
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solar fuels: research and development strategies to accelerate photocatalytic CO2 conversion into hydrocarbon fuels
Eunhee
Gong†
 ,
Shahzad
Ali†
,
Chaitanya B.
Hiragond†
,
Hong Soo
Kim†
,
Niket S.
Powar
,
Dongyun
Kim
,
Hwapyong
Kim
and
Su-Il
In
*
,
Shahzad
Ali†
,
Chaitanya B.
Hiragond†
,
Hong Soo
Kim†
,
Niket S.
Powar
,
Dongyun
Kim
,
Hwapyong
Kim
and
Su-Il
In
*
Department of Energy Science & Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), 333 Techno Jungang-daero, Hyeonpung-eup, Dalseong-gun, Daegu 42988, Republic of Korea. E-mail: insuil@dgist.ac.kr; faraday.in@gmail.com
First published on 16th November 2021
Abstract
Photocatalytic production of solar fuels from CO2 is a promising strategy for addressing global environmental problems and securing future energy supplies. Although extensive research has been conducted to date, numerous impediments to realizing efficient, selective, and stable CO2 reduction have yet to be overcome. This comprehensive review highlights the recent advances in CO2 photoreduction, including critical challenges such as light-harvesting, charge separation, and the activation of CO2 molecules. We present promising strategies for enhancing the photocatalytic activities and discuss theoretical insights and equations for quantifying photocatalytic performance, which are expected to afford a fundamental understanding of CO2 photoreduction. We then provide a thorough overview of both traditional photocatalysts such as metal oxides and state-of-the-art catalysts such as metal–organic frameworks and 2D materials, followed by a discussion of the origin of carbon in CO2 photoreduction as a means to further understand the reaction mechanism. Finally, we discuss the economic viability of photocatalytic CO2 reduction before concluding the review with proposed future research directions.
 Eunhee Gong | Eunhee Gong received her BS degree in Fine Chemical Engineering from Chonnam National University, Republic of Korea, and her master's degree in Energy Science & Engineering from DGIST, Republic of Korea. She is currently a PhD candidate under the supervision of Professor Su-Il In at DGIST. Her current research is focused on the development of nanostructured materials for photocatalytic CO2 conversion. |
 Shahzad Ali | Shahzad Ali received his BS and MS degrees in Chemical Engineering from COMSATS University Islamabad, Pakistan. He holds industrial and research experience related to power and renewable fuels and is currently a PhD candidate under the supervision of Professor Su-Il In at DGIST, Republic of Korea. His current research involves the development of economical systems and materials for photocatalytic CO2 reduction. |
 Chaitanya B. Hiragond | Chaitanya B. Hiragond received his BS in Chemistry from Shivaji University, India, in 2012 and his MS in Organic Chemistry from Bharati Vidyapeeth Deemed University, India, in 2014. He is currently a PhD candidate under the supervision of Professor Su-Il In at DGIST, Republic of Korea. His current research focuses on developing photocatalytic materials for CO2 conversion applications. |
 Hong Soo Kim | Hong Soo Kim received his BS degree from Pusan National University in 2017. He is currently pursuing an integrated MS/PhD degree under the supervision of Professor Su-Il In at DGIST, Republic of Korea. His current research interests are focused on the electrical surface modification of electrode materials that can be used in a variety of applications. |
 Niket S. Powar | Niket S. Powar received his BS in chemistry and MS in Organic Chemistry from Shivaji University, India, in 2013 and 2015, respectively. He subsequently received his MTech in Nanotechnology and Renewable Energy at Amrita Vishwa Vidyapeetham University, India, in 2019. He is currently a PhD candidate under the supervision of Professor Su-Il In at DGIST, Republic of Korea. His current research interest lies in developing gas-phase photocatalysts for CO2 photoreduction to generate value-added hydrocarbon products. |
 Dongyun Kim | Dongyun Kim received his BS degree from the Kumoh National Institute of Technology in 2019 and his MS degree from DGIST in 2021. He is currently working toward his PhD under the supervision of Professor Su-Il In at DGIST, Republic of Korea. His research interest is in the synthesis of photocatalysts for CO2 conversion. |
 Hwapyong Kim | Hwapyong Kim received his BS degree from DGIST, Republic of Korea, in 2018. He is currently working toward his integrated MS/PhD degree under the supervision of Professor Su-Il In at DGIST. His current research interest lies in microbial fuel cells and catalysts for CO2 reduction/hydrogen evolution. |
 Su-Il In | Su-Il In is a professor at DGIST, Republic of Korea. He received his PhD in 2008 from the University of Cambridge, UK. He served as a postdoc at Technical University of Denmark and Penn State University (2008–2012) and later as a visiting scholar at the Department of Environmental Science & Engineering at Caltech, CA, USA (2019–2020). His current research interests are focused on energy and environmental issues, including photo/electrocatalytic CO2 conversion, betavoltaic cells, nano-bio hybrid technology, and microbial fuel cells. |
Broader contextResearch into CO2 reduction has the potential to diminish our dependence on petroleum products and restrain global warming. Fossil fuels account for a large majority of global energy consumption, resulting in excessive emissions of CO2 and other harmful gases. Furthermore, the current global economy and human society are heavily reliant on fossil fuels. Thus, there is an urgent need to develop renewable energy resources to generate energy and chemicals. Photocatalytic CO2 reduction is one promising strategy for obtaining renewable energy and hydrocarbon fuels. However, we must first confront several challenges, such as limited light-harvesting and suboptimal photocatalytic activity. These challenges and potential solutions are the focus of the present review, alongside a discussion of our current theoretical understanding and consideration of the commercial/economic viability of CO2 photoreduction technology. |
1 Introduction
Global energy expenditure has skyrocketed in recent years, leading to the consumption of enormous amounts of fossil fuels such as coal, oil, and natural gas, which currently account for approximately 85% of primary energy supplies.1 Fossil fuel combustion is associated with the excessive emission of harmful gases (e.g., CO2) that contribute to rising atmospheric temperatures, glacial melting, severe storms, ocean compositional changes, biodiversity loss, etc.2,3 Therefore, global warming and its severe negative impacts on the earth have emerged as hotly discussed topics in recent decades. Over the past few years, the global average atmospheric CO2 concentration has increased to over 410 ppm, which is approximately 45% higher than that prior to the Industrial Revolution.4,5 According to the Intergovernmental Panel on Climate Change (IPCC), if this rate of CO2 emission continues, atmospheric CO2 levels will reach 590 ppm by 2100.6 The IPCC proposed the goal of limiting the increase in global average temperature to within 1.5 °C of pre-industrial levels.7,8 Thus, significant efforts have been undertaken worldwide to reduce atmospheric CO2 emissions, such as the well-known Paris Agreement under the United Nations Framework Convention on Climate Change (UNFCCC), in which more than 190 countries affirmed their commitment to decreasing CO2 emissions.4,7 There are three ways to control atmospheric CO2: (i) lowering CO2 emissions, (ii) capture and storage, and (iii) converting it to value-added chemicals. One promising strategy for reducing CO2 levels is using this waste product as a carbon feedstock to produce value-added compounds via catalytic reactions, opening the door to developing an artificial carbon cycle.9 The potential of CO2 to serve as a chemical feedstock has attracted considerable research attention in numerous fields, with a particular focus on photocatalytic,10–12 biocatalytic,13,14 electrochemical,15,16 thermochemical,17,18 and photothermal19,20 CO2 conversion.With the aid of sunlight, plants capture CO2 and convert it into various organic molecules (such as carbohydrates) and oxygen in a slow rate of chemical reaction known as “natural carbon fixation”.21 Because CO2 and water are both readily available on earth, their conversion into chemical fuels mediated by sunlight may provide an eco-friendly alternative to our present energy infrastructure.22
Artificial photocatalysis under ambient conditions offers a promising alternative to thermocatalytic and electrochemical reactions, which are typically conducted at high temperatures and pressures or driven by an external electrical potential. In 1978, Halmann became the first to describe the conversion of CO2 into formic acid, formaldehyde, and methanol using an electrochemical cell with a GaP cathode under Hg arc lamp illumination.23 Numerous attempts at CO2 photoconversion have subsequently been reported; nevertheless, significant problems remain with respect to activity and product selectivity owing to the inertness of CO2 molecules and the complexity of the process.24 CO2 is a thermodynamically stable and chemically inert linear molecule, such that breaking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds requires a substantial amount of energy to drive the process.25 Furthermore, the reduction of CO2 with H2O to form hydrocarbons, such as CH4, is associated with a larger positive change in the Gibbs free energy (818.3 kJ mol−1) than the conversion of H2O into H2 and 1/2O2 (232.2 kJ mol−1) under ordinary conditions.25 Although CO2 conversion is challenging, this waste product can be transformed into other value-added compounds by exploiting appropriate catalysts to overcome the kinetic and thermodynamic barriers and drive the process. In this respect, various photocatalytic materials, such as metal oxides, chalcogenides, carbon-based materials, metal complexes, metal–organic frameworks (MOFs), metal carbides, MXenes, polymers, perovskites, and plasmonic materials, have been frequently applied.4,26–32
O bonds requires a substantial amount of energy to drive the process.25 Furthermore, the reduction of CO2 with H2O to form hydrocarbons, such as CH4, is associated with a larger positive change in the Gibbs free energy (818.3 kJ mol−1) than the conversion of H2O into H2 and 1/2O2 (232.2 kJ mol−1) under ordinary conditions.25 Although CO2 conversion is challenging, this waste product can be transformed into other value-added compounds by exploiting appropriate catalysts to overcome the kinetic and thermodynamic barriers and drive the process. In this respect, various photocatalytic materials, such as metal oxides, chalcogenides, carbon-based materials, metal complexes, metal–organic frameworks (MOFs), metal carbides, MXenes, polymers, perovskites, and plasmonic materials, have been frequently applied.4,26–32
In general, solar light harvesting, charge separation, and surface reaction are the crucial phases in effectively converting CO2 into chemical compounds such as CO, CH4, HCOOH, HCHO, and CH3OH.33 Possible reactions of CO2 reduction to various products and their standard redox potentials are listed in Table 1. A typical first step is single-electron transfer to CO2 to generate CO2˙− with a standard redox potential of −1.90 V vs. normal hydrogen electrode (NHE), which is considered the rate-limiting step owing to its large energy requirement.2 Although proton-assisted multi-electron processes require significantly lower potentials, suitable catalysts must be present to generate the multiple electrons and protons needed to form the various products listed in Table 1.
| Reaction | E 0redox vs. NHE (V) | Main product | Eqn |
|---|---|---|---|
| CO2 + e− → CO2˙− | −1.90 | CO2˙− | (1) |
| CO2 + 2H+ + 2e− → CO + H2O | −0.53 | Carbon monoxide | (2) |
| CO2 + 2H+ + 2e− → HCOOH | −0.61 | Formic acid | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 | Formaldehyde | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 | Methanol | (5) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 | Methane | (6) |
| 2CO2 + 8H+ + 8e− → CH3COOH + 2H2O | −0.31 | Acetic acid | (7) |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | −0.51 | Ethane | (8) |
Several challenges are associated with the aforementioned CO2 photoreduction that have impeded the wider application of photocatalytic technology. In particular, it is difficult to simultaneously realize both light absorption over a broad solar spectrum and reduction–oxidation processes using a single semiconducting material. First, wide-bandgap materials, such as TiO2, ZnO, and CdS, are primarily active in the ultraviolet (UV) region.28,34 Second, although narrow-bandgap semiconductors such as Cu2O are active in the visible–near-infrared (NIR) region,35 their band potentials are unsuitable for simultaneously mediating reduction and oxidation reactions. Consequently, single-component systems have so far proved less satisfactory for photocatalysis, and numerous efforts have been made to overcome the tradeoff between these two beneficial properties of photocatalysts, such as through heterostructure formation.36 Furthermore, spatial charge separation and charge transport from the catalyst surface to the reactants are important factors for reducing CO2. However, it has been reported that the charge recombination process in semiconductors is faster than the surface redox process.37 Charge recombination in semiconductors can often be attributed to Coulombic attraction, a lack of charge trapping states on the catalyst surface, etc.37,38 Thus, if the charge carriers survive recombination, they can participate in the redox reaction. Charge separation, and interfacial charge separation in particular, can be enhanced by metal/non-metal doping, cocatalyst deposition, heterojunction formation, etc. Multicomponent systems have thus been constructed to restrict the recombination of electrons and holes. Apart from these issues, the amount of CO2 adsorption on the catalyst surface greatly influences the catalytic performance. Catalysts with a large surface area provide more active sites for CO2 adsorption. Meanwhile, the selectivity of photocatalytic CO2 reduction is governed by a combination of known and unknown factors. CO2 can be reduced to various products depending upon the availability of electrons (e−) and protons (H+) and the binding strength of the formed product.39 Products with a greater binding strength can receive additional electrons and protons to form highly reduced products. This usually occurs for products arising from the partial reduction of CO2, e.g., the transformation of CO to the highly reduced product CH4.40 In addition to the variation in selectivity based on the degree of reduction, C–C coupling reactions are another crucial aspect governing the selectivity. In this case, the intermediate products, rather than desorbing, preferentially undergo coupling reactions, e.g., ˙CH3 radicals may couple to afford C2H6.5 Under such circumstances, the optoelectronic and structural properties of the photocatalyst must both be considered during catalyst design for optimal selectivity. Furthermore, the stability of the photocatalyst is equally important, especially in terms of scale-up and catalyst reusability.41 The major reported reason for catalyst instability is the strong oxidizing power of photogenerated holes, especially in the case of photocatalysts with a strong valence band (VB), such as TiO2.42 These oxidizing holes or ˙OH radicals generated by water oxidation can oxidize the products/intermediates and photocatalyst metal atoms, leading to dramatically decreased photocatalytic yields.43,44 Various strategies to overcome this issue have been explored, such as heterostructure formation and the use of hole scavengers.45,46 However, these strategies have not yet been reported to provide prolonged stability. These approaches are one of the core subjects of the current review, with a particular focus on surface engineering, band alteration, heterojunction construction, and hybrid formation.
In addition to the aforementioned hurdles, the roles of the various reaction parameters have yet to be fully elucidated, despite numerous efforts during catalyst design. Factors such as reactor type, temperature, pressure, and light source can exert remarkable effects on catalytic activity and stability.47 For example, in batch and flow reactors, the product yield may vary depending upon the reactant feed ratio, catalyst amount, flow rate, etc.47 Furthermore, multi-sun system using a light concentrator affords enhanced photon flux, which can improve CO2 conversion.48 Evaluation of the catalytic activity in terms of efficiency and apparent quantum yield (AQY) is another critical consideration. Various parameters, such as reactor area, incident light, and collected light, should be factored into the efficiency calculation. As the catalytic yields described in the literature have been reported in a variety of units, such as ppm cm−2 h−1 and μmol g−1 h−1, a fair comparison is required.47 Here, we suggest the equation for fair comparison. Next, understanding the complex reaction processes and mechanism of the CO2 conversion process is vital, and quantitative isotopic measurements provide such a toolset for comprehending these aspects.49 In conjunction with the analytical techniques of mass spectroscopy (MS) and nuclear magnetic resonance (NMR), which are frequently applied to study catalytic processes, isotopes, with their similar chemical properties, can help confirm the mechanism or product formation. For example, 13C-labeled CO2 and 2H- or 18O-labeled H2O are often used to investigate the processes of product formation, mitigating the influence of carbon impurities. Such mechanistic studies are also crucial for a comprehensive understanding of these processes.
In the past few years, research on photocatalytic CO2 reduction has blossomed. Several review articles and perspectives have been published on the current status of photocatalytic CO2 reduction, providing an overview of recent advances, material design, present challenges, potential solutions, and so on.4,24,31,39,50–55Fig. 1 shows the number of research papers and review articles published each year between 2010 and 2020 according to data collected from the Web of Science database on June 10, 2021, demonstrating that the scholarly community is becoming increasingly interested in this topic. Several international institutions have also been established that are providing valuable data for research and development efforts, such as the United Nations Environment Programme (UNEP),56 the European Commission,56 the Global Green Growth Institute (GGGI),56,57 and the World Resources Institute (WRI).58 In addition, several international conferences have been organized on a regular basis in recent decades, such as the International Conference on Carbon Dioxide Utilization (ICCDU)59,60 and the International Conference on Greenhouse Gas Control Technologies (GHGT),61 providing a forum for sharing and debating information on CO2 emission, capture, and conversion to improve CO2 reduction research and technologies. Furthermore, climate change has become a top priority for numerous government agencies, academic institutions, and technology companies. Therefore, we believe that CO2 capture and conversion technology will continue to be a promising research topic in the coming decade.
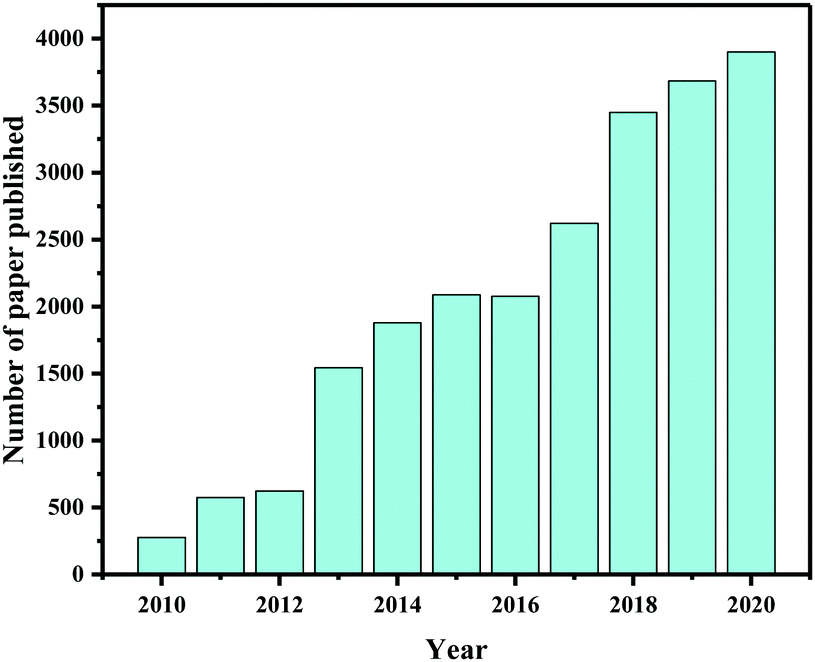 | ||
| Fig. 1 The number of published articles on photocatalytic CO2 conversion each year between 2010 and 2020, retrieved from the Web of Science database using the keywords (topic) of (photo* or solar), (CO2 or carbon dioxide), and (conversion or reduction) with several additional filters such as related field and journals. | ||
Herein, we aim to provide a current overview of photocatalytic CO2 conversion. The review begins with the fundamental aspects and key challenges of photocatalytic CO2 reduction (Section 2) and then discusses the various strategies for overcoming these challenges (Section 3). Next, we summarize the performance of the various reported photocatalysts for CO2 reduction and tabulate their activities and reaction parameters for quick understanding and evaluation (Section 5), after first providing a theoretical basis and the equations needed to calculate photocatalytic yields and efficiencies in a standardized manner (Section 4). We then discuss important insights into the nature of the carbon species involved in CO2 photoreduction that has been gleaned from isotope labeling studies (Section 6). Next, we consider the commercial potential of photocatalytic CO2 reduction based on current and future emission restrictions (Section 7). Finally, we propose likely future research directions based on our current understanding (Section 8). We believe that this review will serve as a valuable guide to researchers involved in the design and development of photocatalytic materials and systems.
2 Fundamentals and challenges of CO2 photoreduction
The solar-light-driven conversion of the ubiquitous waste product CO2 to chemical fuels in the gas phase has become a hot topic of research. Semiconductors are the most suitable materials for catalytic CO2 conversion owing to their ability to simultaneously reduce CO2 and oxidize H2O, and these materials play significant roles in all steps of the catalytic process, including adsorption, activation, dissociation, and product formation. Semiconductors possess two energy bands, namely, the VB, the highest energy band of occupied orbitals, and the conduction band (CB), the lowest energy band of vacant electronic states, which are separated by a quantum mechanically forbidden energy zone referred to as the bandgap (Eg). When a semiconducting material is exposed to sunlight with an energy greater than or equal to the bandgap energy (i.e., hν ≥ Eg), the electrons are excited from the VB to the CB, leaving behind holes in the VB (step I in Fig. 2).62 The free electrons in the CB and holes in the VB transfer from the bulk to the catalyst surface (step II). Meanwhile, owing to the Coulombic force of attraction, a portion of the charges undergo recombination in the bulk before reaching the surface (step III). To complete the energy conversion process, the survived electrons and holes are transferred to surface-adsorbed CO2 and H2O molecules, respectively, resulting in a simultaneous reduction–oxidation reaction to afford the solar fuel (step IV). To achieve the photon-induced uphill CO2 conversion, the CB of the semiconductor should be more negative than the reduction potential of CO2, while the VB should be more positive than the oxidation potential of H2O. Because the recombination of electrons and holes is much faster than the process of charge transfer and consumption at the catalyst interface, the lifetime of the photoexcited electrons must be sufficiently long for completion of the redox reaction. Higher photogenerated electron density at catalyst surfaces can be realized by the active separation of electrons and holes, which can robustly accelerate the rate of a redox reaction, leading to faster hydrocarbon production.39 In addition, the characteristic features of the catalyst govern the product selectivity and catalyst stability; depending upon the reduction potential of the material, number of available electrons, active sites, adsorption of the intermediates, and various other factors (e.g., gas or liquid phase, reaction conditions CO2 in the presence of H2O), the CO2 in the presence of H2O can be converted into a variety of products, such as CO, CH4, and C2H6. An effective catalyst (i) provides maximum solar light absorption to generate electrons and holes, (ii) contains sufficient active sites for CO2 adsorption on its surface, and (iii) efficiently generates electron–hole pairs and mediates their migration to the catalyst surface. Numerous earlier studies focused on these factors in an effort to improve the catalytic performance of CO2 reduction using various types of catalysts and different reaction conditions. However, because the reduction process entails several complex steps, it is not as straightforward as it may initially appear, with particular challenges being (i) limited light absorption, (ii) charge recombination, (iii) adsorption/activation of CO2 molecules, (iv) photostability of the catalyst materials, (v) development of a facile and reasonable synthetic process, and (vi) underlying mechanism/C1 and C2 selectivity (Fig. 2). We will briefly discuss these challenges of photocatalytic CO2 conversion in the remainder of this section.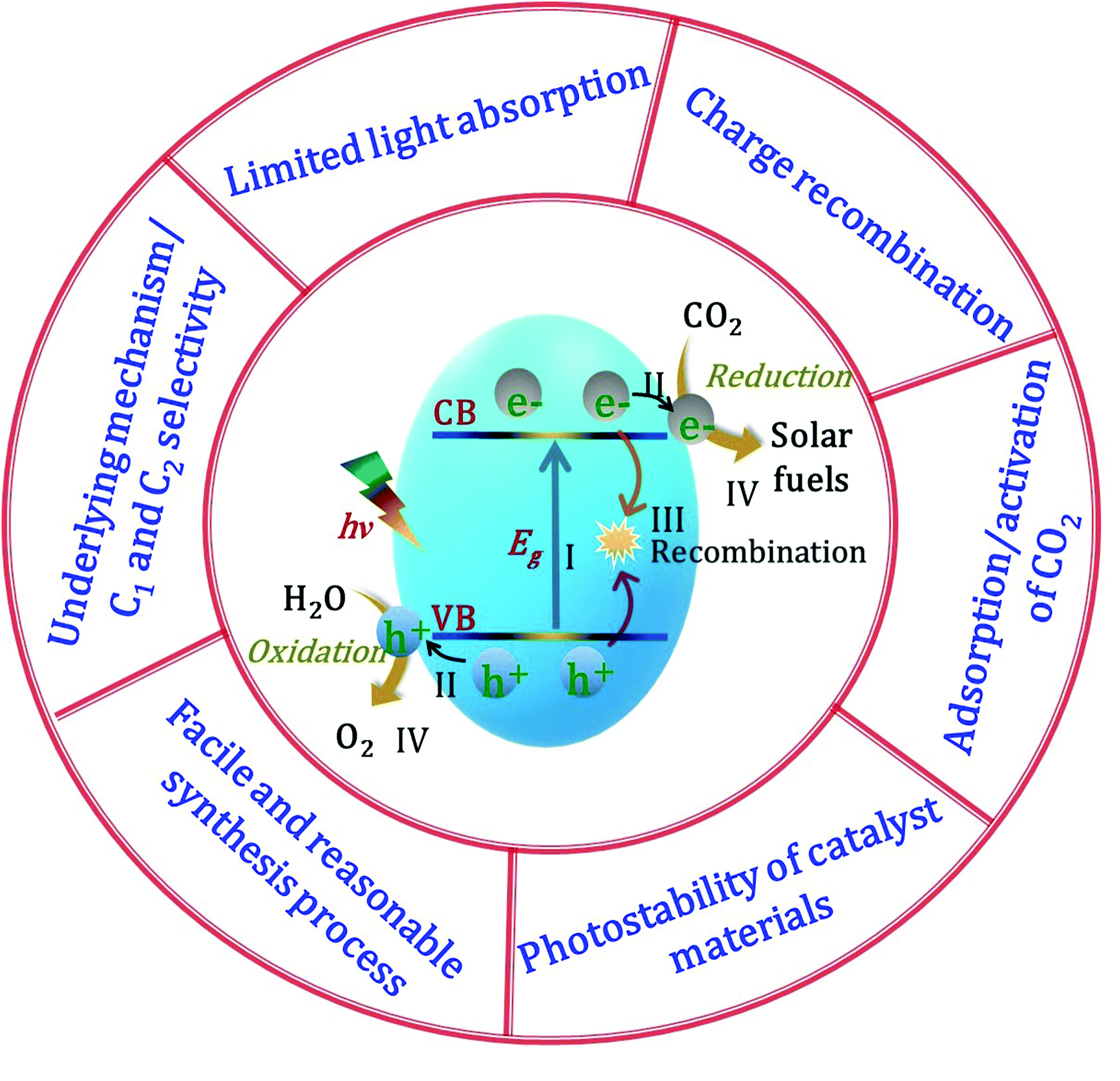 | ||
| Fig. 2 Schematic illustration and challenges of the photocatalytic CO2 conversion process. | ||
2.1 Limited light absorption
The critical processes in CO2 photoreduction reactions are (i) the formation of charge carriers through light absorption and electron excitation and (ii) the reaction of surface electrons with CO2 molecules. As previously discussed, the electronic band structure of the catalyst is a crucial factor in light-driven CO2 conversion, where a catalyst with appropriate redox potentials is required to drive the process smoothly from a thermodynamic standpoint. As depicted in Fig. 3, various semiconducting materials have been explored for this purpose.63–66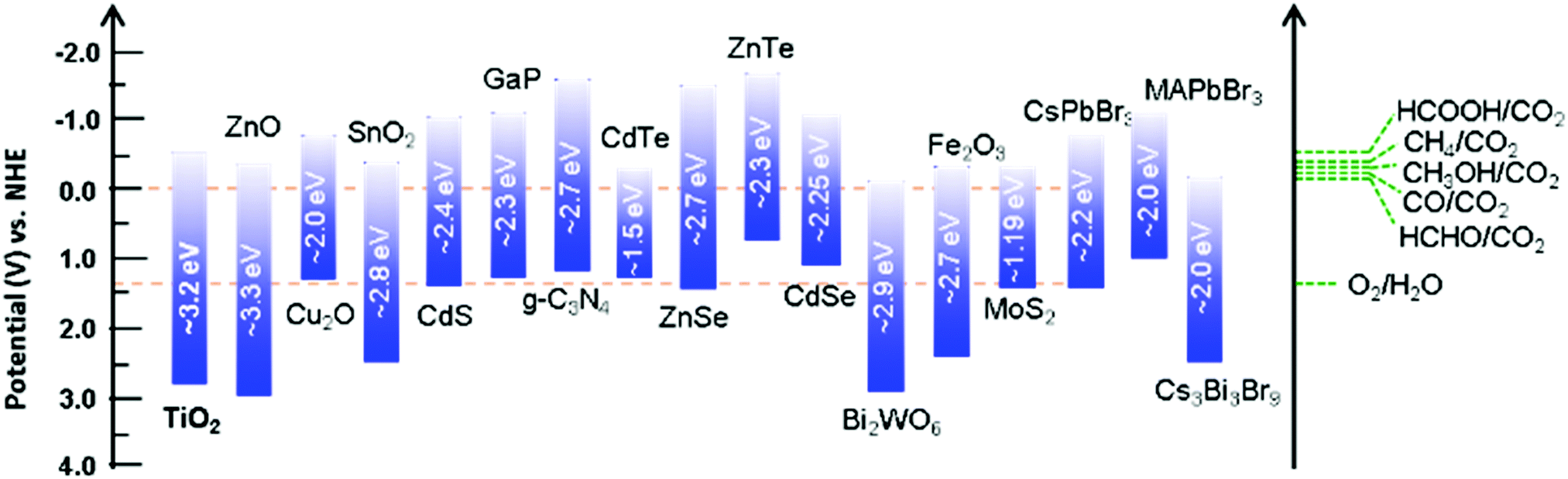 | ||
| Fig. 3 Band potentials and corresponding bandgap energies of various selected photocatalysts relative to the redox potential involved in CO2 photoreduction to various products. | ||
The solar spectrum comprises approximately 5% UV radiation, 43% visible radiation, and 52% infrared (IR) radiation; clearly, the visible and IR regions account for the majority. Thus, catalysts that primarily absorb visible/IR light can be expected to produce the maximum amount of charge to mediate the desired reaction. However, most of the commonly used semiconductors possess unsuitable bandgaps or band potentials to absorb sufficient sunlight for driving the process toward product formation. Although the band potentials of wide-bandgap semiconductors are suitable for mediating the redox process, they are only active in the shorter wavelength region. For example, TiO2 is the most commonly applied semiconductor in this process; however, its wide bandgap (3.2 eV) limits light absorption to the UV region (λ = 390 nm) and pristine TiO2 is inactive with respect to visible-light-induced CO2 conversion.
Various strategies have been investigated in an effort to overcome the limited light absorption ability of TiO2, which will be discussed in greater detail in the following sections (Section 5.1). Some narrow-bandgap semiconductors displaying strong light absorption in the visible region have been reported to convert CO2 into solar fuels; however, such a small bandgap inevitably leads to the simultaneous occurrence of reduction and oxidation processes on the surface and fast charge recombination, such that sacrificial agents are required to scavenge the holes.67 For example, Cu2O is a promising catalyst for CO2 conversion owing to its narrow bandgap (ca. 2.2 eV), which affords efficient photon absorption in the visible region of the solar spectrum. However, this narrow bandgap also causes rapid recombination of photogenerated charges, meaning that pristine Cu2O still exhibits poor catalytic performance; in addition, the holes cause Cu2O to self-oxidize, reducing its photostability.68 Thus, by combining a narrow-bandgap material with a wide-bandgap semiconductor, light absorption in both the UV and visible regions can be effectively increased; such combinations have been widely documented in the literature.43,68
Another viable strategy for improving the photoconversion performance of photocatalysts is broadening their light absorption capability in the IR/NIR region;31 however, few such catalysts have so far been reported, with the exception of WO3,67 B13P2,69etc. The real challenge associated with absorption in the NIR region is its low photonic energy, which can only provide low redox potentials in semiconductors.70 However, considering current trends in nanotechnology, it is anticipated that NIR-active materials displaying superior catalytic properties to conventional semiconductors will ultimately be developed. Therefore, the primary challenge with respect to light absorption is developing catalysts that can function over a wide range of the solar spectrum including the UV, visible, and NIR regions.
2.2 Charge recombination
In photocatalytic CO2 conversion on semiconductors, stable charge separation on the catalyst surface plays a critical role in converting CO2 into value-added products. The relatively low quantum efficiency of most reported photocatalysts can be attributed to the occurrence of charge recombination before the surface reactions of electrons and holes with CO2 and water.71 Thus, slow recombination or fast charge separation of the photoexcited carriers is a key factor in improving solar fuel production via CO2 reduction. Although electrons and holes are separated through the CB and VB, respectively, this process is quite tricky owing to the Coulombic force of attraction between the two species; Durrant and co-workers have demonstrated such recombination in organic solar cells.72 Because there is no driving force to drag the bound electrons and holes apart in pristine semiconductors, they are vulnerable to recombination, which hinders their participation in surface redox reactions.73 Moreover, the non-radiative relaxation of excited electrons to the ground state causes electron–hole recombination on the nanosecond timescale. In contrast, electron transfer at the semiconductor interface is typically two or three orders of magnitude slower than the electron–hole recombination rate. Consequently, the charge dynamics at the surface of semiconductors determine the rate of surface redox reactions and play a crucial role in photocatalytic CO2 reduction. Charge recombination can interfere with charge separation and interfacial charge transfer, making it one of the main limiting factors in the solar-to-energy conversion process.71 Recombination can occur both in the bulk and on the surface of the catalyst. In general, the charge carrier mobility is closely related to the distance from the bulk to the surface, which determines the rate of bulk recombination, while surface recombination can be caused by the absence of sufficient active sites or trapping states on the catalyst surface. As a result, catalysts with efficient charge separation and high intrinsic mobility can prevent recombination. To improve charge separation, nanoengineered materials have been explored that significantly reduce the distance between the point of charge generation and the catalyst surface.Nanostructured semiconductors, because of their high surface-area-to-volume ratio, short charge migration distance, and tunable electronic properties, possess numerous advantages over their bulk counterparts for photocatalysis.74 Previous studies have revealed that the characteristics of a catalyst, such as morphology, crystal structure, and particle size, effectively determine the rates of charge transport and recombination.75,76 The morphology of a catalyst can significantly affect the charge carrier dynamics. For example, because of increased charge separation, 1D nanostructured TiO2 materials such as nanotubes, nanofibers, and nanorods exhibit far superior catalytic activity to TiO2 nanoparticles.77–79 The distance between the point of charge generation and the catalyst interface can also be reduced using 1D nanostructures. Durrant and co-workers performed an interesting study on the relationship between charge recombination and the crystal phase using transient absorption spectroscopy (TAS), which elucidated the charge carrier dynamics in TiO2.38 The authors compared mesoporous TiO2 nanostructures (with a size of 20 nm) and bulk TiO2 (with a size of 50–200 nm) and found that the photogenerated charge carriers produced in the former easily reached the surface, whereas those in the latter did not and remained in the bulk. However, the results also revealed that nanostructuring did not improve the recombination rates, indicating that surface-state-mediated recombination is not a key pathway in the case of TiO2. Instead, charge recombination was dependent on the crystal phase of TiO2, as demonstrated by the rapid charge recombination in rutile TiO2 and superior charge separation capacity over time in anatase TiO2. Similarly, Maity et al. analyzed the bulk charge carrier dynamics in single crystals of rutile and anatase TiO2 and discovered that the anatase phase exhibited a slower recombination rate than the rutile phase.80 Various strategies have been explored over the years to avoid such recombination, including defect formation, doping with metals or non-metals, cocatalyst deposition, heterojunction formation, etc. For instance, a number of studies have demonstrated that the deposition of plasmonic nanoparticles (e.g., Au, Ag, Pt) onto semiconductors can reduce charge recombination by creating a Schottky barrier, resulting in an increased excitons lifetime.81
2.3 Adsorption/activation of CO2
As is well known, CO2 is a thermodynamically stable molecule with linear geometry; therefore, its reduction is challenging. However, it can be transformed into other value-added compounds under appropriate conditions. In this respect, the adsorption and activation of CO2 molecules on catalyst surfaces are the critical kinetic factors in producing solar fuels. There are four key steps in this process: (i) adsorption of CO2 molecules on the catalyst surface, (ii) activation of the CO2 molecules to form partially charged CO2˙− anion radicals or intermediates, (iii) CO bond dissociation to afford another chemical product after reaction with an electron and a proton, and (iv) desorption of the newly formed product from the catalyst surface.82 Zou and co-workers described the possible configurations for the adsorption of CO2 molecules on a catalyst surface (Fig. 4(a)).65 For most metal oxides and sulfides, the C or O atoms of CO2 form weak bonds to single metal sites by hybridization of the 2p and 3d orbitals to generate intermediate products.83 Compared to the highly stable CO bonds of CO2, the weak M–O or M–C bonds can be readily cleaved, enabling the formation of CO or higher hydrocarbons (after protonation). Moreover, the nature of the binding of CO2 molecules on the catalyst surface determines the activity and selectivity of the catalytic reaction. For example, in the case of TiO2, several studies have shown that the binding energy of CO2 on a rutile surface is −0.34 eV, while it increases to −1.08 eV on defective TiO2.84 Therefore, defective TiO2 with oxygen vacancies or Ti3+ states offers more binding sites for CO2 molecules, which act as active sites for the desired reaction.
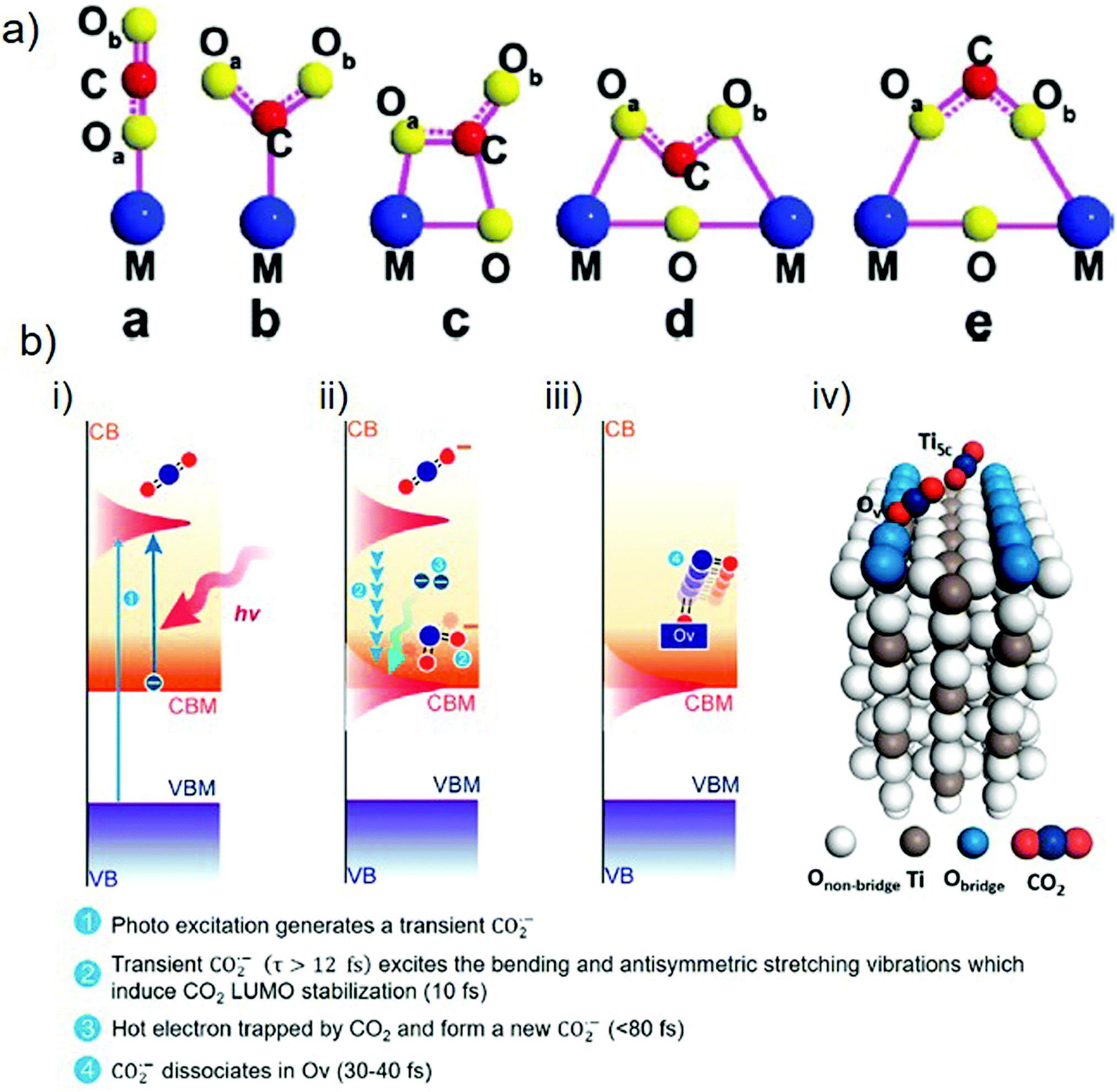 | ||
| Fig. 4 (a) Various configurations of adsorbed CO2 molecules on a photocatalyst surface. Reproduced with permission from ref. 65, Copyright 2020, American Chemical Society. (b) Photocatalytic CO2 reduction over a TiO2(110) surface showing the adsorption and activation of a CO2 molecule: (i)–(iii) stepwise activation of a CO2 molecule over time and (iv) adsorption of CO2 molecules at Ov and Ti5c sites. Reproduced with permission from ref. 82, Copyright 2020, American Chemical Society. | ||
The activation of CO2 molecules is a vital step in the photocatalytic CO2 reduction process. Chu et al. used time-dependent ab initio simulations (nonadiabatic molecular dynamics (NAMD)) to investigate the activation of CO2 molecules on the surface of rutile TiO2(110).82 According to the findings, once the electrons have been excited from the VB to the CB of TiO2, the addition of an electron to the LUMO of the CO2 molecule results in the formation of a transient CO2˙− anion radical possessing a bent geometry (step 1, Fig. 4(b)-(i)). This bending originates from the repulsion between the free electrons of the oxygen atom and the added electron. If CO2˙− has a lifespan of more than 12 fs, with the help of oxygen vacancy, the excitation of vibrational modes (bending and antisymmetric stretching) will stabilize the LUMO of the CO2 molecules to below the conduction band minimum (CBM) of the catalyst within 10 fs (step 2, Fig. 4(b)-(ii)). This form of electronic state alignment can persist for more than 100 fs. Within a timescale of 80 fs, CO2 captures the electrons present on the catalyst surface (step 3, Fig. 4(b)-(ii)) and can subsequently dissociate to afford the product (i.e., CO) within 30–40 fs after the trapping of the electrons (step 4, Fig. 4(b)-(iii)). Because the Ti5c sites have lower adsorption energies for binding CO2 molecules, the results indicate that the association of CO2 with oxygen vacancies is more favorable for the excitation of the antisymmetric stretching mode (Fig. 4(b)-(iv)). The formation of products can differ according to the available electrons and protons participating in the chemical reaction.
Poor CO2 adsorption decreases the amount of CO2 available for the reduction. Hence, the efficiency of CO2 reduction can be significantly improved by enhancing CO2 adsorption on the catalyst surface. In addition to oxygen vacancies, as discussed above, surface functionalization of catalysts with hydroxyl (OH) or amino (NH2) groups can increase CO2 adsorption. These functional groups are most likely to donate electrons to CO2 molecules, resulting in negatively charged HCO2δ− species that enhance CO2 adsorption.85 Increased CO2 adsorption ability has also been realized using catalysts with a large surface area, which provides more active sites for the catalytic reaction. Hiragond et al. reported hierarchical nanostructures that displayed increased CO2 adsorption owing to their unique structures and surface morphologies, including nanofibers, nanotubes/rods, nanosheets, nanoflowers, etc.86 The deposition of alkali or alkaline-earth metals with a greater affinity toward acidic CO2 molecules on catalyst surfaces can also promote the adsorption of CO2 on photocatalysts.48
2.4 Photostability of catalyst materials
Although several solar-active catalysts displaying significant activity for CO2 photoreduction have been reported, most of them suffer from instability. Therefore, photocatalyst stability is a major issue that has severely hampered the practical application of these catalysts. Photocatalyst instability may originate from a number of sources, including transitions from active to inactive photocatalytic oxidation states, buildup of reaction intermediates that are difficult to reduce, oxidation of the products, and morphological changes.42,87 In addition, reverse reactions may also play a role. For example, Punchihewa and co-workers observed the photoreduction of CO2 to formaldehyde and methanol in high yield after 30–45 min; however, the activity decreased after a specific time.88 This decreased activity was caused by a hole-mediated back-reaction that was faster than the CO2 reduction.Under light irradiation, equal amounts of electrons and holes should be formed on the catalyst; however, these electrons and holes may cause photocorrosion due to reduction or oxidation of the catalyst itself. For example, Xu and co-workers reported the degradation of CdS by photogenerated holes. Changes in the oxidation state typically occur when the redox potential of a photocatalyst lies within its bandgap, whereupon the photogenerated electrons and holes can reduce or oxidize the photocatalyst. In the case of Cu2O, the holes were reported to have insufficient oxidizing ability for water oxidation and therefore preferentially oxidized Cu2O, resulting in loss of the active oxidation state.44 Similarly, inefficient utilization of the electrons for photocatalytic CO2 reduction led to the reduction of Cu2O to Cu. In addition, some photocatalysts have very strong oxidizing power, resulting in the generation of active hydroxyl radicals from water, which can subsequently oxidize the intermediates of photocatalytic CO2 reduction to appreciably decrease the reaction yield.42
Catalyst stability is typically investigated by cycling tests in a batch/flow reactor in conjunction with various analytical techniques. Catalysts often become deactivated after repeated cycling for the reasons mentioned above, such that the surface active sites are no longer available to mediate the redox process.89 Li and co-workers studied the deactivation mechanism of a Cu/TiO2 surface using in situ X-ray absorption spectroscopy (XAS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS).46 The air-calcined Cu/TiO2 surface was dominated by the Cu2+ state, whereas the sample after treatment at 200 °C under H2 was rich in Cu+ and oxygen vacancies. The post-treatment sample (Cu/Ti(H2)) displayed 50% higher activity toward CO formation. The authors reported that the deactivation of Cu/Ti(H2) was attributable to the consumption of OH groups and Cu active sites by holes. The Cu/Ti(H2) catalytic activity decreased from a maximum of 7.5 μmol g−1 h−1 to 3.5 μmol g−1 h−1 after 7 h. In situ XAS results revealed that the photooxidation of Cu+ to Cu2+ altered the environment of Cu and led to the decrease in the CO2 photoreduction activity.
Various strategies have been applied to overcome these issues, including the use of hole scavengers to preserve the active oxidation states and intermediates. In addition, a variety of heterostructures and hybrid combinations have been developed to improve the stability of the Cu2O catalyst; for example, the formation of Cu2O/TiO2 heterostructures was reported to protect the Cu2O from photocorrosion.43 Recently, Ali et al. reported that a Z-scheme heterostructure based on reduced titania and Cu2O displayed high photostability over 42 h (seven cycles) with improved catalytic activity for the photoreduction of CO2 to CH4.44 This high stability was attributed to the Z-scheme charge transfer that successfully inhibited the photocorrosion of Cu2O. In addition, thermal and oxidative/reductive treatments (H2O2 or mild acid exposure) have also been applied to eliminate the adsorbed unwanted intermediates. Vacuum annealing can also restore the activity of a catalyst, which is likely attributable to the regeneration of oxygen vacancies or decomposition of adsorbed intermediates on the catalyst surface. The use of cocatalysts or shielding materials such as graphene to prevent oxidation is also an option. These techniques and their respective advantages will be discussed in the following sections.90 Furthermore, Feng and co-workers suggested that increasing the light intensity using a solar concentrator can prevent catalyst deactivation owing to the influence of temperature under multi-sun conditions increasing product desorption.48 Considering the crucial role of catalyst stability in photocatalytic CO2 reduction, more detailed research is necessary to elucidate the deactivation mechanisms.
2.5 Development of facile and reasonable synthetic processes
To date, a variety of photocatalytic materials have been investigated, each with unique size, shape, appearance, physicochemical properties, and so on. All of these properties are influenced by the synthetic strategy adopted to prepare the catalyst. For many years, CO2 conversion has been a central focus of research into semiconducting nanomaterials, especially in the case of TiO2.91 Advanced synthesis techniques can facilitate precise manipulation of the size, morphology, crystal facet, pore network, and structural periodicity. Thus, various rational design and synthesis methodologies have been explored for both single and multicomponent (hybrid) catalysts. For example, bandgap-engineered TiO2 has been reported to be beneficial for CO2 reduction. In addition, various studies have reported the synthesis of reduced titania using thermal treatment with aluminum or magnesium at high temperatures of 500–700 °C.92 However, such high-temperature synthesis methodologies are not feasible for large-scale applications. In this respect, other studies have demonstrated the synthesis of reduced titania with abundant oxygen vacancies or Ti3+ states at lower temperatures (ca. 350 °C) using NaBH4 as a reducing agent.93 Recently, MXenes have emerged as extremely promising materials for catalytic applications. In contrast to conventional 2D nanosheets that are typically obtained via an etching process involving hazardous acids such as hydrofluoric acid (HF), MXenes can also be synthesized in a convenient and facile manner through a hydrothermal approach in NaOH solution or electrochemical etching.94In addition to the methods used to prepare the pristine semiconductors, the techniques used for cocatalyst deposition can also influence catalytic performance. Loading with a cocatalyst is typically accomplished using methods such as galvanic dispersion, photodeposition, wet chemical approaches, and impregnation.92 Our previous studies demonstrated that flawless Schottky junctions between semiconductors and cocatalysts could be obtained using a simple photodeposition process at ambient temperature.5,93,95,96 Most heterojunction combinations have been widely synthesized using in situ hydrothermal, coprecipitation, solvothermal, vacuum annealing, and sonochemical approaches.97 The synthetic procedure may vary depending upon the composition and heterostructure, e.g., p–n junction, core–shell, Z-scheme, and S-scheme. Various studies have demonstrated that simple procedures can afford facile heterostructure combinations under ambient conditions. For example, in our recent study, Cu2O–reduced titania heterojunctions were obtained via a facile, unique low-temperature thermochemical method followed by photodeposition.44 All of these synthetic routes have been frequently applied over the years to prepare catalysts with optimal optoelectronic properties. Nevertheless, the continued development of comprehensive synthetic approaches that meet the requirements of simplicity, cost-effectiveness, high performance (e.g., catalytic activity, selectivity, and physicochemical stability), and scalability remains necessary to satisfy engineering requirements for large-scale applications.
2.6 Underlying mechanism/C1 and C2 selectivity
Understanding the mechanisms of CO2 photoreduction into various products is challenging owing to numerous known and unknown phenomena. Multiple factors, such as the catalyst properties, band potentials, surface defects, CO2 adsorption characteristics, and nature of the active sites and interface, can significantly influence the reaction pathway. The CO2 reduction pathway involves a series of steps after CO2 activation, which are dependent on the number of electrons produced, electron transfer to CO2 molecules, C–O bond breaking, the formation of intermediate species, H2O oxidation to generate protons, coupling of intermediates with protons, new bond formation, etc. There exist various pathways for CO2 reduction to value-added products on a semiconductor surface, including the formaldehyde, carbene, and glyoxal pathways,9,98 as depicted in Fig. 5. As explained above, all of these pathways begin with the adsorption and activation of CO2 molecules on the catalyst surface.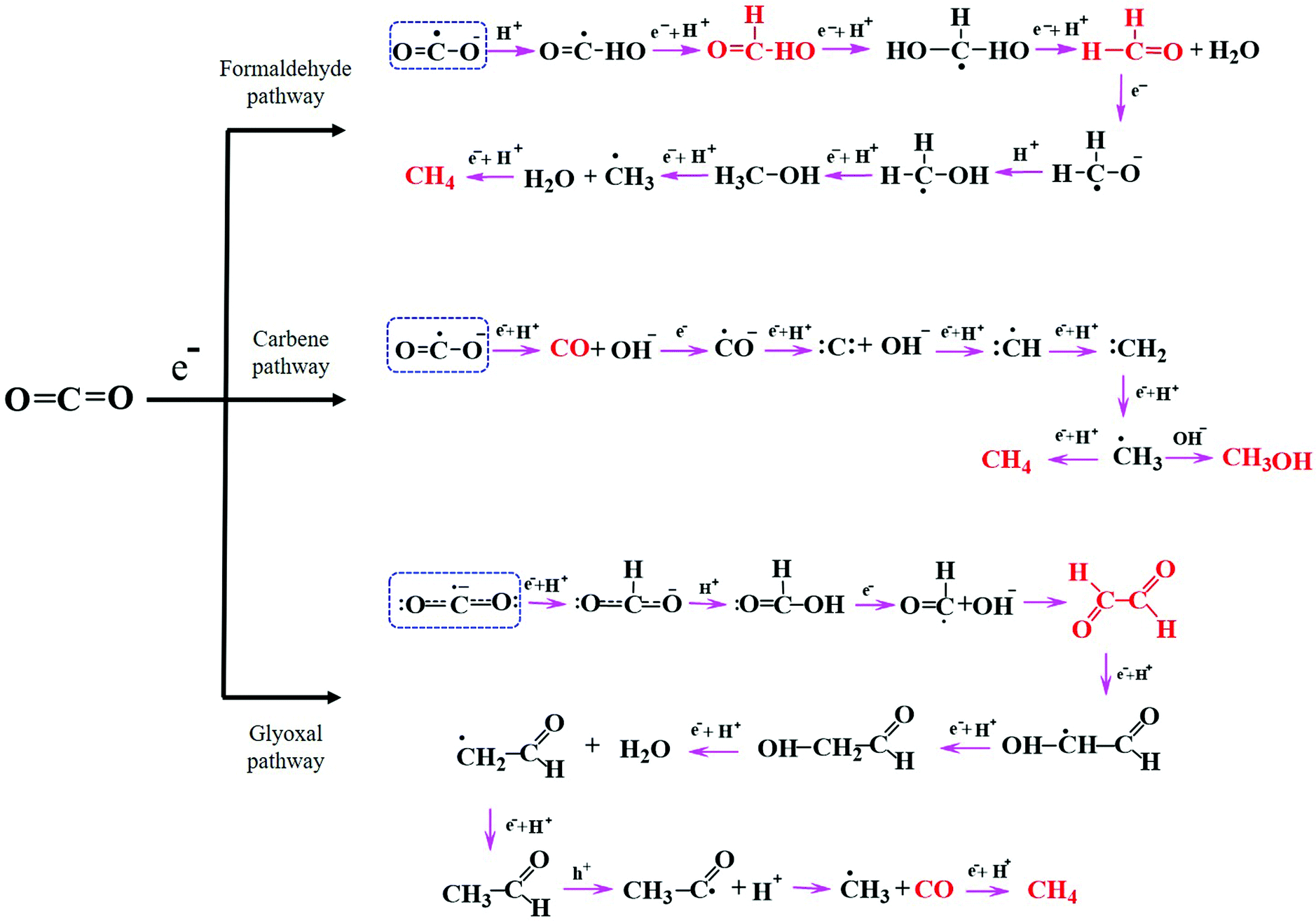 | ||
| Fig. 5 General pathways for photocatalytic CO2 reduction: formaldehyde, carbene, and glyoxal. Adapted from ref. 9, Copyright 2020, Royal Society of Chemistry. | ||
In the formaldehyde pathway, CO2 activation proceeds via binding of one of the O atoms to the active site of the catalyst. One electron transfer to the CO2 molecule leads to the formation of a CO2˙− radical, and subsequent addition of a proton generates a ˙COOH radical intermediate. Next, the consecutive addition of a proton and an electron to ˙COOH yields formic acid. Then, the formic acid accepts two protons to afford formaldehyde and water. It has been reported that the photoconversion of formic acid to formaldehyde has the most significant kinetic barrier in this pathway.99 Methanol and methane can also be produced through this pathway in subsequent steps depending upon the available electrons and protons, where methane formation proceeds through the ˙CH3 radical intermediate.
This route can account for the production of formic acid, formaldehyde, methanol, and methane but not CO, which is one of the most commonly generated products during the CO2 reduction process.
The carbene pathway can lead to CO formation with the consumption of two electrons, where the CO may be either a side product or an intermediate that reacts further to form methane or methanol. Here, the active sites of the catalyst bind with the C atom of CO2. The formation of methanol or methane via the CO intermediate is dependent upon the adsorption strength between CO and the catalyst surface; the CO may either rapidly desorb from the surface or accept electrons and protons to produce subsequent products. The subsequently generated ˙CH3 radical may react with OH− to form methanol or accept a proton and an electron to afford methane. According to the literature, the carbene pathway has been experimentally demonstrated to be the most commonly followed mechanism for the production of CO and other hydrocarbons, with the intermediates easily detectable using various advanced analytical techniques such as electron paramagnetic resonance (EPR).39 The formation of such hydrocarbons via the carbene pathway is well known as a “proton-coupled electron transfer” (PCET), which is kinetically reliant on the partial electron density on the catalyst surface and the concentration of accessible protons. This pathway is complicated because CO2 requires two electrons and protons to generate CO, yet the production of higher hydrocarbons through PCET requires more electrons; for example, the generation of CH4 and CH3OH requires eight and six electrons, respectively. Although significant progress in the photoreduction of CO2 to afford CO or HCOOH has been reported in the literature, there remains a gap in the research when it comes to converting CO2 into higher hydrocarbons such as C2H5OH, C2H4, and C2H6 with high efficiency and selectivity.25
The third potential route for CO2 conversion is the glyoxal pathway, in which the two O atoms of CO2 coordinate to the catalyst active site in a bidentate manner to produce numerous products.100 Initially, the CO2˙− radical interacts with H+ to generate a bidentate formate, which then couples with another H+ to afford formic acid. Subsequent electron and oxygen transfer lead to formyl radicals (HCO˙), which dimerize to generate glyoxal prior to the formation of C2 and C3 products. Similar to the previous two pathways, the combination of ˙CH3 with a proton leads to CH4 formation with the elimination of CO as a byproduct. The ˙CH3 radical can also form C2 products depending upon the presence of other intermediates in the system.
Although various studies have explored these CO2 reduction pathways both theoretically and experimentally, the multiple steps, intermediates, and byproducts involved in the reaction make the process more complex with respect to C2 selectivity. The density of the photogenerated electrons/holes and stabilization of the intermediates influence the C2 selectivity of the reaction; for instance, the stabilization of ˙CH3 radicals is desirable for achieving C2 selectivity. However, this is hampered by (i) rapid hydrogenation of these intermediates to form C1 products and (ii) repulsive forces between the oppositely charged intermediates, which hinder C–C coupling. The creation of adjacent reaction sites displaying opposite charges could be used to weaken the repulsive forces between adsorbed reaction intermediates, especially CO2˙− or CO*, and provide a platform for the coupling of these adsorbates. Therefore, such reaction sites are highly desirable, stabilizing the ˙CH3 radicals, and overcoming the interadsorbate repulsive forces.101 For instance, as shown in Fig. 6, graphene has been reported to provide such sites and mediate the formation of a C2 product when coupled with TiO2.
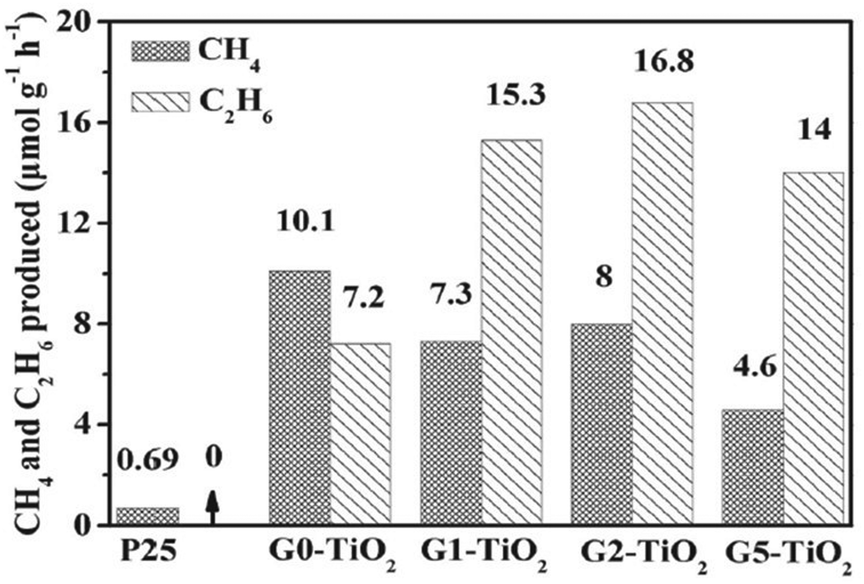 | ||
| Fig. 6 Photocatalytic performance of graphene-wrapped TiO2 showing the formation of C1 and C2 products. Reproduced with permission from ref. 102, Copyright 2013, Wiley-VCH. | ||
3 Strategies to improve CO2 photoreduction
3.1 Energy band structure engineering
As we discussed in Section 2.1, the electronic structure of the photocatalyst is crucial for improving the photocatalytic activity. The photocatalytic performance is highly dependent on the energy band structure (i.e., bandgap and band positions) because the bandgap determines the range of light absorption while the appropriate band positions are required for the photocatalytic redox potentials. Various strategies for tuning the electronic structures of photocatalysts have been reported to date.54 One such strategy is the introduction of defects to improve light absorption. For instance, Mao and co-workers first demonstrated the use of the disorder-engineering approach to prepare “black” TiO2 composed of crystalline TiO2 and disordered surface TiO2, thereby introducing new energy levels.103 Black TiO2 possesses a narrower bandgap (2.18 eV) compared to TiO2 (3.2 eV), which extends the light absorption. Several other approaches for synthesizing reduced TiO2 have since been explored, such as ion implantation,104 the magnesiothermic method,105 and hydride ball milling.106 Here, not only the defects but also the midgap energy band, which refers to the additional electronic states under the CB of TiO2, help to maximize the utilization of solar energy.The electronic structure is highly influenced by band bending, which was first suggested for the metal–semiconductor contact by Schottky and Mott.107,108 When a metal and semiconductor come into contact, the work function difference between the two leads to electron transfer until the Fermi levels are aligned. At equilibrium, the concentration of free charge carriers is different for the metal and semiconductor, resulting in the formation of an electric field at the interface. Consequently, the energy band may bend upward or downward at the interface depending upon the work functions of the metal and semiconductor. For instance, downward band bending was reported for reduced TiO2 at the interface between N-doped graphene oxide and reduced TiO2, which facilitated electron transfer and inhibited electron–hole recombination, resulting in improved photocatalytic activity.96 Besides TiO2, band structure engineering has been conducted for graphitic carbon nitride (g-C3N4), an emerging polymeric photocatalyst, which possesses a moderate bandgap (2.7 eV) and an easily tunable electronic structure.109 The CB of g-C3N4 is composed of C pz orbitals, while the VB consists of the N pz orbitals.110 Approaches for narrowing the bandgap include elemental doping,111 variation of the linking monomer,112 and copolymerization.113
3.2 Non-metal doping and metal cocatalysts
Doping is one method for increasing the photocatalytic CO2 conversion efficiency by creating sub-energy levels within the bandgap, thereby extending the range of light absorption into the visible region. Non-metal dopants such as nitrogen (N), boron (B), carbon (C), sulfur (S), and phosphorus (P) in anatase TiO2 have been investigated.114 For example, substitutional doping of the O atoms in anatase TiO2 with N atoms led to upward bending of the VB edge and a narrower bandgap owing to orbital mixing of the 2p states of O with the 2p states of N. Furthermore, Hashimoto and co-workers reported that N doping of TiO2 resulted in not only mixing of the orbitals but also an isolated N 2p band above the O 2p valence states, thereby improving the visible-light photocatalytic performance of TiO2.115 In addition to N doping, the effects of doping TiO2 with other non-metals, including B,116 C,117 P,118 and S,119 have also been investigated, leading to visible-light-active photocatalysts by reducing the bandgap.Metal cocatalysts have also been extensively applied to improve CO2 photoreduction performance. These can act as electron traps that facilitate electron–hole separation and ultimately improve the photocatalytic activity of semiconductor materials.120 In particular, noble-metal cocatalysts, such as copper (Cu),121 gold (Au),122 silver (Ag),123 palladium (Pd),124 and rhenium (Rh),125 can enhance photocatalytic conversion. For example, Biswas and co-workers developed Pt–TiO2 nanostructured films using a gas-phase deposition method and examined the correlation between the size of the Pt nanoparticles (NPs) and photocatalytic activity (Fig. 7).126 When the size of the Pt NPs was too small, larger energy band separation occurred owing to the quantum confinement effect, inhibiting the electron transfer from TiO2 to Pt. Conversely, when the size of the Pt NPs was too large, the energy band position was similar to that of bulk Pt, thereby acting as a recombination site for the photoexcited electrons and holes.
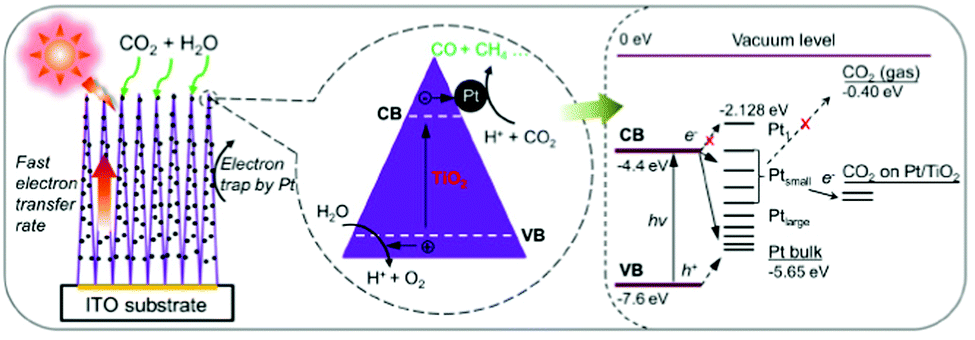 | ||
| Fig. 7 Schematic diagram of CO2 photoreduction mechanism on Pt–TiO2 nanostructured films. The magnified circle (center) shows that the photogenerated electrons can move rapidly inside the highly oriented TiO2 single crystals and flow to the deposited Pt NPs, where the redox reaction occurs to convert CO2 into CO and CH4. The right side of the figure illustrates the energy levels of the Pt–TiO2–CO2 system. H+ and h+ indicate protons and holes, respectively. Reproduced with permission from ref. 126, Copyright 2012, American Chemical Society. | ||
Bimetallic cocatalyst systems have also been explored to improve photocatalytic activity and selectivity. For instance, the combination of Cu and Au has been reported, where Cu provided high affinity for CO2 to mediate the photocatalytic CO2 reduction while Au improved the visible-light absorption owing to its surface plasmonic effects.127 In another example, a Pt@Cu2O core–shell structured bimetallic system was studied, where the Cu2O shell activated the CO2 molecules to enable photocatalytic CO2 conversion in the presence of H2O, while the Pt core acted as an electron trap to extract electrons from TiO2.128 The Cu2O shell on the Pt core also suppressed the reduction reaction of H2O to H2, which otherwise competes with the CO2 reduction reaction, thereby increasing the CO2 conversion activity. Similarly, Long et al. reported the alloying of Cu with Pd to achieve high selectivity for CH4 production by isolating the Cu atoms in a Pd matrix.129 In this case, the Cu NPs served as the active sites for CO2 conversion, but these are susceptible to oxidation under ambient conditions. The alloying approach thus reduced the oxidation of Cu, while the Pd lattice inhibited the H2O reduction reaction. Pd also exhibits strong binding to H atoms, thus suppressing the H2 evolution reaction and affording high selectivity for CH4 formation.
3.3 Heterostructure formation
Efficient photocatalytic CO2 reduction always requires a photocatalyst that can provide large amounts of photogenerated electrons and holes with strong redox potentials. However, by doing so, the optical response of the photocatalyst is compromised because only wide-bandgap photocatalysts can provide strong redox potentials.130 On the contrary, a narrow-bandgap photocatalyst could be applied for enhanced light harvesting, but this requires choosing between a strong oxidation potential and a strong reduction potential. Hence, combining suitable narrow-bandgap photocatalysts could help overcome this tradeoff and realize synergistic effects.131The classification of heterostructures with respect to their band alignment suggests that there exist three main types, namely, straddling alignment (type I), staggered alignment (type II), and broken alignment (type III), as depicted in Fig. 8. In type I heterostructures, the photogenerated charges originating from S1 tend to remain at S2; therefore, no charge separation is achieved. Although this type of charge transfer is beneficial for luminescent materials, it is ineffective for photocatalytic CO2 reduction. In stark contrast, for type III heterostructures, both the CB and VB edges of S1 lie below the CB of S2. Finally, in type II heterostructures, charge separation is achieved but at the expense of lower redox potential. Therefore, type II heterostructures appear to be the most favorable for photocatalytic CO2 reduction and have been reported in various studies. Nonetheless, this is not regarded as an efficient scheme to combine two photocatalysts because the photogenerated charges lose energy during the transfer process.132
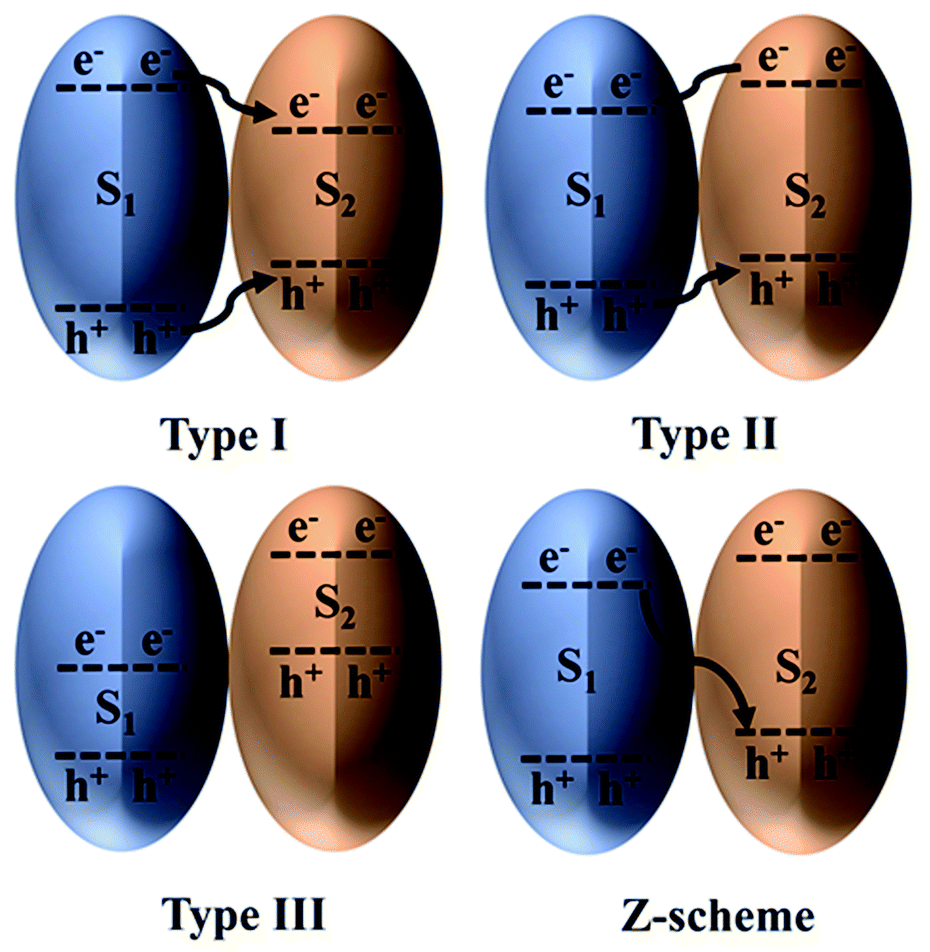 | ||
| Fig. 8 Heterojunction classification based on the charge transfer mechanism. Reproduced with permission from ref. 132, Copyright 2016, Wiley-VCH. | ||
The Z-scheme charge transfer mechanism has the potential to solve the constraints of the aforementioned charge transfer. Under this scheme, the photogenerated charges of both photocatalysts with weak redox potentials recombine with each other while preserving the desired strong charges for photocatalytic CO2 reduction. This type of charge transfer seems justified because of the electrostatic force of attraction between two opposite charges. In addition to these beneficial attributes, this type of charge transfer is also advantageous for improving photocatalyst stability. For example, Cu is susceptible to facile oxidation because its oxidation potential lies within its bandgap. However, by constructing a Cu (i.e., S2) Z-scheme heterostructure with a semiconductor possessing suitable band alignment (i.e., S1), Cu oxidation can be avoided.133
In addition to the direction of the charge transfer, the geometry of the heterostructures also determines their classification. In this regard, interfacial contact between the constituent photocatalysts with high surface area confers beneficial features for the photocatalytic reactions. Various Z-scheme heterostructure geometries have been reported in the literature, which can be classified as (i) core–shell heterostructures, (ii) surface-deposited heterostructures, and (iii) Janus-like systems. The first type has limited exposure of the core to the reaction system, while the second suffers from stability issues due to fast corrosion.43,134,135 However, the synthesis of Janus structures is challenging. In this regard, Ali et al. developed reduced titania–Cu2O Z-scheme heterostructures, somewhat like Janus structures, by using the charge trapping ability of an amorphous shell of reduced titania.44 Under irradiation, the photogenerated electrons accumulated in the amorphous core, causing Cu2+ ions to preferentially accumulate at the edges. The resulting structure not only exposed both photocatalysts to the reactants but also maintained the stability of Cu owing to the higher concentration of electrons.
3.4 Surface modification
Surface characteristics play a major role in CO2 adsorption and activation. Two important types of surface modification are surface functionalization and the deposition of metal complexes. Because CO2 is electrophilic, the introduction of basic groups such as hydroxyl or amine moieties to a photocatalyst can enhance CO2 adsorption.136,137 Some studies have investigated the influence of alkalization on the adsorption of CO2 by TiO2. Treatment with different alkali solutions, such as NaOH, Na2CO3, KOH, and K2CO3, was reported to have various effects on CO2 adsorption.138 The use of NaOH afforded the highest CO2 adsorption owing to the highest amount of free –OH groups. Furthermore, the preparation of layered hydroxide materials rich in surface hydroxyl groups via exfoliation also led to enhanced CO2 uptake.139 Similarly, amine functionalization (–NH species) enables the direct bonding of CO2 molecules. Thus, Liao et al. described the covalent attachment of ethanolamine to ZnO nanosheets via the hydroxyl groups to afford an amine-functionalized surface for the chemisorption of CO2 molecules.140 In addition, He et al. reported that the treatment of TiO2 with HF modulated the ratio of crystal structure facets ((001)/(101)) to improve the dissociation of H2O and reduction of CO2.141The deposition of metal complexes is another approach for surface modification. Early research focused on the combination of semiconductors and mononuclear metal complexes. However, such materials typically exhibit poor stability and insufficient oxidation ability, which has led to the consideration of hybrid materials as preferred alternatives. Nakada et al. revealed that a hybrid material based on g-C3N4 and a Ru complex displayed high efficiency and selectivity for specific hydrocarbons.22,142 By using 2-(1,3-dimethyl-2,3-dihydro-1H-benzimidazol-2-yl)benzoic acid as a sacrificial reducing agent, reverse electron transfer was suppressed to achieve a high quantum yield and selective CO formation.
The morphology/dimensionality of a material surface is another crucial issue that can be used to accelerate the kinetics of photocatalysis.143,144 Low-dimensional materials, defined as nanostructured materials smaller than 100 nm, possess unique optical and electronic properties that originate from quantum confinement and plasmon resonance effects.144–148 Low-dimensional materials can be classified into three categories, namely, (i) zero-dimensional (0D), (ii) one-dimensional (1D), and (iii) two-dimensional (2D) structures.
Zero-dimensional structures, such as nanoparticles, nanospheres, and quantum dots (QDs), are constrained on the nanoscale in all three dimensions. For these structures, high photocatalytic CO2 conversion can be achieved through the excellent light harvesting, satisfactory charge carrier density, and abundant surface sites.147–152 Among such structures, the most extensively used are carbon QDs (CQDs), which exhibit a broad light absorption spectrum due to π-plasmon absorption in the core carbon nanocrystals, thereby enabling the π → π* transition of conjugated carbon atoms in the UV-visible region.153–155 It can be utilized not only in CQDs but also in semiconductor QDs and is advantageous for multi-electron and proton reduction.156,157
One-dimensional structures also possess an attractive morphology that provides excellent charge transfer and extended carrier lifetimes owing to the unique distribution of state density coupled with intrinsically higher reactivity.158–160 In general, 1D structures have high aspect ratios with diameters ranging from 1 to 100 nm, and they include various morphologies such as nanowires, nanotubes, nanobelts, nanoribbons, and nanotubes. As 1D structures contain no grain boundaries, the electron transport distance is greatly reduced, thus improving the electron transfer efficiency and decreasing electron–hole recombination leading to high photocatalytic CO2 conversion (Fig. 9(a)–(c)).159–161 The unique characteristics of 1D structures, including relatively large specific surface areas and good chemical stability, can be used to assemble various heterogeneous surface structures.162–164
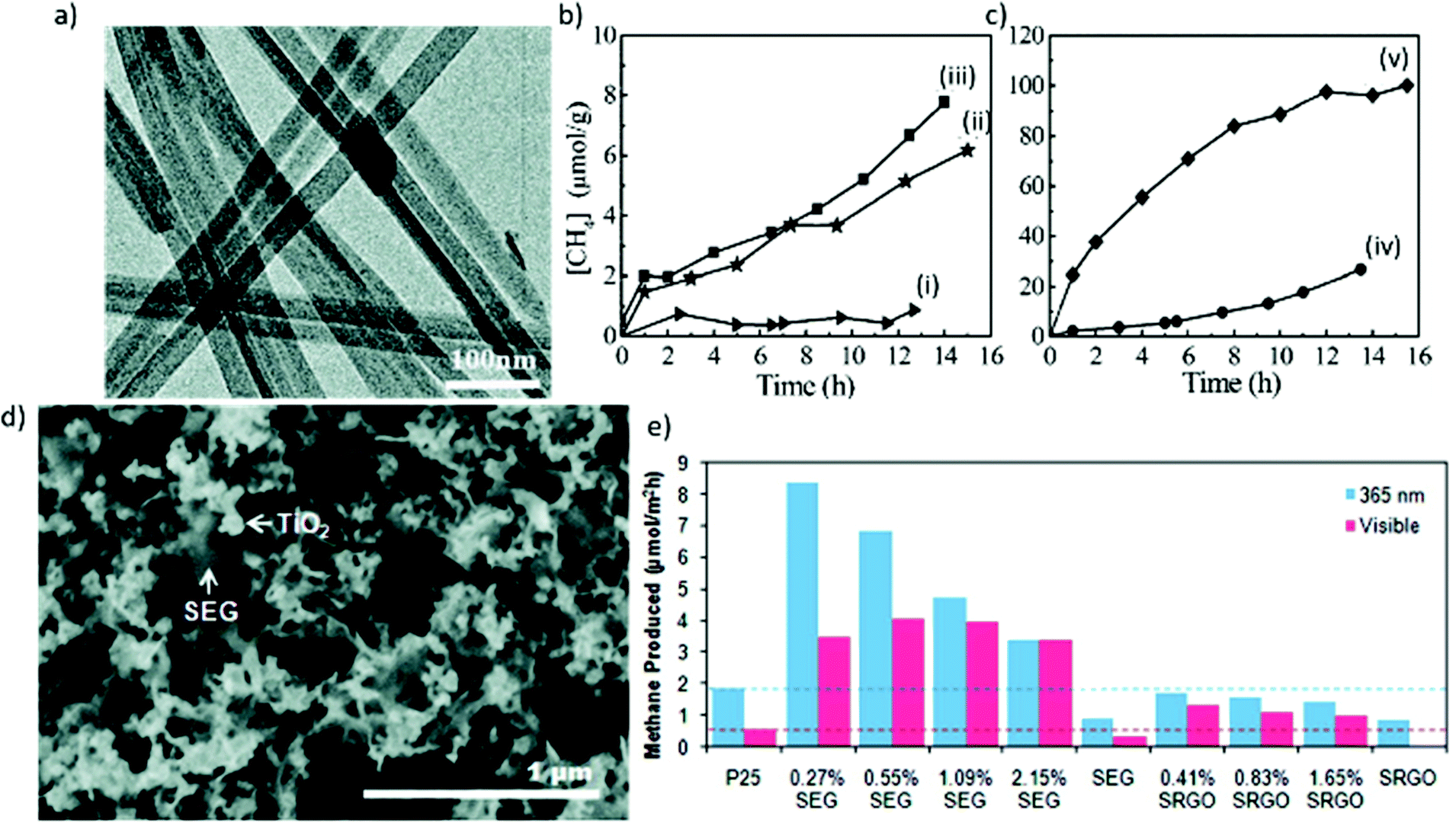 | ||
| Fig. 9 (a) Transmission electron microscopy (TEM) image of Zn2GeO4 nanoribbons, and (b and c) CH4 generation over (i) bulk Zn2GeO2, (ii) nanoribbons, (iii) 1 wt% Pt-loaded nanoribbons, (iv) 1 wt% RuO2-loaded nanoribbons, and (v) 1 wt% RuO2 + 1 wt% Pt-coloaded nanoribbons as a function of light irradiation time. Reproduced with permission from ref. 161, Copyright 2010, American Chemical Society. (d) Scanning electron microscopy (SEM) image of an annealed SEG-P25 nanocomposite, and (e) CO2 photoreduction by SEG-P25 and SRGO-P25 nanocomposites under UV (365 nm) and visible illumination, where SEG and SRGO denote solvent-exfoliated graphene and solvent-reduced graphene oxide, respectively. Reproduced with permission from ref. 165, Copyright 2011, American Chemical Society. | ||
Two-dimensional structures, including nanosheets, nanoflakes, and thin films, have typical thicknesses ranging from several atoms to <100 nm, leading to larger surface-area-to-volume ratios compared to 1D structures. Furthermore, 2D structures display enhanced electron–hole separation, high charge carrier mobility, and reduced recombination.144,166 Among the various 2D structural materials, graphene, which is composed of single-layer carbon nanosheets with a hexagonal packed lattice structure, has received a great deal of attention in the field of photocatalytic CO2 conversion owing to its remarkably high surface area, excellent electrical conductivity (>103 S m−1), and good flexibility.90,167,168 Liang et al. reported that the coupling of graphene with TiO2 showed larger enhancement in photocatalytic CO2 conversion, attributed to superior electric mobility of graphene (Fig. 9(d) and (e)).165
3.5 Reactor design
The design of reactors for photocatalytic CO2 reduction also exerts an important influence on the CO2 conversion efficiency. In addition, the use of different reactor geometries and reaction conditions can render it difficult to compare reaction rates and yields.47,169–171 In general, photocatalytic CO2 reduction is conducted in either batch reactor or continuous-flow reactor systems.In the case of batch reactors, the reduction is typically performed in a pressurized reactor vessel equipped with an optically transparent window and an external temperature controller to adjust the sample temperature. However, there is always the possibility of the readsorption of products and subsequent reverse or side reactions, e.g., re-oxidation to CO2.172,173 Consequently, the yield during batch reactions may decrease owing to the continuous accumulation of the target product as well as byproducts such as oxygen.174,175 In this regard, one effort by Pipelzadeh et al. involved a pressure swing reactor in which the reaction mixture was periodically evacuated from the batch reactor and reinjected.176 This continuous recycling of the products helped overcome the issues of product readsorption and limited mass transfer, increasing the CO production yield to 30–80%. In another effort to improve the photocatalytic yield, a specially designed twin reactor was employed where protons (H+) generated in one compartment were transported to a separate compartment for CO2 reduction, as shown in Fig. 10(a).177,178 This configuration helped overcome the mass transfer limitation and thereby improve the photocatalytic yield. Overall, batch reactor systems make it difficult to compare photocatalytic performance and are often not a suitable option for applications involving prolonged and large-scale reactions.
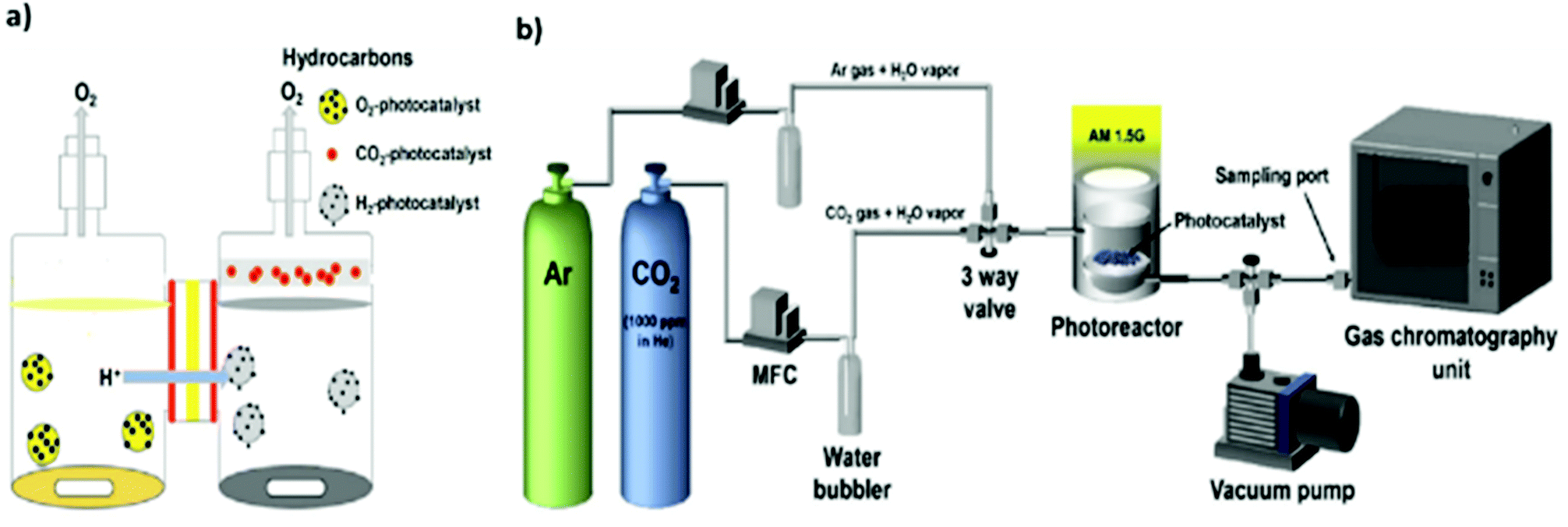 | ||
| Fig. 10 Illustration of two setups for photocatalytic CO2 reduction: (a) twin reactor. Reproduced with permission from ref. 177, Copyright 2018, Elsevier. (b) Continuous-flow reactor. Reproduced with permission from ref. 93, Copyright 2017, Elsevier. | ||
On the other hand, in continuous-flow reactor systems, the reactants and products are moving at a constant flow rate inside the vessel while the reaction is in progress (Fig. 10(b)).5,93 Continuous-flow reactor systems can avoid the problems of batch reactors, such as product readsorption on the photocatalyst surface,172,173 but are also associated with the challenge of a very limited contact time between the reactants and photocatalyst surface owing to the short residence time.47,179,180
As shown in Fig. 11(a), a photoreactor using optical fibers to optimize the light path was developed.181 In contrast to a conventional photoreactor composed of a cylindrical vessel equipped with a quartz window to allow light to enter, optical fibers coated with the catalyst were installed in such a manner as to occupy the maximum possible volume of the reactor, thus improving the contact between the catalyst, reactants, and light and affording high conversion efficiency.182 However, optical fiber photoreactors are difficult to commercialize owing to several disadvantages, including low adhesion strength of catalyst on the fibers, relatively low surface area, and the effective utilization of only approximately 20–30% of the total reactor volume. To compensate for this, a monolith-type photoreactor was also reported as shown in Fig. 11(b).170
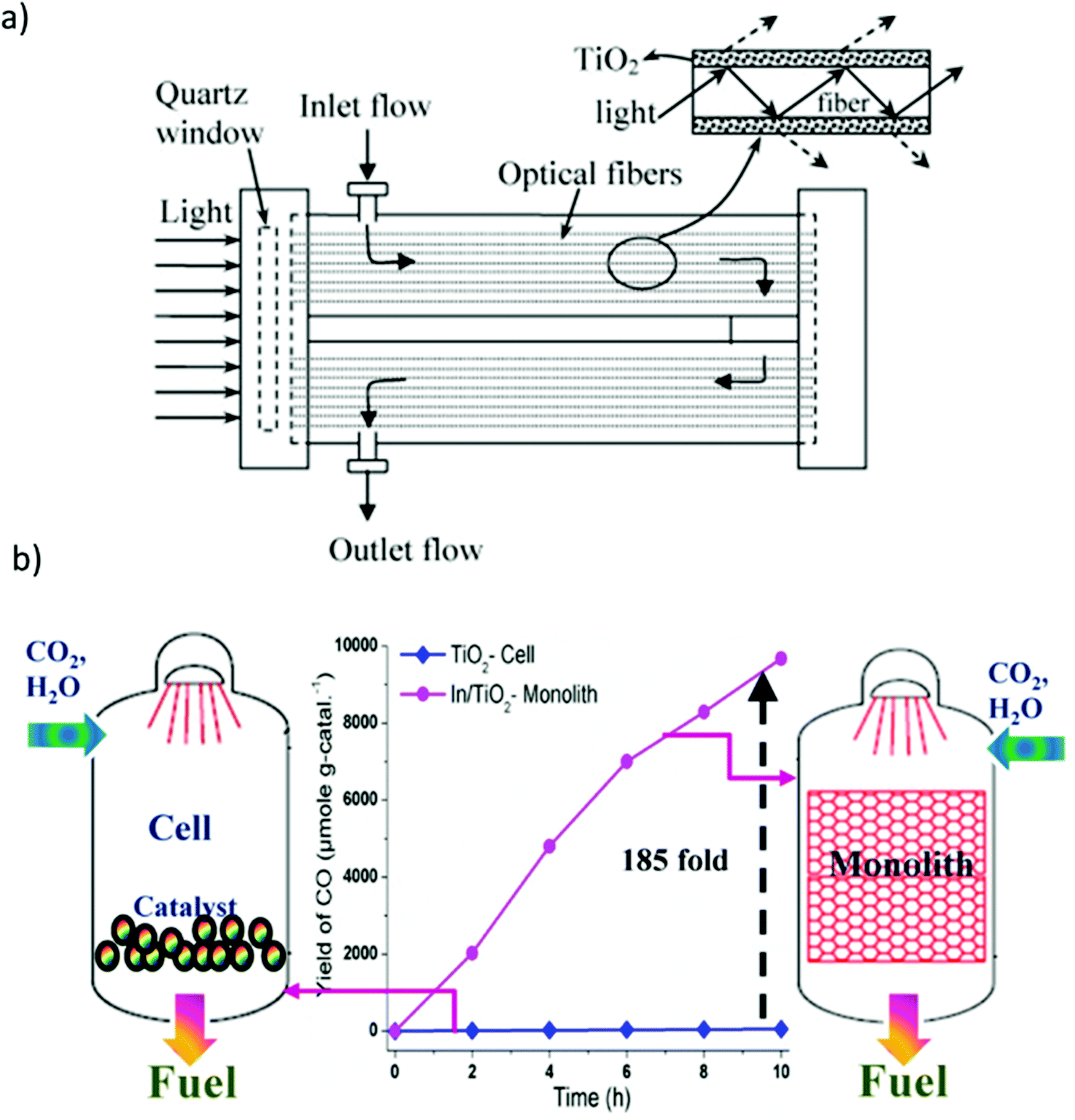 | ||
| Fig. 11 (a) Schematic diagram of an optical fiber reactor, reproduced with permission from ref. 181, Copyright 2007, Elsevier. (b) Comparison of photocatalytic CO2 reduction with H2O using cell- and monolith-type photoreactors. Reproduced with permission from ref. 170, Copyright 2013, Elsevier. | ||
The use of the monolithic catalyst enabled efficient light harvesting and high photon flux owing to its unique structure and high surface-area-to-volume ratio.170,183 Nevertheless, monolith-type photoreactors have the disadvantage that they cannot be used with visible light because of their low penetration depth through the microchannels. Therefore, a combination of a monolithic catalyst and optical fibers can be anticipated to overcome this issue.
In one such effort, Xiong et al. reported the use of a monolithic catalyst with a honeycomb structure through which optical fibers had been inserted.184 This configuration successfully enhanced the photoreaction. In a similar manner, Liou et al. inserted carved polymethylmethacrylate (PMMA) optical fibers into a NiO/InTaO4-coated monolith with a honeycomb structure.185 This reactor afforded an improved product yield when applied to photocatalytic CO2 reduction owing to the large surface area, high photocatalyst loading, and effective light utilization. In another attempt, Cao and co-workers reported a double-chamber reactor as depicted in Fig. 12. Separation of the oxidation and reduction reactions helped to achieve stability of the monolithic two-sided Cu2O/graphene/TNA photocatalyst for up to 60 h.186
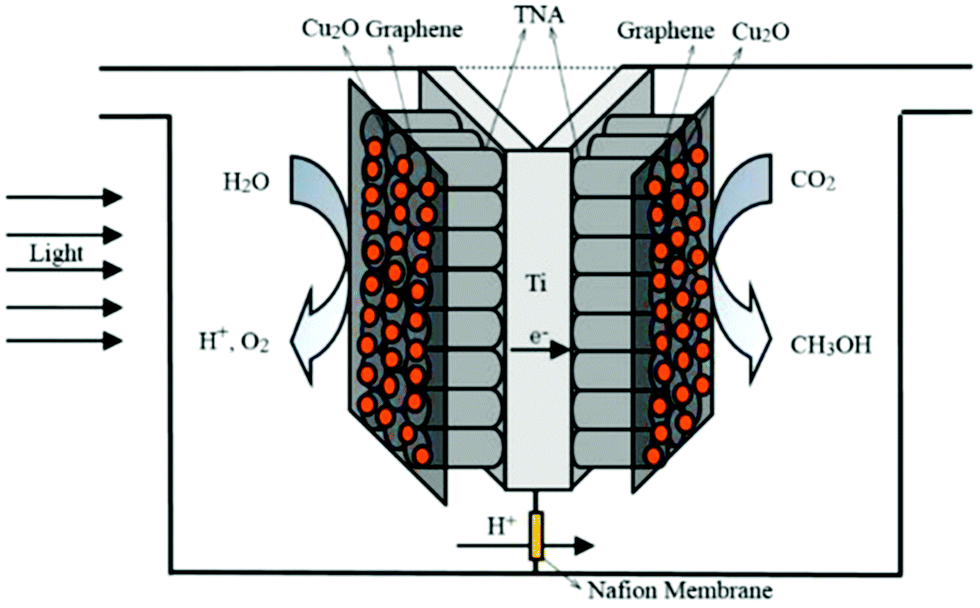 | ||
| Fig. 12 Schematic diagram of a monolithic two-sided Cu2O/graphene/TNA photoreduction reaction system. Reproduced with permission from ref. 186, Copyright 2016, Elsevier. | ||
3.6 Optimization of selectivity
As we discussed in Section 2, the CO2 photoreduction process occurs through a multistep reaction mechanism. Thus, optimization of the product selectivity is always a critical aspect. In general, formaldehyde, carbene, and glyoxal may be formed as intermediate products during the reaction. As such, it is essential for the practical applications of CO2 reduction technology to optimize the selectivity to obtain pure products.98 The main obstacle here is that the reaction mechanism is not yet completely understood at the molecular level. However, several optimization strategies have been reported to date, such as modulating the bandgap, tailoring the surface composition, alkaline treatment of the catalyst, the loading of specific metals at a particular concentration, and improving the interfacial properties.52 As discussed in Section 3.4, the CO2 photoreduction process can be improved by surface modification, and such strategies can also be applied to enhance the selectivity. For example, surface modification with hydrophobic–hydrophilic groups can favor the formation of specific hydrocarbons, in addition to influencing the reaction rate, by altering the affinity of the catalyst surface for H2O molecules.187 He et al. studied the fluorination of anatase TiO2 nanosheets, which introduced Ti3+ species on the catalyst surface that favored the conversion of CO2 to CO2˙−.141 Subsequently, Xing et al. confirmed that the fluorination treatment had no effect on the CO2 adsorption.188 Instead, fluorination treatment induces the built-in electric field by the substitutional F to surface oxygen vacancies. As a result, it increased the rate of CH4 and CO formation. However, the development of more eco-friendly strategies that avoid the use of fluorine would be desirable.Moreover, the photocatalyst acidity or basicity can also contribute to product selectivity. Subrahmanyam et al. studied various metal oxide/metal composites, including TiO2/Pd, CuO/ZnO, and Li2O/TiO2, supported on MgO, Al2O3, or SiO2.189 They found that the basic oxide supported systems displayed reasonable selectivity for the photoreduction of CO2 to C1–C3 compounds. In contrast, the acidic oxide supported catalysts exhibited good selectivity for the generation of C1 compounds. More interestingly, the C1–C3 selectivity remains independent of the confirmed photocatalyst. In conclusion, several strategies may be adopted to optimize the selectivity of CO2 photoreduction, but it remains necessary to understand the underlying reactions at the molecular level, including the heats of formation and the adsorption and desorption energies of the hydrocarbon products.
4 Theoretical insights and equations
4.1 Density functional theory for CO2 photoreduction
Density functional theory (DFT) has attracted enormous attention over the last few decades as a means to understand the kinetics and thermodynamics of reaction mechanisms.190 In the case of CO2 photoreduction, some crucial questions must be answered to overcome the barriers to commercialization, including (i) the nature of the reaction mechanism, (ii) the driving force behind CO2 photoreduction, and (iii) the optimal parameters for achieving the desired photoreduction efficiency. Although these questions seem challenging, it is essential to answer them to establish a robust foundation for CO2 photoreduction technology.89,191To date, the development of semiconductor photocatalysts has been a primary focus of CO2 reduction technology; however, the anticipated efficiency has not yet been realized. Therefore, we need to understand the molecular-level reaction mechanisms responsible for the conversion of CO2 to hydrocarbons. For instance, the carbophilic and oxophilic interactions with the catalyst surface affect the product selectivity.
The kinetic model offers opportunities to design the photoreactor to avoid variable photon flux. In addition, the adsorption and desorption of CO2 molecules on the surface of the photocatalyst also affects other parameters such as light transport, temperature, and pressure.192,193 In the initial stages of research, the microkinetic method was applied to understand the molecular-level interactions of CO2 molecules on the catalyst surface; however, this method does not consider the roles of heat and mass transfer. Thus, with quantum advanced computational modeling, most studies have discussed the reaction kinetics and adsorption energy (Gibbs free energy) of the CO2 molecules, intermediates, and products. Experimental kinetics studies have also been used to elucidate the reaction mechanism at the molecular level.89 The Langmuir–Hinshelwood (LH)-based CO2 photoreduction kinetic model can be used to obtain insights into the reacting reagent species, with both the numerator and denominator terms in eqn (9):
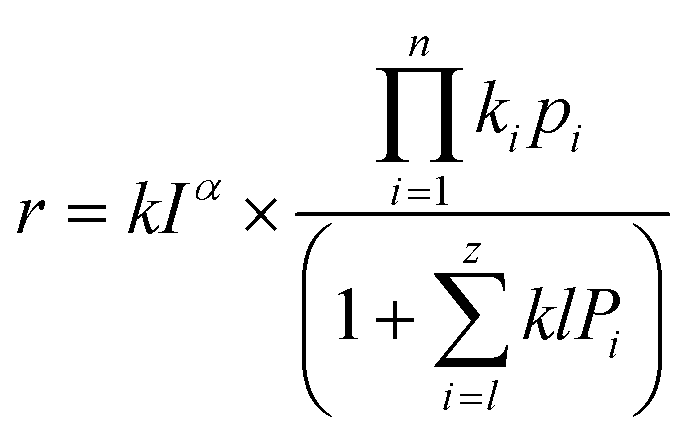 | (9) |
Initially, this theory was primarily used to obtain insights into the electronic structure. Moreover, this theory is mainly dependent upon quantum calculations; it uses the exchange correlation functional to map the interaction electron to the non-interaction electron system, affording the ground state density. Two main types of approximation are used in these calculations, namely, the local density approximation and the generalized gradient approximation (GGA). In the former case, the calculation depends upon the functional electronic density at a given point in space, which leads to a substantial drawback.195,196 Although the local density approximation provides an insight into the atomic energies and energy barriers, it has several disadvantages. Thus, a new approach referred to as the Hubbard model was introduced because the local density and generalized gradient approximations failed to explain the transition metals.197 Various strategies have incorporated the gradient wave (GW) estimation, which yields excellent outcomes for bandgaps, and the Bethe–Salpeter equation (BSE) for absorption spectra.198 These techniques are regularly used because of their unusually low computational expense. The precision of the DFT approach is heavily dependent upon the functionals used. In ab initio studies, the GGA is most commonly performed using the Perdew–Burke–Ernzerhof (PBE) functional. DFT can be used to evaluate the stability of a photocatalyst and the adsorption energies of CO2 and the intermediate products. Here, we discuss some representative results involving theoretical calculations pertaining to CO2 photoreduction.199
Li et al. performed DFT calculations to examine the catalytic activation of CO2 on Cu2O(110) surfaces.200 Cu2O has been identified as a remarkable candidate for CO2 photoreduction owing to its unique electrical and optical properties. Different crystal facets often display distinct catalytic properties; for instance, Cu2O nanocrystals with (100) or (110) surfaces slowly decompose during the reaction, whereas the Cu2O(111) surface has been theoretically shown to possess high stability and rhombic dodecahedral nanocrystals exhibit optimum photocatalytic activity. The DFT study used the Vienna Ab initio Simulation Package (VASP) to simulate the Cu2O(110) surface morphology and calculate the energies during CO2 and CO adsorption. Projector augmented-wave (PAW) atomic pseudopotentials were utilized with a cutoff energy of 400 eV for the plane wave basis set, while the GGA with the PBE parametrization was utilized for the exchange correlation functional. GGA is significantly affected by 3d electrons, so the development energies of 3d transition-metal oxides shows large in fault. Furthermore, to examine the active sites responsible for the catalytic activity, the authors simulated the grazing-incidence X-ray absorption near-edge structure (XANES) by the Grazing-incidence X-ray absorption near-edge structure (GIXANES).200 It is also matched in surface studies due to the limited penetration depth and small X-ray incident angle. In this work, to simulate the surface behavior, only several key layers of the Cu2O(110) section were considered for the slab model. The calculated spectra for the few layers of Cu particles on the ideal surface are presented in Fig. 13(a). The calculated spectra for ideal, O-deficient, and CO2-adsorbed (CO2,O-vac) surfaces utilizing the few layers, along with the corresponding second-derivative spectra, are shown in Fig. 13(b). These spectra were adjusted by coordinating with the edge positions of the surface slab. As shown in Fig. 13(b), the three types of surface displayed similar spectral shapes with small changes in the intrinsic energies. Compared with the ideal surface, the edge position (zero point of the second derivative) of the O-deficient surface displayed a shift toward lower energy of approximately 0.3 eV. After CO2 adsorption, the edge position moved back toward higher energy, which validates the increase of the surface Cu oxidation states because of the charge transfer to the CO2 atom. Hence, aligned spectra of before changing the oxidation state (Cu) and after changing the oxidation both spectra gives the information with rising the energy edges; the changes in the surface oxidation states manifest themselves as changes in the energy of the rising edges.
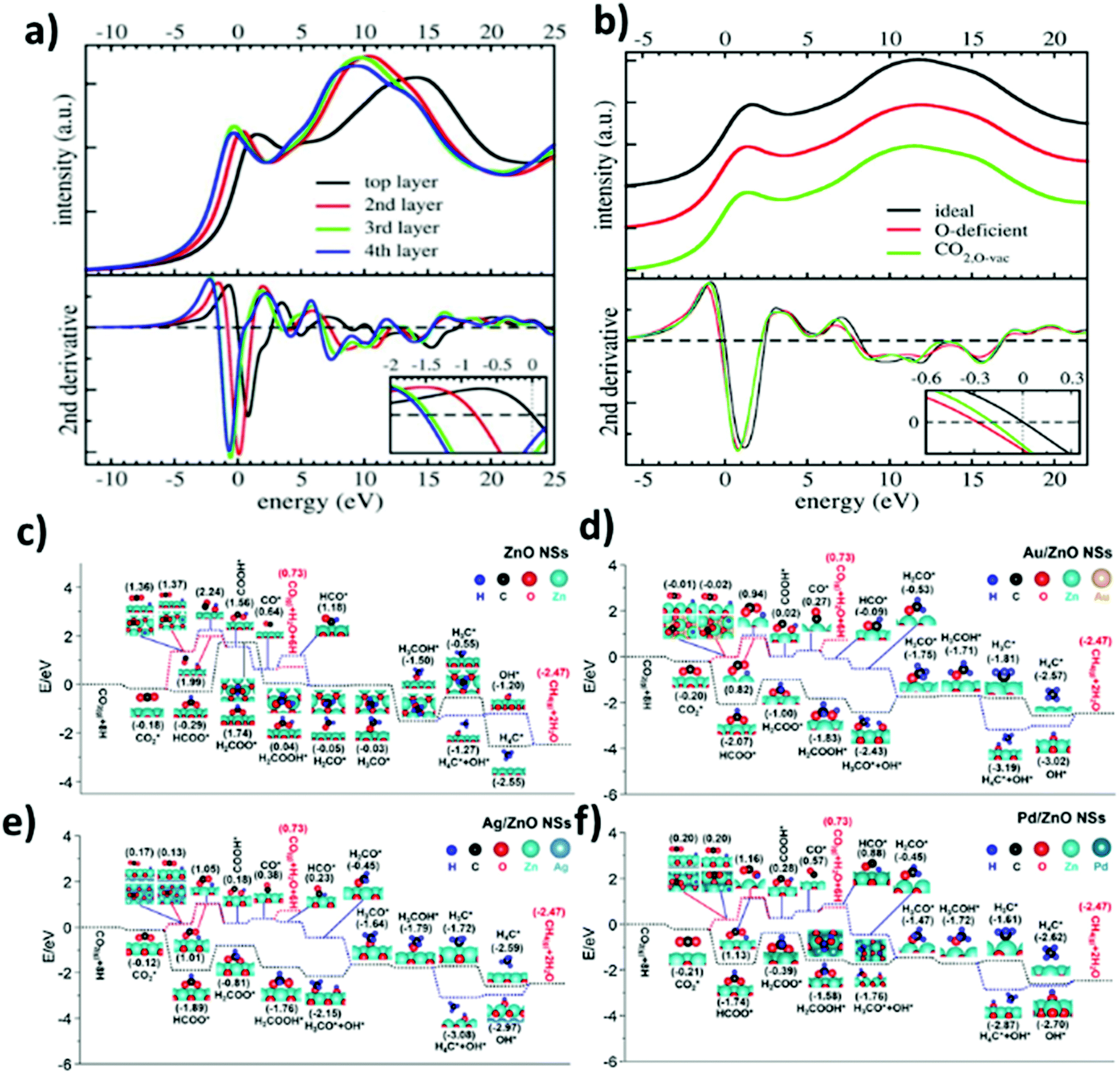 | ||
| Fig. 13 Calculated Cu K-edge XANES spectra for (a) Cu atoms on a Cu2O(110) surface and (b) ideal, O-deficient, and CO2-adsorbed Cu2O(110) surfaces. Reprinted with permission from ref. 200, Copyright 2018, American Chemical Society. (c)–(f) Calculated potential energy (E, eV) diagrams for the CO2 to CH4 reaction on (c) Zn49O49, (d) Zn49O49/Au128(111), (e) Zn49O49/Ag128(111), and (f) Zn36O36/Pd98(111). Reproduced with permission from ref. 201, Copyright 2019, Elsevier. | ||
Tafreshi et al. performed DFT calculations for a Ag3PO4/g-C3N4 nanocomposite to elucidate its electronic properties and photocatalytic activity.202 The VB was composed of the d orbitals of Ag and the p orbitals of O, while the CB consisted of the p orbitals of C and N and the s orbital of Ag. The bandgap decreased from 2.75 eV for pristine Ag3PO4 and 3.13 eV for single-layer g-C3N4 to only 2.52 eV for the Ag3PO4/g-C3N4 nanocomposite. The authors also studied the adsorption geometries and energies of the reaction intermediates for CO2 photoreduction. The heterostructure was found to be thermodynamically favorable for CO2 reduction and displayed high selectivity for CH4. The intermediates HCOOH* and HOCOH* were responsible for the generation of CH4. The most exothermic calculated reaction energy (−2.826 eV) was that for the conversion of trans-COOH to HCOOH*, during the least reaction energy (−0.182 eV) for the hydrogenation of CH2O* to CH2OH* and HCO* to cis-HCOH. The results revealed that the Ag atoms at the interface of Ag3PO4 and C3N4 served as charge recombination centers. In addition, the calculations indicated that the Ag atoms formed midgap states at the interface, leading to a smaller bandgap for this nanocomposite.
Furthermore, Zhao et al. conducted DFT calculations of metal/ZnO nanocomposites based on Au, Ag, or Pd and porous ZnO nanosheets (NSs) to investigate the molecular-level reaction mechanism.201 The calculated potential energy diagrams for CO2 to CH4 conversion on the nanocomposites are presented in Fig. 13(c)–(f). The calculated adsorption energies were 0.29 eV on pristine ZnO NSs, −2.07 eV on Au/ZnO NSs, −1.89 eV on Ag/ZnO NSs, and −1.74 eV on Pd/ZnO NSs. A stable HCO2 hydrogenation intermediate (CO2* + H → HCO2*) bonded with the ZnO nanosheet was considered for understanding the reaction mechanism. The loading of metals on the Zn nanosheets reduced the adsorption energy, confirming the thermodynamic feasibility of the first step of hydrogenation. After that new rate-determining reaction mechanism was studied, which is observed during the dehydroxylation process. The adsorption enthalpies decreased in the order of Au/ZnO NSs (+0.68 eV) > Ag/ZnO NSs (+0.51 eV) > Pd/ZnO NSs (+0.29 eV). Thus, it was concluded that the loading of metal nanoparticles altered the molecular pathway for the conversion of CO2 to CH4.
Overall, the determination of precise reaction intermediates and pathways using DFT calculations remains challenging. However, this approach at least provides supporting evidence for experimental observations. Furthermore, our understanding of the interactions of protons with photocatalyst surfaces is becoming more advanced, although there is still room for improvement. More investigation of the influence of electronically excited states and the solvent on photocatalyst performance is needed. Irrespective of their known limitations, GGA pseudopotentials with basic van der Waals corrections are the principal methodology. Later on, more extensive screening effects will be considered for realizing further improvements in photocatalysts. In addition, to enhance the photocatalytic performance to large scale production, we have to estimate the electronic distribution of catalyst.
4.2 Equations for gas-phase CO2 photoreduction and CO2 electrochemical reduction
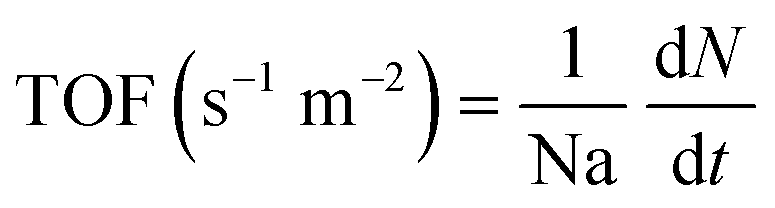 | (10) |
Although this equation is convenient for homogeneous photocatalysts, it is not readily applicable to heterogeneous photocatalysts; because it is difficult to determine the actual active sites and the properties of a heterogeneous catalyst are not directly proportional to its area. In this regard, the efficiency of heterogeneous photocatalysts can be determined in a different way. In photochemistry, for example, the apparent quantum yield (AQY) is often used:5,93,203,206,207
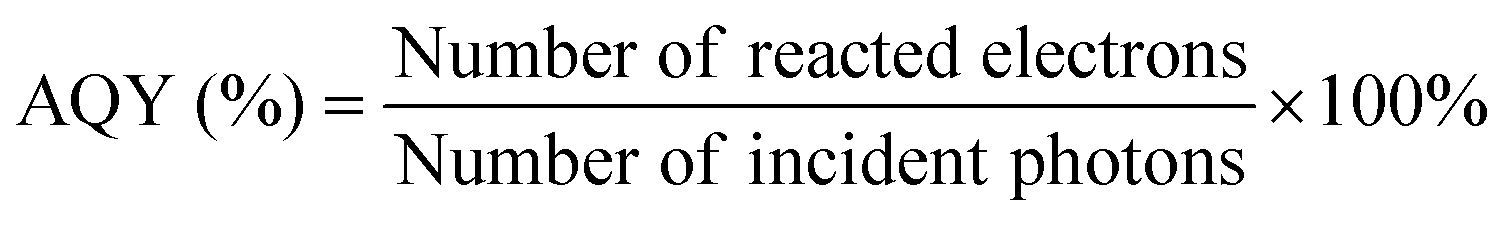 | (11) |
| Number of reacted electrons = number of moles of product (mol) × the number of required electron (8) × NA | (12) |
 | (13) |
| Light absorbed by photocatalyst = H (1000 W m−2) × A (m2) | (14) |
 | (15) |
In these equations, NA is Avogadro's number (6.022 × 1023 mol−1), h is Planck's constant (6.626 × 10−34 J s−1), H is the incident light intensity, A is the irradiation area, and c is the speed of light (3 × 108 m s−1). AQY is defined as the ratio of the number of electrons participating in the photocatalytic reaction to the number of photons absorbed within a specified wavelength range, under the assumption that all photons are absorbed by the photocatalyst.98,203 For example, the number of electrons required for the production of 1 mol of CH4 by photocatalytic CO2 reduction can be calculated using eqn (12). The incident photon flux can also be calculated from eqn (13)–(15). In eqn (14), the value of H for a given reactor can be reliably determined using a reference cell; this value is 1000 W m−2 at AM 1.5. The average photon energy can be determined from the incident light wavelength as expressed in eqn (15).5
Electrocatalysts have also received considerable attention in the field of CO2 reduction for mediating specific redox reactions on an electrode surface.208 Because these electrochemical processes involve electron transfer reactions, the performance is typically measured by the faradaic efficiency (FE):209–211
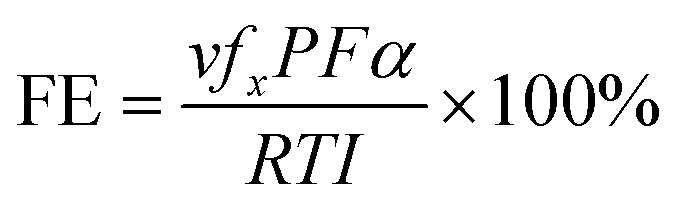 | (16) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and I is the current at the given sampling time.209,212–214
485 C mol−1), and I is the current at the given sampling time.209,212–214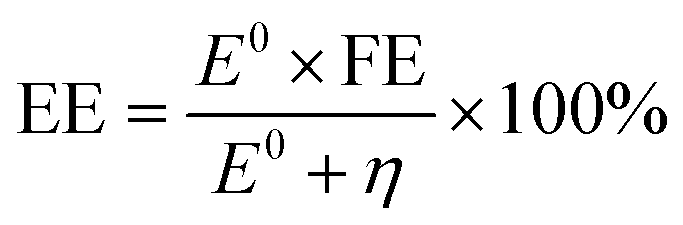 | (17) |
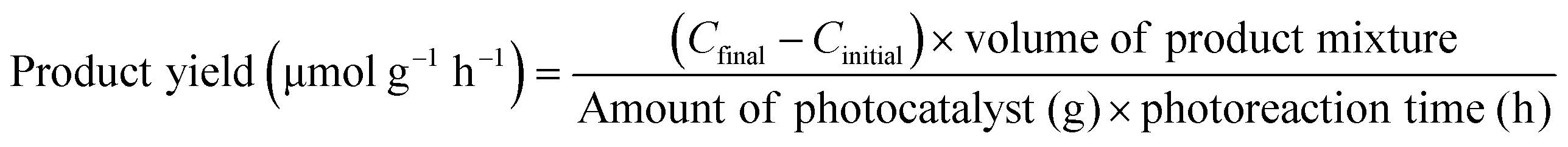 | (18) |
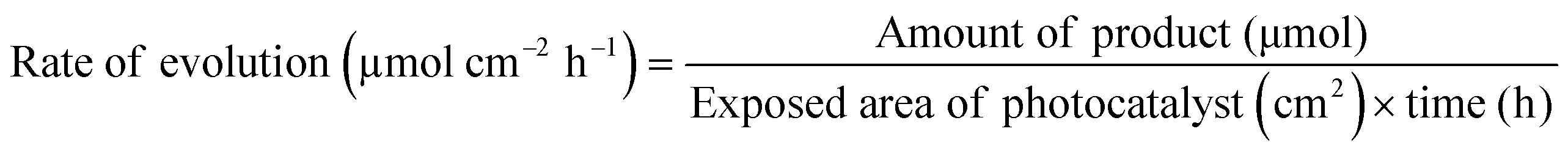 | (19) |
The plot seems a saturation instead of linear graph because the product formation is not linear over time.98 Therefore, the average efficiency of a catalyst depends upon the measurement time. The irradiation time is related to catalytic stability, and it is recommended to use the same reaction time when attempting to compare different catalysts.216
When multiple products are present simultaneously, the selectivity for a particular product such as CH4 or CO can be calculated:93
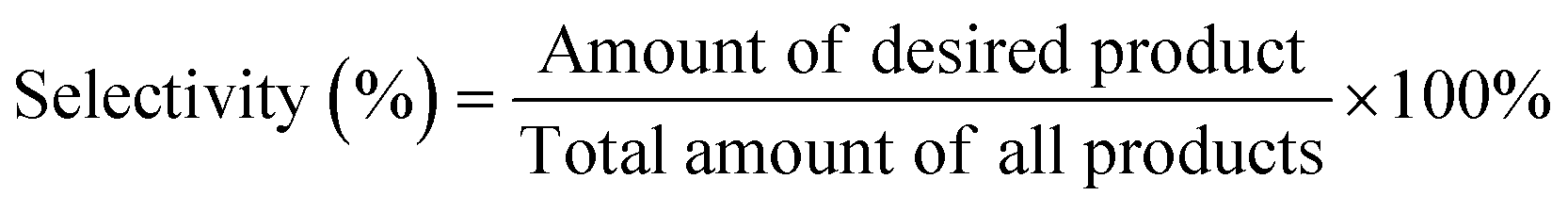 | (20) |
The efficiency can also be expressed by the input vs. output (e.g., in terms of energy).5 In this case, the total mass is usually used as the denominator in eqn (21).98 This can be calculated using the following equations:5,217
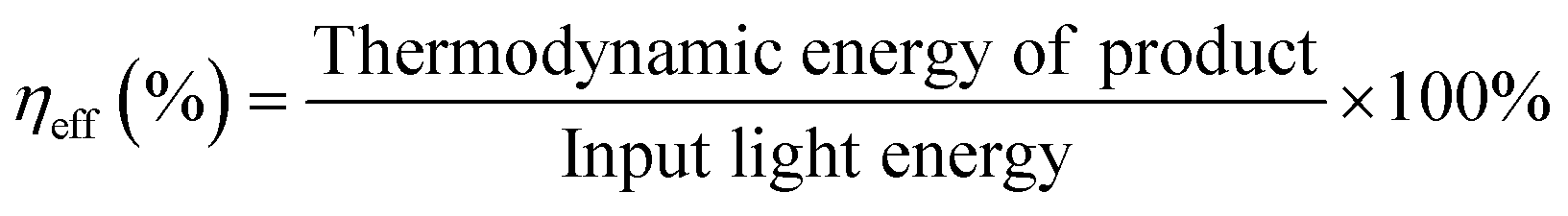 | (21) |
| Thermodynamic energy of product (cm−2 h−1) = [CH4] (mol cm−2 h−1) × ΔH (810 kJ mol−1) | (22) |
| Input light energy (for a 100 mW light source) = 0.100 W cm−2 | (23) |
When the products of CO2 reduction are analyzed by chromatography, the results are measured in ppm. However, when expressed in ppm, it is difficult to immediately understand the actual amount of product. Therefore, many researchers express the amount of product in molar units.44,218 Some recent reports have confusion in calculation, for example, Sorcar et al. reported less accurate assumption and calculation results.5,93,95 Therefore, we suggest better calculation method that extends the application to gas-phase reactions:44
| CH4 yield in μmole = [CH4 yield in ppm] × [moles of the gaseous mixture containing CH4] | (25) |

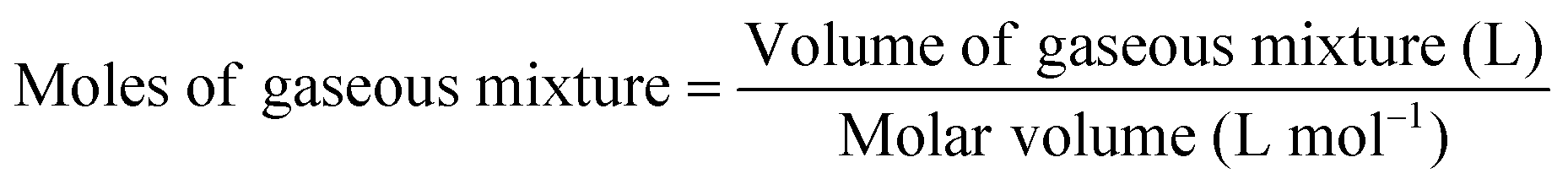
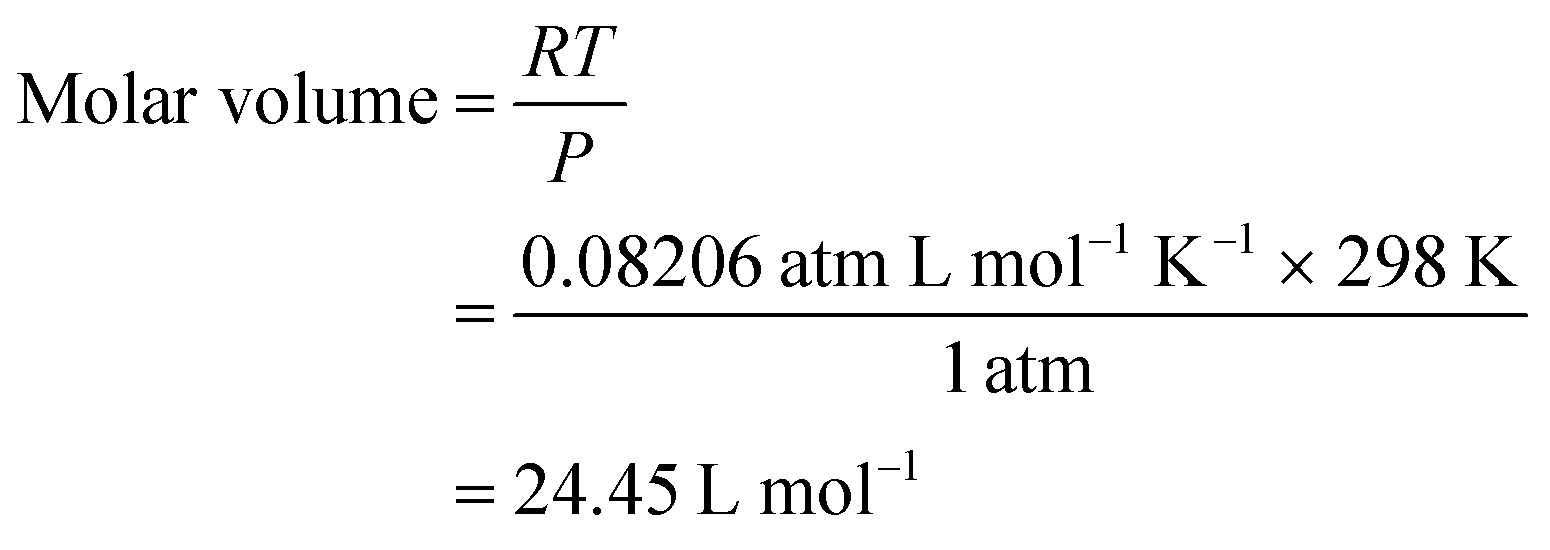
If the performance of photocatalysts can be reliably compared, it will be very beneficial to the field of solar fuel generation. We believe that the equations presented in this review will prove valuable to researchers and encourage further progress in the field.
5 Materials for CO2 photoreduction
5.1 Metal oxides
Metal oxides are widely used in photocatalysis because of their high earth-abundance and exceptional stability under various conditions. Several features of metal oxides are of particular relevance to photocatalysis, such as morphology/composition, light absorption characteristics, and charge transport properties. In 1972, Fujishima and Honda reported the production of hydrogen from TiO2 under light illumination, which was the starting point for metal oxide-based photocatalysts and attracted worldwide attention.219 Since then, numerous studies on metal oxide photocatalysts have been conducted. A comparison of metal oxide-based photocatalysts is presented in Table 2.| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor type | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| TiO2−x | 99.999% CO2, water bubbler (H2O vapor) | 150 W solar simulator (90 mW cm−2) | H2O | 100 mg sample | Flow reactor | CO: 17 μmol g−1 h−1 | 220 |
| CH4: 2 μmol g−1 h−1 | |||||||
| TiO2 | In situ generated CO2 + H2O vapor (NaHCO3 + HCl) | 300 W Xe lamp | H2O | 100 mg sample in Pyrex reactor (200 mL) | Batch reactor | CH4: 1.35 μmol g−1 h−1 | 221 |
| TiO2−x | CO2 + H2O | 300 W Xe lamp (AM 1.5 filter) | H2O | CH4: 1.63 μmol g−1 h−1 | 188 | ||
| TiO2/Ni(OH)2 | In situ generated CO2 + H2O (NaHCO3 + H2SO4) | 350 W Xe lamp (40 mW cm−2) | H2O | 50 mg sample | CH4: 2.20 μmol g−1 h−1 | 222 | |
| CO: 0.71 μmol g−1 h−1 | |||||||
| Pt/TiO2 | CO2 + H2O | 400 W Xe lamp (UV range, 250–388 nm, 19.6 mW cm−2) | H2O | CH4: 1361 μmol g−1 (5 h) | 126 | ||
| PdCu/TiO2 | CO2 + H2O | 300 Xe lamp (λ < 400 nm, 2 mW cm−2) | H2O | 5 mg sample | CH4: 19.6 μmol g−1 h−1 | 129 | |
| Au–Cu nanoalloy supported on TiO2 | 99.995% CO2 + H2O | 100 W Xe lamp | H2O | Sample film | Batch reactor | CH4: 2000 μmol g−1 h−1 | 127 |
| g-C3N4/ZnO | In situ generated CO2 + H2O (NaHCO3 + H2SO4 aqueous solution) | 300 W simulated solar Xe arc lamp | H2O | 100 mg sample | CH3OH: 0.6 μmol g−1 h−1 | 223 | |
| ZnO/Au/g-C3N4 | CO2 (99.999% CO2 + H2O) | 300 W UV lamp | H2O | Film-type sample | CO: 862.1 μmol m−2 h−1 | 224 | |
| ZnO/ZnTe | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp (420 nm cutoff) | H2O | 10 mg sample | CH4: 44.564 μmol g−1 h−1 | 225 | |
| Mo-Doped WO3·0.33H2O | CO2 (400 ppm, N2-based) + H2O (0.5 mL) | 500 W Xe lamp | H2O | 25 mg sample in closed Pyrex reactor (600 mL) | CH4: 5.3 μmol g−1 h−1 | 226 | |
| Ag-Modified ZnGa2O4 | NaHCO3 + CO2 | 400 W high-pressure Hg lamp | H2O | 1.0 g sample | Flow reactor | CO: 155.0 μmol g−1 h−1 H2: 8.5 μmol g−1 h−1 O2: 74.3 μmol g−1 h−1 | 227 |
| Ga2O3 | NaHCO3 + CO2 | UV light (ca. 254 nm, 13 mW cm−2) | H2O | 100 mg sample | CO: 100 μmol g−1 h−1 | 228 | |
| Pt/Zn–β-Ga2O3 nanorods | CO2/H2O (99.999% CO2 + 20 μL H2O) | 15 W UV-C lamps (5.94 mW cm−2) | H2O | 15 mg sample | CH3OH: 0.19 μmol g−1 h−1 | 229 | |
| Pt–RuO2/Zn2GeO4 | High-purity CO2 + 1 mL DI | 300 W Xe lamp | H2O | 100 mg sample | CH4: 100 μmol g−1 (16 h) | 161 | |
| InVO4 | 0.4 mL DI (H2O vapor) High-purity CO2 | 300 W Xe lamp | H2O | 0.1 g sample | CO: 18.28 μmol g−1 h−1 CO: 130 μmol g−1 (7 h) CH4: 2 μmol g−1 (7 h) | 230 | |
| Bi2WO6 (LSPR) | CO2 + H2O | UV-vis light (200 mW cm−2) | H2O | 5 mg sample with 0.2 mL pure water | CH4: 9.9 μmol g−1 h−1 | 231 | |
| Stability: 3 h | |||||||
| CuO–Nb3O8 | 0.5 M KHCO3 aqueous solution, adjusted to pH 12 with NaOH | UV (Hg–Xe lamp, 240–300 nm) | H2O | Dispersed sample in glass reactor (500 mL) | CO: 1.4 μmol (20 h) | 232 | |
| CuOx–ZnO | CO2 + H2O | 300 W Xe lamp (320–780 nm, 100 mW cm−2) | H2O | 5 mg sample | C2H4: 2.7 μmol g−1 h−1 (32.9%) | 233 | |
| CH4: 2.2 μmol g−1 h−1 (26.9%) | |||||||
| CO: 3.3 μmol g−1 h−1 | |||||||
| Stability: 4 cycles (1 cycle = 8 h) | |||||||
| Co3O4 | 0.5 mL DI (water vapor) 99.999% CO2 | 200 W Xe lamp (100 mW cm−2) | H2O | 5 mg sample | CO: 46.3 μmol g−1 h−1 | 234 | |
| Stability: 4 cycles (1 cycle = 5 h) | |||||||
| Cu–Pt/TiO2 | 99.99% CO2 water bubbler | Outdoor sunlight (normalized to a global AM 1.5 value of 100 mW cm−2) | H2O | 2 × 2 cm2 sample | Hydrocarbon: 111 ppm cm−2 h−1 | 78 | |
| MgO/Pt–TiO2 | CO2 4 mL water (H2O vapor) | 100 W Xe lamp (320–780 nm, 60 mW cm−2) | H2O | 20 mg sample | CH4: 11 μmol g−1 h−1 | 235 | |
| CO: 0.03 μmol g−1 h−1 H2: 11 μmol g−1 h−1 | |||||||
| Stability: 3 cycles (1 cycle = 10 h) | |||||||
| TiO2−x | 99.999% CO2 water bubbler (H2O vapor) | UV: 100 W Hg lamp Visible: 450 W Xe lamp with UV filter (400–700 nm) | H2O | 40 mg sample 150 °C | CO: 54.5 μmol g−1 h−1 | 236 | |
| Ag–TiO2 (LSPR) | 99.9999% CO2 + H2O | 6 W UV lamp (47.23 mW m−2) | H2O | 100 mg sample | CH4: 86.5 μmol g−1 (15 h) | 237 | |
| Pt1%-0.50–Graphene/reduced titania | CO2 + H2O | 100 W Xe solar simulator with AM 1.5 filter | H2O | 40 mg sample | Flow reactor | CH4: 37.0 μmol g−1 h−1 | 95 |
| C2H6: 11.0 μmol g−1 h−1 | |||||||
| Cu1.00%–Pt0.35%–Blue titania | CO2 + H2O | 100 W Xe solar simulator with AM 1.5 filter | H2O | 40 mg sample | Flow reactor | CH4: 3.0 mmol g−1 (6 h) | 5 |
| C2H6: 0.15 mmol g−1 (6 h) | |||||||
| Hybrid carbon@TiO2 hollow spheres | In situ generated CO2 + H2O (NaHCO3 + H2SO4) | 300 W Xe arc lamp without filter | H2O | 100 mg sample | CH4: 4.2 μmol g−1 h−1 | 238 | |
| CH3OH: 9.1 μmol g−1 h−1 | |||||||
| Au/TiO2/BiVO4 | CO2 + H2O | 300 W Xe arc lamp (area of 3.5 cm2) | H2O | 0.2 g sample | CH4: 7.5 μmol g−1 h−1 | 239 | |
| CO: 2.5 μmol g−1 h−1 | |||||||
| ZnFe2O4/RGO/In2O3 | CO2 + H2O | 300 W Xe arc lamp (area of 3.5 cm2) | H2O | 0.1 g sample in cylindrical reactor (100 mL) | CH4: 1.95 μmol g−1 h−1 | 240 | |
| CO: 8.85 μmol g−1 h−1 | |||||||
| SnS2/SnO2 | CO2 (400 mL 99.999% CO2 + 0.5 mL H2O) | 300 W Xe lamp | H2O | 4 mg sample | CO: 48.01 μmol g−1 h−1 | 241 | |
| CeO2−x | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp | H2O | 50 mg sample | CO: 1.68 μmol g−1 h−1 | 242 | |
| Au/TiO2−x | CO2 (99.999% CO2 + H2O) | 50 W UV (365 nm, 49.5 mW cm−2) | H2O | 50 mg catalyst | CO: 7.52 μmol g−1 h−1 | 243 | |
| 50 W vis (530 nm, 64.9 mW cm−2) | CH4: 3.57 μmol g−1 h−1 | ||||||
| C2H6: 0.59 μmol g−1 h−1 | |||||||
| PDA–TiO2 | CO2 + H2O (NaHCO3 + H2SO4) | 300 W Xe lamp | H2O | 50 mg catalyst | CH4: 1.50 μmol g−1 h−1 | 244 | |
| TiO2/UiO-66 | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp | H2O | 50 mg catalyst | CH4: 17.9 μmol g−1 h−1 | 245 | |
| CO: 2.0 μmol g−1 h−1 | |||||||
| PbO/TiO2 HPJs | CO2 + H2O (NaHCO3 + H2SO4) | 300 W Xe lamp | H2O | 60 mg catalyst | CH4: 53.21 μmol g−1 h−1 | 246 | |
| CO: 5.99 μmol g−1 h−1 | |||||||
| TiO2/AuCu/ZIF-8 | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp (100 mW cm−2) | H2O | 2 × 2 cm2 film | CO: 83 μmol g−1 h−1 | 247 | |
| CH4: 3.9 μmol g−1 h−1 | |||||||
| Flame-annealed TiO2 | CO2 (99.999% CO2 + H2O) | AM 1.5G (100 mW cm−2) |
H2O | CH4: 156.5 μmol g−1 h−1 | 218 | ||
| CuTCPP/P25m | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp | H2O | 100 mg catalyst | CH4: 19.39 μmol g−1 h−1 | 248 | |
| CO: 2.68 μmol g−1 h−1 | |||||||
| PtRu/TiO2 | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp (80 mW cm−2, 320 < λ < 780 nm) | H2O | 100 mg catalyst | CH4: 38.7 μmol g−1 h−1 | 249 | |
| CO: 2.6 μmol g−1 h−1 | |||||||
| NCQDs/P25 | CO2 (99.999% CO2 + H2O) | 300 W Xe lamp | H2O | 50 mg catalyst | CO: 23.66 μmol g−1 h−1 | 155 | |
| CH4: 15.92 μmol g−1 h−1 | |||||||
| Au/Ag–TiO2 | CO2 + H2 | 35 W Xe lamp (20 mW cm−2) 200 W Hg reflector lamp (150 mW cm−2) | H2 | 10 mg catalyst | CO: 1813 μmol g−1 h−1 | 250 | |
| Au-MMT/TiO2 | CO2 + H2 | UV: 200 W Hg reflector lamp Vis: 100 W Xe lamp | H2 | Monolithic support | UV: CO: 1223 μmol g−1 h−1 | 251 | |
| CH4: 12 μmol g−1 h−1 | |||||||
| Vis: CO: 199 μmol g−1 h−1 | |||||||
| CH4: 42 μmol g−1 h−1 | |||||||
| ZIF-8/TiO2 | CO2 (99.999% CO2 + H2O) | 100 W Xe lamp (100 mW cm−2) | H2O | 100 mg catalyst, 5 bar | CO: 45.16 μmol g−1 h−1 | 176 | |
| Pt-1.0–Reduced titania | 1000 ppm CO2 (He-based) + H2O | 100 W Xe lamp | H2O | 70 mg catalyst | CH4: 1.13 μmol g−1 h−1 | 252 | |
| Pt/TiO2/Au18@SiO2 | CO2 (99.999% CO2 + H2O) | 5 W LED lamp (365 nm/530 nm) | H2O | 32 mg catalyst | CH4: 2.98 μmol g−1 h−1 | 253 | |
| Coordination polymer of Oslo-27-Mg/TiO2 | Humidified CO2 (CO2 + H2O) | 4 W UV lamp (365 nm) | H2O | 10 mg catalyst | CO: 4.09 μmol g−1 h−1 | 254 | |
| CH4: 2.35 μmol g−1 h−1 | |||||||
| Mesoporous TiO2 | CO2 (99.999% CO2 + H2O) | 250 W Xe lamp | H2O | 100 mg catalyst | CH4: 0.192 μmol g−1 h−1 | 255 | |
| CO: 0.145 μmol g−1 h−1 | |||||||
| Cu/TiO2 | CO2 (99.999% CO2 + H2O) | 150 W lamp (90 mW cm−2) | H2O | 50 mg catalyst | CO: 3.8 μmol g−1 h−1 | 40 | |
| CH4: 0.68 μmol g−1 h−1 | |||||||
| Cu/TiO2–SiO2 | CO2 (99.999% CO2 + H2O) | Xe lamp | H2O | 100 mg catalyst | CO: 60 μmol g−1 h−1 | 87 | |
| CH4: 10 μmol g−1 h−1 | |||||||
| Reduced titania–Cu2O | 1000 ppm CO2 (He-based) + H2O | 100 W Xe lamp | H2O | 40 mg catalyst | Flow reactor | CH4: 77 nmol g−1 h−1 | 44 |
| CuO–TiO2−x Nx | CO2 (99.95% CO2 + H2O) | 300 W Xe lamp (100 mW cm−2) | H2O | 100 mg catalyst | CH4: 41.3 ppm g−1 h−1 | 256 |
TiO2 naturally exists in three polymorphs: anatase, brookite, and rutile. Among them, anatase, rutile, and anatase/rutile mixed phase (Degussa, P25) have been extensively studied for CO2 photoreduction owing to their suitable optoelectronic properties. Brookite is the least commonly reported polymorph in photocatalysis as a result of the difficulty associated with obtaining high-purity brookite nanocrystallites.257 In 2012, Andino and co-workers performed first-principles calculations on cluster and periodic slab systems to investigate the interaction between CO2 and the brookite (210) surface.258 The results indicated that perfect brookite is not a suitable catalyst for CO2 photoreduction, whereas the oxygen-deficient brookite (210) surface displayed improved performance. Compared with the oxygen-deficient anatase (101) surface, the oxygen-deficient brookite (210) surface exhibited stronger interactions with CO2, favoring to form bent CO2 molecules. In the same year, this group also experimentally studied the use of defective brookite for CO2 photoreduction and reported that the surface defects (oxygen vacancies and Ti3+) provided additional active sites for CO2 adsorption and activation, leading to improved performance compared to anatase and rutile.220In situ DRIFTS analysis revealed that the surface oxygen vacancies and Ti3+ promoted the formation of the CO2˙− intermediate and facilitated its rapid reaction with H2O to afford higher CO2 reduction activity to generate CH4.
Controlling the crystal facet is also an effective strategy for enhancing photocatalytic activity. Exposing high-energy surfaces, especially reactive crystal facets, has long been considered to increase photocatalytic activity. For instance, anatase TiO2 is usually dominated by the {101} facet, which is thermodynamically stable. In 2008, a pioneering study by Lu and co-workers reported the synthesis of anatase TiO2 with a high percentage of exposed {001} facets, which possess high surface energy and reactivity.259 More recently, Jaroniec and co-workers investigated the photocatalytic CO2 reduction activity of anatase TiO2 with co-exposed {001} and {101} facets.221 DFT calculations of the electronic structures of the two facets revealed that their Fermi levels were located at distinct positions. Therefore, the {001} and {101} surfaces formed surface heterojunctions, which led to efficient photogenerated charge transfer and separation. Anatase TiO2 specimens with different ratios of exposed {101} and {001} facets were prepared by the addition of HF solution, and the optimal ratio for photocatalytic CO2 reduction was determined to be 45:55.
Defective TiO2 (TiO2−x) with oxygen vacancies and Ti3+ was reported to exhibit enhanced visible-light absorption owing to the induced mid-gap band.103 Although the presence of Ti3+ improves the absorption of visible light by narrowing the bandgap, it also reduces the reduction potential, leading to slow kinetics for the photocatalytic CO2 reduction.25 In 2018, Xing et al. reported that fluorination can improve the reduction potential of TiO2−x crystals by replacing surface oxygen vacancies with doped F atoms, resulting in the formation of a built-in electric field (Fig. 14).188 This finding was supported by DFT calculations indicating an upward shift in the Ti3+ energy level upon fluorination, leading to a methane production rate of 1.63 μmol g−1 h−1 (13 times that for untreated TiO2−x). Recently, Sorcar et al. reported facile low-temperature synthesis techniques for reduced TiO2. Reduced TiO2 showed narrow band gap, well-aligned band position for CO2 reduction reaction, and decreased charge recombination, promoting CO2 photoreduction. However, it showed poor reproducibility in CO2 conversion performance and less accuracy in equation.5,93,95
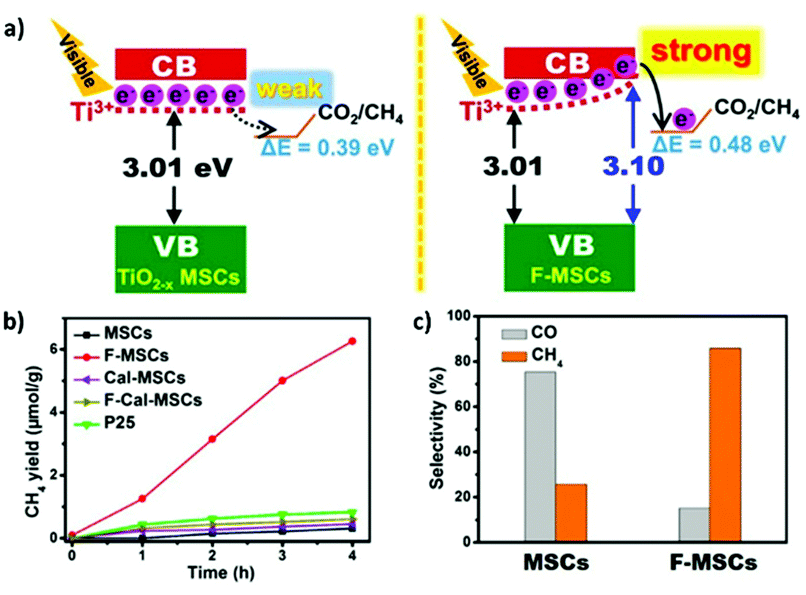 | ||
| Fig. 14 (a) Schematic illustration of the influence of different Ti3+ impurity levels between MSCs (mesoporous single crystal) and F-MSCs (Fluorinated MSCs), where ΔE represents the kinetic overpotential of the reduction process. (b) Time evolution of CH4 production over various samples under solar light irradiation for 4 h (300 W xenon lamp with an AM 1.5 filter, CO2 + H2O). (c) Selectivity of photocatalytic reduction of CO2 and H2O vapor over MSCs and F-MSCs. Reproduced with permission from ref. 188. Copyright 2018, American Chemical Society. | ||
Morphological engineering has also been explored to increase the photocatalytic CO2 reduction efficiency of TiO2. Properly designed structures can help overcome the inherent challenges of photocatalysis such as light absorption. Xu and co-workers found that 1D TiO2 nanofibers displayed remarkable photocatalytic CO2 reduction activity.222 This was ascribed to the 1D morphology enabling increased absorption of reflected and scattered light. Furthermore, the nanofiber structure maximized the number of exposed active sites, ultimately affording high photocatalytic performance. In addition, 2D nanostructured materials have recently attracted substantial attention owing to their remarkable intrinsic properties such as good charge transport and large surface area.260 For example, compared to bulk materials, ultrathin TiO2/g-C3N4 structures provide short electron transfer pathways, leading to superior photocatalytic CO2 reduction activity.
 was greatly decreased by the interfacial contact in the binary photocatalyst, leading to more effective electron transfer during the photocatalytic reaction. Furthermore, Li et al. reported a ZnO/Au/g-C3N4 (3-ZAC) microneedle film displaying local surface plasmon resonance (LSPR) effects.224 The Au NPs added to the interface of ZnO/g-C3N4 acted as an electron transfer bridge and LSPR excited source for the faster separation of electron–hole pairs. Furthermore, Ehsan and He reported the synthesis of a ZnO/ZnTe photocatalyst with a common cation heterostructure through a one-pot hydrothermal approach.225 The ZnO/ZnTe photocatalyst possessed a flower-like nanostructure and displayed the heterojunction characteristics of both p-type ZnTe and n-type ZnO. Through this heterogeneous structure, charge transfer and photocatalytic activity were promoted, allowing the conversion of CO2 into CH4. Meanwhile, Wang et al. studied homogeneous Mo-doped WO3·0.33H2O, which displayed improved photocatalytic activity and selectivity for CO2 reduction to CH4.226 The Mo doping improved the ability of the material to store and localize photogenerated electrons and boosted the transfer of photoexcited electrons, leading to high levels of CH4 production.
was greatly decreased by the interfacial contact in the binary photocatalyst, leading to more effective electron transfer during the photocatalytic reaction. Furthermore, Li et al. reported a ZnO/Au/g-C3N4 (3-ZAC) microneedle film displaying local surface plasmon resonance (LSPR) effects.224 The Au NPs added to the interface of ZnO/g-C3N4 acted as an electron transfer bridge and LSPR excited source for the faster separation of electron–hole pairs. Furthermore, Ehsan and He reported the synthesis of a ZnO/ZnTe photocatalyst with a common cation heterostructure through a one-pot hydrothermal approach.225 The ZnO/ZnTe photocatalyst possessed a flower-like nanostructure and displayed the heterojunction characteristics of both p-type ZnTe and n-type ZnO. Through this heterogeneous structure, charge transfer and photocatalytic activity were promoted, allowing the conversion of CO2 into CH4. Meanwhile, Wang et al. studied homogeneous Mo-doped WO3·0.33H2O, which displayed improved photocatalytic activity and selectivity for CO2 reduction to CH4.226 The Mo doping improved the ability of the material to store and localize photogenerated electrons and boosted the transfer of photoexcited electrons, leading to high levels of CH4 production.
Wang et al. reported a highly crystalline spinel-phase ZnGa2O4 modified with Ag that exhibited high activity and selectivity toward photocatalytic CO evolution.227 The optimized the crystal size and specific surface area of the ZnGa2O4 photocatalyst was synthesized at a calcination temperature of 1123 K for 40 h. Also, by depositing Ag NPs on the ZnGa2O4 sample surface through the chemical reduction method, well-formed metallic Ag NPs with a small size and good dispersion were obtained, thereby improving the selectivity and increasing CO evolution. Similarly, Akatsuka et al. reported the synthesis of a Ga2O3 photocatalyst with coexisting β and γ phases under optimized calcination conditions, which played an important role in the photocatalytic reduction of CO2 and H2O into CO.228 The boundaries between the two phases served as active sites for the CO2 reduction, while defects distributed on the Ga2O3 surface acted as active sites for the water splitting. Furthermore, Yoon et al. studied Pt/Zn-embedded β-Ga2O3 nanorods, which improved the reduction of CO2 into CH3OH owing to the synergistic effect of increased defect sites and high charge transfer.229 In another example, Liu et al. used a binary ethylenediamine (En)/water solvent system to synthesize single-crystalline Zn2GeO4 nanoribbons with lengths of hundreds of micrometers, a thickness of approximately 7 nm (corresponding to five repeating cell units), and aspect ratios (length to width) of up to 10000:1.161 In addition, the photoactivity of Zn2GeO4 was improved in terms of CH4 generation, which was ascribed to the following reasons: (i) a high specific surface area of 22.87 m2 g−1; (ii) improved crystal quality, eliminating the possibility of any grain boundaries and/or other interfaces; (iii) the ultralong longitudinal dimensions of the nanoribbons, which provided sufficiently spacious transport channels for charge separation; and (iv) the ultrathin geometry of the nanoribbons, which allowed charge carriers to move rapidly from the interior to the surface for participation in the photoreduction reaction.
Layered Bi2WO6 is another candidate catalyst and the most studied layered oxide material for photocatalytic applications owing to its suitable bandgap of 2.8 eV, which enables light harvesting in the visible region. Several studies on Bi2WO6 materials in conjunction with other metal NPs, semiconductors, and carbon-based materials have been reported. For example, Kong et al. anchored CQDs on ultrathin Bi2WO6 (UBW) nanosheets using a single-step hydrothermal process.261 This anchoring afforded extended light absorption in the visible-NIR region. In addition, the hybrid photocatalyst displayed several significant advantages for CO2 photoreduction, such as (i) the exposed active facets (001) of UBW improving CO2 adsorption, (ii) CQDs up-converted photoluminescence properties, and (iii) the electron-withdrawing nature of CQDs. Catalytic CO2 conversion was performed in a gas-phase flow reactor system under illumination from a 500 W Xe lamp. The optimized catalyst exhibited photocatalytic CO2 reduction into CH4 with a catalytic activity of 7.19 μmol g−1, which was approximately 9.5 and 3 times greater than those observed for bare Bi2WO6 nanoplatelets and UBW, respectively. In 2019, Kong and co-workers demonstrated a surface-engineered 2D/2D p–n heterojunction catalyst based on Bi2WO6/BiOI (i.e., BWO/BOI).262 Oxygen-vacant BWO (BWO-OV) nanosheets were synthesized by a hydrothermal approach and then self-assembled with BOI. Fig. 15(a) shows the crystal structure of BWO-OV, which displayed intense light absorption in the visible region that extended into the NIR region. Furthermore, the p–n junction heterostructure enhanced the optical absorption over a broad range covering the UV-vis-NIR region (Fig. 15(b)). As a result, the optimized BWO-OV/BOI heterostructure exhibited the highest catalytic activity for CO2 reduction into CH4 (18.32 μmol g−1) compared to other combinations under illumination from a 500 W Xe lamp (Fig. 15(c)). Thus, the coexistence of surface defects and p–n heterojunctions was demonstrated to effectively improve the optoelectronic performance of the catalyst and ultimately the catalytic activity. The mechanism of photocatalytic CO2 reduction to CH4 over BWO-OV/BOI is depicted in Fig. 15(d). The oxygen vacancies over BWO significantly enhanced the optical properties, and the p–n heterojunctions established an internal electric field between BWO and BOI that improved the charge separation and hindered electron–hole recombination.
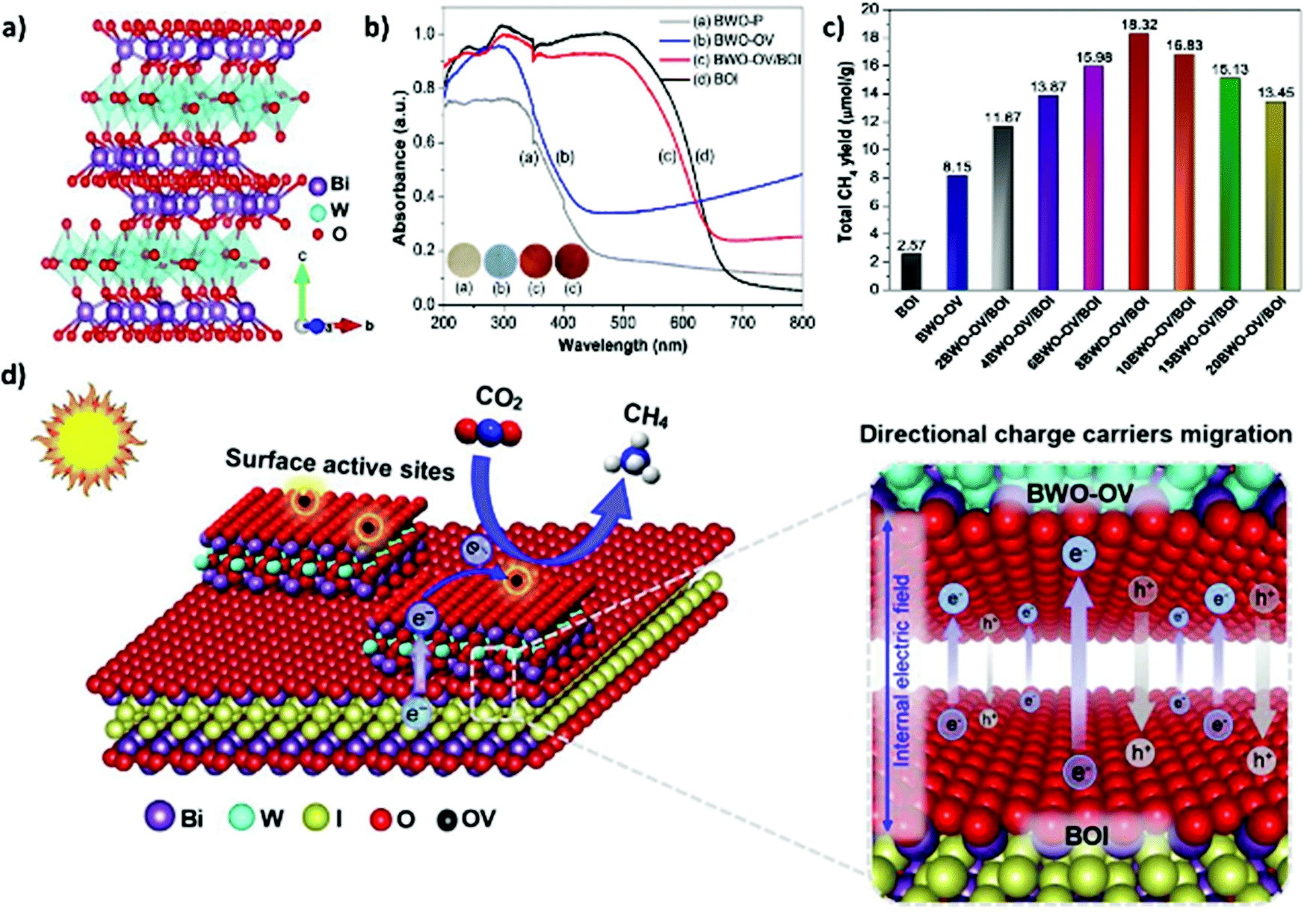 | ||
| Fig. 15 (a) Crystal structure of oxygen-deficient Bi2WO6, (b) UV-vis absorption spectra of various samples, (c) photocatalytic CO2 reduction activity of various catalysts toward CH4 formation, and (d) schematic illustration of the CO2 reduction mechanism on the Bi2WO6/BOI composite. Reproduced with permission from ref. 262, Copyright 2019, Elsevier. | ||
Meanwhile, other researchers have attempted to improve the CO2 conversion efficiency of photocatalysts by using metal oxide materials in conjunction with organic and inorganic compounds. Li et al. studied a novel Z-scheme ternary hierarchical photocatalyst based on ZnFe2O4, In2O3, and reduced graphene oxide (RGO).240 The combination of ZnFe2O4 and In2O3 provided a wide visible-light absorption range and a suitable conduction bandgap position (ca. −1.5 eV), while the addition of RGO promoted charge separation by serving as an electron mediator. The synthesis of ZnFe2O4/RGO/In2O3 hollow tubules was confirmed by both steady-state and time-resolved surface photovoltage spectroscopy, which indicated a prolonged photogenerated charge carrier lifetime and improved charge carrier separation compared to bare ZnFe2O4 and In2O3. The most exciting fact is that neither bare ZnFe2O4 nor ZnFe2O4/In2O3 exhibited ˙OH generation in a series of reaction processes, whereas bare In2O3 and ZnFe2O4/RGO/In2O3 displayed strong signals corresponding to ˙OH as confirmed by EPR analysis. This finding suggests that the photogenerated electrons were transferred from the CB of In2O3 to the VB of ZnFe2O4, resulting in a large quantity of ˙OH groups. These properties allowed the Z-scheme ZnFe2O4/RGO/In2O3 catalyst to exhibit high CO2 conversion. Meanwhile, Yin et al. reported the synthesis of Cu(II)-grafted Nb3O8 nanosheets.232 The small size of the Cu(II) nanoclusters (<3 nm), which were composed of amorphous oxides, promoted the accumulation of excited electrons to drive the efficient multi-electron reduction of oxygen. By grafting the Nb3O8 nanosheets and Cu(II) cocatalysts, which not only increased the specific surface area of the catalyst but also shortened the electron transport distance, CO2 was more effectively converted to CO.
Recently, numerous studies have been conducted on the evolution of C2 (e.g., C2H4) and higher hydrocarbons during the photocatalytic CO2 reduction.263 However, this is associated with many challenges owing to the relatively low efficiency of multi-electron transfer and slow dynamics of C–C coupling. The formation of the key intermediates of *OC–CO and *OC–COH required for the production of C2 and higher hydrocarbons from CO2 over photocatalysts is also very difficult compared to electrocatalytic reactions.264,265 Because photocatalysts produce lower electron densities upon light irradiation.266,267 Wang et al. reported the use of CuOx@p-ZnO to convert CO2 into C2H4.233 The authors synthesized the hybrid material by replacing some of the Zn2+ ions in ZIF-8 with Cu2+ to afford a uniform Cu/Zn atomic ratio as shown in Fig. 16(a)–(f). XAFS analysis revealed the occurrence of surface changes during the photocatalytic reaction, as shown in Fig. 16(g)–(i). During the photocatalytic CO2 reduction, partial reduction of Cu2+ occurred owing to the transfer of electrons from p-ZnO, resulting in the formation of a Cu+ surface layer on the CuOx matrix. CO2 reduction then took place on the CuOx matrix; following the two-electron reduction to CO, a portion of the generated *CO species desorbed to form gaseous CO, while other *CO species remained trapped on the CuOx matrix. This enabled subsequent electron transfer to further reduce the surface-bound *CO into CH4 and C2H4via the intermediates *CHO and *OC–COH, respectively. The results indicated that the hybrid CuOx@p-ZnO catalyst had a lower binding energy than ordinary Cu2O and a high Gibbs free energy (ΔG) for the formation of the *OC–CO intermediate.
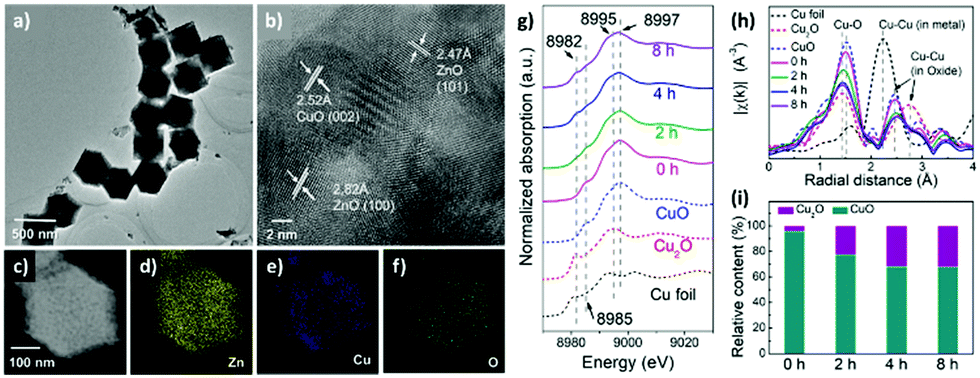 | ||
| Fig. 16 (a) TEM image, (b) high-resolution TEM image, (c) annular dark-field TEM image, and (d)–(f) elemental mapping images of CuOx@p-ZnO. (g) Copper K-edge X-ray absorption near-edge structure (XANES) spectra for pristine Cu foil, Cu2O, CuO, and CuOx@p-ZnO collected after 0, 2, 4, and 8 h of photoreaction. (h) Fourier-transformed k2-weighted (χ(k)) EXAFS spectra and (i) relative contents of CuO and Cu2O on CuOx@p-ZnO after 0, 2, 4, and 8 h of photoreaction. Reproduced with permission from ref. 233, Copyright 2021, American Chemical Society. | ||
5.2 Graphene-based photocatalysts
In recent years, graphene, a valuable carbon-based 2D material composed of a single sheet of sp2-hybridized C atoms arranged in a hexagonal lattice,168 has been the subject of rigorous research owing to its diverse range of potential applications. Graphene exhibits a variety of desirable characteristics, such as electrical conductivity, high surface area (ca. 2600 m2 g−1), and the ability to activate molecules.268–270 The addition of graphene to photocatalysts has proved beneficial for (i) suppressing the recombination of photogenerated electrons and holes, (ii) enhancing CO2 adsorption owing to π–π conjugation between graphene and CO2, (iii) activating CO2 molecules, (iv) improving the corrosion resistance, (v) increasing the surface area, and (vi) enhancing light absorption,167,271–276 all of which are advantageous for photocatalysis.Highly-mobile electrons, which are usually called π electrons, are used to mediate the bonding with other graphene sheets or metals or metal oxides. As a result of this bonding, a strong interaction is developed between the graphene and semiconductor through which the former can readily take away the photogenerated electrons.277–279 The abundant literature on graphene-based photocatalysts indicates that the Fermi level/work function (0 V vs. NHE) of these photocatalysts remains below the CB of many metal oxide-based photocatalysts. Owing to this band alignment, the photogenerated electrons are transferred to the graphene surface, while the holes preferentially remain on the surface of the metal oxide, thereby affording spatial separation of the electrons and holes at the interface. In contrast, only a few studies have considered the role of graphene as a hole conductor.95 Illustrative examples of such charge separation by graphene/TiO2 systems are presented in Fig. 17(a) and (c), in which the presence of graphene remarkably suppressed the charge recombination confirmed by TRPL and PL.
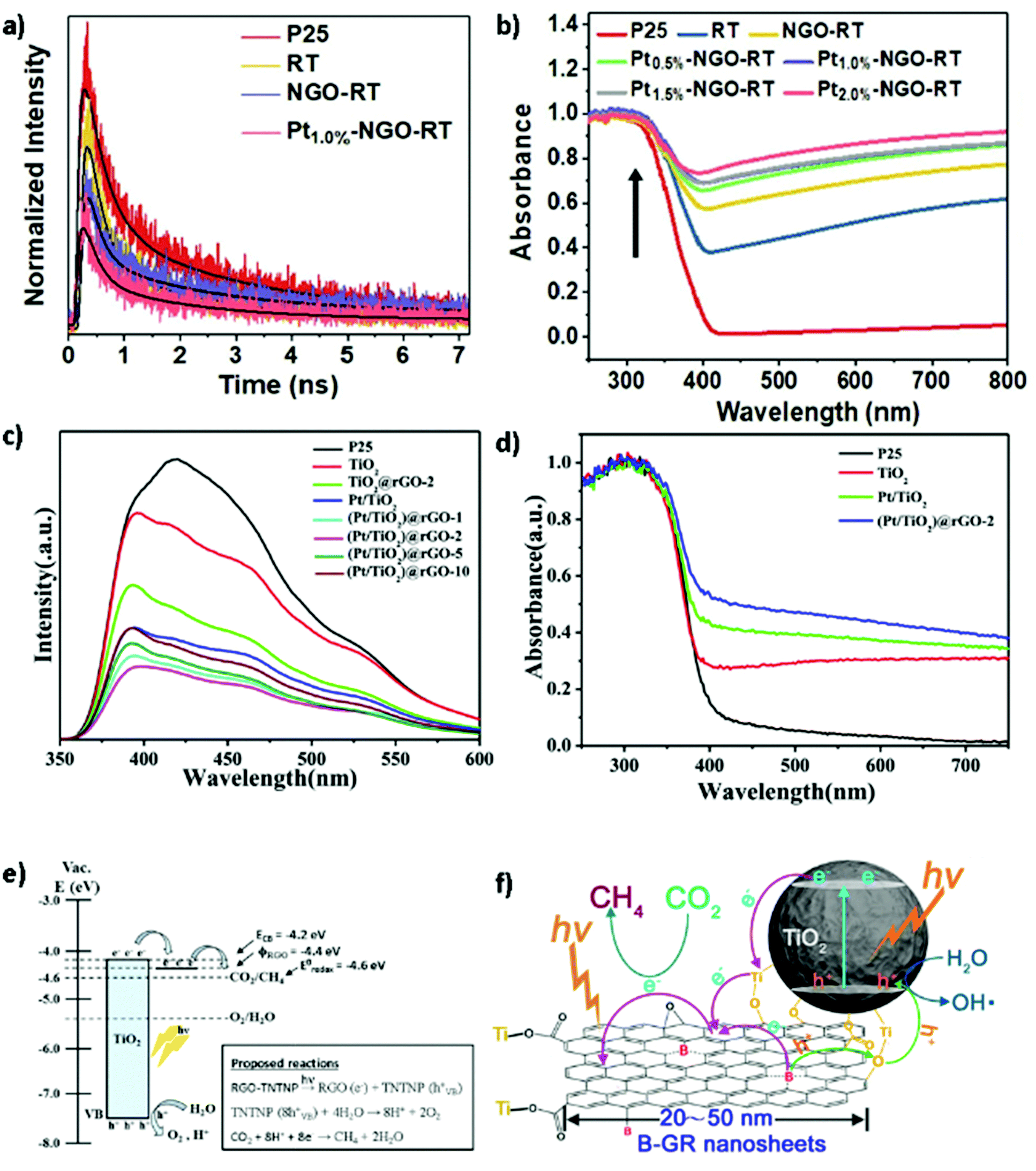 | ||
| Fig. 17 (a) Time-resolved photoluminescence (PL) spectra and (b) UV-vis spectra for graphene added to reduced TiO2 (RT). Reproduced with permission from ref. 96, Copyright 2020, Elsevier. (c) PL spectra and (d) UV-vis spectra for graphene added Pt/TiO2. Reproduced with permission from ref. 280, Copyright 2016, Elsevier. Illustrative examples of (e) a graphene derivative–metal oxide composite with bandgap alignment and proposed proton-assisted multi-electron reactions, reproduced with permission from ref. 281, Copyright 2013, Elsevier, and (f) TiO2/B-GR (boron doped graphene nanosheets) showing the proposed electron–hole transfer reaction, reproduced with permission from ref. 282, Copyright 2013, American Chemical Society. | ||
Graphene absorbs the entire solar spectrum owing to its zero bandgap and black color. However, despite this extended range of light absorption, it is unable to provide the photogenerated charges required for CO2 reduction. To exploit the light harvesting, it is essential to load an appropriate amount of graphene onto the metal oxide semiconductor. Otherwise, it shields the surface of the photocatalyst and thus obstructs the light absorption and other photocatalytic properties as well by curtailing the generation of the photogenerated pairs. This optimal graphene amount engages in electronic interactions with photocatalysts, e.g., TiO2, by which the absorption is enhanced.102,283 Almost all of the studies pertaining to graphene–metal oxide photocatalysts have reported this behavior.284,285 For example, Li and co-workers reported that the addition of graphene enhanced the light absorption of Pt–TiO2, respectively, as shown in Fig. 17(d).280 Similar study was also reported by Hiragond and co-workers for hydrothermally grown graphene over reduced TiO2 (RT), respectively (Fig. 17(b)).96
In addition to the outstanding optoelectronic properties of graphene, its specific surface area is regarded as the maximum among all synthesized materials. Consequently, the use of graphene to prepare photocatalysts can be expected to considerably increase the surface area and therefore the number of exposed reaction sites. These reaction sites of graphene enable π–π interactions with CO2, thus increasing its adsorption. In addition, π–π conjugation can further destabilize and activate CO2, leading to an appreciable enhancement in the CO2 reduction activity.90,286 Under light irradiation, graphene is known to receive electrons from metal oxides, where they reduce the CO2 in the presence of protons, while the leftover holes at the metal oxide oxidize water to generate protons, which is necessary for the CO2 reduction. Fig. 17(e) shows the proposed charge transfer and reaction scheme when graphene is used as a cocatalyst or to form heterostructures. In these studies, various geometries of graphene have been used, including QDs and few layers of 2D sheets. In all of these circumstances, the addition of graphene has proved efficacious for improving the optoelectronic properties of the photocatalysts, where the graphene-based photocatalysts were found to harvest light at longer wavelengths. As a result, abundant photogenerated charges were generated and efficiently utilized for CO2 reduction owing to the additional role of graphene in preventing these charges from undergoing recombination.287
Doped graphene materials, e.g., boron-doped graphene288 or nitrogen-doped graphene,289 have also proved efficacious for CO2 reduction. Boron doping has been found to alter the morphology of the photocatalyst to afford nanoribbons, which facilitates directional charge transfer, while nitrogen doping enhanced the CO2 adsorption. The mechanism of the charge transfer and band alignment for B-GR is depicted in Fig. 17(f).282
Some studies have also reported the use of graphene oxide (GO) for photocatalytic CO2 reduction through bandgap engineering. In this regard, studies by Chen and co-workers described Cu-modified GO, where the Fermi level of the Cu became more negative owing to electron transfer to Cu, while holes accumulated on the graphene surface.290 Therefore, this study completely contradicts the previous reports. Sorcar et al. reported similar results, where the upward band bending of the TiO2 compelled the electrons to remain in the reduced titania while the holes were transferred to GO.95 The performance of graphene-based photocatalysts is summarized in Table 3.
| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor type | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Graphene–Zn0.5Cd0.5S | In situ generated CO2 + H2O (NaHCO3 + HCl) | 300 W Xe arc lamp (400 nm cutoff filter) | H2O | 100 mg sample in reactor (200 mL) | Gas phase | CH3OH: 1.96 μmol g−1 h−1 | 291 |
| Cu2O/graphene/TNA | 300 W Xe arc lamp (400 nm cutoff filter) | Double-chamber reactor (250 mL), two-sided sample (calculated area of 1 cm2), proton exchange reaction for 10 h | Liquid phase | CH3OH: 45 μmol cm−2 h−1 | 186 | ||
| Stability: 6 cycles | |||||||
| AQY = 5.7% | |||||||
| Cu/GO | Moist CO2 | 300 W halogen lamp (100 mW cm−2) | H2O | 100 mg of sample, 300 mL, 2 h | Gas phase | CH3CHO: 3.88 μmol g−1 h−1 | 290 |
| CH3OH: 2.94 μmol g−1 h−1 | |||||||
| G-TNT (titania nanotubes) | 1000 ppm moist CO2 | 100 W Xe arc lamp | H2O | Reactor volume: 15.4 cm3 | Gas-phase batch reactor | CH4: 1.98 ppm cm−2 h−1 | 215 |
| G-Ti0.91O2 hollow spheres | Highly pure moist CO2 | 300 W Xe arc lamp | 10 mg of sample dispersed in 230 mL reactor | Gas-phase batch reactor | CH4: 1.14 μmol g−1 h−1 | 279 | |
| CO: 8.91 μmol g−1 h−1 | |||||||
| RGO-CdS | In situ generated CO2 + H2O (NaHCO3 + HCl) | 300 W Xe arc lamp (λ ≥ 420 nm) | H2O | 100 mg sample in distilled water (10 mL) in glass reactor | Gas phase | CH4: 2.51 g−1 h−1 | 275 |
| AQY = 0.8% | |||||||
| Graphene–TiO2 | Moist pure CO2 | 300 W Xe arc lamp | H2O | 0.1 g sample in reactor (230 mL) | CH4: 8 μmol g−1 h−1 | 102 | |
| C2H6: 16.8 μmol g−1 h−1 | |||||||
| (Pt/TiO2)/rGO | Moist pure CO2 | 300 W Xe arc lamp (320–780 nm) | H2O | 8 h | Continuous-flow gas-phase reactor | CH4: 41.3 μmol g−1 h−1 | 280 |
| CO: 0.4 μmol g−1 h−1 | |||||||
| H2: 5.6 μmol g−1 h−1 | |||||||
| AQY = 1.93% | |||||||
| Pt-G/RBT | 1000 ppm moist CO2 | 100 W Xe arc lamp | H2O | 40 mg of photocatalyst dispersed over ceramic disc, illuminated for 7 h | Continuous-flow gas-phase reactor | CH4: 259 μmol g−1 | 95 |
| C2H6: 77 μmol g−1 | |||||||
| AQY = 7.9% | |||||||
| SEG-P25 nanocomposites | Moist pure CO2 | 100 W Hg vapor lamp (λ = 365 nm) and 60 W daylight bulb | H2O | Teflon reactor (25 mL) | Gas phase | 4.5 times more CH4 than for P25 under UV | 165 |
| 7.2 times more CH4 than for P25 under visible | |||||||
| GR/TiO2 (graphene-supported TiO2 nanocrystals) | Moist pure CO2 (99.999%) | 300 W Xe arc lamp (300 < λ < 400 nm) | H2O | 10 mg photocatalyst in reactor (85 mL) | CH4: 27.8 μmol g−1 h−1 | 272 | |
| CO: 70.8 μmol g−1 h−1 | |||||||
| AQY = 0.0847% | |||||||
| GO–OTiO2 binary composite | Moist pure CO2 (99.999%) | 15 W daylight bulb | H2O | Continuous-flow gas-phase reactor | CH4: 1.718 μmol g−1 h−1 | 292 | |
| GR/g-C3N4 | Highly pure moist CO2 | 15 W daylight bulb | H2O | Under ambient temperature and pressure for 10 h | Continuous-flow gas-phase reactor | CH4: 5.87 μmol g−1 | 293 |
| Pt-NGO-RT | 1000 ppm moist CO2 | 100 W Xe arc lamp | H2O | 40 mg photocatalyst dispersed over ceramic disc, illuminated for 7 h | Gas-phase flow reactor | CH4: 252.0 nmol g−1 | 96 |
| Stability: 5 cycles | |||||||
| Cs4PbBr6/rGO | Ethyl acetate (solvent) | 300 W Xe arc lamp (λ > 420 nm) | H2O | 5 mg sample dispersed in 5 mL of ethyl acetate and 5 μL of water in sealed Pyrex bottle (35 mL), 10 h reaction time | Liquid phase | CO: 11.4 μmol g−1 h−1 | 294 |
| Stability: 6 cycles | |||||||
| CsPbBr3-USGO-α-Fe2O3 (ultrathin and small-size graphene oxide) | Pure CO2 | 300 W Xe arc lamp (400 nm filter) | H2O | 4 mg as-prepared nanomaterial in acetonitrile/deionized water (200: 1 (v/v), 5 mL) in sealed Pyrex bottle (12 mL), 4 h | Liquid phase | CO: 14.6 μmol g−1 h−1 | 295 |
| ZnO/graphene | In situ generated CO2 + H2O (NaHCO3 + H2SO4) | 300 W Xe arc lamp | H2O | 50 mg photocatalyst, 200 mL | Gas-phase batch reactor | CO: 3.38 μmol g−1 h−1 | 296 |
| CH4: 0.59 μmol g−1 h−1 | |||||||
| CH3OH: 0.09 μmol g−1 h−1 | |||||||
| Stability: 5 cycles | |||||||
| Ni@GC | High-purity CO2 (99.999%) | 300 W Xe arc lamp (420 nm long-pass cutoff filter) | Triethanolamine (TEOA) | 95 wt% of the catalyst and 3 mg [Ru(bpy)3]Cl2·6H2O (10 μmol) in 8 mL of acetonitrile, 2 mL of H2O, and 2 mL of triethanolamine, 20 °C, 7 h | Liquid phase | CO: 27.0 μmol g−1 | 297 |
| H2: 9.0 μmol g−1 | |||||||
| Stability: 5 cycles | |||||||
| AgCuInS2–graphene–TiO2 | Pure CO2 gas (filling for 30 min) | 500 W metal halide lamp | Na2SO3 (hole scavenger) | 100 mg photocatalyst dissolved in 50 mL of 0.04 M NaHCO3, under UV light | Liquid-phase batch reactor | CH3OH: 15.21% of reaction mixture | 298 |
| Stability: 4 cycles | |||||||
| AQY = 1.175% | |||||||
| LaYAgO4–Graphene–TiO2 | Carbonated water | 500 W metal halide lamp | 150 mg in 50 mL carbonated water, 48 h | CH3OH: 1945.9 mmol g−1 h−1 (12.27% of reaction mixture) | 299 | ||
| rGO–CuO | CO2 saturated with water/DMF | 20 W white LED bulb (85 W m−2) | H2O/DMF | 24 h | Liquid phase | CH3OH: 1228.0 μmol g−1 h−1 | 300 |
| Stability: 6 cycles | |||||||
| RGO–TiO2 NPs | 1000 ppm moist CO2 | 100 W Xe arc lamp | H2O | 2.0 × 2.0 cm2 photocatalyst film in reactor (15.4 mL) | Gas phase | CH4: 5.67 ppm cm−2 h−1 | 281 |
| N-Doped graphene–CdS | Moist pure CO2 | 350 W Xe arc lamp (420 nm cutoff filter) | 50 mg photocatalyst, 3 h | Gas phase | CO: 2.59 μmol g−1 h−1 | 301 | |
| CH4: 0.33 μmol g−1 h−1 | |||||||
| Stability: 4 cycles |
The combination of graphene with other photocatalytic materials has also been reported to enhance the C2 selectivity. For example, Chen and co-workers reported the formation of CH3CHO over Cu NPs (4–5 nm) anchored on GO, which they ascribed to the effective charge separation at the Cu–GO interface in which the electrons accumulated at Cu while the holes tended to remain on the graphene.290 Sorcar et al. reported a similar type of hole accumulation for their graphene-wrapped blue titania (BT), and they also reported the formation of a C2 product (C2H6).95 They proposed that synergistic effects involving the graphene and electron-enriched Ti3+ states of BT generated ˙CH3 radicals, which underwent “radical substrate reactions” to form C2H6. Zou and co-workers also reported C2H6 formation owing to these synergistic effects for their graphene–TiO2 hybrid material.102
The literature suggests that graphene wrapping is also conducive for improving photocatalyst stability. One possible reason for this could be the oxidation of water on the graphene surface, which avoids the contact of the photocatalyst with both holes and water. Sorcar et al. reported such example, where they used graphene-wrapped reduced BT (G/RBT) deposited with Pt and found that holes were transferred to graphene upon light irradiation.95 During photocatalytic CO2 reduction tests, this material remained stable for six cycles (42 h in total). Another study by Tang and co-workers also reported the efficacy of graphene wrapping for sustaining the photocatalytic performance of Cu2O.178 The authors noted that graphene addition allowed the photocatalysts to exhibit continuous and enhanced CO production for almost 20 h, which was longer than that observed for pristine Cu2O and air-oxidized Cu2O (Fig. 18(a)). One possible reason for this could be the prevention of oxidation of the Cu2O. A similar study by Cao and co-workers reported the role of graphene in mitigating Cu oxidation.186 Upon comparison of the pristine Cu2O/TNA with post-reaction (10 h) samples of Cu2O/TNA and Cu2O/graphene/TNA, the authors observed the oxidation of Cu for the graphene-free samples. Owing to this beneficial effect of the graphene, Cu2O/graphene/TNA exhibited stable performance over 10 consecutive cycles with a performance loss of only 18%, as shown in Fig. 18(b).
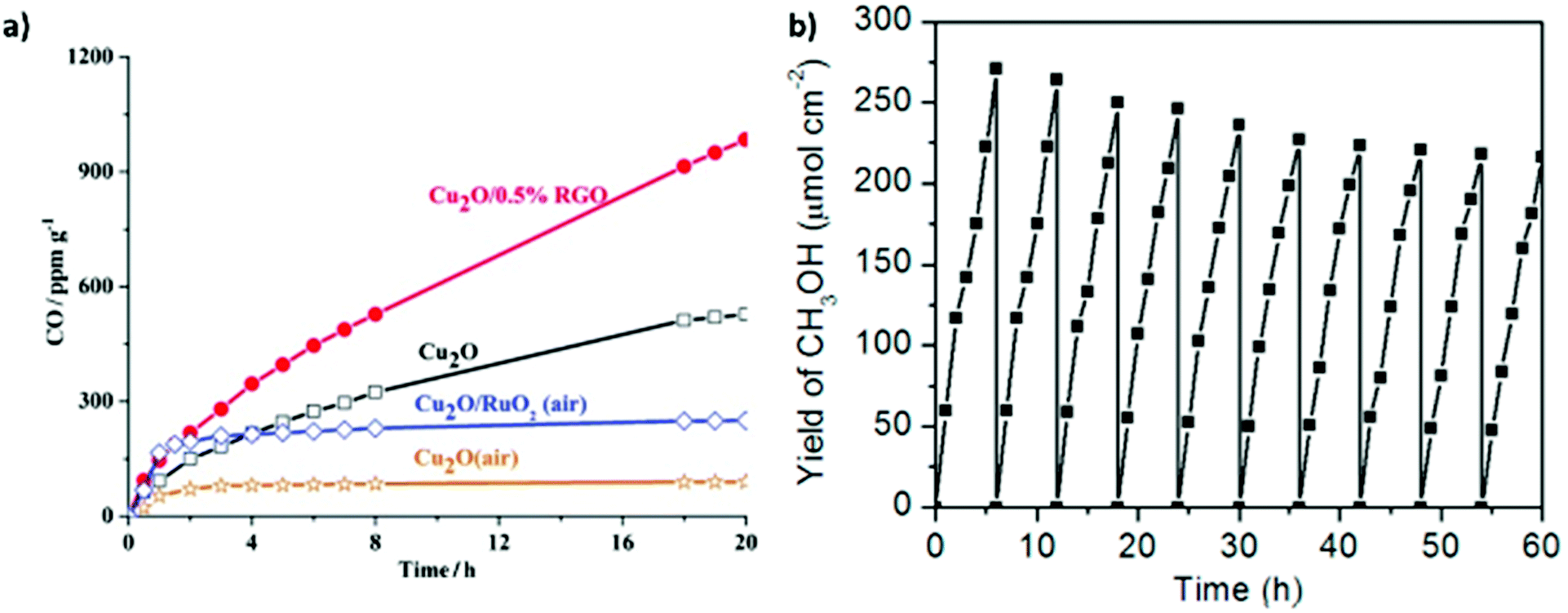 | ||
| Fig. 18 Stability exhibited by (a) Cu2O upon the addition of graphene. Reproduced with permission from ref. 178, Copyright 2014, NCBI and (b) stability exhibited by Cu2O/TNA upon the addition of graphene. Reproduced with permission from ref. 186, Copyright 2016, Elsevier. | ||
5.3 Metal–organic frameworks
Metal–organic frameworks (MOFs) are micro/mesoporous hybrid crystalline materials in which organic linker ligands coordinate and interconnect metal ions or metal cluster nodes. As a result of this coordination network, MOFs possess a porous structure with high pore volume and large surface area. These materials can display physisorption due to van der Waals forces. Thus, MOFs can serve as good absorbents for capturing chemicals such as CO2. To date, MOFs have been applied in numerous research fields, such as gas purification,302 hydrogen storage,303 carbon capture,304 electrocatalysis,305 photocatalysis,306 semiconductors,307 and drug delivery.308Both the organic linker ligands and the metal ions or cluster nodes of MOFs can be tailored for photocatalytic applications. For example, Zecchina and co-workers reported that the metal ions or metal cluster nodes can act as semiconductor QDs, while the organic ligand linkers can play the role of antennas for sensitizing the QDs.309 In terms of functionalizing MOFs, post-synthesis modification (PSM) is the general method for anchoring catalytic sites to MOFs to increase their photocatalytic activity. MOFs can be modified with proper combination of nodes and organic linking groups by anchoring photocatalytically active species. This affords a single photocatalytic reaction site, in contrast to other PSM methods that lead to heterogeneously scattered sites. Catalytically active species supported on MOFs prepared via PSM methods have been designed to meet a variety of catalytic applications. However, MOFs may lose their catalytic selectivity or performance if the anchored catalytic sites on the ligands induce unexpected forms by interactions between the metal complexes and a solid surface.
To solve this problem, Dengrong et al. used MOF-253–Ru(CO)2Cl2, which is constructed by MOF-253 supported Ru carbonyl complex.310 MOF-253 was adopted as a platform for constructing a photocatalytic system that displayed improved charge transfer. MOF-253–Ru(CO)2Cl2 exhibited a photocatalytic CO2 conversion rate of 8.23 μmol g−1 h−1 for HCOO− under visible-light irradiation for 8 h. Ru(bpy)2Cl2 was used to photosensitize MOF-253–Ru(CO)2Cl2 in the visible region through N,N-chelation of the Ru(bpy)2Cl2 by the MOF-253 surface sites to afford immobilized [Ru(bpy)2(X2bpy)]2+ on the MOF surface. Li et al. synthesized a Cu3(BTC)2@TiO2 hybrid photocatalyst possessing the unique structure shown in Fig. 19(a), consisting of a Cu3(BTC)2 octahedral microcrystal core and TiO2 shell. The TiO2 semiconductor shell underwent facile photoexcitation to produce excitons. In addition, the TiO2 shell and Cu3(BTC)2 core afforded a microporous structure that favored the capture of gas molecules in the catalyst core and provided photocatalytic reaction sites. The photoexcited electrons produced on the TiO2 shell were transferred to the interface state of Cu3(BTC)2@TiO2 (Fig. 19(b)), whereupon they activated CO2 on the Cu sites of Cu3(BTC)2. Cu3(BTC)2@TiO2 showed a CH4 production of 2.64 μmol g−1h−1 (Fig. 19(c)). In other words, the core–shell structure of this MOF-based semiconductor was well constructed to produce excitons and provide a microporous core for gas molecule capture.311
 | ||
| Fig. 19 (a) Core–shell structure, (b) charge transfer mechanism, and (c) production rates of CH4 and H2 from CO2 for Cu3(BTC)2@TiO2. Reproduced with permission from ref. 311, Copyright 2014, Wiley-VCH. (d) Synthetic procedure and CO2 reduction process for CsPbBr3/ZIFs. Reproduced with permission from ref. 312, Copyright 2018, American Chemical Society. (e) Schematic illustration of photocatalytic CO2 reduction by PCN-222. Reproduced with permission from ref. 313, Copyright, 2015 American Chemical Society. (f) Synthetic route to PCN-250-Fe3 and PCN-250-Fe2M (M = Mn, Zn, Ni, Co) by the reaction of Fe3 (or Fe2M) clusters and 3,3′,5,5′-azobenzene tetra-carboxylic acid (H4abtc) ligand, and (g) comparison of photocatalytic reaction results for PCN-250-Fe3 and PCN-250-Fe2M in terms of CO production. Reproduced with permission from ref. 314, Copyright 2020, Elsevier. | ||
In another study, Kong and co-workers designed a core–shell halide perovskite@MOF composite with enhanced CO2 reduction activity.312 Coating of the ZIF shell onto the surface of CsPbBr3 was achieved in situ by dispersing CsPbBr3 QDs in a mixture of the metal precursor and imidazole ligand (Fig. 19(d)). This coating and the increased charge separation efficiency were key factors underlying the observed CH4 selectivity. where the improved moisture stability of the CsPbBr3 QDs, CO2 capture ability, and charge separation efficiency contributed to the enhanced photoconversion efficiency of CO2 into CO and CH4. Two different ZIFs were prepared: (i) ZIF-8 using a Zn precursor and (ii) ZIF-67 using a Co precursor. CsPbBr3 was then coated onto the ZIFs, as demonstrated by TEM images, STEM images, and elemental mapping data. The Co-based ZIFs acted as a cocatalyst for CsPbBr3 to improve the optical properties of the hybrid sample CsPbBr3/ZIF-67 compared to pristine CsPbBr3 and the Zn-based composite, resulting in high catalytic activity for CO2 reduction into CO and CH4 with an electron consumption rate of 29.630 μmol g−1 h−1. The catalyst also displayed high stability over six cycles.
Xu et al. synthesized a photocatalytically activated porphyrin-based semiconducting PCN-222 system (Fig. 19(e)) that exhibited superior efficiency to the ligand alone.313 The high CO2 capture ability of the MOF in acetonitrile effectively increased the photocatalytic efficiency. Furthermore, the results of PL spectroscopy and ultrafast transient absorption spectroscopy revealed that highly stable electron trap states of PCN-222 suppressed electron–hole recombination, thus improving the photocatalytic CO2 conversion efficiency.
Dong et al. reported the bimetallic MOF PCN-250-Fe2M (M = Mn, Zn, Ni, Co), which displayed improved photocatalytic activity and selectivity for reducing CO2 into CO compared to the monometallic analogue PCN-250-Fe3.314 PCN-250-Fe3 and PCN-250-Fe2M were synthesized by a solvothermal method (Fig. 19(f)), in which the FeII metal ions of the Fe2IIIFeII metal cluster of PCN-250-Fe3 could be replaced with other MII species (M = Mn, Zn, Ni, Co). In the conversion of CO2 to CO, formation of the carboxyl intermediate (*COOH) is the rate-limiting step for CO2 reduction. DFT calculations indicated that doping with the second metal enhanced the adsorption of CO2 molecules and restrained the hydrogen evolution reaction (HER). Therefore, the photocatalytic efficiency and selectivity for the conversion of CO2 to CO improved for all of the bimetallic PCN-250–Fe2M derivatives compared to PCN-250–Fe3. In particular, the Mn-containing bimetallic catalyst PCN-250–Fe2Mn displayed the highest photocatalytic CO formation rate of 21.51 mmol h−1 g−1 under visible-light irradiation, as shown in Fig. 19(g).
Various approaches have been explored for increasing the photocatalytic efficiency of MOF-based photocatalysts, such as the use of semiconductors and perovskites, regulating the combination of metal ion or complex, and the introduction of trap sites. It is also possible to enhance the efficiency by tuning the light absorption sites through bandgap engineering. Meanwhile, the selectivity can be improved by modifying the CO2 adsorption sites. However, long-term stability is still a key limitation of MOF-based photocatalysts. The photocatalytic CO2 reduction performance of MOF-based photocatalysts is summarized in Table 4.
| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor type | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Sensitized MOF-253–Ru(CO)2Cl2 | Moist CO2 + MeCN/TEOA (10:1, 6 mL) |
Xe lamp (λ = 420–800 nm) | TEOA | 5 mg photocatalyst | Gas phase | HCOO−: 8.23 μmol (8 h) | 310 |
| Cu3(BTC)2@TiO2 | CO2 + H2O (5 mL) | UV irradiation (λ > 400 nm) | H2O | 300 mg photocatalyst in batch reactor (100 mL) | Batch reactor (100 mL) | CH4: 2.64 μmol gTiO2−1 h−1 | 311 |
| CsPbBr3@ZIF-67 | CO2 + H2O (10 μL) | 100 W Xe lamp (150 mW cm−2) | H2O | 4.5 mg photocatalyst in sealed Pyrex reactor (40 mL) | Pyrex reactor (40 mL) | Electron consumption rate: 29.630 μmol g−1 h−1 (3 h) | 312 |
| PCN-222 | MeCN/TEOA (10:1 (v/v), 60 mL) degassed with CO2 |
300 W Xe lamp (λ = 420–800 nm) | TEOA | 50 mg photocatalyst | Gas phase | HCOO−: 3 μmol h−1 | 315 |
| PCN-250-Fe2Mn | CO2 (1 atm) + MeCN/H2O (15:1) with TIPA |
300 W Xe lamp (λ > 420 nm) | H2O | 5 mg photocatalyst | Pyrex reactor (100 mL) | CO: 21.51 mmol g−1 h−1 | 314 |
| AQY = 2.60% |
5.4 Transition-metal dichalcogenides
Two-dimensional materials are emerging nanomaterials that possess interesting electrical and optical properties. These materials may consist of a single layer or several layers but are typically less than 5 nm in thickness. In contrast, the lateral size may be several hundred nanometers. Transition-metal dichalcogenides (TMDCs) are superior 2D materials such as MoS2, WS2, SnS2, MoSe2, and WSe2.316 TMDCs have attracted substantial attention owing to their low cost and excellent catalytic activity, which is comparable to that of noble metals. The TMDC nanostructure contains active sites on both edges and basal planes that provide opportunities for electrocatalysis and photocatalysis.317,318 Optimization of the semiconductor and metallic phases can alter the chemical kinetics, electrical transport, and intrinsic catalytically active sites. The fundamental structural features of TMDCs and strategies for enhancing their catalytic activity were recently reviewed.319 TMDCs can be represented by the general formula MX2, where M is a transition-metal atom belonging to groups IVB–VIIB of the periodic table and X is a chalcogen atom such as S, Se, or Te. The typical structure can be denoted X–M–X, in which a central layer of metal atoms is sandwiched between two layers of chalcogen atoms.320 The monolayer of TMDCs can be stacked owing to van der Waals forces of attraction between each layer. The unit cell structure may possess trigonal prismatic or octahedral geometries. The trigonal prismatic geometry may exist as 2H or 3R polymorphs, which attributes the same metal element but atomic configuration difference in structure. The 2H and 3R phases exhibit hexagonal and rhombohedral symmetry, respectively. In addition, the metallic 1T phase displays tetragonal symmetry with octahedral coordination of the metal atom. The electronic structures of these phases vary depending on the filling of the d orbitals, which alters the energy band structure to afford semiconductor or metallic characteristics.321Overall, these aspects determine the type of structure that can be obtained for a specific application. TMDCs have been extensively used in electrodes for electrochemical and photoelectrochemical CO2 reduction owing to their high electronic conductivity and redox surface area.315,322 In contrast, there have been few reports describing their use as photocatalysts for CO2 reduction. The most important considerations in regard to photocatalysis are exciton generation and charge separation; however, the intrinsic mobility of electrons and holes can be limited in TMDCs.323 Cheng and Liu studied the electron and hole mobility in these materials using density functional perturbation theory and Wannier interpolation of the electron–phonon matrix.324 This study revealed that two types of scattering processes (i.e., longitudinal optical and longitudinal acoustic phonon scattering) limit the charge mobility and are not dependent upon the effective mass of the atoms. However, the charge mobility was found to be influenced by the electrical polarization changes induced by atomic vibration. Furthermore, the two types of scattering processes impede the charge carriers. This investigation indicated that MoS2 and WS2 exhibit higher charge carrier mobility with respect to other chalcogens because they exclusively display acoustic phonon scattering, whereas other chalcogenides exhibit both types of scattering. Therefore, most studies have been performed using MoS2 and WS2. Meier et al. investigated tuning of the bandgap for MoS2 nanoflowers to achieve CO2 conversion to CO.325 The MoS2 nanoflowers were synthesized using single- and three-zone furnaces (SZF and TZF, respectively) with different temperature ramping rates, which led to changes in the bandgap. A high ramping rate influenced the development of the flake surface and edges containing the coordinated atoms, and edges rich in Mo displayed high catalytic activity. However, further increasing the ramping rate resulted in the formation of MoS2 nanosheets, which exhibited reduced photocatalytic activity. Among the various synthetic approaches, the optimized SZF procedure afforded ca. 0.21 μmol gcat−1 h−1 of CO formation, as illustrated in Fig. 20(a). Furthermore, to enhance the CO evolution, MoS2 was treated with H2, which reduced the oxidation state for the Mo, resulting in the highest rate of CO production at 100 °C, as shown in Fig. 20(b).
 | ||
| Fig. 20 (a) CO2-to-CO photoreduction performance of MoS2 nanoflowers synthesized using the SZF and TZF approaches and (b) influence of H2 post-treatment on the CO2 reduction activity. Reproduced with permission from ref. 325, Copyright 2018, American Chemical Society. (c) Photocatalytic CO2 reduction by a TiO2/MoS2 heterostructure. Reproduced with permission from ref. 326, Copyright 2018, Wiley-VCH. (d) Charge separation and transport phenomena in TiO2/MoS2/graphene. Reproduced with permission from ref. 327, Copyright 2018, American Chemical Society. (e) Influence of the S/Se ratio on C1 and C2 product selectivity during CO2 photoreduction. Reproduced with permission from ref. 328, Copyright 2020, American Chemical Society. (f) Photocatalytic performance of SnO2/Ag/MoS2 for CO2 reduction to generate CO and CH4. Reproduced with permission from ref. 329, Copyright 2020, Elsevier. (g) Reaction pathway for CH4 formation on the surface of SiC@MoS2. Reproduced with permission from ref. 330, Copyright 2019, MDPI. | ||
Despite the above efforts, the CO production has remained limited, and MoS2 is typically used as a cocatalyst or composite component to enhance charge transfer. Hence, Tu et al. reported the in situ growth of MoS2 nanosheets on TiO2, resulting in close contact between the two components to improve the interfacial area.331 This close contact afforded nanojunctions that reduced electron–hole recombination. Furthermore, the Mo edges exhibited metallic characteristics with a high d-electron density and stabilized the intermediates through electrostatic attraction to enhance the yield of CH3OH during CO2 reduction. Different morphologies of TiO2 can also be exploited to further improve the charge separation and transport. In this regard, Xu et al. developed the 1D TiO2 and MoS2 heterostructure illustrated in Fig. 20(c).326 They optimized the ratio of TiO2 and MoS2 for photocatalytic CO2 reduction to form CH4 and CH3OH. The pure TiO2 nanofibers exhibited CH3OH production rate of 0.72 μmol g−1 h−1. But, after loading with MoS2, the production of CH4 and CH3OH was enhanced by 2.86 and 2.55 μmol g−1 h−1, respectively. The origin of the improved CO2 photoreduction may be the higher Fermi level of MoS2 with respect to TiO2, enabling the transfer of excited electrons from TiO2 to the MoS2 sheets and then to adsorbed CO2 molecules under UV–vis irradiation.
Nevertheless, the expected photocatalytic performance has not yet been realized. Jung et al. reported the synthesis of a hierarchical structure composed of mesoporous TiO2 on graphene with a few layers of MoS2.327 The mesoporous TiO2 facilitated the adsorption of CO2 and also decreased the electron diffusion length. TiO2 and MoS2 comprised the heterostructure in this hierarchical structure, while graphene assisted in the separation of photoinduced electron–hole pairs. Few-layered MoS2 was used because it has active edges that reduce the electron transfer path and have larger amount of the unsaturated S atoms at the edges, thereby improving the CO2 reduction performance. Moreover, few-layered MoS2 exhibits quantum confinement, leading to the more negative CB shown in Fig. 20(d). Increasing the number of MoS2 layers to more than 5 or 6 resulted in an unsuitable CB alignment for CO2 reduction. Upon applying this structure to CO2 photoreduction, the authors observed CO as the main product and subsequently compared the performance with the non-mesoporous and non-macroporous structures of TiO2 with MoS2 as a heterostructure. The resulting CO yields were 92.33 μmol g−1 h−1 for the mesoporous TiO2/MoS2/graphene structure and 70.09 and 27.09 μmol g−1 h−1 for the non-mesoporous and non-macroporous structures of TiO2, respectively. This study demonstrates the roles of synergistic effects, surface area, and a porous support heterostructure.327 Furthermore, the above discussion shows that CO2 fixation and CB alignment are crucial aspects of CO2 photoreduction.
Long et al. studied the influence of the S/Se ratio in a MoSxSey/TiO2 heterostructure, and found that this parameter changed the band edges and C1 and C2 product selectivity.328 The CO2 photoreduction performance was examined for different S/Se ratios, and photocatalyst formed the CH3CH2OH as a C2 product. Adjusting the S/Se ratio to 2:3 increased the formation of formic acid as a C1 product. The influence of the S/Se ratio on the product selectivity is shown in Fig. 20(e). The metallic character was governed by the S/Se ratio, which thus determined the MoSxSey bandgap. The increase in the CBM of MoSxSey with increasing Se content mirrored the decrease in electronegativity from S to Se, which also led to a diminishing electric potential. Then again, consistently specific photocatalytic CO2 fixation requires an excellently tunable energetic site of the photocatalyst. The S/Se ratio-dependent CBM of MoSxSey enables the selective production in photocatalytic CO2 fixation.
The applications of TMDCs in CO2 reduction are not limited to TiO2 but have also been extended to other metal oxides such as SnO2. Bilawal et al. studied the SnO2/Ag/MoS2 heterostructure to enhance the charge separation and CO2 reduction performance.329 Here, they used MoS2 nanoflowers owing to their high surface area and appropriate conduction band for SnO2, and added Ag NPs to obtain a suitable work function. The resulting heterostructure facilitated the photon-to-electron charge transfer through the cascade band alignment. Fig. 20(f) shows the observed yields of CO and CH4, which varied upon changing the ratio of SnO2 to MoS2/Ag.
The p–n junction of p-MoS2/n-Bi2S3 nanorods has also been studied for CO2 reduction. The treatment of MoS2 at high temperature affords sulfur vacancies that lead to high CO2 adsorption. Kim et al. exploited these sulfur vacancies in MoS2 to obtain a p–n junction catalyst with improved optical properties by heterostructure formation with Bi2S3.330 They observed CO and CH4 yields of 40 and 42.5 μmol g−1, respectively, after 10 h of light illumination. In addition, Wang et al. reported a unique marigold-like SiC@MoS2 nanoflower structure for photocatalytic CO2 reduction.332 In this case, the MoS2 was responsible for H2O oxidation owing to its high hole mobility, while the high electron mobility of SiC contributed to the CO2 reduction. The authors measured the photocatalytic activity under visible light and observed a CH4 production rate of 323 μL g−1 h−1 and O2 evolution rate of 620 μL g−1 h−1. As shown in Fig. 20(g), two paths were available for CO2 reduction, namely, hydrogenation (CO2 → HCOOH → HCHO → CH3OH → CH4) and deoxygenation (CO2 → CO → C˙ → ˙CH3 → CH3OH/CH4). The stability of the SiC@MoS2 photocatalyst was dependent upon the stability of the MoS2 nanosheets. Dai et al. studied a MoS2/Bi2WO6 composite photocatalytic material for CO2 photoreduction.333 The hierarchical flower-like structure displayed significant activity in the visible spectrum, and the MoS2 acted as a cocatalyst. Thus, after 4 h of light illumination, methanol and ethanol were produced as the primary products with yields of 36.7 and 36.6 μmol gcat−1, respectively.
Considerable progress is still needed to realize satisfactory performance from TMDCs, and the stability of exfoliated 2D nanosheets in particular remains a massive challenge. After all, the bulk forms possess indirect bandgaps, and chalcogens based on transition metals do not usually exhibit high charge carrier mobility. Successfully overcoming these challenges may allow 2D TMDCs to find practical applications in photocatalytic CO2 reduction. The photocatalytic activities of TMDCs are summarized in Table 5.
| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor type | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| MoS2 nanoflowers | CO2 + H2O | 1600 W Xe arc lamp | H2O | Reaction temperature maintained at 16 °C | — | CO: 0.22 μmol g−1 h−1 | 325 |
| CH4: 0.17 μmol g−1 h−1 | |||||||
| MoS2–TiO2 | CO2 + 1 M NaHCO3 | 350 W Xe lamp | — | Catalyst treated at 300 °C under Ar for 2 h | — | CH3OH: 10.6 μmol g−1 h−1 | 331 |
| 1D/2D TiO2/MoS2 | CO2 + H2O | 350 W Xe lamp | H2O | 50 mg sample in 10 mL H2O | — | CH3OH: 2.55 μmol g−1 h−1 | 326 |
| CH4: 2.86 μmol g−1 h−1 | |||||||
| Mesoporous TiO2/few-layered MoS2/graphene | CO2 + H2O | 350 W Xe lamp | H2O | Reaction mixture kept for several hours at 40 °C | — | CO: 92.33 μmol g−1 h−1 | 327 |
| TiO2–MoSxSey | CO2 + NaHCO3 | 350 W Xe lamp | — | 0.2 MPa CO2 and 0.1 M NaHCO3 (H2 source), room temperature | — | CH3CH2OH: 704.38 μmol g−1 h−1 | 328 |
| SnO2/Ag/MoS2 | CO2 + H2O | 350 W Xe lamp | H2O | 0.1 g catalyst dispersed in 6 mL H2O | Flow reactor | CO: 9 μmol g−1 h−1 | 329 |
| CH4: 20 μmol g−1 h−1 | |||||||
| Bi2S3/MoS2 | 99.99% CO2 + H2O | Xe lamp (150 mW cm−2) | H2O | Reaction temperature 60 °C | Batch reactor | CO: 40 μmol g−1 (10 h) | 330 |
| CH4: 42.5 μmol g−1 (10 h) | |||||||
| 3D-SiC@2D-MoS2 | CO2 + H2O | 300 W Xe lamp | H2O | 40 mL reaction mixture at 298 K | Schlenk flask reactor | CH4: 323 μL g−1 h−1 | 332 |
| MoS2/Bi2WO6 | 99.99% CO2 + H2O | 350 W Xe lamp | H2O | 50 mg catalyst in 50 mL DI water | — | CH3OH: 36.7 μmol g−1 h−1 | 333 |
| C2H5OH: 36.6 μmol g−1 h−1 | |||||||
| MoS2 nanosheets | 99.99% CO2 + 0.5 M NaHCO3, 0.5 M NaOH, or 0.5 M NaCl | UV light | H2O | 0.1g MoS2 in 50 mL DI water |
— | CH3OH: 27.4/11.2/7.8 μmol g−1 h−1 | 334 |
| CH3CHO: 2.2/2.5/4.8 μmol g−1 h−1 (for NaHCO3/NaOH/NaCl) |
5.5 MXenes
Recently, MXenes, another class of 2D materials, have received enormous attention owing to their high electrical conductivity, large specific surface area, hydrophilicity, and tunable composition.335–339 MXenes possess a lamellar structure with anisotropic properties and can be represented by the general formula Mn+1AXn, where M is a transition metal, n = 1, 2, or 3, A is an A-group in the periodic table (mostly IIIA and IVA), and X is C or N.340 The MAX phase is hexagonally stacked where the tight M layer is inserted with the pure A-group layer, and the X elements fill the octahedral sites.341 The first MXene to be discovered was Ti3C2Tx, where Tx represents a surface terminated group and since then more than 30 other compositions have also been reported.342 Ti3C2 is primarily synthesized by the exfoliation of Ti3AlC2 with HF. This method is expected to afford various compositions.341,342 To date, MXenes have been exploited in applications such as lithium-ion batteries,343 electrochemical supercapacitors,344 and fuel cells.345 In addition to these energy storage applications, they have also been explored as promising photocatalytic materials.346–349 The 2D structure of MXenes affords a high surface area and good pore structure, making these materials an excellent choice for enhanced CO2 adsorption and photocatalytic activity. Moreover, MXenes may serve as efficient cocatalysts in photocatalysis owing to their good electronic conductivity, adjustable bandgap, and strong metallic characteristics.350 The Ran group reported the use of Ti3C2/CdS for H2 generation and demonstrated that Ti3C2 is a promising cocatalyst for photocatalytic applications, providing an alternative to noble metals.351To date, several Ti3C2-based heterostructured photocatalysts have been studied for CO2 conversion. For example, Low et al. prepared TiO2/Ti3C2 by in situ calcination and successfully applied it to photocatalytic CO2 conversion.336 Ultrathin layered 2D Ti3C2 was first synthesized and then combined with TiO2 using simple calcination at various temperatures. The composite exhibited a high surface area with a unique rice crust-like structure. The high electronic conductivity of Ti3C2 was found to be beneficial for the migration of photogenerated electrons from TiO2 to Ti3C2. Photocatalytic CO2 reduction mainly afforded CH4 along with a small amount of other products such as CH3OH and C2H5OH, and the catalytic activity for CH4 formation was approximately 3.1 times higher than that for pristine TiO2. The enhanced catalytic activity was ascribed to the large surface area, improved CO2 adsorption, and the heterogeneous interface between TiO2 and Ti3C2, which improved the charge separation. In another study, Pan et al. presented a functional MXene/CsPbBr3 system for CO2 reduction to CO and CH4, in which the CsPbBr3 was grown in situ on 2D Ti3C2 NSs.352 In this study, HCl–HF solution was used to etch Ti3AlC2 to obtain Ti3C2Tx NSs, upon which cubic CsPbBr3 was grown in situ to afford the final nanocomposites. The PL and time-resolved PL quenching was observed for the composite sample, indicating efficient charge transfer through the CsPbBr3/MXene interface. The optimized composite mediated the reduction of CO2 to CO (26.32 μmol g−1 h−1) and CH4 (7.25 μmol g−1 h−1). Therefore, such perovskite/2D composites can be used to realize efficient photocatalysis.
It is well known that the combination of 2D/2D heterojunctions provides superior structural stability owing to the substantial interfacial contact.336 In this regard, Yu and co-workers reported the heterostructure combination of 2D Bi2WO6 with 2D Ti3C2 NSs.353 The incorporation of Ti3C2 led to enhanced CO2 adsorption owing to the increased surface area. The 2D/2D sandwich-like Ti3C2/Bi2WO6 system displayed excellent charge transfer for CO2 reduction. Under light irradiation, the photoexcited electrons transferred from the CB of Bi2WO6 to Ti3C2 through the ultrathin layered heterostructure and reacted with adsorbed CO2 molecules. Therefore, the photocatalytic CO2 reduction activity toward CH4 and CH3OH was significantly improved. Moreover, DFT calculations revealed that the excellent conductivity of Ti3C2 was beneficial for rapid charge transport. Subsequently, Yang et al. demonstrated that Ti3C2/g-C3N4 nanosheet heterojunctions exhibited improved catalytic activity for CO2 reduction into CO and CH4.335 The intimate contact between two 2D materials also leads to good charge separation. Hence, the catalytic activity of Ti3C2/g-C3N4 for CO2 reduction toward CO and CH4 reached rates of 5.19 and 0.044 μmol h−1 g−1, respectively. This catalytic activity was approximately eightfold higher than that for pristine g-C3N4. Significantly, BET isotherm experiments revealed higher chemical affinity between CO2 molecules and the catalyst surface, which contributed to the improved catalytic performance.
In addition to these binary composites, Wu et al. also described the ternary heterostructure composition Ti3C2Tx/(001)TiO2/C3N4, which was synthesized by in situ oxidation/electrostatic self-assembly.350 The heterojunction composed of Ti3C2Tx/(001)TiO2 NSs with highly exposed {001} TiO2 facets was first synthesized by the in situ oxidation of Ti3C2Tx NSs. Next, the ternary composite with C3N4 was obtained by exploiting the electrostatic attraction between the opposite charges. This 2D/2D heterojunction combination provided broad electron transfer channels that significantly enhanced the charge separation properties, while the incorporation of the 2D MXene into TiO2/C3N4 increased the catalytic activity. The heterojunction catalyst displayed threefold higher CO2 conversion activity compared to pristine TiO2 or C3N4. A similar TiO2/C3N4/Ti3C2 composite was explored in another study by combining 0D Ti3C2 QDs with 2D/2D core–shell TiO2/C3N4 heterojunctions.260 First, ultrathin C3N4 was deposited on the TiO2 nanosheets by the thermal condensation of urea to form a core–shell structure. Then, the as-prepared Ti3C2 QDs were electrostatically assembled on the C3N4 shell. The close attachment of the Ti3C2 QDs on the TiO2/C3N4 is due to van der Waals interactions. Theoretical and experimental results indicated that the charge transfer in the ternary composite occurred via a dual heterojunction involving the S-scheme for TiO2/C3N4 and the Schottky scheme for the C3N4/Ti3C2 QDs. The QDs extracted the electrons from C3N4, enabling S-scheme charge transfer between TiO2 and C3N4. As a result, the composite exhibited a CO evolution rate of 4.39 μmol g−1 h−1.
Therefore, MXenes can be fruitfully applied as cocatalysts for photocatalytic CO2 conversion. In particular, their 2D nature affords a large surface area, providing a promising alternative option to expensive noble metals. Until recently, most studies have focused on the synthesis of MXenes with HF, which may represent one of the primary drawbacks of these systems; the use of such hazardous chemicals may not be desirable for large-scale MXene production. However, various studies are underway to replace such chemicals. For example, Shah and co-workers reported the synthesis of MXenes by electrochemical etching in HCl solution.354 In time, this research may permit the large-scale synthesis of MXenes by environmentally friendly approaches using less hazardous chemicals.
The photocatalytic CO2 reduction performance of MXene-based materials is summarized in Table 6.
| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor type | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Ti3C2/g-C3N4 | CO2 + H2O (H2SO4 + NaHCO3) | 300 W Xe lamp (420 nm cutoff) | H2O | 20 mg catalyst | — | CH4: 0.044 μmol g−1 h−1 | 335 |
| CO: 5.19 μmol g−1 h−1 | |||||||
| TiO2/Ti3C2 | CO2 + H2O (HCl + NaHCO3) | 300 W Xe lamp | — | 50 mg catalyst in Pyrex reactor (200 mL) | — | CH4: 0.22 μmol h−1 | 336 |
| Ti3C2Tx/(001)TiO2/C3N4 | CO2 + H2O (H2O + 15 vol% TEOA) | — | TEOA | 5 mg catalyst on glass in reactor (80 mL) | — | CH4: 1.97 ppm h−1 | 350 |
| CO: 15.0 ppm h−1 | |||||||
| CsPbBr3/Ti3C2Tx | CO2 (ethyl acetate) | 300 W Xe lamp (>420 nm cutoff) | — | — | — | CH4: 7.25 μmol g−1 h−1 | 352 |
| CO: 26.32 μmol g−1 h−1 | |||||||
| Ti3C2/Bi2WO6 | CO2 + H2O (H2SO4 + NaHCO3) | 300 W Xe lamp | — | 100 mg catalyst in Pyrex glass reactor (200 mL) | Batch | CH4: 1.78 μmol g−1 h−1 | 353 |
| CH3OH: 0.44 μmol g−1 h−1 | |||||||
| TiO2/C3N4/Ti3C2 | CO2 + H2O (H2SO4 + NaHCO3) | 350 W Xe lamp | H2O | 30 mg catalyst in two-neck Pyrex reactor | — | CH4: 1.20 μmol g−1 h−1 | 260 |
| CO: 4.39 μmol g−1 h−1 |
5.6 Perovskites
Perovskite materials ranging from oxides to halides have stimulated enormous interest for catalytic applications owing to their exciting features such as cost-effectiveness, tunable bandgaps, high surface area, and surface defects for charge trapping.50 Moreover, in terms of altering the redox potentials, the structural flexibility of perovskites makes them quite different from traditional semiconductors. The ideal perovskite has the general formula ABX3, in which A and B are different cations and X denotes oxide/halide anions surrounded by B cations.CaTiO3 was the first perovskite material discovered, and this formula has since been extended to a variety of new forms, such as A2BX6, A2BB′X6, and so on.355 This structure allows the formation of lattice deficiencies, which can be advantageous for tuning the optoelectronic properties. Besides simple perovskites, layered perovskites and their derivatives display excellent properties such as structural stability and low cost. Previously, oxide perovskites were primarily used as catalyst materials; however, halide perovskites are now one of the most commonly used materials for photocatalytic CO2 conversion. Therefore, we will first discuss recent progress in oxide perovskite photocatalysts before considering halide perovskites.
Depending on the type and composition of the A and B sites, oxide perovskites exhibit different band structures and optoelectronic properties. In addition, replacing or doping the A, B, or O sites with other metal or non-metal elements can change the chemical composition and symmetry of oxide perovskites, which can be beneficial for tuning the material properties, such as band potentials, light absorption, and CO2 adsorption.356 Consequently, the photocatalytic performance of oxide perovskites can be altered by changing their composition, structure, morphology, and heterostructure combination. In 1978, Hemminger and colleagues demonstrated the photo-assisted conversion of CO2 into CH4 on the surface of SrTiO3(111) for the first time without using an electrochemical cell.357 Since this pioneering work, various perovskites have been examined for CO2 conversion applications. However, pure SrTiO3 is not considered a suitable catalyst because of its large bandgap (>3.0 eV) and light absorption mainly in the UV region; thus, surface modification, heterostructure formation, and metal NP deposition have frequently been applied to increase its sensitivity to visible light. For instance, Luo et al. prepared Ti-rich and Sr(OH)2-decorated SrTiO3 catalysts and explored the effects of surface modification on CO2 photoreduction.358
The results revealed that Ti-rich SrTiO3 possesses a narrow bandgap due to a lower Ti 3d ground-state level, which increased light harvesting in the visible region. Therefore, it exhibited high photocatalytic activity toward CO formation (26.4 μmol g−1) compared to pristine SrTiO3 (18.4 μmol g−1) and Sr(OH)2-modified SrTiO3 (13.8 μmol g−1). In another study, oxygen-deficient self-doped SrTiO3−δ was utilized for the photoconversion of CO2 into CH4.359 To obtain Ti3+ states accompanied by oxygen vacancies, the catalyst was synthesized by a combustion process with high-temperature heat treatment. This resulted in improved CO2 adsorption on the surface of the oxygen-deficient catalysts and thus increased CO2 reduction activity. Metal NPs have also been employed in this field owing to their unique features originating from surface plasmon resonance (SPR). For example, Li et al. demonstrated the synergistic effects of metal cocatalysts on oxide perovskites, which significantly enhanced the response to visible light as a result of the plasmonic effect and electron extraction properties of the metal NPs.360 In this study, Rh metal was grafted onto SrTiO3 prior to the loading of Au NPs as a photosensitizer. With 99.4% selectivity for CO formation, the optimized SrTiO3, i.e., Rh(PD)-Au@SrTiO3, demonstrated 153- and 22-fold higher catalytic activity than pristine Rh@SrTiO3 and Au@SrTiO3 samples, respectively.
As discussed earlier, the integration of two semiconducting materials to form a heterojunction combination can be a promising strategy for improving catalytic activity owing to the efficient charge separation via the closely connected interface. In one study, RuO2 NPs were supported on SrTiO3 and the resulting composites were found to exhibit improved catalytic activity for CO2 photoreduction with H2 in a flow reactor system.361 Remarkably, the authors also studied the influence of the photothermal behavior of the catalyst toward CH4 formation at 150 °C. The improved catalytic activity was attributed to the high CO2 adsorption by SrTiO3 and efficient charge separation between RuO2 and SrTiO3. Heterojunction combinations of oxide perovskites with TiO2 (e.g., CaTiO3/TiO2) have also been reported.362 Later, additional oxide perovskites with A site substitution were found to mediate photocatalytic CO2 conversion. For example, Teramura and co-workers studied the photoreduction of CO2 over ATaO3 (A = Li, Na, K) using H2 as a reductant.363 The strong chemisorption of CO2 molecules was observed on the surface of LiTaO3, and the catalytic activity followed the order LiTaO3 > NaTaO3 > KTaO3. Similarly, Zhou et al. investigated photocatalytic CO2 reduction over alkaline tantalates, i.e., MTaO3 (M = Li, Na, K), with 3D hierarchical structure obtained using activated carbonized wood as a template.364 The template provided a high surface area that enhanced the light harvesting and gas diffusion properties. The results indicated that the CO and CH4 formation rates upon CO2 reduction were enhanced by 3.1 and 8.4 times, respectively, by the deposition of the Au cocatalyst on NaTaO3. The formation of CO and CH4 over NaTaO3 was attributed to the suitable band potentials that simultaneously reduced CO2 and oxidized H2O. Both of these studies demonstrated that alkaline NaTaO3 has a synergistic effect on reducing CO2 to CO and CH4. Although CO selectivity was observed under a H2 environment, the presence of H2O led to the formation of both CO and CH4. In another study, the lanthanum-based oxide perovskite LaCoO3 was utilized as a cocatalyst on a Ru complex for the reduction of CO2 to CO under light irradiation.365 The results revealed 20-fold higher activity compared to the system lacking LaCoO3, in addition to 76% selectivity for CO formation and an AQY of 1.36%. Similarly, MnCo2O4 microspheres have been utilized as a stable cocatalyst for the conversion of CO2 to CO.366
The morphology of perovskite nanocrystals is another crucial factor in tuning the properties of the catalyst to improve the catalytic reaction rate. In this regard, Shi et al. prepared Pt-loaded g-C3N4/NaNbO3 nanowires and studied their photocatalytic CO2 reduction behavior.367 The resulting composite displayed improved catalytic activity for CH4 formation (6.4 μmol g−1 h−1) compared to Pt-loaded g-C3N4 and Pt-loaded NaNbO3, which originated from the improved charge separation through the closely connected interface, increased surface area due to the nanowire morphology of NaNbO3, efficient charge transport through the heterostructure, and suitable band potentials for CO2 reduction to CH4. In addition, Kumar and co-workers designed the UV-vis-NIR-active catalyst Ag2CrO4/Ag/BiFeO3@RGO, composed of a Ag-mediated Ag2CrO4/BiFeO3 heterojunction on an RGO matrix.368 Each of the components made a significant contribution to improving the conversion of CO2 into CO and CH4. For example, the BiFeO3 and Ag2CrO4 served as the photoreduction and photooxidation systems, respectively, while the plasmonic Ag NPs mediated the electron donation and the RGO improved the redox capabilities by enhancing the electron mobility. In another study, Wang and colleagues synthesized Ag-deposited H2SrTa2O7 (HST) using a polymerizable complex and an ion-exchange method.369 Owing to the anisotropy of the layered HST perovskite structure, photoinduced electrons and holes gathered on the edges and basal plane. The Ag NPs reduced the kinetic barrier to CO formation and captured more electrons from the perovskite to reduce CO2 to CO, thereby impeding the reduction of H+ to H2. The optimal Ag-loaded sample (Ag/HST) displayed twofold higher reduction ability than pure HST with approximately 60% selectivity for CO formation. In an effort to improve the charge separation, Tu et al. reported a layered ferroelectric perovskite of SrBi4Ti4O15 NSs for CH4 evolution in a gas–solid phase system without the use of any cocatalyst or sacrificial agent.370 The ferroelectric characteristics of SrBi4Ti4O15 (SBTO-1) afforded efficient bulk charge separation and high charge mobility. The catalyst was synthesized via a soft chemical method by adding NaOH as a mineralizer. Post-treatment annealing was then conducted at 350 °C (SBTO) and 650 °C (SBTO-2) to tune the ferroelectric polarization. Notably, SrBi4Ti4O15 displayed significant photocatalytic performance in the reduction of CO2 to CH4 with a rate of 19.8 μmol g−1 h−1, along with a small amount of CO formation (Fig. 21(a) and (b)).
 | ||
| Fig. 21 (a) CH4 and (b) CO evolution over SrBi4Ti4O15 (SBTO) catalysts and several reference samples, (c) in situ DRIFTS analysis showing the intermediate products formed on SBTO catalysts under light irradiation at various time intervals, (d) energy band diagram for an SBTO catalyst, and (e) structure of the SBTO units showing electron–hole separation in different directions. Reproduced with permission from ref. 370, Copyright 2019, Elsevier. (f) Synthetic route to FAPbBr3/Ti3C2, (g)–(i) TEM images of FAPbBr3, Ti3C2, and their heterojunction, (j) dark-field STEM image of FAPbBr3/Ti3C2, (k) photocatalytic CO2 reduction performance of FAPbBr3/x-Ti3C2 samples (x is mg of Ti3C2), (l) structure of FAPbBr3/Ti3C2 heterojunction for CO2 reduction. Reproduced with permission from ref. 371, Copyright 2021, American Chemical Society. (m) CO and CH4 formation over various Bi-based perovskite nanocrystals. Reproduced with permission from ref. 372, Copyright 2019, American Chemical Society. | ||
The catalyst was found to be 8- and 283-fold more active than the reference samples of Bi4Ti3O12 and BiOBr, respectively, with a CH4 selectivity of 93% and an AQY of 1.33% at 365 nm. Time-dependent in situ DRIFTS analysis confirmed the conversion of CO2 into CH4 and CO on the SBTO surface (Fig. 21(c)). According to the calculated band potential value, the negative CB of SrBi4Ti4O15 provided a strong driving force for improving CO2 conversion (Fig. 21(d)). Remarkably, the layered nanocrystal structure with well-aligned distorted polyhedra enhanced the charge separation because the electrons and holes migrated separately to the TiO2 and Bi2O22+ layers (Fig. 21(e)).
Another type of material is metal halide perovskites, which are represented by the general formula ABX3, where A and B are cations and X is an anion.12 Following the successful utilization of methylammonium lead halide perovskites in solar cell and LED applications in 2009, halide perovskites have attracted considerable attention in the field of photocatalysis. To date, several halide perovskites with various compositions have been reported where A = methylammonium (MA), formamidinium (FA), or Cs, B = Pb, Bi, or Sn, and X = Cl, Br, or I. However, similar to pristine metal oxides or any other semiconductors, pristine metal halide perovskites are associated with several drawbacks, such as poor light absorption, poor charge separation, and rapid charge recombination. In an effort to improve these aspects, halide perovskites have been combined with various secondary materials or cocatalysts such as graphene, metal NPs, metal oxides, and metal carbides. In particular, the moisture stability of these perovskites has been significantly improved by embedding them into polymers or metal oxides/complexes. For example, Xu et al. established heterojunctions based on TiO2 nanofibers and CsPbBr3 QDs that promoted electron–hole separation and improved the photoconversion efficiency compared to the pristine perovskite.373 Such hybridization enabled the preparation of heterojunctions with the highest redox ability.
Similarly, some other heterostructure combinations have been reported. For example, in 2017 Xu et al. reported that the rate of electron consumption increased by 25.5% upon combining CsPbBr3 with graphene oxide.374 In addition, Jiang and co-workers described an ingenious ternary heterostructure based on CsPbBr3 nanocrystals and a hierarchical branched ZnO nanowire/macroporous graphene oxide composite.375 In this case, the RGO played an important role by simultaneously adsorbing and activating CO2 molecules via π–π interactions and conjugation, which most likely accelerated the catalytic CO2 conversion. The 3D cross-linked morphology provided a larger active surface area as well as pathways for rapid electron transport and mass transfer. Furthermore, 1D branched ZnO is simple to synthesize, inexpensive, and has ideal energy band potentials for CO2 reduction. In contrast to pristine CsPbBr3, which reduced CO2 to CO and CH4 with reasonable activity, the hybrid composite displayed an increased rate of CH4 formation. Hence, the selectivity for CH4 formation correspondingly increased from 78.2% for CsPbBr3 to 96.7% for the ternary composite. Similarly, Wang et al. synthesized Cs4PbBr6 wrapped with defective RGO hybrids through antisolvent precipitation and applied these materials to CO2 photoconversion.294
The presence of oxygen defects in the rGO nanosheets ultimately extended the lifetime of the electron–hole pairs. Here, COOH and OH moieties acted as anchor points for the hybridization of rGO with CsPbBr3 through forming Pb–O–C bonds. Some additional combinations of metal halide perovskites with graphene oxide have also been reported.376 For example, the MOF nanocomposite CsPbBr3-UiO-66(NH2) has been studied for photocatalytic CO2 conversion and was found to display high catalytic activity for CO formation (98.57 μmol g−1).377 The increased activity and stability were driven by the high surface area, enhanced visible-light absorption, efficient charge separation in the QDs, and presence of the UiO-66(NH2) nanocomposites. The selective formation of CO upon CO2 photoreduction was attributed to the dynamically favorable band potentials of CsPbBr3 and the HOMO/LUMO levels of UiO-66(NH2) for efficiently mediating the 2H+/2e− process. In another work, Kong and co-workers designed a core–shell halide perovskite@MOF composite that exhibited enhanced CO2 reduction activity.312 In this work, coating of the ZIF shell onto the surface of the CsPbBr3 QDs was achieved in situ by dispersing the latter into a solution of the metal ion precursor and imidazole ligand. This coating improved the moisture stability of the CsPbBr3 QDs in addition to the CO2 capture ability and charge separation efficiency, ultimately resulting in enhanced photoconversion efficiency of CO2 to CO and CH4.
Tang et al. conducted a theoretical study on the mechanism of CO2 reduction by performing DFT calculations of Fe- and Co-doped CsPbBr3.378 The results showed that the doped perovskite displayed better adsorption ability of the activated intermediate CO2˙− that led to improved catalytic activity. Free energy calculations suggested that the product selectivity of the pristine perovskite lay toward CO formation, whereas the selectivity shifted toward CH4 formation upon Fe or Co doping. Shyamal et al. also investigated Fe(II)-doped CsPbBr3 perovskite and observed enhanced catalytic activity and selectivity toward CH4 formation.379 The formation of CH4 was drastically improved upon Fe doping, while the pristine perovskite was selective toward CO evolution. The product selectivity of doped CsPbBr3 was related to the adsorption–desorption characteristics, where the more positive adsorption energy of the CH4 molecule enabled it to desorb rapidly from the catalytic surface, which was the major reason for the high selectivity. Similarly, other studies have reported the Co, Ni, and Mn doping of perovskite nanocrystals.380,381 Apart from metal doping, a composite based on CsPbBr3 nanocrystals and 2D Pd NSs has been reported as a highly efficient and stable catalyst for gas-phase photocatalytic CO2 reduction with H2O vapor.382 This combination formed a Schottky contact and improved the electron consumption rate compared with the pristine perovskite nanocrystals. Subsequently, the transition-metal complex Ni(tpy) was immobilized on CsPbBr3 nanocrystals, which provided abundant active sites for capturing CO2 molecules through the metal complexes.383 Meanwhile, the polypyridyl rings efficiently captured and stored electrons for the CO2 reduction process. As a result, the efficiency of CO2 photoreduction to CO/CH4 increased by a factor of 26.
Cesium-based metal halide perovskites are commonly used for photocatalytic CO2 reduction; however, changing the metal component (A site) may alter the optoelectronic properties of the catalyst. In this regard, Wu and co-workers encapsulated CH3NH3PbI3 (MAPbI3) QDs in the pores of the Fe–porphyrin-based MOF PCN-221(Fex) and studied its photocatalytic CO2 reduction behavior to afford CO/CH4.384 The encapsulation improved the perovskite stability and the close contact between these materials shortened the charge transfer distance, resulting in very high catalytic activity. In another case, formamidinium lead bromide (FAPbBr3) perovskite QDs were applied to CO2 photoreduction in various reaction media, where water acted as a proton source and the solvent allowed maximum CO2 saturation.385 Similarly, improved catalytic activity was reported upon Schottky heterojunction formation between FAPbBr3 and Ti3C2 nanosheets.371 The excellent metallic conductivity and high surface area of Ti3C2 were beneficial for improving the optoelectronic properties of the catalyst. Briefly, FAPbBr3 QDs were grown in the presence of Ti3C2 using the hot injection method (Fig. 21(f)). Spectroscopic analysis confirmed the strong interaction between FAPbBr3 and Ti3C2, facilitating separation of the photogenerated electrons through the interface. The formation of FAPbBr3/Ti3C2 could also be observed in the TEM and STEM images (Fig. 21(g)–(j)). Analysis of the optoelectronic properties revealed that the Ti3C2 nanosheets acted as an electron acceptor, allowing for the rapid transfer of photoexcited electrons in FAPbBr3. The electron consumption rate for FAPbBr3/Ti3C2 was reported to be 717.18 μmol g−1 h−1, which was approximately two-fold higher than that for the bare FAPbBr3 sample (Fig. 21(k) and (l)).
Although a variety of metal halide perovskites have shown promise as materials for photocatalytic CO2 reduction, the mass production of lead-based perovskite materials remains problematic owing to the unavoidable issue of lead toxicity, obstructing the long-term viability of this technology. As a result, numerous research efforts have been dedicated to creating environmentally safe lead-free halide perovskite materials for photocatalytic applications. In this regard, Zhou and co-workers studied nanocrystals of the halide double perovskite Cs2AgBiBr6 synthesized via a hot-injection method, which displayed excellent electron consumption (105 μmol g−1) under AM 1.5 G illumination.386 The synthesized nanocrystals were reported to be highly stable in mild polar solvents for more than three weeks, even in the presence of light and humidity. Another recent study used the injection of a precursor at room temperature followed by heating of the reaction mixture to construct 2D multilayered Cs2AgBiX6 (X = Cl, Br, I) nanoplatelets.387 Nanoplatelets possess several desirable characteristics such as exposed facets, well-arranged surface atomic symmetries, and quantum confined photocarriers. The authors synthesized a series of double perovskites by varying the halide composition and combinations, and Cs2AgBiBr6 was found to display efficient photocatalytic performance. To investigate the influence of the nanoplatelet morphology, the catalytic performance of the Cs2AgBiBr6 nanoplatelets was compared to that of simple nanocrystals. The results revealed that the nanoplatelets exhibited higher CO and CH4 production rates than the nanocrystals (eight-fold higher electron consumption rate over 6 h). NPLs have anisotropically confined charge carriers and long diffusion length, resulted in such an improved catalytic performance.
Besides these materials, Cs2AgBiBr6 nanocrystals were combined with g-C3N4 to form Z-scheme and type-II heterojunction systems using toluene and CH2Cl2, respectively.388 Interestingly, by altering the CB of g-C3N4 and the VB of Cs2AgBiBr6, the Z-scheme combination displayed superior photocatalytic CO2 reduction to CH4, whereas the type-II heterojunction system exhibited CO selectivity. In 2020, Lu et al. reported the photoconversion of CO2 upon changing the B site to the aforementioned layered double halide perovskite, i.e., Cs3Sb2Br9.389 The surface-exposed Sb sites led to higher reactivity for CO2 reduction and thus improved catalytic activity. Changing the cation may also lead to different catalytic behavior; for example, Bhosale et al. fabricated Bi-based perovskite photocatalysts (i.e., Cs3Bi2I9, Rb3Bi2I9, and MA3Bi2I9) using a top-down ultrasonication approach.372 They revealed that the cation and crystal structure of the perovskite play important roles in determining the catalytic activity and CO2 reduction pathway using EPR and diffuse-reflectance infrared spectra. The photocatalytic CO2 reduction activities toward CO and CH4 of the perovskite nanocrystals followed the order Cs3Bi2I9 > Rb3Bi2I9 > MA3Bi2I9 > TiO2 (Fig. 21(m)). In addition, the EPR results indicated that Cs3Bi2I9 displayed higher catalytic activity than the other perovskites owing to its greater ability to generate electron–hole pairs.
The studies discussed above suggest unique strategies for obtaining lead-free perovskite-based catalytically active materials for mediating efficient photocatalytic CO2 conversion reactions. The photocatalytic CO2 reduction performance of perovskite-based catalysts is summarized in Table 7.
| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor type | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Ti-Rich SrTiO3 | CO2 (10 mL) + H2O (10 μL) | 300 W Xe lamp (0.190 W cm−2) | H2O | 20 mg sample | Gas phase | CO: 26.4 μmol g−1 (6 h) | 358 |
| SrTiO3−δ | 98% CO2 + 2% H2O | 300 W Xe lamp (420 nm filter) | H2O | 0.3 g sample | Gas phase | CH4: 0.25 μmol m−2 h−1 | 359 |
| Au/Rh@SrTiO3 | CO2 + H2O | 300 W Xe lamp (L42 filter) | H2O | 75 mg sample | Gas-phase batch reactor | CO: 369.2 μmol g−1 h−1 | 360 |
| H2: 69.4 μmol g−1 h−1 | |||||||
| CH4: 2.8 μmol g−1 h−1 | |||||||
| RuO2–SrTiO3 | CO2 + H2 (4:1 (mol/mol)) |
300 W UV-vis Xe lamp (1000 W m−2) | — | 50 mg sample | Gas-phase flow reactor | CH4: 14.6 mmol g−1 h−1 | 361 |
| CaTiO3/TiO2 | CO2 + H2O | 300 W Xe lamp | H2O | 10 mg sample in quartz tube (43 mL) | Gas-phase batch reactor | CO: 11.72 μmol g−1 h−1 | 362 |
| Li/K/Na–TaO3 | CO2 (150 μmol) + H2 (50 μmol) | 200 W Hg–Xe lamp | — | 2.0 g sample in quartz reactor (150 mL) | Gas phase | CO: 0.42 μmol g−1 (10 h) | 363 |
| NaTaO3 | CO2 + H2O (80 kPa of CO2, 2 mL H2O) | 200 W Hg–Xe arc lamp | — | 50 mg sample in Pyrex reaction cell, 2 mL DI water | Gas phase | CO: 173 nmol g−1 h−1 | 364 |
| CH4: 36 nmol g−1 h−1 | |||||||
| LaCoO3 | CO2-saturated MeCN/H2O/TEOA (3:2:1) |
300 W Xe lamp (420 nm cutoff filter) | H2O | 1 mg sample with Ru-complex and TEOA as photosensitizer and electron donor, respectively, reaction controlled at 30 °C | Gas phase | CO: 28.5 μmol (1 h) | 365 |
| AQY = 1.36% | |||||||
| MnCo2O4 | High purity of CO2 in solvent (5 mL, 2:3 H2O/acetonitrile) |
300 W Xe lamp (420 nm cutoff filter) | H2O | 4 μmol sample, Ru-complex, and TEOA in reactor (80 mL) | Gas-phase batch reactor | CO: 27 μmol (1 h) | 366 |
| g-C3N4/NaNbO3 | CO2 + H2O | 300 W Xe arc lamp | H2O | 50 mg sample, 2 mL H2O in Pyrex glass vessel | Gas phase | CO: 0.39 μmol g−1 | 367 |
| Ag2CrO4/Ag/BiFeO3@RGO | 99.99% CO2 + H2O | UV, visible, NIR | H2O | 40 mg sample, 40 mL DI water in Pyrex reactor vessel | Gas phase | CH4: 180 μmol g−1 (8 h) | 368 |
| CO: 38 μmol g−1 (8 h) | |||||||
| SrBi4Ti4O15 | In situ generated CO2 + H2O (NaHCO3 (1.3 g) + H2SO4 (15 mL)) | 300 W Xe lamp (λ > 420 nm) | — | 100 mg sample dispersed in DI H2O and dried at 80 °C | Gas-phase batch reactor | CH4: 19.8 μmol g−1 h−1 | 370 |
| CO: 1.74 μmol g−1 h−1 | |||||||
| AQY = 1.33% (365 nm) | |||||||
| Ag/H2SrTa2O7 | CO2 (95% Ar, 5% CO2) + H2O | 300 W Xe lamp (λ > 200 nm) | — | 200 mg sample | Gas-phase flow reactor | CO: 0.39 μmol g−1 h−1 | 369 |
| TiO2/CsPbBr3 | High-purity CO2 (99.99%) | 300 W Xe arc lamp | H2O | 10 mg sample, 30 mL acetonitrile, 100 μL H2O in quartz/Pyrex hybrid reaction cell | Gas phase | CO: 9.02 μmol g−1 h−1 | 373 |
| CsPbBr3 QDs/GO | Ethyl acetate (10 mL) + CO2 | 100 W Xe lamp (AM 1.5 filter) | — | 4 mg sample in sealed Pyrex bottle (40 mL) | Gas phase | CO: 58.7 μmol g−1 (12 h) | 374 |
| CH4: 29.6 μmol g−1 (12 h) | |||||||
| H2: 1.58 μmol g−1 (12 h) | |||||||
| CsPbBr3/BZNW/MRGO | CO2 | 150 W Xe lamp (AM 1.5 G and 420 nm optical filter, 100 mW cm−2) | H2O | 20 μL distilled water and evaporated at 120 °C for 2 min, Pyrex reaction cell | Gas phase | CH4: 6.29 μmol g−1 h−1 (3 h) | 375 |
| CO: 0.8 μmol g−1 h−1 (3 h) | |||||||
| Cs4PbBr6/rGO | CO2 + ethyl acetate | 300 W Xe lamp (420 nm filter, 100 mW cm−2) 100 mW cm−2) | H2O | 5 mg sample, 5 mL ethyl acetate, and 5 μL water mixed in sealed Pyrex bottle (35 mL) | Gas phase | CO: 11.4 μmol g−1 h−1 | 294 |
| CsPbBr3/USGO/α-Fe2O3 | CO2 | 300 W Xe lamp (420 nm filter, 100 mW cm−2) | H2O | 4 mg sample mixed with acetonitrile/deionized water (200:1 (v/v), 5 mL) in sealed Pyrex bottle (12 mL) |
— | CO: 73.8 μmol g−1 h−1 | 376 |
| CsPbBr3 QDs/UiO-66(NH2) | High-purity CO2 + ethyl acetate | 300 W Xe lamp (420 nm UV cutoff filter) | H2O | 10 mg sample dispersed in H2O/ethyl acetate (1:300 (v/v)), 10 μL H2O in Pyrex reaction cell |
Gas phase | CO: 98.57 μmol g−1 | 377 |
| CH4: 3.08 μmol g−1 (12 h) | |||||||
| CsPbBr3@zeolitic imidazolate | CO2 + H2O vapor (H2O evaporated at 120 °C for 2 min) | 100 W Xe lamp (AM 1.5G filter, 150 mW cm−2) | H2O | 4.5 mg sample (film) in sealed Pyrex bottle (40 mL) | Gas phase | Electron consumption rate: 29.630 μmol g−1 h−1 (3 h) | 312 |
| AQY = 0.035% | |||||||
| Co- and Fe-CsPbBr3 | Theoretical DFT study using DMol3 | 378 | |||||
| Fe(II)–CsPbBr3 | CO2 + H2O (H2O evaporated at 120 °C for 2 min) | 300 W Xe lamp (150 mW cm−2) | H2O | 5 mg sample mixed with CO2 and 10 μL H2O in sealed Pyrex bottle (25 mL) | Gas phase | CO: 6.1 μmol g−1 h−1 (3 h) | 379 |
| CH4: 3.2 μmol g−1 h−1 (3 h) | |||||||
| Co-CsPbBr3/Cs4PbBr6 | CO2 + acetonitrile (methanol as hole scavenger) | 300 W Xe lamp (100 mW m−2) | H2O | 4 mg sample added to acetonitrile/H2O/MeOH (5:15:15 μL) in sealed Pyrex bottle (12 mL) |
— | CO: 1835 μmol g−1 (15 h) | 380 |
| Ni- and Mn-doped CsPbCl3 nanocrystals | CO2 | 300 W Xe lamp (AM 1.5 filter) | H2O vapor | 6 mg sample (film) and 500 μL H2O in sealed Pyrex bottle (120 mL) | Gas phase | CO: 169.37 μmol g−1 h−1 (for Ni) | 381 |
| CO: 152.49 μmol g−1 h−1 (for Mn) | |||||||
| CsPbBr3/Pd nanosheets | CO2 + H2O (H2O evaporated at 120 °C for 3 min) | 150 W Xe lamp (Zolix, AM 1.5 G and 420 nm optical filter, 100 mW cm−2) | H2O vapor | 5.3 mg catalyst (film) and 10 μL water sealed Pyrex bottle (40 mL) | Gas phase | CO: 12.63 μmol g−1 (3 h) | 382 |
| CH4: 10.41 μmol g−1 (3 h) | |||||||
| AQY = 0.033% | |||||||
| [Ni(terpy)2]2+ (Ni(tpy)) CsPbBr3 nanocrystals Ni(tpy)–CsPbBr3 | CO2 + ethyl acetate | 300 W Xe lamp (SolarEdge 700, 100 mW cm−2, λ > 400 nm) | H2O | 5 mg catalyst in ethyl acetate/water solution (5 mL, 49:1 (v/v)) in Pyrex photoreactor (reactor maintained at 25 °C) |
— | CO + CH4: 1724 μmol g−1 (4 h) | 383 |
| Fe/CH3NH3PbI3 (MAPbI3) | CO2 + ethyl acetate | 300 W Xe lamp | H2O | 4 mg catalyst, 5 mL ethyl acetate and H2O (1:0.012 (v/v)) injected into sealed Pyrex bottle (10 mL) |
— | CO + CH4: 1559 μmol g−1 | 384 |
| FAPbBr3 QDs | CO2 | 300 W Xe lamp (100 mW cm−2) | H2O | 4 mg catalyst in 1 mL H2O or ethyl acetate in sealed Pyrex bottle (40 mL) | — | CO: 181.25 μmol g−1 h−1 | 385 |
| FaPbBr3/Ti3C2 | CO2 | 300 W Xe lamp (100 mW cm−2) | H2O | 3 mg catalyst, 0.5 mL H2O in sealed Pyrex bottle (40 mL) | Gas phase | CO: 283.41 μmol g−1 h−1 | 371 |
| CH4: 17.67 μmol g−1 h−1 | |||||||
| H2: 1.33 μmol g−1 h−1 | |||||||
| Cs2AgBiBr6 | Pure CO2 + ethyl acetate (pretreated with 4 A molecular sieves to remove residual water) | 100 W Xe lamp (AM 1.5 G filter) | — | 15 mg catalyst in sealed Pyrex bottle (40 mL) | — | CO: 14.1 μmol g−1 (6 h) | 386 |
| CH4: 9.6 μmol g−1 (6 h) | |||||||
| Cs2AgBiX6 | Dry ethyl acetate + CO2 (99.9%) | 405 nm laser diode | — | 2 mg catalyst, 10 mL ethyl acetate in sealed headspace reactor (20 mL) | — | CO: ca. 25.06 μmol g−1 (6 h) | 387 |
| CH4: ca. 40.06 μmol g−1 (6 h) | |||||||
| AQY = 0.035% | |||||||
| Cs2AgBiBr6@g-C3N4 | Pure CO2 + ethyl acetate + methanol | Xe lamp (80 mW cm−2) | — | 15 mg catalyst, 4 mL ethyl acetate, 1 mL methanol in Schlenk glass bottle (25 mL) | Gas phase | CO + CH4: 2.0 μmol g−1 h−1 | 388 |
| Cs3Sb2Br9 | Pure CO2 | 300 W Xe lamp with AM 1.5 irradiation | — | ca. 50–100 mg catalyst, pre-dried octadecene in water-jacketed Pyrex photoreactor (reaction maintained at 25 °C) | — | CO: 510 μmol g−1 (4 h) | 389 |
| Bi-Based perovskite nanocrystals | 99.99% CO2 + H2O vapor | 32 W UV lamp (305 nm) | H2O | Catalyst in 1 mL trichloromethane and dried by N2 gas flow in quartz reactor (230 mL) | Gas phase | For Cs3Bi2I9 (10 h): | 372 |
| CO: 77.6 μmol g−1 | |||||||
| CH4: 14.9 μmol g−1 | |||||||
| For Rb3Bi2I9 (10 h): | |||||||
| CO: 18.2 μmol g−1 | |||||||
| CH4: 17.0 μmol g−1 | |||||||
| For MA3Bi2I9 (10 h): | |||||||
| CO: 7.2 μmol g−1 | |||||||
| CH4: 9.8 μmol g−1 | |||||||
5.7 Plasmonic materials
The LSPR permits NPs to harvest light it is dependent upon the size of nanoparticles. The resulting interaction of the light and free electrons in the CB of the NPs causes oscillation of the surface electrons with the incident light. LSPR permits the gathering of light photons and creates energized charge transporters and heat. These charge transporters can be utilized to drive chemical reactions. In plasmonic catalysis, the exchange of photoexcited charge transporters from metal NPs to the reactants. The formation of heterostructures based on LSPR and photocatalysts is an exciting approach for CO2 photoreduction.390 Recently, several studies have attempted to apply plasmonic engineering to CO2 photoreduction.391–393 Under light irradiation, free electrons produce an electrical dipole moment by displacing the electrical field to nuclei. Concurrently, the Coulombic attraction between the electrons and nuclei generates a restoring force to produce the resonant oscillation of electrons. This phenomenon is called the quasi-static effect and it significantly enhances light absorption.394,395 In this regard, Kumari et al. reported the use of Ag plasmonic NPs for CO2 reduction under visible-light irradiation.396 They studied discrete adsorbates by in situ surface-enhanced Raman spectroscopy and estimated the product formation energy using DFT simulations.Fig. 22(a) shows the results for a physisorbed CO2 molecule, which was found to lie a considerable distance (3.4 Å) from the Ag surface in the simple structure form with a somewhat out-of-plane geometry for OCO (108.7°). To represent the effect of plasmonic excitation, the authors considered a charge-separated condition of the CO2/metal complex with a −1 charge along the CO2 molecule and a +1 charge on the Ag surface. After geometry relaxation, the CO2 held an electronic charge of −0.4 (balanced by a positive charge of +0.4 on Ag). Reliant upon the binding motif of CO2δ− on the surface, O atoms facing catalyst surface (Fig. 22(b)) or C atom facing catalyst surface (Fig. 22(c)), the structure of adsorbate, free energy, and registered Raman spectrum changed. However, the OCO vibration mode was estimated to be in the range of 1200–1300 cm−1 range, which was not observed in this study. This charged CO2δ− exhibited a bent geometry, the activated type of CO2. Indeed, energy optimization of CO2δ− on Ag in the vicinity of a surface-adsorbed Hδ+ induced the development of a surface-adsorbed HOCO* intermediate (Fig. 22(d) and (e)). However, the O and H atoms pointed away from the surface. The CO vibration stretching mode at 2231 cm−1 (Fig. 22(f)) showed that adsorbed CO is formed.
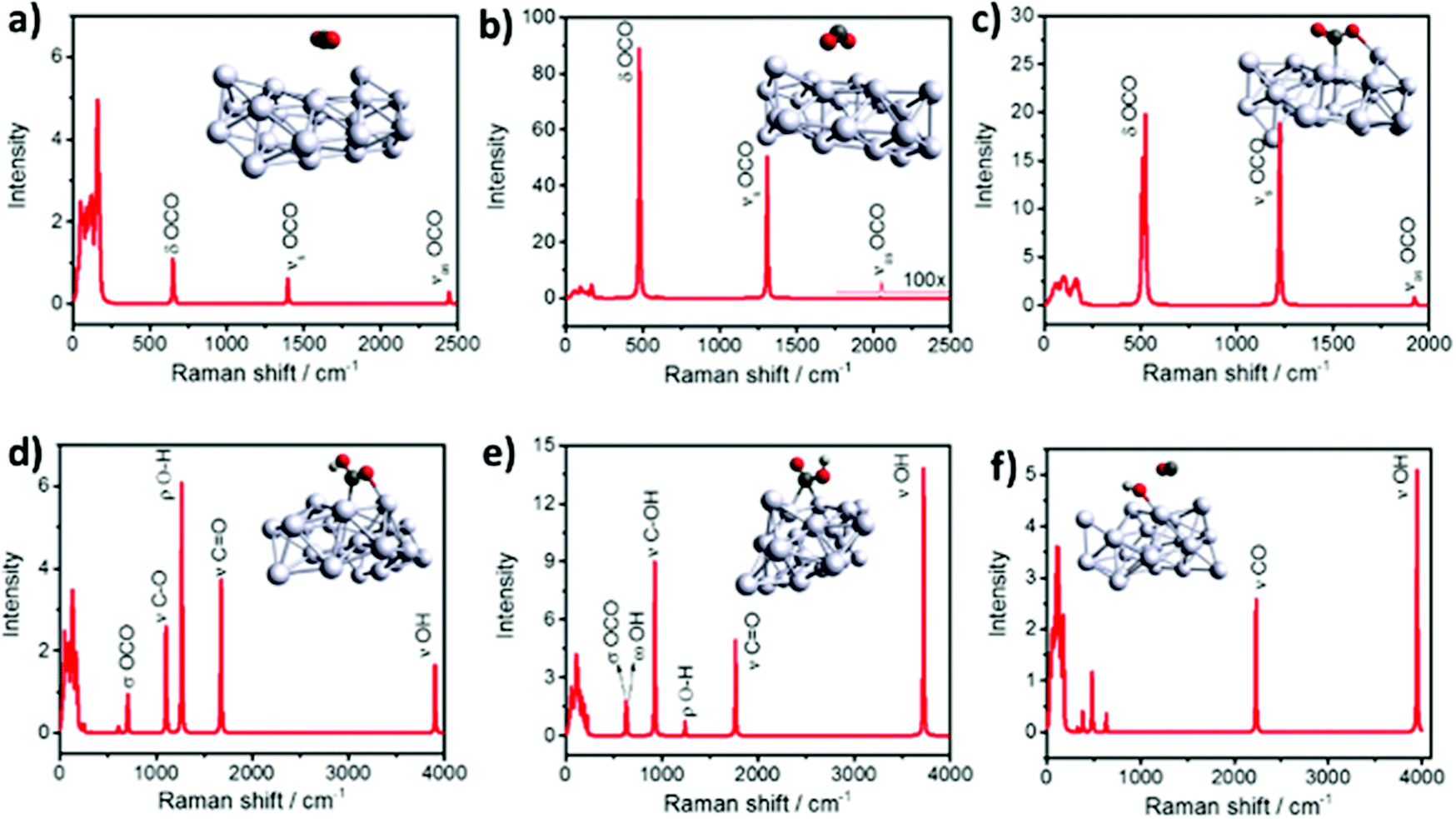 | ||
| Fig. 22 (a) Physisorbed CO2, (b) CO2δ− anion with both O atoms facing the Agδ+ surface, (c) CO2δ− anion binding to the Agδ+ surface via C and O atoms, (d) HOCO* intermediate bound to the Ag surface via both C and O atoms, (e) HOCO* intermediate bound to the Ag surface via a C atom, and (f) surface-bound CO* and OH* formed from dissociation of a HOCO* intermediate with both O atoms facing the Ag surface. Reproduced with permission from ref. 396, Copyright 2018, American Chemical Society. | ||
Furthermore, the decoration of bimetallic Au/Ag NPs on the top of TiO2 nanowires was reported to synergistically enhance the light absorption.250 The surface electrons of Au/Ag become excited and transferred to the CB of TiO2. While Au/Ag NPs could act as electron sink and allow a longer lifetime for photoexcited electrons. Therefore, the authors observed the evolution of CO (1813 μmol gcat−1 h−1) as the primary product with 98% selectivity. In addition, the plasmonic effects of Au in CO2 photoreduction have also been studied. Collado et al. demonstrated that the deposition of small Au nanoparticles on TiO2 resulted in the formation of C1 and C2 products under UV irradiation.397 Upon increasing the amount of Au from 0.5 to 3.0 wt%, the production of CH4 improved with respect to CH3OH, H2, and CO owing to the better charge separation and a number of electrons. Furthermore, Zeng et al. reported the controlled fabrication of a plasmonic Z-scheme Au/TiO2 catalyst for CO2 photoreduction, as shown in Fig. 23(a)–(f).398 This catalyst was synthesized by charge-controlled pulsed anodization and provided tunable product selectivity. The photonic crystals were composed of TiO2 nanotube arrays (referred to as periodically modulated titanium dioxide nanotube arrays (PMTiNTs)) decorated with the gold nanoparticle to form the plasmonic effect. The authors identified two pathways that afforded different selectivity.
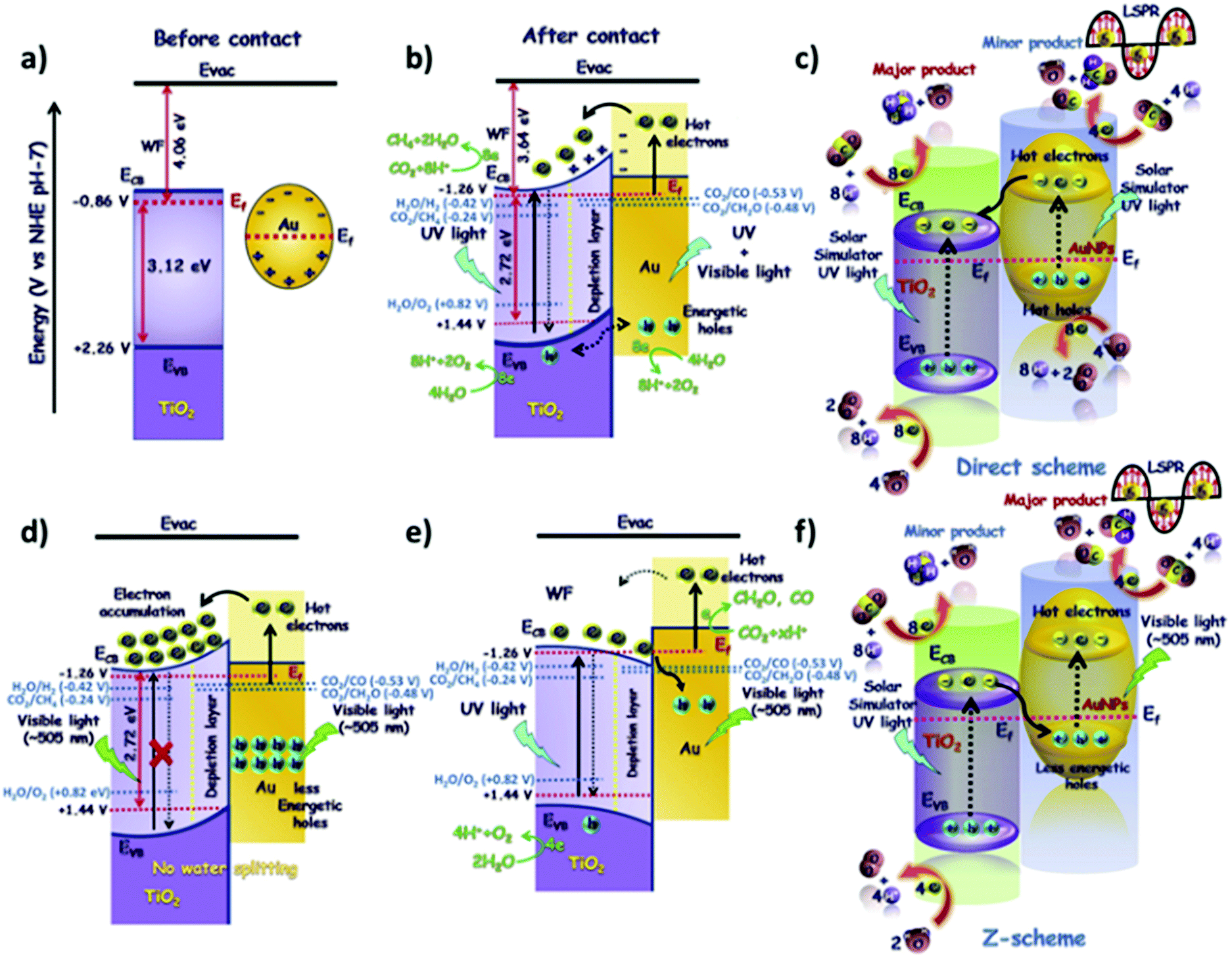 | ||
| Fig. 23 (a) Band energies of TiO2 and Au before contact, (b) Schottky junction formation between Au and TiO2 and their Fermi level alignment during CO2 reduction under simulated sunlight (traditional SPR assisted scheme shown in the blue text), (c) schematic representation of CH4 formation by Au-PMTiNTs under simulated sunlight, (d) hot-electron injection from Au NPs into TiO2 across Schottky barrier under visible light by LSPR, (e) recombination of the accumulated electron in TiO2 originated from the Au NP by LSPR, which forms the plasmonic Z-scheme, and (f) schematic illustration of overall Z-scheme for photocatalytic CO2 photoreduction to CH2O. Reproduced with permission from ref. 398, Copyright 2020, Elsevier. | ||
Under simulated sunlight (AM 1.5G), CH4 and CO were found to be the major and minor products, respectively. This indicated that direct charge transfer of sufficiently energetic electrons to the CB of TiO2 and specific photonic bandgaps avoided the defect-mediated low-energy charge transfer that might produce other hydrocarbons, as shown in Fig. 23(c). In addition, owing to the lower reduction potential of CO2/CH4 with respect to CO2/CO and CO2/CH2O, CH4 formation may be thermodynamically favorable. Furthermore, when the Au NPs were illuminated with 532 nm light, strong absorption was observed owing to the LSPR. However, under visible-light irradiation, a slight positive shift of 25 mV is driven by hot-electron transfer into CB energy of TiO2 across the Schottky junction, followed by absorption and plasmonic dephasing as illustrated in Fig. 23(d); thereby leaving the positively charged holes on the Au NPs. Under simulated sunlight, an accumulation-type interfacial band alignment was formed that promoted strong electron transfer from TiO2 to Au, as shown in Fig. 23(e) and (f).
Thus, a large flux of electrons recombines with holes produced from the Au by plasmonic damping, making the TiO2 photocatalytically active. Although these hot electrons underwent thermalization within picoseconds, gas evolution indicated charge transfer to the CO2 molecules. Thus, the key point of this study is that optically controlled product selectivity is a most delicate technique.
In conclusion, plasmonic photocatalysis provides an opportunity for the optimization of hydrocarbon products during CO2 reduction. However, this field must still face the challenge of understanding the reaction mechanism for various photocatalysts. Its major drawbacks include the high energy input, low yield, and poor catalyst stability. Several in situ spectroscopy techniques may help elucidate the complex reaction mechanisms at the molecular level. Nevertheless, plasmonic photocatalysis has immense potential for realizing high CO2 photoreduction yields in the near future. The photocatalytic CO2 reduction activity of plasmonic materials is summarized in Table 8.
| Catalyst | Feed gas composition | Light source | Reducing agent | Reaction conditions | Reactor | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Ag@Ni/SiO2 | CO2 + H2 + N2 (1:4:1) |
405 nm laser light | H2 | 50 mg sample | — | CH4 selectivity: 55% | 399 |
| Au NP (TiO2/Au) | 99.9999% CO2 + H2O (7:25) |
UV | H2O | 100 mg sample, 50 °C | Flow reactor | CH4: 74.1 μmol g−1 (15 h) | 397 |
| Au/Ag alloy coated on TiO2 | CO2 + H2 | 150 mW cm−2 | H2 | 10 mg sample | Batch reactor | CO: 1053 μmol g−1 h−1 | 250 |
| CH4: 1813 μmol g−1 h−1 | |||||||
| Au/g-C3N4 | 99.999% CO2 + H2O | 300 W Xe lamp | H2O | 50 mg sample | Flow reactor | CO: 13.17 μmol g−1 (2 h) | 400 |
| CH4: 3.10 μmol g−1 (2 h) | |||||||
| Au/TiO2 | 99.99% CO2 + H2 | 252 nm (150 mW cm−2) | H2 | Sample coated ceramic monolith | Monolith flow reactor | CO: 1223 μmol g−1 h−1 | 251 |
| CH4: 42 μmol g−1 h−1 | |||||||
| Pt/Au–SiO2 | CO2 + CH4 | Xe lamp (LA-251, 0.6 W cm−2) | — | 20 mg sample | — | CO: 122.1 μmol g−1 min−1 | 401 |
| CH4: 55.3 μmol g−1 min−1 | |||||||
| Ag@TiO2 core–shell | CO2 + H2O | 300 W Xe | H2O | — | Pyrex glass reactor | CH4: 14.8 μmol g−1 (3 h) | 402 |
| Au@TiO2 yolk–shell hollow spheres | CO2 + H2O | 300 W Xe lamp | H2O | 10 mg sample and 0.4 mL DI water | Flow reactor | CH4: 2.57 μmol g−1 h−1 | 403 |
| C2H6: 1.67 μmol g−1 h−1 | |||||||
| Au–Cu nanoalloy supported on TiO2 | 99.995% CO2 + H2O | 1000 W Xe lamp | H2O | Sample film | Batch reactor | CH4: 2000 μmol g−1 h−1 | 127 |
| Ag–AgCl/C3N4 | 99.999% CO2 + H2O | 15 W daylight map | H2O | Sample coated on glass rods for immobilization | Tubular fixed-bed reactor | CH4: around 10 μmol g−1 (10 h) | 404 |
6 Origin of carbon in CO2 photoreduction
Photocatalysts typically contain a certain amount of deposited carbonaceous species, especially on the surface, and various suggestions have been put forward for their origin. Literature reports have ascribed these species to a combination of the following factors: (i) the attachment of organic materials to the photocatalyst during synthesis, e.g., the use of methanol as a hole scavenger during photodeposition; (ii) CO2 adsorption from the air to form various species such as “C”, “CO32−”, and “HCO3−” as shown in eqn (26) and (27); and (iii) the adsorption of other organic molecules such as HCOOH from the air.405–408 These species have been reported to be adsorbed by reactive surface sites, which are typically oxygen vacancies (Vo) and hydroxyl groups (OH−). The formation mechanism of these species has been reported to consume the surface defects as shown in eqn (26) and (27). In this regard, Zou and co-workers monitored the surface saturation of amorphous zinc germanate (α-ZnGeO) with CO2 using C 1s X-ray photoelectron spectroscopy.408 In addition, Diebold and co-workers observed the selective adsorption of HCOOH from the air by extremely clean TiO2 under a controlled environment.405| CO2 + Ov → C + O2 | (26) |
| CO2 + OH− → HCO3− | (27) |
The carbonaceous species, when illuminated under an inert or CO2− containing atmosphere, decompose to form hydrocarbon products similar to those generated by photocatalytic CO2 reduction. Moreover, in some cases, the yield under an inert atmosphere can even exceed that under CO2, as reported by Xu and co-workers for their Bi2WO6–TiO2 nanosheets (B–T) as shown in Fig. 24(a).407 They also observed a photocatalytic yield under anhydrous conditions, as presented in Fig. 24(b), further confirming the presence of hydrocarbon residues. On this basis, the authors recommended the organic-free synthesis of photocatalytic materials. In a similar study, Mul and co-workers found that Cu(I)/TiO2 synthesized using an organic compound (polyethylene glycol (PEG)) generated more CO compared to the same catalyst synthesized without PEG.409 They thus attributed the CO to carbon deposits originating from the PEG and proposed the mechanism shown in eqn (28) and (29). In addition to organic compounds, surface-adsorbed CO2 and water can also be transformed into products. Under such circumstances, there is a need for the two reactions, i.e., photodecomposition of the carbonaceous species and photocatalytic CO2 reduction, to be evaluated separately.
| CO2 + C → 2CO | (28) |
| H2O + C → CO + H2 | (29) |
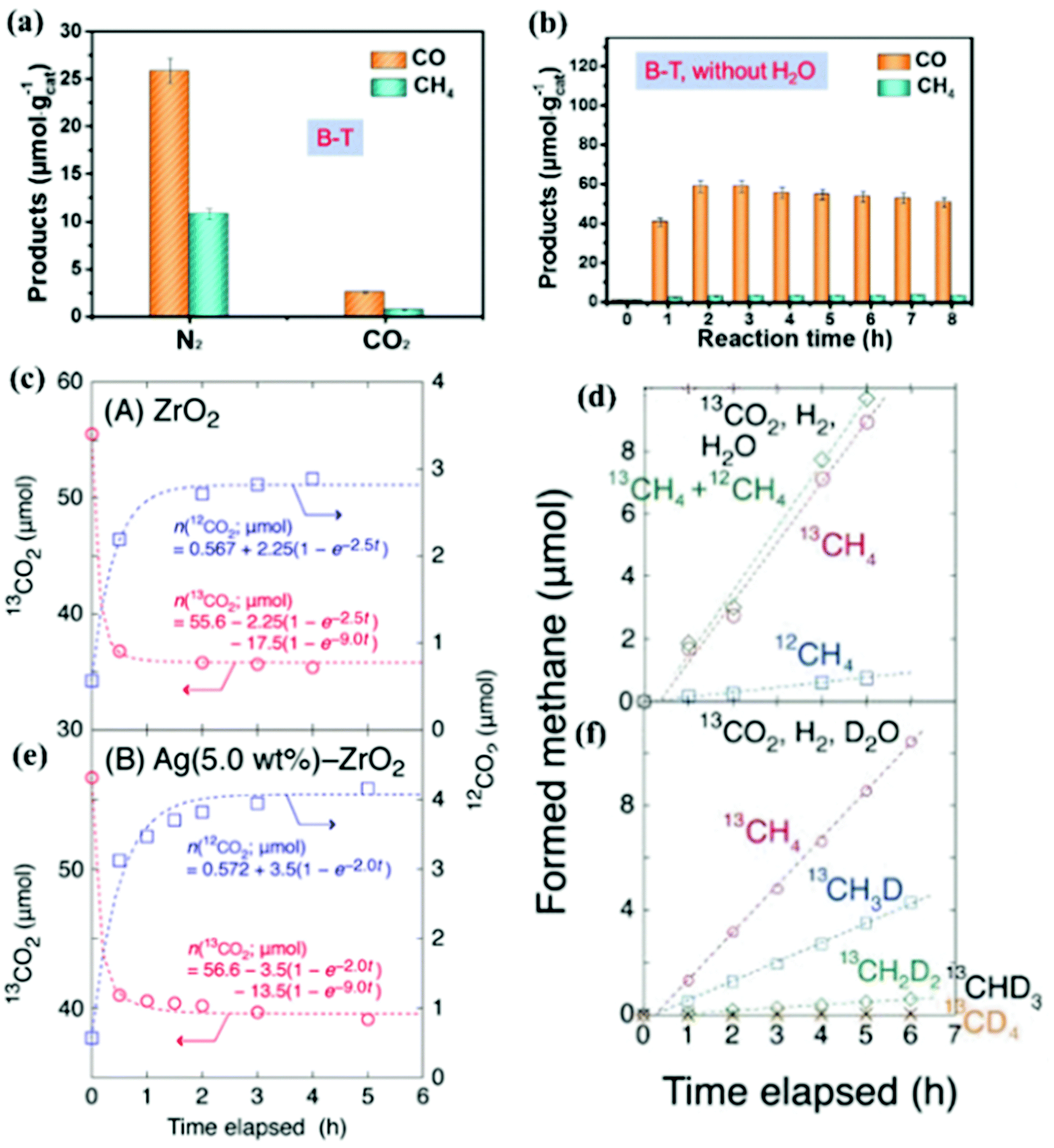 | ||
| Fig. 24 (a) and (b) Photocatalytic yields exhibited by B–T under (a) humid CO2 and N2 and (b) anhydrous N2. Reproduced with permission from ref. 407, Copyright 2018, Elsevier. (c) and (e) 13CO2 exchange reactions under UV-vis light for (c) ZrO2 and (e) Ag–ZrO2. Reproduced with permission from ref. 410, Copyright 2019, American Chemical Society. (d) and (f) Photocatalytic yields from irradiation of Ni–ZrO2 under (d) 13CO2, H2, and H2O, and (f) 13CO2, H2, and D2O. Reproduced with permission from ref. 411, Copyright 2021, Wiley-VCH. | ||
A photocatalyst with pre-adsorbed carbonaceous species may exhibit two types of activity depending upon the reactants consumed. The first is transient activity, in which the photocatalyst exhibits a sudden rise in photocatalytic yield during the initial minutes of photocatalytic CO2 reduction, and the second is steady-state activity, in which the photocatalyst displays a continuous low yield. The former originates from the consumption of pre-adsorbed species such as HCO3− and formate, while the latter results from the real photocatalytic sites mediating CO2 reduction.412 Considering the participation of these two reactions, the overall yield determination could be misleading. Therefore, to evaluate the actual yield, it is essential to estimate the yield derived from the pre-adsorbed carbonaceous species.409,413 In this regard, various treatment methods and spectroscopic techniques could be applied. There have been several reports of methods suitable for removing the carbonaceous/hydrocarbonaceous species, including thermal treatment, prolonged illumination, air/vacuum annealing, and washing with various solvents.252,414,415 As far as the efficacy of such remedial methods is concerned, these species can be reduced using such techniques, but it is hard to completely eliminate them. For example, Mul and co-workers reported that under repeated cycles of irradiation the yield from the carbonaceous species decreased substantially but did not disappear.415 Zou and co-workers presented an interesting explanation for this behavior, noting the formation of oxygen vacancies in their α-ZnGeO catalyst under light irradiation, which upon exposure to the ambient environment readily adsorbed CO2.408 This adsorbed CO2 may be the source of the photocatalytic yield under inert conditions. Therefore, these carbonaceous species are formed continuously and cannot be easily removed. Moreover, the discussed methods may result in alterations to the morphology and reactivity; thus, it is highly desirable to develop methods for solving this issue without altering the beneficial photocatalytic properties.
Isotopic labeling studies with 13CO2 have indicated that carbonate anions intercalated within layered double hydroxide (LDH) tend to exchange atmospheric CO2via dynamic breathing.416 Taking advantage of this phenomenon, 13CO2 can be used as a reactant molecule to trace the actual activity originating from photocatalytic CO2 reduction. In this regard, various studies have reported the utilization of 13CO2 as a tracer molecule, where the resulting products (i.e., 13CO and 13CH4) definitively originate from the 13CO2 photoreduction reaction confirmed by gas chromatography–mass spectrometry (GC-MS). However, most of these studies were performed in batch reactors; although this permits determination of the relative contributions to the product yields, the reaction rate cannot be reliably calculated, which could lead to inaccurate kinetic real reaction rates of photocatalytic 13CO2 reduction by continuously monitoring the yields of 13CO and 13CH4.410,411 In addition, they also explored the exchange of 13CO2 with already adsorbed 12CO2 under irradiation. They found that the exchange of CO2 molecules eventually reached equilibrium, as shown in Fig. 24(c) and (e), and under irradiation this occurred rapidly.410 Considering the presence of pre-adsorbed CO2 and other carbonaceous species, the yield originated from both sources, as shown in Fig. 24(d). They also noted the ratio of product formation rate is agreement with D (Deuterium) ratio in reactant (8.9%), as shown in Fig. 24(f). Under such circumstances, the pre-adsorbed water/hydroxyl groups acted as a source of H+; therefore, in addition to confirming the origin of carbon, the origin modeling of the reactions.5,407
Izumi and co-workers provided insights into the evaluation of the hydrogen are also important, which could be determined by following similar procedures.411 In addition to isotopic analysis of the gaseous products, the intermediate products of 13CO2 over the surface of the photocatalyst could be analyzed by secondary-ion mass spectrometry (SIMS) to further confirm the origin of the products. One such study was performed by Zou and co-workers, in which they confirmed the presence of 13C on the surface of α-Zn–Ge–O owing to its deposition during the reduction of 13CO2.408 Moreover, the participation of both hydrogen and carbon from H2O and CO2, respectively, has been confirmed by NMR studies.42,417 In conclusion, understanding the origin of carbon in photocatalytic CO2 reduction is vital and it has been studied by state-of-art equipment such as GC-MS, SIMS, and NMR.
7 Economic/commercial viability
The worldwide sequestration of CO2 emissions currently accounts for a mere 1% of the total CO2 generated. The resulting continuous increase in atmospheric CO2 concentrations is placing increasing pressure on governments to restrict emissions, and the eventual implementation of regulations for CO2 capture is inevitable.418 When this happens, the CO2 utilization landscape will be realigned and the trend will shift to CO2 capture, storage, and utilization technologies.419 The resulting investments will largely go to technologies that are not viable prior to these regulations. It is anticipated that investors will be primarily interested in technologies that offer maximum benefits by producing valuable chemicals or fuels from CO2. To date, urea and methanol production facilities are the major industrial consumers of CO2, but the scale of these industries is insufficient.420 Hence, finding new opportunities for utilizing CO2 while producing value-added products will surely prove useful.421Thermal power generation and the transportation sector account for two-thirds of CO2 emissions; therefore, finding viable CO2 conversion solutions for these sectors will enhance the scale of CO2 harvesting.422 Various alternative technologies to CO2 reduction, such as photovoltaics, hydroelectricity, and wind power, are becoming increasingly popular because of their renewability and sustainability.423,424 However, considering the current rate of adoption of these technologies, it may take some time to replace fossil energy and reduce its deleterious effects on the environment.425,426 In addition, the energy garnered by these resources cannot be directly applied to the current transportation infrastructure until there is a large-scale shift to electric vehicles.427 Therefore, the major portion of fossil utilities still requires viable, renewable, and eco-friendly solutions.424 Under these circumstances, the continued use of fossil resources will necessitate carbon capture facilities, which will eventually make the use of fossil fuels expensive.428 Thus, reversing the combustion reaction to produce fuels for transportation will eventually lead to mega-scale CO2 harvesting while producing high-value products.419
Photocatalytic CO2 reduction can be considered the reverse of combustion, enabling the transformation of CO2 to solar fuels using sunlight and water under ambient conditions.429 Although the industrial-scale process of thermocatalytic CO2 reduction can also facilitate this transformation, these systems operate at elevated pressures and temperatures and require additional energy inputs and reducing agents such as H2.430 If these are not obtained from renewable resources, the net effects are not promising. Hence, photocatalytic CO2 reduction seems lucrative relative to its peers with respect to commercialization owing to its numerous environmental and economic benefits.431 In this regard, considerable solar-to-fuel efficiencies (STF) have been achieved. For example, Rajh et al. reported an efficiency of 10.1% using their earth-abundant Cu2O photocatalyst.432 Although an STF of 10.1% is sufficient for commercialization, sustaining this efficiency over prolonged periods remains challenging. Therefore, despite the promise of these technologies, their commercialization is still hindered by catalyst instability.
High-value hydrocarbon products such as C2H4 and C2H6 provide another avenue for commercialization. However, the activity and stability remain low.433 C2 products from photocatalytic CO2 reduction have a high market value compared to C1 products.434 The production of longer-chain hydrocarbons with higher market value (e.g., propane) could further enhance the possibilities.435 With the remarkable recent advances in these technologies, there is currently great interest in converting CO2 to C2 and higher hydrocarbon products owing to their greater economic benefits.433 However, realizing C2 selectivity is challenging owing to the complex steps involved in the overall reaction.436 Furthermore, by following this route the CO2 harvesting scale will be low because specialty products are utilized in smaller volumes.419
Government subsidies are the major source of financing for emerging renewable technologies because these technologies are initially not competitive with respect to their well-established rivals; for example, biodiesel is subsidized to compete with mineral diesel.437,438 However, to obtain such financial support, the technology should be at the pilot/bench scale with considerable efficiencies; for instance, KIT in Germany has successfully converted renewable energy to methane with 51.3% conversion efficiency under the “Power to Gas” project and is committed to extending the energy conversion efficiency to 80%.439,440 Another example is high-temperature co-electrolysis to convert CO2 and steam to syngas, which proceeds with an efficiency of up to 70%.419,441 However, to the best of our knowledge, no pilot plant for photocatalytic CO2 reduction has yet been established.
8 Outlook
The conversion of CO2 into value-added chemicals using sunlight is now well established. Although many of the earlier studies into CO2 photoconversion focused on improving the photocatalytic activity, there are also numerous other crucial parameters that warrant serious attention. For example, photocatalyst stability is as important as high activity.186 However, in contrast to the extensive body of research focusing on achieving optimal activity, only limited information exists regarding the possible causes of deactivation.415 These causes include loss of the active oxidation state under irradiation, the buildup of irreducible reaction intermediates, and morphological changes.87,408,442 Various efforts have been made to circumvent these issues, such as the use of hole scavengers, construction of heterostructures to retain the desired oxidation states, and thermal and other treatments to remove the intermediates.252 However, considerable work is still required to circumvent the need for non-renewable hole scavengers and ineffective and energy-intensive photocatalyst regeneration processes.90In this review, various reasons have been discussed which contribute to destabilizing the photocatalyst. The widely identified reasons include losing active oxidation states of photocatalyst and accumulation of the reaction intermediates over the surface of the photocatalyst, i.e., carbon. Therefore, it is mandatory to overcome these limitations to achieve commercial-scale viability. In this regard, photocatalyst is required to re-attain the active oxidation states without being taken out of the reactor. Literature suggests that single metal atom photocatalyst can regain their lost active oxidation states just by exposing them to air, which arises from metal–support interactions.443 Moreover, intense UV irradiation under inert-humid conditions is believed to decompose the reaction intermediates. The protons generated from water oxidation will be utilized to reduce the already present carbonaceous species, and subsequently, activity can be restored. The researchers should focus on such developments which can make the photocatalytic reusable system simpler.
Besides stability, product selectivity is also underexplored, and in this regard, it is highly desirable to develop photocatalysts with the ability to mediate C–C coupling reactions by stabilizing the intermediate products. Several factors influence the product selectivity of CO2 conversions, including the type of materials, band potentials of semiconductors, the intermediate stabilization, etc. Furthermore, the reaction medium has a crucial influence on product selectivity; for example, CH3OH, C2H5OH, and formaldehyde can be generated in an aqueous system, while CO, CH4, and C2H6 production are more feasible in a gas phase system. In the literature, most of the photocatalysts have been reported for C1 products like CO and CH4; however, limited studies have been carried out for C2 selectivity. Literature suggests that metal nanoparticles like Pt, Pd, Au, etc., deposited on semiconductors have selectivity for CH4 formation due to their ability to supply sufficient protons. For the C2+ product, the C–H bonds can be formed by C–O bond cleavage and continuous protonation. The density of photogenerated electrons/holes and the stability of intermediates also impact the reaction's C2 selectivity; for example, stabilization of ˙CH3 radicals is beneficial for achieving C2 selectivity. Theoretically, the breakage of the C–O bonds at the single-metal site is easier than at the dual-metal site because the dual-metal site has a higher charge density due to the charge transfer. Thus, the intermediates become more stable at the dual-metal site.444 To prove the theoretical calculation, in situ/operando XAS analysis can provide the experimental observation on the local coordination environment and oxidation states of the metal center during the reaction. As a result, for C2 selectivity, it is preferable to carefully design catalysts to regulate the reaction intermediates. The combination of graphene with other semiconducting materials has been reported to enhance the C2 selectivity.
To achieve an industry-relevant photocatalytic CO2 reduction, CO2 capture, utilization, and storage also should be considered.445 An effective solution is still needed to avoid such additional costs, despite the use of abundant and cheap reactants. Furthermore, if the photocatalytic CO2 conversion-efficiency is low, then the CO2 concentration in the products may be too high to permit their direct use, thus necessitating separation of the hydrocarbons and CO2.446 One possible solution is passing the gaseous mixture through gas separation membranes, although the installation and operation of these systems would also lead to higher costs.447 Therefore, it would be highly desirable to search for solutions that help avoid these extra processing steps.131
To avoid the construction of CO2-collecting infrastructure, CO2 can be captured from the atmosphere. This abundant resource, if it can be harnessed, would provide many benefits: it avoids the purification and storage of CO2, and in some cases, it contains abundant moisture, thus avoiding the need for humidification.448 However, the use of low concentration CO2 is associated with its own disadvantages, such as low adsorption over a photocatalyst. On the other hand, strategies to minimize the need for post-reaction product purification are also needed, such as the development of photocatalysts that can adsorb CO2 and H2O in higher amounts. Subsequently, the reactant-laden photocatalyst could be subjected to irradiation under controlled conditions to yield a CO2-free product mixture. Therefore, it would be desirable to explore such photocatalysts that can selectively adsorb atmospheric CO2 and H2O with high stability.
Thermal effects can also lead to an enhancement in the yield of the solar products of photocatalytic CO2 reduction.41 One possible way to exploit these effects is the concentration of solar light, which does not require the use of any fossil fuels. However, to enable photothermal reactions, special attention must be paid to the design of photoreactors and photocatalysts with good durability to withstand the harsh conditions. Fortunately, rather than raising the bulk temperature, the photocatalyst surface could be locally heated through LSPR. For example, Izumi and co-workers found that the temperature increased to 392 K owing to Ag LSPR. This dual role of light provides another avenue for enhancing the activity by taking advantage of synergistic effects, but this is only feasible with well-designed photocatalysts and must be explored further.410 Furthermore, the use of renewable hole scavengers such as glycerol, a byproduct of biodiesel manufacture, would also be desirable. In addition, facile photocatalyst regeneration processes must be developed, as in the case of some single-metal-atom catalysts, for which regeneration can be conveniently accomplished simply by exposing the catalyst to the environment.47
Considering the immense potential of CO2 reduction, huge investments are in place, and these will surely escalate in the near future. For instance, the Musk Foundation recently announced the $100 million X PRIZE for carbon removal.449 Similarly, the European Innovation Council has offered a €5 million prize for the development of a bench-scale prototype for artificial photosynthesis.450 Therefore, considering the current and future investments, it can be anticipated that CO2 reduction will eventually find a route to commercialization. In this regard, photocatalytic CO2 reduction could make a strong impression in terms of attracting investment. Such heavy investments in this field will expedite further research and help realize the true potential of photocatalytic CO2 reduction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Ministry of Science and ICT in Korea (2021R1A2C2009459 and 2021M3I3A1085039). This paper is dedicated to Professor Michael R. Hoffman (Caltech, USA) and Professor Wan In Lee (Inha University, Korea), who have contributed to the research and development of photocatalytic fields throughout their lives.References
- Z. Wang, H. Song, H. Liu and J. Ye, Angew. Chem., Int. Ed., 2020, 59, 8016–8035 CrossRef CAS PubMed
.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed
- G. M. Hallegraeff, J. Phycol., 2010, 46, 220–235 CrossRef CAS
- J. He and C. Janáky, ACS Energy Lett., 2020, 5, 1996–2014 CrossRef CAS
- S. Sorcar, Y. Hwang, J. Lee, H. Kim, K. M. Grimes, C. A. Grimes, J.-W. Jung, C.-H. Cho, T. Majima, M. R. Hoffmann and S.-I. In, Energy Environ. Sci., 2019, 12, 2685–2696 RSC
- S. S. Meryem, S. Nasreen, M. Siddique and R. Khan, Rev. Chem. Eng., 2018, 34, 409–425 CrossRef CAS
- U. Nations, 21st Conference of the Parties, United Nations Framework Convention on Climate Change, UNFCCC, Paris, 2015.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and Ib Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- T. Kong, Y. Jiang and Y. Xiong, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2017, 11, 453–460 CrossRef CAS
- C. Hiragond, S. Ali, S. Sorcar and S.-I. In, Catalysts, 2019, 9, 370 CrossRef CAS
- C. B. Hiragond, N. S. Powar and S.-I. In, Nanomaterials, 2020, 10, 2569 CrossRef CAS PubMed
- J. Ye, J. Yu, Y. Zhang, M. Chen, X. Liu, S. Zhou and Z. He, Appl. Catal., B, 2019, 257, 117916 CrossRef CAS
- S. K. Kuk, Y. Ham, K. Gopinath, P. Boonmongkolras, Y. Lee, Y. W. Lee, S. Kondaveeti, C. Ahn, B. Shin, J. Lee, S. Jeon and C. B. Park, Adv. Energy Mater, 2019, 9, 1900029 CrossRef
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS
- M.-Y. Lee, K. T. Park, W. Lee, H. Lim, Y. Kwon and S. Kang, Crit. Rev. Environ. Sci. Technol., 2020, 50, 769–815 CrossRef CAS
- R. C. Pullar, R. M. Novais, A. P. F. Caetano, M. A. Barreiros, S. Abanades and F. A. C. Oliveira, Front. Chem., 2019, 7, 601 CrossRef CAS PubMed
- X.-Y. Wu and A. F. Ghoniem, Prog. Energy Combust. Sci., 2019, 74, 1–30 CrossRef
- J. Jia, P. G. O’Brien, L. He, Q. Qiao, T. Fei, L. M. Reyes, T. E. Burrow, Y. Dong, K. Liao, M. Varela, S. J. Pennycook, M. Hmadeh, A. S. Helmy, N. P. Kherani, D. D. Perovic and G. A. Ozin, Adv. Sci., 2016, 3, 1600189 CrossRef
- X. Meng, T. Wang, L. Liu, S. Ouyang, P. Li, H. Hu, T. Kako, H. Iwai, A. Tanaka and J. Ye, Angew. Chem., Int. Ed., 2014, 53, 11478–11482 CrossRef CAS PubMed
- H. Li, W. Tu, Y. Zhou and Z. Zou, Adv. Sci., 2016, 3, 1500389 CrossRef PubMed
- Y. Zhou, Z. Wang, L. Huang, S. Zaman, K. Lei, T. Yue, Z. Li, B. You and B. Y. Xia, Adv. Energy Mater, 2021, 11, 2003159 CrossRef CAS
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS
- P. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900281 CrossRef PubMed
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol. C, 2015, 24, 16–42 CrossRef CAS
- D. Li, M. Kassymova, X. Cai, S.-Q. Zang and H.-L. Jiang, Coord. Chem. Rev., 2020, 412, 213262 CrossRef CAS
- Y. Y. Lee, H. S. Jung and Y. T. Kang, J. CO2 Util., 2017, 20, 163–177 CrossRef CAS
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CrossRef CAS
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef
- X. Jiao, K. Zheng, Z. Hu, Y. Sun and Y. Xie, ACS Cent. Sci., 2020, 6, 653–660 CrossRef CAS PubMed
- S. Trivedi, D. Prochowicz, A. Kalam, M. M. Tavakoli and P. Yadav, Renewable Sustainable Energy Rev., 2021, 145, 111047 CrossRef CAS
- J. Ran, M. Jaroniec and S. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed
- K. Yang, Z. Yang, C. Zhang, Y. Gu, J. Wei, Z. Li, C. Ma, X. Yang, K. Song, Y. Li, Q. Fang and J. Zhou, Chem. Eng. J, 2021, 418, 129344 CrossRef CAS
- M. Yang, M. Gao, M. Hong and G. W. Ho, Adv. Mater., 2018, 30, 1802894 CrossRef
- X. Xiang, F. Pan and Y. Li, Adv. Compos. Hybrid Mater., 2018, 1, 6–31 CrossRef CAS
- T.-H. Lai, K. Katsumata and Y.-J. Hsu, Nanophotonics, 2021, 10, 777–795 CrossRef CAS
- M. Sachs, E. Pastor, A. Kafizas and J. R. Durrant, J. Phys. Chem. Lett., 2016, 7, 3742–3746 CrossRef CAS
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal., B, 2013, 134, 349–358 CrossRef
- F. Zhang, Y.-H. Li, M.-Y. Qi, Y. M. A. Yamada, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem Catal., 2021, 1, 1–26 CrossRef
- N. M. Dimitrijevic, B. K. Vijayan, O. G. Poluektov, T. Rajh, K. A. Gray, H. He and P. Zapol, J. Am. Chem. Soc., 2011, 133, 3964–3971 CrossRef CAS
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS
- S. Ali, J. Lee, H. Kim, Y. Hwang, A. Razzaq, J.-W. Jung, C.-H. Cho and S.-I. In, Appl. Catal., B, 2020, 279, 119344 CrossRef CAS
- F. Zhang, Y.-H. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2020, 268, 118380 CrossRef CAS
- L. Liu, C. Zhao, J. T. Miller and Y. Li, J. Phys. Chem. C, 2016, 121, 490–499 CrossRef
- S. Ali, M. C. Flores, A. Razzaq, S. Sorcar, C. B. Hiragond, H. R. Kim, Y. H. Park, Y. Hwang, H. S. Kim, H. Kim, E. Gong, J. Lee, D. Kim and S.-I. In, Catalysts, 2019, 9, 727 CrossRef CAS
- S. Sorcar, S. Yoriya, H. Lee, C. A. Grimes and S. P. Feng, Mater. Today Chem., 2020, 16, 100264 CrossRef CAS
- Y. Lin, C. Deng, L. Wu, Y. Zhang, C. Chen, W. Ma and J. Zhao, Energy Environ. Sci., 2020, 13, 2602–2617 RSC
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L. Wu, C. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed
- Z. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef
- D. P. Van Vuuren, E. Stehfest, D. E. H. J. Gernaat, J. C. Doelman, M. Van den Berg, M. Harmsen, H. S. de Boer, L. F. Bouwman, V. Daioglou, O. Y. Edelenbosch, B. Girod, T. Kram, L. Lassaletta, P. L. Lucas, H. Meijl, C. Müller, B. J. Ruijven, S. v. d. Sluis and A. Tabeau, Glob. Environ. Change, 2017, 42, 237–250 CrossRef
-
A. Dechezleprêtre, R. Martin and S. Bassi, Handbook on Green Growth, Edward Elgar Publishing, 2019 Search PubMed
- H. N. Larsen, C. Solli and J. Pettersena, Energy Procedia, 2012, 20, 354–363 CrossRef
-
T. Inui, M. Anpo, K. Izui, S. Yanagida and T. Yamaguchi, Advances in chemical conversions for mitigating carbon dioxide, Elsevier, 1998 Search PubMed
- S.-E. Park, J.-S. Chang and K.-W. Lee, Carbon Dioxide Utilization for Global Sustainability: Proceedings of the 7th International Conference on Carbon Dioxide Utilization, Seoul, Korea, October 12–16, 2003, Elsevier, 2004.
-
B. Eliasson, P. Riemer and A. Wokaun, Greenhouse gas control technologies, Elsevier, 1999 Search PubMed
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu and Y. Yan, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS
- H. Wang, H. Rong, D. Wang, X. Li, E. Zhang, X. Wan, B. Bai, M. Xu, J. Liu, J. Liu, W. Chen and J. Zhang, Small, 2020, 16, 2000426 CrossRef CAS PubMed
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 RSC
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS
- L. Liang, X. Li, Y. Sun, Y. Tan, X. Jiao, H. Ju, Z. Qi, J. Zhu and Y. Xie, Joule, 2018, 2, 1004–1016 CrossRef CAS
- S. Rej, M. Bisetto, A. Naldoni and P. Fornasiero, J. Mater. Chem. A, 2021, 9, 5915–5951 RSC
- L. Shi, X. Ren, Q. Wang, W. Zhou and J. Ye, J. Mater. Chem. A, 2021, 9, 2421–2428 RSC
- X. Wang, F. Wang, Y. Sang and H. Liu, Adv. Energy Mater., 2017, 7, 1700473 CrossRef
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed
- T.-H. Lai, K. Katsumata and Y.-J. Hsu, Nanophotonics, 2020, 10, 777–795 CrossRef
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC
- X. Wang, A. Kafizas, X. Li, S. J. A. Moniz, P. J. T. Reardon, J. Tang, I. P. Parkin and J. R. Durrant, J. Phys. Chem. C, 2015, 119, 10439–10447 CrossRef CAS
- C. Liu, K. Huang, W.-T. Park, M. Li, T. Yang, X. Liu, L. Liang, T. Minari and Y.-Y. Noh, Mater. Horiz., 2017, 4, 608–618 RSC
- X. Li, H. Liu, D. Luo, J. Li, Y. Huang, H. Li, Y. Fang, Y. Xu and L. Zhu, Chem. Eng. J., 2012, 180, 151–158 CrossRef CAS
- O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS PubMed
- P. Reñones, A. Moya, F. Fresno, L. Collado, J. J. Vilatela and A. Víctor, J. CO2 Util., 2016, 15, 24–31 CrossRef
- P. Maity, O. F. Mohammed, K. Katsiev and H. Idriss, J. Phys. Chem. C, 2018, 122, 8925–8932 CrossRef CAS
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS PubMed
- W. Chu, Q. Zheng, O. V. Prezhdo and J. Zhao, J. Am. Chem. Soc., 2020, 142, 3214–3221 CrossRef CAS
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS
- W.-J. Yin, B. Wen, Q. Ge, X.-B. Li, G. Teobaldi and L.-M. Liu, Dalton Trans., 2020, 49, 12918–12928 RSC
- P. Liu, X. Peng, Y.-L. Men and Y.-X. Pan, Green Chem. Eng., 2016, 18, 139–143 RSC
- C. B. Hiragond, S. Ali, S. Sorcar and S.-I. In, Catalysts, 2019, 9, 370 CrossRef CAS
- Y. Li, W. N. Wang, Z. Zhan, M. H. Woo, C. Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS
- K. Tennakone, A. H. Jayatissa and S. Punchihewa, J. Photochem. Photobiol. A Chem., 1989, 49, 369–375 CrossRef CAS
- F. Fresno, I. J. Villar-García, L. Collado, E. Alfonso-González, P. Reñones, M. Barawi and V. A. de la Peña O’Shea, J. Phys. Chem. Lett., 2018, 9, 7192–7204 CrossRef CAS PubMed
- J. Low, J. Yu and W. Ho, J. Phys. Chem. Lett., 2015, 6, 4244–4251 CrossRef CAS
- D. Chen, X. Zhang and A. F. Lee, J. Mater. Chem. A, 2015, 3, 14487–14516 RSC
- L. Andronic and A. Enesca, Front. Chem., 2020, 8, 565489 CrossRef CAS PubMed
- S. Sorcar, Y. Hwang, C. A. Grimes and S.-I. In, Mater. Today, 2017, 20, 507–515 CrossRef CAS
- V. Natu, R. Pai, M. Sokol, M. Carey, V. Kalra and M. W. Barsoum, Chem, 2020, 6, 616–630 CAS
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S.-I. In, Energy Environ. Sci., 2018, 11, 3183–3193 RSC
- C. B. Hiragond, J. Lee, H. Kim, J.-W. Jung, C.-H. Cho and S.-I. In, Chem. Eng. J., 2020, 416, 127978 CrossRef
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed
- Y. Ji and Y. Luo, ACS Catal., 2016, 6, 2018–2025 CrossRef CAS
- W. Ong, L. K. Putri and A. R. Mohamed, Chem. – Eur. J., 2020, 26, 9710–9748 CrossRef CAS PubMed
- E. Vahidzadeh, S. Zeng, A. P. Manuel, S. Riddell, P. Kumar, K. M. Alam and K. Shankar, ACS Appl. Mater. Interfaces, 2021, 13, 7248–7258 CrossRef CAS PubMed
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743–1749 CrossRef CAS
- X. Chen, L. Liu, Y. Y. Peter and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed
- N. Liu, V. Häublein, X. Zhou, U. Venkatesan, M. Hartmann, M. Mačković, T. Nakajima, E. Spiecker, A. Osvet, L. Frey and P. Schmuki, Nano Lett., 2015, 15, 6815–6820 CrossRef CAS PubMed
- A. Sinhamahapatra, J.-P. Jeon and J.-S. Yu, Energy Environ. Sci., 2015, 8, 3539–3544 RSC
- X. Zhou, N. Liu, J. Schmidt, A. Kahnt, A. Osvet, S. Romeis, E. M. Zolnhofer, V. R. R. Marthala, D. M. Guldi, W. Peukert, M. Hartmann, K. Meyer and P. Schmuki, Adv. Mater., 2017, 29, 1604747 CrossRef PubMed
-
N. F. Mott, Mathematical Proceedings of the Cambridge Philosophical Society, Cambridge University Press, 1938, vol. 34, pp. 568–572 Search PubMed
- W. Schottky, Naturwissenschaften, 1938, 26, 843 CrossRef CAS
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 1–9 Search PubMed
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS
- G. Liu, J. Pan, L. Yin, J. T. S. Irvine, F. Li, J. Tan, P. Wormald and H. Cheng, Adv. Funct. Mater., 2012, 22, 3233–3238 CrossRef CAS
- L. Zhang, M. S. Tse, O. K. Tan, Y. X. Wang and M. Han, J. Mater. Chem. A, 2013, 1, 4497–4507 RSC
- N. O. Gopal, H.-H. Lo, T.-F. Ke, C.-H. Lee, C.-C. Chou, J.-D. Wu, S.-C. Sheu and S.-C. Ke, J. Phys. Chem. C, 2012, 116, 16191–16197 CrossRef CAS
- X. Yan, K. Yuan, N. Lu, H. Xu, S. Zhang, N. Takeuchi, H. Kobayashi and R. Li, Appl. Catal., B, 2017, 218, 20–31 CrossRef CAS
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 7610–7627 CrossRef CAS PubMed
- M. Liu, L. Zheng, X. Bao, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, B. Huang and Z. Zheng, Chem. Eng. J., 2021, 405, 126654 CrossRef CAS
- Y. J. Jang, J.-W. Jang, J. Lee, J. H. Kim, H. Kumagai, J. Lee, T. Minegishi, J. Kubota, K. Domen and J. S. Lee, Energy Environ. Sci., 2015, 8, 3597–3604 RSC
- G. Li, Y. Sun, Q. Zhang, Z. Gao, W. Sun and X. Zhou, Chem. Eng. J., 2021, 410, 128397 CrossRef CAS
- N. Blommaerts, N. Hoeven, D. A. Esteban, R. Campos, M. Mertens, R. Borah, A. Glisenti, K. De Wael, S. Bals, S. Lenaerts, S. W. Verbruggen and P. Cool, Chem. Eng. J., 2021, 410, 128234 CrossRef CAS
- Y. Zhu, Z. Xu, Q. Lang, W. Jiang, Q. Yin, S. Zhong and S. Bai, Appl. Catal., B, 2017, 206, 282–292 CrossRef CAS
- W.-N. Wang, W.-J. An, B. Ramalingam, S. Mukherjee, D. M. Niedzwiedzki, S. Gangopadhyay and P. Biswas, J. Am. Chem. Soc., 2012, 134, 11276–11281 CrossRef CAS PubMed
- S. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef PubMed
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., 2013, 125, 5888–5891 CrossRef
- R. Long, Y. Li, Y. Liu, S. Chen, X. Zheng, C. Gao, C. He, N. Chen, Z. Qi, L. Song, J. Jiang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2017, 139, 4486–4492 CrossRef CAS PubMed
- J. C. Wang, L. Zhang, W. X. Fang, J. Ren, Y. Y. Li, H. C. Yao, J. S. Wang and Z. J. Li, ACS Appl. Mater. Interfaces, 2015, 7, 8631–8639 CrossRef CAS PubMed
- G. Y. Yao and Z. Y. Zhao, J. Mater. Chem. C, 2020, 8, 8567–8578 RSC
- L. Guo, Y. Wang and T. He, Chem. Rec., 2016, 16, 1918–1933 CrossRef CAS PubMed
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS
- Q. Yuan, D. Liu, N. Zhang, W. Ye, H. Ju, L. Shi, R. Long, J. Zhu and Y. Xiong, Angew. Chem., 2017, 129, 4270–4274 CrossRef
- Y. Liu, B. Zhang, L. Luo, X. Chen, Z. Wang, E. Wu, D. Su and W. Huang, Angew. Chem., Int. Ed., 2015, 54, 15260–15265 CrossRef CAS
- S. N. Talapaneni, G. Singh, I. Y. Kim, K. AlBahily, A. H. Al-Muhtaseb, A. S. Karakoti, E. Tavakkoli and A. Vinu, Adv. Mater., 2020, 32, 1904635 CrossRef CAS
- X. Meng, S. Ouyang, T. Kako, P. Li, Q. Yu, T. Wang and J. Ye, Chem. Commun., 2014, 50, 11517–11519 RSC
- J. Z. Y. Tan, S. Gavrielides, H. R. Xu, W. A. Thompson and M. M. Maroto-Valer, RSC Adv., 2020, 10, 27989–27994 RSC
- T. Xiong, W. Cen, Y. Zhang and F. Dong, ACS Catal., 2016, 6, 2462–2472 CrossRef CAS
- Y. Liao, Z. Hu, Q. Gu and C. Xue, Molecules, 2015, 20, 18847–18855 CrossRef CAS PubMed
- Z. He, L. Wen, D. Wang, Y. Xue, Q. Lu, C. Wu, J. Chen and S. Song, Energy Fuels, 2014, 28, 3982–3993 CrossRef CAS
- A. Nakada, K. Koike, K. Maeda and O. Ishitani, Green Chem., 2016, 18, 139–143 RSC
- H. A. E. Omr, M. W. Horn and H. Lee, Catalysts, 2021, 11, 418 CrossRef CAS
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 1–17 CrossRef
- J. Zhao, M. A. Holmes and F. E. Osterloh, ACS Nano, 2013, 7, 4316–4325 CrossRef CAS
- A. D. Yoffe, Adv. Phys., 1993, 42, 173–262 CrossRef CAS
- K. A. S. Fernando, S. Sahu, Y. Liu, W. K. Lewis, E. A. Guliants, A. Jafariyan, P. Wang, C. E. Bunker and Y.-P. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 8363–8376 CrossRef CAS PubMed
- M. Zhou, S. Wang, P. Yang, C. Huang and X. Wang, ACS Catal., 2018, 8, 4928–4936 CrossRef CAS
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed
- C. Huang, J. Qiao, R.-N. Ci, X.-Z. Wang, Y. Wang, J.-H. Wang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1244–1257 CAS
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, J. Am. Chem. Soc., 2017, 139, 4250–4253 CrossRef CAS PubMed
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 4246–4249 CrossRef CAS PubMed
- Y. Deng, M. Chen, G. Chen, W. Zou, Y. Zhao, H. Zhang and Q. Zhao, ACS Omega, 2021, 6, 4247–4254 CrossRef CAS PubMed
- H. Li, X. Zhang and D. R. MacFarlane, Adv. Energy Mater., 2015, 5, 1401077 CrossRef
- M. Li, M. Wang, L. Zhu, Y. Li, Z. Yan, Z. Shen and X. Cao, Appl. Catal., B, 2018, 231, 269–276 CrossRef CAS
- H. Wu, X. Li, C. Tung and L. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed
- C. Huang, R. Guo, W. Pan, J. Tang, W. Zhou, X. Liu, H. Qin and P. Jia, Appl. Surf. Sci., 2019, 464, 534–543 CrossRef CAS
- H. Zhu, X. Gao, Y. Lan, D. Song, Y. Xi and J. Zhao, J. Am. Chem. Soc., 2004, 126, 8380–8381 CrossRef CAS PubMed
- A. Selmani, M. Špadina, M. Plodinec, I. Delač Marion, M. G. Willinger, J. Lützenkirchen, H. D. Gafney and E. Redel, J. Phys. Chem. C, 2015, 119, 19729–19742 CrossRef CAS
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS PubMed
- W. Zhou, Z. Yin, Y. Du, X. Huang, Z. Zeng, Z. Fan, H. Liu, J. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed
- J. Huang, Y. Lai, F. Pan, L. Yang, H. Wang, K. Zhang, H. Fuchs and L. Chi, Small, 2014, 10, 4865–4873 CrossRef CAS PubMed
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K.-Q. Zhang, S. S. Al-Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 RSC
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed
- Y. Li, Y.-L. Li, B. Sa and R. Ahuja, Catal. Sci. Technol., 2017, 7, 545–559 RSC
- Q. Xiang, B. Cheng and J. Yu, Angew. Chem., Int. Ed., 2015, 54, 11350–11366 CrossRef CAS PubMed
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. A. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS
- M. Tahir and N. S. Amin, Appl. Catal., A, 2013, 467, 483–496 CrossRef CAS
- S. Delavari and N. A. S. Amin, Appl. Energy, 2016, 162, 1171–1185 CrossRef CAS
- M. Dilla, R. Schlögl and J. Strunk, ChemCatChem, 2017, 9, 696–704 CrossRef CAS
- S. In, M. G. Nielsen, P. C. K. Vesborg, Y. Hou, B. L. Abrams, T. R. Henriksen, O. Hansen and I. Chorkendorff, Chem. Commun., 2011, 47, 2613–2615 RSC
- X. Zhan, C. Yan, Y. Zhang, G. Rinke, G. Rabsch, M. Klumpp, A. I. Schäfer and R. Dittmeyer, React. Chem. Eng., 2020, 5, 1658–1670 RSC
- A. Visan, J. R. van Ommen, M. T. Kreutzer and R. G. H. Lammertink, Ind. Eng. Chem. Res., 2019, 58, 5349–5357 CrossRef CAS
- E. Pipelzadeh, V. Rudolph, G. Hanson, C. Noble and L. Wang, Appl. Catal., B, 2017, 218, 672–678 CrossRef CAS
- Z. Xiong, C.-C. Kuang, K.-Y. Lin, Z. Lei, X. Chen, B. Gong, J. Yang, Y. Zhao, J. Zhang, B. Xia and J. C. S. Wa, J. CO2 Util., 2018, 24, 500–508 CrossRef CAS
- X. An, K. Li and J. Tang, ChemSusChem, 2014, 7, 1086–1093 CrossRef CAS PubMed
- K. Adachi, K. Ohta and T. Mizuno, Sol. Energy, 1994, 53, 187–190 CrossRef CAS
- H. Yamashita, H. Nishiguchi, N. Kamada, M. Anpo, Y. Teraoka, H. Hatano, S. Ehara, K. Kikui, L. Palmisano, A. Sclafani, M. Schiavello and M. A. Fox, Res. Chem. Intermed., 1994, 20, 815–823 CrossRef CAS
- T. V. Nguyen and J. C. S. Wu, Appl. Catal., A, 2008, 335, 112–120 CrossRef CAS
- J. C. S. Wu, T.-H. Wu, T. Chu, H. Huang and D. Tsai, Top. Catal., 2008, 47, 131–136 CrossRef CAS
- A. A. Khan and M. Tahir, J. CO2 Util., 2019, 29, 205–239 CrossRef CAS
- Z. Xiong, Z. Lei, S. Ma, X. Chen, B. Gong, Y. Zhao, J. Zhang, C. Zheng and J. C. S. Wu, Appl. Catal., B, 2017, 219, 412–424 CrossRef CAS
- P.-Y. Liou, S.-C. Chen, J. C. S. Wu, D. Liu, S. Mackintosh, M. Maroto-Valer and R. Linforth, Energy Environ. Sci., 2011, 4, 1487–1494 RSC
- F. Li, L. Zhang, J. Tong, Y. Liu, S. Xu, Y. Cao and S. Cao, Nano Energy, 2016, 27, 320–329 CrossRef CAS
- H. Yamashita, K. Ikeue, T. Takewaki and M. Anpo, Top. Catal., 2002, 18, 95–100 CrossRef CAS
- M. Xing, Y. Zhou, C. Dong, L. Cai, L. Zeng, B. Shen, L. Pan, C. Dong, Y. Chai, J. Zhang and Y. Yin, Nano Lett., 2018, 18, 3384–3390 CrossRef CAS PubMed
- M. Subrahmanyam, S. Kaneco and N. Alonso-Vante, Appl. Catal., B, 1999, 23, 169–174 CrossRef CAS
- S. Poudyal and S. Laursen, Catal. Sci. Technol., 2019, 9, 1048–1059 RSC
- N. G. Moustakas and J. Strunk, Chem. – Eur. J., 2018, 24, 12739–12746 CrossRef CAS PubMed
- B. Ohtani, Phys. Chem. Chem. Phys., 2014, 16, 1788–1797 RSC
- J. Colina-Márquez, F. Machuca-Martínez and G. L. Puma, Environ. Sci. Technol., 2010, 44, 5112–5120 CrossRef PubMed
- W. A. Thompson, E. Sanchez Fernandez and M. M. Maroto-Valer, ACS Sustainable Chem. Eng., 2020, 8, 4677–4692 CrossRef CAS
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048 CrossRef CAS
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671 CrossRef CAS PubMed
- P. Verma and D. G. Truhlar, Theor. Chem. Acc., 2016, 135, 1–15 Search PubMed
- E. E. Salpeter and H. A. Bethe, Phys. Rev., 1951, 84, 1232 CrossRef
-
L. Hedin and S. Lundqvist, Solid state physics, Elsevier, 1970, vol. 23, pp. 1–181 Search PubMed
- L. Li, R. Zhang, J. Vinson, E. L. Shirley, J. P. Greeley, J. R. Guest and M. K. Y. Chan, Chem. Mater., 2018, 30, 1912–1923 CrossRef CAS PubMed
- J. Zhao, B. Liu, L. Meng, S. He, R. Yuan, Y. Hou, Z. Ding, H. Lin, Z. Zhang, X. Wang and J. Long, Appl. Catal., B, 2019, 256, 117823 CrossRef CAS
- S. S. Tafreshi, A. Z. Moshfegh and N. H. de Leeuw, J. Phys. Chem. C, 2019, 123, 22191–22201 CrossRef CAS
- S. E. Braslavsky, A. M. Braun, A. E. Cassano, A. V. Emeline, M. I. Litter, L. Palmisano, V. N. Parmon and N. Serpone, Pure Appl. Chem., 2011, 83, 931–1014 CAS
- S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS
- T. Bligaard, R. M. Bullock, C. T. Campbell, J. G. Chen, B. C. Gates, R. J. Gorte, C. W. Jones, W. D. Jones, J. R. Kitchin and S. L. Scott, ACS Catal., 2016, 6, 2590–2602 CrossRef CAS
- Y.-J. Yuan, Z.-J. Ye, H.-W. Lu, B. Hu, Y.-H. Li, D.-Q. Chen, J.-S. Zhong, Z.-T. Yu and Z.-G. Zou, ACS Catal., 2016, 6, 532–541 CrossRef CAS
- C. T. K. Nguyen, N. Q. Tran, S. Seo, H. Hwang, S. Oh, J. Yu, J. Lee, T. A. Le, J. Hwang, M. Kim and H. Lee, Mater. Today, 2020, 35, 25–33 CrossRef CAS
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed
- X.-M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M.-M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC
- K. Jiang, H. Wang, W.-B. Cai and H. Wang, ACS Nano, 2017, 11, 6451–6458 CrossRef CAS PubMed
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS
- J. Bullock, D. F. Srankó, C. M. Towle, Y. Lum, M. Hettick, M. C. Scott, A. Javey and J. Ager, Energy Environ. Sci., 2017, 10, 2222–2230 RSC
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS
- M. Zubair, H. Kim, A. Razzaq, C. A. Grimes and S. Il In, J. CO2 Util., 2018, 26, 70–79 CrossRef CAS
- T. Maschmeyer and M. Che, Angew. Chem., 2010, 122, 1578–1582 CrossRef
- S. Docao, A. R. Koirala, M. G. Kim, I. C. Hwang, M. K. Song and K. B. Yoon, Energy Environ. Sci., 2017, 10, 628–640 RSC
- P. Kar, S. Zeng, Y. Zhang, E. Vahidzadeh, A. Manuel, R. Kisslinger, K. M. Alam, U. K. Thakur, N. Mahdi, P. Kumar and K. Shankar, Appl. Catal., B, 2019, 243, 522–536 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- L. Liu, H. Zhao, J. M. Andino and Y. Li, ACS Catal., 2012, 2, 1817–1828 CrossRef CAS
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed
- A. Meng, S. Wu, B. Cheng, J. Yu and J. Xu, J. Mater. Chem. A, 2018, 6, 4729–4736 RSC
- W. Yu, D. Xu and T. Peng, J. Mater. Chem. A, 2015, 3, 19936–19947 RSC
- X. Li, H. Jiang, C. Ma, Z. Zhu, X. Song, H. Wang, P. Huo and X. Li, Appl. Catal., B, 2021, 283, 119638 CrossRef CAS
- M. F. Ehsan and T. He, Appl. Catal., B, 2015, 166, 345–352 CrossRef
- H. Wang, L. Zhang, K. Wang, X. Sun and W. Wang, Appl. Catal., B, 2019, 243, 771–779 CrossRef CAS
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, J. Mater. Chem. A, 2015, 3, 11313–11319 RSC
- M. Akatsuka, Y. Kawaguchi, R. Itoh, A. Ozawa, M. Yamamoto, T. Tanabe and T. Yoshida, Appl. Catal., B, 2020, 262, 118247 CrossRef CAS
- H. J. Yoon, J. H. Yang, S. J. Park, C. K. Rhee and Y. Sohn, Appl. Surf. Sci., 2021, 536, 147753 CrossRef CAS
- Q. Han, X. Bai, Z. Man, H. He, L. Li, J. Hu, A. Alsaedi, T. Hayat, Z. Yu, W. Zhang, J. Wang, Y. Zhou and Z. Zou, J. Am. Chem. Soc., 2019, 141, 4209–4213 CrossRef CAS PubMed
- C. Lu, X. Li, Q. Wu, J. Li, L. Wen, Y. Dai, B. Huang, B. Li and Z. Lou, ACS Nano, 2021, 15, 3529–3539 CrossRef CAS PubMed
- G. Yin, M. Nishikawa, Y. Nosaka, N. Srinivasan, D. Atarashi, E. Sakai and M. Miyauchi, ACS Nano, 2015, 9, 2111–2119 CrossRef CAS PubMed
- W. Wang, C. Deng, S. Xie, Y. Li, W. Zhang, H. Sheng, C. Chen and J. Zhao, J. Am. Chem. Soc., 2021, 143, 2984–2993 CrossRef CAS PubMed
- L. Wang, J. Wan, Y. Zhao, N. Yang and D. Wang, J. Am. Chem. Soc., 2019, 141, 2238–2241 CrossRef CAS PubMed
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS
- L. Liu, Y. Jiang, H. Zhao, J. Chen, J. Cheng, K. Yang and Y. Li, ACS Catal., 2016, 6, 1097–1108 CrossRef CAS
- L. Collado, A. Reynal, F. Fresno, M. Barawi, C. Escudero, V. Perez-Dieste, J. M. Coronado, D. P. Serrano, J. R. Durrant and V. A. de la peña O’Shea, Nat. Commun., 2018, 9, 4986 CrossRef PubMed
- W. Wang, D. Xu, B. Cheng, J. Yu and C. Jiang, J. Mater. Chem. A, 2017, 5, 5020–5029 RSC
- J. Bian, Y. Qu, X. Zhang, N. Sun, D. Tang and L. Jing, J. Mater. Chem. A, 2018, 6, 11838–11845 RSC
- J. Li, F. Wei, C. Dong, W. Mu and X. Han, J. Mater. Chem. A, 2020, 8, 6524–6531 RSC
- F. You, J. Wan, J. Qi, D. Mao, N. Yang, Q. Zhang, L. Gu and D. Wang, Angew. Chem., 2020, 132, 731–734 CrossRef
- T. Ye, W. Huang, L. Zeng, M. Li and J. Shi, Appl. Catal., B, 2017, 210, 141–148 CrossRef CAS
- K. Wang, J. Lu, Y. Lu, C. H. Lau, Y. Zheng and X. Fan, Appl. Catal., B, 2021, 292, 120147 CrossRef CAS
- A. Meng, B. Cheng, H. Tan, J. Fan, C. Su and J. Yu, Appl. Catal., B, 2021, 289, 120039 CrossRef CAS
- Y. Ma, Q. Tang, W. Y. Sun, Z. Y. Yao, W. Zhu, T. Li and J. Wang, Appl. Catal., B, 2020, 270, 118856 CrossRef CAS
- J. Jin, S. Chen, J. Wang, C. Chen and T. Peng, Appl. Catal., B, 2020, 263, 118353 CrossRef CAS
- T. Butburee, Z. Sun, A. Centeno, F. Xie, Z. Zhao, D. Wu, P. Peerakiatkhajohn, S. Thaweesak, H. Wang and L. Wang, Nano Energy, 2019, 62, 426–433 CrossRef CAS
- L. Wang, S. Duan, P. Jin, H. She, J. Huang, Z. Lei, T. Zhang and Q. Wang, Appl. Catal., B, 2018, 239, 599–608 CrossRef CAS
- Y. Wei, X. Wu, Y. Zhao, L. Wang, Z. Zhao, X. Huang, J. Liu and J. Li, Appl. Catal., B, 2018, 236, 445–457 CrossRef CAS
- M. Tahir, B. Tahir and N. A. S. Amin, Appl. Catal., B, 2017, 204, 548–560 CrossRef CAS
- M. Tahir, Appl. Catal., B, 2017, 219, 329–343 CrossRef CAS
- A. Razzaq, A. Sinhamahapatra, T.-H. Kang, C. A. Grimes, J.-S. Yu and S.-I. In, Appl. Catal., B, 2017, 215, 28–35 CrossRef CAS
- S. Bera, J. E. Lee, S. B. Rawal and W. I. Lee, Appl. Catal., B, 2016, 199, 55–63 CrossRef CAS
- M. Wang, D. Wang and Z. Li, Appl. Catal., B, 2016, 183, 47–52 CrossRef CAS
- T. Wang, X. Meng, P. Li, S. Ouyang, K. Chang, G. Liu, Z. Mei and J. Ye, Nano Energy, 2014, 9, 50–60 CrossRef CAS
- S. In, D. D. Vaughn and R. E. Schaak, Angew. Chem., Int. Ed., 2012, 51, 3915–3918 CrossRef CAS PubMed
- H. Lin, L. Li, M. Zhao, X. Huang, X. Chen, G. Li and R. Yu, J. Am. Chem. Soc., 2012, 134, 8328–8331 CrossRef CAS PubMed
- M. M. Rodriguez, X. Peng, L. Liu, Y. Li and J. M. Andino, J. Phys. Chem. C, 2012, 116, 19755–19764 CrossRef CAS
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed
- F. He, B. Zhu, B. Cheng, J. Yu, W. Ho and W. Macyk, Appl. Catal., B, 2020, 272, 119006 CrossRef CAS
- X. Y. Kong, W. L. Tan, B.-J. Ng, S.-P. Chai and A. R. Mohamed, Nano Res., 2017, 10, 1720–1731 CrossRef CAS
- X. Y. Kong, W. Q. Lee, A. R. Mohamed and S.-P. Chai, Chem. Eng. J., 2019, 372, 1183–1193 CrossRef CAS
- J. Hao, D. Yang, J. Wu, B. Ni, L. Wei, Q. Xu, Y. Min and H. Li, Chem. Eng., 2021, 423, 130190 CrossRef CAS
- M.-P. Jiang, K.-K. Huang, J.-H. Liu, D. Wang, Y. Wang, X. Wang, Z.-D. Li, X.-Y. Wang, Z.-B. Geng, X.-Y. Hou and S.-H. Feng, Chem, 2020, 6, 2335–2346 CAS
- H. Sheng, M. H. Oh, W. T. Osowiecki, W. Kim, A. P. Alivisatos and H. Frei, J. Am. Chem. Soc., 2018, 140, 4363–4371 CrossRef CAS PubMed
- J. Wu, Y. Huang, W. Ye and Y. Li, Adv. Sci., 2017, 4, 1700194 CrossRef PubMed
- G. A. Ozin, Adv. Mater., 2015, 27, 1957–1963 CrossRef CAS PubMed
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491 CrossRef CAS PubMed
- S. Cui, X. Sun and J. Liu, ChemSusChem, 2016, 9, 1698–1703 CrossRef CAS PubMed
- Z. Xiong, Y. Luo, Y. Zhao, J. Zhang, C. Zheng and J. C. S. Wu, Phys. Chem. Chem. Phys., 2016, 18, 13186–13195 RSC
- K. M. Cho, K. H. Kim, H. O. Choi and H.-T. Jung, Green Chem., 2015, 17, 3972–3978 RSC
- P. Li, Y. Zhou, H. Li, Q. Xu, X. Meng, X. Wang, M. Xiao and Z. Zou, Chem. Commun., 2014, 51, 800–803 RSC
- J. Yu, J. Jin, B. Cheng and M. Jaroniec, J. Mater. Chem. A, 2014, 2, 3407–3416 RSC
- S. Xie, Y. Wang, Q. Zhang, W. Fan, W. Deng and Y. Wang, Chem. Commun., 2013, 49, 2451–2453 RSC
- L. Gu, J. Wang, H. Cheng, Y. Zhao, L. Liu and X. Han, ACS Appl. Mater. Interfaces, 2013, 5, 3085–3093 CrossRef CAS PubMed
- Y. Chen, H. Gao, J. Xiang, X. Dong and Y. Cao, Mater. Res. Bull., 2018, 99, 29–36 CrossRef CAS
- W. Tu, Y. Zhou, Q. Liu, Z. Tian, J. Gao, X. Chen, H. Zhang, J. Liu and Z. Zou, Adv. Funct. Mater., 2012, 22, 1215–1221 CrossRef CAS
- Y. Zhao, Y. Wei, X. Wu, H. Zheng, Z. Zhao, J. Liu and J. Li, Appl. Catal., B, 2018, 226, 360–372 CrossRef CAS
- A. Razzaq, C. A. Grimes and S.-I. In, Carbon, 2016, 98, 537–544 CrossRef CAS
- M. Xing, F. Shen, B. Qiu and J. Zhang, Sci. Rep., 2014, 4, 6341 CrossRef CAS PubMed
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed
- P.-Q. Wang, Y. Bai, P.-Y. Luo and J.-Y. Liu, Catal. Commun., 2013, 38, 82–85 CrossRef CAS
- M. Long, Y. Qin, C. Chen, X. Guo, B. Tan and W. Cai, J. Phys. Chem. C, 2013, 117, 16734–16741 CrossRef CAS
- Y. Tang, X. Hu and C. Liu, Phys. Chem. Chem. Phys., 2014, 16, 25321–25329 RSC
- S. Ali, A. Razzaq and S.-I. In, Catal. Today, 2018, 335, 39–54 CrossRef
- M. Xing, W. Fang, X. Yang, B. Tian and J. Zhang, Chem. Commun., 2014, 50, 6637–6640 RSC
- L.-Y. Lin, Y. Nie, S. Kavadiya, T. Soundappan and P. Biswas, Chem. Eng. J., 2017, 316, 449–460 CrossRef CAS
- I. Shown, H. C. Hsu, Y. C. Chang, C. H. Lin, P. K. Roy, A. Ganguly, C. H. Wang, J. K. Chang, C. I. Wu, L. C. Chen and K. H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed
- P. Madhusudan, S. Wageh, A. A. Al-Ghamdi, J. Zhang, B. Cheng and Y. Yu, Appl. Surf. Sci., 2020, 506, 144683 CrossRef CAS
- L.-L. Tan, W.-J. Ong, S.-P. Chai, B. T. Goh and A. R. Mohamed, Appl. Catal., B, 2015, 179, 160–170 CrossRef CAS
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Chem. Commun., 2015, 51, 858–861 RSC
- X. Wang, K. Li, J. He, J. Yang, F. Dong, W. Mai and M. Zhu, Nano Energy, 2020, 78, 105388 CrossRef CAS
- Y. F. Mu, W. Zhang, G. X. Dong, K. Su, M. Zhang and T. B. Lu, Small, 2020, 16, 1–8 Search PubMed
- L. Wang, H. Tan, L. Zhang, B. Cheng and J. Yu, Chem. Eng. J., 2021, 411, 128501 CrossRef CAS
- X. Lin, S. Wang, W. Tu, H. Wang, Y. Hou, W. Dai and R. Xu, ACS Appl. Energy Mater., 2019, 2, 7670–7678 CrossRef CAS
- Z. Otgonbayar, K. Y. Cho and W. C. Oh, ACS Omega, 2020, 5, 26389–26401 CrossRef CAS PubMed
- Z. Otgonbayar, Y. Liu, K. Y. Cho, C.-H. Jung and W.-C. Oh, Mater. Sci. Semicond. Process., 2021, 121, 105456 CrossRef CAS
- R. Gusain, P. Kumar, O. P. Sharma, S. L. Jain and O. P. Khatri, Appl. Catal., B, 2016, 181, 352–362 CrossRef CAS
- C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 31, 1–6 CrossRef PubMed
- R. B. Lin, S. Xiang, H. Xing, W. Zhou and B. Chen, Coord. Chem. Rev., 2019, 378, 87–103 CrossRef CAS
- Z. Ma, Q. Zhang, W. Zhu, D. Khan, C. Hu, T. Huang, W. Ding and J. Zou, Sustainable Energy Fuels, 2020, 4, 2192–2200 RSC
- M. C. Singo, X. C. Molepo, O. O. Oluwasina and M. O. Daramola, Energy Procedia, 2017, 114, 2429–2440 CrossRef CAS
- L. Zhao, A. Wang, A. Yang, G. Zuo, J. Dai and Y. Zheng, Int. J. Hydrogen Energy, 2020, 45, 31863–31870 CrossRef CAS
- Y. P. Zhu, J. Yin, E. Abou-Hamad, X. Liu, W. Chen, T. Yao, O. F. Mohammed and H. N. Alshareef, Adv. Mater., 2020, 32, 1–8 Search PubMed
- Y. Gao, J. Wu, J. Wang, Y. Fan, S. Zhang and W. Dai, ACS Appl. Mater. Interfaces, 2020, 12, 11036–11044 CrossRef CAS PubMed
- C. Lin, H. He, Y. Zhang, M. Xu, F. Tian, L. Li and Y. Wang, RSC Adv., 2020, 10, 3084–3091 RSC
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2004, 2300–2301 RSC
- D. Sun, Y. Gao, J. Fu, X. Zeng, Z. Chen and Z. Li, Chem. Commun., 2015, 51, 2645–2648 RSC
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H. Jiang, J. Jiang, Q. Zhang and Y. Xie, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. Bin Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed
- H. Dong, X. Zhang, Y. Lu, Y. Yang, Y. P. Zhang, H. L. Tang, F. M. Zhang, Z. Di Yang, X. Sun and Y. Feng, Appl. Catal., B, 2020, 276, 119173 CrossRef CAS
- X. Liu, H. Yang, J. He, H. Liu, L. Song, L. Li and J. Luo, Small, 2018, 14, 1704049 CrossRef PubMed
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. H. Castro Neto, Science, 2016, 353, aac9439 CrossRef CAS PubMed
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed
- Y. Sun, S. Gao and Y. Xie, Chem. Soc. Rev., 2014, 43, 530–546 RSC
- D. Voiry, J. Yang and M. Chhowalla, Adv. Mater., 2016, 28, 6197–6206 CrossRef CAS PubMed
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. S. Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed
- H. Schmidt, F. Giustiniano and G. Eda, Chem. Soc. Rev., 2015, 44, 7715–7736 RSC
- L. Cheng and Y. Liu, J. Am. Chem. Soc., 2018, 140, 17895–17900 CrossRef CAS PubMed
- A. J. Meier, A. Garg, B. Sutter, J. N. Kuhn and V. R. Bhethanabotla, ACS Sustainable Chem. Eng., 2018, 7, 265–275 CrossRef
- F. Xu, B. Zhu, B. Cheng, J. Yu and J. Xu, Adv. Opt. Mater., 2018, 6, 1800911 CrossRef
- H. Jung, K. M. Cho, K. H. Kim, H.-W. Yoo, A. Al-Saggaf, I. Gereige and H.-T. Jung, ACS Sustainable Chem. Eng., 2018, 6, 5718–5724 CrossRef CAS
- D. Long, J. Liu, L. Bai, L. Yan, H. Liu, Z. Feng, L. Zheng, X. Chen, S. Li and M. Lu, ACS Photonics, 2020, 7, 3394–3400 CrossRef CAS
- B. Khan, F. Raziq, M. B. Faheem, M. U. Farooq, S. Hussain, F. Ali, A. Ullah, A. Mavlonov, Y. Zhao and Z. Liu, J. Hazard. Mater., 2020, 381, 120972 CrossRef CAS PubMed
- R. Kim, J. Kim, J. Y. Do, M. W. Seo and M. Kang, Catalysts, 2019, 9, 998 CrossRef CAS
- W. Tu, Y. Li, L. Kuai, Y. Zhou, Q. Xu, H. Li, X. Wang, M. Xiao and Z. Zou, Nanoscale, 2017, 9, 9065–9070 RSC
- Y. Wang, Z. Zhang, L. Zhang, Z. Luo, J. Shen, H. Lin, J. Long, J. C. S. Wu, X. Fu and X. Wang, J. Am. Chem. Soc., 2018, 140, 14595–14598 CrossRef CAS PubMed
- W. Dai, J. Yu, Y. Deng, X. Hu, T. Wang and X. Luo, Appl. Surf. Sci., 2017, 403, 230–239 CrossRef CAS
- R. A. Geioushy, S. M. El-Sheikh, I. M. Hegazy, A. Shawky, S. El-Sherbiny and A.-H. T. Kandil, Mater. Res. Bull., 2019, 118, 110499 CrossRef CAS
- C. Yang, Q. Tan, Q. Li, J. Zhou, J. Fan, B. Li, J. Sun and K. Lv, Appl. Catal., B, 2020, 268, 118738 CrossRef CAS
- J. Low, L. Zhang, T. Tong, B. Shen and J. Yu, J. Catal., 2018, 361, 255–266 CrossRef CAS
- Y. Li, Z. Yin, G. Ji, Z. Liang, Y. Xue, Y. Guo, J. Tian, X. Wang and H. Cui, Appl. Catal., B, 2019, 246, 12–20 CrossRef CAS
- Q. Tang, Z. Zhou and Z. Chen, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 360–379 CAS
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed
- M. W. Barsoum, Prog. Solid State Chem., 2000, 28, 201–281 CrossRef CAS
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed
- Y. Gogotsi and B. Anasori, ACS Nano, 2019, 13, 8491–8494 CrossRef CAS PubMed
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed
- W.-F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC
- O. Mashtalir, K. M. Cook, V. N. Mochalin, M. Crowe, M. W. Barsoum and Y. Gogotsi, J. Mater. Chem. A, 2014, 2, 14334–14338 RSC
- H. Huang, Y. Song, N. Li, D. Chen, Q. Xu, H. Li, J. He and J. Lu, Appl. Catal., B, 2019, 251, 154–161 CrossRef CAS
- Z. Guo, J. Zhou, L. Zhu and Z. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC
- K. Huang, C. Li, H. Li, G. Ren, L. Wang, W. Wang and X. Meng, ACS Appl. Nano Mater, 2020, 3, 9581–9603 CrossRef CAS
- J. Wu, Y. Zhang, P. Lu, G. Fang, X. Li, W. Y. William, Z. Zhang and B. Dong, Appl. Catal., B, 2021, 286, 119944 CrossRef CAS
- J. Ran, G. Gao, F.-T. T. Li, T.-Y. Y. Ma, A. Du and S.-Z. Z. Qiao, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef
- W. Sun, S. A. Shah, Y. Chen, Z. Tan, H. Gao, T. Habib, M. Radovic and M. J. Green, J. Mater. Chem. A, 2017, 5, 21663–21668 RSC
- Z. Zeng, Y. Xu, Z. Zhang, Z. Gao, M. Luo, Z. Yin, C. Zhang, J. Xu, B. Huang, F. Luo, Y. Du and C. Yan, Chem. Soc. Rev., 2020, 49, 1109–1143 RSC
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS
- J. C. Hemminger, R. Carr and G. A. Somorjai, Chem. Phys. Lett., 1978, 57, 100–104 CrossRef CAS
- C. Luo, J. Zhao, Y. Li, W. Zhao, Y. Zeng and C. Wang, Appl. Surf. Sci., 2018, 447, 627–635 CrossRef CAS
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Energy Environ. Sci., 2011, 4, 4211–4219 RSC
- D. Li, S. Ouyang, H. Xu, D. Lu, M. Zhao, X. Zhang and J. Ye, Chem. Commun., 2016, 52, 5989–5992 RSC
- D. Mateo, J. Albero and H. García, Joule, 2019, 3, 1949–1962 CrossRef CAS
- J. Lin, J. Hu, C. Qiu, H. Huang, L. Chen, Y. Xie, Z. Zhang, H. Lin and X. Wang, Catal. Sci. Technol., 2019, 9, 336–346 RSC
- K. Teramura, S. Okuoka, H. Tsuneoka, T. Shishido and T. Tanaka, Appl. Catal., B, 2010, 96, 565–568 CrossRef CAS
- H. Zhou, P. Li, J. Guo, R. Yan, T. Fan, D. Zhang and J. Ye, Nanoscale, 2015, 7, 113–120 RSC
- J. Qin, L. Lin and X. Wang, Chem. Commun., 2018, 54, 2272–2275 RSC
- S. Wang, Y. Hou and X. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4327–4335 CrossRef CAS PubMed
- H. Shi, G. Chen, C. Zhang and Z. Zou, ACS Catal., 2014, 4, 3637–3643 CrossRef CAS
- A. Kumar, G. Sharma, M. Naushad, T. Ahamad, R. C. Veses and F. J. Stadler, Chem. Eng. J., 2019, 370, 148–165 CrossRef CAS
- Y. Wang, M. Liu, W. Chen, L. Mao and W. Shangguan, J. Alloys Compd., 2019, 786, 149–154 CrossRef CAS
- S. Tu, Y. Zhang, A. H. Reshak, S. Auluck, L. Ye, X. Han, T. Ma and H. Huang, Nano Energy, 2019, 56, 840–850 CrossRef CAS
- M. Que, Y. Zhao, Y. Yang, L. Pan, W. Lei, W. Cai, H. Yuan, J. Chen and G. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 6180–6187 CrossRef CAS PubMed
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 1–9 Search PubMed
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed
- Y. Jiang, J.-F. Liao, Y.-F. Xu, H.-Y. Chen, X.-D. Wang and D.-B. Kuang, J. Mater. Chem. A, 2019, 7, 13762–13769 RSC
- Y. Mu, W. Zhang, G. Dong, K. Su, M. Zhang and T. Lu, Small, 2020, 16, 2002140 CrossRef CAS PubMed
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS
- C. Tang, C. Chen, W. Xu and L. Xu, J. Mater. Chem. A, 2019, 7, 6911–6919 RSC
- S. Shyamal, S. K. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7965–7969 CrossRef CAS PubMed
- G.-X. Dong, W. Zhang, Y.-F. Mu, K. Su, M. Zhang and T.-B. Lu, Chem. Commun., 2020, 56, 4664–4667 RSC
- J. Zhu, Y. Zhu, J. Huang, L. Hou, J. Shen and C. Li, Nanoscale, 2020, 12, 11842–11846 RSC
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS
- L. Wu, Y. Mu, X. Guo, W. Zhang, Z. Zhang, M. Zhang and T. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed
- M. Que, Y. Zhao, L. Pan, Y. Yang, Z. He, H. Yuan, J. Chen and G. Zhu, Mater. Lett., 2021, 282, 128695 CrossRef CAS
- L. Zhou, Y. Xu, B. Chen, D. Kuang and C. Su, Small, 2018, 14, 1703762 CrossRef PubMed
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS
- C. Lu, D. S. Itanze, A. G. Aragon, X. Ma, H. Li, K. B. Ucer, C. Hewitt, D. L. Carroll, R. T. Williams, Y. Qiu and S. M. Geyer, Nanoscale, 2020, 12, 2987–2991 RSC
- R. Verma, R. Belgamwar and V. Polshettiwar, ACS Mater. Lett., 2021, 3, 574–598 CrossRef CAS
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS
- X.-C. Ma, Y. Dai, L. Yu and B.-B. Huang, Light Sci. Appl., 2016, 5, e16017 CrossRef CAS PubMed
- M. Wang, M. Ye, J. Iocozzia, C. Lin and Z. Lin, Adv. Sci., 2016, 3, 1600024 CrossRef PubMed
- S. Yu, A. J. Wilson, G. Kumari, X. Zhang and P. K. Jain, ACS Energy Lett., 2017, 2, 2058–2070 CrossRef CAS
- G. Kumari, X. Zhang, D. Devasia, J. Heo and P. K. Jain, ACS Nano, 2018, 12, 8330–8340 CrossRef CAS PubMed
- L. Collado, A. Reynal, J. M. Coronado, D. P. Serrano, J. R. Durrant and V. A. De la Peña O'Shea, Appl. Catal., B, 2015, 178, 177–185 CrossRef CAS
- S. Zeng, E. Vahidzadeh, C. G. VanEssen, P. Kar, R. Kisslinger, A. Goswami, Y. Zhang, N. Mahdi, S. Riddell, A. E. Kobryn, S. Gusarov, P. Kumar and K. Shankar, Appl. Catal., B, 2020, 267, 118644 CrossRef CAS
- I. García-García, E. C. Lovell, R. J. Wong, V. L. Barrio, J. Scott, J. F. Cambra and R. Amal, ACS Sustainable Chem. Eng., 2020, 8, 1879–1887 CrossRef
- H. Li, Y. Gao, Z. Xiong, C. Liao and K. Shih, Appl. Surf. Sci., 2018, 439, 552–559 CrossRef CAS
- H. Song, X. Meng, T. D. Dao, W. Zhou, H. Liu, L. Shi, H. Zhang, T. Nagao, T. Kako and J. Ye, ACS Appl. Mater. Interfaces, 2018, 10, 408–416 CrossRef CAS PubMed
- D. Hong, L.-M. Lyu, K. Koga, Y. Shimoyama and Y. Kon, ACS Sustainable Chem. Eng., 2019, 7, 18955–18964 CrossRef CAS
- W. Tu, Y. Zhou, H. Li, P. Li and Z. Zou, Nanoscale, 2015, 7, 14232–14236 RSC
- L. K. Putri, W.-J. Ong, W. S. Chang and S.-P. Chai, Catal. Sci. Technol., 2016, 6, 744–754 RSC
- J. Balajka, M. A. Hines, W. J. I. DeBenedetti, M. Komora, J. Pavelec, M. Schmid and U. Diebold, Science, 2018, 361, 786–789 CrossRef CAS PubMed
- G. W. Busser, B. Mei, A. Pougin, J. Strunk, R. Gutkowski, W. Schuhmann, M. Willinger, R. Schlögl and M. Muhler, ChemSusChem, 2014, 7, 1030–1034 CrossRef CAS PubMed
- L. Yuan, K. Q. Lu, F. Zhang, X. Fu and Y. J. Xu, Appl. Catal., B, 2018, 237, 424–431 CrossRef CAS
- B. Wang, X. Wang, L. Lu, C. Zhou, Z. Xin, J. Wang, X. K. Ke, G. Sheng, S. Yan and Z. Zou, ACS Catal., 2018, 8, 516–525 CrossRef CAS
- C. C. Yang, Y. H. Yu, B. Van Der Linden, J. C. S. Wu and G. Mul, J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS PubMed
- H. Zhang, T. Itoi, T. Konishi and Y. Izumi, J. Am. Chem. Soc., 2019, 141, 6292–6301 CrossRef CAS PubMed
- H. Zhang, T. Itoi, T. Konishi and Y. Izumi, Angew. Chem., Int. Ed., 2021, 60, 9045–9054 CrossRef CAS PubMed
- M. Borges OrdonÌfo and A. Urakawa, J. Phys. Chem. C, 2019, 123, 4140–4147 CrossRef
- A. Bazzo and A. Urakawa, ChemSusChem, 2013, 6, 2095–2102 CrossRef CAS PubMed
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CrossRef CAS PubMed
- C. C. Yang, J. Vernimmen, V. Meynen, P. Cool and G. Mul, J. Catal., 2011, 284, 1–8 CrossRef CAS
- S. Ishihara, P. Sahoo, K. Deguchi, S. Ohki, M. Tansho, T. Shimizu, J. Labuta, J. P. Hill, K. Ariga, K. Watanabe, Y. Yamauchi, S. Suehara and N. Lyi, J. Am. Chem. Soc., 2013, 135, 18040–18043 CrossRef CAS PubMed
- K. Kamogawa, Y. Shimoda, K. Miyata, K. Onda, Y. Yamazaki, Y. Tamaki and O. Ishitani, Chem. Sci., 2021, 12, 9682–9683 RSC
- A. Rafiee, K. Rajab Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS
- M. A. Sabri, S. Al Jitan, D. Bahamon, L. F. Vega and G. Palmisano, Sci. Total Environ., 2021, 790, 148081 CrossRef CAS PubMed
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59, 7033–7045 CrossRef CAS
- P. Nejat, F. Jomehzadeh, M. M. Taheri, M. Gohari and M. Z. A. Majid, Renewable Sustainable Energy Rev., 2015, 43, 843–862 CrossRef CAS
- H. M. Wee, W. H. Yang, C. W. Chou and M. V. Padilan, Renewable Sustainable Energy Rev., 2012, 16, 5451–5465 CrossRef
- S. Ali, T. Fazal, F. Javed, A. Hafeez, M. Akhtar, B. Haider, M. S. ur Rehman, W. B. Zimmerman and F. Rehman, J. Clean. Prod., 2020, 259, 120729 CrossRef
-
R. G. Newell, D. Raimi and G. Aldana, Resources for the Future, 2019, pp. 8–19 Search PubMed
- T. Covert, M. Greenstone and C. R. Knittel, J. Econ. Perspect., 2016, 30, 117–138 CrossRef
- G. De Pietro, L. J. Gallo Robert Howlett and L. C. Jain, Smart Innovation, Systems and Technologies 98 Intelligent Interactive Multimedia Systems and Services Proceedings of 2018 Conference, 2018.
- E. S. Rubin and H. Zhai, Environ. Sci. Technol., 2012, 46, 3076–3084 CrossRef CAS PubMed
- X. Yang and D. Wang, ACS Appl. Energy Mater, 2018, 1, 6657–6693 CrossRef CAS
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC
- S. Cheng and B. E. Logan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18871–18873 CrossRef CAS PubMed
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C. J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS
- T. Yui, Y. Tamaki, K. Sekizawa and O. Ishitani, Top. Curr. Chem., 2011, 303, 151–184 CrossRef CAS PubMed
- Y. Mi, X. Peng, X. Liu and J. Luo, ACS Appl. Energy Mater., 2018, 1, 5119–5123 CrossRef CAS
- H. Yamashita, K. Ikeue, T. Takewaki and M. Anpo, Top. Catal., 2002, 18, 95–100 CrossRef CAS
- Y. Wang, S. Zhou and H. Huo, Renewable Sustainable Energy Rev., 2014, 39, 370–380 CrossRef
- M. H. Chakrabarti, M. Ali, S. Baroutian and M. Saleem, Process Saf. Environ. Prot., 2011, 89, 165–171 CrossRef CAS
-
F. Graf, Power to gas – state of the art and perspectives. MARCOGAZ-General Assembly: Workshop New Developments, DVGW Research Center at Engler-Bunte-Institut of KIT, 2014 Search PubMed
-
I. P. K. M. Oberdorf, KIT-KIT-Media-Press Releases-Power to Gas: Storing the Wind and Sun in Natural Gas, 2014 Search PubMed
- C. C. Economy, The carbon cycle is broken … Can we close it again? … and how? 2021.
- S. Docao, A. R. Koirala, M. G. Kim, I. C. Hwang, M. K. Song and K. B. Yoon, Energy Environ. Sci., 2017, 10, 628–640 RSC
- B.-H. Lee, E. Gong, M. Kim, S. Park, H. R. Kim, J. Lee, E. Jung, C. W. Lee, J. Bok, Y. Jung, Y. S. Kim, K.-S. Lee, S.-P. Cho, J.-W. Jung, C.-H. Cho, S. Lebègue, K. T. Nam, H. Kim, S.-I. In and T. Hyeon, Energy Environ. Sci., 2021 10.1039/d1ee01574e
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Xhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS
- B. Kim, H. Seong, J. T. Song, K. Kwak, H. Song, Y. C. Tan, G. Park, D. Lee and J. Oh, ACS Energy Lett., 2020, 5, 749–757 CrossRef CAS
- A. M. Yousef, W. M. El-Maghlany, Y. A. Eldrainy and A. Attia, Energy, 2018, 156, 328–351 CrossRef CAS
- S. Luo, Q. Zhang, L. Zhu, H. Lin, B. A. Kazanowska, C. M. Doherty, A. J. Hill, P. Gao and R. Guo, Chem. Mater., 2018, 30, 5322–5332 CrossRef CAS
- A. Goeppert, M. Czaun, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC
- Musk Foundation, $100M Prize for Carbon Removal, 2021.
- European Innovation Council, Fuel from the Sun: Artificial Photosynthesis.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |