
Limonene-in-water Pickering emulsion and on-demand separation using thermo-responsive biodegradable nanoparticles†
Nicolò
Manfredini
 ,
Manuel
Merigo
,
Juri
Ilare
,
Mattia
Sponchioni
* and
Davide
Moscatelli
,
Manuel
Merigo
,
Juri
Ilare
,
Mattia
Sponchioni
* and
Davide
Moscatelli
Department of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Mancinelli 7, 20131 Milano, Italy. E-mail: mattia.sponchioni@polimi.it
First published on 5th April 2021
Abstract
In the last few decades, Pickering emulsions have regained attention due to the possibility of forming stable oil-in-water emulsions with interesting interfacial properties. As an example, the more and more stringent regulations on the products for home and personal care are pushing the market towards the use of biodegradable materials in order to reduce their environmental impact. In this scenario, an appealing opportunity is offered by the use of biodegradable polymeric nanoparticles (NPs) for the stabilization of fragrance oils in water. In this work, modular biodegradable NPs have been synthesized through a combination of ring opening polymerization and reversible addition–fragmentation chain transfer emulsion polymerization and used to produce limonene-in-water Pickering emulsions. This strategy allowed controlling independently the NP size, polymer molecular weight, and hydrophobicity acting on the microstructure of the constituting copolymers. Stable limonene-in-water Pickering emulsions could be obtained, with the size of the oil phase and the wetting by limonene that can be strictly controlled by tuning the NP physico-chemical properties. Finally, the adoption of thermo-responsive polymer chains within the shell of the Pickering emulsifiers enabled the on-demand destabilization of the emulsions and hence the selective dispensing of limonene by simply increasing the temperature.
1. Introduction
Emulsion stabilization by means of solid particles, or Pickering emulsions, has been known since the beginning of the previous century.1,2 Nonetheless, Pickering emulsions were scarcely investigated for a long period of time. Only in the last few decades, a renewed interest towards this technology arose as confirmed from the many types of particles (e.g. silica,3–5 inorganic clays,6–8 barium sulfate,9 polystyrene10,11 and calcium carbonate12,13) that have been used to produce stable emulsions.One of the main reasons for this renewed attention towards Pickering emulsions is the recent advancement in the synthesis of engineered nanoparticles (NPs) able to provide emulsions with interesting interfacial properties and behaviours. Among all the different NPs developed so far, an interesting opportunity to cope with the rigid regulations on the production of nano- and micro-plastics14 is constituted by biodegradable NPs.
In this scenario, many different natural particles have been successfully used to stabilize oil-in-water Pickering emulsions.15–17 However, the production of natural colloids is usually characterized by an intrinsically high batch-to-batch variability and a reduced control over the final properties. In this scenario, a valuable alternative is constituted by synthetic biodegradable polymer NPs. In fact, the incomparable versatility offered by these materials can be combined with the progressive hydrolysis of labile bonds in the polymer chains ensuring the formation of non-toxic and low molecular weight degradation products and in turn no polymer accumulation in the environment. The current trend towards the production of these colloids is based on the synthesis of amphiphilic block copolymers able to self-assemble in water into NPs. These copolymers often comprise a biodegradable and lipophilic polyester portion forming the NP core and a hydrophilic block forming the outer shell responsible for its stabilization.18–20
In this direction, Laredj-Bourezg et al.21 produced biodegradable NPs by nanoprecipitation of poly(caprolactone)-b-poly(ethylene glycol) (PCL-PEG) block copolymers synthesized via ring opening polymerization (ROP) of ε-caprolactone using PEG as an initiator. Although these NPs were successfully employed to obtain oil-in-water Pickering emulsions, the manufacturing procedure intrinsically limits the final latex concentration.22,23 Moreover, one single parameter can be varied in the polymer synthesis, i.e. the length of the poly(caprolactone) chain, through the modulation of the molar ratio between ε-caprolactone and PEG. This evidently reduces the control over the NP properties (i.e. morphology, size, degradation time, wettability) and in turn over those of the final Pickering emulsions.
On the other hand, the combination of ROP with a controlled radical polymerization offers the unique possibility of accessing polymer structures with up to three degrees of freedom and hence a strict control over the NP properties.24
Another aspect of using NPs for the stabilization of emulsions is their strong adherence to the interface. As a matter of fact, the energy required for desorption of these colloids is usually much higher than their thermal energy,25,26 thus preventing NP detachment from the surface. This detachment energy mainly depends on the wettability angle of the particle, the interfacial tension between the two phases and the particle radius, with the bigger particles strongly held at the interphase.25 This strong interaction confers excellent long-term stability to Pickering emulsions, but it may limit their application in those cases where on-demand separation of the emulsion is required (i.e. the release of a drug or fragrance). In the literature, several approaches based on the use of smart materials, i.e. materials able to change their properties in response to an external stimulus, are reported27 to achieve this desirable effect. Among them, the more investigated are Pickering emulsions sensitive to changes in pH,28–31 CO232,33 and temperature.26,34 Thermo-responsive polymers, which undergo phase separation from water in response to a change in temperature,35 are certainly among the most appealing due to the frequent occurrence of temperature gradients. The exploitation of thermal stimuli, either naturally occurring or artificially generated, for the destabilization of Pickering emulsions is an elegant strategy for the on-demand release of the fragrance or other active components within the droplet phase.
In this work, we investigated the possibility of employing thermo-responsive biodegradable NPs to stabilize limonene-in-water Pickering emulsions, with limonene considered because of its widespread use as a fragrance oil. The polymer colloids adopted for this purpose were produced via a combination of ROP and reversible addition–fragmentation chain transfer (RAFT) emulsion polymerization, allowing great control over the NP physico-chemical properties. The synthesis strategy is schematically depicted in Scheme 1. In particular, two oligoester-based macromonomers were synthesized via ROP of ε-caprolactone (CL) using 2-hydroxyethyl methacrylate (HEMA) as an initiator and targeting different chain lengths. Then, two different poly(ethylene glycol)methyl ether methacrylate (EG4)-based macromolecular chain transfer agents (macro-CTA) were synthesized by RAFT polymerization. These macro-CTAs, displaying a lower critical solution temperature (LCST) in water, were finally chain-extended with the lipophilic macromonomers through RAFT emulsion polymerization in order to produce a library of NPs composed of amphiphilic block copolymers. The poly(oligoester) chains are expected to constitute the core of these NPs, with the polyEG4 stabilizing their surface. With this combination of ROP and RAFT polymerization, we could finely control three parameters, namely the length of both the hydrophilic and biodegradable blocks of the copolymer and the length of the macromonomer. After having elucidated the influence of these parameters over the NP physico-chemical properties, these colloids were used to obtain stable limonene-in-water Pickering emulsions. The influence of NP size and composition of the lipophilic core over the Pickering emulsion was systematically evaluated. Finally, the on-demand separation of limonene by application of external heat was investigated.
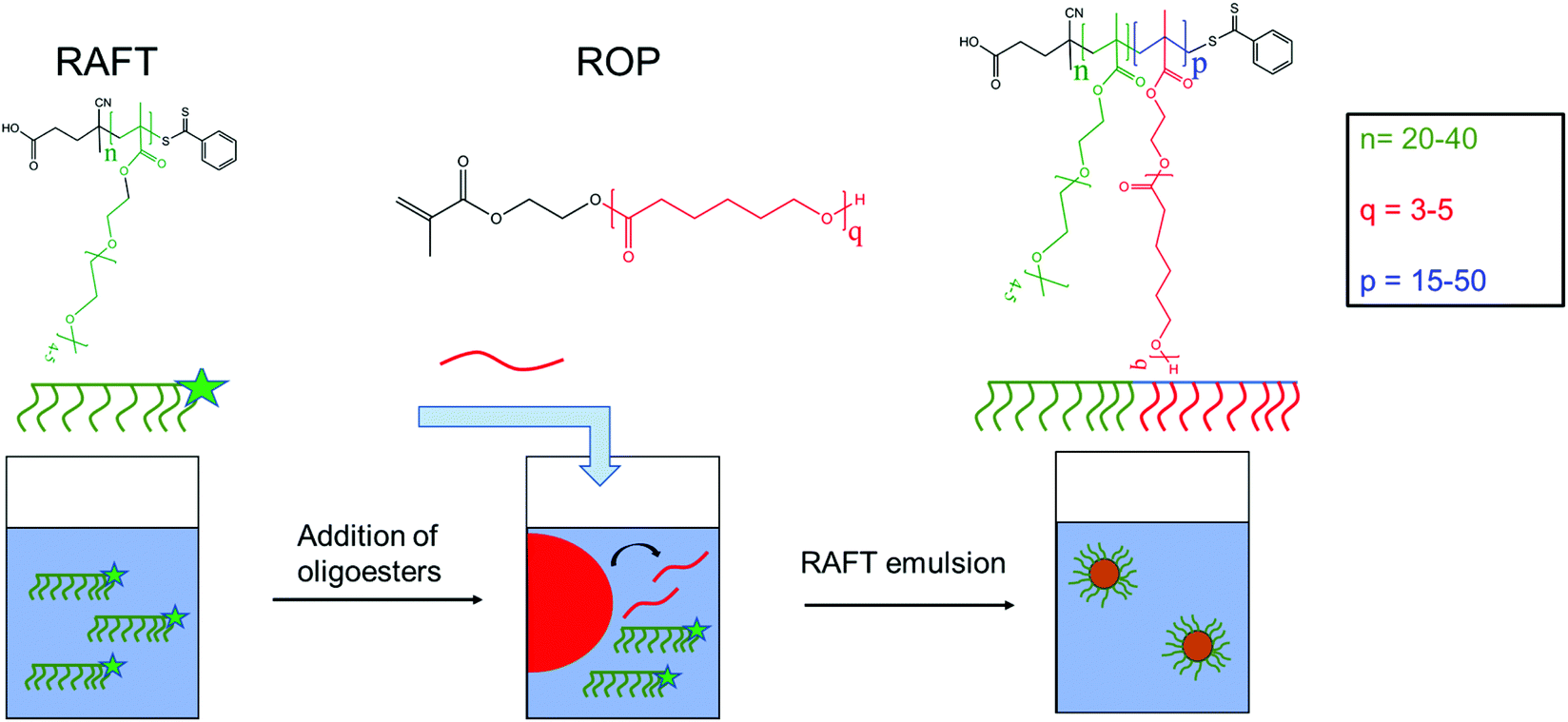 | ||
| Scheme 1 Schematic representation of the production of thermo-responsive and biodegradable NPs with three degrees of freedom, namely n, q and p, through the combination of ROP and RAFT polymerization. | ||
To the best of our knowledge, this is the first study in which thermo-responsive and biodegradable NPs are used to produce stable oil-in-water Pickering emulsions and could pave the way to relevant applications when the on-demand release of the dispersed phase is required.
2. Experimental section
2.1 Materials
ε-Caprolactone (CL, 97%, MW = 114.14, Sigma Aldrich), 2-hydroxyethyl methacrylate (HEMA, 97%, MW = 130.14, Sigma-Aldrich), tin(II) octanoate (Sn(Oct)2, MW = 405.12, Sigma-Aldrich), sodium sulphate (Na2SO4, >99%, Sigma-Aldrich), rhodamine B (Rh, MW = 479.01, Sigma-Aldrich), N,N′-dicyclohexylcarbodiimide (DCC, ≥99%, MW = 206.33, Sigma-Aldrich), 4-(dimethylamino)pyridine (DMAP, ≥99%, MW = 122.17, Sigma-Aldrich), anhydrous dichloromethane (DCM, 99.8%, Sigma-Aldrich), 2,2′-azobis(2-methylpropionamidine)dihydrochloride (V-50, MW = 271.19, 98%, Acros Organics), 4-cyano-4-(phenyl-carbonothioylthio)pentanoic acid (CPA, MW = 279.38, 99%, Sigma-Aldrich), 4-4′-azobis(4-cyanovaleric acid) (ACVA, MW = 280.28, Sigma-Aldrich), deuterated chloroform (CDCl3, 99.8%, MW = 120.38, Sigma-Aldrich), ethanol (EtOH, MW = 46.07, ≥99.8%, Sigma-Aldrich), di-isopropyl ether (Dipe, MW = 102.17, ≥99.8%, Sigma-Aldrich), poly(ethylene glycol)methyl ether methacrylate (EG4, MW = 300, Sigma-Aldrich), and tetrahydrofuran (THF, ≥99.9%, MW = 72.11, Sigma-Aldrich) were of analytical grade purity and used as received.2.2 Synthesis of oligoester-based macromonomers via ROP
Two oligoesters were synthesized via bulk ROP of ε-caprolactone. The polymerization was carried out using Sn(Oct)2 as a catalyst and HEMA as an initiator in order to provide the oligoester with a methacrylate group making it amenable for further radical chemistry. The monomer to initiator molar ratio (q) was set to either 3 or 5 in order to tune the degree of polymerization, while the initiator to Sn(Oct)2 molar ratio was set equal to 200. Hereinafter, the macromonomers will be referred to as HEMACLq.As an example, for the synthesis of HEMACL3 (q = 3), 26.30 mg of Na2SO4, used to keep the system anhydrous, and 26.30 g of CL (0.23 mol) were weighed in a round bottomed flask equipped with a magnetic stirrer and placed in an oil bath at 125 °C under stirring. Meanwhile, 10.00 g of HEMA (77 mmol) and 156 mg of Sn(Oct)2 (0.38 mmol, i.e. HEMA/Sn(Oct)2 = 200) were weighed in a vial and mixed with the aid of a vortex stirrer for 10 minutes. Then, the HEMA/Sn(Oct)2 mixture was withdrawn with a syringe and poured into the flask with CL and Na2SO4 and the reaction was carried out for 2.5 hours at 125 °C under vigorous stirring. The reaction was finally quenched by cooling the flask in a water/ice bath.
All the samples were characterized via proton nuclear magnetic resonance (1H-NMR) analysis on a Bruker Ultrashield 400 MHz spectrometer (the macromonomer structure and the 1H-NMR spectrum are reported in Fig. S1†) dissolving 10 mg of the product in 0.7 mL of CDCl3 in order to calculate the monomer conversion (eqn (S1)†) and the degree of polymerization (eqn (S2)†). The results are reported in Table S1†. The oligomer molecular weight distribution was evaluated via organic gel permeation chromatography (OGPC) on a Jasco LC-2000Plus apparatus (Table S1†). For GPC analysis, the samples were dissolved at a concentration equal to 4 mg mL−1 in THF and filtered through a 0.45 μm polytetrafluoroethylene (PTFE) membrane before injection. The separation was performed in THF at 35 °C with a flow rate of 0.5 mL min−1 and three styrene/divinyl benzene columns (Polymer Standard Service; pore sizes 103, 105, and 106 Å; 300 mm length; and 8 mm internal diameter) and a pre-column (50 mm length and 8 mm internal diameter). The signal was recorded with a refractive index (RI) detector. A calibration curve was applied based on polystyrene standards from 580 to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 250000 Da (Polymer Laboratories).
250000 Da (Polymer Laboratories).
2.3 Synthesis of the fluorescent monomer HEMA-Rh
A fluorescent monomer (HEMA-Rh) was synthesized by DCC-mediated esterification of HEMA and Rhodamine B, following a protocol available in the literature.36 In particular, 50 mg of Rh (0.1 mmol), 135 mg of HEMA (1 mmol), 43 mg of DCC (0.2 mmol, i.e. DCC/Rh = 2 mol mol−1), 2.55 mg of DMAP (0.02 mmol, i.e. DMAP/Rh = 0.2 mol mol−1) and 63 g of anhydrous DCM were weighed in a round bottom flask equipped with a magnetic stirrer. In order to prevent the Rh photo-bleaching, the flask was wrapped in tinfoil paper. The reaction was carried out for 24 hours under vigorous stirring at room temperature. The solution was finally collected and stored at 4 °C.2.4 Synthesis of thermo-responsive macro-CTAs
The thermo-responsive macro-CTAs were synthesized via RAFT solution polymerization of EG4 with ACVA as an initiator, ethanol as a solvent and CPA as a CTA. In particular, two different polymers were obtained by setting the monomer to CPA molar ratio (n) equal to 20 and 40, respectively. This ratio coincides also with the theoretical average degree of polymerization of the resulting macro-CTAs. The initiator to CTA molar ratio (IA) was kept equal to 1/3, while the solution mass concentration was set equal to 20% w/w.As an example, for the synthesis of the macro-CTA with n = 40, hereinafter 40EG4, 10 g of EG4 (33 mmol), 0.232 g of CPA (0.8 mmol) and 0.078 g of ACVA (0.03 mmol) were weighed in a round bottom flask with a magnetic stirrer. Then, 41 g of ethanol were added and the solution was purged with nitrogen for 15 minutes. Finally, the mixture was heated to 65 °C in an oil bath and the reaction was carried out for 24 hours under magnetic stirring.
At the end of the polymerization, the product was double precipitated in a 10-fold excess of di-isopropyl ether and centrifuged at 4000 rpm for 10 minutes. The precipitated polymer was recovered, dried under air and stored at −20 °C. The conversion (eqn (S3)†) and degree of polymerization (eqn (S4)†) were calculated by dissolving 10 mg of polymer in 0.7 mL of CDCl3 and analysing the solution via1H-NMR with 64 scans (see the structure and 1H-NMR spectrum in Fig. S2†).
The molecular weight distribution was evaluated via GPC in the same way as described in section 2.2. All the values obtained are listed in Table S2.†
2.5 Synthesis of thermo-responsive biodegradable NPs
The biodegradable NPs with a thermo-responsive shell were obtained via RAFT emulsion polymerization of HEMACLq using nEG4 as the macro-CTA. In particular, the reaction was carried out at 10% w/w in a mixture of 70/30 v/v water/ethanol using V-50 as an initiator, in a mole ratio of 1/3 mol mol−1 with respect to nEG4. Different HEMACLq/nEG4 mole ratios (p) were tested in order to investigate the influence of a different number of lipophilic units added to the polymer chains over the stabilization of Pickering emulsions. Hereinafter, we will refer to the NP suspensions as nEG4-pCLq in order to highlight the theoretical average degrees of polymerization for the two segments in the constituting block copolymers (n and p) and for the macromonomer employed (q).As an example, for the synthesis of 40EG4-50CL3, 1 g of HEMACL3, 0.189 g of 40EG4, 6.54 g of deionized water and 2.8 g of ethanol were mixed in a round bottom flask equipped with a magnetic stirrer and a rubber cap. The solution was purged with nitrogen for 15 minutes in order to remove the oxygen and heated to 50 °C in an oil bath. Then, 1 mg of V-50 in 0.5 g of deionized water was added to the flask and the reaction was allowed to proceed for 24 hours. The quenching was done by cooling the mixture in ice-cold water. Before usage, the NP suspensions were dialyzed against deionized water for 3 days using regenerated cellulose membranes (Spectra/Por) with a molecular weight cut-off of 3500 Da. The water was changed every 2 h to ensure appropriate sink conditions. In the case of fluorescent NPs, the same procedure was followed with the addition of 1 single unit of HEMA-Rh per chain (HEMA-Rh/HEMACLq = 1/p mol mol−1). The HEMA-Rh solution was weighed in the flask and the DCM was evaporated before the addition of other reactants following the same strategy reported above.
Monomer conversion and the degree of polymerization (Table S3†) were calculated according to eqn (S5) and (S6)† by dissolving 10 mg of dried NPs in 0.7 mL of CDCl3 and analysing the samples via1H-NMR (the polymer structure and 1H-NMR spectrum are shown in Fig. S3†). The molecular weight distribution of the different samples (Table S3†) was evaluated via GPC in the same way as described in section 2.2.
The NP cloud point (Tcp), volume-average diameter (Dv) and polydispersity index (PDI) (Table 1) were measured via dynamic light scattering (DLS) on a Zetasizer Nano ZS at a scattering angle of 173°. The samples were diluted to 0.5% w/w before the analysis and the measurements were performed in triplicate. For the Tcp, a temperature program from 50 to 75 °C and back to 50 °C was implemented and the NP size was measured every 1 °C. The sample was equilibrated at the given temperature for 5 min before the measurement. Tcp was considered as the inflection point of the size vs. temperature curve.37
| Sample | D v [nm] | PDI [−] | Tcp [°C] |
|---|---|---|---|
| 20EG4-15CL3 | 110 ± 1 | 0.11 ± 0.02 | 61 ± 2 |
| 20EG4-25CL3 | 160 ± 5 | 0.20 ± 0.01 | 60 ± 1 |
| 20EG4-50CL3 | 251 ± 10 | 0.15 ± 0.03 | 62 ± 3 |
| 40EG4-15CL3 | 78 ± 4 | 0.11 ± 0.03 | 59 ± 1 |
| 40EG4-25CL3 | 113 ± 8 | 0.14 ± 0.04 | 63 ± 2 |
| 40EG4-50CL3 | 167 ± 10 | 0.16 ± 0.03 | 58 ± 4 |
| 20EG4-25CL5 | 195 ± 5 | 0.21 ± 0.01 | 63 ± 1 |
| 40EG4-15CL5 | 86 ± 3 | 0.11 ± 0.01 | 65 ± 1 |
| 40EG4-25CL5 | 140 ± 2 | 0.11 ± 0.02 | 59 ± 2 |
| 40EG4-50CL5 | 216 ± 9 | 0.09 ± 0.02 | 60 ± 1 |
| 20EG4-25CL3_Rh | 165± 8 | 0.18 ± 0.02 | 60 ± 3 |
The NP degradation was studied by tracking the size and relative scattering intensity (RSI) via DLS according to a protocol reported in the literature.38 Briefly, the NP suspensions were diluted to 0.5% w/w with a 1 M NaOH solution in order to reach pH 14 and accelerate the degradation process.38 DLS measurements were performed at 37 °C by recording the NP size and scattering intensity every 10 minutes. The RSI was computed by normalizing the scattering intensity to the value measured at time 0. All the degradation tests were performed in triplicate.
2.6 Preparation of limonene-in-water pickering emulsions
All the Pickering emulsions were prepared at 50/50 v/v water/limonene. The emulsification process was carried out for 3 minutes with an ultrasonic homogenizer (UP200S, Hielscher Ultrasound Technology), maintaining the amplitude at 80% and fixing the cycle to 10 kHz. In more detail, 2 mL of NP suspension in deionized water at fixed concentrations (5, 2.5, 1 and 0.5% w/w with respect to the water phase) were poured in a glass vial, followed by 2 mL of limonene. The mixture was kept at 0 °C by means of a water/ice bath for the duration of emulsification. The size of the limonene droplets (DD) and their polydispersity index (PDI) were analysed by means of laser diffraction (Malvern Mastersizer 3000 instrument). In particular, the sample was diluted with deionized water until the laser dimming reached 10%. The measurements were performed with a stirring rate of 2000 rpm. The sample was analysed first with blue and then with red light for 15 seconds each. The measurements were repeated 15 times. The formation of a Pickering emulsion was confirmed via transmission electron microscopy (TEM) and optical/fluorescence microscopy. In particular, TEM micrographs were obtained on a Philips CM200 electron microscope at 200 kV, equipped with a field emission gun filament. The NPs were added dropwise onto a 200 mesh carbon-coated copper grid and dried before analysis. A Gatan US 1000 CCD camera was used and 2048 × 2048 pixels images with 256 grey levels were recorded. The optical/fluorescence microscope used was a CELENA® S Digital Imaging System. For the measurement, the samples were placed on a glass microscope slide and analysed. For the fluorescence analysis, a red fluorescent protein filter (Ex530/40, Em605/55) was used.To determine the percentage of limonene emulsified, hereinafter emulsion efficiency (EE), the emulsions were left to stand overnight. Then, the separated limonene was removed with a pipette and weighed. The emulsion efficiency was finally calculated according to eqn (1):
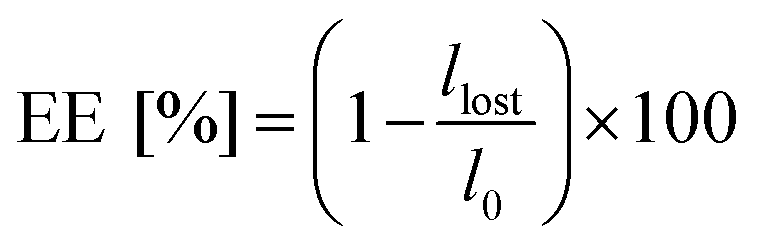 | (1) |
The temperature-mediated release of limonene was evaluated by incubating the emulsions at 70 °C for 2 h. The separated limonene was withdrawn with a pipette and weighed in order to estimate the release according to eqn (2):
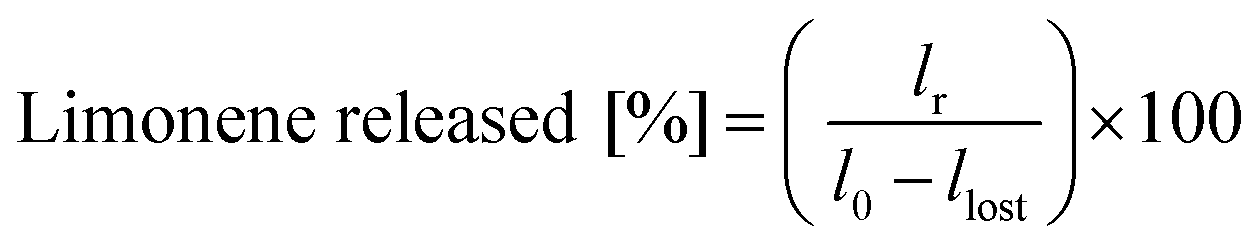 | (2) |
3. Results and discussion
3.1 Synthesis and characterization of modular thermo-responsive and biodegradable NPs
In order to evaluate the possibility of producing oil-in-water emulsions stabilized by thermo-responsive biodegradable NPs as well as to investigate the influence of their physico-chemical properties on the size and stability, ten different NP samples were synthesized through a combination of RAFT polymerization and ROP (Scheme 1).First, oligoesters provided with a reactive methacrylate group were produced via ROP of CL using HEMA as an initiator (the reaction scheme and the 1H-NMR spectrum are reported in Fig. S1†). The use of a controlled polymerization technique (i.e. ROP) allowed us to obtain poorly dispersed oligoesters with controlled molecular weight and high conversions (Table S1†). In particular, we synthesized macromonomers with an average degree of polymerization q = 3.31 and 5.24, very close to the desired target, by modulating the ratio between CL and HEMA.
At the same time, two macro-CTAs with different average molecular weights were synthesized via RAFT polymerization of EG4 (the reaction scheme and the 1H-NMR spectrum are reported in Fig. S2†), achieving good control over the average chain length (n = 25.09 and 43.41, Table S2†). These thermo-responsive macro-CTAs were finally chain-extended with the biodegradable macromonomers via RAFT emulsion polymerization, leading to amphiphilic block copolymers self-assembled into NPs (the reaction scheme and the representative 1H-NMR spectrum are reported in Fig. S3†). As expected from RAFT polymerization, the resulting block copolymers were produced at high monomer conversion (X > 98%), low dispersity (Đ < 1.3) and with an average degree of polymerization (p) close to the targeted value (Table S3†).
The remarkable feature of the RAFT emulsion polymerization is the simultaneous block copolymer synthesis and their self-assembly into NPs, which we produced with a solid content of 10% w/w and low polydispersity (PDI < 0.2, Table 1). The unusual comb-like polymer microstructure is characterized by three parameters n, p and q, which enable the tuning of key NP properties.
In particular, the NP size is affected by all of these parameters. In fact, from a simple geometrical model, the average hydrodynamic diameter (Dn) of the NPs can be related to n, p and q through eqn (3).39
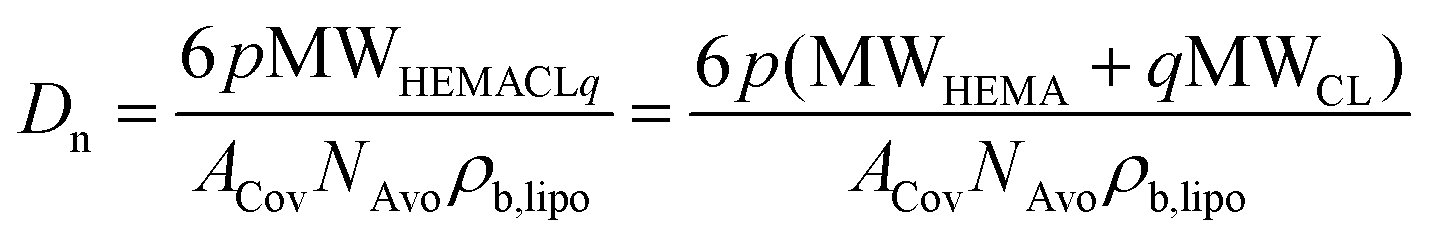 | (3) |
Here, MWHEMA and MWCL are the molecular weigths of HEMA and ε-caprolactone, respectively; ρb,lipo is the density of the lipophilic block, NAvo is the Avogadro number, and ACov is the coverage area, i.e. the area on the NP surface covered by a single hydrophilic chain. By manipulating p, the NP size can be finely controlled, as shown in Fig. 1a and b. In fact, the NP size linearly increases with this parameter at fixed n and q. Moreover, an increase in the length of the oligoester brushes (q) results in larger NPs when p is fixed due to the increase in the molecular weight of the biodegradable macromonomer. Therefore, NPs with a similar size but different hydrophobicities can be produced by independently manipulating p and q, thus enabling the modulation of the NP wetting by limonene, important for the realization of stable Pickering emulsions. Finally, also n has an effect on the NP size, with smaller NPs obtained at a higher n. This phenomenon was already observed in the literature39 and is related to the amphiphilic nature of the PEGylated monomers used for the synthesis of the macro-CTAs. Since these species exhibit a critical micelle concentration (CMC), it is reasonable to assume that the stabilizer is arranged with the hydrophobic backbone tangential to the NP surface and the PEG tethers extended in the aqueous environment. Hence, the longer the macro-CTA (the higher n), the bigger the portion of the NP surface it can cover. Therefore, the coverage area reported in eqn (3) can be expressed as a linear function of n according to eqn (4).39
| ACov = αn + β | (4) |
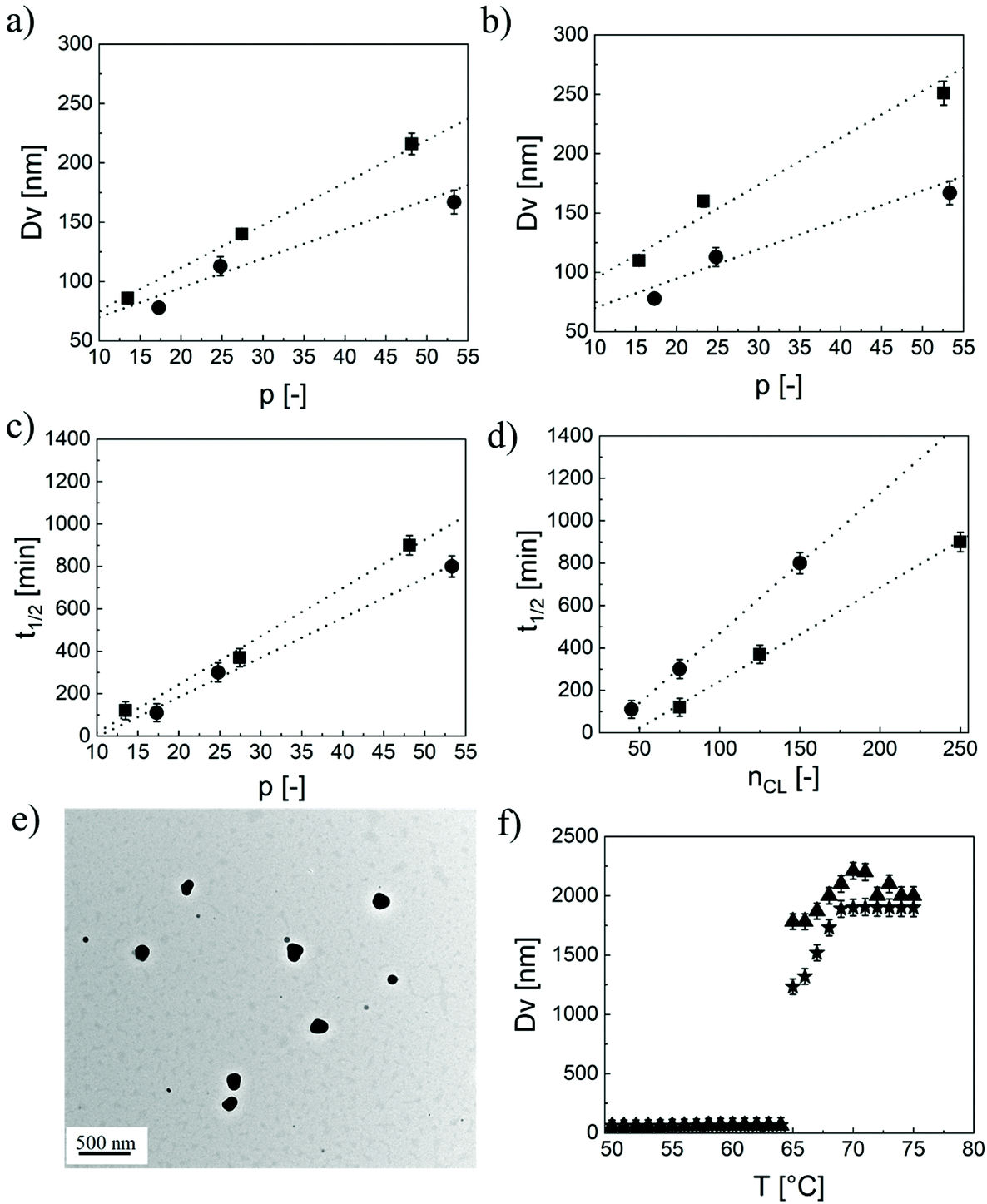 | ||
| Fig. 1 (a) NP size as a function of p for fixed n = 40 and q = 3 (●) or q = 5 (■). (b) NP size as a function of p for fixed q = 3 and n = 20 (■) or n = 40 (●). (c) NP half-life (t1/2) as a function of p for fixed q = 3 (●) or q = 5 (■) and n = 40. (d) t1/2 as a function of the total number of caprolactone units (nCL = p × q) for q = 3 (●) or q = 5 (■) and n = 40. (e) TEM image of 40EG4-50CL3, scale bar = 0.5 μm. (f) NP size measured via DLS at different temperatures for 40EG4-15CL5 during heating (★) and cooling (▲). The temperature was ramped from 50 to 75 °C and vice versa and the NP size was recorded at every 1 °C, with an equilibration time of 5 min. | ||
Another property that we can control with this modular polymer architecture is the NP degradation time. The degradation of these colloids is an important property because it affects their applicability. In fact, the progressive hydrolysis of the lateral ester bonds makes the polymer chains more and more hydrophilic, which may reduce the wettability from limonene, up to the point when the NPs disassemble destabilizing the Pickering emulsion. On the other hand, the degradation prevents the polymer accumulation in the environment, which is an urgent issue affecting the society. Therefore, the possibility of controlling the degradation time is of paramount importance to grant sufficient shelf life and avoid a premature emulsion destabilization without giving up the desirable polymer dissolution upon disposal.
We tracked the degradation by monitoring the relative scattering intensity, i.e. the intensity of the light scattered by these colloids under accelerated degradation conditions (pH = 14) with respect to the value measured at time zero. The RSI is a function of the NP size as well as of their concentration in the medium.40,41 Therefore, it can be reliably considered as an indication of the block copolymer progressive dissolution and of the NP disappearance upon hydrolysis of the ester bonds. As expected, the RSI decreases with time, as clearly shown in Fig. S4a.† This decrease may be attributed to a reduction in either the NP size or concentration. However, caprolactone-based NPs are known to show a bulk-type erosion,38i.e. a degradation that involves the NP core, without affecting the size. This is demonstrated in Fig. S4b,† where we show that the NPs synthesized did not experience any change in their size during the degradation. Therefore, the decrease in the RSI was attributed uniquely to the progressive NP dissolution and consequent reduction in their concentration. For this reason, we evaluated the NP half-life as the time leading to 50% RSI. As shown in Fig. 1c, the half-life linearly increases with p at a fixed q, which is reasonable because of the higher number of hydrophobic units that need to be cleaved before reaching 50% RSI. Then, at a first glance, the degradation appears faster for NPs with q = 3. However, it should be considered that in this case, at a given p, the number of ester bonds to be cleaved in order to lead to the NP dissolution is much lower compared to the case q = 5. For a fair comparison, we then reported in Fig. 1d the half-life as a function of the total caprolactone units nCL = p × q. Here, it is evident that the NP half-life is a function not only of nCL, but also of how the caprolactone units are distributed within the polymer microstructure. Shorter lipophilic blocks composed of longer oligo(caprolactone) pendants (q = 5) degraded faster compared to longer hydrophobic blocks obtained with shorter macromonomers (q = 3). This may be attributed to the statistical nature of the hydrolysis reactions, which in the case of q = 5 can cleave longer oligomers leading to a faster reduction in the NP lipophilicity, followed by a more rapid water diffusion and hence dissolution. This unusual discovery allows decoupling the NP hydrophobicity (and in turn wettability) and degradation time by acting on the caprolactone distribution within the polymer chains. It is worth highlighting that the degradation rate could be kept low with this polymer architecture so that we do not expect influences over the emulsion stability as studied in the following, at least in the timescale of the experiments.
It is worth highlighting that for all the combinations of n, p, and q, the NP morphology remained spherical, as confirmed by the TEM micrograph reported in Fig. 1e for 40EG4-30CL3 as an example and in Fig. S5† for other representative samples. Therefore, the modulation of the NP properties can be imputed only to p, n and q and could not be justified with a change in morphology.
Finally, the thermo-responsive behaviour of these colloids was assessed via DLS. In particular, we expect stable NPs as long as the temperature is below the Tcp. At this temperature, the thermo-responsive shell undergoes a sharp coil-to-globule transition and loses its ability to sterically stabilize the NPs. As a matter of fact, as shown in Fig. 1f, the NPs composed of 40EG4–15CL5 are stable, with an average diameter of 86 nm as long as the temperature is below 65 °C. Above this critical value, the NPs rapidly collapse leading to micrometer-sized aggregates. This transition is completely reversible, with the NPs recovering their initial size upon cooling and only minimal hysteresis was observed between the heating and cooling cycles. A remarkable feature is the rather insensitivity of the Tcp, measured as the inflection point of the size vs. temperature curves, to the structural properties of the block copolymers, as clearly visible in Table 1. This was actually quite expected. In fact, it is reported that the Tcp can be modulated by incorporation of hydrophilic or hydrophobic monomers in the thermo-responsive chain.35,37 On the other hand, when this chain is well compartmentalized, portions of the macromolecule with different nature only slightly affect the transition temperature.37
Overall, highly controllable biodegradable NPs with a reversible aggregation/stabilization behaviour triggered by temperature could be achieved by coupling ROP and RAFT polymerization.
3.2 Limonene-in-water pickering emulsion
Once these highly tunable NPs were obtained and extensively characterized, a study on the impact of their properties over the limonene-in-water Pickering emulsion was performed. Towards this aim, the NPs reported in Table 1 were used as Pickering stabilizers during the formation of the emulsions from 50:50 v/v limonene/water mixtures, following the procedure described in section 2.6 and schematically depicted in Fig. 2a. The effective formation of limonene-in-water emulsions (and not water-in-limonene) is demonstrated through Dupré's equation (eqn (5)).42| Wa = γla + γwa − γlw | (5) |
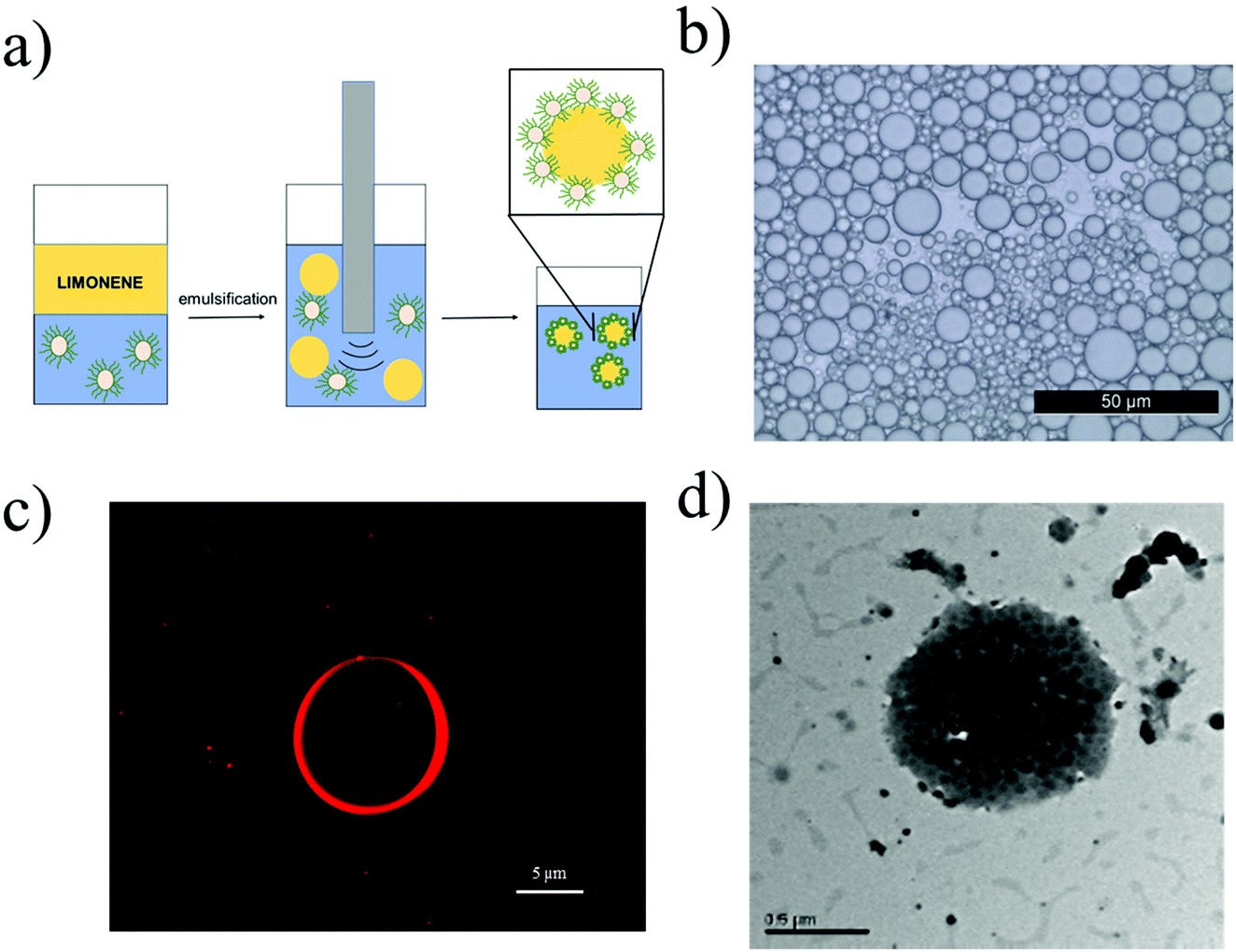 | ||
| Fig. 2 (a) Schematic representation of the emulsification process to form limonene-in-water Pickering emulsions. (b) Microscopy image of the Pickering emulsion obtained from 20EG4-25CL3 at 2.5% w/w. (c) Fluorescence microscopy image of the Pickering emulsion obtained from 20EG4-25CL3-Rh at 2.5% w/w. (d) TEM image of the Pickering emulsion obtained from 40EG4-25CL3 at 5% w/w. | ||
The emulsion stability and dispersity were evaluated by means of an optical microscope (Fig. 2b). Looking at the image, it is possible to see that poorly dispersed emulsions with spherical limonene droplets were obtained. Then, we used a fluorescent microscope to verify the accumulation of the polymer on the droplet surface. In order to do that, fluorescent NPs (20EG4-25CL3-Rh in Table 1) were synthesized following the same procedure described in section 2.5 and adding one unit of the fluorescent monomer HEMA-Rh (section 2.3) in a statistical copolymer with HEMACL3. At the same time, the RAFT polymerization ensures low interchain composition drift, and hence the presence of this fluorescent tag in each polymer chain.43 These NPs were then used in the emulsification of limonene, and the resulting emulsion is shown in Fig. 2c. As visible from the micrograph, the fluorescent NPs cover the limonene droplet forming a corona. The proper formation of emulsions stabilized by the polymer is then demonstrated.
However, one of the risks when producing Pickering emulsions with non-cross-linked NPs is the disruption of the NPs into their unimers during the emulsification process.44,45 In fact, the severe conditions usually adopted to obtain this kind of emulsions may provide enough energy for NP dissociation. If this is the case, the amphiphilic polymer chains originally assembled into NPs may cover the surface of the limonene droplets, which are then stabilized by polymeric surfactants rather than by NPs. In order to verify this eventuality, the emulsions were analysed by means of TEM. As clearly shown in Fig. 2d, the polymer NPs not only survived the emulsification process preserving their spherical morphology, but also adsorbed at the sruface of the limonene droplets providing stability.
To further demonstrate the formation of a Pickering emulsion, we verified the characteristic behaviour of such emulsions compared to their traditional counterparts, which is the relationship between the size of the droplet phase and the polymer concentration. In fact, in a Pickering emulsion, the droplet size (DD) is expected to decrease with the NP concentration according to eqn (6).46
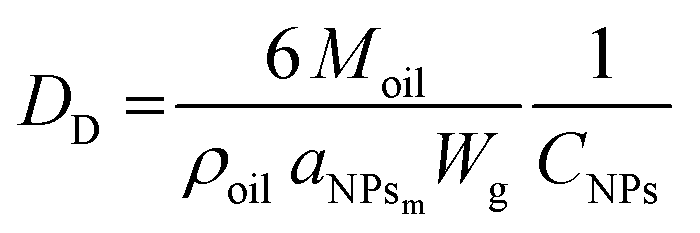 | (6) |
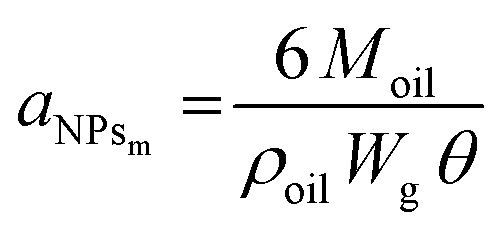 | (7) |
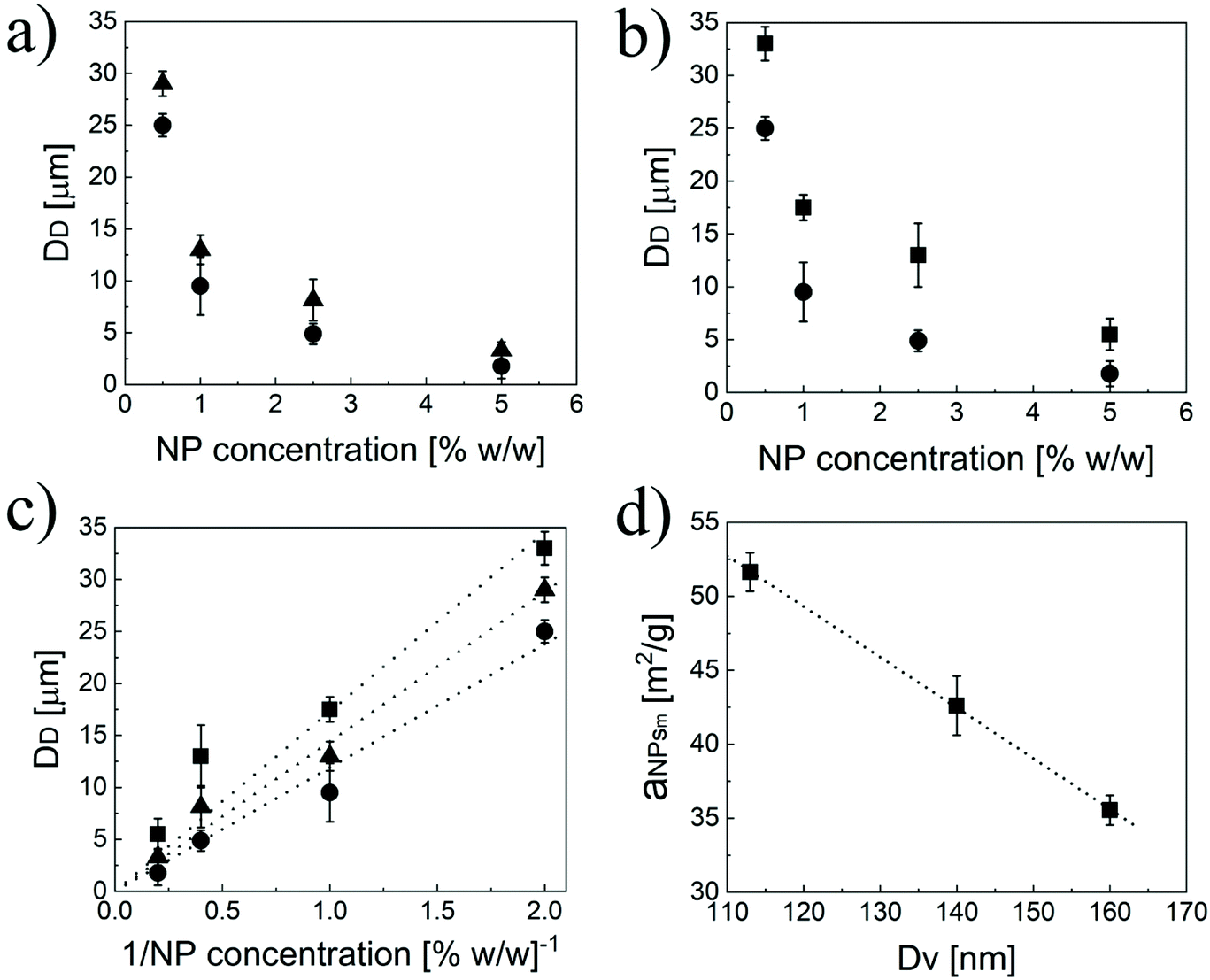 | ||
| Fig. 3 (a) Size of the droplets (DD) of limonene formed at different concentrations of 40EG4-25CL3 (●) and 40EG4-25CL5 (▲). (b) Size of the droplets (DD) of limonene formed at different concentrations of 40EG4-25CL3 (●) and 20EG4-25CL3 (■). (c) Pickering emulsion size as a function of the inverse of NP concentration in the case of 20EG4-25CL3 (■, R2 = 0.89), 40EG4-25CL5(▲, R2 = 0.98), and 40EG4-25CL3 (●, R2 = 0.98), respectively. (d) aNPsm as a function of NP size (R2 = 0.99). | ||
Interestingly, aNPsm is a linear function of the NP size (Fig. 3d), with the bigger NPs covering a smaller portion of the surface at a given mass. Therefore, with the modular NPs reported in this work, it is relatively simple to modulate the area that the unit mass of NPs can cover by tuning n, p and q.
Another aspect to consider is that Pickering emulsions stabilized by NP concentrations lower than 1% w/w are often reported to separate within a few hours due to the incomplete oil coverage.46 However, this is not the case for this system, as confirmed from the small standard deviations reported in Fig. 3 that were obtained from three independent measurements performed in three consecutive days. In order to better investigate this greater stability observed, we calculated the limonene surface coverage (Cs) according to eqn (8).45
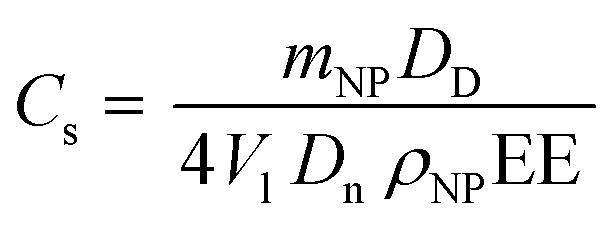 | (8) |
| Sample | m NP [g] | NP [% w/w] | EE [%] | C s [%] |
|---|---|---|---|---|
| 20EG4-25CL3 | 0.1 | 5 | 95 | 40 |
| 20EG4-25CL3 | 0.05 | 2.5 | 94 | 48 |
| 20EG4-25CL3 | 0.02 | 1 | 93 | 26 |
| 20EG4-25CL3 | 0.01 | 0.5 | 91 | 25 |
| 40EG4-25CL3 | 0.1 | 5 | 98 | 18 |
| 40EG4-25CL3 | 0.05 | 2.5 | 97 | 24 |
| 40EG4-25CL3 | 0.02 | 1 | 93 | 20 |
| 40EG4-25CL3 | 0.01 | 0.5 | 92 | 27 |
| 40EG4-25CL5 | 0.1 | 5 | 99 | 26 |
| 40EG4-25CL5 | 0.05 | 2.5 | 98 | 33 |
| 40EG4-25CL5 | 0.02 | 1 | 94 | 22 |
| 40EG4-25CL5 | 0.01 | 0.5 | 92 | 25 |
As can be seen from Table 2, all the Pickering emulsions present Cs significantly lower than 90%, expected for a close-packed 2D layer of hard NPs.48 It is worth mentioning that eqn (8) was obtained from geometrical considerations that assume that the NPs preserve their original size once around the oil phase. However, for systems with a low glass transition temperature (Tg) this assumption may be no longer valid.48 In fact, in these cases it is reasonable assuming that the NPs spread on the droplet thus covering a bigger area than the one calculated mathematically. As a matter of fact, the values listed in Table 2 are in line with what was already reported in the literature for NPs from low Tg polymers.48 Interestingly, no significant change in Cs was noticed, thus suggesting that the Pickering stabilization may occur with a combination of NP spreading and bridging monolayers49 with the latter being probably more significant at lower NP concentrations.
As demonstrated above, the three independent degrees of freedom (i.e. n, p and q) coming from the unusual comb-like structure of the polymer chains allow fine modulation of the properties of the NPs. In turn, these strongly influence the properties of the Pickering emulsions, mainly the size of the dispersed phase and the amount of limonene that can be emulsified. In particular, the average size of the limonene droplets in the continuous phase follows similar trends to the hydrodynamic diameter of the primary NPs. Hence, for fixed n and p, the higher the q, the bigger the droplets (Fig. 3a). Moreover, the higher the n, the smaller the final emulsions (Fig. 3b) when the lipophilicity is fixed (p and q fixed). Additionally, the droplet size linearly increases with p (Fig. 4a) independently of n and q. This behaviour, in line with eqn (6), was not unexpected considering that aNPsm is inversely proportional to the NP diameter (Fig. 3d) and that the NP size is a linear function of p. Therefore, the modulation of p is a useful way to tune the size of the droplets in a Pickering emulsion.
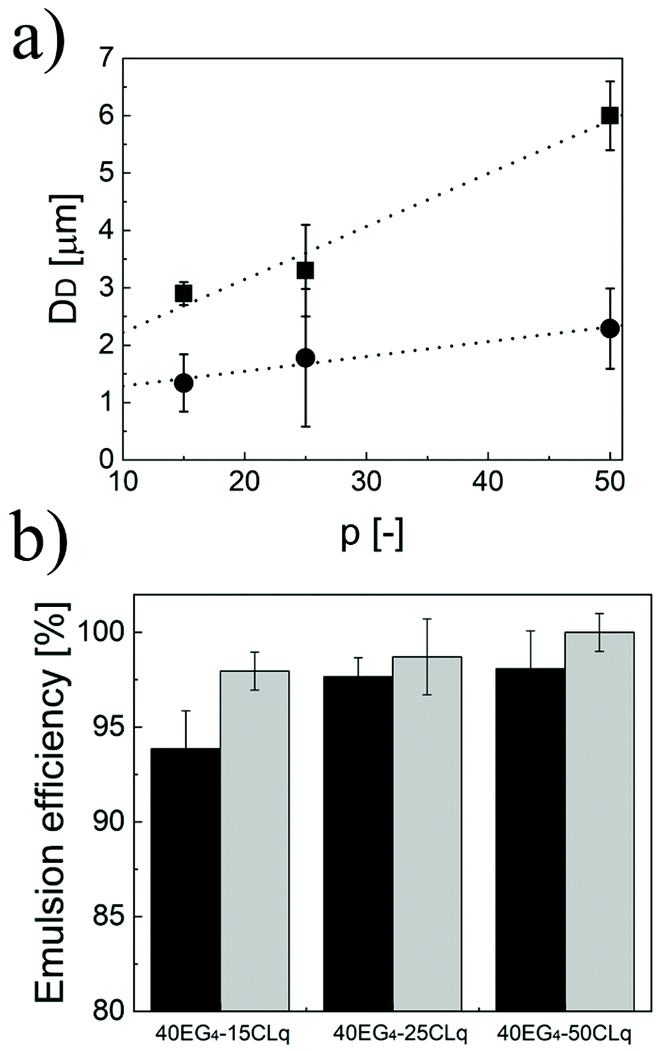 | ||
| Fig. 4 (a) Size of the limonene droplets stabilized by 40EG4-pCL3 (●) and 40EG4-pCL5 (■). (b) Percentage of limonene loaded in the case of NPs with q = 3 (dark grey) and q = 5 (light grey), respectively. | ||
Then, we investigated how n, p and q affect the amount of limonene that can be emulsified in water. In particular, the emulsions formed using these NPs were left to stand overnight and then the separated limonene was removed to evaluate the emulsion efficiency. The results are shown in Fig. 4b. All the NPs led to emulsion efficiencies higher than 93%. In particular, EE increases with the NP lipophilicity. As matter of fact, for a fixed q, the longer p the higher the amount of limonene loaded. Moreover, fixing p and increasing q result in a higher emulsion efficiency as well. These trends can be justified assuming that the lipophilicity of the CL units plays an important role in the NP wetting by limonene droplets, thus favouring its emulsification.
This highlights the high degree of control that it is possible to achieve when preparing Pickering emulsions with highly tunable NPs. Moreover, the three degrees of freedom allow the size and the degradation time38 (and thus the stability) of the Pickering emulsion to be decoupled, paving the way to the development of Pickering emulsions with properties tunable depending on the final application.
An additional peculiarity of the NPs used as emulsifiers is their thermo-responsive behaviour. This feature is added in order to induce the limonene phase separation upon application of thermal stimuli, an application which may find interest for the on-demand release of fragrances. The behaviour we expect for these “smart” emulsions is schematically depicted in Fig. 5a. In particular, as long as the temperature is below the Tcp (values reported in Table 1), the NPs are colloidally stable and in turn can stabilize the Pickering emulsions by adsorption at the limonene–water interface. On the other hand, when the temperature is increased above the Tcp we expect the destabilization and aggregation of the NPs, as already shown in Fig. 1d. This in turn may cause the instability of the emulsion and the coalescence and consequent separation of the limonene droplets.
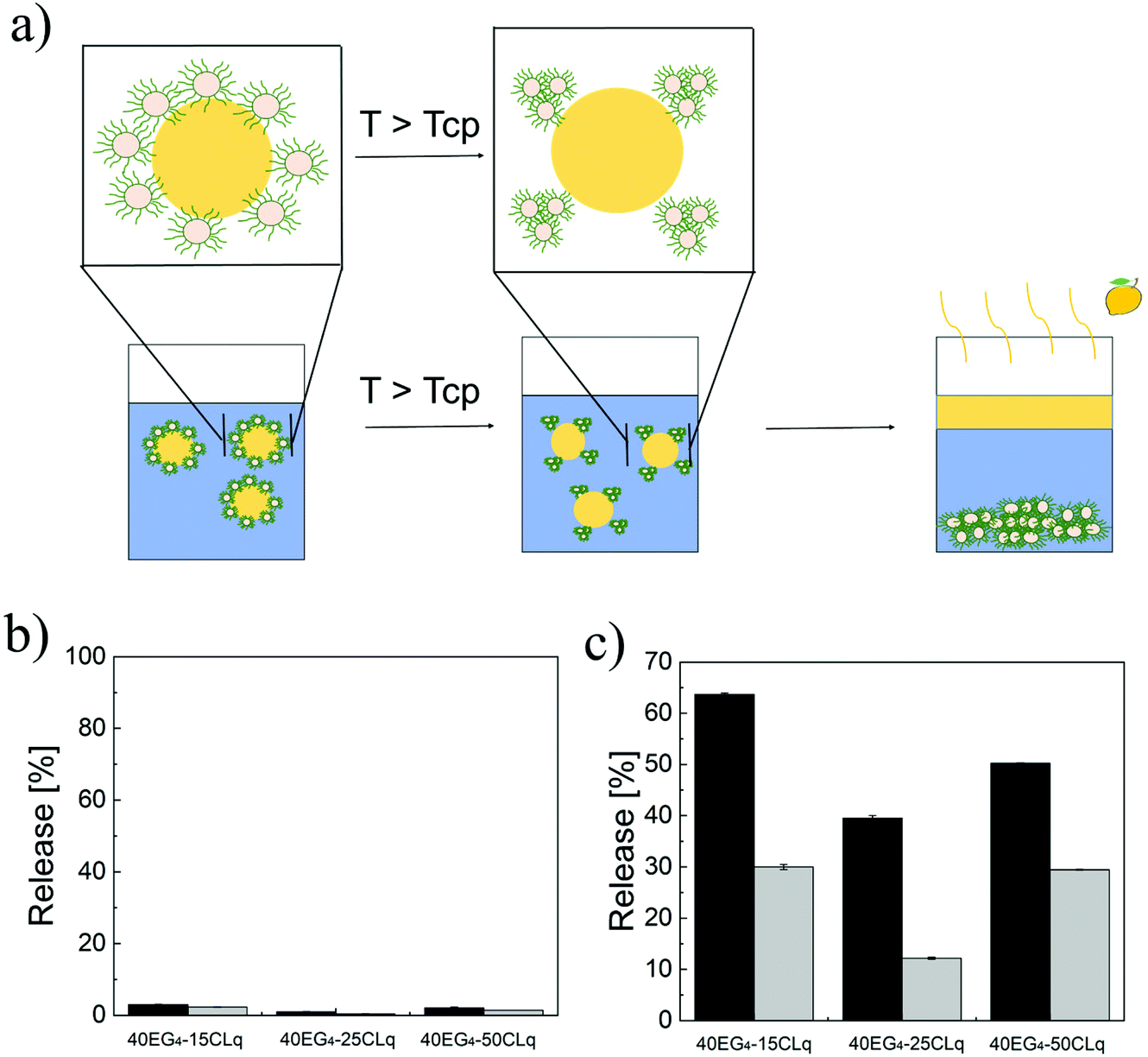 | ||
| Fig. 5 (a) Schematic representation of the expected mechanism of emulsion destabilization and limonene release. Percentage of limonene released in the case of NPs with q = 3 (dark grey) and q = 5 (light grey) at T below (b) and above (c) the Tcp. | ||
Following this expectation, once having demonstrated the formation of stable limonene-in-water Pickering emulsions, the possibility of destabilizing them by simply increasing the environmental temperature was assessed. The emulsions were incubated at 30 °C (<Tcp, Fig. 5b) and 70 °C (>Tcp, Fig. 5c) for 2 h, and the amount of separated limonene was measured. While below the Tcp, the amount of released limonene was lower than 5% for all the emulsions produced, a significantly higher amount of limonene (up to 63%) was released when the temperature was above the Tcp.
Moreover, depending on the NP lipophilicity and size, different amounts of limonene were released. In fact, if the NPs are small (p equal to 15) or the lipophilicity is high (p equal to 50) a higher amount of limonene is released with respect to the NPs with p equal to 25. These results can be justified by considering that the energy required to remove a particle from the interface (E) depends on the NP wetting angle as shown in eqn (9).25,26
| E = πNPrγlw(1 − cosθ)2 | (9) |
It is reasonable to assume that bigger NPs are already more prone to migrate into the limonene phase (i.e. higher degree of lipophilicity) and thus a further change in their wettability caused by the temperature change would cause a relatively easy emulsion destabilization. On the other hand, the smaller the NPs the weaker their energy of adhesion. For this reason, a change in their contact angle would have a more significant impact than a change for bigger NPs.
It is worth mentioning that this selective NP aggregation and limonene release could potentially be implemented as a valuable strategy to recycle the NPs for further Pickering emulsions. However, a fundamental pre-requisite for this approach is the reversibility in the NP phase separation, which in turn is affected by the copolymer microstructure and NP size.50
In summary, thermo-responsive Pickering emulsions were successfully obtained using thermo-responsive biodegradable NPs, thus paving the way to a possible future application of this technology in those fields where the on-demand release of the emulsified phase is required.
4. Conclusions
In this work, we synthesized highly tunable biodegradable NPs by combining two pseudo-living polymerizations, namely RAFT polymerization and ROP. With this strategy we obtained comb-like block copolymers with three degrees of freedom, namely the macromonomer length and the number of hydrophilic and hydrophobic units added to the copolymer chains (i.e. q, n and p, respectively), and the ability to self-assemble in water into NPs. The modulation of these parameters is an appealing strategy to control key NP properties such as size, lipophilicity, degradation time and wettability. Moreover, the adoption of a thermo-responsive stabilizer provided the synthesized NPs with a smart aggregation/redispersion behaviour following the application of thermal heating.These highly engineered colloids were used to produce limonene-in-water Pickering emulsions. Since the size of the droplet phase increased when lowering the NP concentration, we concluded that such NPs survived the emulsification process and actually acted as Pickering emulsifiers, as the TEM micrographs suggested. The properties of these emulsions, such as size, covered surface and limonene loading could be well controlled by manipulating the properties of the primary NPs. In particular, the size of the dispersed phase increased when increasing the NP size, in response to a lower surface covered per unit mass of NPs. Also the emulsion efficiency, which was >93% in each case, could be optimized by increasing the NP wettability acting on p and q. By increasing these parameters the NP wettability was improved, leading to a higher limonene loading. Finally, the possibility of selectively releasing the fragrance by simply increasing the external temperature above the Tcp was demonstrated, thus paving the way to the on-demand fragrance release important in manifold cosmetic and pharmaceutical applications.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are grateful to Maria Francesca Brunella for the TEM images.References
- W. Ramsden, Proc. Royal Soc., 1903, 72, 156–164 CAS.
- S. Pickering, J. Chem. Soc. Trans., 1907, 91, 2001–2021 RSC.
- S. Wang, Y. He and Y. Zou, Particuology, 2010, 8, 390–393 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Phys. Chem. Chem. Phys., 1999, 1, 3007–3016 RSC.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- S. A. F. Bon and P. J. Colver, Langmuir, 2007, 23, 8316–8322 CrossRef CAS PubMed.
- S. Cauvin, P. J. Colver and S. A. F. Bon, Macromolecules, 2005, 38, 7887–7889 CrossRef CAS.
- B. Zheng, B. Zheng, A. J. Carr, X. Yu, D. J. McClements and S. R. Bhatia, Inorganica Chim. Acta, 2020, 508, 119566 CrossRef CAS PubMed.
- S. Abend and G. Lagaly, Clay Miner., 2001, 36, 557–570 CrossRef CAS.
- J. I. Amalvy, G. F. Unali, Y. Li, S. Granger-Bevan, S. P. Armes, B. P. Binks, J. A. Rodrigues and C. P. Whitby, Langmuir, 2004, 20, 4345–4354 CrossRef CAS PubMed.
- J. H. Shin, J. W. Park and H. J. Kim, Appl. Clay Sci., 2019, 182, 105288 CrossRef CAS.
- G. Lagaly, M. Reese and S. Abend, Appl. Clay Sci., 1999, 14, 83–103 CrossRef CAS.
- J. Marto, A. Nunes, A. M. Martins, J. Carvalheira, P. Prazeres, L. Gonçalves, A. Marques, A. Lucas and H. M. Ribeiro, Cosmetics, 2020, 7, 1–12 CrossRef.
- ECHA, Regulation cosmetics available at: https://ec.europa.eu/growth/sectors/cosmetics/legislation_en.
- B. S. Murray, Curr. Opin. Food Sci., 2019, 27, 57–63 CrossRef.
- V. Calabrese, J. C. Courtenay, K. J. Edler and J. L. Scott, Curr. Opin. Green Sustain. Chem., 1907, 12, 83–90 CrossRef.
- F. Zhu, Trends Food Sci. Technol., 2019, 85, 129–137 CrossRef CAS.
- M. Maraldi, R. Ferrari, R. Auriemma, M. Sponchioni and D. Moscatelli, J. Pharm. Sci., 2020, 109(8), 2607–2614 CrossRef CAS PubMed.
- M. Sponchioni, P. Rodrigues Bassam, D. Moscatelli, P. Arosio and U. Capasso Palmiero, Nanoscale, 2019, 11, 16582–16591 RSC.
- A. Zanoni, G. Gardoni, M. Sponchioni and D. Moscatelli, J. CO2 Util., 2020, 40, 101192 CrossRef CAS.
- F. Laredj-Bourezg, Y. Chevalier, O. Boyron and M. A. Bolzinger, Colloids Surf., A, 2012, 413, 252–259 CrossRef CAS.
- D. Moscatelli and M. Sponchioni, in Bioresorbable Polym. Biomed. Appl. From Fundam. to Transl. Med, Elsevier, Academic Press, The Netherlands, 2017, pp. 265–283 Search PubMed.
- U. Capasso Palmiero, J. Ilare, C. Romani, D. Moscatelli and M. Sponchioni, Colloids Surf., B, 2020, 190, 110926 CrossRef CAS PubMed.
- U. Capasso Palmiero, M. Sponchioni, N. Manfredini, M. Maraldi and D. Moscatelli, Polym. Chem., 2018, 9, 4084–4099 RSC.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7(1–2), 21–41 CrossRef CAS.
- S. Tsuji and H. Kawaguchi, Langmuir, 2008, 24, 3300–3305 CrossRef CAS PubMed.
- J. Tang, P. J. Quinlan and K. C. Tam, Soft Matter, 2015, 11, 3512–3529 RSC.
- F. Atyabi, F. Zahir, F. Khonsari, A. Shafiee and F. Mottaghitalab, in Nanostructures for Cancer Therapy, Elsevier, 2017, pp. 541–561 Search PubMed.
- Y. Alkhatib, M. Dewaldt, S. Moritz, R. Nitzsche, D. Kralisch and D. Fischer, Eur. J. Pharm. Biopharm., 2017, 112, 164–176 CrossRef CAS PubMed.
- S. Fujii, Y. Cai, J. V. M. Weaver and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 7304–7305 CrossRef CAS PubMed.
- B. S. Fujii, E. S. Read, B. P. Binks and S. P. Armes, Adv. Mater., 2005, 17(8), 1014–1018 CrossRef.
- C. Liang, Q. Liu and Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 6898–6904 CrossRef CAS PubMed.
- J. Jiang, Y. Zhu, Z. Cui and B. P. Binks, Angew. Chem., 2013, 125, 12599–12602 CrossRef.
- T. Saigal, H. Dong, K. Matyjaszewski and R. D. Tilton, Langmuir, 2010, 26, 15200–15209 CrossRef CAS PubMed.
- M. Sponchioni, U. Capasso Palmiero and D. Moscatelli, Mater. Sci. Eng., C, 2019, 102, 589–605 CrossRef CAS PubMed.
- N. Manfredini, E. Scibona, M. Morbidelli, D. Moscatelli and M. Sponchioni, Ind. Eng. Chem. Res., 2019, 58, 22290–22298 CrossRef CAS.
- M. Sponchioni, R. Ferrari, L. Morosi and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(18), 2919–2931 CrossRef CAS.
- U. Capasso Palmiero, M. Maraldi, N. Manfredini and D. Moscatelli, Biomacromolecules, 2018, 19, 1314–1323 CrossRef CAS PubMed.
- U. C. Palmiero, A. Agostini, S. Gatti, M. Sponchioni, V. Valenti, L. Brunel and D. Moscatelli, Macromolecules, 2016, 49, 8387–8396 CrossRef CAS.
- M. Sponchioni, U. C. Palmiero and D. Moscatelli, Macromol. Chem. Phys., 2017, 218, 1–12 CrossRef.
- M. Sponchioni, L. Morosi, M. Lupi and U. Capasso Palmiero, RSC Adv., 2017, 7, 50981–50992 RSC.
- B. P. Binks, P. D. I. Fletcher, B. L. Holt, P. Beaussoubre and K. Wong, Phys. Chem. Chem. Phys., 2010, 12, 11954–11966 RSC.
- M. Sponchioni, U. Capasso Palmiero, N. Manfredini and D. Moscatelli, React. Chem. Eng., 2019, 4, 436–446 RSC.
- K. L. Thompson, N. Cinotti, E. R. Jones, C. J. Mable, P. W. Fowler and S. P. Armes, Langmuir, 2017, 33, 12616–12623 CrossRef CAS PubMed.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC.
- Y. Chevalier and M. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 CrossRef CAS PubMed.
- M. Destribats, V. Lapeyre, M. Wolfs, E. Sellier, F. Leal-Calderon, V. Ravaine and V. Schmitt, Soft Matter, 2011, 7, 7689–7698 RSC.
- T. S. Horozov and B. P. Binks, Angew. Chem., 2006, 118, 787–790 CrossRef.
- N. Manfredini, M. Tomasoni, M. Sponchioni and D. Moscatelli, Polymers, 2021, 13(7), 1032 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the synthesized copolymers using the 1H NMR spectra to determine the monomer conversion and the degree of polymerization and GPC in order to evaluate their molecular weights and dispersity indexes. See DOI: 10.1039/d1nr00694k |
| This journal is © The Royal Society of Chemistry 2021 |