H. Jiang and L. Liu, Adv. Funct. Mater., 2021, 31, 2009438 CrossRef CAS.
- M. Chen, W. Zhou, A. Wang, A. Huang, J. Chen, J. Xu and C.-P. Wong, J. Mater. Chem. A, 2020, 8, 6828–6841 RSC.
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Eidson, Y. Liang, W. Sun, S. Greenbaum, K. Xu and C. Wang, Joule, 2018, 2, 927–937 CrossRef CAS.
- S. Chen, P. Sun, J. Humphreys, P. Zou, M. Zhang, G. Jeerh, B. Sun and S. Tao, ACS Appl. Mater. Interfaces, 2021, 13, 46634–46643 CrossRef CAS PubMed.
- J. M. Wrogemann, S. Künne, A. Heckmann, I. A. Rodríguez-Pérez, V. Siozios, B. Yan, J. Li, M. Winter, K. Beltrop and T. Placke, Adv. Energy Mater., 2020, 10, 1902709 CrossRef CAS.
- J. Zhu, Y. Xu, Y. Fu, D. Xiao, Y. Li, L. Liu, Y. Wang, Q. Zhang, J. Li and X. Yan, Small, 2020, 16, 1905838 CrossRef CAS PubMed.
- H. Zhang, B. Qin, J. Han and S. Passerini, ACS Energy Lett., 2018, 3, 1769–1770 CrossRef CAS.
- J. Zhao, J. Zhang, W. Yang, B. Chen, Z. Zhao, H. Qiu, S. Dong, X. Zhou, G. Cui and L. Chen, Nano Energy, 2019, 57, 625–634 CrossRef CAS.
- Q. Dou, Y. Wang, A. Wang, M. Ye, R. Hou, Y. Lu, L. Su, S. Shi, H. Zhang and X. Yan, Sci. Bull., 2020, 65, 1812–1822 CrossRef CAS.
- J. Chen, J. Vatamanu, L. Xing, O. Borodin, H. Chen, X. Guan, X. Liu, K. Xu and W. Li, Adv. Energy Mater., 2020, 10, 1902654 CrossRef CAS.
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC.
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- L. Zhang and G. Yu, Angew. Chem., Int. Ed., 2021, 60, 15028–15035 CrossRef CAS PubMed.
- Y. Shang, N. Chen, Y. Li, S. Chen, J. Lai, Y. Huang, W. Qu, F. Wu and R. Chen, Adv. Mater., 2020, 32, 2004017 CrossRef CAS PubMed.
- X. He, B. Yan, X. Zhang, Z. Liu, D. Bresser, J. Wang, R. Wang, X. Cao, Y. Su, H. Jia, C. P. Grey, H. Frielinghaus, D. G. Truhlar, M. Winter, J. Li and E. Paillard, Nat. Commun., 2018, 9, 5320 CrossRef CAS PubMed.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H.-C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- D. Li, L. Cao, T. Deng, S. Liu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 13035–13041 CrossRef CAS PubMed.
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- C. Deng, X. Xie, J. Han, Y. Tang, J. Gao, C. Liu, X. Shi, J. Zhou and S. Liang, Adv. Funct. Mater., 2020, 30, 2000599 CrossRef CAS.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- L. Zhang, B. Zhang, J. Hu, J. Liu, L. Miao and J. Jiang, Small Methods, 2021, 5, 2100094 CrossRef CAS PubMed.
- C. Lee, Y. Yokoyama, Y. Kondo, Y. Miyahara, T. Abe and K. Miyazaki, Adv. Energy Mater., 2021, 11, 2100756 CrossRef CAS.
- Z. Hou, X. Zhang, H. Ao, M. Liu, Y. Zhu and Y. Qian, Mater. Today Energy, 2019, 14, 100337 CrossRef.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- J. Li, L. An, H. Li, J. Sun, C. Shuck, X. Wang, Y. Shao, Y. Li, Q. Zhang and H. Wang, Nano Energy, 2019, 63, 103848 CrossRef CAS.
- S.-B. Wang, Q. Ran, R.-Q. Yao, H. Shi, Z. Wen, M. Zhao, X.-Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef PubMed.
- C. Yan, C. Lv, L. Wang, W. Cui, L. Zhang, K. N. Dinh, H. Tan, C. Wu, T. Wu, Y. Ren, J. Chen, Z. Liu, M. Srinivasan, X. Rui, Q. Yan and G. Yu, J. Am. Chem. Soc., 2020, 142, 15295–15304 CrossRef CAS PubMed.
- Z. Chang, Y. Qiao, H. Deng, H. Yang, P. He and H. Zhou, Joule, 2020, 4, 1776–1789 CrossRef CAS.
- Z. Chang, Y. Qiao, H. Yang, H. Deng, X. Zhu, P. He and H. Zhou, Energy Environ. Sci., 2020, 13, 4122–4131 RSC.
- S. Chen, P. Sun, J. Humphreys, P. Zou, M. Zhang, G. Jeerh and S. Tao, Energy Storage Mater., 2021, 42, 240–251 CrossRef.
- Y. Sun, H. Ma, X. Zhang, B. Liu, L. Liu, X. Zhang, J. Feng, Q. Zhang, Y. Ding, B. Yang, L. Qu and X. Yan, Adv. Funct. Mater., 2021, 31, 2101277 CrossRef CAS.
- S. Cai, X. Chu, C. Liu, H. Lai, H. Chen, Y. Jiang, F. Guo, Z. Xu, C. Wang and C. Gao, Adv. Mater., 2021, 33, 2007470 CrossRef CAS PubMed.
- K. Xu, W. Xu and S. S. Zhang, J. Non-Cryst. Solids: X, 2022, 14, 100088 CAS.
- C.-H. Lin, K. Sun, M. Ge, L. M. Housel, A. H. McCarthy, M. N. Vila, C. Zhao, X. Xiao, W.-K. Lee, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and Y.-C. K. Chen-Wiegart, Sci. Adv., 2020, 6, eaay7129 CrossRef CAS PubMed.
- K. He, M. Sawczyk, C. Liu, Y. Yuan, B. Song, R. Deivanayagam, A. Nie, X. Hu, V. P. Dravid, J. Lu, C. Sukotjo, Y.-P. Lu, P. Král, T. Shokuhfar and R. Shahbazian-Yassar, Sci. Adv., 2020, 6, eaaz7524 CrossRef CAS PubMed.
- D. Chao and S.-Z. Qiao, Joule, 2020, 4, 1846–1851 CrossRef.
- Y.-S. Hu and Y. Lu, ACS Energy Lett., 2020, 5, 3633–3636 CrossRef CAS.
- H. Tian, Z. Li, G. Feng, Z. Yang, D. Fox, M. Wang, H. Zhou, L. Zhai, A. Kushima, Y. Du, Z. Feng, X. Shan and Y. Yang, Nat. Commun., 2021, 12, 237 CrossRef CAS PubMed.
- L. Droguet, A. Grimaud, O. Fontaine and J.-M. Tarascon, Adv. Energy Mater., 2020, 10, 2002440 CrossRef CAS.
- R. F. Service, Science, 2021, 372, 890–891 CrossRef CAS PubMed.
- H. Li, Joule, 2019, 3, 911–914 CrossRef CAS.
- Z. Lin, T. Liu, X. Ai and C. Liang, Nat. Commun., 2018, 9, 5262 CrossRef CAS PubMed.
- Q. Li, Y. Wang, F. Mo, D. Wang, G. Liang, Y. Zhao, Q. Yang, Z. Huang and C. Zhi, Adv. Energy Mater., 2021, 11, 2003931 CrossRef CAS.
- D. Du, Z. Zhu, K.-Y. Chan, F. Li and J. Chen, Chem. Soc. Rev., 2022, 51, 1846–1860 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Peimiao
Zou
a,
Boyao
Sun
a and
Shanwen
Tao
a,
Peimiao
Zou
a,
Boyao
Sun
a and
Shanwen
Tao
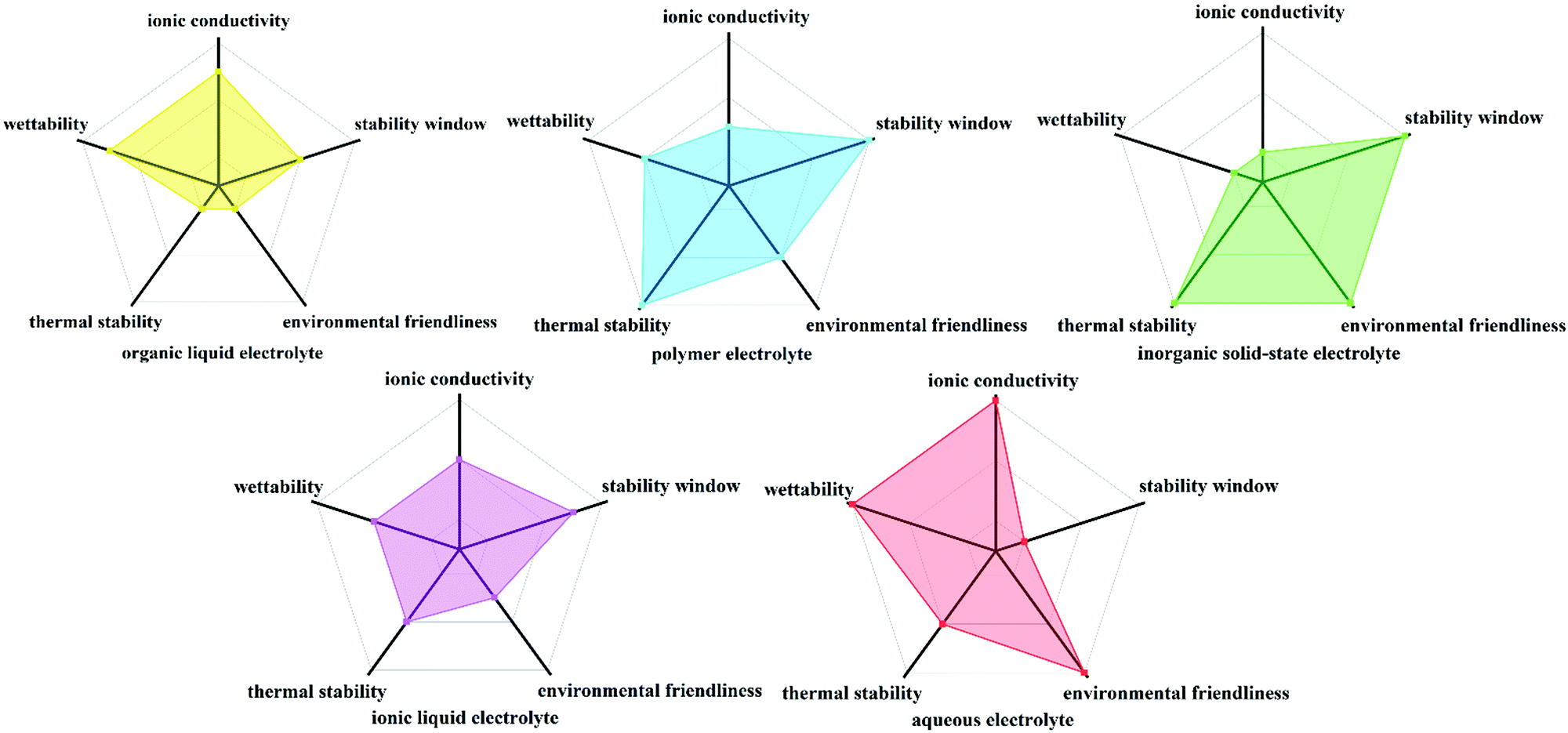
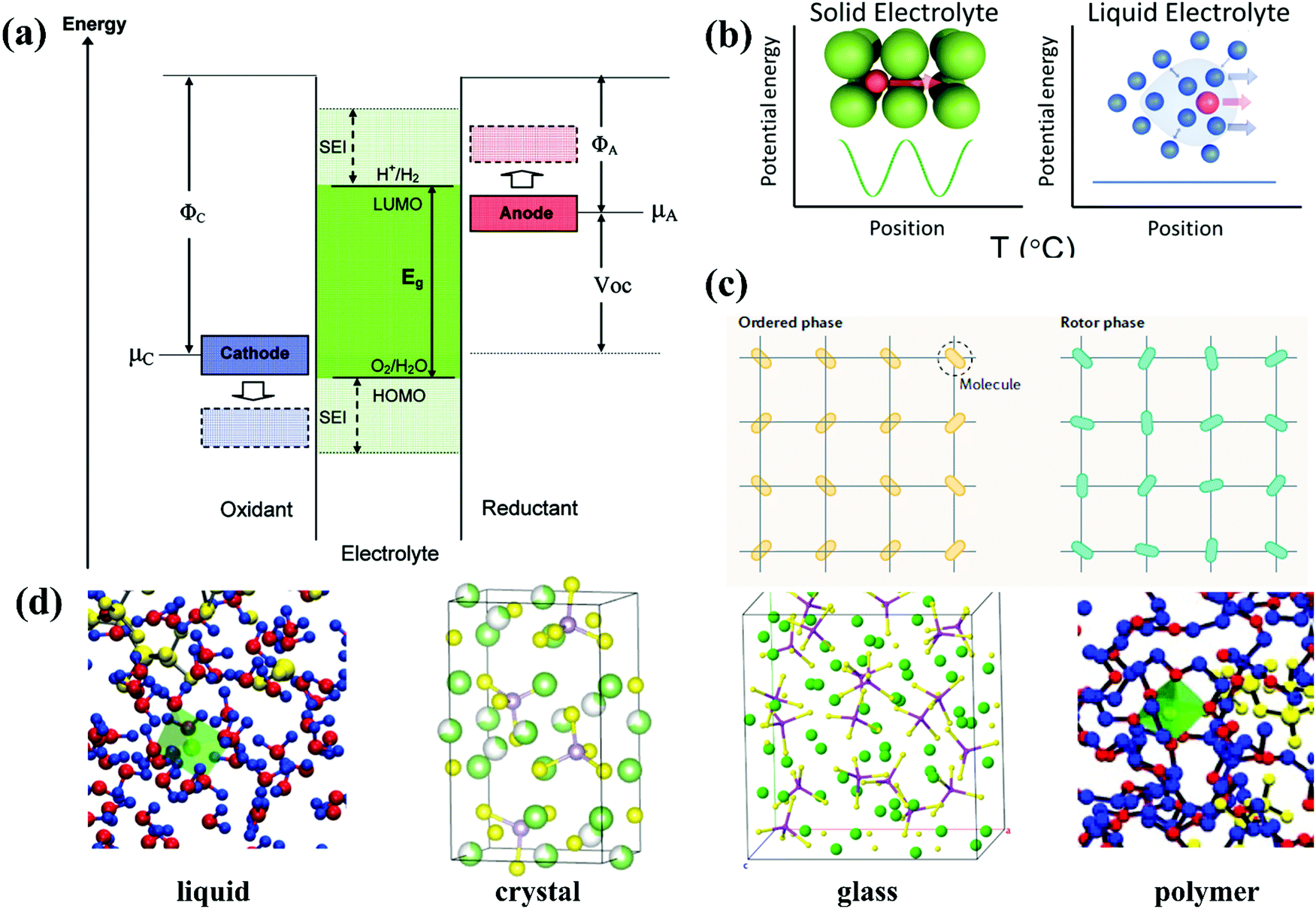
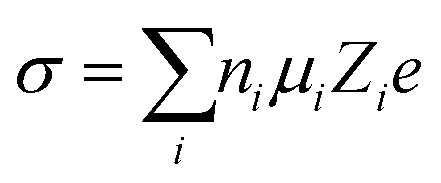
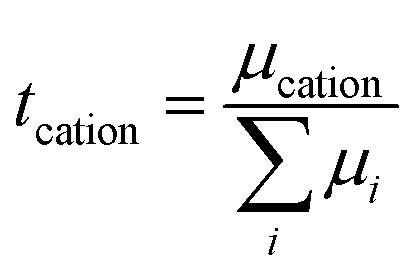
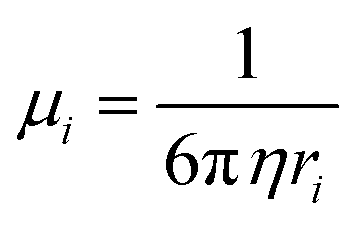
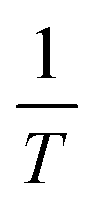 is bent and is best fitted to VTF behavior (after Vogel, Tammann, and Fulcher). To (equilibrium glass-transition temperature) is introduced for amendment, which is related to the (kinetic) measured glass-transition temperature.60 Glass is a promising electrolyte for use in solid-state batteries. Although the Arrhenius relationship and Nernst–Einstein relationship can still be applied to glass electrolytes, cation migration occurs via a mechanism that combines the concerted motion of cations with the large, quasi-permanent reorientation of anions. This effect, known as the ‘paddlewheel’ mechanism, is typically observed in high-temperature crystalline polymorphs. In contrast to the behavior of crystalline materials, the glass paddlewheel dynamics contribute to ion mobility at room temperature.61 The dynamics of ion motions in these materials are often described as being strongly coupled to the structural modes, whereas intermediate degrees of coupling are observed in organic ionic plastic crystals. Owing to the relatively weak interactions between molecules, reorientational processes occur with little hindrance. The orientations of molecules in ordered phases and rotor phases are illustrated in Fig. 2c.62 Recently, in one study, a unified framework was built for all liquid and solid (crystal, polymer and glass) electrolytes based on the concepts of solvation shell for liquids and solvation cage for solids. An LiTFSI aqueous solution, β-Li3PS4 at 537 K, amorphous Li3PS4 glass at 298 K and LiTFSI in polyethylene oxide (PEO) at 363 K were utilized as examples in that work for liquid, crystal, glass and polymer, respectively (Fig. 2d), to study the Li-ion solvation structure.63 The ultimate goal was enabling the rational design of the solvation environment to propel the migration of the working ions in a unified framework.
is bent and is best fitted to VTF behavior (after Vogel, Tammann, and Fulcher). To (equilibrium glass-transition temperature) is introduced for amendment, which is related to the (kinetic) measured glass-transition temperature.60 Glass is a promising electrolyte for use in solid-state batteries. Although the Arrhenius relationship and Nernst–Einstein relationship can still be applied to glass electrolytes, cation migration occurs via a mechanism that combines the concerted motion of cations with the large, quasi-permanent reorientation of anions. This effect, known as the ‘paddlewheel’ mechanism, is typically observed in high-temperature crystalline polymorphs. In contrast to the behavior of crystalline materials, the glass paddlewheel dynamics contribute to ion mobility at room temperature.61 The dynamics of ion motions in these materials are often described as being strongly coupled to the structural modes, whereas intermediate degrees of coupling are observed in organic ionic plastic crystals. Owing to the relatively weak interactions between molecules, reorientational processes occur with little hindrance. The orientations of molecules in ordered phases and rotor phases are illustrated in Fig. 2c.62 Recently, in one study, a unified framework was built for all liquid and solid (crystal, polymer and glass) electrolytes based on the concepts of solvation shell for liquids and solvation cage for solids. An LiTFSI aqueous solution, β-Li3PS4 at 537 K, amorphous Li3PS4 glass at 298 K and LiTFSI in polyethylene oxide (PEO) at 363 K were utilized as examples in that work for liquid, crystal, glass and polymer, respectively (Fig. 2d), to study the Li-ion solvation structure.63 The ultimate goal was enabling the rational design of the solvation environment to propel the migration of the working ions in a unified framework.