
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polysaccharide nanoparticles: from fabrication to applications
Alexander
Plucinski†
,
Zan
Lyu†
 and
Bernhard V. K. J.
Schmidt
*
and
Bernhard V. K. J.
Schmidt
*
School of Chemistry, University of Glasgow, G12 8QQ Glasgow, UK. E-mail: bernhard.schmidt@glasgow.ac.uk
First published on 24th April 2021
Abstract
Polysaccharides have attracted considerable attention in a broad range of applications in recent years, which is due to their remarkable features such as biocompatibility, biodegradability, renewable origin, and facile modification. Considerable research efforts have been focused on developing polysaccharide nanoparticles and to promote their applications in various areas and biomedicine in particular. The present review highlights the properties of common polysaccharides used in nanoparticle formation as well as strategies to fabricate polysaccharide nanoparticles. Furthermore, the combination of polysaccharide nanoparticles and polymers is presented and brought into the context of applications. Finally, applications of polysaccharide nanoparticles as nano-delivery system, Pickering emulsion stabilisers, and material reinforcing agent in the fields of nanomedicine, cosmetics, and food system are highlighted. Moreover, this review describes and critically discusses present limitations and drawbacks in the preparation and use of polysaccharide nanoparticles, revealing directions to develop polysaccharide nanoparticles for further utilisation in various applications in the future.
 Alexander Plucinski | Alexander Plucinski has obtained his chemistry bachelor degree in 2016 and master degree in 2018 at the University of Potsdam. Since 2019, he is a PhD student under the supervision of Dr Bernhard V. K. J. Schmidt at the University of Glasgow. His research focusses on the self-assembly and phase separation of hydrophilic polymers in aqueous environment. |
 Zan Lyu | Zan Lyu received his dual BSc degree from Changzhou University, China and St. Francis Xavier University, Canada in 2019. In 2020, he received his master's degree in Chemistry at the School of Chemistry, University of Glasgow. |
 Bernhard V. K. J. Schmidt | Bernhard V. K. J. Schmidt completed his PhD in 2013 with Prof. Barner-Kowollik at the Karlsruhe Institute of Technology and a PostDoc with Prof. Hawker at the University of California, Santa Barbara. Afterwards he joined the department of Prof. Antonietti at the Max Planck Institute of Colloids and Interfaces as a Group Leader and finished his Habilitation in 2020. Since 2019, he is Lecturer in Synthetic Polymer Chemistry at the University of Glasgow. His research focusses on block copolymer self-assembly, metal–organic framework/polymer hybrids and carbon nitride/polymer hybrid materials. |
1. Introduction
Hydrophilic polymers and water-based polymer systems have gained significant attention over the past decades due to their applications in various interdisciplinary areas e.g. drug-delivery,1 tissue-engineering,2 catalysis,3 membrane technology,4 aggregate formation5,6 or phase separations like aqueous two-phase systems (ATPS).7 Especially, nanoparticle (NP) based technology8 is a significant area in biomedical,9 pharmaceutical,9 cosmetic/cosmeceutical10,11 and food industry.12 As such, NPs have been prepared and utilised in the areas such as targeted substance delivery, emulsion stabilisation, imaging on the molecular scale for diagnostic applications, and material reinforcement.13–18 Moreover, emulsion stabilisers based on NPs have been extensively studied for application as cosmetic formulations,19 in oil-in-water (O/W) emulsions20,21 and in water-in-water (W/W) emulsions.22,23 The transformation of traditional food and agriculture sectors is also significantly promoted by nanotechnology. In addition to transporting bioactive substances like nutrients, numerous novel applications such as smart and active packaging, nanosensors, nanopesticides and nanofertilisers have been invented for improving food quality and agricultural output.12Polysaccharides are a significant class of hydrophilic polymers with natural origin and biocompatibility that find frequent use in water-based polymer systems and in nanotechnology in particular, which is mainly due to their favourable properties in biological systems, e.g. biodegradability, biocompatibility, and low-toxicity. These properties constitute considerable requirements for the utilisation of NPs and thus polysaccharides represent an ideal class of building blocks for NP fabrication.24 For example, in the field of medical therapy, polysaccharide-based NPs have the advantages of high loading efficiencies, fast drug release rates and good targeting ability as well as high stability and low toxicity in physiological environment.9,25,26 In addition to biodegradability and biocompatibility, polysaccharides have gained considerable attention due to their abundance, facile processing and sustainable feedstocks (refer to Section 2).19 The chemical functionalisation of polysaccharides is mainly achieved by using the free carboxyl and hydroxyl groups distributed along the backbone of the polysaccharides. The utilisation of these reactive groups allows the fabrication of suitable polysaccharide derivatives with determined properties (e.g. hydrophobicity, solubility), promoting further use of polysaccharides in specific application areas.25,27,28
Based on common mechanisms such as ionic crosslinking, covalent crosslinking, self-assembly of hydrophobically modified polysaccharides, polyelectrolyte complexation, and forming polysaccharide–drug conjugates (refer to Section 3),25 many researchers have designed different approaches to synthesise polysaccharide-based NPs with controlled size, morphology, and structure (Scheme 1).29 The key point of the choice of the synthetic route is to optimise the final properties of NPs designed for a specific application. In addition, a series of factors such as physicochemical parameters of the polysaccharides used, polysaccharide chemical composition, NPs size, and surface morphology, can be used as a guide during the synthesis process.30 Sometimes, misunderstandings occur because of the misuse of the term NP referring to both NP and nanocrystal, which are indeed different types of materials. NPs are amorphous particles while nanocrystals are crystalline.31 Therefore, polysaccharide nanocrystals will not be discussed detailly in the following sections, but some useful applications will be mentioned.
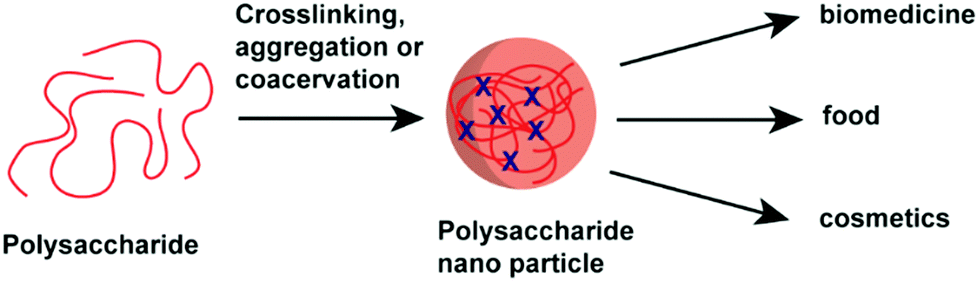 | ||
| Scheme 1 Overview of polysaccharide NP formation and their applications. | ||
As polysaccharide-based NPs have a broad range of possible applications, understanding the mechanism of action is highly important. In this review, the properties of several common polysaccharides (refer to Section 2) and practical methods for preparing their NPs will be discussed (refer to Section 3). Also, the development of polysaccharide/polymer NP systems (refer to Section 4) is discussed, highlighting the advantages of the NP system obtained from a polysaccharide and synthetic polymer combination. Finally, on the basis of the preceding sections, the emphasis of this review is to survey some useful applications of polysaccharide-based NPs in nanomedicine, cosmetics, and food (refer to Section 5). At last, we will summarise the developments in the field of polysaccharide NPs and give an outlook for future directions.
2. Polysaccharides
Polysaccharides are natural polymers consisting of monosaccharide units linked by glycosidic bonds with chitosan (CS), cellulose (CL), starch, hyaluronic acid (HA), and dextran (DEX) being typical examples.25,32 Similar to proteins and glycosaminoglycans, which are also common natural polymers, polysaccharides can be extracted from various sources, for instance, from plant origin, microbial origin and animal origin.25,33 The biological properties and activities of different polysaccharides are diverse due to their different chemical structures (Fig. 1) and ionic nature. Thus, their functions and applications are directly related to their chemical and biochemical specifications.25,28 The properties of common polysaccharides which are widely used in nanomedicine, cosmetics, food, and other areas are summarised in Table 1, and more information is detailed in the following sections.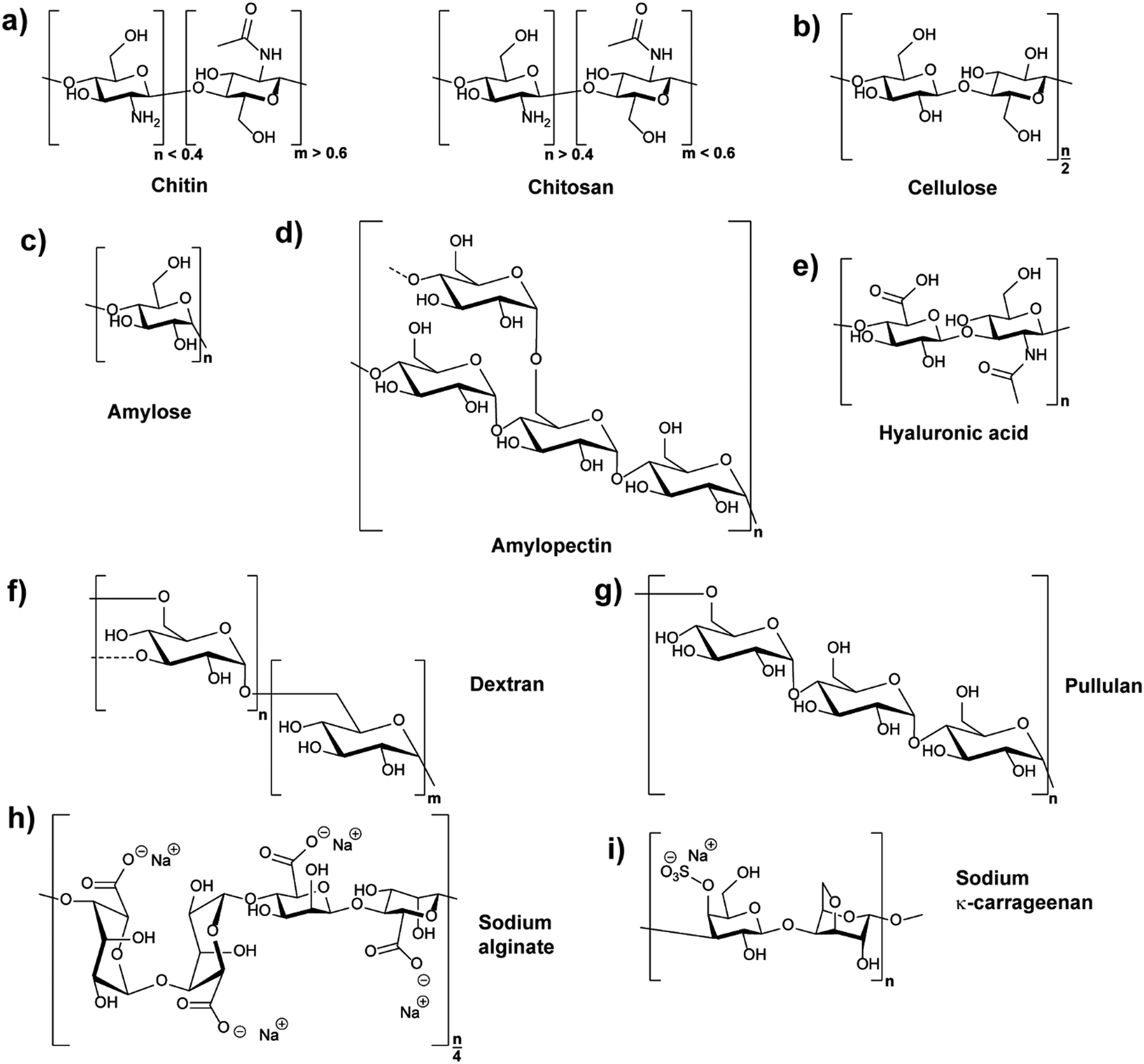 | ||
| Fig. 1 Chemical structures of polysaccharides: (a) chitin (CH) and its derivative from N-deacetylation chitosan (CS), (b) cellulose (CL), (c) and (d) structures of two different polymers coexisting in starch, amylose and amylopectin (dashed lines indicate branches), (e) hyaluronic acid (HA), (f) dextran (DEX) (dashed lines indicate branches), (g) pullulan (PL), (h) sodium alginate (α-L-guluronic acid–α-L-guluronic acid–β-D-mannuronic acid–β-D-mannuronic acid sequence shown), and (i) κ-carrageenan (κ-CRG). | ||
| Polysaccharide | Water solubility | Molar mass range/kDa | Source |
|---|---|---|---|
| Alginate | Soluble (sodium or potassium salt) | 32–40034 | Algae, bacteria |
| Carrageenan (CRG) | Soluble in hot water | 100–100035 | Seaweed |
| Chitin (CH) | Insoluble28 | 1000–250028 | Exoskeleton of non-mammals |
| Chitosan (CS) | Soluble under acidic conditions27 | 100–500 | N-Deacetylation process of chitin |
| Cellulose (CL) | Insoluble36 | 50–200037,38 | Higher plants |
| Dextran (DEX) | Soluble | 3–200039,40 | Bacteria |
| Starch | Insoluble in cold water41 | ∼100 for amylose, and 1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for amylopectin 000 for amylopectin |
Higher plants |
| Hyaluronic acid (HA) | Soluble42 | 20–1000028,43 |
Mammals |
| Pullulan (PL) | Soluble | 100–25044 | Bacteria |
2.1 Chitin and chitosan
Chitin (CH) is the primary component of the crustaceans’ exoskeleton, and it is also the second most abundant natural polysaccharide.25,45 A commonly employed derivative of CH is chitosan (CS), which is a linear polysaccharide obtained from the partial N-deacetylation of CH with a deacetylation degree of over 40% under alkaline conditions and elevated temperature (Fig. 1a).46 CH has a hydrophobic linear structure composed of β-(1,4)-linked N-acetyl-D-glucosamine units, while the structure of CS is mainly based on β-(1,4)-linked glucosamine and N-acetyl glucosamine units.28,47CH is insoluble in water or in other common solvents. However, CS produced from the N-deacetylation process of CH has increased aqueous solubility. Acidic conditions promote the water solubility of CS due to protonation of the basic amino groups.28 In addition, the conversion of CH to CS decreases the molecular weight (MW) from an average range of 1000–2500 kDa to 100–500 kDa. The lower MW of CS resulting from the deacetylation process can also facilitate the water solubility of CS.25,28 The more frequent use of CS in NPs compared to CH can be deduced to the improved water solubility. Furthermore, the amine functions of CS can be employed for chemical reactions, e.g. functionalisation, and CS can be produced in many kinds of forms, such as powder, paste, and film. Frequently, CS is utilised for the synthesis of widely used CS-based nanoparticles (CSNPs).28,33
As the nature of CS favours chemical modifications compared to less reactive CH, the preparation of CS-based nanoparticles (CSNPs) can be conducted by various avenues. Effective chemical modifications enable further options to use the material compared to the natural compound. The chemical modifications are usually carried out at the sites of reactive amino groups of CS, and there is a wide range of examples of commonly used CS derivatives such as thiolated CS, sugar-bearing CS, and carboxyalkyl CS.25,48 These CS derivatives are designed to enhance specific properties of CS. For instance, CS modified by thiol groups can obtain improved mucoadhesive properties, while conjugating hydrophobic moieties (e.g. deoxycholic acid and cholesterol) to CS introduces an avenue to form CSNPs by self-assembly.45,48
CS is widely investigated in many areas of application, for example, drug delivery, tissue engineering, and stabilising cosmetic ingredients.45,49,50 The positive charge density on CS is thought as significant in improving the cell uptake, but it is accompanied with higher toxicity.25,51 To solve this problem, the selection of suitable CS derivatives have attracted growing attention, in order to improve health benefits of CS.51
2.2 Cellulose
Cellulose (CL) is the primary constituent of plant cell walls, and it is the most abundant natural polysaccharide.28 As the base fundamental unit of plants, CL is considered as human/animal/environmental-friendly, biocompatible, and biodegradable.33,52 CL is mainly extracted from plant origin, and its linear polysaccharide structure is formed by β-(1,4)-linked D-glucose units, giving a flat ribbon-like conformation (Fig. 1b).52The CL polymer and polymer fibrils are relatively stable due to their inter- and intra-chain hydrogen bonding network. Thus, the linkage of individual CL chains is stabilised by supramolecular interactions, giving the linear configuration and an axial stiffness in CL fibrils. On the basis of CL biosynthesis and extraction processes, various types of CL-based nanoparticles (CLNPs) and CL nanocomposite materials can be obtained. The main factors which cause the differentiation are the source materials of CL (e.g. wood, plant, and bacteria) and the deconstruction processes during extraction.52,53
For the utilisation of CL, many applications in diverse research areas are described, such as food packaging, drug delivery, films, and reinforcing materials for polymer matrices.33,52 After appropriate surface functionalisation, the surface chemistry of CLNPs enables self-assembly, making CLNPs versatile materials with considerable mechanical properties.52
2.3 Starch
In plant tubers and seed endosperm, starch is the primary storage carbohydrate which can be extracted from many resources such as corn and potato.54 Chemically, two different polysaccharide structures are present in starch, which are amylose (Fig. 1c) and amylopectin (Fig. 1d). Amylose is a linear polymer formed by α-(1,4)-linked D-glucose units while amylopectin is a highly branched polymer composed of short chains of α-(1,4)-linked D-glucose units with branches formed by α-(1,6) linkages at the branch positions.54,55The average MW of amylose is in the range of 105 Da while that of amylopectin is distributed from 106 to 107 Da. For example, rice starch has higher MW compared with those from corn, wheat, and potato, because rice starch has the lowest amylose content amongst them.51 Similar to CL, starch is insoluble in cold water due to the hydrogen bonding effect. Moreover, the high MW of starch decreases the solubility. In relation, amylopectin has higher solubility compared to amylose because of its highly branched structure, which is useful in separating amylose and amylopectin from starch granules.27,51
Starch-based NPs (SNPs) can be prepared by various strategies such as precipitation, solvent evaporation, and emulsion crosslinking. Moreover, different preparation methods will result in variation of SNPs properties (e.g. shape and crystallinity).33,56 Compared with other polysaccharides, starch is unique because it has the property to convert to thermoplastic material in the presence of plasticisers. Also, starch has the advantages of stability, biocompatibility, and biodegradability, making it useful in drug delivery, tissue engineering, and food packaging applications.33,47
2.4 Hyaluronic acid
As a typical extracellular matrix polymer, hyaluronic acid (HA) has broad applications in biomedical and nanotechnological areas. Regarding the structure of HA, it is a hydrophilic linear polysaccharide composed of alternately linked D-glucuronic and N-acetyl-D-glucosamine units via β-1,3 and β-1,4 glycosidic bonds (Fig. 1e).33,43 As HA is a negatively charged polysaccharide, hyaluronic acid-based NPs (HANPs) can be formed by using cationic molecules (e.g. CS) as ionic crosslinkers. Moreover, approaches such as making HA-cargo (e.g. drug) conjugates have also been developed to form HA-based nanocarriers.57,58 HA is mostly extracted from tissues, and it is significant in the areas of tissue repair, wound healing, food industry, and cosmetics. Moreover, HA is not only a good wound healing factor but also an adequate carrier for the delivery of antibiotics that aid in wound healing properties.15,282.5 Dextran
Dextran (DEX) belongs to the first commercial exopolysaccharides produced by bacterial enzymes and is mostly obtained from Leuconostoc mesenteroide. It is branched, and the basic structure of the various types of dextran consists of main chains formed by (1,6)-α-D-glucose with various ratios of linkages and branches (Fig. 1f).59 The ratios of α-(1,6) linkages can vary from 97% to 50%, and the branching can take place mainly in position α-(1,3), and occasionally in position α-(1,2) or α-(1,4).28,59 Because of the reactive hydroxyl chemistries of DEX, it is easy to be functionalised.28 Furthermore, DEX is highly water-soluble and frequently used in ATPS.60,61 Depending on the strain of bacteria and conditions used, properties of DEX such as branching and molecular weight vary considerably. The MW of DEX ranges from 3000 Da to 2000000 Da, and the larger DEX (>60000) are excreted poorly from the kidney and remain in the blood before being completely metabolised.39,62
Non-ionic DEX-based NPs can be obtained by self-assembly, and due to the biocompatibility and biodegradability of DEX, NPs from DEX are discussed as suitable drug/gene nano-vehicles. In addition, the superior water solubility of Dextran-based NPs (DEXNPs) prevents cellular toxicity after completing the drug delivery process.63 Similar to CS and HA, the advantages of DEXNPs can be promoted after suitable chemical modifications or forming DEX–drug conjugates. Typical DEX derivatives such as thiolated DEX, phosphorylated DEX, and dextran sulfate (DS) are all very useful in enhancing the targeting ability of DEXNPs.26,59,63 For example, the chemical modification of DEX to obtain DS enables interactions with lipoproteins and complexation with fibrinogen or DNA. Furthermore, modification with hydrophobic moieties introduces amphiphilicity, which is very important to form self-assembled and water dispersible DEXNPs in the field of drug delivery.26
2.6 Pullulan
Pullulan (PL) is a non-ionic exopolysaccharide which is mainly obtained from the fermentation medium of the fungus-like yeast Aureobasidium pullulans under limiting conditions. PL has been widely used in various areas because it is non-toxic, non-mutagenic, non-carcinogenic, biocompatible, and bio-degradable.64 The structure of PL is based on α-(1,6) linked maltotriose units (Fig. 1g), and the linkage between maltotriose units significantly gives structural flexibility of PL.65,66 PL is easily soluble in water and insoluble in most organic solvents. Compared with other polysaccharides, the PL aqueous solution is stable and has a relatively low viscosity. The MW of PL ranges from 100 kDa to 250 kDa, and its appearance is white or yellowish-white powder.44 PL hydrogel NPs have been employed for gene delivery67 or as composite with CS for vaccine delivery.68,69 Recently, PL was utilised as block in double hydrophilic block copolymers, which were utilised in turn for self-assembly, for example in the formation of completely hydrophilic capsules,70 droplets71,72 or particles.73Similar to DEX-based NPs, PL-based NPs (PLNPs) can be prepared by self-assembly after the hydrophobic modification of PL. In the fields of drug and gene delivery, hydrophobically modified PL has been widely investigated to be a good nanocarrier. Moreover, by mixing hydrophobically modified PL with quantum dots, the formed PLNPs can also play a significant role in medical tracing areas such as tumour imaging.32,66 PL is also being used in the areas of food and cosmetics, for example to extend food shelf life and simplify food processing. For cosmetics, PL can be utilised in lotions, shampoos, and face masks.74
2.7 Alginate
Alginate is a well-known polysaccharide produced by brown algae and bacteria consisting of α-L-guluronic acid and β-D-mannuronic acid building blocks that are linearly linked by 1,4-glycosidic linkages (Fig. 1h).75 Alginate chains consist of blocks of either all α-L-guluronic acid, all β-D-mannuronic acid, or alternating α-L-guluronic acid–β-D-mannuronic acid subunits.76 It is known to be biodegradable, non-toxic and abundant with molar masses between 32 and 400 kDa.34 The polymer is anionic due to the acid functions and frequently used in the form of sodium or potassium salts. A remarkable feature of alginate is its interaction of α-L-guluronic acid–α-L-guluronic acid subunits with divalent cations like calcium ions leading to hydrogel formation.76 These hydrogels are utilised in various medical applications,77e.g. encapsulation of drugs and release78 or as cell growth environment.79 The complexation with Ca2+ can also be employed to form NPs for example via emulsion methods80 or precipitation.81 Also, a combination with cationic CS enables NP formation via coacervation.822.8 Carrageenan
Carrageenans (CRG) are linear sulphated polysaccharides and originate from seaweed.35 The polymer structure consists mainly of 3-linked β-D-galactopyranose and 4-linked α-D-galactopyranose or 4-linked 3,6-anhydro-α-D-galactopyranose units (Fig. 1i with an example structure). CRGs are classified into six types based on the chemical structure (kappa (κ)-, iota (ι)-, lambda (λ)-, mu (μ)-, nu (ν)- and theta (θ)-CRG), i.e. the position of sulfate esters and the presence of anhydro units. Overall, the MW of commercial CRG ranges from 100 to 1000 kDa with solubility in hot water and partially cold water depending on substitution. CRGs form helices in aqueous environment leading to gelation that is exploited in food industry for thickening.83 Due to its polyanionic character, CRG can be used in conjunction with CS to form NPs.84,85 Nevertheless, the biocompatibility and toxicity of CRG is currently under discussion and has to be followed closely for food and biomedical applications in particular.863. Synthesis of polysaccharide nanoparticles
A variety of methods has been proposed to prepare NPs based on natural polysaccharides. The selection of a proper synthesis method depends on the targeted application and its requirements. For example, many factors such as thermal and chemical stability of the active agent, reproducibility of the release kinetic profiles, particle size, and stability of the final product and residual toxicity associated with the final product have to be taken into account before preparing drug delivery nanocarriers87 and NPs for other applications. The avenue for the preparation of NPs is crucial for stability under the intended conditions and in particular the crosslinking chemistry has to be considered. For example, the native charges of polysaccharides can be exploited for the synthesis of polysaccharide NPs88–90 or covalent crosslinking can be implemented.91–93 Mizrahy and Peer discussed the main mechanisms during the synthesis of polysaccharide NPs in their review in detail, which are categorised as chemical (covalent) crosslinking, physical (ionic) crosslinking, polyelectrolyte-complexation and self-assembly (Scheme 2).25 Due to the repeated number of monomer units on the polysaccharide a full network can be achieved. Hence, a complete gelation of the reaction medium has to be suppressed via the preparation conditions, e.g. in dispersed systems.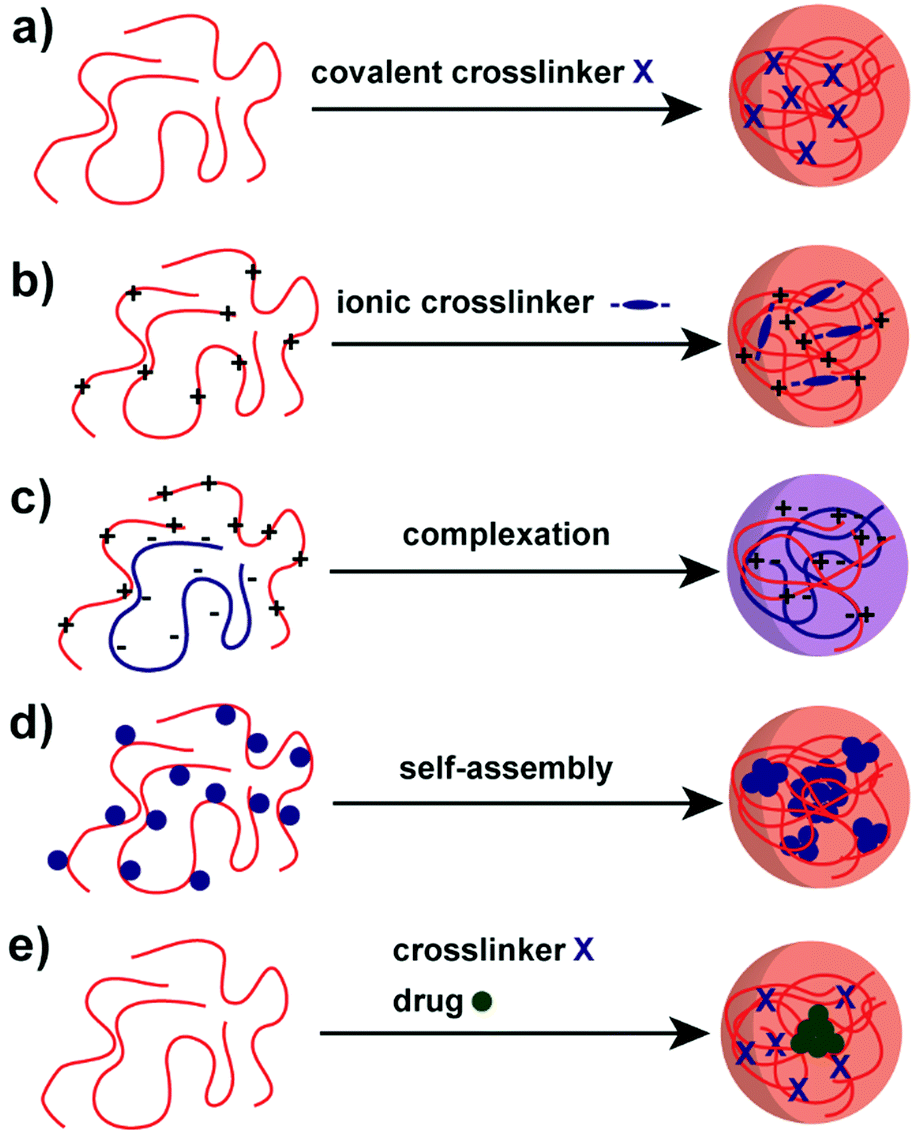 | ||
| Scheme 2 Avenues for the preparation of polysaccharide NPs: (a) covalent crosslinking, (b) ionic crosslinking, (c) complexation of oppositely charged polysaccharides, (d) self-assembly of hydrophobically (blue sphere) modified polysaccharides and (e) polysaccharide–drug (green sphere) conjugate.25 | ||
In addition to the crosslinking process itself, the formation of NPs relies on the formation of materials on the nano scale via various preparative techniques. In this section, important and widely used methods such as nanoprecipitation, complex coacervation, and emulsification will be discussed. Also, the microfluidic technique has proven to be a useful direction for the preparation of polysaccharide NPs, both in emulsion and nanoprecipitation approaches.94 Other related methods developed by researchers will be reviewed as well. The relationship between the properties (e.g. diameter) of the polysaccharide NPs synthesised and the methods utilised will be discussed to showcase how the methods affect the final properties.
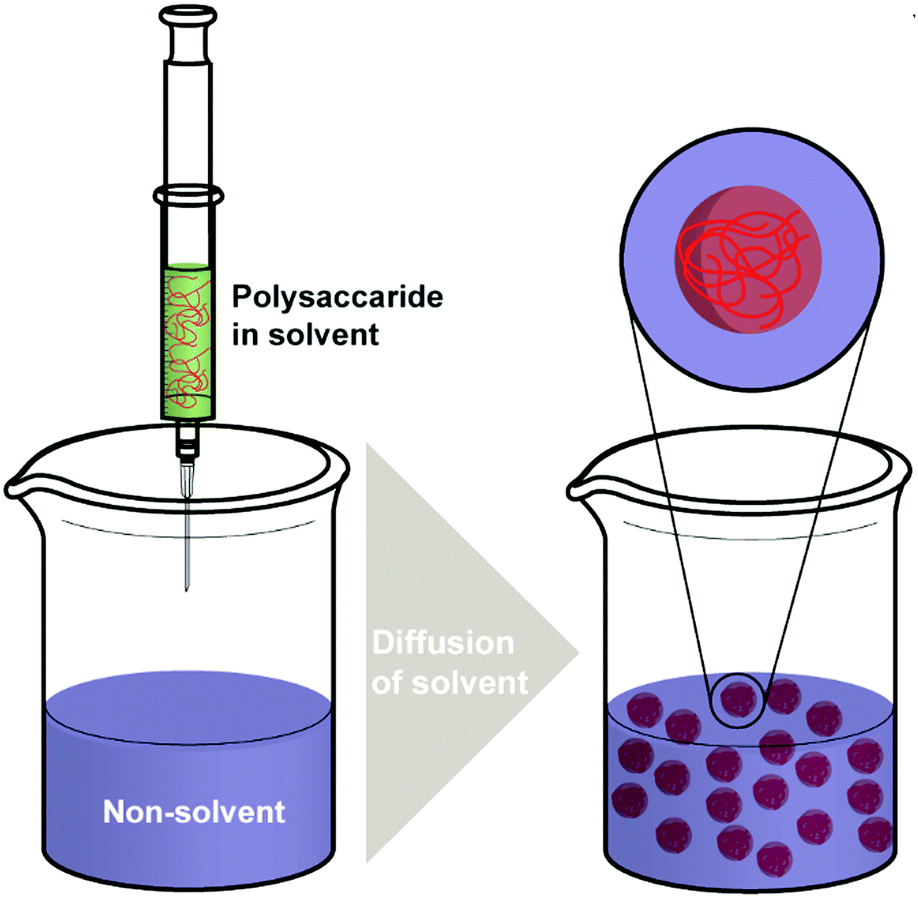 | ||
| Scheme 3 Schematic representation of the nanoprecipitation method. | ||
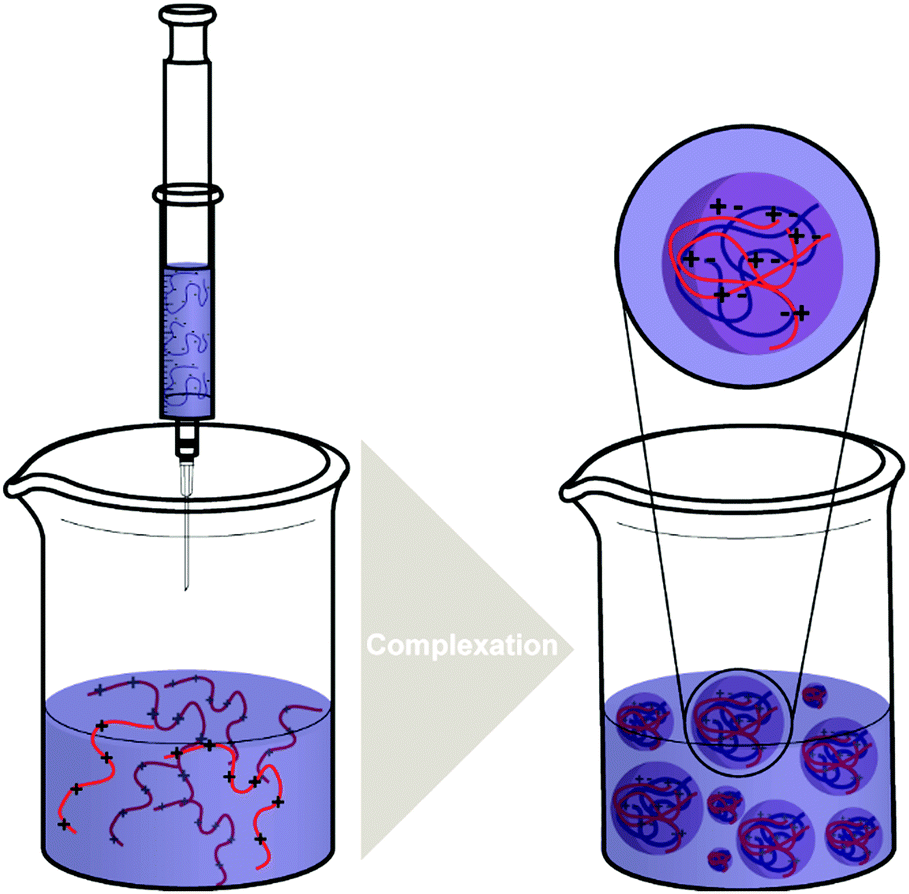 | ||
| Scheme 4 The typical processes of forming polysaccharide NPs such as CS/HA NPs by complex coacervation process. | ||
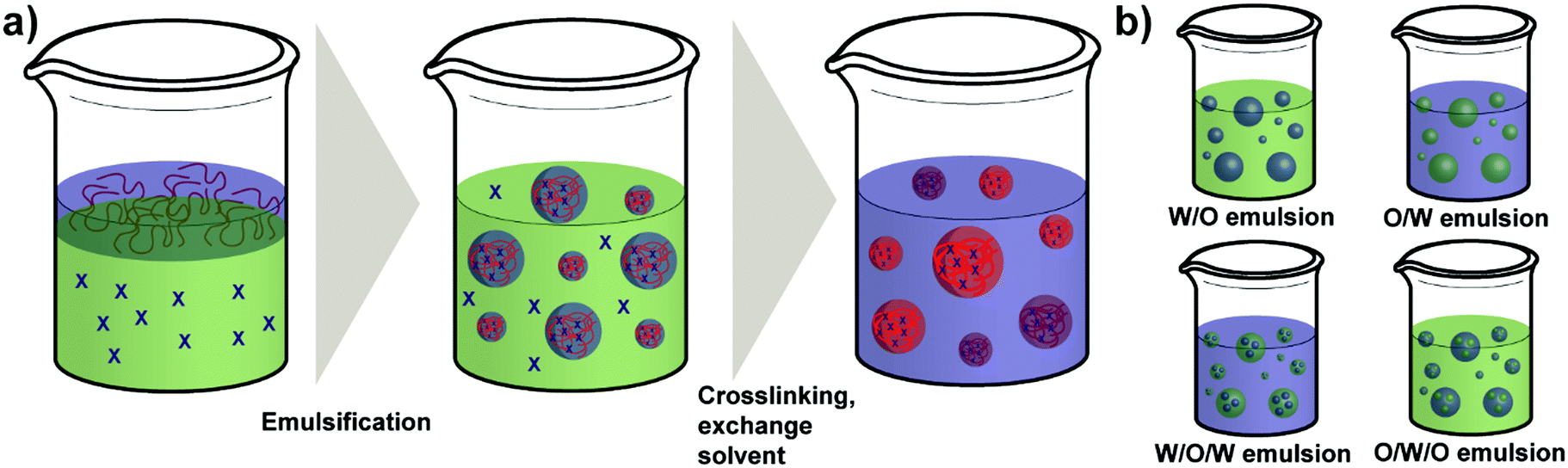 | ||
| Scheme 5 Schematic overview over the emulsion process for polysaccharide NP formation: (a) formation of polysaccharide NPs via water-in-oil emulsion and crosslinking (external gelation) and (b) types of emulsions used to prepare polysaccharide NPs (green: organic solvent; blue: water). | ||
3.1 Crosslinking and aggregation strategies
As for common polymer NPs, one of the main concerns in the fabrication of polysaccharide NPs is the crosslinking of the polysaccharide to form stable particles. Crosslinking has a considerable influence on the polysaccharide NP properties and hence applications, e.g. mechanical properties, swellability or drug-encapsulation/release.Covalent crosslinking introduces irreversible crosslinking points leading to highly stable structures (Scheme 2a) that can withstand chemical stress, e.g. solvent changes or a broad pH range.25 The crosslinker molecules have to be multifunctional in order to react twice with a polysaccharide molecule or with other crosslinkers to form a connection between polysaccharide molecules. Common crosslinkers for polysaccharides are dialdehydes that link via acetal formation. Other options include multifunctional carboxylic acids forming ester or amide bonds. An avenue to introduce degradability or even stimulus response is the utilisation of cleavable crosslinkers.95 One of the issues with covalent crosslinking is the potential toxicity of remaining non-linked small molecules crosslinkers that might hinder biomedical applications.
Another way to introduce crosslinks is via a supramolecular pathway for charged polysaccharides, e.g. in ionic crosslinking (Scheme 2b). For that, oppositely charged small ionic molecules with multiple functionalities are added, e.g. tripolyphosphate (TPP), or ions with multiple vacancies for interaction, for example Ca2+.96 The crosslinking proceeds via electrostatic interactions that connect the crosslinker with the polysaccharide. In contrast to covalent crosslinking, ionic crosslinking features a lower chemical stability, e.g. against changes in pH or ionic strength, and also physical stability, e.g. against temperature changes. However, this defined instability can be exploited to enable degradation on purpose, for example in drug-delivery applications.95
A crosslinking method of high relevance is coacervation or polyelectrolyte complexation of charged polysaccharides. In order to form coacervate structures, oppositely charged polysaccharides and/or polyelectrolytes are employed. In a similar way to ionic crosslinking, electrostatic interactions lead to crosslinking (Scheme 2c). As no toxic small molecule crosslinkers are used, the coacervate approach is considered highly biocompatible at least if the employed polyelectrolytes/polysaccharides are biocompatible. The obtained stability of the NPs relies on charge density and distribution of charges along the chain, as the interaction between both polyelectrolytes/polysaccharides is the main factor for stability.97 Similar to ionic crosslinking, pH, ionic strength and temperature play a crucial role for the stability and preparation of coacervate particles. Polysaccharides like HA, CS and alginate are utilised frequently for coacervate formation. Coacervation/polyelectrolyte complexation can be also utilised in the formation of layer-by-layer assemblies.98,99
Self-assembly of polysaccharide chains can be employed for NPs formation as well, which is another supramolecular avenue.100 Commonly, hydrophilic polysaccharides are functionalised with hydrophobic moieties (bile acids, fatty acids or cholesterol) that will aggregate in aqueous environment leading to micellar structures (Scheme 2d).101,102 As such, NPs with hydrophobic core and hydrophilic polysaccharide shell are obtained. The core can perform as a reservoir for hydrophobic drugs or other molecules. The properties of the micellar NPs can be tailored via the utilised polysaccharide conjugate, e.g. MW or hydrophobic to hydrophilic ratio, but also via the preparation process, e.g. the kinetics of micelle formation or the utilised solvent system. The approach was brought to the next level by Wich and coworkers,103 who synthesised an amphiphilic block copolymer of DEX and hydrophobic acetalated DEX and formed micellar NPs that way. As such, a completely polysaccharide-based amphiphilic block copolymer was presented.
At last, a related structure to micelles should be discussed, namely polysaccharide–drug conjugates (Scheme 2e).48 Instead of adding non-functional hydrophobic molecules to drive supramolecular aggregation, hydrophobic drugs can be conjugated as well to the polysaccharide. As such, polysaccharide NPs can be obtained that serve as drug-delivery vehicles directly. An important characteristic is the connection of drug and polysaccharide that should be cleavable in the body at best at the right point of action.104 In principle, also other methods of aggregation or crosslinking can be combined with polysaccharide–drug conjugates.
3.2 Nanoprecipitation
Nanoprecipitation is one of the first developed techniques for encapsulating drug molecules, and it is also known as solvent displacement or interfacial deposition. Nanoprecipitation has been widely used in the formation of many kinds of NPs such as polymeric NPs, drug NPs, drug-loaded protein NPs and inorganic NPs.105 Though nanoprecipitation has advantages of being simple, reproducible, fast, and economic, it is still challenging to encapsulate water soluble compounds with this method.106In the nanoprecipitation process, two miscible phases are required, which are an aqueous phase and an organic/oil phase.107 A typical procedure (Scheme 3) of nanoprecipitation consists of dissolving hydrophobic solutes in a water-miscible solvent wherein they have high solubility at first. Subsequently, this solution is added to a significant excess amount of an anti-solvent such as water or buffer solutions. Because the solubility of hydrophobic solutes decreases in the aqueous solution, particles of hydrophobic compounds will precipitate. Finally, particles of hydrophobic molecules are obtained after removing the excess solvent by evaporation, dialysis, or lyophilisation.105 The work of Barresi and coworkers showed that mixing of phases has a significant effect on controlling the particle size.108 Briefly, a larger population of smaller NPs can be obtained from good mixing conditions, while poor mixing produced larger NPs. The NP formation also relies considerably on the surface tension difference between aqueous phase (with high surface tension) and organic phase (with low surface tension). The difference in surface tension leads to continuous solvent vortices formation at the interface of both liquids, and the organic solvent diffuses from low surface tension, causing gradual precipitation of the polymer (e.g. polysaccharides) on the organic surface and formation of nanocapsules.109
Zhao and coworkers discussed three widely used nanoprecipitation methods in their work, which are traditional nanoprecipitation, flash nanoprecipitation, and micro-fluidic nanoprecipitation.105 To control the size and morphological structure of the NPs prepared, key parameters such as mixing temperature, solvent/antisolvent ratios, and properties of the chosen polymers are very important. Operation of traditional nanoprecipitation is easy, quick, and affordable. However, it has a lack of control over mixing and has wider size distributions of large particle sizes.105 Flash nanoprecipitation is a simple, rapid and scalable method. Turbulence-based intensive mixing can be achieved within few milliseconds by using flash nanoprecipitation mixers. Therefore, flash nanoprecipitation produces better NP quality with smaller NP size and narrow size distribution.105,110 The limitation of flash nanoprecipitation is that the particles produced do not have adequate stability for some applications.105 Micro-fluidic nanoprecipitation is usually laminar flow-based and operated in continuous flow, its high-speed diffusion-based mixing (at milliseconds or even microseconds) results from a reduced mixing path (down to several tens of nanometres). By changing the configuration of the micromixer or controlling the ratio of flow rates, physicochemical properties of NPs (e.g. size, charge, morphology) can be controlled. However, an issue is system blocking caused by solid particle accumulation. Moreover, small volume and low flow rates in microfluidics result in relatively low productivity.105,111
As a simple and rapid method, nanoprecipitation has been used to prepare various types of polysaccharide NPs. Tay and coworkers synthesised SNPs with sizes in the range between 300 nm and 400 nm by adding dissolved starch solution into excess ethanol under controlled conditions (Fig. 2a).112 The presence of appropriate surfactant was also an important parameter in modulating the shape and size of the produced SNPs (Fig. 2b and c). NPs of hydrophobically modified CL were also synthesised by using nanoprecipitation method. Kulterer et al. prepared cellulose acetate (CA) NPs by developing an efficient nanoprecipitation technique based on using good CA solvents as a modifier for the nonsolvent.113 The size of NPs was reduced by sonication, efficient agitation, and optimisation of process parameters such as tetrahydrofuran (THF) content, temperature and pH value. Spherical NPs of 60 nm with high yield were obtained by adding good polymer solvents (THF) to an anti-solvent (water). A nanoprecipitation process for the formation of DEX NPs was described by Weiss and coworkers.114 Initially, DEX, PL and starch were methacrylated via methacrylic anhydride to enable crosslinking and the hydrophile–lipophile balance (HLB) value measured. In the next step, the modified polysaccharides were dissolved in acetone or THF, precipitated in water and crosslinked under UV light. The obtained particles were subjected to atomic force microscopy (AFM) measurements in order to obtain mechanical properties. Interestingly, the Young's moduli of particles correlated with the HLB value of the utilised polysaccharide, which might be due to the difference in water swelling for particles with different HLB value. Nicolas and coworkers utilised nanoprecipitation in the formation of polysaccharide capsules employing a template.115 Various polysaccharides were employed, e.g. DEX, PL, DS or HA, and crosslinked via isophorone isocyanate during the nanoprecipitation process. Mygliol as templating agent was dissolved together with the crosslinker in acetone and the polysaccharides added in aqueous solution at once. In addition to forming capsules from one polysaccharide component, blends were introduced as well as multilayers.
 | ||
| Fig. 2 Examples of polysaccharide NPs obtained by the nanoprecipitation method observed via Scanning Electron Microscopy (SEM) (scale bar 1 μm): (a) micrograph of precipitated SNPs prepared by addition of 1 mL of 1% starch into 20 mL of ethanol, (b) micrographs of precipitated SNPs prepared by addition of 1 mL of 1% starch into 20 mL of ethanol with 4% of cetrimonium bromide surfactant and (c) micrographs of precipitated SNPs prepared by addition of 1 mL of 1% starch into 20 mL of ethanol with 4% of Tween 80 surfactant. (Reproduced with permission from ref. 112. Copyright Elsevier, 2011.) | ||
Overall, nanoprecipitation is easy to conduct with fast operation processing and simple equipment requirement. However, issues such as low productivity and system blocking during the NPs formation need more attention in future development.105 Thus, improved control for fast processing is required and more complex structures have to be investigated in the future.
3.3 Complex coacervation
The complex coacervation method is carried out by mixing aqueous solutions of two polymers with opposite charges. It is a suitable method for the preparation of NPs due to its simplicity and mild preparation conditions without the need of organic solvents.19 To synthesise and enhance natural polysaccharide NPs, complex coacervation with oppositely charged polyelectrolytes has been widely used.116The mixing of solutions containing oppositely charged polyelectrolytes (polycation and polyanion) (Scheme 4) establishes an equilibrium between the dense liquid phase and the dilute solution phase. The dense liquid phase is also called the coacervate, which is polyelectrolyte-rich and frequently produced from proteins and polysaccharides.117,118 According to the theoretical model of Voorn and Overbeek, the association of polyelectrolytes is significantly determined by the electrostatic interaction and the entropy gain during the complex coacervation process.118,119 Related to this, many important modes of polymer complex coacervation including the Voorn–Overbeek theory were reviewed by Sing.120
An interesting feature of the complex coacervation process of polyelectrolytes, is that the net charge of the NPs formed can be modulated by adding one polyelectrolyte in excess to the other, giving a core–shell structure with the excess component segregated at the outer shell. The physical properties of the polyelectrolyte-complex formed by coacervation can be affected by many factors such as pH of the solution, temperature, ionic strength, charge density, and the molar mass of polyelectrolytes.32,106 For instance, Delair and coworkers found improved stability of the polyelectrolyte-complex formed by DS (polyanion) and CS (polycation) by remaining the charge ratio R (n+/n−) under 0.6.121
For polyelectrolyte-complex formation, CS is the most commonly used cationic polysaccharide. Besides, various negatively charged polysaccharides such as HA, DS, and carboxymethyl cellulose can be combined with CS.32 Chen and coworkers prepared NPs made of oleoyl-carboxymethyl-chitosan (OCMCS) and HA for gene delivery using the coacervation process.122 The results obtained from their work showed that a charge ratio of 5 (n−/n+) and a weight ratio of 4 (OCMCS/HA) were the optimal conditions giving the smallest (165 nm), positively charged (+14.2 mV) and monodisperse NPs (Fig. 3a). Ferreira and coworkers developed a pH sensitive CS/DS NP system for the delivery of insulin using complex coacervation.123 The optimum synthesis conditions had a DS:CS mass ratio of 1.5:1 at pH 4.8, and the stable spherical NPs produced had a mean diameter of 500 nm. It was also found that the insulin released by CS/DS NP system maintained its immunogenic bioactivity, indicating a bright future for the CS/DS NPs as a potential oral delivery system for insulin. Dou and coworkers124 described the formation of complex coacervates as well. At first, CS/DEX nanoparticles were formed via in situ crosslinking and self-assembly. CS and DEX were dissolved in water and radicals formed on polysaccharide backbones via addition of cerium(IV) ions that initiated the formation of poly(methyl acrylate) (PMA) and crosslinking via diallyldisulfide. Due to the formation of hydrophobic grafts an amphiphilic structure was generated that assembled into NPs. In a subsequent step, these particles were combined with HA to obtain a larger super structure via coacervation (Fig. 3b). The formation of multi-particle aggregates or HA-surface modified particles could be tailored via the ratio of HA and particle precursors. Finally, the particles were employed to target cancer cells via the lipid raft-dependent endocytosis pathway.
 | ||
| Fig. 3 Examples of polysaccharide NPs obtained by using the complex coacervation method: (a) oleoyl-carboxymethyl-chitosan (OCMCS)-hyaluronic acid (HA)/DNA NPs with a charge ratio of 5 (n−/n+) and a weight ratio of 4 (OCMCS/HA) (scale bar 500 nm) and (reproduced with permission from ref. 122. Copyright Elsevier, 2013) and (b) TEM image of HA/CS/DEX superstructure NPs formed via coacervation of CS/DEX NPs with HA. (Reproduced with permission from ref. 124. Copyright American Chemical Society, 2020.) | ||
The influence of CS acetylation degree on the synthesis, stability, and performance of CS/HA NPs prepared by electrostatic complex coacervation was studied by Furlani et al.125 A crosslinking mechanism mediated by TPP was involved because the inter-polyelectrolyte interaction between CS and HA alone was not sufficient to form spherical particles. The synthesis of NPs was carried out as follows. First, CS, sodium hyaluronate, and TPP were dissolved individually in deionised water. After solubilisation, HA/TPP solution was prepared by adding TPP solution dropwise to the hyaluronate solution. Then, the formation of NPs was conducted by adding the HA/TPP solution dropwise to CS solution under stirring. The results of their work proved that CS acetylation degree determines the NPs stability, and CS with specific fractions of acetylated units such as 0.16 and 0.63 were found to promote the formation of homogenous NPs. Recently, Tirelli and coworkers studied the formation of CS/HA-based nano particles with respect to the effects of template addition.126 A two-step templating process was compared with a direct polyelectrolyte complexation approach. The two-step templating was performed via ionotropic gelation of CS with TPP and subsequent incubation with HA. It could be shown that HA can quantitatively exchange with TPP in the second step, which was accompanied with aggregation of particles. In contrast to the direct process of CS/HA complexation, larger particles were generated. Furthermore, the utilisation of low MW CS induced the formation of a HA corona.
Another key feature of complex coacervation is the preservation of the structure and property of biochemical drugs including genes and proteins, which is due to the mild conditions required for coacervate formation and their native environment.127 In addition, the complex coacervation process enables the formation of polysaccharide NPs with high loading of biomolecular cargo.128 Although there has been considerable progress in the theory of coacervation,120,129 the prediction of outcomes from the preparation procedure is challenging,130e.g. the control over NPs size and shape. This is mainly due to the various parameters in coacervate preparation like pH, ionic strength, polymer concentration, temperature and polymer type/MW. To improve the performance of NPs in the drug delivery system, the mixing of polysaccharide with other polymers can also be achieved using complex coacervation method.19 As such, access to additional features is possible, e.g. structures with poly(ethylene glycol) (PEG) corona.131
3.4 Emulsion-based methods
As a regular method for preparing polymer NPs,132,133 emulsion-based procedures are also a versatile method used for the synthesis of polysaccharide NPs.19 The term emulsification is defined as a process of forming a metastable dispersion (e.g. oil and water) also called emulsion that would separate into two phases in equilibrium. In biological systems different uses of emulsions are found, for example, lipoprotein forms an emulsion for the delivery and metabolism of fat in the body.30,134Generally, depending on the type of dispersed phase and dispersion medium, emulsions are categorised as O/W direct emulsion and water-in-oil (W/O) inverse emulsion, in which water or oil is the continuous phase, respectively. Besides, more complex multiple emulsions such as oil-in-water-in-oil emulsion and water-in-oil-in-water emulsion can also be obtained by dispersing the O/W or W/O emulsions in oil or water using specific techniques (Scheme 5).134 In addition, W/W emulsions can be formed as well (refer to Section 4.3).135,136 Considering the size of droplets, emulsions formed can be classified as microemulsion, miniemulsion and macroemulsion. Microemulsions are thermodynamically stable with droplet sizes ranging from 10 to 100 nm. Miniemulsions and macroemulsions are both thermodynamically unstable with droplet sizes between 100 nm and 1 μm and larger than 1 μm, respectively.30
Different from the nanoprecipitation method mentioned above, which is a simple one-step method, preparation of organic NPs such as polysaccharide NPs using emulsification method is based on a two-step procedure. At first the emulsion is prepared as mentioned above and subsequently, NPs can be generated from the emulsion via various methods, such as solvent evaporation, solvent diffusion, and reverse salting-out.137 In particular, the formation of polysaccharide NPs by emulsification method is conducted as follows: a solution of polysaccharide is firstly formed by dissolving polysaccharides in deionised water or other solvents. Secondly, the solution is dispersed in an oil phase to fabricate O/W or W/O emulsions by stirring or employing ultrasound. Polysaccharide NPs can finally be prepared by internal or external gelation.19 In external gelation, the crosslinkers diffuse from an external source into the polysaccharide emulsion, i.e. from the continuous phase. While for internal gelation, the crosslinkers are already present inside the droplets before gelation occurs.138
To obtain the emulsification system, low-energy emulsification techniques and high-energy emulsification techniques are commonly used. The former one is based on the expansion of low-energy stirring routes in constant progress. It can be classified as spontaneous emulsification, emulsion inversion point method, and phase inversion temperature method. For high-energy emulsification, high-energy mechanical stirring processes such as sonication and microfluidic methods are employed, the applied stress predominately determines the size of the nanodroplets produced.30 Ravi et al. successfully prepared Lopinavir loaded pullulan acetate NPs (PANPs) for oral delivery using O/W emulsion-solvent-evaporation method in their study.139 Firstly, Lopinavir and pullulan acetate were dissolved in methylene chloride giving the organic phase. Subsequently, to form the emulsification system, the organic phase was added dropwise to an aqueous poly(vinyl alcohol) (PVA) solution under gentle magnetic stirring, where PVA acts as steric stabiliser for the PANPs. After a 15 min high-speed homogenisation, the primary emulsion was centrifuged to obtain the PANPs. Further purification was finally conducted to remove the excess free drug and PVA before storage. According to the results of their work, the size of PANPs prepared was about 197 nm, high entrapment efficiency (∼75%) and monodisperse particles were also obtained. Moreover, a tissue distribution study proved that Lopinavir loaded PANPs could be a remarkable approach in treating HIV infection.
In the study of Park's group, doxorubicin (DOX) loaded HA-based nanocarriers for the intracellular DOX delivery were synthesised by using the O/W emulsion method (Fig. 4a).140 The HA–SS–DDT–DOX NPs formed were based on an amphiphilic HA derivative, the HA–SS–DDT, which was prepared by modifying HA with dodecanethiol (DDT) via disulfide linkage. The primary O/W emulsion was prepared by adding a solution of DOX HCl and triethylamine in chloroform, into the aqueous HA–SS–DDT solution. After evaporation of chloroform, the solution was filtered, dialysed, and lyophilised to obtain HA–SS–DDT–DOX NPs with a diameter around 334 nm. It was found that the synthesised NPs were stable under physiological conditions and DOX was effectively encapsulated with loading efficiency >70%. Moreover, the HA–SS–DDT NPs were effectively taken up by squamous cell carcinoma (SCC7) cells followed by fast release of DOX, indicating the promising future of HA–SS–DDT NPs as a potential DOX carrier (Fig. 4b). Sagis and coworkers utilised sodium alginate to form NPs via an emulsion process.141 Alginate was dissolved in water and the aqueous solution emulsified in an oil with polyglycerol polyricinoleate as surfactant. Then, CaCl2 nanoparticles were added to induce an external gelation process of the alginate via Ca2+/alginate complexation.
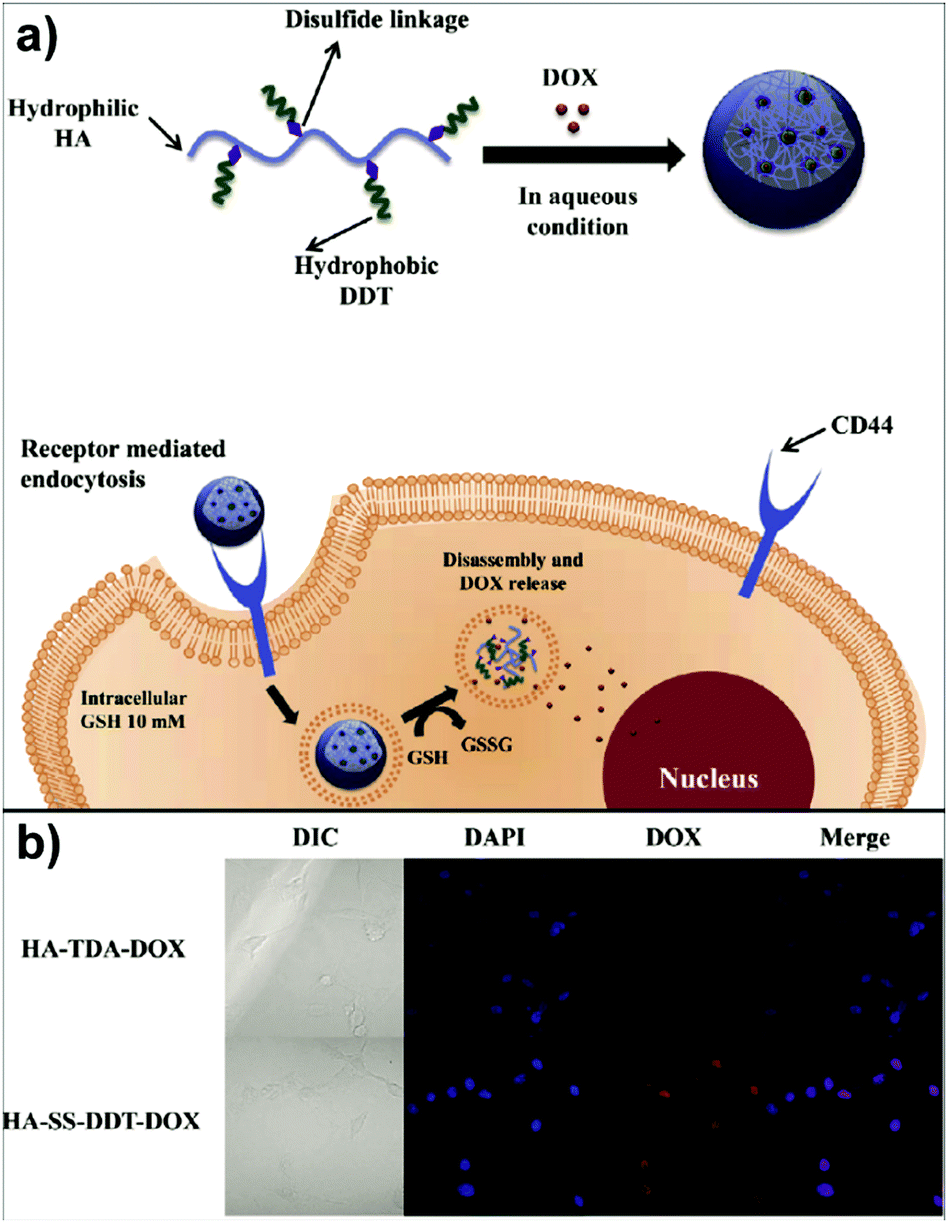 | ||
| Fig. 4 Polysaccharide NPs obtained by using the emulsification method: (a) schematic approach of HA–SS–DDT–DOX utilisation in cancer treatment and (b) fluorescence microscopic images of SCC7 cells treated with HA–TDA–DOX and HA–SS–DDT–DOX showing the release of DOX in cells in the case of HA–SS–DDT–DOX. (Reproduced with permission from ref. 140. Copyright Elsevier, 2014.) | ||
Another kind of HA-based NPs were prepared by Min et al. with the O/W emulsion method.142 In aqueous conditions, HA-5β-cholanic acid conjugates were firstly used to form HANPs by self-assembly. Subsequently, a bioinert and hydrophobic perfluoropentane (PFP) solution was added to the prepared HANPs and emulsified to fabricate PFP-loaded HANPs. The analysis results demonstrated that the PFP-loaded HANPs were stable and robust and had a homogeneous as well as narrow size distribution with an average diameter of 350 nm. Sriamornsak and coworkers described the formation of dextrin NPs via an inverse emulsion approach.143 Water-in-hexane emulsions were employed and dextrin crosslinked with glyoxal. Particle sizes were tailored via sonication time, surfactant type and concentration as well as HLB value. In a similar way, Gupta and coworkers formed nanoparticles from PL with sizes below 50 nm.144
Emulsification methods have a considerable advantage in controlling the size and loading of the synthesised polysaccharide NPs due to the convenient adjustment of emulsion droplet sizes and the option to include cargoes in specific phases of the emulsions. Nevertheless, emulsion methods are more complicated than nanoprecipitation and complex coacervation caused by the additional step of emulsification. Furthermore, the requirements of large amounts of organic solvents limit the utilisation of emulsification method.19
3.5 Other methods
In addition to the three important methods introduced above, there have been other practical methods such as desolvation and dialysis, developed by researchers for preparing polysaccharide NPs, which are also appropriate in producing stable and monodisperse polysaccharide NPs with controllable size.19When using the desolvation method, a desolvation factor such as salts or alcohols is slowly added into the solution of macromolecules to induce polymer precipitation.30 For making drug nanocarriers, the selection of desolvating agents is based on the nature of the drug to be encapsulated inside the polysaccharide NPs.19 Amoabediny and coworkers synthesised curcumin loaded CSNPs and SNPs using the desolvation method in their work.145 The desolvating agent was composed of absolute ethanol and predetermined concentrations of curcumin. The CSNPs and SNPs were formed by ethanol precipitation of an aqueous CS/starch solution. Mean sizes of the synthesised curcumin loaded CSNPs and SNPs were 66 and 61 nm with a maximum loading efficiency of 12% and 14%, respectively. The desolvation method can produce NPs directly in suspension in an aqueous medium, however, a further purification step is required because of the potential toxicity caused by desolvating agents.19
Related to the nanoprecipitation method, a solvent displacement mechanism is used in the dialysis method. Additional tools such as dialysis tubes or semi-permeable membranes are utilised to provide a physical barrier with appropriate molar mass cut off for polymers/polysaccharides.30 Zhang and coworkers synthesised paclitaxel (PTX) loaded HA NPs by using dialysis method.146 HA was modified first by coupling with aminated retinoid acid. Next, PTX solution in ethanol was added into the solution of retinoic acid modified HA (HRA). The obtained solution was then dialysed against distilled water, and the PTX-loaded HRA NPs were finally collected after filtration and lyophilisation. The particle size of the prepared PTX-loaded HRA NPs was around 149 nm, and the inhibition effect showed by PTX-loaded HRA NPs on tumour growth proved its future application in cancer chemotherapy. Compared to nanoprecipitation, the dialysis method relies on a slow exchange of solvent and thus more likely generates thermodynamic products.
Synthesis of NPs using ionic gelation is regularly performed with charged polysaccharides in aqueous medium in very dilute solution. For instance, CSNPs can be produced by ionic gelation because it is positively charged at neutral pH and can form NPs by crosslinking with small molecules (e.g. the monomers to form polymers) with opposite charge via electrostatic interaction.19,30 Alonso and coworkers successfully entrapped DOX into CSNPs through the ionic gelation of CS with TPP.147 CS–DOX complex was firstly prepared by adding DOX into the CS aqueous solution at pH 5.5. Subsequently, a suitable amount of TPP was mixed with the CS–DOX complex to form the DOX loaded CSNPs. According to the results obtained from their study, though the complexation efficiency was low, no complex dissociation was found after the formation of NPs. Moreover, the usefulness of CSNPs as the DOX carrier and to transport it into the cells in its active form was demonstrated.
Spray-drying is a technique notably used in the production of powders, granules or agglomerates from the mixture of drug and excipient solutions as well as suspensions.19,87 A typical spray-drying process is based on drying of aerosol droplets in contact with a hot drying gas to evaporate the moisture and to form the solid product.30 Li et al. described the preparation of NPs formed by modified starch in their study.148 The results showed that state-of-the-art spray drying equipment could lower the size of the produced dried particles by an order of magnitude attaining submicron sizes. Also, there are many factors possible to affect the size of the NPs produced. In the review of Agnihotri et al.,87 various parameters such as the size of the nozzle, spray flow rate, and atomisation pressure are discussed that can be controlled to obtain desired the particle size.
Including what has been introduced above, there are many other practical techniques for polysaccharide NP synthesis that complement the most common methods and cannot be covered in the present review. Overall, further studies are still required to develop more efficient methods/techniques in the future.
4. Polysaccharide nanoparticles and polymers
Developing NP systems formed by polysaccharides and polymers have potential applications in many areas such as drug delivery, Pickering emulsifiers, and improving the properties of polymer-based materials.70,149–151 As such, polysaccharide/polymer NP systems combine the advantages of both polysaccharides and polymers, bringing benefits to overcome difficult challenges that belong to the individual materials (Scheme 6). For example, poly(ethyl acrylate) (PEA) or PMA polymers can introduce flexibility to rigid starch-based materials for further utilisation.152,153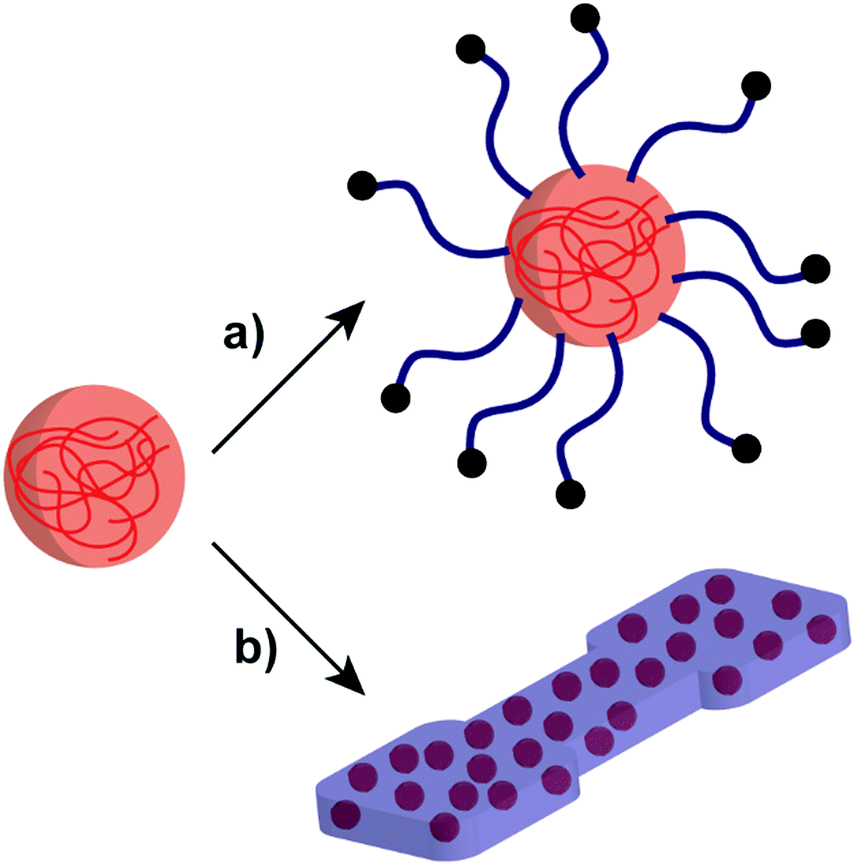 | ||
| Scheme 6 Schematic representation of the polysaccharide NP incorporation with polymer systems: (a) polymer-grafted polysaccharide NP and (b) polysaccharide NPs in a polymer matrix. | ||
This section will mainly focus on the methods of attaching polymers to polysaccharide NPs and inclusion of polysaccharide NPs into polymer structures. The system formed by the inclusion of polysaccharide NPs into the bulk of polymers is also known as a typical core–shell-structured hybrid or composite material. The core–shell structure is based on a sufficient modification of the surface of polysaccharide NPs by synthetic polymers, giving a polysaccharide NP core and a polymer shell.150 Moreover, in some cases, hydrophobic polymer NPs can also be used to form a core modified by outer hydrophilic polysaccharides. In the field of drug/gene delivery, the hydrophobic–hydrophilic core–shell structure plays a very significant role in enhancing the stability, water solubility and biocompatibility of NP structures.154–156 The polymer core–polysaccharide shell structure will not be discussed in detail in this section as it is not directly related to polysaccharide NPs, more related information of how the core–shell structure with a polysaccharide shell is utilised can be found in the literature.157,158
4.1 Polymers attached to polysaccharide NPs
Though polysaccharide NPs have been widely investigated in many research areas, for several applications, their hydrophilicity precludes further use of them. For example, to enhance the properties of hydrophobic polylactide (PLA), starch/PLA composites have been considered as an efficient choice. However, the hydrophobic PLA is immiscible with native hydrophilic starch, causing incompatibility and weak mechanical properties.153 Thus, improved compatibilisation via polymer attachment is a considerable direction.To effectively combine the advantages of polysaccharide NPs and polymers, graft modification/copolymerisation methods are necessary. Two types of grafting reactions can be conducted to form the polymer grafted polysaccharide system, i.e. the “grafting to” and “grafting from” reactions.159 In “grafting to” reactions (Scheme 7a), functionalised polymers are prepared in advance to be reacted with functional groups present on the polysaccharide interface/polysaccharide backbone. Moreover, the polymer chains to be grafted can be fully characterised before attachment. In “grafting from” reactions (Scheme 7b) the polymerisation of monomers takes place directly on the polysaccharide interface started from the polysaccharide backbone, which minimises the steric hindrance. In this case, the full characterisation of the grafted chain becomes challenging if not impossible.160
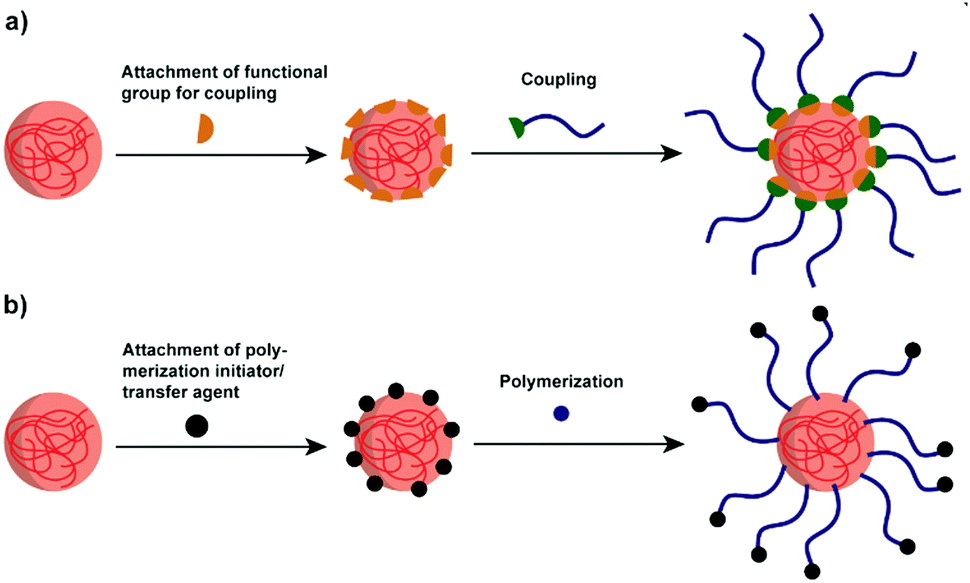 | ||
| Scheme 7 Schematic representation of the two grafting approaches. (a) The “grafting to” approach involving the coupling between the polysaccharide NPs and the pre-formed polymer chains (blue line). (b) The “grafting from” approach relying on polymerisation initiated at the NP surface (initiator/transfer agent: black sphere; monomer: blue sphere). | ||
According to Cunningham and coworkers, SNPs could be modified with synthetic polymers by using the two grafting reactions mentioned above via nitroxide-mediated polymerisation.31,159 The “grafting from” approach was carried out in three main steps under inert conditions, which are SNP-macroinitiator preparation, “grafting from” reaction, and acid hydrolysis (purification). Graft modified SNP with poly(methyl methacrylate-co-styrene) (P(MMA-co-S)), PMA, and poly(acrylic acid) were successfully obtained. In addition, the feasibility of varying the amount of grafted polymer on the SNP surface was demonstrated.31
As well as SNPs, CSNPs were also studied for forming core–shell-structured hybrids. In the work of Tang et al., two types of polymers were investigated to modify the surface of CS nanospheres (spherical CSNPs) via an ATRP “grafting from” process.161 The used polymers were the homopolymer poly(methyl methacrylate) (PMMA) and amphiphilic block copolymer PMMA-b-poly(poly(ethylene glycol) methyl ether methacrylate) (PMMA-b-P(PEGMA)). The obtained structure had a CS nanosphere core and a densely grafted outer PMMA or PMMA-b-P(PEGMA) layer. The CSNP-g-(PMMA-b-P(PEGMA)) core–shell structure described the inclusion of a polysaccharide core in multipolymer layers. According to the results of the stability studies, the block copolymer grafted nanospheres CSNP-g-(PMMA-b-P(PEGMA)) were more stable in both THF and deionised water compared to the Br functionalised CSNP and CSNP-g-PMMA, indicating the potential use for the synthesis of CSNPs based core–shell structure for in vivo biomedical applications and industrial-scale polymerisation.
Yuan et al. grafted poly(hydroxy ethyl methacrylate) (PHEMA) from crosslinked CS microspheres via an ATRP approach.162 There, CS spheres were conjugated with an ATRP initiator and HEMA polymerised. In the following step, the hydroxyl groups from the HMA units were exploited to attach melamine in amide formation via a carbonyl diimidazole activation. As such, microspheres were obtained that featured considerable and rapid Cu(II) adsorption from aqueous solution. Moreover, the microspheres could be recycled via exchange with H+ ions. Another route for the formation of polymer decorated CS microspheres was presented by Peixão et al. cerium(IV) initiation was employed to graft (N-isopropylacrylamide) (NIPAM) directly from the crosslinked microspheres.163 Additionally, the microspheres were embedded with magnetite nanoparticles to introduce magnetic features. The final microspheres featured thermoresponsivity due to PNIPAM and could be easily separated from liquid media via magnetic field. In particular, the system was utilised to remove oil from water, i.e. the microsphere material was added to oily water to capture the oil and separate with a magnet. At ambient temperature a removal efficiency of 85% was observed, while an efficiency of 97% was observed above 40 °C. This difference is due to the fact that the PNIPAM is more hydrophobic above its lower critical solution temperature and thus more susceptible to absorb oil.
Similarly, “grafting to” approaches were also investigated to obtain grafted polysaccharide NPs, e.g. SNP under inert conditions.159 SNP was firstly functionalised with 4-vinylbenzyl chloride (VBC), and in a separate step, the N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)nitroxide] (SG1)-capped P(MMA-co-S) was synthesised. Finally, the prepared SNP-VBC and SG1-capped P(MMA-co-S) were reacted to form graft modified SNP. The “grafting to” approach allows accurate and precise characterisation of the precursors and the grafted SNP. Wang and coworkers synthesised core–shell NPs based on an SNP-based core and a PNIPAM brush shell using surface-initiated single-electron transfer living radical polymerisation (SI-SET-LRP) in their work.150 SNPs were firstly initiator-modified by Br groups using an esterification reaction (Fig. 5a). Subsequently, PNIPAM chains were grafted to initiator-modified SNPs by using SI-SET-LRP. Compared with the initial SNPs, the SNP-g-PNIPAMs revealed an evolution in particle morphology (Fig. 5b–i), proving the successful synthesis.
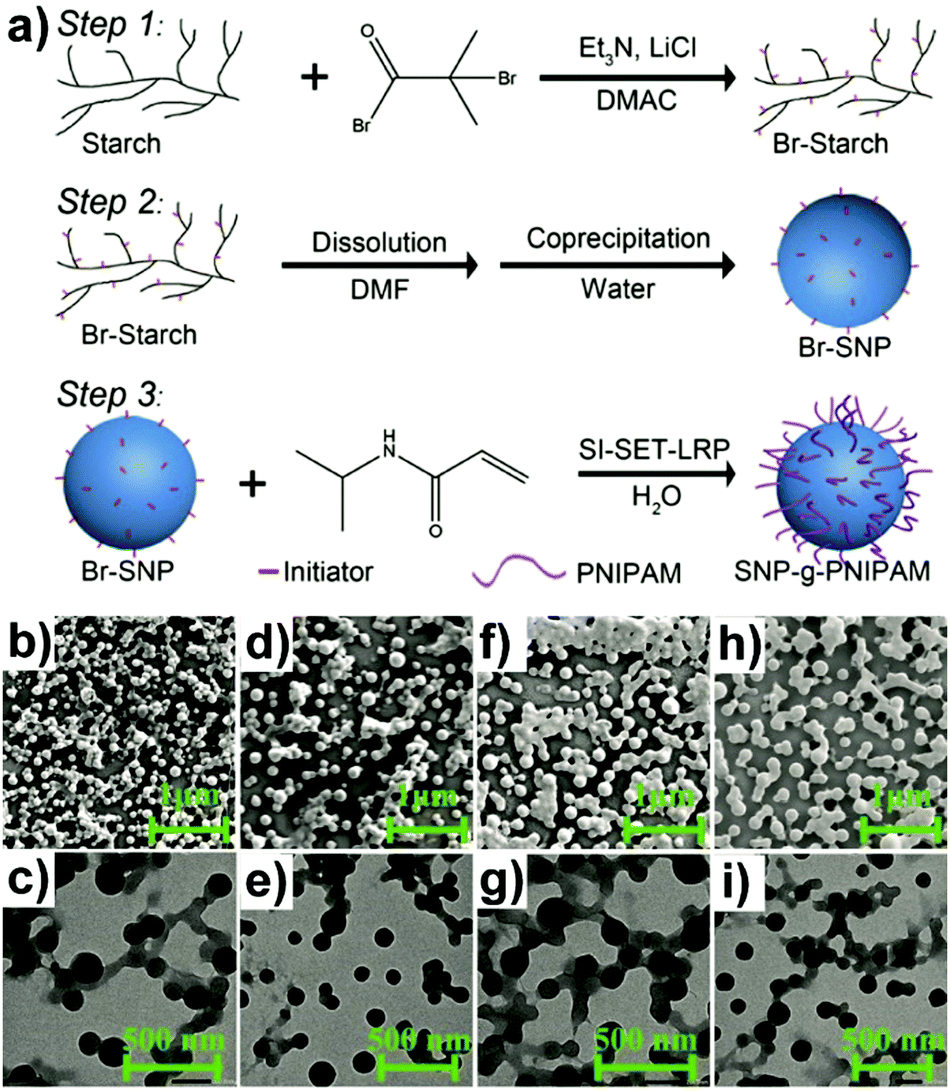 | ||
| Fig. 5 (a) Schematic overview of PNIPAM grafting from SNPs with the degree of substitution of the Br group around 1.1 per saccharide unit, (b) and (c) SNP-g-PNIPAM after 15 min (SEM top: scale bar 1 μm, TEM bottom: scale bar 500 nm), (d) and (e) SNP-g-PNIPAM after 30 min (SEM top, TEM bottom), (f) and (g) SNP-g-PNIPAM after 60 min (SEM top, TEM bottom), (h) and (i) SNP-g-PNIPAM after 120 min (SEM top, TEM bottom). (Reproduced with permission from ref. 150. Copyright American Chemical Society, 2019.) | ||
A “grafting-to” approach was employed by Lin and coworkers to form polysaccharide NPs with heparin core and PEG shell.164 The anionic polysaccharide heparin was combined with poly(lysine) to form NPs via coacervate formation under ultrasound treatment. To enable conjugation with PEG, the heparin chains were modified with tetrazine units. Finally, norbornene end functionalised PEG was added to the NPs to enable grafting. Subsequently, cell interactions with functionalised NPs were investigated, where the grafting could be used to tailor macrophage uptake in vitro.
Another avenue to obtain polysaccharide NPs with polymer-covered surfaces is via self-assembly approaches.165 For example, Park and coworkers grafted PEG and 5β-cholanic acid onto HA.166 Due to the hydrophobic 5β-cholanic acid and the hydrophilic PEG, an amphiphilic structure was obtained that was capable of forming micellar particles in aqueous environment with PEG on the surface. The formed particles featured reduced uptake in the liver, prolonged blood circulation, and improved tumour targetability due to the PEG functionalisation, which was shown via in vivo cell internalisation studies. A similar strategy was implemented by various researchers in the past years to obtain PEGylated particles for applications in the biomedical field, e.g. DOX delivery,167–171 theranostic NPs to target colon cancer,172 as a stabiliser for deacetyl mycoepoxydience nanocrystals,173 gene delivery166,174,175 or sorafenib delivery.176 To enhance the stability of the formed particles, Park and coworkers introduced covalent crosslinking.177 In addition to PEG and 5β-cholanic acid, methacrylamide units were grafted onto HA. The methacrylamide units were crosslinked via UV light after encapsulation of PTX. Sustained release of the drug was observed as well as improved cell internalisation leading to an overall enhanced therapeutic efficacy. Later on, a bioresponsive system was introduced (Fig. 6),178 where HA was grafted with PEG that was attached via a matrix metalloproteinase 9 (MMP9) cleavable connection (pep) as well as conjugated to ovalbumin (OVA) as foreign antigen. In such a way, the PEG corona facilitated efficient transport but was cleaved at the tumour site via MMP9 enabling cell uptake leading to foreign antigen presentation on tumour cells. As such, tumour growth was inhibited considerably.
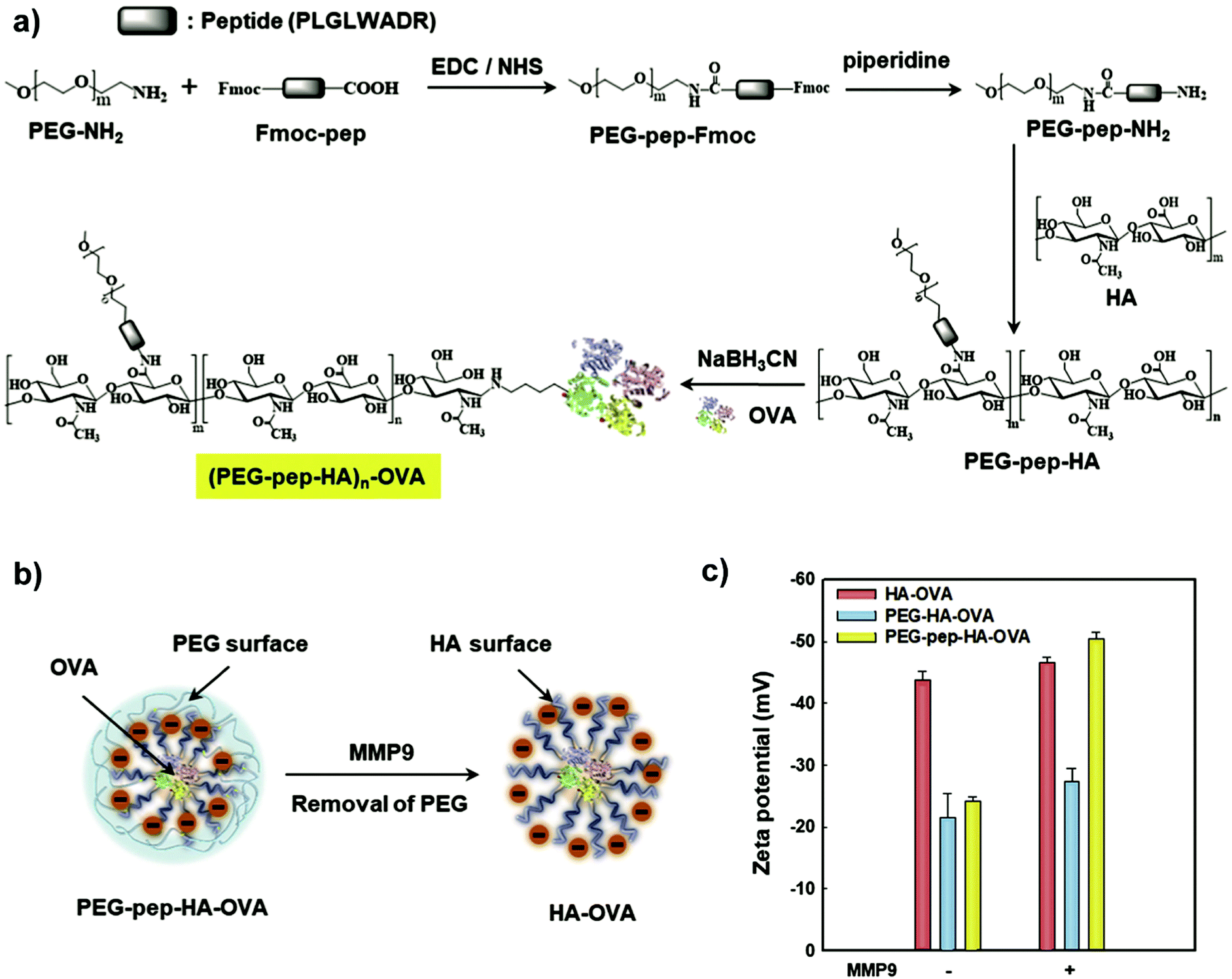 | ||
| Fig. 6 Preparation and characterisation of the PEG–pep–HA–OVA conjugate: (a) synthetic route, (b) schematic illustration of MMP9-responsive removal of PEG from the PEG–pep–HA–OVA and (c) zeta potential values of the PEG–pep–HA–OVA with or without MMP-9. (Reproduced with permission from ref. 178. Copyright Elsevier, 2017.) | ||
PEGylated DEX NPs were synthesised by Xiao and coworkers, who grafted PEG onto DEX in the first step and crosslinked via 3,3′-dithiodipropionic acid in the second step.179 Next, DOX was encapsulated and drug-release studied in vitro, where the disulfide-based crosslinker can be cleaved via a redox stimulus. Pérez-Álvarez and coworkers described the formation of HA-based nanogels crosslinked via PEG spacers in a microemulsion approach.180 The formed nanogels featured pH response due to the HA content, which was revealed by the swelling ratio at different pH. Wich and coworkers fabricated NPs based on spermine functionalised acetalated DEX.181 In a subsequent step, PEG or DEX were grafted onto the NP surface and their stability studied over time. In contrast to PEG grafting, DEX functionalised NPs aggregated over time. With respect to biological activity, PEG grafting led to attenuated particle binding and uptake. DEX grafting showed an opposite effect with enhanced passive targeting of dendritic cells and their stimulation. This combination is desired for vaccination avenues based on dendritic cells aiming to deliver an antigen and an adjuvant to dendritic cells at the same time in order to induce immune response.
Also, CS-based NPs were PEGylated in order to improve circulation in the body and stability. CS-coated nanocapsules were PEGylated by de la Fuente and coworkers.182 Initially, a nanoemulsion method was utilised to form CS capsules with lipophilic core (oleic acid). In the next step, the formed capsules were grafted with PEG via amide bonds. The authors showed that the PEG grafting hindered unspecific protein adsorption, in vitro studies showed low cytotoxicity and considerable internalisation in Vero cells. Bikiaris and coworkers synthesised PEG-grafted CS for the use as protein delivery system.183 For the particle formation, coacervation with poly(glutamic acid) or ionic crosslinking with TPP was utilised. As protein to deliver, bovine serum albumin was encapsulated in the NPs. The observed release characteristics could be tailored via PEG MW, crosslinker content and type. Increased amounts of PEG on the particle surface and increased MW led to a faster release, while crosslinking with the small molecule TPP slowed the release down.
PEGylated CS NPs for the delivery of PTX were studied by Jain and coworkers.184 At first PTX loaded CS NPs were formed via solvent evaporation method together with PVA. In the next step PEG was grafted onto the particles via amide formation and the remaining hydroxyl group at the other end of the PEG chain was used to conjugate transferrin. As such, the particles featured a drug, a stabilising corona and a cancer cell targeting moiety (transferrin). A considerable therapeutic effect was observed in vivo due to sustained release profile, greater intracellular uptake, promoted cell cytotoxicity and prolonged systemic circulation time. Hartmann and coworkers utilised CS NPs for gene delivery.185 At first, CS was grafted with PEG and then combined with unfunctionalised CS as well as small interfering RNA (siRNA) in slightly acidic environment. Due to the negative charge of siRNA and the positive charges of CS, polyplexes were formed, similar to the coacervation technique. The formed NPs featured the cargo, i.e. siRNA in the core and PEG in the shell. The authors studied the effect of PEGylation degree on NP properties. Interestingly, physicochemical properties revealed only minor changes, while biological activity differed significantly pointing towards an optimum between PEG stabilisation of NP transport in the body and activity of the carrier in gene transfection. Also, other cases of PEGylated CS-based NPs were utilised for gene delivery, e.g. for the delivery of siRNA.186–188
A nanoprecipitation avenue was utilised by Ganachaud and coworkers to form core–shell particles.189 A polysaccharide (PL or DEX) was combined with a glycopolymer, i.e. poly(N-[7-(α-D-mannopyranosyloxy)heptyl]methacrylamide-co-glycidyl methacrylate), and mygliol. The polysaccharide and glycopolymer were dissolved in water and precipitated into acetone containing mygliol as a template for the core and crosslinked via isophorone diisocyanate. The technique enabled formation of mono- or multilayered glyco nanocapsules through simultaneous or sequential nanoprecipitation, respectively. The prepared nanocapsules could be loaded with a drug (Camptothecin), covered with biotin or magnetic nanoparticles attached. Due to the incorporation of DEX, enzymatic degradation of the capsules could be achieved via dextranase.
An integrated self-assembly of polysaccharide-based double hydrophilic block copolymers and crosslinking was described by Schmidt and coworkers.190 A completely hydrophilic block copolymer of PL and P(PEGMA) was synthesised and the self-assembly studied in solution showing considerable effect from the length of the P(PEGMA) block. In a subsequent step, PL was crosslinked via sodium trimetaphosphate to obtain stable NPs that could withstand dilution and organic solvent. In a similar way, the block copolymer of PL and poly(N,N-dimethylacrylamide) was studied,91 which showed particle formation. In order to stabilise the structures, crosslinking was performed as well. In this case, the PL block was oxidised with periodate to form aldehyde functions along the backbone that were crosslinked via imine formation with cystamine. As such, NPs were synthesised that could be broken via pH or redox stimulus. Akiyoshi and coworkers described the formation of vesicles based on polymer grafted polysaccharides.191 PL was grafted with poly(propylene oxide) and the thermoresponsivity of the PPO blocks used to turn vesicle formation on or off. The vesicle size could be controlled by polymer concentration.
The examples shown above from previous developments of polysaccharide NP/polymer systems describe how polymer chains can be attached to the surface of polysaccharide NPs and how a combination of polymers and polysaccharides can improve the applicability of polysaccharide NPs. Future directions certainly will consider other polymer shells and incorporation of polymers into polysaccharide NPs as a way to tailor NPs properties. However, issues such as toxicity and poor biodegradability caused by polymers with high molar mass limit the applications of these conjugated systems, which has to be addressed in future research.43
4.2 Inclusion of polysaccharide NPs into polymer structures
The inclusion of a polysaccharide NP core into a polymer shell or matrix (Scheme 6b) has received much attention due to the wide applications in diverse areas.150 The synergistic effect resulting from combining the properties of the core and the shell imparts new functionality to the final obtained material.153Wang et al. prepared core–shell NPs which had a SNP hard core and a PEA outer shell to strengthen the toughness of PLA-based nanocomposites.153 Native starch was firstly esterified with acryloyl chloride and stearyl chloride to introduce hydrophobicity and polymerisable units. After that, a graft emulsion polymerisation was followed using potassium persulfate as the initiator. By using the “grafting from” method, ethyl acrylate monomers were polymerised from the surface of SNPs, forming the SNP-g-PEA core–shell NPs (Fig. 7a–c). Next, the particles were combined with PLA via melt-blending. Compared with pure PLA, elongation at break and notched impact strength were significantly improved after adding 20 wt% SNP-g-PEA core–shell NPs (Fig. 7d and e). The same authors also substituted PEA with poly(glycidyl methacrylate), where the epoxy group can act as a crosslinking point for PLA end groups via ring-opening.192 In a recent follow-up work, the same group showed even enhanced mechanical properties of blended PLA by implementation of two polymer layers around the SNPs, namely PEA and PMMA, formed via emulsion polymerisation.193
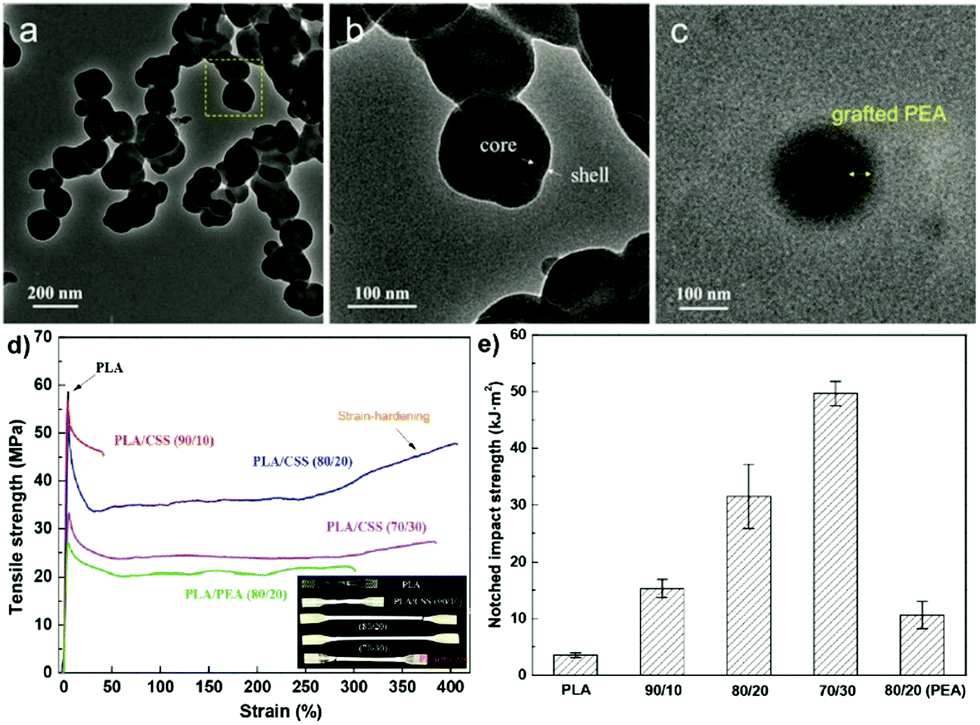 | ||
| Fig. 7 (a–c) TEM images of SNP-g-PEA core–shell NPs, (d) stress–strain curves of PLA and PLA/SNP-g-PEA core–shell NP composite (inset a shows optical photo of the samples), (e) notched impact strengths of PLA and PLA/SNP-g-PEA core–shell NP composite. (Reproduced with permission from ref. 153. Copyright American Chemical Society, 2018.) | ||
By using a similar emulsion polymerisation synthesis method, Liu et al. synthesised SNP-based core–shell NPs with a PMA outer shell to improve the mechanical properties of poly(propylene carbonate) (PPC).152 The benefits resulting from the addition of 20 wt% SNP-g-PMA core–shell NPs to neat PPC were shown via enhanced tensile strength, increased toughness, and increased thermal degradation temperature for the PPC/SNP-g-PMA composite. Moreover, the increased glass transition temperature (Tg) demonstrated that PPC/SNP-g-PMA composite had higher rigidity compared to that of the neat PPC, providing a direction for a more comprehensive modification and a wider application range of PPC. Recently, SNP-g-PMA was utilised as compatibiliser for blends of PPC and PLA.194 The blends were formed via melt-blending and the compatibilisation was investigated via AFM, dynamic scanning microcalorimetry and rheology. Improved mechanical and thermal properties were obtained, e.g. compared to a control blend PPC/PLA (60/40), the elongation at break of PPC/PLA/SNP-g-PMA (60/40/20) was enhanced from 15% to 272% without demoting tensile strength. Additionally, the heat distortion temperature of PPC/PLA/SNP-g-PMA (60/40/20) was increased by 21 °C to 47 °C compared to PPC. Jiang and coworkers presented an avenue to enhance the mechanical properties of poly(propylene) (PP) via incorporation of SNPs.195 Thermoplastic starch acetate and PP were melt-blended, leading to SNP formation in situ due to the addition of poly(ethylene octane) grafted with maleic anhydride. As such, higher notched impact strength and elongation at break of the blend material compared to the plain PP were observed.
For the further use of the inclusion of polysaccharide NPs in polymer structure, Somavarapu and coworkers described a particle-within-particle system formed by CS-based NPs such as CS/TPP NPs incorporated in polymer microparticles.196 By using a modified W/O/W double emulsion method, Ranibizumab was loaded into CSNPs with a size of 17–350 nm that were entrapped in poly(D,L-lactide-co-glycolide) (PLGA) microparticles. The CSNPs loaded PLGA microparticles significantly showed improved Ranibizumab loading and release profile, demonstrating the promising future for this novel system as an adequate nanocarrier. Teng and coworkers presented the fabrication of PLGA and poly(cyclohexane-1,4-diyl acetone dimethylene ketal) microspheres with included HA/CS NPs. A double emulsion process was performed to obtain the composite microspheres. HA/CS NPs were loaded with siRNA as a treatment for rheumatoid arthritis. The deposition of siRNA loaded particles in the polymer matrix prevented siRNA degradation and improved circulation time in vivo. Nevertheless, the observed release was not maintained over 8 days, which is needed as rheumatoid arthritis is a chronical disease requiring long-term treatment. The characteristics of the carrier showed interesting and effective delivery properties and might be useful for the treatment of other diseases but further optimisation is needed.
Berkland and coworkers utilised NPs based on polysaccharides and PLGA for hydrogel formation.197 Alginate and CS containing PLGA NPs were generated via coprecipitation first. In the next step, the oppositely charged NPs were combined to form hydrogels via mixing and charge-based attraction of particles. The gels proved to be sensitive to external force and could be injected. Furthermore, the interactions with human umbilical cord mesenchymal stem cells were studied in vitro and high biocompatibility was indicated.
Overall, polysaccharide NPs can lead to a considerable improvement in properties when combined with a polymer matrix, e.g. mechanical properties but also cargo encapsulation and release. In addition, polysaccharide NPs act as filler material from renewable resources, which is another advantage. More research on polysaccharide/polymer systems is essential to promote them for broader application areas and overcome the challenges when using them.
4.3 Aqueous two-phase system templated polysaccharide microparticles
ATPS are formed from specific mixtures of two hydrophilic polymers or salts at elevated concentration.60,61 A commonly used system is the combination of PEG and DEX. These systems feature a completely water-based environment and very low surface tensions between the two phases.7,198 In recent years, ATPS have been utilised frequently for the separation of biomacromolecules as well as nanoparticles199–201 or as environment for enzymatic catalysis.202 In addition to macroscopic ATPS, dispersed systems, i.e. water-in-water emulsions, have been investigated as well135,136 that can be employed as template for the formation of microparticles in aqueous environment.203 Huck and coworkers synthesised microparticles via microfluidics with droplets containing the ATPS from DEX and poly(ethylene glycol) diacrylate (PEGDA) that were formed in oil.203 The diacrylate phase was photopolymerised to obtain stable particles. In such a way, DEX core/PEGDA shell particles were produced and the particle shape could be tailored via the DEX/PEGDA ratio, e.g. core/shell or particles with socket. Another crosslinking technique was explored by Ono and coworkers, namely amide formation via active esters and amines (Fig. 8).204 In a microfluidic avenue, droplets of PEG/DEX were formed in oil. After phase separation, the PEG phase settled as the shell of the aqueous droplets. Notably, the PEG solution was fed into the system via two streams as two PEG species were employed, i.e. tetra-arm PEG with either NH2 or N-hydroxy succinimide ester functionality that crosslinked immediately after phase separation. In such a way, crosslinking was performed directly without light or other stimuli, revealing PEG/DEX capsules.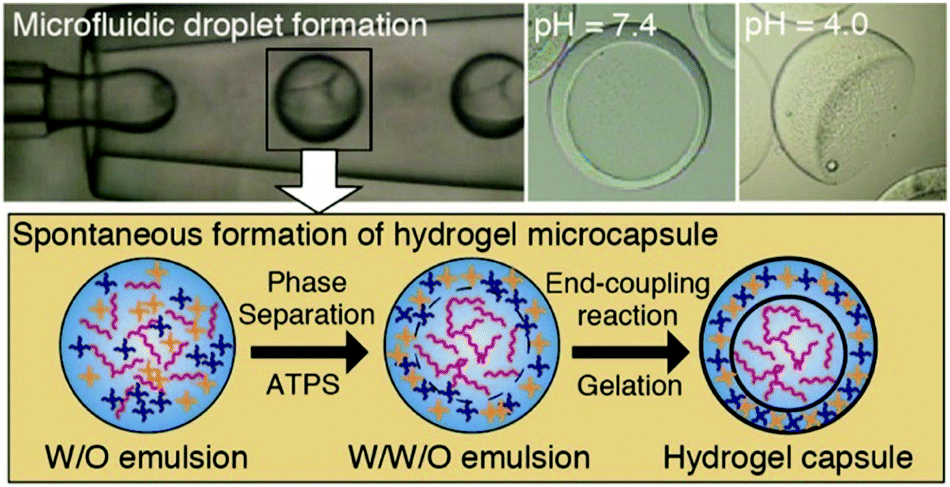 | ||
| Fig. 8 Formation of PEG/DEX capsules via ATPS templating in a microfluidic system. (Reproduced with permission from ref. 204. Copyright American Chemical Society, 2019.) | ||
The fabrication of Janus particles was described by Shum and coworkers.205 Here, also microfluidics were utilised to form droplets containing PEG and DEX in an oil. In order to form half-spherical particles, the partitioning behaviour of agarose in the ATPS was exploited that preferably enriches in the PEG phase. As agarose solidifies at low temperatures, the PEG phase was stabilised against dissolution in water, while the DEX phase dissolved revealing PEG half spheres at temperatures below 17 °C. PNIPAM/alginate based microparticles were presented by Arkedani and coworkers, who used Ba2+ for ionic crosslinking of alginate and microfluidics for droplet formation and thermoresponsive PNIPAM for templating.206 A completely water-based approach was introduced by van Esch and coworkers.207 Three streams were employed in microfluidics, i.e. a PEG-containing stream for the core, a DEX-containing stream for the shell and a PEG-containing stream as continuous phase. PEG-in-DEX droplets were formed in a PEG-solution and the DEX phase crosslinked via photo-induced thiol–yne reaction. As such, permeable capsules were synthesised in a completely aqueous system.
ATPS provide a useful avenue to template the formation of polysaccharide based microparticles in a completely water-based environment. As such, organic solvents can be excluded, which is highly interesting for bio-related applications.
5. Applications of polysaccharide nanoparticles
In various areas of research and application, nano-sized delivery systems based on polysaccharide NPs have been widely used in transporting biomedical “cargoes”, for example, drugs, genes, polypeptides, proteins, vaccines or nucleic acids. Moreover, polysaccharide NPs can be used in other areas of applications such as Pickering emulsion stabilisers/emulsifiers and as reinforcing agents in various materials due to their environmental-friendly properties. For instance, when polysaccharide NPs are used to enhance other biodegradable and renewable polymers, efficient and eco-friendly materials with adequate thermo-mechanical properties can be obtained, which is crucial for the development of materials without any environmental issues and overcoming the disadvantages of the original material, e.g. fragility.19,56For example, Iijima and coworkers utilised polysaccharide NPs as precursors for polysaccharide films that were studied with respect to interactions with biological cells.208 CS was combined with anionic polysaccharides to form NPs and subsequently the dispersions were cast into molds and dried at elevated temperature to obtain particle-based films. Depending on the anionic polysaccharide type cell adhesion could be tailored.
Practical applications of polysaccharide NPs in the fields of nanomedicine, cosmetics, and food systems are summarised in the following sections and discussed according to their suitability and required properties for the respective application.
5.1 Polysaccharide nanoparticles in nanomedicine
In biomedicine and biomaterials engineering, polysaccharide NPs have significant advantages and potential applications due to their biocompatibility and biodegradability.19,209 Especially in the field of nanomedicine, polysaccharide NPs can be exploited due to their favourable properties in biological systems, e.g. due to their mucoadhesive properties and cell uptake.210,211 As such, applications like drug-delivery and tissue engineering are discussed frequently but also other therapeutic developments have been presented in recent years. For example, Park and coworkers described the utilisation of CS-based nanoparticles for photodynamic therapy.212 There, the photosensitiser Pheophorbide A was conjugated to CS via a reactive oxygen species sensitive thioketal linker. The amphiphilic CS-conjugate self-assembled into NPs in aqueous environment leading to a self-quenching of the photosensitiser. The presence of reactive oxygen species in tumour environment resulted in thioketal bond cleavage and release of the photosensitiser. The photosensitiser was activated by near infrared irradiation in turn with a toxic effect. Another approach to photodynamic therapy was described by Wich and coworkers.213 A fully polysaccharide-based block copolymer consisting of DEX and acetalated DEX blocks was synthesised via a copper free click reaction based on thiol exchange leading to disulfide connected blocks. Next, micelles were formed and phthalocyanine zinc as photosensitiser encapsulated, where the block copolymer acted as surfactant for the zinc complex. Delivery of the payload was investigated in vitro, showing the block copolymer stabilised the photosensitiser with increased bioavailability. Upon irradiation with NIR light, a strong concentration dependent toxic effect was observed. It should be mentioned that the disulfide block connection introduced redox responsive degradation of the micellar NPs. In a subsequent contribution, DEX NPs were conjugated with folic acid to improve specific uptake of the photosensitiser into cancer cells (Fig. 9).214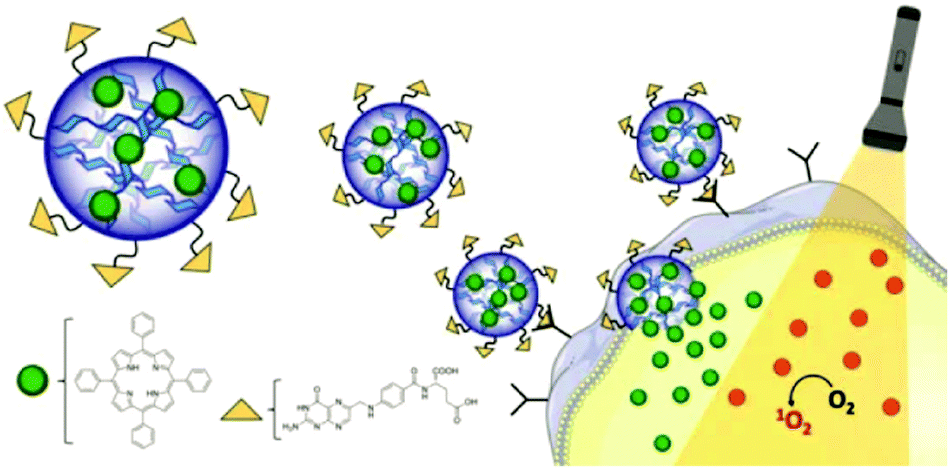 | ||
| Fig. 9 Mechanism of folic acid targeted photodynamic therapy: DEX NPs loaded with tetraphenylporphyrin are taken up via endocytosis and release the photosensitiser for phototoxic effect.214 (Licensed under CC-BY 2019.) | ||
Alonso and coworkers studied CS/DS or CS/HA coacervate NPs loaded with an HIV peptide antigen candidate, i.e. HIV protease cleavage site 5 peptide (PCS5).215 The authors investigated various factors in their NPs and the effect on immune response. For example, the attachment of the antigen via ionic interactions, cleavable or non-cleavable conjugation was investigated as well as the presence of immunomodulatory molecules such as poly(inosinic acid):poly(cytidylic acid) (poly(I:C)). All NP formulations showed a generation of antibody responses. Overall, NPs with PCS5 conjugation and poly(I:C) incorporation featured the strongest activation of antigen-presenting cells. Sacco and coworkers investigated the effect of CS acetylation on the immune cell response.125 CS was combined with short HA in a coacervate formation for the synthesis of NPs. The particles were studied regarding stability at different pH and osmolarity, where only one type of acetylated CS showed stability over a broad range of conditions. In the subsequent immune response studies, the stable formulation induced minor immunogenic response while less stable compositions induced tumour necrosis factor-α production by macrophages. The study revealed that the hydrophilic/hydrophobic balance of polysaccharides is major factor in interactions with biological systems.
Two most relevant applications of polysaccharide nanoparticles in biomedicine, i.e. drug/gene delivery and tissue engineering, are discussed in detail below.
Furthermore, polysaccharide NPs were utilised to improve the solubility of poorly water-soluble drugs, transport the active drug molecules to a targeted pathological site, and release drugs controllably and sustainably for drug/gene delivery.19 Also, various useful theranostic systems based on polysaccharide NPs were developed, as summarised by Lee and coworkers, e.g. fluorescence imaging and drug delivery, magnetic resonance imaging and drug delivery or near-infrared fluorescence imaging and drug delivery.58
The polysaccharides discussed above (Section 2) can effectively be used to develop suitable delivery systems. Aminabhavi and coworkers described diverse pharmaceutical applications of CS particulate systems, including cancer therapy, gene delivery, mucosal delivery, topical delivery, ocular delivery and colon targeted drug delivery.87 Yameen's group developed novel enzyme-responsive DEX based oligoester crosslinked NPs for the controlled release of an anticancer drug, 5-fluorouracil (5-FU).217 The results obtained from their study showed that 5-FU was effectively encapsulated in the DEXNPs (Fig. 10), and the synthesised DEXNPs maintained their stability at pH conditions representative of specific in vivo environments (e.g. stomach, small intestine, and colon). Moreover, as reported by the studies of release kinetics and colon cancer cell viability, it was found that 75% (±4) of the encapsulated 5-FU was released in 12 h in the presence of dextranase and the used HCT116 cell displayed a substantial cell viability decrease. These results demonstrated the potential applications of the studied DEXNPs as a specific 5-FU delivery system in response to the dextranase enzyme. In addition to the DOX-loaded HANPs discussed in Sections 3 and 4, other polysaccharides and their derivatives were also investigated by different research groups for their utilisation in DOX delivery. Based on their studies, CSNPs,48,87,147 CLNPs,218,219 SNPs,220,221 and DEXNPs63,222,223 proved to be potential nanocarriers for antitumour drugs such as DOX in order to enhance their therapeutic effects.
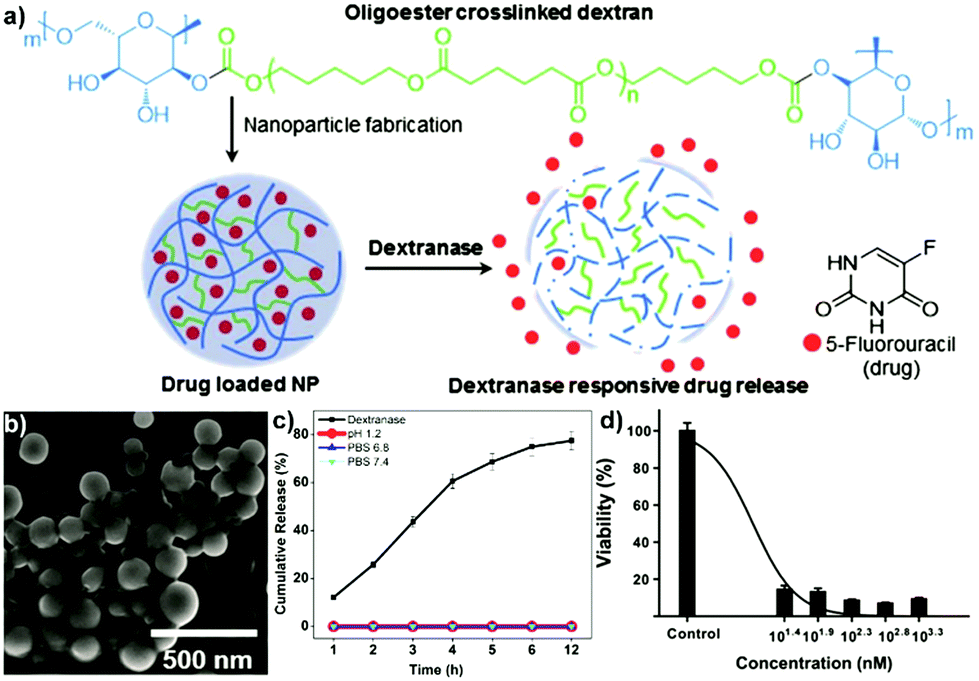 | ||
| Fig. 10 (a) Schematic overview for nanoprecipitation assisted fabrication of DEXNPs in the presence of 5-FU, as well as depiction of dextranase responsive drug release, (b) SEM image of 5-FU loaded DEXNPs, (c) release kinetics of DEXNPs at simulated pH 1.2, PBS 6.8, PBS 7.4 & dextranase (15 U mL−1) and (d) cytotoxicity of DEXNPs against HCT 116 colon cancer cell line after 1 h of exposure of DEXNPs to dextranase. (Reproduced with permission from ref. 217. Copyright Elsevier, 2020.) | ||
Some polymer NPs with polysaccharide shell have also attracted attention to be used as adequate nanocarriers. For encapsulating and protecting Astaxanthin, Xu and coworkers utilised CS oligosaccharides to form a cationic hydrophilic shell around the anionic hydrophobic PLGA NPs.149 The results of their work showed that the core–shell NPs formed had improved water solubility and stability compared to the PLGA NPs without a CS oligosaccharide shell, making the developed NPs suitable for their future applications in transporting hydrophobic nutraceuticals and drugs. A polysaccharide NP based on DS/CS for the delivery of insulin was described by Neufeld and coworkers.224 The NPs were designed for oral dosage and provide improved intestinal absorption. An improved performance compared to oral insulin solution revealing 2–3-fold improvement of oral relative pharmacological availability was observed. Heinze and coworkers described the attachment of a histone deacetylase inhibitor into CL or DEX-based NPs, i.e. valproic acid.225 The acid was attached via esterification and NPs formed via nanoprecipitation or emulsion method. The formed particles could be used for valproate release via enzymatic cleavage of the ester bond, which was studied in vitro and in shell-less Hen's egg tests. Overall, the nanocarriers revealed good biocompatibility. An alginate-based system for the delivery of NO and Ag NPs was described by Seabra and coworkers.226 Alginate NPs were synthesised via gelation with Ca2+ and a precipitation. Furthermore, Ag NPs synthesised from AgNO3 and tea extract and mercaptosuccinic acid (a NO acceptor) were encapsulated into the alginate NPs. The particles could be loaded with NO via nitrosation of mercaptosuccinic acid and utilised subsequently for antibacterial purpose. The particles showed antibacterial activity against three bacteria strains indicating a synergistic effect of NO release and Ag NPs, while not being toxic to Vero cells.
Gene delivery is a frequently investigated application of polysaccharide NPs.227–229 Especially, CS complexation with siRNA or DNA for gene delivery has been studied frequently.230–232 For example, Leong and coworkers synthesised CS-based nanoparticles with DNA loading.233 Thus, CS/DNA NPs were formed via a coacervation process. The particles were PEGylated and conjugated with transferrin to enhance stability and cell uptake. Minimal cytotoxicity was observed and gene transfection in various cell types e.g. HEK293, HeLa or IB-3-1, which could be tailored via the surface functionalisation. Fréchet and coworkers described gene delivery based on DEX NPs.234,235 DEX was partially oxidised via periodate, acetalated and conjugated with spermine by reductive amination. The carrier particles were generated via a double emulsion technique and directly loaded with siRNA. The cationic spermine functionalisation acted as a handle to complex with siRNA for high loading efficiency. Gene transfection was studied via knockdown of luciferase expression in HeLa cells in vitro. As the spermine/siRNA interaction is sensitive to pH, selective release was observed after internalisation in the cell only.
A NP based on PL was described by Letourneur and coworkers (Fig. 11).236 Cationic modified PL was combined with Fucoidan in a coacervation process to form NPs and microRNA adsorbed at the surface. The system promoted microRNA delivery inside cells and selectively targeted towards cells with P-selectin over-expression at the surface of activated platelets and endothelial cells in atherothrombotic-related diseases. Amiji and coworkers studied HA-based gene delivery.237 HA was grafted with hexyl chains to introduce hydrophobicity, PEG to enhance particle stability and poly(ethylene imine) (PEI) to facilitate interaction with siRNA. Tumour necrosis factor-α-specific siRNA was encapsulated and the uptake as well as down regulation of tumour necrosis factor-α in macrophages observed. As such, an improved survival rate was observed in the mouse model. Gene carriers based on alginate/Ca2+ complexation have been described as well. Peng and coworkers formed alginate NPs via W/O microemulsion and crosslinking via Ca2+.80 In order to test gene delivery, GFP-encoding plasmids were encapsulated and the degree of endocytosis by NIH 3T3 cells was investigated. Ca–alginate NPs with an average size around 80 nm in diameter were very efficient gene carriers compared to plasmid DNA (pDNA) condensed by PEI.
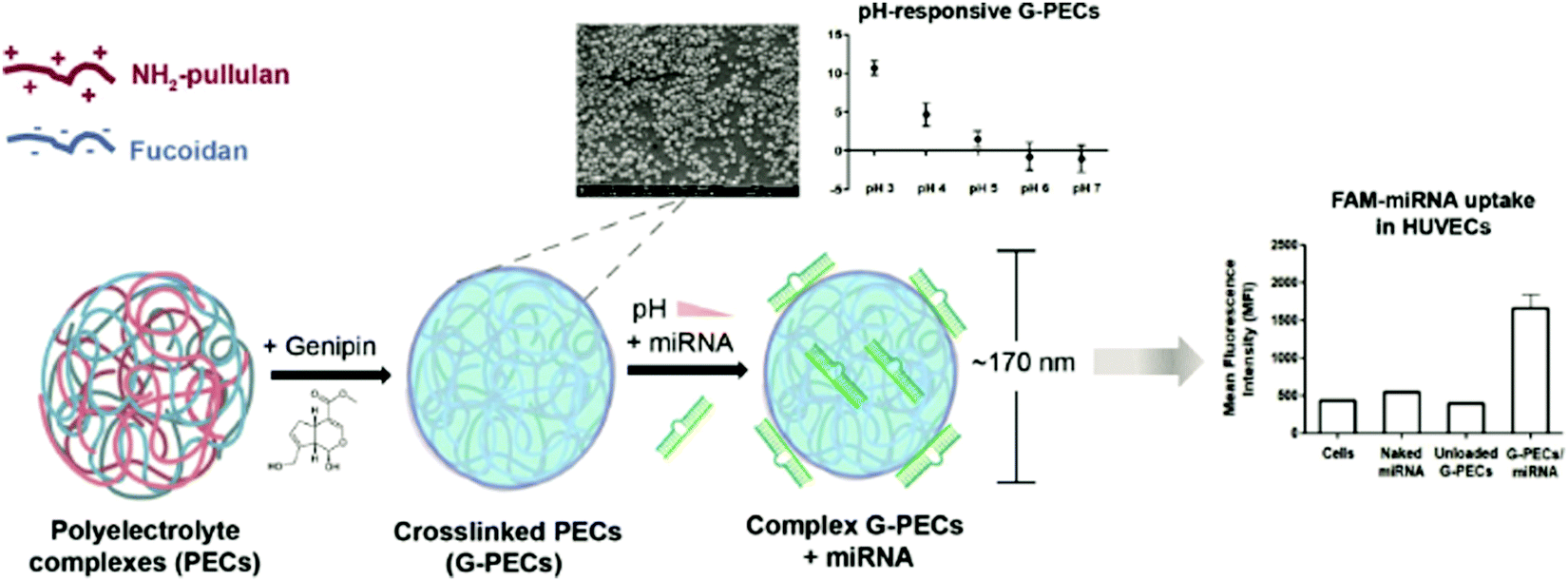 | ||
| Fig. 11 Formation of NH2-PL/fucoidan coacervate NPs and loading with microRNA (miRNA) (left), particle characterisation via SEM and pH response (top) and mean fluorescence intensity (MFI) of fluorescein amidite (FAM) measured by flow cytometry analysis revealing the uptake in human umbilical vein endothelial cells (HUVECs) (right). (Reproduced with permission from ref. 236. Copyright Elsevier, 2021.) | ||
NPs from CRG for drug-delivery purposes have been described as well, where frequently a complexation with CS was used for NP stabilisation. Grenha et al. described the formation of NPs from κ-CRG and CS via a complex coacervation process.238 Particles in a size range from 400 to 700 nm were described with adjustable properties according to the ration of the two polysaccharides. In the next step, loading with OVA as well as release was probed. A sustained and controlled release of OVA over three weeks was observed. A similar structure was described by Chung and coworkers, who used ι-CRG and CS-based NPs for the loading and delivery of insulin via oral administration.239 Abdelghany et al. produced κ-CRG and CS-based NPs for tuberculosis treatment via ethionamide encapsulation.240 The CRG content was varied to study the effect on particle size, zeta potential and drug release. In particular, it was found that CRG stabilised the particles and enhanced ethionamide entrapment. A sustained release was observed and considerable antimycobacterial activity. A CRG-based capsule was described by Tánori and coworkers, who utilised an emulsion process to form capsules from κ-CRG and CS.241 At first olive oil nano emulsion droplets were formed and κ-CRG/CS deposited at the interface via layer-by-layer assembly. The oil droplets were loaded with the lipophilic anti-inflammatory drug Diflunisal and the release studied. A zero-order release profile was observed for three- and four-layer systems, while first order release kinetics were observed for one and two layers as well as a Fickian diffusion for emulsion droplets without polysaccharide coating.
To enhance the cellular delivery of genetic materials, NPs with polysaccharide coating were described as well, for example HA–PEI, HA–PEG, and HA–PEI/HA–PEG were synthesised by Amiji and coworkers in order to encapsulate pDNA.155 The negatively charged HA formed the hydrophilic shell of the NPs, and the combination of HA, PEI and PEG has a considerable effect on gene expression results because of the ability of HA–PEI/HA–PEG NPs to protect pDNA from the environment. In addition, the appropriate pDNA accessibility of HA–PEI/HA–PEG NPs could result in controlled release of pDNA and improved transfection in cells, which might be useful for the delivery of genetic material for cancer therapy. Liu et al. designed a novel core–shell nanoparticle system based on PL and poly(β-amino)ester (PBAE) for the codelivery of pDNA expressing green fluorescent protein (pEGFP) and the model chemotherapy agent methotrexate (MTX).156 The MTX-PL/PBAE/pEGFP NPs prepared had a PL shell and a PBAE/pEGFP core. Also, they were found to exhibit a strong hepatoma-targeting property at both cellular and animal levels, indicating its potential for combination of chemotherapy and gene therapy. Although coating drug/gene-loaded hydrophobic polymer NPs with a hydrophilic polysaccharide shell can improve their stability and water solubility for improved delivery, information of the safety and efficacy of the polysaccharide/polymer systems still requires more research.149
Drug/gene delivery systems based on polysaccharide NPs have great potential in the treatment of cancer and other diseases such as diabetes. However, further study is crucial to overcome several challenges in their use. For example, the drawbacks resulting from the reliance on the EPR effect for passive targeting during the drug/gene delivery: heterogenicity of the EPR effect caused by factors such as variations in NP size can result in uncertainty in delivery efficiency. Moreover, burst release and compromised mechanical properties caused by incomplete degradation of polysaccharide NPs is an issue that should not be ignored.242 Thus, ongoing development of targeted delivery systems is required to improve drug/gene delivery efficiency.
Mandal and coworkers prepared dual growth factor loaded CSNPs by using epidermal growth factor (EGF), fibroblast growth factor (FGF) and CS.245 The increased proliferation of fibroblasts in vitro demonstrated the successful delivery of growth factors from CSNP. Moreover, non-toxicity of growth factors loaded CSNPs and controlled release were observed, and the attachment and growth of fibroblast in EGF/FGF–CSNP system were significantly improved. These results indicated the promising utilisation of this dual growth factors loaded nanoparticle system in tissue engineering area.
In the work of Alonso and coworkers, CS/HA based NPs were synthesised by using TPP ionic gelation for transporting vascular endothelial growth factor (VEGF) and platelet derived growth factor (PDGF-BB).57 The results demonstrated that the CS/HA based nanocarriers were stable in biological media without any cytotoxicity. According to their in vitro release studies, PDGF-BB could be released from CS/HA NPs sustainably in one week, while the release of VEGF was uncontrollable. This report already showed the future of injectable PDGF-BB loaded CS/HA nanoparticle system in the fields of tissue engineering and treating ischemia-related diseases but further research is needed in the control of growth factor release. Wu and coworkers described a novel DEX- and gelatin-based nanoparticle system (DGNPs) for encapsulating and transporting the growth factor bone morphogenetic protein (BMP).246 The mono-dispersed DGNPs formed had a mean diameter of about 54 nm and good swelling and degradation properties in the target media. According to their in vitro release studies, more than 90% of the loaded BMP was maintained for more than 12 days and released in a sustained manner. These positive results proved that the formed DGNPs have the potential to be biodegradable and biocompatible carriers for protein-based growth factors. Goncalves and coworkers presented NPs based on CS and poly(γ-glutamic acid) with diclofenac as non-steroidal anti-inflammatory drug for the treatment of degenerated intervertebral disks.247
Tayebi and coworkers described a microfluidic avenue for the preparation of CS/dexamethasone NPs that were utilised as adjuvants in cell growth (Fig. 12).248 The effect of the adenosine triphosphate-crosslinked particles on the osteogenic differentiation of mesenchymal stem cells was studied. Due to the tailored size of the NPs improved differentiation was observed via enhanced dexamethasone release.
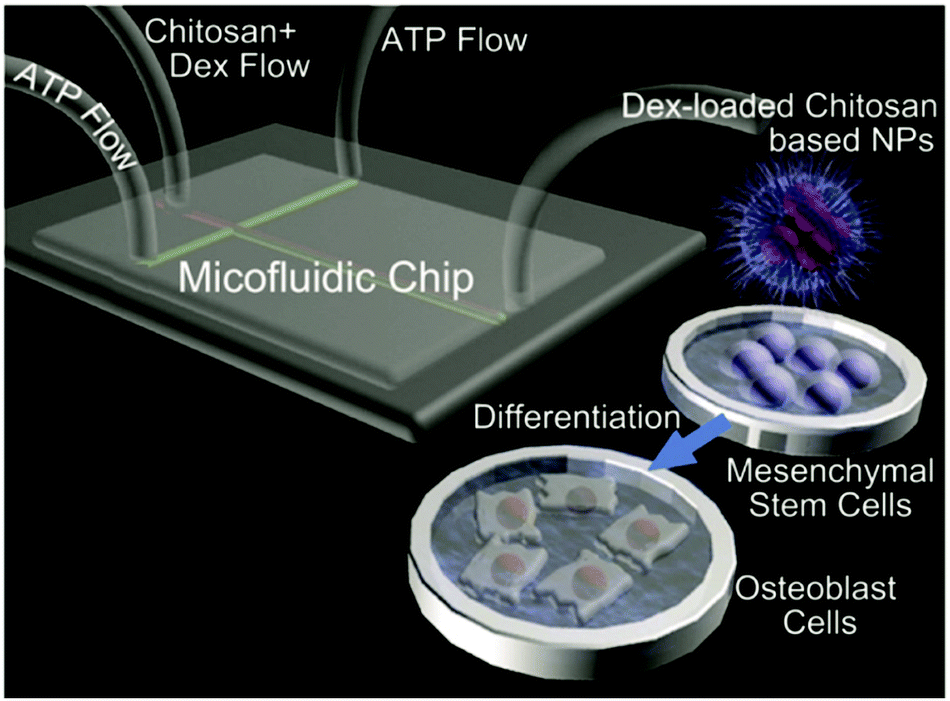 | ||
| Fig. 12 Schematic representation of a cross-junction microfluidic device used to hydrodynamically focus the chitosan–dexamethasone flow using a sheath flow of adenosine triphosphate-containing water for the preparation of CS-based NPs and their use in cell growth. (Reproduced with permission from ref. 248. Copyright Elsevier, 2015.) | ||
It should be noted that polysaccharide NPs with hydrophilic properties can be utilised as adequate growth factor nanocarriers, but several issues (e.g. safety, cost, efficacy) need to be addressed for development of successful growth factor therapies. The preclinical results from in vitro cellular study and in vivo animal model may not perfectly simulate the physiology and pathophysiology in humans, which has to be studied thoroughly to enable future applications.242,244 Thus, further studies are required to enhance the control over polysaccharide NP-governed tissue growth processes.
5.2 Polysaccharide nanoparticles in cosmetics
Cosmetics are consumer products to be placed in contact with the external parts of the human body, with teeth or the mucous membranes of the oral cavity. The primary utilisations of cosmetics include cleaning, change of appearance, and body odour correction.249 The use of polysaccharide-based NPs is a specific branch area of applying nanotechnology in cosmetics. Many researchers have investigated the applications of polysaccharide-based NPs in improving the properties of cosmetic products, for instance, using them as cosmetic ingredients to stabilise Pickering emulsions or deliver useful substances.10Emulsions stabilised with solid NPs are also referred to as Pickering emulsions (Scheme 8). Though surfactants can enhance both the kinetic and thermodynamic stability of the emulsions, their toxicity, high cost, and impractical recovery lead to issues.251,252 However, solid NPs such as polysaccharide NPs used in Pickering emulsions can be environment-friendly and biologically compatible. Besides, Pickering emulsions can impart improved stability against coalescence. These advantages cause that Pickering emulsions can replace the surfactant stabilised emulsion to produce long-term stabilised emulsions to be applied in cosmetics.251,252,254 In the review of Yuan and coworkers, various types of solid particles utilised in forming Pickering emulsions were listed and discussed in detail.250 For example, nanoscale particles derived from natural polysaccharides such as CH nanocrystals,19 CL nanocrystals,255,256 and starch nanocrystals256–258 were found to be potential Pickering emulsion stabilisers due to their superior performance in various areas.
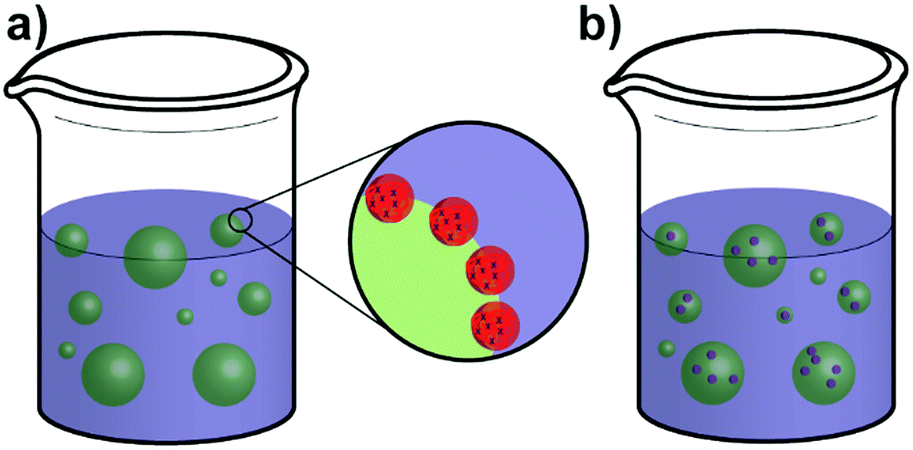 | ||
| Scheme 8 (a) Structure of a polysaccharide NP stabilised Pickering emulsion and (b) Pickering emulsion with cargo loaded droplets. | ||
Biodegradable and antibacterial CS is widely used in cosmetics,259 and CSNPs have been investigated by many researchers for their utilisation in stabilising Pickering emulsions. Tong's group described a simple, straightforward reversible, and long-term stable (>2 months) Pickering emulsion system based on CS.260 At high pH conditions (pH > 6), CS is water-insoluble and the CSNPs were formed in situ by aggregation. These CS aggregates adsorbed at the interface of oil and water to stabilise the O/W emulsions, forming an excellent stabilised Pickering emulsion system. While at low pH conditions (pH < 6), CS is water-soluble and the demulsification phenomenon occurred. The results obtained from this study demonstrated that CS is a practical pH-sensitive emulsifier, and the Pickering emulsions formed by CS could be useful to fabricate a broad range of reversible emulsion systems.
The abundant, low-cost, and biodegradable starch also has potential applications in stabilising cosmetic Pickering emulsions.261,262 However, the broad size distribution of starch particles negatively affects its performance as an emulsifier.250 Therefore, various modifications (e.g. hydrophobic modification) of starch have been investigated to increase the possibility of SNPs to adsorb at the oil–water interface during emulsification.261 Wang and coworkers described a novel particulate emulsifier based on starch–acetic anhydride–phthalic anhydride nanospheres for undecanol- and glyceryl trioctanoate-water emulsions.263,264 According to the results from confocal laser scanning microscopy, the nanospheres were adsorbed at the liquid interface, forming a dense film around the dispersed droplets. Besides, it was found that salt addition could cause emulsion coalescence, and the O/W emulsions could be inverted to W/O emulsions by varying the water/oil volume ratio and pH of aqueous dispersion, respectively. These results demonstrated the promising future of using biocompatible SNPs as practical Pickering emulsions stabilisers.
As the properties of starch depend on its origin, the possibilities of native starches extracted from different sources such as taro, wheat, waxy maize, rice, and potato as Pickering emulsions stabilisers were also studied by other groups.265,266 For instance, Sun's group prepared four different types of SNPs to stabilise the soybean O/W emulsion system.261 Their results showed that the SNPs with near-neutral wettability were synthesised from sweet potato, corn, and tapioca. The near-neutral wettability promoted the adsorption of SNPs at the oil–water interface and the Pickering emulsion stabilisation. In addition, it was also found that the SNPs size could affect the stability of the emulsion, the corn-derived SNPs were less stable than the other two because of the smaller particle size and the caused deviated contact angle from 90°.
A combination of NPs derived from CS and starch for the formation of Pickering emulsions was described by Matos and coworkers.267 Due to their considerable difference in size (nm vs. micron), the order of stabiliser addition had a profound effect on the obtained droplet sizes of oil in water. When CS particles are added first, both particle types perform stabilisation, while in the case of simultaneous addition, stabilisation is predominantly due to the larger starch NP addition that displace CS particles. Wang and coworkers employed starch NPs as Pickering stabiliser for the fabrication of microgels.268 At first starch was hydrophobised via butyl glycidyl ether and palmitate, then NPs were formed via nanoprecipitation. In the next step, an inverse Pickering emulsion was formed with n-hexane as the continuous phase and an aqueous solution of acrylamide, acrylic acid and initiator as dispersed phase. After homogenisation the polymerisation was started at 30 °C and well-defined microgels decorated with starch NPs were obtained. Héroguez and coworkers fabricated Pickering emulsions of medium chain triglyceride or dodecane in water via acetalated DEX NPs (Fig. 13).269 The acetalated DEX NPs were formed via nanoprecipitation and had a diameter around 200 nm. As acetals can be hydrolysed under acidic conditions the particles featured pH responsivity, leading to destabilisation of the emulsion after hydrolysis.
 | ||
| Fig. 13 Cryo-SEM image of the surface of a dodecane droplet in water stabilised by acetalated DEX NPs. (Reproduced with permission from ref. 269. Copyright American Chemical Society, 2020.) | ||
In summary, applying polysaccharide NPs in Pickering emulsions can reduce the tendency of emulsion coalescence and introduce high stability, biocompatibility, and non-toxicity, which will provide support to developing stabilised Pickering emulsions used in different areas such as cosmetics.
Kim et al. successfully encapsulated a cosmetical vitamin A alcohol, e.g. retinol, into CSNPs for cosmetic and pharmaceutical applications.271,272 As such, the solubility issues of retinol in water was solved by incorporating it into CSNPs, and the study showed that the retinol water solubility was increased more than 1600-fold. Inspired by this, Harris et al. prepared CS hydrochloride (HCS)–TPP based NPs for the encapsulation of yerba mate (Ilex paraguariensis (ILE)) extract.273 The results demonstrated that the CSNPs involved encapsulation method is a promising technique to prepare a vehicle for antioxidants protection, e.g. ILE-polyphenols, which are significant ingredients in the nutraceutical and cosmetic area.
The polysaccharide NPs stabilised Pickering emulsion system discussed in Section 5.2.1 can also be used to encapsulate bioactive substances in cosmetics (Scheme 8b). Curcumin has been considered as a cosmetic ingredient due to its antioxidant and anti-inflammatory effects on the skin. The efficiency of curcumin can be enhanced by nanoencapsulation, which provides improved skin penetration and retention.274 Several researchers have investigated the utilisation of CSNPs stabilised Pickering emulsion as a curcumin delivery system. For instance, Li and coworkers encapsulated curcumin in CS–TPP NPs stabilised medium chain triglyceride–water Pickering emulsion system.50 According to the results of stability and in vitro release studies, the half-time for the encapsulated curcumin degradation was about 120 hours, and a sustained curcumin release was obtained. It was also found that the stabilised Pickering emulsion system had high stability against pH change and salts, indicating that the CS–TPP NPs stabilised Pickering emulsion could be an adequate delivery system for bioactive compounds such as curcumin.
The utilisation of CH nanofibril–HA based NPs (CH/HA NPs) as a cosmetic formulation to deliver active ingredients in skin care was discussed by Morganti et al. (Fig. 14).275 In their study, lutein was used as an example of the active ingredient to be delivered. In vitro studies were conducted to evaluate the anti-aging activity of the CH/HA NPs by measuring antioxidant capacity, anti-collagenase activity, and metalloproteinase and pro-inflammatory release. Also, a 60 days double-blind vehicle-controlled study on a subject group of 60 persons affected by photo-aging was carried out to show the in vivo efficacy of the CH/HA NPs. CH/HA NPs were demonstrated to be capable to easily loaded active ingredients, facilitate penetration through the skin layers, and increase their effectiveness and safety as an anti-aging agent. Moreover, the development of biodegradable and biocompatible CH/HA NPs for the targeted delivery of specific ingredients could improve skin aging activity and accelerate wound healing processes.
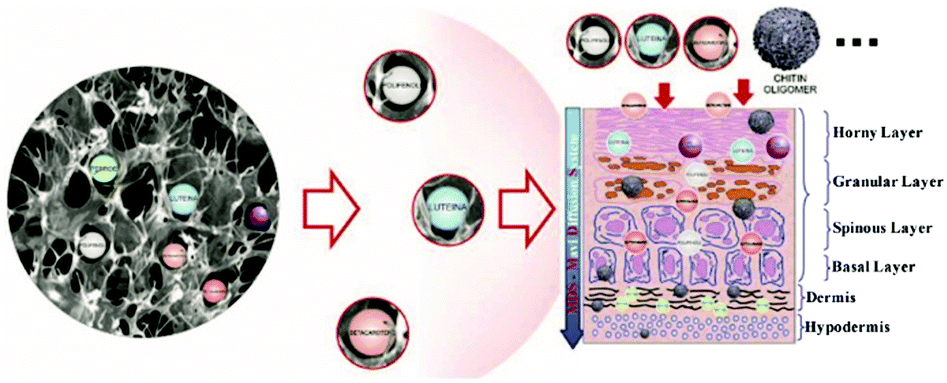 | ||
| Fig. 14 CH/HA NPs have the capacity to disturb the stratum corneum lamellae organisation, increasing the skin penetrability of the active ingredients entrapped, e.g. polyphenols, lutein or β-carotene.275 (Licensed under CC-BY 2014.) | ||
Polysaccharide NP-based delivery systems have not only been utilised in skin care, but also in hair care. Gelfuso and coworkers developed minoxidil sulphate (MXS)-loaded CSNPs for targeted delivery to hair follicles.276 MXS plays a very significant role in treating male and female androgenetic (or pattern) alopecia, however, there are many limitations when using MXS alone, such as low Minoxidil water solubility, high irritation caused by continuous use of the market formulations, and the side effects due to its potent antihypertensive activity. Thus, the targeted delivery of Minoxidil to hair follicles is a significant topic in cosmetics.249 In the work of Gelfuso and coworkers, the MXS loaded CSNPs prepared could accumulate into the hair follicles and release the MXS in a sustained manner. Moreover, there was a two-fold MXS increase in the hair follicles when compared to a control solution, demonstrating that the MXS loaded CSNPs are easy and promising delivery systems to improve the topical treatment of alopecia.
Considerable advantages of polysaccharide NPs based cosmetic delivery systems are their remarkable biodegradability, biocompatibility, and high stability. However, both fungal and bacterial degradation can be a drawback in cosmetic formulations using biodegradable polymers, which has to be addressed in future research directions. In addition, it is necessary to put more emphasis on regulatory status and production processes to reduce the cost and pollution for further advancement.270,275
5.3 Polysaccharide nanoparticles in food systems
In food and agricultural systems, the applications of nanotechnology are growing rapidly. Nanoscale control may lead to the modification and improvement of various macroscale characteristics, such as processability, stability, shelf-life, taste, texture, and other sensory attributes, which enables the design of healthier, tastier, and safer foods by manipulating food ingredients more precisely.277,278 As such, there are many potential applications for polysaccharide-based NPs to be used in food and agricultural systems. Some examples of how polysaccharide NPs can be used in food packaging and food delivery systems will be discussed below.A typical example of developing nanoparticle packaging additives, which simultaneously have intelligent and active properties was described in the study of Kritchenkov et al.280 Their work was on the basis of the preparation and characterisation of polysaccharide based NPs which had a PEG/methylcellulose (MC) core including anthocyanidin and sodium acetate, and a CS/gallotannin-based shell. The PEG/MC core and anthocyanidin/sodium acetate acted as the smart part of the NPs. As such, a system with Tg corresponding in the range of the unacceptable temperature for product storage was designed via the combination of PEG and MC combining a very high and a very low Tg. Anthocyanidin and sodium acetate were mixed to indicate colour change, the neutral colourless anthocyanidin under action of sodium acetate turned purple with increasing temperature of PEG/MC mixture. The active part of the particles was the CS/gallotannin-based shell, which takes the advantage of antibacterial CS and photoprotective and antioxidative gallotannin. The analysis results showed that the prepared SANPs turned from colourless to purple at about 20 °C and 0 °C respectively. Moreover, the SANPs displayed effective antibacterial activity, UV light absorption, antioxidative activity, and non-toxicity, demonstrating the promising application of the prepared polysaccharide-based SANPs in the food packaging area.
The influence of polysaccharide NPs such as CSNPs on the improvement of the physical/chemical properties of food packaging materials such as mechanical strength and thermal stability were investigated by various groups.281,282 For example, the addition of polysaccharide NPs enables reduction of the water vapour permeability (WVP) of food packaging materials, which has attracted frequent attention as it is a vital issue for the development of sustainable packaging materials. NPs are considered to be suitable for reducing WVP because they distort the diffusion pathways for the water vapour through the packaging film.283,284 In the area of mechanical improvements, Mattoso and coworkers described the fabrication of nanocomposite films of CS NPs in pectin matrix (Fig. 15).282 CS NPs were combined with high methyl (HDM) and low methyl (LDM) pectin matrices. Next, films were casted that showed homogeneous, compact, and continuous properties. The addition of CSNPs improved the mechanical properties considerably, e.g. tensile strength increased from 30.81 to 46.95 MPa and from 26.07 to 58.51 MPa for HDM pectin/CSNP and LDM pectin/CSNP, respectively.
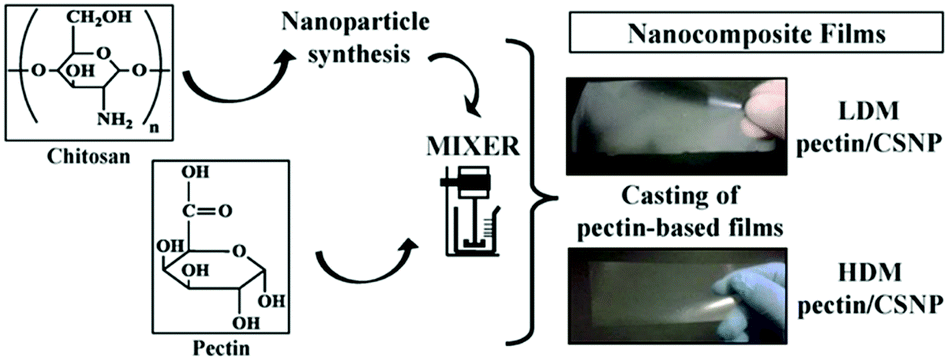 | ||
| Fig. 15 Overview of nanocomposite formation from pectin and CS NPs and casted films. (Reproduced with permission from ref. 282. Copyright Elsevier, 2016.) | ||
Sun and coworkers investigated the effects of taro-derived SNPs content on the properties of corn starch-based packaging films.285 Their work showed that the addition of taro SNPs to the corn starch films increased the tensile strength of films from 1.11 MPa to 2.87 MPa. It was also found that strong interactions between taro SNPs and films improved the thermal stability of starch films. Moreover, the WVP of the films was decreased after addition of taro SNPs, and the degree of the WVP reduction correlated with the concentration of the taro SNPs. These results demonstrated the potential of incorporating taro SNPs into corn starch films for developing biodegradable starch-based films with improved properties. A similar study was conducted by the same group.286 Potato-derived SNPs were incorporated into pea starch-based nanocomposite films. The mechanical properties of the films were found to be enhanced by the addition of potato SNPs, and particularly for the incorporation of 6% potato SNPs, the film tensile strength increased the most (from 8.8 MPa to 15.0 MPa). Compared to control samples, the melting temperature of the nanocomposite films increased with the increased potato SNPs content. For the WVP and water-solubility of the nanocomposite films, the incorporation of 6% potato SNPs led to the most significant reduction of the WVP value but it increased again with higher content values. The phenomenon was assigned to the abundant hydroxyl groups in excessive potato SNPs showing more affinity for water than starch. The lowest water-solubility of the nanocomposite films was also obtained at the potato SNPs content of 6%. The improved reduction of water-solubility was considered to be related to the high tensile strength values and the entanglement between SNPs and starch chains, which decreased the nanocomposites’ solubility. The results discussed above proved that adding potato SNPs into the pea starch films could be applied in developing degradable food packaging, while WVP and water-solubility can be minimised via the optimisation of material composition.
Polysaccharide NPs play a significant role in developing improved food packaging materials. They can combine the advantages of both intelligent packaging and active packaging. Moreover, issues such as high WVP and limited mechanical properties caused by using polysaccharide films can also be overcome by the addition of polysaccharide NPs.282,285,286
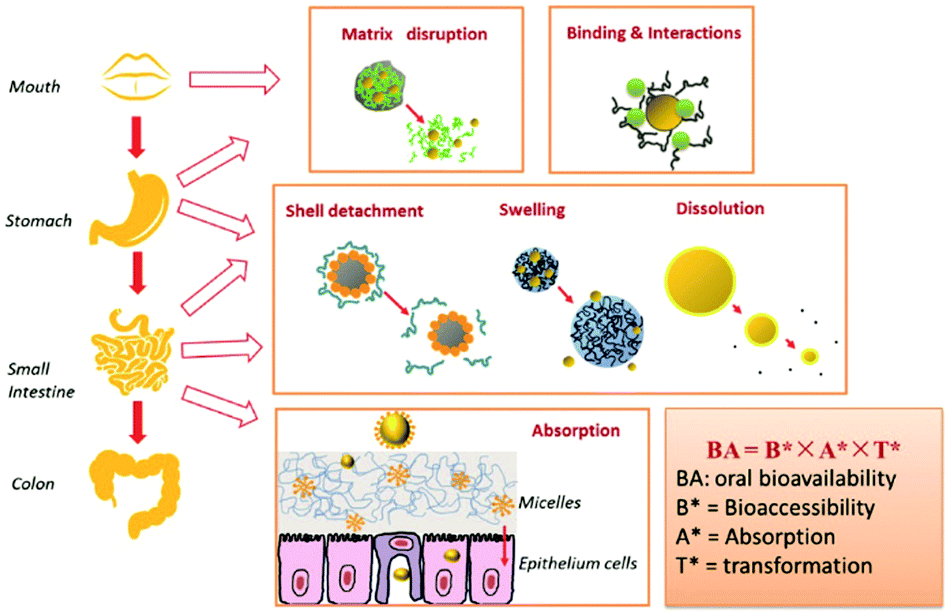 | ||
| Scheme 9 Overview of the design of food ingredient delivery systems to control bioavailability via body–particle interactions. (Reproduced with permission from ref. 290. Copyright John Wiley and Sons, 2017.) | ||
Different examples of suitable nanoscale delivery systems for transporting food constituents were listed and discussed in the review of McClements,287 such as micelles, micro/nanoemulsions, liposomes, microgels, and biopolymer NPs (e.g. protein NPs and polysaccharide NPs). For the delivery systems based on polysaccharides, their properties such as thermal stability, digestibility, and antioxidant capacity, depend significantly on the components used to construct a nanoscale delivery system.
Shimoni's group entrapped a hydrophobic nutraceutical, the water-insoluble isoflavone Genistein, into enzymatically synthesised DEXNPs.291 The dextransucrase generated spherical DEXNPs were prepared under optimised conditions (pH 5.2–6 and sucrose concentration >0.5 M). Although the bioavailability of the DEXNPs was increased due to the smaller particle size, larger surface area, and increased solubility caused by the Genistein inclusion, it was found that the loading efficiency of Genistein was very low. The limitation in Genistein loading was considered to result from the fact that the Genistein was only included in dextran particles without forming complexes with the dextran chains. The study provided a good guide to develop nanosized vehicles to incorporate and stabilise the hydrophobic bioactive compounds in foods, which was useful to establish more platforms for delivering water insoluble bioactive chemicals in food and/or pharmaceuticals.
Developing delivery systems based on polysaccharide NPs can also be combined with the food packaging applications mentioned in Section 5.3.1. Nieto-Suaza et al. prepared curcumin-loaded SNPs as the reinforcing agent to be incorporated into the aloe vera gel-banana starch composite films.292 The results obtained from their work showed that the addition of curcumin-loaded SNPs could enhance several properties of the composite films. For instance, with the increased concentration of the curcumin-loaded SNPs, the important WVP of the composite films decreased. One reason for that phenomenon has been discussed above, which is the distortion of the water diffusion path caused by the addition of SNPs. Besides, the existence of the hydrophobic curcumin also played an important role in reducing WVP because it can limit the interaction between water and the polymeric matrix. Moreover, it was observed that the antimicrobial and antioxidative curcumin could be released from the composite films to different food simulants, such as simulate hydrophobic, simulate hydrophilic, and simulate lipophilic foods. The curcumin release was favoured in simulated lipophilic foods and could be controlled by modifying the polarity of the SNPs by acetylation. The group's work combined the effective utilisations of polysaccharide NPs in food delivery systems and food packaging, indicating the promising future of developing biodegradable composite films that can incorporate bioactive substances in food industries.
Furthermore, there are other practical polysaccharide NPs related food delivery systems widely used in transporting bioactive substances. For example, abundant and antimicrobial CSNPs have been widely used as nanocarriers for transporting tea polyphenols, which are nutraceuticals bringing multi-health benefits including cancer prevention, antioxidant activities, antibacterial activities, and immune enhancement. However, some associated challenges can be caused by factors such as the presence of ions, pH, and enzymes in the gastrointestinal tract. The uncontrollable size of CSNPs resulted from fluctuations of these factors may affect the stability of the tea polyphenols loaded CSNPs.293,294 Gao and coworkers fabricated zein–propylene glycol alginate–tea saponin (TS) ternary complex nanoparticles for the delivery of resveratrol.295 The presence of TS on the particles significantly improved resveratrol loading as well as the environmental stability of resveratrol. The best release characteristics in the small intestine were obtained for a ratio of zein to TS of 4:3 as observed via an in vitro digestion model. As such, the novel protein–polysaccharide–surfactant ternary complex enabled efficient delivery of resveratrol.
McClements and coworkers summarised several examples of forming protein–polysaccharide NPs based delivery systems by conjugating polysaccharide (e.g. CS, DEX) with another common biopolymer protein (e.g. Whey Protein Isolate, OVA).290 The functional properties of proteins can be improved by conjugation to polysaccharides, giving tailored gastrointestinal protection and physicochemical stability. However, regulatory issues may be caused by using conjugates compared to all-natural ingredients, and the production costs may increase due to the additional ingredients and processing operations required to fabricate conjugates.
NPs based on natural polysaccharides can fulfil most of the requirements to be applied in food-grade delivery systems due to their biodegradability and biocompatibility. Despite several advantages like effective encapsulation of bioactive substances and improved stability against enzymes in the gastrointestinal tract, drawbacks such as the aggregation of polysaccharide NPs and burst release caused by small particle size and large surface area need more attention. Therefore, future studies are necessary to overcome these challenges and fabricate polysaccharide nanocarriers with improved properties for delivery systems in food.293,294
6. Conclusion and outlook
Polysaccharide NPs have found considerable attention in the past decade mainly for biomedical applications but also use in cosmetics and food has been implemented. The utilised polysaccharides are obtained from various natural sources making them prime candidates for sustainable nanotechnology. Furthermore, polysaccharides have favourable biological properties and activities depending on their chemical structures. Key features are the remarkable biodegradability, biocompatibility, and low-toxicity of polysaccharides that support their broad applications in biological environment – a key driver for future developments.The basis for polysaccharide NPs are the preparation methods. A variety of synthesis methods for preparing NPs with polysaccharides and their derivatives were presented in here, highlighting the broad range of options in polysaccharide NP synthesis. The choice of a suitable synthesis method depends on the specific application and its requirements. For instance, to fabricate excellent polysaccharide NPs based nanocarriers for in vivo delivery systems, controllable size, charge, surface morphology, loading capacity, and substances release are important. These requirements can mostly be satisfied by choosing one of the common fabrication methods, e.g. nanoprecipitation, complex coacervation, emulsification, or other practical methods. However, there are some limitations of these methods that have to be taken into account, such as the requirement of large amounts of organic solvents for the emulsification method and the potential toxicity issue when using the desolvation method, for example. Although, the fabrication of polysaccharide NPs has been investigated thoroughly in recent years, novel approaches are still sought after. Especially, the replacement of organic solvents with harmless and/or sustainable alternatives could be a considerable direction as well as improved control over particle size and shape with new template methods.
A direction in polysaccharide NPs with considerable growth potential is the combination with polymeric materials. For example, to attach polymers to polysaccharide NPs to form polysaccharide/polymer NP systems, “grafting to” and “grafting from” reactions are applied. In “grafting to” reactions, functionalised and fully characterised polymer is prepared in advance before attachment. While in “grafting from” reactions, monomers polymerise directly on the interface of polysaccharide NPs. After graft modification, polysaccharide–polymer based core–shell NPs are obtained that feature new properties to be tailored via the chemistry of the grafted polymer. The inclusion of a polysaccharide NP core into the polymer shell can introduce improved stability against solvents and stimuli response indicating the potential use of polysaccharide–polymer based core–shell NPs in biomedical applications. Furthermore, inclusion of polysaccharide NPs into polymer materials leads to enhancement of the mechanical properties with possible implications for polymer materials production on an industrial-scale. The field of polysaccharide/polymer hybrid and composite materials is still underrepresented in the literature. Nevertheless, various directions can be foreseen, for example tailoring of polymer surface wettability via polysaccharide NP introduction or control of drug release from polysaccharide NPs via responsive polymer shells.
As effective nano-sized carriers, polysaccharide NPs have broad applications in transporting substances. When utilised in nanomedicine, cosmetics, and food areas, polysaccharide NPs introduce various benefits for the transport of drugs, genes and other hydrophobic and lipophilic substances via efficient protective encapsulation, improving the bioavailability and chemical stability of these substances. In nanomedicine, the EPR effect allows the targeted delivery of drug/gene-loaded polysaccharide NPs to pathological tissues, and various in vitro release studies demonstrated that the loaded drugs and genes could be released in a controlled and sustained manner. These useful results also play a significant role in tissue engineering applications which are based on growth factors delivery. Moreover, it was proven that polysaccharide NPs could be practical for the delivery and controlled-release of cosmetic and food ingredients, showing the promising future of polysaccharide NPs based nanocarriers. Certainly, in the future advanced nanocarriers will be developed that allow more targeted delivery as well as tailored release profiles. Here functionalisation of polysaccharide NPs with targeting moieties and polymers could be an avenue.
On the other hand, polysaccharide NPs can be designed as Pickering emulsion stabilisers and material reinforcing agent in cosmetics and food packaging areas due to their environmental-friendly properties. According to former studies, emulsions stabilised by polysaccharide NPs showed their potential to produce long-term stabilised formulations in cosmetics. After adding polysaccharide NPs to the packaging films, the mechanical properties were enhanced accompanied by lower WVP values, indicating the potential of incorporating polysaccharide NPs into other materials for developing food packaging materials with improved properties. Also, in the stabilisation of emulsions and introduction in food packaging further tailored functionalisation could have a positive effect on overall materials properties, e.g. when interaction of polysaccharide NPs and matrix materials are improved.
Overall, it can be concluded that polysaccharide NPs have proven to be very important future biological nanocarriers, emulsifiers, and material reinforcing additives. However, more research is still required to work out issues when fabricating and using them. For instance, it is important to promote the preclinical results from in vitro cellular studies and in vivo animal studies to the human system. Also, aggregation of polysaccharide NPs and burst release of substances are worth mentioning. Therefore, further deep studies are necessary to promote the applications of polysaccharide NPs and ensure their safety and efficacy in the future.
Abbreviations
| AFM | Atomic force microscopy |
| ATPS | Aqueous two-phase system |
| BMP | Bone morphogenetic protein |
| CA | Cellulose acetate |
| CH | Chitin |
| CL | Cellulose |
| CLNPs | Cellulose-based nanoparticles |
| CRG | Carrageenan |
| CS | Chitosan |
| CSNPs | Chitosan-based nanoparticles |
| DEX | Dextran |
| DEXNPs | Dextran-based nanoparticles |
| DDT | Dodecanethiol |
| DGNPs | DEX- and gelatin-based nanoparticle system |
| DOX | Doxorubicin |
| DS | Dextran sulfate |
| EPR | Enhanced permeation and retention |
| FAM | Fluorescein amidite |
| HA | Hyaluronic acid |
| HANPs | Hyaluronic acid-based nanoparticles |
| HDM | High methyl pectin matrix |
| HRA NPs | NPs formed by coupling HA with aminated retinoid acid (ATRA) |
| HUVEC | Human umbilical vein endothelial cell |
| LDM | Low methyl pectin matrix |
| MC | Methylcellulose |
| MFI | Mean fluorescence intensity |
| MTX | Methotrexate |
| MMP9 | Metalloproteinase 9 |
| MW | Molecular weight |
| MXS | Minoxidil sulphate |
| NP | Nanoparticle |
| OCMCS | Oleoyl-carboxymethyl-chitosan |
| OVA | Ovalbumin |
| O/W | Oil-in-water |
| PBAE | Poly(β-amino)ester |
| PCS-5 | HIV protease cleavage site 5 peptide |
| PDGF-BB | Platelet derived growth factor |
| pDNA | Plasmid DNA |
| PEA | Poly(ethyl acrylate) |
| PEI | Poly(ethylene imine) |
| PEG | Poly(ethylene glycol) |
| PEGDA | Poly(ethylene glycol)diacrylate |
| pEGFP | Plasmid DNA expressing green fluorescent protein |
| pep | MMP9 cleavable connection |
| PFP | Perfluoropentane |
| Poly(I:C) | Poly(inosinic acid):poly(cytidylic acid) |
| PLA | Poly(lactide) |
| PLGA | Poly(D,L-lactide-co-glycolide) |
| PMA | Poly(methyl acrylate) |
| PMMA | Poly(methyl methacrylate) |
| PMMA-b-P(PEGMA) | Poly(methyl methacrylate)-b-poly(poly(ethylene glycol) methyl ether methacrylate) |
| P(MMA-co-S) | Poly(methyl methacrylate-co-styrene) |
| PNIPAM | Poly(N-isopropylacrylamide) |
| PPC | Poly(propylene carbonate) |
| P(PEGMA) | Poly(poly(ethylene glycol) methyl ether methacrylate) |
| PTX | Paclitaxel |
| SANPs | Smart and active nanoparticles |
| SEM | Scanning electron microscopy |
| SG1 | N-tert-Butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)nitroxide] |
| siRNA | Small interfering RNA |
| SI-SET-LRP | Surface-initiated single-electron transfer living radical polymerisation |
| SNPs | Starch-based nanoparticles |
| TEM | Transmission electron microscopy |
| T g | Glass transition temperature |
| THF | Tetrahydrofuran |
| TPP | Tripolyphosphate |
| TS | Tea saponin |
| VBC | 4-Vinylbenzyl chloride |
| VEGF | Vascular endothelial growth factor |
| W/O | Water-in-oil |
| W/W | Water-in-water |
| WVP | Water vapour permeability |
| 5-FU | 5-Fluorouracil |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. S. and A. P. acknowledge funding from the University of Glasgow and the German Research Foundation (grant no. SCHM 3282/3-1).References
- P.-F. Cui, W.-R. Zhuang, X. Hu, L. Xing, R.-Y. Yu, J.-B. Qiao, Y.-J. He, F. Li, D. Ling and H.-L. Jiang, Chem. Commun., 2018, 54, 8218–8221 Search PubMed.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 Search PubMed.
- Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu and S. Liu, Macromolecules, 2007, 40, 3538–3546 Search PubMed.
- N. Kocherginsky, C. L. Tan and W. F. Lu, J. Membr. Sci., 2003, 220, 117–128 Search PubMed.
- S. Matoori and J.-C. Leroux, Mater. Horiz., 2020, 7, 1297–1309 Search PubMed.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 Search PubMed.
- B. V. K. J. Schmidt, Macromol. Chem. Phys., 2018, 219, 1700494 Search PubMed.
- R. K. Ibrahim, M. Hayyan, M. A. AlSaadi, A. Hayyan and S. Ibrahim, Environ. Sci. Pollut. Res., 2016, 23, 13754–13788 Search PubMed.
- R. Seyedebrahimi, S. Razavi and J. Varshosaz, J. Cluster Sci., 2020, 31, 99–108 Search PubMed.
- A. Mihranyan, N. Ferraz and M. Strømme, Prog. Mater. Sci., 2012, 57, 875–910 Search PubMed.
- I. P. Kaur, M. Kapila and R. Agrawal, Ageing Res. Rev., 2007, 6, 271–288 Search PubMed.
- X. He, H. Deng and H.-M. Hwang, J. Food Drug Anal., 2019, 27, 1–21 Search PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 Search PubMed.
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 Search PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 Search PubMed.
- L. Zhang, F. Gu, J. Chan, A. Wang, R. Langer and O. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 Search PubMed.
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485–499 Search PubMed.
- C. Ornelas-Megiatto, P. N. Shah, P. R. Wich, J. L. Cohen, J. A. Tagaev, J. A. Smolen, B. D. Wright, M. J. Panzner, W. J. Youngs and J. M. Fréchet, Mol. Pharmaceutics, 2012, 9, 3012–3022 Search PubMed.
- J. Yang, S. Han, H. Zheng, H. Dong and J. Liu, Carbohydr. Polym., 2015, 123, 53–66 Search PubMed.
- M. V. Tzoumaki, T. Moschakis, V. Kiosseoglou and C. G. Biliaderis, Food Hydrocolloids, 2011, 25, 1521–1529 Search PubMed.
- B. P. Binks and C. P. Whitby, Colloids Surf., A, 2005, 253, 105–115 Search PubMed.
- B. T. Nguyen, T. Nicolai and L. Benyahia, Langmuir, 2013, 29, 10658–10664 Search PubMed.
- M. Pavlovic, A. Plucinski, L. Zeininger and B. V. K. J. Schmidt, Chem. Commun., 2020, 56, 6814–6817 Search PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 Search PubMed.
- S. Mizrahy and D. Peer, Chem. Soc. Rev., 2012, 41, 2623–2640 Search PubMed.
- R. Heo, D. G. You, W. Um, K. Y. Choi, S. Jeon, J.-S. Park, Y. Choi, S. Kwon, K. Kim and I. C. Kwon, Biomaterials, 2017, 131, 15–26 Search PubMed.
- M. Q. Guo, X. Hu, C. Wang and L. Ai, in Polysaccharides: Structure and Solubility, ed. Z. Xu, IntechOpen, 2017, pp. 7–21 Search PubMed.
- A. D. Baldwin and K. L. Kiick, Pept. Sci., 2010, 94, 128–140 Search PubMed.
- M. Gericke, P. Schulze and T. Heinze, Macromol. Biosci., 2020, 20, 1900415 Search PubMed.
- R. Brayner, F. Fiévet and T. Coradin, in Nanomaterials: A Danger or a Promise? A chemical and biological perspective, ed. J. Allouche, Springer-Verlag, 2013, pp. 27–74 Search PubMed.
- J. C. Cazotti, A. T. Fritz, O. Garcia-Valdez, N. M. Smeets, M. A. Dubé and M. F. Cunningham, Carbohydr. Polym., 2020, 228, 115384 Search PubMed.
- S. K. Nitta and K. Numata, Int. J. Mol. Sci., 2013, 14, 1629–1654 Search PubMed.
- S. Raveendran, A. K. Rochani, T. Maekawa and D. S. Kumar, Materials, 2017, 10, 929 Search PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 Search PubMed.
- L. Li, R. Ni, Y. Shao and S. Mao, Carbohydr. Polym., 2014, 103, 1–11 Search PubMed.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 Search PubMed.
- L. Bacakova, K. Novotná and M. Parizek, Physiol. Res., 2014, 63, S29 Search PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 Search PubMed.
- L. Liu, Y. Ding, S. Liu, S. Wang, Y. Fang and M. Lyu, Qual. Assur. Saf. Crops Foods, 2019, 11, 53–59 Search PubMed.
- E. Khalikova, P. Susi and T. Korpela, Microbiol. Mol. Biol. Rev., 2005, 69, 306–325 Search PubMed.
- D. Domene-López, J. C. García-Quesada, I. Martin-Gullon and M. G. Montalbán, Polymers, 2019, 11, 1084 Search PubMed.
- I. Šimkovic, M. Hricovíni, L. Šoltés, R. Mendichi and C. Cosentino, Carbohydr. Polym., 2000, 41, 9–14 Search PubMed.
- G. Tripodo, A. Trapani, M. L. Torre, G. Giammona, G. Trapani and D. Mandracchia, Eur. J. Pharm. Biopharm., 2015, 97, 400–416 Search PubMed.
- R. S. Singh, G. K. Saini and J. F. Kennedy, Carbohydr. Polym., 2008, 73, 515–531 Search PubMed.
- K. Janes, P. Calvo and M. Alonso, Adv. Drug Delivery Rev., 2001, 47, 83–97 Search PubMed.
- A. Singla and M. Chawla, J. Pharm. Pharmacol., 2001, 53, 1047–1067 Search PubMed.
- A. Nešić, G. Cabrera-Barjas, S. Dimitrijević-Branković, S. Davidović, N. Radovanović and C. Delattre, Molecules, 2020, 25, 135 Search PubMed.
- J. H. Park, G. Saravanakumar, K. Kim and I. C. Kwon, Adv. Drug Delivery Rev., 2010, 62, 28–41 Search PubMed.
- S. M. Ahsan, M. Thomas, K. K. Reddy, S. G. Sooraparaju, A. Asthana and I. Bhatnagar, Int. J. Biol. Macromol., 2018, 110, 97–109 Search PubMed.
- B. R. Shah, Y. Li, W. Jin, Y. An, L. He, Z. Li, W. Xu and B. Li, Food Hydrocolloids, 2016, 52, 369–377 Search PubMed.
- T. Kean and M. Thanou, Adv. Drug Delivery Rev., 2010, 62, 3–11 Search PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 Search PubMed.
- M. A. Hubbe, O. J. Rojas, L. A. Lucia and M. Sain, BioResources, 2008, 3, 929–980 Search PubMed.
- K. Chi, H. Wang and J. M. Catchmark, Food Hydrocolloids, 2020, 103, 105696 Search PubMed.
- R. Zafar, K. M. Zia, S. Tabasum, F. Jabeen, A. Noreen and M. Zuber, Int. J. Biol. Macromol., 2016, 92, 1012–1024 Search PubMed.
- A. Dufresne and J. Castaño, Starch/Staerke, 2017, 69, 1500307 Search PubMed.
- Y. Parajó, I. d’Angelo, A. Welle, M. Garcia-Fuentes and M. J. Alonso, Drug Delivery, 2010, 17, 596–604 Search PubMed.
- M. Swierczewska, H. S. Han, K. Kim, J. Park and S. Lee, Adv. Drug Delivery Rev., 2016, 99, 70–84 Search PubMed.
- D. M. Suflet, G. C. Chitanu and J. Desbrières, Carbohydr. Polym., 2010, 82, 1271–1277 Search PubMed.
- Y. Chao and H. C. Shum, Chem. Soc. Rev., 2020, 49, 114–142 Search PubMed.
- C. R. Mace, O. Akbulut, A. A. Kumar, N. D. Shapiro, R. Derda, M. R. Patton and G. M. Whitesides, J. Am. Chem. Soc., 2012, 134, 9094–9097 Search PubMed.
- M. Naessens, A. Cerdobbel, W. Soetaert and E. J. Vandamme, J. Chem. Technol. Biotechnol., 2005, 80, 845–860 Search PubMed.
- A. Banerjee and R. Bandopadhyay, Int. J. Biol. Macromol., 2016, 87, 295–301 Search PubMed.
- N. H. Silva, C. Vilela, A. Almeida, I. M. Marrucho and C. S. Freire, Food Hydrocolloids, 2018, 77, 921–930 Search PubMed.
- C. A. Cozzolino, G. Castelli, S. Trabattoni and S. Farris, Food Packag. Shelf Life, 2016, 8, 50–55 Search PubMed.
- V. D. Prajapati, G. K. Jani and S. M. Khanda, Carbohydr. Polym., 2013, 95, 540–549 Search PubMed.
- M. Gupta and A. K. Gupta, J. Controlled Release, 2004, 99, 157–166 Search PubMed.
- E. Cevher, S. K. Salomon, A. Makrakis, X. W. Li, S. Brocchini and H. O. Alpar, J. Microencapsulation, 2015, 32, 755–768 Search PubMed.
- E. Cevher, S. K. Salomon, S. Somavarapu, S. Brocchini and H. O. Alpar, J. Microencapsulation, 2015, 32, 769–783 Search PubMed.
- J. Willersinn, A. Bogomolova, M. B. Cabré and B. V. K. J. Schmidt, Polym. Chem., 2017, 8, 1244–1254 Search PubMed.
- S. M. Brosnan, H. Schlaad and M. Antonietti, Angew. Chem., Int. Ed., 2015, 54, 9715–9718 Search PubMed.
- R. B. Lira, J. Willersinn, B. V. K. J. Schmidt and R. Dimova, Macromolecules, 2020, 53, 10179–10188 Search PubMed.
- J. Willersinn and B. V. K. J. Schmidt, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3757–3766 Search PubMed.
- S. Farris, I. U. Unalan, L. Introzzi, J. M. Fuentes-Alventosa and C. A. Cozzolino, J. Appl. Polym. Sci., 2014, 131, 40539 Search PubMed.
- J. P. Paques, E. van der Linden, C. J. van Rijn and L. M. Sagis, Adv. Colloid Interface Sci., 2014, 209, 163–171 Search PubMed.
- A. D. Augst, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2006, 6, 623–633 Search PubMed.
- M. S. Hasnain, A. K. Nayak, M. Kurakula and M. N. Hoda, Alginates in Drug Delivery, Elsevier, 2020, pp. 129–152 Search PubMed.
- L. Wells and H. Sheardown, Eur. J. Pharm. Biopharm., 2007, 65, 329–335 Search PubMed.
- M. B. Evangelista, S. X. Hsiong, R. Fernandes, P. Sampaio, H.-J. Kong, C. C. Barrias, R. Salema, M. A. Barbosa, D. J. Mooney and P. L. Granja, Biomaterials, 2007, 28, 3644–3655 Search PubMed.
- J. O. You and C. A. Peng, Macromol. Symp., 2005, 219, 147–153 Search PubMed.
- A. Zahoor, S. Sharma and G. Khuller, Int. J. Antimicrob. Agents, 2005, 26, 298–303 Search PubMed.
- S. K. Motwani, S. Chopra, S. Talegaonkar, K. Kohli, F. J. Ahmad and R. K. Khar, Eur. J. Pharm. Biopharm., 2008, 68, 513–525 Search PubMed.
- R. Yegappan, V. Selvaprithiviraj, S. Amirthalingam and R. Jayakumar, Carbohydr. Polym., 2018, 198, 385–400 Search PubMed.
- A. L. Daniel-da-Silva, L. Ferreira, A. M. Gil and T. Trindade, J. Colloid Interface Sci., 2011, 355, 512–517 Search PubMed.
- S. Rodrigues, A. M. R. da Costa and A. Grenha, Carbohydr. Polym., 2012, 89, 282–289 Search PubMed.
- S. David, C. S. Levi, L. Fahoum, Y. Ungar, E. G. Meyron-Holtz, A. Shpigelman and U. Lesmes, Food Funct., 2018, 9, 1344–1352 Search PubMed.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5–28 Search PubMed.
- C. Alvarez-Lorenzo, B. Blanco-Fernandez, A. M. Puga and A. Concheiro, Adv. Drug Delivery Rev., 2013, 65, 1148–1171 Search PubMed.
- S. P. Strand, S. Lelu, N. K. Reitan, C. de Lange Davies, P. Artursson and K. M. Vårum, Biomaterials, 2010, 31, 975–987 Search PubMed.
- J. Valente, P. Pereira, A. Sousa, J. Queiroz and F. Sousa, Polymers, 2021, 13, 793 Search PubMed.
- J. Willersinn and B. V. K. J. Schmidt, Polym. Chem., 2018, 9, 1626–1637 Search PubMed.
- T. Harifi and M. Montazer, Carbohydr. Polym., 2012, 88, 1125–1140 Search PubMed.
- E. Campos, P. Coimbra and M. Gil, Polym. Bull., 2013, 70, 549–561 Search PubMed.
- E. Chiesa, R. Dorati, S. Pisani, B. Conti, G. Bergamini, T. Modena and I. Genta, Pharmaceutics, 2018, 10, 267 Search PubMed.
- J. Berger, M. Reist, J. M. Mayer, O. Felt, N. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 Search PubMed.
- K. Janes, P. Calvo and M. Alonso, Adv. Drug Delivery Rev., 2001, 47, 83–97 Search PubMed.
- J. Berger, M. Reist, J. M. Mayer, O. Felt and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 35–52 Search PubMed.
- S. Ye, C. Wang, X. Liu, Z. Tong, B. Ren and F. Zeng, J. Controlled Release, 2006, 112, 79–87 Search PubMed.
- L. Richert, P. Lavalle, E. Payan, X. Z. Shu, G. D. Prestwich, J.-F. Stoltz, P. Schaaf, J.-C. Voegel and C. Picart, Langmuir, 2004, 20, 448–458 Search PubMed.
- S. Wang, F. Fontana, M.-A. Shahbazi and H. A. Santos, Chem. Commun., 2021 10.1039/D1CC00811K.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3–27 Search PubMed.
- E. Akiyama, N. Morimoto, P. Kujawa, Y. Ozawa, F. M. Winnik and K. Akiyoshi, Biomacromolecules, 2007, 8, 2366–2373 Search PubMed.
- B. B. Breitenbach, I. Schmid and P. R. Wich, Biomacromolecules, 2017, 18, 2839–2848 Search PubMed.
- D. A. Ossipov, Expert Opin. Drug Delivery, 2010, 7, 681–703 Search PubMed.
- Y. Liu, G. Yang, D. Zou, Y. Hui, K. Nigam, A. P. Middelberg and C.-X. Zhao, Ind. Eng. Chem. Res., 2019, 59, 4134–4149 Search PubMed.
- A. Maaz, W. Abdelwahed, I. Tekko and S. Trefi, Int. J. Acad. Sci. Res., 2015, 3, 1–12 Search PubMed.
- C. J. M. Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. G. Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 Search PubMed.
- F. Lince, D. L. Marchisio and A. A. Barresi, J. Colloid Interface Sci., 2008, 322, 505–515 Search PubMed.
- K. Miladi, S. Sfar, H. Fessi and A. Elaissari, Polymer nanoparticles for nanomedicines, Springer, 2016, pp. 17–53 Search PubMed.
- C. G. Barreras-Urbina, B. Ramírez-Wong, G. A. López-Ahumada, S. E. Burruel-Ibarra, O. Martínez-Cruz, J. A. Tapia-Hernández and F. Rodriguez Felix, Int. J. Food Prop., 2016, 19, 1912–1923 Search PubMed.
- S. Ding, N. Anton, T. F. Vandamme and C. A. Serra, Expert Opin. Drug Delivery, 2016, 13, 1447–1460 Search PubMed.
- S. F. Chin, S. C. Pang and S. H. Tay, Carbohydr. Polym., 2011, 86, 1817–1819 Search PubMed.
- M. R. Kulterer, M. Reischl, V. E. Reichel, S. Hribernik, M. Wu, S. Köstler, R. Kargl and V. Ribitsch, Colloids Surf., A, 2011, 375, 23–29 Search PubMed.
- E. Aschenbrenner, K. Bley, K. Koynov, M. Makowski, M. Kappl, K. Landfester and C. K. Weiss, Langmuir, 2013, 29, 8845–8855 Search PubMed.
- X. Yan, R. A. N. S. Ramos, P. Alcouffe, L. E. Munoz, R. O. Bilyy, F. Ganachaud and J. Bernard, Biomacromolecules, 2019, 20, 3915–3923 Search PubMed.
- M. Leonard, M. R. De Boisseson, P. Hubert, F. Dalencon and E. Dellacherie, J. Controlled Release, 2004, 98, 395–405 Search PubMed.
- E. Kizilay, A. B. Kayitmazer and P. L. Dubin, Adv. Colloid Interface Sci., 2011, 167, 24–37 Search PubMed.
- F. Vecchies, P. Sacco, E. Decleva, R. Menegazzi, D. Porrelli, I. Donati, G. Turco, S. Paoletti and E. Marsich, Biomacromolecules, 2018, 19, 3936–3944 Search PubMed.
- J. T. G. Overbeek and M. Voorn, J. Cell. Comp. Physiol., 1957, 49, 7–26 Search PubMed.
- C. E. Sing, Adv. Colloid Interface Sci., 2017, 239, 2–16 Search PubMed.
- A. Drogoz, L. David, C. Rochas, A. Domard and T. Delair, Langmuir, 2007, 23, 10950–10958 Search PubMed.
- Y. Liu, M. Kong, X. J. Cheng, Q. Q. Wang, L. M. Jiang and X. G. Chen, Carbohydr. Polym., 2013, 94, 309–316 Search PubMed.
- B. Sarmento, A. Ribeiro, F. Veiga and D. Ferreira, Colloids Surf., B, 2006, 53, 193–202 Search PubMed.
- L. Liu, Y. Xu, P. Zhang, J. You, W. Li, Y. Chen, R. Li, B. Rui and H. Dou, Langmuir, 2020, 36, 8580–8588 Search PubMed.
- F. Furlani, P. Sacco, E. Decleva, R. Menegazzi, I. Donati, S. Paoletti and E. Marsich, ACS Appl. Mater. Interfaces, 2019, 11, 9794–9803 Search PubMed.
- A. Gennari, J. M. R. de la Rosa, E. Hohn, M. Pelliccia, E. Lallana, R. Donno, A. Tirella and N. Tirelli, Beilstein J. Nanotechnol., 2019, 10, 2594–2608 Search PubMed.
- N. Eghbal and R. Choudhary, LWT, 2018, 90, 254–264 Search PubMed.
- Y. P. Timilsena, T. O. Akanbi, N. Khalid, B. Adhikari and C. J. Barrow, Int. J. Biol. Macromol., 2019, 121, 1276–1286 Search PubMed.
- C. E. Sing and S. L. Perry, Soft Matter, 2020, 16, 2885–2914 Search PubMed.
- L. Zhou, H. Shi, Z. Li and C. He, Macromol. Rapid Commun., 2020, 41, 2000149 Search PubMed.
- J. R. Magana, C. Sproncken and I. K. Voets, Polymers, 2020, 12, 1953 Search PubMed.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 Search PubMed.
- Q. Cao, T. Heil, B. Kumru, M. Antonietti and B. V. K. J. Schmidt, Polym. Chem., 2019, 10, 5315–5323 Search PubMed.
- K. Sakamoto, R. Lochhead, H. Maibach and Y. Yamashita, Cosmetic science and technology: theoretical principles and applications, Elsevier, 2017 Search PubMed.
- J. Esquena, Curr. Opin. Colloid Interface Sci., 2016, 25, 109–119 Search PubMed.
- J. Zhang, J. Hwang, M. Antonietti and B. V. K. J. Schmidt, Biomacromolecules, 2018, 20, 204–211 Search PubMed.
- C. Vauthier and K. Bouchemal, Pharm. Res., 2009, 26, 1025–1058 Search PubMed.
- B. Lupo, A. Maestro, J. M. Gutiérrez and C. González, Food Hydrocolloids, 2015, 49, 25–34 Search PubMed.
- P. R. Ravi, R. Vats, J. Balija, S. P. N. Adapa and N. Aditya, Carbohydr. Polym., 2014, 110, 320–328 Search PubMed.
- J. M. Shin, S. R. Hwang, R. Heo, G. Saravanakumar and J. H. Park, Polym. Degrad. Stab., 2014, 109, 398–404 Search PubMed.
- J. P. Paques, E. van der Linden, C. J. van Rijn and L. M. Sagis, Food Hydrocolloids, 2013, 31, 428–434 Search PubMed.
- H. S. Min, S. Son, T. W. Lee, H. Koo, H. Y. Yoon, J. H. Na, Y. Choi, J. H. Park, J. Lee and M. H. Han, Adv. Funct. Mater., 2013, 23, 5518–5529 Search PubMed.
- S. Manchun, C. R. Dass and P. Sriamornsak, Carbohydr. Polym., 2014, 101, 650–655 Search PubMed.
- M. Gupta and A. K. Gupta, J. Pharm. Pharm. Sci., 2004, 7, 38–46 Search PubMed.
- A. Maghsoudi, F. Yazdian, S. Shahmoradi, L. Ghaderi, M. Hemati and G. Amoabediny, Mater. Sci. Eng., C, 2017, 75, 1259–1267 Search PubMed.
- J. Yao, L. Zhang, J. Zhou, H. Liu and Q. Zhang, Mol. Pharmaceutics, 2013, 10, 1080–1091 Search PubMed.
- K. A. Janes, M. P. Fresneau, A. Marazuela, A. Fabra and M. A. J. Alonso, J. Controlled Release, 2001, 73, 255–267 Search PubMed.
- X. Li, N. Anton, C. Arpagaus, F. Belleteix and T. F. Vandamme, J. Controlled Release, 2010, 147, 304–310 Search PubMed.
- C. Liu, S. Zhang, D. J. McClements, D. Wang and Y. Xu, J. Agric. Food Chem., 2019, 67, 5113–5121 Search PubMed.
- X. Pei, K. Zhai, C. Wang, Y. Deng, Y. Tan, B. Zhang, Y. Bai, K. Xu and P. Wang, Langmuir, 2019, 35, 7222–7230 Search PubMed.
- A. Plucinski, J. Willersinn, R. B. Lira, R. Dimova and B. V. K. J. Schmidt, Macromol. Chem. Phys., 2020, 221, 2000053 Search PubMed.
- L. Liu, Y. Wang, Q. Hu, T. Li, P. Ma, H. Zhang, M. Du, M. Chen and W. Dong, ACS Sustainable Chem. Eng., 2019, 7, 13081–13088 Search PubMed.
- Y. Wang, Q. Hu, T. Li, P. Ma, S. Zhang, M. Du, M. Chen, H. Zhang and W. Dong, Ind. Eng. Chem. Res., 2018, 57, 13048–13054 Search PubMed.
- H. Kim, M. Shin, S. Han, W. Kwon and S. K. Hahn, Biomacromolecules, 2019, 20, 2889–2903 Search PubMed.
- H. M. Aldawsari, H. K. Dhaliwal, B. M. Aljaeid, N. A. Alhakamy, Z. M. Banjar and M. M. Amiji, Mol. Pharmaceutics, 2018, 16, 128–140 Search PubMed.
- Y. Liu, Y. Wang, C. Zhang, P. Zhou, Y. Liu, T. An, D. Sun, N. Zhang and Y. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 18712–18720 Search PubMed.
- C. Lemarchand, R. Gref and P. Couvreur, Eur. J. Pharm. Biopharm., 2004, 58, 327–341 Search PubMed.
- C. Lemarchand, R. Gref, C. Passirani, E. Garcion, B. Petri, R. Müller, D. Costantini and P. Couvreur, Biomaterials, 2006, 27, 108–118 Search PubMed.
- J. C. Cazotti, A. T. Fritz, O. Garcia-Valdez, N. M. Smeets, M. A. Dubé and M. F. Cunningham, Biomacromolecules, 2020, 21, 4492–4501 Search PubMed.
- A. Dufresne, Molecules, 2010, 15, 4111–4128 Search PubMed.
- F. Tang, L. Zhang, J. Zhu, Z. Cheng and X. Zhu, Ind. Eng. Chem. Res., 2009, 48, 6216–6223 Search PubMed.
- S. Yuan, P. Zhang, Z. Yang, L. Lv, S. Tang and B. Liang, Int. J. Biol. Macromol., 2018, 109, 287–302 Search PubMed.
- M. V. G. Paixão, R. C. S. da Luz and R. de Carvalho Balaban, J. Mol. Liq., 2019, 286, 110792 Search PubMed.
- K. Peuler, N. Dimmitt and C.-C. Lin, Carbohydr. Polym., 2020, 234, 115901 Search PubMed.
- N. V. Rao, J. G. Rho, W. Um, P. K. Ek, V. Q. Nguyen, B. H. Oh, W. Kim and J. H. Park, Pharmaceutics, 2020, 12, 931 Search PubMed.
- K. Y. Choi, K. H. Min, H. Y. Yoon, K. Kim, J. H. Park, I. C. Kwon, K. Choi and S. Y. Jeong, Biomaterials, 2011, 32, 1880–1889 Search PubMed.
- M. Xu, J. Qian, A. Suo, H. Wang, X. Yong, X. Liu and R. Liu, Carbohydr. Polym., 2013, 98, 181–188 Search PubMed.
- H.-J. Cho, I.-S. Yoon, H. Y. Yoon, H. Koo, Y.-J. Jin, S.-H. Ko, J.-S. Shim, K. Kim, I. C. Kwon and D.-D. Kim, Biomaterials, 2012, 33, 1190–1200 Search PubMed.
- K. Y. Choi, H. Y. Yoon, J.-H. Kim, S. M. Bae, R.-W. Park, Y. M. Kang, I.-S. Kim, I. C. Kwon, K. Choi and S. Y. Jeong, ACS Nano, 2011, 5, 8591–8599 Search PubMed.
- C. Teng, Z. Chai, Z. Yuan, L. Ren, C. Lin, Z. Yan, W. He, C. Qin, L. Yang and X. Han, Nanomedicine, 2020, 24, 102105 Search PubMed.
- H. S. Han, J. Lee, H. R. Kim, S. Y. Chae, M. Kim, G. Saravanakumar, H. Y. Yoon, D. G. You, H. Ko and K. Kim, J. Controlled Release, 2013, 168, 105–114 Search PubMed.
- K. Y. Choi, E. J. Jeon, H. Y. Yoon, B. S. Lee, J. H. Na, K. H. Min, S. Y. Kim, S.-J. Myung, S. Lee and X. Chen, Biomaterials, 2012, 33, 6186–6193 Search PubMed.
- J. Zhao, Y. Wang, Y. Ma, Y. Liu, B. Yan and L. Wang, Carbohydr. Polym., 2019, 203, 356–368 Search PubMed.
- S. Ganesh, A. K. Iyer, D. V. Morrissey and M. M. Amiji, Biomaterials, 2013, 34, 3489–3502 Search PubMed.
- S. Ganesh, A. K. Iyer, F. Gattacceca, D. V. Morrissey and M. M. Amiji, J. Controlled Release, 2013, 172, 699–706 Search PubMed.
- L. Mo, J. G. Song, H. Lee, M. Zhao, H. Y. Kim, Y. J. Lee, H. W. Ko and H.-K. Han, Nanomedicine, 2018, 14, 557–567 Search PubMed.
- H. Y. Yoon, H. Koo, K. Y. Choi, I. C. Kwon, K. Choi, J. H. Park and K. Kim, Biomaterials, 2013, 34, 5273–5280 Search PubMed.
- J. M. Shin, S. J. Oh, S. Kwon, V. Deepagan, M. Lee, S. H. Song, H.-J. Lee, S. Kim, K.-H. Song and T. W. Kim, J. Controlled Release, 2017, 267, 181–190 Search PubMed.
- H. Lian, Y. Du, X. Chen, L. Duan, G. Gao, C. Xiao and X. Zhuang, J. Colloid Interface Sci., 2017, 496, 201–210 Search PubMed.
- S. Maiz-Fernández, L. Pérez-Álvarez, L. Ruiz-Rubio, R. Pérez González, V. Sáez-Martínez, J. Ruiz Pérez and J. L. Vilas-Vilela, Polymers, 2019, 11, 742 Search PubMed.
- D. Bamberger, D. Hobernik, M. Konhäuser, M. Bros and P. R. Wich, Mol. Pharmaceutics, 2017, 14, 4403–4416 Search PubMed.
- L. De Matteis, M. Alleva, I. Serrano-Sevilla, S. García-Embid, G. Stepien, M. Moros and J. M. De la Fuente, Mar. Drugs, 2016, 14, 175 Search PubMed.
- S. A. Papadimitriou, D. S. Achilias and D. N. Bikiaris, Int. J. Pharm., 2012, 430, 318–327 Search PubMed.
- M. Nag, V. Gajbhiye, P. Kesharwani and N. K. Jain, Colloids Surf., B, 2016, 148, 363–370 Search PubMed.
- A. Guţoaia, L. Schuster, S. Margutti, S. Laufer, B. Schlosshauer, R. Krastev, D. Stoll and H. Hartmann, Carbohydr. Polym., 2016, 143, 25–34 Search PubMed.
- W. E. Rudzinski, A. Palacios, A. Ahmed, M. A. Lane and T. M. Aminabhavi, Carbohydr. Polym., 2016, 147, 323–332 Search PubMed.
- H. Ragelle, S. Colombo, V. Pourcelle, K. Vanvarenberg, G. Vandermeulen, C. Bouzin, J. Marchand-Brynaert, O. Feron, C. Foged and V. Préat, J. Controlled Release, 2015, 211, 1–9 Search PubMed.
- C. Corbet, H. Ragelle, V. Pourcelle, K. Vanvarenberg, J. Marchand-Brynaert, V. Préat and O. Feron, J. Controlled Release, 2016, 223, 53–63 Search PubMed.
- X. Yan, P. Alcouffe, J. Bernard and F. Ganachaud, Biomacromolecules, 2020, 21, 4591–4598 Search PubMed.
- N. Al Nakeeb, J. Willersinn and B. V. K. J. Schmidt, Biomacromolecules, 2017, 18, 3695–3705 Search PubMed.
- T. Nishimura, S. Shishi, Y. Sasaki and K. Akiyoshi, J. Am. Chem. Soc., 2020, 142, 11784–11790 Search PubMed.
- X. Dong, Z. Wu, Y. Wang, T. Li, X. Zhang, H. Yuan, B. Xia, P. Ma, M. Chen and W. Dong, Polym. Test., 2021, 93, 106926 Search PubMed.
- X. Dong, Z. Wu, Y. Wang, T. Li, X. Zhang, H. Yuan, J. Jiang, B. Xia, P. Ma and M. Chen, Mater. Lett., 2021, 289, 129400 Search PubMed.
- X. Dong, L. Liu, Y. Wang, T. Li, Z. Wu, H. Yuan, P. Ma, D. Shi, M. Chen and W. Dong, Carbohydr. Polym., 2021, 254, 117321 Search PubMed.
- G. Wang, S. Li, Y. Feng, Y. Hu, G. Zhao and W. Jiang, Carbohydr. Polym., 2020, 249, 116795 Search PubMed.
- N. Elsaid, T. L. Jackson, Z. Elsaid, A. Alqathama and S. Somavarapu, Mol. Pharmaceutics, 2016, 13, 2923–2940 Search PubMed.
- Q. Wang, S. Jamal, M. S. Detamore and C. Berkland, J. Biomed. Mater. Res., Part A, 2011, 96, 520–527 Search PubMed.
- Y. Liu, R. Lipowsky and R. Dimova, Front. Chem., 2019, 7, 213 Search PubMed.
- M. Iqbal, Y. Tao, S. Xie, Y. Zhu, D. Chen, X. Wang, L. Huang, D. Peng, A. Sattar and M. A. B. Shabbir, Biol. Proced. Online, 2016, 18, 1–18 Search PubMed.
- O. Akbulut, C. R. Mace, R. V. Martinez, A. A. Kumar, Z. Nie, M. R. Patton and G. M. Whitesides, Nano Lett., 2012, 12, 4060–4064 Search PubMed.
- P.-Å. Albertsson, Nature, 1958, 182, 709–711 Search PubMed.
- M. Pavlovic, A. Plucinski, J. Zhang, M. Antonietti, L. Zeininger and B. V. K. J. Schmidt, Langmuir, 2020, 36, 1401–1408 Search PubMed.
- S. Ma, J. Thiele, X. Liu, Y. Bai, C. Abell and W. T. Huck, Small, 2012, 8, 2356–2360 Search PubMed.
- T. Watanabe, I. Motohiro and T. Ono, Langmuir, 2019, 35, 2358–2367 Search PubMed.
- H. Yuan, Q. Ma, Y. Song, M. Y. Tang, Y. K. Chan and H. C. Shum, Macromol. Chem. Phys., 2017, 218, 1600422 Search PubMed.
- Y. Hu, S. Wang, A. Abbaspourrad and A. M. Ardekani, Langmuir, 2015, 31, 1885–1891 Search PubMed.
- S. Mytnyk, I. Ziemecka, A. G. Olive, J. W. M. van der Meer, K. A. Totlani, S. Oldenhof, M. T. Kreutzer, V. van Steijn and J. H. van Esch, RSC Adv., 2017, 7, 11331–11337 Search PubMed.
- M. Yamazaki and K. Iijima, Polymers, 2020, 12, 435 Search PubMed.
- A. Bianchera and R. Bettini, Expert Opin. Drug Delivery, 2020, 17, 1345–1359 Search PubMed.
- S. Salatin and A. Yari Khosroushahi, J. Cell. Mol. Med., 2017, 21, 1668–1686 Search PubMed.
- S. Chayed and F. M. Winnik, Eur. J. Pharm. Biopharm., 2007, 65, 363–370 Search PubMed.
- S. Uthaman, Y. Kim, J. Y. Lee, S. Pillarisetti, K. M. Huh and I.-K. Park, ACS Appl. Mater. Interfaces, 2020, 12, 28004–28013 Search PubMed.
- B. B. Breitenbach, E. Steiert, M. Konhäuser, L.-M. Vogt, Y. Wang, S. H. Parekh and P. R. Wich, Soft Matter, 2019, 15, 1423–1434 Search PubMed.
- K. Butzbach, M. Konhäuser, M. Fach, D. N. Bamberger, B. Breitenbach, B. Epe and P. R. Wich, Polymers, 2019, 11, 896 Search PubMed.
- T. G. Dacoba, R. W. Omange, H. Li, J. Crecente-Campo, M. Luo and M. J. Alonso, ACS Nano, 2019, 13, 4947–4959 Search PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 Search PubMed.
- M. Abid, M. Naveed, I. Azeem, A. Faisal, M. F. Nazar and B. Yameen, Int. J. Pharm., 2020, 586, 119605 Search PubMed.
- S. Kumari, B. Ram, D. Kumar, S. Ranote and G. S. Chauhan, Mater. Sci. Energy Technol., 2018, 1, 22–28 Search PubMed.
- S. Javanbakht and H. Namazi, Mater. Sci. Eng., C, 2018, 87, 50–59 Search PubMed.
- Y. Li, C. Liu, Y. Tan, K. Xu, C. Lu and P. Wang, Carbohydr. Polym., 2014, 110, 87–94 Search PubMed.
- H. Hu, Y. Li, Q. Zhou, Y. Ao, C. Yu, Y. Wan, H. Xu, Z. Li and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 30833–30844 Search PubMed.
- I. Wasiak, A. Kulikowska, M. Janczewska, M. Michalak, I. A. Cymerman, A. Nagalski, P. Kallinger, W. W. Szymanski and T. Ciach, PLoS One, 2016, 11, e0146237 Search PubMed.
- B. Li, Q. Wang, X. Wang, C. Wang and X. Jiang, Carbohydr. Polym., 2013, 93, 430–437 Search PubMed.
- B. Sarmento, A. Ribeiro, F. Veiga, D. Ferreira and R. Neufeld, Biomacromolecules, 2007, 8, 3054–3060 Search PubMed.
- H. Lindemann, M. Kühne, C. Grune, P. Warncke, S. Hofmann, A. Koschella, M. Godmann, D. Fischer, T. Heinzel and T. Heinze, Macromol. Biosci., 2020, 20, 2000039 Search PubMed.
- A. L. Urzedo, M. C. Gonçalves, M. H. Nascimento, C. B. Lombello, G. Nakazato and A. B. Seabra, Mater. Sci. Eng., C, 2020, 112, 110933 Search PubMed.
- M. S. Huh, E. J. Lee, H. Koo, J. Y. Yhee, K. S. Oh, S. Son, S. Lee, S. H. Kim, I. C. Kwon and K. Kim, Top. Curr. Chem., 2017, 375, 31 Search PubMed.
- S. Salatin and M. Jelvehgari, Pharm Sci., 2017, 23, 84–94 Search PubMed.
- P. R. Wich and J. M. Fréchet, Aust. J. Chem., 2012, 65, 15–19 Search PubMed.
- H. Yasar, D.-K. Ho, C. De Rossi, J. Herrmann, S. Gordon, B. Loretz and C.-M. Lehr, Polymers, 2018, 10, 252 Search PubMed.
- Y. Yang, X. Liu, D. Zhang, W. Yu, H. Xie, J. Zheng and X. Ma, J. Controlled Release, 2011, 152, e160–e161 Search PubMed.
- O. S. Muddineti, A. Shah, S. V. K. Rompicharla, B. Ghosh and S. Biswas, Int. J. Biol. Macromol., 2018, 118, 857–863 Search PubMed.
- H.-Q. Mao, K. Roy, V. L. Troung-Le, K. A. Janes, K. Y. Lin, Y. Wang, J. T. August and K. W. Leong, J. Controlled Release, 2001, 70, 399–421 Search PubMed.
- J. L. Cohen, S. Schubert, P. R. Wich, L. Cui, J. A. Cohen, J. L. Mynar and J. M. Fréchet, Bioconjugate Chem., 2011, 22, 1056–1065 Search PubMed.
- L. Cui, J. L. Cohen, C. K. Chu, P. R. Wich, P. H. Kierstead and J. M. Fréchet, J. Am. Chem. Soc., 2012, 134, 15840–15848 Search PubMed.
- F. C. Moraes, L. M. F. Ramirez, R. Aid, S. Benadda, M. Maire, C. Chauvierre, J. C. Antunes, F. Chaubet and D. Letourneur, Int. J. Pharm., 2021, 597, 120302 Search PubMed.
- V. Y. Kosovrasti, L. V. Nechev and M. M. Amiji, Mol. Pharmaceutics, 2016, 13, 3404–3416 Search PubMed.
- A. Grenha, M. E. Gomes, M. Rodrigues, V. E. Santo, J. F. Mano, N. M. Neves and R. L. Reis, J. Biomed. Mater. Res., Part A, 2010, 92, 1265–1272 Search PubMed.
- P. Sahoo, K. H. Leong, S. Nyamathulla, Y. Onuki, K. Takayama and L. Y. Chung, React. Funct. Polym., 2017, 119, 145–155 Search PubMed.
- S. Abdelghany, M. Alkhawaldeh and H. S. AlKhatib, J. Drug Delivery Sci. Technol., 2017, 39, 442–449 Search PubMed.
- S. Rochín-Wong, A. Rosas-Durazo, P. Zavala-Rivera, A. Maldonado, M. E. Martínez-Barbosa, I. Vélaz and J. Tánori, Polymers, 2018, 10, 760 Search PubMed.
- T. Miao, J. Wang, Y. Zeng, G. Liu and X. Chen, Adv. Sci., 2018, 5, 1700513 Search PubMed.
- C. Qi, X. Yan, C. Huang, A. Melerzanov and Y. Du, Protein Cell, 2015, 6, 638–653 Search PubMed.
- P. Koria, BioDrugs, 2012, 26, 163–175 Search PubMed.
- M. Rajam, S. Pulavendran, C. Rose and A. Mandal, Int. J. Pharm., 2011, 410, 145–152 Search PubMed.
- F.-m. Chen, Z.-w. Ma, G.-y. Dong and Z.-f. Wu, Acta Pharmacol. Sin., 2009, 30, 485–493 Search PubMed.
- G. Q. Teixeira, C. L. Pereira, F. Castro, J. R. Ferreira, M. Gomez-Lazaro, P. Aguiar, M. A. Barbosa, C. Neidlinger-Wilke and R. M. Goncalves, Acta Biomater., 2016, 42, 168–179 Search PubMed.
- M. M. Hasani-Sadrabadi, S. P. Hajrezaei, S. H. Emami, G. Bahlakeh, L. Daneshmandi, E. Dashtimoghadam, E. Seyedjafari, K. I. Jacob and L. Tayebi, Nanomedicine, 2015, 11, 1809–1819 Search PubMed.
- I. Aranaz, N. Acosta, C. Civera, B. Elorza, J. Mingo, C. Castro, M. De los Llanos Gandía and A. Heras Caballero, Polymers, 2018, 10, 213 Search PubMed.
- Y. Yang, Z. Fang, X. Chen, W. Zhang, Y. Xie, Y. Chen, Z. Liu and W. Yuan, Front. Pharmacol., 2017, 8, 287 Search PubMed.
- J. Tang, P. J. Quinlan and K. C. Tam, Soft Matter, 2015, 11, 3512–3529 Search PubMed.
- U. Bains and R. Pal, Appl. Sci., 2019, 9, 4044 Search PubMed.
- K. W. Muhammad, N. Akhtar, M. S. K. Haji, R. Mustafa and G. Murtaza, Lat. Am. J. Pharm., 2014, 33, 731–738 Search PubMed.
- S. Simovic, N. Ghouchi-Eskandar and C. Prestidge, J. Drug Delivery Sci. Technol., 2011, 21, 123–133 Search PubMed.
- W. Wang, G. Du, C. Li, H. Zhang, Y. Long and Y. Ni, Carbohydr. Polym., 2016, 151, 1–8 Search PubMed.
- S. B. Haaj, W. Thielemans, A. Magnin and S. Boufi, ACS Appl. Mater. Interfaces, 2014, 6, 8263–8273 Search PubMed.
- C. Li, P. Sun and C. Yang, Starch/Staerke, 2012, 64, 497–502 Search PubMed.
- K. Wang, Y. Hong, Z. Gu, L. Cheng, Z. Li and C. Li, Int. J. Biol. Macromol., 2020, 155, 273–285 Search PubMed.
- J. Han, F. Chen, C. Gao, Y. Zhang and X. Tang, Int. J. Biol. Macromol., 2020, 157, 202–211 Search PubMed.
- H. Liu, C. Wang, S. Zou, Z. Wei and Z. Tong, Langmuir, 2012, 28, 11017–11024 Search PubMed.
- S. Ge, L. Xiong, M. Li, J. Liu, J. Yang, R. Chang, C. Liang and Q. Sun, Food Chem., 2017, 234, 339–347 Search PubMed.
- X. Pei, K. Zhai, X. Liang, Y. Deng, Y. Tan, P. Wang and K. Xu, Langmuir, 2017, 33, 3787–3793 Search PubMed.
- Y. Tan, K. Xu, C. Liu, Y. Li, C. Lu and P. Wang, Carbohydr. Polym., 2012, 88, 1358–1363 Search PubMed.
- Y. Tan, K. Xu, C. Niu, C. Liu, Y. Li, P. Wang and B. P. Binks, Food Hydrocolloids, 2014, 36, 70–75 Search PubMed.
- P. Shao, H. Zhang, B. Niu and W. Jin, Int. J. Biol. Macromol., 2018, 118, 2032–2039 Search PubMed.
- C. Li, Y. Li, P. Sun and C. Yang, Colloids Surf., A, 2013, 431, 142–149 Search PubMed.
- M. Matos, A. Marefati, R. Bordes, G. Gutiérrez and M. Rayner, Carbohydr. Polym., 2017, 169, 127–138 Search PubMed.
- K. Zhai, F. Zhang, C. Wang, X. Pei, Y. Tan, Y. Bai, B. Zhang, Y. Wang, K. Xu and P. Wang, Polym. Adv. Technol., 2020, 31, 1321–1329 Search PubMed.
- V. Maingret, C. Courrégelongue, V. Schmitt and V. Héroguez, Biomacromolecules, 2020, 21, 5358–5368 Search PubMed.
- A. Ammala, Int. J. Cosmet. Sci., 2013, 35, 113–124 Search PubMed.
- M. P. Lupo, Clin. Dermatol., 2001, 19, 467–473 Search PubMed.
- D.-G. Kim, Y.-I. Jeong, C. Choi, S.-H. Roh, S.-K. Kang, M.-K. Jang and J.-W. Nah, Int. J. Pharm., 2006, 319, 130–138 Search PubMed.
- R. Harris, E. Lecumberri, I. Mateos-Aparicio, M. Mengíbar and A. Heras, Carbohydr. Polym., 2011, 84, 803–806 Search PubMed.
- P. Ganesan and D.-K. Choi, Int. J. Nanomed., 2016, 11, 1987 Search PubMed.
- P. Morganti, M. Palombo, G. Tishchenko, V. E. Yudin, F. Guarneri, M. Cardillo, P. Del Ciotto, F. Carezzi, G. Morganti and G. Fabrizi, Cosmetics, 2014, 1, 140–158 Search PubMed.
- B. N. Matos, T. A. Reis, T. Gratieri and G. M. Gelfuso, Int. J. Biol. Macromol., 2015, 75, 225–229 Search PubMed.
- Q. Huang, H. Yu and Q. Ru, J. Food Sci., 2010, 75, R50–R57 Search PubMed.
- C. Moraru, Q. Huang, P. Takhistov, H. Dogan and J. Kokini, Global issues in food science and technology, Elsevier, 2009, pp. 369–399 Search PubMed.
- A. Bahrami, R. Delshadi, E. Assadpour, S. M. Jafari and L. Williams, Adv. Colloid Interface Sci., 2020, 278, 102140 Search PubMed.
- A. S. Kritchenkov, A. R. Egorov, N. V. Dubashynskaya, O. V. Volkova, L. A. Zabodalova, E. P. Suchkova, A. V. Kurliuk, T. V. Shakola and A. P. Dysin, Int. J. Biol. Macromol., 2019, 134, 480–486 Search PubMed.
- L. Lin, Y. Gu and H. Cui, Food Packag. Shelf Life, 2019, 19, 86–93 Search PubMed.
- M. V. Lorevice, C. G. Otoni, M. R. de Moura and L. H. C. Mattoso, Food Hydrocolloids, 2016, 52, 732–740 Search PubMed.
- A. Arora and G. Padua, J. Food Sci., 2010, 75, R43–R49 Search PubMed.
- S. Shankar, L.-F. Wang and J.-W. Rhim, Food Packag. Shelf Life, 2019, 21, 100363 Search PubMed.
- L. Dai, C. Qiu, L. Xiong and Q. Sun, Food Chem., 2015, 174, 82–88 Search PubMed.
- S. Jiang, C. Liu, X. Wang, L. Xiong and Q. Sun, LWT–Food Sci. Technol., 2016, 69, 251–257 Search PubMed.
- D. J. McClements, J. Food Sci., 2015, 80, N1602–N1611 Search PubMed.
- Y. D. Livney, Curr. Opin. Food Sci., 2015, 3, 125–135 Search PubMed.
- R. F. Gonçalves, J. T. Martins, C. M. Duarte, A. A. Vicente and A. C. Pinheiro, Trends Food Sci. Technol., 2018, 78, 270–291 Search PubMed.
- F. Liu, C. Ma, Y. Gao and D. J. McClements, Compr. Rev. Food Sci. Food Saf., 2017, 16, 76–95 Search PubMed.
- D. Semyonov, O. Ramon, Y. Shoham and E. Shimoni, Food Funct., 2014, 5, 2463–2474 Search PubMed.
- L. Nieto-Suaza, L. Acevedo-Guevara, L. T. Sánchez, M. I. Pinzón and C. C. Villa, Food Struct., 2019, 22, 100131 Search PubMed.
- P. Puligundla, C. Mok, S. Ko, J. Liang and N. Recharla, J. Funct. Foods, 2017, 34, 139–151 Search PubMed.
- J. Liang, H. Yan, P. Puligundla, X. Gao, Y. Zhou and X. Wan, Food Hydrocolloids, 2017, 69, 286–292 Search PubMed.
- Y. Wei, C. Li, L. Dai, L. Zhang, J. Liu, L. Mao, F. Yuan and Y. Gao, Food Funct., 2020, 11, 9973–9983 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |