
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUncovering the interactions driving carotenoid binding in light-harvesting complexes†
Vincenzo
Mascoli
 a,
Nicoletta
Liguori
a,
Lorenzo
Cupellini
b,
Eduard
Elias
a,
Benedetta
Mennucci
b and
Roberta
Croce
*a
a,
Nicoletta
Liguori
a,
Lorenzo
Cupellini
b,
Eduard
Elias
a,
Benedetta
Mennucci
b and
Roberta
Croce
*a
aDepartment of Physics and Astronomy, Institute for Lasers, Life and Biophotonics, Faculty of Sciences, Vrije Universiteit Amsterdam, De Boelelaan 1081, 1081 HV, Amsterdam, The Netherlands. E-mail: r.croce@vu.nl
bDepartment of Chemistry, University of Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy
First published on 15th February 2021
Abstract
Carotenoids are essential constituents of plant light-harvesting complexes (LHCs), being involved in protein stability, light harvesting, and photoprotection. Unlike chlorophylls, whose binding to LHCs is known to require coordination of the central magnesium, carotenoid binding relies on weaker intermolecular interactions (such as hydrogen bonds and van der Waals forces), whose character is far more elusive. Here we addressed the key interactions responsible for carotenoid binding to LHCs by combining molecular dynamics simulations and polarizable quantum mechanics/molecular mechanics calculations on the major LHC, LHCII. We found that carotenoid binding is mainly stabilized by van der Waals interactions with the surrounding chlorophyll macrocycles rather than by hydrogen bonds to the protein, the latter being more labile than predicted from structural data. Furthermore, the interaction network in the binding pockets is relatively insensitive to the chemical structure of the embedded carotenoid. Our results are consistent with a number of experimental data and challenge the role played by specific interactions in the assembly of pigment-protein complexes.
Introduction
In plants and green algae, the light-harvesting complexes (LHCs) are a superfamily of nuclear-encoded 21–29 kDa membrane apoproteins binding chlorophyll (Chl) a, Chl b, and carotenoids.1,2 LHCs primarily work as antennae, collecting light energy in the form of pigment excitation and delivering it to the photosynthetic reaction centers to power photochemistry.3 The structure of LHCs is highly conserved (Fig. 1A) and consists of three transmembrane alpha-helices (A–C) and two short amphipathic helices (D and E) exposed to the thylakoid lumen.2 The N-terminal and C-terminal domains are exposed to the stroma/lumen, respectively. While the central helices are relatively rigid and bind most pigments, the C- and N-terminal portions and the stromal and lumenal loops between helices are more flexible.4–7 The most abundant LHC is trimeric LHCII.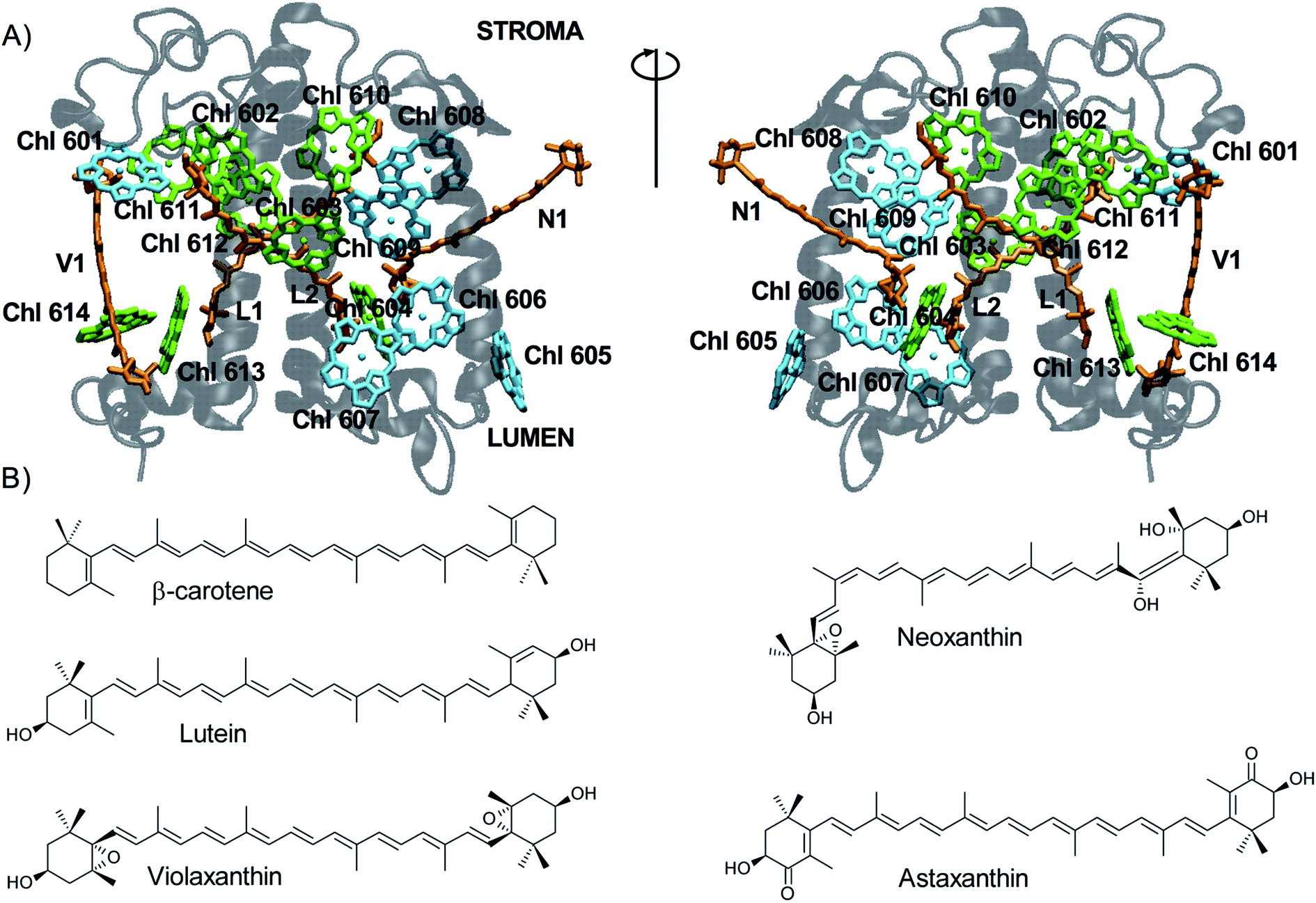 | ||
| Fig. 1 Carotenoids and their binding sites in LHCs. (A) View of LHCII (PDB code: 1RWT)12 from two opposite sides: the protein is shown in transparent gray, Chls a in green, Chls b in cyan, carotenoids in orange (the respective binding sites are labeled). For clarity, only chlorin rings of the Chl pigments are shown. (B) Chemical structure of carotenoids. | ||
LHCs bind up to 15 Chls whose binding sites are highly conserved. Chls consist of a tetrapyrrole ring (1 × 1 nm) coordinating a central Mg2+ ion via its four nitrogen atoms, a fifth isocyclic ring, and a long hydrophobic phytol tail. Most Chls are directly bound to the protein via coordination of their central Mg by side chains of nucleophilic residues (such as His, Glu, Gln, or Asn), while others are coordinated by water molecules, the protein backbone, or a lipid molecule. The Chls form two layers roughly parallel to the membrane plane (Fig. 1A), one closer to the stroma and one to the lumen.2
Carotenoids are accessory pigments consisting of a conjugated polyene chain of variable length that is often terminated by two rings (Fig. 1B). They can be classified as carotenes (which contain only carbon and hydrogen) and xanthophylls (which also contain oxygen). Xanthophylls are generally found in the LHCs, whereas the most important carotene, β-carotene, is mostly found in the photosystem core units.8 All LHCs contain three carotenoid binding sites (named L1, L2 and N1; Fig. 1A). The two central sites – L1 and L2 – form a transmembrane cross close to the two central helices. While L1 always binds the xanthophyll lutein, the occupancy of L2 changes in different LHCs (a second lutein is present in LHCII whereas violaxanthin is found in some minor antennas).9 The N1 site is occupied by neoxanthin (such as in LHCII) or, in some cases, by β-carotene.9–11 The N1 carotenoid is highly bent and partly protrudes into the membrane. A fourth carotenoid can be found in LHCII trimers, located at the interface between monomers at the site V1.12 V1 is occupied by violaxanthin or lutein,13 which can be exchanged for other xanthophyll cycle carotenoids in high light,14,15 and is typically empty in monomeric LHCs.16 The carotenoids in all binding sites but V1 (ref. 13) are involved in light harvesting, and the ones in L1 and L2 also have a key role in photoprotection.17–20
Though it is well established that carotenoids are essential for LHC folding and stability,21,22 the driving forces behind their binding are not fully understood yet. Indeed, while Mg coordination is known to be essential for Chl binding, as confirmed by the possibility to knock-out selected Chls via site-directed mutagenesis,23–26 carotenoid binding is thought to rely on weaker intermolecular forces whose character is far more elusive. As a matter of fact, the only key carotenoid interaction partner identified so far is a conserved Tyr residue (TYR112 in LHCII) stabilizing the binding of neoxanthin in N1.27 One accredited hypothesis is that hydrogen bond (HB) interactions between the –OH groups on the xanthophyll terminal rings and polar/charged protein residues are essential for the stability of carotenoid binding.28 This assumption is motivated by two pieces of evidence: (1) LHCs can only fold in the presence of xanthophylls, but not carotenes,21,29 and (2) extensive structural data suggest that the xanthophylls are often anchored to the protein via HBs involving conserved protein residues.9,12 However, this hypothesis contrasts several experimental results showing that xanthophyll binding is retained when most of these putative key amino acids are mutated.23,30,31 Furthermore, molecular dynamics (MD) simulations have demonstrated the high motility of certain pigment and protein domains, suggesting that the available high-resolution structures of the LHCs might not give a complete view of the interaction patterns between carotenoids and their environment.7,32–35
In order to determine the key elements driving carotenoid binding, we calculated the interaction energies between xanthophylls and the surrounding protein and cofactors for different binding sites of LHCII. The relevance of dynamic effects in the binding properties was assessed by comparing results from MD simulations and the crystal structure. We also performed calculations changing the occupancy of specific carotenoid binding pockets to investigate the influence of carotenoid chemical and structural properties on the binding interaction.
Methods
Molecular dynamics simulations
In this work, 3 distinct LHCII systems were simulated and analyzed, each with an associated set of independent replicas, for a total of 13 molecular dynamics (MD) simulations: 6 replicas of native LHCII (each ∼1 μs long), 3 of LHCII binding violaxanthin in L2 (each ∼1 μs long), and 4 of LHCII with astaxanthin in all binding sites (each ∼800 ns long). The simulations of LHCII wildtype and LHCII binding astaxanthin were already reported in previous works,7,36 whereas the 3 replicas of LHCII with violaxanthin are new.All the simulated systems were prepared starting from the same pre-equilibrated native system described in full detail in Liguori et al.7 Briefly, the initial simulation box of native LHCII included one monomer of native LHCII (binding 6 Chls b, 8 Chls a, 2 luteins, 1 violaxanthin, 1 neoxanthin and 1 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol, DPPG, lipid molecule) embedded in a model lipid membrane composed of 1-palmitoil-2-oleoyl-sn-glycero-3-phosphocholine (POPC, 344 total lipids), solvated with more than 15k water molecules at neutral physiologically relevant salt conditions (10 mM NaCl).37 The initial structure was taken from Liu et al. 2004 (chain A, PDB 1RWT)12 with all the amino acids set in their standard protonation state at pH 7 and including 60 interstitial water molecules. The protocols to reconstruct the missing atoms and embed the pigment-protein complex in the membrane are given in Liguori et al.7
The GROMOS force field was used both for the protein (version 54A7)38 and for the lipids and cofactors (version 53A6).7,39,40 In GROMOS some of the nonpolar hydrogens are implicitly treated and were added for the interaction calculations in a second step, as explained below. The protocols to pre-equilibrate a simulation box containing either native LHCII, or LHCII with astaxanthin, have been reported respectively in ref. 7 and 36. We here report the details of the protocol used to equilibrate LHCII with violaxanthin, which follows the same rationale of the ones used for the other two systems. The pre-equilibrated structure of native LHCII, including the complete simulation box, was used to generate the starting simulation box of LHCII with violaxanthin by replacing the lutein at the L2 site with a molecule of violaxanthin. This structure was first minimized (steepest descent) and then carefully relaxed (10 ps NVT and 40 ns NPT) at 300 K. During the relaxation procedure, isotropic position restraints were applied on the protein backbone and on all of the atoms of the ligands (pigments and DPPG), as necessary to minimize perturbation of the crystal structure before the equilibration run.35 During the 40ns NPT simulation, the restraints started from the high value of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kJ mol−1 nm−2 and were gradually decreased to zero every 10 ns. Starting from this final snapshot, 3 independent replicas were simulated without any position restraint in an NPT ensemble each for about 1 μs. In the simulations of LHCII with astaxanthin the site V1 was left empty.
000 kJ mol−1 nm−2 and were gradually decreased to zero every 10 ns. Starting from this final snapshot, 3 independent replicas were simulated without any position restraint in an NPT ensemble each for about 1 μs. In the simulations of LHCII with astaxanthin the site V1 was left empty.
All the NPT replica simulations were run with an integration step of 2 fs and with constraints on all the bonds (LINCS).41 Long range electrostatics were treated with the Particle Mesh Ewald (PME) scheme and cutoffs of 1 nm and 1.4 nm for the short-range Coulomb and van der Waals (vdW), respectively. Semi-isotropic coupling to a Parrinello–Rahman barostat42 was used to set the pressure to 1 bar, with a 5 ps relaxation time constant and compressibility of 4.5 10−5 bar−1. Coupling to a Nosé–Hoover thermostat43 was employed to maintain the temperature at 300 K, with a 0.5 ps time constant. All the simulations were run with periodic boundary conditions applied.
Interaction energy calculations
Interaction energies were calculated on frames selected every 10 ns from the equilibrated MD trajectories (after the initial ∼400 ns of the unrestrained NPT simulations). For each frame, the simulation box for all following calculations included the protein, all cofactors (Chls, carotenoids, and DPPG), as well as all water, membrane lipid molecules, and ions (Na+ and Cl−) within 45 Å from each atom of each carotenoid (atomic coordinates of the water, lipid molecules, and ions falling within the cutoff but outside the MD simulation box were obtained from the adjacent periodic images of the MD simulation box). At this stage, every frame was converted from the GROMOS united-atom force field used in the MD simulations to an all-atom force field used in the following molecular mechanics (MM) and polarizable quantum mechanics/molecular mechanics (QM/MMPol)44 calculations: the Amber ff14SB45 and lipid14 (ref. 46) force fields were used for protein and lipids, respectively, whereas the pigments were described with ad-hoc parameters.47,48 In the conversion between force fields, all missing atoms (e.g., some of the nonpolar hydrogen atoms) were added via home-made scripts interfaced with the tleap program (Amber). For astaxanthin, a DFT-fitted force field was used.49 The structural parameters (such as dihedrals, angles, bonds) were adapted from the available force-field of the carotenoid zeaxanthin,47 while the parameters for differing atoms were taken from GAFF (Generalized Amber Force Field).Before the interaction energy calculations, the converted frames were optimized via energy minimization at MM level, adopting the following constraints: all atoms belonging to the protein, cofactors, and the water molecules within 2.5 Å from any cofactor were let free in the minimization, whereas for the membrane lipids, ions and the remaining water molecules, only the hydrogen atoms were allowed to move, with all heavy atoms kept frozen. After optimization, the interaction energies between the xanthophylls and their binding pocket were calculated on a smaller system composed of the protein, cofactors, and all water molecules within 2.5 Å from any cofactor. Increasing the latter cut-off to 3.5 Å to include more water molecules increased the electrostatic interaction energy by less than 1% for L1 lutein, and by less than 2% for L2 lutein in LHCII. For each snapshot, the total electrostatic and vdW interaction energies of each carotenoid binding site were calculated at MM level (with the above-described force field). vdW interactions were also calculated between the xanthophylls and selected protein residues or pigments. The total electrostatic interaction was also calculated using the QM/MMPol44 by subtracting the energies from two different calculations: (i) a QM/MMPol energy calculation where the xanthophyll of choice is treated at QM level and all surrounding residues are described through a set of atomic point charges and isotropic polarizabilities, and (ii) a QM energy calculation of the xanthophyll in vacuum. In all QM calculations, we used DFT with the B3LYP functional and the 6-311G(d,p) basis set. Further analyses were performed to identify the major electrostatic interaction partners of each xanthophyll, and extra analyses were targeted to their interaction with water molecules (for instance, identification of hydrogen bonds with the stromal/lumenal –OH groups). Throughout this work, we only show the results from QM/MMPol calculations of electrostatic interactions, while the vdW interactions are always calculated at MM level. Note that the electrostatic interaction energies calculated both at MM and QM/MMPol level display a very good degree of linear correlation (Fig. S1†).
The electrostatic and vdW interactions were calculated using the same procedure on the crystal structure of LHCII (chain A, PDB 1RWT).12 Prior to MM and QM/MMPol calculations, the structures were saturated with all missing hydrogen atoms and subject to MM optimization, with all atoms let free in the energy minimization.
Results
Driving forces in carotenoid binding
Uncorrelated snapshots from various MD trajectories of monomeric LHCII from Liguori et al.7 were used to compute the interaction energies between the carotenoids and their environment. For each binding site, the interaction was decomposed into electrostatic and van der Waals (vdW) contributions. The former was calculated with the multiscale QM/MMPol44 scheme described in the Methods section, whereas the latter, whose quantum mechanical treatment would be computationally unaffordable, was approximated using a classical (AMBER) force field.45–48 We recall that the QM/MMPol scheme allows to account for the implicit effect of the charge distribution of the carotenoid on the environment and vice versa. Tests were performed to verify that the electrostatic interaction calculated with the QM/MMPol scheme and the less expensive AMBER force field were reasonably linearly correlated (Fig. S1†).Fig. 2 shows that, in all binding sites, the total vdW interaction is markedly stronger than the electrostatic interaction (the latter including all possible HBs formed by the xanthophyll –OH groups). Specifically, the two centrally bound luteins (L1 and L2) have total interaction energy of 105–120 kcal mol−1, of which only about 15 kcal mol−1 originate from electrostatic terms. This implies that over 85% of their interaction with protein and cofactors stems from vdW forces. Neoxanthin bound in N1 displays an electrostatic interaction energy comparable to that of the two luteins, whereas the vdW interaction decreases to about 60 kcal mol−1. This results from the reduced interaction surface of neoxanthin inside the protein, as its conjugated chain partly protrudes in the membrane space (Fig. 1A). The V1 binding site is normally empty in LHC monomers and will be neglected in the following analyses (the data in Fig. S2† confirm that the interaction of violaxanthin in V1 of monomeric LHCII is much weaker than that of the other binding sites).
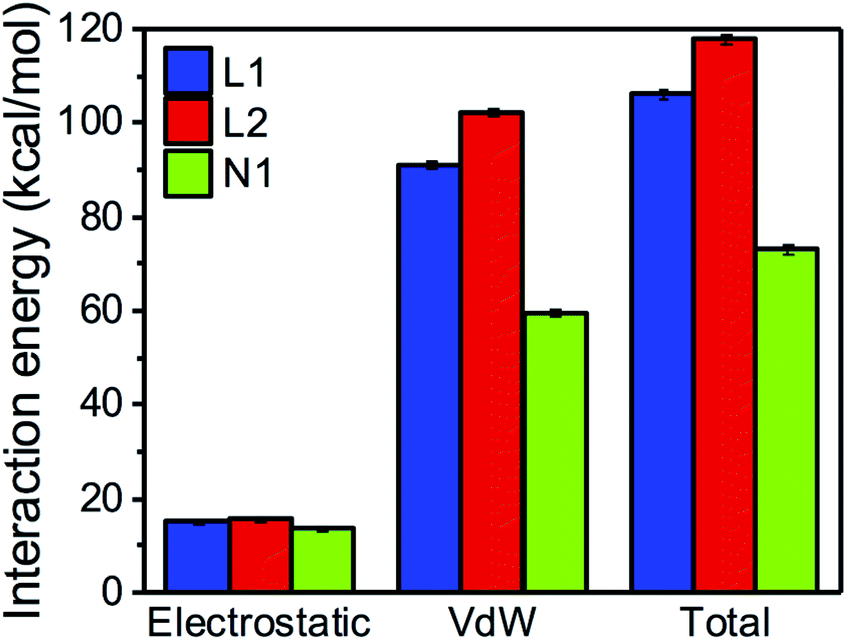 | ||
| Fig. 2 Driving forces in carotenoid binding. Decomposition of interaction energy into electrostatic and vdW terms in different binding sites of LHCII. The value of each bar represents the average over the MD snapshots (cumulating all 6 replicas), while the error bars represent 96% confidence intervals (i.e. 2 times the standard error, with N = 323). | ||
A detailed investigation of the contribution of specific cofactors/protein residues reveals that, for all xanthophylls, the major vdW interaction partners are the surrounding Chl molecules (Fig. 3–5). Indeed, the Chls possess large conjugated rings giving rise to stacking interactions of up to 20 kcal mol−1 with the carotenoid conjugated chain, i.e. much larger than the average energy of an HB (5–10 kcal mol−1). The strongest vdW interaction partners are Chl a610 > a612 > a613 for L1 lutein, Chl a602 > a603 > a604 for L2 lutein, and Chl a604 ∼ b606 > b608 for neoxanthin. These results are explained by the proximity of the π-electrons of the corresponding carotenoid–Chl pairs and are also in qualitative agreement with calculations on the crystal structure (Fig. 3–5, light and dark green bars). The vdW interactions with some protein residues (especially the aromatic and methionine residues) can also be relatively strong (up to 5 kcal mol−1), but always weaker than those with the nearest Chls. The values of vdW interactions between xanthophylls and Chls or aromatic sidechains presented here are comparable to those calculated previously at MP2 (QM) level for spheroidene in LH2 and for β-carotene in Photosystem I.50,51
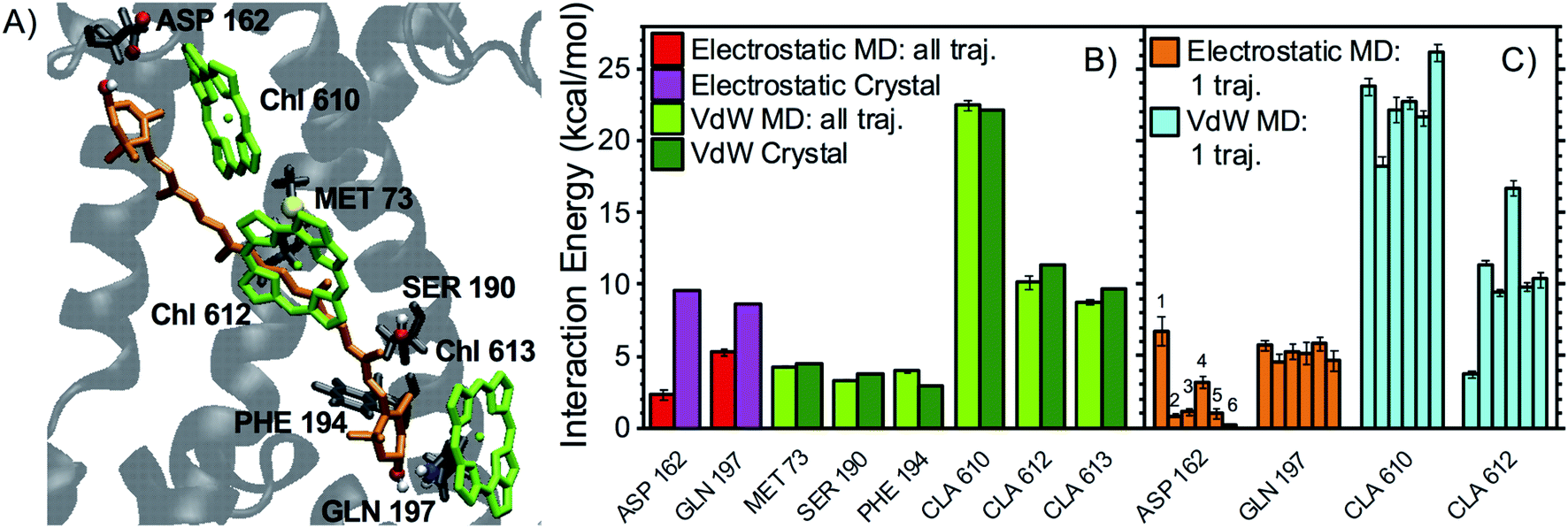 | ||
| Fig. 3 Interaction partners of L1 lutein. (A) Binding pocket of L1 lutein in LHCII (coordinates from the optimized crystal structure) with the main interaction partners highlighted: Chls are in green, lutein in orange, while protein secondary structure and relevant residues are in gray, with the polar atoms of their side chains (as well as those of lutein) highlighted in red, blue, and white, for oxygen, nitrogen, and hydrogen atoms, respectively. (B) Average interaction energies of L1 lutein with selected residues. The red and light-green bars indicate the average electrostatic and vdW energies, respectively, over all cumulated MD snapshots, with their error bars indicating 96% confidence interval (2 times the standard error, with N = 323). The corresponding energies calculated on the crystal structure are shown in magenta and dark green. Only the interactions with an average >3.0 kcal mol−1 over all MD snapshots are shown. Despite averaging below the latter threshold in the MD simulations, the electrostatic interaction energy between L1 lutein and ASP162 is also shown, as this pair gives rise to a strong HB according to the crystal structure and in a substantial fraction of MD snapshots. (C) average electrostatic and vdW interaction energies between L1 lutein and specific residues over each independent MD trajectory (the error bars indicate 96% confidence intervals, i.e. 2 times the standard error, with N = 60, 48, 53, 55, 55, 52 for trajectories 1–6, respectively). | ||
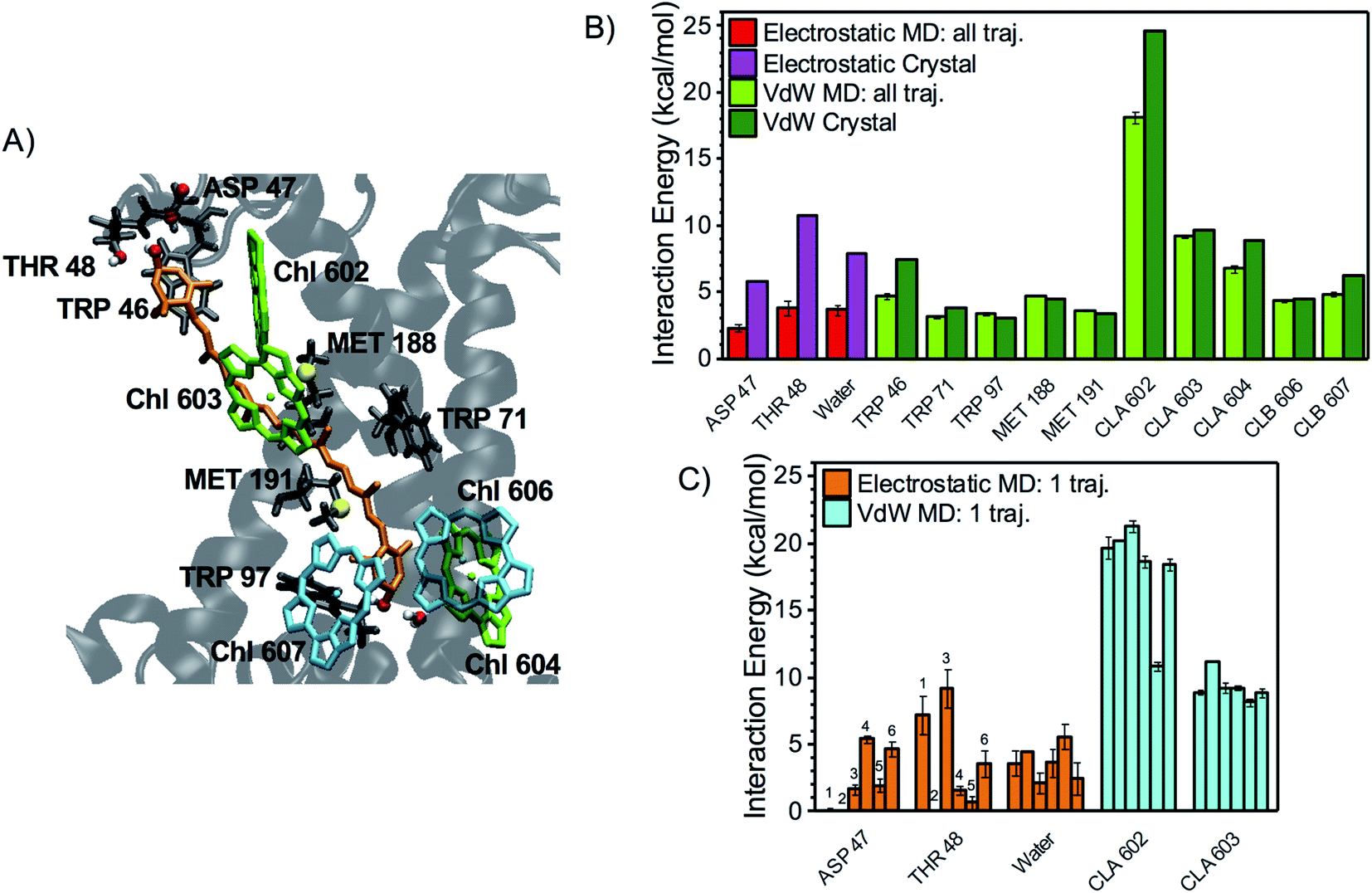 | ||
| Fig. 4 Interaction partners of L2 lutein. (A) Binding pocket of L2 lutein in LHCII (coordinates from the optimized crystal structure) with the main interaction partners highlighted with the same color code used in Fig. 3A. (B) Average interaction energies of L2 lutein with selected residues (color code and error bars as in Fig. 3B). (C) Average electrostatic and vdW interaction energies between L2 lutein and specific residues over each independent MD trajectory (color code and error bars as in Fig. 3C). As for the interaction with water, the sum of the interactions with all water molecules is indicated. However, HBs with water molecules mostly involve the lumenal –OH of lutein, as confirmed by the following statistics: 41% of all MD snapshots display an interaction >3 kcal mol−1 between one water molecule and the lumenal –OH, whereas only 8% of all snapshots display an interaction >3 kcal mol−1 involving the stromal –OH. | ||
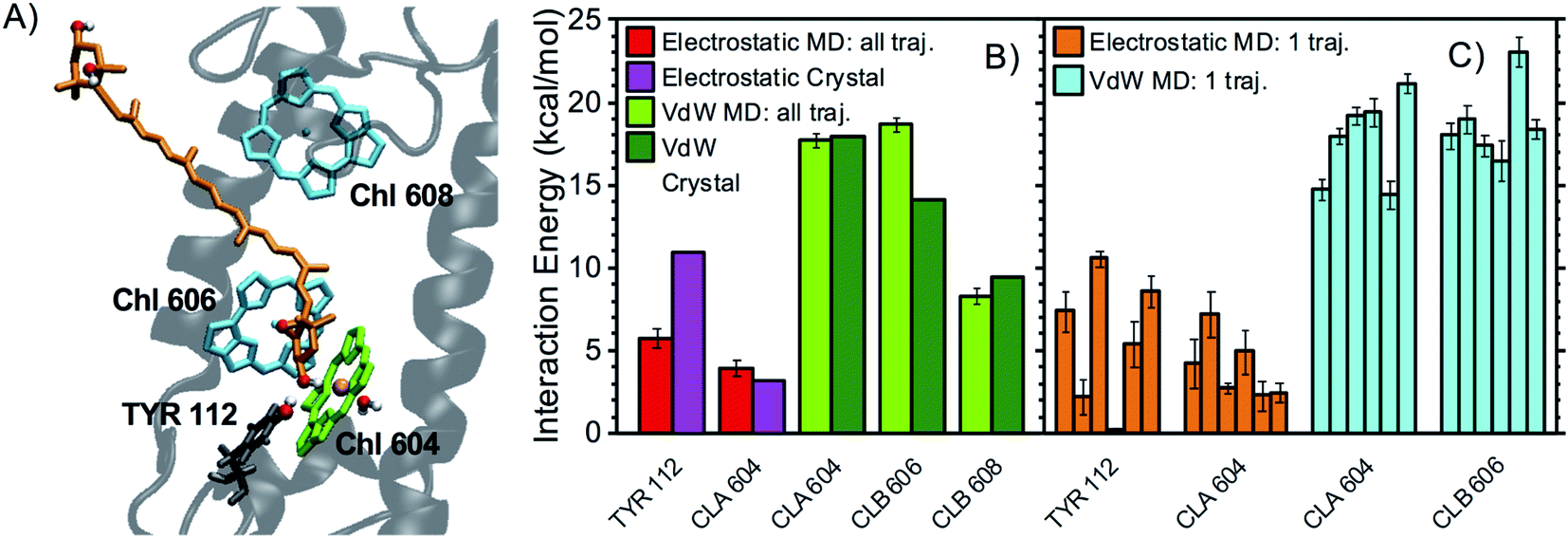 | ||
| Fig. 5 Interaction partners of N1 neoxanthin. (A) Binding pocket of N1 neoxanthin in LHCII (coordinates from the optimized crystal structure) with the main interaction partners highlighted with the same color code used in Fig. 3A. The Mg of Chl a604 is colored in pink. The water molecule coordinating Chl a604 is also shown. (B) Average interaction energies of N1 neoxanthin with selected residues (color code and error bars as in Fig. 3B). (C) Average electrostatic and vdW interaction energies between N1 neoxanthin and specific residues over each independent MD trajectory (color code and error bars as in Fig. 3C). | ||
Fig. 3B, 4B, and 5B, illustrate the main electrostatic interaction partners of the xanthophylls and compare the results from MD simulations with those from the crystal structure. Fig. 3C, 4C, and 5C correspond to single MD trajectories and reveal whether these interactions are stable or labile, whereas Table 1 summarizes the occurrences of such interactions. In nearly all cases, the strongest electrostatic interactions result from HBs between the –OH groups of the xanthophyll terminal rings and polar/charged amino acid sidechains or water molecules. However, while in the MD simulations the vdW interactions approach the values calculated for the crystal structure and are relatively reproducible throughout different trajectories, the HBs are often weaker and more labile than what predicted from a static picture. This is reasonable because the vdW interactions mostly involve the xanthophyll conjugated linear chains, which are buried inside the protein and are therefore relatively static, whereas the HBs involve the terminal rings, which are more peripheral and flexible.7
| L1 partners | % | L2 partners | % | N1 partners | % |
|---|---|---|---|---|---|
| ASP 162 | 28 ± 16 | ASP 47 | 38 ± 17 | TYR 112 | 54 ± 15 |
| GLN 197 | 87 ± 4 | THR 48 | 35 ± 15 | CLA 604 | 34 ± 9 |
| Water (lumen) | 41 ± 9 |
In particular, the most stable HB for L1 lutein is between its lumenal –OH and the –NH2 group of GLN197 (ligand of Chl a613), in agreement with predictions based on the crystal structure. On the stromal side, however, the HB with ASP162 is much more labile than predicted from the structure, indicating that dynamic effects are more relevant in this region of the complex (Fig. 3). Similarly, L2 lutein can form relatively energetic but intermittent HBs with ASP47 and THR48 on the stromal side. The extent of these interactions is substantially reduced in the MDs in comparison to the static structure (Fig. 4). On the lumenal side, an HB with a water molecule, which is also observed in the crystal structure, is sometimes present (giving rise to an interaction >3 kcal mol−1 in over 40% of all MD snapshots). While the carotenoid in L1 forms at least one relatively stable HB (with GLN197 at the lumen side) in all MD trajectories, the carotenoid in L2 seems to lack a stable HB partner. A similar behavior is observed in the other LHCII variants analyzed in this study (see following sections).
The main electrostatic interactions for N1 neoxanthin (Fig. 5) involve the –OH group inside the protein, which can form an HB with the –OH of TYR112, or interact with the Mg of Chl a604. These two interactions are mutually exclusive, indicating that this binding pocket assumes different conformations52 (Fig. 6). In this respect, the crystal structure seems to favor the interaction of neoxanthin with TYR112, while Chl a604 is coordinated by a water molecule (Fig. 5A). This water molecule is lost in the MD snapshots where the Mg of Chl a604 interacts with the –OH of neoxanthin (Fig. 6B). In comparison to the L1 and L2 sites, the N1 neoxanthin also displays a relatively smaller number of strong interaction partners.
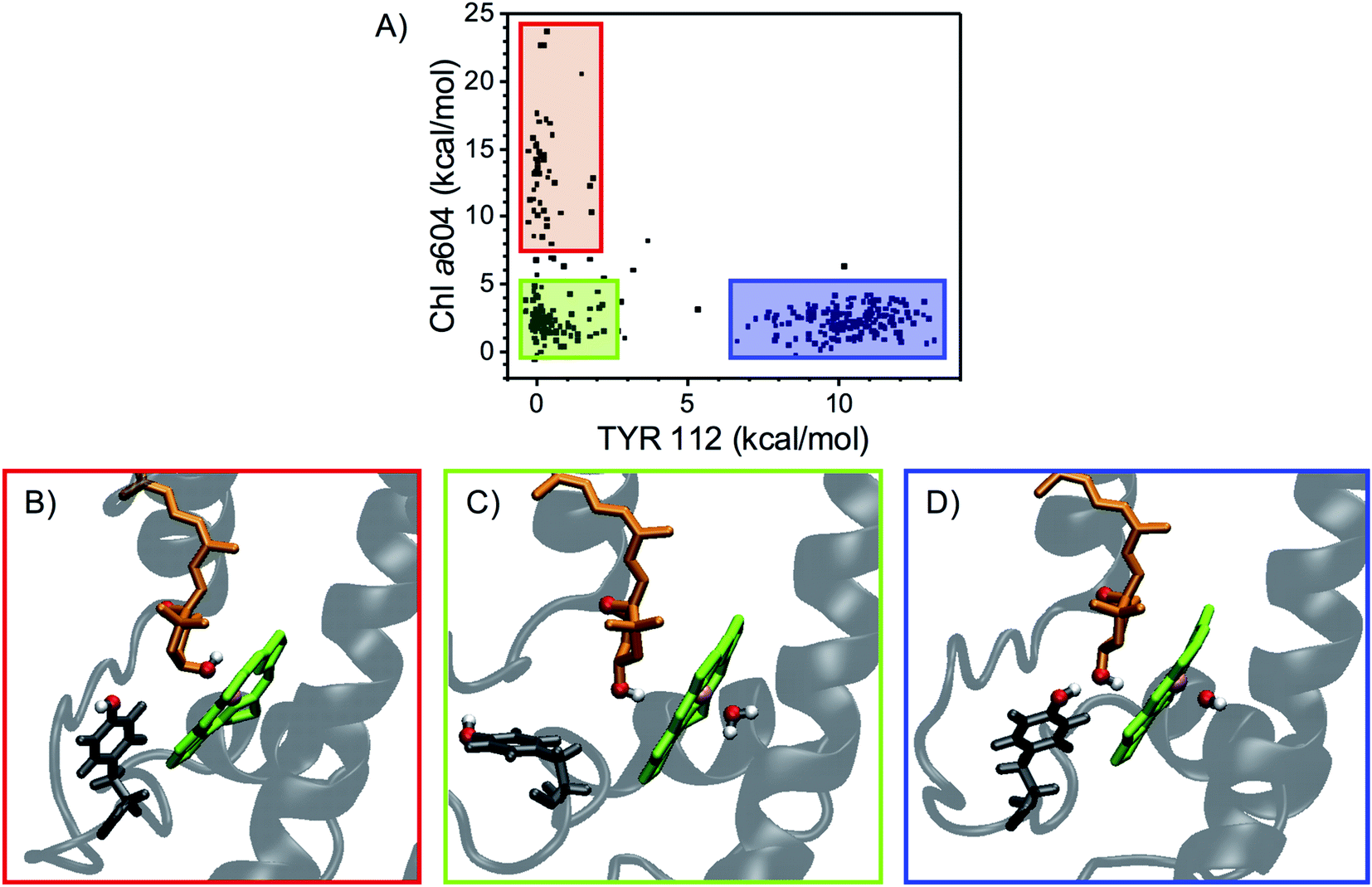 | ||
| Fig. 6 The N1 binding pocket assumes different conformations. (A) Electrostatic interaction energy of N1 neoxanthin of LHCII with either TYR112 (x-axis) or Chl a604 (y-axis). Three different clusters of data points (each point represents a single MD snapshot) can be visualized in the scatter plot, representing either no strong interaction (enclosed in the green rectangle), preferential interaction with Chl a604 (red) or preferential HB to TYR112 (blue). The latter cluster is also the most abundant. (B–D) Conformations of the N1 binding pocket characterized by distinct interaction patterns and corresponding to three MD snapshots representative of the clusters shown in (A), with the same color code adopted for the surrounding rectangles. Neoxanthin is shown in orange, Chl a604 in green, TYR112 in gray, and the protein in transparent gray. The water molecule coordinating Chl a604 is also shown, if present. The polar oxygen and hydrogen atoms are highlighted in red and white, respectively, whereas the Mg of Chl a604 is highlighted in pink. In (B), the –OH group of neoxanthin interacts strongly with the Mg of Chl a604, which lacks the coordinating water molecule. In (C), neoxanthin shows weak interactions with both Chl a604 (which is coordinated by a water molecule) and TYR112. In (D), neoxanthin forms a strong HB with TYR112. | ||
Varying the occupancy of the carotenoid binding sites
To understand whether LHCs adopt different binding strategies to accommodate chemically distinct carotenoids, similar calculations to those presented above for LHCII were performed on two variants of this complex with altered carotenoid content: (i) LHCII with L2 binding violaxanthin instead of lutein (as L2 of LHCII can bind both xanthophylls, though it prefers lutein)16,53 and (ii) LHCII binding astaxanthin in all the internal binding sites. The latter case is interesting because astaxanthin can stably bind to LHCII in vivo54 although it differs from the native xanthophylls from a chemical perspective (it is a keto-carotenoid; Fig. 1B).When bound to L2, violaxanthin displays an increased electrostatic interaction in comparison to lutein (Fig. 7A), as a consequence of its larger affinity for ASP47 at the stroma (Fig. 7B), although the interaction remains intermittent (Table S1†). The vdW interaction energy of violaxanthin in L2 is slightly lower than for lutein (Fig. 7A), although the overall interaction pattern with the nearby residues is very similar (Fig. S3†). When the two contributions are summed, the total interaction energies of the two xanthophylls in L2 do not differ significantly. In the case of astaxanthin bound to L2, the vdW interaction is not significantly different from that of lutein (Fig. 7A). However, the electrostatic interaction is substantially larger due to the increased affinity for water at the lumenal side caused by the additional carbonyl group on the astaxanthin terminal ring (Fig. 7B and Table S1†). In comparison to lutein and violaxanthin, the stromal ring of L2 astaxanthin has a larger preference for THR48 and a lower preference for ASP47, due to the repulsion between the carbonyl oxygen of astaxanthin and the negatively charged sidechain of ASP47.
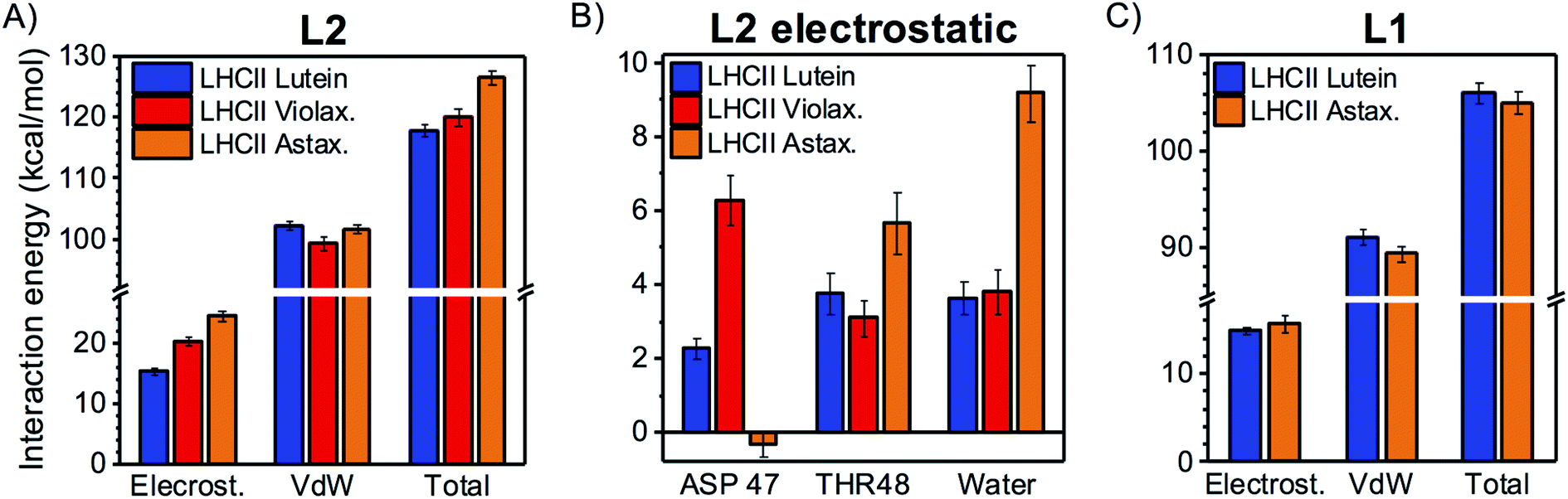 | ||
| Fig. 7 Varying the occupancy of the carotenoid binding sites. (A) Decomposition of interaction energy into electrostatic and vdW terms for the L2 carotenoid of native LHCII (lutein, blue), LHCII with violaxanthin in L2 (red), and LHCII with astaxanthin in all internal binding sites (orange). The value of each bar represents the average over all cumulated MD snapshots, while the error bars represent 96% confidence intervals (i.e. 2 times the standard error, with N = 323 for LHCII, 188 for LHCII with violaxanthin in L2, and 198 for LHCII with astaxanthin). (B) Main HB partners at L2 in different LHCII variants. All fragments with an average interaction >3 kcal mol−1 in at least one of the samples are shown. Positive bars stand for attractive interactions, while negative bars for repulsive ones. As for the interaction between the L2 carotenoid and water, the sum of the interactions with all water molecules is indicated. However, HBs with water molecules mostly involve the lumenal –OH of the xanthophyll, as confirmed by the following statistics: for native LHCII (lutein), 41% (8%) of MD frames display an interaction >3 kcal mol−1 with a water molecule at the lumenal (stromal) ring; for LHCII (violaxanthin), 51% (5%) of frames display an interaction >3 kcal mol−1 with a water molecule at the lumenal (stromal) ring; for LHCII (astaxanthin), 91% (15%) of MD frames display an interaction >3 kcal mol−1 with a water molecule at the lumenal (stromal) ring (over 80% of the HBs with water at the lumenal side involve the carbonyl group of astaxanthin). In the case of astaxanthin, whose rings have two potential HB sites (the –OH group and the carbonyl group), MD frames with n water molecules interacting in the same frame are counted n times in the average. (C) Decomposition of interaction energy into electrostatic and vdW terms for the L1 carotenoid of native LHCII (lutein, blue), and LHCII with astaxanthin (orange). | ||
When replacing lutein with astaxanthin in L1 of LHCII, the total interaction energy is not significantly affected (Fig. 7C). In particular, the additional presence of carbonyl groups on the terminal rings of astaxanthin does not affect the electrostatic interaction, as its HB network remains essentially unchanged (Fig. S4†). Similarly, the replacement of neoxanthin with astaxanthin in N1 does not affect the total interaction energy to a significant extent due to compensating effects in its electrostatic and vdW components (Fig. S5†). The main interaction partners of neoxanthin and astaxanthin in N1 are also very similar (Fig. S6†).
Discussion
Our results suggest that xanthophyll binding to LHCs is mostly driven by vdW rather than electrostatic forces. This downsizes the importance of HBs in the binding thermodynamics. For instance, according to structural data, the L1 and L2 carotenoids in both LHCII12 and the minor antenna CP29 (ref. 9 and 55) are anchored to ASP sidechains through HBs involving their stromal –OH groups. Due to the flexibility of the xanthophyll terminal rings, however, these interactions are shown to be only intermittent or, in some cases, completely absent in the MD simulations, which is consistent with a series of experimental results. Removal of the entire N-terminus segment containing THR47 and ASP48 in LHCII31 or mutation of the corresponding ASP35 in CP29,30 for instance, do not cause the loss of the L2 xanthophyll (lutein in LHCII and violaxanthin in CP29). Similarly, mutation of the ASP residue (ASP180) on the stromal side in CP29 does not cause a loss of L1 lutein.30 Based on our MD simulations, the only persistent HB involving the central xanthophylls is between the lumenal –OH of L1 lutein and the –NH2 group of a conserved GLN residue (which also coordinates Chl a613). However, previous experimental work on both LHCII23,56 and CP29 (ref. 24) showed that, while the presence of this GLN is crucial for Chl a613 binding, it is not required for L1 stability. Although the aforementioned experimental data on LHCs do not provide clear indications in this respect, it is still possible that HBs, while still having a small weight in the total interaction, could be functional for selecting specific carotenoid types in other pigment-protein complexes. Such a possibility is more evident in the single carotenoid binding site found in the reaction center of purple bacteria.57While the HBs mentioned above play only a marginal role in enthalpic terms, it is evident that xanthophyll binding is substantially driven by vdW interactions with the surrounding Chl macrocycles. The latters also represent the most stable interactions, as they mainly involve the xanthophyll linear chains and the more rigid domains inside the complex. The xanthophylls can count on a dense network of vdW contacts and, although exhibiting relatively strong interactions with some of the Chls, their binding does not rely on the presence of specific partners, which is in line with a number of experiments. For instance, the LHCII mutants lacking either Chl a602 or Chl a603 are relatively stable and do not lose L2 lutein,23 even though these two Chls are strong vdW interaction partners for this binding site. Consistently, CP29 does not lose violaxanthin in L2 when Chl a603 is knocked out58 and mutants of either LHCII or CP29 lacking either Chl a612 or Chl a613 are stable and do not lose lutein23,24,56,58 even though both Chl a612 and Chl a613 form strong interactions with L1 lutein. Evidently, the absence of just one Chl is not sufficient for affecting the stability of the two centrally bound xanthophylls. However, in the case of CP24, which constitutively lacks Chl a613, the knockout of Chl a612 translates into a marked loss of lutein.26 This suggests that the presence of at least one of these two Chls might be necessary for L1 stability.
In comparison to L1 and L2, electrostatic terms (including an HB to a conserved TYR residue) have a larger weight in the binding of N1 neoxanthin, which involves a more restricted network of strong interaction partners. This can explain why it was easier to knockout N1 neoxanthin than the two central xanthophylls via single-point mutations. Mutation of the conserved TYR residue, indeed, results in a partial loss of neoxanthin in LHCII, CP26, and CP29.27 Single-point mutants of LHCII and CP29 lacking either Chl 606 or Chl 609 also lose neoxanthin.11,24,56 Both mutations are localized on Helix C and involve the loss of other nearby Chls a/b (including Chls a604 and/or Chl b608), which is consistent with our finding that these Chls are important for N1 stability.
The replacement of the native carotenoids with other xanthophylls does not produce marked changes in the overall interaction patterns in any binding pocket. This is particularly true for the L1 and N1 sites, whereas L2 seems somewhat more capable of tuning its HB network based on the chemical properties of the xanthophyll. The robustness of the L1 interaction network agrees well with the higher degree of conservation (it binds lutein in all of the Lhcb genes)9,10 and “stiffness” observed in MD simulations36 and is consistent with the purportedly predominant role of the L1 carotenoid in photoprotection. Conversely, the somehow higher flexibility exhibited by L2 agrees with the higher variability observed experimentally. Indeed, the L2 sites of different LHCs bind either lutein or violaxanthin. Furthermore, despite preferring lutein, the L2 site of LHCII itself is not entirely selective and can also bind violaxanthin, depending on the composition of the pigment mixture available upon folding.16 In this respect, our results suggest that the interaction energies of lutein and violaxanthin in L2 are not significantly different, implying that the higher selectivity for lutein in LHCII observed experimentally should stem from structural, entropic, or kinetic factors.
In conclusion, while our work makes sense of numerous experimental data showing that the HBs predicted by the crystal structure are not a decisive factor for carotenoid binding, an open question remains: why can LHCs fold only in the presence of xanthophylls but not carotenes (which lack oxygen and cannot form HBs)? Based on our results, we suggest that the –OH groups of xanthophylls are specifically required for assisting the folding kinetics and might be involved in transient interactions that elude those present in the structural data.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
This project was supported by the Netherlands Organization for Scientific Research (NWO) via a Top grant to R. C., and a Veni grant to N. L. L. C. and B. M. acknowledge funding by the European Research Council, under the grant ERC-AdG-786714 (LIFETimeS).Notes and references
- L. Nicol and R. Croce, in Light Harvesting in Photosynthesis, CRC Press, 2018, pp. 59–76 Search PubMed.
- X. Pan, P. Cao, X. Su, Z. Liu and M. Li, Biochim. Biophys. Acta, Bioenerg., 2020, 1861, 148038 CrossRef CAS PubMed.
- R. Croce and H. van Amerongen, Science, 2020, 369, eaay2058 CrossRef CAS PubMed.
- C. Dockter, A. H. Müller, C. Dietz, A. Volkov, Y. Polyhach, G. Jeschke and H. Paulsen, J. Biol. Chem., 2012, 287, 2915–2925 CrossRef CAS PubMed.
- K. Sunku, H. J. M. de Groot and A. Pandit, J. Biol. Chem., 2013, 288, 19796–19804 CrossRef CAS PubMed.
- M. H. Shabestari, C. J. A. M. Wolfs, R. B. Spruijt, H. van Amerongen and M. Huber, Biophys. J., 2014, 106, 1349–1358 CrossRef CAS PubMed.
- N. Liguori, X. Periole, S. J. Marrink and R. Croce, Sci. Rep., 2015, 5, 15661 CrossRef CAS PubMed.
- H. A. Frank and B. Robert, in Light Harvesting in Photosynthesis, ed. R. Croce, R. van Grondelle, H. van Amerongen and I. van Stokkum, CRC Press, 2018, pp. 37–56 Search PubMed.
- X. Su, J. Ma, X. Wei, P. Cao, D. Zhu, W. Chang, Z. Liu, X. Zhang and M. Li, Science, 2017, 357, 815–820 CrossRef CAS PubMed.
- X. Qin, M. Suga, T. Kuang and J. R. Shen, Science, 2015, 348, 989–995 CrossRef CAS PubMed.
- R. Croce, R. Remelli, C. Varotto, J. Breton and R. Bassi, FEBS Lett., 1999, 456, 1–6 CrossRef CAS PubMed.
- Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed.
- S. Caffarri, R. Croce, J. Breton and R. Bassi, J. Biol. Chem., 2001, 276, 35924–35933 CrossRef CAS PubMed.
- B. Demmig-Adams and W. W. Adams, Trends Plant Sci., 1996, 1, 21–26 CrossRef.
- P. Xu, L. Tian, M. Kloz and R. Croce, Sci. Rep., 2015, 5, 13679 CrossRef PubMed.
- R. Croce, S. Weiss and R. Bassi, J. Biol. Chem., 1999, 274, 29613–29623 CrossRef CAS PubMed.
- M. Mozzo, L. Dall'Osto, R. Hienerwadel, R. Bassi and R. Croce, J. Biol. Chem., 2008, 283, 6184–6192 CrossRef CAS PubMed.
- A. V. Ruban, R. Berera, C. Ilioaia, I. H. M. van Stokkum, J. T. M. Kennis, A. A. Pascal, H. van Amerongen, B. Robert, P. Horton and R. van Grondelle, Nature, 2007, 450, 575–578 CrossRef CAS PubMed.
- T. K. Ahn, T. J. Avenson, M. Ballottari, Y.-C. Cheng, K. K. Niyogi, R. Bassi and G. R. Fleming, Science, 2008, 320, 794–797 CrossRef CAS PubMed.
- V. Mascoli, N. Liguori, P. Xu, L. M. Roy, I. H. M. van Stokkum and R. Croce, Chem, 2019, 5, 1–13 Search PubMed.
- F. G. Plumley and G. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 146–150 CrossRef CAS PubMed.
- H. Paulsen, B. Finkenzeller and N. Kühlein, Eur. J. Biochem., 1993, 215, 809–816 CrossRef CAS PubMed.
- R. Remelli, C. Varotto, D. Sandonà, R. Croce and R. Bassi, J. Biol. Chem., 1999, 274, 33510–33521 CrossRef CAS PubMed.
- R. Bassi, R. Croce, D. Cugini and D. Sandonà, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10056–10061 CrossRef CAS PubMed.
- M. Ballottari, M. Mozzo, R. Croce, T. Morosinotto and R. Bassi, J. Biol. Chem., 2009, 284, 8103–8113 CrossRef CAS PubMed.
- F. Passarini, P. Xu, S. Caffarri, J. Hille and R. Croce, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1500–1506 CrossRef CAS PubMed.
- S. Caffarri, F. Passarini, R. Bassi and R. Croce, FEBS Lett., 2007, 581, 4704–4710 CrossRef CAS PubMed.
- E. Pichersky and S. Jansson, in Oxygenic Photosynthesis: The Light Reactions, ed. D. R. Ort and C. F. Yocum, Kluwer Academic Publishers Group, Dordrecht, 1996, pp. 507–521 Search PubMed.
- D. Phillip, S. Hobe, H. Paulsen, P. Molnar, H. Hashimoto and A. J. Young, J. Biol. Chem., 2002, 277, 25160–25169 CrossRef CAS PubMed.
- M. Gastaldelli, G. Canino, R. Croce and R. Bassi, J. Biol. Chem., 2003, 278, 19190–19198 CrossRef CAS PubMed.
- S. Hobe, R. Förster, J. Klingler and H. Paulsen, Biochemistry, 1995, 34, 10224–10228 CrossRef CAS PubMed.
- S. Jurinovich, L. Viani, I. G. Prandi, T. Renger and B. Mennucci, Phys. Chem. Chem. Phys., 2015, 17, 14405–14416 RSC.
- S. Thallmair, P. A. Vainikka and S. J. Marrink, Biophys. J., 2019, 116, 1446–1455 CrossRef CAS PubMed.
- V. Daskalakis, S. Papadatos and T. Stergiannakos, Chem. Commun., 2020, 56, 11215–11218 RSC.
- N. Liguori, R. Croce, S. J. Marrink and S. Thallmair, Photosynth. Res., 2020, 144, 273–295 CrossRef CAS PubMed.
- N. Liguori, P. Xu, I. H. M. van Stokkum, B. van Oort, Y. Lu, D. Karcher, R. Bock and R. Croce, Nat. Commun., 2017, 8, 1994 CrossRef PubMed.
- J. M. Gómez, A. Jiménez, E. Olmos and F. Sevilla, J. Exp. Bot., 2004, 55, 119–130 CrossRef PubMed.
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. Van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS PubMed.
- C. Oostenbrink, T. A. Soares, N. F. A. Van Der Vegt and W. F. Van Gunsteren, Eur. Biophys. J., 2005, 34, 273–284 CrossRef CAS PubMed.
- D. H. De Jong, N. Liguori, T. Van Den Berg, C. Arnarez, X. Periole and S. J. Marrink, J. Phys. Chem. B, 2015, 119, 7791–7803 CrossRef CAS PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- C. Curutchet, A. Muñoz-Losa, S. Monti, J. Kongsted, G. D. Scholes and B. Mennucci, J. Chem. Theory Comput., 2009, 5, 1838–1848 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- C. J. Dickson, B. D. Madej, Å. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould and R. C. Walker, J. Chem. Theory Comput., 2014, 10, 865–879 CrossRef CAS PubMed.
- I. G. Prandi, L. Viani, O. Andreussi and B. Mennucci, J. Comput. Chem., 2016, 37, 981–991 CrossRef CAS PubMed.
- L. Zhang, D. A. Silva, Y. Yan and X. Huang, J. Comput. Chem., 2012, 33, 1969–1980 CrossRef CAS PubMed.
- M. Bondanza, L. Cupellini, F. Lipparini and B. Mennucci, Chem, 2020, 6, 1–17 Search PubMed.
- Y. Wang and X. Hu, J. Am. Chem. Soc., 2002, 124, 8445–8451 CrossRef CAS PubMed.
- Y. Wang, L. Mao and X. Hu, Biophys. J., 2004, 86, 3097–3111 CrossRef CAS PubMed.
- F. Müh, M. E. A. Madjet and T. Renger, J. Phys. Chem. B, 2010, 114, 13517–13535 CrossRef PubMed.
- S. Hobe, H. Niemeier, A. Bender and H. Paulsen, Eur. J. Biochem., 2000, 267, 616–624 CrossRef CAS PubMed.
- P. Xu, V. U. Chukhutsina, W. J. Nawrocki, G. Schansker, L. W. Bielczynski, Y. Lu, D. Karcher, R. Bock and R. Croce, eLife, 2020, 9, e58984 CrossRef CAS PubMed.
- X. Pan, M. Li, T. Wan, L. Wang, C. Jia, Z. Hou, X. Zhao, J. Zhang and W. Chang, Nat. Struct. Mol. Biol., 2011, 18, 309–315 CrossRef CAS PubMed.
- E. Formaggio, G. Cinque and R. Bassi, J. Mol. Biol., 2001, 314, 1157–1166 CrossRef CAS PubMed.
- A. W. Roszak, K. McKendrick, A. T. Gardiner, I. A. Mitchell, N. W. Isaacs, R. J. Cogdell, H. Hashimoto and H. A. Frank, Structure, 2004, 12, 765–773 CrossRef CAS PubMed.
- P. Xu, L. M. Roy and R. Croce, Biochim. Biophys. Acta, Bioenerg., 2017, 1858, 815–822 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00071c |
| This journal is © The Royal Society of Chemistry 2021 |