
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-throughput screening, next generation sequencing and machine learning: advanced methods in enzyme engineering
Rosario
Vanella†
 ab,
Gordana
Kovacevic†
ab,
Vanni
Doffini
ab,
Jaime
Fernández de Santaella
ab and
Michael A.
Nash
*ab
ab,
Gordana
Kovacevic†
ab,
Vanni
Doffini
ab,
Jaime
Fernández de Santaella
ab and
Michael A.
Nash
*ab
aDepartment of Chemistry, University of Basel, 4058 Basel, Switzerland
bDepartment of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland. E-mail: michael.nash@unibas.ch
First published on 24th January 2022
Abstract
Enzyme engineering is an important biotechnological process capable of generating tailored biocatalysts for applications in industrial chemical conversion and biopharma. Typical enhancements sought in enzyme engineering and in vitro evolution campaigns include improved folding stability, catalytic activity, and/or substrate specificity. Despite significant progress in recent years in the areas of high-throughput screening and DNA sequencing, our ability to explore the vast space of functional enzyme sequences remains severely limited. Here, we review the currently available suite of modern methods for enzyme engineering, with a focus on novel readout systems based on enzyme cascades, and new approaches to reaction compartmentalization including single-cell hydrogel encapsulation techniques to achieve a genotype–phenotype link. We further summarize systematic scanning mutagenesis approaches and their merger with deep mutational scanning and massively parallel next-generation DNA sequencing technologies to generate mutability landscapes. Finally, we discuss the implementation of machine learning models for computational prediction of enzyme phenotypic fitness from sequence. This broad overview of current state-of-the-art approaches for enzyme engineering and evolution will aid newcomers and experienced researchers alike in identifying the important challenges that should be addressed to move the field forward.
1. Introduction
The pharmaceutical industry is rapidly moving from small molecule therapeutics towards biologics. Among the various classes of biologics under development, therapeutic enzymes are gaining attention as molecular entities that can catalyze specific chemical reactions inside the body to achieve a therapeutic effect. Therapeutic enzymes can be delivered systemically as full proteins or incorporated into gene therapy vectors to transduce target cells with specific functionality in vivo. Antibody-targeted enzyme prodrug therapy1 and gene-directed enzyme prodrug therapy2 both represent valuable therapeutic strategies with significant potential in the clinic. In all of these envisioned applications, understanding sequence-function relationships of therapeutic enzymes will play a crucial role.There is therefore an urgent need for improved methods for molecular analysis and enhancement of therapeutic enzymes. Naturally occurring enzyme sequences are typically not suitable as biopharmaceuticals due to a general lack of stability, developability, and/or activity. In this context, molecular enhancement by improvement of colloidal stability, catalytic turnover rate, substrate binding affinity, and/or sensitivity to environmental conditions are essential steps in enabling therapeutic enzymes to reach their full potential. The establishment of rapid design, build, test, and learn cycles and the analysis of large-scale sequence-function relationships will be crucial for the advancement of therapeutic enzymes towards clinical translation.
Laboratory directed evolution is by now a well-established paradigm for improving enzyme properties, having been recently awarded a Nobel Prize.3 This process mimics natural evolution by applying selection pressure on a library of genetic variants of a parent enzyme sequence, and propagating proteins with the desired function into subsequent generations, which are then further subjected to diversification and phenotypic screening/selection. Despite the general success of enzyme directed evolution, current technologies only scratch the surface of the vast space of protein sequences. New methods for efficiently exploring productive sequence space, and rapidly screening phenotypes are therefore as important as ever. Recent commoditization of massively parallel DNA sequencing technologies (i.e., next-generation sequencing) are further providing new capabilities for generating large datasets of sequence-function pairings. The purpose of this review article is to explore recent developments within several thematic areas of enzyme engineering with an emphasis on screening methods design and new workflows supported by next generation sequencing and machine learning.
2. Multi-enzyme cascades as readout systems
Many enzymes in nature perform reactions in which products are not easily measured by available instruments at high-throughput. However, a comprehensive toolbox of techniques to measure enzymatic activity is essential for success in a directed evolution campaign. Coupling the initial reaction to one or more auxiliary reactions through a cascade can address this challenge. The most common auxiliary reactions use other enzymes, which in turn produce a measurable change of absorbance or fluorescence. For coupled enzyme reactions systems, typically the auxiliary enzymes need to be in excess compared to the primary enzyme. In this way, the system can be setup such that the rate-limiting step is the reaction performed by the first enzyme. Under these conditions, the overall molecule flux through the pathway reports the activity of the initial enzyme.4 Measuring absorbance or fluorescence through coupled reactions allows continuous monitoring of enzyme activity, and enables simple identification of deviations in activity such as lag period or a falling-off in reaction rate.5,6 In addition, in coupled enzyme assays it is necessary to take into account the suitability of the environmental conditions (temperature, pH) for all involved enzymes in the cascade.As the need for the improved enzymes in various industries has increased over the years, methods for activity detection in enzyme engineering experiments have been fine-tuned. It has become important that enzyme assays have high sensitivity and are adapted to medium- and high-throughput screening campaigns. The numerous examples of coupled assays used in directed evolution and enzyme engineering outlined below have demonstrated the versatility of enzyme cascades in this context.
2.1 Enzyme cascades in library screening and directed evolution
Enzyme cascades can be applied to produce detectable changes in absorbance upon modification of cofactors7 and this process can be used as a platform for enzyme engineering. For example, this strategy was applied in a microwell plate assay to perform directed evolution of lipases and esterases, where the product of these hydrolases (acetic acid) acts as a substrate for an enzyme cascade composed of four different enzymes. As a result of this cascade, an increase in the 340 nm extinction was detectable due to the accumulation of the cofactor NADH.8 Other molecules, such as Ellman's Reagent, have also been used for colorimetric output in a four-step enzyme cascade to assess the activity of S-adenosylmethionine-dependent methyltransferases.9 In another example, Ortiz-Tena and colleagues developed a system where two reactions were performed by five different enzymes, all coordinated into an enzyme-coupled reporter system for the activity of sulfatases.10 In this case, the activity of the sulfatase shifts the equilibrium of the first reaction, generating GDP as a side-product. As a result, pyruvate phosphate dikinase, pyruvate oxidase and horseradish peroxidase (HRP) in a subsequent reaction were coordinated to produce Bindschedler's green dye. The sequential use of an oxidase followed by a peroxidase to create either a dye or a fluorophore is well established and has been combined in multiple ways, proving that robust and sensitive enzyme cascades are a transferable tool among enzyme engineering campaigns.More recently, Begander and colleagues11 developed a similar two-step reaction scheme to assess the enzymatic activity of a D-glycerate dehydratase. In this work, the second reaction uses the same set of enzymes to convert pyruvate into Bindschedler's green dye. It is noteworthy that the key elements of a previously built pathway were successfully transferred to a screening system for a new target enzyme. This demonstrates the capacity of enzyme cascades to widen the applicability of pre-existing screening systems.
Other directed evolution experiments have not only used multi-enzyme cascades to produce a readable output, but also evolved multiple enzymes within a cascade simultaneously. These directed co-evolution experiments targeted two cellulases (an endoglucanase and a β-glucosidase) expressed from a single operon in E. coli. The operon was targeted by error-prone PCR to generate mutagenic libraries. Screening took place as a result of co-expression, where the conversion of the insoluble substrate to oligosaccharides was catalysed by the endoglucanase. Subsequent activity of β-glucosidase produced glucose, which was in turn used as a substrate to the glucose oxidase/HRP cascade that produced a colorimetric dye.12,13 Most interestingly, this experimental setup enabled screening for synergistic effects between individual components in the cascade. The enzymes were evolved individually and simultaneously, with the latter approach proving more effective.
The development of microfluidic high-throughput screening (HTS) methods to detect changes in absorbance is also of special interest for enzyme evolution. The technique allowed in vitro evolution of an L-phenylalanine dehydrogenase by coupling its activity to a reaction that forms a formazan dye through the oxidation of NADH. This work opens a window of opportunity to evolve a wide range of enzymes with HTS methods, which were previously unavailable.14 This system does not use enzyme cascades directly coupled to the reaction of interest, but instead relies on a tetrazolium dye for coupling, an approach that allowed for a 25-fold improvement in detection when compared to direct NADH detection. This shows the power of signal amplification and the applicability of coupled assays within the setting of directed evolution in HTS methods.14
Fluorescence-based detection is generally more sensitive than absorbance-based detection, and much of the recent research has focused on creating fluorescent outputs from enzyme-coupled reporter systems. For example, directed evolution of geraniol synthetase was enabled by the implementation of an enzyme-coupled assay in vivo. Activity of this enzyme resulted in the accumulation of the reduced cofactor NADH, which served as a co-substrate for a secondary reaction catalyzed by diaphorase, resulting in production of the red fluorescent compound resorufin.15 Such a strategy was used for developing a HTS cellulase assay in which expressed variants of cellulases were isolated in droplets together with their encoding genes, the reaction substrate (i.e. carboxymethyl cellulose), and the readout enzymes hexose oxidase and vanadium bromoperoxidase. The former enzyme is a promiscuous oligosaccharide oxidiser which produces H2O2 whilst the latter is the output enzyme producing a positively charged fluorophore in proportion to H2O2 abundance.16,17 Enzyme coupling has also aided the efficient engineering of highly stereoselective cyclohexylamine oxidases using droplet-based HTS methods, where horseradish peroxidase couples the activity of the oxidase with the fluorogenic dye Amplex UltraRed.18
Another approach to enzyme HTS focused on selectively labelling cell surfaces with a fluorophore in order to screen active variants within a library. This strategy relies on cell-surface display of active enzymes and a subsequent enzyme-coupled assay that triggers labelling of the cell surfaces. Some of the earliest examples of applying this HTS method to directed evolution generated an enantioselective esterase by displaying esterases and peroxidases on the cell surface of E. coli. To do so, the different enantiomers were fluorescently labelled and when the esterase was active, the fluorophore from the substrate was released. This enabled the peroxidase to covalently bind it to cell-surface proteins. Finally, the positive variants were sorted using fluorescence activated cell sorting (FACS).19 Further research adapted this technology to the yeast S. cerevisiae and combined it with microfluidics to evolve glucose oxidase (GOx). Cells expressing a library of randomised variants of the enzyme were emulsified in single water-in-oil microdroplets together with the substrate (glucose), a reporter enzyme (HRP) and a fluorescent substrate for the reporter enzyme (fluorescein tyramide), which was covalently linked to the cell surface of yeast when hydrogen peroxide was produced by GOx. After incubation with the enzyme cascade, the oil phase was removed and the labelled cells were analysed using FACS.20 The use of microfluidics inhibited crosstalk and allowed the use of a longer enzyme cascade without requiring the display of both components. However, since the fluorophore had to be covalently linked to the cell-surfaces, lower signal amplification was observed.
Implementing enzyme cascades in evolutionary workflows not only allows detection of a large variety of products but can also amplify signals and provide easily detectable products with a high signal to noise ratio.21 Moreover, the introduction of cascades avoids the accumulation of products in the reaction vessel and can mitigate issues such as product inhibition and product toxicity.22 Furthermore, enzyme cascade readout systems can avoid the requirement of using chemically modified substrate analogues, avoiding bulky fluorescent groups and allowing enzyme variants to be screened on natural substrates.23 Finally, multi-step enzyme cascades offer opportunities to increase screening throughput by in some cases providing an optically readable output that can be evaluated at higher speed and throughput than conventional chemical analysis such as mass spectrometry or liquid chromatography. Enzyme cascades when combined with novel reaction compartmentalization strategies have the potential to enable more efficient evolution workflows. Based on these advantages, the establishment of novel enzyme cascades can broaden the scope of possible enzyme targets that can be screened and studied by directed evolution.
3. Compartmentalization methods in high-throughput screening
An essential feature for directed evolution is maintaining a phenotype–genotype link through the screening process. For pooled screening of binding proteins (e.g., antibodies), labelled target biomarkers can be used to tag cells displaying variants of the binder, however, for enzyme screening the persistent molecular diffusion of substrate and product molecules away from the biocatalyst creates a physical/chemical challenge that must be overcome to achieve fidelity of the genotype–phenotype link. In order to preserve this link, various reaction compartmentalization strategies have been developed throughout the years. One of the most widely utilized compartmentalization methods is simple microtiter plate (MTP) screening, which depending on infrastructure may allow analysis of 104 variants per day. Recently, a fully automated robotic platform was described for MTP library screening of four different enzymes, which increased throughput 2 to 3 fold compared to manual handling of clones.24 However, with the recent rise of ultra-HTS methods, medium-throughput MTP technologies are becoming outdated. Bacterial and eukaryotic cells with their natural membranes can also serve as natural compartments, and these approaches were first exploited for HTS and used in many enzyme directed evolution campaigns, some of which will be explained in detail in the following sections. However, beyond MTP and membrane-separated cells as compartments, artificial reaction compartments in the form of single and double emulsions have emerged for entrapment of cells25 or in vitro translation/transcription (IVTT) machinery to produce the enzyme of interest and physically colocalize genotype and phenotype26 (Fig. 1a and b).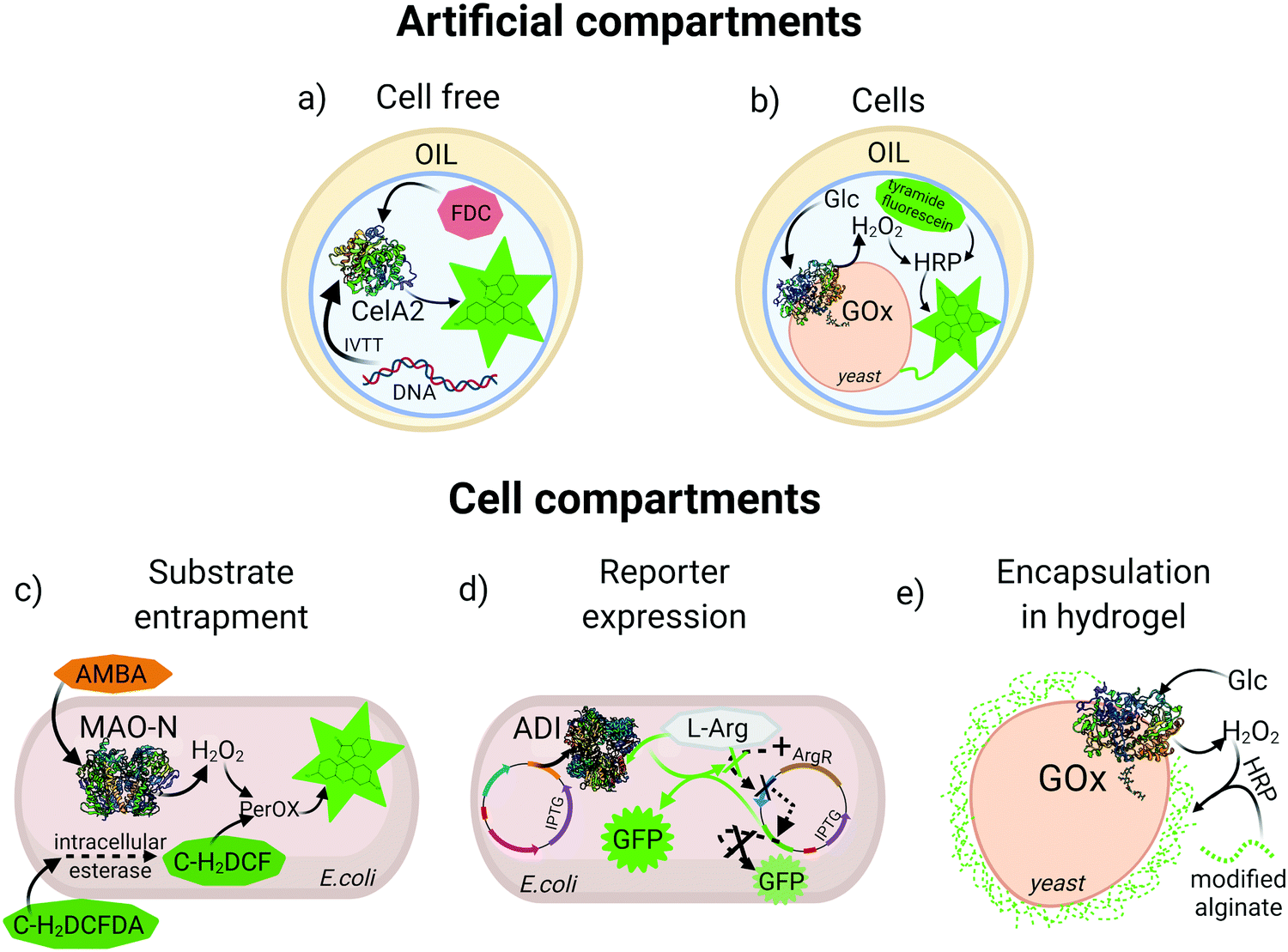 | ||
| Fig. 1 Examples of compartmentalization methods for high-throughput screening. (a) Aqueous droplet entrapping a gene and an in vitro translation/transcription (IVTT) mixture for expression of cellulase A2 (CelA2) which converts fluorescein-di-β-D-cellobioside (FDC) to fluorescein. (b) Aqueous droplet entrapping a yeast cell expressing glucose oxidase (GOx) on the surface which, produces H2O2 for subsequent reaction with horseradish peroxidase (HRP) and covalent labelling of the cell with tyramine-fluorescein. (c) Intracellular expression of monoamine oxidase (MAO-N) oxidizes (S)-(−)-alpha-methylbenzylamine (AMBA) producing H2O2. Carboxy-2,7-dichloro-dihydrofluorescein diacetate (C-H2DCFDA) is cleaved by intracellular esterase, generating carboxy-2,7-dichloro-dihydrofluorescein (C-H2DCF) which is oxidized to fluorescein by an intracellular peroxidase in the presence of H2O2. (d) A GFP reporter is down-regulated by expression of a repressor (ArgR) in the presence of L-arginine (L-Arg), or upregulated with induced expression of arginine deiminase (ADI) that depletes L-Arg. (e) GOx expressed on the yeast surface triggers encapsulation of the cell in a fluorescent alginate hydrogel in presence of HRP and glucose. | ||
Beside these well-established compartments used in HTS, new methods are regularly being developed to increase throughput and sensitivity of the screening process. Some of the more recent examples include screening in microcapillary arrays,27 microbeads,28 or liposomes.29 Microcapillary array screening offers the advantage of cell spatial separation comparable to MTP platforms but with significantly higher throughput. This method uses a sorting method based on a pulsed ultraviolet laser, and extracts cells from a microcapillary with high selectivity and viability. The versatility of this screening method has been shown in the engineering of binders, fluorescent proteins and enzymes. The authors point out several distinctions between high-throughput FACS-based screening and microarray capillary screening. Some of them include the possibility of distinguishing enzyme variants based on reaction kinetics instead of a single point fluorescent intensity, as well as the possibility of direct cell imaging and decoupling cell analysis and sorting.27
3.1 Cells as natural compartments
The use of microbial cells, typically bacteria or yeast, as natural compartments for enzyme directed evolution campaigns has been explored going back many years. The main advantages that cell compartments bring are a direct genotype–phenotype link, ease of manipulation, and ease of recovery of selected clones. Despite these advantages, there are a number of limitations associated with the use of cells as sole natural compartments for high-throughput screening. One limitation is that typically the substrate must readily pass through the cell membrane. Modified substrates can be used for such purposes, but as the saying goes “you get what you screen for”,30 and using a substrate that is structurally as close as possible to the final desired or natural substrate is critical. In these campaigns, conversion of the membrane-permeable substrate into product by a single enzymatic turnover or multi-enzyme reaction cascade should give a readable signal, preferably by modulating cell fitness (e.g., live/die-based selection) or generating a fluorescent signal. Furthermore, the product should not diffuse out of the cell.Recently a FACS-based HTS system using E. coli cells as natural compartments was reported for monoamine oxidase, where all the above-mentioned considerations were made.31 Sadler et al. used an acetylated fluorescein derivative as an indirect reporter probe that could diffuse into the cell, where the intracellular esterases cleaved acetyl groups leaving the probe susceptible to oxidation in a presence of H2O2 and endogenous peroxidases. This approach generated a fluorescent compound (Fig. 1c) that served as the signal for single-cell sorting. This screening method was shown to be versatile, and many different substrates could be screened using the same assay. A similar approach was used for directed evolution of P450 BM3 monooxygenase, in which 7-benzoxy-3-carboxycoumarin ethyl ester underwent intracellular de-esterification and subsequent dealkylation by P450, forming a fluorescent coumarin derivative.32
In developing approaches that leverage appropriate substrate/product pairs, linking fluorescent protein expression to enzyme activity can simplify and increase the throughput of screening assays. One such example was demonstrated in the work of Chen et al., in which the authors developed a screening platform based on ligand-mediated eGFP expression.33 They constructed a 2-vector E. coli expression system, where one vector carried the enzyme of interest (arginine deiminase) and the other vector carried the genes for eGFP (expressed under argG promoter) and argR which acts as a repressor of the argG promoter in presence of arginine. This system relied on the competitive conversion/binding of arginine between arginine deiminase and the arginine repressor. When inactive or low activity enzyme variants were expressed, arginine bound to argR and together they repressed eGFP biosynthesis, while expression of high-activity enzyme variants depleted arginine resulting in an increase in eGFP biosynthesis (Fig. 1d).
One more example of fluorescent protein expression linked to enzyme activity was reported by Sanchez and Ting34 for directed evolution of TEV protease toward increased kcat. They expressed TEV protease in the yeast cell and used a TEV cleavage sequence linked to the transcription factor, which was subsequently released and translocated to the nucleus to start transcription of a reporter protein, citrine. This screening method for TEV protease differed from a previous method reported by Yi et al.,35 who screened a TEV-P library for substrate specificity using yeast surface display. Yi applied endoplasmic reticulum (ER) sequestration, which allowed for simultaneous expression and localization of both enzyme and substrate library in the ER, followed by substrate surface display upon enzyme cleavage.
Another screening strategy relying on yeast surface display was developed for the directed evolution of bond-forming enzymes36,37 such as microbial transglutaminase, an enzyme with potential for antibody–drug conjugate synthesis. Deweid et al.37 displayed microbial transglutaminase on the yeast surface and used intrinsic lysine residues to form an isopeptide bond with a biotinylated oligopeptide. This scheme led to enzyme auto-labeling which enabled the screening of mutant libraries using increased selective pressure by reducing substrate availability.
Fluorescent proteins are not necessarily only used as reporters for enzyme activity, but their applicability as carriers for non-canonical amino acids in library screening of p-cyano-L-phenylalanyl aminoacyl-tRNA synthetase (pCNFRS) has been shown in the work of Kwok et al.38 The authors used a strain-promoted azide–alkyne click (SPAAC) reaction to distinguish superfolder GFP with incorporated p-azido-L-phenylalanine (pAzF), and based on the reaction selectivity they successfully evolved pCNFRS to preferentially incorporate pAzF from the mixture of pAzF and p-cyano-L-phenylalanine (pCNF).
3.2 Cell-free artificial compartments
Artificial reaction compartments in the form of single (water-in-oil) or double (water-in-oil-in-water) emulsions have emerged as an alternative to cellular compartmentalization for the evolution of enzymes. This approach is advantageous for enzymes or substrates that are toxic to the cell.39 These aqueous compartments are able to colocalize genotype and phenotype, and provide the possibility to segregate DNA for translation in vitro to synthesize the enzyme of interest, thus eliminating the need for cell translational machinery.40 One of the first FACS based high-throughput screenings of enzyme libraries in double emulsions was conducted by Mastrobattista et al.26 They evolved the protein Ebg into an enzyme with significant β-galactosidase activity using a fluorogenic fluorescein-based substrate, which after enzyme conversion was entrapped in the aqueous compartment. The separation of droplets by an oil phase is meant to prevent signal cross-talk. However, cross diffusion between droplets for emulsion-based systems is one limitation when choosing a substrate. Differently from cell compartmentalization, here neither the substrate nor product should diffuse across the oil phase. A similar fluorogenic substrate was used for in vitro high-throughput screening of a randomized cellulase library, which yielded a cellulase variant with a 13.3-fold increase in catalytic activity (Fig. 1a).41 In this work, the authors analyzed the influence of different emulsification techniques like stirring, homogenization and membrane extrusion for homogeneity of droplets in size and shape.The challenge of emulsion polydispersity has been addressed using well-controlled microfluidic-based emulsion production methods. Microfluidic systems allow for highly-controlled water-in-oil droplet emulsification, and allows reagent addition by droplet fusion or micro-injection followed by droplet sorting.42 Fallah-Araghi et al. used a microfluidic system to compartmentalize single genes of β-galactosidase and amplify them by PCR before fusing the droplets with an IVTT mix and a fluorogenic substrate. Although microfluidic sorting rates can in some cases be 10-fold lower than typical FACS sorting rates, microfluidics offer a high level of control over the reaction volumes and conditions. Combining microfluidic-based water-in-oil and later water-in-oil-in-water emulsions with FACS sorting can significantly improve the speed of sorting as well as the enrichment factor.43
Microfluidic droplet-based screening relies typically on fluorogenic substrates, or alternatively on fluorescent reporter protein expression, an approach called affinity-fed translation (AFD).44 By using an enzyme that produces an amino acid, it was possible to control the expression of a reporter protein in an aqueous droplet with IVTT. The sensitivity of screening was improved by expressing the enzyme of interest and reporter protein simultaneously. Very recently a novel detection method was also introduced that coupled microfluidic screening and sorting with mass spectrometry,45 which is very powerful as it is chemically generalizable.
3.3 Cells entrapped in artificial compartments
Parallel to the development of cell free artificial compartments for the directed evolution of enzymes, progress in the compartmentalization of whole cells in emulsions has also been made.25 Entrapping whole cells in artificial compartments can produce higher numbers of enzyme molecules per droplet (∼104 compared to ∼102 that are typically obtained using IVTT). With a higher number of molecules, sensitivity of the assay and selection are significantly enhanced, despite the low signal-to-background ratio. Using such an approach, Aharoni et al. evolved serum paraoxonase with negligible thiolactonase activity to a variant with approximately 100-fold increased catalytic activity compared to the wild type enzyme.Analogous to substrate diffusion into the cell, researchers have investigated substrate delivery through the oil phase into aqueous droplets to precisely control the start of the enzymatic reaction and minimize background fluorescence for highly active enzymes.46,47 One of these works developed fluorescence droplet entrapment (FDE) substrates for three classes of enzymes (phosphotriesterases, esterase and glucosidases).47 The authors investigated the hydrophobicity of fluorogenic substrates in terms of permeability through water-in-oil-in-water emulsions, cell membranes, and diffusion outside the inner aqueous droplets using log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D values as an evaluation parameter. In their work, Ostafe et al.46 used a substrate delivery system for glucose oxidase expressed on the yeast surface. Yeast cells were entrapped in water droplets and β-octylglucoside was added to the primary emulsion, where it underwent enzymatic cleavage by externally added β-glucosidase generating glucose. After the glucose became available for glucose-oxidase, cells harboring active variants were covalently labeled with fluorescein-tyramine and extracted from the emulsion droplets. Extraction of the covalently labeled cells simplified the FACS analysis compared to sorting water-in-oil-in-water double emulsions. Further improvements in screening enzyme variants with higher catalytic efficiency instead of the overall activity could be obtained by normalizing fluorescent signals to the expression levels of the enzyme. Normalization was done with either antibody labelling of the enzymes expressed on the cell surface,46 or using co-expression with a reporter gene.48,49
D values as an evaluation parameter. In their work, Ostafe et al.46 used a substrate delivery system for glucose oxidase expressed on the yeast surface. Yeast cells were entrapped in water droplets and β-octylglucoside was added to the primary emulsion, where it underwent enzymatic cleavage by externally added β-glucosidase generating glucose. After the glucose became available for glucose-oxidase, cells harboring active variants were covalently labeled with fluorescein-tyramine and extracted from the emulsion droplets. Extraction of the covalently labeled cells simplified the FACS analysis compared to sorting water-in-oil-in-water double emulsions. Further improvements in screening enzyme variants with higher catalytic efficiency instead of the overall activity could be obtained by normalizing fluorescent signals to the expression levels of the enzyme. Normalization was done with either antibody labelling of the enzymes expressed on the cell surface,46 or using co-expression with a reporter gene.48,49
Microfluidic approaches are also compatible with high-throughput screening and enzyme evolution using whole cells in droplets and emulsions.18,50,51 Cell recovery and post-screening analysis is more straightforward than for IVTT systems, with all the benefits that microfluidic emulsification brings. One of the first successful fluorescent activated droplet sorting (FADS) experiments with whole cells was conducted on a model library of β-glucosidase expressed in E. coli,52 which was later used for sorting of the horseradish peroxidase library expressed on the yeast surface, enabling discovery of an enzyme variant with nearly diffusion-limited catalytic efficiency.53 Besides fluorescent detection, absorbance activated droplet sorting (AADS) was developed and validated for whole cells in microfluidic droplets by sorting a phenylalanine dehydrogenase library.21 Absorbance as a detection method can significantly extend the scope of enzymatic assays that can be used in directed evolution, despite the lower sensitivity (compared to fluorescence) afforded by short microscale path lengths.
3.4 Cell encapsulation in hydrogels
Emerging technology for high-throughput enzyme screening based on whole cells relies on encapsulation in fluorescent hydrogels, as reported by our group and others.54,55 High-throughput screening in hydrogels was firstly introduced by Pitzler and colleagues for the directed evolution of phytase in E. coli cells.54 Their screening system used an enzymatic phytase/glucose oxidase cascade in which H2O2 produced by glucose oxidase reacts with Fe2+ ions to produce hydroxyl radicals. Hydroxyl radicals can then initiate copolymerization of N-vinyl-pyrrolidone, poly(ethyleneglycol)-diacrylate, and fluorescent Polyfluor 570 on the E. coli cell surface, creating a shell around the cell. Using hydrogel encapsulation, the authors were able to differentiate between active and inactive cells, and thereby evolve the phytase and isolate a variant with 31% increased catalytic activity for non-natural fluorescent substrate and 5% increased catalytic activity toward phytic acid. The same technology was used for directed evolution of esterase, lipase and cellulase using glucose derivatives as substrates.56 Even though the applied screening system was shown to be adaptable for different classes of hydrolases, the use of non-natural substrates can lead to false-positive variants and should be taken with caution.In a work from our group, we developed hydrogel-based enzyme activity assays using Fenton chemistry to generate polymerization initiators.57–59 To adapt these approaches to library screening, we developed a hydrogel encapsulation system for screening GOx libraries expressed on the yeast surface for increased enzyme activity and stability.60 Cells expressing active enzyme variants were encapsulated in fluorescent alginate carrying phenol moieties that polymerized in the presence of H2O2 and HRP (Fig. 1e). By screening for variants that could encapsulate the cells following exposure to a denaturing agent, variants with higher stability and activity could be sorted and isolated by FACS. The main advantage of this system is that it allows screening of enzyme libraries in a pooled fashion. Since the radicals generated to initiate the polymerization reaction have limited stability in biological media, the polymerization remains localized to the cell surface. This represents a reaction-based compartmentalization approach and enables one-pot library screening, greatly increasing throughput. Other bottlenecks in throughput including transformation efficiency and FACS determine the ultimate throughput of such systems. This hydrogel high-throughput screening represents the first system used for the direct screening of enzyme stability by flow cytometry, obtaining GOx variants with 13 to 15% increased thermal stability compared to the wild type enzyme. In addition, several advantages were shown when the alginate hydrogel was used for cell encapsulation compared to the previously described method based on Fenton chemistry.54 Alginate hydrogels are thick and robust, protecting the cells from osmotic lysis and allowing size-based filtration of encapsulated cells.55 Besides, the reaction mixture doesn’t require multiple monomer and polymer components, but a single fluorescent polymer. Future work in our group on HRP-mediated alginate polymerization is focusing on screening for alternative reaction chemistries using enzymatic cascades that generate H2O2 as the final reaction product.
4. Next-generation approaches in enzyme engineering
Well-defined library construction methods combined with thorough phenotypic characterization can shed light on structural and biophysical properties of enzymes, report functional consequences of altered residues, and predict their natural evolutionary trajectories. Over the years the optimization of screening methods based on multi-enzyme cascades as readout systems and the development of adequate compartmentalization strategies to guarantee the genotype–phenotype linkage have favoured a higher processivity in testing enzyme variants therefore facilitate a more systematic and complete investigation of enzyme sequence landscapes. Relatively recently the field of enzyme engineering started also to take advantage of the revolutionary advancements in DNA sequencing technologies giving rise to very powerful workflows for the study of enzyme properties by combining the use of high efficiency screening and next-generation DNA sequencing. Below we give a brief summary of prior works that made use of systematic scanning-based mutagenesis approaches coupled with medium to high-throughput screening for the study of enzymes. We also include a further subsection to describe recent studies combining the use of high efficiency screening and next-generation sequencing for the investigation of various enzyme properties.4.1 Systematic investigation of enzyme sequence landscapes
The availability of automated robotic systems paired with controlled mutagenesis protocols allowed, already several years ago, a noticeable boost in mutant libraries quality and screening efficiency leading to a more comprehensive investigation of enzyme sequences and properties. In this class of experiments, enzyme variants are typically tested individually in MTPs at medium throughput.One of the first comprehensive mutagenesis scans on an enzyme was presented by Gray and colleagues, who screened variants of a dehalogenase enzyme using a multi-well plate assay to identify single mutants with higher thermostability.61 Since then, analogous screening workflows were applied to the detection of stability enhancing mutations of other enzymes such as xylanase62,63 and phytase64 and similarly to the study of enantioselectivity of a nitrilase catalyst by combining an MTP assay with mass spectrometry.65 Recently Fulton and colleagues reported a systematic study on a lipase A to determine the effect of single mutations on its detergent tolerance.66 Another leading example of linear scanning of sequence space supported by MTP screening was presented by Van der Meer and colleagues.67,68 In this work, single mutants of the enzyme 4-oxalocrotonate tautomerase (4-OT), a promiscuous catalyst of carbon–carbon bonding reactions, were screened for enhanced Michael-type addition activity and improved enantioselectivity. In addition, selected mutations impacting the enantioselectivity of the catalyst were combined, favoring the expression of multiple mutant variants producing products with further improved enantiopurity.
Typically, the first comprehensive scanning mutagenesis methods applied to enzymes focused mainly on the investigation of catalysts that could provide direct survival advantages or detectable phenotypic changes making the screening of the variant library compatible with high throughput technologies (i.e. plate survival assays and single cell sorting) without any further adaptation required. Pioneering works were the publication of a nearly complete functional map of the T4 lysozyme generated by amber suppression and tested through a plating plaque-forming assay.69 Several other works focused over the years on the study of beta lactamase TEM-1 enzyme that provides resistance against B-lactam antibiotics conveying survival advantage as the selection mechanism.70–72 Other recent examples include mutagenic scanning studies on VIM-2 lactamase that involved scanning-based library construction methods combined with NGS,73,74 as outlined below.
4.2 High-throughput screening and next-generation sequencing, a winning combination
Along with the design and implementation of new tailored high-throughput screening methods, advancements in DNA sequencing technologies have significantly impacted the state of the art in enzyme engineering and in vitro evolution. Deep mutational scanning (DMS) is a method that couples high-throughput screening and high-throughput DNA sequencing technology to enable a thorough investigation of protein fitness landscapes. DMS consists of three main steps. First, systematically designed gene libraries encoding target protein variants are synthesized, validated through sequencing and used for expression of protein variants (Fig. 2a). Next, cells or synthetic compartments carrying the variants are screened using an appropriate method that assays the phenotypic function of the protein. Finally, the DNA library is retrieved from both input and post-screening/selection populations and its contents revealed through massively parallel next-generation sequencing (NGS).75 The frequency of each variant based on the NGS read count is quantified and its enrichment statistic is calculated by comparing its abundance before and after the screening/selection step. Sequences containing mutations with positive effects are expected to be enriched in the post screening/selected population, while enrichment ratios of one or lower are found for sequences with neutral or negative phenotypic effects. Finally, according to the nature of the screening methods adopted, the enrichment ratios are normalized and converted to fitness scores that together generate a comprehensive overview of the protein fitness landscape (Fig. 2b). Collectively, the massive amount of data generated in such an experiment is extremely helpful for decoding complex sequence-function relationships and lays the foundation for the design of informed libraries to accelerate protein engineering work.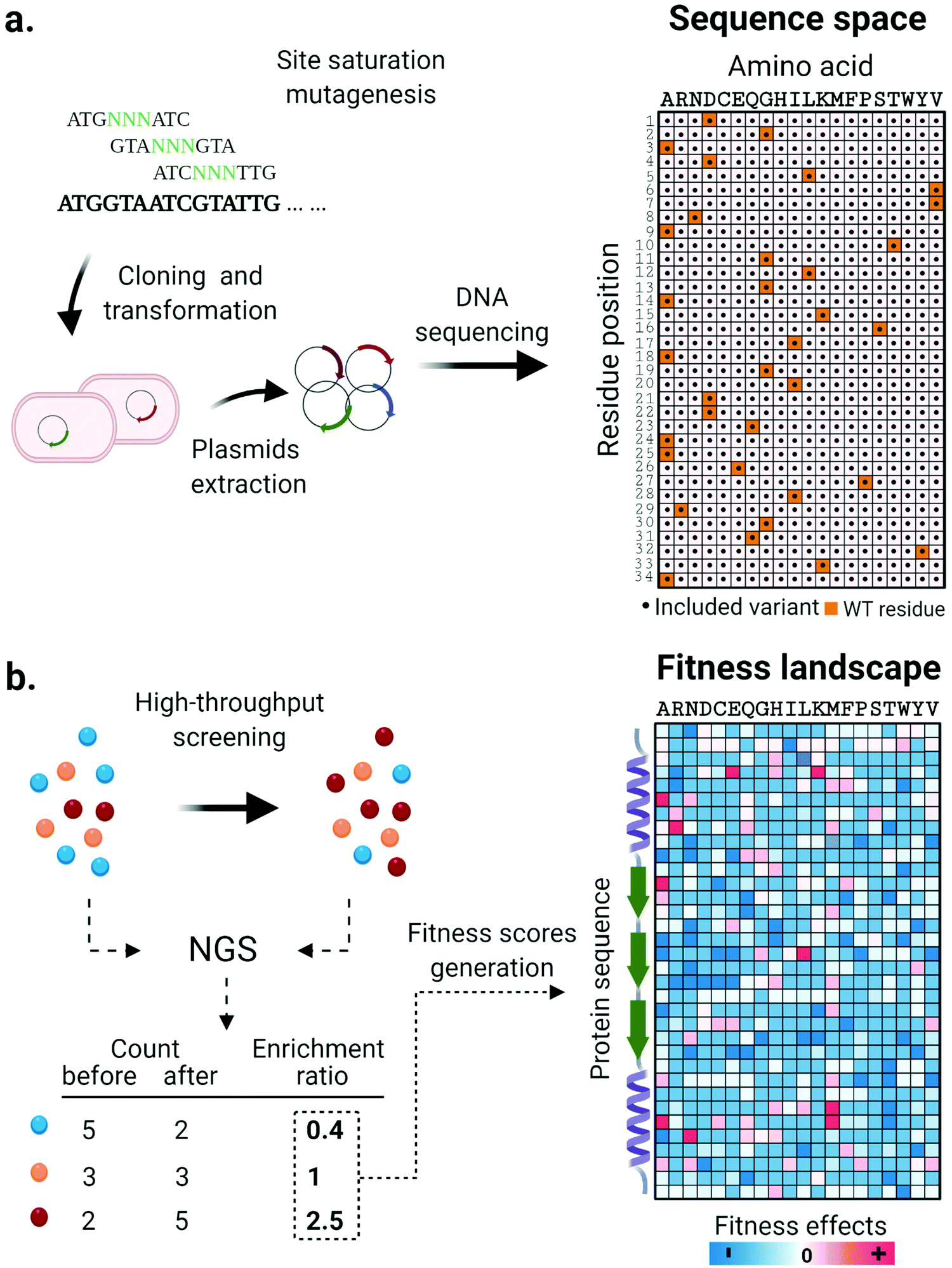 | ||
| Fig. 2 Systematic investigation of enzyme fitness landscapes through deep mutational scanning. (a) A systematically constructed mutant library of a target sequence is generated through site saturation mutagenesis and validated through DNA sequencing. The enzyme variants represented in the library are visualized on a sequence space map. (b) Enzyme variants are expressed and tested using high-throughput screening or selection methods. The DNA material is extracted and an enrichment value is calculated for each variant by comparing its abundance in the population before and after screening/selection. Depending on the screening method, enrichment factors are converted into fitness scores in various ways. Finally, the effect of each single amino acid mutation on the properties of the target enzymes is represented in a thoroughly informative fitness landscape map. Hotspots indicated regions of productive sequence space that can be used in future library designs. | ||
DMS requires the analysis of several thousands of variants in a single campaign. For the evolution of binders, this approach has become quite common, relying on methods such as cell surface, ribosome and phage display as expression platforms to enable DMS. However, for enzymatic reactions the comparatively lower throughput of enzyme screening assays has resulted in fewer reports of the application of DMS. For some enzyme screening methods, the requisite library sizes for DMS (typically on the order of 104 variants) are still too large.
Nevertheless, almost a decade ago the first investigation of an enzyme fitness landscape supported by deep sequencing appeared in literature.76 The authors targeted TEM1 beta lactamase and screened mutant libraries of the enzyme through selection on agar plates containing a fixed concentration of antibiotic. Sequencing of selected variants revealed positions with different inclinations to accept amino acid changes without impacting the enzyme activity. Along the same lines, Firnberg and colleagues reported mutational scanning of TEM-1 by screening a nearly comprehensive single-mutant library at 13 different ampicillin concentrations, thereby generating a detailed overview of the effects of each amino acid substitution on the overall protein fitness at different levels of selective pressure.77 Moreover, the authors analyzed and reported effects of silent DNA mutations on the stability and functionality at both RNA and protein levels, a novel aspect addressed later on in other works.78,79 Starita and colleagues adopted an auto-ubiquitination assay of phage displayed variants to explore the fitness landscape of E3 ubiquitin ligase.80 By deep-sequencing the library before and after selection, the authors found 25 single amino acid mutations that enhanced activity. Many mutations were located far from the catalytic site, and would have been difficult to predict using classical focused or random approaches for enzyme engineering at active site and first shell residues.
In work combining microfluidic approaches and DMS, Romero and colleagues developed a microdroplet encapsulation method for single cells expressing glucosidase variants that they coupled with fluorescence sorting. This DMS work revealed the effect of amino acid mutations on enzyme fitness and, by screening the mutant library following thermal stress, the authors discovered mutations enhancing enzyme thermostability.81
Coupling survival or growth rate of the host (i.e., selection) is also readily combined with DMS-based readout methods for characterizing phenotypic fitness. Klesmith and colleagues developed a microbial strain able to grow on levoglucosan as the sole organic source by linking the activity of the investigated enzyme, levoglucosan kinase, to the fitness of the host organism.82 Similarly, the activity of an RNA guided endonuclease was linked to the survival of microbial cells through inducible expression of a toxic DNA gyrase inhibitor, which served as a selection system that was then used for DMS of a CAS9 enzyme.83
DMS was also specifically applied to the study of sequence determinants that impact protein solubility. TEM-1 and levoglucosan kinase activities were abolished through site directed mutagenesis and the effects of mutations on translation and folding of the proteins addressed. By coupling these new data to previous findings on activity for the same catalysts the authors built a fitness landscape including both properties and confirmed that shared mutations impacting positively both activity and solubility in an enzyme are rare.84
With the large datasets provided by DMS, researchers have begun to address more fundamental questions about enzyme function and evolution, such as how substrate choice can impact evolutionary trajectories of enzymes or how the evolutionary trajectory is influenced by the strength of the selective pressure. Along this line of research, Melnikov and colleagues demonstrated how the fitness landscape of an enzyme varies significantly depending on the nature of the selection system. They constructed 6 parallel fitness landscapes of a Tn5 transposon-derived kinase that confers resistance to antibiotics and used 6 structurally distinct substrates at increasing concentrations for the selection.85 The authors identified protein residues responsible for orthogonal activity on different substrates without any additional support from structural or in silico analyses. Furthermore, TEM-1 beta lactamase was again used as a model enzyme by Stiffler and colleagues to study the connection between evolvability and robustness of a fitness landscape. By exposing a comprehensive single mutant library of TEM-1 to selective pressure at increasing concentrations of its natural substrate ampicillin as well as on a new substrate cefotaxime, the authors concluded that the robustness of a sequence (i.e. its capacity to accept and tolerate mutations without impacting the function) strongly depends on the strength of the selection used and that its divergent evolvability towards the use of new substrates is facilitated at lighter selective conditions rather than under strong selective pressure.86 This intuitively makes sense under the consideration that most mutations are neutral or deleterious.
A key interest among protein biochemists is to gain a deeper understanding of how enzymes encode substrate specificity. Several directed evolution campaigns have been successful in tuning or changing substrate specificity of catalysts. Nevertheless, these works typically explore mutations in the vicinity of the substrate binding pocket and ignore mutations at distal residues. In recent work, Wrenbeck and colleagues applied DMS to study the fitness landscape of an amide hydrolase, linking its activity to the growth rate of the host cells and screening a nearly comprehensive single mutant enzyme library using three different amides as substrates.78 This work showed that mutations beneficial for a specific substrate are often not proximal to the catalytic site, once more demonstrating the advantages of systematic scanning methods such as DMS in comparison to random or rational approaches. Furthermore the authors concluded that screening of an enzyme mutant library using different substrates produces unique fitness landscapes with profound differences, emphasized even more when molecules with significant structural divergence are used.
Currently, DMS supported by NGS and high-throughput screening platforms represent the most advanced pipelines to engineer enzymes and explore enzyme sequence and function. Nevertheless, the application of these methods to the study of enzymes suffers from a mismatch in throughput, with state-of-the-art NGS throughput surpassing the best and fastest screening methods by several orders of magnitude. In fact, while we have witnessed over the past years steady advancements in quality and efficiency of sequencing technologies, no striking breakthroughs have been registered for the development of equally important high-throughput screening for many categories of enzymes. This throughput bottleneck has motivated the development of computational and machine learning approaches that can be trained on limited experimental data and interpolate accurate phenotypes from input sequences, as described below.
5. Machine learning in enzyme engineering
Machine learning and big data analysis techniques are computational methods suited to address the challenge of navigating the vastness of protein sequence space. For this reason enzyme engineers have started to regularly use those methods to improve the outcome of directed evolution and enzyme engineering campaigns.87–97 Machine learning can improve the efficiency of downstream experimental studies thereby adding value and complimenting purely experimental bioengineering approaches. Moreover, the exponential increase in DNA sequencing throughput presents a significant opportunity to combine state-of-the-art computation and machine learning with large biological datasets in an attempt to learn sequence-function maps in protein sequence space.The generic pipeline of a protein engineering campaign supported by machine learning consists of the generation of experimental data representing sequence-phenotype pairs, and training a statistical or machine learning model to predict phenotypes from input sequences never assayed before. The phenotypic property of interest can be chosen from several features, including catalytic properties,98,99 substrate affinity, stability,100–102 and expression level in the host organism. Prominent examples of machine learning assisted protein engineering include membrane channel engineering,103,104 protein structure prediction105,106 and protein–protein interactions.107–109 Although we cannot hope to comprehensively cover this exciting and rapidly developing field in this review, we would like to outline below some basic considerations in applying machine learning with a focus on appropriate methodology for enzyme engineering.
In order to apply machine learning to enzyme engineering, the amino acid sequences need to be converted and represented by numerical arrays. Different methods are available to accomplish this task, from the tabulations of single amino acid physical parameters (solubility, charges, pKas, etc.) to combinations of such parameters such as in AAindex,110 a collection of indices, mutation matrices and statistical protein contact potentials; or T-scale,111 which uses principal component analysis to reduce the dimensionality of topological and structural data of 135 amino acids. Another successful and well established method, because of its simplicity, is to convert each amino acid into bit-based vectors that can be more (one-hot encoding) or less sparse (binary numbers). Representations of amino acids can even be actively learned in so-called embeddings.112
Next, a model typology needs to be selected/chosen according to the nature of the problem. If the fitness function studied behaves in a continuous and ungroupable space, a regression model may be appropriate. Otherwise, if the discrimination between different fitness categories of the studied population is well defined, the model of choice would be a classifier. An approach could be to start with the implementation of simple linear models and move to more complex ones if non-linearity is needed to describe the system under study.
Another aspect to consider for the establishment of the ML workflow is the quantity of data that needs to be processed. Kernel based methods such as support vector machines are well suited for handling hundreds of data points while neural networks are more suitable if the amount of data is in the order of hundreds of thousands or even millions of experimental sequences.
A key step consists of the separation of the data into different sets. The set referred to as the training set includes the majority of the data (usually 70–80%) and is used to train the model and learn the best parameters in order to predict the response variable of interest. The second set referred to as the validation set is the second most populated one (20–10%) and it allows a balance of the complexity of the model, known as the hyper-parameters. This is crucial to avoid underfitting, where the true behavior of a system is not described sufficiently, as well as overfitting, where the model fits the training set extremely well but fails to generalise to other points. Last but not least, it is important to save a small portion of data (∼10%), which must never be used during the training, to test the goodness of the model to interpolate and, eventually, extrapolate to unexplored sequence space. After the generation of a working trained model, in silico screening of a large number of different candidate sequences not present in the initial dataset can be used to evaluate and rank candidates. Subsequently, the best candidates discovered in silico can be synthesised in the lab and characterized, and eventually included in future iterations of model training. This approach can ultimately lead to massive savings in resources, money and time. For further explanations on model heuristics and exemplary case studies, we refer the reader to the work of Yang and colleagues.113
An early theoretical work from Fox114 reported application of partial least square (PLS) regression coupled with genetic algorithms to improve directed evolution outcomes. Similarly another PLS based algorithm was implemented in more recent work by Cadet and colleagues94 where researchers processed the data with Fast Fourier Transformation (FFT) and used PLS to develop a predictive model for the improvement of epoxide hydrolase.
The technique used by Romero and coworkers115 involved optimizing thermostable and active P450 enzymes using a Gaussian process. Specifically, they fit a model using a library containing 261 sequences. Using this model, they were able to identify and synthesize a variant with a ∼9 °C improvement in thermal denaturation temperature when compared to a previously engineered variant obtained by classical directed evolution. The Gaussian process has the advantage of including uncertainty in regions that were not explored experimentally, however it is computationally expensive and training time scales poorly for large datasets.
6. Conclusions and outlook
Enzymes will continue to play an important role in biocatalytic production of high-value chemicals and directly as biopharmaceutical therapeutics in the years to come. The extremely high-level specificity and catalytic activity of biological enzymes as compared to non-biological catalysts enables the salient features of living systems, and these unique properties remain enticing for scientists and engineers to harness for artificial purposes.We have identified several trends representing state-of-the-art modern methods for enzyme engineering and evolution including concepts such as novel reaction cascades for readout, approaches to reaction compartmentalization to achieve a genotype–phenotype link, and systematic scanning mutagenesis and deep mutational scanning approaches for generating mutability maps. Improvements in ultrahigh-throughput enzyme screening technology and DNA sequencing have significantly increased the amount of experimental data that can be obtained from directed evolution experiments, however, big data is extremely costly and time consuming to obtain. The availability of computational tools such as machine learning to extract the most value from these datasets will acquire more importance with time as screening capacity, DNA sequencing throughput and computational power increase. In such a scenario, machine learning approaches will considerably improve directed enzyme evolution by substantially lowering time and resources needed to achieve a desired activity level, or by significantly increasing the performance of enzymes that are engineered at a fixed cost level. By manipulating genes encoding catalytic enzymes, protein engineers can push these molecules to new levels of fitness and stability and help them reach their full potential.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Basel, ETH Zürich, the Swiss National Science Foundation (Project 200021_191962), the Swiss Nanoscience Institute, and the National Center for Competence in Research (NCCR) Molecular Systems Engineering.References
- M. Rooseboom, J. N. M. Commandeur and N. P. E. Vermeulen, Pharmacol. Rev., 2004, 56, 53–102 CrossRef CAS PubMed.
- G. U. Dachs, J. Tupper and G. M. Tozer, Anticancer Drugs, 2005, 16, 349–359 CrossRef CAS PubMed.
- E. Gibney, R. Van Noorden, H. Ledford, D. Castelvecchi and M. Warren, Nature, 2018, 562, 176 CrossRef CAS PubMed.
- O. P. Malhotra, P. K. Ambasht, P. Prabhakar, A. K. Lal and A. M. Kayastha, Biochem. Educ., 1996, 24, 56–59 CrossRef.
- J. H. Wilkinson, J. Clin. Pathol. Suppl., 1970, 4, 14–21 CrossRef CAS PubMed.
- W. Van Roy, G. Woronoff, A. M. Jimenez Valencia, T. Stakenborg and W. A. Clarke, Biochem. Eng. J., 2020, 161, 107699 CrossRef CAS.
- D. Böttcher, P. Zägel, M. Schmidt and U. T. Bornscheuer, in Metagenomics: Methods and Protocols, ed. W. R. Streit and R. Daniel, Springer New York, New York, NY, 2017, pp. 197–204 Search PubMed.
- U. Bornscheuer, M. Baumann, US Pat., 20040219625:A1, 2004.
- C. L. Hendricks, J. R. Ross, E. Pichersky, J. P. Noel and Z. S. Zhou, Anal. Biochem., 2004, 326, 100–105 CrossRef CAS PubMed.
- J. G. Ortiz-Tena, B. Rühmann and V. Sieber, Anal. Chem., 2018, 90, 2526–2533 CrossRef CAS PubMed.
- B. Begander, A. Huber, M. Döring, J. Sperl and V. Sieber, Int. J. Mol. Sci., 2020, 21(1), 335 CrossRef CAS PubMed.
- M. Liu, J. Gu, W. Xie and H. Yu, Chem. Commun., 2013, 49, 7219–7221 RSC.
- M. Liu, W. Xie, H. Xu, J. Gu, X. Lv, H. Yu and L. Ye, Biotechnol. Lett., 2014, 36, 1801–1807 CrossRef CAS PubMed.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS PubMed.
- J.-L. Lin, H. Ekas, K. Markham and H. S. Alper, Biochem. Eng. J., 2018, 139, 95–100 CrossRef CAS.
- R. Ostafe, R. Prodanovic, U. Commandeur and R. Fischer, Anal. Biochem., 2013, 435, 93–98 CrossRef CAS PubMed.
- R. Ostafe, R. Prodanovic, W. Lloyd Ung, D. A. Weitz and R. Fischer, Biomicrofluidics, 2014, 8, 041102 CrossRef PubMed.
- A. Debon, M. Pott, R. Obexer, A. P. Green, L. Friedrich, A. D. Griffiths and D. Hilvert, Nat. Catal., 2019, 2, 740–747 CrossRef CAS.
- S. Becker, H. Höbenreich, A. Vogel, J. Knorr, S. Wilhelm, F. Rosenau, K.-E. Jaeger, M. T. Reetz and H. Kolmar, Angew. Chem., Int. Ed., 2008, 47, 5085–5088 CrossRef CAS PubMed.
- R. Prodanovic, R. Ostafe, A. Scacioc and U. Schwaneberg, Comb. Chem. High Throughput Screening, 2011, 14, 55–60 CrossRef CAS PubMed.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7383–E7389 CrossRef CAS PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- A. Aharoni, K. Thieme, C. P. C. Chiu, S. Buchini, L. L. Lairson, H. Chen, N. C. J. Strynadka, W. W. Wakarchuk and S. G. Withers, Nat. Methods, 2006, 3, 609–614 CrossRef CAS PubMed.
- M. Dörr, M. P. C. Fibinger, D. Last, S. Schmidt, J. Santos-Aberturas, D. Böttcher, A. Hummel, C. Vickers, M. Voss and U. T. Bornscheuer, Biotechnol. Bioeng., 2016, 113, 1421–1432 CrossRef PubMed.
- A. Aharoni, G. Amitai, K. Bernath, S. Magdassi and D. S. Tawfik, Chem. Biol., 2005, 12, 1281–1289 CrossRef CAS PubMed.
- E. Mastrobattista, V. Taly, E. Chanudet, P. Treacy, B. T. Kelly and A. D. Griffiths, Chem. Biol., 2005, 12, 1291–1300 CrossRef CAS PubMed.
- B. Chen, S. Lim, A. Kannan, S. C. Alford, F. Sunden, D. Herschlag, I. K. Dimov, T. M. Baer and J. R. Cochran, Nat. Chem. Biol., 2016, 12, 76–81 CrossRef CAS PubMed.
- B. Zhu, T. Mizoguchi, T. Kojima and H. Nakano, PLoS One, 2015, 10, e0127479 CrossRef PubMed.
- A. Uyeda, T. Watanabe, Y. Kato, H. Watanabe, T. Yomo, T. Hohsaka and T. Matsuura, ChemBioChem, 2015, 16, 1797–1802 CrossRef CAS PubMed.
- C. Schmidt-Dannert and F. H. Arnold, Trends Biotechnol., 1999, 17, 135–136 CrossRef CAS PubMed.
- J. C. Sadler, A. Currin and D. B. Kell, Analyst, 2018, 143, 4747–4755 RSC.
- A. J. Ruff, A. Dennig, G. Wirtz, M. Blanusa and U. Schwaneberg, ACS Catal., 2012, 2, 2724–2728 CrossRef CAS.
- F. Cheng, T. Kardashliev, C. Pitzler, A. Shehzad, H. Lue, J. Bernhagen, L. Zhu and U. Schwaneberg, ACS Synth. Biol., 2015, 4, 768–775 CrossRef CAS PubMed.
- M. I. Sanchez and A. Y. Ting, Nat. Methods, 2020, 17, 167–174 CrossRef CAS PubMed.
- L. Yi, M. C. Gebhard, Q. Li, J. M. Taft, G. Georgiou and B. L. Iverson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7229–7234 CrossRef CAS PubMed.
- B. M. Dorr, H. O. Ham, C. An, E. L. Chaikof and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13343–13348 CrossRef CAS PubMed.
- L. Deweid, L. Neureiter, S. Englert, H. Schneider, J. Deweid, D. Yanakieva, J. Sturm, S. Bitsch, A. Christmann, O. Avrutina, H.-L. Fuchsbauer and H. Kolmar, Chemistry, 2018, 24, 15195–15200 CrossRef CAS PubMed.
- H. S. Kwok, O. Vargas-Rodriguez, S. V. Melnikov and D. Söll, ACS Chem. Biol., 2019, 14, 603–612 CrossRef CAS PubMed.
- E. G. Worst, M. P. Exner, A. De Simone, M. Schenkelberger, V. Noireaux, N. Budisa and A. Ott, Bioorg. Med. Chem. Lett., 2015, 25, 3658–3660 CrossRef CAS PubMed.
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652–656 CrossRef CAS PubMed.
- G. Körfer, C. Pitzler, L. Vojcic, R. Martinez and U. Schwaneberg, Sci. Rep., 2016, 6, 26128 CrossRef PubMed.
- A. Fallah-Araghi, J.-C. Baret, M. Ryckelynck and A. D. Griffiths, Lab Chip, 2012, 12, 882–891 RSC.
- A. Zinchenko, S. R. A. Devenish, B. Kintses, P.-Y. Colin, M. Fischlechner and F. Hollfelder, Anal. Chem., 2014, 86, 2526–2533 CrossRef CAS PubMed.
- G. Woronoff, M. Ryckelynck, J. Wessel, O. Schicke, A. D. Griffiths and P. Soumillion, ChemBioChem, 2015, 16, 1343–1349 CrossRef CAS PubMed.
- D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore, S. Sun and R. T. Kennedy, Angew. Chem., Int. Ed., 2020, 132, 4500–4507 CrossRef.
- R. Ostafe, R. Prodanovic, J. Nazor and R. Fischer, Chem. Biol., 2014, 21, 414–421 CrossRef CAS PubMed.
- F. Ma, M. Fischer, Y. Han, S. G. Withers, Y. Feng and G. Y. Yang, Anal. Chem., 2016, 88, 8587–8595 CrossRef CAS PubMed.
- G. Kovačević, R. Ostafe, A. M. Balaž, R. Fischer and R. Prodanović, J. Biosci. Bioeng., 2019, 127, 30–37 CrossRef PubMed.
- J. Santos-Aberturas, M. Dörr, G. S. Waldo and U. T. Bornscheuer, Chem. Biol., 2015, 22, 1406–1414 CrossRef CAS PubMed.
- R. Prodanović, W. L. Ung, K. I. Đurđić, R. Fischer, D. A. Weitz and R. Ostafe, Molecules, 2020, 25(10), 2418 CrossRef PubMed.
- T. Beneyton, S. Thomas, A. D. Griffiths, J.-M. Nicaud, A. Drevelle and T. Rossignol, Microb. Cell Fact., 2017, 16, 18 CrossRef PubMed.
- J.-C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison, J. J. Agresti, D. R. Link, D. A. Weitz and A. D. Griffiths, Lab Chip, 2009, 9, 1850–1858 RSC.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4004–4009 CrossRef CAS PubMed.
- C. Pitzler, G. Wirtz, L. Vojcic, S. Hiltl, A. Böker, R. Martinez and U. Schwaneberg, Chem. Biol., 2014, 21, 1733–1742 CrossRef CAS PubMed.
- R. Vanella, A. Bazin, D. T. Ta and M. A. Nash, Chem. Mater., 2019, 31, 1899–1907 CrossRef.
- N. Lülsdorf, C. Pitzler, M. Biggel, R. Martinez, L. Vojcic and U. Schwaneberg, Chem. Commun., 2015, 51, 8679–8682 RSC.
- K. H. Malinowska and M. A. Nash, Curr. Opin. Biotechnol, 2016, 39, 68–75 CrossRef CAS PubMed.
- K. H. Malinowska, T. Rind, T. Verdorfer, H. E. Gaub and M. A. Nash, Anal. Chem., 2015, 87, 7133–7140 CrossRef CAS PubMed.
- K. H. Malinowska, T. Verdorfer, A. Meinhold, L. F. Milles, V. Funk, H. E. Gaub and M. A. Nash, ChemSusChem, 2014, 7, 2825–2831 CrossRef CAS PubMed.
- R. Vanella, D. T. Ta and M. A. Nash, Biotechnol. Bioeng., 2019, 116, 1878–1886 CrossRef CAS PubMed.
- K. A. Gray, T. H. Richardson, K. Kretz, J. M. Short, F. Bartnek, R. Knowles, L. Kan, P. E. Swanson and D. E. Robertson, Adv. Synth. Catal., 2001, 343, 607–617 CrossRef CAS.
- N. Palackal, Y. Brennan, W. N. Callen, P. Dupree, G. Frey, F. Goubet, G. P. Hazlewood, S. Healey, Y. E. Kang, K. A. Kretz, E. Lee, X. Tan, G. L. Tomlinson, J. Verruto, V. W. K. Wong, E. J. Mathur, J. M. Short, D. E. Robertson and B. A. Steer, Protein Sci., 2004, 13, 494–503 CrossRef CAS PubMed.
- C. Dumon, A. Varvak, M. A. Wall, J. E. Flint, R. J. Lewis, J. H. Lakey, C. Morland, P. Luginbühl, S. Healey, T. Todaro, G. DeSantis, M. Sun, L. Parra-Gessert, X. Tan, D. P. Weiner and H. J. Gilbert, J. Biol. Chem., 2008, 283, 22557–22564 CrossRef CAS PubMed.
- J. B. Garrett, K. A. Kretz, E. O’Donoghue, J. Kerovuo, W. Kim, N. R. Barton, G. P. Hazlewood, J. M. Short, D. E. Robertson and K. A. Gray, Appl. Environ. Microbiol., 2004, 70, 3041–3046 CrossRef CAS PubMed.
- G. DeSantis, K. Wong, B. Farwell, K. Chatman, Z. Zhu, G. Tomlinson, H. Huang, X. Tan, L. Bibbs, P. Chen, K. Kretz and M. J. Burk, J. Am. Chem. Soc., 2003, 125, 11476–11477 CrossRef CAS PubMed.
- A. Fulton, V. J. Frauenkron-Machedjou, P. Skoczinski, S. Wilhelm, L. Zhu, U. Schwaneberg and K.-E. Jaeger, ChemBioChem, 2015, 16, 930–936 CrossRef CAS PubMed.
- J.-Y. van der Meer, H. Poddar, B.-J. Baas, Y. Miao, M. Rahimi, A. Kunzendorf, R. van Merkerk, P. G. Tepper, E. M. Geertsema, A.-M. W. H. Thunnissen, W. J. Quax and G. J. Poelarends, Nat. Commun., 2016, 7, 10911 CrossRef CAS PubMed.
- J.-Y. van der Meer, L. Biewenga and G. J. Poelarends, ChemBioChem, 2016, 17, 1792–1799 CrossRef CAS PubMed.
- D. Rennell, S. E. Bouvier, L. W. Hardy and A. R. Poteete, J. Mol. Biol., 1991, 222, 67–88 CrossRef CAS PubMed.
- H. Jacquier, A. Birgy, H. Le Nagard, Y. Mechulam, E. Schmitt, J. Glodt, B. Bercot, E. Petit, J. Poulain, G. Barnaud, P.-A. Gros and O. Tenaillon, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13067–13072 CrossRef CAS PubMed.
- W. Huang, J. Petrosino, M. Hirsch, P. S. Shenkin and T. Palzkill, J. Mol. Biol., 1996, 258, 688–703 CrossRef CAS PubMed.
- T. Palzkill and D. Botstein, Proteins, 1992, 14, 29–44 CrossRef CAS PubMed.
- J. Z. Chen, D. M. Fowler and N. Tokuriki, eLife, 2020, 9, e56707 CrossRef CAS PubMed.
- J. Z. Chen, D. M. Fowler and N. Tokuriki, bioRxiv, 2021.04.14.439889 DOI:10.1101/2021.04.14.439889.
- D. M. Fowler and S. Fields, Nat. Methods, 2014, 11, 801–807 CrossRef CAS PubMed.
- Z. Deng, W. Huang, E. Bakkalbasi, N. G. Brown, C. J. Adamski, K. Rice, D. Muzny, R. A. Gibbs and T. Palzkill, J. Mol. Biol., 2012, 424, 150–167 CrossRef CAS PubMed.
- E. Firnberg, J. W. Labonte, J. J. Gray and M. Ostermeier, Mol. Biol. Evol., 2014, 31, 1581–1592 CrossRef CAS PubMed.
- E. E. Wrenbeck, L. R. Azouz and T. A. Whitehead, Nat. Commun., 2017, 8, 15695 CrossRef CAS PubMed.
- M. S. Faber, E. E. Wrenbeck, L. R. Azouz, P. J. Steiner and T. A. Whitehead, Mol. Biol. Evol., 2019, 36, 2764–2777 CrossRef CAS PubMed.
- L. M. Starita, J. N. Pruneda, R. S. Lo, D. M. Fowler, H. J. Kim, J. B. Hiatt, J. Shendure, P. S. Brzovic, S. Fields and R. E. Klevit, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1263–E1272 CrossRef CAS PubMed.
- P. A. Romero, T. M. Tran and A. R. Abate, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7159–7164 CrossRef CAS PubMed.
- J. R. Klesmith, J.-P. Bacik, R. Michalczyk and T. A. Whitehead, ACS Synth. Biol., 2015, 4, 1235–1243 CrossRef CAS PubMed.
- J. M. Spencer and X. Zhang, Sci. Rep., 2017, 7, 16836 CrossRef PubMed.
- J. R. Klesmith, J.-P. Bacik, E. E. Wrenbeck, R. Michalczyk and T. A. Whitehead, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2265–2270 CrossRef CAS PubMed.
- A. Melnikov, P. Rogov, L. Wang, A. Gnirke and T. S. Mikkelsen, Nucleic Acids Res., 2014, 42, e112 CrossRef PubMed.
- M. A. Stiffler, D. R. Hekstra and R. Ranganathan, Cell, 2015, 160, 882–892 CrossRef CAS PubMed.
- R. J. Fox and G. W. Huisman, Trends Biotechnol., 2008, 26, 132–138 CrossRef CAS PubMed.
- S. Mazurenko, Z. Prokop and J. Damborsky, ACS Catal., 2020, 10, 1210–1223 CrossRef CAS.
- G. Li, Y. Dong and M. Reetz, Adv. Synth. Catal., 2019, 361, 2377–2386 CAS.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS PubMed.
- N. E. Siedhoff, U. Schwaneberg and M. D. Davari, Methods Enzymol., 2020, 643, 281–315 CAS.
- N. Singh, S. Malik, A. Gupta and K. R. Srivastava, Emerging Top. Life Sci., 2021, 5, 113–125 CrossRef PubMed.
- G. Qu, A. Li, Z. Sun, C. G. Acevedo-Rocha and M. T. Reetz, Angew. Chem., Int. Ed. Engl., 2020, 59(32), 13204–13231 CrossRef CAS PubMed.
- F. Cadet, N. Fontaine, G. Li, J. Sanchis, M. Ng Fuk Chong, R. Pandjaitan, I. Vetrivel, B. Offmann and M. T. Reetz, Sci. Rep., 2018, 8, 16757 CrossRef PubMed.
- J. Liao, M. K. Warmuth, S. Govindarajan, J. E. Ness, R. P. Wang, C. Gustafsson and J. Minshull, BMC Biotechnol., 2007, 7, 16 CrossRef PubMed.
- M. Scherer, S. J. Fleishman, P. R. Jones, T. Dandekar and E. Bencurova, Front. Bioeng. Biotechnol., 2021, 9, 673005 CrossRef PubMed.
- R. Lipsh-Sokolik, D. Listov and S. J. Fleishman, Protein Sci., 2021, 30, 151–159 CrossRef CAS PubMed.
- B. M. Bonk, J. W. Weis and B. Tidor, J. Am. Chem. Soc., 2019, 141, 4108–4118 CrossRef CAS PubMed.
- R. Ostafe, N. Fontaine, D. Frank, M. Ng Fuk Chong, R. Prodanovic, R. Pandjaitan, B. Offmann, F. Cadet and R. Fischer, Biotechnol. Bioeng., 2020, 117, 17–29 CrossRef CAS PubMed.
- G. Li, K. S. Rabe, J. Nielsen and M. K. M. Engqvist, ACS Synth. Biol., 2019, 8, 1411–1420 CrossRef CAS PubMed.
- K. Yoshida, S. Kawai, M. Fujitani, S. Koikeda, R. Kato and T. Ema, Sci. Rep., 2021, 11, 11883 CrossRef PubMed.
- G. Li, Y. Qin, N. T. Fontaine, M. Ng Fuk Chong, M. A. Maria-Solano, F. Feixas, X. F. Cadet, R. Pandjaitan, M. Garcia-Borràs, F. Cadet and M. T. Reetz, ChemBioChem, 2021, 22, 904–914 CrossRef CAS PubMed.
- C. N. Bedbrook, K. K. Yang, J. E. Robinson, E. D. Mackey, V. Gradinaru and F. H. Arnold, Nat. Methods, 2019, 16, 1176–1184 CrossRef CAS PubMed.
- C. N. Bedbrook, K. K. Yang, A. J. Rice, V. Gradinaru and F. H. Arnold, PLoS Comput. Biol., 2017, 13, e1005786 CrossRef PubMed.
- A. W. Senior, R. Evans, J. Jumper, J. Kirkpatrick, L. Sifre, T. Green, C. Qin, A. Žídek, A. W. R. Nelson, A. Bridgland, H. Penedones, S. Petersen, K. Simonyan, S. Crossan, P. Kohli, D. T. Jones, D. Silver, K. Kavukcuoglu and D. Hassabis, Nature, 2020, 577, 706–710 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- T. Sun, B. Zhou, L. Lai and J. Pei, BMC Bioinf., 2017, 18, 277 CrossRef PubMed.
- D. M. Mason, S. Friedensohn, C. R. Weber, C. Jordi, B. Wagner, S. Meng, P. Gainza, B. E. Correia and S. T. Reddy, bioRxiv, 2019, 617860 Search PubMed.
- P. Gainza, F. Sverrisson, F. Monti, E. Rodolà, D. Boscaini, M. M. Bronstein and B. E. Correia, Nat. Methods, 2020, 17, 184–192 CrossRef CAS PubMed.
- S. Kawashima, P. Pokarowski, M. Pokarowska, A. Kolinski, T. Katayama and M. Kanehisa, Nucleic Acids Res., 2008, 36, D202–D205 CrossRef CAS PubMed.
- F. Tian, P. Zhou and Z. Li, J. Mol. Struct., 2007, 830, 106–115 CrossRef CAS.
- K. K. Yang, Z. Wu, C. N. Bedbrook and F. H. Arnold, Bioinformatics, 2018, 34, 2642–2648 CrossRef CAS PubMed.
- K. K. Yang, Z. Wu and F. H. Arnold, Nat. Methods, 2019, 16, 687–694 CrossRef CAS PubMed.
- R. Fox, J. Theor. Biol., 2005, 234, 187–199 CrossRef CAS PubMed.
- P. A. Romero, A. Krause and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E193–E201 CrossRef CAS PubMed.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |