
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted cancer phototherapy using phthalocyanine–anticancer drug conjugates
Christopher C.
Rennie
 and
Robert M.
Edkins
*
and
Robert M.
Edkins
*
WestCHEM Department of Pure and Applied Chemistry, Thomas Graham Building, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: robert.edkins@strath.ac.uk
First published on 17th August 2022
Abstract
Phototherapy, the use of light to selectively ablate cancerous tissue, is a compelling prospect. Phototherapy is divided into two major domains: photodynamic and photothermal, whereby photosensitizer irradiation generates reactive oxygen species or heat, respectively, to disrupt the cancer microenvironment. Phthalocyanines (Pcs) are prominent phototherapeutics due to their desirable optical properties and structural versatility. Targeting of Pc photosensitizers historically relied on the enhanced permeation and retention effect, but the weak specificity engendered by this approach has hindered bench-to-clinic translation. To improve specificity, antibody and peptide active-targeting groups have been employed to some effect. An alternative targeting method exploits the binding of anticancer drugs to direct the photosensitizer close to essential cellular components, allowing for precise, synergistic phototherapy. This Perspective explores the use of Pc–drug conjugates as targeted anticancer phototherapeutic systems with examples of Pc–platin, Pc–kinase, and Pc–anthracycline conjugates discussed in detail.
 Christopher C. Rennie | Christopher C. Rennie is from Aberdeen (Scotland). He completed his undergraduate MChem degree at the University of Strathclyde. During this time, he undertook research projects on the synthesis of minor-groove binders and immunomodulatory compounds under the supervision of Prof. Colin Suckling. He joined the group of Dr Robert Edkins as a PhD student at the University of Strathclyde in 2019. His research interests include the synthesis of novel chromophores for photomedical applications. |
 Robert (Bob) Edkins | Robert (Bob) Edkins studied for his MChem degree and PhD at Durham University, supervised by Prof. Andrew Beeby. Bob held an Alexander von Humboldt Fellowship at Julius-Maximilians-Universität Würzburg with Prof. Todd Marder and then a Royal Commission for the Exhibition of 1851 Fellowship at the University of Oxford hosted by Prof. Stephen Faulkner. In 2018, Bob started as a Chancellor's Fellow and Lecturer in Chemistry at the University of Strathclyde where his group focuses on the synthesis, photophysics, and applications of dyes for various bio-imaging modalities and photosensitizers for phototherapy. |
1. Phthalocyanines as phototherapeutic anticancer agents
Cancer is one of the leading healthcare challenges facing humanity today with global cancer-related deaths approaching 10 million per annum.1 With this harrowing statistic, it is obvious that novel cancer-combating approaches are required. Standard oncological treatment still predominantly relies upon surgery, chemotherapy, or radiotherapy; however, the invasiveness of surgery and the non-specific nature of chemo- and radiotherapies can cause adverse side effects and damage to healthy tissue, along with growing prevalence of resistance to chemotherapeutics.2Over the last few decades, phototherapy has become an emerging treatment for selective tumour ablation.3–5 Phototherapy can be classified into two distinct forms: photodynamic therapy (PDT) and photothermal therapy (PTT), both of which utilize a photosensitizer to elicit a cancer-killing effect. PDT occurs through either a type I or type II process where electron transfer results in the formation of radical-based reactive oxygen species (ROS) or energy transfer generates singlet oxygen, respectively, with both causing localized oxidative damage to superficial or deep-seated tumours.6,7 The photophysical basis for the different forms of PDT can be best understood using a Jablonksi diagram, as shown in Fig. 1. PTT proceeds through a localized increase in cellular temperature, or hyperthermia, to induce apoptosis or necrosis of the tumour microenvironment.8,9
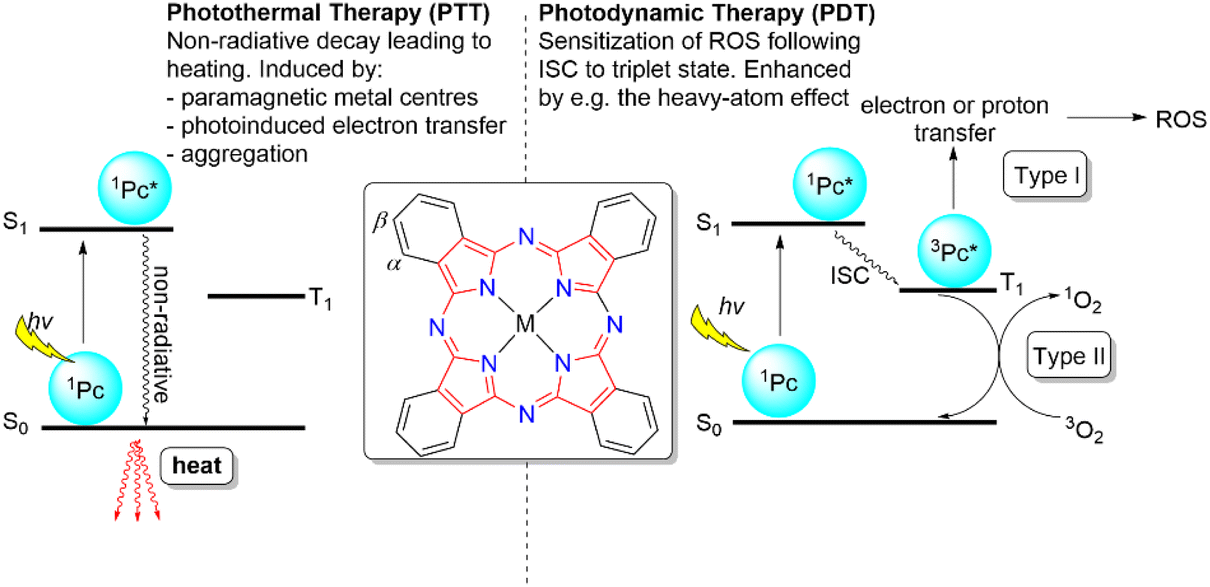 | ||
| Fig. 1 Different phototherapies that use Pc photosensitizers and their mechanisms of action depicted using Jablonski diagrams. Left: photothermal therapy mechanism; right: photodynamic therapy mechanism; centre: structure of Pc and labelling of α- and β-sites. ISC = intersystem crossing. | ||
A photosensitizer must meet the following criteria to be considered suitable for either phototherapeutic modality: strong red or near-infrared (NIR) absorption to allow deep penetration of light into biological tissue, negligible dark toxicity and few side-effects but high cytotoxicity under light irradiation, good solubility and stability in biological media, preferential accumulation in cancerous tissue, and suitable clearance rate.3 For PDT, this is augmented by the need of the photosensitizer to have a high triplet quantum yield (ΦT) and subsequent high singlet-oxygen quantum yield (ΦΔ) when considering the more typical type-II approach,10,11 while for PTT the photosensitizer must facilitate efficient photothermal conversion via nonradiative decay pathways (Fig. 1) to produce a sufficient increase in cellular temperature (e.g. to >45 °C) to induce cell death.12,13 Many types of nanomaterial and molecular photosensitizer have been investigated for both types of phototherapy.14–17 While nanomaterials have been shown to be effective photosensitizers for phototherapies, their relatively limited tunability, poor batch-to-batch reproducibility, wide size distributions, morphology-dependent responses, and unknown long-term biological effects potentially make molecular photosensitizers a more appealing solution.12,13
Porphyrinoids encompass a broad family of compounds that have received significant attention for phototherapy, with phthalocyanines (Pcs) (Fig. 1) in particular meeting all of the aforementioned requirements for a photosensitizer, most notably their strong and tuneable red-to-NIR absorption and biocompatibility.18,19 The ease with which their π-delocalized electronic structure can be controlled through introduction of appropriate substituents or by varying the central coordinated element makes it possible to tailor Pcs towards either PDT or PTT by modulating appropriate radiative and nonradiative processes.20 Further structural modification enables targeting and thus, the Pc structure and resulting photophysics are ideal for phototherapeutics.
Four Pc photosensitizers have been developed as far as to have been evaluated at various stages of clinical trials for PDT: silicon Pc-4, photocyanine, CGP55847, and photosens (Fig. 2).10 Pc-4, designed and studied by Kenney, Oleinick and coworkers,21 reached phase I clinical trials for treatment of lymphoma and non-melanomatous skin cancer and is currently undergoing trials to improve delivery mechanisms.21 Photocyanine reached phase II clinical trials for the treatment of human hepatocellular carcinoma in clinical trials.22 CGP55847 reached phase II clinical trials for treatment of solid human tumours before being abandoned.21 Photosens, is currently undergoing clinical trials for treatment of pleural mesothelioma, basal cell carcinoma, lung, head, neck, gastric, breast and oesophageal cancer metastases.21,23 The discovery of neurotoxicity in rabbit models with Photosens was modulated by reducing the number of sulfonic acid moieties on the chromophore from four to two, allowing a compromise between solubility and off target effects.10
 | ||
| Fig. 2 Pc-based photosensitizers that have reached phased clinical trials. For photosens, R = H or SO3Na as a mixture of di-, tri- and tetrasulfonated compounds. Photocyanine and photosens are a statistical mixture of isomers with substituents on either β-position of the indicated isoindole units of the Pc. | ||
All Pcs that have advanced to clinical trials thus far have seemingly relied on the enhanced permeability and retention (EPR) effect to target cancerous tissue, where higher molecular-weight drugs (i.e. those that are non-Lipinski compliant) typically show a mild preference for accumulation in tumours rather than healthy tissue due to differences in vasculature and lymphatic structure.24 Cancer cells proliferate quicker than healthy cells, resulting in aggregated tumours of an unusual morphology, large fenestrations, and defective endothelial cells.25 These poorly aligned aggregates lack a smooth muscle layer, innervation with lumen, impaired functional receptors for angiotensin II, whilst also lacking adequate lymphatic drainage, all of which contribute to EPR of nanomedicines.25 Danhier has shown that this passive EPR effect is not an effective targeting mechanism for nanomedicines, as evidenced by lack of clinical translation from murine to human models, and thus, throws into question whether more effective targeting paradigms should be explored for all therapeutics that exploit the EPR effect.24 Photosensitizers with active-targeting moieties that reliably direct the chromophore towards the tumour microenvironment are desirable as they could offer a more successful approach (Fig. 3).
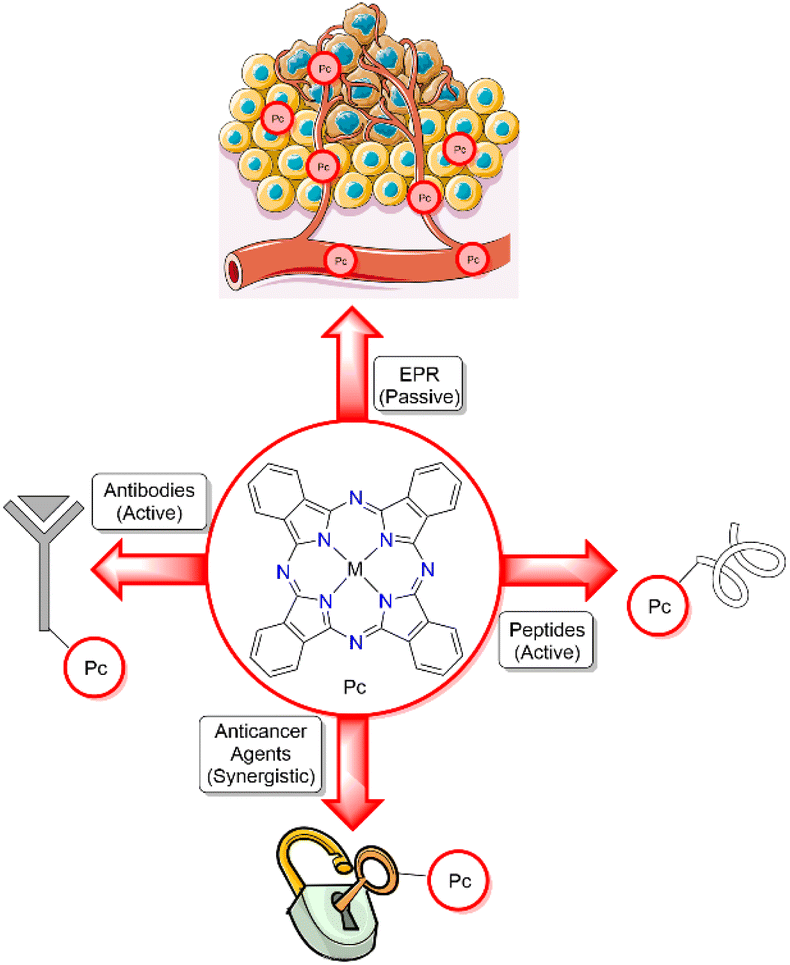 | ||
| Fig. 3 Different targeting modalities for Pc photosensitizers. Top: EPR effect; right: peptide active targeting; left: antibody active targeting; bottom: anticancer agent targeting.26 | ||
Conjugation of naturally inspired targeting groups, including: peptides,27 antibodies,28 aptamers,29 and lectins,30 to Pcs can be an effective strategy for enhancing selective uptake, while sometimes having shortcomings such as synthetic difficulty and instability.31 This topic has been extensively reviewed elsewhere16,32,33 and will not be further considered here. An alternative active-targeting approach of conjugation of Pcs with known anticancer agents has been developed. Capitalizing on established target binding of anticancer drugs, the Pc can be selectively bound and retained at the desired site of action to elicit specific oxidative or thermal damage. During the preparation of this manuscript a review describing the utility of porphyrnoid–drug conjugates more broadly was published.34 This perspective highlights recent reports of Pc–anticancer drug conjugates for targeted phototherapy of cancer and how different conjugation strategies affect activities with comparisons of their photophysical properties (absorption spectra, fluorescence quantum yields, and singlet-oxygen generation), as well as their photocytotoxic properties (phototoxic index (PI), the ratio of dark and light IC50 values).
2. Phthalocyanine–platin conjugates
Platinum-containing anticancer drugs, often referred to as platins, are on the World Health Organization's list of essential medicines, where they are deemed a necessity due to their broad-spectrum anticancer activity and economic price.35,36 The platins used clinically are typically Pt(II) complexes with two or three nitrogen-donor ligands and chloro (e.g. cisplatin) or oxygen-donor (e.g. carboplatin, nedaplatin, oxaliplatin) ancillary ligands (Fig. 4). These ancillary ligands can be displaced to facilitate binding to DNA.35 Octahedral Pt(IV) prodrugs (e.g. ethacraplatin) have been developed to combat growing resistance mechanisms and off-target toxicity that have been observed with square-planar Pt(II) derivatives.36 The higher coordination number of Pt(IV) complexes further allows the preparation of complexes with additional active-targeting groups or dual-acting prodrugs through axial functionalization.36 These Pt(IV) complexes are reduced in situ to the active Pt(II) form by biological reductants.36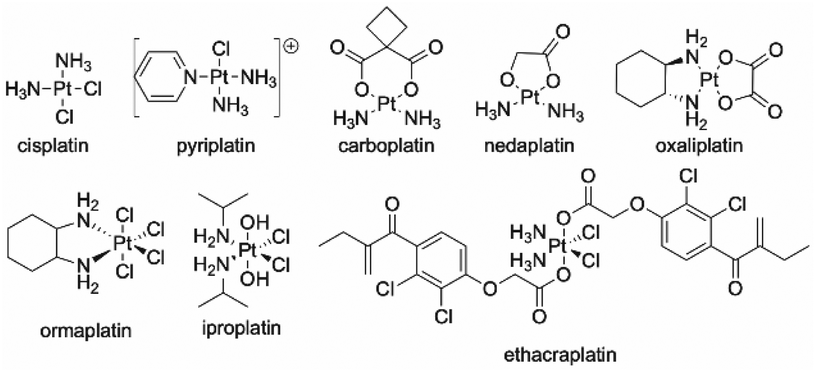 | ||
| Fig. 4 Common platin agents employed in chemotherapy. Top row: Pt(II) agents; bottom row: Pt(IV) prodrugs. | ||
The anticancer activity of the platin family is a product of these drugs’ ability to form intra- or interstrand Pt–DNA complexes.36 Using cisplatin as an example, upon crossing the cell membrane (through active or passive transport), sequential ligand exchange occurs to form the monoaqua [Pt(NH3)Cl(OH2)]+ and subsequently the diaqua [Pt(NH3)(OH2)2]2+ Pt(II) complexes, aided by the trans-effect of the amine ligands.35 The resulting net positive charge attracts the complex towards the negatively charged sugar-phosphate backbone of DNA, where the nucleophilic N7 sites of purine bases (typically guanine) displace the aqua-ligands to form the Pt–DNA complex (Fig. 5).35 The Pt–DNA adduct prevents replication and transcription pathways for the cell, leading to programmed cell death (apoptosis).35–37
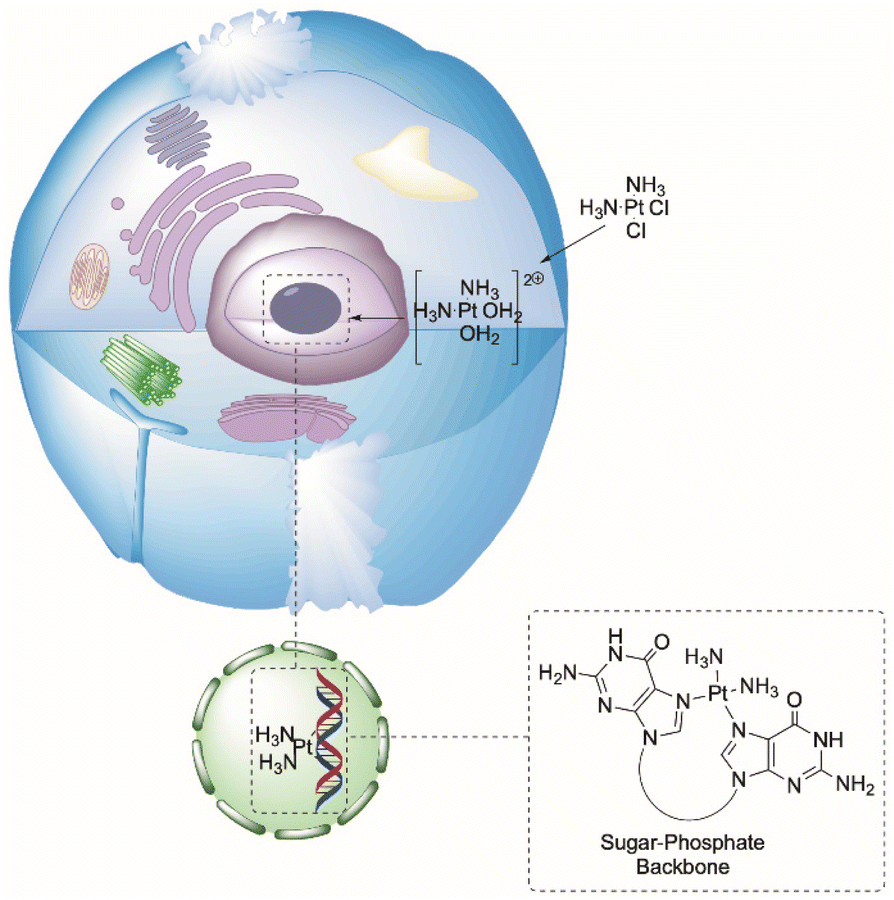 | ||
| Fig. 5 Mechanism of action for platins using cisplatin as a representative example. Cisplatin crosses the cell membrane into the cell, undergoes ligand exchange, enters the nucleoli, and coordinates at the N7 sites of guanosine DNA bases.26 | ||
Given the anticancer mechanism of platins, they have themselves been employed as active-targeting moieties to anchor Pc photosensitizers onto DNA in cancer cells, so that upon photoirradiation, localized DNA damage leads to apoptosis (Fig. 6). The first report of a Pc–platin conjugate was by Guo et al., who used a Si(IV)Pc scaffold decorated with either meta- or para-linked pyriplatin derivatives as axial ligands (P1 and P2, respectively) (Fig. 7) to allow efficacious DNA anchoring through displacement of the chloro ligand to form Pc–platin–DNA adducts (Fig. 7).38 Irradiation of the DNA-anchored photosensitizer allowed specific DNA damage by photodynamic action with the accompanying covalent attachment preventing further replication and transcription, making the synergistic treatment especially effective.38 The Pc–pyriplatin conjugates P1 and P2 displayed markedly lower cytotoxicity in comparison to unconjugated cisplatin or pyriplatin, demonstrating low dark toxicity of the conjugate, a crucial requirement for a practical photosensitizer.38 Even at a high concentration of 100 μM, the Pc–pyriplatin constructs only achieved inhibition ratios of 45–55% in the dark, which was drastically lower than the free pyriplatin that had 97% inhibition at this concentration.38 Upon red-light irradiation (600–710 nm), the Pc–pyriplatin conjugates P1 and P2 had a 25-fold and 7-fold increase, respectively, in cytotoxicity at a concentration of 1.0 μM, increasing from 4 and 13% to 95 and 99% respectively for P1 and P2.38 Since the maximum effective range of singlet oxygen is approximately 20 nm, the close proximity of the anchored Pc to a DNA strand afforded a beneficial PDT modality.38–40 The use of both the platin and the Pc photosensitizer therefore showed combinational benefits, as the platin allowed DNA targeting with accompanying chemotherapy, while the Pc endows the Pt(II) complex with red-light induced photodynamic action.38 Guo et al. further demonstrated a theragnostic approach, as incubation of the Pc–platin complex in cancerous tissue allowed confocal fluorescence microscopy to be performed. This technique enabled visualization of the distribution of the Pc–platin within the cell, providing valuable insight into the uptake of the conjugates into the nucleoli from the cytoplasm.38
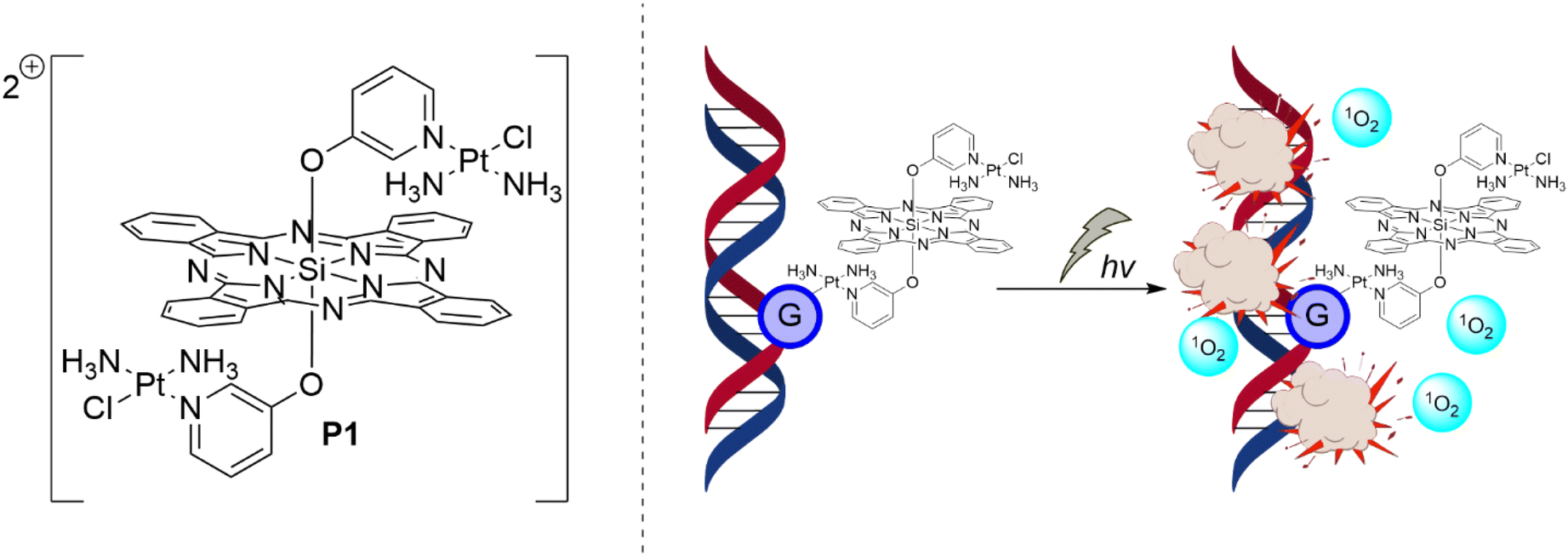 | ||
| Fig. 6 Schematic PDT mechanism for Pc–platin conjugate P1, where anchoring to DNA leads to highly effective damage by singlet oxygen formed upon photoirradiation of the Pc photosensitizer once coordinated to the DNA strand(s). | ||
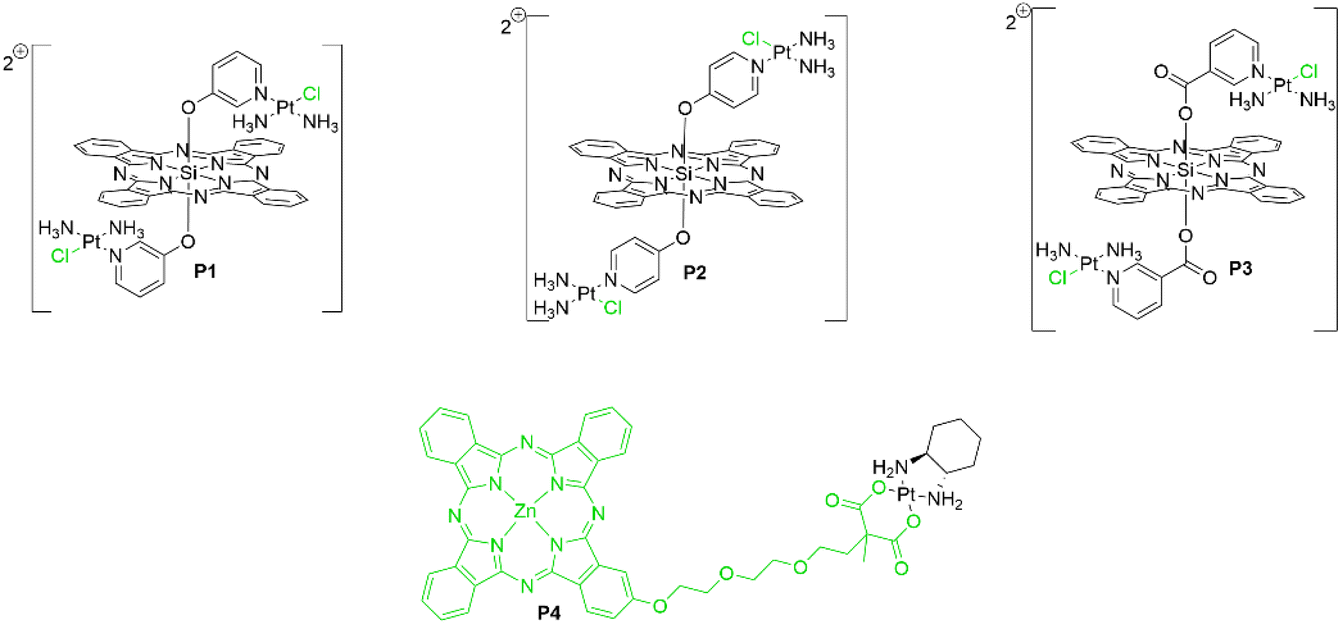 | ||
| Fig. 7 Pc–platin conjugates with the ligands displaced upon aquation highlighted in green. | ||
Hartman et al. elaborated upon this design by encapsulating the related cationic Pc P3 in an anionic hyaluronate nanoparticle (Fig. 7).39 This nanoparticle formulation of P3 targets CD44 receptors that are commonly overexpressed in cancer cell lines.39 The nanoparticle undergoes cellular uptake before endosomal escape and subsequent enzymatic degradation to release the free Pc–platin conjugate P3.39 Confocal fluorescence microscopy showed preferential accumulation of P3 in the mitochondria instead of the nucleus of MDA-MB-231 human breast adenocarcinoma cells.39 Repeating the fluorescence microscopy experiment with HEK293T human embryonic kidney cells further illustrated the selective uptake for cancerous cells due to the absence hyaluronate acid receptors in healthy human kidney cells.39 Localization of the complex in the mitochondria rather than nucleosomes has several advantages, such as the greater oxygenation of the mitochondria and the lack of mitochondrial DNA nuclear excision repair mechanisms that may otherwise cleave the Pc–Pt–DNA adduct.39 This allows substantial damage to the mitochondrial DNA by the abovementioned platin-anchoring system to allow dual chemo–photodynamic therapy, ablating the organelle required for energy generation and preventing extensive tumour proliferation.39
Ng et al. synthesized Pc–platin conjugate P4 (Fig. 7), wherein the platin moiety is attached to the periphery of the macrocycle and, due to the nature of the conjugate design, the platin complex is liberated from the Pc upon crossing the cell membrane and aquation.40 It was determined that using both moieties lowered the IC50 value five-fold compared to using the ZnPc complex alone, with P4 having an IC50 value of 0.11 μM under 672 nm irradiation and a modest dark toxicity of 78.5 μM (PI = 710).40 In comparison, the free Pc had light and dark IC50 values of 0.55 μM and >100 μM, respectively (PI = 180) (Table 1).40 Hence, demonstrating a cooperative effect between the two cytotoxic components with IC50 values markedly lower for the conjugate than the individual constituents. The authors also attributed the low cytotoxicity to the reduced aggregation of the extended π-system of the chromophore coupled with high cellular uptake to allow efficient generation of ROS.40P4 had enhanced cellular uptake relative to the free ZnPc model system and showed preferential accumulation within lysosomes, allowing highly localized, intracellular oxidative damage upon irradiation.40
| Conjugated | λ maxabs (nm) | Light IC50 (μM) | Dark IC50 (μM) | Phototoxicity index | Φ Δ | Φ f | Ref. | |
|---|---|---|---|---|---|---|---|---|
| a Not determined. b Formulated as a hyaluronate nanoparticle for additional active targeting. | ||||||||
| P1 | Yes | 688 | —a | —a | —a | 0.24 | 0.21 | 38 |
| P2 | Yes | 688 | —a | — | —a | 0.22 | 0.19 | 38 |
P3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Yes | 744 | 0.05 | 77 | 1500 | 0.24 | —a | 39 |
| P3 | Yes | 690 | 0.09 | 146 | 1600 | 0.22 | 0.20 | 39 |
| P4 | No | 672 | 0.11 | 78.5 | 710 | 0.56 | 0.22 | 40 |
| P5xi | No | 698 | 103.8 | 301.5 | 2.9 | 0.60 | 0.17 | 43 |
| Free Pc | No | 672 | 0.55 | >100 | >180 | 0.57 | 0.25 | 40 |
It has thus been shown that anchored derivative P3 provides is more effective than non-anchored P4, with the anchored system having a lower IC50 value under irradiation than P4 leading to a higher PI value. This observation can be rationalized as the beneficial effect of the Pc being tethered in close proximity to DNA for P3, facilitating specific and localized oxidative damage, in conjunction with a chemotherapeutic effect attributed to the platin moiety. In contrast, the Pc of the non-anchored P4 will have been displaced during the aquation step once the complex has entered the cell membrane and with the short effective range of singlet oxygen, it is apparent that having the photosensitizer located within close proximity allows for more efficient and selective damage to the cancerous cell.38–40 This was supported through confocal florescence microscopy, which showed that the free Pc complex, cleaved during the aquation step, was preferentially localizing in lysosome organelles and not in the nucleus or mitochondria, unlike conjugates P1–P3.40
Nyokong and coworkers synthesized covalent Pc–platin conjugates with varying Pt(II) moieties attached to the Pc periphery (Fig. 8).40–43 Although due to the insolubility of the compounds only P5xi could be tested in aqueous media with a light IC50 value of 103.8 μM and a dark IC50 of 301.5 μM in HEp2 cells, leading to a PI value of 2.9 (Table 1).43 These Pc–platin conjugates work akin to P4 in that the Pc moiety would be displaced upon crossing the cell membrane, splitting the conjugate into its constituent parts.
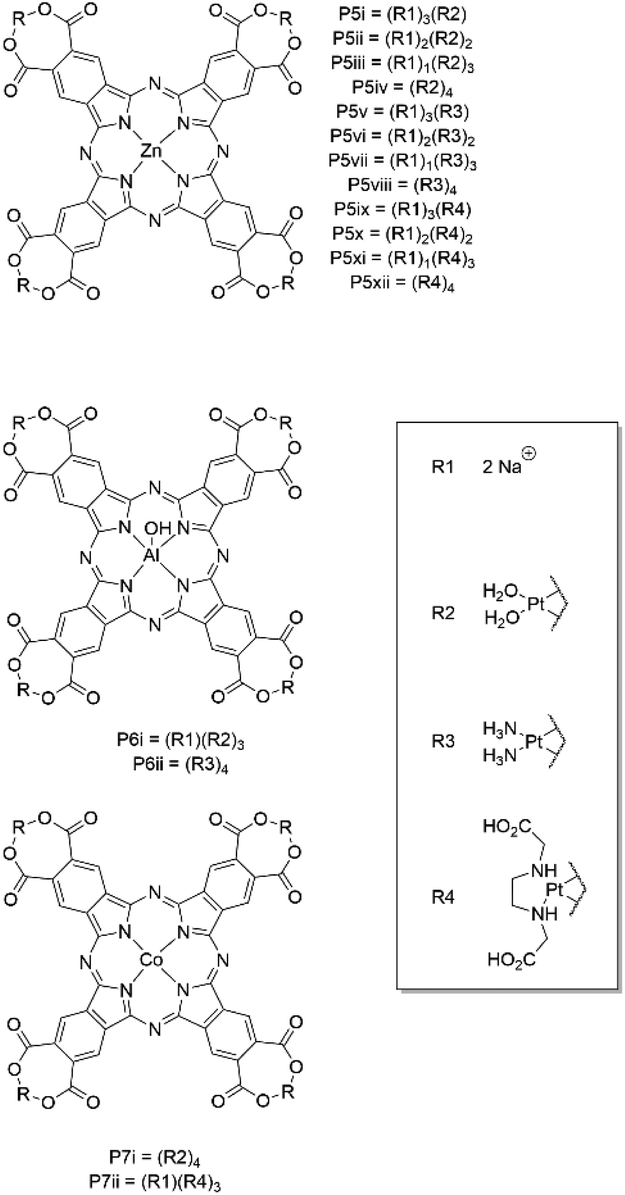 | ||
| Fig. 8 Pc–platin conjugates developed by Nyokong and coworkers. | ||
It has been observed that the combination of ROS and platin complexes can exacerbate off-target effects (e.g. cell dysfunction, healthy tissue damage, amplified renal damage, and ototoxic damage) due to ROS inhibiting the antioxidant defence mechanisms within the cell, preventing cytoplasmic glutathione from detoxifying healthy cells of the free platin.35 We suggest this may be overcome by promoting nonradiative decay in place of intersystem crossing, thus inducing a PTT response instead of PDT, with no generation of ROS.20 This is achievable through use of paramagnetic metals or photoinduced electron transfer (PeT) substituents onto the Pc architecture.20 Thus far, there has only been a single example of a Pc–platin molecule that could undergo a PTT mechanism. Dolotova and Kaliya successfully synthesized a CoPc–platin conjugate that would deactivated through nonradiative pathways after 678 nm photoexcitation due to the paramagnetism of the square-planar Co(II)Pc (P7i).44 Unfortunately the compound was not tested for its photothermal performance against any cancerous cell lines as solubility of the conjugate was poor in aqueous media.44 The solubility of the conjugate was attempted to be improved through using a modified amine ligand in place of the aqua-ligands but the compound was still insoluble and could not be biologically tested.45 No emission data was recorded meaning it was difficult to determine if the radiative decay pathway had been quenched fully and how effective the compound would be as a photothermal agent.
The examples of Pc–platin conjugates in section 2 demonstrate how platins can be used as effective targeting groups for Pc-based PDT photosensitizers by increasing specificity for tumour uptake and limiting dark toxicity. Pc–platin conjugates have lower IC50 values under irradiation than Pcs alone, showing the additional synergistic effect of the platin chemotherapeutic targeting vector. It has been evidenced that platins could be a simple and efficient targeting group for Pcs while often also enabling higher ΦT and ΦΔ values due to the heavy-atom effect.
3. Phthalocyanine–kinase inhibitor conjugates
Kinase inhibitors are rapidly becoming one of the most valuable assets for targeting malignant tumours.46,47 The development of kinase inhibitors such as imatinib has turned terminal diseases like chronic myelogenous leukaemia and gastrointestinal cancer into manageable conditions with improved patient outcomes.46,47 With the ever expanding family of kinase inhibitors (Fig. 9), and their broad-spectrum anticancer activity, it is anticipated that many other cancers that currently have poor patient prognosis could become controllable.46,47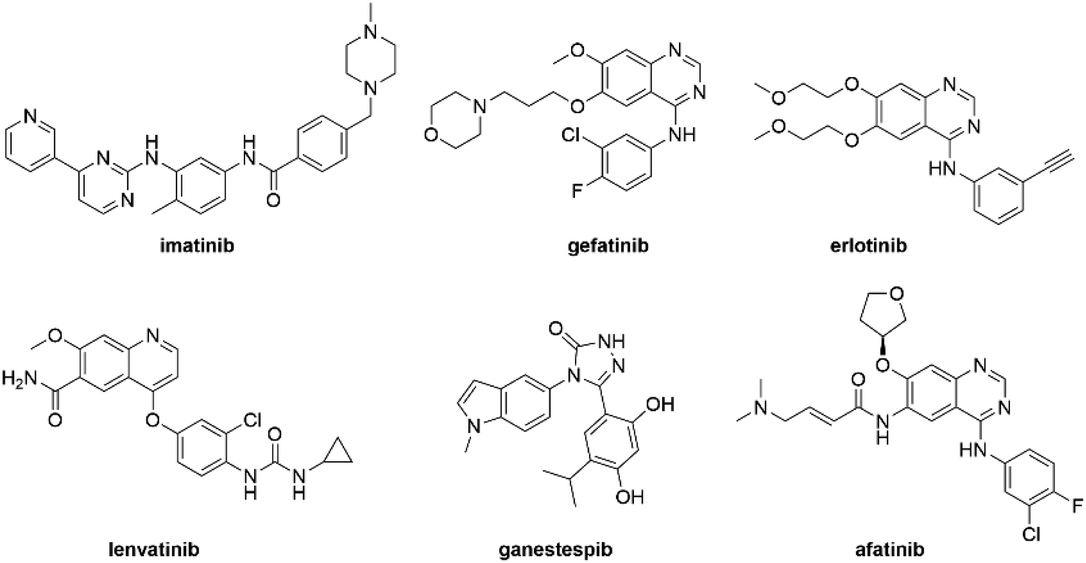 | ||
| Fig. 9 Common kinase inhibitors employed for their anticancer effects. | ||
The phosphorylation process facilitated by kinases regulates most aspects of the lifecycle of a cell. However, when defective, kinase enzymes phosphorylate abnormally and this enables extensive progression of various cancers.46,47 With 518 different kinases encoded into the human genome that all rely on the recognition of adenosine triphosphate (ATP) cofactors, achieving selective inhibition of a single kinase may appear challenging. However, selectivity can be achieved by exploiting differences between hydrophobic pockets and proximal amino-acid residues in the ATP-binding domain.46,47 The majority of kinase inhibitors resemble the binding displayed by ATP (Fig. 10): the purine base of ATP is buried deep within the binding site of the enzyme and is locked in place through hydrogen-bonding interactions within the hinge region. Covalent kinase inhibitors containing electrophilic moieties, in addition to the purine pharmacophore, are being investigated that provide an overall more potent anticancer agent.48 Covalent binding of the kinase inhibitor through nucleophilic attack of an amino acid residue within the active site prevents the inhibitor being reversibly displaced by endogenous ATP. One example is afatinib that includes an acrylamide moiety that undergoes a Michael addition with a proximal cysteine residue within the active site. This design is likely to become more prevalent with the synthesis of new kinase inhibitors.48
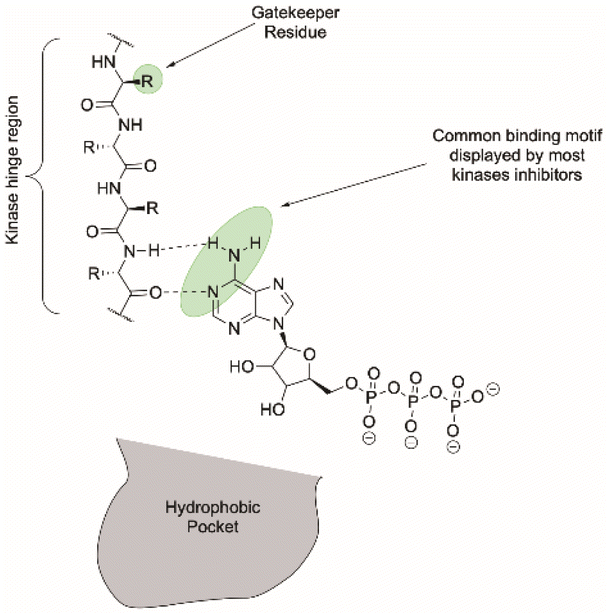 | ||
| Fig. 10 Binding of ATP within the kinase active site, highlighting key components that allow specificity.49 | ||
Several Pc–kinase inhibitor conjugates have been reported that demonstrate the benefit of a kinase-targeting moiety. Greater phototherapeutic efficacy can be attained, as the kinase inhibitor directs the photosensitizer within range of a faulty enzyme. However, it is critical that conjugation to the Pc does not tamper with the ATP-resembling pharmacophore otherwise binding is compromised.
The first example of a Pc–kinase conjugate was reported by Xue et al., who synthesized a Pc–erlotinib conjugate that displayed high affinity for HepG2 cancer cells.50 The Pc–erlotinib conjugate was synthesized with an oligoethylene glycol spacer that allowed the distance between the erlotinib and Pc moieties to be tuned, modulating the proximity of the photosensitizer to the kinase active site.50 The conjugates displayed impressive ΦΔ values of 0.66 (K1α-3, n = 2) and 0.57 (K1α-5, n = 4) (Fig. 11), respectively, with both K1α-3 and K1α-5 having a similar ΦF value of 0.27 (Table 2), allowing the opportunity for fluorescence-based theragnostic modalities.50 Fluorescence molecular tomography in vivo showed strong preference of the Pc–erlotinib conjugates for rapidly proliferating cancerous tumours with little off-target binding. The combination of high specificity for epidermal growth factor receptors (EGFRs, a type of receptor tyrosine kinase) and low light IC50 values demonstrates strong anticancer effects, in contrast to the Pc alone, which displayed little selectivity for either cancerous or healthy tissue.50 The trend in the IC50 values indicated that the closer the photosensitizer was to the kinase enzyme (i.e. the shorter chain length between photosensitizer and inhibitor), the greater the potency of the conjugate towards HepG2 and A431 tumour cells, with IC50 values under irradiation being 0.01 μM (K1α-3) and 0.04 μM (K1α-5) with both showing dark IC50 values >50 μM, giving an impressive PI of >5000 and >1000, respectively.50
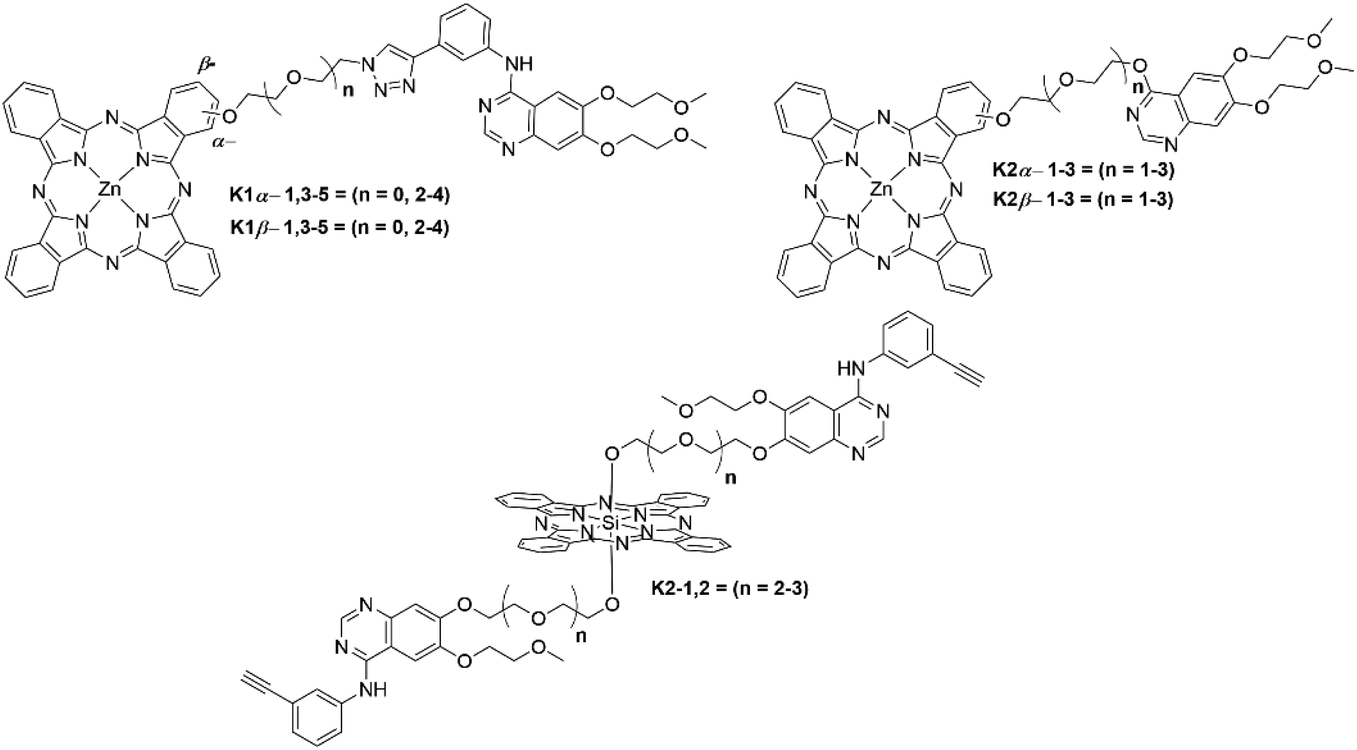 | ||
| Fig. 11 Phthalocyanines that employ a kinase inhibitor as a targeting vector. Where relevant, the position of attachment of the linker (α- or β-position of the Pc) is indicated in the name; however, each is a single isomer. | ||
| λ maxabs (nm) | Φ Δ | Φ F | Light IC50 (nM) | Dark IC50 (nM) | Phototoxicity index | Ref | |
|---|---|---|---|---|---|---|---|
| a Not determined. | |||||||
| K1α-1 | 678 | 0.63 | 0.26 | 12.9 | >50000 |
>3880 | 50 |
| K1α-3 | 678 | 0.66 | 0.26 | 9.61 | >50000 |
>5200 | 50 |
| K1α-4 | 678 | 0.63 | 0.26 | 27.8 | >50000 |
>1800 | 50 |
| K1α-5 | 678 | 0.57 | 0.26 | 44.5 | >50000 |
>1120 | 50 |
| K1β-1 | 672 | 0.53 | 0.27 | 34.0 | >50000 |
>1470 | 50 |
| K1β-3 | 672 | 0.54 | 0.27 | 44.8 | >50000 |
>1120 | 50 |
| K1β-4 | 672 | 0.53 | 0.27 | 41.9 | >50000 |
>1190 | 50 |
| K1β-5 | 672 | 0.50 | 0.27 | 91.8 | >50000 |
>545 | 50 |
| K2α-1 | 678 | 0.56 | 0.17 | 3.70 | >1000 | >270 | 51 |
| K2α-2 | 677 | 0.50 | 0.22 | 13.8 | >1000 | >72.5 | 51 |
| K2α-3 | 678 | 0.55 | 0.22 | 6.70 | >1000 | >149 | 51 |
| K2β-1 | 672 | 0.44 | 0.25 | 13.2 | >1000 | >75.7 | 51 |
| K2β-2 | 672 | 0.46 | 0.25 | 16.7 | >1000 | >59.9 | 51 |
| K2β-3 | 672 | 0.53 | 0.25 | 8.70 | >1000 | >115 | 51 |
| K2-1 | 675 | 0.26 | 0.34 | 8.00 | —a | —a | 52 |
| K2-2 | 673 | 0.34 | 0.43 | 27.0 | —a | —a | 52 |
Xue et al. made analogous Pc–erlotinib conjugates to determine whether the position of conjugation of erlotinib to the Pc (α, β, or axial position) and the linker length affected their activity. Specificity is key for targeting a druggable site, and subtle modifications to the structure of the conjugate can result in altered activity.51 From a photophysical perspective, varying the conjugation site and linker length displayed minimal effects, with the K1α-series exhibiting slightly higher ΦΔ values (0.57–0.66) than the K1β-series (0.50–0.54) while the ΦF values remained constant between the two structural families (0.26–0.27) (Table 2).51 This translated to lower IC50 values with erlotinib conjugated to the α-position compared to the β-conjugated analogues due to the marginally higher ΦΔ values.51 In comparison to the free ZnPc (light IC50 value = 43.3 nM), all erlotinib conjugates linked to the α-position had a lower IC50 value when the linker length was n < 5, but for the β-conjugates the length of the linker had to be n < 2 for them to outcompete the parent ZnPc photosensitizer.51 Confocal fluorescence microscopy in vitro evidenced pronounced localization within the lysosomes and mitochondria for all K1 conjugates.51
Further studies by Xue et al. investigated the site of conjugation on the erlotinib inhibitor (K2-series).52 This was to test whether the modification of the erlotinib (i.e. removal of the 3-ethynylaniline group) would affect the targeting to the ATP binding site. Initial molecular-docking studies showed the hydrogen-bonding interactions of the pharmacophore were still present with additional hydrogen-bonding sites observed within the ethylene glycol chain connecting Pc to erlotinib. The binding energy to the EGFR was calculated to be −9.97 kcal mol−1 for K2α-1 (Fig. 12), suggesting the 3-ethynylaniline group had little effect on binding to the receptor in comparison to an analogue containing the 3-ethynylaniline moiety (binding energy = −9.62 kcal mol−1).52 The distance between the modified erlotinib and Pc moieties was further investigated by variation in oligioethylene linker lengths and position of conjugation to Pc. All compounds showed similar photophysical properties (Table 2), but K1α-3 from the preliminary studies was determined to be the optimum structure as displayed by its high PI > 5200.52 The K2 series all showed impressive PI values (Table 2) but could not outperform their K1 predecessors due to their lower dark IC50 values translating into lower PI values.52 Thus, experimentally showing that the loss of the 3-ethynylaniline group had a significant effect on the dark toxicity.52 Fluorescence intensity studies showed the K2-series still maintained a high preference for cancerous cells with overexpressed EGFR content (HepG2 cells) over the low EGFR content of human lung fibroblast (HELF) cells even with the modified erlotinib targeting vector. Demonstrating the strong targeting potential of erlotinib and variants towards cancerous cells with high EGFR content as removal of the vector (i.e. the Pc alone) displayed no selectively for targeting HepG2 cells.52
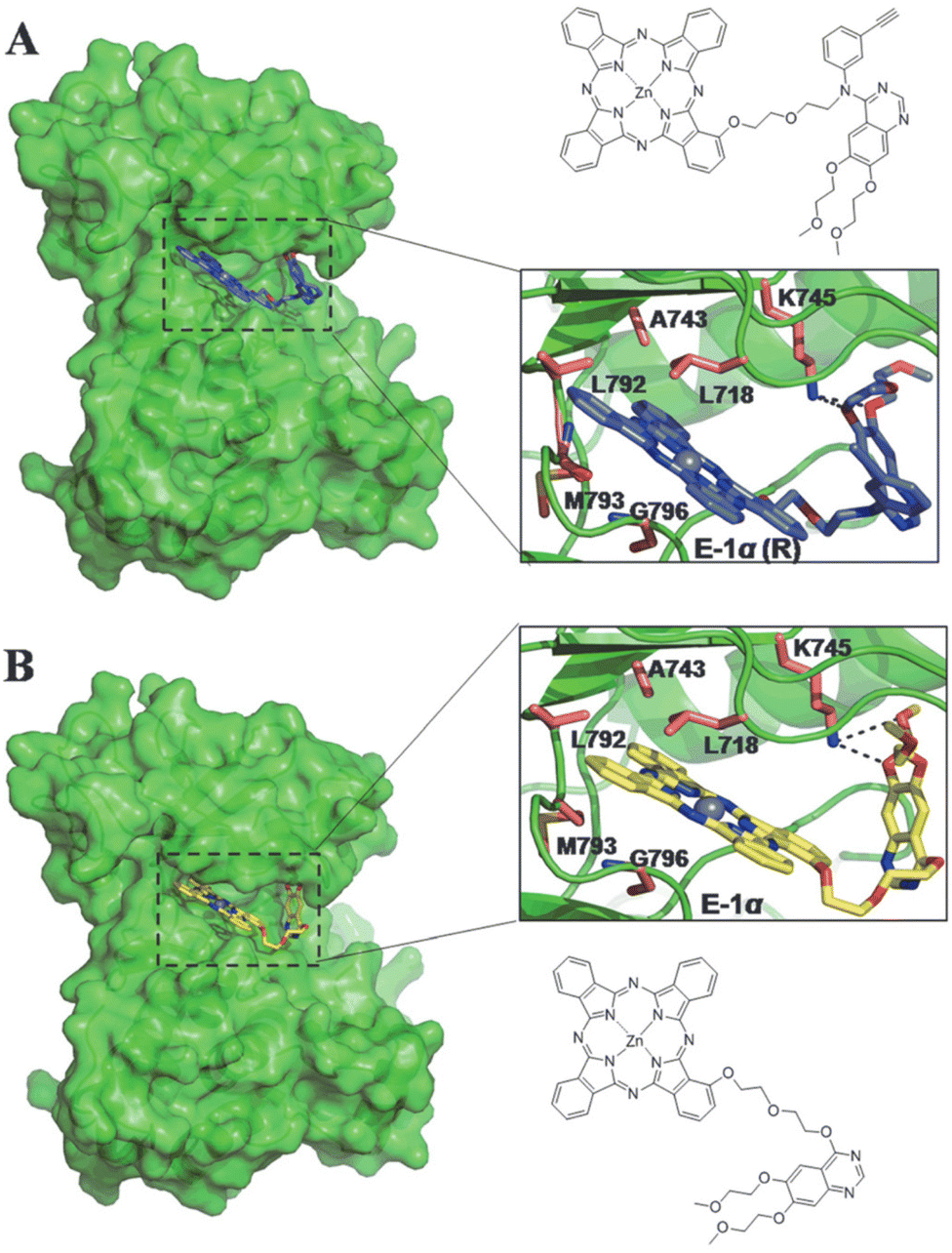 | ||
| Fig. 12 The calculated binding of a modified hypothetical Pc–erlotinib conjugate (A) vs experimentally tested conjuagte K2α-1 (B) to evaluate the role of the ethynylaniline group in binding to an EGFR (receptor tyrosine kinase). Colour code: enzyme in green; Pc–kinase inhibitors shown in blue and yellow stick form; side chains involved with ligand binding in salmon; oxygen atoms in red; nitrogen atoms in blue; and hydrogen bonds displayed by black dashed lines. Reproduced with permission from ref. 52. | ||
Xue et al. further experimented with axially conjugated SiPc–erlotinib conjugates (Fig. 11) that, like their predecessors, showed improved targeting of HepG2 cells over the non-conjugated Pc photosensitizer. Fluorescence microscopy indicated preferential localization of the conjugates within lysosomes and mitochondria.53 Higher ΦF values were observed with the SiPc–erlotinib conjugates (ΦF = 0.34–0.43) than their ZnPc–erlotinib homologues, at the expense of singlet-oxygen sensitization (ΦΔ = 0.26–0.39) (Table 2). The change in the photophysical properties could be attributed to the reduced aggregation of the SiPc–erlotinib conjugate, with respect to the ZnPc–erlotinib conjugates, due to the axially ligated erlotinib preventing π-stacking. It is also possible that reduced spin–orbit coupling going from Zn to Si could result in reduced singlet-oxygen sensitization.53 As displayed with the ZnPc conjugates, the SiPc conjugates showed a similar tend with shorter chain lengths (i.e. the closer the Pc was to the binding domain of the enzyme) resulting in lower light IC50 values on comparison of n = 2 and n = 3 SiPc–erlotinib conjugates (Table 2).53 These studies show that linker length and site of conjugation (i.e., whether at the α, β, or axial positions) cannot be overlooked when considering how the Pc–kinase inhibitor (or other anticancer drug) conjugate interacts with the target site.
Although the majority of Pc–kinase inhibitor conjugates have focused on modifications of erlotinib, there have been successes with other kinase inhibitors. Zhao et al. reported conjugation of ganetespib (K3) to the β-position of a ZnPc photosensitizer via a short alkyl spacer (Fig. 13).54 The Pc–ganetespib conjugate maintained the traditional Q-band displayed by Pc macrocycles with an absorption maximum situated in the red region at 671 nm.54K3 was demonstrated to bind to extracellular Hsp90 protein in vivo with sufficient ROS production to prevent cell proliferation and induced apoptosis in a more efficient manner than the individual constituents.54
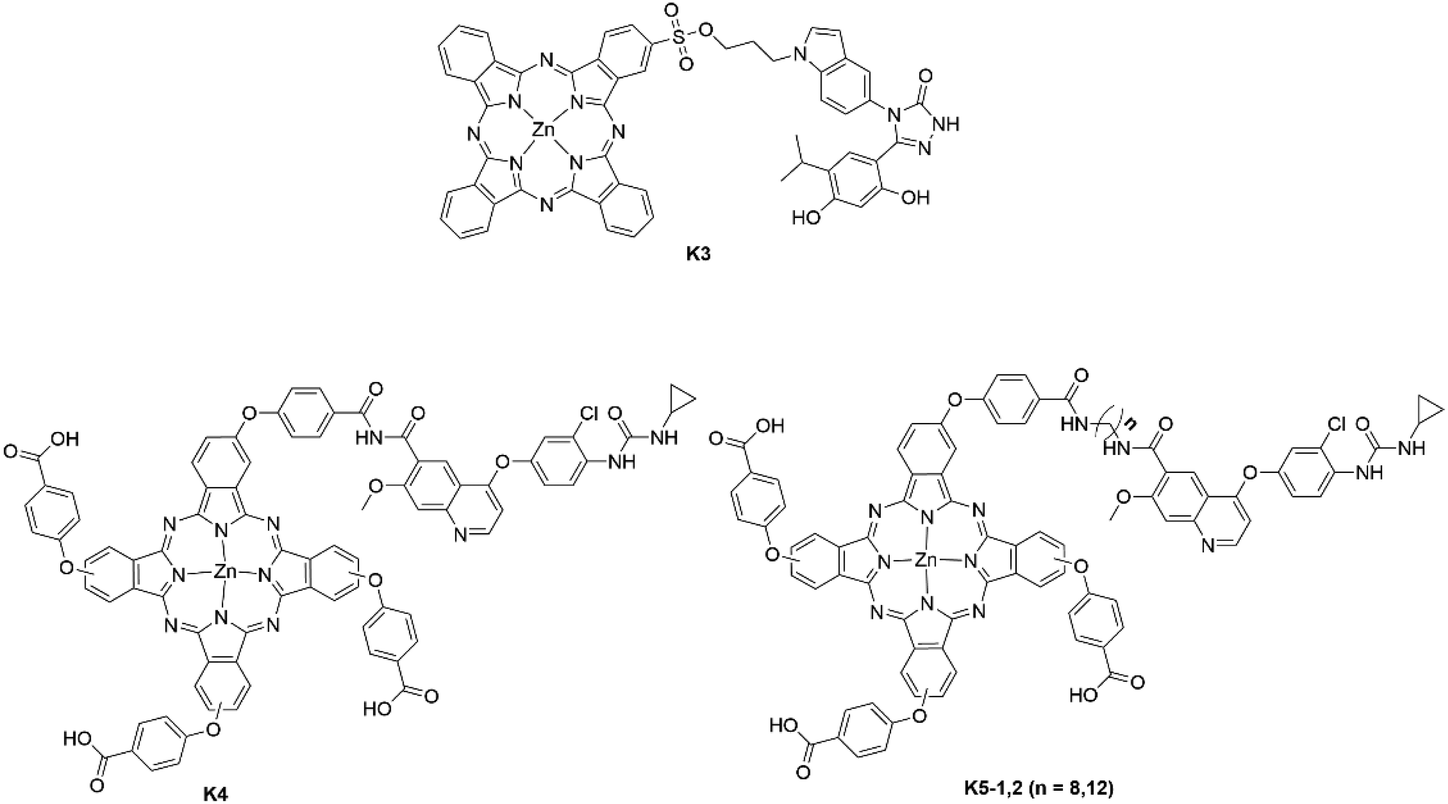 | ||
| Fig. 13 Top: Pc–ganetespib conjugate; bottom: Pc–lenvatinib conjugates. K4 and the K5 series are statistical mixtures of isomers with all combinations of substitution in the β-positions of the isoindole units of the Pcs. | ||
Zhao and co-workers also developed a series of Pc–lenvatinib conjugates with varying length alkyl linkers (n = 0 (K4), n = 8 (K5-1) and n = 12 (K5-2)) (Fig. 13).55 All three conjugates displayed reduced dark cytotoxicity in comparison to free lenvatinib and equimolar ZnPc/lenvatinib, which significantly increased with 660–670 nm irradiation. K5-1 also showed good accumulation within cancerous cells and a modest light IC50 value of 19.4 μM, a 3.5-fold decrease compared to free lenvatinib against 4T1 cells; the light IC50 value against MCF-7 cells was further improved for K5-1 with a value of 14.5 μM, a 4.5-fold decrease compared to free lenvatinib.55 The IC50 against MCF-7/ADR cells also demonstrated the ability of K5-1 to overcome multidrug resistance through glutathione depletion with a light IC50 value of 47.3 μM in comparison to 329 μM for free lenvatinib.55 The authors used the combination index (CI) to determine whether the designed conjugate was behaving in a synergistic manner (CI < 1 for synergistic behaviour; CI = 1 for additive behaviour; CI > 1 for antagonistic behaviour). The CI index demonstrated the synergism of the two cytotoxic components with a CI value < 1 against all the tested cell lines using K5–1.55 The authors also demonstrated the potency of the conjugate in vivo by formulating the K5–1 conjugate into PEG2000-PLA20000 nanoparticles to improve solubility, thus allowing in vivo experiments to be performed. The fluorescence of the Pc allowed a theragnostic paradigm to be successfully employed in vivo and almost complete tumour ablation within tumour-bearing mice with no obvious signs of off-target toxicity observed. In contrast to the previous kinase inhibitor examples described, the authors did not consider Pc regiochemistry in their conjugate design. The different isomers within the isomeric Pc mixture may have different photophysical and biological properties, binding energies, and overall efficacy, ultimately making such a mixture more challenging to translate.
Due to the major role kinase enzymes play in the development of various cancers, the use of kinase inhibitors as targeting vectors for Pcs is a useful synergistic platform for dual chemo- and phototherapy to enhance the specificity of the treatment. The key finding has been that the conjugation site of the kinase inhibitor on the Pc is vital to maintain the highly specific binding of the kinase inhibitor and, thus, for the Pc–kinase inhibitor to elicit a phototherapeutic effect.
4. Phthalocyanine–anthracycline conjugates
Anthracyclines are anticancer agents extracted from Streptomyces bacteria that are renowned for their broad-spectrum activity.56 Anthracyclines have been central to chemotherapy since their inception in the 1960s with the most notable examples being doxorubicin (DOX), daunorubicin, epirubicin, and idarubicin (Fig. 14). All anthracyclines have a similar core structural motif of a quinone containing fused tetracyclic structure linked to a modified amino-sugar residue.57 The potency and broad-spectrum anticancer activity of anthracyclines is desirable, although accompanied by dose-dependent and cumulative cardiotoxicity that limits their utility.58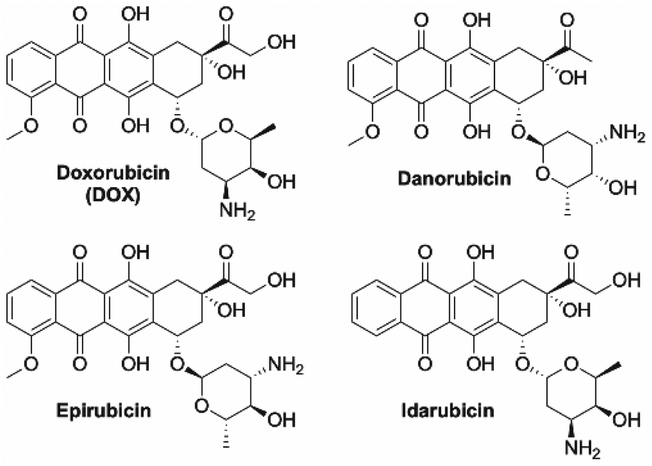 | ||
| Fig. 14 Common anthracyclines agents used in chemotherapy treatments. | ||
There is still much debate regarding the mechanism of action of anthracyclines, even though they have been used clinically for almost sixty years. The two currently accepted mechanisms are as follows: (1) intercalation of the drug into the DNA double helix, facilitated by π–π interactions of the planar conjugated structure and the base pairs, preventing DNA replication, and (2) reduction of the quinone moiety, causing oxidative damage via the formation of ROS to which nucleic acids are highly susceptible.57,59
The potential for anthracyclines to partially quench the photodynamic action of Pcs due to their electron-accepting structure suggests conjugation of the two moieties may decrease the cytotoxic effect of a Pc–anthracycline conjugate.60 Huang et al. synthesized both a traditional Pc–doxorubicin (DOX) conjugate (A1) and a prodrug conjugate (A2) with a cleavable peptide linker that liberates DOX upon exposure to the fibroblast activation protein (FAP) that is commonly overexpressed in cancerous cell lines to establish the benefits of each approach (Fig. 15).60 Before exposure of A2 to FAP, both the photosensitizing ability of the Pc and the anticancer effect of DOX were reduced until the molecule was guided towards a cancer cell and the Pc–DOX conjugate cleaved into its constituent parts.60 The liberation of DOX endowed the Pc with increased ΦΔ and ΦF values whilst simultaneously increasing the cytotoxicity of both species.60 Huang and co-workers established a prodrug approach was an effective synergistic platform in vivo that can limit the off-target effects attributed to both DOX and Pc.60 The ΦΔ values were 0.40 and 0.30 for A1 and A2, respectively.60 Both compounds had respectable ΦF values of 0.12 and 0.10 respectively, with the cleaved Pc photosensitizer having ΦΔ and ΦF values of 0.60 and 0.23, respectively (Table 3).60 Localization studies were performed in vitro with fluorescence microscopy, which demonstrated that both the non-cleavable conjugate and the prodrug conjugate preferentially localized in the mitochondria, although when incubated with FAP, the DOX constituent was evidenced in the nucleus.60 IC50 values for A1 and A2 interestingly showed that the cleavable peptide linker had both a greater light toxicity and a lower dark toxicity than the traditional conjugate (light IC50 = 0.42 vs. 0.56 μM and dark IC50 = 20 vs. 6.4 μM), demonstrating the efficacy of the traditional conjugate in comparison to the prodrug (PI = 47.7 (A1) vs. 11.4 μM (A2)) (Table 3).60 This can be rationalized as the photosensitizer being anchored to the mitochondrial DNA, allowing specific oxidative damage, and thus single strand DNA breaks with the cooperative anticancer effect of the anthracycline moiety. The light IC50 value of prodrug conjugate A2 dropped to 0.13 μM in the presence of FAP; however, so the dark IC50 value also fell to 1.14 μM with added FAP (PI = 8.8).60 This is disadvantageous as a low dark toxicity is a key requirement for an effective photosensitizer. On evaluation of the data, the traditional Pc–drug conjugate, which is bound and retained at the desired site of action, produces an overall more effective synergistic effect than the prodrug approach.
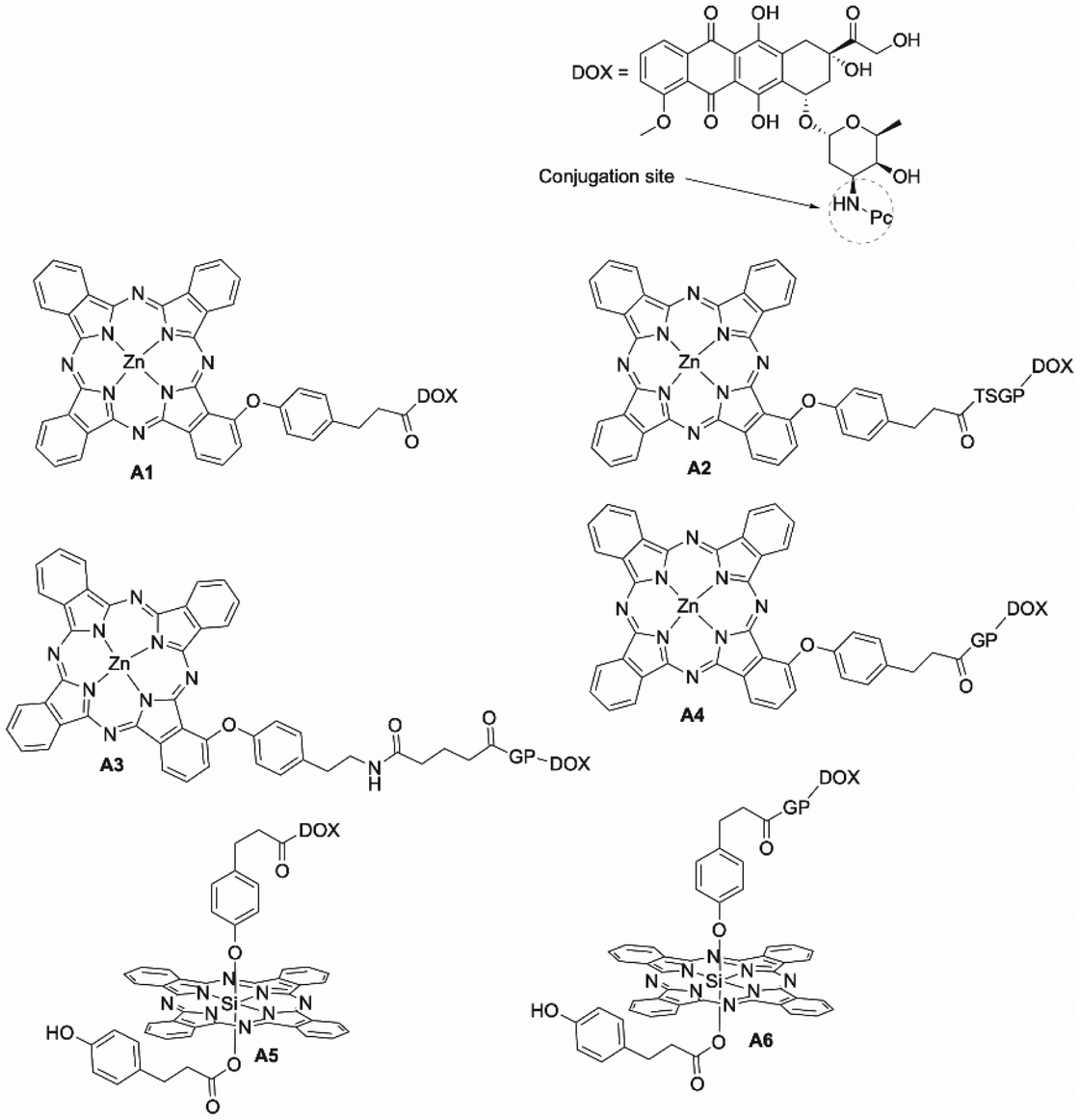 | ||
| Fig. 15 Pc–doxorubicin (DOX) conjugates. T = threonine, S = serine, G = glycine, P = proline. | ||
| Traditional or prodrug | λ maxabs(nm) | Φ Δ | Φ F | Light IC50 (μM) | Dark IC50 (μM) | Phototoxicity index | Ref. | |
|---|---|---|---|---|---|---|---|---|
| a Values for A2 + FAP were not reported. Values listed for Pc with the peptide unit only. | ||||||||
| A1 | Traditional | 676 | 0.40 | 0.12 | 0.42 | 20 | 47.7 | 60 |
| A2 | Prodrug | 674 | 0.30 | 0.10 | 0.56 | 6.4 | 11.4 | 60 |
| A2 + FAP | — | 674a | 0.60a | 0.23a | 0.13 | 1.14 | 8.8 | 60 |
| A3 (1b) | Prodrug | 675 | 0.27 | 0.10 | — | — | — | 61 |
| A4 (1c) | Prodrug | 674 | 0.40 | 0.12 | — | — | — | 61 |
| A5 (2b) | Traditional | 676 | 0.20 | 0.01 | — | — | — | 61 |
| A6 (3c) | Traditional | 681 | 0.12 | 0.12 | — | — | — | 61 |
| A7 (3b) | Prodrug | 682 | 0.09 | 0.10 | — | — | — | 61 |
Huang expanded on this work by comparing both the prodrug and the traditional conjugates whilst also investigating the effects of chain length between the two components and the location of DOX on the Pc macrocycle (axial vs. peripheral) (A3–A6).61 FAP responsiveness experiments showed that the prodrugs undergo facile enzymolysis of the linker to release the DOX unit.61 In the absence of in vitro or in vivo testing it is difficult to accurately state which linker length or difference between axial or peripheral location of the DOX moiety provided the best overall synergistic effect. The photophysical data indicated that the presence of the DOX moiety did reduce ΦΔ values of both the prodrug and traditional conjugates, with the free Pc displaying higher ΦΔ values on all occasions (Table 3).61 The traditional conjugates with shorter chain lengths and peripheral conjugation sites displayed greater ΦΔ and ΦF values,61 although it is not known if this translates to better PDT performance.
The development of highly effective Pc–DOX conjugates displaying limited dark toxicity in conjunction with a strong PDT response still requires additional exploration. Specifically, the optimal structure where the DOX unit does not quench the photosensitizing ability of the Pc moiety and the conjugate also retains a high PI still has to be attained. The design of the agents could be improved by alternative conjugation sites, as conjugation of the photosensitizer to the amino moiety of the sugar residue prevents important ionic-bond formation between the cationic protonated amino moiety (under physiological conditions) and the anionic sugar-phosphate backbone. More generally, this illustrates that important binding moieties should not be tampered with when designing photosensitizer–anticancer conjugates. In future work there may be more success in designing these forms of conjugates as photothermal agents as this could negate the problem of quenched sensitization of singlet oxygen by DOX.
5. Miscellaneous Pc–anticancer agent conjugates
This subsection contains other notable Pc–anticancer drug conjugates that follow one of the following strategies: (1) target estrogen receptors through conjugation to tamoxifen, (2) target the nucleus through conjugation to histone deacetylase inhibitors or DNA alkylating agent chlorambucil, (3) disrupt vasculature through conjugation to combretastatin, chalcones, and taxanes, or (4) contain coumarin and perphenazine targeting vectors.The role of estrogen receptors (ERs) in the formation and progression of many malignant breast tumours is well documented. Estrogen receptor-positive (ER+) breast cancer is typically treated with one of three classes of endocrine therapeutics: selective estrogen modulators (SERM), aromatase inhibitors, or selective estrogen-receptor degraders.62 SERMs work through competitive inhibition of overexpressed ERs in place of the steroidal hormone 17β-estradiol (E2), initiating apoptosis by preventing binding of associated transcription cofactors, which thus stops abnormal cell proliferation and differentiation.63
Liu and coworkers conjugated the SERM tamoxifen to the α-position of a Pc photosensitizer through an oligoethylene glycol spacer of varying length (Fig. 16). The hypothesis was that this would tether the Pc to overexpressed ERs, which in turn would lead to selective oxidative damage and disruption of downstream cell-signalling pathways.62,63 The conjugate displayed a high ΦΔ value of 0.61 with a ΦF value of 0.17 coupled with a Q-band absorption maximum of 678 nm (Table 4).63 Binding of the conjugate to ERs was confirmed through in vitro experiments where it was shown that inhibition of ERs was dependent upon exogenous E2 concentration, while the Pc derivative without tamoxifen behaved independently.63 This had a direct impact on the photocytotoxicities of the conjugates, where photodynamic efficiency decreased with increasing concentrations of exogenous E2.63In vivo experiments further confirmed the specificity towards ER+ tumour tissue through fluorescence tomography of BALB/c nude mice supporting MCF-7 tumours. The fluorescence intensity was notably higher for cancerous tissue due to preferential localization of the Pc–tamoxifen derivative in cells with a high ER content opposed to healthy tissue, and in contrast to the Pc photosensitizer alone.63 The conjugate displayed an impressive light IC50 value of 13.8 nM with little dark cytotoxicity observed until concentrations of the conjugate exceeded 12.5 μM (PI > 906) whilst maintaining the antiproliferative abilities and targeting of tamoxifen when incubated with MCF-7 cancer cells in vitro.63 The light IC50 value was lower than the free tamoxifen drug (IC50 = 21 μM), displaying the advantages of a synergistic approach.63 The IC50 values of the conjugate could not be maintained when using the individual (non-conjugated) Pc and tamoxifen components, showing that the close proximity of the Pc and tamoxifen moieties promotes the synergistic effect.63
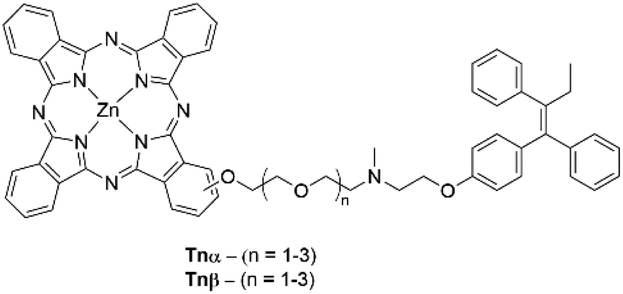 | ||
| Fig. 16 Pc–tamoxifen conjugates that target ER+ breast cancers. Linkers of varying length between the two units are either conjugated at the α or β position of the Pc, as indicated in the naming scheme. | ||
| Chain length, n | λ maxabs (nm) | Φ Δ | Φ F | Light IC50 (nM) | Dark IC50 (nM) | Phototoxicity index | Ref. | |
|---|---|---|---|---|---|---|---|---|
| T2α | 2 | 678 | 0.61 | 0.17 | 13.8 | 12500 |
906 | 63 |
| T1α | 1 | 678 | 0.59 | 0.17 | 80.5 | >1000 | >12.4 | 64 |
| T3α | 3 | 678 | 0.63 | 0.17 | 23.8 | >1000 | >42.0 | 64 |
| T1β | 1 | 672 | 0.51 | 0.21 | 16.1 | >1000 | >62.1 | 64 |
| T2β | 2 | 672 | 0.53 | 0.21 | 52.2 | >1000 | >19.2 | 64 |
| T3β | 3 | 672 | 0.56 | 0.22 | 89.5 | >1000 | 11.2 | 64 |
Xue et al. extended on the work performed by Liu et al. by creating a similar series of Pc–tamoxifen derivatives,64 focusing on the site of conjugation to tamoxifen on the Pc (α- or β-position), and the length of the oligoethylene glycol linker between the two moieties (Fig. 16).64 All derivatives showed the typical red absorption of the Pc moiety with Q-band maxima of 678 nm and 672 nm for the α- and β-derivatives respectively.64 The α-derivatives showed higher ΦΔ values at the expense of ΦF (Table 4), with ΦΔ values increasing with the length of the linker.64 All the conjugates displayed better targeting towards ER+ MCF-7 cells than estrogen receptor-negative (ER–) MDA-MB-231 cells, showing the conjugate could successfully target cells overexpressing ER.64 Confocal fluorescence microscopy experiments indicated preferential localization of the conjugates within lysosomes over mitochondria or nuclei cellular compartments.64 Testing the photodynamic activities and antiproliferative abilities of the conjugates gave the general trend that the α-conjugated series generally exhibited lower light IC50 values than their β-substituted counterparts (Table 4) with all conjugates showing no dark cytotoxicity up to 1 μM.64 The higher potency of the α-series is thought to be due to the better singlet-oxygen sensitization ability.64
Histone deacetylase inhibitors (HDACi) have become attractive drugs for preventing cancer progression and improving prognoses. HDAC enzymes play a key role in epigenetic modulation and are known to have varied effects due to the non-uniformity of their mechanism of action but typically they can induce apoptosis, prevent angiogenesis and facilitate immunomodulation.65 Aru et al. synthesised a SiPc–HDACi compound, with the HDACi axially ligated to the SiPc (NT-1) (Fig. 17). The compound had an absorption maximum of 672 nm with ΦΔ and ΦF values of 0.68 and 0.05, respectively.65 The SiPc–HDACi conjugate had light IC50 values of 42.0 μM (HUVECs), 9.2 μM (MCF-7) and 37.3 μM (MDA-MB-231), which was lower than the control Pc with no axially ligated HDACi 45.6 μM (HUVEC), 69.1 μM (MCF-7), and 89.3 μM (MDA-MB-231).65 Subcellular localization studies of the conjugate displayed retention within the nuclei, nucleoli and the nuclear membranes of the cell.65 Thus, the HDACi moiety brings the SiPc close to DNA strands to cause extensive localized photodamage, resulting in selective apoptosis of cancerous cells.
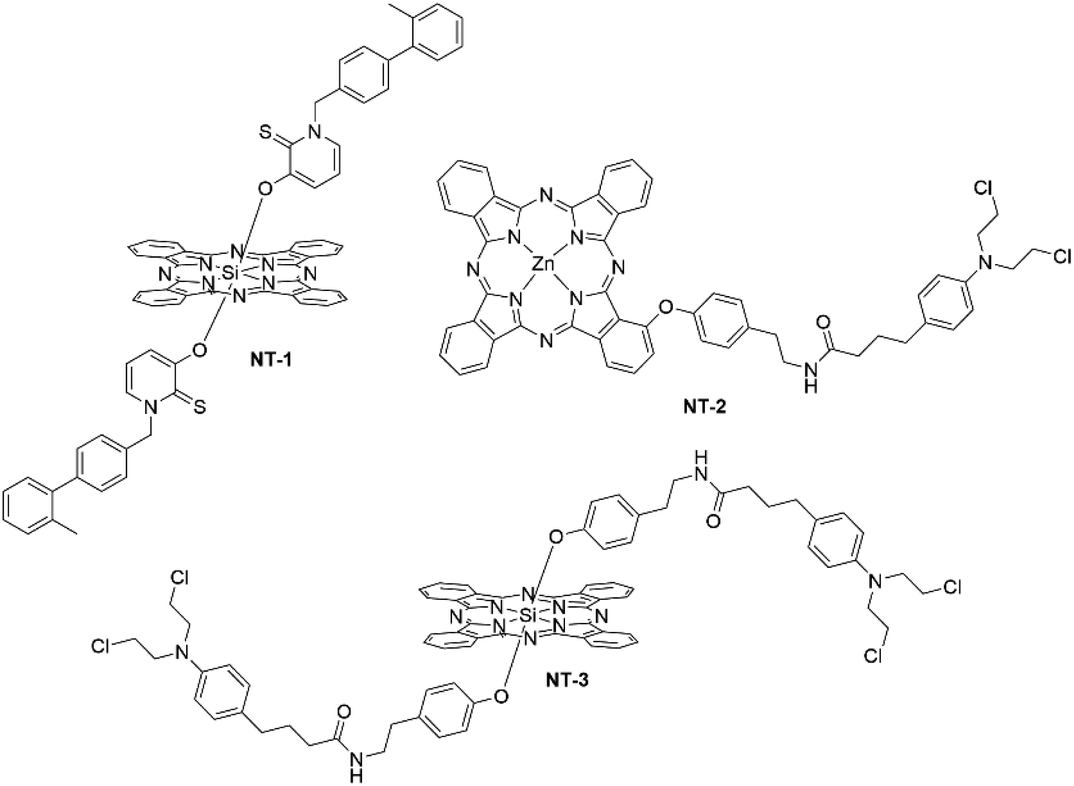 | ||
| Fig. 17 Miscellaneous nuclear-targeting Pc–drug conjugates. | ||
Chlorambucil, like the platins, is an alkylating agent that works by reaction with nucleophilic DNA sites to form inter- and intrastrand crosslinks that prevent DNA replication and cause overall loss of base-pairing fidelity. Huang et al. synthesized a Pc–chlorambucil conjugate and compared the effects of ligation of the chlorambucil conjugate either peripherally or axially to ZnPc or SiPc (NT-2 and NT-3, respectively) (Fig. 17).66 The molecules showed absorption maxima of 674 nm and 680 nm for the respective ZnPc and SiPc.66 Due to the spin–orbit coupling attributed to the Zn atom, the ZnPc conjugate had an impressive ΦΔ value of 0.63, whilst the SiPc had a reduced ΦΔ value of 0.05.66 The ΦF values of the derivatives also showed the ZnPc to have higher ΦΔ values of 0.22 in comparison to the SiPcs ΦΔ value of 0.05.66 Significant aggregation of NT-3 led to a high nonradiative rate and thus lower than expected fluorescence for a SiPc (ΦF = 0.04). IC50 values of the derivatives were obtained against HepG2 cells.66 The ZnPc-conjugate derivative had an impressive light IC50 value of 0.20 μM with a dark value of 85.6 μM, giving a PI value of 428.66 Whilst this is an adequate value it could not outcompete the sole Pc photosensitizer without the chlorambucil moiety (light IC50 value = 0.031 μM, dark IC50 = 33.8 μM, PI = 1090).66 This trend also extended onto the SiPc derivative with the light IC50 value of 17.47 μM and a dark value of 25.19 μM (PI = 1.44) as the Si–chlorambucil conjugate could also not outcompete the SiPc photosensitizer (light IC50 = 0.009 μM dark IC50 = 33.2 μM and PI = 3689).66 The authors theorized that due to the extensive aggregation profile of the Pc–chlorambucil conjugates, they were not efficiently uptaken by the HepG2 cells, unlike their sole Pc analogues.66 Subcellular localization studies showed preference for all the conjugates to localize within the mitochondria.66 This is a rare example of the synergism of the two components not being evidenced, and reinforces the difficulty of predicting such synergism.66
Vascular-disrupting agents (VDAs), such as vadimezan, fosbretabulin, and combretastatin A-4, are widely used chemotherapeutic agents.67 Acute administration of VDAs fully disrupts the vasculature of deep-seated tumours, preventing blood flow and inducing comprehensive necrosis of large malignant masses.67 The mechanism of action of VDAs relies on binding to tubulin to disrupt the cytoskeleton of proliferating endothelial cells through microtubule depolymerization and subsequent induction of the necrosis pathway.67
You and coworkers exploited the activity of the VDA combretastatin A-4 through ligation to a SiPc photosensitizer via an amino-acrylate linker (Fig. 18). The hypothesis was that the linker would be cleaved upon generation of singlet-oxygen to release the free drug. A second conjugate with a non-cleavable alkyl linker was also synthesized (Com-1).68 Both compounds showed low dark cytotoxicity that increased upon laser excitation, with initial dark IC50 values equal to 173 nM and 916 nM for the cleavable and non-cleavable linkers respectively, compared to the dark IC50 value of 9 nM for combretastatin A-4 alone.68 Upon illumination the IC50 values dropped to 6 nM (PI = 30) and 34 nM (PI = 27) for the cleavable and non-cleavable linker, respectively.68 It was also demonstrated that both the cleavable and non-cleavable conjugates had different mechanisms of action. The Pc–combretastatin A-4 conjugate, with the non-cleavable linker, did not effectively bind to tubulin, and thus, had a PDT effect only, whilst the analogue with a cleavable linker had a synergistic effect, as demonstrated by bystander effects in vitro.68 Both drugs were detected by fluorescence optical imaging techniques in the proximity of the tumours indicating the subtle targeting effect of the combretastatin moiety.68 It is also worth considering that conjugation to the phenolic-OH may be inhibiting binding of combretastatin to microtubules, and so an alternative conjugation site might improve activity and targeting.
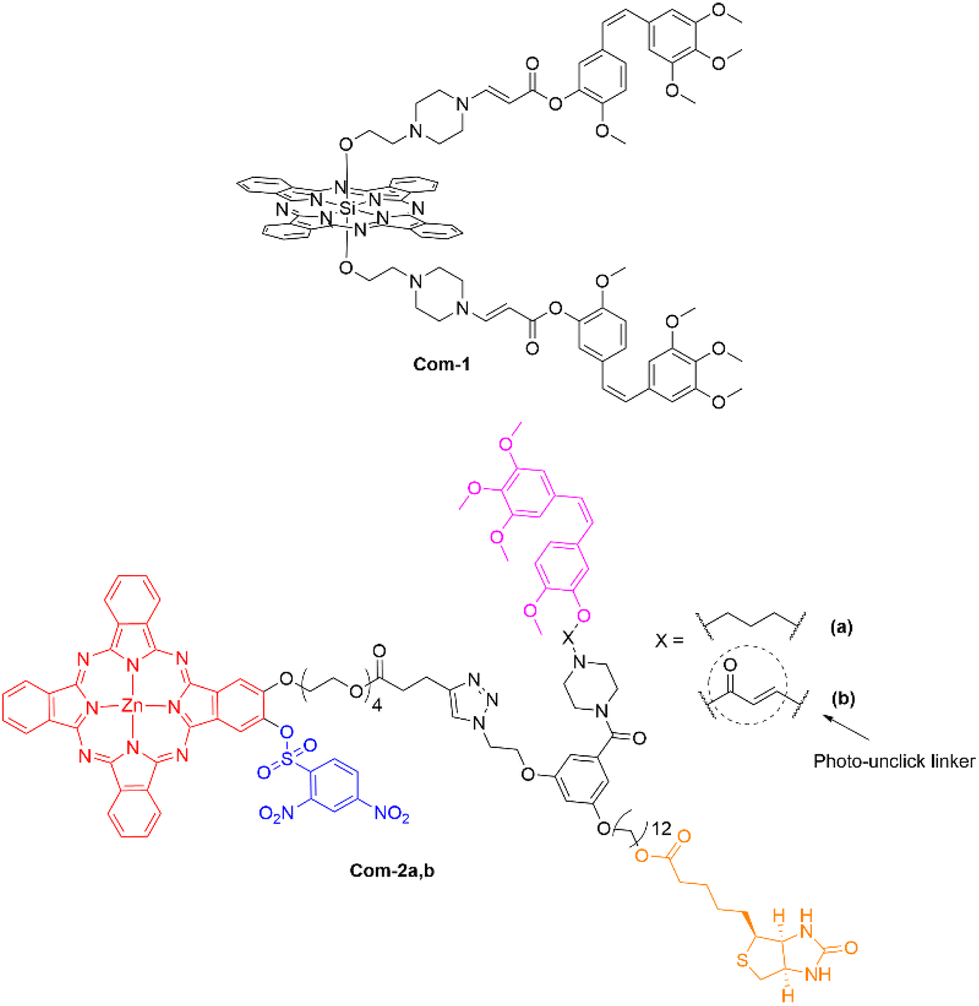 | ||
| Fig. 18 Pc–combretastatin A-4 conjugates that allow efficacious targeting of tubulin. | ||
Ng and coworkers synthesized a similar multifunctional ZnPc–combretastatin A-4 (Comb-2) conjugate with a singlet-oxygen cleavable amino acrylate linker (Fig. 18).69 However, this construct also contained an additional active-targeting biotin moiety to allow targeting of HepG2 cells that are biotin-receptor positive.69 Since the addition of an extra targeting ligand doesn't explicitly rely on the targeting of the combretastatin A-4 derivative, the design is considered outside of the scope of this review, but is nonetheless an effective approach.
Dumoulin et al. further experimented with VDAs by attaching a chalcone unit onto a ZnPc scaffold (Fig. 19).70 The authors employed an A3B design in which three isoindole units have a triethylene glycol monomethyl ether group to improve the hydrophilicity of the molecule with the fourth unit containing a chalcone connected via a tetraethylene glycol spacer.70 The chalcone still retained its antivascular capabilities after conjugation to the Pc chromophore, as supported by a HUVEC migration assay.70 The Pc also retained its impressive photophysical properties with a NIR absorption maximum of 704 nm and ΦΔ value of 0.55.70 The conjugate itself showed a dark IC50 value of 16.8 μM, with the chalcone unit enhancing the cytotoxicity in comparison to the Pc alone, which had an IC50 value >50 μM.70 Upon photoirradiation the conjugate showed an LD50 of 0.51 μM in comparison to the sole ZnPc that had an LD50 value of 3.32 μM.70 Dumoulin et al. also synthesized a tetrachalcone Pc linked by tetraethylene glycol spacers but no biological data was recorded for this conjugate.70,71 The antivascular activity of the chalcone was lower than expected, which was attributed to low bioavailability. To extend on their design and employ a possible “prodrug” approach, three Pc–chalcone conjugates with varying linkers were synthesized.72 One linker was a standard ether functionality that would resist metabolic cleavage as a control to prove their hypothesis that the chalcone had to be released to exert its vascular disruption capabilities.72 They also employed the Huisgen cycloaddition with an alkyne attached to the phthalonitrile and the azide attached to the chalcone and vice versa with the idea that the resulting triazoles formed form the reaction would be subject to different release conditions upon acidic or reductive environments.72 The authors did not record any photophysical or biological data with these compounds but did demonstrate that alternative Pc–chalcone designs were possible.
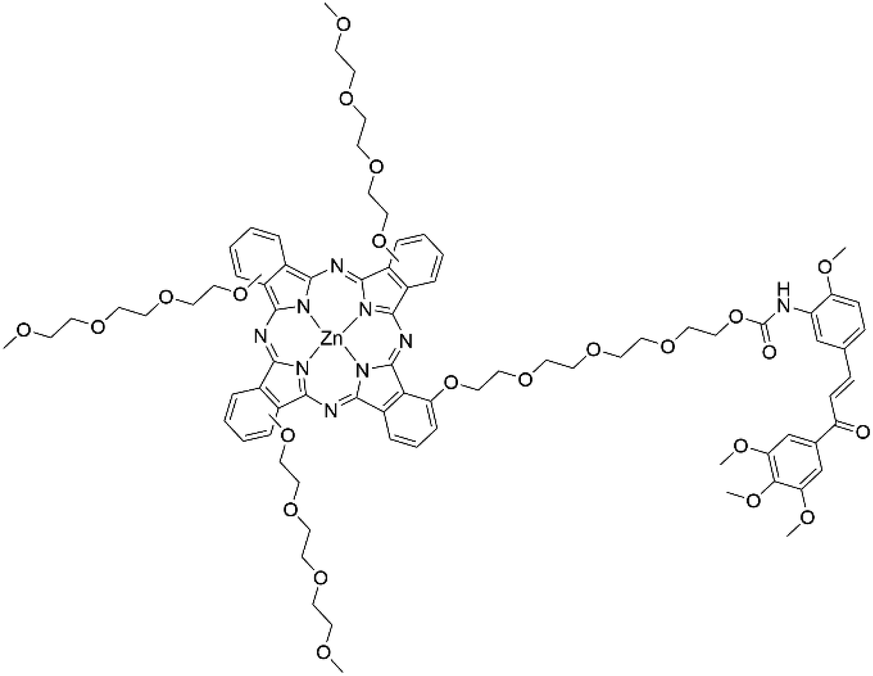 | ||
| Fig. 19 A Pc–chalcone conjugate used as a vascular disrupting agent. A statistical mixture of isomers is indicated, whereby the triethylene glycol methyl ether groups can be at either of the α positions of the isoindole units. | ||
Paclitaxel (PTX) is a broad-spectrum anticancer agent belonging to the taxane family that has found utility in the treatment of ovarian, breast, pancreatic, and non-small cell lung cancer but is also associated with significant off-target cytotoxicity.73,74 PTX operates through disruption of microtubule function by stabilizing tubulin-containing guanosine diphosphate and therefore promoting tubulin polymerization and suppressing microtubule depolymerization mechanisms, and in turn, preventing cell mitosis and cancer-cell proliferation.73,74
You et al. conjugated PTX at the axial position of a SiPc photosensitizer (PTX1/2), with both a prodrug and traditional linker, to form a potent synergistic agent (Fig. 20).73 The prodrug form was designed in a fashion that the PTX moiety would be released from the photosensitizer on the generation of singlet oxygen and its subsequent reaction with an amino acrylate linker.73 The PTX moiety was conjugated to the photosensitizer through its 2′-OH group that is critical for PTX binding to tubulin, thus, attenuating the activity of PTX in the conjugate form and establishing a prodrug modality.73 Both Pc–PTX conjugates displayed Q-band absorption maxima at 672 nm with emission wavelengths of 678 and 677 nm for the prodrug and non-cleavable conjugate, respectively.73 As expected, in the dark, both the prodrug and traditional conjugate had reduced tubulin polymerization effects with respect to free PTX due to conjugation to the 2′-OH group significantly affecting the binding.73 Under irradiation, however, the prodrug showed a significant increase in polymerization effects relative to the dark measurement due to the PTX moiety being cleaved upon generation of singlet oxygen, whilst tubulin polymerization effects induced by the non-cleavable conjugate did not change when irradiated.73 Dark and phototoxicity studies demonstrated the benefits of the design, with the prodrug displaying an IC50 value of 3.9 nM in the light and 910 nM in the dark (PI = 230), whilst the traditional conjugate had IC50 values of 24 nM upon irradiation and a dark IC50 value of 1279 nM (PI = 53).73 Both conjugates showed a significantly reduced dark IC50 value compared to free PTX (IC50 = 4.7 nM), meaning the conjugates showed a 190- and 270-fold reduction in activity in the dark with respect to free PTX. The prodrug Pc–PTX conjugate had a lower light IC50 value than PTX alone.73 You and coworkers further elaborated on their “photo-unclick” prodrug design by replacing one of the PTX units with a folate targeting moiety linked with an extended PEG chain to improve the hydrophilicity of the molecule that could also target folate positive receptor cells (Fig. 18).74 All conjugates showed similar photophysical characteristics with Q-band absorption maxima at 647 nm for all derivatives irrespective of chain length (PTX4a–d with n = 1000, 2000, 3500, and 5000).74 Conjugates with n = 1000, 2000 and 3500 showed the best potency with light IC50 values of 124 nM, 137 nM and 132 nM, respectively, with conjugates PEGn = 0 or 5k being the worst performers with respective values of 415 nM and 406 nM.74 This could be rationalized by the 1k–3.5k PEG chains having the best cellular uptake in FR-positive SKOV-3 cells to illicit their cancer-killing ability more effectively than with no PEG chain or PEGn = 5k, this argument was also supported when in vivo optical imaging showed that the chain length of 2k had better tumour localization than no PEG chain or a PEGn = 5k.74
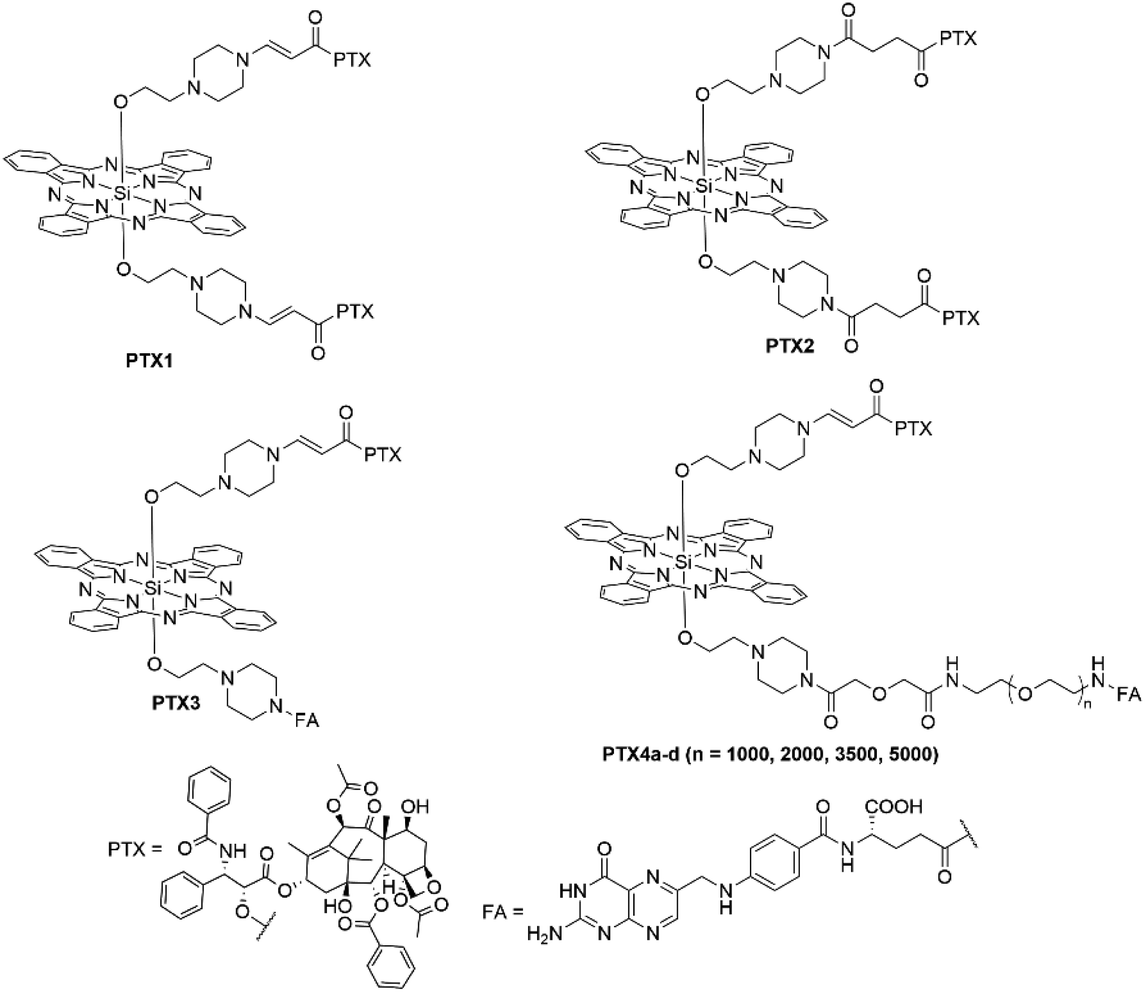 | ||
| Fig. 20 Pc–paclitaxel conjugates exhibiting both traditional and “photo-unclick” linkers. PTX = paclitaxel; FA = folic acid. | ||
Coumarins are attractive pharmacophores for the development of new medicine due to their diverse antimicrobial, anti-inflammatory and anticancer activities.75 Xue et al. created four novel Pc-coumarin conjugates and tested their efficacy against HepG2 cells.76 The authors employed the use of 7-hydroxycoumarin and 7-hydroxy-4-trifluoromethylcoumarin (Fig. 21) as the cytotoxic agents, and conjugated one derivative to either the α- or β-position of a ZnPc with an oligioethylene linker. Both α-derivatives had an absorption maximum of 677 nm while the β-analogues had a Q-band at 672 nm.76 The general trend was that the α-derivatives had a higher ΦΔ values at the expense of ΦF (Table 5).76 On evaluation of the light IC50 values it showed that the 7-hydroxycoumarin analogue had a greater activity against HepG2 cells when conjugated to the β-position and vice versa for the 7-hydroxy-4-trifluoromethylcoumarin all of which had impressive PI values (Table 5).76 Confocal laser-scanning microscopy indicated the subcellular localization of these compounds.76 Interestingly, the different derivatives had different localization. With the 7-hydroxycoumarin conjugated in the α-position preferring the cell nuclei.76 The other three conjugates localize in mitochondria or the lysosome organelles with little preference for either.76
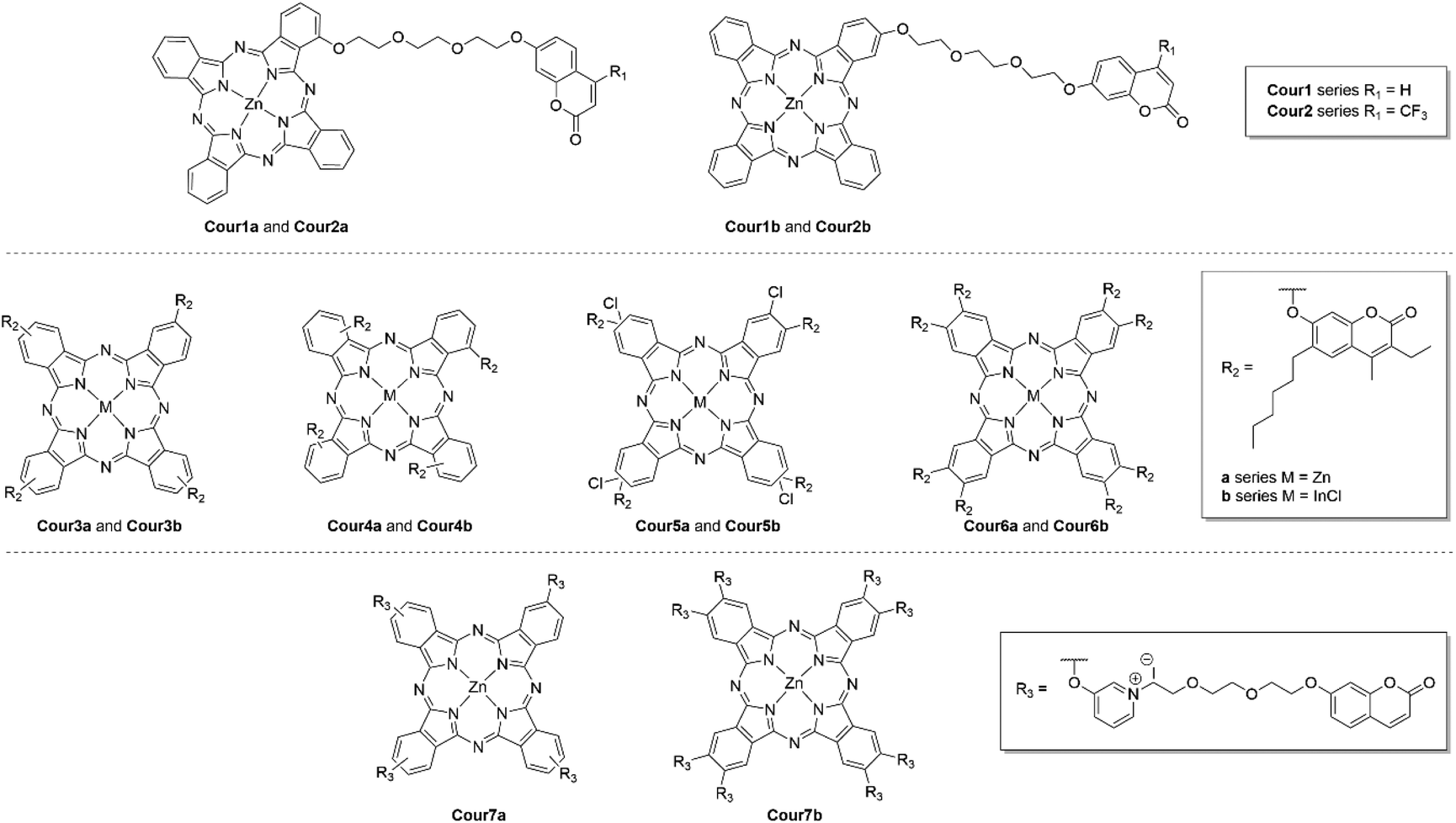 | ||
| Fig. 21 Pc–coumarin conjugates. A statistical distribution of conjugation sites to the two α-positions (Cour4 series) or two β-positions (Cour3 series and Cour7a) of each of the isoindole units of the Pcs is indicated where relevant. The Cour5 series also exists as mixtures of isomers but with a statistical distribution of the two different β-substituents on the isoindole rings of the Pc. | ||
| λ maxabs (nm) | Φ Δ | Φ F | Light IC50 (μM) | Dark IC50 (μM) | Phototoxicity index | Ref. | |
|---|---|---|---|---|---|---|---|
| Cour1a | 677 | 0.64 | 0.25 | 0.044 | 4.43 | 111 | 76 |
| Cour1b | 672 | 0.57 | 0.27 | 0.018 | >10 | 556 | 76 |
| Cour2a | 677 | 0.54 | 0.20 | 0.014 | >10 | 714 | 76 |
| Cour2b | 672 | 0.53 | 0.20 | 0.021 | >10 | 476 | 76 |
| Cour3a | 679 | 0.92 | 0.036 | — | — | — | 77 |
| Cour3b | 684 | 0.88 | 0.014 | — | — | — | 77 |
| Cour4a | 691 | 0.95 | 0.032 | — | — | — | 77 |
| Cour4b | 709 | 0.99 | 0.009 | — | — | — | 77 |
| Cour5a | 682 | 0.38 | 0.18 | — | — | — | 77 |
| Cour5b | 687 | 0.46 | 0.002 | — | — | — | 77 |
| Cour6a | 679 | 0.84 | 0.020 | — | — | — | 77 |
| Cour6b | 688 | 0.96 | 0.005 | — | — | — | 77 |
| Cour7a | 683 | 0.52 | 0.17 | — | — | — | 78 |
| Cour7b | 681 | 0.64 | 0.15 | — | — | — | 78 |
Bulut et al. also created a series of 7-hydroxy-3-ethyl-6-hexyl-4-methylcoumarin compounds conjugated to either a Zn or In(Cl)Pc, with variation in the site of conjugation with respect to the Pc and additional substitution (Fig. 21).77 The Q-band was bathochromically shifted for the In(Cl)Pcs and when the coumarin was conjugated to the α-position.77 All Pcs had high ΦΔ values (Table 5), with the InCl central moiety providing a greater spin–orbit coupling to promote the sensitization singlet oxygen due to the heavy-atom effect.77 The authors did not conduct any biological testing of the compounds but their impressive ΦΔ values suggests they could make excellent PDT agents, although some of the derivatives were not regiochemically pure with respect to Pc.77
Voskuhl et al. utilized a umbelliferone (a coumarin) conjugated to onto the β-position of a ZnPc via a quatenrized pyridyl group with an oligoethylene spacer to create a highly water soluble derivative with the capacity to be effective not only against HepG2 cancer cells but also Gram-negative and Gram-positive bacteria in an “all in one” type photosensitizer (Fig. 21).78 All conjugates retained their red-light absorption with the general trend of the β4-substituted derivatives having a slightly bathachromically shifted Q-band with respect to their β8-substituted derivatives (Table 5).78 Both derivatives had respectable ΦΔ values of 0.52 and 0.64 and ΦF values of 0.17 and 0.15, respectively, for the β4- and the β8-substituted derivatives.78 Confocal laser-scanning microscopy showed that all derivatives preferentially localized within the nucleus of HepG2 cells.78 Under irradiation the compounds caused 80 and 60% cell death for the β4- and β8-substituted derivatives, respectively.78
Huang et al. created the first synergistic Pc–anticancer drug conjugate that operates via a PTT mechanism.79 The authors synthesized a SiPc with an axially ligated perphenazine group.79 Perphenazine is a common antipsychotic drug used in the management of schizophrenia and other psychotic illnesses but has shown anticancer efficacy recently.79 In aqueous solution, the compound self-assembles into a nanoparticle with the self-assembly attributed to both π–π interactions of the Pc and hydrogen-bond interactions between phenolic hydroxyl group.79 The nanoparticle assembly had a Q-band maximum situated at approximately 700 nm.79 The SiPc derivative used a mixture of type-I PDT and a PTT response to elicit its cancer-killing effect, making the compound especially effective in commonly observed hypoxic tumour conditions.79 The compound efficiently produced highly cytotoxic superoxide, even in hypoxic environments, and displayed a negligible ΦΔ value.79 The compound showed an efficient temperature change of ∼30 °C after 685 nm irradiation for 10 minutes, which is high enough to induce apoptosis and necrosis of the tumour site.79 The compound was found to have a photothermal efficiency of 18.3%. The combined PDT and PTT effects led to light IC50 values of 0.08 μM and 0.09 μM in normoxic and hypoxic tumour conditions, respectively.79 The nanostructure showed excellent tumour accumulation, as proven by fluorescence and thermal imaging of the compound in H22 tumour-bearing mice, and the compound displayed a 64% tumour suppression after irradiation.79
6. Summary and outlook
Photodynamic and photothermal therapy show potential for localized light-activated cancer treatment with limited off-target effects compared to traditional chemotherapeutics. Phthalocyanines have especially favourable properties in this context, including red-to-NIR absorption, a tailorable structure, and efficient singlet-oxygen generation. Many Pc photosensitizers have now been developed for cancer phototherapy and have been shown to be effective, but arguably the next stage of their evolution is the incorporation of active-targeting moieties to enhance their specificity and potency. This is especially important given the unreliability of passive targeting through the EPR effect. The synthetic difficulty of conjugation to biologically inspired targeting vectors, such as antibodies and aptamers, as well as their instabilities, has led to the emergence of a new class of active-targeting groups based on known anticancer agents. Using these known anticancer drugs in conjunction with Pc conjugation leverages their proven binding of key oncological targets, whilst facilitating additional, and sometimes synergistic, anticancer effects of their own.This review explores the development of phthalocyanine–anticancer drug conjugates for phototherapy with a particular focus on conjugates with platins, kinase inhibitors, and anthracyclines as promising classes. Careful choice of linker and site of conjugation on both the Pc and the anticancer drug are shown to be critical to achieve effective targeting, as conjugation can unintentionally inhibit the activity or binding of the anticancer agent: this is often a result of the most easily conjugatable site also being critical to activity. Prodrug approaches have been used successfully in constructing efficacious Pc–anticancer drug conjugates with high phototoxicity indices; for example, the toxicity of a Pc–PTX conjugate was limited until photosensitization of singlet oxygen by the Pc lead to immolation of linker and release of free PTX, or where in a Pc–DOX conjugate both the phototherapeutic and chemotherapeutic effects are mutually reduced by quenching and blocking of the binding site, respectively, until reaction of the linker with the overexpressed enzyme FAP induces both effects. Although prodrug approaches are apt for the purpose, the best conjugate design appears to be the one that allows the Pc–drug conjugate to be bound and retained at the desired site of action, to elicit damage to essential cellular compartments, by either oxidative or thermal means. This was displayed by platins and kinases, with the latter being considered the “gold standard” due to their high PI values. Going forward, a photothermal effect also should be considered in such situations when an anticancer drug quenches the sensitization of singlet oxygen or the tumour site is hypoxic due to the oxygen independence of PTT.
In conclusion, effective targeting of a photosensitizer with anticancer drug conjugates within short range of a specific biological target, such as DNA or a specific receptor, improves the efficiency of phototherapy by concentrating and localizing the effect at the target site and can additionally lead to synergistic effects. Thus, the combination of phototherapy and chemotherapy is a compelling prospect. With cancer cases continually on the rise, the synergism of the two modalities may provide more specific and stronger cancer-killing effects than the individual constituents, ultimately improving patient prognosis.
Author contributions
CCR: conceptualization, writing – original draft, writing – review & editing. RME: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RME and CCR thank the University of Strathclyde for a Research Excellence Award PhD studentship for CCR.References
- W. Chen, Z. Sun and L. Lu, Angew. Chem., Int. Ed., 2021, 60, 5626–5643 CrossRef CAS PubMed.
- R. Baskar, K. A. Lee, R. Yeo, K.-W. Yeoh, R. Baskar and M. Phil, Int. J. Med. Sci., 2012, 9, 193–199 CrossRef PubMed.
- L. B. Josefsen and R. W. Boyle, Met.-Based Drugs, 2008, 2008, 1–24 CrossRef PubMed.
- X. Li, S. Lee and J. Yoon, Chem. Soc. Rev., 2018, 47, 1174–1188 RSC.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–115331 CrossRef CAS PubMed.
- Y. You, Org. Biomol. Chem., 2018, 16, 4044–4060 RSC.
- H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. S. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC.
- J. Nam, S. Son, L. Ochyl, R. Kuai, A. Schwendeman and J. J. Moon, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandry, D. K. P. Ng, T. Torres and F. Dumoulin, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- Q. Zhang, J. He, Y. Li, Z. Liu, B. Zhou and Y. Liu, RSC Med. Chem., 2020, 11, 427–437 RSC.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- S. Li, Q. Deng, Y. Zhang, X. Li, G. Wen, X. Cui, Y. Wan, Y. Huang, J. Chen, Z. Liu, L. Wang, C.-S. Lee, S. Li, X. Li, X. Cui, Y. Wan, J. Chen, C. Lee, Q. Deng, Y. Huang, Z. Liu, Y. Zhang, G. Wen and L. Wang, Adv. Mater., 2020, 32, 2001146–2001154 CrossRef CAS PubMed.
- B. Sun, R. Chang, S. Cao, C. Yuan, L. Zhao, H. Yang, J. Li, X. Yan and J. M. Van Hest, Angew. Chem., Int. Ed., 2020, 59, 20582–20588 CrossRef CAS PubMed.
- S. Liu, X. Pan and H. Liu, Angew. Chem., 2020, 132, 943–953 Search PubMed.
- K. Wang, Y. Xiang, W. Pan, H. Wang, N. Li and B. Tang, Chem. Sci., 2020, 11, 8055–8072 RSC.
- H. Huang, S. Long, D. Huang, J. Du, J. Fan, X. Peng and R. Li, Chem. Commun., 2021, 57, 9100–9103 RSC.
- T. Furuyama, K. Satoh, T. Kushiya and N. Kobayashi, J. Am. Chem. Soc., 2014, 136, 765–776 CrossRef CAS PubMed.
- K. Li, W. Dong, Q. Zhu, G. Lv, M. Xie, X. Sun, L. Qiu and J. Lin, J. Photochem. Photobiol., B, 2019, 190, 1–7 CrossRef CAS PubMed.
- X. Li, X. H. Peng, B. De Zheng, J. Tang, Y. Zhao, B. Y. Zheng, M. R. Ke and J. D. Huang, Chem. Sci., 2018, 9, 2098–2104 RSC.
- J. He, H. E. Larkin, Y.-S. Li, B. D. Rihter, S. I. A. Zaidi, M. A. J. Rodgers, H. Mukhtar, M. E. Kenney and N. L. Oleinick, Photochem. Photobiol., 1997, 65, 581–586 CrossRef CAS PubMed.
- J. Shao, J. Xue, Y. Dai, H. Liu, N. Chen, L. Jia and J. Huang, Eur. J. Cancer, 2012, 48, 2086–2096 CrossRef CAS PubMed.
- C. M. Allen, W. M. Sharman and J. E. Van Lier, J. Porphyrins Phthalocyanines, 2001, 5, 161–169 CrossRef CAS.
- F. Danhier, J. Controlled Release, 2016, 244, 108–121 CrossRef CAS PubMed.
- R. Li, K. Zheng, C. Yuan, Z. Chen and M. Huang, Nanotheranostics, 2017, 1, 346–357 CrossRef PubMed.
- Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License ( https://creativecommons.org/licenses/by/3.0/).
- M. R. Ke, S. L. Yeung, W. P. Fong, D. K. P. Ng and P. C. Lo, Chem. – Eur. J., 2012, 18, 4225–4233 CrossRef CAS PubMed.
- W. M. Darwish, N. A. Bayoumi, H. M. El-Shershaby and N. M. Allahloubi, J. Photochem. Photobiol., B, 2020, 203, 111777–111784 CrossRef CAS PubMed.
- S. Alpugan, D. Topkaya, D. Atilla, V. Ahsen, J. H. Niazi and F. Dumoulin, J. Porphyrins Phthalocyanines, 2017, 21, 887–892 CrossRef CAS.
- G. Obaid, I. Chambrier, M. J. Cook and D. A. Russell, Photochem. Photobiol. Sci., 2015, 14, 737–747 CrossRef CAS PubMed.
- M. Paradís-Bas, J. Tulla-Puche and F. Albericio, Chem. Soc. Rev., 2016, 45, 631–654 RSC.
- Z. Iqbal, J. Chen, Z. Chen and M. Huang, Curr. Drug Metab., 2015, 16, 816–832 CrossRef CAS PubMed.
- J. Sandland and R. W. Boyle, Bioconjugate Chem., 2019, 30, 975–993 CrossRef CAS PubMed.
- V. F. Otvagin, N. S. Kuzmina, E. S. Kudrishova, A. V. Nyuchev, A. E. Gavryushin and A. Y. Fedorov, J. Med. Chem., 2022, 65, 1695–1734 CrossRef CAS PubMed.
- C. Yu, Z. Wang, Z. Sun, L. Zhang, W. Zhang, Y. Xu and J.-J. Zhang, J. Med. Chem., 2020, 63, 13397–13412 CrossRef CAS PubMed.
- Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2523–2544 RSC.
- K. Liu, X. Liu, Q. Zeng, Y. Zhang, L. Tu, T. Liu, X. Kong, Y. Wang, F. Cao, S. A. G. Lambrechts, M. C. G. Aalders and H. Zhang, ACS Nano, 2012, 6, 4054–4062 CrossRef CAS PubMed.
- J. Mao, Y. Zhang, J. Zhu, C. Zhang and Z. Guo, Chem. Commun., 2009, 908–910 RSC.
- K. Mitra, M. Samsó, C. E. Lyons and M. C. T. Hartman, J. Mater. Chem. B, 2018, 6, 7373–7377 RSC.
- J. T. F. Lau, P.-C. Lo, W.-P. Fong and D. K. P. Ng, J. Med. Chem., 2012, 55, 5446–5454 CrossRef CAS PubMed.
- R. A. Bulgakov, N. A. Kuznetsova, O. V. Dolotova, L. I. Solovieva, J. Mack, W. J. U. Chidawanyika, O. L. Kaliya and T. Nyokong, J. Porphyrins Phthalocyanines, 2012, 16, 1217–1224 CrossRef CAS.
- N. Malinga, O. Dolotova, R. Bulgakov, E. Antunes and T. Nyokong, Dyes Pigm., 2012, 95, 572–579 CrossRef CAS.
- R. A. Bulgakov, N. A. Kuznetsova, O. V. Dolotova, E. N. Shevchenko, A. D. Plyutinskay, O. L. Kaliya and T. Nyokong, Macroheterocycles, 2012, 5, 350–357 CrossRef.
- O. V. Dolotova and O. L. Kaliya, Russ. J. Coord. Chem., 2007, 33, 111–115 CrossRef CAS.
- O. Dolotova and O. L. Kaliya, J. Porphyrins Phthalocyanines, 2011, 15, 632–638 CrossRef CAS.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- P. Cohen and D. R. Alessi, ACS Chem. Biol., 2013, 8, 96–104 CrossRef CAS PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- F.-L. Zhang, Q. Huang, K. Zheng, J. Li, J.-Y. Liu and J.-P. Xue, Chem. Commun., 2013, 49, 9570–9572 RSC.
- F.-L. Zhang, Q. Huang, K. Zhang, J. Li, J.-Y. Liu, M.-D. Huang and J.-P. Xue, ChemMedChem, 2014, 10, 312–320 CrossRef PubMed , 1504–1511.
- J. Chen, H. Ye, M. Zhang, J. Li, J. Liu and J. Xue, Chin. J. Chem., 2016, 34, 983–988 CrossRef CAS.
- J.-J. Chen, Y.-Z. Huang, M.-R. Song, Z.-H. Zhang and J.-P. Xue, ChemMedChem, 2017, 12, 1504–1511 CrossRef CAS PubMed.
- L. Huang, G. Wei, X. Sun, Y. Jian, Z. Huang, Y. Huang, Y. Shen, X. Xu, Y. Liao and C. Zhao, Eur. J. Med. Chem., 2018, 151, 294–303 CrossRef CAS PubMed.
- G. Wei, L. Huang, Y. Jiang, Y. Shen, Z. Huang, Y. Huang, X. Sun and C. Zhao, Eur. J. Med. Chem., 2019, 169, 53–64 CrossRef CAS PubMed.
- D. Toroz and I. R. Gould, Nat. Sci. Rep., 2019, 9, 2155–2167 CrossRef CAS PubMed.
- S.-K. Pang, RSC Adv., 2016, 6, 74426–74435 RSC.
- J. V. Mcgowan, R. Chung, A. Maulik, I. Piotrowska, J. M. Walker and D. M. Yellon, Cardiovasc. Drugs Ther., 2017, 31, 63–75 CrossRef CAS PubMed.
- C. Pérez, N. Busto and M. Leal, J. Phys. Chem. B, 2014, 118, 1288–1295 CrossRef PubMed.
- M.-R. Ke, S.-F. Chen, X.-H. Peng, Q.-F. Zheng, B.-Y. Zheng, C.-K. Yeh and J.-D. Huang, Eur. J. Med. Chem., 2017, 127, 200–209 CrossRef CAS PubMed.
- X.-H. Peng, S.-F. Chen, C.-H. Xu, B.-Y. Zheng, M.-R. Ke and J.-D. Huang, ChemistrySelect, 2018, 3, 5405–5411 CrossRef CAS.
- B. Rozeboom, N. Dey and P. De, Am. J. Cancer Res., 2019, 9, 2821–2831 CAS.
- F.-L. Zhang, M.-R. Song, G.-K. Yuan, H.-N. Ye, Y. Tian, M.-D. Huang, J.-P. Xue, Z.-H. Zhang and J.-Y. Liu, J. Med. Chem., 2017, 60, 6693–6703 CrossRef CAS PubMed.
- F. L. Zhang, N. Huang, H. L. Weng and J. P. Xue, J. Porphyrins Phthalocyanines, 2019, 23, 1073–1083 CrossRef CAS.
- B. Aru, A. Günay, E. Şenkuytu, G. Yanlkkaya Demirel, A. G. Gürek and D. Atilla, ACS Omega, 2020, 5, 25854–25867 CrossRef CAS PubMed.
- X. H. Peng, S. F. Chen, B. Y. Zheng, B. D. Zheng, Q. F. Zheng, X. S. Li, M. R. Ke and J. D. Huang, Tetrahedron, 2017, 73, 378–384 CrossRef CAS.
- D. W. Siemann, Cancer Treat. Rev., 2011, 37, 63–74 CrossRef CAS PubMed.
- M. Bio, P. Rajaputra, G. Nkepang and Y. You, J. Med. Chem., 2014, 57, 3401–3409 CrossRef CAS PubMed.
- S. Y. Y. Ha, Y. Zhou, W.-P. Fong and D. K. P. Ng, J. Med. Chem., 2020, 63, 8512–8523 CrossRef CAS PubMed.
- S. Tuncel, A. Trivella, D. Atilla, K. Bennis, H. Savoie, F. Albrieux, L. Delort, H. Billard, V. Dubois, V. Ahsen, F. Caldefie-Che, C. Richard, R. W. Boyle, S. Ducki and F. Dumoulin, Mol. Pharmaceutics, 2013, 10, 3706–3716 CrossRef CAS PubMed.
- S. Tuncel, J. Fournier-Dit-Chabert, F. Albrieux, V. Ahsen, S. Ducki and F. Dumoulin, Org. Biomol. Chem., 2012, 10, 1154–1157 RSC.
- F. Aribi, C. Vey, D. Topkaya, S. T. Kostakoglu, J. Fournier-Dit-Chabert, S. I. Büyükeksi, G. C. Taskln, S. Alpugan, F. Albrieux, A. G. Gürek, M. Cucca, K. Bennis, D. Atilla, V. Ahsen, S. Ducki and F. Dumoulin, J. Porphyrins Phthalocyanines, 2016, 20, 497–504 CrossRef CAS.
- P. Thapa, M. Li, M. Bio, P. Rajaputra, G. Nkepang, Y. Sun, S. Woo and Y. You, J. Med. Chem., 2016, 59, 3204–3214 CrossRef CAS PubMed.
- P. Thapa, M. Li, R. Karki, M. Bio, P. Rajaputra, G. Nkepang, S. Woo and Y. You, ACS Omega, 2017, 2, 6349–6360 CrossRef CAS PubMed.
- E. K. Akkol, Y. Genç, B. Karpuz, E. Sobarzo-Sánchez and R. Capasso, Cancers, 2020, 12, 1959–1984 CrossRef CAS PubMed.
- X. Q. Zhou, L. B. Meng, Q. Huang, J. Li, K. Zheng, F. L. Zhang, J. Y. Liu and J. P. Xue, ChemMedChem, 2015, 10, 304–311 CrossRef CAS PubMed.
- M. Özdemir, B. Karapinar, B. Yalçin, Ü. Salan, M. Durmuş and M. Bulut, Dalton Trans., 2019, 48, 13046–13056 RSC.
- A. Sowa, A. Höing, U. Dobrindt, S. K. Knauer, A. Galstyan and J. Voskuhl, Chem. – Eur. J., 2021, 27, 14672–14680 CrossRef CAS PubMed.
- Y. Y. Zhao, L. Zhang, Z. Chen, B. Y. Zheng, M. Ke, X. Li and J. D. Huang, J. Am. Chem. Soc., 2021, 143, 13980–13989 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |