
Cyanogroup functionalized sub-2 nm ultrafine Pt nanonetworks reinforce electrocatalytic hydrogen evolution in a broad pH range†
Qicheng
Liu
 ,
Xuan
Wang
,
Jiaqi
Liu
,
Xinyi
Zhou
,
Qingwei
Meng
,
Xinrui
Zhou
,
Dongmei
Sun
* and
Yawen
Tang
*
,
Xuan
Wang
,
Jiaqi
Liu
,
Xinyi
Zhou
,
Qingwei
Meng
,
Xinrui
Zhou
,
Dongmei
Sun
* and
Yawen
Tang
*
Jiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Centre of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: sundongmei@njnu.edu.cn; tangyawen@njnu.edu.cn
First published on 12th July 2021
Abstract
Surface functionalization is an effective way to improve the activity of electrocatalysts by adjusting the adsorption and desorption capacity of intermediate species on noble metal surfaces. Herein, density functional theory (DFT) calculations demonstrate that cyanogroup functionalization can effectively reduce the d-band centre (εd) of the Pt (111) plane, thus resulting in a hydrogen evolution reaction (HER) performance improvement. Cyanogroup functionalized Pt ultrafine nanonetworks (CN–Pt UNs, sub-2 nm) are synthesized via a facile one-pot heating method with high yield and abundant defects/grain boundaries as the highly active sites. Owing to the optimized Pt electronic properties and ultrathin network nanostructure, the overpotentials of CN–Pt UNs at 10 mA cm−2 are merely 6 mV and 8 mV in 1 M HClO4 and 1 M KOH, respectively, and are superior to those of commercial 20 wt% Pt/C in a wide pH range. This work offers a new perspective towards the rational design of noble metal catalysts for boosting electrocatalysis by surface cyanogroup functionalization.
Hydrogen, as a pure and sustainable energy source, has significant theoretical and applied research value due to the increasingly serious environmental pollution and energy crisis.1–6 Electrochemical water splitting is an effective and clean way to produce hydrogen in a cathode when compared with the conventional hydrogen production processes through fossil fuels.7–13 Regrettably, the HER can hardly be extended to large scale applications due to unfavourable reaction kinetics and the high cost of the most optimal catalyst Pt.14–19 Hence, it is essential to develop Pt-based electrocatalysts with abundant highly active sites through modifying the geometric and electronic structures of Pt.20,21 To cope with this challenge, numerous Pt-based nanocatalysts with different morphologies, especially three-dimensional (3D) nanonetworks, have been designed and fabricated to expose more active sites.22–25 3D network nanostructures can not only hamper dissolution, aggregation, and Ostwald ripening during long-term operation, in comparison to zero-dimensional (0D) commercial Pt nanoparticles, but also effectually facilitate mass/electron transfer.26–29
Surface functionalization of organic groups is also an effective way to improve the catalytic activity of Pt-based catalysts by changing the adsorption/desorption capacity of the reaction intermediates on Pt sites.30–33 Surface modification of specific groups can tune the electronic interaction and d-band centre of electrocatalysts, accordingly changing the chemisorption strength of intermediates and further modulating the kinetic and thermodynamic factors of electrocatalytic reactions.34–37 For instance, ethylenediamine-functionalized Pt nanowires exhibited an excellent activity for the selective generation of N-hydroxylanilines owing to electronic effects,38 and chlorophenyl Pt nanocrystals revealed enhanced reactivity for the oxygen reduction reaction (ORR) compared with naked Pt nanocrystals due to steric effects.39 Dusan Strmcnik et al. also reported that the ORR performance of the Pt (111) surface can be highly enhanced by CNads modification. The introduced CN-groups can prevent OH* adsorption on the metal surface and prevent catalyst poisoning.40
Inspired by the above analysis, we investigated the impact of cyanogroup functionalization on the Pt electronic structure and corresponding HER performance through DFT calculations in advance. Next, we synthesized a cyano-modified Pt ultrafine nanonetwork (CN–Pt UN) electrocatalyst via a simple one-pot method. The HER performance of the CN–Pt UNs was compared to those of commercial Pt/C and other Pt-based control samples in a wide pH range. Specifically, for the CN–Pt UNs, the overpotentials at 10 mA cm−2 are only 6 mV and 8 mV in 1 M HClO4 and 1 M KOH, respectively, immensely surpassing those of commercial 20 wt% Pt/C (37 mV and 16 mV).
DFT calculations were primarily performed to investigate the effect of cyanogroup modification on the Pt electronic structure and the adsorption/desorption capacity of H* on the Pt (111) surface. Fig. S1† shows an established model of the CN–Pt UNs. As illustrated in the differential charge density of CN–Pt UNs (Fig. 1a), electrons transfer from the Pt (111) facets to the modified cyanogroup on the catalyst surface. To further determine the electron distribution on CN–Pt UNs, the Bader charge population was calculated. As shown in Fig. 1b, the surface charge of Pt sites increases to 0.113, while that of the N sites decreases to −1.059 under the inductive effect. The formation of a charge gap between the cyanogroup and Pt (111) facet is beneficial to tuning the adsorption strength of H* on Pt sites during the HER.41 The orbital-decomposed partial density of states (PDOS) curves in Fig. 1c and d display that after the cyanogroup modification onto the Pt (111) facet, the electron density becomes closer to the Fermi level, which exhibits the possibly enhanced adsorption of H*, therefore promoting the Volmer step. In addition, the d-band centre was calculated to downshift from −2.1704 (Pt) to −2.1728 eV (CN–Pt UNs) relative to the Fermi level, revealing the lower occupancy of antibonding states after the cyanogroup modification, which was favourable for the adsorption of H* and might promote the Volmer step in the HER.42,43
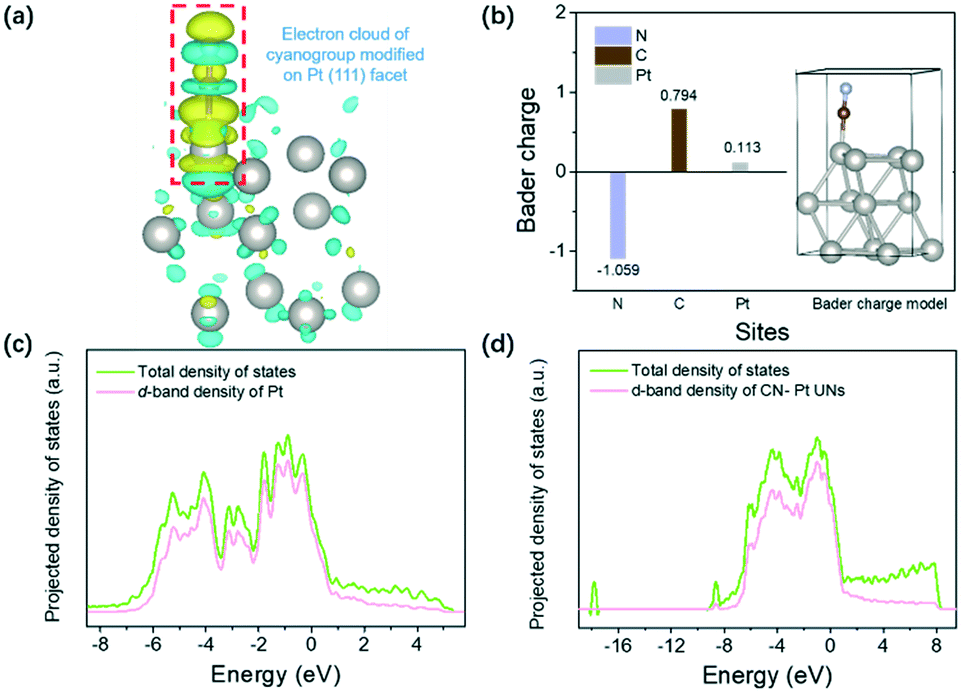 | ||
| Fig. 1 (a) Differential charge density of CN–Pt UNs, where the iso-surface value is 0.003 e bohr−3. The light yellow and blue regions in the diagram represent the gain and loss of electrons, respectively. (b) Bader charge population of the different elements in CN–Pt UNs. Orbital-decomposed partial density of states of (c) Pt and (d) CN–Pt UNs in the nanocontact model. | ||
To verify the theoretical hypothesis, we prepared the CN–Pt UNs through a one-step wet-chemical method, using cyanoacetic acid (CAA) as the morphology guide agent and sodium cyanoborohydride (SC) as the reducing agent (Fig. 2a). The growth mechanism of CN–Pt UNs was explored via a series of control experiments. As the important components, the addition of CAA and SC is key to the successful formation of the network nanostructure and cyanogroup modification. Based on the FT-IR spectra displayed in Fig. 2b and S2,† the absorption peak of Pt–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N appears at 2162 cm−1, revealing the successful cyanogroup coordination with Pt in CN–Pt UNs. In contrast, no characteristic peaks could be detected in the range of 2100–2400 cm−1 in the FT-IR spectra of SC–Pt and CAA–Pt, indicating that the cyanogroup modification could only be successful when CAA and SC were introduced into the reaction system at the same time.
N appears at 2162 cm−1, revealing the successful cyanogroup coordination with Pt in CN–Pt UNs. In contrast, no characteristic peaks could be detected in the range of 2100–2400 cm−1 in the FT-IR spectra of SC–Pt and CAA–Pt, indicating that the cyanogroup modification could only be successful when CAA and SC were introduced into the reaction system at the same time.
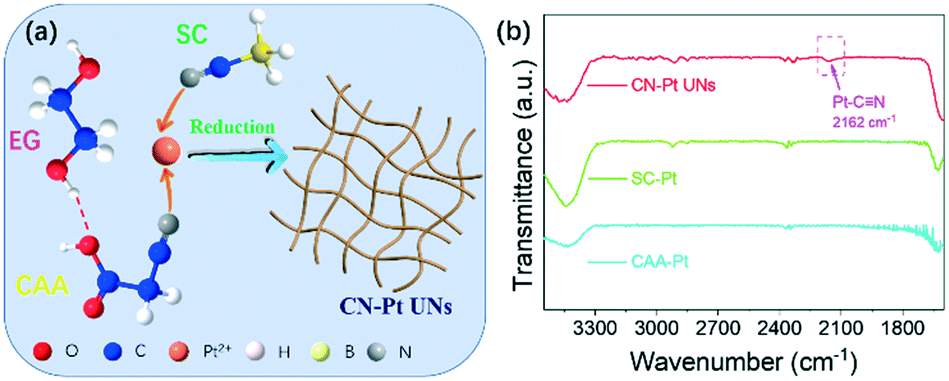 | ||
| Fig. 2 (a) Schematic diagram of CN–Pt UNs; (b) FT-IR spectra of CN–Pt UNs, SC–Pt NPs and CAA–Pt NNs. | ||
For the morphology control, the resulting SC–Pt nanoparticles (NPs) merely consisted of irregular nanoparticles in the absence of CAA (Fig. S5a†), while CAA–Pt nanonetworks (NNs) with a bigger particle size were acquired using NaBH4 as the alternative reductant (Fig. S5b†). This implies that the intermolecular hydrogen bonds formed between the carboxyl group in CAA and the alcoholic hydroxyl group in the solvent actuate the oriented self-assembly of Pt nanoparticles to generate the network morphology.44,45 Besides, SC, being a milder reducing agent than NaBH4 under alkaline conditions, is superior in regulating the morphology of CN–Pt UNs, favouring the configuration of ultrafine nanonetworks. Moreover, we also explored the influence of solvents on the final products. Diethylene glycol (DEG), triethylene glycol (TEG), deionized water, and N,N-dimethylformamide (DMF) were separately selected as the contrast solvents for morphological comparison. When using DEG and TEG as substitute solvents, the acquired Pt catalysts can exhibit a similar ultrafine network nanostructure (Fig. S6†) to that of CN–Pt UNs, further attesting the great importance of intermolecular hydrogen bonds generated between the alcoholic hydroxyl groups and CAA. In contrast, only Pt nanoparticles (Fig. S7†) can be acquired in the case of the solvent being changed to water or DMF, which might be due to the failure of forming hydrogen bonds with suitable strength in these solvents.
Transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) were performed to examine the morphology and nanostructure of the as-prepared CN–Pt UNs in detail. Representative TEM (Fig. 3a) and STEM (Fig. 3b) images of the products clearly reveal that the Pt nanonetworks consisted of continuous and wave-like ultrafine nanowires (nearly 2 nm) with high yield and uniformity. The lattice spacing was measured to be about 0.223 nm in the HRTEM image (Fig. 3c), which could be assigned to the Pt (111) facet of the face-centred cubic (fcc) structure. It is worth noting that the ultrafine nanowires present a rough surface and an uneven diameter, and contain rich defects and distinct grain boundaries, as marked by the red arrows and dotted lines in Fig. 3c. This would offer adequate highly active sites on the CN–Pt UN surface, which play a crucial role in the ultimate electrocatalytic performance improvement.26,46,47 The homogeneous distribution of the Pt and N elements in the energy dispersive X-ray spectroscopy (EDS) elemental mapping images (Fig. 3d–f) further evidenced the sufficient cyanogroup modification on the CN–Pt UN surface, corresponding to the FT-IR spectral results (Fig. 2b).
 | ||
| Fig. 3 Morphological and elemental characterization of CN–Pt UNs: (a) TEM image, (b) HAADF-STEM image, (c) HRTEM images, and (d–f) EDS elemental mapping images. | ||
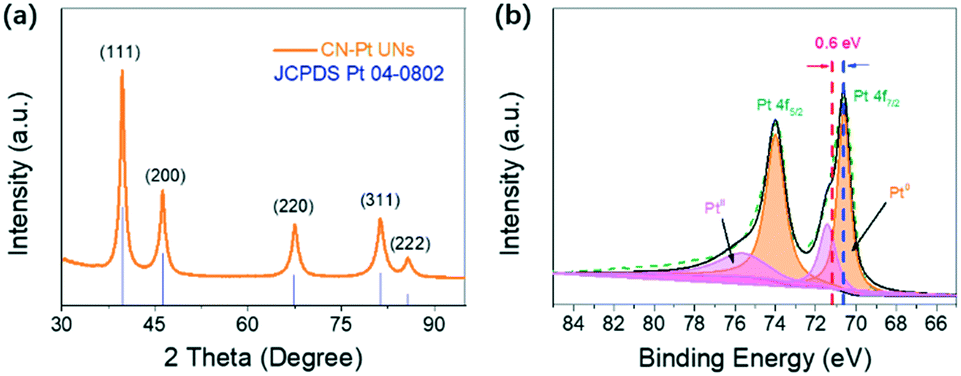 | ||
| Fig. 4 (a) XRD pattern of CN–Pt UNs; (b) Pt 4f XPS spectrum of CN–Pt UNs. | ||
The powder X-ray diffraction (XRD) pattern (Fig. 4a) was obtained to determine the crystal structure of CN–Pt UNs. Five obvious diffraction peaks located at 39.5°, 46.0°, 67.3°, 81.1°, and 85.6° are in accordance with the (111), (200), (220), (311), and (222) facets of fcc Pt (JCPDS no. 04-0802), respectively. X-ray photoelectron spectroscopy (XPS) was further utilized to analyse the near-surface elemental composition and chemical states. As shown in Fig. S3,† the peak of N 1s at 397.4 eV indirectly proves the existence of cyanogroups on the surface of CN–Pt UNs. In the high-resolution XPS spectrum of Pt (Fig. 4b), the peaks located at 73.9 and 70.6 eV correspond to Pt 4f5/2 and Pt 4f7/2 of the Pt metallic state (Pt0), respectively. The dominant Pt0 peaks show the successful reduction of Pt in the CN–Pt UNs, and the weak oxidation state peaks can be attributed to the exposure to air. It is remarkable that the Pt0 peaks of CN–Pt UNs exhibit a negative shift of 0.6 eV compared to that of pure metallic Pt (71.2 eV), which is probably ascribed to the effective cyanogroup modification.48
Significantly, the negative shift of the Pt binding energy implied a downward shift of the Pt d-band centre of CN–Pt UNs according to the Hammer–Nørskov reactivity model, corresponding well to the DFT results in Fig. 1.49–51 The Brunauer–Emmett–Teller (BET) surface area of CN–Pt UNs was measured to be 62.34 m2 g−1. A typical hysteresis loop between 0.6 and 0.95 p/p0 in the N2 adsorption–desorption isotherms (Fig. S4†) further indicates the vast microporous/mesoporous structures of CN–Pt UNs, endowing the catalyst satisfactory structural stability and facilitating mass transport and electron transfer in the reaction process.52
Based on the compositional and structural advantages, the HER performance of CN–Pt UNs was firstly evaluated in a N2-saturated acidic electrolyte (1 M HClO4), with commercial 20 wt% Pt/C, SC–Pt NPs, and CAA–Pt NNs as the reference samples. The HER polarization curves are shown in Fig. 5a and the corresponding overpotentials at different current densities are summarized in Fig. 5b. Undoubtedly, CN–Pt UNs exhibited the best HER activity with merely 6 and 34 mV overpotentials at 10 and 50 mA cm−2, respectively, which are significantly lower than those of commercial 20 wt% Pt/C (37 and 59 mV) and the other two control samples, indicating that CN–Pt UNs have remarkable HER activity in either ordinary or high-current environments. Fig. 5c shows that the Tafel slope of CN–Pt UNs (42.3 mV dec−1) is similar to that of commercial Pt/C (43.6 mV dec−1) and the other control samples, indicating a Volmer–Tafel dominated HER mechanism, and the recombination process of chemisorbed hydrogen is the rate-determining step.53 According to the integral calculation of the peak area of hydrogen adsorption and desorption, the electrochemical active surface area (ECSA) of CN–Pt UNs under acidic conditions was 10.71 m2 g−1. Whereafter, the long-term electrochemical stability of CN–Pt UNs was examined through chronoamperometry tests. Compared to the rapid decay current density of commercial 20 wt% Pt/C, CN–Pt UNs can maintain 70% current density at a constant voltage of 10 mA cm−2 for a 24 hour test. In the meantime, the prominent electrochemical stability was also confirmed via accelerated durability tests (ADTs). After a continuous cyclic voltammetry (CV) scan of 1000 cycles, the overpotential of CN–Pt UNs occurs a slightly negative shift of 32 mV (vs. 37 mV for commercial Pt/C, Fig. S8a†), and few morphological changes or agglomeration (Fig. S9a†) and slight crystalline structure changes (Fig. S10†) were observed.
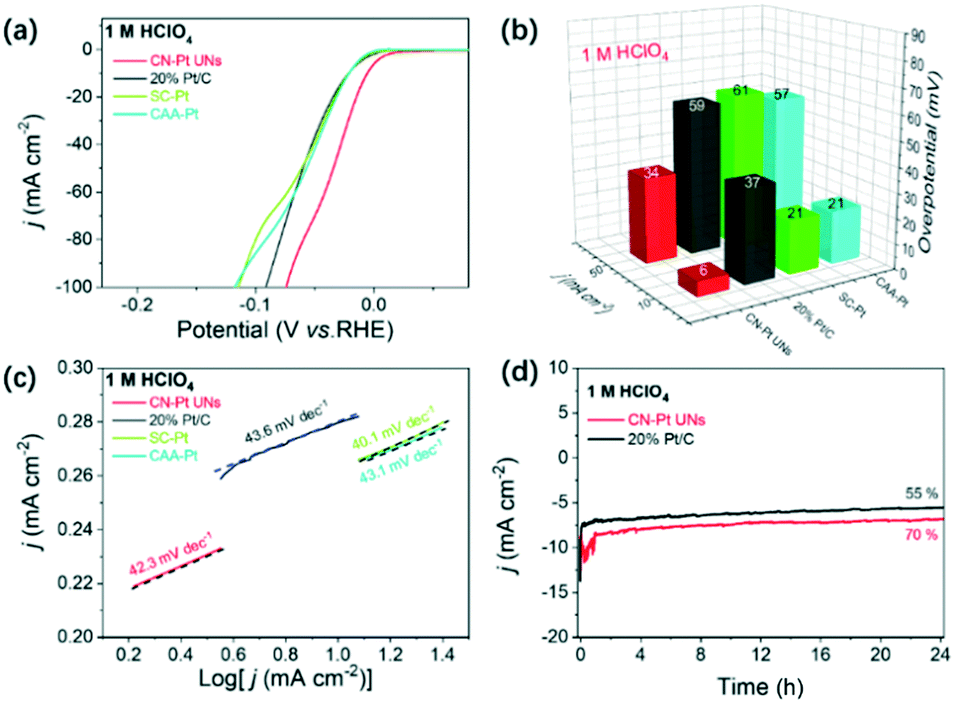 | ||
| Fig. 5 Comparison of the HER performance of CN–Pt UNs and the other catalysts in 1 M HClO4: (a) HER polarization curves, (b) overpotentials at 10 mA cm−2 and 50 mA cm−2, (c) Tafel curves, and (d) time-dependent current density curves in 1 M HClO4. | ||
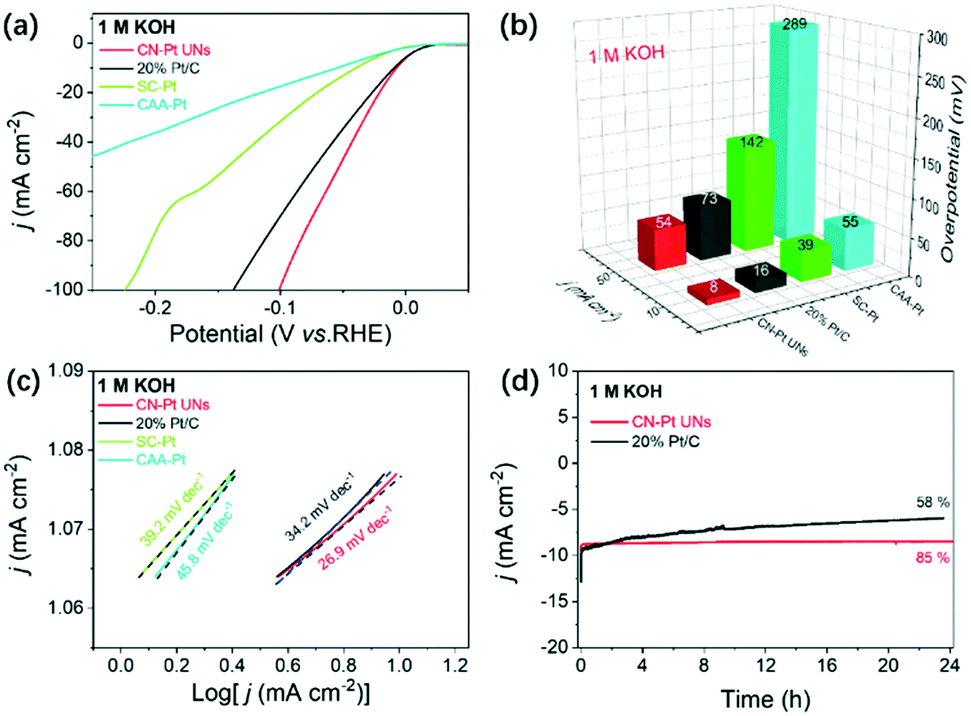
Strikingly, the CN–Pt UNs also exhibited a compelling HER performance in an alkaline environment (1 M KOH), significantly more favourable than that of the commercial 20 wt% Pt/C, SC–Pt NPs, and CAA–Pt NNs (Fig. 6a). As shown in Fig. 6b, both overpotentials of CN–Pt UNs at 10 mA cm−2 and 50 mA cm−2 (8 mV and 16 mV) are obviously better than those of commercial Pt/C (16 mV and 73 mV). There is no doubt that CN–Pt UNs also exhibit an extremely better HER performance than CAA–Pt NNs and SC–Pt NPs in alkaline electrolytes. Impressively, the Tafel slope of CN–Pt UNs is calculated to be only 39.2 mV dec−1, much lower than those of commercial 20 wt% Pt/C (34.2 mV dec−1), SC–Pt NPs (39.2 mV dec−1), and CAA–Pt NNs (45.8 mV dec−1), revealing the superior HER catalytic kinetics of CN–Pt UNs (Fig. 6c). Meanwhile, the extremely low Tafel slope of CN–Pt UNs in 1 M KOH also suggests a Volmer–Tafel HER reaction pathway.54 Similarly, the ECSA of CN–Pt UNs under alkaline conditions was calculated to be 10.11 m2 g−1. Also, the catalytic durability of CN–Pt UNs in 1 M KOH was estimated via amperometric i–t curves and ADTs. As expected, in Fig. 6d, CN–Pt UNs can maintain 85% current density at a constant voltage of 10 mA cm−2 for 24 hours, memorably stabler than commercial Pt/C (maintain 58% current density). After 1000 cycles of non-Faraday CV test, the HER polarization curve of CN–Pt UNs only negative shifts of 10 mV, exceeding the 16 mV of commercial Pt/C (Fig. S8b†). Also, there was hardly a morphological and crystalline structure change in CN–Pt UNs after the ADTs, indicating their superior electrochemical stability in 1 M KOH (Fig. S9b and S10†). It is worth noting that the superior HER performance of CN–Pt UNs to those of SC–Pt NPs and CAA–Pt NNs both under acidic and alkaline conditions prove that cyanogroup modification is an extremely crucial factor for the HER performance improvement in a wide pH range.
 | ||
| Fig. 6 Comparison of the HER performance of CN–Pt UNs and the other catalysts in 1 M KOH: (a) HER polarization curves, (b) overpotentials at 10 mA cm−2 and 50 mA cm−2, (c) Tafel curves, and (d) time-dependent current density curves in 1 M KOH. | ||
The impressive HER performance of CN–Pt UNs can be attributed to the unique geometric shape and modified Pt electronic structure: (I) the ultrafine nanonetwork provided plenty of accessible structural defects and distinct grain boundaries,55 which can act as catalytic hot spots in the HER process. Simultaneously, the porous 3D structure favoured mass transport and electron transfer, and promoted stability, which would effectively prevent particle agglomeration and Ostwald ripening during the HER.52 (II) The cyanogroup functionalization was conducive to the downshift of the Pt d-band centre relative to the Fermi level, thus tuning the adsorption strength of H* on Pt sites and facilitating the Volmer step in the HER, as predicted by the DFT calculations.
Conclusion
In conclusion, DFT calculations were firstly carried out to demonstrate that cyanogroup modification can effectively regulate the Pt electronic structure, thus promoting the Volmer step in the HER process. Ultrafine Pt nanonetworks with cyanogroup modification were synthesised via a facile one-pot wet-chemical method. Based on the sufficiently exposed active sites and the cyanogroup introduction, the as-prepared CN–Pt UNs exhibited superior HER activity and durability in a wide pH range to those of commercial 20 wt% Pt/C and other control samples, further supporting the theoretical calculations. This work highlights a new way to adjust the geometric and electronic structure (cyanogroup modification) of noble metals to markedly enhance the HER performance and beyond.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 21875112 and Grant No. 22072067) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20171473). The authors also thank the support from the National and Local Joint Engineering Research Centre of Biomedical Functional Materials, a project sponsored by the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Tianhe-2 Computational Resources Platform in Lvliang, Shaanxi Province.Notes and references
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 8, 4174–4192 RSC.
- J. Callejas, C. Read, C. Roske, N. Lewis and R. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- J. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, 23 CrossRef.
- H. Wang, W. Fu, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 6926–6956 RSC.
- Z. Bai, L. Yang, J. Zhang, L. Li, J. Lv, C. Hu and J. Zhou, Catal. Commun., 2010, 11, 919–922 CrossRef CAS.
- R. Yang, Y. Wang, G. Gao, L. Chen, Y. Chen, S. Li and Y. Lan, Small Structures, 2021 DOI:10.1002/sstr.202100012.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2019, 31, 1802880 CrossRef.
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- D. Kuznetsov, M. Naeem, P. Kumar, P. Abdala, A. Fedorov and C. Müller, J. Am. Chem. Soc., 2020, 142, 7883–7888 CrossRef CAS.
- L. Huang, D. Chen, G. Luo, Y. Lu, C. Chen, Y. Zou, C. Dong, Y. Li and S. Wang, Adv. Mater., 2019, 31, 1901439 CrossRef PubMed.
- Z. Wu, D. Nie, M. Song, T. Jiao, G. Fu and X. Liu, Nanoscale, 2019, 11, 7506–7512 RSC.
- M. Song, Z. Zhang, Q. Li, W. Jin, Z. Wu, G. Fu and X. Liu, J. Mater. Chem. A, 2019, 7, 3697–3703 RSC.
- Z. Chen, H. Qing, K. Zhou, D. Sun and R. Wu, Prog. Mater. Sci., 2020, 108, 100618 CrossRef CAS.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- G. Zhou, M. Li, Y. Li, H. Dong, D. Sun, X. Liu, L. Xu, Z. Tian and Y. Tang, Adv. Funct. Mater., 2019, 30, 1905252 CrossRef.
- Q. Liu, C. Fan, X. Zhou, J. Liu, S. Jiang, S. Wang, X. Wang and Y. Tang, New J. Chem., 2020, 44, 21021–21025 RSC.
- S. Bai, C. Wang, M. Deng, M. Gong, Y. Bai, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 12120–12124 CrossRef CAS PubMed.
- C. Zhang, X. Liang, R. Xu, C. Dai, B. Wu, G. Yu, B. Chen, X. Wang and N. Liu, Adv. Funct. Mater., 2021, 31, 2008298 CrossRef CAS.
- C. Xie, W. Chen, S. Du, D. Yan, Y. Zhang, J. Chen, B. Liu and S. Wang, Nano Energy, 2020, 71, 104653 CrossRef CAS.
- Z. Li, M. Li, X. Wang, G. Fu and Y. Tang, Nanoscale Adv., 2021, 3, 1813–1829 RSC.
- Y. Yao, X. Gu, D. He, Z. Li, W. Liu, Q. Xu, T. Yao, Y. Lin, H. Wang, C. Zhao, X. Wang, P. Yin, H. Li, X. Hong, S. Wei, W. Li, Y. Li and Y. Wu, J. Am. Chem. Soc., 2019, 51, 19964–19968 CrossRef.
- M. Zhu, B. Huang, Q. Shao, Y. Pi, Y. Qian and X. Huang, ChemCatChem, 2018, 10, 3214–3218 CrossRef CAS.
- X. Jiang, G. Fu, X. Wu, Y. Liu, M. Zhang, D. Sun, L. Xu and Y. Tang, Nano Res., 2018, 11, 499–510 CrossRef CAS.
- M. Li, Z. Li, G. Fu and Y. Tang, Small, 2021, 17, 2007179 CrossRef CAS.
- Y. Feng, L. Bu, S. Guo, J. Guo and X. Huang, Small, 2016, 12, 4464–4470 CrossRef CAS.
- P. Song, X. Cui, Q. Shao, Y. Feng, X. Zhu and X. Huang, J. Mater. Chem. A, 2017, 5, 24626–24630 RSC.
- K. Jiang, L. Bu, P. Wang, S. Guo and X. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 15061–15067 CrossRef CAS PubMed.
- B. Qin, H. Yu, X. Gao, D. Yao, X. Sun, W. Song, B. Yi and Z. Shao, J. Mater. Chem. A, 2018, 6, 20374–20382 RSC.
- G. Fu, X. Yan, Z. Cui, D. Sun, L. Xu, Y. Tang, J. B. Goodenough and J. M. Lee, Chem. Sci., 2016, 7, 5414–5420 RSC.
- G. Fu, X. Jiang, M. Gong, Y. Chen, Y. Tang, J. Lin and T. Lu, Nanoscale, 2014, 6, 8226–8234 RSC.
- X. Jiang, Y. Liu, J. Wang, Y. Wang, Y. Xiong, Q. Liu, N. Li, J. Zhou, G. Fu, D. Sun and Y. Tang, Nano Res., 2018, 12, 323–329 CrossRef.
- X. Jiang, Y. Xiong, Y. Wang, J. Wang, N. Li, J. Zhou, G. Fu, D. Sun and Y. Tang, J. Mater. Chem. A, 2019, 7, 5248–5257 RSC.
- X. Wang, L. Bai, J. Lu, X. Zhang, D. Liu, H. Yang, J. Wang, P. Chu, S. Ramakrishna and X. Yu, Angew. Chem., Int. Ed., 2019, 58, 19060–19066 CrossRef CAS PubMed.
- Q. Jia, E. Liu, L. Jiao, J. Li and S. Mukerjee, Curr. Opin. Electrochem., 2018, 12, 209–217 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- K. Ojha, S. Saha, P. Dagar and A. Ganguli, Phys. Chem. Chem. Phys., 2018, 20, 6777–6799 RSC.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang and Z. Zhao, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed.
- Z. Zhou, X. Kang, Y. Song and S. Chen, Chem. Commun., 2012, 48, 3391–3393 RSC.
- D. Strmcnik, M. Escudero-Escribano, K. Kodama, V. Stamenkovic, A. Cuesta and N. Markovic, Nat. Chem., 2010, 2, 880–885 CrossRef CAS PubMed.
- X. Chen, Z. Xiong, L. Andrews and Y. Gong, Inorg. Chem., 2020, 59, 6489–6495 CrossRef CAS PubMed.
- X. Liu, X. Lv, P. Wang, Q. Zhang, B. Huang, Z. Wang, Y. Liu, Z. Zheng and Y. Dai, Electrochim. Acta, 2020, 333, 135488 CrossRef CAS.
- J. Zhang, Y. Liu, C. Sun, P. Xi, S. Peng, D. Gao and D. Xue, ACS Energy Lett., 2018, 3, 779–786 CrossRef CAS.
- G. Yang, Y. Wang, L. Xu, Y. Li, L. Li, Y. Sun, Z. Yuan and Y. Tang, Chem. Eng. J., 2020, 379, 122230 CrossRef CAS.
- G. Fu, R. Wu, C. Liu, J. Lin, D. Sun and Y. Tang, RSC Adv., 2015, 5, 18111–18115 RSC.
- X. Huang, Z. Zhao, Y. Chen, C. Chiu, L. Ruan, Y. Liu, M. Li, X. Duan and Y. Huang, Nano Lett., 2014, 14, 3887–3894 CrossRef CAS PubMed.
- Z. Teng, M. Li, Z. Li, Z. Liu, G. Fu and Y. Tang, Mater. Today Energy, 2021, 19, 100596 CrossRef CAS.
- Y. Wang, H. Zhuo, X. Zhang, Y. Li, J. Yang, Y. Liu, X. Dai, M. Li, H. Zhao, M. Cui, H. Wang and J. Li, J. Mater. Chem. A, 2019, 7, 24328–24336 RSC.
- P. Zhang, X. Dai, X. Zhang, Z. Chen, Y. Yang, H. Sun, X. Wang, H. Wang, M. Wang, H. Su, D. Li, X. Li and Y. Qin, Chem. Mater., 2015, 27, 6402–6410 CrossRef CAS.
- M. Gong, G. Fu, Y. Chen, Y. Tang and T. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 7301–7308 CrossRef CAS PubMed.
- M. Ganduglia-Pirovano, V. Natoli, M. Cohen, J. Kudrnovský and I. Turek, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 8892 CrossRef CAS PubMed.
- Q. Shao, K. Lu and X. Q. Huang, Small Methods, 2019, 3, 1800545 CrossRef.
- D. Kweon, M. Okyay, S. Kim, J. Jeon, H. Noh, N. Park, J. Mahmood and J. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- B. Tang, Z. Yu, Y. Zhang, C. Tang, H. Seng, Z. Seh, Y. Zhang, S. Pennycook, H. Gong and W. Yang, J. Mater. Chem. A, 2019, 7, 13339–13346 RSC.
- J. Li, M. Qin, J. Zhang, Y. Du, D. Sun and Y. Tang, Chem. Res. Chin. Univ., 2019, 40, 988–994 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ce00796c |
| This journal is © The Royal Society of Chemistry 2021 |