
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented collateral sensitivity for cisplatin-resistant lung cancer cells presented by new ruthenium organometallic compounds†
Ricardo G.
Teixeira
a,
Dimas C.
Belisario
b,
Xavier
Fontrodona
c,
Isabel
Romero
 c,
Ana Isabel
Tomaz
a,
M. Helena
Garcia
a,
Chiara
Riganti
b and
Andreia
Valente
*a
c,
Ana Isabel
Tomaz
a,
M. Helena
Garcia
a,
Chiara
Riganti
b and
Andreia
Valente
*a
aCentro de Química Estrutural and Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade de Lisboa, Campo Grande, 1749-016 Lisboa, Portugal. E-mail: amvalente@ fc.ul.pt
bDepartment of Oncology, University of Torino, Torino, Italy
cDepartament de Química and Serveis Tècnics de Recerca, Universitat de Girona, C/M. Aurèlia Campmany, 69, E-17003 Girona, Spain
First published on 12th February 2021
Abstract
Platinum-based therapies continue to be the main regimen used to treat non-small cell lung cancers (NSCLC), where multidrug resistance plays a key role in treatment failure and strategies to overcome this limitation are urgently sought for. In view to contribute to the development of this field, two sets of new organometallic Ru(II) compounds with general formula [Ru(η5-C5H4R′)(bipyR)(PPh3)][CF3SO3], where R′ = CHO or CH2OH and bipyR = 2,2′-bipyridine (1, 5), 4,4′-dimethyl-2,2′-bipyridine (2, 6), 4,4′-di(hydroxymethyl)-2,2′-bipyridine (3, 7) and 4,4′-dibiotin ester-2,2′-bipyridine (4), were synthesized and fully characterized. All compounds were tested against four types of NSCLC cell lines (A549, NCI-H228, Calu-3 and NCI-H1975), and four of them (1, 2, 4 and 6) presented a strong activity against cisplatin-resistant NSCLC cells. They were also able to increase cisplatin cytotoxicity up to 1390-fold (when administrated at nontoxic doses) by inhibiting MRP1 and P-gp transporters. Given the role of MRP1 in cisplatin resistance, in particular in lung cancer where cisplatin is the first-line treatment, the finding that these compounds are inducers of collateral sensitivity is of particular relevance. As far as we are aware, these are the first ruthenium-based compounds with such a mechanism of action, taking advantage of an “Achilles’ heel” and acting as MDR-selective compounds.
Introduction
Cancer is a leading cause of death worldwide, with an estimated 9.6 million deaths in 2018. Among all types of cancer, lung cancer is the most common and most lethal disease, due to late diagnosis and intrinsic and/or acquired resistance. In particular, non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers and its treatment relies on surgery, chemotherapy, radiation, immunotherapy or combinations thereof.1 The first line treatment for locally advanced stage NSCLC is simultaneous chemotherapy with different chemotherapeutics and radiation.1 A multi-chemotherapeutics approach is frequently used and includes a platinum-based agent (such as cisplatin or carboplatin) and other drugs with different mechanisms of action such as a taxane or a vinca alkaloid.2 Yet, due to drug resistance these therapeutic options have only limited success.During the last years several research groups have developed new metallodrugs/metallodrugs formulations aiming at overcoming several forms of multidrug resistance.3–12 However, in what concerns lung cancer, promising examples are still scarce. One of the strategies used has been the co-delivery of valproic acid (VPA), a histone deacetylase inhibitor, with a metallodrug.13,14 For example, Mao and co-workers have developed VPA-functionalized cyclometalated iridium(III) complexes through a hydrolysable ester bond.13 The results have shown a significant increased activity for the new conjugates (vs. VPA alone or a mixture of complexes + VPA) validating the strategy adopted. Also, the conjugates were able to overcome cisplatin resistance in human lung carcinoma cells (A549R). In another work, Du, Meng and co-workers, used L-cysteine decorated Zr-based metal–organic frameworks (MOFs) to co-deliver cisplatin and VPA.14 Immunoblot and immunofluorescence analyses showed that the new MOFs were able to downregulate the expression of vascular endothelial growth factor (VEGF) and improved the sensitivity to cisplatin of resistant A549/CDDP. The in vivo experiments confirmed that chemotherapy using these MOFs combined with microwave thermal therapy significantly improved the therapeutic effect of cisplatin resistant lung cancer. In a different approach, a family of CoII and CoIII tris(bipyridine) compounds has shown an important cytotoxicity against a panel of cancer cell lines that included taxol-resistant, cisplatin-resistant and p53-deficient cancer cells.15 Overall, there was no direct correlation between the oxidation state of the complexes and their cytotoxicity, yet the methyl substituent at the bipyridine (vs. non-substituted or methoxy) seemed to impart a favorable response in cisplatin-resistant (A2780, SGC-7901, OV2008 and C13 vs. resistant phenotype), in taxol-resistant (MCF-7, HCT-8, and A549 vs. resistant phenotype) and in p53-deficient apoptosis-resistant (HCT116 p53+/+vs. HCT116 p53−/−) cancer cells. It was further elucidated that compound [Co(4,4′-dimethyl-2,2′-bipyridine)3]3+ was able to inhibit P-gp, which is associated to taxol-resistance, and suppressed ∼50% of tumor growth in a lung cancer xenograft model. Indeed, one of the most important mechanisms of cell resistance is the overexpression of drug transporters, such as P-gp.2 In this frame, there are a few reports on metal complexes that are able to inhibit ABC pumps. For example, Choudhuri and coworkers developed a series of [M(N-(2-hydroxy acetophenone)glycinate)(H2O)n] complexes, where the M is FeIII (n = 3),16 NiII (n = 1),17 CuII (n = 3)18 or ZnII (n = 1).19 In particular, the CuII complex was shown to directly interact with P-gp showing potential to reverse P-gp mediated drug resistance.18 Interestingly, it did not compete for the substrate binding or to verapamil-, vinblastine- and progesterone-binding sites.
Ruthenium complexes are nowadays considered promising alternatives to the metal-based drugs in clinical use with some compounds still in clinical trials, namely KP1019 sodium salt NKP1339/IT-139 (sodium trans-[tetrachlorobis(indazole)ruthenate(III)])20,21 and TLD1433 (Ru(II) polypyridyl complex; ClinicalTrials.gov Identifier: NCT03053635).22 In the scope of ruthenium MDR modulators, i.e., compounds able to block the drug efflux, only a few examples are known. Among them, the compound [Ru(η6-p-cymene)Cl2(N-(Anthracen-9-yl)-imidazole)] (Fig. 1, left), developed by Juillerat-Jeanneret, Dyson and coworkers, showed good cytotoxicity against A549 lung, HT29 colon, and T47D breast carcinoma and inhibited P-gp close to the levels of the reference inhibitor verapamil.23 In this regard, we have been also engaged in the last years in the development of ruthenium(II) compounds with ATP Binding Cassette (ABC) efflux pumps inhibitory properties. Notably, during structure–activity studies on compounds from the [RuII(η5-C5H4R)(2,2′-bipyridine-4,4′-R′)(PPh3)]+ family, one compound (LCR134 for R = H, R′ = biotin ester, Fig. 1, right) has shown remarkable ability to inhibit P-gp (even better than the reference inhibitor verapamil)24 and another stood out as a possible MRP1 inhibitor (RT11 for R = CH3 and R′ = CH3) (Fig. 1, bottom).25 In addition, both compounds (LCR134 and RT11) were also cytotoxic for breast (MCF-7 and MDA-MB-231) and ovarian (A2780/A2780cisR) cancer cells, respectively, exhibiting a rare dual behavior as both cytotoxic agents and ABC pump inhibitors. It was further observed that the substituent at the bipyridine is key to provide inhibitory properties to this family of compounds. Thus, in the continuation of these studies other substituents were added at the cyclopentadienyl ring (–CHO or –CH2OH) allowing the synthesis of a new family of compounds. The results disclosed in this work will highlight the importance that specific chemical groups have on the inhibitory properties of ABC pumps and how this might contribute to improve the activity of the compounds and to sensitize NSCLC cells to cisplatin.
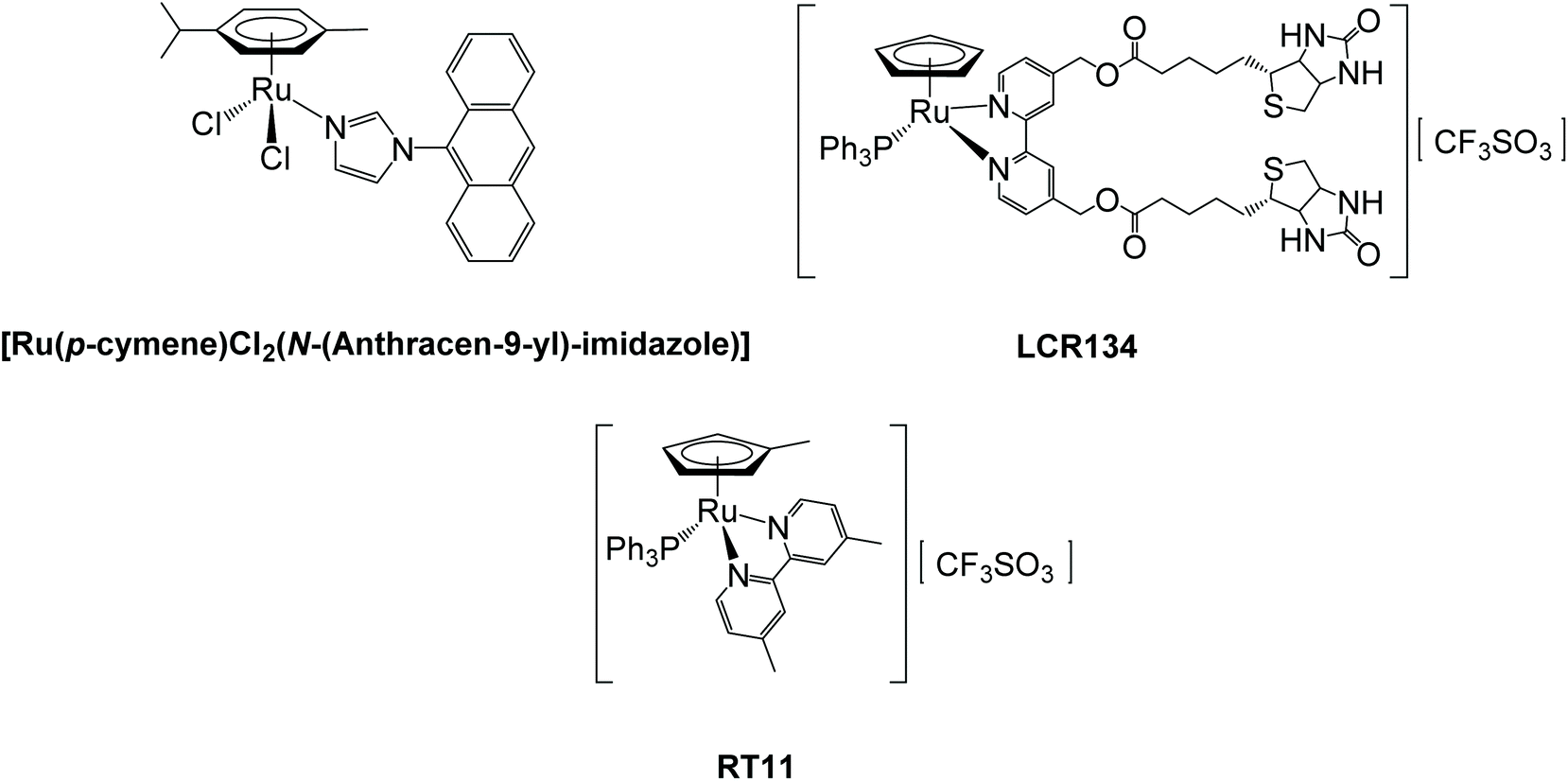 | ||
| Fig. 1 Ruthenium ABC pump inhibitors. | ||
Results and discussion
Synthesis and characterization
For the synthesis of the new compounds, we first optimized the method described by Bai Wei et al.26 to prepare the precursor [Ru(η5-C5H4CHO)(PPh3)2Cl], by slightly changing the purification protocol avoiding chromatography (cf. Experimental section). Using this complex as starting material, three new cationic compounds with general formula [Ru(η5-C5H4CHO)(bipy)(PPh3)][CF3SO3], where bipy is 2,2′-bipyridine (1), 4,4′-dimethyl-2,2′-bipyridine (2) and 4,4′-bis(hydroxymethyl)-2,2′-bipyridine (3), were synthesized in moderate to good yield by chloride abstraction with silver trifluoromethanesulfonate (Fig. 2, left). However, the reaction with 4,4′-dibiotin ester-2,2′-bipyridine (bipy-biotin) originated a secondary product very difficult to eliminate from the mixture even by column chromatography or repeated recrystallization. In order to overcome this difficulty, the ‘chemistry-on-the-complex’ concept27 was used. Thus, complex 3 was used as a platform to directly conjugate the envisaged vitamin via carbodiimide-based activation method using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (Scheme S1†).28 This proved to be an efficient alternative method to afford complex [Ru(η5-C5H4CHO)(bipy-biotin)(PPh3)][CF3SO3] (4) with a high yield (88%). Finally, we reduced the formyl group in the cyclopentadienyl ring of the previous four compounds into their hydroxymethyl analogues of general formula [Ru(η5-C5H4CH2OH)(bipyR)(PPh3)][CF3SO3] (where bipyR = 2,2′-bipyridine (5), 4,4′-dimethyl-2,2′-bipyridine (6), 4,4′-di(hydroxymethyl)-2,2′-bipyridine (7) and 4,4′-dibiotin ester-2,2′-bipyridine (8)) (Fig. 2, right). This reduction reaction was successfully performed for complexes 1–3 in THF in a one-step high yield procedure, using sodium borohydride as reduction agent (Scheme S2†). Yet, the reduction reaction of complex 4 did not lead to the expected hydroxymethyl analogue and the main product of the reaction contained the methylated-bipyridyl counterpart (confirmed by NMR and single crystal X-ray diffraction). Thus, under the reduction reaction conditions, the bipy-biotin ester bonds were found to be very sensitive and ended by being cleaved. Therefore, another approach was to reduce the formyl group at the precursor prior to the coordination of the biotinylated ligand, since the presence of three equally reactive primary alcohol groups at compound 7 prevented the use of the previously proposed ‘chemistry-on-the-complex’ approach. Unfortunately, all purification attempts of complex 8 were unsuccessful and therefore the characterization and, consequently, biological activity of complex 8 was not evaluated in this study. Thus, the seven new compounds were fully analyzed by 1H, 13C and 31P-NMR, FTIR and UV-vis spectroscopies, ESI-MS and their purity confirmed by elemental analysis. In addition, single crystals of six compounds (1, 2, 3, 5, 6 and 7) were successfully obtained and studied using single-crystal X-ray crystallography.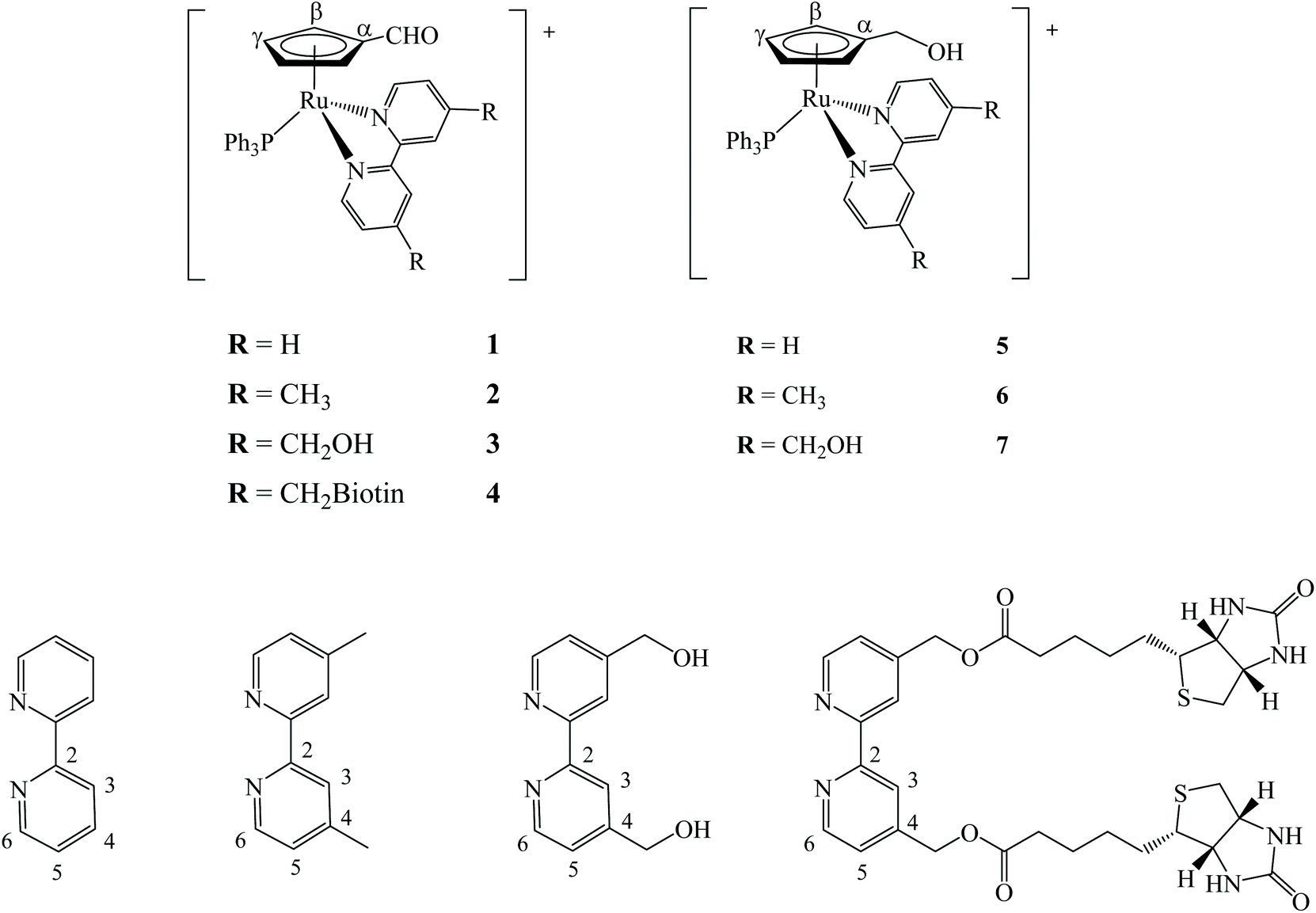 | ||
| Fig. 2 Chemical structures of compounds 1–8. All compounds were isolated as CF3SO3− salts. | ||
1H, 13C{1H} and 31P{1H} NMR characterization of the new ruthenium-based compounds fully supported the proposed formulations (see ESI†). 2D NMR techniques (COSY, HMQC and HMBC) were used to assist on the assignment of all signals. Overall, analysis of the 1H NMR spectra shows that Sigma coordination of the bipyridyl derivatives to the ruthenium-cyclopentadienyl scaffold induces a common deshielding effect of the monosubstituted η5-cyclopentadienyl protons along with a deshielding of H6 (ΔH6 up to 0.82 ppm) and a shielding of H3 (ΔH3 up to −0.41 ppm) bipyridyl protons, as it was reported for related ruthenium cationic compounds.29 Additionally, other resonances in the aromatic region (6.95 ppm < δ < 7.42 ppm) were ascribed to the aromatic ortho, meta and para protons of the triphenylphosphane co-ligand. As expected, compounds 5, 6 and 7 exhibit shielded resonances for the hydroxymethylcyclopentadienyl protons when compared to their formylcyclopentadienyl analogues, which is in good agreement with the electron donating character of the newly introduced pendant group. Detailed spectroscopic data concerning APT-13C{1H} and 31P{1H}-NMR experiments are included in the Experimental section and are in accordance with the effects discussed in this 1H NMR analysis.
The solid state FTIR of the complexes (KBr pellets) of the novel organometallic ruthenium-cyclopentadienyl compounds 1–7 shows the bands for the νC–H stretching vibration of the phosphane, cyclopentadienyl and bipyridyl ligands in the range 3100–3050 cm−1 and bands for νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ranging from 1489 to 1390 cm−1. In addition, the presence of the triflate counter-ion was observed at 1250–1260 cm−1. Characteristic stretching vibrations of the formyl and hydroxymethyl appended groups were also found at the expected region at ∼1670 cm−1 and ∼3420 cm−1, respectively.
C ranging from 1489 to 1390 cm−1. In addition, the presence of the triflate counter-ion was observed at 1250–1260 cm−1. Characteristic stretching vibrations of the formyl and hydroxymethyl appended groups were also found at the expected region at ∼1670 cm−1 and ∼3420 cm−1, respectively.
Optical absorption spectra of complexes 1–7 were recorded at room temperature using ∼10−4 to ∼10−6 M solutions in dichloromethane and dimethylsulfoxide. Table S1† presents molar absorptivity coefficient (ε) values and the correspondent wavelength (λmax) for the bands observed. Fig. 3A shows the spectra of compounds 1 and 5 which are representative of the behavior of formyl- or hydroxymethylcyclopentadienyl series of compounds (which exhibit very similar spectral features), whereas Fig. 3B illustrates the solvatochromic phenomenon observed for compound 3. The same trend is observed in the electronic spectra of all complexes with two very intense absorption bands at high energy values (λ = 240–330 nm) that are ascribed to electronic transitions occurring in the organometallic fragment ({[Ru(η5-C5H4R)((PPh3)]+} R = CHO or CH2OH) and the coordinated ligands. In the visible range, one or two medium-strength absorption bands assigned to metal-to-ligand charge transfer transitions (MLCT) from Ru 4d orbitals to the π* orbitals of the phosphane and bipyridyl ligands (λ = 330–550 nm), as previously reported for related compounds.25,30 The charge transfer character of these bands is corroborated by solvatochromic studies in DMSO where a clear blue-shift is observed with the increase of polarity of the solvent for compound 3 (379 nm in CH2Cl2vs. 366 nm in DMSO).
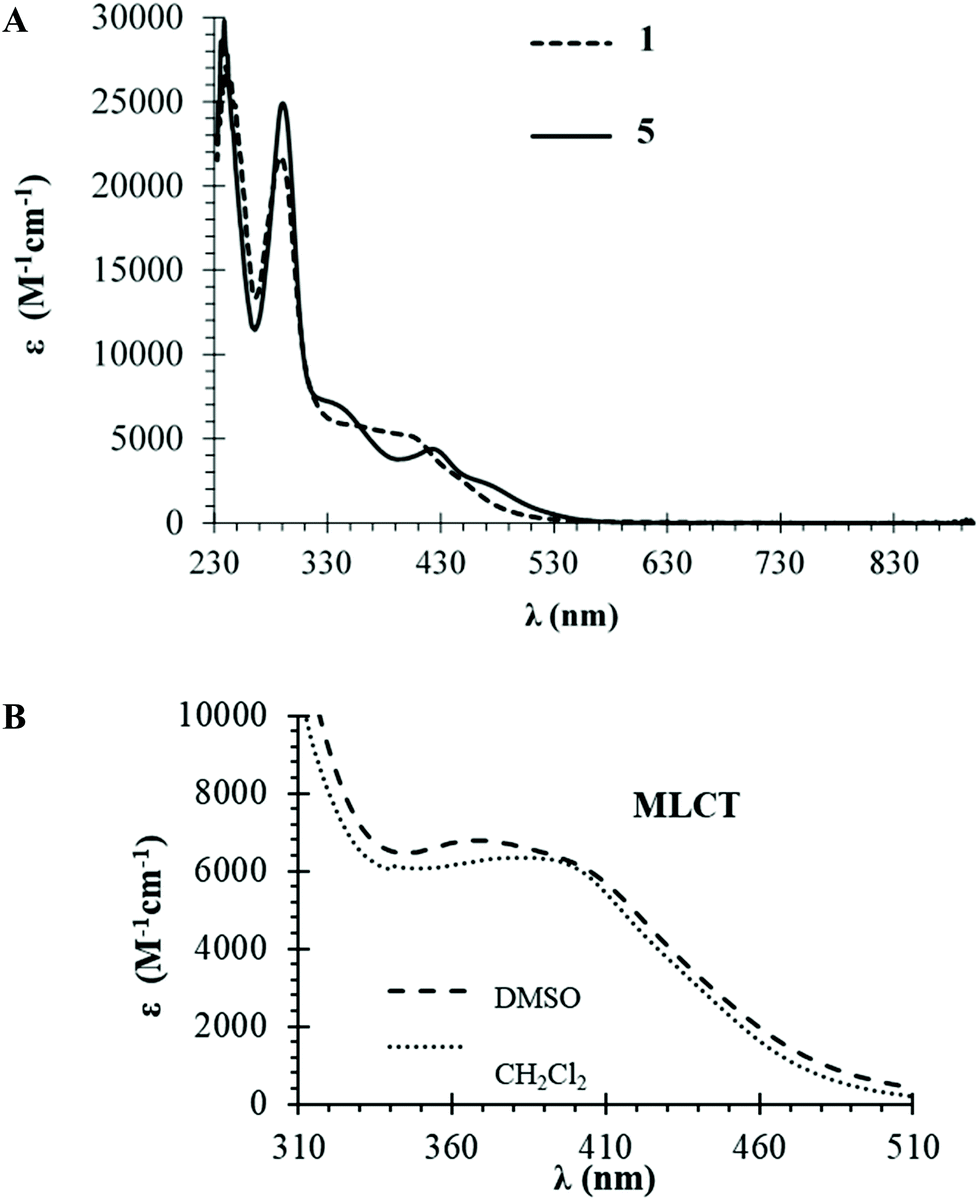 | ||
| Fig. 3 Electronic absorption spectra: (A) UV-visible spectrum in dichloromethane for compounds 1 (––) and 5 (….) as representative of each series of compounds; (B) solvatochromic effect for compound 3. | ||
The crystal structures of complexes 1–3 and 5–7 have been solved by X-ray diffraction analysis. Fig. 4 displays the ORTEP diagrams of their molecular structures whereas the main crystallographic data and selected bond distances and angles can be found in the ESI† section (Tables S2–S3†). All compounds crystallize in the monoclinic system, space group C12/c1 (1), P121/c1 (3, 7) or P121/n (5, 6) with exception of compound 2 which crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In all cases the corresponding unit cells of the complexes display two enantiomers in the racemic crystal.
. In all cases the corresponding unit cells of the complexes display two enantiomers in the racemic crystal.
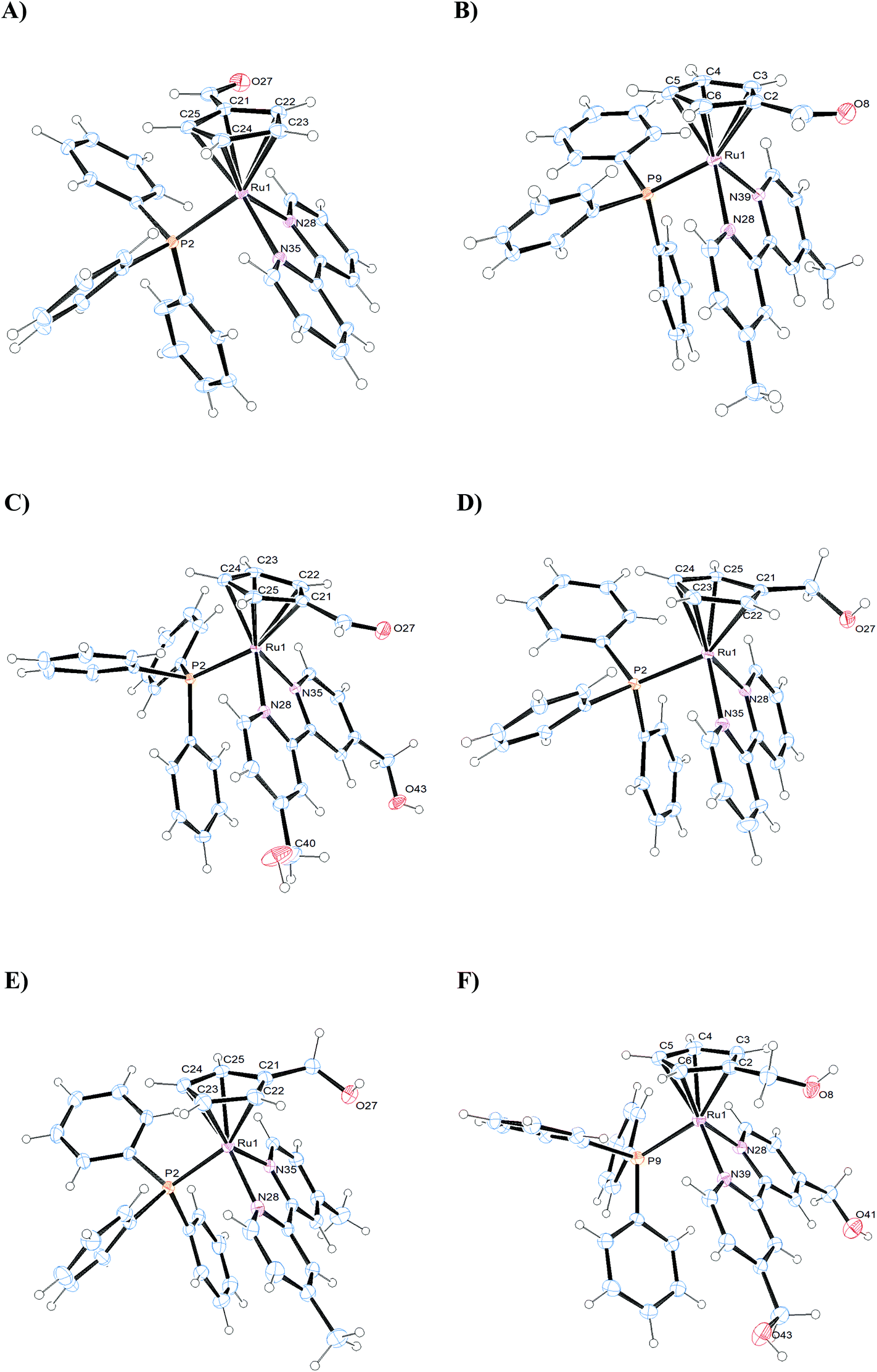 | ||
| Fig. 4 ORTEP plots and labeling schemes for the cations of complexes 1–3 (A–C) and 5–7 (D–F). | ||
All complexes adopt the classical pseudo-octahedral three-legged piano stool geometry and, hence, the cyclopentadienyl arene rings exhibit the usual π-bonded η5 coordination mode, whereas the bipyridyl ligands are coordinated in a bidentate fashion. The sixth coordination site is occupied by the triphenylphosphane ligands. The distances between Ru and the centroids of the cyclopentadienyl moiety in all the complexes, are within a narrow interval (1.828–1.841 Å), similar to other cyclopentadienyl ruthenium complexes described in the literature.29
The Ru–P bond distances in the complexes are all in the same range (2.3086(11)–2.3351(3) Å), similar to the Ru–Nbipy bond distances, which are in the same order for all the compounds (2.0685(11)–2.1112(3) Å). The Ru–C distances are in general higher than Ru–Nbipy, probably due to the σ-donor character of the cyclopentadienyl rings. Moreover, the distances between Ru and the monosubstituted carbon on the η5-cyclopentadienyl ring in complexes 1–3 (Ru–C21 = 2.1901(13) 1; Ru–C2 = 2.212(3) 2; Ru–C21 = 2.1820(17) 3 Å) are in general slightly shorter than in complexes 5–7 (Ru–C21 = 2.230(2) 5; Ru–C2 = 2.213(3) 6; Ru–C21 = 2.235(3) 7 Å). This behavior could be due to the presence of the –CH2OH substituent onto the arene ring in the latter, which presents an electron-withdrawing character a lower than that of the –CHO substituent. In the case of complex 1 (see Fig. S29†) an intramolecular hydrogen bond is formed between one pyridyl H atom and the O atom from the substituent –CHO (H-bond distances are H29–O27 = 2.580 Å), since the O atom of –CHO is orientated toward the bipyridine ligand.
The angles N–Ru–N and C–Ru–C in the complexes, show the geometrical restrictions imposed by the bipyridyl and cyclopentadienyl ligands, and the values found are similar to other compounds described in the literature.25,29
It is worth mentioning the packing structure of the compounds, where additional intermolecular hydrogen bonds can be observed between oxygen atoms of the triflate anions and the hydrogen atoms of the triphenylphosphane (1) or the bipyridine ligands (2, 3); in addition, intermolecular hydrogen bonds are displayed between the oxygen atom of the –CH2OH substituent and the hydrogen atoms of the bipyridine ligand of a neighboring molecule (5, 6, 7). In the case of 7 additional intermolecular hydrogen bonds are observed between O atom of the triflate anions and H atoms of the bipyridine ligand (Fig. S30†).
Stability in organic and aqueous media
All compounds were evaluated over time by monitoring their UV-Vis profile in 100% DMSO and 2–3% DMSO/DMEM in order infer about their stability in conditions mimicking the biological assays. These studies were performed during 24 h at room temperature. UV-Vis absorption spectra of the complexes in cellular media exhibit one strong absorption band in the UV range and two broad absorption bands in the visible range, similarly to their correspondent spectra in organic solvents (Fig. S31†). No significant changes in the absorption spectra were observed over time, indicating adequate stability of all complexes for the biological assays. Additionally, we evaluated the behavior of compounds 1–4 by NMR to check the stability of the formyl substituent. The 1H NMR spectra measured at 1 h, 2 h, 4 h and 24 h in 65% DMEM/35% DMSO (Fig. S32†) confirmed the stability of the formyl substituent over time.Biological evaluation of the compounds
In order to access the biological activity of the new compounds, four non-small cell lung cancer cell lines (A549, NCI-H228, Calu-3, NCI-H1975 cells) with different expression levels of P-gp and MRP1 transporters were used (Fig. S33†), i.e. the two main transporters involved in cisplatin efflux.31 Cells were incubated with complexes 1–7 and cisplatin and cell viability was assessed after a 72 h period and their cytotoxicity (IC50 values) was determined using the WST-1 assay.Analyzing the different IC50 obtained for cisplatin (Table 1), we observed that the higher the level of MRP1 in the cells analyzed (A549 and NCI-H228), the greater their resistance to cisplatin (i.e., a higher IC50 value is obtained), suggesting that MRP1 plays a key role in the efflux of cisplatin in our cellular models.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Cisplatin | |
|---|---|---|---|---|---|---|---|---|
| Viability of cells measured after 72 h incubation with increasing concentrations (0–100 μM) of each compound, measured with a spectrophotometric assay. Data are means ± SD (n = 4). | ||||||||
| A549 | 10.8 ± 1.3 | 12.4 ± 3.6 | >100 | 15.4 ± 2.6 | >100 | 12.5 ± 2.1 | >100 | >100 |
| NCI-H228 | 4.3 ± 0.7 | 3.8 ± 1.4 | >100 | 16.5 ± 1.3 | >100 | 7.8 ± 1.2 | >100 | >100 |
| Calu-3 | 24.7 ± 4.1 | 4.9 ± 1.6 | >100 | 28.9 ± 0.8 | >100 | 5.9 ± 1.2 | >100 | 63.4 ± 8.7 |
| NCI-H1975 | 91.8 ± 10.4 | >100 | >100 | >100 | >100 | >100 | >100 | 3.8 ± 1.1 |
Based on the IC50 values obtained, the resistance to cisplatin varies following the rank order: A549 > NCI-H228 > Calu-3 > NCI-H1975 cells.
The cytotoxicity of cisplatin, commonly evaluated with the MTT assay, often results in lower IC50 values in NSCLC cells32 than those measured in the present work with the WST-1 assay. The different methods as well as the incubation time of the dye in the MTT assay (ranging from 1 to 4 h in different experimental works) can explain such discrepancies.
In a first experimental set, we evaluated the cell viability of the cell lines in the presence of increasing concentrations of the new ruthenium compounds (Table 1).
According to the IC50 values obtained for the Ru compounds it can be noticed that four compounds – 1, 2, 4 and 6 – were more active. For the panel of compounds tested herein, the presence of the substituent –CH2OH seems to be detrimental for the activity of the overall compound, whether placed in the cyclopentadienyl ring (in 5 and 7) or in the bipyridine ligand (in 3). The formyl substituent in the cyclopentadienyl ring seems advantageous, with compounds 1, 2 and 4 being active. The methyl substituent in the bipyridine co-ligand (complexes 2 and 6) is, in this panel of complexes, highly beneficial for the activity, rendering active complexes. Indeed, compounds 1, 2, 4 and 6, were stronger killers of A549 and NCI-H228 cells, i.e. the wo cell lines showing the highest resistance to cisplatin among those analyzed, suggesting that they could be inducers of collateral sensitivity of drug resistant cells.
This is not the first time that metal-based compounds are reported to exert a selective cytotoxicity against drug resistant cells: for instance, cobalt-based complexes15 and ferrocene-based compounds33 overcome resistance to taxol-resistant tumors. Ruthenium-based compounds have been described as potent anti-cancer drugs,34–37 but their potential as collateral sensitivity inducers and/or MDR-reversing agents remains poorly known.
To verify if the compounds can improve the sensitivity to cisplatin, in particular in the cell lines with the highest degree of resistance to this drug, we incubated cells with increasing concentrations of cisplatin together with 1, 2, 4 and 6 at 1 μM, a concentration equivalent to IC25 of the Ru compounds for all cell lines. Working at these poorly toxic concentrations, it is indeed easier to evaluate the effect of the compounds as enhancers of cisplatin toxicity, not as cytotoxic drugs. The ratio IC50(cisplatin)/IC50(cisplatin + Ru compound), i.e. the Resistance factor (Rf) reported in Table 2, was considered an index of sensitization toward cisplatin38 and indicates how many folds the compound enhances the efficacy of cisplatin.
| 1 | 2 | 4 | 6 | |
|---|---|---|---|---|
| Viability of cells measured after 72 h incubation with increasing concentrations (0–100 μM) of cisplatin, co-incubated with 1 μM of each compound, measured with a spectrophotometric assay. Values are data from Rf = IC50(cisplatin)/IC50(cisplatin + Ru compound) (n = 4). | ||||
| A549 | 71.4 | 555.6 | 1250 | 243.9 |
| NCI-H228 | 333.3 | 1389.9 | 588.2 | 344.8 |
| Calu-3 | 26.3 | 126 | 33.2 | 78.7 |
| NCI-H1975 | 2.9 | 0.6 | 1.6 | 0.7 |
All the compounds strongly sensitized A549 and NCI-H228 cell lines, showing a moderate sensitization effect in the mild cisplatin-resistant Calu-3 cells, and losing their effect on the cisplatin-sensitive NCI-H1975 cells.
Of note, A549 and NCI-H228 cells were the cell lines with the highest expression of MRP1, whereas in Calu-3 the most abundant transporter was P-gp. Thus, we decided to investigate if the Ru compounds can directly modulate the activity of these transporters by measuring the ATPase activity of cell extracts after incubation with the compounds (Fig. 5). This assay indicates if the compounds, once entered within the cells, bind the ABC transporters and interfere with their catalytic activity that is based on the hydrolysis of two ATPs associated to the efflux of the drug. Therein, a reduction in the ATPase activity of MRP1 or P-gp may suggest that the compounds inhibit the efflux activity of these transporters, e.g. by acting as inhibitors, substrates or negative allosteric modulators, as previously reported.39
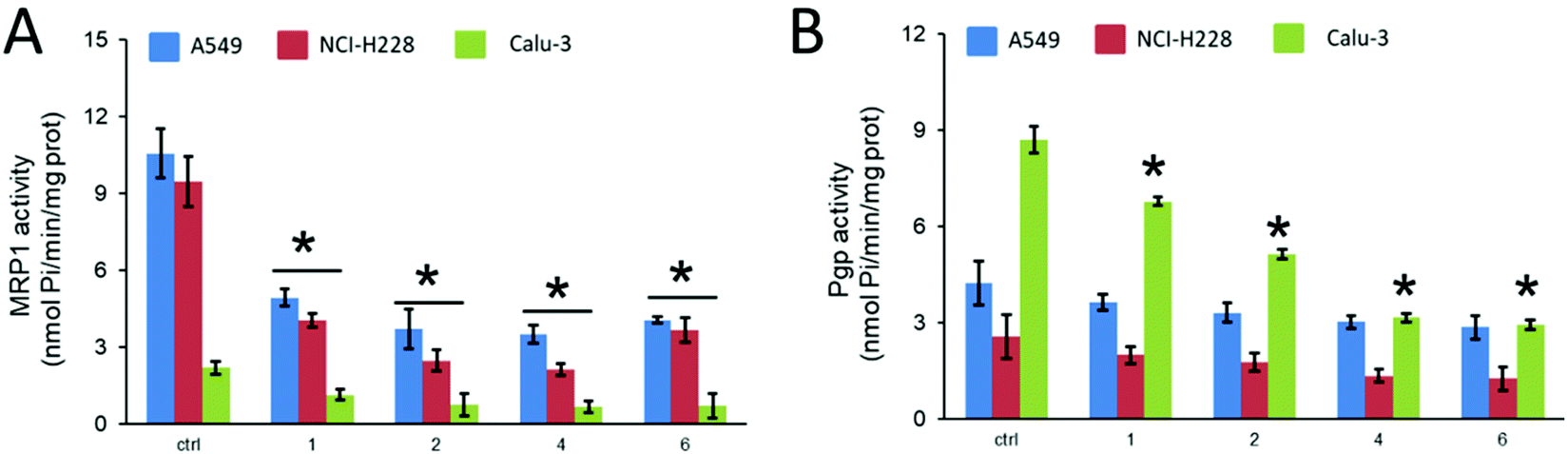 | ||
| Fig. 5 MRP1 (A) and P-gp (B) ATPase activity, measured spectrophotometrically on the proteins immune-purified from cells treated without (ctrl) or with 1 μM of compounds 1, 2, 4 and 6 for 24 h. Data are means ± SD (n = 3). *p < 0.05: vs. ctrl. | ||
MRP1 and P-gp activities differed between the cell lines, in agreement with the expression levels of the protein, as observed for the controls. Interestingly, all Ru compounds inhibited MRP1 ATPase activity (Fig. 5A). They also inhibited P-gp activity in Calu-3 cells, that expresses P-gp to a greater extent (Fig. 5B). We could not detect any significant decrease of P-gp ATPase activity in A549 and NCI-H228 cells, most likely as a consequence of the lower amount of P-gp expressed. From the four complexes tested, compounds 4 and 6 seem to be the most efficient in inhibiting P-gp activity in Calu-3 cell extracts.
These results provided evidence that the selected ruthenium compounds increased the sensitivity to cisplatin in the resistant cell lines by directly impairing the catalytic activity of the main drug efflux transporters expressed the panel of lung cancer cell lines chosen. We are now investigating if this inhibition is exerted on other ABC transporters and by which molecular mechanisms. Considering our previous results on anti-cancer ruthenium cyclopentadienyl compounds, it seems that a trend is gaining some consistency: concerning their cytotoxicity, hydroxylated [Ru(η5-C5H4R)(PPh3)(bipy)]+ (R = H, CH3, CHO or CH2OH) based compounds are less active or inactive in any of the cell lines tested (lung, breast or ovarian). This could possibly be related to their behavior as substrates of ABC pumps as we have previously observed for [Ru(η5-C5H5)(4,4′-di(hydroxymethyl)-2,2′-bipyridine)(PPh3)]+ (pmc79) and [Ru(η5-C5H4CH3)(4,4′-di(hydroxymethyl)-2,2′-bipyridine)(PPh3)]+ (RT12) compounds that were possibly P-gp substrates.25,28RT12 was also a poor substrate for MRP1, MRP2 and BCRP. On the contrary, [Ru(η5-C5H5)(4,4′-dibiotin ester-2,2′-bipyridine)(PPh3)]+ (LCR134) and [Ru(η5-C5H4CH3)(4,4′-dimethyl-2,2′-bipyridine)(PPh3)]+ (RT11) were found to be P-gp and MRP1/MRP2 inhibitors, respectively.25,28 We will now focus on understanding how these structural changes influence the ability of the compounds to act as inhibitors at the molecular level.
Conclusions
Two sets of new piano-stool compounds with the general formula [Ru(η5-C5H4CHO)(bipyR)(PPh3)][CF3SO3] and [Ru(η5-C5H4CH2OH)(bipyR)(PPh3)][CF3SO3], where bipyR = 2,2′-bipyridine (1, 5), 4,4′-dimethyl-2,2′-bipyridine (2, 6), 4,4′-di(hydroxymethyl)-2,2′-bipyridine (3, 7) and 4,4′-dibiotin ester-2,2′-bipyridine (4), have been synthesized and completely characterized by analytical and spectroscopic techniques. In addition, the molecular structure of compounds 1–3 and 5–7 was unveiled by X-ray diffraction studies. All compounds were evaluated as prospective chemotherapeutics for lung cancer, and their activity assessed in a panel of different human lung cells. From this set of compounds, four of them (1, 2, 4 and 6) have shown strong activity against cisplatin-resistant non-small cell lung cancer cell lines, disclosing them as highly promising agents for the treatment of lung cancer.These four complexes were further tested for their ability to sensitize cisplatin-resistant cells. Complexes 1, 2, 4 and 6 increased cisplatin cytotoxicity up to 1390-fold by inhibiting MRP1 and P-gp transporters. This is of particular interest since, besides exerting an important role in cisplatin resistance,40 MRP1 has been proposed as a potential mediator of collateral sensitivity.41–43 Few compounds targeting MRP1 are inducers of collateral sensitivity44 but the research in this field is in great expansion. In lung cancer, where cisplatin is the first-line treatment and MRP1 is one of the main responsible for resistance to this drug,45 finding MRP1-targeting agents, inducing collateral sensitivity is absolutely preeminent. In this context, complexes 1, 2, 4 and 6 emerge as valuable prospective agents for lung cancer chemotherapy, specially compounds 4 and 6, both quite active as cytotoxic agents and able to inhibit both MRP1 and P-gp, suggesting an important ability to overcome drug resistance mechanisms.
Methods
Materials
All chemicals were purchased from commercial sources and used without further purification (unless otherwise stated).Instrumentation and methods (Experimental section)
Synthesis of the ruthenium complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) was refluxed for 3 h. After that period, the reaction mixture was cooled to room temperature and concentrated to dryness and the aqueous phase discarded. The brown residue was washed with water (5 mL × 2), light petroleum ether (5 mL × 3), a mixture of light petroleum ether/diethyl ether (v/v = 3/3 × 3) and hexane (5 mL × 3). The experimental data obtained is in agreement with the previous literature.
:2, 15 mL) and filtered. The filtrate was concentrated to dryness and the residues obtained were washed with n-hexane (15 mL × 3) and recrystallized from acetone/n-hexane (1 and 2) or dichloromethane/ether (3), to give dark orange-brown crystalline solids.
:3) was refluxed for 3 h. After that period, the reaction mixture was cooled to room temperature and concentrated to dryness and the aqueous phase discarded. The brown residue was washed with water (5 mL × 2), light petroleum ether (5 mL × 3), a mixture of light petroleum ether/diethyl ether (v/v = 3/3 × 3) and hexane (5 mL × 3). The experimental data obtained is in agreement with the previous literature.
:2, 15 mL) and filtered. The filtrate was concentrated to dryness and the residues obtained were washed with n-hexane (15 mL × 3) and recrystallized from acetone/n-hexane (1 and 2) or dichloromethane/ether (3), to give dark orange-brown crystalline solids.
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.35 (broad, 2H, H6), 9.24 (s, 1H, (η5-C5H4C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) O), 8.20 (d, 2H, 3JHH = 8.0, 3), 7.97 (t, 2H, 3JHH = 7.6, 4), 7.46 (m, 5H, pPPh3 + 5), 7.36 (m, 6H, mPPh3), 7.12 (t, 6H, 3JHH = 8.4, oPPh3), 5.80 (broad, 2H, β-η5-C5H4CHO), 4.93 (broad, 2H, γ-η5-C5H4CHO).
O), 8.20 (d, 2H, 3JHH = 8.0, 3), 7.97 (t, 2H, 3JHH = 7.6, 4), 7.46 (m, 5H, pPPh3 + 5), 7.36 (m, 6H, mPPh3), 7.12 (t, 6H, 3JHH = 8.4, oPPh3), 5.80 (broad, 2H, β-η5-C5H4CHO), 4.93 (broad, 2H, γ-η5-C5H4CHO).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 189.1 (η5-C5H4![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HO), 156.8 (6), 138.1 (4), 133.9 (d, 2JCP = 11, HoPPh3), 131.8 (d, 1JCP = 43.8, qPPh3), 131.4 (d, 4JCP = 1.9, HpPPh3), 129.5 (d, 3JCP = 9.8, HmPPh3), 126.7 (5), 124.4 (3), 100.8 (α-η5-C5H4CHO), 85.2 (β-η5-C5H4CHO), 79.1 (γ-η5-C5H4CHO).
HO), 156.8 (6), 138.1 (4), 133.9 (d, 2JCP = 11, HoPPh3), 131.8 (d, 1JCP = 43.8, qPPh3), 131.4 (d, 4JCP = 1.9, HpPPh3), 129.5 (d, 3JCP = 9.8, HmPPh3), 126.7 (5), 124.4 (3), 100.8 (α-η5-C5H4CHO), 85.2 (β-η5-C5H4CHO), 79.1 (γ-η5-C5H4CHO).
31P{1H} NMR [(CD3)2CO, δ/ppm]: 48.81 (s, PPh3).
FTIR [KBr, cm−1]: 3105–3055 (νC–H aromatic rings), 1667 (νCO, η5-C5H4CHO), 1433 (νCC aromatic rings), 1262 (νCF3SO3 counter ion).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 289 (24.49), 362 (6.46), 415 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 289 (21.71), 357 (Sh), 409 (Sh), 457 (Sh).
Elemental analysis calc. for C35H28F3N2O4PRuS (761.71): C, 55.2, H, 3.7; N, 3.7; S, 4.2. Found: C, 54.9; H, 3.5; N, 3.6; S, 4.3.
ESI-MS: [1-CF3SO3]+ calc. for [C34H28N2OPRu]+: 613.10 found: 613.05.
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.25 (s, 1H, (η5-C5H4CO)), 9.16 (broad, 2H, 6), 8.04 (s, 2H, 3), 7.47 (m, 3H, pPPh3), 7.36 (m, 6H, mPPh3), 7.30 (br s, 2H, 5), 7.12 (t, 6H, 3JHH = 8.4, oPPh3), 5.75 (broad, 2H, β-(η5-C5H4CHO), 4.85 (broad, 2H, γ-(η5-C5H4CHO), 2.48 (s, 6H, C3).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 189.0 (η5-C5H4CO), 156.5 (2), 156.0 (6), 150.5 (4), 134.0 (d, 2JCP = 11, HoPPh3), 132.2 (d, 1JCP = 43, qPPh3), 131.3 (d, 4JCP = 2, HpPPh3), 129.5 (d, 3JCP = 9, HmPPh3), 127.7 (5), 125.1 (3), 103.0 (α-η5-C5H4CHO), 84.8 (β-η5-C5H4CHO), 78.6 (γ-η5-C5H4CHO), 20.8 (H3).
31P{1H} NMR [(CD3)2CO, δ/ppm]: 48.94 ppm (s, ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) Ph3).
Ph3).
FTIR [KBr, cm−1]: 3071–3050 cm−1 (νC–H aromatic rings), 2922 cm−1 (νC–H alkanes), 1670 (νCO, η5-C5H4CHO), 1440 (νCC aromatic rings), 1258 cm−1 (νCF3SO3 counterion).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 289 (26.27), 371 (7.21), 412 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 290 (26.10), 366 (6.91), 412 (Sh).
Elemental analysis calc. for C37H32F3N2O4PRuS (789.76): C, 56.3, H, 4.1; N, 3.6; S, 4.1. Found: C, 54.8; H, 3.9; N, 3.4; S, 4.0.
ESI-MS: [2-CF3SO3]+ calc. for [C36H32N2OPRu]+: 641.13 found: 641.02.
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.26 (m, 3H, (η5-C5H4CHO) + 6), 8.09 (s, 2H, 3), 7.43 (m, 5H, pPPh3 + 5), 7.35 (m, 6H, mPPh3), 7.12 (t, 6H, 3JHH = 8, oPPh3), 5.78 (broad, 2H, β-η5-C5H4CHO), 4.87–4.80 (m, 8H, γ-η5-C5H4CHO + C2OH + CH2O).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 189.0 (η5-C5H4HO), 156.5 (2), 156.2 (6), 154.9 (4), 134.0 (d, 2JCP = 11, HoPPh3), 132.1 (d, 1JCP = 44, qPPh3), 131.3 (d, 4JCP = 2, HpPPh3), 129.5 (d, 3JCP = 10, HmPPh3), 123.9 (5), 121.3 (3), 103.0 (α-η5-C5H4CHO), 89.4 (β-η5-C5H4CHO), 78.7 (γ-η5-C5H4CHO), 62.4 (H2OH).
31P NMR [(CD3)2CO, δ/ppm]: 48.94 (s, Ph3).
FTIR [KBr, cm−1]: 3464 (νO–H), 3084–2916 (νC–H aromatic rings); 2884–2857 (νC–H alkanes), 1678 (νCO, η5-C5H4CHO), 1440 (νCC aromatic rings), 1246 cm−1 (νCF3SO3 counter ion), 1221 cm−1 (νC–O).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 290 (27.15), 366 (7.27), 416 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 277 (Sh), 289 (28.43), 379 (7.43), 451 (Sh).
Elemental analysis calc. for C37H32F3N2O6PRuS (821.76): C, 54.1; H, 3.9; N, 3.4; S, 3.9. Found: C, 54.1; H, 3.9; N, 3.4; S, 3.9.
ESI-MS: [3-CF3SO3]+ calc. for [C36H32N2O3PRu]+: 673.12 found: 673.02.
Yield: 88% (135 mg).
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.35 (broad, 2H, 6), 9.26 (s, 1H, η5-C5H4CO), 8.15 (s, 2H, 3), 7.48 (m, 3H, 3JHH = 7.6, 4), 7.46 (m, 5H, pPPh3 + 5), 7.36 (m, 6H, mPPh3), 7.12 (t, 6H, 3JHH = 8, oPPh3), 6.11 (broad s, 2H, NH) 5.90 (d, 2H, NH), 5.83 (broad, 2H, β-η5-C5H4CHO), 5.28 (m, 4H, bipy-C2Biotin), 4.93 (broad, 2H, γ-η5-C5H4CHO), 4.50 (SCH2-CBiotin), 4.32 (m, 2H, CBiotin), 3.22 (m, 2H, S-CBiotin), 2.94 (m, 2H, SC2Biotin), 2.67 (m, 2H, H), 2.52 + 1.75–1.49 (3× m, 16H, C2C2C2C2Biotin).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 189.3 (η5-C5H4HO), 173.5 (O, bipy-Biotin ester), 164.1 (CO, bipy-Biotin, urea), 157.1 (C1), 156.6 (C2), 148.7 (C4), 133.9 (2JCP = 11, Cortho-PPh3), 131.9 (1JCP = 40, Cq, PPh3), 131.5 (4JCP = 1, Cpara-PPh3), 129.6 (3JCP = 10, Cmeta-PPh3), 123.9 (d, 4JCP = 6, 5), 122.6 (d, 4JCP = 8, 3), 107.5 (α-η5-C5H4CHO), 85.1 (2 signals, s, Cβ-η5C5H4CHO), 79.1 (2 signals, s, Cγ-η5-C5H4CHO), 64.0 (2 signals, s, bipy-H2Biotin), 62.6 (2 signals, s, HBiotin), 60.9 (2 signals, s, SCH2-HBiotin), 56.7 (2 signals, s, SHBiotin), 41.1 (2 signals, s, SH2Biotin), 34.3 + 29.2* + 25.6 (sets of 2 signals, H2H2H2H2Biotin).
31P NMR [(CD3)2CO, δ/ppm]: 48.70 (s, Ph3).
FTIR [KBr, cm−1]: 3073 (νC–H aromatic rings), 2928–2859 (νC–H alkanes), 1736–1620 (νCO η5-C5H4CHO, urea, ester), 1433 (νCC aromatic rings), 1260 cm−1 (νCF3SO3 counter ion).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 293 (47.69), 358 (11.94), 414 (Sh) 468 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 291 (12.59), 345 (Sh), 416 (2.39), 479 (Sh).
Elemental analysis calc. for C57H60F3N6O10PRuS3 (1274.35): C, 53.7; H, 4.8; N, 6.6; S, 7.6. Found: C, 52.8; H, 5.2; N, 6.7; S, 7.0.
ESI-MS: [4-CF3SO3]+ calc. for [C56H60N6O7PRuS2]+: 1125.27 found: 1125.09.
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.49 (broad, 2H, 6), 8.17 (d, 2H, 3JHH = 8, 3), 7.88 (t, 2H, 3JHH = 8, 4), 7.41–7.32 (m, 11H, pPPh3 + mPPh3 + 5), 7.11 (t, 6H, 3JHH = 8, oPPh3), 4. 94 (broad, 2H, β-η5-C5H4CH2OH), 4.62 (broad, 2H, γ-η5-C5H4CH2OH), 4.11 (s, 3H, η5-C5H4C2OH + η5-C5H4CH2O).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 157.0 (d, JCP = 2, 6), 156.7 (2), 136.9 (4), 133.8 (d, 2JCP = 11, HoPPh3), 132.6 (d, 1JCP = 41, qPPh3), 130.9 (d, 4JCP = 2, HpPPh3), 129.3 (d, 3JCP = 10, HmPPh3), 126.1 (5), 124.1 (3), 104.9 (α-η5-C5H4CH2OH), 76.7 + 76.5 (β-η5-C5H4CH2OH + γ-η5-C5H4CH2OH), 57.7 (η5-C5H4H2OH).
31P NMR [(CD3)2CO, δ/ppm]: 51.25 (s, Ph3).
FTIR [KBr, cm−1]: 3412 (νO−H), 3075–3055 (νC–H aromatic rings), 2962–2853 (νC–H alkanes), 1435 (νCC aromatic rings), 1256 (νCF3SO3 counter ion).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 292 (16.59), 344 (Sh), 411 (2.93), 483 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 291 (24.89), 348 (Sh), 423 (4.38), 486 (Sh).
Elemental analysis calc. for C35H30F3N2O4PRuS (763.73): C, 55.0; H, 4.0; N, 3.7; S, 4.2. Found: C, 53.6; H, 3.9; N, 3.5; S, 4.2.
ESI-MS: [5-CF3SO3]+ calc. for [C34H30N2OPRu]+: 615.11 found: 615.02.
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.29 (broad, 2H, 6), 8.01 (s, 2H, 3), 7.41 (m, 3H, pPPh3), 7.32 (m, 6H, mPPh3), 7.20 (broad, 2H, 5), 7.12 (t, 6H, 3JHH = 8, oPPh3), 4.88 (broad, 2H, β-η5-C5H4CH2OH), 4.56 (broad, 2H, γ-η5-C5H4CH2OH), 4.10 (s, 3H, η5-C5H4C2OH + η5-C5H4CH2O), 2.45 (s, 6H, C3).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 156.4 (2), 156.1 (d, JCP = 2, 6), 149.1 (4), 133.9 (d, 2JCP = 11, HoPPh3), 133.0 (d, 1JCP = 41, qPPh3), 130.8 (d, 4JCP = 2, HpPPh3), 129.6 (d, 3JCP = 9, HmPPh3), 127.1 (5), 124.6 (3), 104.5 (d, 2JCP = 6, αη5-C5H4CH2OH), 76.2 (β-η5-C5H4CH2OH), 76.1 (d, 2JCP = 2, γ-η5-C5H4CH2OH), 57.8 (H2OH), 20.8 (H3).
31P NMR [(CD3)2CO, δ/ppm]: 51.28 [s, PPh3].
FTIR [KBr, cm−1]: 3415 (νO−H), 3071–3050 cm−1 (νC–H aromatic rings), 2922 cm−1 (νC–H alkanes), 1440 (νCC aromatic rings), 1258 cm−1 (νCF3SO3 counterion).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 291 (4.11), 351 (Sh), 418 (6.86), 472 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 287 (29.20), 345 (Sh), 417 (5.28), 470 (Sh).
Elemental analysis calc. for C37H34F3N2O4PRuS (791.78): C, 56.1; H, 4.3; N, 3.5; S, 4.1. Found: C, 55.4; H, 4.1; N, 3.3; S, 4.0.
ESI-MS: [6-CF3SO3]+ calc. for [C36H34N2OPRu]+: 643.15 found: 643.00.
1H NMR [(CD3)2CO, Me4Si, δ/ppm]: 9.39 (broad, 2H, 6), 8.07 (s, 2H, 3), 7.41 (m, 3H, pPPh3), 7.32 (m, 8H, mPPh3 + 5), 7.12 (t, 6H, 3JHH = 8, oPPh3), 4.91 (broad, 2H, β-η5-C5H4CH2OH), 4.79 (s, 6H, C2OH + CH2O), 4.60 (broad, 2H, γ-η5-C5H4CH2OH), 4.11 (m, 3H, η5-C5H4C2OH + η5-C5H4CH2O).
APT-13C{1H} NMR [(CD3)2CO, δ/ppm]: 156.4 (2), 156.3 (d, 3JCP = 2, 6), 153.7 (4), 133.8 (d, 2JCP = 11, HoPPh3), 132.8 (d, 1JCP = 40, qPPh3), 130.8 (d, 4JCP = 2, HpPPh3), 129.3 (d, 3JCP = 10, HmPPh3), 123.4 (5), 120.9 (3), 104.5 (α-η5-C5H4CH2OH), 76.3 + 76.2 (β-η5-C5H4CH2OH + γ-η5-C5H4CH2OH), 62.5 (H2OH), 57.8 (η5-C5H4H2OH).
31P NMR [(CD3)2CO, δ/ppm]: 51.27 (s, Ph3).
FTIR [KBr, cm−1]: 3486–3470 (νO–H), 2922–2864 (νC–H alkanes), 1440 (νCC aromatic rings), 1261 (νCF3SO3 counter ion), 1234 (νC–O).
UV-vis [DMSO, λmax/nm (ε × 103/M−1 cm−1)]: 292 (5.67), 348 (Sh), 416 (1.02), 478 (Sh).
UV-vis [CH2Cl2, λmax/nm (ε × 103/M−1 cm−1)]: 291 (4.11), 351 (Sh), 418 (6.86), 472 (Sh).
Elemental analysis calc. for C37H34F3N2O6PRuS (823.78): C, 53.9; H, 4.1; N, 3.4; S, 3.9. Found: C, 52.3; H, 4.0; N, 3.1; S, 3.0.
ESI-MS: [7-CF3SO3]+ calc. for [C36H34N2O3PRu]+: 675.14 found: 675.01.
X-ray structure analysis
The X-ray intensity data were measured on a D8 QUEST ECO three-circle diffractometer system equipped with a Ceramic X-ray tube (Mo Kα, λ = 0.71076 Å) and a doubly curved silicon crystal monochromator, using APEX346 software package. The frames were integrated with the Bruker SAINT.47 Data were corrected for absorption effects using the Multi-Scan method (SADABS).48 The structures were solved and refined using the Bruker SHELXTL.49The crystallographic data as well as details of the structure solution and refinement procedures are reported in ESI.† CCDC 2042407–2042412† contain the supplementary crystallographic data for this paper.
Stability studies
For the stability studies, all complexes were dissolved in 100% DMSO and a sample containing each compound in 2% DMSO/DMEM at 90–180 μM was prepared. Their electronic spectra were recorded in the range allowed by the solvent mixture at set time intervals. Samples were stored at room temperature and protected from light between measurements. The variation percentage between measurements were calculated by the following expression: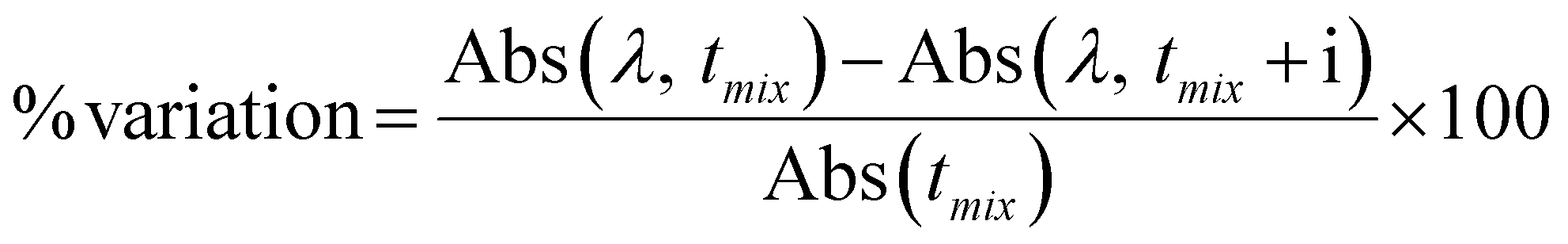
Cell lines
Human non-small cell lung cancer cells A549, NCI-H228, Calu-3, NCI-H1975 were purchased from ATCC (Manassas, VA). Cells were grown in RPMI-1640 medium, supplemented with 10% v/v FBS and 1% penicillin–streptomycin, at 37 °C, 5% CO2, in a humidified atmosphere.Immunoblotting
Cells were rinsed with ice-cold lysis buffer (50 mM Tris, 10 mM EDTA, 1% v/v Triton-X100), supplemented with the protease inhibitor cocktail set III (80 μM aprotinin, 5 mM bestatin, 1.5 mM leupeptin, 1 mM pepstatin; Calbiochem, San Diego, CA), 2 mM phenylmethylsulfonyl fluoride and 1 mM Na3VO4, then sonicated (10 bursts of 1 s, amplitude 40%; Hielscher UP200S, Ultrasound Sonicator, GmbH, Teltow, Germany) and centrifuged at 13000g for 10 min at 4 °C. 20 μg protein extracts were subjected to SDS-PAGE and probed with the following antibodies: anti-P-gp (1:250, rabbit polyclonal #sc-8313, Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-MRP1 (1:500, mouse clone MRPm5, Abcam, Cambridge, UK), anti-β-tubulin (1:1000, mouse clone D10, Santa Cruz Biotechnology Inc.). This procedure was followed by the exposure to a peroxidase-conjugated secondary antibody (Bio-Rad Laboratories). The membranes were washed with Tris-buffered saline-Tween 0.1% v/v solution, and the proteins were detected by enhanced chemiluminescence (Bio-Rad Laboratories).
Cytotoxic activity
Cells were seeded in 96 well plates. In a first experimental set, cells were incubated for 72 h with DMSO as a solvent or cisplatin at the following concentrations: 1 nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM. In a second experimental set, cells were incubated with DMSO or each compound at the following concentrations: 1 nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM. In the third experimental set, cells were incubated for 72 h with 1 μM of the selected compound plus cisplatin at the following concentrations: 1 nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM. Cell viability was evaluated using the WST-1 assay (Sigma-Merck), as per manufacturer's instructions, using a Packard EL340 microplate reader (Bio-Tek Instruments, Winooski, VT). The absorbance units of the untreated cells were considered 100%; the absorbance units of the other experimental conditions were expressed as percentage versus untreated cells. IC50 and IC25 were defined as the concentrations of cisplatin of compound that killed 50% and 25% cells, respectively. The Resistance Factor was the ratio between IC50 of cisplatin alone/IC50 of cisplatin + compound.ATPase activity
The P-gp ATPase activity was measured in membrane vesicles as described previously.39 Cells were washed with Ringer's solution (148.7 mM NaCl, 2.55 mM K2HPO4, 0.45 mM KH2PO4, 1.2 mM MgSO4; pH 7.4), lysed on crushed ice with lysis buffer (10 mM Hepes/Tris, 5 mM EDTA, 5 mM EGTA, 2 mM dithiothreitol; pH 7.4) supplemented with 2 mM phenylmethylsulfonyl fluoride, 1 mM aprotinin, 10 μg ml−1 pepstatin, 10 μg ml−1 leupeptin, and subjected to nitrogen cavitation at 1200 psi for 20 min. Samples were centrifuged at 300g for 10 min in the pre-centrifugation buffer (10 mM Tris/HCl, 25 mM sucrose; pH 7.5), overlaid on a sucrose cushion (10 mM Tris/HCl, 35% w/v sucrose, 1 mM EDTA; pH 7.5) and centrifuged at 14000g for 10 min. The interface was collected, diluted in the centrifugation buffer (10 mM Tris/HCl, 250 mM sucrose; pH 7.5) and subjected to a third centrifugation at 100000g for 45 min. The vesicle pellet was re-suspended in 0.5 ml centrifugation buffer and stored at −80 °C until the use, after the quantification of the protein content. 1 mg of total protein were immunoprecipitated with the anti-P-gp or anti-MRP1 antibody. 100 μg of each immunpurified protein was incubated for 30 min at 37 °C with 50 μl of the reaction mix (25 mM Tris/HCl, 3 mM ATP, 50 mM KCl, 2.5 mM MgSO4, 3 mM dithiothreitol, 0.5 mM EGTA, 2 mM ouabain, 3 mM NaN3; pH 7.0). The reaction was stopped by adding 0.2 ml ice-cold stopping buffer (0.2% w/v ammonium molybdate, 1.3% v/v H2SO4, 0.9% w/v SDS, 2.3% w/v trichloroacetic acid, 1% w/v ascorbic acid). After 30 min incubation at room temperature, the absorbance of the phosphate hydrolyzed from ATP was measured at 620 nm, using a Packard EL340 microplate reader. The absorbance was converted into nmoles hydrolyzed phosphate (Pi) per min per mg proteins, according to the titration curve previously prepared.
Statistical analysis
All data in the text and figures are provided as means ± SD. The results were analyzed by a one-way analysis of variance (ANOVA) and Tukey's test. p < 0.05 was considered significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Portuguese Foundation for Science and Technology (Fundação para a Ciência e Tecnologia, FCT) within the scope of Projects UIDB/00100/2020 (Centro de Química Estrutural) and PTDC/QUI-QIN/28662/2017. R. G. Teixeira thanks FCT for his Ph.D. Grant (SFRH/BD/135830/2018). A. Valente acknowledges the CEECIND 2017 Initiative (CEECIND/01974/2017). C.R. thanks the Associazione Italiana per la Ricerca sul Cancro (AIRC; grant IG21408). This article is based upon work from COST Action 17104 STRATAGEM supported by COST (European Cooperation in Science and Technology; www.cost.eu).References
- A. Aupérin, C. Le Péchoux, E. Rolland, W. J. Curran, K. Furuse, P. Fournel, J. Belderbos, G. Clamon, H. C. Ulutin, R. Paulus, T. Yamanaka, M. C. Bozonnat, A. Uitterhoeve, X. Wang, L. Stewart, R. Arriagada, S. Burdett and J. P. Pignon, Meta-Analysis of Concomitant versus Sequential Radiochemotherapy in Locally Advanced Non - Small-Cell Lung Cancer, J. Clin. Oncol., 2010, 28(13), 2181–2190, DOI:10.1200/JCO.2009.26.2543.
- V. S. Iglesias, L. Giuranno, L. J. Dubois, J. Theys and M. Vooijs, Drug Resistance in Non-Small Cell Lung Cancer: A Potential for NOTCH Targeting?, Front. Oncol., 2018, 8, 267, DOI:10.3389/fonc.2018.00267.
- S. C. Marker, A. P. King, R. V. Swanda, B. Vaughn, E. Boros, S. B. Qian and J. J. Wilson, Exploring Ovarian Cancer Cell Resistance to Rhenium Anticancer Complexes, Angew. Chem., Int. Ed., 2020, 59(32), 13391–13400, DOI:10.1002/anie.202004883.
- C. Licona, J. B. Delhorme, G. Riegel, V. Vidimar, R. Cerón-Camacho, B. Boff, A. Venkatasamy, C. Tomasetto, P. Da Silva Figueiredo Celestino Gomes, D. Rognan, J. N. Freund, R. Le Lagadec, M. Pfeffer, I. Gross, G. Mellitzer and C. Gaiddon, Anticancer Activity of Ruthenium and Osmium Cyclometalated Compounds: Identification of ABCB1 and EGFR as Resistance Mechanisms, Inorg. Chem. Front., 2020, 7(3), 678–688, 10.1039/c9qi01148j.
- I. Landini, A. Lapucci, A. Pratesi, L. Massai, C. Napoli, G. Perrone, P. Pinzani, L. Messori, E. Mini and S. Nobili, Selection and Characterization of a Human Ovarian Cancer Cell Line Resistant to Auranofin, Oncotarget, 2017, 8(56), 96062–96078, DOI:10.18632/oncotarget.21708.
- W. H. Ang, A. De Luca, C. Chapuis-Bernasconi, L. Juillerat-Jeanneret, M. Lo Bello and P. J. Dyson, Organometallic Ruthenium Inhibitors of Glutathione-S-Transferase P1–1 as Anticancer Drugs, ChemMedChem, 2007, 2(12), 1799–1806, DOI:10.1002/cmdc.200700209.
- P. R. Florindo, D. M. Pereira, P. M. Borralho, P. J. Costa, M. F. M. Piedade, C. M. P. Rodrigues and A. C. Fernandes, New [(H5-C5H5)Ru(N-N)(PPh3)][PF6] Compounds: Colon Anticancer Activity and GLUT-Mediated Cellular Uptake of Carbohydrate-Appended Complexes, Dalton Trans., 2016, 45(30), 11926–11930, 10.1039/c6dt01571a.
- H. Peng, H. Jin, H. Zhuo and H. Huang, Enhanced Antitumor Efficacy of Cisplatin for Treating Ovarian Cancer in Vitro and in Vivo via Transferrin Binding, Oncotarget, 2017, 8(28), 45597–45611, DOI:10.18632/oncotarget.17316.
- I. N. Stepanenko, A. Casini, F. Edafe, M. S. Novak, V. B. Arion, P. J. Dyson, M. A. Jakupec and B. K. Keppler, Conjugation of Organoruthenium(Ii) 3-(1h-Benzimidazol-2 Yl)Pyrazolo[3, 4-b]Pyridines and Indolo[3, 2-d]Benzazepines to Recombinant Human Serum Albumin: A Strategy to Enhance Cytotoxicity in Cancer Cells, Inorg. Chem., 2011, 50(24), 12669–12679, DOI:10.1021/ic201801e.
- Y. R. Zheng, K. Suntharalingam, T. C. Johnstone, H. Yoo, W. Lin, J. G. Brooks and S. J. Lippard, Pt(IV) Prodrugs Designed to Bind Non-Covalently to Human Serum Albumin for Drug Delivery, J. Am. Chem. Soc., 2014, 136(24), 8790–8798, DOI:10.1021/ja5038269.
- G. Spengler, A. Kincses, B. Rácz, B. Varga, G. Watanabe, R. Saijo, H. Sekiya, E. Tamai, J. Maki, J. Molnár and M. Kawase, Benzoxazole-Based Zn(II) and Cu(II) Complexes Overcome Multidrug-Resistance in Cancer, Anticancer Res., 2018, 38(11), 6181–6187, DOI:10.21873/anticanres.12971.
- Y. Liu, Z. Zhou, X. Lin, X. Xiong, R. Zhou, M. Zhou and Y. Huang, Enhanced Reactive Oxygen Species Generation by Mitochondria Targeting of Anticancer Drug to Overcome Tumor Multidrug Resistance, Biomacromolecules, 2019, 20(10), 3755–3766, DOI:10.1021/acs.biomac.9b00800.
- R. R. Ye, J. J. Cao, C. P. Tan, L. N. Ji and Z. W. Mao, Valproic Acid-Functionalized Cyclometalated Iridium(III) Complexes as Mitochondria-Targeting Anticancer Agents, Chem. – Eur. J., 2017, 23(60), 15166–15176, DOI:10.1002/chem.201703157.
- W. Guo, Z. Chen, L. Tan, Q. Wu, X. Ren, C. Fu, Y. Du, J. Ren and X. Meng, L-Cysteine, Decorated Nanoscale Metal-Organic Frameworks Delivering Valproic Acid/Cisplatin for Drug-Resistant Lung Cancer Therapy, Chem. Commun., 2020, 56(27), 3919–3922, 10.1039/c9cc09712k.
- B. Y. K. Law, Y. Q. Qu, S. W. F. Mok, H. Liu, W. Zeng, Y. Han, F. Gordillo-Martinez, W. K. Chan, K. M. C. Wong and V. K. W. Wong, New Perspectives of Cobalt Tris(Bipyridine) System: Anti-Cancer Effect and Its Collateral Sensitivity towards Multidrug-Resistant (MDR) Cancers, Oncotarget, 2017, 8(33), 55003–55021, DOI:10.18632/oncotarget.18991.
- A. Ganguly, S. Basu, P. Chakraborty, S. Chatterjee, A. Sarkar, M. Chatterjee and S. K. Choudhuri, Targeting Mitochondrial Cell Death Pathway to Overcome Drug Resistance with a Newly Developed Iron Chelate, PLoS One, 2010, 5(6), e11253, DOI:10.1371/journal.pone.0011253.
- K. Banerjee, M. K. Biswas and S. K. Choudhuri, A Newly Synthesized Nickel Chelate Can Selectively Target and Overcome Multidrug Resistance in Cancer through Redox Imbalance Both in Vivo and in Vitro, J. Biol. Inorg. Chem., 2017, 22(8), 1223–1249, DOI:10.1007/s00775-017-1498-4.
- R. D. Ghosh, P. Chakraborty, K. Banerjee, A. Adhikary, A. Sarkar, M. Chatterjee, T. Das and S. K. Choudhuri, The Molecular Interaction of a Copper Chelate with Human P-Glycoprotein, Mol. Cell. Biochem., 2012, 364(1–2), 309–320, DOI:10.1007/s11010-012-1232-z.
- R. D. Ghosh, S. Das, A. Ganguly, K. Banerjee, P. Chakraborty, A. Sarkar, M. Chatterjee, A. Nanda, K. Pradhan and S. K. Choudhuri, An in Vitro and in Vivo Study of a Novel Zinc Complex, Zinc N-(2-Hydroxyacetophenone)Glycinate to Overcome Multidrug Resistance in Cancer, Dalton Trans., 2011, 40(41), 10873–10884, 10.1039/c1dt10501a.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, NKP-1339, the First Ruthenium-Based Anticancer Drug on the Edge to Clinical Application, Chem. Sci., 2014, 5(8), 2925–2932, 10.1039/c3sc53243g.
- E. Alessio and L. Messori, Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry, Molecules, 2019, 24, 1–20 CrossRef.
- J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, A Novel Class of Ruthenium-Based Photosensitizers Effectively Kills in Vitro Cancer Cells and in Vivo Tumors, Photochem. Photobiol. Sci., 2015, 14(11), 2014–2023, 10.1039/c4pp00438h.
- C. A. Vock, W. H. Ang, C. Scolaro, A. D. Phillips, L. Lagopoulos, L. Juillerat-Jeanneret, G. Sava, R. Scopelliti and P. J. Dyson, Development of Ruthenium Antitumor Drugs That Overcome Multidrug Resistance Mechanisms, J. Med. Chem., 2007, 50(9), 2166–2175, DOI:10.1021/jm070039f.
- L. Côrte-Real, B. Karas, P. Gírio, A. Moreno, F. Avecilla, F. Marques, B. T. Buckley, K. R. Cooper, C. Doherty, P. Falson, M. H. Garcia and A. Valente, Unprecedented Inhibition of P-Gp Activity by a Novel Ruthenium-Cyclopentadienyl Compound Bearing a Bipyridine-Biotin Ligand, Eur. J. Med. Chem., 2019, 163, 853–863, DOI:10.1016/j.ejmech.2018.12.022.
- L. Côrte-Real, R. G. Teixeira, P. Gírio, E. Comsa, A. Moreno, R. Nasr, H. Baubichon-Cortay, F. Avecilla, F. Marques, M. P. Robalo, P. Mendes, J. P. P. Ramalho, M. H. Garcia, P. Falson and A. Valente, Methyl-Cyclopentadienyl Ruthenium Compounds with 2,2′-Bipyridine Derivatives Display Strong Anticancer Activity and Multidrug Resistance Potential, Inorg. Chem., 2018, 57, 4629–4639, DOI:10.1021/acs.inorgchem.8b00358.
- W. Bai, S. K. S. Tse, K. H. Lee, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Synthesis and Characterization of MH⋯HOR Dihydrogen Bonded Ruthenium and Osmium Complexes (H5-C5H4CH 2OH)MH(PPh3)2 (M = Ru, Os), Sci. China: Chem., 2014, 57(8), 1079–1089, DOI:10.1007/s11426-014-5143-6.
- T. Mede, M. Jäger and U. S. Schubert, “chemistry-on-the-Complex”: Functional RuII Polypyridyl-Type Sensitizers as Divergent Building Blocks, Chem. Soc. Rev., 2018, 47(20), 7577–7627, 10.1039/c8cs00096d.
- L. Côrte-Real, B. Karas, P. Gírio, A. Moreno, F. Avecilla, F. Marques, B. T. Buckley, K. R. Cooper, C. Doherty, P. Falson, M. H. Garcia and A. Valente, Unprecedented Inhibition of P-Gp Activity by a Novel Ruthenium-Cyclopentadienyl Compound Bearing a Bipyridine-Biotin Ligand, Eur. J. Med. Chem., 2019, 163, 853–863, DOI:10.1016/j.ejmech.2018.12.022.
- L. Côrte-Real, B. Karas, A. R. Brás, A. Pilon, F. Avecilla, F. Marques, A. Preto, B. T. Buckley, K. R. Cooper, C. Doherty, M. Helena Garcia and A. Valente, Ruthenium-Cyclopentadienyl Bipyridine-Biotin Based Compounds: Synthesis and Biological Effect, Inorg. Chem., 2019, 58(14), 9135–9149, DOI:10.1021/acs.inorgchem.9b00735.
- L. Côrte-Real, M. Paula Robalo, F. Marques, G. Nogueira, F. Avecilla, T. J. L. Silva, F. C. Santos, A. Isabel Tomaz, M. Helena Garcia and A. Valente, The Key Role of Coligands in Novel Ruthenium(II)-Cyclopentadienyl Bipyridine Derivatives: Ranging from Non-Cytotoxic to Highly Cytotoxic Compounds, J. Inorg. Biochem., 2015, 150, 148–159, DOI:10.1016/j.jinorgbio.2015.06.015.
- M. M. Gottesman, T. Fojo and S. E. Bates, Multidrug Resistance in Cancer: Role of Atp-Dependent Transporters, Nat. Rev. Cancer, 2002, 2(1), 48–58, DOI:10.1038/nrc706.
- N. Sarin, F. Engel, G. V. Kalayda, M. Mannewitz, J. Cinatl, F. Rothweiler, M. Michaelis, H. Saafan, C. A. Ritter, U. Jaehde and R. Frötschl, Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Associated with an Abrogation of Cisplatin-Induced G2/M Cell Cycle Arrest, PLoS One, 2017, 12(7), 1–26, DOI:10.1371/journal.pone.0181081.
- A. Podolski-Renić, S. Bsze, J. Dinić, L. Kocsis, F. Hudecz, A. Csámpai and M. Pešić, Ferrocene-Cinchona Hybrids with Triazolyl-Chalcone Linkers Act as pro-Oxidants and Sensitize Human Cancer Cell Lines to Paclitaxel, Metallomics, 2017, 9(8), 1132–1141, 10.1039/c7mt00183e.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Structure-Activity Relationships for Ruthenium and Osmium Anticancer Agents-towards Clinical Development, Chem. Soc. Rev., 2018, 47(3), 909–928, 10.1039/c7cs00332c.
- R. G. Kenny and C. J. Marmion, Toward Multi-Targeted Platinum and Ruthenium Drugs - A New Paradigm in Cancer Drug Treatment Regimens?, Chem. Rev., 2019, 119(2), 1058–1137, DOI:10.1021/acs.chemrev.8b00271.
- G. Tamasi, A. Merlino, F. Scaletti, P. Heffeter, A. A. Legin, M. A. Jakupec, W. Berger, L. Messori, B. K. Keppler and R. Cini, {Ru(CO)X}-Core Complexes with Benzimidazole Ligands: Synthesis, X-Ray Structure and Evaluation of Anticancer Activity in Vivo, Dalton Trans., 2017, 46(9), 3025–3040, 10.1039/c6dt04295c.
- T. S. Morais, A. Valente, A. I. Tomaz, F. Marques and M. H. Garcia, Tracking Antitumor Metallodrugs: Promising Agents with the Ru(II)- and Fe(II)-Cyclopentadienyl Scaffolds, Future Med. Chem., 2016, 8(5), 527–544 CrossRef CAS.
- J. Kopecka, S. Porto, S. Lusa, E. Gazzano, G. Salzano, A. Giordano, V. Desiderio, D. Ghigo, M. Caraglia, G. De Rosa and C. Riganti, Self-Assembling Nanoparticles Encapsulating Zoledronic Acid Revert Multidrug Resistance in Cancer Cells, Oncotarget, 2015, 6(31), 31461–31478, DOI:10.18632/oncotarget.5058.
- J. Kopecka, G. Salzano, I. Campia, S. Lusa, D. Ghigo, G. De Rosa and C. Riganti, Insights in the Chemical Components of Liposomes Responsible for P-Glycoprotein Inhibition, Nanomedicine, 2014, 10(1), 77–87, DOI:10.1016/j.nano.2013.06.013.
- J. F. Lu, D. Pokharel and M. Bebawy, MRP1 and Its Role in Anticancer Drug Resistance, Drug Metab. Rev., 2015, 47(4), 406–419, DOI:10.3109/03602532.2015.1105253.
- L. Dury, R. Nasr, D. Lorendeau, E. Comsa, P. Falson, A. Di Pietro, H. Baubichon-Cortay, I. Wong, X. Zhu, K. F. Chan, T. H. Chan and L. Chow, Flavonoid Dimers Are Highly Potent Killers of Multidrug Resistant Cancer Cells Overexpressing MRP1, Biochem. Pharmacol., 2017, 124, 10–18, DOI:10.1016/j.bcp.2016.10.013.
- K. W. Tan, A. Sampson, B. Osa-Andrews and S. H. Iram, Calcitriol and Calcipotriol Modulate Transport Activity of ABC Transporters and Exhibit Selective Cytotoxicity in MRP1-Overexpressing Cells, Drug Metab. Dispos., 2018, 46(12), 1856–1866, DOI:10.1124/dmd.118.081612.
- C. C. Gana, K. M. Hanssen, D. M. T. Yu, C. L. Flemming, M. S. Wheatley, G. Conseil, S. P. C. Cole, M. D. Norris, M. Haber and J. I. Fletcher, MRP1 Modulators Synergize with Buthionine Sulfoximine to Exploit Collateral Sensitivity and Selectively Kill MRP1-Expressing Cancer Cells, Biochem. Pharmacol., 2019, 168(July), 237–248, DOI:10.1016/j.bcp.2019.07.009.
- G. Szakács, M. D. Hall, M. M. Gottesman, A. Boumendjel, R. Kachadourian, B. J. Day, H. Baubichon-Cortay and A. Di Pietro, Targeting, the Achilles Heel of Multidrug-Resistant Cancer by Exploiting the Fitness Cost of Resistance, Chem. Rev., 2014, 114(11), 5753–5774, DOI:10.1021/cr4006236.
- C. Riganti, R. Giampietro, J. Kopecka, C. Costamagna, F. S. Abatematteo, M. Contino and C. Abate, Mrp1-Collateral Sensitizers as a Novel Therapeutic Approach in Resistant Cancer Therapy: An in Vitro and in Vivo Study in Lung Resistant Tumor, Int. J. Mol. Sci., 2020, 21(3333), 1–17, DOI:10.3390/ijms21093333.
- APEX3 V2018 1-0. APEX3 v2018 1-0. BRUKER AXS., 2018.
- SAINT V8.38A Bruker AXS. SAINT V8.38A Bruker AXS., 2017.
- SADABS-2016/2 - Bruker AXS Area Detector Scaling and Absorption Correction. SADABS-2016/2 - Bruker AXS area detector scaling and absorption correction.
- G. M. Sheldrick, A Short History of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64(1), 112–122, DOI:10.1107/S0108767307043930.
Footnote |
| † Electronic supplementary information (ESI) available: Schemes forsynthesis of compounds 1–7; characterization of compounds 1–7 (ESI-MS and NMR spectra and UV-vis data); crystallographic data and structural refinement details for X-ray data for 1, 2, 3, 4, 5, and 6; stability curves in DMSO/DMEM; P-gp and MRP1 expression in non-small cell lung cancer cell lines (immunoblotting). CCDC 2042407–2042412. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi01344g |
| This journal is © the Partner Organisations 2021 |