
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gating the photoactivity of azobenzene-type ligands trapped within a dynamic system of an M4L6 tetrahedral cage, an M2L2 metallocycle and mononuclear MLn complexes†
Piotr
Cecot‡
 ab,
Anna
Walczak‡
ab,
Grzegorz
Markiewicz
b and
Artur R.
Stefankiewicz
*ab
ab,
Anna
Walczak‡
ab,
Grzegorz
Markiewicz
b and
Artur R.
Stefankiewicz
*ab
aFaculty of Chemistry, Adam Mickiewicz University, ul. Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: ars@amu.edu.pl
bCenter for Advanced Technologies, Adam Mickiewicz University, Poznań, ul. Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
First published on 20th October 2021
Abstract
Complexation of transition metal ions by a doubly chelating bis(diimine)-type ligand incorporating a photoresponsive azobenzene linker yielded two types of structurally distinct metallosupramolecular architectures, an [M4L6]8+ tetrahedral cage and an [M2L2]4+ metallocycle. In solution, these complexes are open for reversible interconversions between each other by varying the M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L ratio, or switching into a dynamic library of [M(L′/L′′)n]2+ mononuclear species upon addition of a competing monoamine. While the unbound ligand presents the reversible photoactivity of the azo bond, its complexes are photochemically inert, due to the inherent topology of these assemblies resulting from the restrictions of coordinate bond formation.
:L ratio, or switching into a dynamic library of [M(L′/L′′)n]2+ mononuclear species upon addition of a competing monoamine. While the unbound ligand presents the reversible photoactivity of the azo bond, its complexes are photochemically inert, due to the inherent topology of these assemblies resulting from the restrictions of coordinate bond formation.
Introduction
Multistimuli-induced reversibility of chemical bonds is a key challenge of modern chemistry.1–5 When achieved, one can gain control over the global topology, physicochemical properties and ultimately potential functions of dynamic assembly, for instance, in catalysis to prevent product inhibition or in medicine to release an active compound at a specific site. Structural switching can be achieved by changing the proportion of the added reagents e.g. metal ions and/or via external stimuli, e.g. temperature or light.6–9 The controlled modulation of more than two dynamic bonds (molecular and/or supramolecular) within a single system remains challenging and is limited to only a few examples.10–14 Azobenzene derivatives are a well-known class of photoactive compounds that can undergo reversible isomerization upon light irradiation and/or thermal activation. This property has been used for the creation of many advanced photoswitchable systems.15 The structural and topological control can as well be achieved through component exchange and/or metal-ion coordination, and one of the most useful units for this is the 2-pyridine-imine N,N-chelating center. It owes its popularity to the reversibility of coordinate and imine bonds16 as well as the convenient synthesis of highly complex metallosupramolecular structures.17–19Several principles for designing appropriate ligands for multi-walled coordination cages were proposed by, inter alia, the Raymond,20 Fujita, Ballester, Stang, Clever and Nitschke groups.21–24 The structure of these complexes is subject to the influence of several factors, including ligand shape, host–guest interactions, external environment, and the metal-ion:ligand ratio.25–31 Thus, depending on the degree of freedom of rotation around the bonds and their stability, one can completely change the structures or adjust their topology to adapt to the guest molecule or other environmental stimuli.
In earlier work, we presented a photochemical study of acyclic azobenzene-substituted metal ion complexes.32 Although the coordination indeed affected the trans → cis conversions, the complexes obtained retained the partial photoactivity of the azo bonds, since these were present in unconstrained arms of the ligands. Recently,33 the retention of photosensitivity was also shown for a Pd2L4 type complex based on a bis(pyridylazo)terphenyl ligand. Gated photochromism is much less common but can be achieved in several ways,34,35 including, inter alia, the complexation process. Yamamura et al. found that the formation of both macromonocyclic and macrobicyclic complexes of B(III) and Ti(IV), respectively, with catecholate co-ligands led to a complete loss of photosensitivity and locking of the ligand azo unit in its trans configuration. The opposite case was found with terpyridine units in place of catecholates, where the cis form of the ligand was locked in the mononuclear Fe(II) complex.36,37 To the best of our knowledge no dynamic photochromic system, in which the photosensitivity of the azo bond can be locked by coordination within an M2L2 metallocycle, an M4L6 tetrahedral cage and a library of ML2/ML3 complexes, has been reported.
Herein, we describe the dynamic system of structurally distinct metallosupramolecular assemblies of acyclic and cyclic topologies formed between transition metal ions (Zn2+, Fe2+, and Cd2+) and mono- and doubly-chelating azobenzene ligands. In solution, these species are labile, allowing reversible switching between them driven by the changes of the M:L ratio and/or via transimination of the ligands upon addition of competitive amines. While the ligand itself exhibits the typical photochemical activity of azobenzene units, its complexes present gated photochromism upon irradiation, due to the coordination driven locking of the azo bonds within the rigid metallosupramolecular structures.
Results and discussion
Synthesis
Ligand trans-L was synthesized by the condensation of commercially available pyridine-2-carbaldehyde and trans-4,4′-diaminoazobenzene, following a previously reported procedure (Scheme 1, see the ESI† for details).32 As expected, the ligand was isolated in its thermodynamically favoured pure trans–azo form, as confirmed by 1H NMR spectroscopy (vide infra). To a suspension of trans-L in acetonitrile, solutions of Zn2+, Cd2+ or Fe2+ salts were added, yielding two structurally different complexes, depending on the metal:ligand ratio. [M4L6]8+ tetrahedral cages were obtained when trans-L was mixed with Zn2+ and Fe2+ salts in a 2:3 ratio, while a 1:1 ratio yielded [M2L2]4+ metallocycles for Zn2+ and Cd2+ salts (Scheme 1, see the ESI† for details).
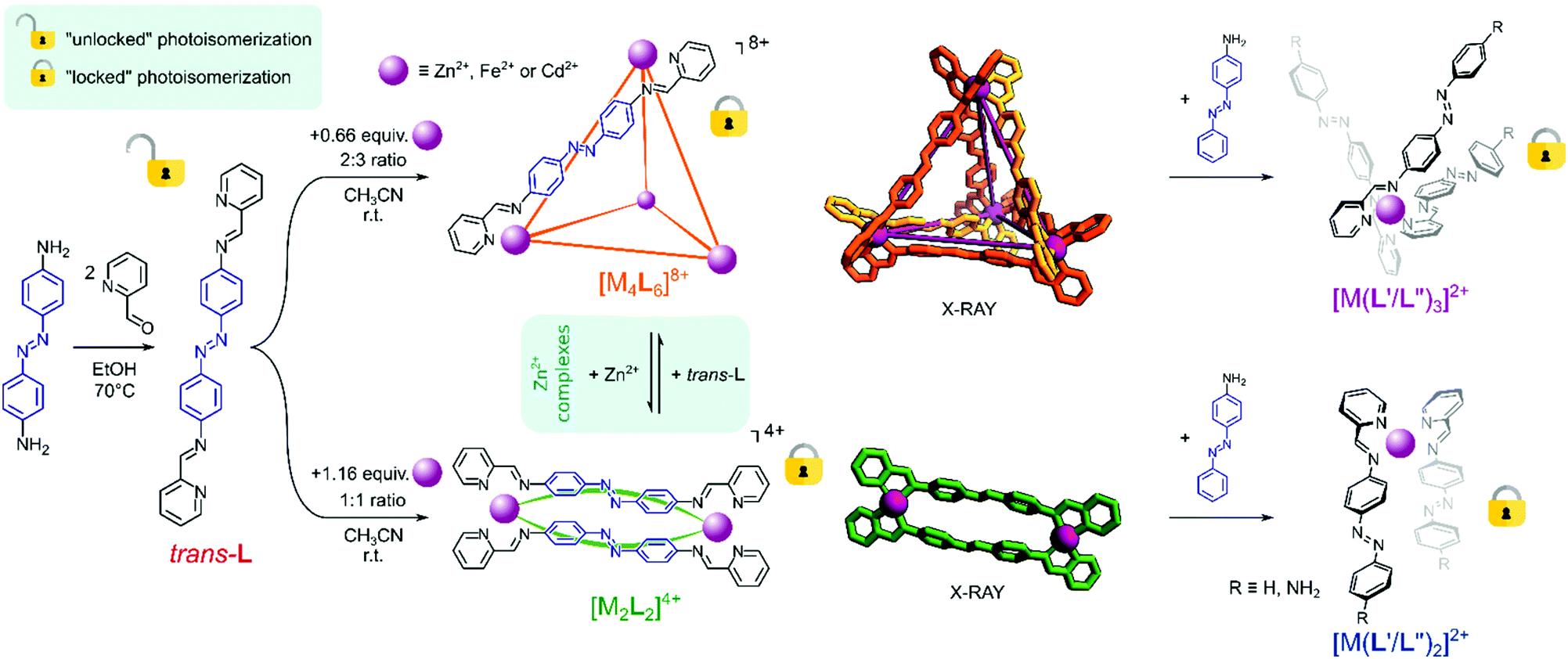 | ||
| Scheme 1 a) Synthesis of trans-L and its [M4L6](anion)8 and [M2L2](anion)4 complexes, followed by reversible interconversion between [M4L6]8+ and [M2L2]4+ complexes and their switching into a dynamic library of mononuclear [M(L′/L′′)n]2+ species upon transimination of the ligand trans-L with 4-aminoazobenzene. Counterions are omitted for clarity; transimination schemes for a (b) tetrahedral cage and (c) metallocycle. | ||
Solid state study
Slow vapor diffusion of Et2O into an acetonitrile solution of [Zn4L6](ClO4)8 yielded orange crystals, which were characterized by single-crystal X-ray diffraction. The structural determination showed that it crystallizes in the tetragonal space group P42/n with the cation as the S4 isomer (ΔΔΛΛ or ΛΛΔΔ), composed of four equivalent Zn2+ cations coordinated by six trans-L molecules (Scheme 1 and Fig. S26a†). Unfortunately, the crystals diffracted weakly, especially in high-angle regions; therefore, the structure was optimized using DFT b3lyp/6-31g(d) computation to locate the missing electron densities (Fig. S26b, see the ESI† for optimization details). The same crystallization technique with the material obtained from the reaction in a 1:1 stoichiometry yielded single crystals of the metallocycle [Cd2L2](ClO4)4 (Fig. S26c†). The complex crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Each of the two Cd(II) centres is coordinated by two pyridine-imine units and two ClO4− counterions, which complement both the coordination sphere and the charge of the central Cd2+ cation.
. Each of the two Cd(II) centres is coordinated by two pyridine-imine units and two ClO4− counterions, which complement both the coordination sphere and the charge of the central Cd2+ cation.
Solution study
After characterization in the solid state, we focused our studies on solutions to establish the stability of the complexes obtained and to perform photochemical measurements. The 1H NMR spectra of the ligand trans-L, [Zn4L6](NTf2)8, and [Zn2L2](NTf2)4 are shown in Fig. 1. In solution, the ligand exists exclusively as the trans-azo isomer, as evidenced by a single set of resonances, with the most indicative being the single imine proton resonance at 8.67 ppm (ref. 38) (cis-L form was found upon photoisomerization – vide infra). The ligand trans-L is non-planar when coordinated and has axial chirality, so its incorporation within the tetrahedral cage structure, along with the four chiral coordination centres (each one with octahedral geometry) could yield a set of diastereoisomeric cages. Considering only the four metal ion centres, three isomers of the [M4L6]8+ cation are possible: S4 (ΔΔΛΛ or ΛΛΔΔ), C3 (ΔΔΔΛ or ΛΛΛΔ) and T (ΔΔΔΔ or ΛΛΛΛ),20,25 but, as described above, the species found in the crystal is the pure S4 isomer, where the chirality of the ligand units appears to be determined by the nature of the metal ion centres that are bridged, so that there are no complications in the stereochemistry arising from this factor.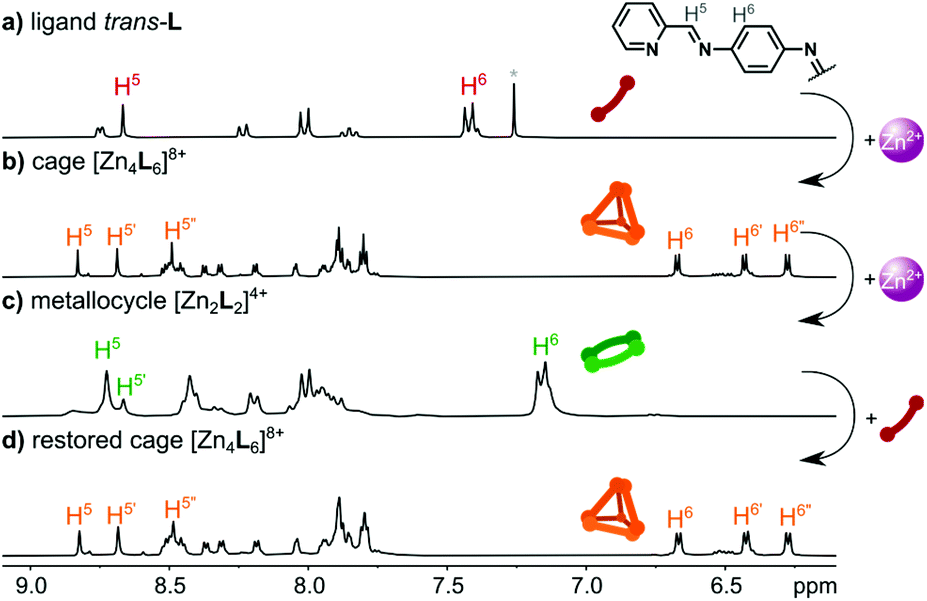 | ||
| Fig. 1 1H NMR spectra of (a) ligand trans-L (300 MHz, CDCl3); (b) [Zn4L6](NTf2)8 cage (300 MHz, CD3CN); and (c) [Zn2L2](NTf2)4 metallocycle (300 MHz, CD3CN). (d) Interconversion of [Zn2L2](NTf2)4 into [Zn4L6](NTf2)8 upon addition of 0.5 equiv. of trans-L. | ||
This is consistent with the fact that the solution 1H NMR spectrum of the cage (Fig. 1b) can be resolved into sets of peaks of the S4, C3 and T isomers. The [Zn4L6]8+ cage in acetonitrile solution exists predominantly as the S4 isomer, as proven by the splitting of the H5 imine signals into three well-defined singlets, and splitting and shielding of the H6 phenylene signal into three doublets. The molar ratio of the S4:C3:T isomers in acetonitrile solution was found to be 77:20:3 (Fig. S17 and Table S1†), showing that the S4 form is the favoured isomer of [Zn4L6]8+ both in solution and the solid state. This isomeric ratio is not affected by the counterion used (ClO4−, BF4−, OTf−, NTf2−) (Table S1 and Fig. S18, see the ESI† for details). ESI–TOF–MS analysis performed for the cage complex revealed a peak centred at 1103.4846 m/z, assigned to the {[Fe4L6](OTf)5}3+ ion, a tetranuclear species with the stoichiometry expected for tetrahedral cage–type structures of [M4L6](anion)8 complexes (Fig. S11†).
The formation of the cage [Zn4L6]8+ and the metallocycle [Zn2L2]4+ was further supported by diffusion ordered NMR spectroscopy (Fig. S7, S15 and Table S3†), which showed the formation of discrete products with diffusion coefficients of D = 4.76 × 10–6 cm2 s−1 (r = 13.4 Å) and D = 5.50 × 10−6 cm2 s−1 (r = 11.6 Å), respectively. These radii correspond well to the sizes of the [Zn4L6]8+ and [Zn2L2]4+ ions found in the crystal lattices (Fig. S35†).
Topological switching
To determine whether the interconversion between [Zn2L2]4+ and [Zn4L6]8+ is possible, a suspension of trans-L in CD3CN was titrated with Zn(NTf2)2. As shown in Fig. S20,† a gradual increase in the Zn2+ concentration up to a 2:3 M:L ratio led to the exclusive formation of the [Zn4L6]8+ complex. However, when the M:L ratio exceeded 2:3, the cage began to convert into the [Zn2L2]4+ metallocycle, yielding at first the cage:metallocycle mixture, then pure [Zn2L2]4+ after the addition of excess Zn2+ (Fig. 1c). Finally, the solution of metallocycle [Zn2L2](NTf2)4 was treated with a suspension of trans-L, to observe the direct return of [Zn2L2]4+ into the [Zn4L6]8+ species. As shown in Fig. 1d, [Zn2L2]4+ fully converted into [Zn4L6]8+ after the addition of 0.5 equiv. of trans-L. The subsequent addition of 0.5 equiv. of Zn(NTf2)2 led to the restoration of the [Zn2L2]4+ structure (Fig. S21†).
These experiments showed that the complexation of trans-L is a dynamic process with an outcome that is directly related to the M:L stoichiometry. The observed interconversions are governed by the relatively strong chelation of Zn(II) by the N,N donor sites of the ligand, the [Zn2L2]4+ metallocycle being the least entropically disfavoured species where both Zn(II) centres are bis(chelated), and [Zn4L6]8+ the smallest species where all the Zn(II) centres are tris(chelated). The lability of Zn(II) means that a variation in the composition of the reaction mixture can be used to enable the isolation of each one of the complexes obtained. In the gas phase, the availability of two coordination sites not occupied by a chelate unit in the metallocyclic complex appears to favour its fragmentation, explaining why multiple attempts to observe [M2L2]4+ species or their solvates using the ESI-MS technique have failed (see the ESI† for details).
Photoactivity
The photochemical behaviour of trans-L and the complexes [Zn4L6](NTf2)8 and [Zn2L2](NTf2)4 was investigated by electronic absorption and 1H NMR spectroscopy (Fig. 2). The ligand trans-L shows behaviour typical of azobenzene derivatives that undergo reversible trans → cis photoisomerization of the azo bond induced by UV light. Irradiation of trans-L solutions in acetonitrile (UV-vis) or chloroform (NMR) with near-UV light (λmax = 395 nm) results in its partial isomerization to cis-L, as indicated by a gradual decrease in the absorption intensity of the π → π* azo band (λmax = 380 nm) in the absorption spectrum (Fig. S27a†), and appearance of a new set of shielded resonances in the 1H NMR spectrum (Fig. 2a). Plotting those changes as a function of irradiation time revealed that the reaction follows reversible first-order kinetics, with a (k = ktrans→cis + kcis→trans) rate constant of 0.74 s−1 (Fig. S27b†). The reaction equilibrates at 62% of cis-L in the photostationary state (K = 1.63) with isomerization rate constants of ktrans→cis = 0.46 s−1 and kcis→trans = 0.28 s−1.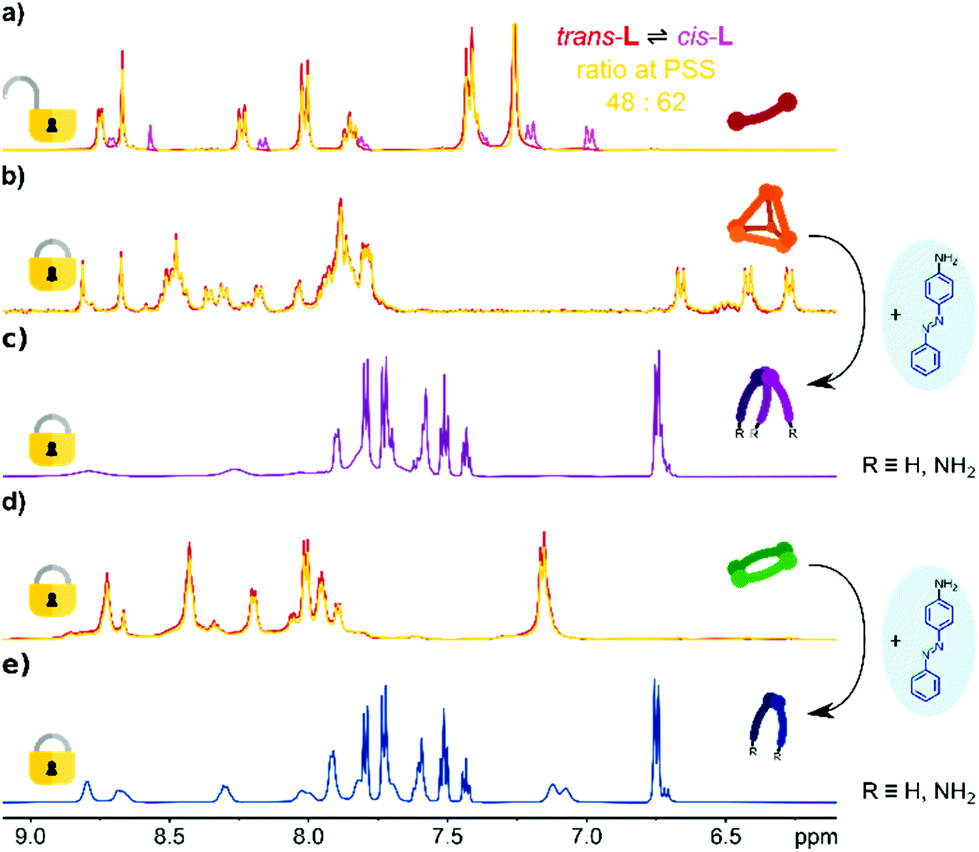 | ||
| Fig. 2 1H NMR spectra (600 MHz) of (a) ligand trans-L in CDCl3 before (red) and after irradiation (yellow); signals of cis-L marked in magenta. (b) [Zn4L6](NTf2)8 cage in CD3CN before (red) and after irradiation (yellow). (c) Conversion of the [Zn4L6](NTf2)8 cage in CD3CN into a library of [M(L′/L′′)3]2+ species after addition of 4-aminoazobenzene (for full titration spectra, see Fig. S22†). (d) [Zn2L2](NTf2)4 metallocycle in CD3CN before (red) and after irradiation (yellow). (e) Conversion of the [Zn2L2](NTf2)4 metallocycle in CD3CN into a library of [M(L′/L′′)2]2+ species after addition of 4-aminoazobenzene (for full titration spectra, see Fig. S23 and S24†). | ||
Upon heating the cis/trans-L mixture converts back from PSS to a pure trans-L isomer. The thermally driven reaction clearly follows first-order kinetics with k′cis→trans thermal = 2.4 10−3 s−1 and t1/2 = 288 s at 333 K (Fig. S34a and 34b†). Both photochemical and thermal reactions were found to be fully reversible in irradiation/heating cycles (Fig. S30 and S31†).
In contrast to our previous study,32 where the photoactive moieties were placed in the outer coordination sphere, the azobenzene units of both the [M4L6]8+ cage and the [M2L2]4+ metallocycle are tied up within the cyclic and rigid coordination structures. Taking into account that the trans → cis isomerization of the azo bond leads to a significant shortening of the L molecule, and a subsequent change in the angle between two chelating centres, photoisomerization of the bound ligand in the complexes would be expected to lead to rearrangement into species of quite different topology. Indeed, in the past irradiation has been shown to lead to complete dissociation of a metallocage species derived from a ligand incorporating two diazo units linking simple pyridine donors.33
The absorption spectra of both [Zn4L6](NTf2)8 and [Zn2L2](NTf2)4 show similar profiles to that of the trans-L spectrum, with λmax values at 362 and 372 nm, respectively. Irradiation of their solutions under identical conditions to those found to isomerize trans-L, gave, however, unexpected results. Namely, both cage and metallocycle showed high resistance to photoisomerization, yielding only <10% conversions in the photostationary state achieved after prolonged photoirradiation (Fig. S28 and S29†). Given that the measurements were made on very dilute solutions (required for absorption spectroscopy), and the formation of both [Zn4L6]8+ and [Zn2L2]4+ complexes is reversible, it was concluded that the observed conversions most likely originated from the released trans-L molecules, not from the complexes. Indeed, when the irradiated samples of [Zn4L6]8+ and [Zn2L2]4+ were heated to induce a backward cis → trans conversion, an abnormal release of trans-L was observed. In contrast to the isolated L, the first-order backward isomerisation is affected by the disassembly processes so the conversion exceeds 100% of trans-L (Fig. S34c and 34d†).
To omit the problem with the partial disassembly, we followed the isomerization by 1H NMR spectroscopy, which allows the use of higher concentrations and observation of all species in the solution. As shown in Fig. 2b, UV irradiation of the [Zn4L6](NTf2)8 cage does not lead to any observable changes. All resonances remain unchanged, proving that the cage is photochemically stable, in terms of azo bond photoisomerization. Similar results were obtained for [Zn2L2](NTf2)4 (Fig. 2d), proving that the metallocyclic structure is also photochemically locked. Having the photoactive ligand L, and its photochemically inert complexes, we decided to check if it is possible to introduce the cis-L into assemblies prior to the complexation. However, when the mixture of trans/cis-L was treated with Zn2+ salt we observed a gradual decrease in the absorbance, without indicative red/blue shifts for the selective formation of metallosupramolecular assemblies. Coordination also does not stimulate the reverse isomerization of the azo unit, even when the mixture with an excess of a metal salt is allowed to stand for 24 h for a spontaneous cis → trans backward conversion (Fig. S32†).
Transimination
Having the photoinactive cyclic complexes, we decided to examine whether they could be converted into acyclic products via the transimination reaction and thus possibly modulate the photosensitivity of the azo units. Addition of the competing 4-aminoazobenzne into a solution of [Zn4L6](NTf2)8 in CD3CN yielded, through a transimination reaction, a library of mononuclear [M(L′/L′′)3]2+ octahedral complexes bearing azobenzene units (Scheme 1 and Fig. 2c). Similar changes were observed for the [Zn2L2](NTf2)4 metallocycle, which was also converted into a library of mononuclear complexes, although due to the limited number of ligands formed, the reaction yielded [M(L′/L′′)2]2+ species (Scheme 1 and Fig. 2e). Our previous work32 has shown that the mononuclear [MLn]i+ complexes bearing azobenzene moieties retain some partial photoactivity of the azobenzene unit despite the metal ion complexation. However, when the dynamic libraries of [M(L′/L′′)3]2+ and [M(L′/L′′)3]2+ complexes were subjected to UV radiation, we did not observe a significant increase in their photoactivity (trans → cis conversions were found to be about 10% for both libraries; Fig. S33†).Conclusions
In conclusion, a new dynamic metallosupramolecular system consisting of photoactive ligands and transition metal ions in which photoactivity can be restricted by topology changes has been obtained. Depending on the M:L ratio, two types of cyclic complexes, i.e. [M4L6]8+ tetrahedral cages and [M2L2]4+ metallocycles, have been generated. We have shown that the self-assembly of both tetrahedral cages and metallocycle species is a dynamic process, allowing reversible interconversion between both structures steered by the M:L ratio. What is more, the transimination reaction observed upon addition of monoamine into [M2L2]4+ and [M4L6]8+ solutions yielded dynamic libraries of the mononuclear [M(L′/L′′)2]2+ and [M(L′/L′′)3]2+ complexes, respectively. Photochemical studies have shown that while the uncoordinated ligand displays typical behaviour of photoactive azobenzene derivatives, its macrocyclic complexes resist isomerization due to the locking of the azo bonds within the rigid metallosupramolecular topologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. S. and A. W. thank the National Science Centre (grant SONATA BIS 2018/30/E/ST5/00032) for financial support. We thank Prof. J. M. Harrowfield and Prof. J. R. Nitschke for helpful discussions. This work was supported by the INNCHEM (grant no. POWR.03.02.00-00-I023/17) co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development. The calculations were carried out at the Poznań Supercomputing and Networking Center (grant no. 401). A. W. and G. M. are supported by the Foundation for Polish Science (FNP).Notes and references
- J. Hu, S. K. Gupta, J. Ozdemir and M. H. Beyzavi, Applications of Dynamic Covalent Chemistry Concept towards Tailored Covalent Organic Framework Nanomaterials: A Review, ACS Appl. Nano Mater., 2020, 3, 6239–6269 CrossRef CAS PubMed
.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Recent advances in dynamic covalent chemistry, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC
- J. R. Nitschke, Supramolecular and dynamic covalent reactivity, Chem. Soc. Rev., 2014, 43, 1798–1799 RSC
- J. M. Lehn, From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry, Chem. Soc. Rev., 2007, 36, 151–160 RSC
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed
- M. Schmittel, Dynamic Functional Molecular Systems: From Supramolecular Structures to Multi–Component Machinery and to Molecular Cybernetics, Isr. J. Chem., 2019, 59, 197–208 CrossRef CAS
- P. Tecilla and D. Bonifazi, Configurational Selection in Azobenzene-Based Supramolecular Systems Through Dual-Stimuli Processes, ChemistryOpen, 2020, 9, 529–544 CrossRef PubMed
- E. Moulin, L. Faour, C. C. Carmona-Vargas and N. Giuseppone, From Molecular Machines to Stimuli-Responsive Materials, Adv. Mater., 2020, 32, 1906036 CrossRef CAS PubMed
- R. Gostl, A. Senf and S. Hecht, Remote-controlling chemical reactions by light: towards chemistry with high spatio-temporal resolution, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC
- H. M. Seifert, K. Ramirez Trejo and E. V. Anslyn, Four Simultaneously Dynamic Covalent Reactions. Experimental Proof of Orthogonality, J. Am. Chem. Soc., 2016, 138, 10916–10924 CrossRef CAS PubMed
- J. Septavaux, C. Tosi, P. Jame, C. Nervi, R. Gobetto and J. Leclaire, Simultaneous CO2 capture and metal purification from waste streams using triple-level dynamic combinatorial chemistry, Nat. Chem., 2020, 12, 202–212 CrossRef CAS PubMed
- S. Lascano, K. D. Zhang, R. Wehlauch, K. Gademann, N. Sakai and S. Matile, The third orthogonal dynamic covalent bond, Chem. Sci., 2016, 7, 4720–4724 RSC
- L. Rocard, A. Berezin, F. De Leo and D. Bonifazi, Templated Chromophore Assembly by Dynamic Covalent Bonds, Angew. Chem., Int. Ed., 2015, 54, 15739–15743 CrossRef CAS PubMed
- R. J. Sarma, S. Otto and J. R. Nitschke, Disulfides, imines, and metal coordination within a single system: interplay between three dynamic equilibria, Chem. – Eur. J., 2007, 13, 9542–9546 CrossRef CAS PubMed
- J. Dokic, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, Quantum chemical investigation of thermal cis-to-trans isomerization of azobenzene derivatives: substituent effects, solvent effects, and comparison to experimental data, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef CAS PubMed
- M. E. Belowich and J. F. Stoddart, Dynamic imine chemistry, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC
- D. Zhang, T. K. Ronson and J. R. Nitschke, Functional Capsules via Subcomponent Self-Assembly, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed
- S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Molecular containers in complex chemical systems, Chem. Soc. Rev., 2015, 44, 419–432 RSC
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Stimuli-Responsive Metal-Ligand Assemblies, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed
- D. L. Caulder and K. N. Raymond, Supermolecules by Design, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Covalent post-assembly modification in metallosupramolecular chemistry, Chem. Soc. Rev., 2018, 47, 626–644 RSC
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular coordination: self-assembly of finite two- and three-dimensional ensembles, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed
- P. Ballester, M. Fujita and J. Rebek Jr., Molecular containers, Chem. Soc. Rev., 2015, 44, 392–393 RSC
- M. Han, D. M. Engelhard and G. H. Clever, Self-assembled coordination cages based on banana-shaped ligands, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC
- W. Meng, J. K. Clegg, J. D. Thoburn and J. R. Nitschke, Controlling the transmission of stereochemical information through space in terphenyl-edged Fe4L6 cages, J. Am. Chem. Soc., 2011, 133, 13652–13660 CrossRef CAS PubMed
- A. M. Johnson, M. C. Young and R. J. Hooley, Reversible multicomponent self-assembly mediated by bismuth ions, Dalton Trans., 2013, 42, 8394–8401 RSC
- J. K. Clegg, J. Cremers, A. J. Hogben, B. Breiner, M. M. J. Smulders, J. D. Thoburn and J. R. Nitschke, A stimuli responsive system of self-assembled anion-binding Fe4L68+ cages, Chem. Sci., 2013, 4, 68–76 RSC
- A. M. Johnson, C. A. Wiley, M. C. Young, X. Zhang, Y. Lyon, R. R. Julian and R. J. Hooley, Narcissistic self-sorting in self-assembled cages of rare Earth metals and rigid ligands, Angew. Chem., Int. Ed., 2015, 54, 5641–5645 CrossRef CAS PubMed
- S. J. Wezenberg, Light-switchable Metal-Organic Cages, Chem. Lett., 2020, 49, 609–615 CrossRef CAS
- A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Fluorescent sensors: A bright future for cages, Coord. Chem. Rev., 2021, 434, 213820 CrossRef
- R. G. Siddique, K. S. A. Arachchige, H. A. Al-Fayaad, A. J. Brock, A. S. Micallef, E. T. Luis, J. D. Thoburn, J. C. McMurtrie and J. K. Clegg, The kinetics and mechanism of interconversion within a system of [Fe2L3]4+ helicates and [Fe4L6]8+ cages, Chem. Commun., 2021, 57, 4918–4921 RSC
- G. Markiewicz, A. Walczak, F. Perlitius, M. Piasecka, J. M. Harrowfield and A. R. Stefankiewicz, Photoswitchable transition metal complexes with azobenzene-functionalized imine-based ligands: structural and kinetic analysis, Dalton Trans., 2018, 47, 14254–14262 RSC
- S. Fu, Q. Luo, M. Zang, J. Tian, Z. Zhang, M. Zeng, Y. Ji, J. Xu and J. Liu, Light-triggered reversible disassembly of stimuli-responsive coordination metallosupramolecular Pd2L4 cages mediated by azobenzene-containing ligands, Mater. Chem., 2019, 3, 1238–1243 CAS
- M. Lohse, K. Nowosinski, N. L. Traulsen, A. J. Achazi, L. K. von Krbek, B. Paulus, C. A. Schalley and S. Hecht, Gating the photochromism of an azobenzene by strong host-guest interactions in a divalent pseudo[2]rotaxane, Chem. Commun., 2015, 51, 9777–9780 RSC
- G. Baggi, L. Casimiro, M. Baroncini, S. Silvi, A. Credi and S. J. Loeb, Threading-gated photochromism in [2]pseudorotaxanes, Chem. Sci., 2019, 10, 5104–5113 RSC
- M. Yamamura, Y. Okazaki and T. Nabeshima, Photoisomerization locking of azobenzene by formation of a self-assembled macrocycle, Chem. Commun., 2012, 48, 5724–5726 RSC
- M. Yamamura, K. Yamakawa, Y. Okazaki and T. Nabeshima, Coordination-driven macrocyclization for locking of photo- and thermal cis→trans isomerization of azobenzene, Chem. – Eur. J., 2014, 20, 16258–16265 CrossRef CAS PubMed
- Y. Jung, J. Nam, J.-H. Kim and W.-D. Jang, Hydrophilic–hydrophobic phase transition of photoresponsive linear and macrocyclic poly(2-isopropyl-2-oxazoline)s, RSC Adv., 2017, 7, 10074–10080 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2060413–2060414. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qi01063h |
| ‡ These authors contributed equally in this work. |
| This journal is © the Partner Organisations 2021 |