
Effect of precise linker length, bond density, and broad temperature window on the rheological properties of ethylene vitrimers†
Bhaskar
Soman
ab and
Christopher M.
Evans
 *ab
*ab
aDepartment of Materials Science and Engineering and Urbana, Illinois 61801, USA. E-mail: cme365@illinois.edu
bFrederick Seitz Materials Research Laboratory University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 24th November 2020
Abstract
Dynamic networks which undergo topology conserving exchange reactions, sometimes called vitrimers, show properties intermediate to thermosets and thermoplastics. The dynamic nature of the networks results in complex rheological properties and has attracted much attention in the past decade for self-healing, malleable and recyclable polymers. Here, we investigate a series of precise, high crosslink density telechelic ethylene vitrimers as a function of temperature and crosslink density. The networks show a rubbery plateau at high frequencies and a terminal flow regime at lower frequencies. With increasing crosslink density, the rubbery plateau modulus shows a monotonic increase and the terminal flow shifts to lower frequencies. The plateau modulus at high frequency increases as a function of temperature, as expected for a conserved network topology. When plotted against inverse temperature, the zero shear viscosities show a characteristic Arrhenius behavior, and the activation energy monotonically increases with crosslink density. Crossover frequency and shift factors (from time temperature superposition) also show Arrhenius behavior with activation energies in good agreement with those determined from zero shear viscosity. A positive deviation from this Arrhenius trend is observed beginning as high as 100 K above the glass transition temperature for C6 and C8 networks. Further investigations of such networks are critical for the development of sustainable and recyclable replacements for commercial plastics.
Introduction
Dynamic polymer networks including supramolecular polymers,1–9 covalent adaptable networks10–14 and vitrimers15–44 are being studied as self-healing, recyclable and sustainable polymers making them attractive materials to replace many commodity or functional plastics. Their unique properties are a consequence of the dynamic crosslinks which undergo reversible exchange reactions and allow for local rearrangements of network strands resulting in stress relaxation and flow. On short time scales the networks appear permanent and show a rubbery plateau, whereas on longer timescales they flow. The rheological properties of dynamic networks depend on the network architecture, crosslink density, crosslink chemistry, solvent, bond exchange kinetics and inherent segmental dynamics. While supramolecular polymers formed by secondary interactions such as hydrogen bonding, metal–ligand coordination or host–guest interactions and have been studied for almost half a century, vitrimers differ in one key aspect where the crosslink junction is formed by dynamic covalent bonds which exchange in a topology conserving manner. The term vitrimer was coined by Leibler30 based on an Arrhenius temperature dependence of viscosity for polyester networks which is reminiscent of vitreous silica. It has been noted recently that this definition is not unique to dynamic networks with conserved exchange reactions and is also frequently observed in dissociative dynamic networks.45 Thus, there is still a need to understand under what conditions the mechanism of the exchange reaction will play a key role to the function or properties of dynamic networks. The rekindled interest in these networks in the last decade has led to numerous articles on vitrimers for shape memory polymers,35–37 shockwave dissipation,38 flame retardants,39 and solid electrolytes.40 While initial work was focused on stress relaxation times and self-healing as a function of bond chemistry and polymer backbone, recent work has moved to investigating the effects of crosslink density,41,42 defects43 and solvent concentration44 on the rheological properties of vitrimers.Two areas of vitrimer physics which have received little attention are the roles of extremely broad temperature windows and precise spacing between dynamic crosslinks. While the initial work of Leibler looked at a 180 °C temperature range,30 subsequent works have been largely limited to a less than 50 °C window.45 It is important to investigate wider windows because many polymer are processed well above their use temperatures, and it is unclear if the Arrhenius dependence will persist. Du Prez and coworkers found two distinct Arrhenius slopes in vinylogous urethane vitrimers17 attributed to a change in the dominant exchange mechanism with temperature. Additionally, many polymer are tested far above Tg where bond exchange is antipacted to control the macroscopic stress relaxation. It has been suggested15 that viscosity will show increasing deviations from Arrhenius behavior when approaching Tg, but little experimental data exists in this regime.
The crosslink density in vitrimers has been investigated showing how the stress relaxation times and creep are impacted.31–34 In supramolecular or dissociative networks, scaling relationships have been derived46,47 and measured experimentally48 but similar works do not yet exist for vitrimeric materials. Additionally, in prior works there is a distribution of molecular weights between crosslinks and the role of exact carbon spacers is unknown. Recent work from our group on permanent networks showed that odd–even effects can emerge in amorphous materials when the linkers are precise, and Tg jumps up and down as the carbon number systematically increases from 4 to 12.49 Winey and coworkers50 have also shown the importance of precision spacing of ionic groups along a polyethylene backone which can crystallize and form favorable structures for proton conduction. In the context of vitrimers, an understanding of how precise spacing of dynamic bonds impacts the macroscopic rheology is lacking.
Here, we report ethylene vitrimers synthesized by step growth polymerization of telechelic alkane diols and boric acid. A series of precise networks with varying crosslink densities were synthesized with exactly 6, 8, 10 and 12 carbons (C6–C12) between boronic ester crosslinking junctions. All networks have sub-ambient glass transition temperatures and are malleable at room temperature. Oscillatory shear rheology shows an increasing plateau modulus as a function of temperature, providing phenomenological evidence that the topology is conserved in the networks. Increasing crosslink density leads to a monotonic increase in the rubbery modulus and a delay of the terminal relaxation. The zero-shear viscosity, shift factors and crossover frequencies show characteristic Arrhenius behavior when measured far above Tg. The activation energies (Ea) calculated from the fits are in good agreement with each other and systematically increase with crosslink density. The values are lower than reported for prior boronic ester vitrimers,51 which we attribute to the telechelic nature of the network. With decreasing temperature, the zero-shear viscosities show positive deviations from Arrhenius behavior as the glass transition is approached indicating an increasing role of segmental dynamics on the rheology. We also discuss the importance of broad temperature measurements in the context of determining topology freezing temperatures of dynamic networks.
Results and discussion
Dynamic PE networks were synthesized by the step growth polymerization of precise telechelic alkane diols and boric acid. Networks with exactly 6, 8, 10 and 12 carbons (C6, C8, C10, C12) between boronic ester crosslinking junctions were prepared (Fig. 1a). Fourier transform infrared spectroscopy (FTIR) of the networks was performed at 80 °C to confirm the absence of the broad OH peak in the 3000 cm−1 to 3500 cm−1 range corresponding to unreacted species of the diols and boric acid. Within the sensitivity of the instrument, no OH peak is detected indicating that the network formation reached high conversion (Fig. 1b). The emergence of a sharp peak at 1300 cm−1 corresponds to the asymmetric B–O stretch52 indicating the formation of boronic ester crosslinks.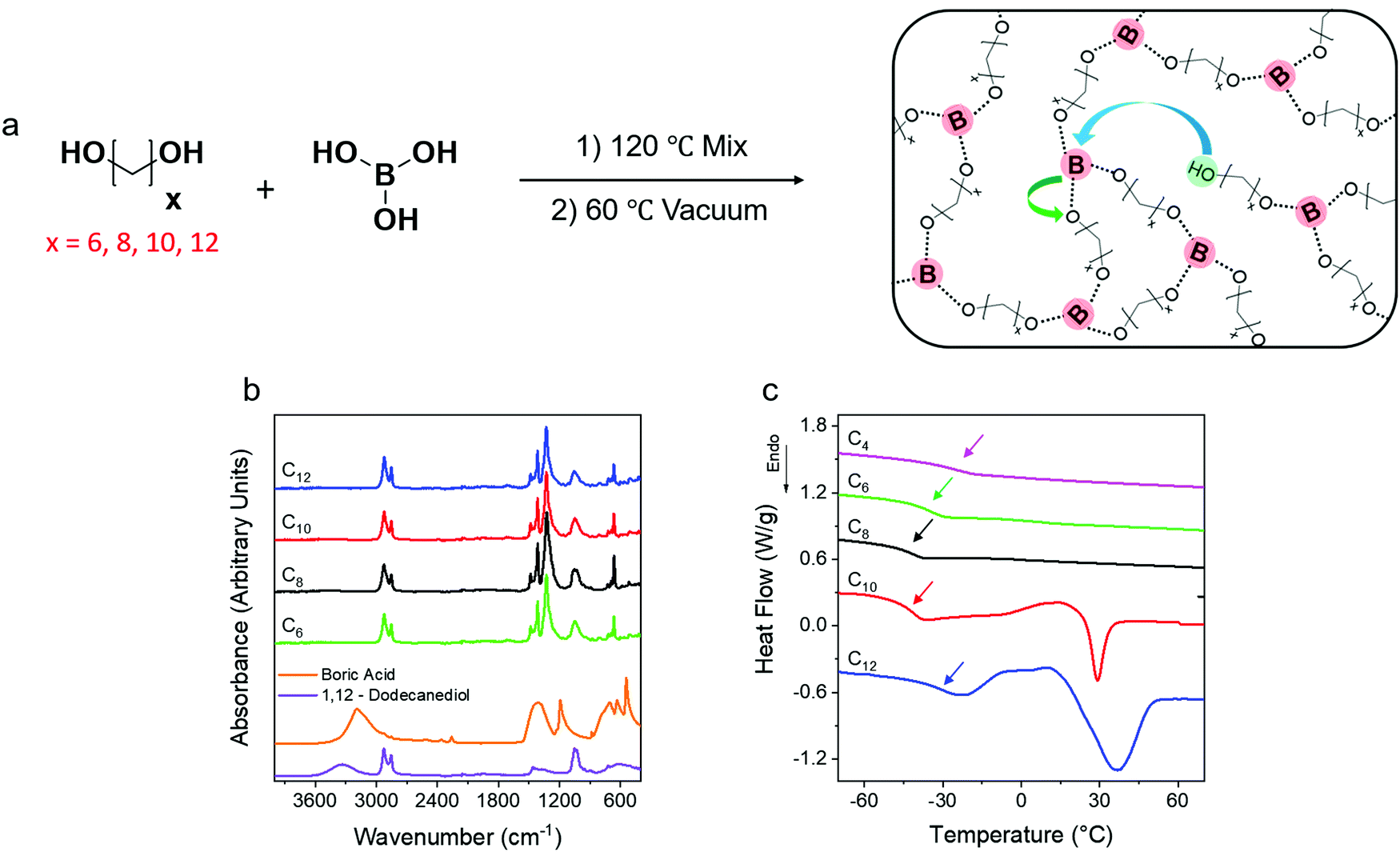 | ||
| Fig. 1 (a) Step growth polymerization of telechelic alkane diols and boric acid produces ethylene vitrimers. By controlling the length of the diols, networks with precise spacing between crosslinks were synthesized. The arrows in the schematic represent boronic transesterification reactions that result in local rearrangement of network strands and gives the network its dynamic properties. (b) ATR-FTIR spectra of the vitrimers show no free OH peak (3000–3500 cm−1) indicating a high reaction conversion as there are no detectable unreacted diols. (c) Heating of the networks after quenching to −80 °C reveals a non-monotonic increase in Tg (arrows) with increasing linker length or decreasing crosslink density. Following the Tg, cold crystallization and melting transition were observed in C10 and C12 networks. | ||
Thermal properties of the networks were investigated using differential scanning calorimetry (DSC) by first rapidly quenching from 150 °C to −80 °C and then heating at 20 °C min−1. The C6 and C8 networks show no melting transitions while the C10 and C12 networks show cold crystallization followed by a melting transition with peak melting temperatures (Tm) in the 25–30 °C range. The glass transition temperatures (Tg) of the networks are −27 °C (C4), −36 °C (C6), −43 °C (C8), −41 °C (C10) and −32 °C (C12) (Fig. 1c). The increase in crosslink density is expected to lead to a monotonic increase in Tg,49 but a reproducible non-monotonic trend is observed. This is correlated with the appearance of crystallization during quenching and cold crystallization on heating in the C10 and C12 networks (Fig. S1, ESI†). No crystallization is observed in the heating or cooling cycles for C4, C6 and C8 networks. The crystalline domains may provide greater area for the rigid amorphous fraction to form which hinders segmental dynamics and raises Tg. All subsequent measurements were performed above any melting transitions to avoid competing phenomena.
Oscillatory shear rheology was used to probe the storage (G′) and loss (G′′) modulus of the PE networks. Since the C10 and C12 networks are crystalline at room temperature, all experiments were performed above 40 °C to prevent the interference of crystallization on rheological measurements. Frequency sweeps were performed at 10 °C intervals from 40 °C to 140 °C. The ethylene networks show a solid like (G′ > G′′) rubbery response at high frequencies and the plateau modulus at 40 °C varies from ∼4 to 14 MPa depending on crosslink density (Fig. 2a). This is expected of a rubbery network53 and similar results have been shown in other experimental works on dynamic networks.54 Although some of the data overlap at low frequency, the modulus at 20 Hz changes proportionally to the carbon spacer as expected (Fig. 2b). At lower frequencies a terminal flow regime with characteristic slopes of ∼2 for G′ and ∼1 for G′′ (Fig. S2, ESI†) is seen for all temperature. The terminal relaxation shifts to lower frequencies with increasing crosslink density, as has been previously reported for other telechelic systems.41,55 The modulus measured at a constant frequency of 20 Hz also increases with temperature, as expected for systems with conserved network topology due to entropic elasticity.52
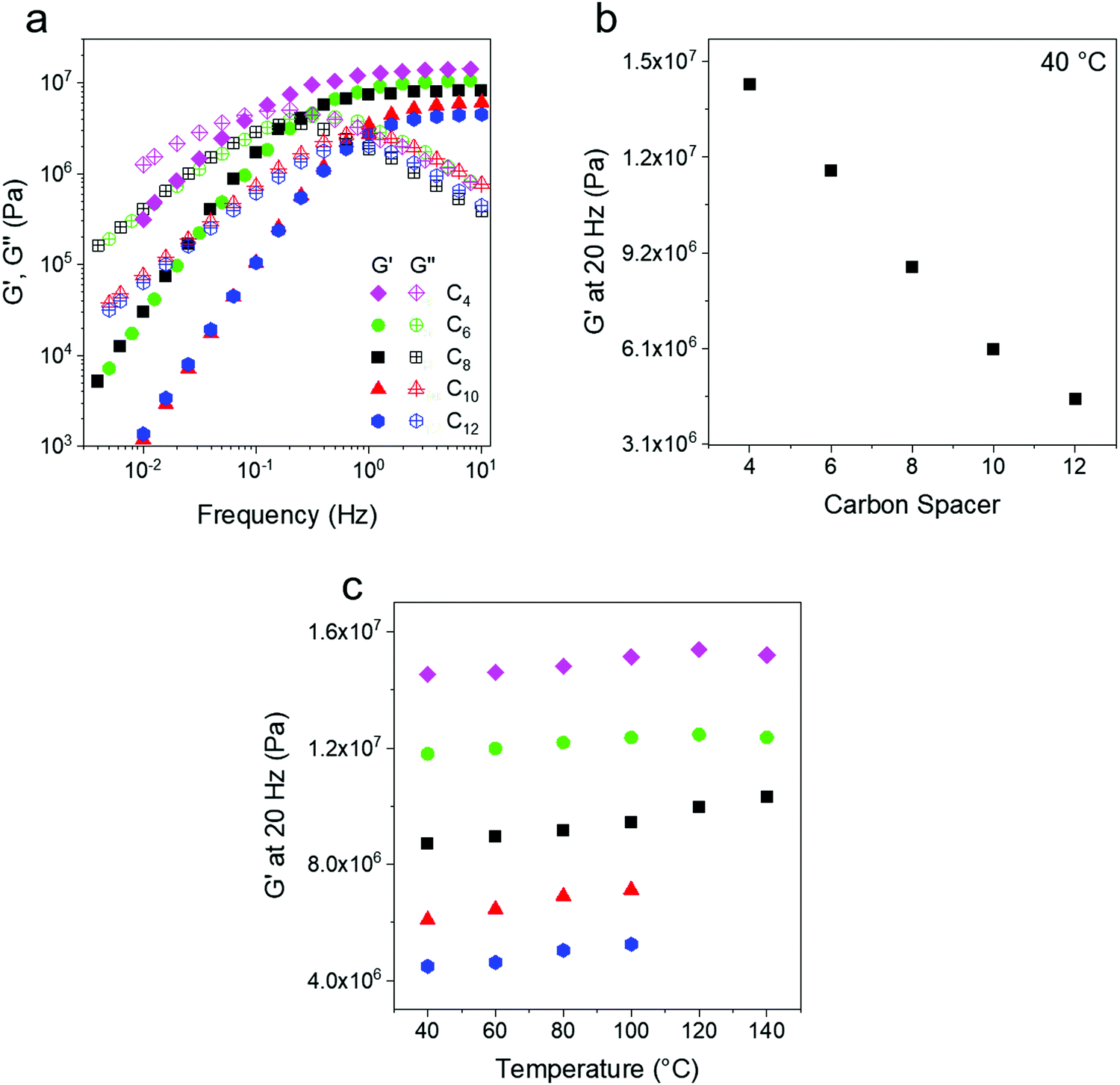 | ||
| Fig. 2 (a) Storage and loss moduli of ethylene vitrimers at 40 °C. A monotonic increase in rubbery plateau modulus at high frequency is observed with increasing crosslinking density. The terminal relaxation shifts to lower frequency as the crosslink density increases. (b) A comparison of the rubbery modulus with increasing linker length (decreasing crosslink density) shows a linear decrease as anticipated. (c) The rubbery modulus of the networks also shows a slight increase with increasing temperature as expected for networks with conserved topology. Symbols are the same as in (a). | ||
Master curves of the storage and loss modulus (Fig. 3) are made by horizontally shifting individual frequency sweeps onto a reference curve (Tref = 140 °C) using the time temperature superposition (TTS) principle. On a seven order of magnitude log scale, the plateau appears constant; however, a closer inspection reveals that the modulus does increase with temperature, particularly at higher frequency (Fig. 2c and Fig. S3, ESI†). Boronic transesterification exchange reactions proceed in a topology conserving manner as shown by several groups51,56,57 and thus this increase is expected. In prior work, polyethylene oxide dynamic networks with the same boronic ester bonds as the present work also showed an increase in modulus on heating.40 This trend is in contrast to dissociative dynamic networks where modulus decreases with temperature.13,45
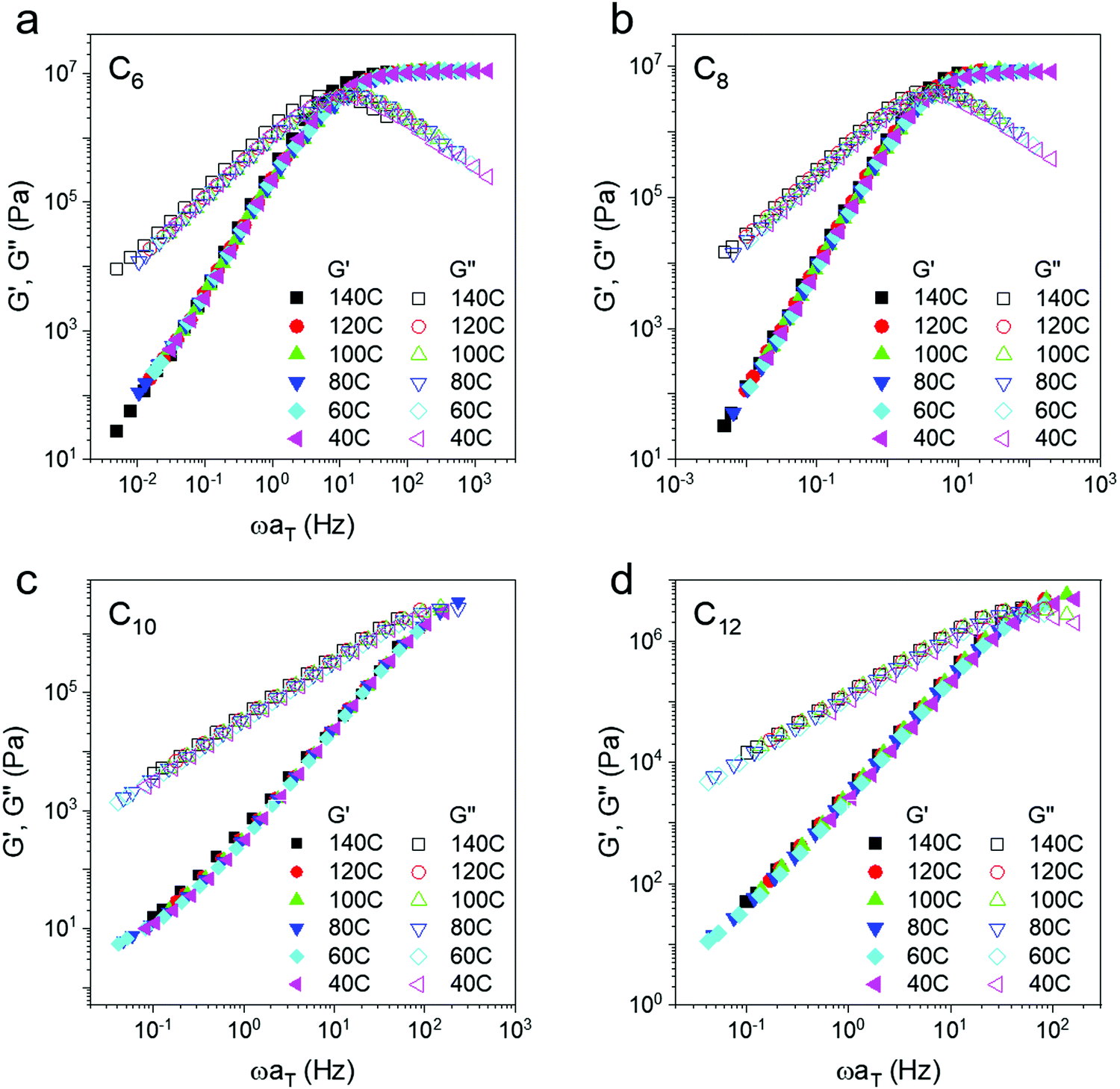 | ||
| Fig. 3 Horizontally shifted master curves (Tref = 140 °C) of the networks. The high frequency rubbery modulus remains constant with increasing temperature, intermediate to expectations for permanent and dissociative dynamic networks. | ||
From the horizontal shifting of frequency sweeps, shift factors (aT) were obtained and plotted for all networks and show a systematic increase in the slope, and thus activation energy, from an Arrhenius fit (Fig. 4a). Next, the zero shear viscosity of the networks was calculated from 140 to 0 °C using the low frequency slope of the loss modulus,58 and a monotonic increase with increasing crosslink density is observed (Fig. 4b). Complex viscosity was also calculated and compared to this method, with both approaches yielding identical results within error (Fig. S4, ESI†). Viscosity plotted against inverse temperature was fit to an Arrhenius expression, and shows quantitative agreement with the values from aT data (Table 1) indicating that they both reflect the same macroscopic flow controlled by dynamic bond exchange. A third metric, the crossover frequency (ωcrossover), was also examined and defined where G′ = G′′ corresponding to the transition from a viscoelastic solid to a viscoelastic liquid. In the case of C10 and C12, a crossover frequency could not be measured at temperatures above 60 °C because even at the highest frequencies G′′ > G′. For C4, C6 and C8 networks crossover frequencies were obtained as a function of temperature and are shown in Fig. 4c. Although ωcrossover was only obtained in three networks, it is also in good agreement with zero shear viscosity and shift factor in reflecting the activation energy associated with flow. In the case of vitrimers which obey time-temperature superposition, the choice of analysis metric does not appear to matter for determining activation energies. The Ea values for our telechelic networks are all lower than what has been reported for boronic ester vitrimers with pendent dynamic bonds along the backbone by Guan and coworkers who measured Ea = 52.7 kJ mol−1.51 Sokolov and coworkers have noted that in associating polymers, telechelic structures lead to lower bond breaking energies59 which is consistent with our findings. Additionally, we observe a major increase in Ea with increased crosslink density suggesting that exchange processes are easier in more open networks (Table 1). From the shift factor data Ea ∼ N−0.4, and it will be important to push to higher linker lengths to determine where the value becomes molecular weight independent. It is also unclear at present how linker polydispersity will affect these values.
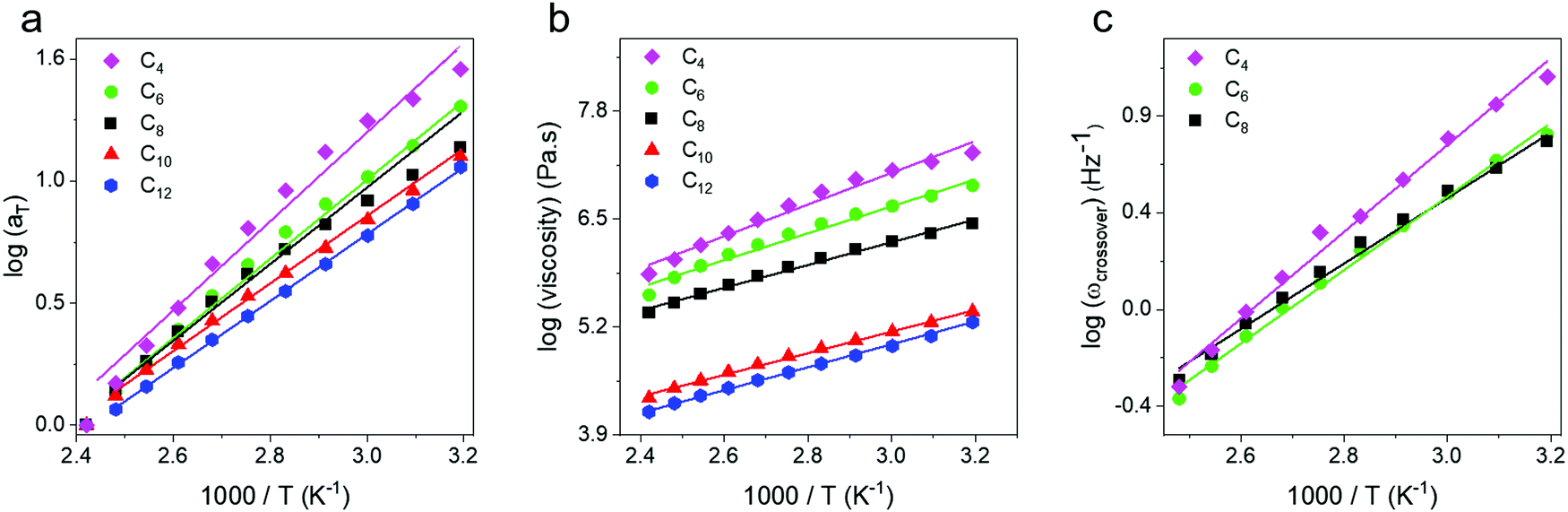 | ||
| Fig. 4 (a) Shift factors, (b) zero shear viscosity and (c) crossover frequency as a function of inverse temperature. All quantities show an Arrhenius behavior in the 100 °C temperature window. | ||
In addition to Tg and Tm, a hypothetical topology freezing temperature (Tv) has been suggested for vitrimers where the bond exchange occurs on slow enough timescales that the network is frozen and η = 1012 Pa s,60 the same criterion invoked for the glass to rubber transition. The position of the Tv with respect to the Tg and Tm may provide insights regarding the relative importance of bond exchange or segmental dynamics in controlling stress relaxation in different temperature regimes.61,62 The Tv of our networks was obtained by extrapolating the zero shear viscosities to a value of 1012 Pa s (Fig. 5a). We note this massive extrapolation is not rigorous but was used to mimic protocols in the vitrimers literature.62,63 We view Tv as a potentially ambiguous temperature as it shares the same criteria as the glass transition (viscosity = 1012 Pa s). Additionally, depending on the modulus the Tv could correspond to timescales of relaxation spanning 108 to 103 s for networks ranging from ultrasoft (10 kPa) to glassy (1 GPa) through the Maxwell relation η = Gτ. A topology freezing transition based on a relaxation time would seem more informative than a temperature, but it is unclear what timescale is appropriate.
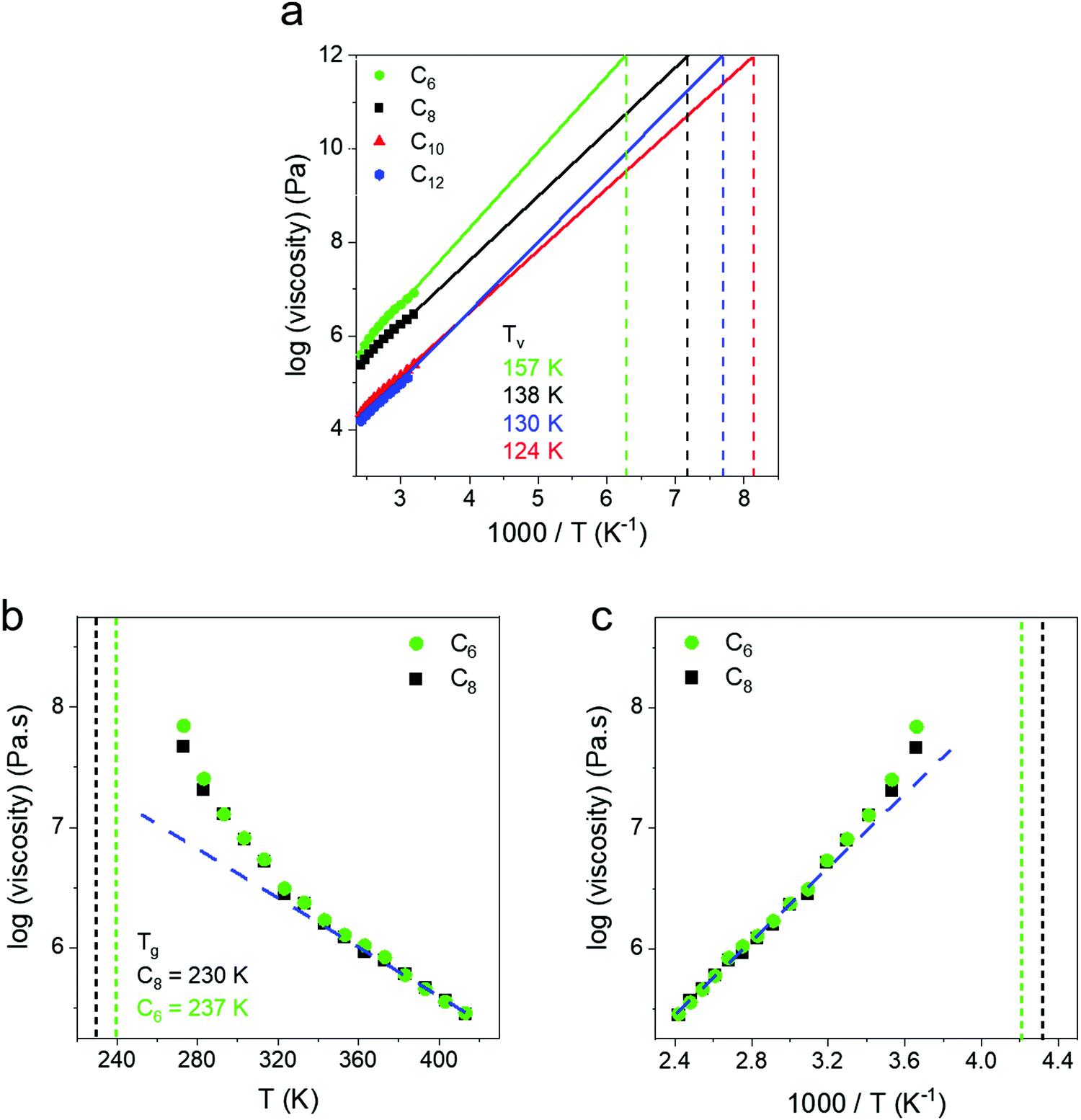 | ||
| Fig. 5 (a) The topological freezing temperature (Tv) is defined as the temperature at which the melt viscosity equals a value of 1012 Pa s. We obtained the Tv by extrapolating the Arrhenius zero shear viscosity fit to a value of 1012 Pa s. (b) Zero shear viscosity vs. temperature and (c) inverse temperature. As the Tg is approached a positive deviation from the Arrhenius behavior is observed. The deviation indicates a transition from a regime controlled by exchange reactions to a regime where slow segmental dynamics begin to contribute to viscosity. | ||
The Tv values were determined as −116 °C (C6), −135 °C (C8), −149 °C (C10) and −143 °C (C12) which are all well below Tg. Thus, upon cooling the Tg is expected to intervene and lead to an increase in viscosity above the extrapolation of an Arrhenius fit. Such a deviation is observed at ∼Tg + 100 °C, and corresponds to a transition from a regime controlled by exchange reactions to a regime where segmental dynamics begin to contribute to an increased viscosity (Fig. 5b). This can be clearly seen in C6 and C8 as their zero shear viscosities show a positive deviation from the Arrhenius trend on a 1000 T−1 plot (Fig. 5c). These networks remain amorphous on cooling (Fig. S1, ESI†), while C10 and C12 crystallize and thus viscosity measurements were not made below a temperature of 40 °C. Our findings are in contrast to the initial work of Leibler who found a single Arrhenius dependence of zero-shear viscosity from Tg to Tg + 180 °C, presumably because their Tv was higher than Tg.30 In the present work, the extrapolation based on high temperature gives a different Tv than would be determined from the lower temperature data, and emphasizes the importance of measuring relaxation over a broad temperature window. Understanding where deviations begin depending on the combination of dynamic bond and polymer chemistry will be an important direction for future research.
Conclusion
A series of precise telechelic ethylene vitrimers was investigated to understand the effect of temperature and precise crosslink density on their rheological properties. The dynamic nature of the networks manifests as a rubbery plateau at high frequencies and a low frequency terminal relaxation. Increasing crosslink density (C12 to C6) leads to a monotonic increase in the rubbery modulus and a delay of the terminal relaxation. The zero shear viscosity shows a characteristic Arrhenius temperature dependence and monotonically increases with increasing crosslink density. Similar Arrhenius fits are made for the crossover frequency and shift factors, and the activation energies calculated from all metrics are in good agreement with each other. Telechelic networks show a lower activation energy than pendant networks with boronic esters.51 At high temperature, the rheological properties of the networks are dominated by the exchange reactions while a deviation from Arrhenius behavior is observed upon cooling. The additional increase in viscosity is attributed to the impending Tg and slowing segmental dynamics which begin to contribute as Tg + 100 K. These broad temperature window studies on model systems provide key insights to the understanding and application of vitrimers for a range of applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding to support this work was provided by the Department of Materials Science and Engineering at the University of Illinois Urbana-Champaign. We also acknowledge the use of facilities at the Materials Research Laboratory.References
- J.-M. Lehn, Supramolecular polymer chemistry—scope and perspectives, Polym. Int., 2002, 51(10), 825–839 CrossRef CAS.
- S. Seiffert and J. Sprakel, Physical chemistry of supramolecular polymer networks, Chem. Soc. Rev., 2012, 41(2), 909–930 RSC.
- R. F. M. Lange, M. Van Gurp and E. W. Meijer, Hydrogen-bonded supramolecular polymer networks, J. Polym. Sci., Part A: Polym. Chem., 1999, 37(19), 3657–3670 CrossRef CAS.
- X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, A Multiresponsive, Shape-Persistent, and Elastic Supramolecular Polymer Network Gel Constructed by Orthogonal Self-Assembly, Adv. Mater., 2012, 24(3), 362–369 CrossRef CAS.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Supramolecular Polymer Materials: Chain Extension of Telechelic Polymers Using a Reactive Hydrogen-Bonding Synthon, Adv. Mater., 2000, 12(12), 874–878 CrossRef CAS.
- S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, A Dual-Responsive Supramolecular Polymer Gel Formed by Crown Ether Based Molecular Recognition, Angew. Chem., Int. Ed., 2011, 50(8), 1905–1909 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, A Healable Supramolecular Polymer Blend Based on Aromatic π–π Stacking and Hydrogen-Bonding Interactions, J. Am. Chem. Soc., 2010, 132(34), 12051–12058 CrossRef CAS.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, A Mechanically Strong, Highly Stable, Thermoplastic, and Self-Healable Supramolecular Polymer Hydrogel, Adv. Mater., 2015, 27(23), 3566–3571 CrossRef CAS.
- Q. Zhang, Y.-X. Deng, H.-X. Luo, C.-Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D.-H. Qu, Assembling a Natural Small Molecule into a Supramolecular Network with High Structural Order and Dynamic Functions, J. Am. Chem. Soc., 2019, 141(32), 12804–12814 CrossRef CAS.
- M. Delahaye, J. M. Winne and F. E. Du Prez, Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry, J. Am. Chem. Soc., 2019, 141(38), 15277–15287 CrossRef CAS.
- B. M. Richardson, D. G. Wilcox, M. A. Randolph and K. S. Anseth, Hydrazone covalent adaptable networks modulate extracellular matrix deposition for cartilage tissue engineering, Acta Biomater., 2019, 83, 71–82 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–yne Cross-Linking, Angew. Chem., Int. Ed., 2020, 59(9), 3609–3617 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Covalent adaptable networks: smart, reconfigurable and responsive network systems, Chem. Soc. Rev., 2013, 42(17), 7161–7173 RSC.
- K. M. Lindenmeyer, R. D. Johnson and K. M. Miller, Self-healing behaviour of furan–maleimide poly(ionic liquid) covalent adaptable networks, Polym. Chem., 2020, 11, 5321–5326 RSC.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: permanent organic networks with glass-like fluidity, Chem. Sci., 2016, 7(1), 30–38 RSC.
- A. Gablier, M. O. Saed and E. M. Terentjev, Rates of transesterification in epoxy-thiol vitrimers, Soft Matter, 2020, 16(22), 5195–5202 RSC.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, Fluorinated Vitrimer Elastomers with a Dual Temperature Response, J. Am. Chem. Soc., 2018, 140(41), 13272–13284 CrossRef CAS.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- S. Kaiser, S. Wurzer, G. Pilz, W. Kern and S. Schlögl, Stress relaxation and thermally adaptable properties in vitrimer-like elastomers from HXNBR rubber with covalent bonds, Soft Matter, 2019, 15(30), 6062–6072 RSC.
- R. Long, H. J. Qi and M. L. Dunn, Modeling the mechanics of covalently adaptable polymer networks with temperature-dependent bond exchange reactions, Soft Matter, 2013, 9(15), 4083–4096 RSC.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Tuning the Viscosity Profile of Ionic Vitrimers Incorporating 1,2,3-Triazolium Cross-Links, Adv. Funct. Mater., 2017, 27(45), 1–10 CrossRef.
- R. G. Ricarte, F. Tournilhac, M. Cloître and L. Leibler, Linear Viscoelasticity and Flow of Self-Assembled Vitrimers: The Case of a Polyethylene/Dioxaborolane System, Macromolecules, 2020, 53(5), 1852–1866 CrossRef CAS.
- F. Snijkers, R. Pasquino and A. Maffezzoli, Curing and viscoelasticity of vitrimers, Soft Matter, 2017, 13(1), 258–268 RSC.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Fast processing of highly crosslinked, low-viscosity vitrimers, Mater. Horiz., 2020, 7(1), 104–110 RSC.
- Y. Zhou, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Poly(butylene terephthalate)/Glycerol-based Vitrimers via Solid-State Polymerization, Macromolecules, 2017, 50(17), 6742–6751 CrossRef CAS.
- Y. Zhou, R. Groote, J. G. P. Goossens, R. P. Sijbesma and J. P. A. Heuts, Tuning PBT vitrimer properties by controlling the dynamics of the adaptable network, Polym. Chem., 2019, 10(1), 136–144 RSC.
- H. Li, B. Zhang, K. Yu, C. Yuan, C. Zhou, M. L. Dunn, H. J. Qi, Q. Shi, Q.-H. Wei and J. Liu, Influence of treating parameters on thermomechanical properties of recycled epoxy-acid vitrimers, Soft Matter, 2020, 16(6), 1668–1677 RSC.
- C. He, P. R. Christensen, T. J. Seguin, E. A. Dailing, B. M. Wood, R. K. Walde, K. A. Persson, T. P. Russell and B. A. Helms, Conformational Entropy as a Means to Control the Behavior of Poly(diketoenamine) Vitrimers In and Out of Equilibrium, Angew. Chem., Int. Ed., 2020, 59(2), 735–739 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis, Science, 2017, 356(6333), 62 CrossRef.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965 CrossRef CAS.
- T. Stukenbroeker, W. Wang, J. M. Winne, F. E. Du Prez, R. Nicolaÿ and L. Leibler, Polydimethylsiloxane quenchable vitrimers, Polym. Chem., 2017, 8(43), 6590–6593 RSC.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Reprocessable Acid-Degradable Polycarbonate Vitrimers, Macromolecules, 2018, 51(2), 389–397 CrossRef CAS.
- M. Hayashi and R. Yano, Fair Investigation of Cross-Link Density Effects on the Bond-Exchange Properties for Trans-Esterification-Based Vitrimers with Identical Concentrations of Reactive Groups, Macromolecules, 2020, 53(1), 182–189 CrossRef CAS.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Vitrimers Designed Both To Strongly Suppress Creep and To Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments, Macromolecules, 2018, 51(15), 5537–5546 CrossRef CAS.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Inverse vulcanized polymers with shape memory, enhanced mechanical properties, and vitrimer behavior, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS.
- F. Ji, X. Liu, D. Sheng and Y. Yang, Epoxy-vitrimer composites based on exchangeable aromatic disulfide bonds: reprocessibility, adhesive, multi-shape memory effect, Polymer, 2020, 122514 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, A fully bio-based epoxy vitrimer: self-healing, triple-shape memory and reprocessing triggered by dynamic covalent bond exchange, Mater. Des., 2020, 186, 108248 CrossRef CAS.
- J. Lee, B. B. Jing, L. E. Porath, N. R. Sottos and C. M. Evans, Shock Wave Energy Dissipation in Catalyst-Free Poly (dimethylsiloxane) Vitrimers, Macromolecules, 2020, 53, 4741–4747 CrossRef CAS.
- L. Zhou, G. Zhang, Y. Feng, H. Zhang, J. Li and X. Shi, Design of a self-healing and flame-retardant cyclotriphosphazene-based epoxy vitrimer, J. Mater. Sci., 2018, 53(9), 7030–7047 CrossRef CAS.
- B. B. Jing and C. M. Evans, Catalyst-Free Dynamic Networks for Recyclable, Self-Healing Solid Polymer Electrolytes, J. Am. Chem. Soc., 2019, 141(48), 18932–18937 CrossRef CAS.
- Y. Liu, Z. Tang, J. Chen, J. Xiong, D. Wang, S. Wang, S. Wu and B. Guo, Tuning the mechanical and dynamic properties of imine bond crosslinked elastomeric vitrimers by manipulating the crosslinking degree, Polym. Chem., 2020, 11(7), 1348–1355 RSC.
- R. Hajj, A. Duval, S. Dhers and L. Avérous, Network Design to Control Polyimine Vitrimer Properties: Physical Versus Chemical Approach, Macromolecules, 2020, 53, 3796–3805 CrossRef CAS.
- S. Ciarella, F. Sciortino and W. G. Ellenbroek, Dynamics of Vitrimers: Defects as a Highway to Stress Relaxation, Phys. Rev. Lett., 2018, 121(5), 58003 CrossRef CAS.
- M. Vatankhah-Varnoosfaderani, S. Hashmi, F. J. Stadler and A. GhavamiNejad, Mussel-inspired 3D networks with stiff-irreversible or soft-reversible characteristics – It's all a matter of solvent, Polym. Test., 2017, 62, 96–101 CrossRef CAS.
- B. R. Elling and W. R. Dichtel, Reprocessable cross-linked polymer networks: are associative exchange mechanisms desirable?, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS.
- G. Marrucci, S. Bhargava and S. L. Cooper, Models of shear-thickening behavior in physically crosslinked networks, Macromolecules, 1993, 26(24), 6483–6488 CrossRef CAS.
- L. Leibler, M. Rubinstein and R. H. Colby, Dynamics of reversible networks, Macromolecules, 1991, 24(16), 4701–4707 CrossRef CAS.
- D. Xu and S. L. Craig, Scaling Laws in Supramolecular Polymer Networks, Macromolecules, 2011, 44(13), 5465–5472 CrossRef CAS.
- C. Shen, Q. Zhao and C. M. Evans, Ion specific, odd–even glass transition temperatures and conductivities in precise network polymerized ionic liquids, Mol. Syst. Des. Eng., 2019, 4(2), 332–341 RSC.
- E. B. Trigg, T. W. Gaines, M. Maréchal, D. E. Moed, P. Rannou, K. B. Wagener, M. J. Stevens and K. I. Winey, Self-assembled highly ordered acid layers in precisely sulfonated polyethylene produce efficient proton transport, Nat. Mater., 2018, 17(8), 725–731 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds, J. Am. Chem. Soc., 2015, 137(20), 6492–6495 CrossRef CAS.
- M. K. Smith and B. H. Northrop, Vibrational properties of boroxine anhydride and boronate ester materials: model systems for the diagnostic characterization of covalent organic frameworks, Chem. Mater., 2014, 26(12), 3781–3795 CrossRef CAS.
- H. M. James and E. Guth, Theory of the elastic properties of rubber, J. Chem. Phys., 1943, 11(10), 455–481 CrossRef CAS.
- T. Yan, K. Schröter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, What Controls the Structure and the Linear and Nonlinear Rheological Properties of Dense, Dynamic Supramolecular Polymer Networks?, Macromolecules, 2017, 50(7), 2973–2985 CrossRef CAS.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Polybutadiene Vitrimers Based on Dioxaborolane Chemistry and Dual Networks with Static and Dynamic Cross-links, Macromolecules, 2019, 52(18), 7102–7113 CrossRef CAS.
- G. Springsteen and B. Wang, A detailed examination of boronic acid–diol complexation, Tetrahedron, 2002, 58(26), 5291–5300 CrossRef CAS.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters, Macromolecules, 2015, 48(7), 2098–2106 CrossRef CAS.
- R. L. Thompson, A. A. Alicke and P. R. de Souza Mendes, Model-based material functions for SAOS and LAOS analyses, J. Non-Newtonian Fluid Mech., 2015, 215, 19–30 CrossRef CAS.
- K. Xing, M. Tress, P.-F. Cao, F. Fan, S. Cheng, T. Saito and A. P. Sokolov, The Role of Chain-End Association Lifetime in Segmental and Chain Dynamics of Telechelic Polymers, Macromolecules, 2018, 51(21), 8561–8573 CrossRef CAS.
- H. Fang, W. Ye, Y. Ding and H. H. Winter, Rheology of the Critical Transition State of an Epoxy Vitrimer, Macromolecules, 2020, 53, 4855–4862 CrossRef CAS.
- N. J. Van Zee and R. Nicolaÿ, Vitrimers: permanently crosslinked polymers with dynamic network topology, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- F. Gamardella, F. Guerrero, S. De la Flor, X. Ramis and A. Serra, A new class of vitrimers based on aliphatic poly(thiourethane) networks with shape memory and permanent shape reconfiguration, Eur. Polym. J., 2020, 122, 109361 CrossRef CAS.
- G. R. Lopez, L. Granado, G. l. Coquil, A. S. Lárez-Sosa, N. Louvain and B. Améduri, Perfluoropolyether (PFPE)-based vitrimers with ionic conductivity, Macromolecules, 2019, 52(5), 2148–2155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01544j |
| This journal is © The Royal Society of Chemistry 2021 |