
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrediction and mitigation of mutation threats to COVID-19 vaccines and antibody therapies†
Jiahui
Chen‡
a,
Kaifu
Gao‡
a,
Rui
Wang‡
 a and
Guo-Wei
Wei
*abc
a and
Guo-Wei
Wei
*abc
aDepartment of Mathematics, Michigan State University, MI 48824, USA. E-mail: weig@msu.edu
bDepartment of Electrical and Computer Engineering, Michigan State University, MI 48824, USA
cDepartment of Biochemistry and Molecular Biology, Michigan State University, MI 48824, USA
First published on 13th April 2021
Abstract
Antibody therapeutics and vaccines are among our last resort to end the raging COVID-19 pandemic. They, however, are prone to over 5000 mutations on the spike (S) protein uncovered by a Mutation Tracker based on over 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 genome isolates. It is imperative to understand how mutations will impact vaccines and antibodies in development. In this work, we first study the mechanism, frequency, and ratio of mutations on the S protein which is the common target of most COVID-19 vaccines and antibody therapies. Additionally, we build a library of 56 antibody structures and analyze their 2D and 3D characteristics. Moreover, we predict the mutation-induced binding free energy (BFE) changes for the complexes of S protein and antibodies or ACE2. By integrating genetics, biophysics, deep learning, and algebraic topology, we reveal that most of the 462 mutations on the receptor-binding domain (RBD) will weaken the binding of S protein and antibodies and disrupt the efficacy and reliability of antibody therapies and vaccines. A list of 31 antibody disrupting mutants is identified, while many other disruptive mutations are detailed as well. We also unveil that about 65% of the existing RBD mutations, including those variants recently found in the United Kingdom (UK) and South Africa, will strengthen the binding between the S protein and human angiotensin-converting enzyme 2 (ACE2), resulting in more infectious COVID-19 variants. We discover the disparity between the extreme values of RBD mutation-induced BFE strengthening and weakening of the bindings with antibodies and angiotensin-converting enzyme 2 (ACE2), suggesting that SARS-CoV-2 is at an advanced stage of evolution for human infection, while the human immune system is able to produce optimized antibodies. This discovery, unfortunately, implies the vulnerability of current vaccines and antibody drugs to new mutations. Our predictions were validated by comparison with more than 1400 deep mutations on the S protein RBD. Our results show the urgent need to develop new mutation-resistant vaccines and antibodies and to prepare for seasonal vaccinations.
000 genome isolates. It is imperative to understand how mutations will impact vaccines and antibodies in development. In this work, we first study the mechanism, frequency, and ratio of mutations on the S protein which is the common target of most COVID-19 vaccines and antibody therapies. Additionally, we build a library of 56 antibody structures and analyze their 2D and 3D characteristics. Moreover, we predict the mutation-induced binding free energy (BFE) changes for the complexes of S protein and antibodies or ACE2. By integrating genetics, biophysics, deep learning, and algebraic topology, we reveal that most of the 462 mutations on the receptor-binding domain (RBD) will weaken the binding of S protein and antibodies and disrupt the efficacy and reliability of antibody therapies and vaccines. A list of 31 antibody disrupting mutants is identified, while many other disruptive mutations are detailed as well. We also unveil that about 65% of the existing RBD mutations, including those variants recently found in the United Kingdom (UK) and South Africa, will strengthen the binding between the S protein and human angiotensin-converting enzyme 2 (ACE2), resulting in more infectious COVID-19 variants. We discover the disparity between the extreme values of RBD mutation-induced BFE strengthening and weakening of the bindings with antibodies and angiotensin-converting enzyme 2 (ACE2), suggesting that SARS-CoV-2 is at an advanced stage of evolution for human infection, while the human immune system is able to produce optimized antibodies. This discovery, unfortunately, implies the vulnerability of current vaccines and antibody drugs to new mutations. Our predictions were validated by comparison with more than 1400 deep mutations on the S protein RBD. Our results show the urgent need to develop new mutation-resistant vaccines and antibodies and to prepare for seasonal vaccinations.
1 Introduction
The expeditious spread of the coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to 95932739 confirmed cases and 2054853 fatalities as of January 20, 2021. In the 21st century, three major outbreaks of deadly pneumonia have been caused by β-coronaviruses: SARS-CoV (2002), Middle East respiratory syndrome coronavirus (MERS-CoV) (2012), and SARS-CoV-2 (2019).1 Similar to SARS-CoV and MERS-CoV, SARS-CoV-2 causes respiratory infections, and the transmission of viruses occurs among family members or in healthcare settings at the early stages of the outbreak. However, SARS-CoV-2 has an unprecedented scale of infection. Considering the high infection rate, high prevalence rate, long incubation period,2 asymptomatic transmission,3,4 and potential seasonal pattern5 of COVID-19, the development of specific antiviral drugs, antibody therapies, and effective vaccines is of paramount importance. Traditional drug discovery takes more than ten years, on average, to bring a new drug to the market.6 However, developing potent SARS-CoV-2 specific antibodies and vaccines is a relatively more efficient and less time-consuming strategy to combat COVID-19 for the ongoing pandemic.7 Antibody therapies and vaccines depend on the host immune system. Recently, studies have been working on the host–pathogen interaction, host immune responses, and the pathogen immune evasion strategies,8–13 which provide insight into understanding the mechanism of antibody therapies and vaccine development.
The immune system is a host defense system that protects the host from pathogenic microbes, eliminates toxic or allergenic substances, and responds to an invading pathogen.14 It has the innate immune system and adaptive immune system as two major subsystems. The innate system provides an immediate but non-specific response, while the adaptive immune system provides a highly specific and effective immune response. Once the pathogen breaches the first physical barriers, such as the epithelial cell layers, secreted mucus layer, and mucous membranes, the innate system will be triggered to identify pathogens by pattern recognition receptors (PRRs), which is expressed on dendritic cells, macrophages, or neutrophils.15 Specifically, PPRs identify pathogen-associated molecular patterns (PAMPs) located on pathogens and then activate complex signaling pathways that introduce inflammatory responses mediated by various cytokines and chemokines, which promote the eradication of the pathogen.16,17 Notably, the transmission of SARS-CoV-2 even occurs in asymptomatic infected individuals, which may delay the early response of the innate immune response.8 Another important line of host defense is the adaptive immune system. B lymphocytes (B cells) and T lymphocytes (T cells) are special types of leukocyte that are the acknowledged cellular pillars of the adaptive immune system.18 Two major subtypes of T cells are involved in the cell-mediated immune response: killer T cells (CD8+ T cells) and helper T cells (CD4+ cells). The killer T cells eradicate cells invaded by pathogens with the help of major histocompatibility complex (MHC) class I. MHC class I molecules are expressed on the surface of all nucleated cells.19 The nucleated cells will firstly degrade foreign proteins via antigen processing when viruses infect them. Then, the peptide fragments will be presented by MHC class I, which will activate killer T cells to eliminate these infected cells by releasing cytotoxins.20 Similarly, helper T cells cooperate with MHC class II, a type of MHC molecule that is constitutively expressed on antigen-presenting cells, such as macrophages, dendritic cells, monocytes, and B cells.21 Helper T cells express T cell receptors (TCR) to recognize antigen bound to MHC class II molecules. However, helper T cells do not have cytotoxic activity. Therefore, they cannot kill infected cells directly. Instead, the activated helper T cells will release cytokines to enhance the microbicidal function of macrophages and the activity of killer T cells.22 Notably, an unbalanced response can result in a “cytokine storm,” which is the main cause of the fatality of COVID-19 patients.23 Correspondingly, a B cell gets involved in the humoral immune response and identifies pathogens by binding to foreign antigens with its B cell receptors (BCRs) located on its surface. The antigens that are recognized by antibodies will be degraded to peptides in B cells and displayed by MHC class II molecules. As mentioned above, helper T cells can recognize the signal provided by MHC class II and upregulate the expression of the CD40 ligand, which provides extra stimulation signals to activate antibody-producing B cells,24 making millions of copies of antibodies (Ab) that recognize the specific antigen. Additionally, when the antigen first enters the body, the T cells and B cells will be activated, and some of them will be differentiated to long-lived memory cells, such as memory T cells and memory B cells. These long-lived memory cells will play a role in quickly and specifically recognizing and eliminating a specific antigen that encountered the host and initiated a corresponding immune response in the future.25 The vaccination mechanism is to stimulate the primary immune response of the human body, which will activate T cells and B cells to generate the antibodies and long-lived memory cells that prevent infectious diseases, which is one of the most effective and economical means of combating COVID-19 at this stage.
As mentioned above, secreted by B cells of the adaptive immune system, antibodies can recognize and bind to specific antigens. Conventional antibodies (immunoglobulins) are Y-shaped molecules that have two light chains and two heavy chains.26 Each light chain is connected to the heavy chain via a disulfide bond, and heavy chains are connected through two disulfide bonds in the mid-region known as the hinge region. Each light and heavy chain contains two distinct regions: the constant region (stem of the Y) and variable region (“arms” of the Y).27 An antibody binds the antigenic determinant (also called epitope) through the variable regions in the tips of the heavy and light chains. There is an enormous amount of diversity in the variable regions. Therefore, different antibodies can recognize many different types of antigenic epitope. To be specific, there are three complementarity determining regions (CDRs) that are arranged non-consecutively in the tips of each variable region. CDRs generate most of the variations between antibodies, which determine the specificity of individual antibodies. In addition to conventional antibodies, camelids also produce heavy-chain-only antibodies (HCAbs). HCAbs, also referred to as nanobodies, or VHHs, contain a single variable domain (VHH) that makes up the equivalent antigen-binding fragment (Fab) of conventional immunoglobulin G (IgG) antibodies.28 This single variable domain can typically acquire affinity and specificity for antigens comparable to conventional antibodies. Nanobodies can easily be constructed into multivalent formats and have higher thermal stability and chemostability than most antibodies do.29 Another advantage of nanobodies is that they are less susceptible to steric hindrances than large conventional antibodies.30
Considering the broad specificity of antibodies, seeking potential antibody therapies has become one of the most feasible strategies to fight against SARS-CoV-2. In general, an antibody therapy is a form of immunotherapy that uses monoclonal antibodies (mAb) to target pathogenic proteins. The binding of an antibody and pathogenic antigen can facilitate an immune response, direct neutralization, radioactive treatment, the release of toxic agents, and cytokine storm inhibition (aka immune checkpoint therapy). The SARS-CoV-2 entry into a human cell is facilitated by the process of a series of interactions between its spike (S) protein and the host receptor angiotensin-converting enzyme 2 (ACE2), primed by host transmembrane protease serine 2 (TMPRSS2).31 As such, most COVID-19 antibody therapeutic developments focus on the SARS-CoV-2 spike protein antibodies that were initially generated from the patient immune response and T-cell pathway inhibitors that block T-cell responses. A large number of antibody therapeutic drugs are in clinical trials. Fifty-five S protein antibody structures are available in the Protein Data Bank (PDB), offering a great resource for mechanistic analysis and biophysical studies.
Currently, most of the antibody therapy developments focus on the use of antibodies isolated from patients' convalescent plasma to directly neutralize SARS-CoV-2,32–34 although there are efforts to alleviate the cytokine storm. A more effective and economical means to fight against SARS-CoV-2 is the vaccine,35 which is the most anticipated approach for preventing the COVID-19 pandemic. A vaccine is designed to stimulate effective host immune responses and provide active acquired immunity by exploiting the body's immune system, including the production of antibodies, and is made of an antigenic agent that resembles a disease-causing microorganism, or surface protein, or genetic material that is needed to generate the surface protein. For SARS-CoV-2, the first choice of surface proteins is the spike protein. There are four types of COVID-19 vaccine, as shown in Fig. 1. (1) Virus vaccines use the virus itself in a weakened or inactivated form. (2) Viral-vector vaccines are designed to genetically engineer a weakened virus, such as measles or adenovirus, to produce coronavirus S proteins in the body. Both replicating and non-replicating viral-vector vaccines are being studied now. (3) Nucleic-acid vaccines use DNA or mRNA to produce SARS-CoV-2 S proteins inside host cells to stimulate the immune response. (4) Protein-based vaccines are designed to directly inject coronavirus proteins, such as S protein or membrane (M) protein, or their fragments, into the body. Both protein subunits and viral-like particles (VLPs) are under development for COVID-19.36 Among these technologies, nucleic-acid vaccines are safe and relatively easy to develop.36 However, they have not been approved for any human use before.
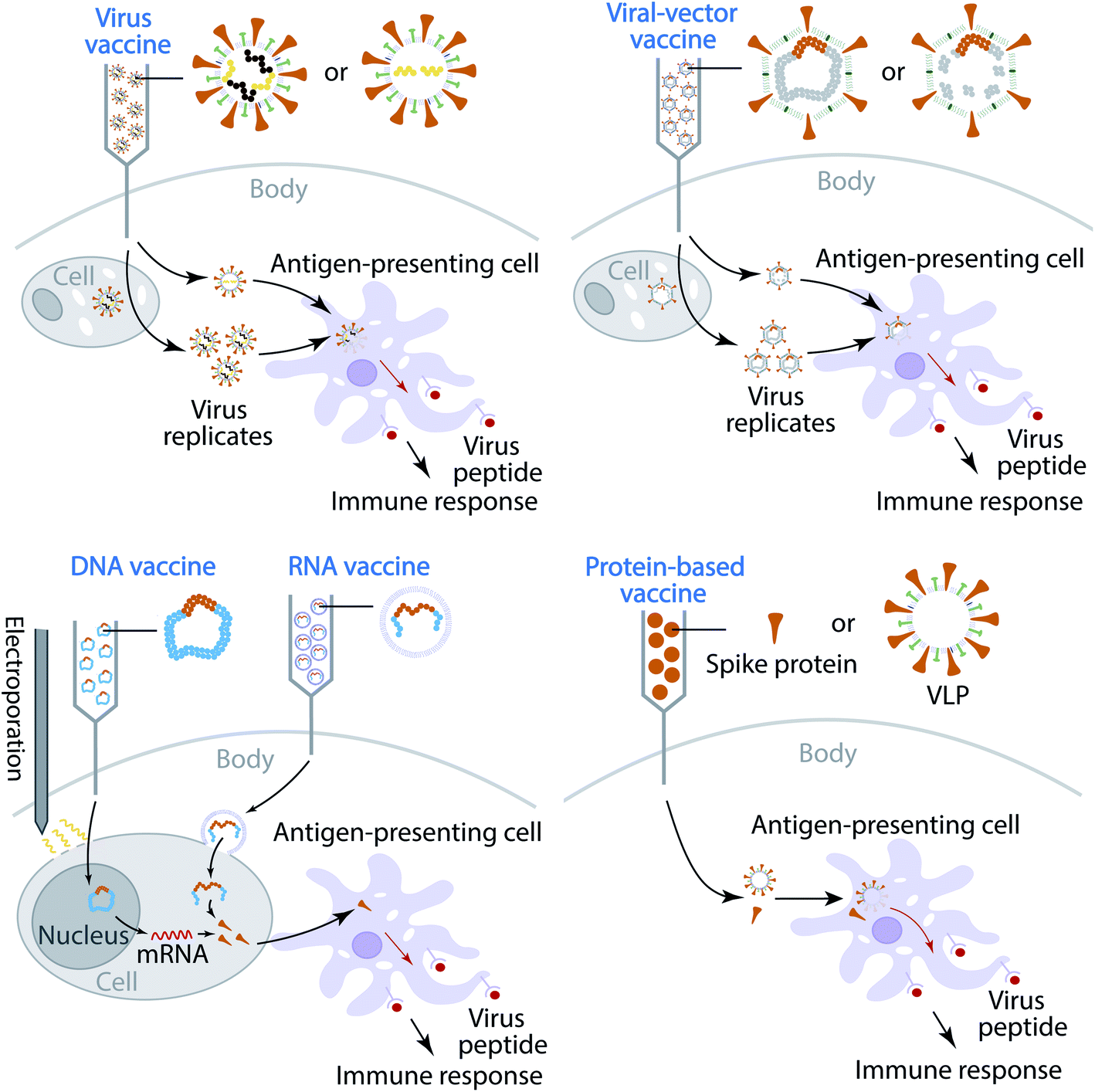 | ||
| Fig. 1 Illustration of four types of COVID-19 vaccine that are currently in development. | ||
However, the general population's safety concerns are the major factors that hinder the rapid approval of vaccines and antibody therapies. A major potential challenge is an antibody-dependent enhancement, in which the binding of a virus to suboptimal antibodies enhances its entry into host cells. All vaccine and antibody therapeutic developments are currently based on the reference viral genome reported on January 5, 2020.37 SARS-CoV-2 belongs to the Coronaviridae family and the Nidovirales order, which has been shown to have a genetic proofreading mechanism regulated by non-structure protein 14 (NSP14) in synergy with NSP12, i.e., RNA-dependent RNA polymerase (RdRp).38,39 Therefore, SARS-CoV-2 has a higher fidelity in its transcription and replication process than other single-stranded RNA viruses, such as the flu virus and HIV. However, the S protein of SARS-CoV-2 has been undergoing many mutations, as reported in ref. 40 and 41. As of January 20, 2021, a total of 5003 unique mutations on the S protein have been detected on 203346 complete SARS-CoV-2 genome sequences. Among them, 462 mutations were on the receptor-binding domain (RBD), the most popular target for antibodies and vaccines. Therefore, it is of paramount importance to establish a reliable paradigm to predict and mitigate the impact of SARS-CoV-2 mutations on vaccines and antibody therapies. Moreover, the efficacy of a given COVID-19 vaccine depends on many factors, including the SARS-CoV-2 biological properties associated with the vaccine, mutation impacts, vaccination schedule (dose and frequency), idiosyncratic response, and assorted factors such as ethnicity, age, gender, and genetic predisposition. The effect of COVID-19 vaccination also depends on the fraction of the population that accepts vaccines. It is essentially unknown at this moment how these factors will unfold for COVID-19 vaccines.
There is no doubt that any preparation that leads to an improvement in the COVID-19 vaccination effect will be of tremendous significance to human health and the world economy. Therefore, in this work, we integrate genetic analysis and computational biophysics, including artificial intelligence (AI), as well as additional enhancement from advanced mathematics to predict and mitigate mutation threats to COVID-19 vaccines and antibody therapies. We perform single nucleotide polymorphism (SNP) calling41 to identify SARS-CoV-2 mutations. For mutations on the S protein, we analyze their mechanism,42 frequency, ratio, and secondary structural traits. We construct a library of 56 existing antibody structures by January 1, 2021 from the PDB and analyze their two-dimensional (2D) and three-dimensional (3D) characteristics. We further predict the mutation-induced binding free energy (BFE) changes of antibody and S protein complexes using a topology-based network tree (TopNetTree),43 which is a state-of-the-art model that integrates deep learning and algebraic topology.44–46 In this work, TopNetTree is trained with newly available deep mutation datasets on the S protein, ACE2, and some antibodies and its predictions are validated with thousands of experimental data points. Our studies indicate that most mutations will significantly disrupt the binding of essentially all known antibodies to the S protein. Therefore, vaccines and antibody drugs that were developed based on the early SARS-CoV-2 genome will be seriously compromised by mutations. Additionally, we show that most known mutations will strengthen the binding between the S protein and ACE2, which gives rise to more infectious variants. Our studies also reveal that SARS-CoV-2 is at an advanced stage of evolution with respect to its ability to infect humans. Although the human immune system is able to produce antibodies that are optimized with respect to a pathogen, the antibodies, once produced, are very vulnerable to attack by mutants.
2 Mutations on the spike protein
As a fundamental biological process, mutagenesis changes the organism's genetic information and serves as a primary source for many kinds of cancer and heritable diseases, which is a driving force for evolution.47,48 Generally speaking, virus mutations are introduced by natural selection, replication mechanism, cellular environment, polymerase fidelity, gene editing, random genetic drift, recent epidemiological features, host immune responses, etc.49,50 Notably, understanding how mutations have changed the SARS-CoV-2 structure, function, infectivity, activity, and virulence is of great importance for coming up with life-saving strategies in virus control, containment, prevention, and medication, especially in the development of antibodies and vaccines. Genome sequencing, SNP calling, and phenotyping provide an efficient means to parse mutations from a large number of viral samples40 (see the ESI (S1†)). In this work, we retrieved more than 200000 complete SARS-CoV-2 genome sequences from the GISAID database51 and created a real-time interactive SARS-CoV-2 Mutation Tracker to report more than 26000 unique single mutations along with their mutation frequency on SARS-CoV-2 as of January 20, 2021. Fig. 2 is a screenshot of our online Mutation Tracker. It describes the distribution of mutations on the complete coding region of SARS-CoV-2. The y-axis shows the natural log frequency for each mutation at a specific position. A reader can download the detailed mutation SNP information from our Mutation Tracker website.
 | ||
| Fig. 2 The distribution of genome-wide SARS-CoV-2 mutations on 26 proteins. The y-axis represents the natural log frequency for each mutation on a specific position of the complete SARS-CoV-2 genome. While only a few landmark positions are labeled with gene (protein) names, the relative positions of other genes (proteins) can be found in our Mutation Tracker (https://users.math.msu.edu/users/weig/SARS-CoV-2_Mutation_Tracker.html). | ||
As mentioned before, the S protein has become the first choice for antibody and vaccine development. Among the 203346 complete genome sequences, 5003 unique single mutations are detected on the S protein. The number of unique mutations (NU) is determined by counting the same type of mutation in different genome isolates only once, while the number of non-unique mutations (NNU, i.e., frequency) is calculated by counting the same type of mutation in different genome isolates repeatedly. Table 1 lists the distribution of 12 SNP types among unique and non-unique mutations on the S protein of SARS-CoV-2 worldwide. It can be seen that C > T and A > G are the two dominant SNP types, which may be due to the innate host immune response via APOBEC and ADAR gene editing.42
604 non-unique mutations on the S gene of SARS-CoV-2 worldwide. NU is the number of unique mutations and NNU is the number of non-unique mutations. RU and RNU represent the ratios of 12 SNP types among unique and non-unique mutations
| SNP type | Mutation type | N U | N NU | R U | R NU |
|---|---|---|---|---|---|
| A > T | Transversion | 454 | 5236 | 9.07% | 1.12% |
| A > C | Transversion | 341 | 2571 | 6.82% | 0.55% |
| A > G | Transition | 700 | 199015 |
13.99% | 42.56% |
| T > A | Transversion | 356 | 1614 | 7.12% | 0.35% |
| T > C | Transition | 779 | 19313 |
15.57% | 4.13% |
| T > G | Transversion | 277 | 1940 | 5.54% | 0.41% |
| C > T | Transition | 542 | 158898 |
10.83% | 33.98% |
| C > A | Transversion | 313 | 10301 |
6.26% | 2.20% |
| C > G | Transversion | 156 | 968 | 3.12% | 0.21% |
| G > T | Transversion | 435 | 34421 |
8.69% | 7.36% |
| G > C | Transversion | 225 | 6090 | 4.50% | 1.30% |
| G > A | Transition | 425 | 27237 |
8.49% | 5.82% |
Moreover, 144 non-degenerate mutations occurred on the S protein RBD, which are relevant to the binding of SARS-CoV-2 S protein and most antibodies as well as ACE2. Additionally, the 218 mutations that occurred on the S protein N-terminal domain (NTD) (residue id: 14 to 226) are relevant to the binding of another two antibodies (4A8 and FC05) and SARS-CoV-2 S protein.
Furthermore, since antibody CDRs are random coils, the complementary antigen-binding domains must involve random coils as well. Table 2 lists the statistics of non-degenerate mutations on the secondary structures of SARS-CoV-2 S protein. Here, the secondary structures are mostly extracted from the crystal structure of 7C2L,52 and the missing residues are predicted by RaptorX-Property.53 We can see that for both unique and non-unique cases, the average mutation rates on the random coils of the S protein have the highest values. Particularly, the 23403 A > G-(D614G) mutation on the random coils has the highest frequency of 192284. If we do not consider the 23403 A > G-(D614G) mutations, then the unique and non-unique average rates on the random coils of S protein still have the highest values (2.81 and 212.01), indicating that mutations are more likely to occur on the random coils. Consequently, the natural selection of mutations may tend to disrupt antibodies.
| Secondary structure | Length | N U | N NU | ARU | ARNU |
|---|---|---|---|---|---|
| Helix | 249 | 516 | 9535 | 2.07 | 38.29 |
| Sheet | 276 | 711 | 20422 |
2.58 | 73.99 |
| Random coils | 748 | 2100 | 350659 |
2.81 | 468.80 |
| Whole spike | 1273 | 3327 | 380616 |
2.61 | 298.99 |
3 SARS-CoV-2 antibodies
In this work, we consider 56 3D structures available from the PDB (https://www.rcsb.org) before January 1, 2021. These 56 structures include 51 structures of antibodies binding to S protein RBD, 4 structures of antibodies having binding domains outside the S protein RBD, and an ACE2-S protein complex. Among the four structures having binding domains outside the RBD, there are three distinct antibodies not binding to the RBD, namely 4A8,52 FC05,54 and 2G12.55 This is because FC05 has two sets of structures (PBD IDs 7CWU and 7CWS) that differ from each other by their components on the RBD (i.e., H014 and P17). Some antibodies are given as combinations of other unique ones. Among the 51 antibodies on the RBD, there are only 42 unique ones, including MR17-K99Y as a mutant of MR17.563.1 3D antibody structure alignment on the S protein
We present the 3D alignment of 45 structures of SARS-CoV-2 S protein with ACE2 and antibodies (excluding the mutant MR17-K99Y of MR17) in Fig. 3. ACE2 in Fig. 3(a) is a reference. Fig. 3(a)–(j) list 42 single antibodies binding to the RBD, and Fig. 3(k) includes the other 3 alignments of 4A8, FC05, and 2G12 whose binding domains are outside the RBD. Fig. 3(m) presents a 3D structure of a single chain of S protein. The PDB IDs of these complexes can be found in Fig. 4.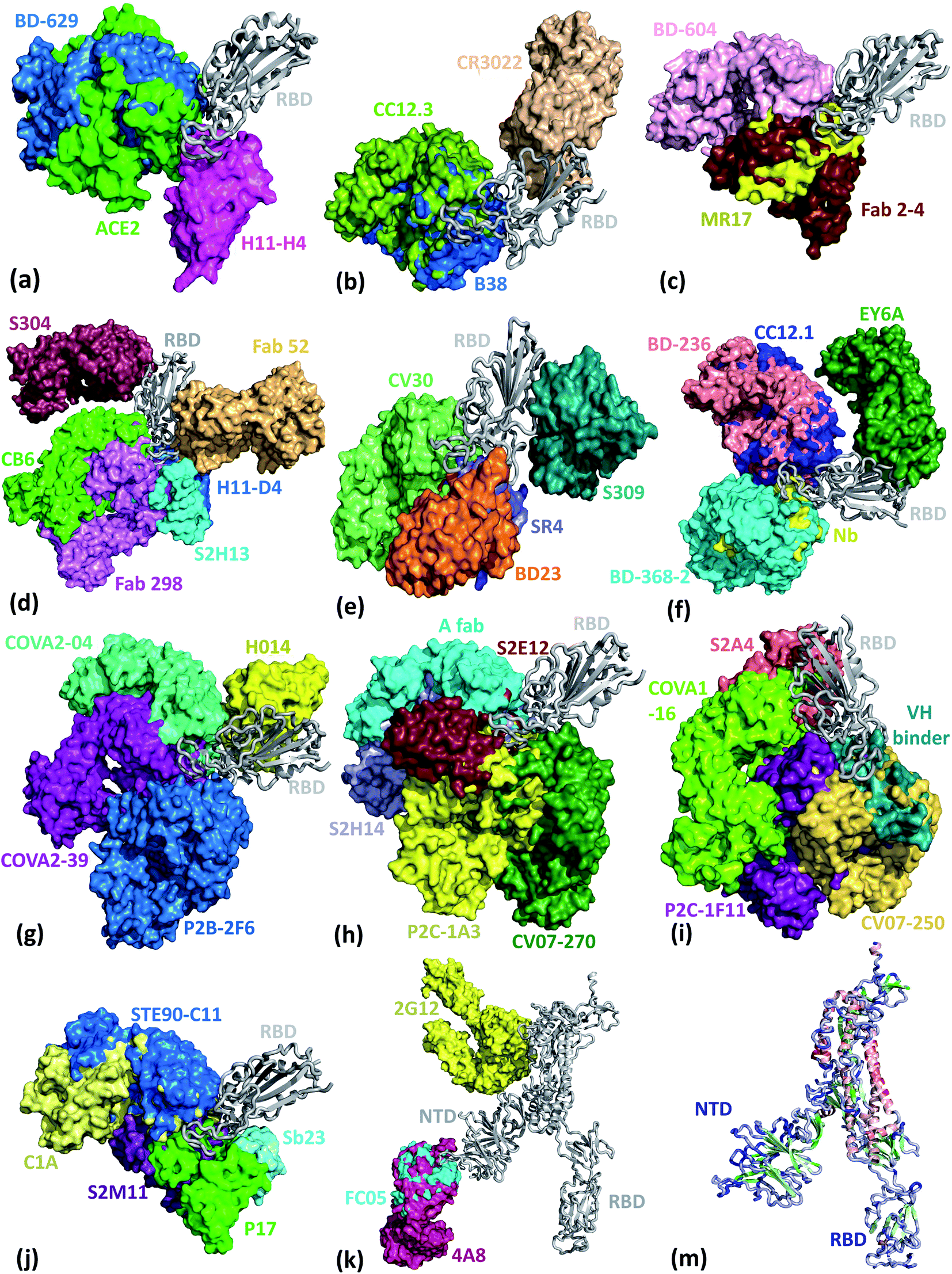 | ||
| Fig. 3 Aligned structures of 46 complexes of the S protein and ACE2 and single antibodies. (a)–(j) The 3D alignment of the available unique 3D structures of SARS-CoV-2 S protein RBD in binding complexes with 42 antibodies (MR17-K99Y is excluded because its binding mode is the same as that of MR17). (k) The 3D alignment of the three antibodies binding outside RBD. (m) The 3D structure of S protein RBD. The red, green, and blue colors represent helix, sheet, and random coils of RBD, respectively. The darker color represents the higher mutation frequency on a specific residue. The structures are (a) ACE2 (6M0J),57 BD-629 (7CH5), H11-H4 (6ZBP); (b) CC12.3 (6XC4),58 B38 (7BZ5),59 CR3022 (6XC3);58 (c) BD-604 (7CH4), MR17 (7C8W),56 Fab 2-4 (6XEY);56 (d) S304 (7JW0),60 CB6 (7C01),61 Fab 52 (7K9Z),62 S2H13 (7JV6),60 H11-D4 (6YZ5),63 Fab 298 (7K9Z);62 (e) CV30 (6XE1),64 BD23 (7BYR),65 SR4 (7C8V),56 S309 (6WPS);66 (f) CC12.1 (6XC2),58 EY6A (6ZCZ),67 BD-236 and nanobody (Nb) (7CHE),68 BD-368-2 (7CHH);68 (g) H014 (7CAH),69 COVA2-04 (7JMO),70 COVA2-39 (7JMP),70 P2B–2F6 (7BWJ);71 (h) P2C-1A3 (7CDJ), CV07-270 (6XKP),72 S2H14 (7JX3),60 A fab (7CJF), S2E12 (7K45);73 (i) CV07-250 (6XKQ),72 P2C–1F11 (7CDI), VH binder (7JWB),74 S2A4 (7JVA),60 COVA1-16 (7JMW);75, (j) C1A (7KFV),76 STE90-C11 (7B3O),77 Sb23 (7A29),78 S2M11 (7K43),73 P17 (7CWM);79; and (k) 4A8 (7C2L),52 FC05 (7CWU),54 and 2G12 (7L06).55 | ||
 | ||
| Fig. 4 Illustration of the contact positions of the antibody and ACE2 paratope with SARS-CoV-2 S protein RBDs on RBD 2D sequences. The corresponding PDB IDs are given in parentheses. | ||
Fig. 3 reveals, except for Fab 52,62 S309,57 CR3022,63 EY6A,67 4A8,52 FC05,54 and 2G12,55 all the other 38 antibodies have their binding sites spatially clashing with that of ACE2. Notably, the paratopes of H014 (ref. 69) and S304 (ref. 60) do not overlap with that of ACE2 directly, but in terms of 3D structures, their binding sites still overlap. This suggests that the bindings of 39 antibodies are in direct competition with that of ACE2. Theoretically, this direct competition reduces the viral infection rate. Such antibodies with strong binding ability will directly neutralize SARS-CoV-2 without the need for antibody-dependent cell cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or other immune mechanisms.
The paratopes of S309, Fab 52, CR3022, and EY6A on the RBD are away from that of ACE2, leading to the absence of binding competition.66,67,80 One study shows that the ADCC and ADCP mechanisms contribute to the viral control conducted by S309 in infected individuals.66 For Fab 52, it was suggested that its mechanism could involve S protein destabilization.62 For CR3022, one research indicates that it neutralizes the virus in a synergistic fashion.81 For EY6A, the hypothesis is that glycosylation of ACE2 accounts for at least part of the observed crosstalk between ACE2 and EY6A.67 More radical examples are 4A8, FC05, and 2G12. 4A8 binds to the NTD of the S protein (Fig. 3(h)), which is quite far from the RBD. It is speculated that 4A8 may neutralize SARS-CoV-2 by restraining the conformational changes of the S protein, which is very important for the SARS-CoV-2 cell entry.52 FC05 is combined with P17 or H014 to form a cocktail.542G12 binds to the S protein S2 domain.55 Any antibody or drug that can inhibit the serine protease TMPRSS2 priming of the S protein priming can effectively stop the viral cell entry.31
3.2 2D residue contacts between antibodies and the S protein RBD
Fig. 3 provides a visual illustration of antibody and ACE2 competitions. It remains to be known in the residue detail what has happened to these competitions. To better understand the antibody and S protein interactions, we study the residue contacts between antibodies and the S protein. We include the ACE2 as a reference but excluding antibodies MR17-K99Y as well as 4A8, FC05, and 2G12 that bind to other domains.In Fig. 4, the paratopes of 42 individual antibodies (excluding MR17-K99Y) and ACE2 were aligned on the S protein RBD 2D sequence, and their contact regions are highlighted. From the figure, one can see that, except for Fab 52, S309, CR3022, EY6A, H014, and S304, all the other 36 antibodies have their antigenic epitopes overlapping with the ACE2, especially on the residues from 486 to 505 of the RBD. Although the paratopes of H014 and S304 do not overlap with that of ACE2 directly, their binding sites still overlap in 3D structures. Therefore, these 38 antibodies competitively bind against ACE2 as revealed in Fig. 3.
3.3 Antibody sequence alignment and similarity analysis
The next question is whether there is any connection or similarity between the antibody paratopes in our library, particularly for those antibodies that share the same binding sites. To better understand this perspective, we carry out multiple sequence alignment (MSA) to further study the similarities and differences among existing antibodies. Many antibodies are very similar to each other and can be classified into several clusters using the CD-HIT suite.82 The first and largest cluster includes COVA2-04, CC12.1, BD-236, BD-604, B38, EY6A, S304, P2C-1A3, A fab, C1A, STE90-C11, and CB6. Their identity scores to CB6 are 90.48%, 94.74%, 93.59%, 93.35%, 94.77%, 92.52%, 90.62%, 90.51%, 91.18%, 94.08%, and 93.00%, respectively. The second cluster contains BD-629, CC12.3, P2C-1F11, and CV30. Their identity scores to CV30 are 95.41%, 96.32%, and 97.68%, respectively. The third cluster has CV07-270 and COVA2-39, and the pairwise identity score is 90.18%. The fourth cluster is composed of H11-H4, H11-D4, and Nb, and their identity scores to Nb are 99.25% and 95.52%, respectively. They are all nanobodies. The fifth cluster has Fab 298 and COVA1-16, and the pairwise identity score is 90.80%. Their alignment plots are given in the ESI (Fig. S1–S5†).The above similarity indicates that the adaptive immune systems of individuals have a common way to generate antibodies. On the other hand, the existence of five distinct clusters, as well as antibodies 4A8, FC05, and 2G12 suggests the diversity in the immune response. Note that we have also included ACE2 in our MSA as a reference, but none of the existing antibodies are similar to ACE2 because they were created from entirely different mechanisms.
4 Mutation impacts on SARS-CoV-2 antibodies
To investigate the influences of existing S protein mutations on the binding free energy (BFE) of S protein and antibodies, we consider 462 mutations that occurred on the S protein RBD, which are relevant to the binding of SARS-CoV-2 S protein and antibodies as well as ACE2. Additionally, 540 mutations occurred on the NTD of the S protein (residue id: 14 to 226) which are relevant to the binding of SARS-COV-2 S protein and antibody 4A8 (PDB: 7C2L). We predict the free energy changes following existing mutations using our TopNetTree model.43 The mutations on the RBD are considered for the predictions of BFE changes. Our predictions are built from the X-ray crystal structure of SARS-CoV-2 S protein and ACE2 (PDB 6M0J),57 and various antibodies (PDBs 6WPS,666XC2,586XC3,586XC4,586XC7,586XE1,646XEY,836XKP,726XKQ,726YLA,636YZ5, 6Z2M, 6ZBP, 6ZCZ,676ZER,677A29,787B3O, 7BWJ,717BYR,657BZ5,597C01,617C2L,527C8V,567C8W,567CAH,697CAH,697CAN,567CDI, 7CDJ, 7CH4,687CH5,687CHB,687CHE,687CHF,687CHH,687CJF, 7CWM,797CWN797JMO,707JMP,707JMW,757JV6,607JVA,607JVC,607JW0,607JWB,747JX3,607K43,737K45,737K9Z,627KFV,767KFW,767KFX,76 and 7KFY76). The BFE change following mutation (ΔΔG) is defined as the subtraction of the BFE of the mutant type from the BFE of the wild type: ΔΔG = ΔGW − ΔGM, where ΔGW is the BFE of the wild type and ΔGM is the BFE of the mutant. Therefore, a negative BFE change means that the mutation decreases affinities, making the protein–protein interaction less stable.Four antibody–S protein complexes are examined in this section. Next, we present a library of mutation-induced BFE changes for all mutations and 51 antibodies, as well as ACE2. The statistical analysis of mutation impacts on antibodies is discussed.
4.1 Single antibody–S protein complex analysis
For four antibody–S protein complexes, since there are too many mutations, we only consider those mutations whose frequencies are greater than 10. We first present the BFE changes (ΔΔG) of the SARS-CoV-2 S protein binding domain with antibody 4A8 in Fig. 5, which is one of the three complexes that are not on the RBD in our collections of S protein and antibody complexes. A total 141 of 540 mutations on residue ID from 14 to 226 have frequencies larger than 10. Most mutations have small BFE changes (from −0.5 kcal mol−1 to 0.5 kcal mol−1) in their binding free energies, while 28 mutations have negative BFE changes less than −0.5 kcal mol−1. Notably, 53 out of 141 mutations on the binding domain have positive BFE changes, which means that the mutations increase affinities and would make the S protein–4A8 interactions more stable. However, the majority (63%) of mutations have negative BFE changes, including high-frequency mutations R102I and W152C with frequencies of 89 and 356, respectively. Since the largest positive and negative BFE changes are 0.37 and −2.06 kcal mol−1 (−3.1 if low frequency mutations are counted), respectively, the prediction indicates that antibody 4A8 isolated from 10 convalescent patients at the early stage of the pandemic52 is an optimized product of the human immune system with respect to the original S protein. It is also noted that many mutations on the binding domain, such as W152L, S247N, and Y248H, have significant negative free energy changes. The mutations on the binding domain with large negative BFE changes reveal that the binding of antibody 4A8 and S protein will be potentially disrupted. | ||
| Fig. 5 Illustration of SARS-CoV-2 mutation-induced binding free energy changes for the complexes of S protein and 4A8 (PDB: 7C2L). The blue color in the structure plot indicates a positive BFE change while the red color indicates a negative BFE change, and toning indicates the strength. Here, mutations R102I, W152C, W152L, S247N, and Y248H could potentially disrupt the binding of antibody 4A8 and S protein. | ||
Next, we study the BFE changes (ΔΔG) induced by 80 mutations on the SARS-CoV-2 S protein RBD for the antibody Fab 2-4 (PDB: 6XEY) in Fig. 6. Antibody Fab 2-4 shares a similar binding domain with ACE2 and thus is a potential candidate for the direct neutralization of SARS-CoV-2. Most mutations induce small changes in the binding free energies, while mutations E484K, E484Q, F486L, and F490S have large negative BFE changes. Overall, 38 out of 80 mutations on the RBD lead to negative BFE changes, which means 48% of mutations will potentially weaken the binding between antibody Fab 2-4 and S protein. For positive BFE changes, the largest value is only 0.55 kcal mol−1 and the average of positive BFE changes is 0.16 kcal mol−1. However, many mutations with negative BFE changes have a very large magnitude, indicating that antibody Fab 2-4 was an immune product optimized with respect to the original un-mutated S protein. In general, the mutations on S protein weaken the Fab 2-4 binding with S protein and make it less competitive with ACE2 as most mutations strengthen the S protein and ACE2 binding. It is interesting to note that mutation E484K is the so-called South Africa variant. It indeed has a strong vaccine-escape effect.
 | ||
| Fig. 6 Illustration of SARS-CoV-2 mutation-induced binding free energy changes for the complexes of S protein and Fab 2-4 (PDB: 6XEY). The blue color in the structure plot indicates a positive BFE change while the red color indicates a negative BFE change, and toning indicates the strength. Here, mutations E484K, E484Q, F486L, and F490S could potentially disrupt the binding of antibody Fab 2-4 and the S protein. | ||
In Fig. 7, we illustrate the mutation-induced BFE changes for antibody MR17 (PDB: 7C8W), which shares the binding domain with ACE2 as well. One can notice that five mutations, L452R, E484K, F486L, F490S, and S494L, have BFE changes less than −1 kcal mol−1 as well as high frequencies. The rest of the mutations have a small magnitude of changes. 27 out of 80 mutations have positive BFE changes with the largest value less than 0.25 kcal mol−1. Our results indicate that antibody MR37 is likely to be isolated from patients at the early stage and thus, it was optimized based on an early version of the SARS-CoV-2 virus. Mutations L452R, E484K, F486L, F490S, and S494L will reduce its competitiveness with ACE2 (Fig. 7).
 | ||
| Fig. 7 Illustration of SARS-CoV-2 mutation-induced binding free energy changes for the complexes of S protein and MR17 (PDB: 7C8W). Blue in the structure plot indicates a positive BFE change while red indicates a negative BFE change, and toning indicates the strength. Here, mutations L452R, E484K, F486L, F490S, and S494L could potentially disrupt the binding of antibody MR17 and the S protein. | ||
 | ||
| Fig. 8 Illustration of SARS-CoV-2 mutation-induced binding free energy changes for the complexes of S protein and S309 (PDB: 6WPS). The blue color in the structure plot indicates a positive BFE change while the red color indicates a negative BFE change, and toning indicates the strength. Here, mutations E340A, N354D, and K356R could potentially weaken the binding of antibody S309 and the S protein. | ||
Finally, we consider the BFE change predictions for the antibody S309 and S protein complex, whose receptor binding motif (RBM) does not overlap with the RBM of ACE2 (see Fig. 3(e)). The BFE changes induced by 80 mutations are predicted. Among them, 38 changes are positive. Similar to the aforementioned antibodies, most of the mutations lead to small changes in their binding affinity magnitude but three mutations, E340A, N354D, and K356R, induce moderate negative changes. Interestingly, none of the 80 RBD mutations have a major impact on S309. Although mutation R403K might disrupt S309, it does weaken many other antibody bindings with the S protein. While antibodies play a variety of functions in the human immune system, such as neutralization of infection, phagocytosis, antibody-dependent cellular cytotoxicity, etc., their binding with antigens is crucial for these functions. Our analysis of BFE changes following mutations on the S protein suggests that some antibodies will be less affected by mutations, which is important for developing vaccines and antibody therapies.
4.2 Mutation impact library
In this section, we build a library of mutation-induced BFE changes for all mutations and all antibodies as well as ACE2. In principle, we could create a library of all possible mutations for all antibodies, as we did for ACE2.84 Here, we limit our effort to all existing mutations. Antibody 4A8 on the NTD has been discussed above. We consider antibodies on the RBD.Based on our earlier analysis, three types of SARS-CoV-2 S protein secondary structural residue have different mutation rates. Among them, the random coils are major components of the RDB and the NTD, as shown in Fig. 3. Most RBD mutations (287 of 462) occur on the residues whose secondary structures are coil, while 93 out of 462 mutations are on the helix, and 82 out of 462 mutations are on the sheet. Therefore, mutations on the RBD are split into three categories based on their locations in secondary structures of helix, sheet, and coil. In Fig. 9, we present the BFE changes for the complexes of the S protein and antibodies or ACE2 induced by mutations on the helix residues of the S protein RBD. The frequency for each mutation is also presented. Most mutations on helix residues lead to negative BFE changes (pink squares), which weaken the bindings, while some mutations induce positive BFE changes (green squares). It is noted that most mutations lead to the strengthening of the S protein and ACE2 binding, which is consistent with the natural selection rule. Mutations N406G, I418N, N422K, D442H, Y505S, and Y505C give rise to a strong weakening effect on most antibodies. The N439K mutation having the highest frequency, shows a positive BFE change on ACE2 but negative changes on most antibodies. Mutation D405Y appears to strengthen most antibodies.
 | ||
| Fig. 9 Illustration of the SARS-CoV-2 helix-residue mutation induced BFE changes for the complexes of S protein and 51 antibodies or ACE2. Positive changes strengthen the binding while negative changes weaken the binding. Mutation frequency is given for each mutation. Grey color indicates that PDB structures do not include residues induced by those mutations. | ||
In Fig. 10, we present the BFE changes for the S protein and antibody (ACE2) complexes following sheet residue mutations of the S protein RBD. Like the last case, most mutations lead to positive BFE changes for ACE2, indicating infectivity strengthening. There are many disruptive mutations, such as R355W, F401I, F401C, I402F, C432G, I434K, A435P, O493P, V510E, V512G, and L513P, that will weaken most antibody and S protein complexes. On the other hand, most mutations strengthen certain antibodies but weaken other ones, which allows the effectiveness of antibody cocktails for better protection. The binding of antibody H014 and the S protein is strengthened by many mutations, particularly S375F, K378O, R403K, and Y453F. Among them, Y453F is an infectivity-strengthening mutation with a relatively high frequency.
 | ||
| Fig. 10 Illustration of SARS-CoV-2 sheet-residue mutation induced BFE changes for the complexes of S protein and 51 antibodies or ACE2. Positive changes strengthen the binding while negative changes weaken the binding. Mutation frequency is presented for each mutation. Grey color indicates that PDB structures does not include residues induced by those mutations. | ||
Fig. 11–13 present the BFE changes for the S protein and antibody (ACE2) complexes following coil residue mutations of the S protein RBD. Overall, most mutations on coil residues lead to mild negative BFE changes. However, mutations V350F, W353R, I401N, G416V, G431V, Y449D, Y449S, C480R, P491R, P491L, Y495C, and O506P will weaken most antibody bindings to the S protein. Some residues, like A348, N460, and P521, can produce many binding-strengthening mutations for most antibodies and ACE2. For the high-frequency mutation S447N in Fig. 13, the BFE changes are mild on ACE2 and antibodies. Additionally, the N501Y mutation, one of the typical mutations in the UK B.1.1.7 variant, strengthens the infectivity but induces mixed reactions to antibodies as shown in Fig. 13.
 | ||
| Fig. 11 Illustration of SARS-CoV-2 coil-residue mutation induced BFE changes for the complexes of S protein and 51 antibodies or ACE2. Positive changes strengthen the binding while negative changes weaken the binding. Mutation frequency is presented for each mutation. Grey color indicates that PDB structures do not include residues induced by those mutations. | ||
 | ||
| Fig. 12 Illustration of SARS-CoV-2 coil-residue mutation induced BFE changes for the complexes of S protein and 51 antibodies or ACE2 (continued from Fig. 11). Positive changes strengthen the binding while negative changes weaken the binding. Mutation frequency is presented for each mutation. Grey color indicates that PDB structures do not include residues induced by those mutations. | ||
 | ||
| Fig. 13 Illustration of SARS-CoV-2 coil-residue mutation induced BFE changes for the complexes of S protein and 51 antibodies or ACE2 (continued from Fig. 12). Positive changes strengthen the binding while negative changes weaken the binding. Mutation frequency is presented for each mutation. Grey color indicates that PDB structures do not include residues induced by those mutations. | ||
4.3 Statistical analysis of mutation impacts on COVID-19 antibodies
First, we perform a statistical analysis of all mutation-induced BFE changes studied in the last section. Most mutations induce binding-weakening BFE changes. The total rate of negative BFE changes is 71% (i.e., 16661 out of 23512); for coil residues, 67% BFE changes are negative, while for helix and sheet residues, 72% and 80% BFE changes are negative, respectively. However, for ACE2, 300 out of 462 mutations (i.e., 65%) on the RBD produce positive or binding-strengthening BFE changes, showing the effect of the natural selection of mutations. In contrast, at most 200 out of 462 mutations on the RBD give rise to negative BFE changes for antibodies. More specifically, 11 antibodies have less than 100 positive BFE changes while 41 antibodies have less than 200 positive BFE changes. Interestingly, in our prediction, 4 out of the 43 single antibodies have less than 100 positive BFE changes, while 7 out of the 9 antibody cocktails have less than 100 positive BFE changes. Although antibody cocktails have mild negative BFE changes, it turns out that they have high affinities to S protein and the BFE changes are mild for positive ones as well.
Fig. 14 indicates the BFE change extreme values (maximal in cyan and minimal in pink) and average values (positive in blue and negative in red) of the complexes of S protein and ACE2 or antibodies following mutations. The maximal BFE changes of the helix, sheet, and coil residues are 1.44 kcal mol−1, 1.94 kcal mol−1, and 1.00 kcal mol−1, respectively, while the minimal BFE changes are −3.87 kcal mol−1, −3.9 kcal mol−1, and −4.38 kcal mol−1, respectively. The disparity in their maximal and minimal values indicates the relatively optimal nature of the S protein and antibody binding complexes. It means that the human immune system has the ability to produce optimized antibodies for a given antigen. However, antibodies, once generated, are prone to infection by new mutants. The disparity shown in Fig. 14 also means that the SARS-CoV-2 was at an advanced stage of evolution with respect to human infection. There is not much room for SARS-CoV-2 to improve its infectivity by single-site mutations.
 | ||
| Fig. 14 Illustration of SARS-CoV-2 mutation-induced maximal and minimal BFE changes in cyan and pink for the complexes of S protein and 51 antibodies or ACE2, and average of positive and negative BFE changes in blue and red. Here, the maximal change strengthens the binding while the minimal change weakens the binding for each complex. | ||
Many antibody cocktails, such as CR3022/H11-D4, CC12.1/CR3022, BD-236/BD368-2, BD604/BD368-2, S309/S2H14/S304, and Fabs 298/52, are relatively less sensitive to the current S protein mutations. However, some other antibodies, such as H11-D4, CV30, CC12.3, and S2H13, can be dramatically affected by SARS-CoV-2 mutations. Importantly, ACE2 is also impacted by mutations and has the largest positive BFE change on average.
5 Mutation impacts on COVID-19 vaccines
The increasing number of infection cases and deaths, the global spread situation, and the lack of prophylactics and therapeutics give rise to an urgent need for the prevention of COVID-19. Vaccination is the most effective and economical means to control pandemics.35 Currently, 248 vaccines are in various clinical trial stages, as reported in an online COVID-19 Treatment And Vaccine Tracker (https://covid-19tracker.milkeninstitute.org/#vaccines_intro). Broadly speaking, there are four types of coronavirus vaccine in development: virus vaccines, viral-vector vaccines, nucleic-acid vaccines, and protein-based vaccines, as shown in Fig. 1. The first type of vaccine is the virus vaccine, which injects weakened or inactivate viruses into the human body. A virus is conventionally weakened by altering its genetic code to reduce its virulence and elicit a stronger immune response. A biotechnology company, Codagenix, is currently working on a “codon optimization” technology to weaken viruses, and its weakened virus vaccine is in development.85 Unlike a weakened virus, an inactivated virus cannot replicate in the host cell. A virus is inactivated by heating or using chemicals, which induces neutralizing antibody titers and has been proven to be safe.86 At this stage, both Sinopharm, which works with the Beijing Institute of Biological Products and Wuhan Institute of Biological Products, and Sinovac, which works with Institute Butantan and Bio Farma, are developing inactive SARS-CoV-2 vaccines that are in phase III clinical trials.The second type of vaccine is the viral-vector vaccine, which is genetically engineered so that it can produce coronavirus surface proteins in the human body without causing diseases. There are two subtypes of viral-vector vaccine: the non-replicating viral vector and the replicating viral vector. On February 25, 2021, the World Health Organization (WHO) granted an emergency use listing (EUL) for a vaccine developed by AstraZeneca and the University of Oxford, which is a non-replicating viral vector vaccine. Moreover, there are 3 non-replicating viral vector vaccines in phase III trials as well. They work by taking a chimpanzee virus and coating it with the S proteins of SARS-CoV-2. The chimp virus causes a harmless infection in humans, but the spike proteins will activate the immune system to recognize signs of a future SARS-CoV-2 invasion. Notably, booster shots may be needed to retain long-lasting immunity. Furthermore, at this stage, only one replicating viral-vector vaccine is in phase II. The University of Hong Kong, in cooperation with Xiamen University and Wantai Biological Pharmacy, is developing such a replicating viral vaccine, which tends to be safe and provoke a strong immune response.
The third type of vaccine is nucleic acid vaccines, which include two subtypes: DNA-based vaccines and RNA-based vaccines. At least 40 teams are currently working on nucleic-acid vaccines since they are safe and easy to develop. The DNA-based vaccine works by inserting genetically engineered blueprints of the viral gene into small DNA molecules such as plasmids for injection. Moreover, the electroporation technique is employed to create pores in membranes to increase DNA uptake into cells. The injected DNA will produce mRNA by transcription with the help of the nucleus in human cells. Such an mRNA will translate viral proteins (mostly spike proteins), which are dutifully produced by cells in response to the genes, alarm the immune system, and should produce immunity. Currently, there is one DNA-based vaccine in phase III. Similar to DNA-based vaccines, RNA-based vaccines provide immunity through the introduction of RNA, which is encased in a lipid coat to ensure that it enters into cells. Two RNA-based vaccines have been granted authorization for emergency use in many countries. One is designed by BioNTech, which cooperates with Pfizer, and the other one is from Moderna.
The fourth type of vaccine is the protein-based vaccine, which aims to inject viral proteins directly to human bodies to trigger immune readiness. The protein subunit vaccine is one of the subtypes of the protein-based vaccine. More than 80 teams are working on vaccines with viral protein subunits, such as spike proteins and membrane (M) proteins. Another subtype of the protein-based vaccine is the virus-like particle (VLP) vaccine. VLP vaccines closely resemble viruses. However, they are not infectious since they do not contain viral genetic material. Their non-replicating properties provide a safer alternative to weakened virus vaccines; the HPV vaccine or newer flu vaccines are VLP vaccines. Currently, 22 teams are working on VLP vaccines for future prevention of COVID-19.
5.1 Secondary structures of antigenic determinants
Since the structural basis of antibody CDRs, or paratopes, is random coils, we hypothesize that CDRs favor antigenic random coils as complementary epitopes, i.e., antigenic determinants.87,88Fig. 15 depicts the 3D structure of S protein, where the random coils are drawn with green strings, and the other secondary structure is described with the purple surface. It shows that the RBD and the NTD mostly consist of random coils. The RBD is the antigenic determinant of 43 structurally known SARS-CoV-2 antibodies; meanwhile, the NTD is the binding domain of antibodies 4A8 and FC05 and antibody 2G12 also binds to the S2 domain with random coils, which confirms our hypothesis. More detailed analysis considered the random coil percentages of antibodies' paratopes which are summarized in Table S1 of the ESI.† It reveals that antibodies predominantly contact residues in random coils of S protein. Most of the antibody paratopes had greater than 90% random coil content. | ||
| Fig. 15 The 3D rotational structure of SARS-CoV-2 S protein. The random coils of S protein are drawn with green strings and the other secondary structure is described with a purple surface. (a) 3D structure of S protein. (b) 3D structure of S protein that is rotated 90° based on (a). (c) 3D structure of S protein that is rotated 180° based on (a). (d) 3D structure of S protein that is rotated 270° based on (a). | ||
Fig. 16 shows the secondary structure of the S protein. The red, blue, and green colors represent helix, sheet, and random coils of S protein. It can be seen that the S protein mostly consists of random coils, which means that there are many other potential antigenic epitopes on the S protein for antibody CDRs. We believe that the emphasis on direct binding competition with ACE2 in the past66,67,80 has led to the neglecting of many important antibodies that do not bind to the RBD. Therefore, we suggest that researchers pay more attention to antibodies that do not bind to the RBD.
 | ||
| Fig. 16 The secondary structure of S protein. The red, green, and blue colors represent helix, sheet, and random coils of S protein. | ||
5.2 Statistical estimation of mutation impacts on COVID-19 vaccines
Vaccine efficacy is an essential issue for the control of the COVID-19 pandemic. The S protein is one of the most popular surface proteins for vaccine development. However, mutations have accumulated on the S protein of SARS-CoV-2, which may reduce the vaccine efficacy. As we found in section 2, mutations are more likely to happen on the random coils of S protein, which may have a devastating effect on vaccines in development.As shown in Fig. 14, mutations could considerably weaken the binding between the S protein and antibodies and thus pose a direct threat to reduce the efficacy of vaccines. However, there are a few obstacles in determining the exact impacts of mutations on COVID-19 vaccines. Firstly, the four types of vaccine platform can produce very different virus peptides, resulting in different immune responses, as well as antibodies. Secondly, even for a given vaccine platform, different peptides may be produced due to different immune responses caused by gender difference, age difference, race difference, etc. Therefore, in this work, we proposed to understand the impact of SARS-CoV-2 mutations on COVID-19 vaccines by statistical analysis. By evaluating the binding affinity changes induced by 51 existing SARS-CoV-2 antibodies, as shown in Fig. 9 to 13, we can identify vaccine escape mutants that will strengthen the binding between the S protein and ACE2 while disrupting the binding between the S protein and antibodies. Table 3 lists a collection of the most disruptive mutations. However, this list is not complete. There are many other antibody disrupting mutations as shown in Fig. 9 to 13. For example, the infectivity-strengthening South Africa mutant E484K can cause dramatically disruptive effects on many antibodies such as H11-D4, Fab 2-4, H11-H4, COVA2-39, BD368-2, etc. but it also enhances the binding of other antibodies, such as B38, CV30, CC21.1, Sb23, Fabs 298 52, etc. The infectivity-strengthening mutation N501Y in UK B.1.1.7 variants has a disruptive effect only on a few known antibodies, including B38, CC12.3, S2M11, NAB, S309, S2H12, S304, C1A-B12, STE90-C11, etc.
| Location | Mutants |
|---|---|
| Helix | E406G, I418N, Y421D, N422K, D442H, Y505S |
| Sheet | R355W, F400I, F400C, I402F, C432G, I434K, A435P, Q493P, V510E, V512G, L513P |
| Coils | V350F, W353R, I410N, G416V, G431V, Y449D, Y449S, L461H, S469P, C480R, P491R, P491L, Y495C, Q506P |
In a nutshell, by setting up a SARS-CoV-2 antibody library with the statistical analysis based on the mutation-induced binding free energy changes, we can estimate the impacts of SARS-CoV-2 mutations on COVID-19 vaccines, which will provide a way to infer how a specific mutation will pose a threat to vaccines. This approach works better when more antibody structures become available.
Another important factor in prioritization is mutation frequency. Fig. 9–13 have provided frequency information from our SNP calling. Once a mutation is identified as a potential threat, it can be incorporated into the next generation of vaccines in a cocktail approach. In principle, all four types of vaccine platform allow the accommodation of new viral strains.
6 Validation
Although the details of the methods used in this work are presented in the ESI,† we provide a validation of our deep learning prediction model, TopNetTree,43 which is crucial to the credibility of this work. Specifically, we demonstrate the prediction performance of S protein mutation induced BEF changes on CTC-445.2 compared to the experimental deep mutation enrichment data.89 More detailed descriptions of methods and datasets are provided in the ESI.†Fig. 17 presents a comparison between experimental deep mutation enrichment data on the RBD and machine learning predicted RBD-mutation-induced BFE changes for the SARS-CoV-2 S protein and CTC-445.2 complex. In the heatmaps of Fig. 17, one can see that the predicted BFE changes have a very high correlation with the experimental enrichment ratio data. Both enrichment ratios and BFE changes describe the affinity strength of the protein–protein interaction induced by mutations. The high similarity between these heatmaps demonstrates the reliability of our machine learning predictions of BFE changes following mutations on the S protein RBD.
 | ||
| Fig. 17 A comparison between experimental deep mutation enrichment data and TopNetTree predictions for the SARS-CoV-2 S protein RBD and CTC-445.2 complex (7KL9 (ref. 89)). Top left: deep mutational scanning heatmap showing the average effect on the enrichment for single site mutants of the RBD when assayed by yeast display for binding to CTC-445.2.89 Top right: the RBD colored by average enrichment at each residue position bound to CTC-445.2. Bottom: machine learning predicted BFE changes for the CTC-445.2 and S protein complex induced by single site mutations on the RBD. | ||
7 Conclusion
The coronavirus disease 2019 (COVID-19) pandemic has gone out of control globally. There is no specific medicine or effective treatment for this viral infection at this point. Vaccination is widely anticipated to be the endgame for taming the viral rampage. Another promising treatment that is relatively easy to develop is antibody therapies. However, both vaccines and antibody therapies are prone to more than 26000 unique mutations recorded in the Mutation Tracker.
We present the most comprehensive analysis and prediction of mutation threats to vaccines and antibody therapies. First, we identify existing mutations on the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) protein, which is the main target for both vaccines and antibody therapies. We analyze the mechanism, frequency, and ratio of mutations along with the secondary structures of the S protein. Additionally, we build a library of 55 antibodies with structures available from the Protein Data Bank (PDB) and analyze their two-dimensional (2D) and three-dimensional (3D) characteristics by employing computational biophysics. We further predict the mutation-induced binding free energy (BFE) changes of S protein and antibody complexes using a model called TopNetTree based on deep learning and algebraic topology. The performance of our model has been extensively validated by its prediction of experimental deep mutation data. Our significant findings are as follows. First, we reveal that none of the known mutations are safe to all antibodies. On average, most mutations (i.e., 71%) will weaken the binding between the S protein and antibodies, which implies that vaccines will also be compromised by existing mutations. Additionally, we identify 31 antibody disrupting mutants that dramatically weaken the binding between the S protein and most known antibodies. Moreover, we find that most RBD mutations (i.e., 64.9%) will enhance the binding strength between the S protein and angiotensin-converting enzyme 2 (ACE2), which implies that most existing mutations will strengthen the SARS-CoV-2 infectivity. This result is consistent with the natural selection of mutations and our earlier findings.84 Finally, we discover that the maximal BFE change magnitudes of binding-strengthening mutations are much smaller than those of binding-weakening mutations for all antibodies, which shows that current human antibodies were optimized with respect to the original S protein and are prone to the S protein mutations. Our findings indicate the pressing need to keep developing mutation-resistant vaccines and antibody drugs and to be ready for seasonal vaccinations.
8 Data availability
Detailed mutation information is available for download at Mutation Tracker.Author contributions
Conceptualization: Guo-Wei Wei. Data curation: Jiahui Chen, Kaifu Gao, Rui Wang. Formal analysis: Jiahui Chen, Guo-Wei Wei. Funding acquisition: Guo-Wei Wei. Investigation: Jiahui Chen, Guo-Wei, Wei. Methodology: Jiahui Chen, Rui Wang. Project administration: Guo-Wei Wei. Resources: Jiahui Chen, Kaifu Gao, Rui Wang. Software: Jiahui Chen, Rui Wang. Supervision: Guo-Wei Wei. Validation: Jiahui Chen, Guo-Wei Wei. Visualization: Jiahui Chen, Kaifu Gao, Rui Wang, Guo-Wei Wei. Writing – original draft: Jiahui Chen, Kaifu Gao, Rui Wang, Guo-Wei Wei. Writing–review & editing: Jiahui Chen, Guo-Wei Wei.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by NIH grant GM126189, NSF grants DMS-2052983, DMS-1761320, and IIS-1900473, NASA grant 80NSSC21M0023, Michigan Economic Development Corporation, George Mason University award PD45722, Bristol-Myers Squibb 65109, and Pfizer. The authors thank The IBM TJ Watson Research Center, The COVID-19 High Performance Computing Consortium, NVIDIA, and MSU HPCC for computational assistance. RW thanks Dr Changchuan Yin for useful discussions.References
- R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang and N. Zhu, et al., Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding, Lancet, 2020, 395(10224), 565–574 CrossRef CAS.
- M. D. Shin, S. Shukla, Y. H. Chung, V. Beiss, S. K. Chan, O. A. Ortega-Rivera, D. M. Wirth, A. Chen, M. Sack and J. K. Pokorski, et al., COVID-19 vaccine development and a potential nanomaterial path forward, Nat. Nanotechnol., 2020, 1–10 Search PubMed.
- M. Day, Covid-19: four fifths of cases are asymptomatic, China figures indicate, 2020 Search PubMed.
- Q.-X. Long, X.-J. Tang, Q.-L. Shi, Q. Li, H.-J. Deng, J. Yuan, J.-L. Hu, W. Xu, Y. Zhang and F.-J. Lv, et al., Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections, Nat. Med., 2020, 26(8), 1200–1204 CrossRef CAS PubMed.
- S. M. Kissler, C. Tedijanto, E. Goldstein, Y. H. Grad and M. Lipsitch, Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period, Science, 2020, 368(6493), 860–868 CrossRef CAS PubMed.
- 12-year trip, https://www.medicinenet.com/script/main/art.asp?articlekey=9877.
- E. M. Bloch, S. Shoham, A. Casadevall, B. S. Sachais, B. Shaz, J. L. Winters, C. van Buskirk, B. J. Grossman, M. Joyner and J. P. Henderson, et al., Deployment of convalescent plasma for the prevention and treatment of COVID-19, J. Clin. Invest., 2020, 130(6), 2757–2765 CrossRef CAS PubMed.
- E. Prompetchara, C. Ketloy and T. Palaga, Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic, Asian Pac. J. Allergy Immunol., 2020, 38(1), 1–9 CAS.
- F. Wu, A. Wang, M. Liu, Q. Wang, J. Chen, S. Xia, Y. Ling, Y. Zhang, J. Xun, L. Lu, et al., in Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications, 2020, medRxiv Search PubMed.
- J.-Y. Li, C.-H. Liao, Q. Wang, Y.-J. Tan, R. Luo, Y. Qiu and X.-Y. Ge, The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway, Virus Res., 2020, 286, 198074 CrossRef CAS PubMed.
- A. Tufan, A. A. Güler and M. Matucci-Cerinic, COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs, Turk. J. Med. Sci., 2020, 50(SI-1), 620–632 CrossRef CAS PubMed.
- Y. Liang, M.-L. Wang, C.-S. Chien, A. A. Yarmishyn, Y.-P. Yang, W.-Y. Lai, Y.-H. Luo, Y.-T. Lin, Y.-J. Chen and P.-C. Chang, et al., Highlight of Immune Pathogenic Response and Hematopathologic Effect in SARS-CoV, MERS-CoV, and SARS-CoV-2 infection, Front. Immunol., 2020, 11, 1022 CrossRef CAS PubMed.
- M. Catanzaro, F. Fagiani, M. Racchi, E. Corsini, S. Govoni and C. Lanni, Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2, Signal Transduction Targeted Ther., 2020, 5(1), 1–10 CrossRef PubMed.
- D. D. Chaplin, Overview of the immune response, J. Allergy Clin. Immunol., 2010, 125(2), S3–S23 CrossRef PubMed.
- H. Kumar, T. Kawai and S. Akira, Pathogen recognition by the innate immune system, Int. Rev. Immunol., 2011, 30(1), 16–34 CrossRef CAS PubMed.
- O. Takeuchi and S. Akira, Pattern recognition receptors and inflammation, Cell, 2010, 140(6), 805–820 CrossRef CAS PubMed.
- H. Kumar, T. Kawai and S. Akira, Pathogen recognition in the innate immune response, Biochem. J., 2009, 420(1), 1–16 CrossRef CAS PubMed.
- Z. Pancer and M. D. Cooper, The evolution of adaptive immunity, Annu. Rev. Immunol., 2006, 24, 497–518 CrossRef CAS PubMed.
- E. W. Hewitt, The MHC class I antigen presentation pathway: strategies for viral immune evasion, Immunology, 2003, 110(2), 163–169 CrossRef CAS PubMed.
- J. T. Harty, A. R. Tvinnereim and D. W. White, CD8+ T cell effector mechanisms in resistance to infection, Annu. Rev. Immunol., 2000, 18(1), 275–308 CrossRef CAS PubMed.
- J. P.-Y. Ting and J. Trowsdale, Genetic control of MHC class II expression, Cell, 2002, 109(2), S21–S33 CrossRef CAS.
- B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, P. Walter and K. Roberts, et al. , Molecular Biology of the Cell, 2015 Search PubMed.
- B. Hu, S. Huang and L. Yin, The cytokine storm and COVID-19, J. Med. Virol., 2021, 93(1), 250–256 CrossRef CAS PubMed.
- I. S. Grewal and R. A. Flavell, CD40 and CD154 in cell-mediated immunity, Annu. Rev. Immunol., 1998, 16(1), 111–135 CrossRef CAS PubMed.
- S. Crotty and R. Ahmed, Immunological memory in humans, in Seminars in immunology, Elsevier, 2004, vol. 16, pp. 197–203 Search PubMed.
- F. W. Putnam, Y. S. Liu and T. L. Low, Primary structure of a human IgA1 immunoglobulin. IV. streptococcal IgA1 protease, digestion, Fab and Fc fragments, and the complete amino acid sequence of the alpha 1 heavy chain, J. Biol. Chem., 1979, 254(8), 2865–2874 CrossRef CAS.
- W. Wang, S. Singh, D. L. Zeng, K. King and S. Nema, Antibody structure, instability, and formulation, J. Pharm. Sci., 2007, 96(1), 1–26 CrossRef CAS PubMed.
- C. Hamers-Casterman, T. Atarhouch, S. Muyldermans, G. Robinson, C. Hammers, E. B. Songa, N. Bendahman and R. Hammers, Naturally occurring antibodies devoid of light chains, Nature, 1993, 363(6428), 446–448 CrossRef CAS PubMed.
- R. H. J. Van der Linden, L. G. J. Frenken, B. De Geus, M. M. Harmsen, R. C. Ruuls, W. Stok, L. De Ron, S. Wilson, P. Davis and C. T. Verrips, Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1431(1), 37–46 CrossRef CAS.
- A. Forsman, E. Beirnaert, M. M. I. Aasa-Chapman, B. Hoorelbeke, K. Hijazi, W. Koh, V. Tack, A. Szynol, C. Kelly and A. McKnight, et al., Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120, J. Virol., 2008, 82(24), 12069–12081 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu, A. Nitsche, M. A. Muller, C. Drosten and S. Pohlmann, SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor, Cell, 2020, 181(2), 271–280 CrossRef CAS PubMed.
- X. Cao, COVID-19: immunopathology and its implications for therapy, Nat. Rev. Immunol., 2020, 20(5), 269–270 CrossRef CAS PubMed.
- L. Chen, J. Xiong, L. Bao and Y. Shi, Convalescent plasma as a potential therapy for COVID-19, Lancet Infect. Dis., 2020, 20(4), 398–400 CrossRef CAS PubMed.
- C. Shen, Z. Wang, F. Zhao, Y. Yang, J. Li, J. Yuan, F. Wang, D. Li, M. Yang and L. Xing, et al., Treatment of 5 critically ill patients with COVID-19 with convalescent plasma, Jama, 2020, 323(16), 1582–1589 CrossRef CAS PubMed.
- J. Zhang, H. Zeng, J. Gu, H. Li, L. Zheng and Q. Zou, Progress and prospects on vaccine development against SARS-CoV-2, Vaccines, 2020, 8(2), 153 CrossRef CAS PubMed.
- E. Callaway, The race for coronavirus vaccines: a graphical guide, Nature, 2020, 580(7805), 576 CrossRef CAS PubMed.
- F. Wu, S. Zhao, B. Yu, Y.-M. Chen, W. Wang, Z.-G. Song, Yi Hu, Z.-Wu Tao, J.-H. Tian and Y.-Y. Pei, et al., A new coronavirus associated with human respiratory disease in China, Nature, 2020, 579(7798), 265–269 CrossRef CAS PubMed.
- M. Sevajol, L. Subissi, E. Decroly, B. Canard and I. Imbert, Insights into RNA synthesis, capping, and proofreading mechanisms of SARS-coronavirus, Virus Res., 2014, 194, 90–99 CrossRef CAS.
- F. Ferron, L. Subissi, A. T. S. De Morais, N. T. Tuyet Le, M. Sevajol, L. Gluais, E. Decroly, C. Vonrhein, G. Bricogne and B. Canard, et al., Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(2), E162–E171 CrossRef CAS.
- R. Wang, Y. Hozumi, C. Yin, and G.-W. Wei, Decoding SARS-CoV-2 transmission, evolution and ramification on COVID-19 diagnosis, vaccine, and medicine, 2020, arXiv preprint arXiv:2004.14114 Search PubMed.
- R. Wang, Y. Hozumi, C. Yin and G.-W. Wei, Decoding SARS-CoV-2 Transmission and Evolution and Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine, J. Chem. Inf. Model., 2020, 32530284 Search PubMed.
- R. Wang, Y. Hozumi, Y.-H. Zheng, C. Yin and G.-W. Wei, Host immune response driving SARS-CoV-2 evolution, Viruses, 2020, 12(10), 1095 CrossRef CAS PubMed.
- M. Wang, Z. Cang and G.-W. Wei, A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation, Nature Machine Intelligence, 2020, 2(2), 116–123 CrossRef.
- G. Carlsson, Topology and data, Bulletin of the American Mathematical Society, 2009, 46(2), 255–308 CrossRef.
- H. Edelsbrunner, D. Letscher, and A. Zomorodian. Topological persistence and simplification, in Proceedings 41st annual symposium on foundations of computer science, IEEE, 2000, pp. 454–463 Search PubMed.
- K. Xia and G.-W. Wei, Persistent homology analysis of protein structure, flexibility, and folding, International journal for numerical methods in biomedical engineering, 2014, 30(8), 814–844 CrossRef PubMed.
- T. G. Kucukkal, M. Petukh, L. Li and E. Alexov, Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins, Curr. Opin. Struct. Biol., 2015, 32, 18–24 CrossRef CAS.
- P. Yue, Z. Li and J. Moult, Loss of protein structure stability as a major causative factor in monogenic disease, J. Mol. Biol., 2005, 353(2), 459–473 CrossRef CAS PubMed.
- R. Sanjuán and P. Domingo-Calap, Mechanisms of viral mutation, Cell. Mol. Life Sci., 2016, 73(23), 4433–4448 CrossRef PubMed.
- N. D. Grubaugh, W. P. Hanage and A. L. Rasmussen, Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear, Cell, 2020, 182(4), 794–795 CrossRef CAS PubMed.
- Y. Shu and J. McCauley, GISAID: Global initiative on sharing all influenza data-from vision to reality, Eurosurveillance, 2017, 22(13), 30494 CrossRef PubMed.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang, M. Hao, Z. Zhang, P. Fan, Y. Dong and Y. Yang, et al., A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2, Science, 2020, 369(6504), 650–655 CrossRef CAS.
- S. Wang, W. Li, S. Liu and J. Xu, RaptorX-Property: a web server for protein structure property prediction, Nucleic Acids Res., 2016, 44(W1), W430–W435 CrossRef CAS PubMed.
- N. Wang, Y. Sun, R. Feng, Y. Wang, Y. Guo, L. Zhang, Y.-Q. Deng, L. Wang, Z. Cui and L. Cao, et al., Structure-based development of human antibody cocktails against SARS-CoV-2, Cell Res., 2021, 31(1), 101–103 CrossRef CAS PubMed.
- P. Acharya, W. Williams, R. Henderson, K. Janowska, K. Manne, R. Parks, M. Deyton, J. Sprenz, V. Stalls, M. Kopp, et al., in A glycan cluster on the SARS-CoV-2 spike ectodomain is recognized by Fab-dimerized glycan-reactive antibodies, 2020, bioRxiv Search PubMed.
- D. Li, T. Li, H. Cai, H. Yao, B. Zhou, Y. Zhao, W. Qin, C. A. J. Hutter, Y. Lai and J. Bao, et al., in Potent synthetic nanobodies against SARS-CoV-2 and molecular basis for neutralization, 2020, bioRxiv Search PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang and L. Zhang, et al., Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor, Nature, 2020, 1–6 Search PubMed.
- M. Yuan, H. Liu, N. C. Wu, C.-C. D. Lee, X. Zhu, F. Zhao, D. Huang, W. Yu, Y. Hua and H. Tien, et al., Structural basis of a shared antibody response to SARS-CoV-2, Science, 2020, 369(6507), 1119–1123 CrossRef CAS PubMed.
- Y. Wu, F. Wang, C. Shen, W. Peng, D. Li, C. Zhao, Z. Li, S. Li, Y. Bi and Y. Yang, et al., A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2, Science, 2020, 368(6496), 1274–1278 CrossRef CAS PubMed.
- L. Piccoli, Y.-J. Park, M. A. Tortorici, N. Czudnochowski, A. C. Walls, M. Beltramello, C. Silacci-Fregni, D. Pinto, L. E. Rosen and J. E. Bowen, et al., Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology, Cell, 2020, 183(4), 1024–1042 CrossRef CAS.
- R. Shi, C. Shan, X. Duan, Z. Chen, P. Liu, J. Song, T. Song, X. Bi, C. Han and L. Wu, et al., A human neutralizing antibody targets the receptor binding site of SARS-CoV-2, Nature, 2020, 1–8 Search PubMed.
- E. Rujas, I. Kucharska, Y. Z. Tan, S. Benlekbir, H. Cui, T. Zhao, G. A. Wasney, P. Budylowski, F. Guvenc and J. C. Newton, et al., in Multivalency transforms SARS-CoV-2 antibodies into broad and ultrapotent neutralizers, 2020, bioRxiv Search PubMed.
- J. Huo, A. Le Bas, R. R. Ruza, H. M. E. Duyvesteyn, H. Mikolajek, T. Malinauskas, T. K. Tan, P. Rijal, M. Dumoux and P. N. Ward, et al., in Structural characterisation of a nanobody derived from a naïve library that neutralises SARS-CoV-2, Nature Protfolio, 2020 Search PubMed.
- N. K. Hurlburt, Yu-H. Wan, A. B. Stuart, J. Feng, A. T. McGuire, L. Stamatatos and M. Pancera, in Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation 2020, bioRxiv Search PubMed.
- Y. Cao, B. Su, X. Guo, W. Sun, Y. Deng, L. Bao, Q. Zhu, X. Zhang, Y. Zheng and C. Geng, et al., Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients B cells, Cell, 2020, 182(1), 73–84 CrossRef CAS PubMed.
- D. Pinto, Y.-J. Park, M. Beltramello, A. C. Walls, M. A. Tortorici, S. Bianchi, S. Jaconi, K. Culap, F. Zatta and A. De Marco, et al., in Structural and functional analysis of a potent sarbecovirus neutralizing antibody, 2020, bioRxiv Search PubMed.
- D. Zhou, H. M. E. Duyvesteyn, C.-P. Chen, C.-G. Huang, T.-H. Chen, S.-R. Shih, Y.-C. Lin, C.-Yu Cheng, S.-H. Cheng and Y.-C. Huang, et al., Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient, Nat. Struct. Mol. Biol., 2020, 1–9 Search PubMed.
- S. Du, Y. Cao, Q. Zhu, P. Yu, F. Qi, G. Wang, X. Du, L. Bao, W. Deng and H. Zhu, et al., Structurally resolved SARS-CoV-2 antibody shows high efficacy in severely infected hamsters and provides a potent cocktail pairing strategy, Cell, 2020, 183(4), 1013–1023 CrossRef CAS PubMed.
- Z. Lv, Y.-Q. Deng, Q. Ye, L. Cao, C.-Y. Sun, C. Fan, W. Huang, S. Sun, Y. Sun and L. Zhu, et al., Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody, Science, 2020, 369(6510), 1505–1509 CrossRef CAS PubMed.
- N. C. Wu, M. Yuan, H. Liu, C.-C. D. Lee, X. Zhu, S. Bangaru, J. L. Torres, T. G. Caniels, P. J. M. Brouwer and M. J. Van Gils, et al., in An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain, 2020, BioRxiv Search PubMed.
- B. Ju, Q. Zhang, J. Ge, R. Wang, J. Sun, X. Ge, J. Yu, S. Shan, B. Zhou and S. Song, et al., Human neutralizing antibodies elicited by SARS-CoV-2 infection, Nature, 2020, 1–8 Search PubMed.
- J. Kreye, S. Momsen Reincke, H.-C. Kornau, E. Sánchez-Sendin, V. M. Corman, H. Liu, M. Yuan, N. C. Wu, X. Zhu and C.-C. D. Lee, et al., A therapeutic non-self-reactive SARS-CoV-2 antibody protects from lung pathology in a covid-19 hamster model, Cell, 2020, 183(4), 1058–1069 CrossRef CAS PubMed.
- M. A. Tortorici, M. Beltramello, F. A. Lempp, D. Pinto, H. V. Dang, L. E. Rosen, M. McCallum, J. Bowen, A. Minola and S. Jaconi, et al., Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms, Science, 2020, 370(6519), 950–957 CrossRef CAS PubMed.
- C. J. Bracken, S. A. Lim, P. Solomon, N. J. Rettko, D. P. Nguyen, B. S. Zha, K. Schaefer, J. R. Byrnes, J. Zhou and I. Lui, et al., Bi-paratopic and multivalent VH domains block ACE2 binding and neutralize SARS-CoV-2, Nat. Chem. Biol., 2021, 17(1), 113–121 CrossRef PubMed.
- H. Liu, N. C. Wu, M. Yuan, S. Bangaru, J. L. Torres, T. G. Caniels, J. Van Schooten, X. Zhu, C.-C. D. Lee and P. J. M. Brouwer, et al., Cross-neutralization of a SARS-CoV-2 antibody to a functionally conserved site is mediated by avidity, Immunity, 2020, 53(6), 1272–1280 CrossRef CAS.
- S. A. Clark, L. E. Clark, J. Pan, A. Coscia, L. G. A. McKay, S. Shankar, R. I. Johnson, A. Griffiths and J. Abraham, in Molecular basis for a germline-biased neutralizing antibody response to SARS-CoV-2, 2020, bioRxiv Search PubMed.
- F. Bertoglio, V. Fühner, M. Ruschig, P. Alexander Heine, U. Rand, T. Klünemann, D. Meier, N. Langreder, S. Steinke and R. Ballmann, et al., in A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients by phage display is binding to the ACE2-RBD interface and is tolerant to known RBD mutations, 2020, bioRxiv Search PubMed.
- T. F. Custódio, H. Das, D. J. Sheward, L. Hanke, S. Pazicky, J. Pieprzyk, M. Sorgenfrei, M. A. Schroer, A. Yu Gruzinov and C. M. Jeffries, et al., Selection, biophysical and structural analysis of synthetic nanobodies that effectively neutralize SARS-CoV-2, Nat. Commun., 2020, 11(1), 1–11 CrossRef PubMed.
- H. Yao, Y. Sun, Y.-Q. Deng, N. Wang, Y. Tan, N.-N. Zhang, X.-F. Li, C. Kong, Y.-P. Xu and Q. Chen, et al., Rational development of a human antibody cocktail that deploys multiple functions to confer Pan-SARS-CoVs protection, Cell Res., 2021, 31(1), 25–36 CrossRef CAS PubMed.
- X. Tian, C. Li, A. Huang, S. Xia, S. Lu, Z. Shi, L. Lu, S. Jiang, Z. Yang and Y. Wu, et al., Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody, Emerging Microbes Infect., 2020, 9(1), 382–385 CrossRef CAS.
- J. T. Meulen, E. N. Van Den Brink, L. L. M. Poon, W. E. Marissen, C. S. W. Leung, F. Cox, C. Y. Cheung, A. Q. Bakker, J. A. Bogaards and E. Van Deventer, et al., Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants, PLoS Med., 2006, 3(7) Search PubMed.
- Y. Huang, B. Niu, Y. Gao, L. Fu and W. Li, CD-HIT Suite: a web server for clustering and comparing biological sequences, Bioinformatics, 2010, 26(5), 680–682 CrossRef CAS PubMed.
- L. Liu, P. Wang, M. S. Nair, J. Yu, M. Rapp, Q. Wang, Y. Luo, J. F.-W. Chan, V. Sahi and A. Figueroa, et al., Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike, Nature, 2020, 584(7821), 450–456 CrossRef CAS PubMed.
- J. Chen, R. Wang, M. Wang and G.-W. Wei, Mutations strengthened SARS-CoV-2 infectivity, J. Mol. Biol., 2020, 432(19), 5212–5226 CrossRef CAS PubMed.
- W.-H. Chen, U. Strych, P. J. Hotez and M. E. Bottazzi, The SARS-CoV-2 vaccine pipeline: an overview, Current tropical medicine reports, 2020, 1–4 Search PubMed.
- J. Lin, J.-S. Zhang, N. Su, J.-G. Xu, N. Wang, J.-T. Chen, X. Chen, Y.-X. Liu, H. Gao and Y.-P. Jia, et al., Safety and immunogenicity from a phase I trial of inactivated severe acute respiratory syndrome coronavirus vaccine, Antiviral Ther., 2007, 12(7), 1107 CAS.
- Y. Li, X. Liu, Y. Zhu, X. Zhou, C. Cao, X. Hu, H. Ma, H. Wen, X. Ma and J.-B. Ding, Bioinformatic prediction of epitopes in the Emy162 antigen of Echinococcus multilocularis, Exp. Ther. Med., 2013, 6(2), 335–340 CrossRef CAS PubMed.
- J. V. Kringelum, M. Nielsen, S. Berg Padkjær and O. Lund, Structural analysis of B-cell epitopes in antibody: protein complexes, Mol. Immunol., 2013, 53(1–2), 24–34 CrossRef CAS PubMed.
- T. W. Linsky, R. Vergara, N. Codina, J. W. Nelson, M. J. Walker, W. Su, C. O. Barnes, T.-Y. Hsiang, K. Esser-Nobis and K. Yu, et al., De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2, Science, 2020, 370(6521), 1208–1214 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: (S1) Methods; (S2) multiple sequence alignments of antibodies and pairwise identity scores; (S3) random coil percentages of antibody paratopes; and (S4) additional analysis of antibody–S protein complexes. See DOI: 10.1039/d1sc01203g |
| ‡ The first three authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |