
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutodetachment dynamics of 2-naphthoxide and implications for astrophysical anion abundance†
Eleanor K.
Ashworth
 a,
Cate S.
Anstöter
b,
Jan R. R.
Verlet
*b and
James N.
Bull
*a
a,
Cate S.
Anstöter
b,
Jan R. R.
Verlet
*b and
James N.
Bull
*a
aSchool of Chemistry, Norwich Research Park, University of East Anglia, Norwich NR4 7TJ, UK. E-mail: james.bull@uea.ac.uk
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: j.r.r.verlet@durham.ac.uk
First published on 3rd March 2021
Abstract
Astrochemical modelling has proposed that 10% or more of interstellar carbon could be tied up as polycyclic aromatic hydrocarbon (PAH) molecules. Developing reliable models of the interstellar carbon lifecycle requires calibration data obtained through laboratory studies on relevant chemical and physical processes, including on the photo-induced and electron-induced dynamics of potential interstellar PAHs. Here, the excited state dynamics of the S1(ππ*) state of 2-naphthoxide are investigated using frequency-, angle-, and time-resolved photoelectron imaging. Frequency-resolved photoelectron spectra taken over the S1(ππ*) band reveal low electron kinetic energy structure consistent with an indirect, vibrational mode-specific electron detachment mechanism. Time-resolved photoelectron imaging using a pump photon energy tuned to the 0–0 transition of the S1(ππ*) band (hν = 2.70 eV) and a non-resonant probe photon provides the excited state autodetachment lifetime at τ = 130 ± 10 fs. There is no evidence for internal conversion to the ground electronic state or a dipole-bound state. These results imply that 2-naphthoxide has no resilience to photodestruction through the absorption of visible radiation resonant with the S1(ππ*) band, and that electron capture by the S1(ππ*) state, which is formally a shape resonance, is not a doorway state to a stable interstellar anion.
1 Introduction
The list of circumstellar and interstellar molecules has been steadily increasing over the last few decades, standing at 204 in the “2018 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules”.1 While polycyclic aromatic hydrocarbon (PAH) molecules are proposed to be ubiquitous in the form of free molecules and adsorbed onto icy dust grains,2–4 meteorites, and other interplanetary particles,5,6 there is limited direct evidence for their existence despite astrochemical models proposing that 10% or more of interstellar carbon could be in the form of PAH molecules.7–9 One of the most compelling indications for small to medium sized PAHs (e.g. two to ten rings) in space comes from the identification of the C60+ molecule as a carrier for several diffuse interstellar bands.10,11Interestingly, of the molecules known to exist in space through assignment of astronomical spectra, only six are anions: C2nH− (n = 2–4) and C2n−1N− (n = 1–3). Theoretical modelling has indicated that larger carbonaceous anions, such as longer-chain polyynes and PAHs, including those with oxygenation, should be present in space.12–15 However, there is a dearth of knowledge on the ultrafast photon- and electron-induced photophysics of isolated PAHs, particularly anionic PAHs; these processes ultimately define the formation, destruction, and photoresilience mechanisms of PAH molecules in space. Determination of these properties is important for defining whether certain molecules or classes of molecules can exist in space and thus inform on the interstellar carbon balance.16,17
The 2-naphthoxide species, 2-Np− shown in Fig. 1, is one of the simplest polycyclic aromatic hydrocarbon (PAH) anions possessing heteroatom functionalisation. This class of PAH molecules are often invoked as probable interstellar carbonaceous species.2 For example, laboratory studies on pristine PAHs encapsulated in icy matrices irradiated with ultraviolet light or impacted with high-energy particles show formation of alcohol, ketone, and carboxylic acid functional groups,14,15,18,19 indicating that oxygenated PAHs, most likely as closed-shell molecules, should contribute to the interstellar carbon balance. Other experiments which subjected PAHs to interstellar conditions found extensive hydrogenation, oxygenation, and hydroxylation, ultimately leading to more complex organics.20 In a recent contribution, one of the present authors reported a photodetachment study on 2-Np− thermalised to T ≈ 300 K,21 providing an action spectrum of the S1(ππ*) band as well as identifying several other weak spectral features spanning the electron detachment threshold, which were attributed to a progression of dipole-bound states. Two recent investigations using cryogenic photoelectron and photodetachment techniques have reported near-threshold spectra of 2-Np−, and also identified dipole-bound states.22,23
 | ||
| Fig. 1 Structure of 2-naphthoxide, 2-Np−. | ||
In this paper, we report a frequency-, angle-, and time-resolved photoelectron imaging study of 2-Np− thermalised to T ≈ 300 K. The key questions that this study addresses are: (1) What is the lifetime of the S1(ππ*) state when exciting at the 0–0 transition? (2) What are the decay dynamics of the excited state, and is there any internal conversion to recover the ground electronic state? (3) What do the above-threshold dynamics tell us about the photoresiliance and propensity for stable anion formation in space?
2 Experimental methods
Photoelectron spectroscopy measurements using the frequency- and angle-resolved photoelectron imaging strategy24–26 were performed using an instrument that combines electrospray ionisation, ion trapping, time-of-flight mass-to-charge separation, and velocity-map imaging detection.27,28 Electrosprayed 2-Np− (starting with 99% 2-naphthol, Sigma-Aldrich) from methanol were accumulated in a radio frequency ring-electrode ion trap. The trapped anions were unloaded at 10 Hz repetition rate for the frequency-resolved experiments and 333 Hz for the time-resolved experiments into a colinear time-of-flight optics assembly that accelerated the ions along a 1.3 m flight region toward a continuous-mode penetrating field velocity-mapping assembly.28 Light from a nanosecond-pulsed laser system (Continuum Horizon OPO pumped by a Continuum Surelite II Nd:YAG laser, 10 Hz, unfocused, <0.5 mJ cm−1 pulse−1) or femtosecond laser system was timed to interact with a mass-selected ion packet at the centre of the velocity-map imaging stack. Ejected electrons were velocity-mapped onto a dual (chevron) multichannel plate detector, followed by a P43 phosphor screen, which was monitored with a charge-coupled device camera. Velocity-map images were accumulated with a 500 ns multi-channel plate gate. The velocity-mapping resolution was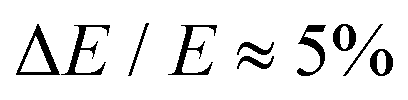 , and the electron kinetic energy (eKE) scale was calibrated from the spectrum of I−. Velocity-map image reconstructions used antialiasing and polar onion-peeling algorithms,29 providing the photoelectron spectra and associated photoangular distributions quantified by the conventional β2 anisotropy parameter.30
, and the electron kinetic energy (eKE) scale was calibrated from the spectrum of I−. Velocity-map image reconstructions used antialiasing and polar onion-peeling algorithms,29 providing the photoelectron spectra and associated photoangular distributions quantified by the conventional β2 anisotropy parameter.30
For the time-resolved measurements, femtosecond laser pulses were derived from a Spectra-Physics Ti:sapphire oscillator and regenerative amplifier. The hν = 2.70 eV (459 ± 5 nm, ≈20 μJ) pump pulses were produced through fourth-harmonic generation (two successive BBO crystals) of the idler output from an optical parametric amplifier (Light Conversion TOPAS-C). The hν = 1.55 eV (800 nm, ≈100 μJ) probe pulse corresponds to the fundamental output of the femtosecond laser. Pump and probe pulses were delayed relative to each other (Δt) using a motorised delay line. Both pulses were combined colinearly using a dichroic mirror and were loosely focused into the interaction region using a curved metal mirror. The pump–probe cross correlation was ≈60 fs.
3 Results and discussion
3.1 Frequency- and angle-resolved photoelectron imaging
Several photoelectron spectra of 2-Np− are shown in Fig. 2a, including a spectrum associated with excitation of the dipole-bound state (see inset). The selected photoelectron spectra were taken at photon energies corresponding to vibronic features in the photodetachment action spectrum (Fig. 2b), which has been reproduced from ref. 21. Photoelectron spectra taken at other photon energies are given in the ESI.† The photoelectron spectra all show two clear vibrations at low-eKE (35 ± 5 meV and 66 ± 5 meV) with β2 ≈ 0 (isotropic electron ejection), consistent with an indirect electron detachment mechanism.31 Significantly, delay of the detector acquisition gate by 50 ns relative to the OPO light pulse resulted in no photoelectron signal, demonstrating that all photoelectrons are ejected over a timescale shorter than 50 ns.25,32 This result confirms that there is no low-eKE signal associated with thermionic emission due to recovery of the ground electronic state followed by statistical electron ejection, which usually occurs on the microsecond to millisecond timescale. The low-eKE vibrational structure has spacing ≈35 meV (282 cm−1), which is significantly reduced compared with the spacing in the dipole-bound state progression in Fig. 2b at ≈420 cm−1. These low-eKE vibrations are discussed in detail in Section 3.3.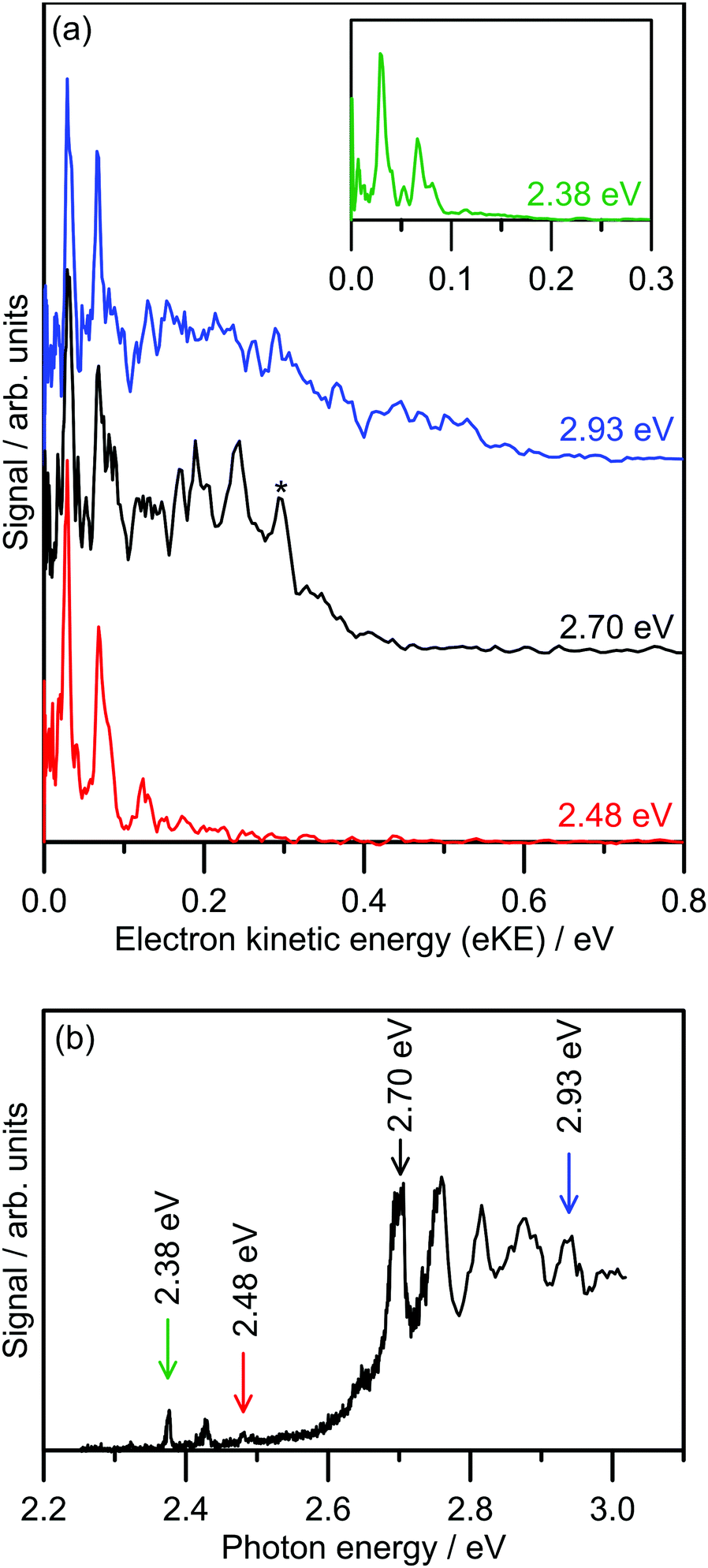 | ||
| Fig. 2 Photoelectron spectroscopy of 2-Np−: (a) example photoelectron spectra; the spectrum in the inset is associated with exciting the dipole-bound state at hν = 2.38 eV, (b) photodetachment spectrum at T ≈ 300 K taken from ref. 21. The ADE is 2.4037 ± 0.0005 eV (taken from ref. 23). The feature denoted by * in the black trace in (a) is attributed to detachment to the ground vibrational state of the 2-naphthoxy neutral (2A′′). | ||
The photoelectron spectra shown in Fig. 2a show a prompt detachment feature – an eKE distribution that tracks the increase in photon energy – which contains contributions from direct photodetachment and prompt autodetachment without significant nuclear relaxation. Because photoexcitation cross-sections for the S1(ππ*) and S2(ππ*) state will be much larger than cross-sections for direct photodetachment, the prompt detachment feature will be dominated by the prompt autodetachment contribution. The prompt detachment feature is most obvious when comparing all of the photoelectron spectra given in the ESI.† The higher photon energy spectra, e.g. hν = 2.93 eV in Fig. 2a and other spectra in the ESI,† exhibit three spectral features: (i) the low-eKE vibrational structure as described above, (ii) the prompt detachment feature, and (iii) a delayed autodetachment band (e.g. the fixed 0.1 ≤ eKE ≤ 0.4 eV window in the hν = 2.93 eV spectrum and other spectra in the ESI†). The delayed autodetachment band has several possible origins, including changing Franck–Condon factors with photon energy, fast nuclear relaxation followed by autodetachment, and at higher photon energies could have a contribution from excitation of the S2(ππ*) state followed by internal conversion and autodetachment from the S1(ππ*) state. β2 values over the prompt detachment and delayed autodetachment features are approximately zero, consistent with isotropic electron ejection.
The hν = 2.70 eV photoelectron spectrum and others given in the ESI† show vibrational structure on the prompt detachment feature with spacing of 60 ± 10 meV (480 ± 80 cm−1). This vibrational structure is consistent with that observed in the photodetachment action spectrum (Fig. 2b), which was assigned to the principal Franck–Condon active mode for excitation of the S1(ππ*) state (calculated ν32(a′) = 437 cm−1) through Franck–Condon-Herzberg–Teller simulations.21 It is also worth noting that this vibrational structure is consistent with the principal Franck–Condon modes for direct photodetachment characterised by an earlier cryogenic photoelectron spectroscopy experiment,22 involving ν32(a′) = 422 cm−1 (neutral), which is an in-plane CCC bending mode and has most displacement localised on the oxygen-containing benzyl ring. In the present photoelectron spectra, the vibrational structure over the prompt detachment band in the photoelectron spectra is presumably associated with prompt autodetachment through electron shake-off by the ν32 mode of the anion excited state.
3.2 Time-resolved photoelectron imaging
The electron detachment dynamics following excitation at the 0–0 transition of the S1(ππ*) band of 2-Np− were characterised using time-resolved photoelectron imaging (Fig. 3). This experiment involved exciting 2-Np− with a pump photon at hν = 2.70 eV (459 ± 5 nm) followed by a probe photon at hν = 1.55 eV (800 nm) photon at a series of time delays, Δt. Fig. 3a shows a time-resolved photoelectron spectrum at Δt = 50 fs, i.e., pump–probe spectrum minus pump-only spectrum. There are two features, photodepletion signal at low-eKE, which is associated with the low-eKE signal in the frequency-resolved spectrum, and pump–probe signal in the 1.3 ≤ eKE ≤ 2.0 eV window. With increasing Δt, the pump–probe signal decays, without any significant change in spectral shape. Concomitantly, the photodepletion signal at low-eKE recovers – this data is noisier than the pump–probe appearance signal due to subtracting a small depletion signal from a large background, and is not shown. The integrated pump–probe signal in the 1.3 ≤ eKE ≤ 2.0 eV window with Δt is shown in Fig. 3b, showing that the photoexcited population has decayed within 1 ps. The time-resolved signal can be well described by a single exponential decay function, with lifetime τ = 130 ± 10 fs, convoluted with a Gaussian cross-correlation function (width ≈60 fs).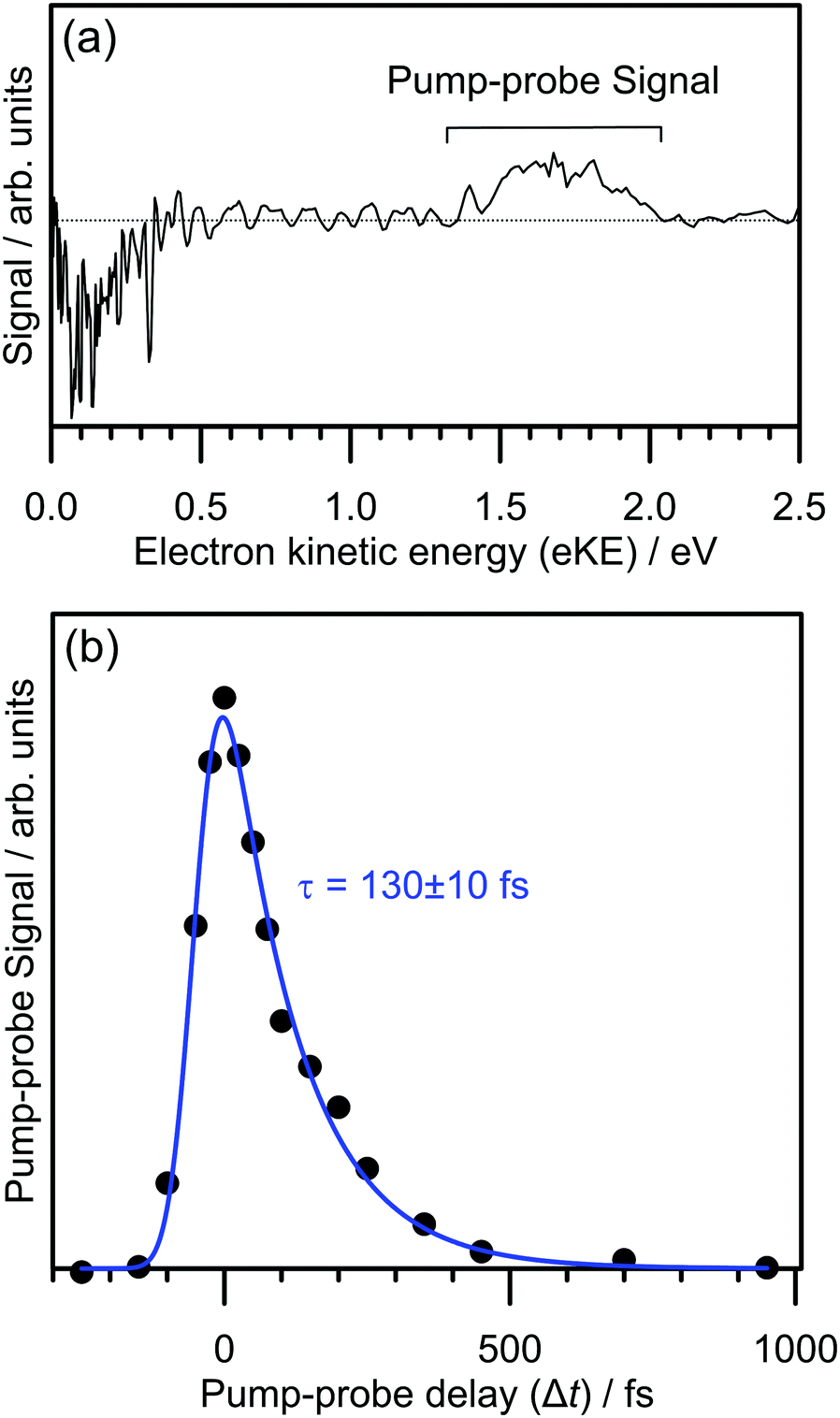 | ||
| Fig. 3 Time-resolved photoelectron spectroscopy of 2-Np− with the pump photon energy tuned to the 0–0 transition of the S1(ππ*) band (hν = 2.70 eV): (a) example time-resolved spectrum for Δt = 50 fs with a 1.55 eV (800 nm) probe. Photodepletion signal at low-eKE approximately equals the pump–probe signal in the 1.3 ≤ eKE ≤ 2.0 eV window (true for all Δt). (b) Pump–probe signal with Δt. The blue trace is the fit of the experimental data, returning the decay lifetime of τ = 130 ± 10 fs. | ||
The time-resolved photoelectron spectra and velocity-map images do not show any evidence for internal conversion to a dipole-bound state, which would be evident as a sharp feature in the time-resolved spectra at an eKE close to the probe photon energy and present as an anisotropic ring in the velocity-map images.31,33 It is worth noting that although the relative photodetachment cross-section (i.e. probe cross-section) from the dipole-bound state and the S1(ππ*) state are not known, time-resolved imaging experiments on other systems in which a photoexcited valence-localized state internally converts to a dipole-bound state show that population of the dipole-bound state should be evident in the time-resolved spectra, even if only a small fraction (e.g. <10%) of photoelectron signal in the pump-only photoelectron spectrum is attributed to internal conversion to the dipole-bound state.31 We conclude that there is no substantial amount of internal conversion from the S1(ππ*) state to the dipole-bound state. Regardless of the detailed mechanism, the time-resolved experiments show that the excited state population almost exclusively decays by autodetachment, and, therefore, no ground state of the anion is recovered by internal conversion as has been observed for several other anions.25,32,34 Based on the action spectrum, the vibrational period of the dominant nuclear motion (if exciting above the 0–0 transition) involves the 437 cm−1 mode, which has a vibrational period of 76 fs. Hence, with an autodetachment lifetime of 130 fs, only a few vibrations can take place on the excited state before the electron is ejected.
3.3 Autodetachment dynamics leading to the low-eKE vibrational structure
The frequency-resolved and time-resolved photoelectron data show that 2-Np− photoexcited over the S1(ππ*) band decays through ultrafast autodetachment. A striking observation is that the low-eKE vibrational structure appears to be present at all photon energies considered in this study, including when the OPO was tuned to excite the dipole-bound state(s). Three possible explanations for the low-eKE vibrational structure are (Fig. 4):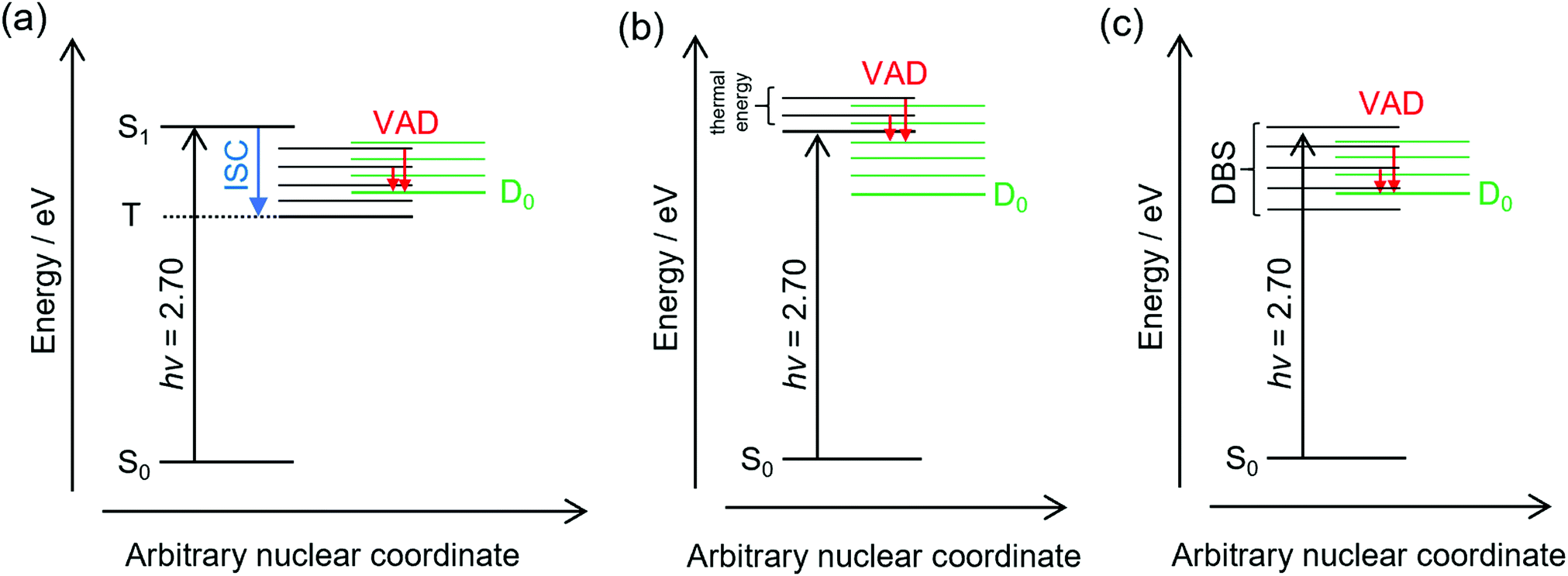 | ||
| Fig. 4 Possible mechanisms giving rise to the low-eKE vibrational structure following excitation of the S1(ππ*) band of 2-Np−: (a) ultrafast intersystem crossing to a triplet state (T) followed by T → D0 mode-specific vibrational autodetachment, (b) nuclear motion on the S1(ππ*) state, e.g. by the thermally populated ν33 mode, leads to S1 → D0 mode-specific vibrational autodetachment (electron shake-off), and (c) dipole-bound states are excited over an extended range and undergo DBS → D0 mode-specific vibrational autodetachment. The discussion concludes that mechanism (b) is the most likely explanation when exciting over the S1(ππ*) band, and mechanism (c) can explain the low-eKE vibrational structure when directly exciting the dipole-bound state. Key: ISC – intersystem crossing, VAD – mode-specific vibrational autodetachment, DBS – manifold of dipole-bound states. | ||
(a) Some fraction of the photoexcited population undergoes an ultrafast intersystem crossing to a triplet state (T), which is situated below the detachment threshold. The triplet state then undergoes a mode-specific autodetachment. The triplet state may have a very low photodetachment cross-section and thus not be observed in the time-resolved experiments using a 800 nm probe. A recent study on a related oxygenated PAH anion has implied an ultrafast intersystem crossing,35 although the study hypothesised that an intermediate dipole-bound state facilitated triplet formation; there was no evidence for internal conversion to a dipole-bound state in the present study. We also note that our earlier study using ion mobility spectrometry coupled with laser spectroscopy on carboxylate substituted versions of 2-Np− infer triplet formation through the observation of fluorine-substitution photoreactions following excitation of the S1(ππ*) band but not the S2(ππ*) band.21 Those experiments indicate that the excited state in the carboxylate substituted versions of 2-Np− must survive for nanoseconds based on the buffer gas pressure and the rate for ion–molecule collisions leading to products. No such photoreactions were observed for 2-Np−. Ultimately, we conclude that assignment of the low-eKE vibrational structure to mode-specific autodetachment from a triplet state is unlikely.
(b) Some fraction of the photoexcited population on the S1(ππ*) state undergoes a mode-specific autodetachment induced by a low-frequency mode, which is presumably thermally populated at T ≈ 300 K in the hν = 2.70 eV experiment since we are exciting at the 0–0 transition. The nuclear motion induced by population of the low-frequency mode leads to a modulation of the vertical detachment threshold and consequently a ‘shake off’ of the electron. The low-eKE vibrational structure with spacing of ≈35 meV (282 cm−1) could potentially correspond to the ν33(a′) = 313 cm−1 mode calculated for the S1(ππ*) state.21 This mode is the lowest frequency a′-symmetry mode involving an in-plane bend. However, it is difficult to reconcile the observation of similar low-eKE structure in the photoelectron spectra for photon energies hν < 2.7 eV (e.g. 2.38 eV, Fig. 2a), where the S1(ππ*) state is not excited and only the dipole-bound state(s) is accessible.
(c) A small fraction of the anions are excited directly to the dipole-bound state, i.e. there is a progression of dipole-bound states. The dipole-bound state autodetaches through mode-specific vibrational autodetachment invoked by modes that strongly couple the dipole-moment to the continuum.31,33,36,37 These modes would be similar to the modes of the final neutral ground state. The lowest frequency mode of a′ symmetry for the D0 electronic state was calculated at ν33 = 310 cm−1 and is moderately infra-red active. This vibrational mode satisfies the criterion for efficient electron ejection or ‘shake off’ when directly exciting the dipole-bound state – modulation of the dipole-bound state orbital by the vibrational motion – and would generate a similar (s-wave) low-eKE spectrum to mode-specific autodetachment proposed in mechanism (b).33,36–41 This detachment mechanism usually involves Δv = −1, where v is a vibrational level, although anharmonicity associated with low-frequency bending modes has shown that Δv = −2 also occurs in this system.23 The proposed low frequency mode, ν33, is likely to be populated at T ≈ 300 K according to Boltzmann statistics. It is worth noting that the recent cryogenic study of 2-Np− by Qian et al.23 performed photoelectron spectroscopy following resonant excitation of 38 levels of the dipole-bound state. They found that seven Franck–Condon-forbidden out-of-plane bending modes were involved in mode-specific autodetachment, including the lowest two frequency a′′ modes at ν48 = 102 cm−1 and ν47 = 171 cm−1. Furthermore, both ν33 and ν32 modes are major features in the resonant photoelectron spectra for various excited dipole-bound levels. Whatever the active modes leading to mode-specific vibrational autodetachment, mechanism (c) readily explains the low-eKE vibrational structure in the hν = 2.38 eV spectrum, although mechanism (c) is unlikely at hν = 2.70 eV (0–0 transition of the S1(ππ*) band) since there was no evidence for a dipole-bound state in the time-resolved measurements. Unfortunately, time-resolved experiments with a pump photon energy tuned to directly excite the dipole-bound state and monitor the autodetachment dynamics were unsuccessful due to the low photoexcitation cross-section. However, such an experiment has been recently performed on cryogenically cooled phenoxide,41 determining lifetimes ranging between 33.5 and 1.4 ps depending on the dipole-bound state vibrational level (the lifetime of the dipole ground vibrational state will be substantially longer). Lifetimes for dipole-bound state levels of 2-Np− are presumably shorter due to the increased density of states and, in the present study, initial ion temperature.
Ultimately, the consistent interpretation for photon energies over the S1(ππ*) band of 2-Np− at T ≈ 300 K is provided by mechanism (b), which involves partial nuclear relaxation of the S1(ππ*) state followed by vibrational electron shake-off by the low-frequency ν33(a′) mode. A similar electron shake off occurs by the corresponding mode of the neutral molecular core when directly exciting the dipole-bound state, consistent with mechanism (c).
3.4 Astrophysical implications
The astrochemistry community frequently assumes that interstellar anions are formed through either dissociative electron attachment or radiative electron attachment.12,17 The dissociative electron attachment mechanism is less likely to be important for unsubstituted PAH species due to strong molecular bonding and an electron detachment threshold that is lower in energy than σ* states, which could lead to excited state dissociation processes, or lower in energy than ground electronic state bond dissociation energies, which could lead to statistical dissociation processes. Anion formation through radiative electron attachment involves the parent neutral molecule capturing a free electron by either a dipole-bound state42 or an above-threshold resonance,25e.g. the S1(ππ*) state in 2-Np−, which is formally classified as a shape resonance since it is situated in the detachment continuum. After electron capture, the ground electronic state must be formed through internal conversion followed by infra-red emission to lower the total internal energy to a level below the electron detachment threshold.32,43,44 Efficient formation of a stable anion requires that the rate of infra-red emission exceeds or is competitive with the rates for anion destruction, e.g. thermionic emission. Note that radiative electron capture requires the parent (radical) neutral species to be present in space. Because radicals are typically unstable toward photodissociation,12,45 the formation mechanism is more likely in dark regions of space compared with photon-dominated regions (PDRs).It is worth mentioning that in the present experiment we optically prepared the same excited states that could be occupied through an electron capture mechanism.24,37 From an experimental point of view, our approach has two key advantages over an electron capture experiment: (i) the ability to directly probe the dynamics using femtosecond laser strategies, and (ii) mass-selectivity through separation of anions using time-of-flight mass spectrometry.
As we outlined in the introduction, while small oxygenated PAH molecules are likely interstellar species, the present experiments on 2-Np− have shown that population of either the lowest energy dipole-bound state or the S1(ππ*) state does not result in formation of a stable anion, rather that the anion excited states decay through autodetachment. The radiative electron capture mechanism is therefore not important for this species. We acknowledge that the present study considered ions initially thermalised to T ≈ 300 K, which is substantially warmer than typical interstellar molecule conditions of T = 10–20 K.46 However, we expect similar results for cold anions since the S1(ππ*) state is situated in the detachment continuum and is unbound to the electron detachment threshold, and because there was no evidence for internal conversion from the dipole-bound state. The situation is different for other PAH-based anions known to undergo ultrafast internal conversion to the ground electronic state anion after excitation to a resonance,25,32 attributed to the presence of lower-lying bound excited states; 2-Np− has no such states. Known interstellar polyyne anions, e.g. C6H− and C8H−, have similar bound electronic states.47 That being said, we have shown that other molecules such as the radical anion of para-benzoquinone,24 which has no valence-bound excited electronic states, may exhibit a small fraction thermionic emission when excited to the lowest lying above-threshold resonances. Ultimately, we conclude that 2-Np− is unlikely to be formed in the interstellar medium through radiative electron attachment and has no resilience to photodestruction through the absorption of visible radiation resonant with the S1(ππ*) band.
4 Conclusions
This study has shown that photoexcitation at the 0–0 transition of the S1(ππ*) band of 2-naphthoxide thermalised to T ≈ 300 K leads to an ultrafast autodetachment with lifetime 130 ± 10 fs. Low-eKE vibrational structure in the photoelectron spectra that are evident at all photon energies over the S1(ππ*) band are consistent with an ultrafast, mode-specific autodetachment mechanism. There was no evidence for internal conversion to a dipole-bound state or the ground electronic state. Significantly, the lack of internal conversion to the ground electronic state implies that the anion has no photoresiliance toward absorption of visible radiation by the S1(ππ*) band, and presumably also UV radiation that would excite higher-lying excited states. Future studies will investigate the influence of the number of rings and molecular rigidity on the propensity for ultrafast internal conversion between electronic states in oxygenated PAHs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from University of East Anglia start-up funds (to JNB) and the European Research Council Starting Grant scheme (306536 to JRRV).Notes and references
- B. A. McGuire, Astrophys. J., Suppl. Ser., 2018, 239, 17 CrossRef CAS.
- A. G. G. M. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289 CrossRef CAS.
- E. E. Hardegree-Ullman, M. S. Gudipati, A. C. A. Boogert, H. Lignell, L. J. Allamandola, K. R. Stapelfeldt and M. Werner, Astrophys. J., 2014, 784, 172 CrossRef.
- A. G. G. M. Tielens, EAS Publ. Ser., 2011, 46, 3 CrossRef.
- L. J. Allamandola, S. A. Sandford and B. Wopenka, Science, 1987, 237, 56 CrossRef CAS.
- S. J. Clemett, C. R. Maechling, R. N. Zare, P. D. Swan and R. M. Walker, Science, 1993, 262, 721 CrossRef CAS PubMed.
- R. Ruiterkamp, T. Halasinski, F. Salama, B. H. Foing, L. J. Allamandola, W. Schmidt and P. Ehrenfreund, Astron. Astrophys., 2002, 390, 1153 CrossRef CAS.
- G. Lagache, H. Dole, J.-L. Puget, P. G. Pérez-González, E. L. Floc'h, G. H. Rieke, C. Papovich, E. Egami, A. Alonso-Herrero, C. W. Engelbracht, K. D. Gordon, K. A. Misselt and J. E. Morrison, Astrophys. J., Suppl. Ser., 2004, 154, 112 CrossRef CAS.
- C. W. Bauschlicher, A. Ricca, C. Boersma and L. J. Allamandola, Astrophys. J., Suppl. Ser., 2018, 234, 32 CrossRef.
- E. K. Campbell, M. Holz, D. Gerlich and J. P. Maier, Nature, 2015, 523, 322 CrossRef CAS PubMed.
- M. A. Cordiner, H. Linnartz, N. L. J. Cox, J. Cami, F. Najarro, C. R. Proffitt, R. Lallement, P. Ehrenfreund, B. H. Foing, T. R. Gull, P. J. Sarre and S. B. Charnley, Astrophys. J., 2019, 875, L28 CrossRef CAS.
- T. J. Millar, C. Walsh and T. A. Field, Chem. Rev., 2017, 117, 1765 CrossRef CAS.
- D. McElroy, C. Walsh, A. J. Markwick, M. A. Cordiner, K. Smith and T. J. Millar, Astron. Astrophys., 2013, 550, A36 CrossRef.
- M. P. Bernstein, J. P. Dworkin, S. A. Sandford and L. J. Allamandola, Adv. Space Res., 2002, 30, 1501 CrossRef.
- M. P. Bernstein, J. E. Elsila, J. P. Dworkin, S. A. Sandford, L. J. Allamandola and R. N. Zare, Astrophys. J., 2002, 576, 1115 CrossRef CAS.
- T. P. Snow and A. N. Witt, Science, 1995, 270, 1455 CrossRef CAS PubMed.
- S. Petrie and D. K. Bohme, Mass Spectrom. Rev., 2006, 26, 258 CrossRef PubMed.
- M. P. Bernstein, S. A. Sandford, L. J. Allamandola, J. S. Gillette, S. J. Clemett and R. N. Zare, Science, 1999, 283, 1135 CrossRef CAS PubMed.
- A. M. Cook, A. Ricca, A. L. Mattioda, J. Bouwman, J. Roser, H. Linnartz, J. Bregman and L. J. Allamandola, Astrophys. J., 2015, 799, 14 CrossRef.
- M. S. Gudipati and R. Yang, Astrophys. J., 2012, 756, L24 CrossRef.
- J. N. Bull, J. T. Buntine, M. S. Scholz, E. Carrascosa, L. Giacomozzi, M. H. Stockett and E. J. Bieske, Faraday Discuss., 2019, 217, 34 RSC.
- S. J. Kregel and E. Garand, J. Chem. Phys., 2018, 149, 074309 CrossRef PubMed.
- C.-H. Qian, G.-Z. Zhu, Y.-R. Zhang and L.-S. Wang, J. Chem. Phys., 2020, 152, 214307 CrossRef CAS PubMed.
- C. W. West, J. N. Bull, E. Antonkov and J. R. R. Verlet, J. Phys. Chem. A, 2014, 118, 11346 CrossRef CAS.
- J. N. Bull, C. W. West and J. R. R. Verlet, Chem. Sci., 2015, 6, 1578 RSC.
- C. S. Anstöter, J. N. Bull and J. R. R. Verlet, Int. Rev. Phys. Chem., 2016, 35, 509 Search PubMed.
- G. M. Roberts, J. Lecointre, D. A. Horke and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2010, 12, 6226 RSC.
- D. A. Horke, G. M. Roberts, J. Lecointre and J. R. R. Verlet, Rev. Sci. Instrum., 2012, 83, 063101 CrossRef PubMed.
- G. M. Roberts, J. L. Nixon, J. Lecointre, E. Wrede and J. R. R. Verlet, Rev. Sci. Instrum., 2009, 80, 053104 CrossRef CAS.
- R. N. Zare, Mol. Photochem., 1972, 4, 1 CAS.
- J. N. Bull, C. S. Anstöter and J. R. R. Verlet, Nat. Commun., 2019, 10, 5820 CrossRef CAS PubMed.
- J. N. Bull, C. W. West and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2015, 17, 32464 RSC.
- J. N. Bull, C. W. West and J. R. R. Verlet, Chem. Sci., 2016, 7, 5352 RSC.
- D. A. Horke, Q. Li, L. Blancafort and J. R. R. Verlet, Nat. Chem., 2013, 5, 711 CrossRef CAS PubMed.
- G.-Z. Zhu, L. F. Cheung, Y. Liu, C.-H. Qian and L.-S. Wang, J. Phys. Chem. Lett., 2019, 10, 4339 CrossRef CAS PubMed.
- C. S. Anstöter and J. R. R. Verlet, J. Phys. Chem. Lett., 2020, 11, 6456 CrossRef PubMed.
- C. S. Anstöter, G. Mensa-Bonsu, P. Nag, M. Ranković, T. P. Ragesh Kumar, A. N. Boichenko, A. V. Bochenkova, J. Fedor and J. R. Verlet, Phys. Rev. Lett., 2020, 124, 203401 CrossRef PubMed.
- J. Simons, J. Am. Chem. Soc., 1981, 103, 3971 CrossRef CAS.
- H.-T. Liu, C.-G. Ning, D.-L. Huang, P. D. Dau and L.-S. Wang, Angew. Chem., Int. Ed., 2013, 52, 8976 CrossRef CAS PubMed.
- J. Simons, J. Phys. Chem. A, 2020, 124, 8778 CrossRef CAS PubMed.
- D. H. Kang, S. An and S. K. Kim, Phys. Rev. Lett., 2020, 125, 093001 CrossRef CAS PubMed.
- P. J. Sarre, Mon. Not. R. Astron. Soc., 2000, 313, L14 CrossRef CAS.
- J. N. Bull, M. S. Scholz, E. Carrascosa, M. K. Kristiansson, G. Eklund, N. Punnakayathil, N. de Ruette, H. Zettergren, H. T. Schmidt, H. Cederquist and M. H. Stockett, J. Chem. Phys., 2019, 151, 114304 CrossRef.
- M. H. Stockett, J. N. Bull, J. T. Buntine, E. Carrascosa, E. K. Anderson, M. Gatchell, M. Kaminska, R. F. Nascimento, H. Cederquist, H. T. Schmidt and H. Zettergren, Eur. Phys. J. D, 2020, 74, 150 CrossRef CAS.
- M. Khamesian, N. Douguet, S. F. dos Santos, O. Dulieu, M. Raoult, W. J. Brigg and V. Kokoouline, Phys. Rev. Lett., 2016, 117, 123001 CrossRef PubMed.
- E. A. Bergin and M. Tafalla, Annu. Rev. Astron. Astrophys., 2007, 45, 339 CrossRef CAS.
- T. Pino, M. Tulej, F. Güthe, M. Pachkov and J. P. Maier, J. Chem. Phys., 2002, 116, 6126 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photoelectron spectra recorded at other photon energies. See DOI: 10.1039/d1cp00261a |
| This journal is © the Owner Societies 2021 |