
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeting protein–protein interactions in the DNA damage response pathways for cancer chemotherapy
Kerry Silva
McPherson
and
Dmitry M.
Korzhnev
 *
*
Department of Molecular Biology and Biophysics, University of Connecticut Health Center, Farmington, CT 06030, USA. E-mail: korzhniev@uchc.edu; Fax: +1 860 679 3408; Tel: +1 860 679 2849
First published on 21st June 2021
Abstract
Cellular DNA damage response (DDR) is an extensive signaling network that orchestrates DNA damage recognition, repair and avoidance, cell cycle progression and cell death. DDR alteration is a hallmark of cancer, with the deficiency in one DDR capability often compensated by a dependency on alternative pathways endowing cancer cells with survival and growth advantage. Targeting these DDR pathways has provided multiple opportunities for the development of cancer therapies. Traditional drug discovery has mainly focused on catalytic inhibitors that block enzyme active sites, which limits the number of potential drug targets within the DDR pathways. This review article describes the emerging approach to the development of cancer therapeutics targeting essential protein–protein interactions (PPIs) in the DDR network. The overall strategy for the structure-based design of small molecule PPI inhibitors is discussed, followed by an overview of the major DNA damage sensing, DNA repair, and DNA damage tolerance pathways with a specific focus on PPI targets for anti-cancer drug design. The existing small molecule inhibitors of DDR PPIs are summarized that selectively kill cancer cells and/or sensitize cancers to front-line genotoxic therapies, and a range of new PPI targets are proposed that may lead to the development of novel chemotherapeutics.
 Kerry Silva McPherson | Kerry Silva McPherson is a PhD candidate studying biomedical sciences at the University of Connecticut Health Center. She researches the assembly of protein complexes and designs drugs targeting protein–protein interactions within the translesion synthesis pathway in the laboratory of Dr Dmitry M. Korzhnev. She received an MS in Cell and Molecular Biology researching metastasis of triple-negative breast cancer under Dr Christina I. Zito at the University of New Haven, Connecticut, USA, and a BA in biotechnology from Assumption College in Worcester, Massachusetts, USA. |
 Dmitry M. Korzhnev | Dmitry M. Korzhnev, PhD, is an Associate Professor of Molecular Biology and Biophysics at the University of Connecticut Health Center. His NIH and NSF funded research program focuses on structural biology of multi-protein assemblies acting in DNA replication and damage response, and targeting these assemblies with small molecules for anti-cancer drug design. His post-doctoral training at the University of Toronto, Canada and Göteborg University, Sweden focused on structural biology and NMR methodology development. He graduated with PhD in biophysics and MS in applied physics and mathematics from Moscow Institute of Physics and Technology. |
1. Introduction
The DNA damage response (DDR) is an extensive signaling network that combines cellular pathways collectively responsible for detection and recognition of DNA damage, DNA remodeling and repair, DNA damage bypass during replication, cell cycle control and cell fate decisions in response to DNA alternations.1–5 This network includes the ever-growing list of over 450 genes encoding proteins that orchestrate the appropriate response to numerous forms of DNA damage. The DDR is redundant and tightly coordinated both in time and in space with multiple factors responsible for the choice of DNA repair mechanism and crosstalk between the pathways. An appropriate and timely DDR is critical for maintaining genomic integrity and accurate passage of genetic information to the next generation of cells. Dysregulation and mutation of DDR genes may cause genome instability, uncontrolled cell growth, and avoidance of cell death, all of which are hallmarks of cancer.2–7The DDR pathways and their therapeutic targeting have been subject to extensive investigation and review.1–13 DNA damage caused by various genotoxic agents elicits a complex response, which is contingent on the type of DNA lesion and the cell cycle status.4,14 The DDR is initiated by sensor proteins recognizing specific DNA substrates, which alert DDR kinases to transduce the signal. The master DDR kinases belong to the phosphatidylinositol 3-kinase-related kinase (PIKK) family and include DNA protein kinase (DNA-PK), ataxia telangiectasia and Rad3-related (ATR), and ataxia telangiectasia mutated (ATM).15 These kinases coordinate the DDR by phosphorylating substrates such as checkpoint kinases, DNA repair proteins, and apoptotic regulators. Checkpoint kinases are activated to freeze the cell cycle to allow time for DNA repair, while apoptosis or senescence can be initiated if the DNA damage is too detrimental for repair. The downstream DNA repair pathways are subsequently employed to reverse DNA damage, including double-stranded break (DSB) repair via non-homologous end-joining (NHEJ) or homologous recombination (HR), nucleotide excision repair (NER), base excision repair (BER), mismatch repair (MMR), and interstrand cross-link (ICL) repair through the Fanconi Anemia (FA) pathway.3–5 In addition to repair pathways, the DDR also utilizes DNA damage tolerance (DDT) pathways such as translesion synthesis (TLS), which allows bypass replication over DNA lesions at the price of increased mutagenesis.16–19
Alteration or loss of DDR capabilities is a hallmark of many types of cancer, which is often compensated by the use of alternative DDR pathways that can be exploited pharmaceutically to selectively kill cancer cells.4–13 Furthermore, genotoxic front-line chemotherapeutics induce DNA damage that can be repaired and/or avoided by specific DDR pathway(s), which can be targeted for the development of adjuvant drugs to enhance efficacy of existing treatments.12,13,20 Numerous DDR proteins have been proposed as chemotherapeutic targets, some of which have been inhibited successfully by drugs undergoing clinical testing or that have already received approval.8–11
The majority of drug discovery has traditionally focused on proteins that are deemed “druggable”, including enzymes (kinases, proteases, phosphodiesterases), ion channels, and G-protein coupled receptors,21 while protein–protein interactions (PPIs) were long considered “undruggable” due to their large, flat, and mostly hydrophobic interfaces.22–29 Typically, “druggable” proteins feature active sites within deep cavities with surface areas of 300 to 500 Å2 capable of binding soluble small molecule inhibitors. As of 2015, about 30% of approved anti-cancer drugs targeted protein kinases, which represent only an estimated 4% of DDR proteins.6 However, catalytic inhibition might not be the optimal strategy for drug development targeting DDR pathways,12 as catalytic inhibitors often have poor selectivity that may result in deleterious side effects.25,26 For example, DNA polymerases share similar active sites, making the design of inhibitors with high selectivity challenging.13 Furthermore, many proteins within DDR pathways do not have catalytic function and play roles in cellular localization, complex formation, and substrate recognition.6 To circumvent this issue and expand the range of drug targets, researchers are turning towards the development of a new class of therapeutics, inhibitors of DDR PPIs.12,13 Thus, clinically relevant PPI inhibitors such as tirofiban, an integrin disrupter to treat stroke patients,24,30 and Mdm2–p53 PPI inhibitors to treat cancer,31,32 spark optimism that disrupting PPIs is a promising avenue for drug design.
In this review, we will outline the process of structure-based design of small molecule PPI inhibitors and discuss the rationale for targeting PPIs in the DDR pathways for cancer therapy. Then, we will describe how disrupting various PPIs that mediate assembly of multi-protein DDR complexes, facilitate recruitment and activation of DDR factors, control DNA repair and damage tolerance, regulate cell cycle, and decide cell fate can pave the way for the development of novel cancer treatments. We will overview recent progress and provide specific examples of targeting the DDR network for anti-cancer drug design, as well as highlight potential new avenues for PPI inhibition within the DDR pathways that may lead to the development of novel chemotherapies.
2. Structure based design of PPI inhibitors
In contrast to forward pharmacology, where potential drugs are tested in cells or in vivo, and afterwards the therapeutic mechanism is investigated,33 targeting protein–protein interactions (PPIs) with small molecules generally requires a structure based drug design (SBDD) approach, a combination of structural and computational biology to design drugs with high specificity for a target molecule.34–36 While structure determination of protein complexes may be demanding, the SBDD approach benefits from extensive structural data already available in the Protein Data Bank (PDB)37,38 and robust computational methods for modeling structures of homologous proteins.39,40 As of 2020, the Protein Data Bank included 167![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 518 structures primarily solved by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, a 90078 increase in the last decade. Additionally, the advancements in cryo-EM technology now enables structure determination of protein complexes above 64 kDa at resolution as low as 1.8 Å,41–43 substantially increasing the number of macromolecular structure depositions in the PDB. Ideally, SBDD for a PPI would utilize the structure of a protein in complex with its binding partner. However, if such a structure is unavailable, molecular docking driven by experimental restraints can be used to produce a computational model.44,45 SBDD targeting a PPI is also possible using an apo-protein structure, albeit less optimal.25
518 structures primarily solved by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, a 90078 increase in the last decade. Additionally, the advancements in cryo-EM technology now enables structure determination of protein complexes above 64 kDa at resolution as low as 1.8 Å,41–43 substantially increasing the number of macromolecular structure depositions in the PDB. Ideally, SBDD for a PPI would utilize the structure of a protein in complex with its binding partner. However, if such a structure is unavailable, molecular docking driven by experimental restraints can be used to produce a computational model.44,45 SBDD targeting a PPI is also possible using an apo-protein structure, albeit less optimal.25
A flowchart of the SBDD protocol for PPI inhibitors is shown in Fig. 1,34–36 which includes (i) structure determination of a protein complex and identification of potential target site(s) within the PPI interface (target identification), (ii) biochemical high-throughput screening (HTS)46,47 and structure-based virtual screening (SBVS)48,49 for candidate small molecule inhibitors (hit identification), (iii) experimental validation of small molecule binding to the target protein, PPI disruption, and structural characterization of small-molecule protein interaction (hit validation), and (iv) optimization of validated hits using computational, structural and binding studies to improve the ligand potency (lead optimization). Optimization is an iterative process, which requires multiple rounds of hit validation and lead optimization until desirable binding and pharmacokinetics properties are achieved.
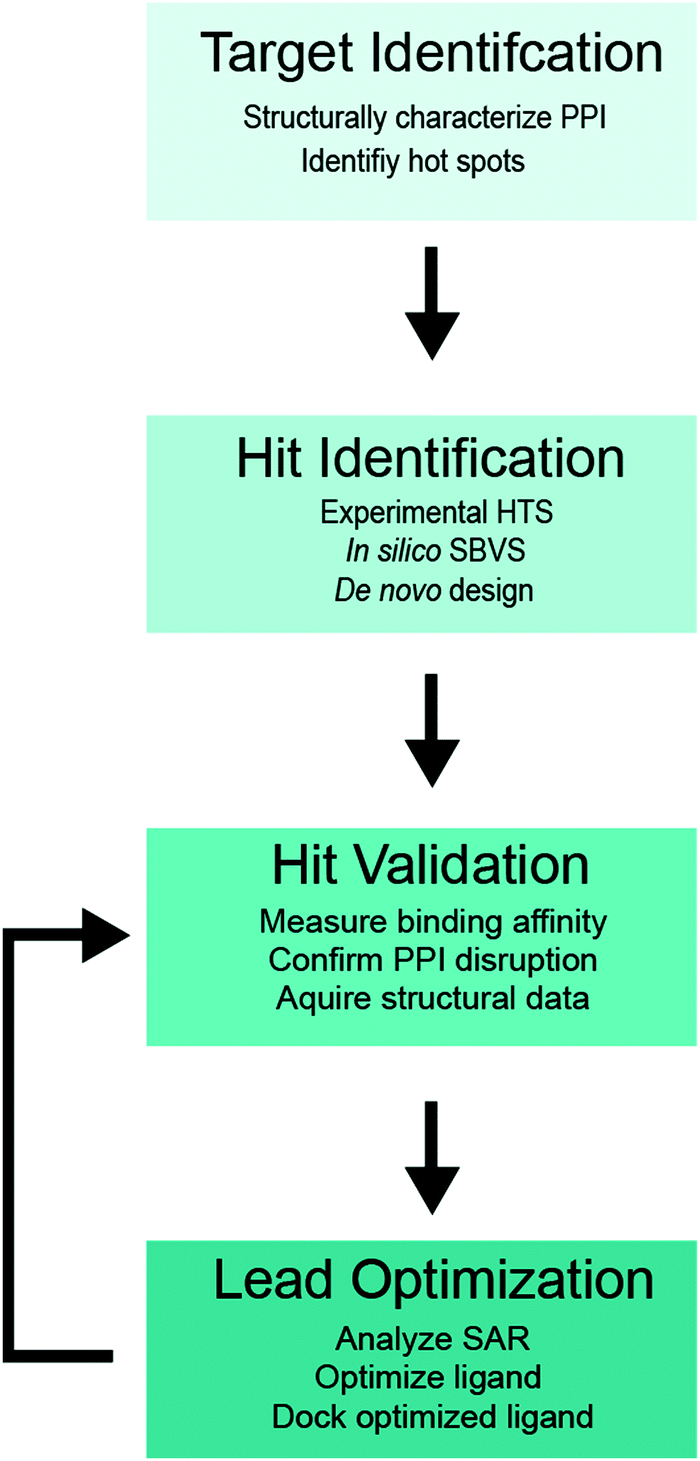 | ||
| Fig. 1 SBDD flowchart for PPI inhibitors. | ||
2.1. Target identification
PPI interfaces usually cover 1000–3000 Å2 of protein surface area, which is much greater than the footprint of a typical drug-like small molecule, and on average 28 residues participate in binding.24,50 However, in most cases only a handful of critical residues within the PPI interface are responsible for the lion's share of binding free energy, appropriately termed “hot spots”. A strategy for PPI inhibition generally involves targeting these “hot spots” rather than blocking the entity of the interaction interface.23–25,50–52 Structural analysis of the PPI interfaces and identification of “hot spot” residues are the first steps in the SBDD protocol for a PPI of interest (Fig. 1). PPI interfaces often include core and rim areas, as well as support amino acids that are partially exposed in the apo-protein and become buried after binding. The core residues that create topological features on the PPI interface such as hydrophobic cavities or pockets are the primary sites for targeting with small molecules, while polar or charged rim residues that outline the core region, or support residues that fortify the PPI but contribute little binding free energy are less likely to participate in a “hot spot”.50,51 Since PPI “hot spots” are generally more hydrophobic than catalytic sites, inhibitors targeting PPIs also tend to be hydrophobic. Due to their decreased cellular uptake and solubility, hydrophobic ligands have smaller bioavailability, which should be considered throughout the design process.24,25,52A combination of biochemical, structural and computational data can be used to identify “hot spots” on the PPI interface. Mutational analyses in immunoprecipitation, yeast 2-hybrid (Y2H) assays, and other binding studies can indicate potential “hot spot” residues, although these data should be interpreted with caution, as mutations may disrupt protein structure and/or alter dynamics.25,52 Therefore, structural data on a protein complex is invaluable for determining “hot spot” residues and cavities within the PPI interface that can be targeted with small molecules. Analyzing the interface topography and measuring the size of cavities on the PPI interface is plausible based on a static structure using software such as CAST.53 However, the detailed studies of binding energetics should also take into account the plasticity of the PPI interface, as proteins often undergo conformational changes upon binding consistent with an induced fit rather than a lock and key mechanism. Molecular dynamics (MD) simulations provide a powerful computational tool for “hot spot” identification, which enables calculation of binding energy contributions for individual residues within the PPI interface.25,54–56 MD simulations can also aid in identifying cavities of an appropriate size (250–900 Å2) to bind small molecules, as illustrated by the successful development of inhibitors targeting a hidden cavity in HIV integrase revealed by MD.57
An important consideration for choosing the PPI target is the demonstrated ability of PPI inhibition to abolish the function of the protein and/or cellular pathway (i.e. functional druggability of PPI), which can be established through cellular or in vivo studies.12 The loss of protein function can be validated in cellular assays by silencing the corresponding gene using siRNA or gene editing, while functional druggability of a PPI can be demonstrated through the inability of the PPI-disrupting mutants to functionally complement a knockout or knockdown gene in cellular and/or in vivo models. This functional target validation is usually performed in independent studies carried out before the intended design of PPI inhibitors.
2.2. Hit identification
Once the PPI target is selected, compound hits can be identified by biochemical high throughput screening (HTS)46,47 or in silico structure-based virtual screening (SBVS)48,49 of extensive libraries of drug-like compounds (Fig. 1). Alternatively, fragment-based drug design (FBDD) methods offer an advantage of probing large chemical space with a limited number of small (<200 Da) organic molecules to identify hit fragments weakly interacting with the target protein, which can be further combined and assembled into more potent compounds with drug-like characteristics.58–60HTS is an established technology for identification of new lead compounds for drug development.46,47 A variety of experimental methods, including NMR spectroscopy,61–65 isothermal titration calorimetry (ITC),66,67 surface plasmon resonance (SPR),68 fluorescence polarization (FP) and intensity (FI) measurements,69,70 Förster resonance energy transfer (FRET),71–73 yeast 2-hybrid (Y2H) assays,74–76 microscale thermophoresis (MST),77,78 and others can be used to probe direct binding of compounds to a target protein and/or displacement of binding partner from a protein complex. Displacement assays provide a direct measure of PPI disruption by compounds and are preferable in HTS for PPI inhibitors.
Many of the above methods such as FP, Y2H and FRET have been successfully implemented in HTS format to identify small molecule inhibitors of PPIs.69,72,76 For example, measurement of FP changes for a small molecule fluorophore upon binding to a macromolecule or for a fluorescently tagged peptide upon displacement from a protein–peptide complex by an inhibitor has become a common method in HTS due to its sensitivity and low protein concentration requirements.69,70 Y2H assays performed in specialized yeast strains are advantageous for HTS because they select small molecules not only for PPI disruption but also for bioavailability. However, Y2H assays may suffer from low membrane permeability of inhibitors.74–76 Another favored method for HTS is FRET, which measures distance dependent fluorescence energy transfer between donor and acceptor fluorophores attached to interacting proteins and can be used for displacement assays in vitro or in cells.71–73 FRET is often chosen for HTS because in vitro assays require small amounts of protein,71 while cellular assays may be performed in physiologically relevant cell types.72 However, similar to FP, a disadvantage of FRET-based HTS is the use of protein tags that may affect protein binding, cellular localization, or expression levels.
SBVS is a powerful computational approach that enables rapid screening of virtual libraries of millions of drug-like compounds through their docking to a target site on a protein with known three-dimensional structure.48,49 SBVS can be performed using software packages such as DOCK,79 FlexX,80 SEED,81 Glide,82 GOLD83 and AutoDock84 by docking each small molecule in a library of virtual compounds to a target site on a protein to determine optimal position(s) and binding pose(s) of the compounds, and then sorting the compounds according to binding energy scoring function to select best hits.56,60 These in silico screening protocols are robust and cost effective, but have a tendency to yield false positives, which necessitates synthesis and biochemical evaluation of the best hits to test their ability to bind the target protein and disrupt the PPI.
Alternative to HTS and SBVS of full-sized drug-like compounds, which may result in very few hits, is FBDD that uses in silico or experimental screens to identify small organic molecules (<200 Da) with weak (high μM to mM range) binding affinities to a target protein.58–60 Following identification of such fragments, the hits are experimentally and computationally validated, and combined piece by piece into de novo designed drug-like compounds with increased binding affinity. While some of the methods for studying protein–ligand interactions are not suitable to probe sub-mM binding, the method of choice for fragment screening in the FBDD approach is ligand-based NMR, which may also be used during the subsequent validation step of SBDD.61,62
2.3. Hit validation
To eliminate false positives from HTS and SBVS screens and identify key structure–activity relationships (SAR) necessary for further development of the identified scaffolds as drug leads, the SBDD protocol usually includes a thorough hit validation step (Fig. 1).34–36 This step generally includes verification of the compound binding to the target protein and measurements of binding affinity, confirming disruption of a target PPI, precise mapping of the compound binding site on a target protein, and high-resolution structure determination of a protein in complex with the identified compound.Protein–ligand binding studies and displacement assays to show PPI disruption by the identified small molecules can be performed with experimental techniques described in the previous section. However, preferred for binding studies are the quantitative methods that can demonstrate direct binding of the hit compound to the protein and measure affinity of this interaction, including NMR,61–65 ITC,66,67 SPR68 and MST.77,78 Provided that dissociation constant (Kd) for the PPI is known, the binding affinity of the PPI inhibitor can be estimated from the concentration dependent displacement data (e.g. FP69,70 or FRET71–73) via the exact formula or the Cheng–Prusoff equation.85,86 Each method for validation of protein-compound binding presents advantages and challenges. For example, ITC measures heat released during titration of a ligand into protein (or vice versa) and determines not only the binding affinity, but also the binding enthalpy and stoichiometry.66,67 The protein is neither immobilized nor labeled, however, solubility is often a limitation to this technique. SPR can measure the binding affinity and binding kinetics for a ligand flown over the protein immobilized on a chip surface, and utilizes less protein than ITC, mitigating the solubility limitation.68 However, SPR requires immobilization of a protein which may affect binding. MST can measure the binding affinity by monitoring diffusion of a fluorescently tagged protein at various concentrations of added ligand in a temperature gradient created by an infrared laser in a capillary.77 MST has gained popularity because it requires small amounts of protein (pm–nm concentrations) and immobilization of the protein is not required. The downside of MST is the necessity to fluorescently label the protein, although the label-free MST has been proposed utilizing intrinsic fluorescence of protein aromatic residues.78
NMR spectroscopy is a versatile method commonly used throughout the drug design process, which enables high-resolution mapping of the protein–ligand interface and accurate measurements of the binding affinity.61–65 This information is usually derived from protein-based NMR titration series,61–63 in which the ligand is gradually added to solution of an isotopically (15N and/or 13C) labeled protein, monitored by 1H–15N or 1H–13C correlation spectra recorded at each step of the titration. The ligand binding induces NMR chemical shift changes for protein residues in the vicinity of the binding site, providing accurate mapping of the binding interface, while protein chemical shifts measured vs ligand concentration allow extraction of dissociation constant (Kd). Protein-based NMR can detect weak protein–ligand interactions with affinities in high μM to mM range, and, therefore, is a suitable choice for identification of fragments that weakly bind to a protein at early stages of FBDD. Limitations of protein-based NMR include the necessity for protein isotopic labeling, NMR resonance assignments, and high concentrations (>50 μM).61–63 Conversely, protein–ligand interactions can be studied by ligand-based NMR wherein an unlabeled protein is added into a small molecule solution monitored by 1D 1H or 19F spectra of the ligand, which can pinpoint atoms within the ligand that interact with the protein.61–65 Common techniques in ligand-based NMR include Water–ligand observed via gradient spectroscopy (WaterLOGSY)61–64 and saturation transfer difference (STD)61–63,65 experiments, which exploit nuclear Overhauser effect (NOE) magnetization transfer between the protein or transiently bound water and the ligand and can detect binding with Kd in the μM–mM range. Since ligand-based NMR requires little protein (0.5–50 μM), it is also practical to utilize this technique for HTS hit identification.61,62
Structure determination of protein–small molecule complexes by X-ray crystallography is an integral step in the SBDD hit validation commonly used to verify ligand binding to the target interface and identify critical SAR for hit compounds.87,88 Crystallization of a ligand–protein complex is usually performed by crystal soaking whereby an inhibitor is incubated with protein crystals, or co-crystallization whereby an inhibitor is mixed with protein solution prior to crystallization. Each of these methods has advantages and limitations. During soaking, the ligand might perturb the crystal and adulterate diffraction or fail to bind to the interface buried within the crystal lattice, while during co-crystallization, the protein–ligand mixture might not crystallize in the same conditions as the apo-protein, so that a new crystallization buffer may need to be identified.87 If diffracting crystals are obtained, extra caution must be taken during the structure refinement to establish the presence of a ligand, as crystallization protocols often result in apo-protein crystals. Furthermore, even when the ligand is present, partial occupancy and flexibility may contribute to ambiguity in the electron density. Therefore, researchers should always check that the SAR is chemically reasonable, as steric clashes or lack of contacts that contribute to binding might indicate an error in ligand modeling.87,88 Recently, structure determination by cryo-EM has gained popularity to identify SAR for ligand protein interactions.41,42,89 Cryo-EM requires less protein than X-ray crystallography and may be preferred if the protein is very large or a cryo-EM structure already exists.
2.4. Lead optimization
Structural and binding data acquired during the validation step of SBDD are subsequently used to optimize the identified hit compounds to become the inhibitor leads (Fig. 1). The goal of this optimization is to utilize the available SAR to introduce modifications in the identified small molecule scaffolds to improve potency and/or binding affinity, while also addressing potential pharmacokinetic (PK) liabilities.34–36 To optimize binding properties, chemical groups may be added to facilitate favorable interactions with the protein, or removed if unfavorable contacts or clashes are observed. Docking protocols and MD can be used to predict contributions to binding energy for individual groups of the original and modified ligand, as well as probe flexibility of the protein and inhibitor.56,60 Rigidity is an important factor to be considered during optimization, as rigid molecules tend to exhibit favorable affinities due to less entropy lost upon binding.90 During optimization, multiple factors contributing to drug-like properties such as solubility, membrane permeability, metabolic stability and others should be considered to design compounds with favorable physicochemical and absorption, distribution, metabolism, excretion and toxicity (ADMET) profiles.91,92 As noted previously, optimization is an iterative process. Once changes are made to the initial hit, the new molecules should be validated and the SAR reassessed.3. Targeting the DDR for cancer treatment
The DDR is frequently altered in cancers by either genetic mutation or aberrant expression of DDR genes.2–7 A dysregulated DDR enables unregulated growth of cancer cells, increased viability during stress, avoidance of cell death, and genome instability. A deficiency in a specific DDR pathway is often compensated by a dependency on an alternative pathway essential for cancer cell survival, which provides therapeutic opportunity to selectively kill cancer cells.4–13 Therefore, targeting DDR pathways has become a popular strategy for the development of anti-cancer therapy.8–13 This section will introduce the general modes of killing cancer cells via DDR inhibition, while the following sections will overview the major DDR pathways and discuss current and potential pharmaceutical targets within the DDR network with the specific focus on druggable PPIs.3.1. Synthetic lethality
Synthetic lethality is a phenomenon whereby two genes are simultaneously disrupted to induce cell death, while mutation of only one gene does not.93,94 Cancer cells that have lost the function of one DDR gene may rely on another gene for survival. By identifying a deficient gene, a second gene can be pharmaceutically inhibited to cause a synthetic lethal phenotype, thereby selectively killing cancer cells that harbor a mutation. This concept has been successfully applied to develop poly(ADP-ribose)polymerase (PARP) inhibitors (PARPi) that specifically target BRCA deficient cancers with defective HR.95–98 PARPs are a family of proteins that recognize specific types of DNA damage best known for their roles in DNA single-strand break repair (SSBR).4,99–101 PARP inhibition causes accumulation of SSBs and their replication-dependent conversion to deleterious double-strand breaks (DSBs) that require functional HR for repair. The HR deficient cells are unable to efficiently cope with excessive DSBs during S-phase leading to cell death. The clinical approval of PARP inhibitors such as Niraprib, Olaparib, Talazorparib, and Rucaparib as monotherapies for BRCA deficient breast and ovarian cancers provides strong evidence that inducing synthetic lethality is a powerful approach for killing cancer cells with robust specificity.93.2. Sensitization to DNA damaging agents and averting resistance
The mainstream treatment for the majority of cancers remains to be the use of genotoxic agents that induce DNA damage.102–106 The elevated levels of DNA damage and replication stress characteristic of cancer cells can be further enhanced by genotoxic chemotherapies such as platinating and alkylating agents, causing replication catastrophe, induction of apoptosis and cell death.102–106 Cisplatin and other platinating agents covalently bind DNA bases, causing inter- and intra-strand crosslinks,102–105 while alkylating agents such as cyclophosphamide form bulky DNA adducts.106 These modifications impede replicative DNA polymerases, cause replication fork stalling and collapse, and trigger apoptosis or senescence. On the other hand, ionizing radiation (IR) and topoisomerase 2 inhibitors such as etoposide, camptothecin, and doxorubicin kill cancer cells by inducing DNA DSBs.10,107,108 DNA damage induced by genotoxic agents may be repaired and/or tolerated by various DDR pathways, which can be targeted to enhance efficacy of front-line therapies.12,13,20 Furthermore, resistance to these therapies can develop due to a hyperactive DDR, so that targeting DDR pathways can also restore chemosensitivity of drug-resistant cancers. For example, upregulation of ATM, ATR, DNA-PK, and DSB repair proteins may cause radio- or chemo-resistant phenotypes, which can be reversed by inhibiting the apical DDR kinases and DSB repair pathways to sensitize cells to topoisomerase inhibitors and radiotherapy.20,109,110 Resistance to platinating and alkylating agents may be facilitated by an increase in DNA damage repair pathways like NER and BER that repair the covalently modified DNA bases.111,112 Additionally, TLS can promote resistance to genotoxic chemotherapy due to its ability to avoid DNA damage during replication.113–116 TLS is extremely error-prone and can further the genomic instability of cancer cells by introducing mutations that contribute to chemo-resistant phenotypes.16–19 Therefore, TLS inhibition was proposed as a strategy to sensitize cancers to genotoxic chemotherapy and avert chemoresistance by preventing mutagenesis.13,117,1183.3. Targeting cell cycle dependent pathways
Another motive for DDR inhibition is to target cell cycle dependent DDR pathways that promote rapid division of cancer cells. The DDR is active throughout the cell cycle, but some repair pathways are more active during S, G2 and M phases.4,14 Since uncontrolled growth is a hallmark of cancer, targeting pathways necessary for proliferation opens an avenue for selectively attacking cancer cells. Thus, TLS and HR are predominantly utilized during S-phase for the replicative bypass of DNA lesions and DSB repair, and can be inhibited for this mode of cytotoxicity.10–13 The apical kinase ATR mainly used in S-phase is another cell-cycle dependent chemotherapeutic target.10,11,153.4. Inducing cell death programs
Stabilizing apoptotic pathways will reverse cancer's ability to evade cell death. The tumor suppressor, p53, discussed in further detail later in this review, is a transcription factor that regulates transcription of genes involved in numerous processes, including cell cycle arrest and cell death via senescence and apoptosis.119,120 The importance of p53 for guarding cells against the malignant transformation cannot be overstated, as the TP53 gene encoding the tumor suppressor is by far the most frequently mutated gene in cancers.119,120 The cellular levels of p53 are normally kept low though its ubiquitination by the E3 ligase, Mdm2, which tags p53 for proteasomal degradation.119–121 However, under conditions of genotoxic stress, p53 is stabilized in the cell and its levels increase to trigger cell cycle arrest and DNA repair, or apoptosis and cell death. Considering the importance of p53 for cancer etiology, cellular stabilization of p53 and other apoptotic promoting factors has emerged as a promising strategy for selectively killing cancer cells or sensitizing cancers to existing chemotherapies.32,122–1274. Inhibition of apical PIKK kinases
Three related protein kinases within the PIKK family, ATR, ATM and DNA-PK, are primarily responsible for orchestrating the appropriate response to DNA damage (Fig. 2).15 While each of the three kinases have unique phosphorylation substrates, they also share considerable functional redundancy.15,128–130 ATM, ATR, and the DNA-PK catalytic subunit (DNA-PKcs) have similar structural organization and are comprised of four structured domains, including HEAT repeats, a FRAP-ATM-TRRAP (FAT) domain, a PIKK regulatory domain, and a kinase domain.15ATR is a central kinase in the response to DNA replication stress, which is activated by ssDNA coated by the replication protein A (RPA) that binds and stabilizes ssDNA regions accumulated following DNA damage at stalled replication forks.15 The ATR-interacting protein (ATRIP) binds RPA, and recruits the ATR/ATRIP complex,131,132 while the activator proteins TopBP1 and ETAA1 bind ATR via their ATR-activation domains and stimulate its kinase activity to initiate DDR.133–135 ATR has hundreds of phosphorylation substrates, many that overlap with ATM. Substrates of note include checkpoint kinase 1 (Chk1), which facilitates cell cycle arrest, and RPA and SMARCAL1, which stabilize the replication fork.15,128ATM regulates the cellular response to DNA DSBs, and is recruited to DSB ends by the MRE11-Rad50-NBS1 (MRN) complex via a PPI between the N-terminus of NBS1 and ATM.132,136,137 ATM has hundreds of phosphorylation substrates such as histone H2AX, p53, checkpoint kinase 2 (Chk2), and DSB repair proteins, 53BP1 and BRCA1.15,128,129DNA-PK is a regulator of DSB repair by NHEJ, which is comprised of two subunits, the catalytic subunit DNA-PKcs and Ku. Ku is a heterodimer of Ku70 and Ku80 subunits that recognizes and binds DSB ends,138,139 thus recruiting and activating DNA-PKcs.15,140 DNA-PK has numerous phosphorylation substrates, including itself, H2AX, p53, and various NHEJ proteins.15,130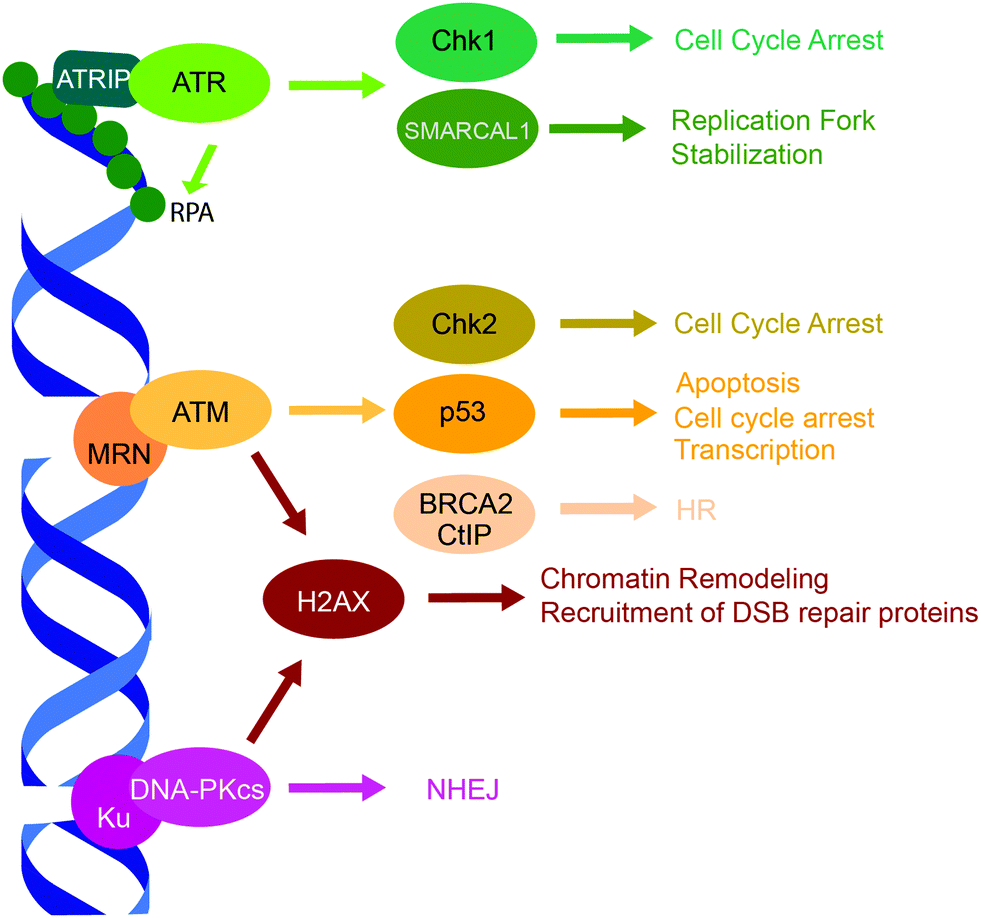 | ||
| Fig. 2 PIKK family DDR kinases, ATR, ATM, and DNA-PK, and their representative substrates in a DDR signaling cascade. | ||
4.1. Pharmaceutical targeting of PIKK kinases
Targeting the master DDR PIKK family kinases provides multiple avenues for anti-cancer drug design. First, PIKK inhibition amplifies genotoxic chemo- and radio-therapy sensitivity.11,15 Second, synthetic lethal relationships exist between the PIKK family members and other proteins commonly affected in cancer, such as between ATM and ATR, ATM and DNA-PK, ATR and Chk1, and ATM and p53, which can be exploited for the development of selective cancer therapeutics.11,15,141 Additionally, uncontrolled growth of cancer cells is often dependent on ATR signaling, and inhibition of ATR was shown to be more cytotoxic to cancer cells than normal cells.142 Multiple inhibitors of the PIKK family have been designed and tested for their use as anti-cancer agents, and those undergoing clinical trials have been extensively reviewed by Cleary et al.9 and Brown et al.11 ATR, ATM, and DNA-PK inhibitors targeting the active site or the ATP-binding pocket often lack selectivity against a specific kinase.9,11 Furthermore, this mode of inhibition may result in accumulation of catalytic dead kinases at the chromatin, causing more genomic instability than catalytic inhibition itself. This steric hindrance of kinases at chromatin may result in the increased toxicity of PIKK inhibitors.143,1444.2. PPI targets for PIKK inhibition
Disruption of PPIs that mediate recruitment and activation of the PIKK family kinases may provide a viable strategy for the development of selective and less toxic PIKK inhibitors. ATR is recruited to RPA-coated ssDNA through PPIs with ATRIP and activated through PPIs with partners such as TopBP115,131,132 and ETAA1,15,133–135 while the MRN complex (MRE11-Rad50-NBS1) and the Ku70/Ku80 heterodimer recruit and activate ATM15,132,136,137 and DNA-PK,15,140 respectively. ATRIP, Ku80, and NBS1 have motifs in their C-terminus with a consensus sequence of EExXXXDDL (where ‘X’ is any residue, ‘x’ is any residue or a gap) that bind to HEAT repeats of ATR, DNA-PKcs, and ATM.132 Structural data is available to aid the SBDD for this PPI, albeit at low resolution.145–151 Two cryo-EM structures have been reported for the ATR–ATRIP complex in H. sapiens at 4.7 Å and S. cerevisiae (Mec1-Ddc2) at 3.9 Å (PDB: 5YZ0 and 5X6O),145–147 which may aid SBDD of inhibitors against this PPI. The PPI stoichiometry and interfaces for human and yeast ATR–ATRIP complexes are compared and discussed in Baretić et al.147 Two cryo-EM structures of the DNA-PK holoenzyme are available at 6.6 Å and 5.8 Å resolution,148–150 revealing that the Ku/DNA-PKcs complex contains interfaces between the M-HEAT repeat of DNA-PKcs and Ku70, the N-HEAT repeat of DNA-PKcs and the bridge region formed by Ku70/Ku80, and M-HEAT and the Ku80 C-terminal domain (Ku80-CTD). These structural studies also confirmed the previous finding that the Ku80-CTD is a DNA-PKcs binder.152 While no structure of the NBS1-ATM has been reported, a 4.7 Å cryo-EM structure revealed the homo-dimerization interface of ATM, which may provide insight into PPIs necessary for ATM activity (PDB: 5NP0).147,151 Overall, the PIKK PPIs may provide potential targets for anti-cancer drug design, however, the limited low-resolution structural data available at this time present challenges for developing highly specific small molecule inhibitors of these PPIs.5. Targeting checkpoint kinases and cell fate regulators
Cell cycle arrest and cell death pathways such as apoptosis or senescence are coordinated by numerous downstream targets of the PIKK family DDR kinases (Fig. 2).15,119,120,153 Mutations associated with these proteins are prevalent in cancer and they have been extensively studied as chemotherapeutic targets.9,154 Although copious DDR proteins are involved in cell cycle stalling and determining cell fate, here we focus on the three key DDR regulators of cell fate and cell cycle, the tumor suppressor p53119,120 and the checkpoint kinases Chk1 and Chk2.1535.1. Targeting p53–Mdm2
P53 is a tumor suppressor and transcription factor that transcriptionally regulates cellular responses to DNA damage, including cell cycle arrest, apoptosis and senescence.119,120 P53 levels in a cell are regulated by a ubiquitin ligase Mdm2 that tags p53 for proteasomal degradation.119–121 In response to DNA damage, p53 is stabilized either through its direct phosphorylation by ATM or other protein kinases, or through phosphorylation of Mdm2. These phosphorylation events disrupt the interaction of p53 and Mdm2, and prevent p53 ubiquitination and prosomal degradation (Fig. 3A). Therefore, a number of small molecule inhibitors of the Mdm2–p53 PPI that stabilize p53 and induce apoptosis have been developed as anticancer agents, several of which progressed to clinical trials (Fig. 3B).32,122–125 Additionally, several small molecule re-activators of p53 variants mutated in cancers have been developed and tested in clinical trials.122,126 Finally, an alternative strategy for p53 stabilization via inhibition of the p53 deubiquitinating enzyme, USP7, has been also explored.127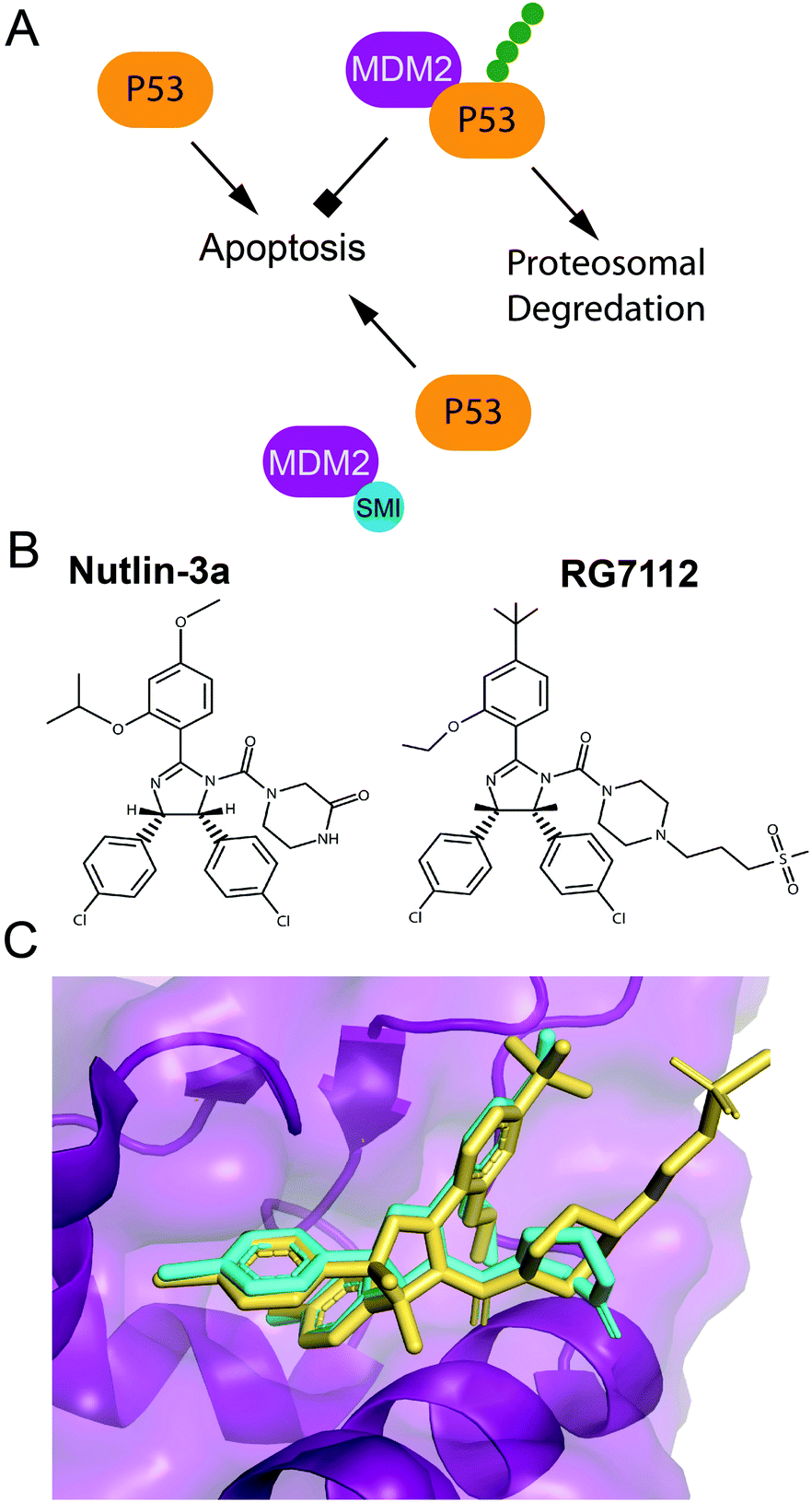 | ||
| Fig. 3 Pharmaceutical disruption of the Mdm2–p53 PPI. (A) Schematic of p53 stabilization by a small molecule inhibitor. (B) Nutlin-3 and RG7112 structures. (C) Nutlin-3 (cyan) (PDB: 4J3E) and RG7112 (yellow) (PDB: 4IPF) in complex with the p53 binding site of Mdm2. | ||
A 2.60 Å resolution crystal structure of the N-terminal domain of Mdm2 bound to the 15-residue transactivation domain (TAD) peptide of p53 (PDB: 1YCR) revealed that p53 residues F19, W23, and L26 bind inside a hydrophobic pocket on Mdm2.155 This structure was utilized in SBDD by several research groups to create potent p53 stabilizing inhibitors that disrupt the Mdm2–p53 PPI, which are reviewed extensively (Fig. 3B and C).32,122–125 Nutlins were the first group of Mdm2–p53 PPI inhibitors, which bind to Mdm2's hydrophobic pocket and mimic the FWL motif of p53.31 A nutlin analog, RG7112, was the first to be clinically tested but resulted in hematological toxicity.156,157 Seven Mdm2–p53 PPI inhibitors are currently in clinical trials.124,125
5.2. Chk1 and Chk2 inhibition
The downstream targets of ATR and ATM are serine/threonine checkpoint kinases, Chk1 and Chk2, that regulate the intra-S and G2/M checkpoints and the G1/S checkpoint, respectively, to initiate cell cycle arrest and DNA repair (Fig. 2).15,153 Like the PIKK family kinase inhibitors, early inhibitors of Chk1 and Chk2 had poor selectivity, while later inhibitors exhibited higher specificity. Over 70 Chk1-inhibitor structures have been reported, a few of which have progressed to clinical trials, reviewed in Dent et al.158 and Ronco et al.154 Fewer Chk2 inhibitors have been reported, and thus far, inhibition of Chk1 is favored as a chemotherapeutic strategy.154 Chk1 inhibition may sensitize BRCA deficient cells to PARPis,159 and Chk1 and ATR have a synthetic lethal relationship.154 However, a number clinical trials of Chk1 inhibitor were terminated due to toxicity, and no trials so far have progressed to phase III, suggesting the mechanism of Chk1 inhibition might warrant improvement.158Similar to the PIKK family kinases, targeting Chk1 PPIs may provide a novel strategy to developing inhibitors with lower toxicity. For example, Chk1 activation and ATR–Chk1 interaction within a multi-protein complex formed on ssDNA are mediated by Chk1 PPIs with S/T-phosphorylated ExxxLC(S/T)GxFE repeats of an adaptor protein, Claspin.160 Small molecule inhibitors of this PPI would impede Chk1 activation, and since Claspin expression is elevated in many cancers, could have advantageous clinical applications.161,162 A recent 1.8 Å resolution X-ray crystal structure of Chk1 in complex with a high-affinity phosphorylated Claspin motif (PDB: 7AKO) provides a structural basis for the development of inhibitors against this PPI.163 Another PPI of interest is the Chk2 dimer interface. Chk2 phosphorylation by ATM induces Chk2 dimerization and promotes its autophosphorylation and activation of kinase activity.164,165 Li-Fraumeni syndrome, a predisposition to cancer, is caused by I157T mutation, which disrupts dimerization and autoactivation of Chk2.164 A 3.25 Å resolution dimeric crystal structure of Chk2 (PDB: 3I6W) reveals an expansive dimer interface with connections in the forkhead association (FHA) domain, the kinase domain, and the loop between FHA and kinase domains.165 I157T mutation abolished dimerization in vitro, identifying a hotspot on the dimer interface, while W97A mutation drastically reduced Chk2 dimerization, suggesting another potential hot spot that can be targeted with small molecule inhibitors.
6. Targeting DSB repair
6.1. DSB repair pathways
DSBs are extremely cytotoxic and, if unrepaired, can cause chromosomal translocations and cell death. Substrate recognition by sensor DNA binding proteins, including Ku and the MRN complex, and subsequent intracellular signaling initiates DSB repair by one of two major pathways, non-homologous end-joining (NHEJ) or homologous recombination (HR) (Fig. 2).3–5,166–169 The choice of DSB repair pathway is dictated by the extent of DNA end resection at the break.166 While classical NHEJ does not require extensive DNA end processing, HR is dependent on end resection to create ssDNA overhangs. Paramount for NHEJ is the protection of blunt ends and negative regulation of end resection, which is achieved through phosphorylation of p53-binding protein 1 (53BP1) by ATM followed by RAP1-interacting factor 1 (RIF1) dependent recruitment of the shieldin complex to DSB ends.170–175 The shieldin complex directly binds the CST complex, which recruits the Polα primase to remodel ssDNA ends into blunt ends needed for NHEJ.170,171 Proteins that promote NHEJ and HR generally act antagonistically to each other, with a reduction in NHEJ pathway choice promoting HR and vice versa.166,176 HR is commonly employed during S and G2-phases of the cell cycle and is a high-fidelity pathway, while NHEJ repairs the majority of DSBs throughout the cell cycle and is error-prone.3–5,166–169 The two secondary DSB repair pathways, alternative non-homologous end-joining (alt-NHEJ) and single-strand annealing (SSA), are error-prone and require DNA end resection.166,167NHEJ is very efficient in repairing DNA DSBs and plays the primary role in protecting genome integrity despite its error-prone nature (Fig. 4).3–5,167,168 DSB ends to be repaired by NHEJ are recognized by the Ku70/Ku80 heterodimer (Ku) that binds to the DSB ends and activates DNA-PKcs.138,139 Together, Ku and DNA-PKcs form the DNA-PK complex that binds, phosphorylates and activates various NHEJ proteins. Ku serves as a scaffold for the recruitment of proteins that remodel the DNA break ends such as ARTEMIS, a pivotal NHEJ nuclease, and X-family DNA polymerases λ and μ. X-Ray Repair Cross Complementing 4 (XRCC4), XRCC4-like factor (XLF), Paralog of XRCC4 and XLF (PAXX), and DNA ligase IV (Lig IV), in conjunction with Ku align and ligate the two DNA ends.
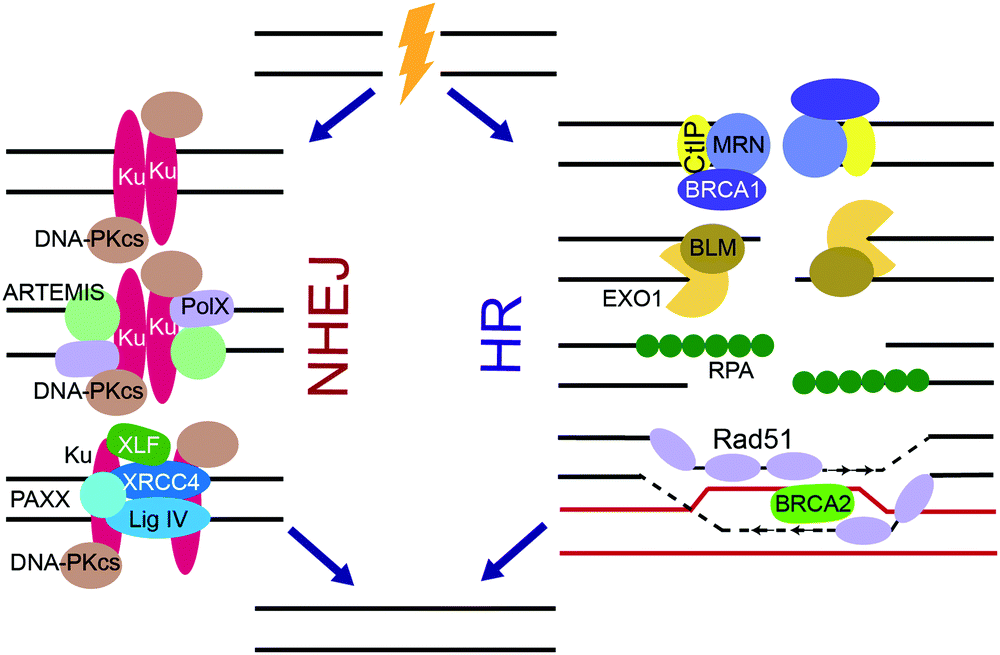 | ||
| Fig. 4 HR and NHEJ, the two major NHEJ pathways. For NHEJ, DSB ends are recognized by Ku, which binds and activates DNA-PKcs. DNA nucleases and X-family polymerases Pol λ or μ remodel DSB ends. The DSBs are ligated by the ligation complex containing Lig IV, XLF, PAXX, and XRCC4. For HR, ends are resected by the MRN/BRCA1/CtIP complex and then various nucleases, resulting in long stretches of RPA-coated ssDNA. RPA is replaced by Rad51 in a BRCA2 dependent manner, and Rad51 facilitates strand invasion of a homologous DNA sequence, which is used as a template for DNA synthesis. After synthesis, the resulting HR intermediates are resolved and the ends are ligated. | ||
HR provides a high-fidelity alternative to NHEJ during S and G2-phases, which can repair DNA DSBs in an error-free manner by using a sister chromatid as template (Fig. 4).3–5,169 When a DSB is to commit to HR repair pathway choice, the DSB end resection is controlled by ATM trough CtBP interacting protein (CtIP), which forms a multi-protein complex with MRN and BRCA1. The MRN complex, which is initially recruited to DSBs by PARP, resects the DSB ends to form long stretches of ssDNA. Next, Bloom syndrome helicase (BLM) unwinds the DNA ends, while exonuclease 1 (EXO1), the Sgs1-Top3-Rmi1 (STR) complex, and endonuclease DNA2 perform further resection to the DNA ends, forming large 3′ ssDNA overhangs. RPA bound to the overhangs is subsequentially displaced by the recombinase, Rad51,177,178 leading to assembly of Rad51 filaments on ssDNA mediated by BRCA2. Rad51 invades duplex DNA and seeks out a homologous sequence, usually a sister chromatid. Rad51 facilitates base pairing of the 3′ overhang with the template DNA, and the template DNA displaces from its complementary strand forming a displacement loop. Polδ replicates the template strand, extending the 3′ overhang. After replication, the newly synthesized 3′ end ligates to the 5′ end of the DSB, thus resolving the DSB.
Alt-NHEJ and SSA are auxiliary homology-driven DSB repair mechanisms that require DNA end resection.166,167,179–182 Both pathways are extremely error prone and may cause gross genome rearrangements, and thus are implicated in oncogenesis. To join DNA ends, alt-NHEJ uses <25 bp microhomology, while SSA requires longer homology regions. In alt-NHEJ, the MRN complex accumulates at DSBs in a PARP1-dependent manner and resects DNA ends to form 15–100 bp ssDNA overhangs. DNA polymerase, Polθ, aids in remodeling of the DSB and is responsible for the mutagenic insertions caused by alt-NHEJ, while ligases I or III ligate the remodeled ends together. Alt-NHEJ is hyperactive in HR deficient cells and inhibition of Polθ is synthetic lethal with many HR related genes such as BRCA1 and BRCA2.179–181 Another mutagenic homology-driven DSB repair pathway, SSA, utilizes direct annealing of the resected ssDNA regions with homologous sequence by the Rad52 protein, with the resulting DNA flaps removed by XPF/ERCC1.166,167,182
6.2. NHEJ inhibition
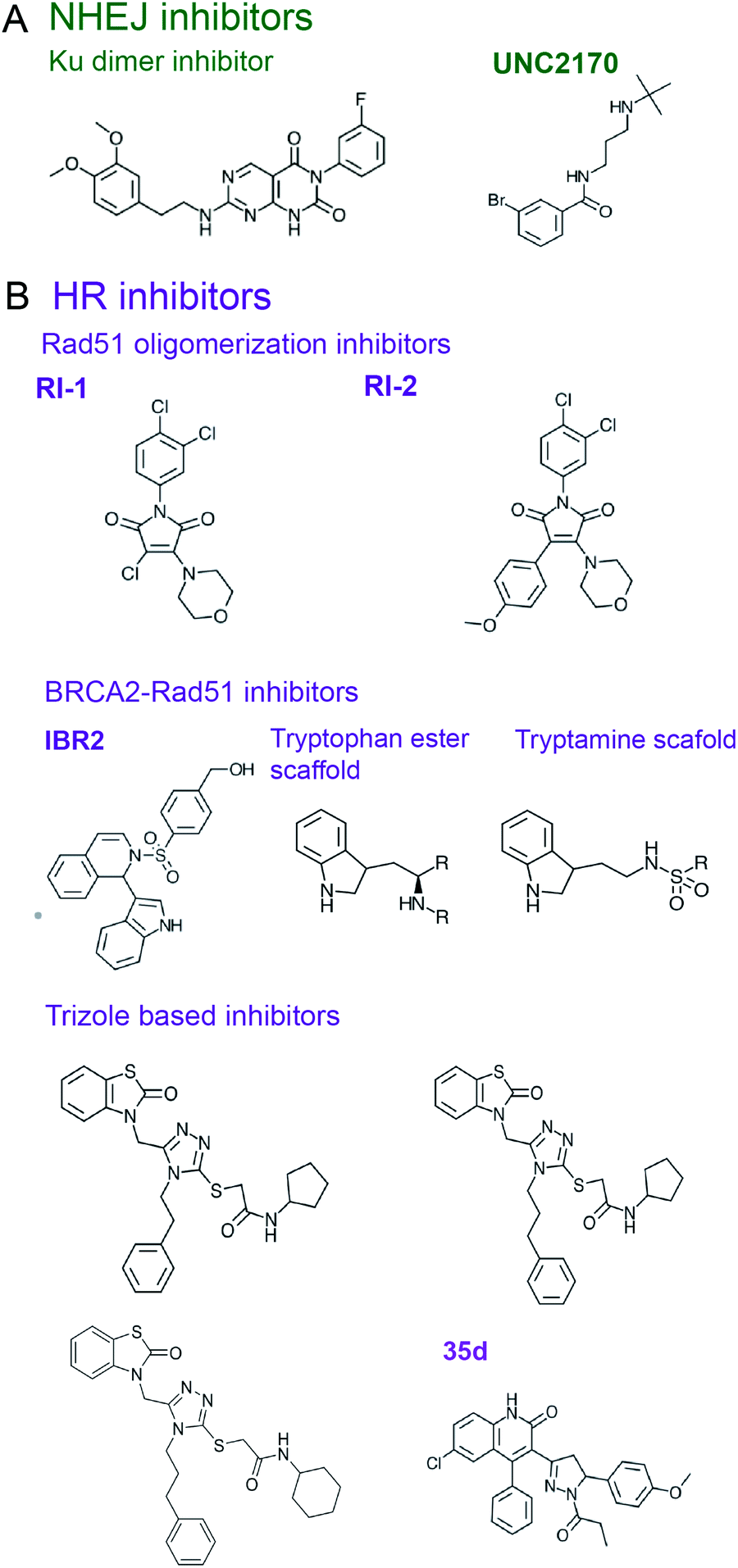 | ||
| Fig. 5 Inhibitors of DSB repair by (A) NHEJ and (B) HR. | ||
As a scaffold, Ku mediates various PPIs to recruit NHEJ proteins, which can be explored as potential targets for the development of small molecule NHEJ inhibitors. Thus, Ku binds PAXX, a protein that supports end ligation and is synthetic lethal with XLF, whose mutants confer radio- and chemo-sensitivity.167,188 PAXX's C-terminus (residues 177–204) binds Ku, and PAXX point mutants V199A and F201A disrupted the PAXX-Ku PPI and sensitized cells to IR treatment, identifying a PPI ‘hot spot’.188 Ku also binds the C-terminus of XLF, which forms a homodimer within the ligation complex. Deletion of the XLF C-terminal domain not only disrupts the XLF-Ku PPI, but also mitigates recruitment of XRCC4 to DSB ends, suggesting this PPI is crucial to the ligation complex.189 As seen in the crystal structures of Ku bound to an XLF peptide, the last four residues of XLF (GLFS) bind a hydrophobic pocket of Ku80 (PDB: 6ERH, 6ERG).190 Mutations of L297 in this Ku-binding motif (KBM) of XLF disrupted its localization to Ku and also decreased XLF-XRCC4 filament formation.
XRCC4 PPIs necessary for NHEJ's ligase complex formation, which consists of XRCC4, XLF, PAXX and LigIV (or Lig4) and aids in tethering DSB ends, provide potential targets for NHEJ inhibitor design.167 Thus, XRCC4–Lig4 PPI is essential in DSB end ligation, with deficiency in either of the binding partners or deletion of the Lig4-binding region of XRCC4 conferring radiosensitivity.191 Crystal structures were reported for XRCC4(1–203) bound to Lig4 C-terminal BRCT domains (residues 654–911) (PDB 3II6),192 and for XRCC4(1–213) bound to Lig4's XRCC4 interaction region (XIR) (residues 755–782) (PDB: 1IK9),193 revealing the Lig4 N-terminus forms a coiled-coiled homodimer. The structures identified an extensive interface between XRCC4 and Lig4-BRCT, encompassing both Lig4-BRCT1 and BRCT2 domains and their connector region, which can be further explored for the design of inhibitors of this PPI. XRCC4-XLF is another PPI essential for the NHEJ's ligation complex whose disruption was shown to sensitize cells to IR.194 A 5.5 Å crystal structure of the complex XRRC4(1–157)–XLF(1–224) (PDB: 3Q4F) reveals a filamentous structure formed by the two proteins with a PPI interface mediated by the N-terminal regions of XRCC4 and XLF.195 A more recent 3.9 Å XRCC4(1–140)–XLF(1–224) (PDB: 3SR2) revealed that Leu115 in XLF, termed the Leu-lock, is a hot-spot of this PPI.196
53BP1 is a scaffold protein, which plays a central role in NHEJ initiation and HR suppression, whose PPIs hold promise as targets for NHEJ inhibitors.197,198 53BP1 accumulates at chromatin near DSBs via interactions with modified histones, H2AK15-Ub and H4K20me2, through its focus formation region (FFR) and tandem Tudor domains, respectively.197–200 A small molecule that prevents 53BP1 accumulation at chromatin201 was rationally designed using crystal and an NMR structures of 53BP1 bound to a H4K20me2 peptide (PDB: 2IG0 and 2LVM).200,202 A fragment-like small molecule inhibitor, UNC2170, was designed to bind the lysine methyl binding cage of the 53BP1 Tudor domain formed by Tyr1502, Tyr1523, Phe1519, Trp149 (co-crystal structure, PDB: 4RG2) (Fig. 5A).201 This inhibitor attenuated class-switch recombination (CSR), but was not tested for its radiosensitization ability.
Shieldin complex is a protector of DSB ends that promotes NHEJ and counters HR, providing another target to be considered for the NHEJ inhibitor design.170–175 Loss of shieldin proteins sensitizes DT40 and U2OS cells to radiomimetic agents, topoisomerase inhibitors, and IR.203 The structure of the shieldin complex comprised of Rev7, Shld1, Shld2 and Shld3 subunits is not yet available, however, a 3.45 Å crystal structure (PDB: 6KTO) has been reported for a Rev7 dimer bound to Shld2(1–64) and Shld3(1–52) fragments, which is a hub for the shieldin complex assembly.204 Rev7 dimer-breaking mutants attenuated NHEJ in CH12-F3 cells,174 suggesting disruption of Rev7 dimer with small molecules may attenuate NHEJ. In shieldin, the two Rev7 protomers interact with the two Shld3 Rev7-binding motifs (RBM, defined as Pxxx(A/P)P, where x is any residue205,206) that are also found in Rev7 interactors from other pathways.204,207,208 An inhibitor was identified that blocks the Rev7 PPI with an RBM motif from the Rev3 subunit of TLS DNA polymerase Polζ (reviewed below),209 suggesting that disruption of Rev7 PPIs with Shld3-RBMs might be a viable strategy for NHEJ inhibition.
6.3. HR inhibition
Inhibition of HR proteins can lead to radio- and genotoxic chemosensitivity,169,210 while synthetic lethal relationships of HR genes also exist that can be exploited for cancer treatment.97,98 HR has also been reported to facilitate resistance to alkylating therapy in glioblastoma cells more so than NHEJ.211 Together, this opens multiple avenues for targeting HR proteins for cancer therapy.MRN is a hexameric complex, composed of 2 copies of each MRE11, RAD50, and NBS1.212,213 MRE11 has DNA binding and nuclease activity, Rad50 hydrolyzes ATP to regulate nuclease activity, and NBS1 is a scaffold. The nuclease function of MRN promotes HR, however, MRN may also aid in NHEJ by tethering DSB ends.
Current inhibitors of MRN act against MRE11's nuclease activity.214,215 However, disruption of PPIs in the MRN complex could also provide a potential chemotherapeutic strategy. Mutations within PPI regions of NBS1, the scaffold protein of MRN, are correlated with cancer susceptibility,216 and knock down of NBS1 leads to IR sensitivity,217 alluding to the importance of NBS1 PPI for DSB repair. NBS1 contains an FHA domain, two tandem BRCT domains, a MRE11 interaction region (MIR, residues 474–531), and an ATM interaction motif mentioned above, all of which facilitate PPIs.212,213 For example, the structure of the N-terminal FHA and the two tandem BRCT domains of S. pombe NBS1 bound to a Thr-phosphorylated CtIP peptide (PDB: 3HUF) provides insights into MRN-CtIP recognition.218 Integral to the MRN complex is the MRE11 dimer stabilized by the MRE11-NBS1 PPI. The details of this PPI are available from a crystal structure of S. pombe MRE11/NBS1-MIR complex (PDB: 4FBQ).219 Furthermore, the human MRE11 core complex crystal structure (PDB: 3T1I) identified key residues in the MRE11 homodimerization interface.220 A crystal structure of the dimer zinc-hook domain from P. furiosus Rad50 (PDB: 1L8D) reveals that the highly conserved CXXC motif coordinates dimerization, whose mutations sensitize yeast cells to IR.221 The structures of P. furiosus Rad50 in complex with MRE11's Rad50 binding domain (PDB: 3QKR, 3QKS) were also determined providing details of this PPI.222
Rad51 is the ssDNA binding protein and recombinase responsible for strand invasion and homology searching in HR,177,178 which is often upregulated in radio- and chemo-resistant cancers.8 Several small molecule compounds were identified that inhibit Rad51–ssDNA binding, preventing the nucleoprotein filament formation (reviewed by Budke et al.210). In addition, inhibitors were designed to block Rad51 oligomerization, a process essential to form Rad51 filaments. Thus, the chemical inhibitor RI-1 covalently binds C319 on the Rad51 oligomerization interface, reduces HR in cells, and increases cell death in response to genotoxic chemotherapy (Fig. 5B).223 An improved analogue RI-2 was proposed, which circumvents high reactivity and low specificity of RI-1 and reversibly inhibits Rad51 oligomerization. RI-2 was also shown to sensitize cells to cross-linking agents.224
Another approach to disrupting Rad51 nucleoprotein filament formation is inhibition of the BRCA2-Rad51 PPI. BRCA2 loads Rad51 onto RPA-coated ssDNA, resulting in RPA displacement.3–5,169,225 BRCA2 harbors eight ∼35 amino-acid long BRC repeats featuring an FxxA motif in the region spanning residues 990–2100, with BRC repeats 1–4 binding to free Rad51 and BRC repeats 5–8 interacting with ssDNA-bound Rad51.226 Pellegrini et al.227 reported a crystal structure of the ATPase domain of Rad51 in complex with the BRC4 repeat of BRCA2 (PDB: 1N0W), revealing an extensive PPI interface that involves 28 residues of BRC4. A small molecule compound, IBR2, which has a phenylsulfonyl indolyl isoquinoline moiety, was identified as a disrupter of Rad51-BRCA2 PPI using a reverse Y2H-HTS assay and was validated to compete for Rad51 binding to BRC repeats by SPR (Fig. 5B).228 Docking studies suggest IBR2 inhibits Rad51–BRCA2 PPI by blocking the hydrophobic pocket on Rad51 that accommodates the BRC FxxA motif. Furthermore, IBR2 disrupts Rad51 oligomerization, induces its proteasomal degradation, attenuates HR, and induces apoptosis in cells. IBR2 treatment in a chronic myeloid leukemia mouse model prolonged overall survival.228
Scott et al.229 used FBDD to identify two additional scaffolds, containing a tryptamine or a tryptophan ester moiety, that interrupt Rad51 association with the BRC FxxA motif by binding to Rad51's pocket described by Pellegrini et al.227 (Fig. 5B). The best fragments bind Rad51 with low μM range affinities and require further building and optimization to create drug leads.229 The same crystal structure227 was used by Cavalli and co-workers to conduct a virtual screen and identify four small molecule inhibitors of Rad51–BRCA2 PPI that bind to the FxxA pocket on Rad51.230–232 Two triazole-based compound hits identified in the screen were validated to disrupt Rad51–BRC4 PPI in an ELISA assay with EC50 of 53 and 25 μM, with the later working synergistically with PARPi to kill pancreatic cancer cells.230 Subsequent optimization led to a compound with EC50 of 8 μM which, however, did not induce synthetic lethality with PARPi.231 Going forth, the same group performed a virtual screen for another binding pocket on Rad51 that binds the BRCA2-BRC4 LFDE motif, which led to identification of dihydroquinoline pyrazoline derivatives as inhibitors of this PPI. Subsequent optimization to improve the Rad51–BRCA2 PPI disruption resulted in a compound 35d (Fig. 5B), which exhibited an EC50 of 19 μM in the ELISA assay, had a binding affinity for Rad51 of 80 nM, inhibited HR by 54% in pancreatic cancer cells with a 20 μM concentration, and displayed synergy with PARPi in pancreatic cells with a concentration of 15 μM.232
6.4. Alt-NHEJ and SSA inhibition
Alt-NHEJ , also termed Polθ-mediated end joining,179–181 has recently garnered popularity as a potential chemotherapeutic target.181 Polθ is synthetic lethal with ATM and various HR genes. Deficiency in Polθ causes sensitivity to ionizing radiation, DSB inducing drugs, and PARP inhibitors. Furthermore, Polθ overexpression is correlated with poor prognosis in cancer patients.179 Polθ has Rad51 binding motifs and is reported to bind to Rad51 in immunoprecipitation assays. It is not yet established if this PPI is necessary for alt-NHEJ function and therefore more investigation is needed.180SSA is another pathway of chemotheraputic interest,166,167,182 as Rad52 enhances the synthetic lethality of PARPi and BRCA deficiency.233,234 Thus, inhibitors of Rad52 were identified using fluorescence-quenching HTS assay for Rad52's ssDNA annealing activity, which selectively kill BRCA deficient cells.2357. Targeting NER, BER and MMR
More frequent than DSBs, which is the most catastrophic DNA damage, are less severe DNA lesions such as chemically altered nucleotides or mispaired bases counteracted by several major DNA repair mechanisms.3–5 Thus, intra-strand cross links, pyrimidine dimers and bulky adducts are primarily repaired by nucleotide excision repair (NER),3–5,236–238 small base modifications are mitigated by base excision repair (BER),3–5,239–241 and nucleotide mismatches are corrected by mismatch repair (MMR).3–5,242–244 In addition, a combination of NER, HR and TLS (discussed below) is used to unhook and repair inter-strand cross-links (ICL) through a complex Fanconi Anemia (FA) pathway.245,246 In this section, we briefly overview the three major DNA repair pathways and discuss their targeting with small molecule PPI inhibitors.7.1. NER, BER and MMR pathways
NER is employed to remove bulky lesions that distort the DNA helix and includes two sub-pathways, the global genome NER (gg-NER) and the transcription coupled NER (TC-NER).3–5,236–238 The first step of the gg-NER is recognition of DNA damage by the Xeroderma Pigmentosum complementation group C protein (XPC) in complex with HR23B (Rad23B) and Centrin2 (CETN2) (Fig. 6A). In addition, UV-induced cyclobutane pyrimidine dimers (CPDs) can be recognized by ultraviolet radiation-DNA damage binding protein 1 and 2 (DDB1 and DDB2), which then recruit XPC. A ten-subunit complex, the transcription initiation factor IIH (TFIIH), recognizes XPC bound to DNA and unwinds the DNA region around the lesion, exposing ssDNA. RPA binds the undamaged DNA strand, while xeroderma pigmentosum group A protein (XPA) binds chemically alerted nucleotides and recruits the endonuclease complex, XPF-ERCC1. The XPF-ERCC1 complex and XPG nick the DNA on the 5′ and 3′ end of the lesion, respectively, creating an exposed ssDNA gap within the double helix. DNA polymerases (such as Polδ, Polε, or Polκ) with the aid of PCNA and replication factor C (RFC) carry out DNA replication to fill the gap, and Lig1 or XRCC1-Lig3 ligases seal the final nick. The second NER sub-pathway, TC-NER, is initiated by stalling of the RNA polymerase II (PolII) at a bulky DNA lesion. Cockayne syndrome protein B (CSB) binds to the stalled RNA polymerase and recruits cockayne syndrome protein A (CSA) which, in turn, recruits additional factors to remodel chromatin, allowing backtracking of the stalled RNA polymerase II followed by recruitment of TFIIH. Then, the NER pathway commences as described above.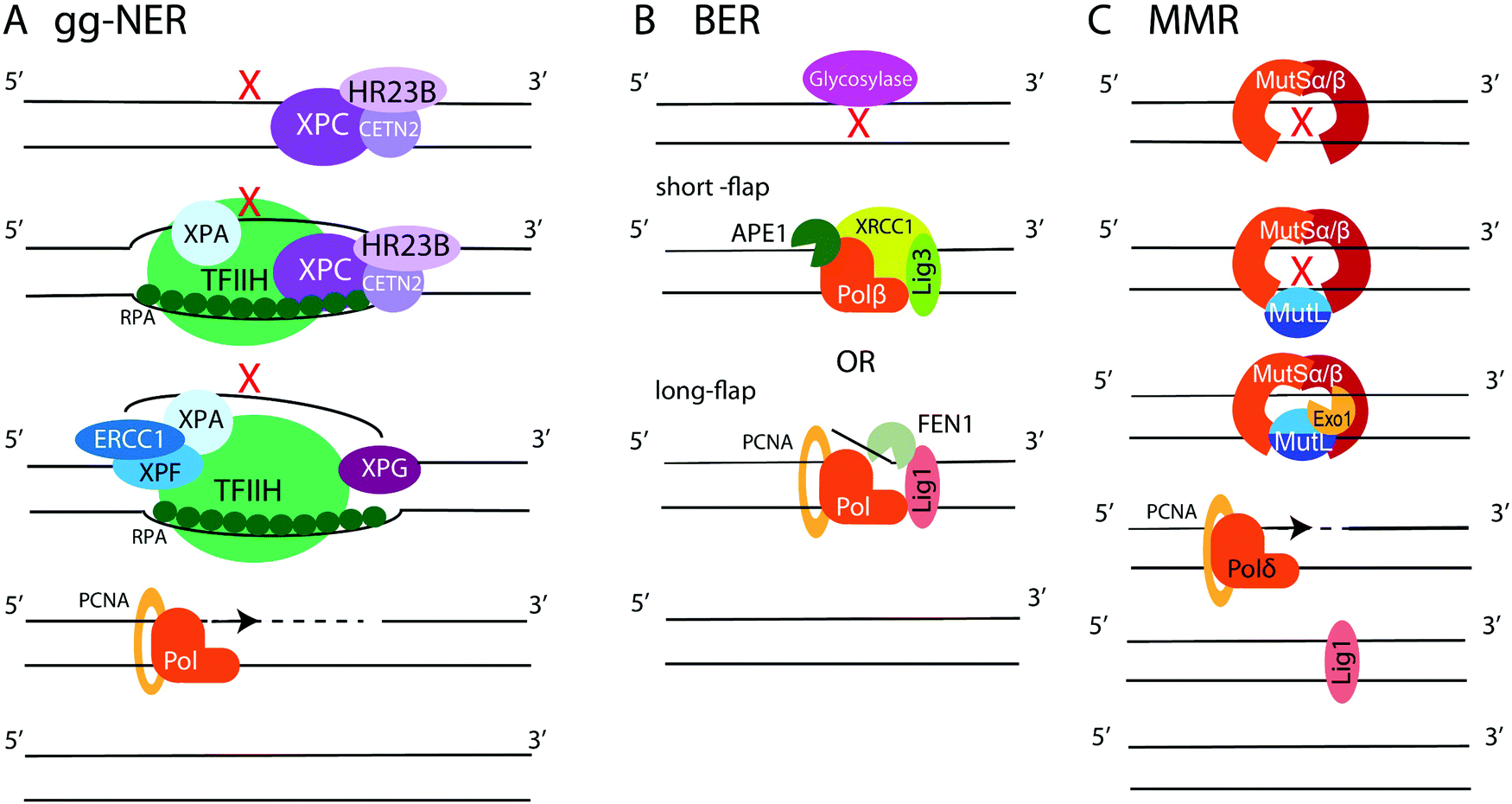 | ||
| Fig. 6 Schematic of NER, BER, and MMR. (A) gg-NER begins with recognition of a helix-distorting lesion by XPC/HR23B/CETN2, which recruits TFIIH to unwind DNA around the lesion. RPA binds the undamaged ssDNA strand, while XPA/XPF/ERCC1 recognizes the damage and, along with XPG associated with TFIIH, cuts DNA fragment around the lesion. A polymerase fills the gap and XRCC1-Lig3 ligates the newly synthesized DNA. (B) BER. One of 11 DNA glycosylases recognizes DNA damage and excises modified base, creating abasic site. In the short-patch BER, APE1 nicks DNA next to abasic site, and Polβ fills a single nucleotide gap and removes abasic sugar by its dRP lyase activity. In the long-parch BER. Polβ or Polδ/ε synthetizes longer DNA stretch, creating a flap removed by FEN1. The final nick is sealed by XRCC1-Lig3 or Lig1. (C) MMR. A mismatch or a deletion-insertion loop is recognized by MutSα or MutSβ. MutL binds MutS and recruits Exo1, which excises the mismatch. The gap is filled by Polδ and nick ligated by Lig1. | ||
BER repairs single-base DNA damage that has not resulted in significant distortion of the DNA helix. At least 11 DNA glycosylases in human cells detect and excise modified and flipped-out DNA bases, creating abasic sites and initiating either short- or long-patch BER (Fig. 6B).3–5,239–241 In short-patch repair, the AP endonuclease (APE1) nicks the backbone 5′ to the abasic site and recruits DNA polymerase Polβ to fill a single nucleotide gap and remove abasic sugar via its deoxyribose phosphate (dRP) lyase activity. This is followed by sealing of the final nick by XRCC1-Lig3 or Lig1 ligases. In long-patch repair, Polβ or a replicative polymerase Polδ or Polε (during S or G2 phases) synthetizes 2–10 nucleotide DNA fragment to fill the gap, displacing several nucleotides from the opposite strand. The generated ssDNA flap is removed by flap endonuclease 1 (FEN1), followed by ligation of the final nick by Lig1. Additionally, ssDNA break repair (SSBR) is performed in a manner similar to BER.100,101,241 First, the SSB is detected by PARP1. Then, APE1, polynucleotide kinase/phosphatase (PNKP), aprataxin, and tyrosyl-DNA phosphodiesterase 1 (TDP1) modify the DNA break ends. Lastly, the exposed ssDNA is filled in and ligated using the BER protein complexes.
MMR specializes on the repair of base-pair mismatches and loops caused by insertions-deletions (Fig. 6C).3–5,242–244 The heterodimer MSH2/MSH6 (MutSα) scans DNA for mis-incorporated bases, while the MSH2/MSH3 (MutSβ) heterodimer seeks out insertion-deletion loops. When MutSα or MutSβ detects a mismatch, another heterodimer, MLH1/PMS2 (MutLα), is recruited to coordinate repair. MutLα aided by PCNA and RFC recruits the endonuclease Exo1, which carries out mismatch excision creating an RPA-coated ssDNA gap. The gap is filled by Polδ, and Lig1 performs the final ligation step. Mutations in MMR proteins are implicated in Lynch syndrome, a predisposition to colon cancer.247
7.2. Targeting NER PPIs
NER is involved in repairing DNA damage caused by genotoxic chemotherapies, particularly front-line platinating and alkylating agents, and therefore provides a promising target for cancer sensitization to these drugs.102–106,237 Multiple PPIs are essential to the NER pathway that could potentially be inhibited for chemotherapeutic development.
XPA
is a major NER scaffold protein with an extensive network of binding partners, including DNA, RPA, DDB1/2, TFIIH, ERCC1, PCNA, and ATR (reviewed by Sugitani et al.248). Mutation to the XPA gene causes Xeroderma pigmentosum (XP), a syndrome of light sensitivity and a high risk of cancer, suggesting XPA is crucial to the NER pathway.249 XPA binds ERCC1 to initiate the 5′ incision.250 Mutations in XPA that disrupt the XPA-ERCC1 PPI limit NER activity,251 and decreased expression of either protein leads to UV and chemotherapeutic sensitivity.252 Solution NMR structure is available of the ERCC1 complex in complex with an XPA peptide, permitting a SBDD for this PPI (PDB: 2JNW).253 Researchers at the University of Alberta successfully conducted SBVS of a 50000 small molecule library against 10 ERCC1 models to identify inhibitors of the XPA-ERCC1 PPI, followed by docking and MD simulations to narrow down the hits.254 One of the identified inhibitors, NERI01, is predicted to facilitate six hydrogen bonds with ERCC1. NERI01 was validated to bind ERCC1 in fluorescent quenching assay and sensitized colon cancer cells to UV radiation (Fig. 7).
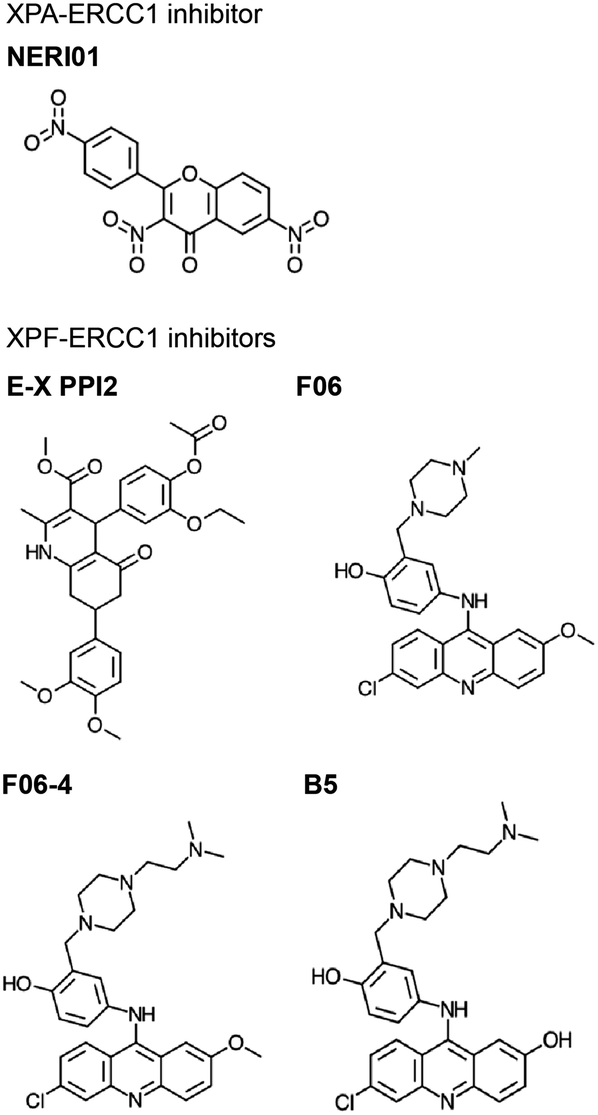 | ||
| Fig. 7 Inhibitors of NER PPIs that target XPA-ERCC1 (top) and XPF-ERCC1 (bottom) complex formation. | ||
XPF-ERCC1 is another complex necessary for the 5′ incision in NER, which also participates in HR and NHEJ.255 XPF-ERCC1 is a major chemotherapeutic target with a few inhibitors identified.256–260 XPF and ERCC1 form a heterodimer via mainly hydrophobic interactions between their double helix–hairpin–helix (HhH2) C-terminal regions (PDB: 2A1J, 1Z00, 6SXA, 6SXB).261–263 Researchers at the University of Edinburgh utilized in silico SBVS to target pockets on the XPF binding site for ERCC1, resulting in identification of 29 compound hits, 4 of which were confirmed to bind XPF by SPR.256 The most potent hit, E-X PPI2, impeded NER in cells with an IC50 of 20 μM (Fig. 7). E-X PPI2 also sensitized melanoma cells to cisplatin and disrupted XPF-ERCC1 dimerization in ovarian cancer cells. In another study, researchers at the University of Lyon performed in silico SBVS and identified 73 hits predicted to bind XPF's ERCC1 interface.257 SBVS hits were validated via methylthiazoletetrazolium (MTT) cytotoxicity assays to test for synergy with cisplatin, mitomycin C (MMC), and UVC irradiation. Compounds that exhibited synergy were then tested for their ability to bind to XPF's C-terminus by SPR and fluorescence quenching experiments. In these assays, compound F06 was the strongest binder, and also disrupted the XPF-ERCC1 PPI in cells (Fig. 7). However, the authors raised concerns about potency and safety, admitting its further optimization is needed to be considered a drug lead. In the follow-up study, optimization of F06 was carried out via docking, pharmacophore modeling, and MD, resulting in seven F06 derivatives.258 One of the analogues, F06-4 (Fig. 7), sensitized colon cancer cells to UV radiation and cyclophosphamide, exhibited IC50 of 0.33 μM (improved 5-fold over 1.86 μM for F06), and displayed favorable physicochemical and ligand efficiency profiles, suggesting it may be effective in vivo.259 A second round of F06 optimization culminated in the design of B5, which inhibited nuclease activity of the XPF-ERCC1 complex with an IC50 of 0.49 μM (Fig. 7).258 Taken together, these studies indicate that further optimization of the F06 scaffold is a promising avenue to develop effective chemotherapeutic sensitizers that inhibit XPF-ERCC1 PPI in NER.
TFIIH is a protein complex made of ten subunits.237,264 A 4.4 Å resolution Cryo-EM structure of TFIIH was reported (PDB: 5OF4), revealing overall architecture of the complex.265 In addition to its central role in NER, TFIIH also functions as a transcription initiator, so targeting this complex should be executed in a manner specific to NER to avoid toxicity. The N-terminal pleckstrin homology (PH) domain of the p62 subunit of TFIIH binds and recruits the 3′ nuclease, XPG. Truncation of this domain reduces incision and excision during NER, but does not affect transcription, suggesting this PPI is specific to NER.266 Solution NMR structure of the p62-XPG yeast homolog, Tfb1-Rad2 complex, is available (PDB: 2LOX).267 Although the human p62-XPG PPI has not been structurally characterized, apo and DNA-bound structures of XPG are available (PDB: 6TUW, 6TUX),268 as well as solution NMR structures of the p62 PH domain (PDB: 1PFJ),266,269 potentially allowing a SBDD of inhibitors of the p62-XPG PPI.
7.3. Targeting BER PPIs
BER is employed to remove several types of lesions induced by genotoxic chemotherapy, especially alkylation therapy,3–5,239–241 and mitigate oxidative DNA damage elevated in the highly oxidative tumor environment.270,271 Inhibition of BER DNA polymerase Polβ is synthetic lethal in cancers that are deficient in MMR genes,110 while XRCC1 deficiency is synthetic lethal with PARP and causes hypersensitivity to genotoxic chemotherapy.272 Overexpression of BER components are correlated with poor survival in gastric cancer.273 Together, these data highlight potential benefits of BER inhibition by targeting key PPIs of this pathway for treatment of multiple forms of cancer.XRCC1 is a BER scaffold central to the assembly of enzymatic complexes in BER and SSBR, which interacts with DNA glycosylases, APE1, Polβ, DNA ligases, and PARP1.239–241,274 XRCC1 has three domains, the N-terminal domain (X1NTD) that binds polβ, the central BRCT domain (X1BRCTa) that binds PARP1, and the C-terminal BRCT domain (X1BRCTb) that binds Lig3.
The XRCC1–Polβ PPI is important for cell resistance to alkylating agents.275 Although Polβ catalytic inhibitors are available, they are only moderately efficient and little data suggests they are specific,276,277 indicating that PPI inhibition may be a viable alternative. X-ray crystal structures were solved for both reduced and oxidized X1NTD in complex with the Polβ catalytic domain (PDB: 3K75, 3LQC).278 NMR chemical shift mapping of the X1NTD–Polβ interface is also available, which deviates from that observed by crystallography.279 The crystal structure revealed an extensive X1NTD–Polβ PPI interface involving 40 residues of Polβ. The PPI is strengthened by oxidation that stimulates a disulfide bond formation between C2–C12 of X1NTD, which is thought to be a molecular switch that increases BER activity during oxidative stress.278 Mutational analysis of the PPI interface identified potential hot-spot residues in X1NTD that can be targeted with small molecules.280 These studies provided plentiful structural data for SBDD of XRCC1–Polβ PPI inhibitors.
The XRCC1–Lig3 complex, which is utilized for ligation in both BER and NER, is another attractive PPI for targeting with small molecule inhibitors. XRCC1 and Lig3 interact through their C-terminal BRCT domains, with X-ray crystal structures available for the BRCT-mediated heterodimer (PDB: 3PC8, 3QVG, 6WH1), and for BRCT domain homodimers of both proteins (PDB: 3PC6, 3PC7, 6WH2).281,282 The dimerization interface is similar in hetero- and homo-dimers, suggesting the proteins are competitive binding partners. The available structural data primes XRCC1–Lig3 for possible SBDD of small molecule PPI inhibitors.
XRCC1-APE1 is another BER PPI of pharmaceutical interest. APE1 inhibition was proposed as a strategy to potentiate cytotoxicity of alkylating agents.283 The APE1 binding region of XRCC1 was mapped to the BRCTa domain and a hinge connecting NTD and BRCTa.284,285 Although the crystal structures of APE1 are available (PDB: 1DE8, 1DE9, 1DEW),286 the structure of XRCC1–APE1 complex is yet to be determined.
7.4. Targeting MMR
Few reports suggest that targeting MMR can provide a viable chemotherapeutic strategy, as loss of MMR is mutagenic and can lead to an oncogenic transformation.12,287,288 Although some studies report that loss of MMR can sensitize cells to DSB-inducing agents,289 other reports suggest that loss of MMR can cause resistance.290 MutSα (MSH2/MSH6 heterodimer) expression promotes sensitivity to cisplatin treatment, while its inhibition would lead to cisplatin resistance.291 Loss of MMR also causes resistance to alkylating therapy.292 Intriguingly, MMR deficiency is associated with a robust response to immunotherapy; however, current hypotheses predict this is due to microsatellite instability that has accumulated over time, rather than a direct consequence of MMR deficiency.293 Although plenty of PPIs are imperative to this pathway, there is little rationale that their inhibition has non-toxic chemotherapeutic potential.8. Targeting TLS
8.1. TLS mechanisms
Translesion synthesis (TLS) is a mechanism of cellular DNA damage tolerance (DDT) that allows bypass replication over sites of DNA damage without the need of immediate repair.16–19 When a replicative polymerase, Polδ or Polε, stalls at a DNA lesion, Rad6/Rad18-dependent mono-ubiquitination of PCNA at K164 signals recruitment of TLS DNA polymerases, which take over replication and synthetize a stretch of DNA across from the lesion (Fig. 8).294,295 TLS DNA polymerases also participate in post-replication gap filling, which is most prevalent during G2/M.296–298 Some DNA lesions can be bypassed by a single TLS DNA polymerase, as exemplified by the accurate and efficient replication over TT-CPDs by Polη.299,300 However, replicative bypass of most DNA lesions occurs by a two-step Rev1/Polζ-dependent TLS,301,302 which involves recruitment of multiple TLS DNA polymerases through multivalent PPIs with PCNA and Rev1.19 Y-family TLS polymerases bind PCNA trough their PCNA-interacting protein box (PIP-box) motifs (Polη, Polι, and Polκ),303 or a BRCT domain (Rev1).304,305 These PPIs are enhanced by ubiquitin binding domains of TLS enzymes (UBM in Polι/Rev1, UBZ in Polη/Polκ) that interact with ubiquitin moiety on ub-PCNA.306,307 The multi-protein TLS complex is additionally stabilized by the Rev1 C-terminal domain (Rev1-CT), a critical TLS scaffold that binds the Rev7 subunit of TLS polymerase Polζ (Rev3/Rev7/PolD2/PolD3 complex308,309) and Rev1-interacting regions (RIRs) of Polη, Polι, Polκ, and PolD3.310–316 In the first step of Rev1/Polζ-dependent TLS, an inserter Y-family polymerase, Polη, Polι, Polκ or Rev1, inserts a nucleotide across the lesion. In the second step, an extender polymerase, often Polζ, continues DNA replication past the lesion-distorted DNA primer–template (Fig. 8).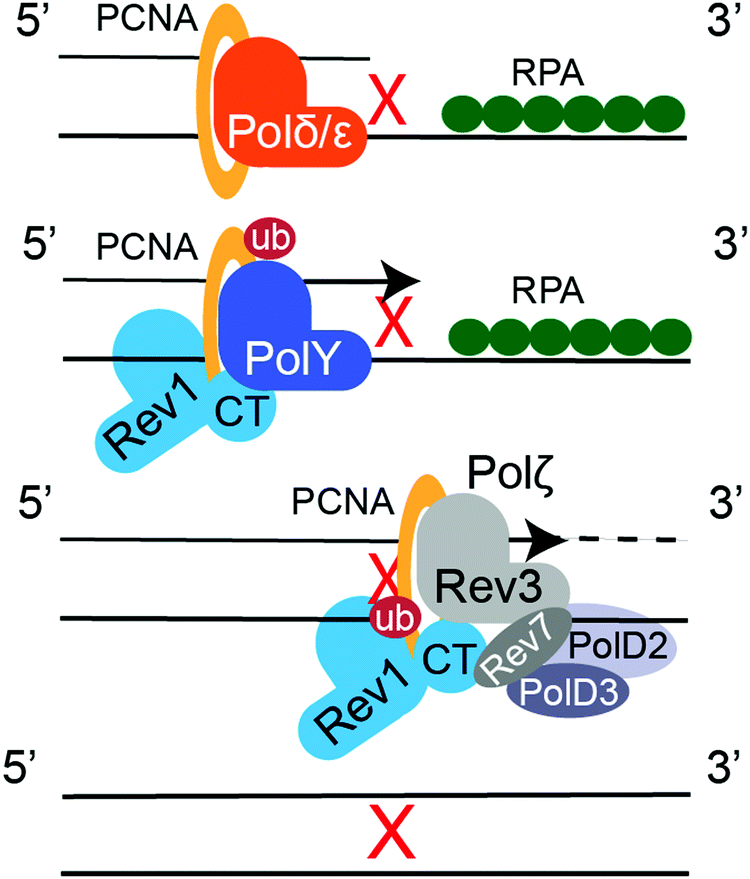 | ||
| Fig. 8 Two step Rev1/Polζ-dependent TLS. After PCNA monoubiquitination by Rad6/Rad18, an inserter Y-family TLS polymerase replaces a replicative polymerase, Polδ or Polε, and inserts nucleotides across the DNA lesion. The extender TLS polymerase Polζ continues replication past the site of DNA damage. | ||
8.2. Inhibitors of TLS PPIs
TLS polymerases are implicated in the replicative bypass of DNA adducts formed by genotoxic platinating and alkylating agents, increasing cancer cell survival after first-line chemotherapy.113–116 Furthermore, TLS polymerases are extremely error-prone, and induce mutagenesis that allows cancer cells to adapt and develop drug resistance.113,114 TLS inhibitors may potentially serve as combination drugs to enhance efficacy of first-line genotoxic therapy and reduce mutagenesis, delaying the onset of chemoresistance.13,117,118 Catalytic inhibition of TLS DNA polymerases is difficult due to structural similarity of active sites. A few non-selective TLS polymerase catalytic inhibitors have been identified that also bind to replicative polymerases.317–319 Therefore, PPI disrupters may be the preferred route for TLS inhibition.Ub-PCNA PPIs that recruit TLS polymerases to sites of DNA damage are critical to TLS function. Researches at St. Jude's Children's Hospital designed an inhibitor, T2 amino alcohol (T2AA), that disrupts PCNA PPIs with the PIP-box motifs of replicative DNA polymerases and other replication-related proteins (e.g. PDB: 1AXC320), and inhibits DNA replication (Fig. 9A).321 This compound also disrupted PCNA-Polη PPI in vitro and localization of TLS polymerases to PCNA in cells, suggesting it acts to attenuate TLS.322 In another study from this research group, two structurally related inhibitors, 1 and 2, were identified in an AlphaScreen323 that bind to Rev1-UBM2 and inhibit Rev1 PPI with ub-PCNA (Fig. 9A).324 Binding of the compounds to Rev1–UBM2 was confirmed by STD-NMR and protein-based 1H–15N HSQC NMR experiments. Compound 1 restricted Rev1 recruitment, limited ICL repair, and sensitized U2OS cells to cisplatin. The same research group also identified a small molecule, MLAF50, as a binding partner of Rev1–UBM2 and disrupter of its PPI with ub-PCNA (Fig. 9A), confirmed that the compound binds Rev1–UBM2 and is displaced from the complex by ubiquitin using NMR, and demonstrated that MLAF50 displaces Rev1 from chromatin in U20S cells.325
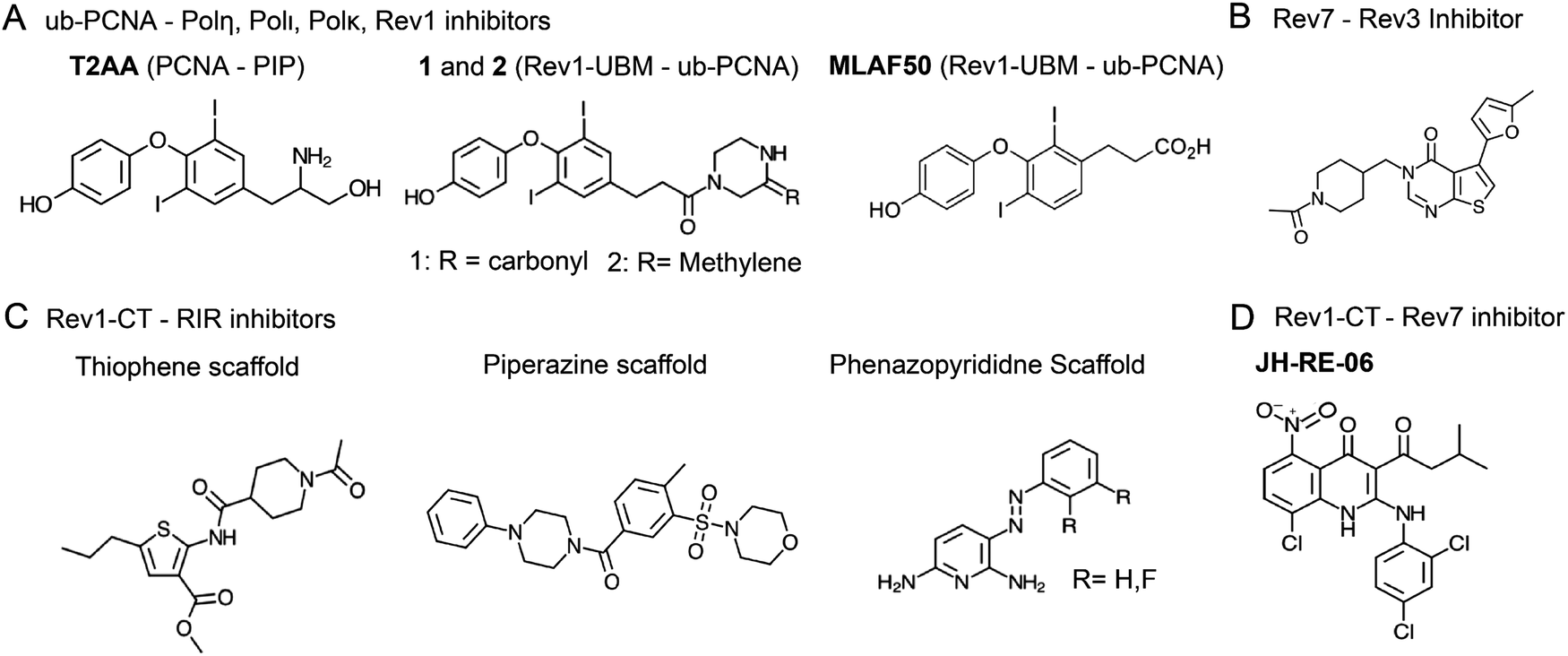 | ||
| Fig. 9 Inhibitors of TLS PPIs between (A) Y-family TLS polymerases and ub-PCNA, (B) Rev7 and Rev3-RBM, (C) Rev1-CT and RIR motifs, and (D) Rev1-CT and Rev7. | ||
Rev7-Rev3 PPI is essential for assembly and function of the ‘extender’ B-family TLS polymerase Polζ.19 Rev7 is an accessory subunit of Polζ that forms a homodimer interacting with the two Rev7-binding motifs (RBM1 and RBM2; consensus sequence Pxxx(A/P)P205,206) of Polζ's Rev3 catalytic subunit. X-ray crystal structures are available for a dimer-breaking R124A Rev7 mutant bound to Rev3-RBM1 and RBM2 peptides (PDB: 3ABD, 3ABE, 6BC8), revealing a binding mechanism in which a ‘safety-belt’ loop of Rev7 wraps around the RBM peptide.205,326 The above research group at St. Jude's Children's Hospital performed a HTS AlphaScreen assay323 and identified a compound with a furan ring moiety capable of disrupting Rev7 PPI with the Rev3-RBM1 peptide.209 An optimized analog of this compound (Fig. 9B) was shown to bind Rev7 by NMR, and sensitized HeLa cells cisplatin. It should be noted that Rev7 also interacts with proteins from several other pathways, including Shld3 (see section 6.2 on NHEJ inhibition), chromosome associated maintenance protein (CAMP), and Ras-associated nuclear protein (RAN), in a similar manner to Rev3,204,207,208 suggesting that the Rev7/RBM PPI inhibitors may have a complex mechanism of action.
Rev1-CT PPIs with RIR motifs of Polη, Polι, Polκ, and PolD3 (a Polζ subunit), and with the Rev7 subunit of Polζ are the scaffolding interactions necessary for assembly and function of the multi-protein TLS complex.19 RIR motifs were also identified in proteins from other pathways, including the BER scaffold XRCC1.327 Rev1-CT is a four-helix bundle that has two independent binding interfaces for TLS DNA polymerases, the N-terminal interface for RIR motifs and the C-terminal interface for Rev7.311–315 Several X-ray crystal and solution NMR structures are available for apo Rev1-CT and its complexes with Polη, Polκ and PolD3 RIR motifs (PDB: 2LSJ, 2LSG, 2LSK, 2LSY, 2N1G, 4FJO),311–314 as well as a crystal structure of the triple Rev1-CT/Rev7/Rev3-RBM1 complex (PDB: 3VU7),316 revealing details of Rev1-CT PPIs.
RIR motifs (nFFhhhh, where n is a N-capping residue, h is a helix-forming residue310,311), upon binding, form an α-helix that inserts side chains of FF residues into a pocket on Rev1-CT,311–314 which provides a druggable ‘hot-spot’ for the development of PPI inhibitors. In a series of works, researchers at the University of Connecticut identified multiple small molecule TLS inhibitors targeting Rev1-CT/RIR PPIs.328–332 Two initial scaffolds, thiophene and piperazine, identified in an FP-based HTS assay disrupted the Rev1-CT PPI with a fluorescently tagged FAM-Polκ-RIR peptide (Fig. 9C).328 The thiophene scaffold compound was shown to bind the RIR-interface of Rev1-CT by NMR, sensitized fibrosarcoma cells to cisplatin, and reduced cisplatin-induced mutagenesis. An extended HTS screen using 10000 compounds from the ChemBridge DIVERSet library identified multiple Rev1-CT/RIR inhibitors, which were grouped into five clusters based on structural similarity with the two clusters corresponding to the initial thiophene and piperazine scaffolds.329 The following studies reported a SBVS, which identified several new chemotypes that disrupt Rev1-CT/RIR PPI,330 and a SBDD utilizing structural knowledge of the Rev1-CT/RIR interface, which identified a phenazopyridine scaffold mimicking the RIR FF pair as an inhibitor of this PPI (Fig. 9C).331 An extensive experimental validation of the identified inhibitors was performed, including FP- or FI-based displacement assays, protein-based and 19F ligand-based NMR binding studies, and cellular assays showing that several scaffolds enhance cisplatin sensitivity of cultured cells. Recently, a 2.5 Å resolution X-ray crystal structure has been determined for the most potent phenazopyridine compound bound to Rev1-CT (in the context of Rev1-CT/Rev7/Rev3-RBM1 complex; PDB: 6WS5), which guided the design of second-generation PAP derivatives exhibiting low μM binding affinities to Rev1-CT that are improved by an order of magnitude relative to the first-generation compounds.332
The second PPI interface of Rev1-CT binds the Rev7 subunit of Polζ, an interaction integral to the Polζ recruitment to DNA.315,316 Researchers at Duke University and Massachusetts Institute of Technology utilized an ELISA assay HTS and identified, a 1,4-dihydroquinolin-4-one derivative, JH-RE-06, as a disruptor of Rev7/Rev1-CT PPI (Fig. 9D).333,334JH-RE-06 binds Rev1-CT with a Kd of 0.42 μM in ITC assays, and disrupts Rev1-CT/Rev7 PPI with an IC50 of 0.78 μM in an AlphaScreen assay.323 A 1.50 Å crystal structure of the inhibitor bound to Rev1-CT (PDB: 6C8C) revealed a unique mechanism of PPI inhibition, in which JH-RE-06 binds the Rev7 interaction site on Rev1-CT and induces dimerization of Rev1-CT enclosing the inhibitor within the dimer interface.333 The inhibitor increased cytotoxicity and reduce mutagenesis in cells treated with cisplatin, and also improved cisplatin efficacy in a mouse model, halting tumor growth with the combination treatment of JH-RE-06 and cisplatin.333 In a subsequent work, the authors demonstrated that JH-RE-06 enhances tumor response to chemotherapy by inducing senescence.334
Additional TLS PPIs that may be targeted with small molecules and their implications in cisplatin resistance have been extensively studied. For example, a triple Rev7 mutant, K44A,R124A,A135D, that abrogates Rev7 dimerization was unable to induce cisplatin resistance in Rev7 knockout cells, indicating the Rev7 dimerization interface is a possible target for chemotherapeutic intervention.326 In addition to Rev3 and Rev7, Polζ contains two accessory subunits, PolD2 and PolD3, that form a heterodimer interacting with a Fe–S cluster in the Rev3 C-terminus.308,309 PolD2 and PolD3 that are also subunits of the replicative DNA polymerase, Polδ, mediate PPIs and have no catalytic activity. Mutation of Rev3's Fe–S cluster that interacts with PolD2 decreases mutagenesis, suggesting disruption of this PPI may be sufficient to attenuate TLS.
Rad6–Rad18 complex responsible for PCNA ubiquitination is important, but not essential for TLS.335 Overexpression of Rad6 ubiquitin conjugating enzyme is correlated with cisplatin resistance in triple-negative breast cancer.336 Therefore, Rad6–Rad18 system may provide another possible target for chemotherapeutic intervention. Catalytic inhibitors of Rad6 have been described that reduce Rad6-ubiquitin thioester formation.337,338 Potential targets for the Rad6–Rad18 PPI inhibitors may include interfaces of Rad6 with the Rad18 N-terminal ring domain, and with the Rad18 C-terminal Rad6-binding domain (PDB: 2YBF).339
9. Conclusions
Attacking cancer's aberrant DDR with small molecule inhibitors is a promising route to creating new chemotherapies, as exemplified by the development of PARP inhibitors that received clinical approval for treatment of BRCA deficient cancers.9 To date, most inhibitors have been designed against traditionally “druggable” proteins with the majority targeting the enzyme active sites. This greatly restricts the number and quality of drug targets and often results in drugs with poor selectivity. The DDR signaling network provides a vast number of protein targets that mediate various crucial PPIs, which can be targeted with small molecules with high efficiency and specificity. Genes encoding these proteins are essential for cancer cell survival and often exhibit synthetic lethal relationships with other DDR genes, providing abundant opportunities for the development of new drugs that selectively kill cancer cells and/or sensitize cancers to existing therapies. The examples discussed in this review highlight the immense potential of targeting DDR PPIs for the development of novel cancer chemotherapies. The discussion, however, covered only a few essential PPI targets in several major DDR pathways, providing a sparce overview of numerous therapeutic opportunities within a vast DDR PPI network. The detailed picture of this network is only now beginning to emerge, suggesting multiple new PPIs targets for anti-cancer drug development will be identified in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH R01CA233959 grant to M. Kyle Hadden and Dmitry M. Korzhnev.References
- E. C. Friedberg, G. C. Walker, W. Siede and R. D. Wood, DNA repair and mutagenesis, American Society for Microbiology Press, 2005 Search PubMed.
- S. P. Jackson and J. Bartek, The DNA-damage response in human biology and disease, Nature, 2009, 461, 1071–1078 CrossRef CAS.
- A. Sancar, L. A. Lindsey-Boltz, K. Unsal-Kacmaz and S. Linn, Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints, Annu. Rev. Biochem., 2004, 73, 39–85 CrossRef CAS PubMed.
- A. Ciccia and S. J. Elledge, The DNA damage response: making it safe to play with knives, Mol. Cell, 2010, 40, 179–204 CrossRef CAS.
- N. Chatterjee and G. C. Walker, Mechanisms of DNA damage, repair, and mutagenesis, Environ. Mol. Mutagen., 2017, 58, 235–263 CrossRef CAS PubMed.
- L. H. Pearl, A. C. Schierz, S. E. Ward, B. Al-Lazikani and F. M. Pearl, Therapeutic opportunities within the DNA damage response, Nat. Rev. Cancer, 2015, 15, 166–180 CrossRef CAS PubMed.
- P. Bouwman and J. Jonkers, The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance, Nat. Rev. Cancer, 2012, 12, 587–598 CrossRef CAS PubMed.
- S. R. Hengel, M. A. Spies and M. Spies, Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy, Cell Chem. Biol., 2017, 24, 1101–1119 CrossRef CAS PubMed.
- J. M. Cleary, A. J. Aguirre, G. I. Shapiro and A. D. D’Andrea, Biomarker-guided development of DNA repair inhibitors, Mol. Cell, 2020, 78, 1070–1085 CrossRef CAS PubMed.
- M. J. O’Connor, Targeting the DNA damage response in cancer, Mol. Cell, 2015, 60, 547–560 CrossRef PubMed.
- J. S. Brown, B. O’Carrigan, S. P. Jackson and T. A. Yap, Targeting DNA repair in cancer: Beyond PARP inhibitors, Cancer Discovery, 2017, 7, 20–37 CrossRef CAS PubMed.
- N. Fujii, Potential strategies to target protein-protein pnteractions in the DNA damage response and repair pathways, J. Med. Chem., 2017, 60, 9932–9959 CrossRef CAS PubMed.
- D. M. Korzhnev and M. K. Hadden, Targeting the translesion synthesis pathway for the development of anti-cancer chemotherapeutics, J. Med. Chem., 2016, 59, 9321–9336 CrossRef CAS PubMed.
- D. Branzei and M. Foiani, Regulation of DNA repair throughout the cell cycle, Nat. Rev. Mol. Cell Biol., 2008, 9, 297–308 CrossRef CAS PubMed.
- A. N. Blackford and S. P. Jackson, ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response, Mol. Cell, 2017, 66, 801–817 CrossRef CAS.
- L. S. Waters, B. K. Minesinger, M. E. Wiltrout, S. D'Souza, R. V. Woodruff and G. C. Walker, Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance, Microbiol. Mol. Biol. Rev., 2009, 73, 134–154 CrossRef CAS PubMed.
- J. E. Sale, A. R. Lehmann and R. Woodgate, Y-family DNA polymerases and their role in tolerance of cellular DNA damage, Nat. Rev. Mol. Cell Biol., 2012, 13, 141–152 CrossRef CAS PubMed.
- M. F. Goodman and R. Woodgate, Translesion DNA polymerases, Cold Spring Harbor Perspect. Biol., 2013, 5, a010363 Search PubMed.
- A. A. Rizzo and D. M. Korzhnev, The Rev1-Polζ translesion synthesis mutasome: Structure, interactions and inhibition, Enzymes, 2019, 45, 139–181 CAS.
- M. Toulany, Targeting DNA double-strand break repair pathways to improve radiotherapy response, Genes, 2019, 10, 25 CrossRef PubMed.
- A. L. Hopkins and C. R. Groom, The druggable genome, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS PubMed.
- H. Jubb, A. P. Higueruelo, A. Winter and T. L. Blundell, Structural biology and drug discovery for protein-protein interactions, Trends Pharmacol. Sci., 2012, 33, 241–248 CrossRef CAS PubMed.
- M. R. Arkin and J. A. Wells, Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream, Nat. Rev. Drug Discovery, 2004, 3, 301–317 CrossRef CAS.
- M. R. Arkin, Y. Tang and J. A. Wells, Small-molecule inhibitors of protein-protein interactions: progressing toward the reality, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- E. Cukuroglu, H. B. Engin, A. Gursoy and O. Keskin, Hot spots in protein-protein interfaces: towards drug discovery, Prog. Biophys. Mol. Biol., 2014, 116, 165–173 CrossRef CAS.
- M. Aeluri, S. Chamakuri, B. Dasari, S. K. Guduru, R. Jimmidi, S. Jogula and P. Arya, Small molecule modulators of protein-protein interactions: selected case studies, Chem. Rev., 2014, 114, 4640–4694 CrossRef CAS.
- J. C. Fuller, N. J. Burgoyne and R. M. Jackson, Predicting druggable binding sites at the protein-protein interface, Drug Discovery Today, 2009, 14, 155–161 CrossRef CAS PubMed.
- L. Mabonga and A. P. Kappo, Protein-protein interaction modulators: advances, successes and remaining challenges, Biophys. Rev., 2019, 11, 559–581 CrossRef CAS PubMed.
- X. Ran and J. E. Gestwicki, Inhibitors of protein-protein interactions (PPIs): an analysis of scaffold choices and buried surface area, Curr. Opin. Chem. Biol., 2018, 44, 75–86 CrossRef CAS PubMed.
- G. D. Hartman, M. S. Egbertson, W. Halczenko, W. L. Laswell, M. E. Duggan, R. L. Smith, A. M. Naylor, P. D. Manno, R. J. Lynch and G. Zhang, et al., Non-peptide fibrinogen receptor antagonists. 1. Discovery and design of exosite inhibitors, J. Med. Chem., 1992, 35, 4640–4642 CrossRef CAS.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- S. Wang, Y. Zhao, A. Aguilar, D. Bernard and C. Y. Yang, Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: Progress and challenges, Cold Spring Harbor Perspect. Biol., 2017, 7, a026245 CrossRef PubMed.
- A. Vogt and J. S. Lazo, Chemical complementation: a definitive phenotypic strategy for identifying small molecule inhibitors of elusive cellular targets, Pharmacol. Ther., 2005, 107, 212–221 CrossRef CAS.
- A. C. Anderson, The process of structure-based drug design, Chem. Biol., 2003, 10, 787–797 CrossRef CAS PubMed.
- R. L. M. van Montfort and P. Workman, Structure-based drug design: aiming for a perfect fit, Essays Biochem., 2017, 61, 431–437 CrossRef PubMed.
- M. Batool, B. Ahmad and S. Choi, A structure-based drug discovery paradigm, Int. J. Mol. Sci., 2019, 20, 2783 CrossRef CAS PubMed.
- S. K. Burley, H. M. Berman, C. Bhikadiya, C. Bi, L. Chen, L. Di Costanzo, C. Christie, J. M. Duarte, S. Dutta, Z. Feng, S. Ghosh, D. S. Goodsell, R. K. Green, V. Guranovic, D. Guzenko, B. P. Hudson, Y. Liang, R. Lowe, E. Peisach, I. Periskova, C. Randle, A. Rose, M. Sekharan, C. Shao, Y.-P. Tao, Y. Valasatava, M. Voigt, J. Westbrook, J. Young, C. Zardecki, M. Zhuravleva, G. Kurisu, H. Nakamura, Y. Kengaku, H. Cho, J. Sato, J. Y. Kim, Y. Ikegawa, A. Nakagawa, R. Yamashita, T. Kudou, G.-J. Bekker, H. Suzuki, T. Iwata, M. Yokochi, N. Kobayashi, T. Fujiwara, S. Velankar, G. J. Kleywegt, S. Anyango, D. R. Armstrong, J. M. Berrisford, M. J. Conroy, J. M. Dana, M. Deshpande, P. Gane, R. Gáborová, D. Gupta, A. Gutmanas, J. Koča, L. Mak, S. Mir, A. Mukhopadhyay, N. Nadzirin, S. Nair, A. Patwardhan, T. Paysan-Lafosse, L. Pravda, O. Salih, D. Sehnal, M. Varadi, R. Vařeková, J. L. Markley, J. C. Hoch, P. R. Romero, K. Baskaran, D. Maziuk, E. L. Ulrich, J. R. Wedell, H. Yao, M. Livny and Y. E. Ioannidis, Protein Data Bank: the single global archive for 3D macromolecular structure data, Nucleic Acids Res., 2019, 47, D520–D528 CrossRef PubMed.
- P. D. B. RCSB, PDB Statistics: Overall Growth of Released Structures Per Year, https://www.rcsb.org/stats/growth/growth-released-structures, Accessed 11/13/2020, 2020.
- M. A. Marti-Renom, A. C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, Comparative protein structure modeling of genes and genomes, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS.
- M. T. Muhammed and E. Aki-Yalcin, Homology modeling in drug discovery: Overview, current applications, and future perspectives, Chem. Biol. Drug Des., 2019, 93, 12–20 CrossRef CAS.
- A. Merk, A. Bartesaghi, S. Banerjee, V. Falconieri, P. Rao, M. I. Davis, R. Pragani, M. B. Boxer, L. A. Earl, J. L. S. Milne and S. Subramaniam, Breaking cryo-EM resolution barriers to facilitate drug discovery, Cell, 2016, 165, 1698–1707 CrossRef CAS.
- J. P. Renaud, A. Chari, C. Ciferri, W. T. Liu, H. W. Rémigy, H. Stark and C. Wiesmann, Cryo-EM in drug discovery: achievements, limitations and prospects, Nat. Rev. Drug Discovery, 2018, 17, 471–492 CrossRef CAS PubMed.
- M. Khoshouei, M. Radjainia, W. Baumeister and R. Danev, Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate, Nat. Commun., 2017, 8, 16099 CrossRef CAS PubMed.
- C. Dominguez, R. Boelens and A. M. Bonvin, HADDOCK: a protein-protein docking approach based on biochemical or biophysical information, J. Am. Chem. Soc., 2003, 125, 1731–1737 CrossRef CAS PubMed.
- G. C. P. van Zundert, J. Rodrigues, M. Trellet, C. Schmitz, P. L. Kastritis, E. Karaca, A. S. J. Melquiond, M. van Dijk, S. J. de Vries and A. Bonvin, The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS PubMed.
- M. Entzeroth, H. Flotow and P. Condron, Overview of high-throughput screening, Current protocols in pharmacology, 2009, ch. 9, unit 9.4 Search PubMed.
- K. H. Bleicher, H. J. Bohm, K. Muller and A. I. Alanine, Hit and lead generation: beyond high-throughput screening, Nat. Rev. Drug Discovery, 2003, 2, 369–378 CrossRef CAS.
- T. Cheng, Q. Li, Z. Zhou, Y. Wang and S. H. Bryant, Structure-based virtual screening for drug discovery: a problem-centric review, AAPS J., 2012, 14, 133–141 CrossRef CAS PubMed.
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe Jr, Computational methods in drug discovery, Pharmacol. Rev., 2014, 66, 334–395 CrossRef PubMed.
- E. D. Levy, A simple definition of structural regions in proteins and its use in analyzing interface evolution, J. Mol. Biol., 2010, 403, 660–670 CrossRef CAS PubMed.
- M. Rosell and J. Fernández-Recio, Hot-spot analysis for drug discovery targeting protein-protein interactions, Expert Opin. Drug Discovery, 2018, 13, 327–338 CrossRef CAS.
- T. Clackson and J. A. Wells, A hot spot of binding energy in a hormone-receptor interface, Science, 1995, 267, 383–386 CrossRef CAS PubMed.
- W. Tian, C. Chen, X. Lei, J. Zhao and J. Liang, CASTp 3.0: computed atlas of surface topography of proteins, Nucleic Acids Res., 2018, 46, W363–w367 CrossRef CAS PubMed.
- J. K. Morrow and S. Zhang, Computational prediction of protein hot spot residues, Curr. Pharm. Des., 2012, 18, 1255–1265 CrossRef CAS PubMed.
- S. Schlee, K. Straub, T. Schwab, T. Kinateder, R. Merkl and R. Sterner, Prediction of quaternary structure by analysis of hot spot residues in protein-protein interfaces: the case of anthranilate phosphoribosyltransferases, Proteins, 2019, 87, 815–825 CrossRef CAS PubMed.
- P. Śledź and A. Caflisch, Protein structure-based drug design: from docking to molecular dynamics, Curr. Opin. Struct. Biol., 2018, 48, 93–102 CrossRef PubMed.
- J. R. Schames, R. H. Henchman, J. S. Siegel, C. A. Sotriffer, H. Ni and J. A. McCammon, Discovery of a novel binding trench in HIV integrase, J. Med. Chem., 2004, 47, 1879–1881 CrossRef CAS PubMed.
- P. J. Hajduk and J. Greer, A decade of fragment-based drug design: strategic advances and lessons learned, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS PubMed.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A 'rule of three' for fragment-based lead discovery?, Drug Discovery Today, 2003, 8, 876–877 CrossRef PubMed.
- V. Zoete, A. Grosdidier and O. Michielin, Docking, virtual high throughput screening and in silico fragment-based drug design, J. Cell. Mol. Med., 2009, 13, 238–248 CrossRef CAS PubMed.
- M. J. Harner, A. O. Frank and S. W. Fesik, Fragment-based drug discovery using NMR spectroscopy, J. Biomol. NMR, 2013, 56, 65–75 CrossRef CAS PubMed.
- T. Sugiki, K. Furuita, T. Fujiwara and C. Kojima, Current NMR techniques for structure-based drug discovery, Molecules, 2018, 23, 148 CrossRef PubMed.
- A. Furukawa, T. Konuma, S. Yanaka and K. Sugase, Quantitative analysis of protein-ligand interactions by NMR, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 96, 47–57 CrossRef CAS PubMed.
- R. Huang and I. K. H. Leung, Protein-small molecule interactions by WaterLOGSY, Methods Enzymol., 2019, 615, 477–500 CAS.
- A. Viegas, J. Manso, F. L. Nobrega and E. J. Cabrita, Saturation-transfer difference (STD) NMR: A simple and fast method for ligand screening and characterization of protein binding, J. Chem. Educ., 2011, 88, 990–994 CrossRef CAS.
- S. Leavitt and E. Freire, Direct measurement of protein binding energetics by isothermal titration calorimetry, Curr. Opin. Struct. Biol., 2001, 11, 560–566 CrossRef CAS PubMed.
- T. Wiseman, S. Williston, J. F. Brandts and L. N. Lin, Rapid measurement of binding constants and heats of binding using a new titration calorimeter, Anal. Biochem., 1989, 179, 131–137 CrossRef CAS.
- A. Olaru, C. Bala, N. Jaffrezic-Renault and H. Y. Aboul-Enein, Surface plasmon resonance (SPR) biosensors in pharmaceutical analysis, Crit. Rev. Anal. Chem., 2015, 45, 97–105 CrossRef CAS.
- Y. Du, Fluorescence polarization assay to quantify protein-protein interactions in an HTS format, Methods Mol. Biol., 2015, 1278, 529–544 CrossRef CAS.
- X. Huang and A. Aulabaugh, Application of fluorescence polarization in HTS assays, Methods Mol. Biol., 2016, 1439, 115–130 CrossRef CAS.
- R. L. Cornea, S. J. Gruber, E. L. Lockamy, J. M. Muretta, D. Jin, J. Chen, R. Dahl, T. Bartfai, K. M. Zsebo, G. D. Gillispie and D. D. Thomas, High-throughput FRET assay yields allosteric SERCA activators, J. Biomol. Screening, 2013, 18, 97–107 CrossRef PubMed.
- D. R. Stroik, S. L. Yuen, K. A. Janicek, T. M. Schaaf, J. Li, D. K. Ceholski, R. J. Hajjar, R. L. Cornea and D. D. Thomas, Targeting protein-protein interactions for therapeutic discovery via FRET-based high-throughput screening in living cells, Sci. Rep., 2018, 8, 12560 CrossRef.
- R. B. Sekar and A. Periasamy, Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations, J. Cell Biol., 2003, 160, 629–633 CrossRef CAS PubMed.
- S. Fields and O. Song, A novel genetic system to detect protein-protein interactions, Nature, 1989, 340, 245–246 CrossRef CAS PubMed.
- P. James, J. Halladay and E. A. Craig, Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast, Genetics, 1996, 144, 1425–1436 CrossRef CAS PubMed.
- J. H. Wong, M. Alfatah, M. F. Sin, H. M. Sim, C. S. Verma, D. P. Lane and P. Arumugam, A yeast two-hybrid system for the screening and characterization of small-molecule inhibitors of protein-protein interactions identifies a novel putative Mdm2-binding site in p53, BMC Biol., 2017, 15, 108 CrossRef.
- M. Asmari, R. Ratih, H. A. Alhazmi and S. El Deeb, Thermophoresis for characterizing biomolecular interaction, Methods, 2018, 146, 107–119 CrossRef CAS PubMed.
- S. A. Seidel, P. M. Dijkman, W. A. Lea, G. van den Bogaart, M. Jerabek-Willemsen, A. Lazic, J. S. Joseph, P. Srinivasan, P. Baaske, A. Simeonov, I. Katritch, F. A. Melo, J. E. Ladbury, G. Schreiber, A. Watts, D. Braun and S. Duhr, Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions, Methods, 2013, 59, 301–315 CrossRef CAS.
- W. J. Allen, T. E. Balius, S. Mukherjee, S. R. Brozell, D. T. Moustakas, P. T. Lang, D. A. Case, I. D. Kuntz and R. C. Rizzo, DOCK 6: Impact of new features and current docking performance, J. Comput. Chem., 2015, 36, 1132–1156 CrossRef CAS.
- M. Rarey, B. Kramer, T. Lengauer and G. Klebe, A fast flexible docking method using an incremental construction algorithm, J. Mol. Biol., 1996, 261, 470–489 CrossRef CAS.
- N. Majeux, M. Scarsi and A. Caflisch, Efficient electrostatic solvation model for protein-fragment docking, Proteins, 2001, 42, 256–268 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Improved protein-ligand docking using GOLD, Proteins, 2003, 52, 609–623 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- Z. X. Wang, An exact mathematical expression for describing competitive binding of two different ligands to a protein molecule, FEBS Lett., 1995, 360, 111–114 CrossRef CAS.
- Y. Cheng and W. H. Prusoff, Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
- L. Maveyraud and L. Mourey, Protein X-ray crystallography and drug discovery, Molecules, 2020, 25, 1030 CrossRef CAS PubMed.
- D. R. Cooper, P. J. Porebski, M. Chruszcz and W. Minor, X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery, Expert Opin. Drug Discovery, 2011, 6, 771–782 CrossRef CAS PubMed.
- T. Ceska, C. W. Chung, R. Cooke, C. Phillips and P. A. Williams, Cryo-EM in drug discovery, Biochem. Soc. Trans., 2019, 47, 281–293 CrossRef CAS PubMed.
- C. E. Chang, W. Chen and M. K. Gilson, Ligand configurational entropy and protein binding, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1534–1539 CrossRef CAS PubMed.
- H. van de Waterbeemd and E. Gifford, ADMET in silico modelling: towards prediction paradise?, Nat. Rev. Drug Discovery, 2003, 2, 192–204 CrossRef CAS.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, The role of ligand efficiency metrics in drug discovery, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- J. C. Lucchesi, Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanfgaster, Genetics, 1968, 59, 37–44 CrossRef CAS PubMed.
- W. G. Kaelin Jr, The concept of synthetic lethality in the context of anticancer therapy, Nat. Rev. Cancer, 2005, 5, 689–698 CrossRef PubMed.
- H. Farmer, N. McCabe, C. J. Lord, A. N. Tutt, D. A. Johnson, T. B. Richardson, M. Santarosa, K. J. Dillon, I. Hickson, C. Knights, N. M. Martin, S. P. Jackson, G. C. Smith and A. Ashworth, Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy, Nature, 2005, 434, 917–921 CrossRef CAS PubMed.
- H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, N. J. Curtin and T. Helleday, Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase, Nature, 2005, 434, 913–917 CrossRef CAS PubMed.
- T. Helleday, The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings, Mol Oncol., 2011, 5, 387–393 CrossRef CAS PubMed.
- C. J. Lord and A. Ashworth, PARP inhibitors: Synthetic lethality in the clinic, Science, 2017, 355, 1152–1158 CrossRef CAS PubMed.
- V. Schreiber, F. Dantzer, J. C. Ame and G. de Murcia, Poly(ADP-ribose): novel functions for an old molecule, Nat. Rev. Mol. Cell Biol., 2006, 7, 517–528 CrossRef CAS PubMed.
- K. W. Caldecott, Single-strand break repair and genetic disease, Nat. Rev. Genet., 2008, 9, 619–631 CrossRef CAS.
- K. W. Caldecott, DNA single-strand break repair, Exp. Cell Res., 2014, 329, 2–8 CrossRef CAS PubMed.
- Z. H. Siddik, Cisplatin: mode of cytotoxic action and molecular basis of resistance, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- D. B. Zamble and S. J. Lippard, Cisplatin and DNA repair in cancer chemotherapy, Trends Biochem. Sci., 1995, 20, 435–439 CrossRef CAS.
- D. Wang and S. J. Lippard, Cellular processing of platinum anticancer drugs, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS PubMed.
- L. Kelland, The resurgence of platinum-based cancer chemotherapy, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- H. Lajous, B. Lelièvre, E. Vauléon, P. Lecomte and E. Garcion, Rethinking alkylating(-like) agents for solid tumor management, Trends Pharmacol. Sci., 2019, 40, 342–357 CrossRef CAS PubMed.
- A. C. Begg, F. A. Stewart and C. Vens, Strategies to improve radiotherapy with targeted drugs, Nat. Rev. Cancer, 2011, 11, 239–253 CrossRef CAS PubMed.
- Y. Pommier, Drugging topoisomerases: lessons and challenges, ACS Chem. Biol., 2013, 8, 82–95 CrossRef CAS PubMed.
- S. Matt and T. G. Hofmann, The DNA damage-induced cell death response: a roadmap to kill cancer cells, Cell. Mol. Life Sci., 2016, 73, 2829–2850 CrossRef CAS PubMed.
- J. A. Nickoloff, D. Jones, S. H. Lee, E. A. Williamson and R. Hromas, Drugging the cancers addicted to DNA repair, J. Natl. Cancer Inst., 2017, 109, djx059 CrossRef PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Molecular mechanisms of cisplatin resistance, Oncogene, 2012, 31, 1869–1883 CrossRef CAS.
- J. N. Sarkaria, G. J. Kitange, C. D. James, R. Plummer, H. Calvert, M. Weller and W. Wick, Mechanisms of chemoresistance to alkylating agents in malignant glioma, Clin. Cancer Res., 2008, 14, 2900–2908 CrossRef CAS PubMed.
- K. Nojima, H. Hochegger, A. Saberi, T. Fukushima, K. Kikuchi, M. Yoshimura, B. J. Orelli, D. K. Bishop, S. Hirano and M. Ohzeki, Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells, Cancer Res., 2005, 65, 11704–11711 CrossRef CAS PubMed.
- K. Xie, J. Doles, M. T. Hemann and G. C. Walker, Error-prone translesion synthesis mediates acquired chemoresistance, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20792–20797 CrossRef CAS PubMed.
- F. Wu, X. Lin, T. Okuda and S. B. Howell, DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance, Cancer Res., 2004, 64, 8029–8035 CrossRef CAS PubMed.
- X. Lin, T. Okuda, J. Trang and S. B. Howell, Human REV1 modulates the cytotoxicity and mutagenicity of cisplatin in human ovarian carcinoma cells, Mol. Pharmacol., 2006, 69, 1748–1754 CrossRef CAS PubMed.
- K. Yamanaka, N. Chatterjee, M. T. Hemann and G. C. Walker, Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy?, PLoS Genet., 2017, 13, e1006842 CrossRef PubMed.
- S. M. Patel, R. C. Dash and M. K. Hadden, Translesion synthesis inhibitors as a new class of cancer chemotherapeutics, Expert Opin. Invest. Drugs, 2021, 30, 13–24 CrossRef CAS PubMed.
- B. Vogelstein, D. Lane and A. J. Levine, Surfing the p53 network, Nature, 2000, 408, 307–310 CrossRef CAS PubMed.
- A. Hafner, M. L. Bulyk, A. Jambhekar and G. Lahav, The multiple mechanisms that regulate p53 activity and cell fate, Nat. Rev. Mol. Cell Biol., 2019, 20, 199–210 CrossRef CAS PubMed.
- Y. Haupt, R. Maya, A. Kazaz and M. Oren, Mdm2 promotes the rapid degradation of p53, Nature, 1997, 387, 296–299 CrossRef CAS PubMed.
- M. J. Duffy, N. C. Synnott, S. O’Grady and J. Crown, Targeting p53 for the treatment of cancer, Semin. Cancer Biol., 2020 DOI:10.1016/j.semcancer.2020.07.005.
- A. Beloglazkina, N. Zyk, A. Majouga and E. Beloglazkina, Recent small-molecule inhibitors of the p53-MDM2 protein-protein interaction, Molecules, 2020, 25, 1211 CrossRef CAS PubMed.
- M. Espadinha, V. Barcherini, E. A. Lopes and M. M. M. Santos, An update on MDMX and dual MDM2/X inhibitors, Curr. Top. Med. Chem., 2018, 18, 647–660 CrossRef CAS PubMed.
- W. Wang, J. J. Qin, M. Rajaei, X. Li, X. Yu, C. Hunt and R. Zhang, Targeting MDM2 for novel molecular therapy: Beyond oncology, Med. Res. Rev., 2020, 40, 856–880 CrossRef CAS PubMed.
- V. J. N. Bykov, S. E. Eriksson, J. Bianchi and K. G. Wiman, Targeting mutant p53 for efficient cancer therapy, Nat. Rev. Cancer, 2018, 18, 89–102 CrossRef CAS PubMed.
- A. Pozhidaeva and I. Bezsonova, USP7: Structure, substrate specificity, and inhibition, DNA Repair, 2019, 76, 30–39 CrossRef CAS PubMed.
- S. Matsuoka, B. A. Ballif, A. Smogorzewska, E. R. McDonald 3rd, K. E. Hurov, J. Luo, C. E. Bakalarski, Z. Zhao, N. Solimini, Y. Lerenthal, Y. Shiloh, S. P. Gygi and S. J. Elledge, ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage, Science, 2007, 316, 1160–1166 CrossRef CAS PubMed.
- Y. Shiloh, The ATM-mediated DNA-damage response: taking shape, Trends Biochem. Sci., 2006, 31, 402–410 CrossRef CAS PubMed.
- N. Jette and S. P. Lees-Miller, The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis, Prog. Biophys. Mol. Biol., 2015, 117, 194–205 CrossRef CAS PubMed.
- L. Zou and S. J. Elledge, Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes, Science, 2003, 300, 1542–1548 CrossRef CAS PubMed.
- J. Falck, J. Coates and S. P. Jackson, Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage, Nature, 2005, 434, 605–611 CrossRef CAS PubMed.
- A. Kumagai, J. Lee, H. Y. Yoo and W. G. Dunphy, TopBP1 activates the ATR-ATRIP complex, Cell, 2006, 124, 943–955 CrossRef CAS PubMed.
- M. Sokka, D. Koalick, P. Hemmerich, J. E. Syväoja and H. Pospiech, The ATR-activation domain of TopBP1 is required for the suppression of origin firing during the S phase, Int. J. Mol. Sci., 2018, 19, 2376 CrossRef PubMed.
- P. Haahr, S. Hoffmann, M. A. Tollenaere, T. Ho, L. I. Toledo, M. Mann, S. Bekker-Jensen, M. Räschle and N. Mailand, Activation of the ATR kinase by the RPA-binding protein ETAA1, Nat. Cell Biol., 2016, 18, 1196–1207 CrossRef CAS PubMed.
- T. Uziel, Y. Lerenthal, L. Moyal, Y. Andegeko, L. Mittelman and Y. Shiloh, Requirement of the MRN complex for ATM activation by DNA damage, EMBO J., 2003, 22, 5612–5621 CrossRef CAS PubMed.
- J. H. Lee and T. T. Paull, ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex, Science, 2005, 308, 551–554 CrossRef CAS PubMed.
- J. A. Downs and S. P. Jackson, A means to a DNA end: the many roles of Ku, Nat. Rev. Mol. Cell Biol., 2004, 5, 367–378 CrossRef CAS PubMed.
- P. Frit, V. Ropars, M. Modesti, J. B. Charbonnier and P. Calsou, Plugged into the Ku-DNA hub: The NHEJ network, Prog. Biophys. Mol. Biol., 2019, 147, 62–76 CrossRef CAS PubMed.
- B. K. Singleton, M. I. Torres-Arzayus, S. T. Rottinghaus, G. E. Taccioli and P. A. Jeggo, The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit, Mol. Cell. Biol., 1999, 19, 3267–3277 CrossRef CAS PubMed.
- O. L. Kantidze, A. K. Velichko, A. V. Luzhin, N. V. Petrova and S. V. Razin, Synthetically lethal interactions of ATM, ATR, and DNA-PKcs, Trends Cancer, 2018, 4, 755–768 CrossRef CAS PubMed.
- D. W. Schoppy, R. L. Ragland, O. Gilad, N. Shastri, A. A. Peters, M. Murga, O. Fernandez-Capetillo, J. A. Diehl and E. J. Brown, Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR, J. Clin. Invest., 2012, 122, 241–252 CrossRef CAS PubMed.
- D. Menolfi, W. Jiang, B. J. Lee, T. Moiseeva, Z. Shao, V. Estes, M. G. Frattini, C. J. Bakkenist and S. Zha, Kinase-dead ATR differs from ATR loss by limiting the dynamic exchange of ATR and RPA, Nat. Commun., 2018, 9, 5351 CrossRef PubMed.
- D. Menolfi and S. Zha, ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: inhibition≠deletion, Cell Biosci., 2020, 10, 8 CrossRef PubMed.
- Q. Rao, M. Liu, Y. Tian, Z. Wu, Y. Hao, L. Song, Z. Qin, C. Ding, H. W. Wang, J. Wang and Y. Xu, Cryo-EM structure of human ATR-ATRIP complex, Cell Res., 2018, 28, 143–156 CrossRef CAS PubMed.
- X. Wang, T. Ran, X. Zhang, J. Xin, Z. Zhang, T. Wu, W. Wang and G. Cai, 3.9 Å structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP, Science, 2017, 358, 1206–1209 CrossRef CAS PubMed.
- D. Baretic, T. Maia de Oliveira, M. Niess, P. Wan, H. Pollard, C. M. Johnson, C. Truman, E. McCall, D. Fisher, R. Williams and C. Phillips, Structural insights into the critical DNA damage sensors DNA-PKcs, ATM and ATR, Prog. Biophys. Mol. Biol., 2019, 147, 4–16 CrossRef CAS PubMed.
- X. Yin, M. Liu, Y. Tian, J. Wang and Y. Xu, Cryo-EM structure of human DNA-PK holoenzyme, Cell Res., 2017, 27, 1341–1350 CrossRef CAS.
- H. Sharif, Y. Li, Y. Dong, L. Dong, W. L. Wang, Y. Mao and H. Wu, Cryo-EM structure of the DNA-PK holoenzyme, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7367–7372 CrossRef CAS PubMed.
- G. Watanabe, M. R. Lieber and D. Williams, Structural step forward for NHEJ, Cell Res., 2017, 27, 1304–1306 CrossRef CAS PubMed.
- D. Baretić, H. K. Pollard, D. I. Fisher, C. M. Johnson, B. Santhanam, C. M. Truman, T. Kouba, A. R. Fersht, C. Phillips and R. L. Williams, Structures of closed and open conformations of dimeric human ATM, Sci. Adv., 2017, 3, e1700933 CrossRef PubMed.
- D. Gell and S. P. Jackson, Mapping of protein-protein interactions within the DNA-dependent protein kinase complex, Nucleic Acids Res., 1999, 27, 3494–3502 CrossRef CAS PubMed.
- J. Smith, L. M. Tho, N. Xu and D. A. Gillespie, The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer, Adv. Cancer Res., 2010, 108, 73–112 CrossRef CAS.
- C. Ronco, A. R. Martin, L. Demange and R. Benhida, ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells, MedChemComm, 2017, 8, 295–319 RSC.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain, Science, 1996, 274, 948–953 CrossRef CAS PubMed.
- A. Aguilar, J. Lu, L. Liu, D. Du, D. Bernard, D. McEachern, S. Przybranowski, X. Li, R. Luo, B. Wen, D. Sun, H. Wang, J. Wen, G. Wang, Y. Zhai, M. Guo, D. Yang and S. Wang, Discovery of 4-((3’R,4’S,5’R)-6′′-Chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2′′-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3′′-indoline]-5’-carboxamido)bicyclo[2.2.2]octane-1-carboxylic acid (AA-115/APG-115): A potent and orally active murine double minute 2 (MDM2) inhibitor in clinical development, J. Med. Chem., 2017, 60, 2819–2839 CrossRef CAS PubMed.
- B. Vu, P. Wovkulich, G. Pizzolato, A. Lovey, Q. Ding, N. Jiang, J. J. Liu, C. Zhao, K. Glenn, Y. Wen, C. Tovar, K. Packman, L. Vassilev and B. Graves, Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development, ACS Med. Chem. Lett., 2013, 4, 466–469 CrossRef CAS PubMed.
- P. Dent, Investigational CHK1 inhibitors in early phase clinical trials for the treatment of cancer, Expert Opin. Invest. Drugs, 2019, 28, 1095–1100 CrossRef CAS PubMed.
- B. Haynes, J. Murai and J. M. Lee, Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition, Cancer Treat. Rev., 2018, 71, 1–7 CrossRef CAS PubMed.
- A. Kumagai and W. G. Dunphy, Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1, Nat. Cell Biol., 2003, 5, 161–165 CrossRef CAS PubMed.
- V. A. J. Smits, E. Cabrera, R. Freire and D. A. Gillespie, Claspin - checkpoint adaptor and DNA replication factor, FEBS J., 2019, 286, 441–455 CrossRef CAS PubMed.
- D. Azenha, M. C. Lopes and T. C. Martins, Claspin: From replication stress and DNA damage responses to cancer therapy, Adv. Protein Chem. Struct. Biol., 2019, 115, 203–246 CAS.
- M. Day, S. Parry-Morris, J. Houghton-Gisby, A. W. Oliver and L. H. Pearl, Structural basis for recruitment of the CHK1 DNA damage kinase by the CLASPIN scaffold protein, Structure, 2021, 29, 531–539 CrossRef CAS PubMed.
- J. K. Schwarz, C. M. Lovly and H. Piwnica-Worms, Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation, Mol. Cancer Res., 2003, 1, 598–609 CAS.
- Z. Cai, N. H. Chehab and N. P. Pavletich, Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase, Mol. Cell, 2009, 35, 818–829 CrossRef CAS PubMed.
- R. Ceccaldi, B. Rondinelli and A. D. D’Andrea, Repair pathway choices and consequences at the double-strand break, Trends Cell Biol., 2016, 26, 52–64 CrossRef CAS PubMed.
- H. H. Y. Chang, N. R. Pannunzio, N. Adachi and M. R. Lieber, Non-homologous DNA end joining and alternative pathways to double-strand break repair, Nat. Rev. Mol. Cell Biol., 2017, 18, 495–506 CrossRef CAS PubMed.
- B. Zhao, E. Rothenberg, D. A. Ramsden and M. R. Lieber, The molecular basis and disease relevance of non-homologous DNA end joining, Nat. Rev. Mol. Cell Biol., 2020, 21, 765–781 CrossRef CAS PubMed.
- W. D. Wright, S. S. Shah and W. D. Heyer, Homologous recombination and the repair of DNA double-strand breaks, J. Biol. Chem., 2018, 293, 10524–10535 CrossRef CAS PubMed.
- D. Setiaputra and D. Durocher, Shieldin – the protector of DNA ends, EMBO Rep., 2019, 20, e47560 CrossRef PubMed.
- Z. Mirman, F. Lottersberger, H. Takai, T. Kibe, Y. Gong, K. Takai, A. Bianchi, M. Zimmermann, D. Durocher and T. de Lange, 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in, Nature, 2018, 560, 112–116 CrossRef CAS PubMed.
- H. Dev, T. W. Chiang, C. Lescale, I. de Krijger, A. G. Martin, D. Pilger, J. Coates, M. Sczaniecka-Clift, W. Wei, M. Ostermaier, M. Herzog, J. Lam, A. Shea, M. Demir, Q. Wu, F. Yang, B. Fu, Z. Lai, G. Balmus, R. Belotserkovskaya, V. Serra, M. J. O'Connor, A. Bruna, P. Beli, L. Pellegrini, C. Caldas, L. Deriano, J. J. L. Jacobs, Y. Galanty and S. P. Jackson, Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells, Nat. Cell Biol., 2018, 20, 954–965 CrossRef CAS PubMed.
- R. Gupta, K. Somyajit, T. Narita, E. Maskey, A. Stanlie, M. Kremer, D. Typas, M. Lammers, N. Mailand, A. Nussenzweig, J. Lukas and C. Choudhary, DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity, Cell, 2018, 173, 972–988.e923 CrossRef CAS PubMed.
- H. Ghezraoui, C. Oliveira, J. R. Becker, K. Bilham, D. Moralli, C. Anzilotti, R. Fischer, M. Deobagkar-Lele, M. Sanchiz-Calvo, E. Fueyo-Marcos, S. Bonham, B. M. Kessler, S. Rottenberg, R. J. Cornall, C. M. Green and J. R. Chapman, 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ, Nature, 2018, 560, 122–127 CrossRef CAS PubMed.
- S. M. Noordermeer, S. Adam, D. Setiaputra, M. Barazas, S. J. Pettitt, A. K. Ling, M. Olivieri, A. Álvarez-Quilón, N. Moatti, M. Zimmermann, S. Annunziato, D. B. Krastev, F. Song, I. Brandsma, J. Frankum, R. Brough, A. Sherker, S. Landry, R. K. Szilard, M. M. Munro, A. McEwan, T. Goullet de Rugy, Z. Y. Lin, T. Hart, J. Moffat, A. C. Gingras, A. Martin, H. van Attikum, J. Jonkers, C. J. Lord, S. Rottenberg and D. Durocher, The shieldin complex mediates 53BP1-dependent DNA repair, Nature, 2018, 560, 117–121 CrossRef CAS PubMed.
- C. Escribano-Díaz, A. Orthwein, A. Fradet-Turcotte, M. Xing, J. T. Young, J. Tkáč, M. A. Cook, A. P. Rosebrock, M. Munro, M. D. Canny, D. Xu and D. Durocher, A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice, Mol. Cell, 2013, 49, 872–883 CrossRef PubMed.
- P. Baumann and S. C. West, Role of the human RAD51 protein in homologous recombination and double-stranded-break repair, Trends Biochem. Sci., 1998, 23, 247–251 CrossRef CAS.
- S. K. Godin, M. R. Sullivan and K. A. Bernstein, Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication, Biochem. Cell Biol., 2016, 94, 407–418 CrossRef CAS PubMed.
- R. D. Wood and S. Doublié, DNA polymerase θ (POLQ), double-strand break repair, and cancer, DNA Repair, 2016, 44, 22–32 CrossRef CAS PubMed.
- R. Ceccaldi, J. C. Liu, R. Amunugama, I. Hajdu, B. Primack, M. I. Petalcorin, K. W. O'Connor, P. A. Konstantinopoulos, S. J. Elledge, S. J. Boulton, T. Yusufzai and A. D. D'Andrea, Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair, Nature, 2015, 518, 258–262 CrossRef CAS PubMed.
- Z. Wang, Y. Song, S. Li, S. Kurian, R. Xiang, T. Chiba and X. Wu, DNA polymerase θ (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse, J. Biol. Chem., 2019, 294, 3909–3919 CrossRef CAS PubMed.
- R. Bhargava, D. O. Onyango and J. M. Stark, Regulation of single-strand annealing and its role in genome maintenance, Trends Genet., 2016, 32, 566–575 CrossRef CAS PubMed.
- I. S. Ayene, L. P. Ford and C. J. Koch, Ku protein targeting by Ku70 small interfering RNA enhances human cancer cell response to topoisomerase II inhibitor and gamma radiation, Mol. Cancer Ther., 2005, 4, 529–536 CrossRef CAS PubMed.
- Y. Nimura, T. Kawata, K. Uzawa, J. Okamura, C. Liu, M. Saito, H. Shimada, N. Seki, A. Nakagawara, H. Ito, T. Ochiai and H. Tanzawa, Silencing Ku80 using small interfering RNA enhanced radiation sensitivity in vitro and in vivo, Int. J. Oncol., 2007, 30, 1477–1484 CAS.
- O. Osipovich, S. K. Durum and K. Muegge, Defining the minimal domain of Ku80 for interaction with Ku70, J. Biol. Chem., 1997, 272, 27259–27265 CrossRef CAS PubMed.
- J. R. Walker, R. A. Corpina and J. Goldberg, Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair, Nature, 2001, 412, 607–614 CrossRef CAS PubMed.
- E. Weterings, A. C. Gallegos, L. N. Dominick, L. S. Cooke, T. N. Bartels, J. Vagner, T. O. Matsunaga and D. Mahadevan, A novel small molecule inhibitor of the DNA repair protein Ku70/80, DNA Repair, 2016, 43, 98–106 CrossRef CAS PubMed.
- T. Ochi, A. N. Blackford, J. Coates, S. Jhujh, S. Mehmood, N. Tamura, J. Travers, Q. Wu, V. M. Draviam, C. V. Robinson, T. L. Blundell and S. P. Jackson, DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair, Science, 2015, 347, 185–188 CrossRef CAS PubMed.
- K. Yano, K. Morotomi-Yano, K. J. Lee and D. J. Chen, Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF, FEBS Lett., 2011, 585, 841–846 CrossRef CAS PubMed.
- C. Nemoz, V. Ropars, P. Frit, A. Gontier, P. Drevet, J. Yu, R. Guerois, A. Pitois, A. Comte, C. Delteil, N. Barboule, P. Legrand, S. Baconnais, Y. Yin, S. Tadi, E. Barbet-Massin, I. Berger, E. Le Cam, M. Modesti, E. Rothenberg, P. Calsou and J. B. Charbonnier, XLF and APLF bind Ku80 at two remote sites to ensure DNA repair by non-homologous end joining, Nat. Struct. Mol. Biol., 2018, 25, 971–980 CrossRef CAS PubMed.
- U. Grawunder, D. Zimmer, P. Kulesza and M. R. Lieber, Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo, J. Biol. Chem., 1998, 273, 24708–24714 CrossRef CAS PubMed.
- P. Y. Wu, P. Frit, S. Meesala, S. Dauvillier, M. Modesti, S. N. Andres, Y. Huang, J. Sekiguchi, P. Calsou, B. Salles and M. S. Junop, Structural and functional interaction between the human DNA repair proteins DNA ligase IV and XRCC4, Mol. Cell. Biol., 2009, 29, 3163–3172 CrossRef CAS PubMed.
- B. L. Sibanda, S. E. Critchlow, J. Begun, X. Y. Pei, S. P. Jackson, T. L. Blundell and L. Pellegrini, Crystal structure of an XRCC4-DNA ligase IV complex, Nat. Struct. Biol., 2001, 8, 1015–1019 CrossRef CAS PubMed.
- S. Roy, S. N. Andres, A. Vergnes, J. A. Neal, Y. Xu, Y. Yu, S. P. Lees-Miller, M. Junop, M. Modesti and K. Meek, XRCC4's interaction with XLF is required for coding (but not signal) end joining, Nucleic Acids Res., 2012, 40, 1684–1694 CrossRef CAS PubMed.
- V. Ropars, P. Drevet, P. Legrand, S. Baconnais, J. Amram, G. Faure, J. A. Márquez, O. Piétrement, R. Guerois, I. Callebaut, E. Le Cam, P. Revy, J. P. de Villartay and J. B. Charbonnier, Structural characterization of filaments formed by human XRCC4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12663–12668 CrossRef CAS PubMed.
- M. Hammel, M. Rey, Y. Yu, R. S. Mani, S. Classen, M. Liu, M. E. Pique, S. Fang, B. L. Mahaney, M. Weinfeld, D. C. Schriemer, S. P. Lees-Miller and J. A. Tainer, XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair, J. Biol. Chem., 2011, 286, 32638–32650 CrossRef CAS PubMed.
- S. Panier and S. J. Boulton, Double-strand break repair: 53BP1 comes into focus, Nat. Rev. Mol. Cell Biol., 2014, 15, 7–18 CrossRef CAS PubMed.
- Z. Mirman and T. de Lange, 53BP1: a DSB escort, Genes Dev., 2020, 34, 7–23 CrossRef CAS PubMed.
- A. Fradet-Turcotte, M. D. Canny, C. Escribano-Diaz, A. Orthwein, C. C. Leung, H. Huang, M. C. Landry, J. Kitevski-LeBlanc, S. M. Noordermeer, F. Sicheri and D. Durocher, 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark, Nature, 2013, 499, 50–54 CrossRef CAS PubMed.
- M. V. Botuyan, J. Lee, I. M. Ward, J. E. Kim, J. R. Thompson, J. Chen and G. Mer, Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair, Cell, 2006, 127, 1361–1373 CrossRef CAS PubMed.
- M. T. Perfetti, B. M. Baughman, B. M. Dickson, Y. Mu, G. Cui, P. Mader, A. Dong, J. L. Norris, S. B. Rothbart, B. D. Strahl, P. J. Brown, W. P. Janzen, C. H. Arrowsmith, G. Mer, K. M. McBride, L. I. James and S. V. Frye, Identification of a fragment-like small molecule ligand for the methyl-lysine binding protein, 53BP1, ACS Chem. Biol., 2015, 10, 1072–1081 CrossRef CAS PubMed.
- J. Tang, N. W. Cho, G. Cui, E. M. Manion, N. M. Shanbhag, M. V. Botuyan, G. Mer and R. A. Greenberg, Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination, Nat. Struct. Mol. Biol., 2013, 20, 317–325 CrossRef CAS PubMed.
- S. Gao, S. Feng, S. Ning, J. Liu, H. Zhao, Y. Xu, J. Shang, K. Li, Q. Li, R. Guo and D. Xu, An OB-fold complex controls the repair pathways for DNA double-strand breaks, Nat. Commun., 2018, 9, 3925 CrossRef PubMed.
- L. Liang, J. Feng, P. Zuo, J. Yang, Y. Lu and Y. Yin, Molecular basis for assembly of the shieldin complex and its implications for NHEJ, Nat. Commun., 2020, 11, 1972 CrossRef CAS PubMed.
- K. Hara, H. Hashimoto, Y. Murakumo, S. Kobayashi, T. Kogame, S. Unzai, S. Akashi, S. Takeda, T. Shimizu and M. Sato, Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase ζ and REV1, J. Biol. Chem., 2010, 285, 12299–12307 CrossRef CAS PubMed.
- J. Tomida, K.-I. Takata, S. S. Lange, A. C. Schibler, M. J. Yousefzadeh, S. Bhetawal, S. Y. R. Dent and R. D. Wood, REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase ζ, Nucleic Acids Res., 2015, 43, 1000–1011 CrossRef CAS PubMed.
- K. Hara, S. Taharazako, M. Ikeda, H. Fujita, Y. Mikami, S. Kikuchi, A. Hishiki, H. Yokoyama, Y. Ishikawa, S. I. Kanno, K. Tanaka and H. Hashimoto, Dynamic feature of mitotic arrest deficient 2-like protein 2 (MAD2L2) and structural basis for its interaction with chromosome alignment-maintaining phosphoprotein (CAMP), J. Biol. Chem., 2017, 292, 17658–17667 CrossRef CAS PubMed.
- X. Wang, N. Pernicone, L. Pertz, D. Hua, T. Zhang, T. Listovsky and W. Xie, REV7 has a dynamic adaptor region to accommodate small GTPase RAN/Shigella IpaB ligands, and its activity is regulated by the RanGTP/GDP switch, J. Biol. Chem., 2019, 294, 15733–15742 CrossRef CAS PubMed.
- M. L. Actis, N. D. Ambaye, B. J. Evison, Y. Shao, M. Vanarotti, A. Inoue, E. T. McDonald, S. Kikuchi, R. Heath, K. Hara, H. Hashimoto and N. Fujii, Identification of the first small-molecule inhibitor of the REV7 DNA repair protein interaction, Bioorg. Med. Chem., 2016, 24, 4339–4346 CrossRef CAS PubMed.
- B. Budke, W. Lv, A. P. Kozikowski and P. P. Connell, Recent developments using small molecules to target RAD51: How to best modulate RAD51 for anticancer therapy?, ChemMedChem, 2016, 11, 2468–2473 CrossRef CAS PubMed.
- N. Berte, A. Piée-Staffa, N. Piecha, M. Wang, K. Borgmann, B. Kaina and T. Nikolova, Targeting homologous recombination by pharmacological inhibitors enhances the killing response of glioblastoma cells treated with alkylating drugs, Mol. Cancer Ther., 2016, 15, 2665–2678 CrossRef CAS PubMed.
- A. Syed and J. A. Tainer, The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair, Annu. Rev. Biochem., 2018, 87, 263–294 CrossRef CAS PubMed.
- L. Bian, Y. Meng, M. Zhang and D. Li, MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment, Mol. Cancer, 2019, 18, 169 CrossRef CAS PubMed.
- A. Dupré, L. Boyer-Chatenet, R. M. Sattler, A. P. Modi, J. H. Lee, M. L. Nicolette, L. Kopelovich, M. Jasin, R. Baer, T. T. Paull and J. Gautier, A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex, Nat. Chem. Biol., 2008, 4, 119–125 CrossRef PubMed.
- A. Shibata, D. Moiani, A. S. Arvai, J. Perry, S. M. Harding, M. M. Genois, R. Maity, S. van Rossum-Fikkert, A. Kertokalio, F. Romoli, A. Ismail, E. Ismalaj, E. Petricci, M. J. Neale, R. G. Bristow, J. Y. Masson, C. Wyman, P. A. Jeggo and J. A. Tainer, DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities, Mol. Cell, 2014, 53, 7–18 CrossRef CAS PubMed.
- A. Rosinha, J. Assis, F. Dias, A. Nogueira, D. Pereira, J. Mauricio, A. L. Teixeira and R. Medeiros, DNA repair system and renal cell carcinoma prognosis: under the influence of NBS1, Med. Oncol., 2015, 32, 255 CrossRef PubMed.
- Y. Zhang, C. U. Lim, E. S. Williams, J. Zhou, Q. Zhang, M. H. Fox, S. M. Bailey and H. L. Liber, NBS1 knockdown by small interfering RNA increases ionizing radiation mutagenesis and telomere association in human cells, Cancer Res., 2005, 65, 5544–5553 CrossRef CAS PubMed.
- R. S. Williams, G. E. Dodson, O. Limbo, Y. Yamada, J. S. Williams, G. Guenther, S. Classen, J. N. Glover, H. Iwasaki, P. Russell and J. A. Tainer, Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair, Cell, 2009, 139, 87–99 CrossRef CAS PubMed.
- C. B. Schiller, K. Lammens, I. Guerini, B. Coordes, H. Feldmann, F. Schlauderer, C. Möckel, A. Schele, K. Strässer, S. P. Jackson and K. P. Hopfner, Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling, Nat. Struct. Mol. Biol., 2012, 19, 693–700 CrossRef CAS PubMed.
- Y. B. Park, J. Chae, Y. C. Kim and Y. Cho, Crystal structure of human Mre11: understanding tumorigenic mutations, Structure, 2011, 19, 1591–1602 CrossRef CAS PubMed.
- K. P. Hopfner, L. Craig, G. Moncalian, R. A. Zinkel, T. Usui, B. A. Owen, A. Karcher, B. Henderson, J. L. Bodmer, C. T. McMurray, J. P. Carney, J. H. Petrini and J. A. Tainer, The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair, Nature, 2002, 418, 562–566 CrossRef CAS PubMed.
- G. J. Williams, R. S. Williams, J. S. Williams, G. Moncalian, A. S. Arvai, O. Limbo, G. Guenther, S. SilDas, M. Hammel, P. Russell and J. A. Tainer, ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair, Nat. Struct. Mol. Biol., 2011, 18, 423–431 CrossRef CAS PubMed.
- B. Budke, H. L. Logan, J. H. Kalin, A. S. Zelivianskaia, W. Cameron McGuire, L. L. Miller, J. M. Stark, A. P. Kozikowski, D. K. Bishop and P. P. Connell, RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells, Nucleic Acids Res., 2012, 40, 7347–7357 CrossRef CAS PubMed.
- B. Budke, J. H. Kalin, M. Pawlowski, A. S. Zelivianskaia, M. Wu, A. P. Kozikowski and P. P. Connell, An optimized RAD51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity, J. Med. Chem., 2013, 56, 254–263 CrossRef CAS PubMed.
- R. B. Jensen, A. Carreira and S. C. Kowalczykowski, Purified human BRCA2 stimulates RAD51-mediated recombination, Nature, 2010, 467, 678–683 CrossRef CAS PubMed.
- A. Carreira and S. C. Kowalczykowski, Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10448–10453 CrossRef PubMed.
- L. Pellegrini, D. S. Yu, T. Lo, S. Anand, M. Lee, T. L. Blundell and A. R. Venkitaraman, Insights into DNA recombination from the structure of a RAD51-BRCA2 complex, Nature, 2002, 420, 287–293 CrossRef CAS PubMed.
- J. Zhu, L. Zhou, G. Wu, H. Konig, X. Lin, G. Li, X. L. Qiu, C. F. Chen, C. M. Hu, E. Goldblatt, R. Bhatia, A. R. Chamberlin, P. L. Chen and W. H. Lee, A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia, EMBO Mol. Med., 2013, 5, 353–365 CrossRef CAS PubMed.
- D. E. Scott, A. G. Coyne, A. Venkitaraman, T. L. Blundell, C. Abell and M. Hyvönen, Small-molecule inhibitors that target protein-protein interactions in the RAD51 family of recombinases, ChemMedChem, 2015, 10, 296–303 CrossRef CAS PubMed.
- F. Falchi, E. Giacomini, T. Masini, N. Boutard, L. Di Ianni, M. Manerba, F. Farabegoli, L. Rossini, J. Robertson, S. Minucci, I. Pallavicini, G. Di Stefano, M. Roberti, R. Pellicciari and A. Cavalli, Synthetic lethality triggered by combining olaparib with BRCA2-Rad51 disruptors, ACS Chem. Biol., 2017, 12, 2491–2497 CrossRef CAS PubMed.
- M. Roberti, F. Schipani, G. Bagnolini, D. Milano, E. Giacomini, F. Falchi, A. Balboni, M. Manerba, F. Farabegoli, F. De Franco, J. Robertson, S. Minucci, I. Pallavicini, G. Di Stefano, S. Girotto, R. Pellicciari and A. Cavalli, Rad51/BRCA2 disruptors inhibit homologous recombination and synergize with olaparib in pancreatic cancer cells, Eur. J. Med. Chem., 2019, 165, 80–92 CrossRef CAS PubMed.
- G. Bagnolini, D. Milano, M. Manerba, F. Schipani, J. A. Ortega, D. Gioia, F. Falchi, A. Balboni, F. Farabegoli, F. De Franco, J. Robertson, R. Pellicciari, I. Pallavicini, S. Peri, S. Minucci, S. Girotto, G. Di Stefano, M. Roberti and A. Cavalli, Synthetic lethality in pancreatic cancer: Discovery of a new RAD51-BRCA2 small molecule disruptor that inhibits homologous recombination and synergizes with olaparib, J. Med. Chem., 2020, 63, 2588–2619 CrossRef CAS PubMed.
- Z. Feng, S. P. Scott, W. Bussen, G. G. Sharma, G. Guo, T. K. Pandita and S. N. Powell, Rad52 inactivation is synthetically lethal with BRCA2 deficiency, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 686–691 CrossRef CAS PubMed.
- K. Sullivan-Reed, E. Bolton-Gillespie, Y. Dasgupta, S. Langer, M. Siciliano, M. Nieborowska-Skorska, K. Hanamshet, E. A. Belyaeva, A. J. Bernhardy, J. Lee, M. Moore, H. Zhao, P. Valent, K. Matlawska-Wasowska, M. Muschen, S. Bhatia, R. Bhatia, N. Johnson, M. A. Wasik, A. V. Mazin and T. Skorski, Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells, Cell Rep., 2018, 23, 3127–3136 CrossRef CAS PubMed.
- F. Huang, N. Goyal, K. Sullivan, K. Hanamshet, M. Patel, O. M. Mazina, C. X. Wang, W. F. An, J. Spoonamore, S. Metkar, K. A. Emmitte, S. Cocklin, T. Skorski and A. V. Mazin, Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors, Nucleic Acids Res., 2016, 44, 4189–4199 CrossRef CAS PubMed.
- W. L. de Laat, N. G. Jaspers and J. H. Hoeijmakers, Molecular mechanism of nucleotide excision repair, Genes Dev., 1999, 13, 768–785 CrossRef CAS PubMed.
- J. A. Marteijn, H. Lans, W. Vermeulen and J. H. Hoeijmakers, Understanding nucleotide excision repair and its roles in cancer and ageing, Nat. Rev. Mol. Cell Biol., 2014, 15, 465–481 CrossRef CAS PubMed.
- G. Spivak, Nucleotide excision repair in humans, DNA Repair, 2015, 36, 13–18 CrossRef CAS PubMed.
- H. E. Krokan and M. Bjørås, Base excision repair, Cold Spring Harbor Perspect. Biol., 2013, 5, a012583 Search PubMed.
- A. B. Robertson, A. Klungland, T. Rognes and I. Leiros, Base excision repair: the long and short of it, Cell. Mol. Life Sci., 2009, 66, 981–993 CrossRef CAS PubMed.
- N. A. Moor and O. I. Lavrik, Protein-protein interactions in DNA base excision repair, Biochemistry, 2018, 83, 411–422 CAS.
- P. Modrich and R. Lahue, Mismatch repair in replication fidelity, genetic recombination, and cancer biology, Annu. Rev. Biochem., 1996, 65, 101–133 CrossRef CAS PubMed.
- T. A. Kunkel and D. A. Erie, DNA mismatch repair, Annu. Rev. Biochem., 2005, 74, 681–710 CrossRef CAS PubMed.
- J. Jiricny, The multifaceted mismatch-repair system, Nat. Rev. Mol. Cell Biol., 2006, 7, 335–346 CrossRef CAS PubMed.
- G. L. Moldovan and A. D. D’Andrea, How the Fanconi anemia pathway guards the genome, Annu. Rev. Genet., 2009, 43, 223–249 CrossRef CAS PubMed.
- J. Niraj, A. Farkkila and A. D. D’Andrea, The Fanconi anemia pathway in cancer, Annu. Rev. Cancer Biol., 2019, 3, 457–478 CrossRef PubMed.
- D. Cunningham, W. Atkin, H. J. Lenz, H. T. Lynch, B. Minsky, B. Nordlinger and N. Starling, Colorectal cancer, Lancet, 2010, 375, 1030–1047 CrossRef.
- N. Sugitani, R. M. Sivley, K. E. Perry, J. A. Capra and W. J. Chazin, XPA: A key scaffold for human nucleotide excision repair, DNA Repair, 2016, 44, 123–135 CrossRef CAS PubMed.
- A. R. Lehmann, DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy, Biochimie, 2003, 85, 1101–1111 CrossRef CAS PubMed.
- D. L. Croteau, Y. Peng and B. Van Houten, DNA repair gets physical: mapping an XPA-binding site on ERCC1, DNA Repair, 2008, 7, 819–826 CrossRef CAS PubMed.
- L. Li, C. A. Peterson, X. Lu and R. J. Legerski, Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair, Mol. Cell. Biol., 1995, 15, 1993–1998 CrossRef CAS PubMed.
- C. Welsh, R. Day, C. McGurk, J. R. Masters, R. D. Wood and B. Köberle, Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines, Int. J. Cancer, 2004, 110, 352–361 CrossRef CAS PubMed.
- O. V. Tsodikov, D. Ivanov, B. Orelli, L. Staresincic, I. Shoshani, R. Oberman, O. D. Schärer, G. Wagner and T. Ellenberger, Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA, EMBO J., 2007, 26, 4768–4776 CrossRef CAS PubMed.
- K. H. Barakat, L. P. Jordheim, R. Perez-Pineiro, D. Wishart, C. Dumontet and J. A. Tuszynski, Virtual screening and biological evaluation of inhibitors targeting the XPA-ERCC1 interaction, PLoS One, 2012, 7, e51329 CrossRef CAS PubMed.
- M. Faridounnia, G. E. Folkers and R. Boelens, Function and interactions of ERCC1-XPF in DNA damage response, Molecules, 2018, 23, 3205 CrossRef PubMed.
- E. M. McNeil, K. R. Astell, A. M. Ritchie, S. Shave, D. R. Houston, P. Bakrania, H. M. Jones, P. Khurana, C. Wallace, T. Chapman, M. A. Wear, M. D. Walkinshaw, B. Saxty and D. W. Melton, Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance, DNA Repair, 2015, 31, 19–28 CrossRef CAS PubMed.
- L. P. Jordheim, K. H. Barakat, L. Heinrich-Balard, E. L. Matera, E. Cros-Perrial, K. Bouledrak, R. El Sabeh, R. Perez-Pineiro, D. S. Wishart, R. Cohen, J. Tuszynski and C. Dumontet, Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells, Mol. Pharmacol., 2013, 84, 12–24 CrossRef CAS PubMed.
- F. Gentile, A. H. Elmenoufy, G. Ciniero, D. Jay, F. Karimi-Busheri, K. H. Barakat, M. Weinfeld, F. G. West and J. A. Tuszynski, Computer-aided drug design of small molecule inhibitors of the ERCC1-XPF protein-protein interaction, Chem. Biol. Drug Des., 2020, 95, 460–471 CrossRef CAS PubMed.
- A. H. Elmenoufy, F. Gentile, D. Jay, F. Karimi-Busheri, X. Yang, O. M. Soueidan, C. Weilbeer, R. S. Mani, K. H. Barakat, J. A. Tuszynski, M. Weinfeld and F. G. West, Targeting DNA repair in tumor cells via inhibition of ERCC1-XPF, J. Med. Chem., 2019, 62, 7684–7696 CrossRef CAS PubMed.
- T. M. Chapman, K. J. Gillen, C. Wallace, M. T. Lee, P. Bakrania, P. Khurana, P. J. Coombs, L. Stennett, S. Fox, E. A. Bureau, J. Brownlees, D. W. Melton and B. Saxty, Catechols and 3-hydroxypyridones as inhibitors of the DNA repair complex ERCC1-XPF, Bioorg. Med. Chem. Lett., 2015, 25, 4097–4103 CrossRef CAS PubMed.
- O. V. Tsodikov, J. H. Enzlin, O. D. Schärer and T. Ellenberger, Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11236–11241 CrossRef CAS PubMed.
- K. Tripsianes, G. Folkers, E. Ab, D. Das, H. Odijk, N. G. Jaspers, J. H. Hoeijmakers, R. Kaptein and R. Boelens, The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair, Structure, 2005, 13, 1849–1858 CrossRef CAS PubMed.
- M. Jones, F. Beuron, A. Borg, A. Nans, C. P. Earl, D. C. Briggs, A. P. Snijders, M. Bowles, E. P. Morris, M. Linch and N. Q. McDonald, Cryo-EM structures of the XPF-ERCC1 endonuclease reveal how DNA-junction engagement disrupts an auto-inhibited conformation, Nat. Commun., 2020, 11, 1120 CrossRef CAS PubMed.
- E. Compe and J. M. Egly, TFIIH: when transcription met DNA repair, Nat. Rev. Mol. Cell Biol., 2012, 13, 343–354 CrossRef CAS PubMed.
- B. J. Greber, T. H. D. Nguyen, J. Fang, P. V. Afonine, P. D. Adams and E. Nogales, The cryo-electron microscopy structure of human transcription factor IIH, Nature, 2017, 549, 414–417 CrossRef CAS PubMed.
- V. Gervais, V. Lamour, A. Jawhari, F. Frindel, E. Wasielewski, S. Dubaele, J. M. Egly, J. C. Thierry, B. Kieffer and A. Poterszman, TFIIH contains a PH domain involved in DNA nucleotide excision repair, Nat. Struct. Mol. Biol., 2004, 11, 616–622 CrossRef CAS PubMed.
- J. Lafrance-Vanasse, G. Arseneault, L. Cappadocia, H. T. Chen, P. Legault and J. G. Omichinski, Structural and functional characterization of interactions involving the Tfb1 subunit of TFIIH and the NER factor Rad2, Nucleic Acids Res., 2012, 40, 5739–5750 CrossRef CAS PubMed.
- R. González-Corrochano, F. M. Ruiz, N. M. I. Taylor, S. Huecas, S. Drakulic, M. Spínola-Amilibia and C. Fernández-Tornero, The crystal structure of human XPG, the xeroderma pigmentosum group G endonuclease, provides insight into nucleotide excision DNA repair, Nucleic Acids Res., 2020, 48, 9943–9958 CrossRef PubMed.
- M. Okuda, T. Ekimoto, J. I. Kurita, M. Ikeguchi and Y. Nishimura, Structural and dynamical insights into the PH domain of p62 in human TFIIH, Nucleic Acids Res., 2021, 49, 2916–2930 CrossRef PubMed.
- M. Dizdaroglu, Oxidatively induced DNA damage and its repair in cancer, Mutat. Res., Rev. Mutat. Res., 2015, 763, 212–245 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Oxidative stress, inflammation, and cancer: how are they linked?, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- R. Ali, M. Alabdullah, A. Alblihy, I. Miligy, K. A. Mesquita, S. Y. Chan, P. Moseley, E. A. Rakha and S. Madhusudan, PARP1 blockade is synthetically lethal in XRCC1 deficient sporadic epithelial ovarian cancers, Cancer Lett., 2020, 469, 124–133 CrossRef CAS PubMed.
- F. Ni, H. Tang, C. Wang, Z. Wang, F. Yu, B. Chen and L. Sun, Berzosertib (VE-822) inhibits gastric cancer cell proliferation via base excision repair system, Cancer Manage. Res., 2019, 11, 8391–8405 CrossRef CAS PubMed.
- R. E. London, The structural basis of XRCC1-mediated DNA repair, DNA Repair, 2015, 30, 90–103 CrossRef CAS PubMed.
- H. K. Wong and D. M. Wilson 3rd, XRCC1 and DNA polymerase beta interaction contributes to cellular alkylating-agent resistance and single-strand break repair, J. Cell. Biochem., 2005, 95, 794–804 CrossRef CAS PubMed.
- D. Arian, M. Hedayati, H. Zhou, Z. Bilis, K. Chen, T. L. DeWeese and M. M. Greenberg, Irreversible inhibition of DNA polymerase β by small-molecule mimics of a DNA lesion, J. Am. Chem. Soc., 2014, 136, 3176–3183 CrossRef CAS PubMed.
- A. S. Jaiswal, H. Panda, B. K. Law, J. Sharma, J. Jani, R. Hromas and S. Narayan, NSC666715 and its analogs inhibit strand-displacement activity of DNA polymerase β and potentiate temozolomide-induced DNA damage, senescence and apoptosis in colorectal cancer cells, PLoS One, 2015, 10, e0123808 CrossRef PubMed.
- M. J. Cuneo and R. E. London, Oxidation state of the XRCC1 N-terminal domain regulates DNA polymerase beta binding affinity, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6805–6810 CrossRef CAS PubMed.
- A. Marintchev, M. A. Mullen, M. W. Maciejewski, B. Pan, M. R. Gryk and G. P. Mullen, Solution structure of the single-strand break repair protein XRCC1 N-terminal domain, Nat. Struct. Biol., 1999, 6, 884–893 CrossRef CAS PubMed.
- A. Marintchev, M. R. Gryk and G. P. Mullen, Site-directed mutagenesis analysis of the structural interaction of the single-strand-break repair protein, X-ray cross-complementing group 1, with DNA polymerase beta, Nucleic Acids Res., 2003, 31, 580–588 CrossRef PubMed.
- M. J. Cuneo, S. A. Gabel, J. M. Krahn, M. A. Ricker and R. E. London, The structural basis for partitioning of the XRCC1/DNA ligase III-α BRCT-mediated dimer complexes, Nucleic Acids Res., 2011, 39, 7816–7827 CrossRef CAS PubMed.
- M. Hammel, I. Rashid, A. Sverzhinsky, Y. Pourfarjam, M. S. Tsai, T. Ellenberger, J. M. Pascal, I. K. Kim, J. A. Tainer and A. E. Tomkinson, An atypical BRCT-BRCT interaction with the XRCC1 scaffold protein compacts human DNA Ligase IIIalpha within a flexible DNA repair complex, Nucleic Acids Res., 2021, 49, 306–321 CrossRef PubMed.
- R. Abbotts and S. Madhusudan, Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer, Cancer Treat. Rev., 2010, 36, 425–435 CrossRef CAS PubMed.
- A. E. Vidal, S. Boiteux, I. D. Hickson and J. P. Radicella, XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions, EMBO J., 2001, 20, 6530–6539 CrossRef CAS PubMed.
- S. Marsin, A. E. Vidal, M. Sossou, J. Menissier-de Murcia, F. Le Page, S. Boiteux, G. de Murcia and J. P. Radicella, Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1, J. Biol. Chem., 2003, 278, 44068–44074 CrossRef CAS PubMed.
- C. D. Mol, T. Izumi, S. Mitra and J. A. Tainer, DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected], Nature, 2000, 403, 451–456 CrossRef CAS PubMed.
- X. Lin, H. K. Kim and S. B. Howell, The role of DNA mismatch repair in cisplatin mutagenicity, J. Inorg. Biochem., 1999, 77, 89–93 CrossRef CAS PubMed.
- S. A. Martin, C. J. Lord and A. Ashworth, Therapeutic targeting of the DNA mismatch repair pathway, Clin. Cancer Res., 2010, 16, 5107–5113 CrossRef CAS PubMed.
- S. Jacob, M. Aguado, D. Fallik and F. Praz, The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells, Cancer Res., 2001, 61, 6555–6562 CAS.
- A. Fedier, V. A. Schwarz, H. Walt, R. D. Carpini, U. Haller and D. Fink, Resistance to topoisomerase poisons due to loss of DNA mismatch repair, Int. J. Cancer, 2001, 93, 571–576 CrossRef PubMed.
- A. Sawant, A. Kothandapani, A. Zhitkovich, R. W. Sobol and S. M. Patrick, Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links, DNA Repair, 2015, 35, 126–136 CrossRef CAS PubMed.
- J. Stritzelberger, L. Distel, R. Buslei, R. Fietkau and F. Putz, Acquired temozolomide resistance in human glioblastoma cell line U251 is caused by mismatch repair deficiency and can be overcome by lomustine, Clin. Transl. Oncol., 2018, 20, 508–516 CrossRef CAS PubMed.
- D. Ciardiello, P. P. Vitiello, C. Cardone, G. Martini, T. Troiani, E. Martinelli and F. Ciardiello, Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy, Cancer Treat. Rev., 2019, 76, 22–32 CrossRef CAS PubMed.
- C. Hoege, B. Pfander, G.-L. Moldovan, G. Pyrowolakis and S. Jentsch, RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO, Nature, 2002, 419, 135–141 CrossRef CAS PubMed.
- E. C. Friedberg, A. R. Lehmann and R. P. Fuchs, Trading places: how do DNA polymerases switch during translesion DNA synthesis?, Mol. Cell, 2005, 18, 499–505 CrossRef CAS PubMed.
- Y. Daigaku, A. A. Davies and H. D. Ulrich, Ubiquitin-dependent DNA damage bypass is separable from genome replication, Nature, 2010, 465, 951–955 CrossRef CAS PubMed.
- G. I. Karras and S. Jentsch, The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase, Cell, 2010, 141, 255–267 CrossRef CAS PubMed.
- N. Diamant, A. Hendel, I. Vered, T. Carell, T. Reißner, N. de Wind, N. Geacinov and Z. Livneh, DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity, Nucleic Acids Res., 2012, 40, 170–180 CrossRef CAS PubMed.
- R. E. Johnson, S. Prakash and L. Prakash, Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta, Science, 1999, 283, 1001–1004 CrossRef CAS PubMed.
- C. Masutani, M. Araki, A. Yamada, R. Kusumoto, T. Nogimori, T. Maekawa, S. Iwai and F. Hanaoka, Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity, EMBO J., 1999, 18, 3491–3501 CrossRef CAS PubMed.
- S. Shachar, O. Ziv, S. Avkin, S. Adar, J. Wittschieben, T. Reißner, S. Chaney, E. C. Friedberg, Z. Wang and T. Carell, Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals, EMBO J., 2009, 28, 383–393 CrossRef CAS PubMed.
- Z. Livneh and S. Shachar, Multiple two-polymerase mechanisms in mammalian translesion DNA synthesis, Cell Cycle, 2010, 9, 729–735 CrossRef CAS PubMed.
- G. L. Moldovan, B. Pfander and S. Jentsch, PCNA, the maestro of the replication fork, Cell, 2007, 129, 665–679 CrossRef CAS PubMed.
- C. Guo, E. Sonoda, T.-S. Tang, J. L. Parker, A. B. Bielen, S. Takeda, H. D. Ulrich and E. C. Friedberg, REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo, Mol. Cell, 2006, 23, 265–271 CrossRef CAS PubMed.
- Y. Pustovalova, M. W. Maciejewski and D. M. Korzhnev, NMR mapping of PCNA interaction with translesion synthesis DNA polymerase Rev1 mediated by Rev1-BRCT domain, J. Mol. Biol., 2013, 425, 3091–3105 CrossRef CAS PubMed.
- M. Bienko, C. M. Green, N. Crosetto, F. Rudolf, G. Zapart, B. Coull, P. Kannouche, G. Wider, M. Peter, A. R. Lehmann, K. Hofmann and I. Dikic, Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis, Science, 2005, 310, 1821–1824 CrossRef CAS PubMed.
- P. L. Kannouche, J. Wing and A. R. Lehmann, Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage, Mol. Cell, 2004, 14, 491–500 CrossRef CAS PubMed.
- A. G. Baranovskiy, A. G. Lada, H. M. Siebler, Y. Zhang, Y. I. Pavlov and T. H. Tahirov, DNA polymerase δ and ζ switch by sharing accessory subunits of DNA polymerase δ, J. Biol. Chem., 2012, 287, 17281–17287 CrossRef CAS PubMed.
- R. E. Johnson, L. Prakash and S. Prakash, Pol31 and Pol32 subunits of yeast DNA polymerase δ are also essential subunits of DNA polymerase ζ, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12455–12460 CrossRef CAS PubMed.
- E. Ohashi, T. Hanafusa, K. Kamei, I. Song, J. Tomida, H. Hashimoto, C. Vaziri and H. Ohmori, Identification of a novel REV1-interacting motif necessary for DNA polymerase κ function, Genes Cells, 2009, 14, 101–111 CrossRef CAS PubMed.
- A. Pozhidaeva, Y. Pustovalova, S. D'Souza, I. Bezsonova, G. C. Walker and D. M. Korzhnev, NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase η, Biochemistry, 2012, 51, 5506–5520 CrossRef CAS PubMed.
- Y. Pustovalova, M. T. Magalhães, S. D’Souza, A. A. Rizzo, G. Korza, G. C. Walker and D. M. Korzhnev, Interaction between the Rev1 C-Terminal domain and the PolD3 subunit of Polζ suggests a mechanism of polymerase exchange upon Rev1/Polζ-dependent translesion synthesis, Biochemistry, 2016, 55, 2043–2053 CrossRef CAS PubMed.
- J. Wojtaszek, J. Liu, S. D'Souza, S. Wang, Y. Xue, G. C. Walker and P. Zhou, Multifaceted recognition of vertebrate Rev1 by translesion polymerases ζ and κ, J. Biol. Chem., 2012, 287, 26400–26408 CrossRef CAS PubMed.
- J. Wojtaszek, C.-J. Lee, S. D'Souza, B. Minesinger, H. Kim, A. D. D'Andrea, G. C. Walker and P. Zhou, Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) ζ, and Pol κ, J. Biol. Chem., 2012, 287, 33836–33846 CrossRef CAS PubMed.
- Y. Pustovalova, I. Bezsonova and D. M. Korzhnev, The C-terminal domain of human Rev1 contains independent binding sites for DNA polymerase η and Rev7 subunit of polymerase ζ, FEBS Lett., 2012, 586, 3051–3056 CrossRef CAS PubMed.
- S. Kikuchi, K. Hara, T. Shimizu, M. Sato and H. Hashimoto, Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins, J. Biol. Chem., 2012, 287, 33847–33852 CrossRef CAS PubMed.
- D. Dorjsuren, D. M. Wilson 3rd, W. A. Beard, J. P. McDonald, C. P. Austin, R. Woodgate, S. H. Wilson and A. Simeonov, A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases, Nucleic Acids Res., 2009, 37, e128 CrossRef PubMed.
- K. Yamanaka, D. Dorjsuren, R. L. Eoff, M. Egli, D. J. Maloney, A. Jadhav, A. Simeonov and R. S. Lloyd, A comprehensive strategy to discover inhibitors of the translesion synthesis DNA polymerase kappa, PLoS One, 2012, 7, e45032 CrossRef CAS PubMed.
- A. Ketkar, M. K. Zafar, L. Maddukuri, K. Yamanaka, S. Banerjee, M. Egli, J. Y. Choi, R. S. Lloyd and R. L. Eoff, Leukotriene biosynthesis inhibitor MK886 impedes DNA polymerase activity, Chem. Res. Toxicol., 2013, 26, 221–232 Search PubMed.
- J. M. Gulbis, Z. Kelman, J. Hurwitz, M. O'Donnell and J. Kuriyan, Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA, Cell, 1996, 87, 297–306 CrossRef CAS.
- C. Punchihewa, A. Inoue, A. Hishiki, Y. Fujikawa, M. Connelly, B. Evison, Y. Shao, R. Heath, I. Kuraoka, P. Rodrigues, H. Hashimoto, M. Kawanishi, M. Sato, T. Yagi and N. Fujii, Identification of small molecule proliferating cell nuclear antigen (PCNA) inhibitor that disrupts interactions with PIP-box proteins and inhibits DNA replication, J. Biol. Chem., 2012, 287, 14289–14300 CrossRef CAS PubMed.
- A. Inoue, S. Kikuchi, A. Hishiki, Y. Shao, R. Heath, B. J. Evison, M. Actis, C. E. Canman, H. Hashimoto and N. Fujii, A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin, J. Biol. Chem., 2014, 289, 7109–7120 CrossRef CAS PubMed.
- A. Yasgar, A. Jadhav, A. Simeonov and N. P. Coussens, AlphaScreen-Based assays: ultra-high-throughput screening for small-molecule inhibitors of challenging enzymes and protein-protein interactions, Methods Mol. Biol., 2016, 1439, 77–98 CrossRef CAS PubMed.
- M. Vanarotti, B. J. Evison, M. L. Actis, A. Inoue, E. T. McDonald, Y. Shao, R. J. Heath and N. Fujii, Small-molecules that bind to the ubiquitin-binding motif of REV1 inhibit REV1 interaction with K164-monoubiquitinated PCNA and suppress DNA damage tolerance, Bioorg. Med. Chem., 2018, 26, 2345–2353 CrossRef CAS PubMed.
- M. Vanarotti, C. R. Grace, D. J. Miller, M. L. Actis, A. Inoue, B. J. Evison, S. Vaithiyalingam, A. P. Singh, E. T. McDonald and N. Fujii, Structures of REV1 UBM2 Domain Complex with Ubiquitin and with a Small-Molecule that Inhibits the REV1 UBM2-Ubiquitin Interaction, J. Mol. Biol., 2018, 430, 2857–2872 CrossRef CAS PubMed.
- A. A. Rizzo, F. M. Vassel, N. Chatterjee, S. D'Souza, Y. Li, B. Hao, M. T. Hemann, G. C. Walker and D. M. Korzhnev, Rev7 dimerization is important for assembly and function of the Rev1/Polζ translesion synthesis complex, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8191–E8200 CrossRef CAS PubMed.
- S. A. Gabel, E. F. DeRose and R. E. London, XRCC1 interaction with the REV1 C-terminal domain suggests a role in post replication repair, DNA Repair, 2013, 12, 1105–1113 CrossRef CAS PubMed.
- V. Sail, A. A. Rizzo, N. Chatterjee, R. C. Dash, Z. Ozen, G. C. Walker, D. M. Korzhnev and M. K. Hadden, Identification of small molecule translesion synthesis inhibitors that target the Rev1-CT/RIR protein-protein interaction, ACS Chem. Biol., 2017, 12, 1903–1912 CrossRef CAS PubMed.
- Z. Ozen, R. C. Dash, K. R. McCarthy, S. A. Chow, A. A. Rizzo, D. M. Korzhnev and M. K. Hadden, Small molecule scaffolds that disrupt the Rev1-CT/RIR protein-protein interaction, Bioorg. Med. Chem., 2018, 26, 4301–4309 CrossRef CAS PubMed.
- R. C. Dash, Z. Ozen, K. R. McCarthy, N. Chatterjee, C. A. Harris, A. A. Rizzo, G. C. Walker, D. M. Korzhnev and M. K. Hadden, Virtual pharmacophore screening identifies small-molecule inhibitors of the Rev1-CT/RIR protein-protein interaction, ChemMedChem, 2019, 14, 1610–1617 CrossRef CAS PubMed.
- R. C. Dash, Z. Ozen, A. A. Rizzo, S. Lim, D. M. Korzhnev and M. K. Hadden, Structural approach to identify a lead scaffold that targets the translesion synthesis polymerase Rev1, J. Chem. Inf. Model., 2018, 58, 2266–2277 CrossRef CAS PubMed.
- K. S. McPherson, A. M. Zaino, R. C. Dash, A. A. Rizzo, Y. Li, B. Hao, I. Bezsonova, M. K. Hadden and D. M. Korzhnev, Structure-based drug design of Pphenazopyridine derivatives as inhibitors of Rev1 interactions in translesion synthesis, ChemMedChem, 2021, 16, 1126–1132 CrossRef CAS PubMed.
- J. L. Wojtaszek, N. Chatterjee, J. Najeeb, A. Ramos, M. Lee, K. Bian, J. Y. Xue, B. A. Fenton, H. Park, D. Li, M. T. Hemann, J. Hong, G. C. Walker and P. Zhou, A small molecule targeting mutagenic translesion synthesis improves chemotherapy, Cell, 2019, 178, 152–159.e111 CrossRef CAS PubMed.
- N. Chatterjee, M. A. Whitman, C. A. Harris, S. M. Min, O. Jonas, E. C. Lien, A. Luengo, M. G. Vander Heiden, J. Hong, P. Zhou, M. T. Hemann and G. C. Walker, REV1 inhibitor JH-RE-06 enhances tumor cell response to chemotherapy by triggering senescence hallmarks, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 28918–28921 CrossRef CAS PubMed.
- A. Hendel, P. H. Krijger, N. Diamant, Z. Goren, P. Langerak, J. Kim, T. Reissner, K. Y. Lee, N. E. Geacintov, T. Carell, K. Myung, S. Tateishi, A. D'Andrea, H. Jacobs and Z. Livneh, PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells, PLoS Genet., 2011, 7, e1002262 CrossRef CAS PubMed.
- B. Haynes, A. Gajan, P. Nangia-Makker and M. P. Shekhar, RAD6B is a major mediator of triple negative breast cancer cisplatin resistance: Regulation of translesion synthesis/Fanconi anemia crosstalk and BRCA1 independence, Biochim. Biophys. Acta, Mol. Basis Dis., 2020, 1866, 165561 CrossRef CAS PubMed.
- M. A. Sanders, G. Brahemi, P. Nangia-Makker, V. Balan, M. Morelli, H. Kothayer, A. D. Westwell and M. P. V. Shekhar, Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification, and functional characterization, Mol. Cancer Ther., 2013, 12, 373–383 CrossRef CAS PubMed.
- M. A. Sanders, B. Haynes, P. Nangia-Makker, L. A. Polin and M. P. Shekhar, Pharmacological targeting of RAD6 enzyme-mediated translesion synthesis overcomes resistance to platinum-based drugs, J. Biol. Chem., 2017, 292, 10347–10363 CrossRef CAS PubMed.
- R. G. Hibbert, A. Huang, R. Boelens and T. K. Sixma, E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5590–5595 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |