
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled delivery of gold nanoparticle-coupled miRNA therapeutics via an injectable self-healing hydrogel†
Casper F. T.
van der Ven
 abcd,
Mark W.
Tibbitt
de,
João
Conde
fg,
Alain
van Mil
abh,
Jesper
Hjortnaes
ab,
Pieter A.
Doevendans
bh,
Joost P. G.
Sluijter
ab,
Elena
Aikawa‡
*ci and
Robert S.
Langer‡
*dj
abcd,
Mark W.
Tibbitt
de,
João
Conde
fg,
Alain
van Mil
abh,
Jesper
Hjortnaes
ab,
Pieter A.
Doevendans
bh,
Joost P. G.
Sluijter
ab,
Elena
Aikawa‡
*ci and
Robert S.
Langer‡
*dj
aRegenerative Medicine Center, University Medical Center Utrecht, Uppsalalaan 8, 3584 CT Utrecht, the Netherlands
bDepartment of Cardiology, Experimental Cardiology Laboratory, Circulatory Health Laboratory, University Medical Center Utrecht, Utrecht University, Heidelberglaan 100, 3584 CX Utrecht, the Netherlands
cCenter of Excellence in Cardiovascular Biology, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Woman's Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, Boston 02115, MA, USA
dDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge 02142, MA, USA
eMacromolecular Engineering Laboratory, Department of Mechanical and Process Engineering, ETH Zurich, Sonneggstrasse 3, 8092 Zurich, Switzerland
fNOVA Medical School, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, 1169-056 Lisboa, Portugal
gCentre for Toxicogenomics and Human Health, Genetics, Oncology and Human Toxicology, NOVA Medical School, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, 1169-056 Lisboa, Portugal
hNetherlands Heart Institute, Moreelsepark 1, 3511 EP Utrecht, the Netherlands
iCenter for Interdisciplinary Cardiovascular Sciences, Division of Cardiovascular Medicine, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, 3 Blackfan Circle, Boston 02115, MA, USA. E-mail: eaikawa@bwh.harvard.edu
jDepartment of Chemical Engineering, Massachusetts Institute of Technology, 25 Ames Street, Cambridge 02142, MA, USA. E-mail: r.langer@mit.edu
First published on 24th November 2021
Abstract
Differential expression of microRNAs (miRNAs) plays a role in many diseases, including cancer and cardiovascular diseases. Potentially, miRNAs could be targeted with miRNA-therapeutics. Sustained delivery of these therapeutics remains challenging. This study couples miR-mimics to PEG-peptide gold nanoparticles (AuNP) and loads these AuNP-miRNAs in an injectable, shear thinning, self-assembling polymer nanoparticle (PNP) hydrogel drug delivery platform to improve delivery. Spherical AuNPs coated with fluorescently labelled miR-214 are loaded into an HPMC-PEG-b-PLA PNP hydrogel. Release of AuNP/miRNAs is quantified, AuNP-miR-214 functionality is shown in vitro in HEK293 cells, and AuNP-miRNAs are tracked in a 3D bioprinted human model of calcific aortic valve disease (CAVD). Lastly, biodistribution of PNP-AuNP-miR-67 is assessed after subcutaneous injection in C57BL/6 mice. AuNP-miRNA release from the PNP hydrogel in vitro demonstrates a linear pattern over 5 days up to 20%. AuNP-miR-214 transfection in HEK293 results in 33% decrease of Luciferase reporter activity. In the CAVD model, AuNP-miR-214 are tracked into the cytoplasm of human aortic valve interstitial cells. Lastly, 11 days after subcutaneous injection, AuNP-miR-67 predominantly clears via the liver and kidneys, and fluorescence levels are again comparable to control animals. Thus, the PNP-AuNP-miRNA drug delivery platform provides linear release of functional miRNAs in vitro and has potential for in vivo applications.
Introduction
Since their discovery, microRNAs (miRNAs) have become a focus of (bio)medical research. The research community has elucidated the role of miRNAs in the etiology and progression of various diseases, including cardiovascular disease,1–3 cancer,4,5 and hepatitis C.6 The discovery of their central role in several pathological conditions has provided the basis for using miRNAs in the treatment of these diseases. For example, miR-34 has been tested in phase I trials to treat multiple types of cancer (ClinicalTrials.gov: NCT01829971, NCT02862145) and miR-122 has been explored for the treatment of hepatitis C.6 The latter successfully completed phase II, whereas the former two trials were terminated prematurely due to serious adverse events, including enterocolitis, hypoxia/systemic inflammatory response syndrome, colitis/pneumonitis, hepatic failure, and cytokine release syndrome/respiratory failure that could be attributed to the treatment.7 The mixed outcomes of these trials highlight the potential of miRNA therapeutics and the present challenge in their successful delivery to the targeted cells and tissues.Prior to clinical testing and application, there are several hurdles for RNA therapeutics to overcome regarding their delivery, retention, stability, and degradation. To prevent endosomal degradation, miRNAs were functionalized with fusogenic pH-sensitive peptides that enabled endosomal escape.8 When miRNAs are injected directly, washout is high and retention is low.9 Carrier vehicles and modifications have been explored to reduce the washout and improve the delivery of miRNAs. We previously described non-viral vectors to improve delivery, including lipids,10 microbubbles,11 polymers,12 and inorganic materials,13 as well as modifications to improve both biostability and binding stability.14 Injectable hydrogels are promising candidates to improve and localize the delivery of therapeutic miRNA molecules while allowing minimally invasive application.
Injectable hydrogels have been designed with natural or synthetic polymers, including ECM,15 collagen, fibrin, alginate, functionalized poly(ethylene glycol) (PEG), or poly(N-isopropylacrylamide) (pNIPAM).16 Traditionally, hydrogels have been based on covalent cross-linking methods that require initiation by temperature,17 light,18 or a change in pH.19–21 For biomedical applications, including drug delivery, these covalent cross-linking methods can provide an additional hurdle, as the cross-linking reaction is not instantaneous, and drugs can be lost during network formation. Additionally, for some materials it requires an external stimulus or an instrument, such as a UV light source. To simplify the application of hydrogels and avoid the challenges associated with covalent cross-linking, non-covalent cross-linking allows for spontaneous gel formation without an external trigger. In addition, non-covalent hydrogels often exhibit shear-thinning (the ability to flow upon application of stress) and self-healing (reformation of the gel upon relaxation of the external stress) properties. Shear-thinning facilitates injection and minimally invasive delivery, thus improving clinical application.
Injectable hydrogels have been developed using leucine zipper domains,22 dock-and-lock proteins,23 and host–guest interactions.24 A pH cross-linkable hydrogel was developed for miRNA delivery based on non-covalent ureido-pyrimidinone (UPy) cross-linking. Near complete release of miRNA molecules from UPy gels was achieved after two days.21 This release could be extended by modification of the miRNA molecules with cholesterol groups. We recently developed a class of shear-thinning and self-healing hydrogels based on polymer–nanoparticle (PNP) interactions.25,26 These properties arise from the reversible, non-covalent interactions between the polymer and the nanoparticles within the gel, employing a hydroxypropylmethylcellulose derivative (HPMC-C12) and core–shell nanoparticles [poly(ethylene glycol)-block-poly(lactic acid) (PEG-b-PLA) nanoparticles].
This research set out to improve both the biostability and the delivery of miRNA. In order to improve biostability and reduce degradation, the miRNA molecules were attached to gold nanospheres (AuNP) that were functionalized with poly(ethylene-glycol) (PEG).13,27–29 In addition, influenza hemagglutinin (HA1) peptide was added to stimulate endosomal escape intracellularly by destabilizing the endosomal membrane.
These functionalized AuNP (AuNP-miR) were loaded into a biocompatible, shear-thinning, and self-healing injectable hydrogel, based on the PNP gel platform, to improve minimally invasive, local, and sustained delivery. This research shows (1) linear release of AuNP-miRs from the PNP hydrogel over multiple days, and (2) that AuNP-miRs retain functionality in vitro. Additionally, it shows that (3) AuNP-miRs retain functionality by employing an in vitro miRNA reporter assay and a complex model system, specifically in a 3D in vitro model of human calcific aortic valve disease.30 Lastly, it shows (4) the biodistribution of AuNP-miRs after subcutaneous injection in vivo. Together, this data demonstrates an innovative approach to achieve controlled release of functional miRNA from an injectable, self-healing PNP hydrogel.
Results & discussion
Production of AuNP-miR-loaded PNP hydrogels
To engineer an injectable biomaterial for controlled release of miRNAs, we prepared AuNP-miRNAs within PNP hydrogel formulations. We synthesized the PNP hydrogel components and functionalized the AuNPs with PEG, cel-miR-67 or has-miR-214, and HA1-peptide (Fig. S1†). Cel-miR-67 was used as a negative control as it has minimal sequence identity with murine or human miRNAs and it is not naturally present in human cells; thus, it should not have any biological effect. The HA1 peptide, a fusogenic peptide (influenza hemagglutinin HA1 peptide, N-YPYDVPDYA-C23) was used to increase miRNA uptake by destabilizing the endosomal membrane stimulating endosomal discharge by a pH-responsive machinery.13 This peptide was functionalized on the surface of the AuNPs via carbodiimide chemistry assisted by N-hydroxisuccinimide using an EDC/NHS coupling reaction between the carboxylated PEG spacer and the amine terminal group of the peptide. HPMC-C12 and PEG-b-PLA polymers were synthesized as previously described.25 PEG-b-PLA polymers were formulated into NPs via nanoprecipitation and characterized by dynamic light scattering (DLS). PEG-b-PLA NPs with a diameter (Dh) of ∼88 nm and dispersity (Đ) of 0.12 were used to formulate the PNP hydrogel. Based on prior work, NPs of this diameter form gels by enabling polymer bridging over several NPs in contrast to polymer wrapping around single NPs.31 PNP hydrogels were formulated at 1 wt% HPMC-C12 and 10 wt% PEG-b-PLA NPs (Fig. 1a).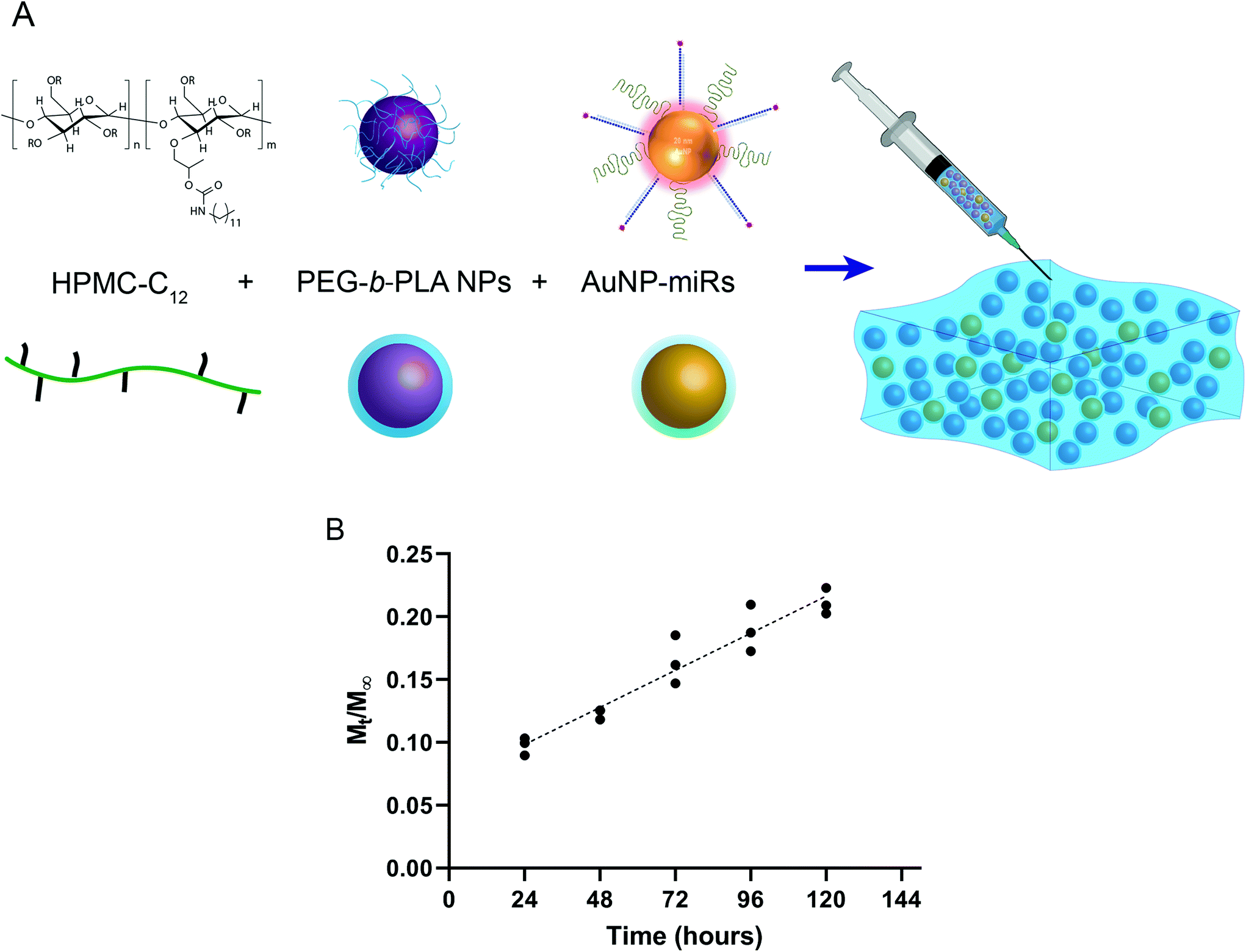 | ||
| Fig. 1 (A) schematically represents the structure of the PNP hydrogel and the functionalized AuNP-miRs. (B) Depicts in vitro release profile of AuNP-miRs from the PNP hydrogel at 37 °C demonstrating the cumulative sustained release of AuNP-miRs in a linear pattern of up to ∼20% [Mt/M∞; the cumulative fractional mass released at time t (n = 3)]. R2 = 0.9875, Mt/M∞ = 0.0012 t + 0.0688. | ||
Linear release of AuNP-miRs from PNP hydrogels
To monitor the release of AuNP-miRs from the PNP hydrogels, non-modified AuNPs and AuNP-miR-67 were loaded into the PNP hydrogel in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (PEG-b-PLA:HPMC-C12:AuNP; final concentration AuNP-miR-67 0.34 nM) and incubated at 37 °C, 5% CO2. The supernatant was collected and replaced at 24 h intervals to quantify the concentration of AuNPs in the supernatant based on a calibrated absorbance measurement (Fig. S2†). Sustained linear release of AuNP-miR-67 was observed up to ∼20% of the loaded AuNPs over the course of 5 days (Fig. 1b), demonstrating extended release compared to other hydrogel-based drug-delivery systems.14,20,31–33,35 Following an initial burst release of ∼10% of the loaded AuNP over the first 24 h, ∼3% of the loaded AuNP were released per day over the course of 5 days. The observed linear release was consistent with an erosion-based release that has been observed for NP release from PNP hydrogels.25
:1:1 ratio (PEG-b-PLA:HPMC-C12:AuNP; final concentration AuNP-miR-67 0.34 nM) and incubated at 37 °C, 5% CO2. The supernatant was collected and replaced at 24 h intervals to quantify the concentration of AuNPs in the supernatant based on a calibrated absorbance measurement (Fig. S2†). Sustained linear release of AuNP-miR-67 was observed up to ∼20% of the loaded AuNPs over the course of 5 days (Fig. 1b), demonstrating extended release compared to other hydrogel-based drug-delivery systems.14,20,31–33,35 Following an initial burst release of ∼10% of the loaded AuNP over the first 24 h, ∼3% of the loaded AuNP were released per day over the course of 5 days. The observed linear release was consistent with an erosion-based release that has been observed for NP release from PNP hydrogels.25
AuNP-miR-214 demonstrates in vitro mRNA targeting activity
Next, we verified the mRNA targeting capacity of miR-214 after conjugation to the AuNPs. HEK293 cells were transfected with a miR-214 target luciferase reporter34 and incubated with AuNP-miR-214s. Compared to a non-transfected control and an inactive AuNP-miR-67 control, AuNP-miR-214 significantly reduced the Luciferase signal (∼42%) after 48 h, demonstrating preserved miR-214 bioactivity after coupling to the AuNP (Fig. 2). As a positive control, transfection with unmodified miR-214 mimics using Lipofectamine resulted in a knockdown of ∼67% relative to the control groups. The higher transfection efficiency of Lipofectamine compared to AuNP-miR-214 is expected, considering the thorough optimization of commercially available Lipofectamine.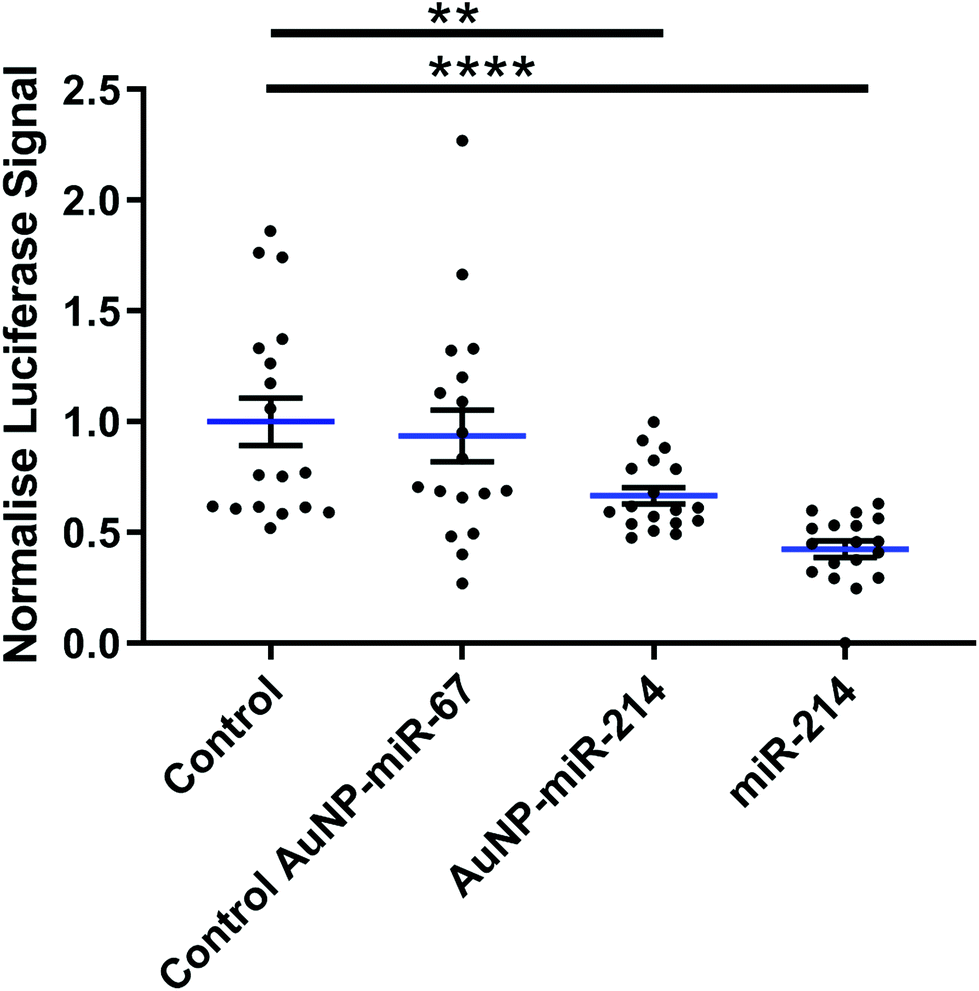 | ||
| Fig. 2 AuNP-miR-214 significantly reduces Luciferase activity. AuNP-miR-214 were added to HEK293 cells introduced to the pMIR-REPORT-QKI-3′ UTR Luciferase vector in vitro. Compared to an untransfected control and an inactive control AuNP-miR-67, AuNP-miR-214 suppresses the Luciferase signal (∼42%) after 48 h. Transfection with miR-214 mimic using Lipofectamine results in knockdown of ∼67% relative to the control groups, compared to ∼42% in the AuNP-miR-214 transfection. Mean ± SEM; * p < 0.05, ** p < 0.01, *** p < 0.001; n = 6, experiment conducted in triplicate. | ||
Suppression of a target gene up to 90% has been observed for small interfering RNA (siRNA) released from alginate or collagen hydrogels in a green fluorescent protein (GFP) expressing HEK293 cell line,33 though over a longer time scale of 6 days. 80% knockdown of GFP signal was achieved with miRNA released from a PEG hydrogel over a period of 42 days.36 This suggests that an extended testing period for AuNP-miR-214 should be considered for translational use.
AuNP-miR-67 infiltrate haVICs in a 3D bioprinted in vitro CAVD model
In order to test AuNP-miR uptake in a more complex tissue model, we tracked fluorescently labelled AuNP-miR-67 in a 3D model of CAVD. Cel-miR-67 was used as a negative control as it has minimal sequence identity with murine or human miRNAs and it is not naturally present in human cells; thus, it should not have any biological effect. Cy5.5-labelled AuNP-miR-67 were added to the cell culture media of a 3D bioprinted model of CAVD containing human aortic valve interstitial cells (haVICs).30 In this model, VICs cultured in NM maintain a quiescent phenotype and VICs exposed to OM produce microcalcifications (Fig. S3†). Labeling nuclei with Hoechst (blue), cytoplasm with CellTracker Green (green), and lysosomes with LysoTracker Red (red) aided in demonstrating that AuNP-miR-67 (white) were taken up by the cells in lysosomes (Fig. 3a), indicated by co-localization (pink arrows) of CellTracker Red and the Cy5.5-labelled AuNP-miR-67 (Fig. 3b). We hypothesize that AuNP-miRs are taken up via endocytosis, indicated by co-localization (pink arrows) of CellTracker Red and white AuNP-miR-67, and that they can escape the lysosomes, indicated by the presence of both lysosomes (red arrows) and AuNP-miRs (white arrows).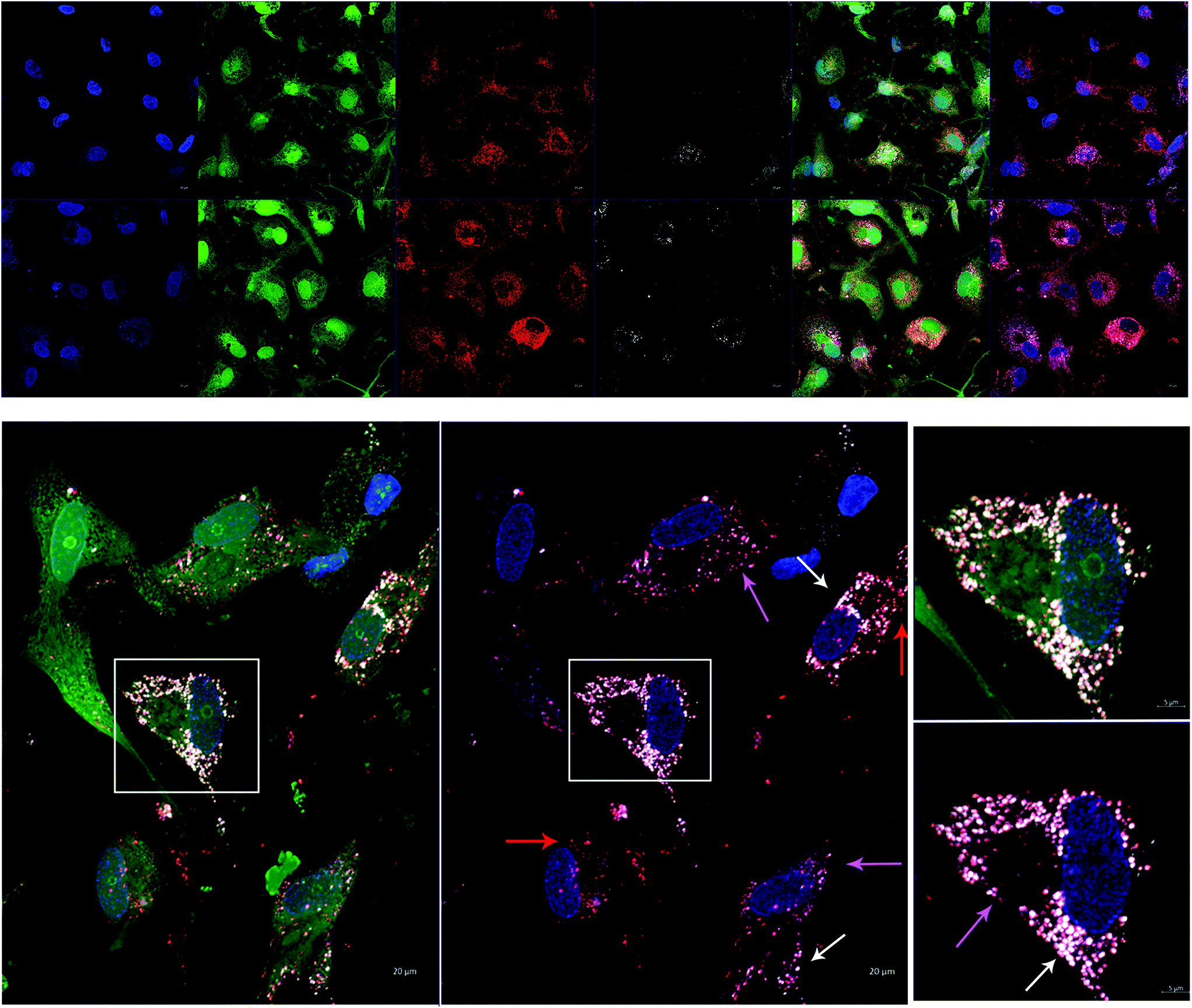 | ||
| Fig. 3 Infiltration of AuNP-miR-67 into human aortic valve interstitial cells (haVICs) in a 3D bioprinted model of calcific aortic valve disease (CAVD). Nuclei were labelled with Hoechst (blue), haVICs were labelled with CellTracker Green (green), and lysosomes were labelled with LysoTracker Red (red). Co-localization of lysosomes (red) and Cy5.5-labelled AuNP-miR-67 (white) demonstrated that AuNP-miRs were taken up by the cells in lysosomes (scale bar = 20 μm, scale bar insert = 5 μm, t = 48 h, n = 3). | ||
AuNP-miR-214 increase alkaline phosphatase expression in a 3D CAVD model
Additionally, we employed the 3D bioprinted in vitro human CAVD model to test AuNP-miR-214 functionality.30 Osteogenic medium (OM) has been shown to stimulate the formation of calcium minerals through osteogenic differentiation of haVICs.30 miR-214 was found to increase calcification of haVICs in vitro in traditional 2D cell culture.37 Here, we demonstrate that compared to non-transfected controls 30 nM AuNP-miR-214 transfection significantly increased ALP activity, a phospholytic enzyme associated with early calcification in CAVD,38 in haVICs within a 3D-bioprinted in vitro model of human CAVD cultured in normal medium (NM) (Fig. 4). Compared to NM, OM increased ALP activity (left column), confirming earlier results.39 Compared to NM, addition of AuNP-miR-214 significantly increased ALP activity (Fig. 4a top row; Fig. 4b). The difference between the effect of AuNP-miR-214 and OM on haVICs was likely due to the relatively low concentration of AuNP-miR-214 and the prolonged exposure to OM. These findings further support a role for miR-214 in the development of CAVD by stimulating ALP and encourage further investigation.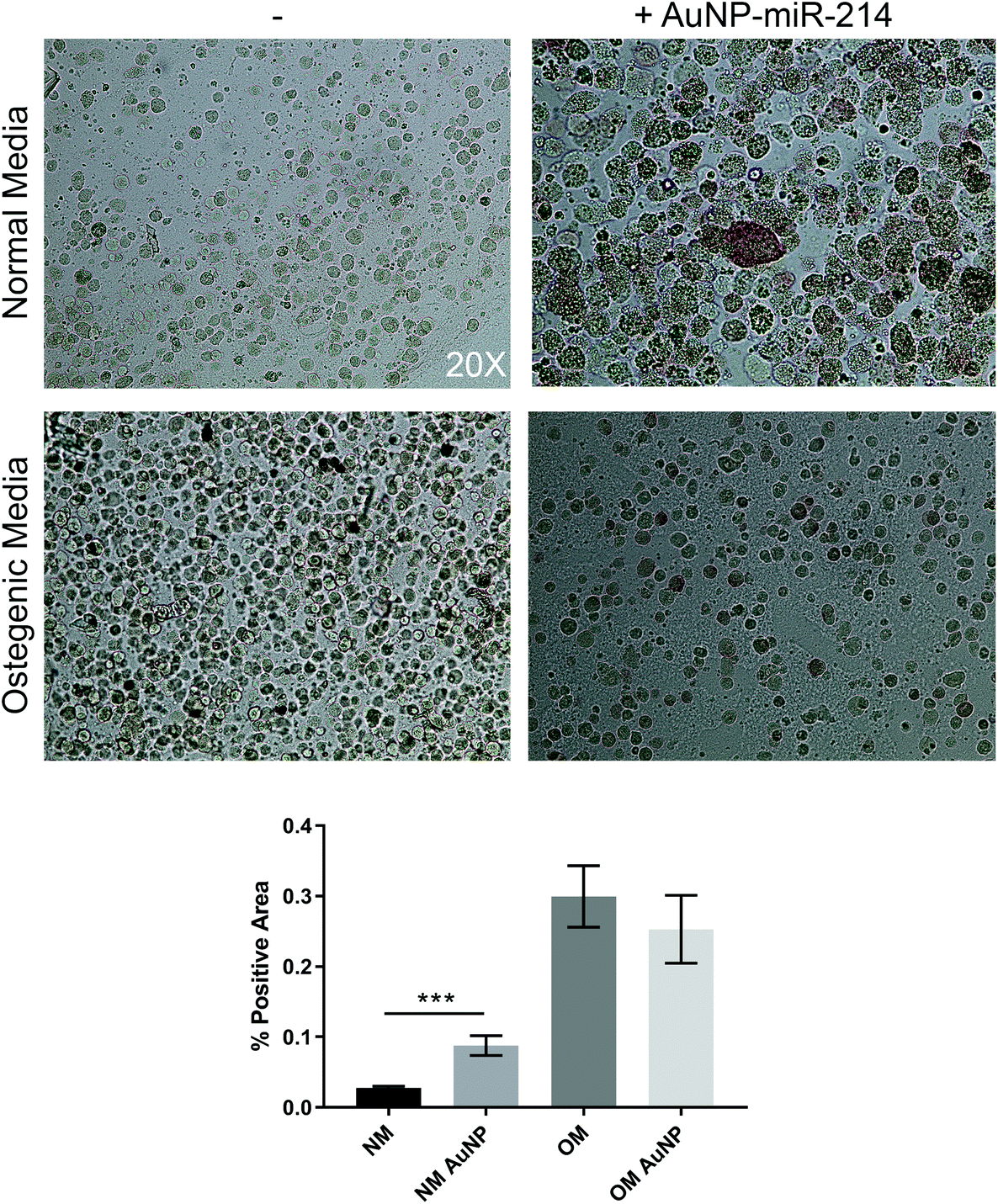 | ||
| Fig. 4 AuNP-miR-214 increase alkaline phosphatase expression (red) after 48 h in human aortic valve interstitial cells (haVICs) comparable to osteogenic stimulation in 3D human calcific aortic valve disease model. Brightfield microscopy, mean ± SEM, *** p < 0.001, n = 3, 20× magnification. | ||
The involvement of miR-214 in CAVD is a relatively recent discovery, and its precise role in CAVD requires further clarification. miR-214 involvement in CAVD was first identified in porcine AV endothelial cells (paVECs).40 Specifically, micro-array analysis and ex vivo validation of healthy ventricular and aortic paVECs RNA yielded differential expression levels of miR-214 as a result of oscillatory shear due to disturbed blood flow. Compared to the ventricularis side, increased expression of miR-214 in the fibrosa side of pAVs resulted in thickening and calcification. Later, in a comparison between valves of healthy controls and patients with calcific aortic stenosis, miR-214 was increased in CAVD patients and osteocalcin, osteopontin, Runx2, and osterix were identified and validated as its targets.41 Furthermore, in excised AVs from patients with CAVD, increased miR-214 expression was found to play a role in suppressing the apoptosis repressor with caspase recruitment domain ARC.42 In addition, it was demonstrated that miR-214 stimulates the formation of calcific nodules in haVICs in vitro via the MyD88/NF-κB inflammatory pathway.37 Contradictory to the aforementioned results, in larger micro-array studies miR-214 was found low in valves from patients with CAVD compared to non-diseased control valves.43–45 Furthermore, dynamic stretch on porcine AVs ex vivo demonstrated that miR-214 was significantly downregulated during late stage calcification, and addition of miR-214 mimic in static stretch conditions resulted in lower levels of calcification. Therefore, the role of miR-214 in the development of CAVD is complex and needs to be further elucidated.46
AuNP biodistribution following subcutaneous implantation in PNP hydrogels
We further investigated the biodistribution of AuNP-miRs following delivery from the PNP hydrogel in vivo. In general, parenteral administration of therapeutics or drug delivery systems via injection can be achieved easily and in a minimally invasive manner. However, traditional intravenous injections result in relatively short residence time in the body. On the other hand, surgical implantation of a material-based controlled release system provides longer-lasting effects but is more invasive. Injectable hydrogels with controlled release provide an attractive method to administer drugs locally, in a minimally invasive manner, and with extended biological effect. For example, hyaluronic acid-based PNP hydrogels were recently administered to cardiac tissue via catheter-based delivery.47 Therefore, we employed AuNP-miR-loaded PNP hydrogels for administration of an AuNP-miR releasing depot following minimally invasive subcutaneous injection. AuNP-miR-67-loaded PNP hydrogel was injected subcutaneously in nine C57BL/6 mice to assess biodistribution. Cel-miR-67 was used as a negative control as it has minimal sequence identity with murine or human miRNAs and it is not naturally present in human cells; thus, it should not have any biological effect. To monitor the biodistribution of the AuNP-miR, fluorescent signals of the Cy5.5 labelled miR-67 were measured in the lungs, liver, spleen, kidney, and skin surrounding the injection site in three adult male wildtype C57BL/6 mice on days 1, 4, and 11 after injection (Fig. 5). Three additional animals were injected with unloaded PNP hydrogel as negative control and aforementioned tissues were harvested and monitored on day 11. Image quantification of the fluorescent signal of the labelled AuNP-miR-67 demonstrated accumulation in the lungs, spleen, liver and kidney on day 1 and suggests clearance via the liver and kidney by day 11, as demonstrated by decreasing fluorescence that approached the values of control animals.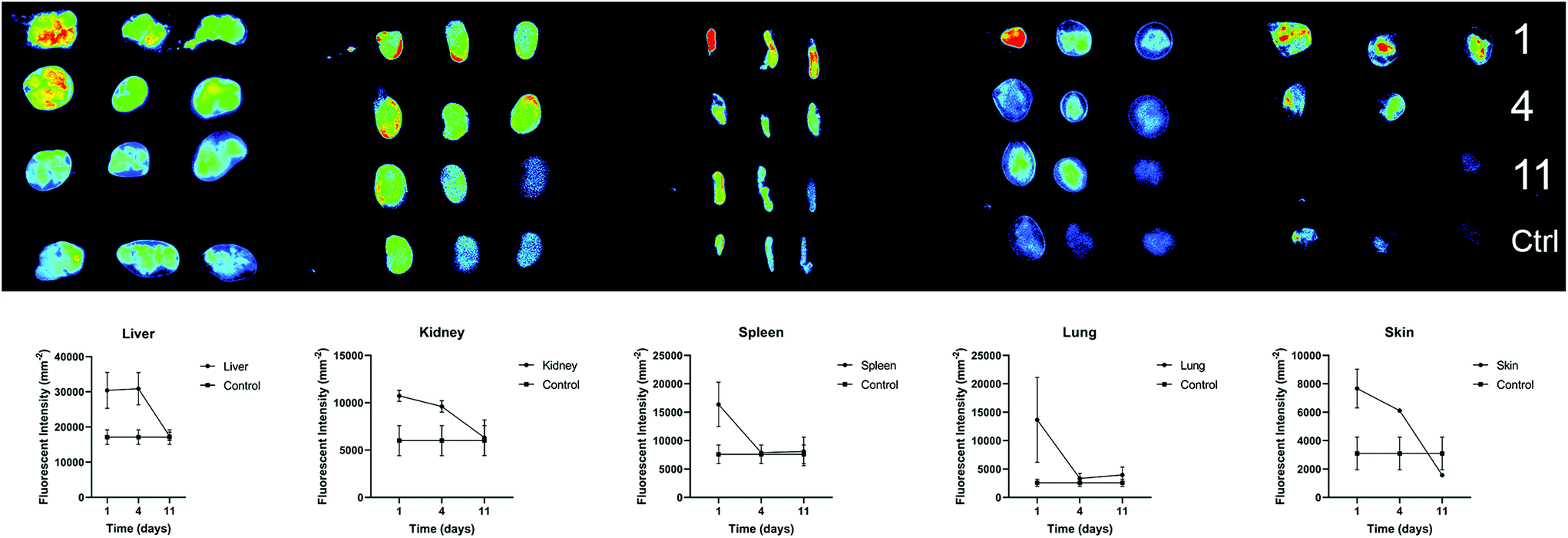 | ||
| Fig. 5 To assess biodistribution of AuNP-miR-67 n = 9 C57BL/6 mice were injected subcutaneously with PNP-AuNP-miR-67. (Top) Fluorescent signals of the Cy5.5 labelled AuNP-miR-67 were measured in the lungs, liver, spleen, kidney, and skin surrounding the injection site in n = 3 animals on days 1, 4, and 11 after injection. N = 3 mice were injected with unloaded PNP hydrogel as a control and aforementioned tissues were harvested on day 11. Intensity of the fluorescent signal of labelled AuNP-miR-67 demonstrates accumulation in the lungs, spleen, liver and kidney on day 1 and clearance via the kidneys after 11 days, as demonstrated by decreasing fluorescent signal comparable to control animals. (Bottom) Quantified fluorescent signal in liver, kidney, lung, spleen, and skin surrounding the injection site on days 1, 4, and 11 demonstrates clearance over 11 days. N = 3 animals per time point. Mean ± SEM. | ||
Experimental section
PNP hydrogel synthesis
PNP hydrogels were synthesized as previously described.25 In short:AuNP-miR synthesis
AuNP-miRs were synthesized as previously described.13 In summary:000g, 30 min, 4 °C) removed excess PEG, which was quantified by the Ellman's Assay. The excess of thiolated chains in the supernatant was quantified by interpolating a calibration curve set by reacting α-Mercapto-ω-carboxy PEG solution (200 μL) in phosphate buffer (100 μL, 0.5 M, pH 7) with 5,5′-dithio-bis(2-nitrobenzoic) acid (DTNB, 7 μL, 5 mg mL−1) in phosphate buffer (0.5 M, pH 7) and measuring the absorbance at 412 nm after 10 min reaction. The linear range for the PEG chain obtained by this method is 0–0.1 mg mL−1 (Abs at 412 nm = 8.0353 × [PEG, mg mL−1] + 0.0486). The number of exchanged chains is given by the difference between the amount determined by this assay and the initial amount incubated with the AuNP. There is a point at which the AuNP becomes saturated with a thiolated layer and is not able to take up more thiolated chains – maximum coverage per AuNP, which was 0.03 mg mL−1 of PEG for these AuNP. The AuNP were functionalized with 50% PEG layer in order to leave space for binding the thiolated miRNAs and HA1 peptide (Fig. S1†).
000g, 20 min, 4 °C), and re-suspended in DEPC-water. This procedure was repeated 3 times. The number of miRNA per AuNP was determined by quantification of the excess miRNA oligos in the supernatants collected during synthesis via the emission spectra of Cy5.5 (excitation/emission, 688 nm/707 nm) dye in a microplate reader (Varioskan Flash Multimode Reader; Thermo Scientific). All nanoparticle samples and standard solutions of the thiolated-miRNAs were kept at the same pH and ionic strength for all measurements. Fluorescence emission was converted to molar concentrations by interpolation from a standard linear calibration curve prepared with known concentrations of miRNA (Fig. S4†).
000g for 30 min at 4 °C and washed three times with Milli-Q water.
HA1 quantification was performed with the Pierce® BCA Protein Assay kit (Thermo Scientific) according to manufacturer’ instructions. Briefly, each standard (0.025 mL) and unknown sample (the supernatants; 0.025 mL) was mixed with the BCA™ Working Reagent (50:1, BCA reagent A:BCA reagent B; 0.2 mL) to each tube. The reaction mixture was incubated at 60 °C for 30 min. After incubation, the tubes were cooled down to room temperature and the absorbance measured at 562 nm. The standard curve was used to determine the HA1 concentration of each unknown sample (supernatant). The calibration curve for a working range (0–125 μg mL−1) is given by the following equation Abs 562 nm = 0.0036 × [HA1 peptide, μg mL−1] + 0.8016, R2 = 0.9939 for HA1 peptide (Fig. S4†).
Release of AuNP-miRs from PNP hydrogel
AuNP-miR-214 were loaded into PNP hydrogels and incubated at 37 °C, 5% CO2 with MilliQ water in 1.5 mL Eppendorf tubes. Supernatant was collected at regular 24 h intervals and the absorbance of the supernatant was measured at 524 nm on a Tecan Infinite 200 (Tecan Group Ltd. Männedorf, Switzerland) plate reader from 400 nm to 700 nm. The calibration curve based on a standard series of AuNP and AuNP-miRs (0.0–1.0 nM) is given by the following equations: Abs 524 nm = 0.299 × [AuNP] + 0.0044, R2 = 0.9984, and Abs 524 nm = 1.4239 × [AuNP-miRs] – 0.0048, R2 = 0.9986, respectively.HEK293 cell culture
Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; ThermoFisher Gibco, supplemented with 10% FBS (ThemoFischer Gibco), 1% Pen/Strep (ThermoFisher Gibco 15070063), 1% Non-Essential Amino Acids NEAA (ThermoFisher Gibco11140050), 1% Sodium Pyruvate (ThermoFisher Gibco, 11360070), on 2% gelatin coated until confluent. For experiments, cells were seeded at 25000 cells per well in media (100 μL) in a 96 well plate.
To assess the suppression efficiency of AuNP-miR-214, HEK293 cells were co-transfected with pMIR-REPORT-QKI-3′ UTR Luciferase vector (100 ng) and a pMIR-REPORT β-Gal control plasmid (100 ng) in OptiMEM for 4 h to evaluate transfection efficiency. Subsequently, pre-miR-214 (60 nM) was delivered using Lipofectamine 3000 (ThermoFisher, L3000015). AuNP-miR-214 and AuNP-miR-67, as a scramble control microRNA, were added directly to the OptiMEM. Luciferase and β-Gal activity was assessed after 48 h with the Steady-Glo Luciferase Assay Kit (Promega E2520) and β-Gal assay buffer (200 mM sodium phosphate, 2 mM magnesium chloride, 100 mM beta-mercaptoethanol, 1.33 mg mL−1ortho-nitrophenyl-β-galactoside (ONPG) in water), respectively. Luciferase activity was assessed by measuring luminescence and β-Gal activity was determined by measuring absorbance at 405 nm and 570 nm. Six biological replicates were transfected per conditions. Experiments were conducted in triplicate.
3D Calcific aortic valve disease (CAVD) model
A 3D in vitro model of human CAVD was employed to image AuNP-miRs uptake into cells. We previously published a detailed description and validation of this model.30 Briefly, human aortic valves were obtained from patients undergoing valve replacement surgery at Brigham and Women's Hospital (Boston, MA, USA) as a result of aortic valve (AV) calcific stenosis. Leaflets were obtained and utilized in accordance with protocols approved by the Institutional Review Board (IRB protocol #2011P001703/PHS). Valvular interstitial cells (VICs) were isolated as previously described.48 In short, CAVD AV leaflets were cut into 5 mm × 5 mm pieces, incubated in collagenase (Roche 10103586001; 10 mL) solution at 37 °C, 5% CO2 for 12 h, and homogenized with a serological pipette. The digested tissue was centrifuged, the supernatant aspirated, and the pellet resuspended in VIC cell culture media [DMEM supplemented with 10% FBS (ThermoFisher, Gibco) and 1% P/S (ThermoFisher, Gibco); 5 mL]. The cells were then centrifuged a second time, resuspended in VIC cell culture media (10 mL), and plated in a T75 culture flask. Media was replenished every 48 h. VICs of passage five were used for further experiments. VICs were incorporated in hydrogel pre-polymers by mixing VICs and media (to a final concentration of 10 × 106 cells per mL in the gel) with a GelMA (10 wt%), HAMA (3 wt%), and LAP solution (5 wt%) at 37 °C to form a GelMA (5%), HAMA (1%), and LAP (0.3%) hydrogel. GelMA and HAMA were synthesized as previously described.49,50Constructs were designed in Tinkercad (AutoDesk, Inc., San Rafael, CA, USA), and encoded using Repetier-Host (version 2.0.0; Hot-World GmbH & Co. KG, Willich, Germany), and Sublime Text 3 (Sublime HQ, Pty Ltd., Darlinghurst, NSW, Australia). 3D bioprinting was performed using the Inkredible + (Cellink, Cambridge, MA, USA). Pluronic gel (Pluronic F-127; Allevi, Philadelphia, PA, USA) was printed as a cylindrical mold (outer diameter = 9.0 mm, inner diameter = 8.6 mm, height = 1.5 mm) using a stainless steel needle nozzle (JG27-0.25HPX; Jensen Global Inc., Santa Barbara, CA, USA) at a fill density of 95%, layer height of 0.1 mm, printing speed of 4 mm s−1, and printing pressure of 320 kPa. The second extruder was filled with the required hydrogel pre-polymer compositions and heated to 37 °C. A hydrogel disc was then printed inside the mold from the second extruder using a 23 g stainless steel nozzle (Fisnar 5901005, Ellsworth Adhesives, Germantown, WI, USA) by opening the valve of the second extruder for 40 ms at 15–20 kPa. Cross-linking the pre-polymers for 90 s with 365 nm UV light produced 8.6 mm × 1.0 mm hydrogel discs. The UV light was calibrated to an intensity of 2.5 mW cm−2 using a radiometer (85009, Sper Scientific Direct, Scottsdale, AZ, USA). After printing, the Pluronic gel was dissolved by washing in cold PBS. One day after printing, hydrogels were switched to normal media (NM; 10% FBS, 1% P/S) or osteogenic media (NM supplemented with 10 nM dexamethasone, 10 ng mL−1 ascorbic acid, and 10 mM β-glycerolphosphate) as previously described,51 for up to 14 days. Media was changed every 48 h for all constructs. Cells were transfected with miR-214 (30 nM) by addition of AuNP-miR-214 to the cell culture media 48 h prior to imaging.
CellTracker/LysoTracker and confocal imaging
To trace the AuNP-miRs into the cells in the 3D CAVD model LysoTracker Red (ThermoFisher, L7528; 50 nM), CellTracker Green (ThermoFisher, C2925; 10 mM) dyes, and Hoechst 33342 (ThermoFisher, H1399) were added according to the manufacturer's protocol. CellTracker Green was incubated overnight at 37 °C, 5% CO2, LysoTracker Red was incubated for 1 h at 37 °C, 5% CO2, Hoechst (1:5000) was added and incubated for 30 min at 37 °C, 5% CO2, prior to imaging. Z-stack images of the constructs were taken on a ZEISS LSM 880 Confocal Microscope with AiryScan.
Animal procedures
All animal procedures were performed according to MIT Animal Care and Use Committee approved protocols. Adult male C57BL/6 mice were injected subcutaneously on the back with PNP-AuNP-miR-67 gels (100 μL; HPMC-C12 1 wt%; PEG-b-PLA NPs 10 wt%; AuNP-miR-67 85 nM) using a 26G syringe. At 1, 4, and 11 days following injection 3 animals per time point were sacrificed and lung, spleen, liver, kidneys and the tissue surrounding the injection site were harvested, flash-frozen, and stored until further analysis. Fluorescence reflectance of tissues was measured using a Kodak Image Station 4000 mm Pro.Statistics
Quantitative data is given as mean ± standard error. The number of independent experiments is given as n. Statistical analyses were performed with GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA), Student's t-tests were performed for two-group comparisons, and for multiple group comparisons one-way or two-way ANOVA with Tukey's post-hoc HSD tests were used as appropriate. P-Values < 0.05 were considered statistically significant.Conclusion
In conclusion, our data demonstrate that AuNP-miRs can be delivered via a minimally invasive, injectable self-assembling hydrogel, expressing controlled release. We showed that approximately 20% of AuNP-miRs were released from the PNP hydrogel over 5 days. Additionally, we observed that AuNP-miR-214 remained functional after delivery in both a 2D in vitro miR-214 target luciferase reporter assay and in a 3D bioprinted human CAVD model. Furthermore, we demonstrated that AuNP-miRs were cleared after 11 days in mice following subcutaneous injection of the PNP-AuNP-miR hydrogel delivery system. Without any cell-targeting ligands, these subcutaneously administered AuNP-miRs were taken up by the spleen, lungs, and liver, and were disposed via renal excretion. Additional targeting ligands and site- specific delivery could tailor this platform to specific cells or tissues, especially in combination with RNAi molecules adapted to a specific tissue, cell type, or disease.Author contributions
Conceptualization: CV, MWT, JC, EA. Data curation: – Formal Analysis: CV, MWT, JC. Funding acquisition: CV, PD, JS, EA, RL. Investigation: CV, MWT, JC. Methodology: CV, MWT, JC, AM. Project administration: EA, RL. Resources: AM, JH, JS, EA, RL. Software: – Supervision: AM, JH, JS, EA, RL. Validation: CV, MWT, JC. Visualization: CV, MWT, JC. Writing – original draft: CV, MWT, JC. Writing – review & editing: AM, JH, PD, JS, EA, RL.Conflicts of interest
J.C. is a co-founder and shareholder of TargTex S.A.Acknowledgements
This research was funded by the National Institutes of Health (NIH) R01 grants R01HL 141719, R01HL136431 and R01HL147095 (E.A.); the National Institutes of Health grant EB000244 (to R.L.); the Netherlands CardioVascular Research Initiative (CVON: The Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science) and Vrienden UMC Utrecht (C.V., J.S.); an unrestricted grant from CELLINK to Vrienden UMC Utrecht (C.V., J.S.); the Harvard Catalyst Advanced Microscopy Pilot grants (C.V., E.A.); and the NIH Ruth L. Kirschstein National Research Service Award F32HL122009 (M.W.T.). J.C. acknowledges the European Research Council Starting Grant (ERC-StG-2019-848325). This work was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102) and financial contributions from Harvard University and its affiliated academic healthcare centers. We thank the Harvard Center for Biological Imaging for infrastructure and support, in particular Dr D. Richardson and S. Terclavers. The authors would like to thank Elia Guzzi for assistance with hydrogel preparation.References
- A. Eulalio, M. Mano, M. Dal Ferro, L. Zentilin, G. Sinagra, S. Zacchigna and M. Giacca, Nature, 2012, 492, 376 CrossRef CAS PubMed.
- C. Wahlquist, D. Jeong, A. Rojas-Muñoz, C. Kho, A. Lee, S. Mitsuyama, A. van Mil, W. J. Park, J. P. Sluijter, P. A. Doevendans, R. F. Hajjar and M. Mercola, Nature, 2014, 508, 531 CrossRef CAS PubMed.
- A. Aguirre, N. Montserrat, S. Zacchigna, E. Nivet, T. Hishida, M. N. Krause, L. Kurian, A. Ocampo, E. Vazquez-Ferrer, C. Rodriguez-Esteban, S. Kumar, J. J. Moresco, J. R. Yates, 3rd, J. M. Campistol, I. Sancho-Martinez, M. Giacca and J. C. Izpisua Belmonte, Cell Stem Cell, 2014, 15, 589 CrossRef CAS PubMed.
- H. Hosseini, M. M. S. Obradović, M. Hoffmann, K. L. Harper, M. S. Sosa, M. Werner-Klein, L. K. Nanduri, C. Werno, C. Ehrl, M. Maneck, N. Patwary, G. Haunschild, M. Gužvić, C. Reimelt, M. Grauvogl, N. Eichner, F. Weber, A. D. Hartkopf, F. A. Taran, S. Y. Brucker, T. Fehm, B. Rack, S. Buchholz, R. Spang, G. Meister, J. A. Aguirre-Ghiso and C. A. Klein, Nature, 2016, 540, 552 CrossRef CAS PubMed.
- J. Su, S. M. Morgani, C. J. David, Q. Wang, E. E. Er, Y. H. Huang, H. Basnet, Y. Zou, W. Shu, R. K. Soni, R. C. Hendrickson, A. K. Hadjantonakis and J. Massagué, Nature, 2020, 577, 566 CrossRef CAS PubMed.
- M. H. van der Ree, J. M. de Vree, F. Stelma, S. Willemse, M. van der Valk, S. Rietdijk, R. Molenkamp, J. Schinkel, A. C. van Nuenen, U. Beuers, S. Hadi, M. Harbers, E. van der Veer, K. Liu, J. Grundy, A. K. Patick, A. Pavlicek, J. Blem, M. Huang, P. Grint, S. Neben, N. W. Gibson, N. A. Kootstra and H. W. Reesink, Lancet, 2017, 389, 709 CrossRef CAS.
- D. S. Hong, Y. K. Kang, M. Borad, J. Sachdev, S. Ejadi, H. Y. Lim, A. J. Brenner, K. Park, J. L. Lee, T. Y. Kim, S. Shin, C. R. Becerra, G. Falchook, J. Stoudemire, D. Martin, K. Kelnar, H. Peltier, V. Bonato, A. G. Bader, S. Smith, S. Kim, V. O'Neill and M. S. Beg. 2020 Apr 2. Epub ahead of print.
- S. Oliveira, I. van Rooy, O. Kranenburg, G. Storm and R. M. Schiffelers, Int. J. Pharm., 2007, 331, 211 CrossRef CAS PubMed.
- A. van Mil, P. A. Doevendans and J. P. Sluijter, Mini-Rev. Med. Chem., 2009, 9, 235 CrossRef CAS PubMed.
- O. S. Fenton, K. J. Kauffman, J. C. Kaczmarek, R. L. McClellan, S. Jhunjhunwala, M. W. Tibbitt, M. D. Zeng, E. A. Appel, J. R. Dorkin, F. F. Mir, J. H. Yang, M. A. Oberli, M. W. Heartlein, F. DeRosa, R. Langer and D. G. Anderson, Adv. Mater., 2017, 29(33) DOI:10.1002/adma.201606944.
- R. F. Kwekkeboom, J. P. Sluijter, B. J. van Middelaar, C. H. Metz, M. A. Brans, O. Kamp, W. J. Paulus and R. J. Musters, J. Controlled Release, 2016, 222, 18 CrossRef CAS PubMed.
- A. Kargaard, J. P. G. Sluijter and B. Klumperman, J. Controlled Release, 2019, 316, 263 CrossRef CAS PubMed.
- J. Conde, N. Oliva, Y. Zhang and N. Artzi, Nat. Mater., 2016, 15, 1128 CrossRef CAS PubMed.
- C. F. T. van der Ven, P. J. Wu, M. W. Tibbitt, A. van Mil, J. P. G. Sluijter, R. Langer and E. Aikawa, Clin. Sci., 2017, 131, 181 CrossRef CAS PubMed.
- M. J. Hernandez, R. Gaetani, V. M. Pieters, N. W. Ng, A. E. Chang, T. R. Martin, E. van Ingen, E. A. Mol, J. P. G. Sluijter and K. L. Christman, Adv. Ther. (Weinh), 2018, 1(3), 1800032 CrossRef PubMed.
- N. Bayat, Y. Zhang, P. Falabella, R. Menefee, J. J. Whalen 3rd, M. S. Humayun and M. E. Thompson, Sci. Transl. Med., 2017, 9(419), eaan3879 CrossRef PubMed.
- D. Q. Wu, T. Wang, B. Lu, X. D. Xu, S. X. Cheng, X. J. Jiang, X. Z. Zhang and R. X. Zhuo, Langmuir, 2008, 24, 10306 CrossRef CAS PubMed.
- J. A. Burdick and K. S. Anseth, Biomaterials, 2002, 23, 4315 CrossRef CAS PubMed.
- Y. S. Pek, A. C. A. Wan, A. Shekaran, L. Zhou and J. Y. A. Ying, Nat. Nanotechnol., 2008, 3, 671 CrossRef CAS PubMed.
- M. M. Bastings, S. Koudstaal, R. E. Kieltyka, Y. Nakano, A. C. Pape, D. A. Feyen, F. J. van Slochteren, P. A. Doevendans, J. P. Sluijter, E. W. Meijer, S. A. Chamuleau and P. Y. Dankers, Adv. Healthcare Mater., 2014, 3, 70 CrossRef CAS PubMed.
- M. H. Bakker, E. van Rooij and P. Y. W. Dankers, Chem. – Asian J., 2018, 13, 3501 CrossRef CAS PubMed.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389 CrossRef PubMed.
- H. D. Lu, M. B. Charati, I. L. Kim and J. A. Burdick, Biomaterials, 2012, 33, 2145 CrossRef CAS PubMed.
- C. B. Rodell, A. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125 CrossRef CAS PubMed.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS PubMed.
- O. S. Fenton, M. W. Tibbitt, E. A. Appel, S. Jhunjhunwala, M. J. Webber and R. Langer, Biomacromolecules, 2019, 20, 4430 CrossRef CAS PubMed.
- A. Gilam, J. Conde, D. Weissglas-Volkov, N. Oliva, E. Friedman, N. Artzi and N. Shomron, Nat. Commun., 2016, 7, 12868 CrossRef PubMed.
- J. Conde, N. Oliva and N. Artzi, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, E1278–E1287 CrossRef CAS PubMed.
- J. Conde, A. Ambrosone, V. Sanz, Y. Hernández, V. Marchesano, F. Tian, H. Child, C. C. Berry, M. R. Ibarra, P. V. Baptista, C. Tortiglione and J. M. de la Fuente, ACS Nano, 2012, 6, 8316–8324 CrossRef CAS PubMed.
- D. C. van der Valk, C. F. T. van der Ven, M. C. Blaser, J. M. Grolman, P. J. Wu, O. S. Fenton, L. H. Lee, M. W. Tibbitt, J. L. Andresen, J. R. Wen, A. H. Ha, F. Buffolo, A. van Mil, C. V. C. Bouten, S. C. Body, D. J. Mooney, J. P. G. Sluijter, M. Aikawa, J. Hjortnaes, R. Langer and E. Aikawa, Nanomaterials, 2018, 8, 296 CrossRef PubMed.
- E. A. Appel, M. W. Tibbitt, J. M. Greer, O. S. Fenton, K. Kreuels, D. G. Anderson and R. Langer, ACS Macro Lett., 2015, 4, 848 CrossRef CAS.
- E. A. Mol, Z. Lei, M. T. Roefs, M. H. Bakker, M. J. Goumans, P. A. Doevendans, P. Y. W. Dankers, P. Vader and J. P. G. Sluijter, Adv. Healthcare Mater., 2019, 8(20), e1900847 CrossRef PubMed.
- M. D. Krebs, O. Jeon and E. Alsberg, J. Am. Chem. Soc., 2009, 131, 9204 CrossRef CAS PubMed.
- A. Van Mil, S. Grundmann, M. J. Goumans, Z. Lei, M. I. Oerlemans, S. Jaksani, P. A. Doevendans and J. P. Sluijter, Cardiovasc. Res., 2012, 93, 655 CrossRef CAS PubMed.
- N. Segovia, M. Pont, N. Oliva, V. Ramos, S. Borrós and N. Artzi, Adv. Healthcare Mater., 2015, 4, 271 CrossRef CAS PubMed.
- M. K. Nguyen, O. Jeon, M. D. Krebs, D. Schapira and E. Alsberg, Biomaterials, 2014, 35, 6278 CrossRef CAS PubMed.
- D. Zheng, Y. Zang, H. Xu, Y. Wang, X. Cao, T. Wang, M. Pan, J. Shi and X. Li, Clin. Res. Cardiol., 2019, 108, 691 CrossRef CAS PubMed.
- E. Aikawa, M. Nahrendorf, D. Sosnovik, V. M. Lok, F. A. Jaffer, M. Aikawa and R. Weissleder, Circulation, 2007, 115, 377 CrossRef CAS PubMed.
- J. Hjortnaes, C. Goettsch, J. D. Hutcheson, G. Camci-Unal, L. Lax, K. Scherer, S. Body, F. J. Schoen, J. Kluin, A. Khademhosseini and E. Aikawa, J. Mol. Cell Cardiol., 2016, 94, 13 CrossRef CAS PubMed.
- S. Rathan, C. J. Ankeny, S. Arjunon, Z. Ferdous, S. Kumar, J. Fernandez Esmerats, J. M. Heath, R. M. Nerem, A. P. Yoganathan and H. Jo, Sci. Rep., 2016, 6, 25397 CrossRef CAS PubMed.
- H. X. Xu, Y. Wang, D. D. Zheng, T. Wang, M. Pan, J. H. Shi, J. H. Zhu and X. F. Li, Clin. Lab., 2017, 63, 1163 CAS.
- M. I. Jan, R. A. Khan, T. Ali, M. Bilal, L. Bo, A. Sajid, A. Malik, N. Urehman, N. Waseem, J. Nawab, M. Ali, A. Majeed, H. Ahmad, S. Aslam, S. Hamera, A. Sultan, M. Anees, Q. Javed and I. Murtaza, Arch. Biochem. Biophys., 2017, 633, 50 CrossRef CAS PubMed.
- S. Coffey, M. J. Williams, L. V. Phillips, I. F. Galvin, R. W. Bunton and G. T. Jones, Sci. Rep., 2016, 6, 36904 CrossRef CAS PubMed.
- R. Song, D. A. Fullerton, L. Ao, K. S. Zhao, T. B. Reece, J. C. Jr. Cleveland and X. Meng, J. Am. Heart Assoc., 2017, 6, e005364 Search PubMed.
- H. Wang, J. Shi, B. Li, Q. Zhou, X. Kong and Y. Bei, BioMed Res. Int., 2017, 2017, 4820275 Search PubMed.
- E. Aikawa and P. A. Libby, Circulation, 2017, 135, 1951 CrossRef PubMed.
- A. N. Steele, L. M. Stapleton, J. M. Farry, H. J. Lucian, M. J. Paulsen, A. Eskandari, C. E. Hironaka, A. D. Thakore, H. Wang, A. C. Yu, D. Chan, E. A. Appel and Y. J. Woo, Adv. Healthcare Mater., 2019, 8(5), e1801147 CrossRef PubMed.
- R. A. Gould and J. T. Butcher, J. Visualized Exp., 2010,(46), 2158 Search PubMed.
- J. W. Nichol, S. T. Koshy, H. Bae, C. M. Hwang, S. Yamanlar and A. Khademhosseini, Biomaterials, 2010, 31, 5536 CrossRef CAS PubMed.
- J. A. Burdick, C. Chung, X. Jia, M. A. Randolph and R. Langer, Biomacromolecules, 2005, 6, 386 CrossRef CAS PubMed.
- J. D. Hutcheson, C. Goettsch, S. Bertazzo, N. Maldonado, J. L. Ruiz, W. Goh, K. Yabusaki, T. Faits, C. Bouten and G. Franck, Nat. Mater., 2016, 15, 335 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available See DOI: 10.1039/d1nr04973a |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |