
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational design and experimental characterisation of a stable human heparanase variant†
Cassidy
Whitefield‡
 a,
Nansook
Hong‡
a,
Joshua A.
Mitchell
a and
Colin J.
Jackson
*ab
a,
Nansook
Hong‡
a,
Joshua A.
Mitchell
a and
Colin J.
Jackson
*ab
aResearch School of Chemistry, Australian National University, Canberra, ACT, 2601, Australia. E-mail: colin.jackson@anu.edu.au
bAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, Australian National University, Canberra, ACT 2601, Australia
First published on 15th February 2022
Abstract
Heparanase is the only human enzyme known to hydrolyse heparin sulfate and is involved in many important physiological processes. However, it is also unregulated in many disease states, such as cancer, diabetes and Covid-19. It is thus an important drug target, yet the heterologous production of heparanase is challenging and only possible in mammalian or insect expression systems, which limits the ability of many laboratories to study it. Here we describe the computational redesign of heparanase to allow high yield expression in Escherchia coli. This mutated form of heparanase exhibits essentially identical kinetics, inhibition, structure and protein dynamics to the wild type protein, despite the presence of 26 mutations. This variant will facilitate wider study of this important enzyme and contributes to a growing body of literature that shows evolutionarily conserved and functionally neutral mutations can have significant effects on protein folding and expression.
Introduction
Heparan sulfate (HS) consists of 1–4 linked disaccharide units that are negatively charged and structurally heterogeneous due to variable sulfation, deacetylation and epimerization during biosynthesis.1 HS is often covalently linked to proteins and peptides to form heparan sulfate proteoglycans (HSPGs).2 HSPGs are themselves a major component of the extracellular matrix (ECM) and basement membranes, forming a protective barrier by interacting with other major components of the ECM such as collagen, fibronectin and laminin. Their structural diversity and negative charge attract various cationic proteins and water, forming porous hydrogels that are able to store bioactive molecules including growth factors,3,4 chemokines5 and enzymes.6Heparanase (HPSE) is the only mammalian enzyme that is known to hydrolyse HS.7–10 In adults, HPSE is normally expressed at low levels, found only in platelets, immune cells and the placenta,11–13 but increased expression of HPSE has been observed in many disease states, including cancer and Covid-19.14–16 When overexpressed, HPSE catalyses the hydrolysis of HS, resulting in weakening of the ECM barrier, which can promote inflammation,17,18 cancer cell invasion, growth and migration,19,20 as well as angiogenesis.21,22 HPSE is also associated with tumour initiation by up-regulating pro-inflammatory mediators.23,24 Moreover, animal studies have shown HPSE genetic knock-outs improve cancer prognosis and increased survival without significant side effects.25,26
Owing to its roles in many diseases, HPSE has been a drug target for many years. For instance, HPSE expression promotes resistance to chemotherapy, whereas targeting HPSE with inhibitors can overcome chemoresistance and tumour relapse.27 Indeed, many groups have attempted to produce drug-like HPSE inhibitors over recent decades.1,26,28,29 However, HPSE production currently relies on complex and expensive eukaryotic expression systems such as mammalian7,8,30 and insect cells.31,32 While some prokaryotic HPSE expression methods have been reported,33,34 they have not been sufficiently robust for widespread adoption. HPSE has many features that are known to reduce soluble expression prokaryotic systems, such as Escherichia coli, including multiple disulfide bonds and large positive regions on the surface,35–37 as well as N-glycosylation.22,38 Moreover, HPSE is natively expressed as a pre-proheparanase which undergoes proteolytic cleavage of a signal peptide then a linker segment, resulting in an active heterodimer composed of 8 kDa and 50 kDa subunits (Fig. 1)39 Thus, in prokaryotic expression systems the 8 kDa and 50 kDa subunits have to be expressed separately and assemble into a heterodimeric complex.33,34
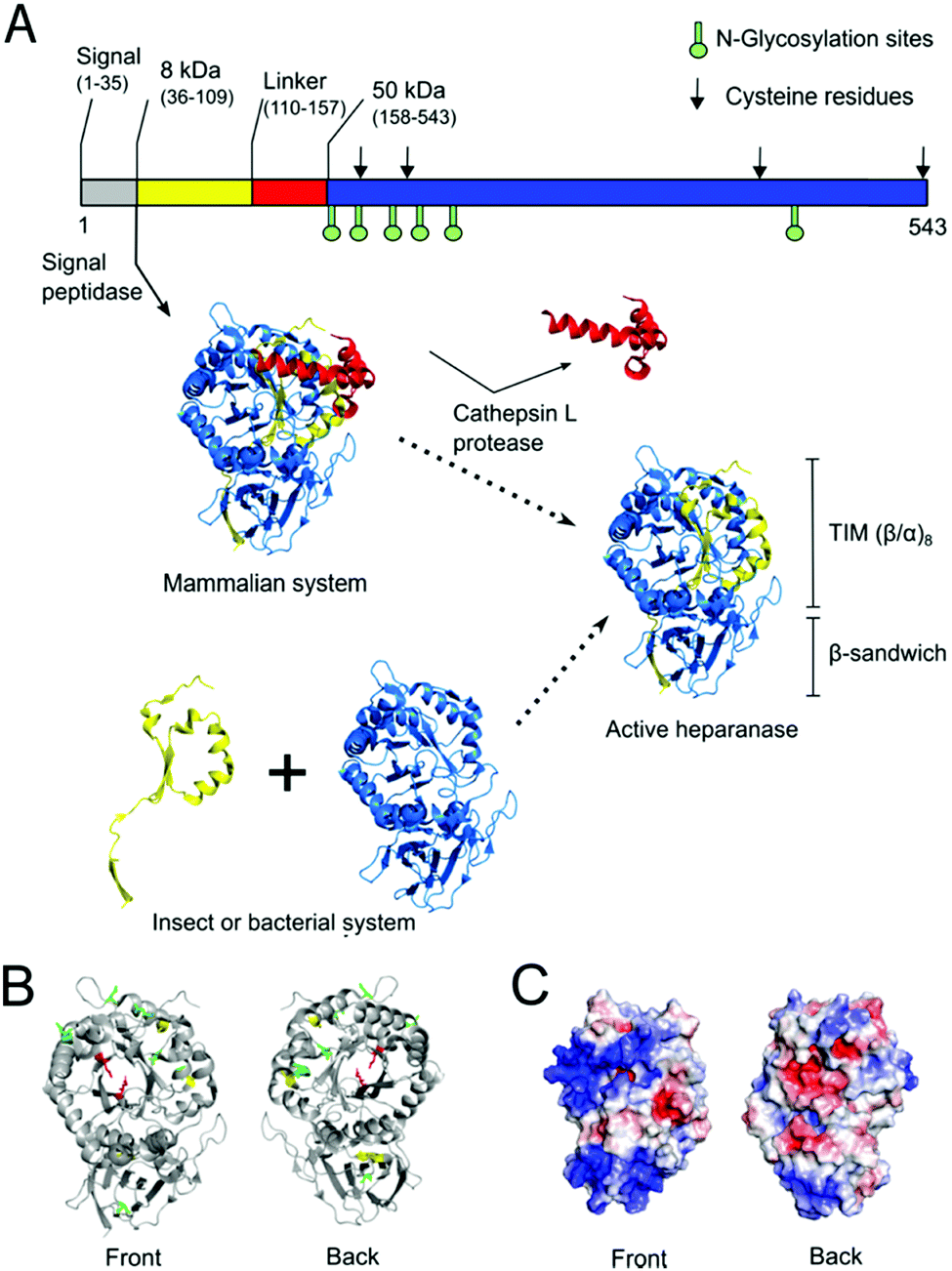 | ||
| Fig. 1 Native maturation and folding of HPSE in mammalian cells compared with heterologous production in insect or bacterial systems. (A) In mammalian cells, pre-proheparanase (Met1–Ile543) undergoes successive cleavage events of the N-terminal signal peptide (Met1–Ala35) and linker (Ser110–Gln157, red cartoon) segments to produce mature HPSE. The resulting heterodimer assembly of two subunits (8 kDa subunit (Gln36–Glu109, yellow cartoon) and 50 kDa subunit (Lys158–Ile543, blue cartoon)) consists of a TIM barrel (β/α)8 and β-sandwich fold. The sequence of HPSE is shown on top as a bar representation in which glycosylation sites are shown as green sticks and cysteine residues are indicated as black arrows. In non-mammalian systems active protein must instead be produced via co-expression of the two subunits.31 (B) Crystal structure of human HPSE expressed in an insect expression system (PDB ID: 5E8M) is shown as grey cartoons (bottom-left). Six N-glycosylation sites (Asn162, 178, 200, 217, 238, 459) are shown as green sticks and four cysteines (Cys179, 211, 437, 542) are shown as yellow sticks whereby two of them form a disulfide bond (Cys437-542) at the β-sandwich domain. Catalytic residues (Glu343, Glu225) at the TIM face are shown as red sticks. (C) Electrostatic potential surface was calculated using amino acid residues in the crystal structure by APBS (glycans were not included in the calculation). This shows two large positively-charged patches at the TIM domain and at the β-sandwich domain, which may promote aggregation in the nucleic acid rich micro environment.37 | ||
There are many experimental and computational methods that have been developed to improve enzyme function and stability, such as bioinformatics-based approaches like consensus design40,41 or ancestral sequence reconstruction,42,43 or forcefield-based approaches like Rosetta44 or FoldX.45 However, both approaches have limitations. The Protein Repair One Stop Shop (PROSS) algorithm combines forcefield-based Rosetta modelling and phylogenetic sequence information to create variants with improved stability.46 Here, we describe the use of PROSS to generate the first stable human HPSE variant to be expressed in E. coli. We demonstrate that it has significantly increased solubility, very similar catalytic activity and identical inhibition by competitive inhibitors, when compared to wild type human HPSE produced from mammalian cells. Our results are supported by an X-ray crystal structure and molecular dynamics simulations, which demonstrate that the introduced mutations stabilise HPSE with almost no effect on the three-dimensional structure or dynamics. This mutant HPSE should significantly reduce the costs and technical barriers to the development of HPSE inhibitors and its widespread study.
Results
Computational design of a soluble HPSE variant
We first tested bacterial expression of human HPSE (wild type) by cloning two subunits (8 kDa and 50 kDa, Tables S1 and S2, ESI†) into the dual expression vector (pETDuet-1). To optimize the chances of obtaining soluble, properly folded protein, we co-expressed the protein with chaperones (trigger factor and GroEL/GroES),47–49 and used E.coli Shuffle T7 Express50 cells, which allow disulfide bonds to form in the cytosol. Under these conditions, the 50 kDa subunit was totally insoluble, while the 8 kDa subunit was partially soluble (Fig S1, ESI†).Given that the molecular structure of HPSE has recently been solved,31,32 it is now an appropriate candidate to be engineered to allow expression in simple and inexpensive expression systems, such as E. coli. Recently the PROSS algorithm46 has demonstrated its utility in designing stable variants of challenging proteins for soluble and functional expression in bacteria.51–53 Unlike conventional consensus mutagenesis approaches, in which poorly conserved residues are mutated back to their consensus identity (from a multiple sequence alignment),54 PROSS combines this approach with computational modelling with Rosetta,55 generating a set variants, each containing multiple mutations that ideally act together to increase stability.46 We therefore used PROSS to redesign the insoluble 50 kDa subunit based on the crystal structure of the insect cell expressed human HPSE (PDB ID: 5E9C). The substrate binding site and the heterodimer interface residues were restrained to maintain function and preserve the interaction with the 8 kDa subunit. Seven variants with accumulated mutations were generated (Fig. S2, ESI†), which were subsequently synthesized and sub-cloned into multiple cloning site 2 of pETDuet-1 vector. The 8 kDa subunit with a N-terminal poly histidine tag was sub-cloned into multiple cloning site 1. All variants were tested (Fig. S3, ESI†) and the most soluble design, containing 26 amino acid substitutions was identified and purified (HPSE P6), using Ni2+-NTA, heparin and size exclusion chromatography. This resulted in pure, homogeneous, heterodimeric HPSE with a final yield of 4 mg from 1 L E. coli culture (Fig. 2). Notably, PROSS is not infallible; many of the designs did not produce soluble protein, which emphasises the need to test multiple different variants.
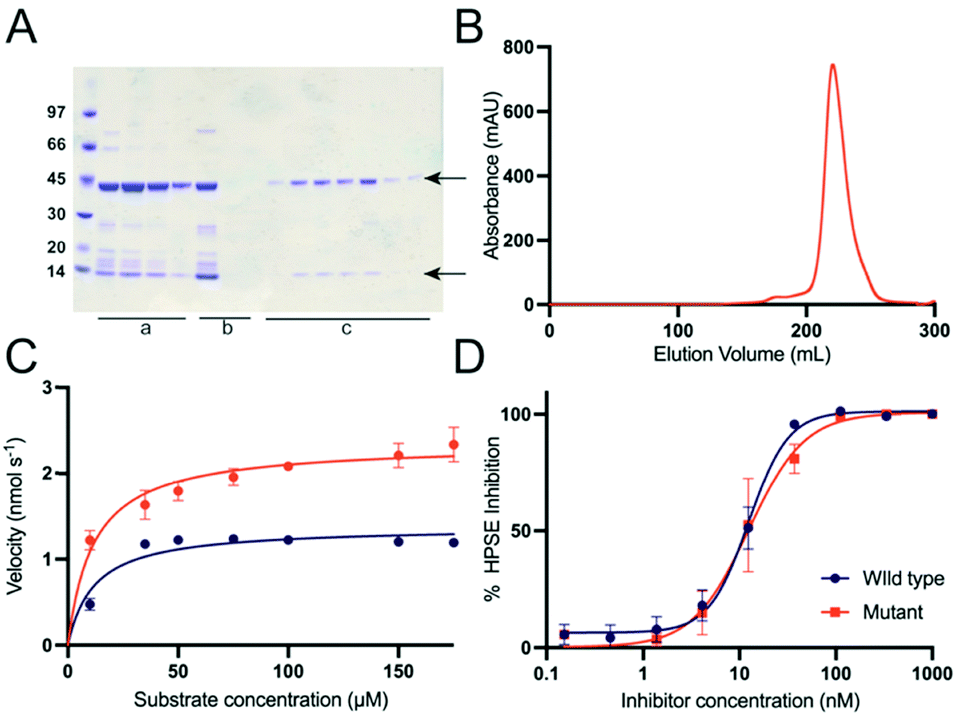 | ||
| Fig. 2 Expression, purification and activity of the successful HPSE P6 (A) Ni-NTA elution fractions (lanes a), heparin column flow in and flow through fractions (lanes b) and size exclusion elution fractions (lanes c). LMW protein marker (GE healthcare) is on the first lane. The sizes corresponding to the two subunits of the HPSE sit at 44 kDa, and 8 kDa, noting that the size of the large subunit is smaller than the previously reported value of 50 kDa due to the lack of glycosylation. (B) Size exclusion chromatography (HiLoad 26/600 Superdex 200 pg, GE Healthcare) shows one major peak corresponding to monomeric HPSE. (C) Kinetics HPSE P6 and HPSE WT, where catalytic rate (kcat) of HPSE P6 was 70% higher (kcat 2.94 ± 0.13) compared to (kcat 1.72 ± 0.07) for WT. The binding affinity (KM 11.6 ± 2.8 μM and 11.83 ± 2.7 μM respectively) was the same. (D) Pentosan polysulfate was used to compare the design with the human HPSE expressed in mammalian cell. This was measured using colorimetric method with fondaparinux. Error bars represent standard error from a minimum of three measurements. | ||
HPSE P6 exhibits wild-type like activity and response to inhibitors
Given the large number of mutations and loss of glycosylation sites, it was important to test whether these changes had any effect on the activity of the protein. The catalytic activity of purified HPSE P6 was tested by colorimetric assay using fondaparinux56 (Arixtra), a synthetic analogue of HS. Although, the catalytic rate (kcat) of HPSE P6 was slightly (less than 2-fold) higher (kcat = 2.94 ± 0.13 s−1) compared to HPSE WT (kcat = 1.72 ± 0.07 s−1), the binding affinity (KM = 11.6 ± 2.8 μM and 11.83 ± 2.7 μM respectively) was the same, demonstrating that the introduced mutations have no effect on the interaction between enzyme and substrate (Fig. 2C, Table 1). In fact, the slight increase in kcat is most likely due to the higher purity of the HPSE P6, compared to the commercially available HPSE WT. The loss of the six glycosylation sites no effect on the activity of the enzyme, suggesting these sites may be important for protein solubility in mammalian systems.| Parameter | HPSE WT | HPSE P6 |
|---|---|---|
| K M (μM) | 11.8 ± 2.7 | 11.6 ± 2.8 |
| k cat | 1.72 ± 0.07 | 2.94 ± 0.13 |
| IC50 (nM) | 12.5 ± 1.3 | 12.4 ± 2.5 |
Having established the enzyme kinetic parameters are comparable to HPSE WT, we then tested whether the HPSE P6 variant would interact identically with heparan sulfate mimetic inhibitors; in this case pentosan polysulfate57 (Fig. 2C and D). As with the enzyme kinetics, the inhibitory response to the model inhibitor pentosan polysulfate was near identical between HPSE WT and HPSE P6, with an IC50 of 12.46 ± 1.26 nM and 12.43 ± 2.47 nM, respectively (Fig. 2D).
HPSE P6 is thermostable and structurally isomorphous to HPSE WT
The thermal stability HPSE P6 was measured using circular dichroism (CD) by observing the loss of helicity at 222 nm over 20–90 °C. This revealed that HPSE P6 is somewhat thermostable, undergoing a transition to an unfolded state with a Tm value of 63.6 ± 0.19 °C, (Fig. 3A). This Tm value is similar to other engineered variants of human proteins produced through the use of PROSS,46,53 and significantly exceeds the normal temperature range that human proteins are exposed to (∼37 °C).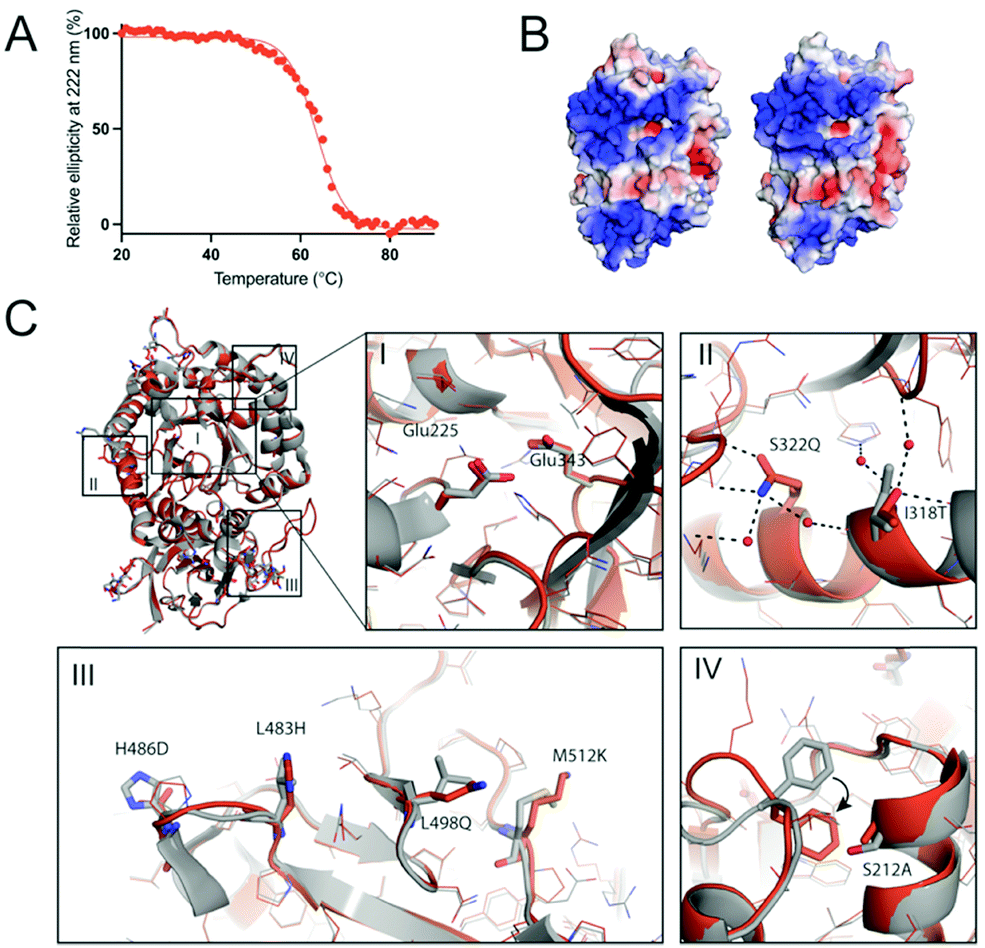 | ||
| Fig. 3 Thermal stability and structural insight of the designed HPSE. (A) The ellipticity at 222 nm was measured using circular dichroism over 20–90 °C, resulting in the melting temperature of 63.6 °C, similar to the values of other engineered proteins by the PROSS algorithm.46,51,53 (B) The front views of wild type (WT) and designed HPSE (HPSE P6) are calculated using APBS58 and visualized using PyMol. (C) The superimposed structures of wild type (grey) and HPSE P6 (orange, mutated residues are shown as sticks) are shown as overall (top-left) and detailed views (I–IV). Overall (top-left) and active site view (I) show closely aligned Cα backbones (RMSD of 0.645 Å) and the side chain conformations in the active site. Overall, the mutations reduce the hydrophobicity and increase polarity (II and III), to introduce new hydrogen bonds (black dotted lines, II) and to increase the hydrophobic packing (IV). The Phe258 side chain folds into the hydrophobic packing area (shown as black arrow) with a nearby mutation Ser212Ala causes a loss of an interaction with Thr257. PDB ID: 5E8M (WT), 7RG8 (HPSE P6). | ||
To understand how the 26 mutations in HPSE P6 result in enhanced protein folding and stability, we solved the crystal structure of HPSE P6 at 1.30 Å resolution (Table S3, ESI†). The protein crystallised in the P212121 space group within 1 day, forming rod shaped crystals. This compares to WT HPSE crystallising in the P21 space groups, after 1–3 days.
Despite 26 mutations, the crystal structure of HPSE P6 shows almost identical overall backbone and active site structures to HPSE WT expressed in an insect system (with the exception of the absence of any glycosylation). The Cα RMSD was 0.645 Å, with an alignment score of 0.017 (Fig. 3C).
Many subtle changes were observed due to the 26 mutations. Firstly, surface polarity, which is known to positively contribute to folding and stability,59 was increased by substitutions to surface leucine and alanine residues to more polar or smaller amino acids (e.g. Leu197Gly, Leu354Gly, Leu498Gln, Leu230Arg, Ala195Ser) (Fig. 3C). Secondly, additional stabilising interactions, including increased hydrogen bonding networks and hydrophobic packing were introduced, which stabilise the folded state. For example, new hydrogen bonding interactions were introduced by Leu483His, lle318Thr, Lys477Gln and Ser322Gln and new hydrophobic interactions were introduced by Ser530Ala, Ser292Ala and Arg307Leu in the partially solvent exposed areas. Interestingly, we observed indirect conformational change of Phe258 by Ser212Ala (Fig. 3C.iv). Finally, the disulfide bond (Cys437–Cys542) was possibly stabilized by introduction of proline at the position 540 (Ala540Pro) on the loop (Fig. S4, ESI†).
In previous applications of PROSS, it was noted that large positively charged patches, which could promote aggregation in the nucleic acid rich micro environment,37 were eliminated or reduced.60,61 Here, in the case of HPSE, the electrostatic potential surface of HPSE WT shows two large positive patches around the active site and the β-sandwich domain (Fig. 3B). For HPSE P6, the theoretical isoelectric point was the same as the wild type (pI 9.4), and the electrostatic surface potential shows that while one of the large positive patches around the β-sandwich domain was slightly diminished by two lysine mutations (Lys427Asp and Lys477Gln), the electrostatic potential around the active site at the TIM face was maintained as the area was constrained during the design process (Fig. 3B).
Molecular dynamic simulations to account stabilization
It has previously been shown that the dynamics and function of similar proteins can be very different despite ground state structures appearing very similar in terms of Cα RMSD.62 Crystallographic B-factors are commonly used to probe differences in the conformational flexibility of proteins within a crystal lattice, although this approach can be limited by the existence of crystallographic artifacts, whereby flexible regions on the protein surface could be stabilized by interactions with the lattice symmetry mates. Comparison between the B-factors of HPSE WT and HPSE P6 reveal the overall trend in terms of regions with high or low B-factors is conserved, although a decrease in the overall B factors of the P6 variant in the TIM (β/α)8 domain fluctuations, mostly around the surface loops of the active site (Fig. S4A, ESI†). However, this analysis is confounded by the higher resolution, lower Wilson B-factor and different crystal packing of the HPSE P6 variant.To complement the structural analysis, we also performed molecular dynamics simulations to examine the effects of these mutations on the conformational sampling and motions of the protein. To identify whether the dynamic range of HPSE P6 is the same as the HPSE WT, a total simulation time of 1 μs per protein was completed. Principal component analysis was conducted to visualize motions that represent the major fluctuations of the system. Principal components 1 and 2 of the HPSE WT and HPSE P6 (10.4% and 9.0%) overlap, demonstrating that the breathing motion of the active site is conserved (Fig. 4A). The third major component, which only contributes 6.5% of the total movement of the protein, shows slight differences, being comprised predominately of the movement of surface-exposed loops. No other principal component showed any difference between the two proteins (up to 20 components).
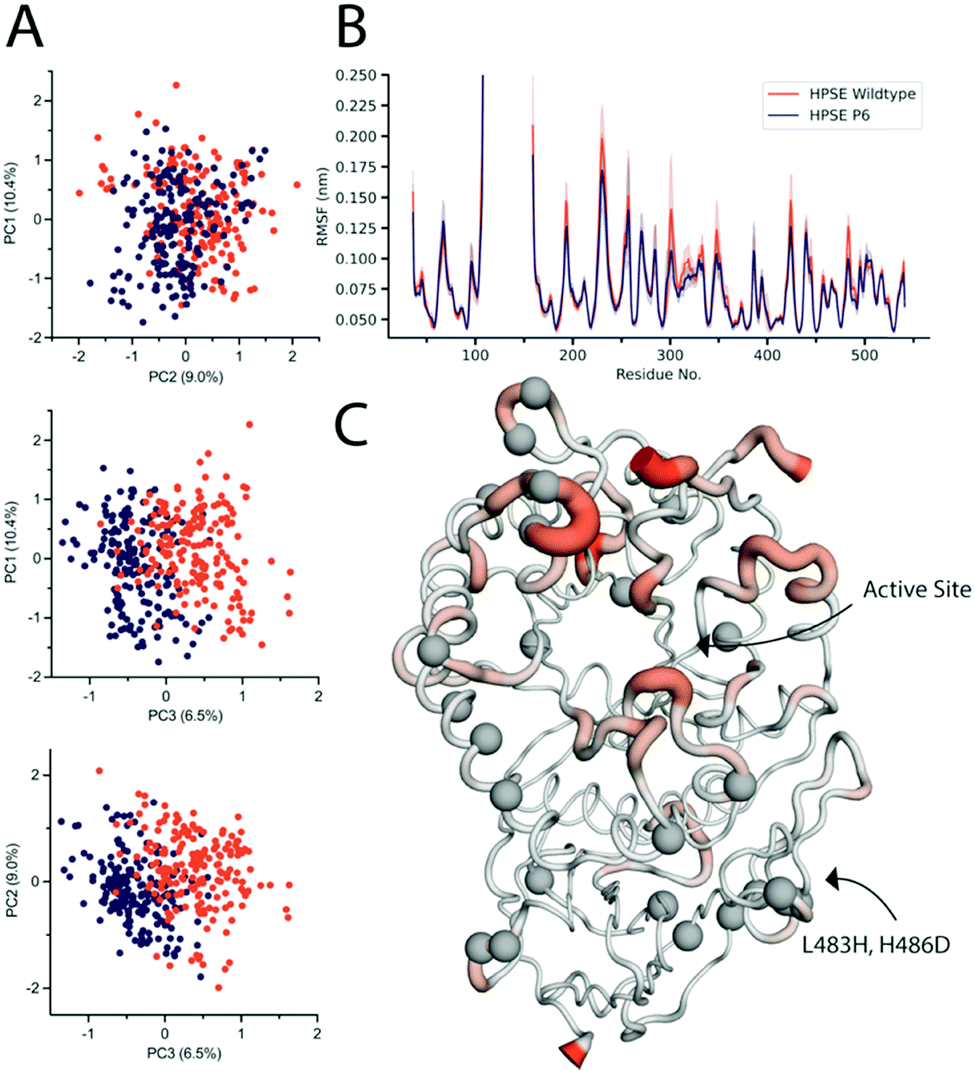 | ||
| Fig. 4 Molecular dynamic simulations of the wild type the pross design (A) PCA of the two proteins comparing PC1, PC2 and PC3, showing PC3 to have slight differences between the two proteins. (B) RMSF plot of the 95% CI of the wild type, and the mutant average RMSF showing that the mutant stays within the 95% CI, suggesting similar fluctuations. (C) HPSE P6 with average RMSF overlaid. Large RMSF is represented in orange putty, mostly seen around the active site. Mutations are represented as grey spheres. | ||
Root mean square fluctuations were also analysed to identify the displacement of amino acids throughout the course of the simulation (Fig. 4B). The average RMSF and their 95% confidence intervals, (where 95% of the residue's displacement occurs in that region) are overlaid for both proteins. This demonstrates that HPSE WT and P6 fluctuations overlap closely. There were very few differences overall, where the most consistent change is a decrease in magnitude of surface loops for HPSE P6 in comparison to WT simulations. Even though these residues have a very slightly decreased magnitude, the RMSF still has the same overall shape.
The only residues with RMSF values outside of the 95% CI are residues 488–495. This is a surface loop on the β-sandwich domain with two introduced mutations; Leu483His and His486Asp. These mutations allow an increase of hydrogen bonding causing a slight rigidification of this loop (Fig. 4C). Overall, the conservation of the protein dynamics despite 26 mutations is striking and unexpected. Indeed, the great majority of these mutations (identified as grey spheres in Fig. 4C) do not cause any significant difference on the dynamics of the protein. This analysis is also fully consistent with the functional data, which revealed almost to effect of the mutations on activity or inhibition.
Discussion
Despite the widespread interest in HPSE as an important enzyme in human physiology and a drug target, the difficulty related to obtaining large quantities of pure recombinant protein has limited the ability of many groups to study the protein. Bacterial expression systems, such as E. coli, are widely accessible, allow protein to be expressed in high yields and at low cost. While prokaryotic HPSE expression methods have been reported,33,34 none have been repeated in the literature or have been widely adopted. Here, a stable version of human HPSE has been computationally designed, which allowed the mature heterodimeric enzyme to be expressed at reasonable yield in soluble form in E.coli. The subsequent characterization of this designed version (HPSE P6) showed the enzyme to behave essentially identically to HPSE WT in terms of its interactions with substrates (Table 1) and inhibitors (Fig. 1). Thus, this computationally designed HPSE P6 variant should be a useful surrogate for the wild-type enzyme in structural biology, inhibitor screening and kinetic analyses.It is notable that despite 26 mutations, the enzyme is essentially structurally isomorphous to the wild-type, with no significant changes to the C-α backbone or side chain rotamer sampling, especially in the vicinity of the active site. Additionally, the dynamics of the enzyme were also largely identical to the wild-type enzyme, suggesting that there were no significant changes to the relative stabilities of different conformational substates. This reinforces the functional neutrality of many mutations and the power of bioinformatics inspired approaches such as consensus design and PROSS; these mutations were acquired through phylogenetic analysis i.e., they are known to be tolerated in related enzymes. Indeed, while their individual effects might be small, the summation of the effects can become considerable. However, the route to HPSE P6 was not simple or trivial; P6 was the only design of the seven that we tested that was effective. Thus, while the combinatorial effects of the mutations can be powerful, the unpredictable effects of the mutations, and their epistatic interactions, make it imperative that a range of designs are trialled.
Our structural analysis of HPSE P6 shows that many of the mutations appear to have effects that can be rationalised in terms of our understanding of how proteins fold: increasing surface polarity, forming additional non-covalent interactions such as hydrogen bonds, increased packing within the hydrophobic core, etc. The lack of major structural changes, such as strong salt bridges or significant changes to internal cavities, which are characteristic of rational or computationally designed stabilising mutations, meant that the structural dynamics of the protein, and thus its catalytic function, was largely unperturbed. This study is thus an interesting example of protein stabilisation: on the one hand, 26 mutations could be considered to be a large number of mutations, but the counter argument is that 26 functionally neutral mutations that have almost no effect on the structure and dynamics is in fact a very conservative method for stabilizing a protein, in comparison to a smaller number of mutations that might have a larger effect on the structure, dynamics and function of the enzyme.
Experimental
Stability design
Chain A of the crystal structure of the ligand bound human HPSE (PDB ID: 5E9C) was submitted to the PROSS stability design algorithm46 on the web server (http://pross.weizmann.ac.il), with constrained residues, which have contacts with the ligand (Dp4) and with the chain B. This generated 7 mutants.Cloning
The linear 8 kDa (Gln36–Glu109) and 50 kDa (Lys158–Ile543) subunits of the human HPSE were E. coli codon optimized and synthesized by IDT (Australia). The seven PROSS designs were E. coli codon optimized and synthesized by Twist bioscience. The 8 kDa subunit was amplified and sub-cloned into the multiple cloning site 1 of the linearized pETDuet-1 vector (Novagen) through the BamHI and NotI restriction sites (Fast Digest,Thermo) by Gibson one-step isothermal assembly.63 The resulting plasmid DNA was linearized using NdeI and XhoI restriction enzymes (Fast Digest, Thermo) and designs were inserted into the multiple cloning site 2 by Gibson assembly.63 The ligated DNA was transformed to E. coli TOP10 cells and the plasmid DNA was extracted and sent to Garvan Institute (Australia) for Sanger sequencing to confirm the sequences.Protein expression and purification
The wild type and the 7 designs were transformed in E. coli Shuffle T7 Express cells (NEB), together with different combinations of chaperones in a pACYC vector and spread on an Agar plate with ampicillin and chloramphenicol. 1% overnight seed culture from a single colony was inoculated into 1 L of LB medium supplemented with ampicillin (100 mg L−1) and chloramphenicol (34 mg L−1), then incubated at 37 °C for 5 hours. Overexpression was induced by adding IPTG to a final concentration of 0.05 mM and the culture was further incubated for 3 hours at 37 °C. The cell pellets were resuspended in buffer A (20 mM HEPES pH 8, 300 mM NaCl, 5 mM β-mercaptoethanol, 10% glycerol, 0.05% Tween, 20 mM Imidazole) with Turbonuclease (Sigma) and lysed by sonication (Omni Sonic Ruptor 400 Ultrasonic homogenizer). The lysate was filtered (0.45 μm) and loaded onto Ni-NTA column (GE healthcare) and eluted with 100% buffer B (buffer A + 500 mM Imidazole). The peak eluent was diluted 5 times with buffer C (20 mM HEPES pH 7.4, 200 mM NaCl, 1 mM DTT, 10% glycerol, 0.05% Tween20) and loaded to heparin affinity column (GE healthcare) and eluted with 100% buffer D (buffer C + 1.5 M NaCl). The peak eluent was loaded onto a size exclusion column (HiLoad 26/600 Superdex 200 pg, GE healthcare) and eluted in a buffer E (20 mM HEPES pH 7.4, 200 mM NaCl, 10% glycerol, 1 mM DTT, 0.05% Tween20). The final concentration of the monomeric heparinase from the gel filtration was estimated by absorbance at 280 nm using NanoDrop One (Thermo) and the yield was more than 2 mg per litre of LB culture.Colorimetric assay using fondaparinux
Assays were conducted using the colorimetric assay designed by Hammond et al.56 Bovine serum albumin-coated 96 well microplates were used for all assays and were prepared by incubation of the plates with 1% BSA dissolved in phosphate-buffered saline (PBS) with 0.05% Tween-20 (PBST) at 37 °C for 75 minutes. The plates were then washed three times with PBST, dried and stored at 4 °C. Assay mixtures contain 40 mM sodium acetate buffer (pH 5.0), 0.8 nM HPSE in 0.01% Tween 20 sodium acetate buffer and 100 mM fondaparinux (GlaxoSmithKline) with or without increasing concentrations of inhibitor. Plates were incubated at 37 °C for 2–20 hours before the reaction was stopped with 100 μL of 1.69 mM 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3d-benzene disulfonate (WST-1) in 0.1 M NaOH. The plates were resealed and developed at 60 °C for 60 minutes, and the absorbance was measured at 584 nm. Kinetics were carried out with a standard curve constructed with D-galactose as the reducing sugar standard, prepared in the same buffer and volume over the range of 0–2 μM. All curve fitting to calculate IC50 values and Michaelis–Menten constants, was done using GraphPad Prism software (version 8.1).Circular dichroism analysis
The size exclusion fraction was directly used to measure the CD using the Chirascan CD spectrometer (Applied Photophysics). The thermal stability of the protein (0.15 mg mL−1) was measured in a range of temperatures 20–90 °C by monitoring the ellipticity at 222 nm using a cuvette with 1 mm path length. Data analysis was performed using GraphPad Prism, within which the mid-point of the melting curve was calculated using Boltzmann sigmoid equation.Structure determination and refinement
Well diffracted single crystals were obtained by the hanging-drop vapor-diffusion method at 18 °C by combining the protein (6–8 mg mL−1) and the well solution (1.9 M (NH4)2SO4) with a ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 μL. Crystals appeared within a week and continued to grow for 1–2 months. The crystal was cryoprotected with additional 30% glycerol to the mother liquor before flash freezing in liquid nitrogen. Crystallographic data were collected at 100 K at the Australian Synchrotron (MX2,64 0.9537 Å). The obtained diffraction data were indexed and integrated with XDS.65 Resolution estimation and data truncation were performed using aimless program in CCP466 on the basis of the datasets overall half-dataset correlation, a CC1/2 value of 0.3.67 All structures were solved by molecular replacement using the Molrep program in CCP466 using the structure deposited under PDB accession code 5E9M as a starting model. The models were refined using phenix.refine,68 and the model was subsequently optimized by iterative model building with the program COOT v0.8.69 The alternative conformations were modelled based on mFo–DFc density and the occupancies and B-factors were determined using phenix.refine.68 The structures were then evaluated using MolProbity70 in Phenix. Details of the refinement statistics were produced by Phenix v1.1771 and summarized in Table S3 (ESI†). The structures were visualized and analysed using PyMol v2.372 or Maestro,73 whereby APBS58 program in PyMol was used to calculated the electrostatic potential and protein alignment program in Maestro was used to calculate the Cα RMSD.
:1.5 μL. Crystals appeared within a week and continued to grow for 1–2 months. The crystal was cryoprotected with additional 30% glycerol to the mother liquor before flash freezing in liquid nitrogen. Crystallographic data were collected at 100 K at the Australian Synchrotron (MX2,64 0.9537 Å). The obtained diffraction data were indexed and integrated with XDS.65 Resolution estimation and data truncation were performed using aimless program in CCP466 on the basis of the datasets overall half-dataset correlation, a CC1/2 value of 0.3.67 All structures were solved by molecular replacement using the Molrep program in CCP466 using the structure deposited under PDB accession code 5E9M as a starting model. The models were refined using phenix.refine,68 and the model was subsequently optimized by iterative model building with the program COOT v0.8.69 The alternative conformations were modelled based on mFo–DFc density and the occupancies and B-factors were determined using phenix.refine.68 The structures were then evaluated using MolProbity70 in Phenix. Details of the refinement statistics were produced by Phenix v1.1771 and summarized in Table S3 (ESI†). The structures were visualized and analysed using PyMol v2.372 or Maestro,73 whereby APBS58 program in PyMol was used to calculated the electrostatic potential and protein alignment program in Maestro was used to calculate the Cα RMSD.
Molecular dynamics
Molecular dynamic simulations were performed using the GROMACS 2018.8 engine with parameters from the Charm22* force field.74,75 All chain termini were capped with neutral acetyl or methylamide groups. Protonation states were assigned with the PDB2PQR server for pH 5.0.58 Completed structures were solvated with a TIP3P water model76 using a rhombic dodecahedron simulation box with a minimum distance of 12 Å between the protein and simulation box, followed by the addition of 200 mM NaCl to the aqueous phase and sufficient ions to neutralise the system charge. Simulation systems of WT and PROSS 6 were relaxed using the standard steepest descent minimization using at least 10000 steps before being equilibrated for 1 ns in the isothermal–isobaric (NPT) ensemble to stabilize the system. Ten replicates of each system were simulated for 100 ns under NPT. Periodic boundary conditions were used, and long-range electrostatics were calculated using the particle-mesh Ewald method with a cutoff of 1.2 nm.77 Non-bonded interactions were evaluated using a Verlet cut-off scheme. The temperature in all simulations was set to 300 K and controlled via the Bussi–Donadio–Parrinello stochastic velocity rescaling thermostat;78 the initial velocities of all particles were pseudo-randomly generated. Pressure coupling was handled with the Berendsen barostat during equilibration and the Parrinello–Rahman barostat for production.79,80 The LINCS (Linear Constraint Solver) algorithm was used to constrain bonds involving hydrogen in conjunction with an integration time step of 2 fs.81 Constraints were applied to the starting configuration of the production run. Analyses of simulations were preformed using the tools provided in the GROMACS package. Data was collected from the last 90 ns of each production simulation, as RMSF had stabilised by this time.
Principal component analysis
Principal component analysis was performed using the MDTraj python library and the scikit-learn machine learning tool.82,83 Using the aligned and concatenated trajectory, a merged dataset was created, from which the WT and P6 systems were projected. Data was plotted in Graphpad prism.Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under accession code PDB 7RG8.Conclusions
This study describes the production of a new variant of HPSE, which is functionally identical to the wild-type protein in terms of activity, inhibition, structure and dynamics that is easily expressed in E. coli and crystallises within a day, yielding high resolution crystals. This protein should make the study of HPSE function and the development of inhibitors significantly easier and less expensive. It is notable that the large number of mutations in HPSE P6 were functionally neutral. This contributes to a growing understanding of the relationship between protein sequence and folding, where evolutionarily conserved and functionally neutral consensus-like mutations can be understood to significantly affect the efficiency of protein folding and expression and protein thermostability.Author contributions
C. W., N. H. and C. J. J. conceived the study and analysed data. C. W designed variants. N. H. conducted cloning and expression trials. C. W. expressed, purified and crystallised HPSE P6. C. W and N. H. processed crystallography data. N. H. analysed crystal structures. C. W. and J. A. M. performed and analysed molecular dynamics simulations and principal component analysis. C. W., N. H. and C. J. J. wrote the manuscript. All authors provided intellectual input and edited and approved the final manuscript.Conflicts of interest
CJ has received funding from Beta Therapeutics to work on heparanase inhibitors.Acknowledgements
We acknowledge the ARC Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012), the ARC Centre of Excellence in Synthetic Biology (CE200100029) and an ARC Linkage Grant (LP160101552). We thank the staff of the MX2 beamline at the Australian Synchrotron, part of ANSTO, which made use of the Australian Cancer Research Foundation (ACRF) detector. The table of contents entry was created with http://BioRender.com.References
- S. Rivara, F. M. Milazzo and G. Giannini, Future Med. Chem., 2016, 8, 647–680 CrossRef CAS PubMed.
- J. R. Bishop, M. Schuksz and J. D. Esko, Nature, 2007, 446, 1030–1037 CrossRef CAS PubMed.
- D. Aviezer and A. Yayon, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12173–12177 CrossRef CAS PubMed.
- T. F. Zioncheck, L. Richardson, J. Liu, L. Chang, K. L. King, G. L. Bennett, P. Fugedi, S. M. Chamow, R. H. Schwall and R. J. Stack, J. Biol. Chem., 1995, 270, 16871–16878 CrossRef CAS PubMed.
- A. Amara, O. Lorthioir, A. Valenzuela, A. Magerus, M. Thelen, M. Montes, J. L. Virelizier, M. Delepierre, F. Baleux, H. Lortat-Jacob and F. Arenzana-Seisdedos, J. Biol. Chem., 1999, 274, 23916–23925 CrossRef CAS PubMed.
- S. Eisenberg, E. Sehayek, T. Olivecrona and I. Vlodavsky, J. Clin. Invest., 1992, 90, 2013–2021 CrossRef CAS PubMed.
- I. Vlodavsky, Y. Friedmann, M. Elkin, H. Aingorn, R. Atzmon, R. Ishai-Michaeli, M. Bitan, O. Pappo, T. Peretz, I. Michal, L. Spector and I. Pecker, Nat. Med., 1999, 5, 793–802 CrossRef CAS PubMed.
- M. D. Hulett, C. Freeman, B. J. Hamdorf, R. T. Baker, M. J. Harris and C. R. Parish, Nat. Med., 1999, 5, 803 CrossRef CAS PubMed.
- P. H. Kussie, J. D. Hulmes, D. L. Ludwig, S. Patel, E. C. Navarro, A. P. Seddon, N. A. Giorgio and P. Bohlen, Biochem. Biophys. Res. Commun., 1999, 261, 183–187 CrossRef CAS PubMed.
- M. Toyoshima and M. Nakajima, J. Biol. Chem., 1999, 274, 24153–24160 CrossRef CAS PubMed.
- R. Goshen, A. A. Hochberg, G. Korner, E. Levy, R. Ishai-Michaeli, M. Elkin, N. De Groot and I. Vlodavsky, Mol. Hum. Reprod., 1996, 2, 679–684 CrossRef CAS PubMed.
- L. Gutter-Kapon, D. Alishekevitz, Y. Shaked, J. P. Li, A. Aronheim, N. Ilan and I. Vlodavsky, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7808–E7817 CrossRef CAS PubMed.
- I. Vlodavsky, A. Eldor, A. Haimovitz-Friedman and Y. Matzner, Invasion and metastasis.
- B. Buijsers, C. Yanginlar, A. de Nooijer, I. Grondman, M. L. Maciej-Hulme, I. Jonkman, N. A. F. Janssen, N. Rother, M. de Graaf, P. Pickkers, M. Kox, L. A. B. Joosten, T. Nijenhuis, M. G. Netea, L. Hilbrands, F. L. van de Veerdonk, R. Duivenvoorden, Q. de Mast and J. van der Vlag, Front. Immunol., 2020, 11 DOI:10.3389/FIMMU.2020.575047.
- I. Vlodavsky, Z. Fuks, M. Bar-Ner, Y. Ariav and V. Schirrmacher, Cancer Res., 1983, 43(6), 2704–2711 CAS.
- M. Nakajima, T. Irimura, D. Di Ferrante, N. Di Ferrante and G. L. Nicolson, Science, 1983, 220, 611–613 CrossRef CAS PubMed.
- M. Waterman, O. Ben-Izhak, R. Eliakim, G. Groisman, I. Vlodavsky and N. Ilan, Mod. Pathol., 2007, 20, 8–14 CrossRef CAS PubMed.
- I. Vlodavsky, R. V. Iozzo and R. D. Sanderson, Matrix Biol., 2013, 32, 220–222 CrossRef CAS PubMed.
- I. Vlodavsky, P. Beckhove, I. Lerner, C. Pisano, A. Meirovitz, N. Ilan and M. Elkin, Cancer Microenviron., 2012, 5, 115–132 CrossRef CAS PubMed.
- C. P. Baburajeev, C. D. Mohan, S. Rangappa, D. J. Mason, J. E. Fuchs, A. Bender, U. Barash, I. Vlodavsky, Basappa and K. S. Rangappa, BMC Cancer, 2017, 17, 235 CrossRef CAS PubMed.
- E. A. McKenzie, Br. J. Pharmacol., 2007, 151, 1–14 CrossRef CAS PubMed.
- I. Vlodavsky, O. Goldshmidt, E. Zcharia, R. Atzmon, Z. Rangini-Guatta, M. Elkin, T. Peretz and Y. Friedmann, Semin. Cancer Biol., 2002, 12, 121–129 CrossRef CAS PubMed.
- F. Sanchez-Muñoz, A. Dominguez-Lopez and J. K. Yamamoto-Furusho, World J. Gastroenterol. WJG, 2008, 14, 4280 CrossRef PubMed.
- L. M. Coussens, B. Fingleton and L. M. Matrisian, Science, 2002, 295, 2387–2392 CrossRef CAS PubMed.
- E. Zcharia, J. Jia, X. Zhang, L. Baraz, U. Lindahl, T. Peretz, I. Vlodavsky and J.-P. Li, PLoS One, 2009, 4, e5181 CrossRef PubMed.
- C. D. Mohan, S. Hari, H. D. Preetham, S. Rangappa, U. Barash, N. Ilan, S. C. Nayak, V. K. Gupta, Basappa, I. Vlodavsky and K. S. Rangappa, iScience, 2019, 15, 360–390 CrossRef CAS PubMed.
- V. C. Ramani, F. Zhan, J. He, P. Barbieri, A. Noseda, G. Tricot and R. D. Sanderson, Oncotarget, 2016, 7, 1598 CrossRef PubMed.
- L. Jia and S. Ma, Eur. J. Med. Chem., 2016, 121, 209–220 CrossRef CAS PubMed.
- V. Masola, G. Zaza, G. Gambaro, M. Franchi and M. Onisto, Semin. Cancer Biol., 2020, 62, 86–98 CrossRef CAS PubMed.
- M. Toyoshima and M. Nakajima, J. Biol. Chem., 1999, 274, 24153–24160 CrossRef CAS PubMed.
- L. Wu, C. M. Viola, A. M. Brzozowski and G. J. Davies, Nat. Struct. Mol. Biol., 2015, 22, 1016–1022 CrossRef CAS PubMed.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C.-L. Kuo, M. Artola, W. Dai, C. van Elk, M. van Eijk, G. A. van der Marel, J. D. C. Codée, B. I. Florea, J. M. F. G. Aerts, H. S. Overkleeft and G. J. Davies, Nat. Chem. Biol., 2017, 13, 867–873 CrossRef CAS PubMed.
- S. Winkler, D. Schweiger, Z. Wei, E. Rajkovic and A. J. Kungl, Carbohydr. Res., 2014, 389, 72–77 CrossRef CAS PubMed.
- A. Pennacchio, A. Capo, S. Caira, A. Tramice, A. Varriale, M. Staiano and S. D’Auria, Biotechnol. Appl. Biochem., 2018, 65, 89–98 CrossRef CAS PubMed.
- J. Warwicker, S. Charonis and R. A. Curtis, Mol. Pharm., 2013, 11, 294–303 CrossRef PubMed.
- R. M. Kramer, V. R. Shende, N. Motl, C. N. Pace and J. M. Scholtz, Biophys. J., 2012, 102, 1907–1915 CrossRef CAS PubMed.
- P. Chan, R. A. Curtis and J. Warwicker, Sci. Rep., 2013, 3, 3333 CrossRef PubMed.
- S. Simizu, K. Ishida, M. K. Wierzba and H. Osada, J. Biol. Chem., 2004, 279, 2697–2703 CrossRef CAS PubMed.
- F. Levy-Adam, H. Q. Miao, R. L. Heinrikson, I. Vlodavsky and N. Ilan, Biochem. Biophys. Res. Commun., 2003, 308, 885–891 CrossRef CAS PubMed.
- D. Li, A. M. Damry, J. R. Petrie, T. Vanhercke, S. P. Singh and C. J. Jackson, Biochemistry, 2020, 59, 1398–1409 CrossRef CAS PubMed.
- N. Amin, A. D. Liu, S. Ramer, W. Aehle, D. Meijer, M. Metin, S. Wong, P. Gualfetti and V. Schellenberger, Protein Eng., Des. Sel., 2004, 17, 787–793 CrossRef CAS PubMed.
- J. H. Whitfield, W. H. Zhang, M. K. Herde, B. E. Clifton, J. Radziejewski, H. Janovjak, C. Henneberger and C. J. Jackson, Protein Sci., 2015, 24, 1412 CrossRef CAS PubMed.
- M. A. Spence, J. A. Kaczmarski, J. W. Saunders and C. J. Jackson, Curr. Opin. Struct. Biol., 2021, 69, 131–141 CrossRef CAS PubMed.
- B. Borgo and J. J. Havranek, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1494–1499 CrossRef CAS PubMed.
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33(2), 382–388 CrossRef PubMed.
- A. Goldenzweig, M. Goldsmith, S. E. Hill, O. Gertman, P. Laurino, Y. Ashani, O. Dym, T. Unger, S. Albeck, J. Prilusky, R. L. Lieberman, A. Aharoni, I. Silman, J. L. Sussman, D. S. Tawfik and S. J. Fleishman, Mol. Cell, 2016, 63, 337–346 CrossRef CAS PubMed.
- J. W. Lamppa, S. A. Tanyos and K. E. Griswold, J. Biotechnol., 2013, 164, 1–8 CrossRef CAS PubMed.
- F. Georgescauld, K. Popova, A. J. Gupta, A. Bracher, J. R. Engen, M. Hayer-Hartl and F. U. Hartl, Cell, 2014, 157, 922–934 CrossRef CAS PubMed.
- S. Haldar, R. Tapia-Rojo, E. C. Eckels, J. Valle-Orero and J. M. Fernandez, Nat. Commun., 2017, 8, 668 CrossRef PubMed.
- J. Lobstein, C. A. Emrich, C. Jeans, M. Faulkner, P. Riggs and M. Berkmen, Microb. Cell Fact., 2012, 11, 56 CrossRef CAS PubMed.
- X. Brazzolotto, A. Igert, V. Guillon, G. Santoni and F. Nachon, Molecules, 2017, 22, 1828 CrossRef PubMed.
- O. Khersonsky, R. Lipsh, Z. Avizemer, Y. Ashani, M. Goldsmith, H. Leader, O. Dym, S. Rogotner, D. L. Trudeau, J. Prilusky, P. Amengual-Rigo, V. Guallar, D. S. Tawfik and S. J. Fleishman, Mol. Cell, 2018, 72, 178–186.e5 CrossRef CAS PubMed.
- I. Campeotto, A. Goldenzweig, J. Davey, L. Barfod, J. M. Marshall, S. E. Silk, K. E. Wright, S. J. Draper, M. K. Higgins and S. J. Fleishman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 998–1002 CrossRef CAS PubMed.
- M. Sternke, K. W. Tripp and D. Barrick, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11275–11284 CrossRef CAS PubMed.
- T. A. Whitehead, A. Chevalier, Y. Song, C. Dreyfus, S. J. Fleishman, C. De Mattos, C. A. Myers, H. Kamisetty, P. Blair, I. A. Wilson and D. Baker, Nat. Biotechnol., 2012, 30, 543–548 CrossRef CAS PubMed.
- E. Hammond, C. P. Li and V. Ferro, Anal. Biochem., 2010, 396, 112–116 CrossRef CAS PubMed.
- C. Freeman and R. C. Parish, Biochem. J., 1998, 330, 1341–1350 CrossRef CAS PubMed.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen and K. Liles, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
- G. Raghunathan, S. Sokalingam, N. Soundrarajan, B. Madan, G. Munussami and S.-G. Lee, Mol. BioSyst., 2013, 9, 2379–2389 RSC.
- I. Campeotto, A. Goldenzweig, J. Davey, L. Barfod, J. M. Marshall, S. E. Silk, K. E. Wright, S. J. Draper, M. K. Higgins and S. J. Fleishman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 998–1002 CrossRef CAS PubMed.
- A. R. Lambert, J. P. Hallinan, R. Werther, D. Głów and B. L. Stoddard, Structure, 2020, 28, 760–775.e8 CrossRef CAS PubMed.
- E. Campbell, M. Kaltenbach, G. J. Correy, P. D. Carr, B. T. Porebski, E. K. Livingstone, L. Afriat-Jurnou, A. M. Buckle, M. Weik, F. Hollfelder, N. Tokuriki and C. J. Jackson, Nat. Chem. Biol., 2016, 12, 944–950 CrossRef CAS PubMed.
- D. G. Gibson, in Synthetic Biology, Part B, ed. C. B. T.-M. in E. Voigt, Academic Press, 2011, vol. 498, pp. 349–361 Search PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. A. Karplus and K. Diederichs, Science, 2012, 336, 1030–1033 CrossRef CAS PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty, M. Mustyakimov, T. C. Terwilliger, A. Urzhumtsev, P. H. Zwart and P. D. Adams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 352–367 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- P. V. Afonine, R. W. Grosse-Kunstleve, V. B. Chen, J. J. Headd, N. W. Moriarty, J. S. Richardson, D. C. Richardson, A. Urzhumtsev, P. H. Zwart and P. D. Adams, J. Appl. Crystallogr., 2010, 43, 669–676 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC Search PubMed.
- Schrodinger Release 2020-3, Maestro,Schrödinger, LLC, New York, NY, 2020.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. Piana, K. Lindorff-Larsen and D. E. Shaw, Biophys. J., 2011, 100, L47–L49 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- M. Parrinello and A. Rahman, Phys. Rev. Lett., 1980, 45, 1196–1199 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- R. T. McGibbon, K. A. Beauchamp, M. P. Harrigan, C. Klein, J. M. Swails, C. X. Hernández, C. R. Schwantes, L. P. Wang, T. J. Lane and V. S. Pande, Biophys. J., 2015, 109, 1528–1532 CrossRef CAS PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00239b |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |