
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein degradation profile reveals dynamic nature of 20S proteasome small molecule stimulation†
Rachel A.
Coleman
a,
Rodrigo
Mohallem
 b,
Uma K.
Aryal
b and
Darci J.
Trader
*a
b,
Uma K.
Aryal
b and
Darci J.
Trader
*a
aDepartment of Medicinal Chemistry and Molecular Pharmacology, Purdue University, 575 West Stadium Avenue, West Lafayette, Indiana 47907, USA. E-mail: dtrader@purdue.edu
bPurdue Proteomics Facility, Bindley Bioscience Center and Department of Comparative Pathobiology, Purdue University, West Lafayette, Indiana 47907, USA
First published on 5th January 2021
Abstract
Small molecules have been discovered to stimulate the 20S core particle (CP) of the proteasome to degrade proteins. However, the impact a 20S CP stimulator can have on the regulation of protein levels has not been fully characterized. Previous studies have focused on using one kind of stimulator to enhance the degradation of specific 20S CP substrates. We present here a study that utilizes several 20S CP stimulators to determine how each can affect the degradation of proteins in a biochemical assay with purified proteins and of an overexpressed GFP-fusion protein in cells. We also evaluate the effects of two stimulators on the whole cellular proteome in HEK-293T cells using label-free quantitative proteomic analysis for a broader understanding on their impact. Our studies demonstrate that 20S CP stimulation is likely to promote the degradation of significantly disordered proteins; however, the specific effect on the regulation of protein levels appears to be dependent on the mechanism of action of each stimulator due to the dynamic nature of the 20S CP. Our results reveal the potential of tailoring small molecule stimulators to influence the degradation of certain protein types and 20S CP substrates.
Introduction
The most common cellular pathway regulating the proteolysis of misfolded and damaged proteins is the ubiquitin-dependent proteasome system (UPS).1,2 For this degradation pathway, ubiquitin-tagged proteins are recognized and degraded by the 26S proteasome, comprised of the 19S regulatory particle (19S RP) and 20S core particle (20S CP) (Fig. 1A). The 20S CP alone can accept and degrade proteins in a ubiquitin-independent system (UIPS).3,4 In this case, proteins are not ubiquitinated and must be disordered enough to enter the catalytic core without being denatured by the 19S RP (Fig. 1A). The UIPS has been shown to play an important role in the degradation of oxidatively damaged proteins during times of cellular stress.5–8 It has also been hypothesized that the 20S CP alone is responsible for degradation “by default” in which proteins that contain a high degree of intrinsic disorder are degraded.9 A number of endogenous substrates of the 20S CP have been identified, all of which are considered intrinsically disordered proteins or contain intrinsically disordered regions.10–14 These proteins often function as part of a complex with other proteins to prevent their “default” degradation by the 20S CP. The stimulation and enhancement of proteasome activity has recently emerged as an exciting new field of study, and this pathway has been suggested as a possible therapeutic avenue for disease states marked by accumulation of damaged or disordered proteins, such as aging, Parkinson's, and Alzheimer's disease.15–24 Specifically, stimulation of the 20S CP has recently been shown to be promoted by small molecules, leading to a more rapid degradation of disordered proteins.16–19 While these studies have focused on particular proteins that are known 20S CP substrates, it is currently unclear how many, or what types of proteins may be affected by enhancing this degradation process.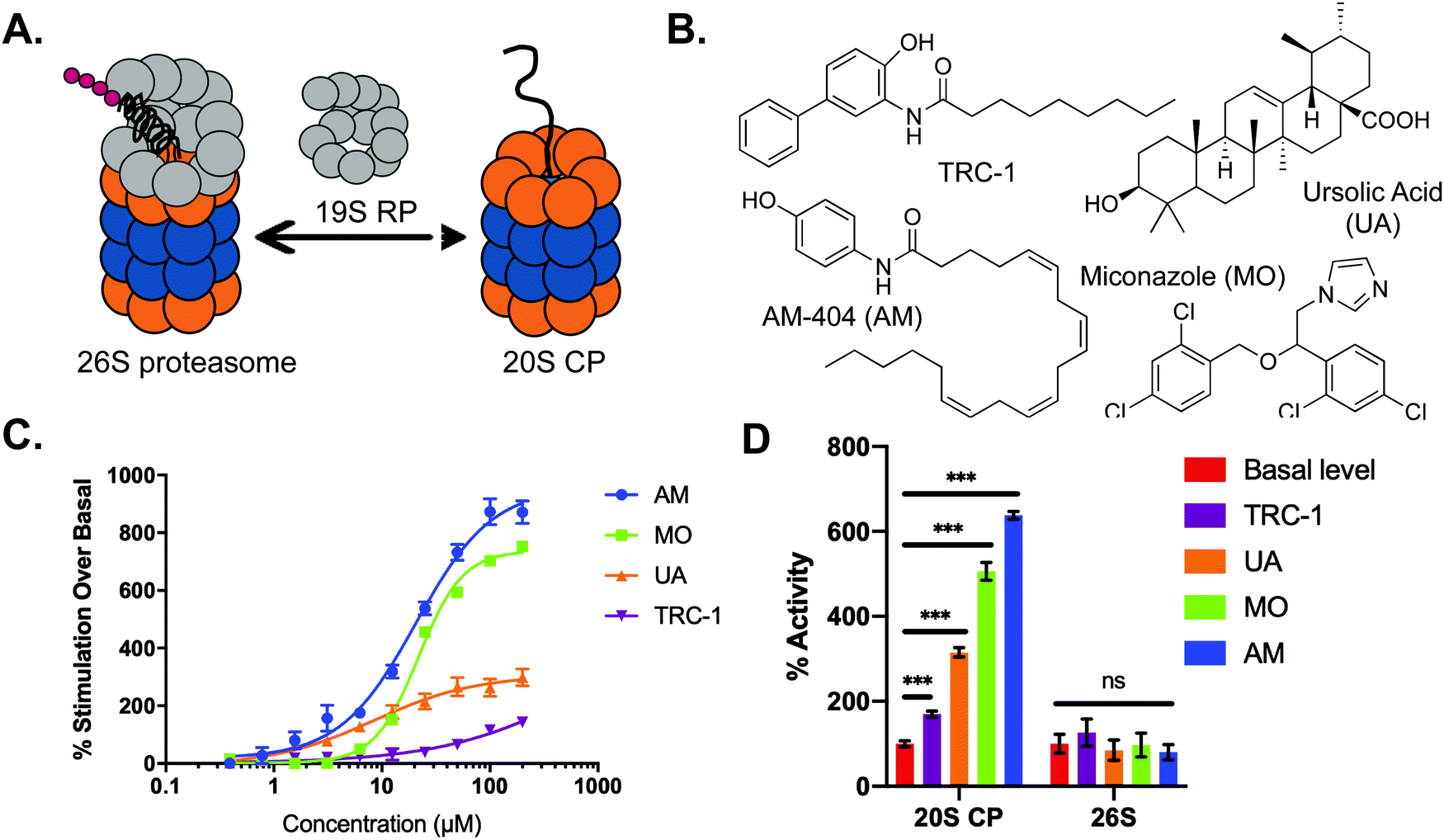 | ||
| Fig. 1 (A) 26S and 20S CP isoforms of the proteasome. (B) Structures of the small molecule stimulators of the 20S CP utilized for this study. (C) The effect of each of the stimulators in (B) was analyzed using our FRET assay to determine a dose–response impact on 20S CP activity. (D) The FRET assay was also used to examine how each stimulator impacts the activity of the 26S proteasome (A). ***p < 0.001. | ||
To answer this question, we chose to evaluate several small molecule stimulators that have been shown to affect 20S CP activity to various degrees in our FRET assay.25 Using these stimulators, we analyzed their effect on the 20S CP-mediated degradation of 15 different purified proteins to determine what types of proteins are more rapidly turned over in the presence of various 20S CP stimulators. We then utilized a fluorescent peptide probe and GFP-fusion proteins to demonstrate the efficacy of our small molecule stimulators in HEK-293T cells. To gain a deeper understanding of the impact of 20S CP stimulators in a cellular environment, we performed an untargeted label-free quantitative proteomic analysis of HEK-293T cells treated with a 20S CP stimulator and compared these results to cells also treated with a 20S CP inhibitor. Our results indicate that small molecule stimulators of the 20S CP are likely to increase the degradation of highly disordered proteins. However, the impact of a stimulator on the regulation of proteins by the 20S CP appears to be dependent on the mechanism of 20S CP enhancement. Taken altogether, our study demonstrates the potential of tailoring small molecule stimulators of the 20S CP to induce a rapid degradation of particular target proteins.
Results and discussion
Characterizing the activities of small molecule stimulators of the 20S CP
To begin our study, we chose four small molecule stimulators of the 20S CP that have various impacts on 20S CP activity: AM-404 (AM), an AM-404 derivative (TRC-1), ursolic acid (UA), and miconazole (MO) (Fig. 1B and C). While the effective concentration to induce 50% of the maximum stimulatory activity of the 20S CP (EC50) achieved by UA and TRC-1 differ greatly, the EC50s of AM and MO are both between 20–25 μM. Because AM and MO were shown to have the greatest stimulatory activity in our FRET assay, we chose to use 25 μM for every molecule in the subsequent analyses. At this concentration, the molecules stimulate the activity of the 20S CP 70% (TRC-1), 215% (UA), 406% (MO), and 538% (AM) over the control level of activity, providing a range of activities that can be used for further experiments, Fig. 1D.To evaluate how each of these molecules influence 20S CP activity, we chose to look at the chymotrypsin-like activity using the Suc-LLVY-AMC reporter, which is specific for the beta-5 subunit of the 20S CP (Fig. S1, ESI†). Prior to the addition of the small molecule stimulators, the 20S CP was pre-treated with DMSO (control), 250 μM sodium dodecyl sulfate (SDS), or 10 nM bortezomib (Btz), a beta-5 subunit inhibitor. Using this reporter, UA displayed the greatest stimulatory activity, increasing 20S CP activity more than 300% over the basal level control. Compared to the results of the FRET assay, AM and MO were much less active, stimulating activity only 132% and 60%, respectively. TRC-1 was shown to have no effect on the activity of the beta-5 subunit in this assay. Pre-treatment with SDS, a known 20S CP stimulator, resulted in an apparent additive effect when UA was included; however, when the other stimulators were added, the activity of the strongest stimulator was most prevalent. For example, inclusion of TRC-1 or MO did not affect the activity of SDS alone, whereas the presence of SDS had a minimal impact the stimulatory activity of AM (Fig. S1A, ESI†). Including 10 nM Btz decreased all observed activities by approximately 40–50%, indicating that the observed effects of our small molecules were due to proteolytic cleavage by the 20S CP. The apparent differences in how each stimulator impacted the hydrolysis of Suc-LLVY-AMC and the FRET peptide reporter point to distinct mechanisms of action for each stimulator.
Since we are most interested in stimulators of the 20S CP, we first confirmed that these molecules do not significantly affect the activity of the 26S proteasome. Previously reported 20S CP stimulators, such as SDS (Fig. S1B, ESI†), have been shown to lack stimulatory activity on the 26S proteasome.15,16,18 We therefore repeated the FRET assay to evaluate the impact of each of our stimulators on the 26S proteasome (Fig. 1A). None of the molecules we tested were able to significantly impact 26S-mediated hydrolysis of the FRET peptide reporter, Fig. 1D. As the 20S CP is not bound to a regulatory particle, this seems to suggest that the presence of a regulatory particle, which is present in the 26S proteasome, may prevent or disrupt the activity of these molecules. With this understanding, we then evaluated how the various stimulatory activities of these molecules shown in our activity assays translate to the 20S CP-mediated degradation of a protein substrate.
Stimulated 20S CP-mediated degradation of proteins in vitro
It has been widely reported that the 20S CP is capable of degrading proteins with minimal structure, allowing only mostly disordered or damaged proteins to enter the gate to be hydrolyzed.7,26 However, it is currently unknown if a small molecule stimulator will only cause the 20S CP to degrade endogenous substrates more quickly, or if the inclusion of a stimulator will result in the acceptance of proteins not normally degraded by the 20S CP. To systematically determine the types of proteins the 20S CP can degrade in the absence or presence of a small molecule stimulator, we chose a diverse pool of proteins that varied in length/size and amount of disorder as calculated by the PONDR VSL2 algorithm.27 Utilizing a similar approach to what has been done previously, we evaluated protein degradation in a biochemical assay using purified human 20S CP (5 nM) to which was added 200 ng of the protein and 25 μM of a 20S CP stimulator.15 After incubating for 2 h at 37 °C, the 20S CP activity was quenched with 4× SDS gel loading buffer. The amount of the protein of interest was then determined after performing an SDS–PAGE gel and analyzing band intensities, Fig. 2B and Appendix I. These intensities were then compared between samples to evaluate the degree of protein degradation in the presence of each stimulator, Table S1 (ESI†). The statistical analysis of these results is shown in Table S2 (ESI†).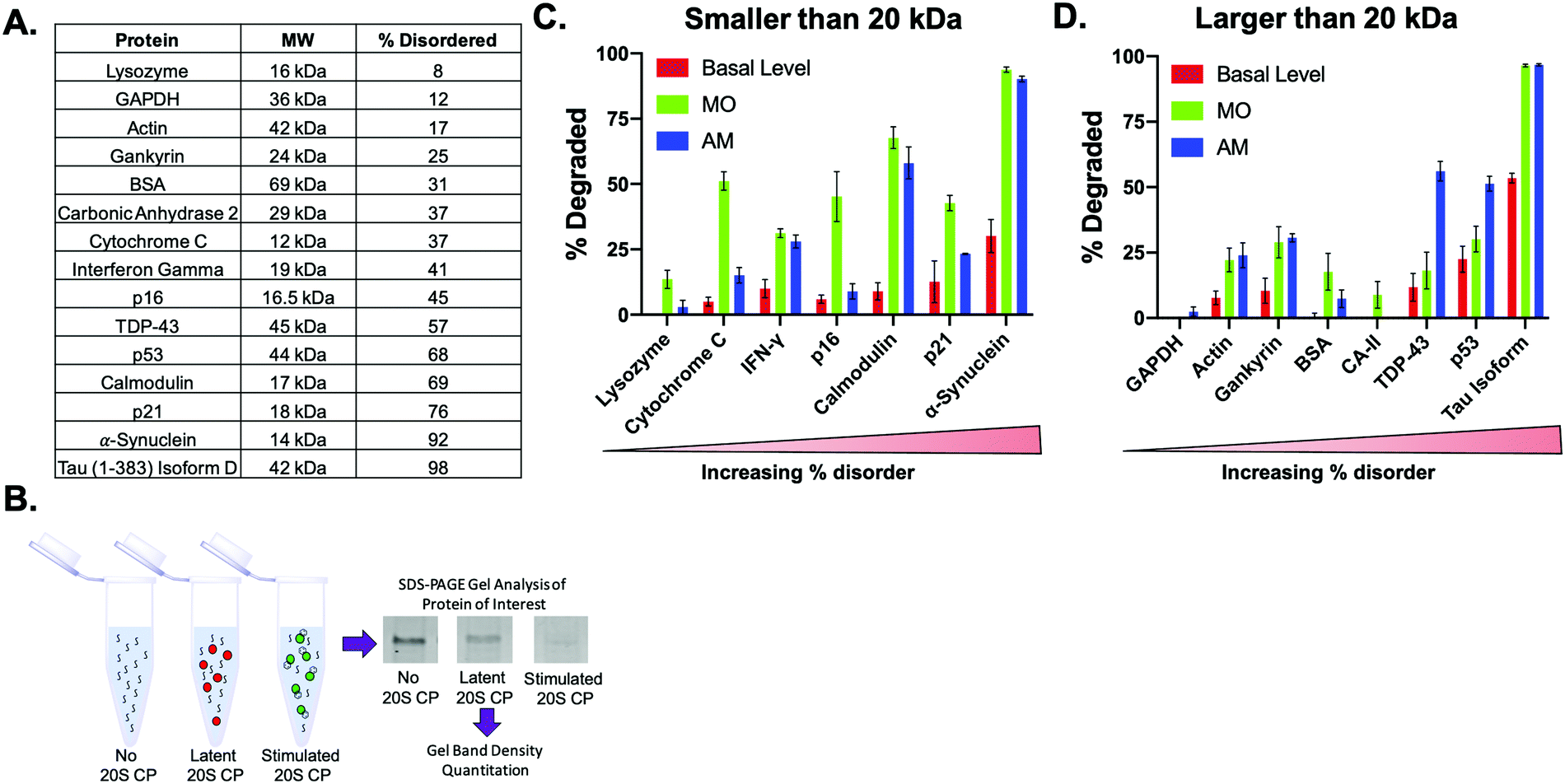 | ||
| Fig. 2 (A) Table of proteins utilized in biochemical protein degradation assay. These proteins varied in size and % disorder. (B) Overview of the protein degradation assay. Results of the assay for miconazole and AM-404 are shown in (C) for proteins smaller than 20 kDa and (D) protein larger than 20 kDa. For each graph, the proteins are shown in order of increasing disorder. | ||
After analyzing the degradation of each protein in the absence and presence of the stimulators, we separated the data into two sets based on protein size: proteins smaller than 20 kDa and proteins larger than 20 kDa. The basal level of degradation in each data set followed the trend observed previously: proteins with a greater degree of disorder are more easily degraded by the 20S CP.
Proteins that were not degraded by the 20S CP in our assay were still not significantly degraded by the 20S CP after the addition of three of our stimulators, Fig. 2C and D and Fig. S2A and B (ESI†). Four of the proteins we tested: lysozyme, GAPDH, BSA, and carbonic anhydrase (CA-II) were not degraded by the 20S CP alone. Only the addition of MO changed the calculated degradation of lysozyme, BSA, and CA-II, whereas TRC-1, UA, and AM had no effect on the degradation of these proteins. The levels of GAPDH remained the same in all cases. In addition, only the degradation of α-synuclein and Tau, which are approximately 92% and 98% disordered, respectively, was significantly increased by all four stimulators. These two proteins had the highest level of basal degradation, indicating that proteins that are easily accepted by the 20S CP are the most likely to be impacted by a 20S CP stimulator. Since the 20S CP is best known for degrading intrinsically disordered proteins and oxidatively damaged proteins, which are common pathological proteins in diseases marked by protein accumulation, our results here add credibility to the prospect of 20S CP stimulation as a therapeutic avenue in these disease states.
We also observed that each small molecule stimulator had a different impact on the degradation of proteins less readily degraded by the 20S CP. MO was shown to more greatly impact the degradation of smaller proteins, indicating that this molecule may enhance the acceptance of proteins by the 20S CP (Fig. 2C). For this assay, there was a greater concentration of the smaller proteins, so an observed increase in degradation could be a result of increased acceptance of these proteins. An increase in acceptance is also corroborated by the observed degradation of the proteins that were not degraded by the 20S CP alone in the 2 h incubation time period. The impact of MO on the degradation of smaller proteins compared to larger proteins is an unexpected result and potentially offers an undiscovered mechanism of 20S CP stimulation. With the exception of p21, AM appears to more significantly increase the degradation of proteins that are greater than 50% disordered, such as TDP-43, p53, calmodulin, α-synuclein, and Tau, compared to more structured proteins. Similarly, UA and TRC-1 increased the degradation of α-synuclein and Tau much more than that of other proteins (Fig. S2A and B, ESI†). Because the 20S CP is known to degrade intrinsically disordered proteins, it is not surprising that α-synuclein and Tau, which are both greater than 90% disordered, would be the most sensitive to 20S CP stimulation, even in the presence of our least active stimulators, UA and TRC-1.
The results of this assay indicate that the impact of a small molecule stimulator of the 20S CP on the degradation of a particular protein is likely dependent on the particular mechanism of action of the stimulator and is not necessarily related to the potency of the stimulator observed in biochemical activity assays. While our degradation assay provides an easy, straightforward method of analyzing 20S CP-mediated protein degradation of a particular protein, there are a few limitations. It is possible that our small molecules can bind to our proteins of interest and affect its degradation in this manner, since there is significantly more small molecule than protein in solution. In addition, this assay mimics a restricted environment, unlike that of a cell. In this environment, the 20S CP may degrade some proteins that are not endogenous cellular substrates, due to cellular location or the action of regulatory proteins. However, these initial results are exciting because we determine that not all stimulators modulate protein degradation in the same way, indicating the potential for selective modification of 20S CP activity.
Monitoring stimulated 20S CP-mediated degradation in cellulo using a fluorescent probe
In order to gain a greater understanding of how a 20S CP stimulator would impact proteasome activity in HEK-293T cells, we chose to utilize TAS3 (Fig. 3A), a selective proteasome activity reporter shown to be sensitive to proteasome stimulation in cells.28 HEK-293T cells were pre-incubated with DMSO (control, 1 h), bortezomib (Btz, a 20S CP inhibitor, 1 h), TRC-1, UA, MO, or AM (all stimulators, 30 min) before performing the assay. Due to this short incubation time period, we used a much higher concentration of Btz (10 μM) than has been used for longer term studies. After this incubation period, the media was removed, and the TAS3 probe with the appropriate control or compound was added to the cells. Because our molecules are not covalent modulators, it is important to keep the cells in the presence of the compound being tested throughout the assay. The fluorescence intensity of the probe was monitored over time to determine if the stimulators were capable of enhancing proteasome-mediated hydrolysis of the probe in cells. The results reveal a significant increase in fluorescence intensity compared to the control in the MO sample, Fig. 3B and C. AM was also able to enhance probe hydrolysis but to a lesser extent than MO. TRC-1 marginally increased activity over the control, and UA had no impact. Since MO and AM were shown to be the most effective in cells, we chose to continue our studies with only these stimulators.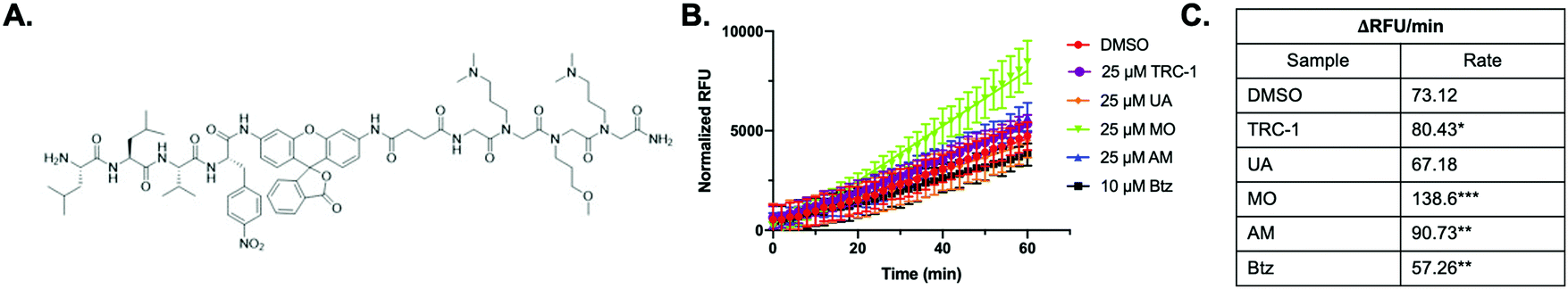 | ||
| Fig. 3 (A) Structure of TAS3 peptide probe. (B) The relative fluorescence units (RFU) recorded on the plate reader were plotted against time. Btz was included as a control and was able to decrease proteasome activity by approximately 22% (**p < 0.01). MO and AM were shown to increase activity 90% (***p < 0.001) and 26% (**p < 0.01), respectively. TRC-1 had a 10% increase (*p < 0.05), and UA had no effect on TAS3 probe hydrolysis. (C) The change in fluorescence units per time for each of the samples in (B). | ||
Degradation of GFP-fusion proteins in cellulo
To evaluate how MO and AM affect the degradation of an overexpressed protein in HEK-293T cells, we chose to analyze the degradation of three proteins from our biochemical assay (actin, p53, and α-synuclein) fused to GFP (Fig. 4A and Appendix II). We chose to use the GFP-fusions because this allowed us to use the anti-GFP antibody, rather than a separate antibody for each of the proteins, limiting the variability during the analysis. These fusion proteins were transiently transfected in HEK-293T cells, which were then treated with DMSO, 25 μM AM, or 25 μM MO. Rather than looking for the disappearance of a protein, we analyzed the amount of free GFP that accumulates by western blotting cell lysates with an anti-GFP antibody.15 We then determined the amount of the protein fused to the GFP that had been degraded by using a ratio of the amount of free GFP to that of the fusion protein. The effect of the stimulator on degradation was then determined by comparing this ratio to that of the basal level control (DMSO). Because we did not use cycloheximide to stop protein synthesis, the levels of the overexpressed fusion protein were not expected to be significantly different between samples. For this assay, we made the assumption that GFP degradation by the 26S proteasome or through the autophagy pathway would be consistent for all samples.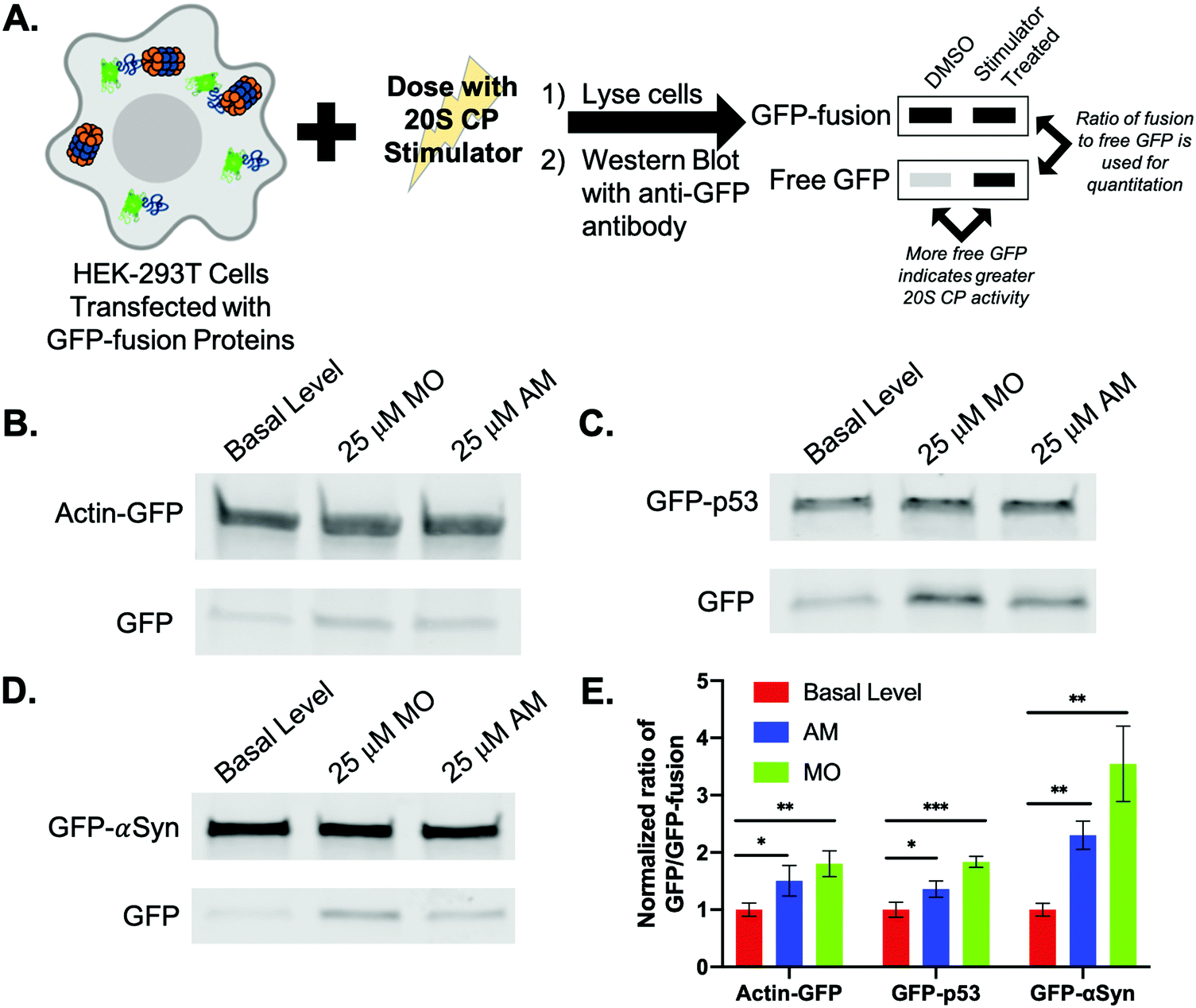 | ||
| Fig. 4 (A) HEK-293T cells were transiently transfected to express three different GFP-fusion proteins: actin (B), p53 (C), or α-synuclein (D). The cells were treated with DMSO (basal level), 25 μM MO, or 25 μM AM for 24 h. (E) Following quantitative analysis of the western blot, we determined that MO and AM were able to increase the degradation of these proteins to different extents. | ||
After transfecting HEK-293T cells to express the actin-GFP fusion and dosing with the small molecule stimulators, both MO and AM were able to significantly increase the degradation of actin (Fig. 4B). While both of these small molecules did increase the degradation of actin in our biochemical assay by 2.9-fold with MO and 3.1-fold with AM, the fold-change in degradation observed in our cellular assay is much lower for both of these compounds (Fig. S3A, ESI†), indicating the importance of using more than one technique to determine if a protein's degradation rate can be affected by a 20S CP stimulator.
We also chose to look at a fusion of GFP and p53, which is a known endogenous substrate of the 20S CP.10 However, p53 is an essential cell cycle and tumor-suppressor protein; increasing its degradation could increase a cell's susceptibility to becoming cancerous.29 We were therefore interested in seeing how our small molecule stimulators affected the levels of a p53-fusion protein in an overexpression model. Similar to actin, both MO and AM were able to increase the degradation of p53, with MO having a greater effect, Fig. 4C. This is opposite from what was shown in the biochemical assay, in which AM was the only small molecule that increased p53 degradation to a significant extent of 2.3-fold over basal level (Fig. S3B, ESI†). As our cellular assay is using an overexpression model, it is unclear how our small molecules would impact the degradation of endogenous p53.
The last fusion we used was GFP-α-synuclein (GFP-αSyn, Fig. 4D). α-Synuclein, as previously mentioned, is a highly intrinsically disordered protein and is also a known endogenous substrate of the 20S CP.14 In our biochemical assay, we showed that the degradation of α-synuclein was significantly enhanced by all stimulators. This percent increase in degradation in the presence of both MO and AM was greater than the increase in degradation observed for both actin and p53. After analysis of the GFP-α-synuclein fusion, the degradation of α-synuclein in the presence of MO or AM was greatly and significantly increased over the DMSO treated samples to a similar extent to what was shown in our biochemical assay (Fig. S3C, ESI†).
The increases in the degradation of actin and p53, which are approximately the same size, were similar for the MO-treated samples, while α-synuclein degradation was largely enhanced in the presence of MO, Fig. 4E. These results corroborate that of the biochemical assay, in which MO had the greatest impact on smaller proteins and those that are highly disordered. Similarly, AM was shown to have a more significant impact on proteins that had a greater degree of disorder in the biochemical assay. This is also observed in our cellular assay, in which AM was shown to more greatly increase the degradation of α-synuclein, a highly intrinsically disordered protein, compared to actin and p53, Fig. 4E. While the degrees of protein degradation differed in our biochemical and cellular assays, the trends we observed for each small molecule stimulator remain consistent.
Proteomics analysis of HEK-293T cells treated with 20S CP stimulators
While the studies with purified proteins and overexpressed proteins in cells have provided interesting insight into the types of proteins that can be affected by a 20S CP stimulator, these studies do not indicate the extent to which a 20S CP stimulator would impact total protein levels in a cell. To determine this, we performed label-free quantitative proteomic analysis of HEK-293T cells in the presence of MO or AM. These samples were then compared to the DMSO control and to samples treated with both 10 nM Btz and MO/AM to evaluate which changes in endogenous protein levels in the stimulator-treated cells were due to proteasome activity. Although HEK-293T cells are not known to be particularly reliant on the activity of the 20S CP isoform, our study with the GFP-fusion proteins indicates that 20S CP activity in these cells can be modulated to impact protein degradation after 24 h of treatment.After treatment, the cells were washed several times with PBS prior to cell lysis, and the protein was obtained for further analysis. LC-MS/MS-based label-free quantitation provides a convenient way to assess a high number of proteins in each sample and determine how protein levels change after treatment. After analysis, we were able to detect 2200–3200 proteins in each sample (Fig. S4, ESI†). Each treated sample was compared to the DMSO-treated control to examine the fold-change of each individual protein level and to determine which proteins were significantly impacted. First, we examined the proteins that increased or decreased in each stimulator or inhibitor treated sample as compared to the DMSO samples (Fig. 5A and Fig. S4, ESI†) Those proteins that had a log2(fold-change) of at least 0.5 with a p-value less than 0.05 were considered to be significantly different than the DMSO control. As anticipated, Btz caused many changes in protein levels, affecting 29% of proteins, Fig. 5A. Both MO and AM had a much smaller impact, changing 11% and 5.8% of proteins, respectively. Based on this analysis, it is important to note that stimulating the 20S CP does not impact nearly as many protein levels as the proteasome inhibitor, Btz. This result is not surprising, as Btz will inhibit all forms of the proteasome, having a drastic impact on many protein levels and cellular systems, whereas the 20S CP stimulators should be affecting only the 20S CP isoform of the proteasome. To determine which cellular pathways were being impacted by each of these treatments, we performed a Gene Ontology biological process analysis of the proteins that were significantly upregulated or downregulated in the Btz-, MO-, or AM-treated samples (Fig. 5B). This analysis again shows that Btz treatment has a much broader impact on cellular pathways than treatment with either MO or AM. Importantly, Btz was shown to upregulate many of the proteasome subunit protein levels (Fig. S5, ESI†), which has previously been reported by several groups.30,31 While both MO and AM were shown to downregulate proteins involved in the peptide biosynthetic process (GO:0043043) and response to heat (GO:0009408), which were both upregulated with Btz treatment, the extent to which either of these pathways was affected and the effect on other pathways was distinct for each stimulator. Similarly, both AM and MO were shown to upregulate proteins involved in the cellular amino acid metabolic process (GO:0006520), but the other upregulated pathways differed for each stimulator treatment. More potent stimulators are required to further elucidate the effects an increase in 20S CP activity can have on these pathways.
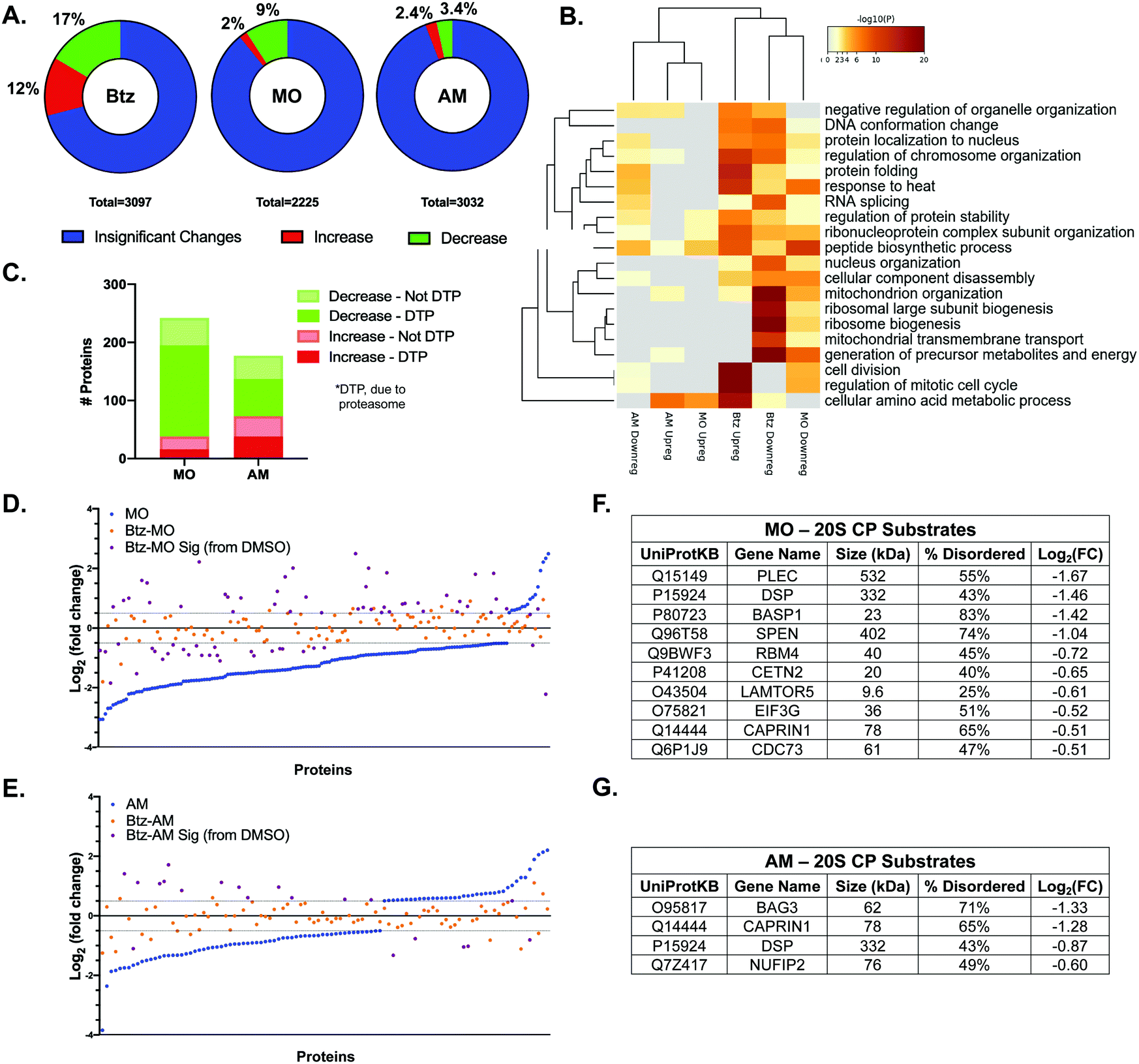 | ||
| Fig. 5 (A) Percent of protein levels that increased or decreased in the Btz (left), MO (middle), or AM (right) treated samples compared to a DMSO treated control. (B) Gene ontology analysis of the proteins shown to be upregulated or downregulated in each of the treatments shown in (A). (C) We compared the changes in protein levels due to MO/AM treatment with the changes in the same protein levels in the Btz-MO/AM treated samples to determine which protein level changes were due to proteasome activity. The log2(fold change) of proteins that significantly changed and were shown to be regulated by proteasome activity in the MO (D) and AM (E) treated samples. Each dot represents a particular protein and the impact of MO/AM (blue) or Btz-MO/AM dual treatment (orange) on the protein levels compared to DMSO. The purple dots represent proteins in the Btz-MO/AM samples that were significantly changed (log2(FC) > 0.5; p < 0.05) compared to DMSO. Known 20S CP substrates in the MO (F) or AM (G) treated samples shown to decrease due to proteasome activity. | ||
To determine which changes in protein levels were due to proteasome activity in the MO- or AM-treated samples, we compared how the proteins shown to be upregulated or downregulated in these samples changed in the corresponding dual-treatment with Btz (Btz-MO or Btz-AM). For this analysis, we looked for an opposite or reduced change in protein levels since the proteasome inhibitor Btz should reduce or prevent the effect caused by the proteasome stimulators, MO or AM, as demonstrated in our biochemical 20S CP activity assay (Fig. S1, ESI†). For the Btz-MO and Btz-AM samples, we looked specifically at the proteins shown to change with MO or AM treatment; a reduction in the log2(fold change) of at least 0.5 or, for the proteins that were minimally upregulated or downregulated, a 50% decrease in the effect observed in the MO or AM samples were considered to be significantly regulated (Fig. 5C–E). While the log2(fold change) of at least 0.5 is standard, a cut-off of 50% was chosen because the inclusion of 10 nM Btz in the biochemical 20S CP assay resulted in a similar decrease in the effect of MO or AM on 20S CP activity. From this comparison, 173 proteins (71.5%, Table S3, ESI†) of the 242 proteins shown to change in the MO treatment were due to proteasome activity, whereas only 102 proteins (57.6%, Table S4, ESI†) of the 177 proteins were related to proteasome activity in the AM treatment. This seems to indicate that MO is a more specific stimulator of the 20S CP than AM and has fewer off-target effects that result in significant changes in protein levels.
A previous proteomics evaluation of HeLa cells identified a number of proteins as 20S CP substrates.32 Using this list, we wanted to determine how many known 20S CP substrates decreased due to proteasome activity in the MO- and AM-treated samples. This evaluation revealed enhanced degradation of 12 known 20S CP substrates with MO treatment and 4 substrates with AM treatment (Fig. 5F and G). Only two of the known substrates were shown to decrease in both treatments, again indicating distinct impacts on protein degradation with each stimulator. Similar to what was performed for the biochemical protein degradation assay, we chose to look at the size and percent disorder of each of the substrates in Fig. 5F and G. Almost all of the substrates were shown to be greater than 40% disordered, though there was not a clear correlation between the percent disorder and log2(fold change). MO did impact more lower molecular weight proteins (5 proteins < 50 kDa) compared to AM (no proteins < 50 kDa); however, the levels of very large proteins (>100 kDa) were shown to decrease in both treatments. This analysis reinforces that the degradation of proteins with a high degree of structural disorder is more likely to be enhanced in the presence of a 20S CP stimulator compared to less disordered proteins. However, each stimulator appears to have a distinct effect on protein regulation by the 20S CP.
Conclusion
The findings of this study demonstrate, for the first time, that small molecule stimulators of the 20S CP can have different impacts on protein degradation, though proteins with a high degree of structural disorder are likely to be affected even by weak small molecule stimulators. This reveals the dynamic nature of the 20S CP, as small molecule stimulators can have a variety of mechanisms of action to change protein degradation activity. Further studies are necessary to identify and understand these mechanisms of action and how they might be manipulated to more fully control the population of proteins that undergo enhanced degradation in the presence of a 20S CP stimulator. However, this study reveals that the amount of 20S CP stimulation and the types of proteins affected can likely be controlled by structural modification of stimulators with varying mechanisms of action.Conflicts of interest
Prof. Trader is a share holder and consultant for Booster Therapeutics GmbH. Other authors declare no conflict of interest.Acknowledgements
This work was supported through a start-up package from Purdue University School of Pharmacy, the Purdue University Center for Cancer Research NIH grant (P30 CA023168), the American Cancer Society Institutional Research Grant (IRG-14-190-56) to the Purdue University Center for Cancer Research, and a grant from the NIH-NIGMS (R21GM131206). Proteomic experiments including sample preparation, mass spectrometry data collection and analysis was performed at the Purdue Proteomics Facility, Bindley Bioscience Center, Purdue University.References
- O. Coux, K. Tanaka and A. L. Goldberg, Structure and functions of the 20S and 26S proteasomes, Annu. Rev. Biochem., 1996, 65, 801–847, DOI:10.1146/annurev.bi.65.070196.004101.
- D. Nandi, P. Tahiliani, A. Kumar and D. Chandu, The ubiquitin-proteasome system, J. Biosci., 2006, 31(1), 137–155, DOI:10.1007/BF02705243.
- J. Erales and P. Coffino, Ubiquitin-independent proteasomal degradation, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843(1), 216–221, DOI:10.1016/j.bbamcr.2013.05.008.
- G. Ben-Nissan and M. Sharon, Regulating the 20S proteasome ubiquitin-independent degradation pathway, Biomolecules, 2014, 4(3), 862–884, DOI:10.3390/biom4030862.
- C. T. Aiken, R. M. Kaake, X. Wang and L. Huang, Oxidative stress-mediated regulation of proteasome complexes, Mol. Cell. Proteomics, 2011, 10(5), R110.006924, DOI:10.1074/mcp.M110.006924.
- T. Grune, B. Catalgol, A. Licht, G. Ermak, A. M. Pickering, J. K. Ngo and K. J. A. Davies, HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress, Free Radical Biol. Med., 2011, 51(7), 1355–1364, DOI:10.1016/j.freeradbiomed.2011.06.015.
- K. J. Davies, Degradation of oxidized proteins by the 20S proteasome, Biochimie, 2001, 83(3–4), 301–310, DOI:10.1016/s0300-9084(01)01250-0.
- A. M. Pickering and K. J. A. Davies, Degradation of damaged proteins – the main function of the 20S proteasome, Prog. Mol. Biol. Transl. Sci., 2012, 109, 227–248, DOI:10.1016/B978-0-12-397863-9.00006-7.
- G. Asher, N. Reuven and Y. Shaul, 20S proteasomes and protein degradation “by default”, BioEssays News Rev. Mol. Cell. Dev. Biol., 2006, 28(8), 844–849, DOI:10.1002/bies.20447.
- G. Asher, P. Tsvetkov, C. Kahana and Y. Shaul, A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors P53 and P73, Genes Dev., 2005, 19(3), 316–321, DOI:10.1101/gad.319905.
- G. Asher, Z. Bercovich, P. Tsvetkov, Y. Shaul and C. Kahana, 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1, Mol. Cell, 2005, 17(5), 645–655, DOI:10.1016/j.molcel.2005.01.020.
- C. M. Wiggins, P. Tsvetkov, M. Johnson, C. L. Joyce, C. A. Lamb, N. J. Bryant, D. Komander, Y. Shaul and S. J. Cook, BIM(EL), an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation, J. Cell Sci., 2011, 124(Pt 6), 969–977, DOI:10.1242/jcs.058438.
- T. Grune, D. Botzen, M. Engels, P. Voss, B. Kaiser, T. Jung, S. Grimm, G. Ermak and K. J. A. Davies, Tau protein degradation is catalyzed by the ATP/ubiquitin-independent 20S proteasome under normal cell conditions, Arch. Biochem. Biophys., 2010, 500(2), 181–188, DOI:10.1016/j.abb.2010.05.008.
- G. K. Tofaris, R. Layfield and M. G. Spillantini, Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome, FEBS Lett., 2001, 509(1), 22–26 CrossRef CAS.
- D. J. Trader, S. Simanski, P. Dickson and T. Kodadek, Establishment of a suite of assays that support the discovery of proteasome stimulators, Biochim. Biophys. Acta, 2017, 1861(4), 892–899, DOI:10.1016/j.bbagen.2017.01.003.
- R. A. Coleman and D. J. Trader, Development and application of a sensitive peptide reporter to discover 20S proteasome stimulators, ACS Comb. Sci., 2018, 20(5), 269–276, DOI:10.1021/acscombsci.7b00193.
- R. A. Coleman, C. S. Muli, Y. Zhao, A. Bhardwaj, T. R. Newhouse and D. J. Trader, Analysis of chain length, substitution patterns, and unsaturation of AM-404 derivatives as 20S proteasome stimulators, Bioorg. Med. Chem. Lett., 2019, 29(3), 420–423, DOI:10.1016/j.bmcl.2018.12.030.
- C. L. Jones, E. Njomen, B. Sjögren, T. S. Dexheimer and J. J. Tepe, Small molecule enhancement of 20S proteasome activity targets intrinsically disordered proteins, ACS Chem. Biol., 2017, 12(9), 2240–2247, DOI:10.1021/acschembio.7b00489.
- E. Njomen, P. A. Osmulski, C. L. Jones, M. Gaczynska and J. J. Tepe, Small molecule modulation of proteasome assembly, Biochemistry, 2018, 57(28), 4214–4224, DOI:10.1021/acs.biochem.8b00579.
- A. F. Kisselev, D. Kaganovich and A. L. Goldberg, Binding of hydrophobic peptides to several non-catalytic sites promotes peptide hydrolysis by all active sites of 20S proteasomes evidence for peptide-induced channel opening in the α-rings, J. Biol. Chem., 2002, 277(25), 22260–22270, DOI:10.1074/jbc.M112360200.
- M. Giżyńska, J. Witkowska, P. Karpowicz, R. Rostankowski, E. S. Chocron, A. M. Pickering, P. Osmulski, M. Gaczynska and E. Jankowska, Proline- and arginine-rich peptides as flexible allosteric modulators of human proteasome activity, J. Med. Chem., 2019, 62(1), 359–370, DOI:10.1021/acs.jmedchem.8b01025.
- K. A. Opoku-Nsiah and J. E. Gestwicki, Aim for the core: suitability of the ubiquitin-independent 20S proteasome as a drug target in neurodegeneration, Transl. Res., 2018, 198, 48–57, DOI:10.1016/j.trsl.2018.05.002.
- R. A. Coleman and D. J. Trader, All about the core: a therapeutic strategy to prevent protein accumulation with proteasome core particle stimulators, ACS Pharmacol. Transl. Sci., 2018, 1(2), 140–142, DOI:10.1021/acsptsci.8b00042.
- E. Njomen and J. J. Tepe, Proteasome activation as a new therapeutic approach to target proteotoxic disorders, J. Med. Chem., 2019, 62(14), 6469–6481, DOI:10.1021/acs.jmedchem.9b00101.
- R. A. Coleman and D. J. Trader, A sensitive high-throughput screening method for identifying small molecule stimulators of the core particle of the proteasome, Curr. Protoc. Chem. Biol., 2018, 10(4), e52, DOI:10.1002/cpch.52.
- T. Wenzel and W. Baumeister, Conformational constraints in protein degradation by the 20S proteasome, Nat. Struct. Biol., 1995, 2(3), 199–204, DOI:10.1038/nsb0395-199.
- B. Xue, R. L. Dunbrack, R. W. Williams, A. K. Dunker and V. N. Uversky, PONDR-FIT: a meta-predictor of intrinsically disordered amino acids, Biochim. Biophys. Acta, 2010, 1804(4), 996–1010, DOI:10.1016/j.bbapap.2010.01.011.
- B. L. Zerfas, R. A. Coleman, A. F. Salazar-Chaparro, N. J. Macatangay and D. J. Trader, Fluorescent probes with unnatural amino acids to monitor proteasome activity in real-time, ACS Chem. Biol., 2020, 15, 2588–2596, DOI:10.1021/acschembio.0c00634.
- L. A. Donehower, M. Harvey, B. L. Slagle, M. J. McArthur, C. A. Montgomery, J. S. Butel and A. Bradley, Mice deficient for P53 are developmentally normal but susceptible to spontaneous tumours, Nature, 1992, 356(6366), 215–221, DOI:10.1038/356215a0.
- M. Matondo, M. Marcellin, K. Chaoui, M.-P. Bousquet-Dubouch, A. Gonzalez-de-Peredo, B. Monsarrat and O. Burlet-Schiltz, Determination of differentially regulated proteins upon proteasome inhibition in AML cell lines by the combination of large-scale and targeted quantitative proteomics, Proteomics, 2017, 17(7), 1600089, DOI:10.1002/pmic.201600089.
- S. Uttenweiler-Joseph, D. Bouyssié, D. Calligaris, P. G. Lutz, B. Monsarrat and O. Burlet-Schiltz, Quantitative proteomic analysis to Decipher the differential apoptotic response of Bortezomib-treated APL cells before and after retinoic acid differentiation reveals involvement of protein toxicity mechanisms, Proteomics, 2013, 13(1), 37–47, DOI:10.1002/pmic.201200233.
- N. Myers, T. Olender, A. Savidor, Y. Levin, N. Reuven and Y. Shaul, The disordered landscape of the 20S proteasome substrates reveals tight association with phase separated granules, Proteomics, 2018, 18(21–22), 1800076, DOI:10.1002/pmic.201800076.
Footnote |
| † Electronic supplementary information (ESI) available: Raw data for analysis of the stimulators in the biochemical and cell-based assays. Additionally, conditions for the proteomics studies are described. See DOI: 10.1039/d0cb00191k |
| This journal is © The Royal Society of Chemistry 2021 |